
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA photoinduced electron-transfer strategy for switchable fluorescence and phosphorescence in lanthanide-based coordination polymers†
Yu-Juan
Ma‡
 ,
Fei
Xu‡
,
Xin-Ye
Ren
,
Fan-Yao
Chen
,
Jie
Pan
,
Jin-Hua
Li
*,
Song-De
Han
* and
Guo-Ming
Wang
*
,
Fei
Xu‡
,
Xin-Ye
Ren
,
Fan-Yao
Chen
,
Jie
Pan
,
Jin-Hua
Li
*,
Song-De
Han
* and
Guo-Ming
Wang
*
College of Chemistry and Chemical Engineering, Key Laboratory of Shandong Provincial Universities for Functional Molecules and Materials, Qingdao University, Qingdao, Shandong 266071, P. R. China. E-mail: gmwang_pub@163.com; hansongde@qdu.edu.cn; jinhuali1978@163.com
First published on 2nd October 2024
Abstract
Smart optical materials with tunable fluorescence and room temperature phosphorescence (RTP) exhibit promising application prospects in the field of intelligent switches, information security, etc. Herein, a tetraimidazole derivative was grafted to one-dimensional lanthanum-diphosphonate through H-bonds, generating a coordination polymer (CP), (H4-TIBP)·[La2Li(H2-HEDP)4(H-HEDP)]·3H2O (termed La; TIBP = 3,3,5,5-tetra(imidazole-1-yl)-1,1-biphenyl; H4-HEDP = 1-hydroxyethylidene-1,1-diphosphonic acid) with a three-dimensional supramolecular structure. La shows dynamic fluorescence from blue to red and switchable monotonous yellowish-green RTP, which can be manipulated by reversible photochromism. It is worth noting that Eu3+/Tb3+-doped CPs exhibit time-resolved (red to yellow) and monotonous green afterglow, respectively, which can be attributed to multiple emissions with different decay rates. The dynamic and multicolor luminescence endows these CPs with potential for application in the domains of optical communications, multi-step encryption, and anti-counterfeiting. This work not only integrates color-adjustable fluorescence, switchable RTP, and photochromism in one material, but also realizes the manipulation of the resultant optical performances via photochromism, paving the pathway for the design and synthesis of smart optical materials.
Introduction
Room temperature phosphorescence (RTP) materials, which have the characteristics of millisecond lifetimes and large Stokes shifts, have great promise for versatile applications in communications, security encryption, bioimaging, and information storage.1–5 Based on the composition, RTP materials can be divided into three categories: inorganics,6,7 organics,8–18 and hybrids.19–22 The emissions in organics and hybrids both originate from organic molecules, whereas hybrids were created to optimize the RTP properties of organics due to the heavy atom effect of metals/halogens and the rigid environment constructed by coordination bonds and intermolecular interactions, and both of them are collectively referred to as molecule-based RTP materials. To date, hundreds of molecule-based RTP materials have been reported, but most present static and monotonous afterglow,2 which limits their application in intelligent fields.Smart stimulus-responsive RTP materials always exhibit adjustable afterglow (i.e., color, lifetime, quantum yield, etc.) under external stimuli, including heat, light, force, and chemicals, which triggers dynamic RTP.23–28 Particularly, the light stimulus is of interest because of its low cost, cleanliness, remote control, and easy switching. However, most photo-responsive materials are based on the consumption of triplet oxygen, which is relatively uncontrollable and random.29,30 Photochromism with two stable forms has been employed to regulate RTP properties, and a molecule-based photochromic system is designable and accessible by (1) introducing photoactive functional groups, such as spiropyrans,31 azo,32 diarylethenes,33,34etc.; (2) constructing feasible electron transfer (ET) pathways by selecting suitable electron donor/acceptor (ED/EA) pairs.35–38 For example, Ma et al. reported that the RTP colors could be remotely regulated by controlling the isomerization of the energy acceptor;39 Yan and coworkers achieved a series of metal–organic frameworks (MOFs) with switchable long-lived RTP controlled by reversible ET-induced photochromism;40 our group realized bidirectional regulation of photo-switchable RTP based on photo-generated radicals.41 However, research on similar materials is still scarce due to the competitive relationship between photochromism and luminescence42,43 but such a competitive relationship also provides opportunities to develop photo-controllable RTP based on reversible photochromism.
Considering the above factors, coordination polymers (CPs) can serve as promising candidates for the development of photo-controllable RTP, and two basic procedures are indispensable, (1) selecting an excellent phosphor containing heteroatoms to realize long-lived RTP in CPs by virtue of the heavy atom effect and anchoring phosphor in suitable CPs to mimic the freezing environment; (2) creating allowable ET paths between EDs and EAs to achieve ET-induced photochromism. Therefore, we chose 3,3,5,5-tetra(imidazole-1-yl)-1,1-biphenyl (TIBP) as the phosphor and EA, and 1-hydroxyethylidene-1,1-diphosphonic acid (H4-HEDP) as EDs, which are co-assembled with rare-earth metal and produced a new CP, (H4-TIBP)·[La2Li(H2-HEDP)4(H-HEDP)]·3H2O (La). The heterometallic phosphonate chains act as EDs and also provide a rigid environment for phosphor, while protonated TIBP moieties serve as the phosphor and EAs. La is colorless and shows blue steady-state photoluminescence (PL) under UV light and yellowish-green afterglow after turning off the UV lamp. After continuous photo-stimulus, the photoactivated sample (termed Laa) shows obvious ET-triggered photochromism from colorless to pale yellow, accompanied by fluorescence change from blue to red and afterglow disappearing. Substituting partial La3+ ions with other lanthanide ions, we successfully synthesized isostructural Eu3+/Tb3+-doped CPs, which were endowed with the characteristic emissions of Eu3+/Tb3+. Based on the photochromism-induced dynamic PL performances, as a proof of concept, we also demonstrated the application prospects of these crystals in the fields of optical communications, multiple encryption, and anti-counterfeiting.
Results and discussion
Single-crystal X-ray diffraction (SCXRD) characterization shows that La crystallizes in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group and its asymmetric unit contains two La3+ ions, one Li+ ion, five partially deprotonated diphosphonate units (four H2-HEDP units and one H-HEDP unit), two protonated TIBP (H4-TIBP) moieties with a site-occupancy factor of 1/2, and three guest water molecules (Fig. S1†). All metal ions are coordinated by O-atoms of phosphonate. La1 and La2 show octa-/hepta-coordinated modes to generate biaugmented trigonal prism and capped trigonal prism configurations, respectively, whereas Li1 displays the [LiO4] tetrahedron configuration (Fig. 1a, Table S2†). The distances of La–O and Li–O are in the range of 2.414(2)–2.625(3) Å and 1.918(7)–2.082(7) Å, respectively. The angles centered on La3+ and Li+ are between 64.37(8)°–150.90(9)° and 85.6(3)°–133.4(4)° (Tables S3 and S4†). The La3+ ions coordinate with diphosphonate ligands (H2-HEDP) forming a one-dimensional structure, while the Li+ ions binding a terminal-coordinated diphosphonate (H-HEDP) further connect with the former constructing the final anionic chain (Fig. 1b). The protonated TIBP balances the charges of anionic chains while serving as a mediator to connect four adjacent chains through H-bonds (N–H⋯O: 1.79–2.09 Å) to produce a three-dimensional (3D) supramolecular structure (Fig. 1c and d). Also, the lattice provides a rigid environment for TIBP which is beneficial for phosphorescence.19,40
space group and its asymmetric unit contains two La3+ ions, one Li+ ion, five partially deprotonated diphosphonate units (four H2-HEDP units and one H-HEDP unit), two protonated TIBP (H4-TIBP) moieties with a site-occupancy factor of 1/2, and three guest water molecules (Fig. S1†). All metal ions are coordinated by O-atoms of phosphonate. La1 and La2 show octa-/hepta-coordinated modes to generate biaugmented trigonal prism and capped trigonal prism configurations, respectively, whereas Li1 displays the [LiO4] tetrahedron configuration (Fig. 1a, Table S2†). The distances of La–O and Li–O are in the range of 2.414(2)–2.625(3) Å and 1.918(7)–2.082(7) Å, respectively. The angles centered on La3+ and Li+ are between 64.37(8)°–150.90(9)° and 85.6(3)°–133.4(4)° (Tables S3 and S4†). The La3+ ions coordinate with diphosphonate ligands (H2-HEDP) forming a one-dimensional structure, while the Li+ ions binding a terminal-coordinated diphosphonate (H-HEDP) further connect with the former constructing the final anionic chain (Fig. 1b). The protonated TIBP balances the charges of anionic chains while serving as a mediator to connect four adjacent chains through H-bonds (N–H⋯O: 1.79–2.09 Å) to produce a three-dimensional (3D) supramolecular structure (Fig. 1c and d). Also, the lattice provides a rigid environment for TIBP which is beneficial for phosphorescence.19,40
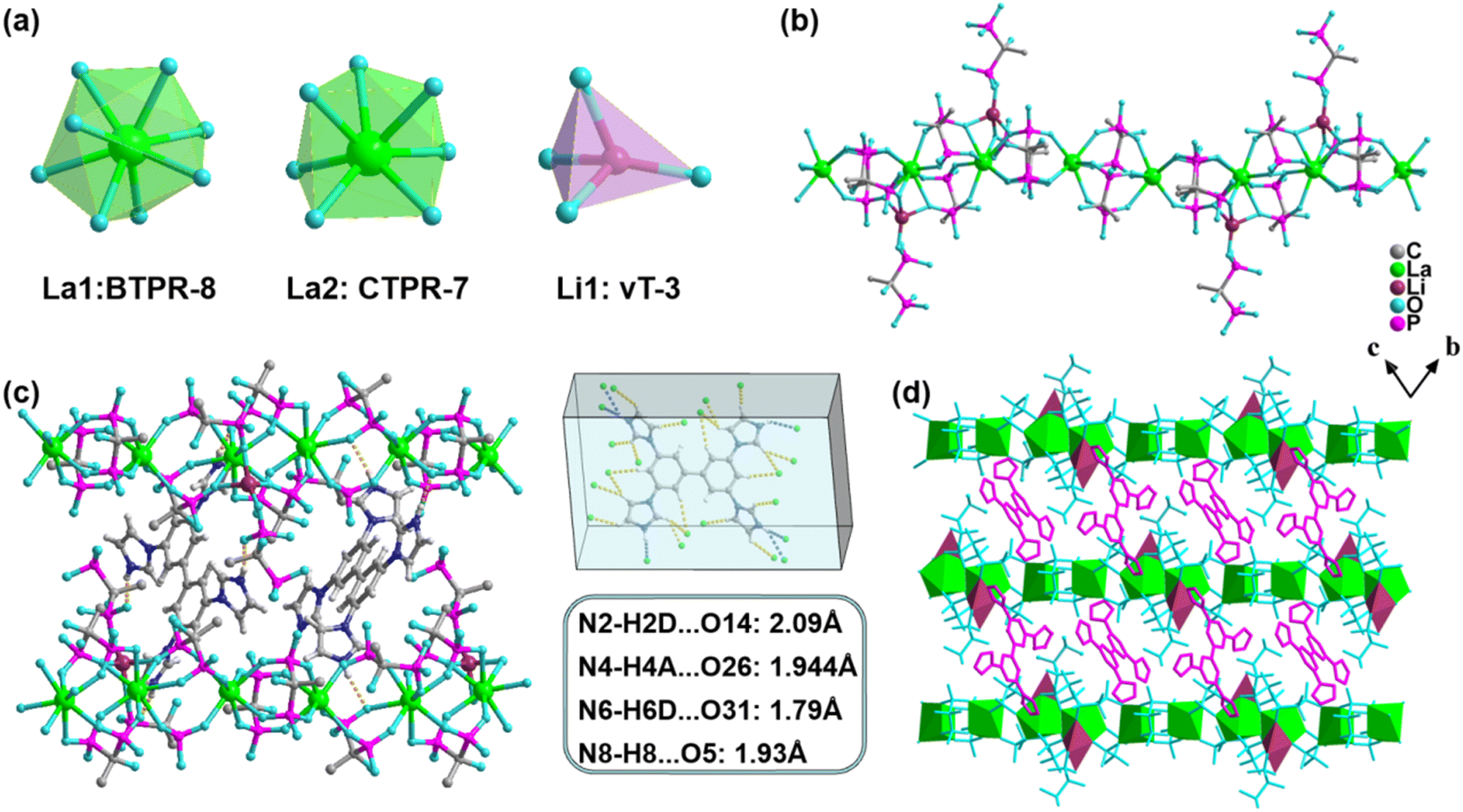 | ||
| Fig. 1 (a) The geometric configurations of La1, La2, and Li1 (BTPR-8: biaugmented trigonal prism; CTPR-7: capped trigonal prism; vT-3: vacant tetrahedron); (b) heterometallic phosphonate chain; (c) H-bonds between protonated TIBP units and phosphonate chains (orange dashed); (d) 3D supermolecular structure. | ||
La exhibits coloration from colorless to pale yellow within 3 min after photo-stimulus with a Xe-lamp (300 W). To investigate the photochromic process, the irradiation time-varied solid UV-vis absorption spectra were obtained. As shown in Fig. 2a and S2,† a new absorption peak at 250–450 nm can be observed after successive photo-stimulus, which can be ascribed to the yield of TIBP· radicals. The photo-induced radicals were further confirmed by solid-state electron paramagnetic resonance (EPR) measurements. The EPR spectra reveal a sharp signal at g = 1.9931 after photo-stimulus (Fig. 2b), while no signal is observed before, clearly indicating the generation of radicals. Detailed structural analysis was conducted to illustrate the photochromic mechanism. The rigid protonated TIBP guests are potential EAs, while the anionic heterometallic phosphonate hosts with O-abundant characteristics are potential EDs.35,38 The closest distance (2.602 Å) between NTIBP and OHEDP is advantageous to ET in the presence of H-bonds (N–H⋯O). Under UV light, La absorbs light energy and converts it into chemical energy, promoting the ET from OHEDP to NTIBP (Fig. 2c). Therefore, the photocoloration of La can be interpreted as photo-driven ET between EDs and EAs. The powder XRD and IR spectra (Fig. S3 and S4†) suggest that no apparent structural change is observable after coloration. Similar to ET-induced photochromic CPs, the Laa sample could relax to its original state after heating treatment at 120 °C for 3 h, which can be due to the oxidative quenching of TIBP· radicals at high temperatures.40,44–46 Also, thermogravimetric analysis (TGA) reveals that La can be stable up to 200 °C, which confirms the stability during the heating relaxation process (Fig. S5†).
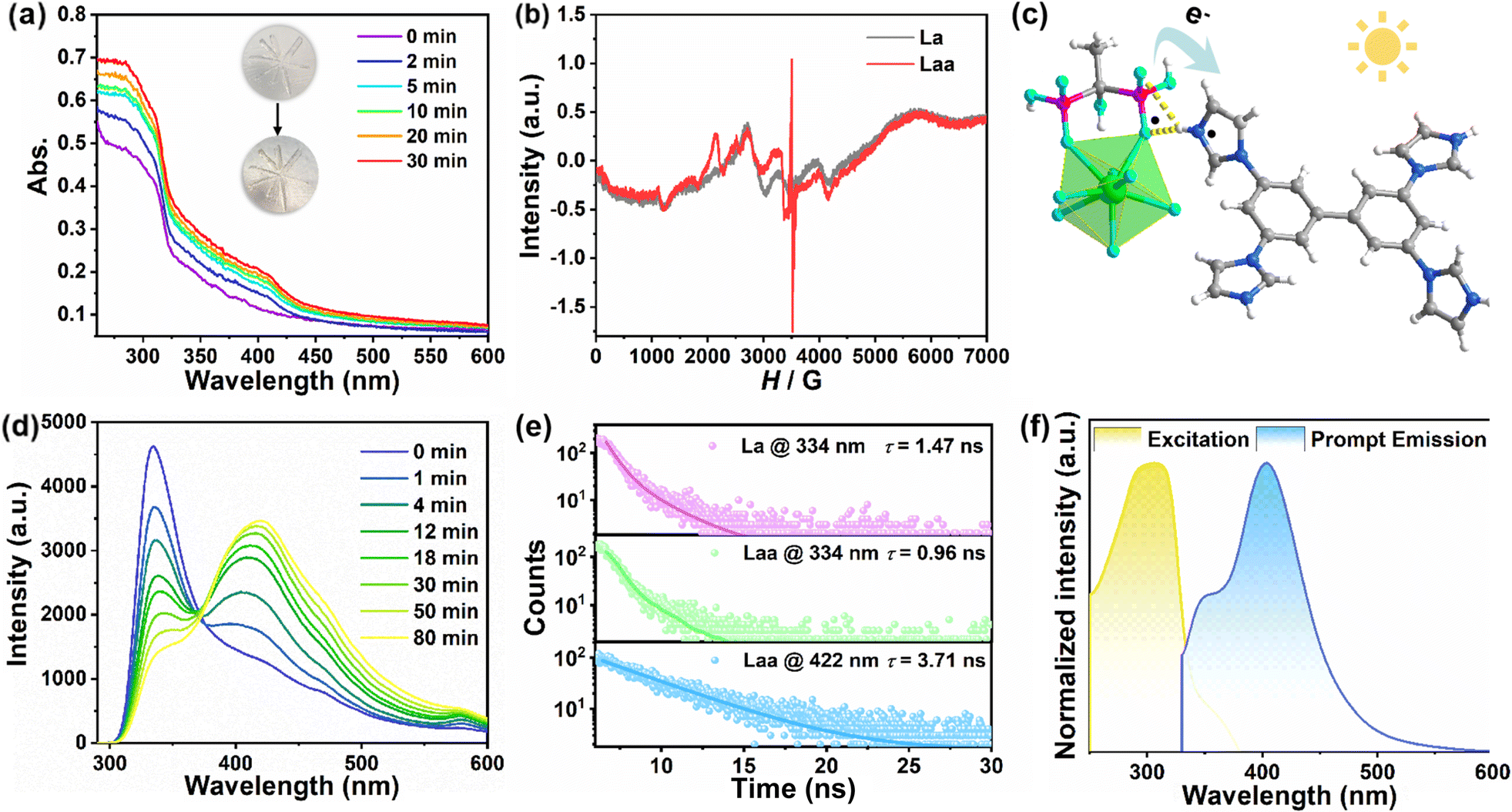 | ||
| Fig. 2 (a) The irradiation time-dependent UV-vis absorption spectra; (b) EPR spectra before and after irradiation; (c) the schematic diagram of the ET process; (d) the irradiation time-dependent fluorescence spectra excited by 280 nm UV light; (e) the decay curves monitored in 334 nm and 422 nm; and (f) the prompt excitation and emission spectra of TIBP. | ||
The reversible photochromism induces dynamic dual fluorescence in La. The prompt PL spectra in Fig. 2d show one emission peak located at 334 nm with a shoulder in the range of 365–500 nm when excited by 280 nm UV light. The decay curves obtained at 334 nm suggest that the lifetime is 1.47 ns (Fig. 2e), indicating that the emission can be ascribable to fluorescence. After consecutive photoactivation, the emission peak at 334 nm is gradually quenched, while the shoulder peak centered at 422 nm appeared and gradually increased, showing the redshift of fluorescence from violet to blue (Fig. S6†). The decay curve obtained at 422 nm suggests the lifetime is 3.71 ns, which is also rooted in the singlet state. To understand the redshift of fluorescence, we obtained the PL spectrum of TIBP as shown in Fig. 2f, which shows a dominating emission band at 405 nm with a shoulder centered at 350 nm. The similar emissions between TIBP and Laa inspire us to speculate as follows, (1) the PL in La mainly originates from the TIBP ligand;19,40 (2) the light induces the weakening protonation of TIBP, and the increasing electron cloud density, which leads to the fluorescence of Laa tending towards the unprotonated TIBP.35,38
When excited by 365 nm UV light, La exhibits blue emission while Laa changes to pink, so a series of excitation wavelength-varied emission spectra were obtained. As shown in Fig. S7,†La and Laa both exhibit wide excitation wavelength-dependent PL properties due to their multiple emissions involving singlet and triplet excitons. Surprisingly, the PL spectra of Laa show two new emissions at 580 nm and 631 nm when excited by 335–375 nm light, so a series of PL spectra with different irradiation times were obtained (λex = 370 nm). As shown in Fig. 3a and S8,† one emission peak is located at 430 nm with a lifetime of 3.99 ns. As the irradiation time increases, the intensity at 430 nm decreases and the lifetime shortens to 2.79 ns, and two new peaks appear at 580 nm and 631 nm which increase gradually, which result in a red-shift in CIE coordinates (Fig. 3b). The decay curves prove that the lifetimes are 10.54 and 10.88 ns, respectively, suggesting that these two emissions may originate from intramolecular charge transfer (CT) rather than from rare earths.47 The characteristic peaks of a series of Eu3+/Tb3+-doped CPs in subsequent experiments are completely different from the two peaks mentioned above, which also confirms this point. In addition, the absorption band at 250–300 nm can be due to the π–π* transition of aromatic rings, whereas the band at 300–400 nm is due to the n–π* transition involving the ligand to ligand CT process (Fig. 2a and S2†). This also proves that the enhanced ligand to ligand CT process in the photoactivated sample may induce two new emissions.
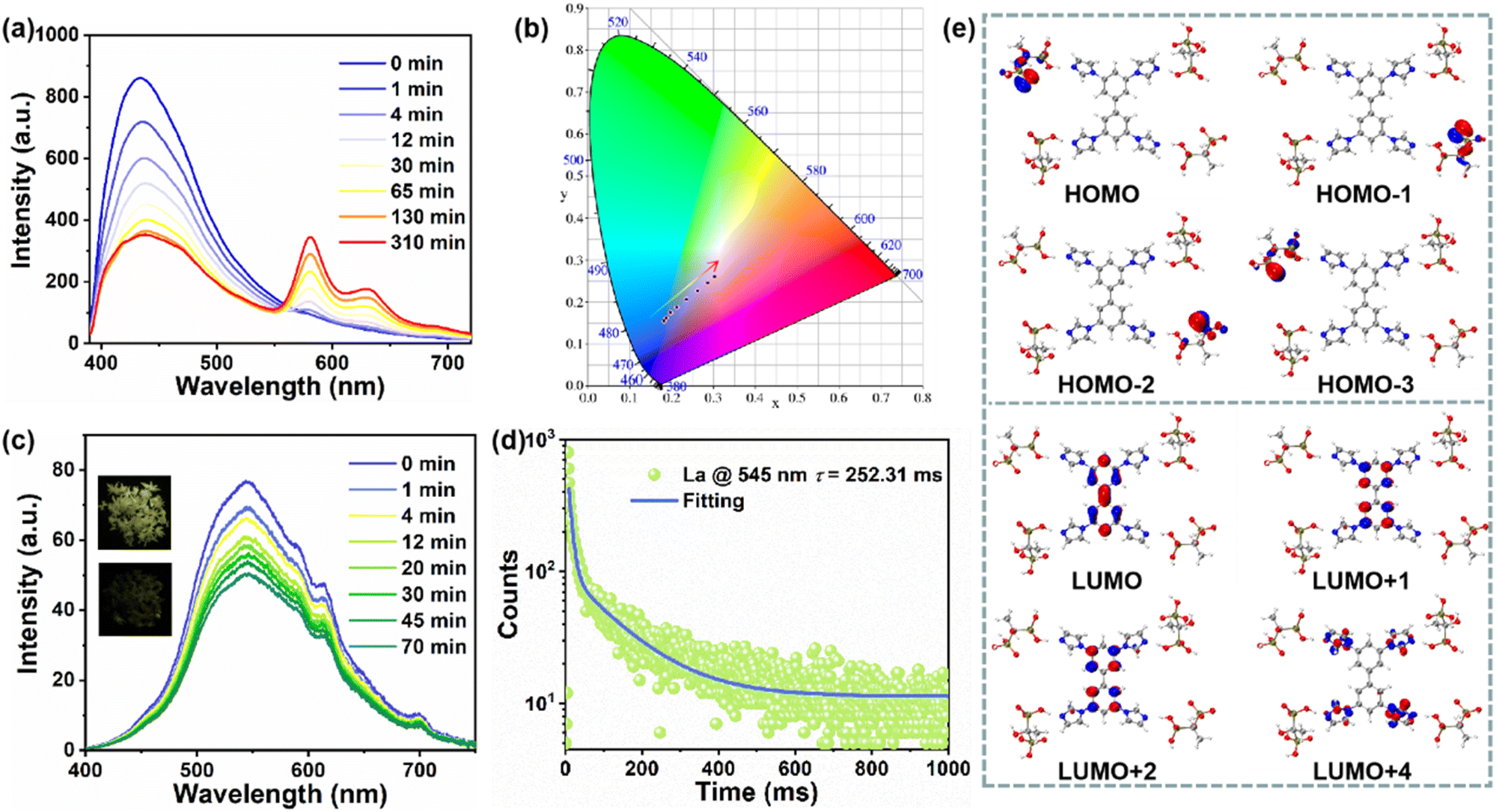 | ||
| Fig. 3 (a and b) The irradiation time-dependent fluorescent spectra excited by 370 nm UV light and their corresponding CIE coordinates; (c) the irradiation time-dependent delayed PL spectra of La (inset: photographs of La and Laa after turning off the 365 nm UV lamp); (d) long-lived decay curves of La; (e) calculated molecular orbitals. | ||
The reversible photochromism also endows La with photo-switchable afterglow. As shown in Fig. 3c and S9,†La exhibits yellowish green afterglow after ceasing the 365 nm UV light, which almost disappears after coloration, so the delayed PL emission spectra with different irradiation times were measured. Before irradiation, the emission is at around 545 nm with a lifetime of 252.31 ms (Fig. 3d), which is mainly donated by TIBP ligands and can be assigned to RTP due to its long-lived lifetime and large Stokes shift (Fig. S10†). The temperature-varied delayed PL spectra in Fig. S11† show that the emission intensity increases with the temperature decreasing, further suggesting that the long-lived emission can be assigned to phosphorescence. The RTP intensity is gradually decreased as the irradiation time increases, corresponding to the diminishing afterglow until it disappears, which is consistent with previous findings.40,43
To understand the photophysical process, we built a model based on La and calculated the highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs) based on the density functional theory (DFT) calculations. As shown in Fig. 3e and S12,† the HOMOs are mainly localized on HEDP, while the LUMOs are mainly populated on the benzene or imidazole group of TIBP, suggesting the occurrence of charge transfer from HEDP to TIBP (LLCT), which mainly serves the photoemission process.
Utilizing the similarity in coordination configurations and differences in characteristic emissions of rare earths, we doped a series of amounts of Eu3+ and Tb3+ (molar ratio: 1%, 2%, 5%, 10%, and 20%) into La, and obtained a series of isostructural CPs to achieve multicolor fluorescence and RTP (Fig. S13†).48,49 For Eu-doped CPs, the fluorescence goes from blue to pink while afterglow changes from yellowish-green to orange, and orange-red with the doping proportion increasing (Fig. 4a and S14†). Employing La0.9Eu0.1 as an example, scanning electron microscope (SEM) and elemental mapping images reveal a microrod morphology with a uniform distribution of La and Eu elements on the crystal surface (Fig. 4b and S15†), and the Eu3+ content is determined to be 12.91%. To further confirm the ratio between metal centers, we also performed X-ray fluorescence (XRF) spectroscopy, with the more precise results summarized in Table S7.† The experimental XRF data indicate that La0.9Eu0.1 contains a lower Eu3+ content of 8.57%. The prompt and delayed PL spectra of La0.9Eu0.1 both exhibit four new sharp peaks at 592, 616, 655, and 701 nm, corresponding to the 5D0–7FJ (J = 1–4) transitions of Eu3+. When excited by 280 nm UV light, the emission intensity at 334 nm decreases while other emission intensities increase with prolonged irradiation (Fig. S16a†). However, the prompt and delayed spectra in Fig. 4c and S16b† show that all emission intensities decrease as the irradiation time increases, suggesting that its PL properties can also be switched by ET-induced photochromism (Fig. S17†). These different phenomena also confirm the simultaneous occurrence of energy transfer from S1 to Eu3+ and from T1 to Eu3+. It is worth mentioning that La0.9Eu0.1 exhibits time-resolved afterglow change from orange to yellow. To charity the multi-color afterglow, the decay curves monitored in main peaks were exported. As shown in Fig. 4d, the lifetime at 545 nm is 296.45 ms, while that at 592, 616, and 701 nm is 41.97, 60.06, and 50.44 ms, suggesting that (1) the lifetime of characteristic transitions is significantly longer than that of traditional rare-earth-based materials, which results in the orange afterglow; (2) the decay rate at 545 nm is slower than others, so the afterglow changes from orange to yellow with time evolution. To capture the time-resolved afterglow, a series of time-resolved emission spectra (TRES) excited by 365 nm UV light were recorded. The delayed spectrum and TRES show that Eu3+-dominated emission (592, 616, 655, and 701 nm) is significantly stronger than the emission at 545 nm (from TIBP) when delayed by 1 ms and 10 ms, corresponding to orange-red afterglow (Fig. 4c, e and f). As aforementioned, Eu3+-dominated emission decays rapidly while TIBP-dominated emission decays slowly, so as the delay time increases (>100 ms), the intensities of TIBP- and Eu3+-dominated peaks trend to contribute equally, corresponding to yellow afterglow (Fig. S18†).
 | ||
| Fig. 4 (a) The photographs of La0.9Eu0.1 and La0.9Tb0.1 before and after turning off the UV lamp; (b) SEM and elemental mapping images of La0.9Eu0.1; (c) the irradiation time-dependent delayed PL spectra of La0.9Eu0.1 excited by 365 nm UV light; (d) long-lived decay curves of La0.9Eu0.1; (e and f) the time-resolved emission spectra (TRES) and the corresponding emission spectra; (g) the photophysical processes of La0.9Eu0.1. | ||
Different from Eu3+-doped CPs, the fluorescence of Tb3+-doped CPs does not exhibit obvious changes, but their afterglows are brighter (Fig. 4a and S19†). For La0.9Tb0.1, SEM and elemental mapping images also reveal a microrod morphology with a uniform distribution of La and Tb elements on the crystal surface (Fig. S20†), and the Tb3+ content is determined to be 12.23%. The XRF analysis shows that La0.9Tb0.1 contains a higher Tb3+ content of 19.61% (Table S7†). The prompt and delayed PL spectra both show four new sharp peaks at 490, 545, 585, and 622 nm, corresponding to the 5D4–7FJ (J = 3–6) transitions of Tb3+.When excited by 280 nm UV light, the Tb3+-dominated emissions remain unchanged with prolonged irradiation (Fig. S21a†). However, the prompt and delayed spectra in Fig. S21b and S22† show that all emission intensities decrease as the irradiation time increases, corresponding to photo-switchable PL properties (Fig. S23†). The long-lived decay curves suggest that their corresponding lifetimes are 49.28, 138.30, 47.15, and 62.27 ms (Fig. S24†). Therefore, it can be inferred that the brighter second-level afterglow is the result of the combination of the triplet state of TIBP and the antenna effect of Tb3+. However, a series of Tb3+-doped CPs show a monotonous green afterglow, which can be due to the characteristic emission of Tb3+ being similar to TIBP-originated RTP.
Benefiting from the photochromism and photo-controllable fluorescence, La microrods show photo-modulated optical waveguide properties, which are of great significance for the information security of photonic communications. As depicted in Fig. 5a and c, both endpoints of the rod-shaped crystal exhibit stronger emission under unfocused UV light, which means the optical waveguide properties. Therefore, detailed spatial-resolved PL microscopy images were obtained by changing the local positions on this microrod. Under a 375 nm laser beam, La exhibits bright blue emission, and the intensity at the tip gradually decreases with increasing propagation distance, which indicates that some of the photons generated by the excitation are effectively locked in the microrod and propagated from the excitation position to the tip. The optical waveguide loss performance can be evaluated by fitting the optical loss coefficient (R): Itip/Ibody = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp (−RD) (Itip/Ibody = the intensity at the tips/excited positions; D = the distance between exciting positions and tips). The R of La is 4.83 × 10−4 dB μm−1, which is significantly lower than that of most of the optical waveguide materials reported so far, indicating that La possesses excellent optical waveguide performance. This superior performance can be due to La's high crystallinity, distinctive shape and smooth surface, which can effectively minimize the optical loss caused by scattering from the domain boundary. Since La shows dynamic photo-induced PL emission, the optical waveguide properties after irradiation were also detected (Fig. 5b and d). Laa exhibits similar optical waveguide performance while the emission changes from blue to red under the 375 nm laser beam. The emission intensity gradually decreases as the excitation spot moves along the crystal, and the R value of Laa is 2.24 × 10−3 dB μm−1. Laa exhibits inferior optical waveguide performance compared to La, which can be due to two primary factors: (1) Laa exhibits stronger self-absorption (Fig. 2a). During photonic propagation within the Laa microrod, a portion of photons is lost through self-absorption, resulting in higher optical loss. (2) As depicted in the irradiation time-dependent fluorescence spectra (Fig. 3a), the emission intensity of Laa is lower than that of La. Also, the main emission peak (580 and 631 nm) of Laa exhibits a longer fluorescence lifetime (Fig. S8†), which competes with the quantum yield. However, both the R value and the waveguide color show significant changes before and after irradiation, suggesting that the optical waveguide behavior can be regulated by reversible photochromism.
exp (−RD) (Itip/Ibody = the intensity at the tips/excited positions; D = the distance between exciting positions and tips). The R of La is 4.83 × 10−4 dB μm−1, which is significantly lower than that of most of the optical waveguide materials reported so far, indicating that La possesses excellent optical waveguide performance. This superior performance can be due to La's high crystallinity, distinctive shape and smooth surface, which can effectively minimize the optical loss caused by scattering from the domain boundary. Since La shows dynamic photo-induced PL emission, the optical waveguide properties after irradiation were also detected (Fig. 5b and d). Laa exhibits similar optical waveguide performance while the emission changes from blue to red under the 375 nm laser beam. The emission intensity gradually decreases as the excitation spot moves along the crystal, and the R value of Laa is 2.24 × 10−3 dB μm−1. Laa exhibits inferior optical waveguide performance compared to La, which can be due to two primary factors: (1) Laa exhibits stronger self-absorption (Fig. 2a). During photonic propagation within the Laa microrod, a portion of photons is lost through self-absorption, resulting in higher optical loss. (2) As depicted in the irradiation time-dependent fluorescence spectra (Fig. 3a), the emission intensity of Laa is lower than that of La. Also, the main emission peak (580 and 631 nm) of Laa exhibits a longer fluorescence lifetime (Fig. S8†), which competes with the quantum yield. However, both the R value and the waveguide color show significant changes before and after irradiation, suggesting that the optical waveguide behavior can be regulated by reversible photochromism.
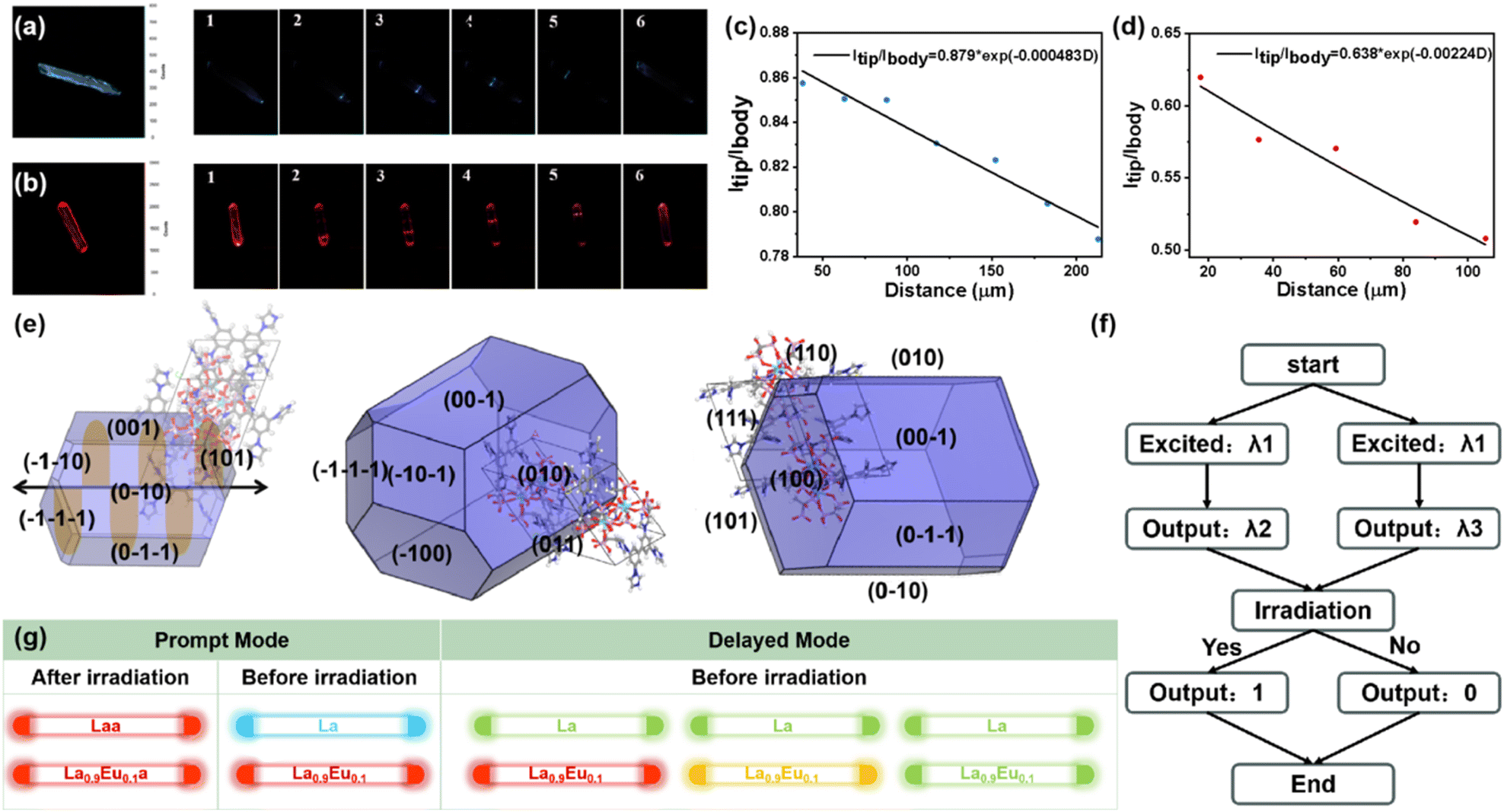 | ||
| Fig. 5 PL images of microrods La (a) and Laa (b) excited with focused 370 nm light at different positions; the optical loss coefficients of La (c) and Laa (d); (e) the morphology simulation and corresponding crystal faces of La based on the BFDH method; (f) illustration of the optical logic gates; (g) predicted multi-mode 1D active waveguide properties. | ||
To elucidate the relationships between 1D photonic properties and morphologies, the morphology simulation and prediction for La was conducted on the basis of Bravais–Friedel–Donnay–Harker (BFDH) theory (Fig. 5e).50,51 The simulated morphology suggests four fast-growing crystal facets of {0 1 0}, {0 0 1}, {0 1 1}, and {1 0 0} with surface areas of 27.27, 24.80, 13.42, and 16.16%, respectively (Table S5†), and La grows into a 1D microrod crystal along the direction of the black arrow (Fig. S25†).
By virtue of the dynamic optical waveguide behavior driven by photochromism, we designed an advanced secure optical logic gate prototype using La and Laa (Fig. 5f). We input an excitation wavelength (λ1) into a flat waveguide coupler to activate the optical logic gate system, which can collect two wavelengths at the output port (λ2, λ3). If the emission in the tip is blue, the output port is “0”; otherwise the output is “1”. In this manner, a binary number output could be achieved within the 1D microcrystal. Based on the photo-controllable fluorescence and RTP in La, we reasonably speculate that the Eu3+/Tb3+-doped CPs would exhibit optical waveguide behavior with multicolor emission, whether in prompt or delayed modes. As shown in Fig. 5g, La not only exhibits bi-mode optical waveguide properties (blue-red) in prompt mode, but is also predicted to present waveguide properties with yellowish-green emission in delayed mode. However, La0.9Eu0.1 would exhibit completely different waveguide behaviors, which show red emissions before and after irradiation in prompt mode, and changes to orange and yellowish-green over time in delayed modes. Therefore, utilizing such smart and diverse waveguide behavior will be promising for designing more advanced optical logic gate systems.52
The dynamic fluorescence and RTP properties provide La and Eu3+/Tb3+-doped CPs with opportunities for applications in multi-step encryption and anti-counterfeiting fields. For example, we designed a house pattern that was filled with as-synthesized samples, in which the roof and door were filled with La0.9Eu0.1, the wall and tree trunk were filled with La, the chimney was filled with Laa, and leaves were filled with La0.9Tb0.1 (Fig. 6a). When the pattern is exposed to daylight, all houses are white, which is easily imitated and forged. When the pattern is exposed to UV light, and then turned off for 0.1 s, 1 s and 2 s, the pattern presents different emissions, which achieves temporal- and spatial-resolved multiple anti-counterfeiting. Meanwhile, the more emission states there are, the higher the difficulty for forging, confirming the application prospects of these CPs in the field of anti-counterfeiting.
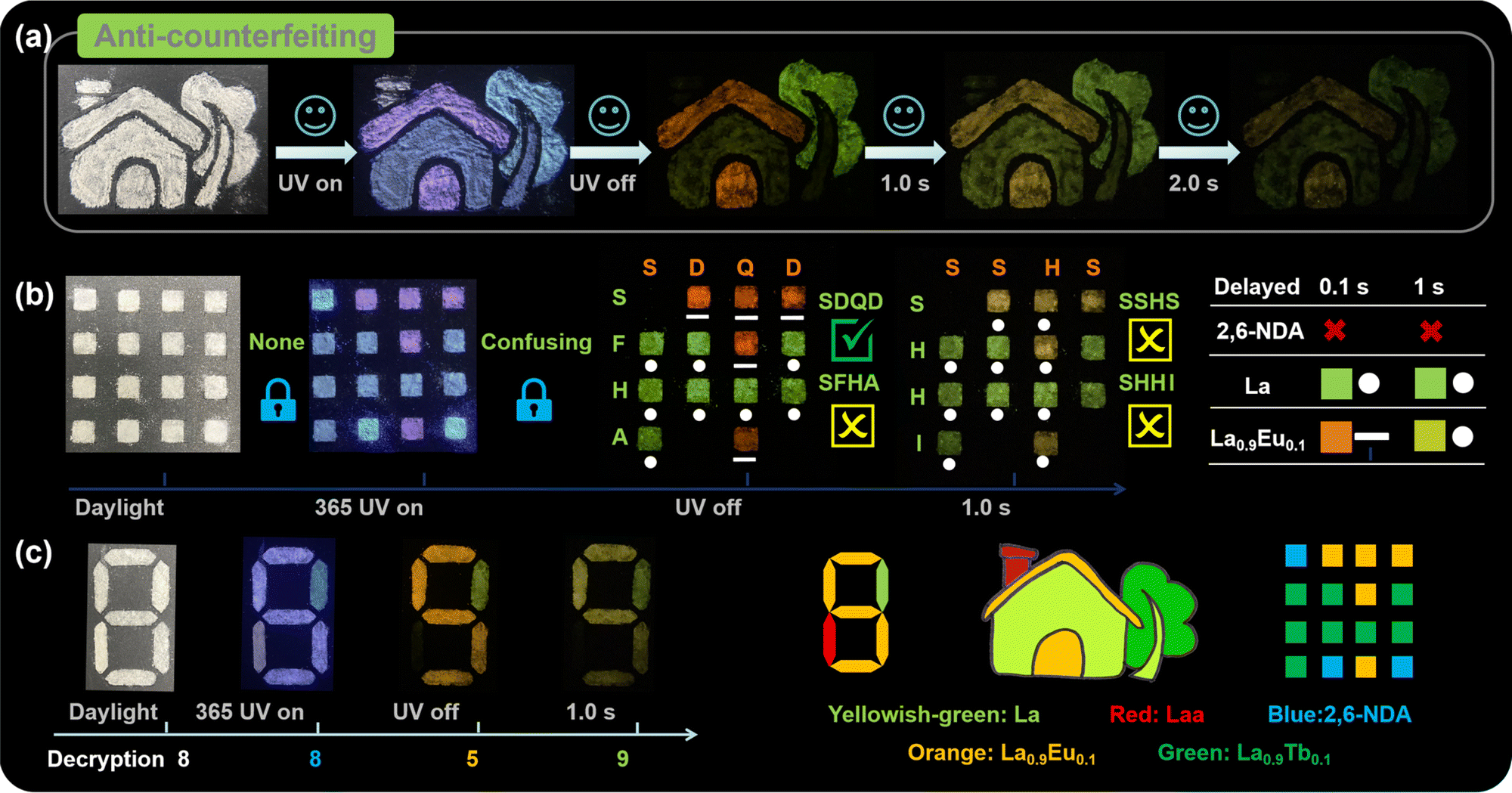 | ||
| Fig. 6 (a) The house pattern under daylight, UV light, and after turning off for 0.1 s, 1 s, and 2 s; (b) the model for Morse code application; (c) the digit model. | ||
Inspired by the time-resolved emission of La0.9Eu0.1 and its similar emission to La, we also designed a Morse code application employing 2,6-naphthalene dicarboxylic acid (NDA) as a costar. As shown in Fig. 6b and S26,† all blocks are white under daylight, and show multi-color emissions under 365 nm UV light, so we cannot obtain any effective information. After turning off the UV light, the blocks filled with NDA disappear first, while La exhibits yellowish-green afterglow marked as “·” and Eu-doped CPs exhibit red-orange afterglow marked as “–”. The pattern can be horizontally decrypted as “SDQD”, which is the correct decoding letter representing “Shandong Qingdao”. Simultaneously, the “SFHA” in the vertical direction can confuse enemies. Also, the correct information “SDQD” can disappear in time. After turning off the UV lights for 1 s, the pattern changes to “SSHS” and “SHHI” in the horizontal and vertical directions, respectively. Based on the as-synthesized CPs, more diverse and useful Morse code can be designed and hopefully applied in practice.
These CPs are also promising for application in advanced data encryption and smart display. As depicted, an “8” pattern can also be designed as a digital encryption using La, Laa, and La0.9Eu0.1 (Fig. 6c). Under daylight, the “8” pattern appears white. Under 365 nm UV light, all parts show indistinguishable emissions, so the pattern displays as “8”. When the UV light is extinguished, the emission of Laa disappears, while La and La0.9Eu0.1 show yellow-green and orange afterglow, respectively, so the digit “5” can be recognized with orange afterglow. After 1 s, the pattern changes to “9” with yellow-green afterglow. Therefore, the whole digital password “8859” can be obtained.
Experimental
Materials and measurements
IR spectra were obtained on a MAGNA-560 (Nicolet) FT-IR spectrometer. The room -temperature photoluminescence spectra were recorded on an F-4700 fluorescence spectrometer. The time-resolved emission spectra (TRES) were obtained on an FS5 fluorescence spectrometer. The temperature-dependent emission spectra and all decay curves were obtained on an FLS 1000 fluorescence spectrometer. The solid-state UV-vis spectra were measured on a Puxi Tu-1901 spectrophotometer with a BaSO4 reference. Electron paramagnetic resonance (EPR) was recorded on a Bruker E500 spectrometer. A scanning electron microscope (SEM, Hitachi, S2400) equipped with an energy dispersive spectroscopy (EDS) detector was used to investigate the microstructures and elements. Chemical compositions of lanthanum oxide products were investigated by using a PANalytical Axios X-ray fluorescence spectrometer. The experimental powder X-ray diffraction (PXRD) analyses were conducted on a Rigaku D/max-2550 diffractometer with Cu-Kα radiation (λ = 1.5418 Å). The simulated PXRD curve was derived from the SCXRD data and Mercury software. The Xe-lamp for time-dependent solid UV-vis absorption characterization is a Perfect Light PLS-SXE 300. The photosource for the luminescence characterization is the intrinsic light of the instrument.Synthesis of La and Laa
A mixture of La2O3 (0.03 g, 0.092 mmol), TIBP (0.02 g, 0.05 mmol), H4-HEDP·H2O (0.10 g, 0.45 mmol), LiF (0.10 g, 4.00 mmol), 2 mL CH3OH and 4 mL H2O was sealed in a Teflon-lined autoclave and heated at 120 °C for 6 days, and colorless crystals (termed La) were obtained and washed with absolute ethanol. Elemental analysis for C34H57N8O38P10La2Li (%). Calculated: C, 22. 94; H, 3.23; N, 6.29. Found: C, 23. 47; H, 3. 58; N, 6. 02. IR of La (KBr pellets, cm−1): 3407(s), 3155(s), 2851(m), 2365(w), 1618(m), 1545(w), 1467(w), 1363(w), 1129(s), 1067(s), 920(s), 800(w), 650(w), 560(m). After sufficient illumination of the above crystals, light yellow crystals were obtained (termed Laa). IR of Laa (KBr pellets, cm−1): 3395(s), 3155(s), 2970(m), 2932(w), 2859(m), 2744(w), 2645(w), 1618(m), 1543(w), 1468(w), 1365(w), 1125(s), 1076(s), 918(s), 857(w), 800(w), 646(w), 557(m).Synthesis of Eu3+/Tb3+-doped CPs
Replace La2O3 with different doping proportions of Eu2O3 or Tb2O3 (1%, 2%, 5%, 10%, and 20%), and other processes are consistent with the synthesis of La. Elemental analysis for C34H57N8O38P10La1.8Eu0.2Li (%). Calculated: C, 22. 90; H, 3.22; N, 6.28. Found: C, 23. 01; H, 3. 52; N, 6. 33. IR of La0.9Eu0.1 (KBr pellets, cm−1): 3435(s), 2977(w), 2927(w), 2852(w), 1625(s), 1465(w), 1417(w), 1122(m), 1086(m), 1043(m), 926(w), 808(w), 643(w), 561(w). Elemental analysis for C34H57N8O38P10La1.8Tb0.2Li (%). Calculated: C, 23.30; H, 3.54; N, 6.02. Found: C, 22. 97; H, 3. 50; N, 5.89. IR of La0.9Tb0.1 (KBr pellets, cm−1): 3435(s), 2977(w), 2927(w), 2854(w),1618(s), 1468(w), 1422(w), 1125(m), 1072(m), 926(w), 804(w), 646(w), 557(w).X-ray crystallography
The single-crystal X-ray diffraction data of La and Laa were collected on a Rigaku Oxford Diffraction at 293(2) K with Mo-Kα radiation (λ = 0.71073 Å). The SHELX-2016 software was employed to solve the structure.53 Detailed crystallographic data for La and Laa are summarized in Table S1,† and the selected bond lengths and angles are listed in Tables S3 and S4.† Full crystallographic data have been deposited with CCDC 2206527 for La and 2206528 for Laa.Calculation details
The calculations were conducted using the density functional theory (DFT) method in Gaussian16.54 B3LYP-D3/def2-SVP was applied to get the Frontier Molecular Orbital.55–58 Wavefunction analyses were executed using Multiwfn.59 Visualization of the frontier molecular orbitals was achieved using the VMD software.60Conclusions
In summary, a series of La3+-based and Eu3+/Tb3+-doped CPs were successfully assembled by using metal-diphosphonate and rigid tetraimidazole with the heterometallic phosphonate chains as electron-donating hosts and protonated tetraimidazole moieties as electron-accepting guests. Under photo-stimulus, these CPs characterized with an ED-EA skeleton not only show obvious ET-facilitated photochromism, but also display photochromism-induced color-adjustable fluorescence and switchable afterglow, which can be due to photo-generated radicals. It is worth mentioning that Eu3+-doped CPs exhibit time-resolved afterglow, while Tb3+-doped CPs exhibit brighter green afterglow, which inspires us that reasonable doping would be an effective strategy for constructing multi-color and efficient RTP materials. The variable fluorescence and RTP in these CPs provide these materials with more optical communication applications worth exploring, such as La microrods show photo-modulated bi-mode switchable optical waveguide properties (blue to red) with a low optical loss coefficient. In addition, a series of CPs also present promising application prospects in the fields of multi-step encryption and anti-counterfeiting. This work demonstrates photo-adjustable fluorescence and RTP by regulating photochromism, and provides a doping strategy to obtain phosphorescence with different surprising properties, affording a theoretical and experimental basis for the development of intelligent optical materials.Data availability
The data supporting this article have been included as part of the Supplementary Information. Crystallographic data for La and Laa has been deposited at the CCDC under 2206527 and 2206528.Author contributions
Y.-J. Ma and F. Xu contributed equally to this work. Y.-J. Ma, J.-H. Li, S.-D. Han and G.-M. Wang conceived the main idea and designed the experiments. F. Xu, X.-Y. Ren and J. Pan synthesized and characterized the coordination polymers. Y.-J. Ma, F. Xu, X.-Y. Ren and F.-Y. Chen performed and analyzed the photophysical measurements. Y.-J. Ma, J.-H. Li, S.-D. Han and G.-M. Wang analyzed all data. Y.-J. Ma, F. Xu, J.-H. Li, S.-D. Han and G.-M. Wang wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (22071125 and 22071126), the Natural Science Foundation of Shandong Province (ZR2021MB064), the Youth Innovation Team Project of Shandong Provincial Education Department (No. 2023KJ234), and the Qingdao University discipline cluster interdisciplinary joint research project (FZ2024201).Notes and references
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef.
- X. Yang, G. I. N. Waterhouse, S. Lu and J. Yu, Chem. Soc. Rev., 2023, 52, 8005–8058 RSC.
- T. Maldiney, A. Bessière, J. Seguin, E. Teston, S. K. Sharma, B. Viana, A. J. J. Bos, P. Dorenbos, M. Bessodes, D. Gourier, D. Scherman and C. Richard, Nat. Mater., 2014, 13, 418–426 CrossRef PubMed.
- X. Zhang, J. Liu, B. Chen, X. He, X. Li, P. Wei, P. F. Gao, G. Zhang, J. W. Y. Lam and B. Z. Tang, Matter, 2022, 5, 3499–3512 CrossRef.
- X.-K. Ma, W. Zhang, Z. Liu, H. Zhang, B. Zhang and Y. Liu, Adv. Mater., 2021, 33, 20074 Search PubMed.
- G. Cai, L. Giordano, C. Richard and B. Viana, Nanomaterials, 2023, 13, 2175 CrossRef PubMed.
- Y. Cao, X. Wang, Z. Zhang and Y. Wang, J. Phys. Chem. Lett., 2021, 12, 958–965 CrossRef CAS PubMed.
- D. B. Clapp, J. Am. Chem. Soc., 1939, 61, 523–524 CrossRef CAS.
- Z. Mao, Z. Yang, Z. Fan, E. Ubba, W. Li, Y. Li, J. Zhao, Z. Yang, M. P. Aldred and Z. Chi, Chem. Sci., 2019, 10, 179–184 RSC.
- Z. Wu, F. Dinkelbach, F. Kerner, A. Friedrich, L. Ji, V. Stepanenko, F. Würthner, C. M. Marian and T. B. Marder, Chem.–Eur. J., 2022, 28, e202200525 CrossRef CAS.
- M. Singh, K. Shen, W. Ye, Y. Gao, A. Lv, K. Liu, H. Ma, Z. Meng, H. Shi and Z. An, Angew. Chem., Int. Ed., 2024, 63, e202319694 CrossRef CAS.
- T. Zhang, X. Ma, H. Wu, L. Zhu, Y. Zhao and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 11206–11216 CrossRef CAS PubMed.
- S. Xu, W. Wang, H. Li, J. Zhang, R. Chen, S. Wang, C. Zheng, G. Xing, C. Song and W. Huang, Nat. Commun., 2020, 11, 4802 CrossRef CAS PubMed.
- X. Zhang, K. C. Chong, Z. Xie and B. Liu, Angew. Chem., Int. Ed., 2023, 62, e202310335 CrossRef CAS.
- S. Feng, Q. Huang, S. Yang, Z. Lin and Q. Ling, Chem. Sci., 2021, 12, 14451–14458 RSC.
- Z. Lin, R. Kabe, N. Nishimura, K. Jinnai and C. Adachi, Adv. Mater., 2018, 30, 1803713 CrossRef PubMed.
- Y. Zhang, X. Chen, J. Xu, Q. Zhang, L. Gao, Z. Wang, L. Qu, K. Wang, Y. Li, Z. Cai, Y. Zhao and C. Yang, J. Am. Chem. Soc., 2022, 144, 6107–6117 CrossRef CAS PubMed.
- P. Pattanayak, A. Nandi, R. Patra and P. Purkayastha, Adv. Opt. Mater., 2024, 12, 2302155 CrossRef CAS.
- H. Liu, W. Ye, Y. Mu, H. Ma, A. Lv, S. Han, H. Shi, J. Li, Z. An, G. Wang and W. Huang, Adv. Mater., 2021, 34, 2107612 CrossRef PubMed.
- P. Gao, K. Zhang, D. Ren, H. Liu, H. Zhang, H. Fu, L. Ma and D. Li, Adv. Funct. Mater., 2023, 33, 2300105 CrossRef CAS.
- P.-Y. Fu, B.-N. Li, Q.-S. Zhang, J.-T. Mo, S.-C. Wang, M. Pan and C.-Y. Su, J. Am. Chem. Soc., 2022, 144, 2726–2734 CrossRef CAS PubMed.
- P. Leo, G. Orcajo, J. A. García, A. M. Ortuño, J. M. Cuerva, D. Briones, G. Calleja, A. Rodríguez-Diéguez, R. Sanz, J. Cepeda and F. Martínez, J. Mater. Chem. C, 2021, 9, 5544–5553 RSC.
- Q. Zhou, C. Yang and Y. Zhao, Chemistry, 2023, 9, 2446–2480 CrossRef CAS.
- Y. Zhou, P. Zhang, Z. Liu, W. Yan, H. Gao, G. Liang and W. Qin, Adv. Mater., 2024, 36, 2312439 CrossRef CAS PubMed.
- Z. Chai, C. Wang, J. Wang, F. Liu, Y. Xie, Y.-Z. Zhang, J.-R. Li, Q. Li and Z. Li, Chem. Sci., 2017, 8, 8336–8344 RSC.
- C. Xing, B. Zhou, D. Yan and W.-H. Fang, CCS Chem., 2023, 5, 2866–2876 CrossRef.
- Y. Tian, J. Yang, Z. Liu, M. Gao, X. Li, W. Che, M. Fang and Z. Li, Angew. Chem., Int. Ed., 2021, 60, 20259–20263 CrossRef CAS PubMed.
- W. Li, Q. Huang, Z. Mao, X. He, D. Ma, J. Zhao, J. W. Y. Lam, Y. Zhang, B. Z. Tang and Z. Chi, Nat. Commun., 2022, 13, 7423 CrossRef CAS PubMed.
- M. Gmelch, H. Thomas, F. Fries and S. Reineke, Sci. Adv., 2019, 5, eaau7310 CrossRef PubMed.
- S. Feng, Y. Ma, S. Wang, S. Gao, Q. Huang, H. Zhen, D. Yan, Q. Ling and Z. Lin, Angew. Chem., Int. Ed., 2022, 61, e202116511 CrossRef CAS PubMed.
- A. S. Burova, O. V. Venidiktova, M. A. Savelyev, V. A. Barachevsky, T. V. Bukreeva and T. N. Borodina, Mater. Lett., 2021, 303, 130558 CrossRef CAS.
- L. Stricker, M. Bçckmann, T. M. Kirse, N. L. Doltsinis and B. J. Ravoo, Chem.–Eur. J., 2018, 24, 8639–8647 CrossRef CAS PubMed.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef CAS PubMed.
- Z. Li, H. Chen, B. Li, Y. Xie, X. Gong, X. Liu, H. Li and Y. Zhao, Adv. Sci., 2019, 6, 1901529 CrossRef PubMed.
- S.-D. Han, J.-X. Hu and G.-M. Wang, Coord. Chem. Rev., 2022, 452, 214304 CrossRef.
- X.-Q. Yu, M.-S. Wang and G.-C. Guo, Adv. Funct. Mater., 2023, 33, 2212907 CrossRef.
- H.-Y. Li, Y.-L. Wei, X.-Y. Dong, S.-Q. Zang and T. C. W. Mak, Chem. Mater., 2015, 27, 1327–1331 CrossRef.
- Y.-J. Ma, J.-X. Hu, S.-D. Han, J. Pan, J.-H. Li and G.-M. Wang, J. Am. Chem. Soc., 2020, 142, 2682–2689 CrossRef PubMed.
- L. Ma, Q. Xu, S. Sun, B. Ding, Z. Huang, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2022, 61, e202115748 CrossRef PubMed.
- Y.-J. Ma, X. Fang, G. Xiao and D. Yan, Angew. Chem., Int. Ed., 2021, 61, e202114100 CrossRef PubMed.
- D.-X. Feng, Y. Mu, J. Li, S.-D. Han, J.-H. Li, H.-L. Sun, M. Pan, J.-X. Hu and G.-M. Wang, Adv. Funct. Mater., 2023, 33, 2305796 CrossRef CAS.
- Y. Yang, J. Wang, D. Li, J. Yang, M. Fang and Z. Li, Adv. Mater., 2021, 33, 2104002 CrossRef CAS PubMed.
- D.-D. Yang, H.-W. Zheng, Y.-H. Fang, Q.-F. Liang, Q.-Z. Han, Y.-S. Shi and X.-J. Zheng, Inorg. Chem., 2022, 61, 7513–7522 CrossRef CAS PubMed.
- X.-S. Xing, R.-J. Sa, P.-X. Li, N.-N. Zhang, Z.-Y. Zhou, B.-W. Liu, J. Liu, M.-S. Wang and G.-C. Guo, Chem. Sci., 2017, 8, 7751–7757 RSC.
- X.-Y. Ren, F.-Y. Chen, C.-H. Zhang, Z.-G. Liang, X.-Y. Yu, S.-D. Han and G.-M. Wang, Chem.–Eur. J., 2024 DOI:10.1002/chem.202402581.
- S.-L. Li, M. Han, Y. Zhang, G.-P. Li, M. Li, G. He and X.-M. Zhang, J. Am. Chem. Soc., 2019, 141, 12663–12672 CrossRef CAS PubMed.
- X. Xu, L. Mo, Y. Li, X. Pan, G. Hu, B. Lei, X. Zhang, M. Zheng, J. Zhuang, Y. Liu and C. Hu, Adv. Mater., 2021, 33, 2104872 CrossRef PubMed.
- Y. Yang, K.-Z. Wang and D. Yan, Chem. Commun., 2017, 53, 7752–7755 RSC.
- C. Yang, F. Artizzu, K. Folens, G. Du Laing and R. Van Deun, J. Mater. Chem. C, 2021, 9, 7154–7162 RSC.
- J. D. H. Donnay and D. Harker, Am. Mineral., 1937, 22, 446–467 Search PubMed.
- R. Docherty, G. Clydesdale, K. J. Roberts and P. Bennema, J. Phys. D: Appl. Phys., 1991, 24, 89–99 CrossRef.
- B. Zhou and D. Yan, Chem. Sci., 2022, 13, 7429–7436 RSC.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji and X. Li, Gaussian 16 (Version B01), Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154119 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data for La and Laa have been deposited. CCDC 2206527 and 2206528. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04632c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |