
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotosensitizer-free singlet oxygen generation via a charge transfer transition involving molecular O2 toward highly efficient oxidative coupling of arylamines to azoaromatics†
Shivendra
Singh
and
Tushar Kanti
Mukherjee
 *
*
Department of Chemistry, Indian Institute of Technology Indore, Indore 453552, Madhya Pradesh, India. E-mail: tusharm@iiti.ac.in
First published on 2nd August 2024
Abstract
Photosensitizer (PS)-mediated generation of singlet oxygen, O2 (a1Δg) is a well-explored phenomenon in chemistry and biology. However, the requirement of appropriate PSs with optimum excited state properties is a prerequisite for this approach which limits its widespread application. Herein, we report the generation of O2 (a1Δg) via direct charge-transfer (CT) excitation of the solvent–O2 (X3Σg−) collision complex without any PS and utilize it for the catalyst-free oxidative coupling of arylamines to azoaromatics under ambient conditions in aqueous medium. Electron paramagnetic resonance (EPR) spectroscopy revealed the formation of O2 (a1Δg) upon direct excitation with 370 nm light. The present approach shows broad substrate scope, remarkably fast reaction kinetics (90 and 40 min under an open and O2 atm, respectively), high selectivity (100%), and excellent yields (up to 100%), and works well for both homo- and hetero-coupling of arylamines. The oxidative coupling of arylamines was found to proceed through the generation of amine radicals via electron transfer (ET) from amines to O2 (a1Δg). Notably, electron-rich amines show higher yields of azo products compared to electron-deficient amines. Detailed mechanistic investigations using various spectroscopic tools revealed the formation of hydrazobenzene as an intermediate along with superoxide radicals which subsequently transform to hydrogen peroxide. The present study is unique in the way that molecular O2 simultaneously acts as a light-absorbing chromophore (solvent–O2 complex) as well as an efficient oxidant (O2 (a1Δg)) in the same reaction. This is the first report on the efficient, selective, and sustainable synthesis of azo compounds in aqueous medium under an ambient atmosphere without any PCs/PSs and paves the way for further in-depth understanding of the chemical reactivity of O2 (a1Δg) generated directly via CT excitation of the solvent–O2 complex toward various photochemical and photobiological transformations.
Introduction
Molecular oxygen (O2) plays key roles in many organic transformations due to the generation of various reactive oxygen species (ROS) including singlet O2 (a1Δg) and superoxide ions (O2˙−).1–8 Among these, O2 (a1Δg) is an efficient and the most utilized ROS and finds applications in other fields such as water disinfection9 and photodynamic therapy (PDT).10 As a consequence of its widespread applications, various sensitive O2 (a1Δg) detection techniques have been developed over the years.11–13 Earlier, Mulliken first predicted three energetically closely lying electronic states of O2 on the basis of electronic configuration and later assigned them as the triplet ground state (X3Σg−) and two excited singlet states, a1Δg and b1Σg+.14 Deactivation of O2 (a1Δg) mainly happens either via radiative or non-radiative pathways and these include the radiative transition from the singlet to triplet ground state via phosphorescence at ∼1270 nm and non-radiative quenching by organic molecules, solvents, or photosensitizers.15Conventionally, O2 (a1Δg) is generated using a photosensitizer (PS) upon appropriate light irradiation.15–17 The dominant pathway involves initial excitation of the PS to the singlet state 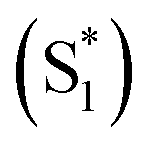 followed by intersystem crossing (ISC) to the triplet excited state
followed by intersystem crossing (ISC) to the triplet excited state 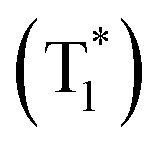 and finally O2 (a1Δg) is produced via energy transfer from the
and finally O2 (a1Δg) is produced via energy transfer from the 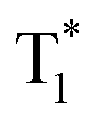 state of the PS to ground state O2 (X3Σu−) (Scheme 1A). While energy exchange between PS and O2 (X3Σu−) results in the formation of both O2 (a1Δg) and O2 (b1Σg+), due to rapid spin-allowed deactivation of b1Σg+ to a1Δg, only O2 (a1Δg) persists in the reaction mixture.15 The requisition of an ideal PS imposes several criteria such as high molar absorption coefficient at the excitation wavelength, triplet state energy greater than 95 kJ mol−1 to allow efficient energy transfer to ground state O2 (X3Σu−), appreciable triplet state quantum yield, long triplet state lifetime, and high photostability.17 Previously, a wide range of PSs such as porphyrins and their derivatives,18–21 nanoparticles,22–24 organic molecules,25,26 metal–organic frameworks (MOFs),27 and metal complexes28,29 have been utilized to produce O2 (a1Δg) for various applications. In recent years, heavy-atom-free non-porphyrinoid PSs have emerged as an alternative class of PSs for PDT applications.30 While significant efforts have been made to understand and improve the efficiency of O2 (a1Δg) generation via PS-based systems, the requirement of suitable PSs often raises several challenges. These include unwanted photobleaching of PSs, finite excited state lifetimes of PSs, optimum singlet–triplet energy gap, the presence of other quenchers, competing chemical reactions, and environmental fluctuations. All these limiting factors may influence the efficiency and quantum yield of O2 (a1Δg) generation.
state of the PS to ground state O2 (X3Σu−) (Scheme 1A). While energy exchange between PS and O2 (X3Σu−) results in the formation of both O2 (a1Δg) and O2 (b1Σg+), due to rapid spin-allowed deactivation of b1Σg+ to a1Δg, only O2 (a1Δg) persists in the reaction mixture.15 The requisition of an ideal PS imposes several criteria such as high molar absorption coefficient at the excitation wavelength, triplet state energy greater than 95 kJ mol−1 to allow efficient energy transfer to ground state O2 (X3Σu−), appreciable triplet state quantum yield, long triplet state lifetime, and high photostability.17 Previously, a wide range of PSs such as porphyrins and their derivatives,18–21 nanoparticles,22–24 organic molecules,25,26 metal–organic frameworks (MOFs),27 and metal complexes28,29 have been utilized to produce O2 (a1Δg) for various applications. In recent years, heavy-atom-free non-porphyrinoid PSs have emerged as an alternative class of PSs for PDT applications.30 While significant efforts have been made to understand and improve the efficiency of O2 (a1Δg) generation via PS-based systems, the requirement of suitable PSs often raises several challenges. These include unwanted photobleaching of PSs, finite excited state lifetimes of PSs, optimum singlet–triplet energy gap, the presence of other quenchers, competing chemical reactions, and environmental fluctuations. All these limiting factors may influence the efficiency and quantum yield of O2 (a1Δg) generation.
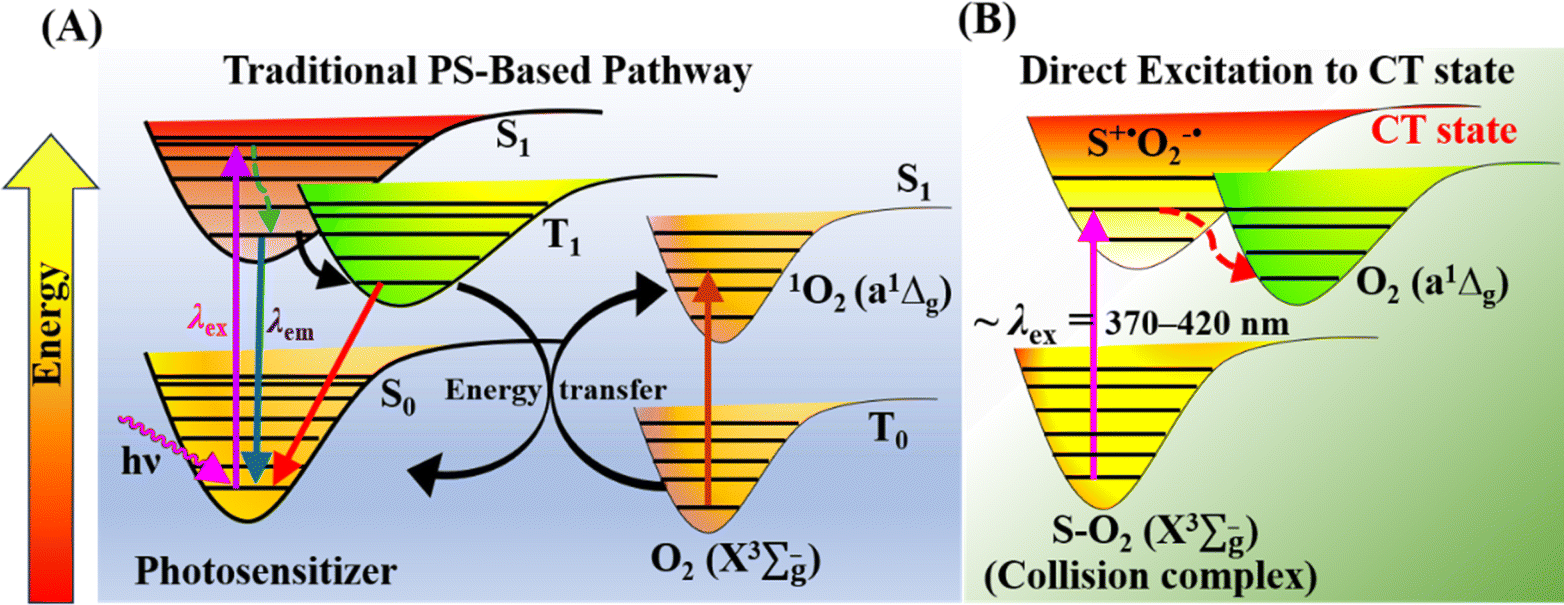 | ||
| Scheme 1 Generation of O2 (a1Δg) via (A) traditional PS-based approach and (B) direct excitation to the CT state. | ||
An alternative route for O2 (a1Δg) generation was proposed previously via direct charge-transfer (CT) excitation involving molecular O2 and organic solvents without any PS (Scheme 1B).31–36 Initially, Tsubomura and Mulliken observed significant changes in the absorption bands of different solvents in the presence of dissolved O2 upon irradiation.37 The structureless absorption tail observed at longer wavelengths in different oxygen-saturated solvents was attributed to the CT interaction between solvent molecules as electron donors and O2 (X3Σu−) as an electron acceptor. Later, seminal studies of Ogilby and coworkers demonstrated the formation of O2 (a1Δg) upon irradiation of solvent–O2 collision complexes.31–36 It has been shown that common solvents such as water, methanol, benzene, mesitylene, cyclohexane, acetonitrile, etc. under open atmosphere conditions exhibit a structureless long absorption tail due to transition from the solvent–O2 collision complex to a solvent–O2 charge transfer (CT) state. Subsequent decay of this CT state results in the production of O2 (a1Δg). The long wavelength absorption tail appears in a broad wavelength range between 300 and 400 nm, which is solvent-dependent.37 The quantum yield of O2 (a1Δg) produced upon direct CT band excitation was reported to be ∼0.2 in organic solvents.33 In recent work, Bregnhøj and Ogilby utilized a nonlinear two-photon excitation process using visible light (600–730 nm) to produce an appreciable amount of O2 (a1Δg).35 Two competing decay channels of the CT state were proposed previously in water.34,35 In the first channel, the CT state relaxes to yield solvated O2 (a1Δg), while the second channel produces O2 (a1Δg) via photosensitization of ground state O2 (X3Σu−) by triplet state water produced upon relaxation. Notably, it has been shown that the nature of the solvent influences the partitioning between these two decay channels.34,35 Although these previous studies thoroughly investigated the fundamental photophysical processes involving direct CT excitation and subsequent quantification of O2 (a1Δg) in various solvents with appreciable yield, the photogenerated O2 (a1Δg) has not been utilized yet for any chemical transformation. We seek to address whether the O2 (a1Δg) generated through direct CT excitation in a neat solvent can be utilized for a multistep oxidative coupling reaction. In the present study, we have utilized the O2 (a1Δg) generated through direct CT excitation in water as an oxidant to drive the oxidative coupling of arylamines to azoaromatics without any PSs/photocatalysts (PCs).
Azo compounds are an important class of functional organic molecules that have enormous potential in molecular materials38 and industrial applications.39 Over the past few decades, several synthetic strategies have been formulated for the synthesis of azoaromatics.40 In recent times, efforts were made to convert nitroarenes and aromatic amines to azoaromatics using various metal nanoparticles (NPs) as catalysts;41–50 however, most of these methodologies suffer from poor selectivity, slow reaction kinetics, requirement of high temperature, low to moderate yields, and/or inert atmosphere. Similarly, photocatalytic reductive coupling of nitroarenes to azoaromatics with simultaneous production of nitrosobenzene or azoxyaromatics has been demonstrated using various PCs such as supported Au NPs,51 Cu NPs,52 and Au NP functionalized organic cages.53 On the other hand, photocatalytic oxidative coupling (POC) using molecular O2 as an oxidant has been utilized recently for the synthesis of azo compounds using Ir-based photocatalysts.54 Very recently, using a common organic photocatalyst, eosin Y, we formulated a highly efficient and selective POC of arylamines at the charged aqueous interfaces via utilizing a non-diffusive ultrafast electron transfer process at the micellar surface.55 Motivated by our previous study, we seek to explore this oxidative coupling reaction under mild reaction conditions without any external PCs/PSs. Notably, most of the earlier studies utilized costly metal-based PCs for the synthesis of azo compounds (Table S1†). To the best of our knowledge, the present study is the first of its kind to demonstrate the utilization of O2 (a1Δg) generated directly via CT excitation toward an industrially important oxidative coupling reaction without any external PCs/PSs.
Results and discussion
PS-free generation and detection of singlet oxygen
First, we recorded the absorption spectra of different solvents such as water, methanol, benzene, mesitylene, and acetonitrile saturated with either N2 or O2 to monitor the CT features in the UV-visible region. Notably, a long absorption tail (>350 nm) was clearly evident in all O2-saturated solvents compared to those in N2-saturated solvents (Fig. 1A–C and S1†). The differences in the spectral signature between N2 and O2 saturated solvents suggest the active role of O2 in the appearance of this long absorption tail. Earlier, Tsubomura and Mulliken assigned this O2-dependent absorption feature to a transition from a ground state solvent–O2 (X3Σu−) contact complex to a solvent–O2 (X3Σu−) CT state (sol˙+O2˙−).37 Moreover, it has also been shown that this long structureless absorption tail can be reversibly removed by purging the solvents with N2 gas. The molar absorption coefficient (ε) of this CT transition in the benzene–O2 complex is reported to be ∼100 M−1 cm−1.37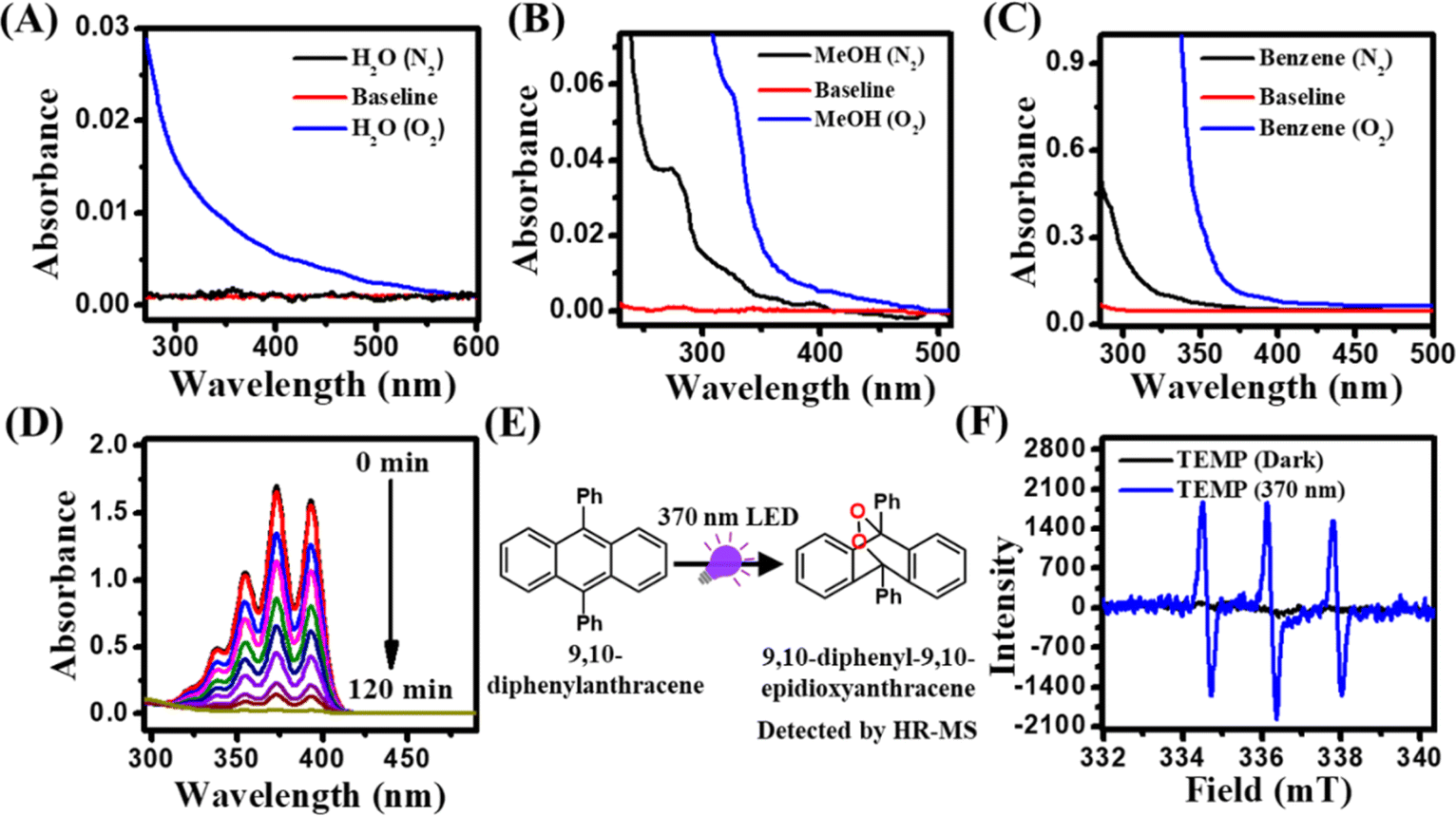 | ||
| Fig. 1 UV-vis spectra of (A) water, (B) methanol, and (C) benzene under N2 and O2 atmosphere. (D) Changes in the absorption spectra of DPA in benzene under the irradiation of a 370 nm LED under an open atmosphere. (E) Schematic showing the conversion of DPA to 9,10-diphenyl-9,10-epidioxyanthracene under the irradiation of a 370 nm LED in benzene under an open atmosphere. (F) Changes in the EPR spectra of TEMP (50 mM) under dark and 370 nm LED irradiation for 30 min in aqueous medium. | ||
Previously, it has been shown that these excited CT states subsequently undergo decay or back electron transfer to generate O2 (a1Δg) via mixing of various solvent–O2 states.31–36 To authenticate the O2 (a1Δg) formation in our present system, we utilized 9,10-diphenylanthracene (DPA) as a model probe for the detection of singlet oxygen under the irradiation of a 370 nm LED. It has been well-documented in the literature that anthracene and its derivatives are efficient probes for the detection of singlet oxygen.56,57 The experiment was carried out in benzene with 200 μM DPA under open air conditions. DPA in benzene exhibits characteristic multiple absorption peaks due to different vibronic transitions (Fig. 1D). Upon irradiation at 370 nm, the absorbance of DPA decreased gradually over time. This decrease in the absorbance indicates the possible transformation of DPA to its endoperoxide form (9,10-diphenyl-9,10-epidioxyanthracene).56,57 The formation of 9,10-diphenyl-9,10-epidioxyanthracene was further confirmed using HR-MS measurement (Fig. 1E). A characteristic HR-MS peak for sodium salt of 9,10-diphenyl-9,10-epidioxyanthracene was observed at 385.1188 g mol−1, indicating the formation of O2 (a1Δg) under the irradiation of 370 nm light (Fig. S2†). To further authenticate the formation of O2 (a1Δg), we performed EPR measurement using 2,2,6,6-tetramethylpiperidine (TEMP) as a singlet oxygen probe in an aqueous medium.2 EPR spectra were recorded under dark and 370 nm irradiation. While under dark conditions, no noticeable peaks for O2 (a1Δg) were observed, three characteristic peaks appeared in the EPR spectrum of the sample kept under 370 nm light irradiation for 30 min, suggesting the formation of TEMP-trapped O2 (a1Δg) (Fig. 1F).58 Taken together, these observations authenticate the formation of O2 (a1Δg) upon CT band excitation of the solvent–O2 complex under open air conditions without any external PSs. Next, we seek to address whether this O2 (a1Δg) can be utilized directly as an efficient oxidant to drive the oxidative coupling of arylamines.
Oxidative coupling of arylamines to azoaromatics
We envisaged that the O2 (a1Δg) generated via direct CT band excitation of the solvent–O2 complex might act as an efficient oxidant to oxidize arylamine to facilitate the oxidative coupling under ambient conditions without any PCs/PSs. Previously, it has been shown that the presence of a base is essential for the conversion of arylamines to azoaromatics as proton abstraction is a critical step in the reaction pathway.54,55 Herein, we utilized p-toluidine as a model amine substrate and K3PO4 as base to check the feasibility of the coupling reaction in aqueous medium under 370 nm light irradiation without any PCs. The optimized reaction conditions for the conversion of p-toluidine to 1,2-bis(4-methylphenyl)diazene in the presence of 2 equiv. of K3PO4 under 370 nm light irradiation are tabulated in Table 1. Remarkably, a 100% conversion of p-toluidine to 1,2-bis(4-methylphenyl)diazene was observed within 90 min of reaction under 370 nm irradiation and open air conditions (Table 1, entry 1). Here it should be noted that a much slower reaction kinetics with reaction time of 24 and 4 h have been reported previously using Ir-based and eosin Y (EY) PCs, respectively.54,55 The absence of any desired azo product under dark conditions suggests the essential role of light in the conversion (Table 1, entry 2).|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Conversiona [%] |
| a Determined from GC-MS measurements. b Standard conditions: 0.05 mmol p-toluidine, 2 equiv. of K3PO4, 5 mL of H2O, under an ambient air atmosphere with 370 nm (44 W) illumination for 90 min. | ||
| 1 | Standardb | 100 |
| 2 | No light | 0 |
| 3 | N2 atmosphere | 0 |
| 4 | No K3PO4 | 0 |
| 5 | 1 equiv. of K3PO4 | 86 |
| 6 | 2 equiv. of K2CO3 | 100 |
| 7 | 2 equiv. of DABCO | 100 |
| 8 | 2 equiv. of piperidine | 100 |
| 9 | Methanol as solvent | 90 |
| 10 | Acetonitrile as solvent | 38 |
| 11 | 60 °C | 0 |
To know the role of atmospheric O2, we performed the reaction under a N2 atmosphere upon 370 nm irradiation. Importantly, no azo product was formed under a N2 atmosphere, authenticating the direct role of O2 in the reaction mechanism (Table 1, entry 3). Furthermore, the conversion yield was found to depend strongly on the concentrations of K3PO4 used, suggesting that the proton abstraction is also an essential step for the present reaction (Table 1, entries 4 and 5). Interestingly, organic bases such as DABCO and piperidine were also found to be equally effective for the coupling reaction and show 100% conversion within 90 min of reaction (Table 1, entries 6–8). Replacement of water with methanol as solvent showed 90% conversion efficiency within 90 min of reaction (Table 1, entry 9 and Fig. S3A†). On the other hand, acetonitrile as solvent yielded very slow kinetics and low conversion yield compared to that in water and methanol under standard reaction conditions (Table 1, entry 10 and Fig. S3B†). To check the influence of temperature, we performed the reaction at 60 °C under dark conditions. Notably, no azo product was formed at higher temperature in the absence of 370 nm light (Table 1, entry 11), indicating the negligible role of temperature in the reaction mechanism. Moreover, the conversion yield was found to depend on the wavelength of light used. While 370 nm excitation light resulted in 100% conversion yield, 440 and 525 nm light sources showed 13 and 0% conversions upon 90 min of irradiation (Fig. 2A). It should be noted that p-toluidine in water does not absorb 370 nm light (Fig. S4†). In addition, the coupling reaction was found to be more efficient under an O2 atmosphere and showed 100% conversion within 40 min of irradiation (Fig. 2B). Therefore, we propose that the essential role of 370 nm light irradiation is to provide sufficient energy for the CT band excitation of the water–O2 complex in the reaction mixture to generate O2 (a1Δg).
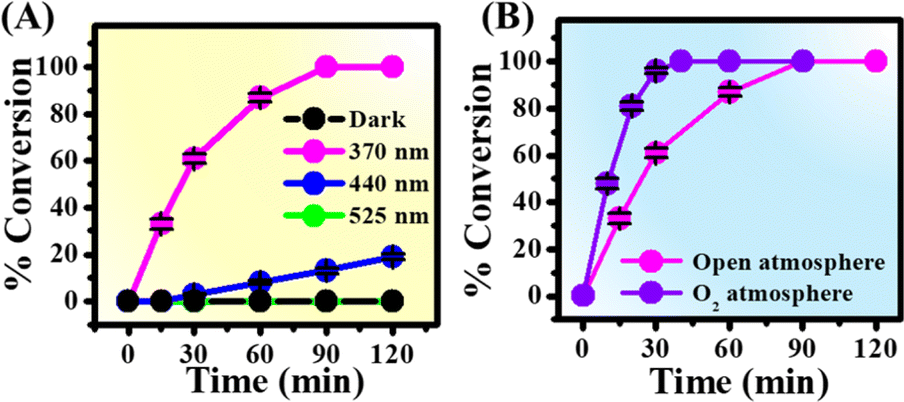 | ||
| Fig. 2 Time-dependent conversions of p-toluidine under standard reaction conditions as a function of (A) wavelength of light and (B) atmospheric conditions. | ||
Our present PC-free approach shows a broad substrate scope with excellent conversion yields for a wide range of substituted amines including electron-rich and -deficient amine moieties (Scheme 2). Azo compounds were first isolated and purified using solvent extraction and column chromatography and subsequently characterized using GC-MS and NMR measurements. Arylamines with electron-donating groups (rxns (1)–(6) in Scheme 2A) showed over 90% conversion yield in just 90 min, which is significantly better than the earlier reported studies.54,55 Unsubstituted aniline exhibited a conversion yield of 97% under standard reaction conditions (rxn (7) in Scheme 2A). However, p-chloroaniline showed a conversion yield of 73% (rxn (8) in Scheme 2A), possibly due to the electron-withdrawing nature of chlorine.54,55 Excellent yields were also obtained with arylamines having both electron-donating and -withdrawing substituents (rxns (9)–(11) in Scheme 2A). In addition, 5,6,7,8-tetrahydronaphthalen-1-amine, a fused ring amine substrate also showed an excellent yield of the azo product with a conversion yield of 78% (rxn (12) in Scheme 2A). In contrast, no azo product was obtained for p-nitroaniline possibly due to the high electron-withdrawing nature of the nitro group (rxn (13) in Scheme 2A), which is similar to that observed previously.55
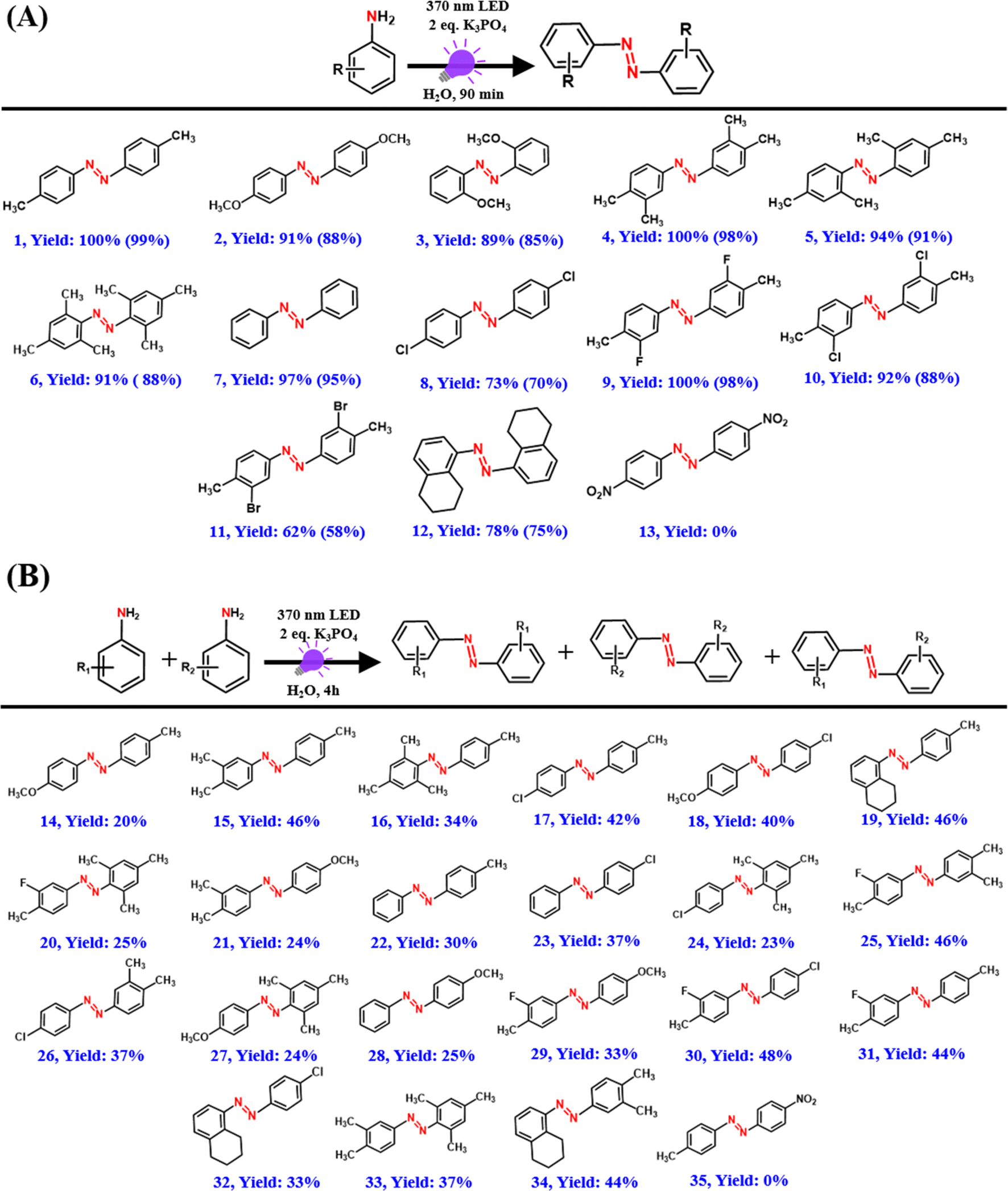 | ||
| Scheme 2 Substrate scope for (A) homo-coupling and (B) hetero-coupling reactions under standard conditions. Values in parentheses are isolated yields obtained after column chromatography. | ||
Our present approach can also be extended for the hetero-coupling of different amines to their corresponding azo compounds with the formation of all three possible products as revealed from the GC-MS measurements (Scheme 2B and Fig. S5–S25†). The hetero-coupling of amines was carried out for an extended period (4 h) under the standard conditions mainly to have 100% conversion of reactants to products for all the cases. No hetero-coupled product was obtained for the hetero-coupling of p-nitroaniline with p-toluidine possibly due to the highly electron-deficient amine group of p-nitroaniline (Scheme 2B and Fig. S26†). This reactivity trend of arylamines correlates well with the previous reports and suggests the possible role of the electron transfer process.54,55 Importantly, except for the desired azo product, no unwanted side products such as nitro, nitroso, or azoxy compounds were observed during the course of the reaction (Fig. S27†).
To know whether any oxygenated intermediates such as nitroso or nitro compounds are involved in the reaction mechanism, we performed control experiments with an equal mixture of aniline and nitrosobenzene as starting substrates under standard reaction conditions. The product distributions were monitored using GC-MS measurements. Notably, the reaction with an equal mixture of aniline and nitrosobenzene under standard reaction conditions yielded both azobenzene and azoxybenzene with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 conversion ratio (Scheme 3A). On the other hand, the reaction with nitrosobenzene alone yielded nitrobenzene and azoxybenzene with a 11:1 conversion ratio (Scheme 3A). These product distributions are quite different than that observed with aniline alone as the substrate under standard reaction conditions (rxn (7) in Scheme 2A). Therefore, these findings rule out the possible involvement of oxygenated intermediates in the reaction mechanism. Furthermore, the PC- and PS-free approach of our present methodology makes it highly facile to scale it up to the gram scale (Scheme 3B), which is highly promising for the large-scale industrial production of azo compounds.
:1 conversion ratio (Scheme 3A). On the other hand, the reaction with nitrosobenzene alone yielded nitrobenzene and azoxybenzene with a 11:1 conversion ratio (Scheme 3A). These product distributions are quite different than that observed with aniline alone as the substrate under standard reaction conditions (rxn (7) in Scheme 2A). Therefore, these findings rule out the possible involvement of oxygenated intermediates in the reaction mechanism. Furthermore, the PC- and PS-free approach of our present methodology makes it highly facile to scale it up to the gram scale (Scheme 3B), which is highly promising for the large-scale industrial production of azo compounds.
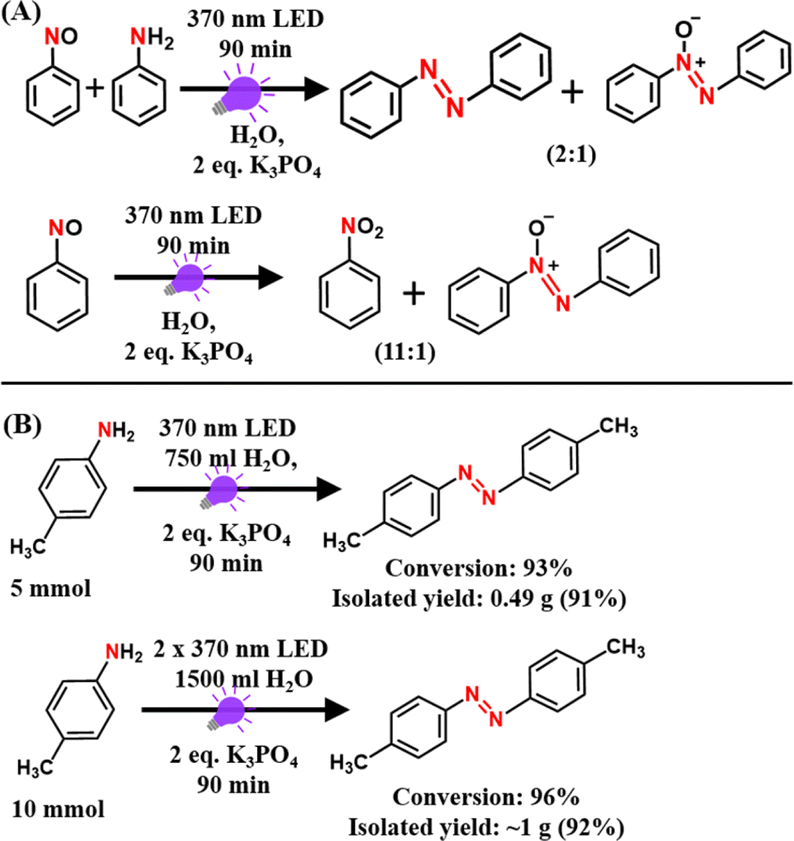 | ||
| Scheme 3 (A) Control reactions with 0.05 mmol nitrosobenzene in the presence and absence of 0.05 mmol aniline under standard reaction conditions. (B) Scalability experiments for the conversion of p-toluidine to its corresponding azo compound. | ||
Mechanistic pathway
We performed several control experiments to understand the mechanism behind this PC- and PS-free synthesis of azo compounds. To rule out the possibility of direct excitation of amine substrates, we recorded the absorption spectra of different arylamines (Fig. S28†). However, none of the amines were found to absorb light ∼370 nm except 5,6,7,8-tetrahydronaphthalen-1-amine and p-nitroaniline, which showed conversion yields of 78 and 0%, respectively.Our findings indicate that most of these amine substrates do not absorb the excitation light during the reaction. Furthermore, no appreciable changes in the long wavelength region were observed in the absorption spectrum of p-toluidine in the presence of K3PO4, which ruled out the possibility of any light-absorbing complex formation between amine substrates and base (Fig. S29†). Therefore, the only light-absorbing species in the reaction mixture is the water–O2 complex which absorbs 370 nm light to undergo CT excitation.31–37 Our findings revealed that this CT state subsequently produced O2 (a1Δg) upon relaxation. Previously, it has been reported that amines are good quenchers of O2 (a1Δg).15 Generally, amines with low ionization potential readily transfer their electrons to O2 (a1Δg) and get converted into a radical cation. To check this possibility, we monitored the formation of amine radicals using 2,6-di-tert-butyl-4-methylphenol (BHT) as a radical scavenger.55 The reaction was set up with 0.05 mmol p-toluidine and 2 equiv. K3PO4 in 5 mL 1:1 MeOH–H2O mixture due to the low solubility of BHT in aqueous medium. Subsequently, after the addition of 1 equiv. BHT, the reaction mixture was irradiated with 370 nm light for 90 min (Fig. 3A). The product was isolated and characterized using HR-MS and GC-MS measurements. A characteristic HR-MS peak for the p-toluidine–BHT adduct was observed at 326.2441 g mol−1 (Fig. S30†).
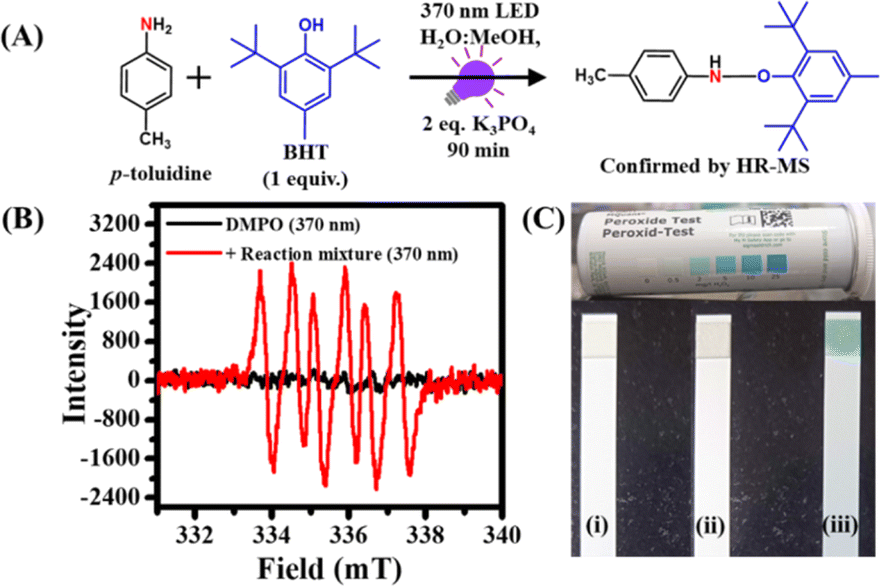 | ||
| Fig. 3 (A) Schematic of the radical trap experiment during the oxidative coupling of 0.05 mmol p-toluidine. (B) Changes in the EPR spectra of DMPO (50 mM) in the absence and presence of the reaction mixture under the irradiation of a 370 nm LED in the presence of 0.05 mmol p-toluidine and 2 equiv. K3PO4. (C) Daylight photographs of peroxide test strips under different conditions (i) control blank strip, (ii) before the reaction, and (iii) after 90 min of reaction with 0.05 mmol p-toluidine under standard reaction conditions. | ||
Moreover, the presence of BHT resulted in only a trace amount of the azo product even after 90 min of irradiation. These observations clearly validate the electron transfer mechanism from amines to O2 (a1Δg) and authenticates the formation of amine radicals. On the other hand, to confirm the formation of superoxide radicals after the electron transfer to O2 (a1Δg), we performed EPR measurements using DMPO as a probe under standard reaction conditions (Fig. 3B).59 The EPR spectrum of pure DMPO under 370 nm irradiation showed no noticeable EPR peaks. In contrast, a characteristic EPR spectrum of superoxide radicals with six well-resolved peaks was observed upon addition of DMPO into the reaction mixture under 370 nm irradiation (Fig. 3B), which matches well with the earlier literature.59 This observation authenticates the generation of superoxide radicals through single electron transfer from amines to O2 (a1Δg) during the reaction. Altogether, these results confirm the formation of amine radicals and superoxide ions via single electron transfer.
The oxidative coupling of arylamines involves the radical coupling of amines to generate hydrazobenzene as an active intermediate.54,55 To check the formation of hydrazobenzene under standard reaction conditions, we performed LC-MS measurement after 10 min of irradiation under a 370 nm LED. Notably, a characteristic LC-MS peak for 1,2-bis(4-methylphenyl)hydrazine was observed at an m/z value of 213.2037 g mol−1 (Fig. S31†). Finally, to know whether H2O2 is generated in the reaction mixture from superoxide radicals,54,55 we utilized H2O2 sensing strips. These colorless strips were dipped into the reaction mixtures before and after the reaction (Fig. 3C(i)). No color change of the strip was observed upon dipping it inside the reaction mixture before light irradiation (Fig. 3C(ii)). However, a distinct color change of the sensing strip from colorless to blue was noticed after the irradiation of the reaction mixture with 370 nm light (Fig. 3C(iii)). This clearly indicates the formation of H2O2 in the reaction mixture.
In accordance with our findings, we propose a plausible reaction mechanism for the oxidative coupling of arylamines to azoaromatics under 370 nm light irradiation (Scheme 4). The solvent (S)–O2 (X3Σu−) ground state complex absorbs 370 nm light to undergo excitation at the CT state (S˙+O2˙−).31–37 This CT state subsequently relaxed to yield O2 (a1Δg). Owing to its strong oxidizing nature, the photogenerated O2 (a1Δg) readily accepts a single electron from the amine substrates to form superoxide radicals and amine radicals. These amine radicals form the hydrazobenzene intermediate via a coupling reaction after proton abstraction by a base. After another cycle of electron transfer in the presence of O2 (a1Δg) and subsequent proton abstraction, these hydrazobenzene intermediates ultimately converted to the desired azo products with the generation of 2 equiv. of H2O2 as the byproduct. Therefore, the overall reaction proceeds via the production of 4 electrons and 4 protons as reported previously in bulk acetonitrile and micellar medium in the presence of PCs.54,55 The estimated quantum yield for the photooxidative conversion of p-toluidine to its corresponding azo compound under our standard reaction conditions is estimated to be ∼0.15, which substantiates that the present reaction does not follow the radical chain mechanism as proposed previously for photocatalytic Diels–Alder cycloaddition reactions.60 To the best of our knowledge, this is the first report where the PS-free generation of O2 (a1Δg) has been utilized to develop a catalyst-free approach for the selective synthesis of azo compounds in the aqueous medium (Table S1†). The present study paves the way for the development of other sustainable chemical transformations via utilizing a direct CT transition involving molecular O2 in a diverse range of solvents.
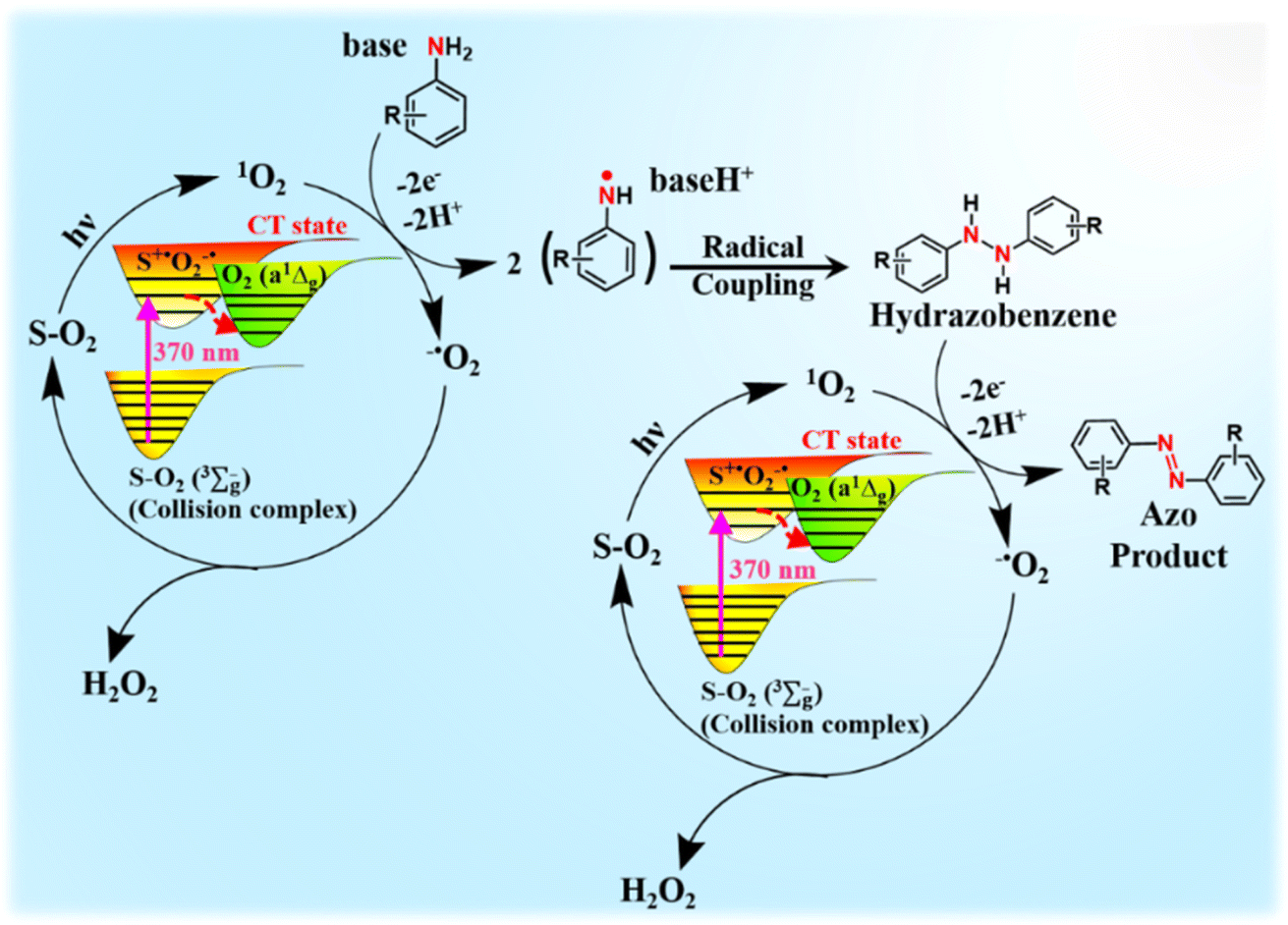 | ||
| Scheme 4 Plausible reaction mechanism for the oxidative coupling of arylamines via PS-free singlet oxygen. | ||
Conclusions
In the present study, we have developed an efficient, sustainable, and viable synthetic approach for the oxidative coupling of arylamines to azoaromatics under ambient conditions in aqueous medium without any PCs/PSs. The present approach is based on the efficient generation of O2 (a1Δg) via CT excitation of the water–O2 complex at 370 nm. The formation of O2 (a1Δg) has been authenticated using various spectroscopic measurements including the EPR technique. The photogenerated O2 (a1Δg) has been utilized as a sustainable oxidant for the oxidative coupling of arylamines in the presence of a base under an ambient atmosphere. The reaction proceeds via the formation of amine radicals which undergo a coupling reaction to yield hydrazobenzene as an intermediate. These hydrazobenzene intermediates finally transform into azo products after another cycle of electron transfer and proton abstraction steps with the formation of H2O2 as the only byproduct. Our methodology shows broad substrate scope with fast kinetics (90 min) excellent yields (up to 100%), and 100% selectivity under an ambient atmosphere in aqueous medium, and can be easily scaled up to the gram scale without any unwanted toxic byproducts. The present study lays the foundations for further in-depth understanding of the hidden role of O2 (a1Δg) generated directly via CT excitation of the solvent–O2 complex toward a diverse range of chemical and biochemical transformations.Data availability
The additional data supporting this article have been included as part of the ESI.†Author contributions
T. K. M. and S. S. conceptualized the research problem and designed the experiments. S. S. performed the experiments. S. S. and T. K. M. analyzed the data. All authors contributed to the final version of the manuscript (writing – review & editing).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Indian Institute of Technology (IIT) Indore for providing financial support and infrastructure. T. K. M. is grateful to the Science and Engineering Research Board (SERB) for a generous research grant (CRG/2023/003004). The authors acknowledge SIC, IIT Indore, and SAIF, IIT Bombay for instrumental facilities. S. S. acknowledges the Prime Minister's Research Fellowship (PMRF), India for research fellowship.Notes and references
- D. S. Ovoshchnikov, B. G. Donoeva and V. B. Golovko, ACS Catal., 2015, 5, 34–38 CrossRef CAS.
- S. Suleman, Y. Zhang, Y. Qian, J. Zhang, Z. Lin, Ö. Metin, Z. Meng and H. -L. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202314988 CrossRef CAS PubMed.
- L. Guo, E. Cui, H. Li, C. -H. Tung and Y. Wang, ACS Sustainable Chem. Eng., 2021, 9, 16670–16677 CrossRef CAS.
- Y. -Z. Chen, Z. U. Wang, H. Wang, J. Lu, S. -H. Yu and H. -L. Jiang, J. Am. Chem. Soc., 2017, 139, 2035–2044 CrossRef CAS PubMed.
- A. Sagadevan, K. C. Hwang and M. -D. Su, Nat. Commun., 2017, 8, 1812 CrossRef PubMed.
- A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS PubMed.
- D. A. Singleton, C. Hang, M. J. Szymanski, M. P. Meyer, A. G. Leach, K. T. Kuwata, J. S. Chen, A. Greer, C. Foote and K. N. Houk, J. Am. Chem. Soc., 2003, 125, 1319–1328 CrossRef CAS PubMed.
- M. Hayyan, M. A. Hashim and I. M. Al Nashef, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- S. Loeb, R. Hofmann and J. -H. Kim, Environ. Sci. Technol. Lett., 2016, 3, 73–80 CrossRef CAS.
- J. S. Nam, M. -G. Kang, J. Kang, S. -Y. Park, S. J. C. Lee, H. -T. Kim, J. K. Seo, O. -H. Kwon, M. H. Lim, H. -W. Rhee and T.-H. Kwon, J. Am. Chem. Soc., 2016, 138, 10968–10977 CrossRef CAS PubMed.
- M. Niedre, M. S. Patterson and B. C. Wilson, Photochem. Photobiol., 2002, 75, 382–391 CrossRef CAS PubMed.
- C. G. Hübner, A. Renn, I. Renge and U. P. Wild, J. Chem. Phys., 2001, 115, 9619–9622 CrossRef.
- S. Oelckers, T. Ziegler, I. Michler and B. Röder, J. Photochem. Photobiol., B, 1999, 53, 121–127 CrossRef CAS PubMed.
- R. S. Mulliken, Phys. Rev., 1928, 32, 186–222 CrossRef CAS.
- C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685–1757 CrossRef CAS PubMed.
- J. F. Lovell, T. W. B. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839–2857 CrossRef CAS PubMed.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- P. Maillard, P. Krausz, C. Giannotti and S. Gaspard, J. Organomet. Chem., 1980, 197, 285–290 CrossRef CAS.
- G. S. Cox, D. G. Whitten and C. Giannotti, Chem. Phys. Lett., 1979, 67, 511–515 CrossRef CAS.
- C. Tanielian and C. Wolff, J. Phys. Chem., 1995, 99, 9825–9830 CrossRef CAS.
- R. Schmidt and C. Tanielian, J. Phys. Chem. A, 2000, 104, 3177–3180 CrossRef CAS.
- R. Vankayala, A. Sagadevan, P. Vijayaraghavan, C. -L. Kuo and K. C. Hwang, Angew. Chem., Int. Ed., 2011, 50, 10640–10644 CrossRef CAS PubMed.
- R. Ho-Wu, S. Y. Yau and T. Goodson III, J. Phys. Chem. B, 2017, 121, 10073–10080 CrossRef CAS PubMed.
- D. Li, X. Li, T. Zhao, H. Liu, S. Jiang, Q. Zhang, H. Ågren and G. Chen, ACS Nano, 2020, 14, 12596–12604 CrossRef CAS PubMed.
- A. A. Gorman and M. A. J. Rodgers, J. Am. Chem. Soc., 1986, 108, 5074–5078 CrossRef CAS.
- J. Bendig, R. Schmidt and H. -D. Brauer, Chem. Phys. Lett., 1993, 202, 535–541 CrossRef CAS.
- A. Schlachter, P. Asselin and P. D. Harvey, ACS Appl. Mater. Interfaces, 2021, 13, 26651–26672 CrossRef CAS PubMed.
- J. A. Jackson, C. Turro, M. D. Newsham and D. G. Nocera, J. Phys. Chem., 1990, 94, 4500–4507 CrossRef CAS.
- J. Li and T. Chen, Coord. Chem. Rev., 2020, 418, 213355 CrossRef CAS.
- V. -N. Nguyen, Y. Yan, J. Zhao and J. Yoon, Acc. Chem. Res., 2021, 54, 207–220 CrossRef CAS PubMed.
- R. D. Scurlock and P. R. Ogilby, J. Am. Chem. Soc., 1988, 110, 640–641 CrossRef CAS.
- E. A. Gooding, K. R. Serak and P. R. Ogilby, J. Phys. Chem., 1991, 95, 7868–7871 CrossRef CAS.
- R. D. Scurlock and P. R. Ogilby, J. Phys. Chem., 1989, 93, 5493–5500 CrossRef CAS.
- P. -G. Jensen, J. Arnbjerg, L. P. Tolbod, R. Toftegaard and P. R. Ogilby, J. Phys. Chem. A, 2009, 113, 9965–9973 CrossRef CAS PubMed.
- M. Bregnhøj and P. R. Ogilby, J. Phys. Chem. A, 2019, 123, 7567–7575 CrossRef PubMed.
- M. Bregnhøj, M. Westberg, B. F. Minaev and P. R. Ogilby, Acc. Chem. Res., 2017, 50, 1920–1927 CrossRef PubMed.
- H. Tsubomura and R. S. Mulliken, J. Am. Chem. Soc., 1960, 82, 5966–5974 CrossRef CAS.
- H. Murakami, A. Kawabuchi, K. Kotoo, M. Kunitake and N. Nakashima, J. Am. Chem. Soc., 1997, 119, 7605–7606 CrossRef CAS.
- A. Bafana, S. S. Devi and T. Chakrabarti, Environ. Rev., 2011, 19, 350–371 CrossRef CAS.
- E. Merino, Chem. Soc. Rev., 2011, 40, 3835–3853 RSC.
- A. Corma, P. Concepción and P. Serna, Angew. Chem., Int. Ed., 2007, 46, 7266–7269 CrossRef CAS PubMed.
- A. Grirrane, A. Corma and H. García, Science, 2008, 322, 1661–1664 CrossRef CAS PubMed.
- C. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2010, 49, 6174–6177 CrossRef CAS PubMed.
- A. Grirrane, A. Corma and H. Garcia, Nat. Protoc., 2010, 5, 429–438 CrossRef CAS PubMed.
- S. Cai, H. Rong, X. Yu, X. Liu, D. Wang, W. He and Y. Li, ACS Catal., 2013, 3, 478–486 CrossRef CAS.
- B. Dutta, S. Biswas, V. Sharma, N. O. Savage, S. P. Alpay and S. L. Suib, Angew. Chem., Int. Ed., 2016, 55, 2171–2175 CrossRef CAS PubMed.
- M. Wang, J. Ma, M. Yu, Z. Zhang and F. Wang, Catal. Sci. Technol., 2016, 6, 1940–1945 RSC.
- Y. Zou, M. Zhang, F. Cao, J. Li, S. Zhang and Y. Qu, J. Mater. Chem. A, 2021, 9, 19692–19697 RSC.
- S. Han, Y. Cheng, S. Liu, C. Tao, A. Wang, W. Wei, H. Yu and Y. Wei, Angew. Chem., Int. Ed., 2021, 60, 6382–6385 CrossRef CAS PubMed.
- J. Qin, Y. Long, F. Sun, P. -P. Zhou, W. D. Wang, N. Luo and J. Ma, Angew. Chem., Int. Ed., 2022, 61, e202112907 CrossRef CAS PubMed.
- H. Zhu, X. Ke, X. Yang, S. Sarina and H. Liu, Angew. Chem., Int. Ed., 2010, 49, 9657–9661 CrossRef CAS PubMed.
- X. Guo, C. Hao, G. Jin, H. -Y. Zhu and X. -Y. Guo, Angew. Chem., Int. Ed., 2014, 53, 1973–1977 CrossRef CAS PubMed.
- B. Mondal and P. S. Mukherjee, J. Am. Chem. Soc., 2018, 140, 12592–12601 CrossRef CAS PubMed.
- J. D. Sitter and A. K. Vannucci, J. Am. Chem. Soc., 2021, 143, 2938–2943 CrossRef CAS PubMed.
- S. Singh, V. Agarwal, T. K. Sarma and T. K. Mukherjee, Green Chem., 2023, 25, 9109–9114 RSC.
- H. H. Wasserman, J. R. Scheffer and J. L. Cooper, J. Am. Chem. Soc., 1972, 94, 4991–4996 CrossRef CAS.
- I. Jahović, Y. Yang, T. K. Ronson and J. R. Nitschke, Angew. Chem., Int. Ed., 2023, 62, e202309589 CrossRef PubMed.
- Y. Yamakoshi, N. Umezawa, A. Ryu, K. Arakane, N. Miyata, Y. Goda, T. Masumizu and T. Nagano, J. Am. Chem. Soc., 2003, 125, 12803–12809 CrossRef CAS PubMed.
- L. Ren, M. M. Yang, C. H. Tung, L. Z. Wu and H. Cong, ACS Catal., 2017, 7, 8134–8138 CrossRef CAS.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04115a |
| This journal is © The Royal Society of Chemistry 2024 |