DOI:
10.1039/D4SC04735D
(Edge Article)
Chem. Sci., 2024,
15, 17927-17936
Charting the coordinative landscape of the 18F–Sc/44Sc/177Lu triad with the tri-aza-cyclononane (tacn) scaffold†
Received
16th July 2024
, Accepted 27th September 2024
First published on 3rd October 2024
Abstract
The widely established PET isotope 18F does not have a therapeutic partner. We have recently established that the Sc–F bond can be formed under aqueous, high yielding conditions, paving the way to providing 18F as diagnostic partners to 47Sc and 177Lu radiotherapeutics. Here, we synthesized a library of tacn-based chelators comprised of 10 structurally unique permutations incorporating acetate, methyl-benzylamide and picolinate donor arms. The chelator library encompasses chelators ranging from 6- to 9-dentate, and produces complex changes ranging from +3 to −1. The corresponding Sc–F/Sc and Lu chelate complexes were characterized using computational, spectroscopic and potentiometric methods, followed by optimization of radiolabeling with 18F, 44Sc and 177Lu and concluded by in vivo validation. We identify characterization benchmarks that chart the coordinative landscape of radiochelation approaches for this unusual triad. Our screening identifies two ligand systems, H2L111 and H3L201 as ideal, readily functionalizable constructs for prospective, targeted theranostic applications with 18F/44Sc/177Lu.
Introduction
The short-lived isotope 18F (Eβ+avg = 250 keV, t1/2 = 109 min) is currently the most frequently and readily utilized positron emission tomography (PET) isotope worldwide, for both diagnostic and research studies, frequently preceding administration of therapeutic isotopes for the management of disease with radiotherapy.1 The lack of a suitable therapeutic isotopologue for 18F, can render 18F PET probes suboptimal diagnostic partners to chemically dissimilar, radiometal-based radiotherapies.2 Additionally, conventional radiofluorination strategies involve C–F bond formation in anhydrous, aprotic solvents at high temperatures, which can limit the radiolabeling of thermally and chemically sensitive (bio)molecules.3 To address this issue, the use of prosthetic groups4 and the formation of B–F, Si–F and Al–F bonds has been successfully explored to access mild radiochemical 18F labeling strategies compatible with aqueous solvents and proteins.5–8
However, none of these approaches provide access to a chemically identical theranostic pair, where the same chemical construct can be employed to incorporate 18F and a radiometal such as the widely commercially successful radiotherapeutic isotope 177Lu (Eβ−avg = 134 keV, t1/2 = 159.6 h), or the more experimental Sc-triad: 43Sc (Eβ+avg = 476 keV, t1/2 = 3.9 h), 44Sc (Eβ+avg = 632 keV, t1/2 = 4 h) or 47Sc (Eβ−avg = 162 keV, t1/2 = 80.4 h).9–12 Establishing such a theranostic triad or tetrad could take advantage of the growing, global low energy cyclotron infrastructure and provide accelerated development and translation of theranostic agents. Previously, we established that the displacement of a labile, inner-sphere water of the 7-coordinate Sc(mpatcn) (herein listed as [ScF(L201)]–) and Sc(picaga) chelate with a 18F− ion produces in vivo compatible and 18F–Sc/47Sc(picaga)-conjugates that show no statistical difference with respect to their in vivo biodistribution.13
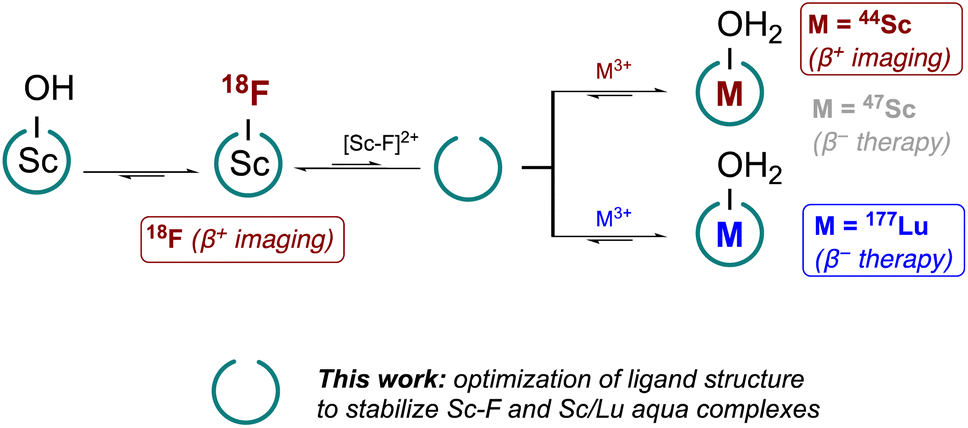
However, questions and challenges remained after our pilot study: (1) what are the relevant characteristics of chelators that can stabilize Sc–F, Sc and Lu chelates under macroscopic and radiotracer level conditions? Can we establish benchmarks using analytical characterization methods to predict compatibility with the theranostic F/Sc/Lu triad? (2) Can we uncover coordination complexes that improve access to bifunctional constructs by simplifying synthesis? (3) Can we improve radiochemical labeling approaches to increase compatibility with currently employed radiopharmaceutical practices and accelerate clinical translation (Fig. 1)?
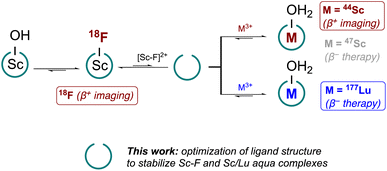 |
| | Fig. 1 Overview of desired reactivity with optimized tacn ligand scaffold to enable chelation of radioisotopes of Sc3+, Lu3+ and the [Sc–F]2+ core. Reactions are indicated as equilibria with favored product/reactivity indicated. | |
Here, we address these questions by probing a series of tri-aza-cyclononane (tacn) based chelators. The compound library, a permutation of acetate, methyl-benzylamide and picolinate donor arms, is synthetically accessible by sequential alkylations. Following characterization, a combination of computational, multi-nuclear spectroscopic, spectrophotometric, and radiochemical methods was employed to predict and identify the formation of in vivo compatible radiochemical coordination complexes. The subsequent in vivo analysis identified optimal chelator denticity and charge equilibration properties that will enable simplified, targeted imaging and therapy approaches with a single bifunctional chelator using 18F, 44Sc and 177Lu isotopes.
Results and discussion
Ligand design and chemical synthesis
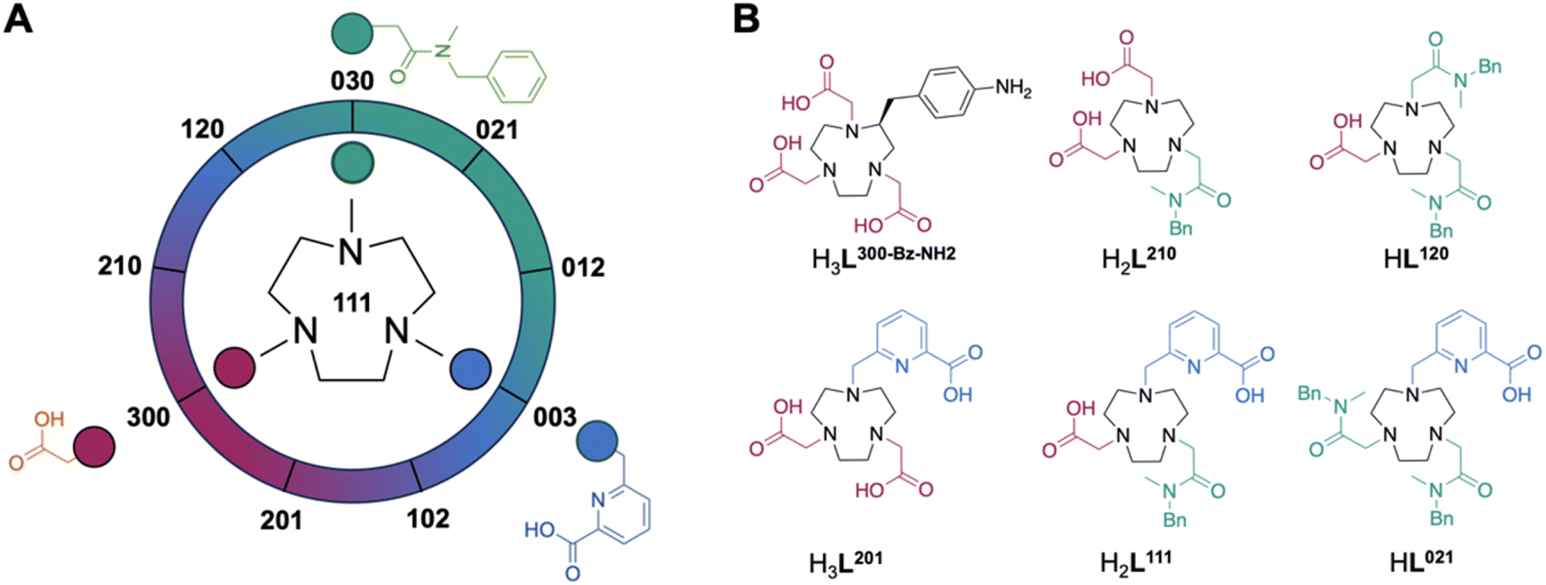
Our previous work showed that the 7-coordinate ligand mpatcn formed complexes with all three isotopes of interest, with the bifunctional analogue picaga exhibiting good in vivo performance for targeted imaging and therapy applications. However, we did not systematically explore how the ligand denticity, donor group basicity and resultant charge would affect the corresponding chelator's ability to stabilize all three types of complexes. In accordance with previous work by us and others, preferred ligand systems for Sc3+ and Lu3+ overwhelmingly incorporate hard to borderline hard donors such as aliphatic amines, acetates and picolinate donors. To access a diverse (not exhaustive) chelator library with respect to hardness and denticity, we selected acetate, methyl-benzylamide and picolinate donor arms and employed sequential alkylation with the corresponding, acetate protected alkyl bromide species (ESI, Schemes S1 and S2†). While acetate and picolinate donor groups exhibit only one coordinative mode, amide donors can coordinate by carbonyl-O or deprotonated amido-N, especially in presence of Lewis acidic metal ions such as Sc3+. To avoid coordinative ambiguity, we incorporate methyl-benzylamide which enforces coordination through the carbonyl-O exclusively.
Chromatographic separation following deprotection of the acetate donor groups of the acetyl and picolyl groups affords the free chelators which can be directly employed to form the coordination complex species of interest. 10 permutations are possible if all positions are alkylated (Fig. 2A, Table 1). Of these, four are 6-coordinate, three are 7-coordinate, two are 8-coordinate and one is 9-coordinate. Previous literature on aqueous Sc chelation indicates a pronounced preference for the formation of 8-coordinate complex species in aqueous environments, with 6- and 7-coordinate complexes forming ternary complexes with solvent or buffer molecules.14 9-coordinate Sc-coordination complexes are rare, indicating that the 8- and 9-coordinate ligand systems will not accommodate the coordination of a ternary F− ligand. All non-radioactive complexes were formed from MCl3·6H2O (M = Sc3+ or Lu3+) in presence of ammonium acetate buffer to mimic radiochemical labelling conditions and keep the solution pH optimal for complexation. Subsequent fluorination to produce the Sc–F complex was achieved by treatment of the corresponding Sc-complexes [Sc(L201)], [Sc(L111)]+, and [Sc(L021)]2+ with 5 equivalents of NH4F or CsF, followed by precipitation of excess fluoride via redissolution of the soluble Sc–F complex in acetonitrile to remove insoluble, residual fluoride salts. Of note, while mass spectrometric evidence of the formation of the ScF complex of 6-coordinate ligand systems was obtained, no significant amounts of product could be isolated. Absence of the 6-coordinate Sc complexes coincided with appearance of precipitate in the reaction mixture attributed to formation of insoluble ScF3.
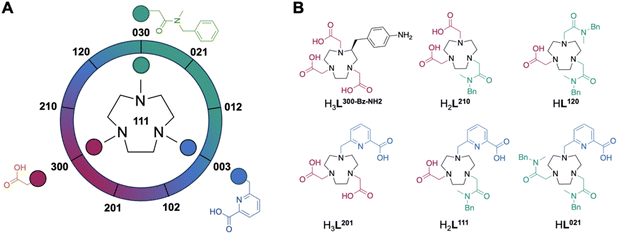 |
| | Fig. 2 (A) general ligand design scheme incorporating acetate, methyl-benzylamide and picolinate donor arms in different permutations to produce 10 ligand systems discussed herein. The ligand nomenclature system is designed so that the three digits represent the number of acetyl, amido, and picolyl groups, respectively. Numbering for 9 of the ligands is shown around the wheel with the 10th ligand, L111, being depicted in the center. (B) Selected 6- and 7-coordinate chelator structures with corresponding color-coded donor arms and representative nomenclature. | |
Table 1 Summary of naming convention, Sc/Lu complex charges (m), ScF complex charges (n) and ligand coordination number (CN) of all synthesized ligand systems of the permutational tacn library described in this work
| Ligand (HnLxxx) |
H3L300 |
H2L210 |
HL120 |
L030
|
H3L201 |
H2L111 |
HL021 |
HL102 |
H2L012 |
H2L003 |
| [M(Lxxx)]m |
0 |
+1 |
+2 |
+3 |
0 |
+1 |
+2 |
0 |
+1 |
0 |
| [ScF(Lxxx)]n |
−1 |
0 |
+1 |
+2 |
−1 |
0 |
+1 |
−1 |
0 |
−1 |
| Ligand CN |
6 |
6 |
6 |
6 |
7 |
7 |
7 |
8 |
8 |
9 |
Solid state characterization
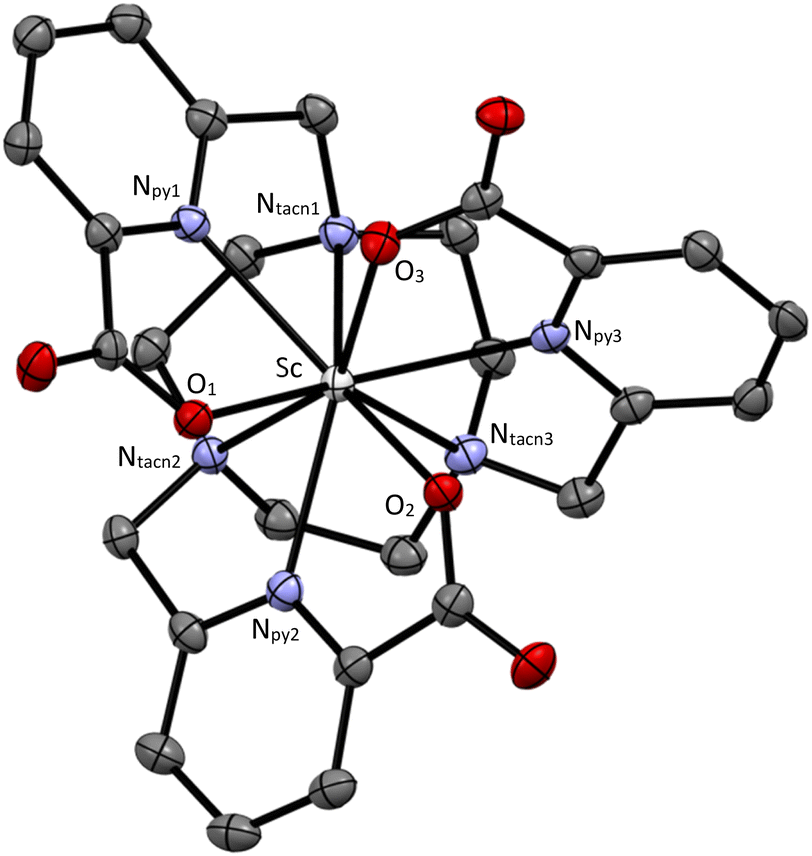
Crystallization attempts were only successful with [Sc(L003)]·3H2O (CCDC: 2369298, Fig. 3, ESI Tables S12–S20†). The Sc atom is at the core of a 9-coordinate tricapped trigonal prismatic complex isostructural with several Ln analogues (Ln = Nd, Eu, Gd, Tb, Yb, and Lu) reported in the literature.15–17 The coordination environments of high denticity Sc3+ and lanthanide atoms in these complexes are similar despite differences in the ionic radii of these metals (Sc3+ = 0.745 Å, Ln3+ = 0.861–1.032 Å). The structural similarity between Sc3+ and Lu3+ complexes is noteworthy due to the clinical use of 177Lu for targeted radiation therapy. As expected, the metal–ligand bond distances for this series of complexes exhibit a positive correlation with the ionic radii of the metal center (i.e. longer metal–ligand distances correspond to metals with larger ionic radii) when comparing structures determined at the same data collection temperature (Table 2).
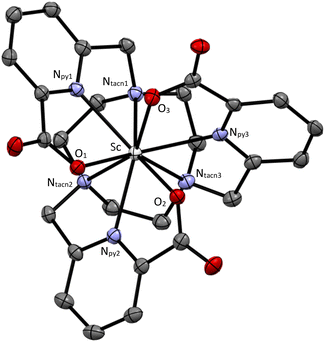 |
| | Fig. 3 Single crystal X-ray diffraction representation of the Sc(L003)-Λ(λ,λ,λ) complex at 50% thermal ellipsoid probability. Hydrogen atoms and solvent molecules omitted for clarity. | |
Table 2 Metal-to-ligand donor bond distances, ligand donor interplane distances, tacn macrocycle torsion angles and interplane torsion angles (dihedral angles)
|
|
Sc(L003)-Λ(λ,λ,λ) |
Lu(L003)-Λ(λ,λ,λ) |
Difference |
|
Mean bond distances [Å]
|
| M–Ntacn |
2.564 |
2.613 |
+0.049 |
| M–Npy |
2.426 |
2.494 |
+0.068 |
| M–O |
2.206 |
2.309 |
+0.103 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
Mean plane distances [Å]
|
| Ntacn–Npy |
2.014 |
2.068 |
+0.054 |
| Ntacn–O |
3.401 |
3.490 |
+0.089 |
| Npy–O |
1.387 |
1.423 |
+0.036 |
| Ntacn–M |
1.960 |
2.006 |
+0.046 |
|
|
Mean torsion [°]
|
| Ntacn–Ntacn |
−45.95 |
−46.22 |
−0.27 |
| Ntacn–Npy |
43.70 |
42.06 |
−1.64 |
| Ntacn–O |
103.83 |
100.39 |
−3.44 |
| Npy–O |
60.15 |
58.37 |
−1.78 |
Spectroscopic solution phase characterization
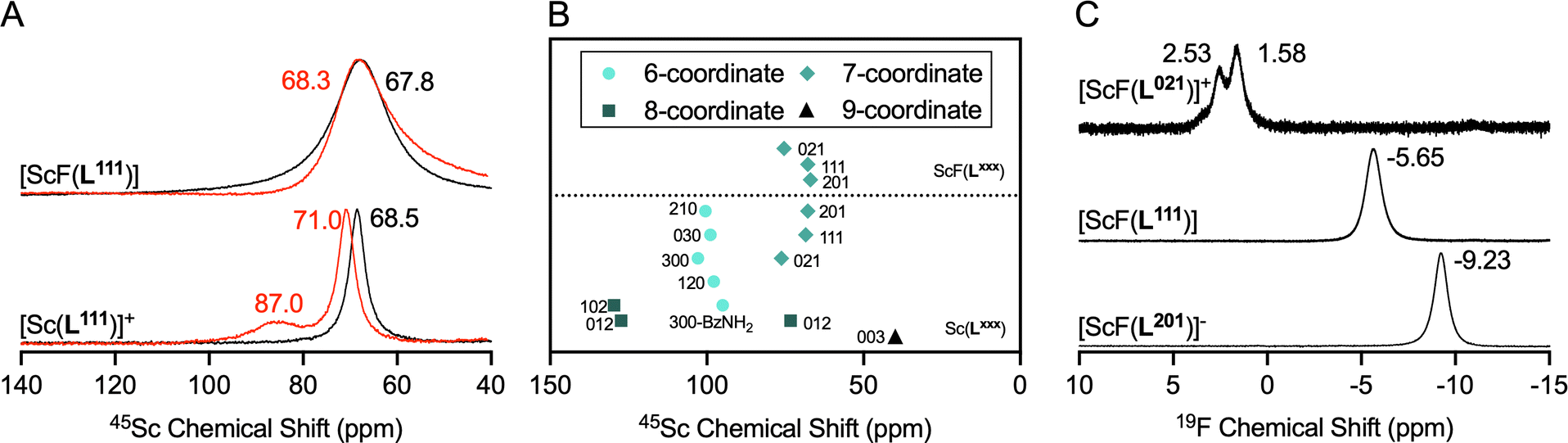
Additionally, the complexes were characterized using 1H, 13C{1H}, 45Sc and 19F NMR spectroscopy. While 1H and 13C{1H} spectra were characteristic of the asymmetric and dynamic aza-macrocycle solution environment, 45Sc and 19F spectral data provided clearer insight into solution chemical features of the corresponding complexes.18 Acquisition of spectral data in MeOD limits the formation of ternary Sc complexes with solvent and buffer molecules such as acetate and water. Literature on 45Sc spectral data for aqueous coordination complexes is sparse, as the 45Sc quadrupole produces broad peaks in asymmetric environments and therefore confers little structural information when considered in isolation. However, the compound library described here provides a data set large enough that specific trends emerge (Fig. 4A and B). Specifically, we find that chemical shift regions are characteristic for coordinative numbers and solvent access. While not observed to date for mononuclear, solution-phase systems, this correlates well with 45Sc NMR spectroscopy data collected on solid state structures.19–21 In MeOD, Sc complexes producing 45Sc chemical shifts ranging from 65 to 75 ppm are enveloped by a 7-coordinate ligand. Chemical shifts ranging from 95 to 105 ppm are produced when Sc is in a 6-coordinate ligand environment with acetate buffer molecules occupying the inner sphere. Under the same conditions, 8-coordinate ligand systems without inner sphere coordination are found above 125 ppm. Of note, even in MeOD, the 8-coordinate [Sc(L012)]+ produces two characteristic signals (127 and 73 ppm respectively), indicative of multiple solution species, with 8-coordinate and a potential 7-coordinate species in dynamic equilibrium. Displacement of the inner-sphere water ligand with F− to form [ScF(L201)]–, [ScF(L111)] and [ScF(L021)]+ results in an upfield shift of the 45Sc chemical shift of <5 ppm and significant line broadening of the 45Sc resonance. For the Sc–F complexes, 19F NMR provides additional relevant insight. Due to the quadrupolar coupling of the 19F and 45Sc nuclei, corresponding spectral features are broad, but appear in a characteristic range from −9.3, −3.8 to 2.5 ppm for [ScF(L201)]–, [ScF(L111)] and [ScF(L021)]+, respectively. The chemical shift range is typical for metal-bound F− species and the observed trend reflects increasing de-shielding of the fluoride ion in accordance with increasing positive charge.22 The formation of coordinative isomers is observed in all spectra, but indeed produces two distinct signals for [ScF(L021)]+ where isomer interconversion due to presence of the methyl-benzylamide groups is further slowed. Taken together, the heteronuclear spectroscopy data offers unprecedented insight into the coordination environment of these complexes, providing clear delineation of desirable complex hallmarks of 65–75 ppm, characteristic for 7-coordinate ligand systems.
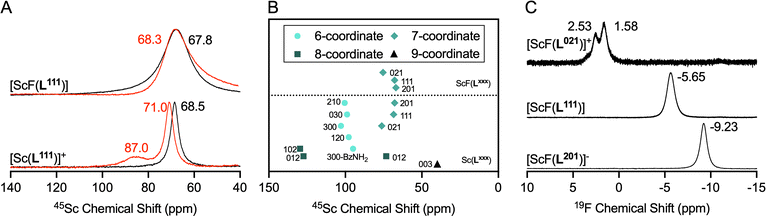 |
| | Fig. 4 NMR-spectroscopic characterization of Sc and ScF complex series synthesized as part of this work. (A) 45Sc-NMR spectra of [ScF(L111)] (top) and [Sc(L111)]+ (bottom) in D2O (red trace), and MeOD (black trace). (B) Summary of 45Sc spectral data in MeOD of the entire complex series shows characteristic chemical shift ranges for all complexes investigated, grouped as follows, from highest to lowest field: 9-coordinate symmetric species (triangle), 7-coordinate species (diamonds), 6-coordinate species (spheres) and 8-coordinate species (squares); notably, [ScF(L012)]+ exhibits two distinct solution species. (C) 19F-NMR spectra of [ScF(L021)]+, [ScF(L111)] and [ScF(L201)]–, indicating increase in electronic shielding as the negative complex charge increases. | |
To gain further insight into structural parameters that may be predictive of the formation of stabilized ScF complexes, we conducted DFT structure optimization and determination of the bond dissociation energy (BDE) of [Sc(OH2)(LXXX)], [Sc(OH)(LXXX)] and [ScF(LXXX)] complexes where inner-sphere coordination was readily accessible (6- and 7-coordinate complex systems). For all complexes, Δ- and Λ-configurations were calculated at the B3LYP-D3(BJ)/cc-pVDZ level of theory with SMD solvation and considering a single inner sphere ternary ligand (F−, OH− or OH2), Table 3 (further reading and parameters can be found within the Supportive Information, Sections 1.5 and 4). All ligand systems which incorporate the methyl-benzylamido donor were truncated to the bis-methylamide species to decrease isomeric complexity induced by the formation of E/Z isomers. In accordance with our previous computational characterization of the [ScF(L201)]– complex, all investigated complex systems exhibit BDE values above those obtained for the corresponding hydroxo complex species by 30–50 kJ mol−1, and greater than 200 kJ mol−1 above the aqua complex. Within the 6- and 7-coordinate complex series evaluated, a trend emerges with respect to relative Sc–F bond lengths: increasing complex charge results in a shortened Sc–OH2 and Sc–F bond lengths and increasing BDE values. Computed Sc–OH2 bond lengths are elongated by 0.1–0.2 Å when compared with crystal structure data of the few 7-coordinate Sc chelate systems reported, ranging from 2.24 (AAZTA)14 to 2.12 (py2macrodipa)23 and 2.14 (py-macrodipa)24 and indicate increased inner sphere crowding in small-cavity macrocycle systems. The same relative trends are observed for Sc–OH2, albeit restriction of the inner sphere ternary ligand binding pocket may diminish H2O coordination, while the F− ternary ligand can be accommodated throughout the entire ligand series due to its smaller relative size at a virtually unchanged bond length. This behavior correlates well with observations made for the formation of aqua complexes across the lanthanide series, where contraction and increase of steric bulk of the ligand environment eventually excludes ternary complex formation.25 The level of theory used in these calculations has been previously show to perform well under most tests.26,27 Additionally, RMSD comparison between the XRD and DFT optimized structure for [Sc(L003)] yields an RMSD of 0.1946 (Table S3†).
Table 3 DFT optimized, structural parameters for 6- and 7-coordinate ligand systems investigated, including relevant bond lengths and calculated bond dissociation energies (BDE). Ternary coordination number values refer to the species employed for corresponding DFT calculations
| Complex |
X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OH2 OH2 |
BDE (kJ mol−1) |
Calculated bond length [Å] |
Ternary CN |
| XOH− |
XF− |
Sc–F |
Sc–OH2 |
| Δ-ScXL300 |
54 |
219 |
270 |
1.967 |
2.349 |
7 |
| Δ-ScXL300 |
54 |
218 |
271 |
1.967 |
2.350 |
7 |
| Δ-ScXL210 |
60 |
236 |
285 |
1.957 |
2.334 |
7 |
| Λ-ScXL210 |
60 |
236 |
285 |
1.957 |
2.334 |
7 |
| Δ-ScXL120 |
63 |
250 |
299 |
1.952 |
2.322 |
7 |
| Λ-ScXL120 |
60 |
247 |
296 |
1.952 |
2.324 |
7 |
| Δ-ScXL201 |
27 |
189 |
229 |
1.958 |
2.354 |
8 |
| Λ-ScXL201 |
27 |
189 |
229 |
1.958 |
2.354 |
8 |
| M-Δ-ScXL111 |
34 |
206 |
250 |
1.951 |
2.311 |
8 |
| M-Λ-ScXL111 |
34 |
199 |
244 |
1.954 |
2.337 |
8 |
| P-Δ-ScXL111 |
34 |
199 |
244 |
1.954 |
2.337 |
8 |
| P-Λ-ScXL111 |
35 |
206 |
251 |
1.951 |
2.311 |
8 |
| Δ-ScXL021 |
40 |
219 |
263 |
1.949 |
2.324 |
8 |
| Λ-ScXL021 |
39 |
218 |
262 |
1.948 |
2.324 |
8 |
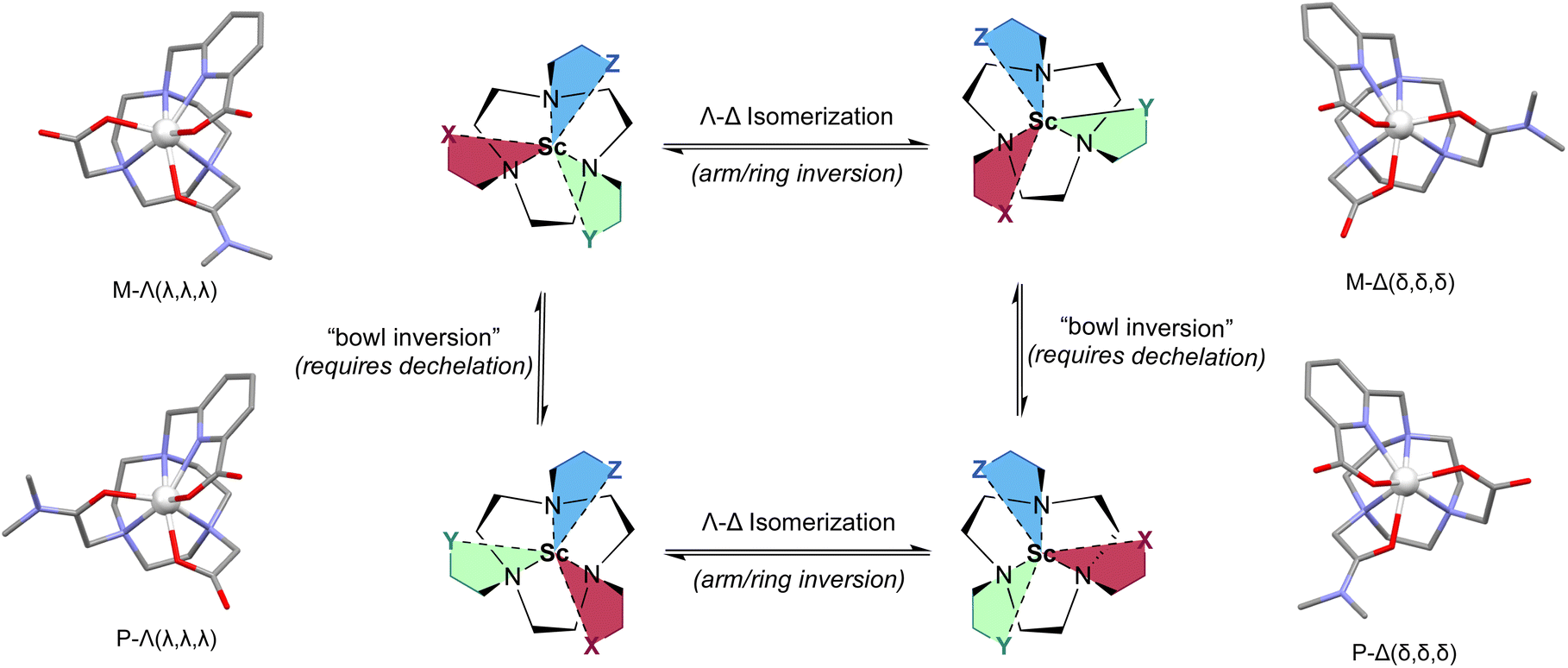
Finally, as the metal center acts as a chiral center, the formation of Δ- and Λ-configurational isomers can induce differential selectivity for the formation of the corresponding ternary complex; specifically, [ScF(L111)] provided different relative BDE and bond length values for the Δ- and Λ-isomers of the plus (P)- and minus (M)-isomeric forms. The formation of metallo-atropisomers is observed upon chelation with the achiral H2L111 due to the resulting three-dimensional structure of the complex. Fig. 5 depicts the resulting four diastereomers (combination of Δ/Λ- and P/M-isomers). To define the P- and M-isomers, the tacn-based ligand is depicted as having three different pendant arms: X, Y and Z with priority determined by the rules dictating traditional carbon-based R/S isomers. In this case the acetate pendant arm is given highest priority (X), the amide is given Y, and the picolinate is depicted as Z. Then following the pendant arms around the macrocycle X–Y–Z, a clockwise direction gives the P-isomer and counterclockwise gives the M-isomer. The complexity of the 1H NMR spectra for [Sc(L111)]+, ScF(L111), and [Lu(L111)]+ as well as the observation of multiple signals in their respective HPLC traces is likely a result of this high degree of isomerization (additional E- and Z-isomers of the amide functional group are also observed for all three complexes and the H2L111, see ESI, Fig. S25, S31 and S38†). Metallo-atropisomers are frequently observed with poly-functionalized azamacrocyclic chelates, however rapid interconversion of the respective isomers generally precludes isolation of the corresponding species.28
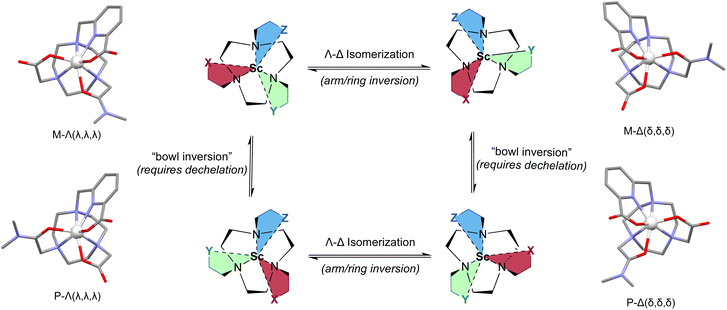 |
| | Fig. 5 Description and depiction of 4 distinct metallo-atropoisomers of [Sc(L111)]+ and their interconversion enthalpies/isomerization equilibria. Structures are depicted schematically (center), and corresponding [Sc(L111)]+ complexes (DFT-optimized) are shown adjacent to each isomer with isomeric descriptors. Specifically, M/P refers to directionality induced by the orientation of the arm substituents, whereas Δ- and Λ-refers to directionality of the arm substituents and the macrocycle induced by metal complexation. | |
Here, we hypothesize that interconversion between Δ- and Λ-isomers of Sc3+ complexes; given the calculated ±8 kJ mol−1 difference in energy observed using DFT between these isomers, a Sc3+-[L111]2- interaction energy of ∼−890 kJ mol−1 is likely too large to overcome for interconversion of P- and M-isomers. Thus, we hypothesized the all four metal-based isomers of [Sc(L111)]+, ScF(L111), and [Lu(L111)]+ would be observed using chiral separation approaches. Indeed, the corresponding characterization of [Sc(L111)]+ using chiral chromatography resolved 4 individual species (with a ratio of 35:14:34:17) and providing identical mass spectrometric signature (ESI, Fig. S23, Table S4†).
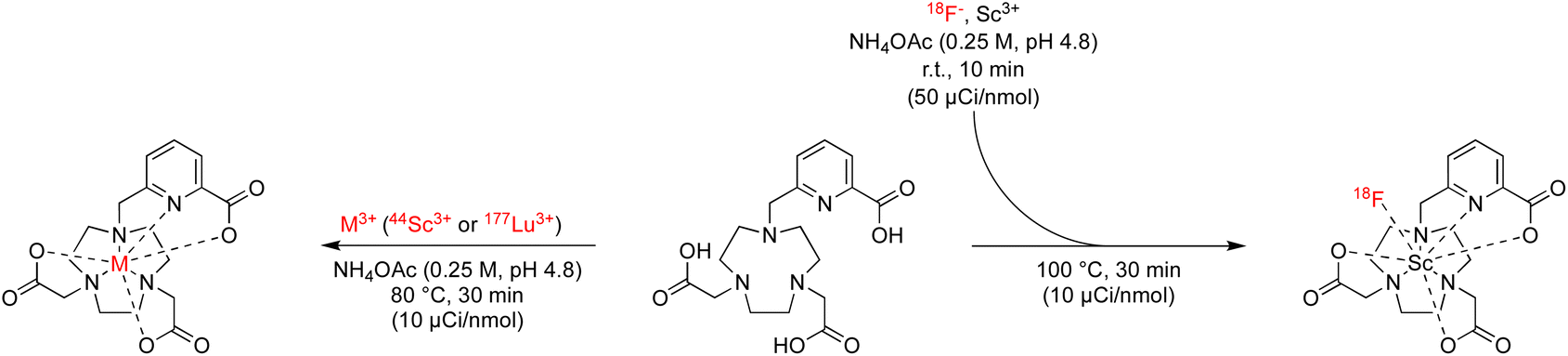
Radiochemical labeling studies and formulation stability
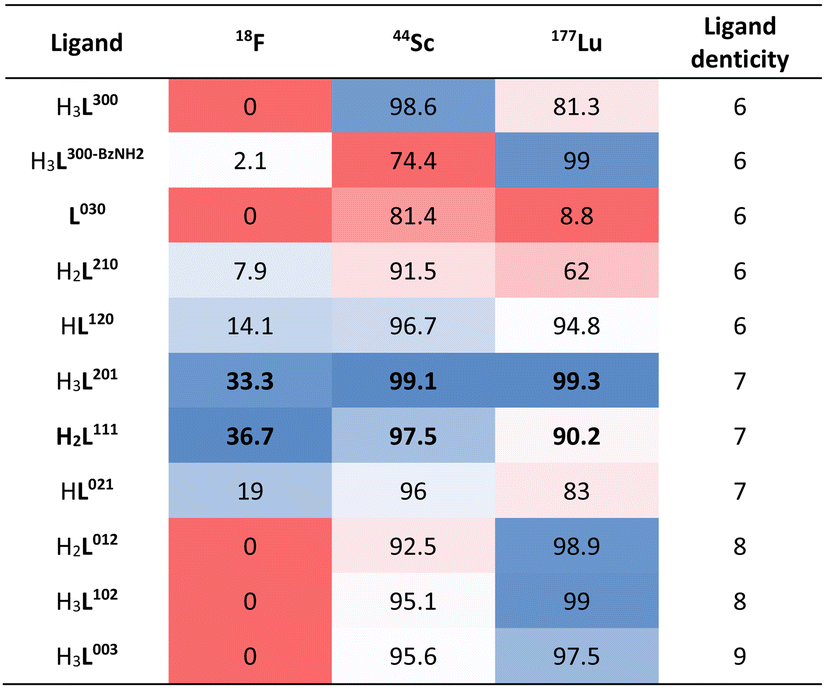
In accordance with our goal to identify coordinative structures that could efficiently incorporate and coordinatively stabilize the radioisotopes 18F, 44Sc and 177Lu, we conducted radiochemical labeling studies next. Following previously established radiochemical labelling approaches (Scheme 1),13 we radiofluorinated by pre-formation of the [18F][ScF]2+ species which was subsequently mixed with the ligand at 100 nmol mCi−1 molar activity and incubated at 80 °C for 30 min at pH 4.8 (1 M NH4Ac buffer). Radiochemical conversions were determined using iTLC or radioHPLC measurements (ESI, Fig. S24–S39†), and radiochemical purity and formulation stability affirmed using radioHPLC. Under these conditions the majority of 44Sc and 177Lu complexes form readily, with high radiochemical conversions and purity of 80% and above. The main discrepancies were observed among the two nuclides with respect their tolerance for amide donors and lower denticity ligand systems (Table 4). Lower ligand denticity and amide donation is generally better tolerated by Sc, which exhibits more covalent bonding character and smaller ionic radius, while Lu shows a preference for ligand systems with greater negative charge for charge equilibration of the trivalent metal cation. Large discrepancies in labeling efficiency were observed for 18F: specifically, 6-coordinate systems produced lower radiochemical conversions ranging from 0 to 14%, with 7-coordinate systems affording significantly higher conversions of 19–37%. Indeed, radiochemical conversions correlated well with computational data, which predicts strengthening of the Sc–F and weakened Sc–OH2 bond for [ScF(L201)]– and [ScF(L111)] systems. Poor charge equilibration and increased steric bulk likely limits attainable radiochemical conversions for [ScF(L021)]+. In accordance with our prediction that 8- and 9-coordinate ligand systems did not accommodate inner-sphere ternary ligands, no formation of the radiofluorinated product was observed for the for [Sc(L102)], [Sc(L012)]+ and [Sc(L003)] complexes. Complexes that could be efficiently separated using preparative HPLC and exhibited sufficient formulation stability upon reanalysis, were employed for subsequent in vivo experiments.
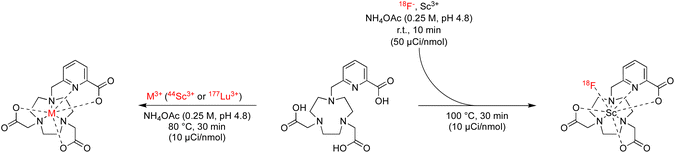 |
| | Scheme 1 Reaction scheme and conditions for radiochemical labeling conversion screen with 18F, 44Sc, 177Lu isotopes. | |
Table 4 Radiochemical labeling conversions at 100 nmol mCi−1 molar activity, 80 °C, 30 min at pH 4.8. Color coding indicates relative radiochemical conversion ranging from red (low relative conversion) to blue (high relative conversion). Columns are color coded independently from one another
In vivo biodistribution and metabolite analysis
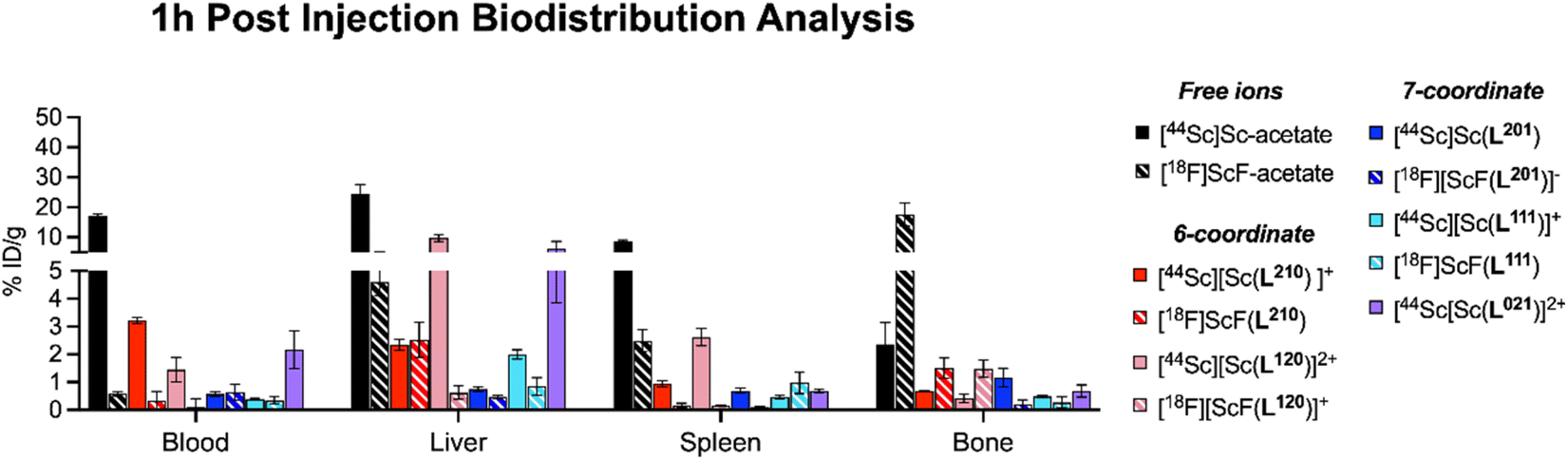
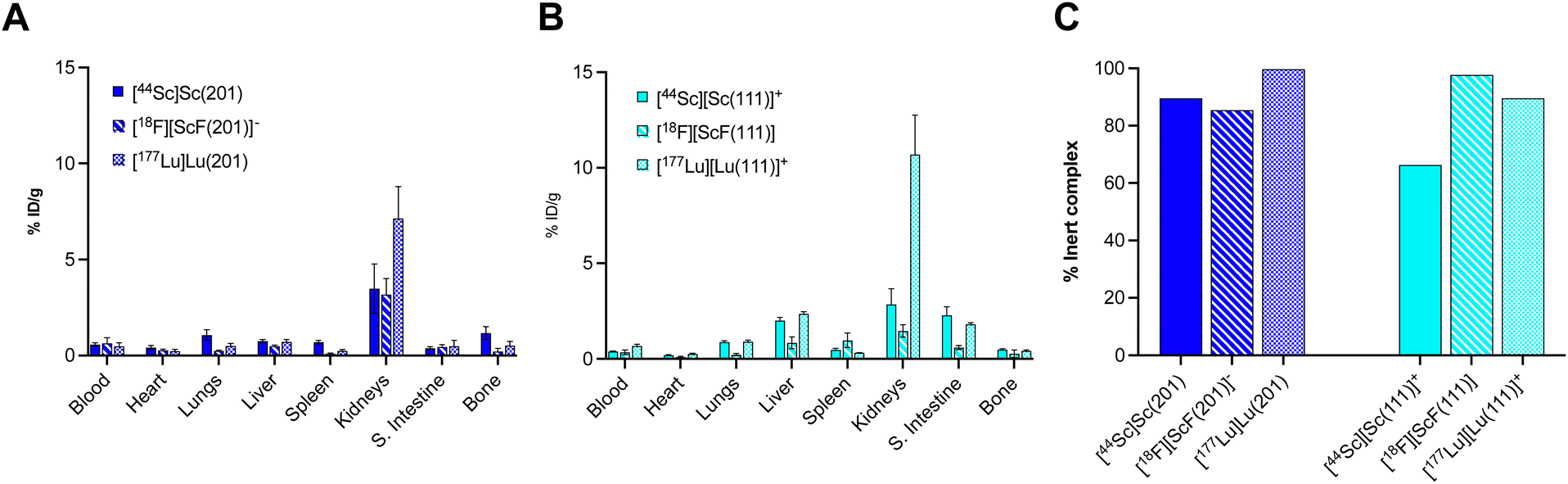
While in vitro stability studies can give some indication about the relative inertness of radiochelate complexes to plasma protein binding, transchelation and metal ion selectivity, in vivo biodistribution and urine metabolite analysis have proven the most reliable predictors of in vivo performance in our hands. Importantly, biodistribution profiles must be compared to those of the corresponding non-chelated isotopes to determine off-target deposition due to de- or transchelation. To this end, Sc18F and 44Sc complexes were synthesized, radio-chromatographically separated, and reformulated in phosphate buffered saline (PBS) prior for injection. Formulated complexes were injected intravenously into cohorts of balb/C naïve mice, ex vivo biodistribution and urine metabolite analysis was conducted 1 hour post injection, taking into consideration the short circulation time of small molecular complexes. Detailed biodistribution analysis of all organs can be found within the ESI (Tables S4–S9†). A graphical summary of a key set of organs (blood, liver, spleen, bone) is shown graphically in Fig. 6, together with comparative data sets of the corresponding free ion distributions of Sc3+ and F−. Our control data sets show pronounced blood, liver and splenic uptake as typical hallmarks of Sc3+ release, while elevated bone uptake is characteristic of labile F−. Among the tested chelate systems, the 6-coordinate donor environment resulted in biodistribution profiles indicative of dechelation, while 7-coordinate system exhibited improved inertness, with low deposition of radioactivity across all relevant off-target organs. Of note, attempts to reformulate [18F][ScF(L021)]+ in PBS following chromatographic separation were not successful, indicating diminished complex inertness. Based on the biodistribution screen, H3L201 and H2L111 remained of interest for further investigation. Subsequently, we formed the corresponding [177Lu][Lu(L201)] and [177Lu][Lu(L111)]+ complexes and conducted matching biodistribution analysis. Results indicate good correlation of 18F, 44Sc and 177Lu profiles (Fig. 7), evident for most organs except the kidneys, which is elevated for both 177Lu complexes. The reason for this behavior is unclear, considering the corresponding 44Sc complex exhibits identical chemical properties. Elevated liver and intestinal uptake for the H2L111 series are a consequence of the increased lipophilicity compared to the H3L201 series, as the metabolite analysis does not exhibit significantly diminished inertness of the rare earth complexes when compared across compound series. Of note, the two neutral complexes [177Lu][Lu(L201)] and [18F][ScF(L111)] exhibit highest inertness in isolated urine metabolites, indicating that interaction of cationic or anionic complexes with blood serum proteins may induce accelerated destabilization. For both complex series, inertness remained favorable and biodistribution profiles indicated no significant off target accumulation, motivating future investigation of conjugate systems to determine the chelators' suitability of targeted imaging and therapy applications.
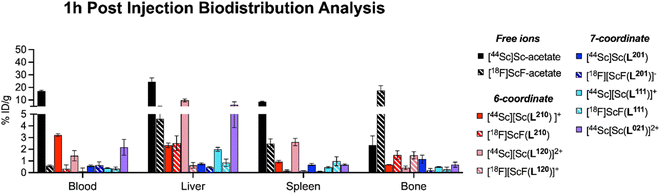 |
| | Fig. 6 Summary of 1 h post injection biodistribution analysis of Sc18F and 44Sc complexes in direct comparison with the distribution profile of corresponding free ion species in key organs blood, liver, spleen and bone indicate that 7-coordinate chelates result in improved complex inertness and a more favorable in vivo distribution profile. | |
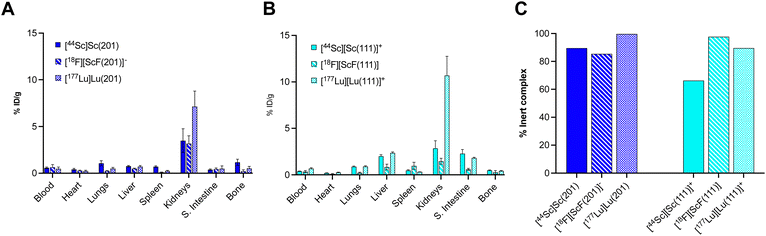 |
| | Fig. 7 (A) Biodistribution analysis 1 hour post injection of [Sc(L201)], [ScF(L201)]– and [Lu(L201)]. (B) Biodistribution analysis 1 hour post injection of [Sc(L111)]+, [ScF(L111)] and [Lu(L111)]+. (C) Quantitation of intact metabolite in the urine for each investigated species as determined by radioHPLC analysis. | |
pH-dependent speciation for lead chelates
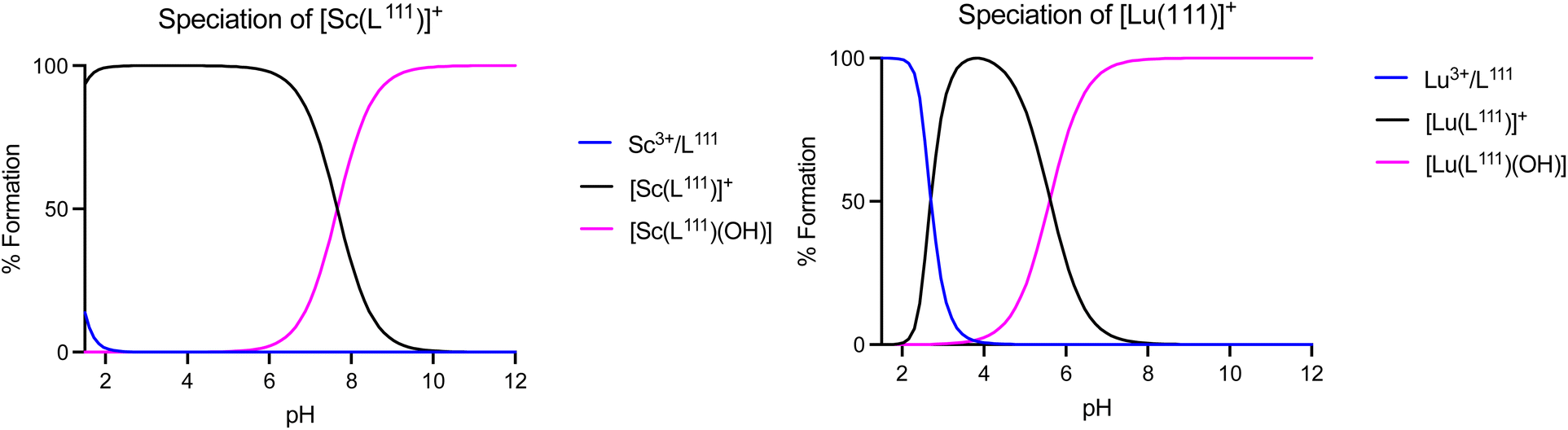
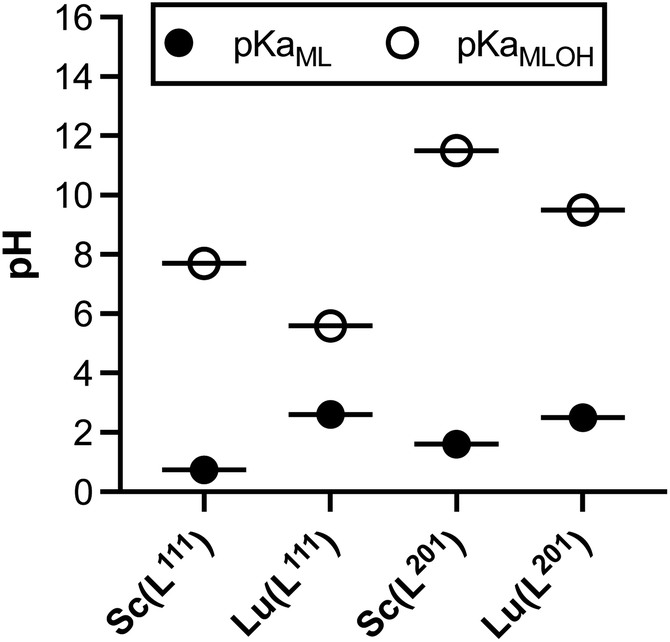
To further compare and contrast the pH dependent speciation of Sc3+ and Lu3+ complexes with H3L201 and H2L111, spectrophotometric and NMR-assisted speciation studies were performed. Specifically, ligand and complex samples were prepared at 23 different pH values (2–12). Corresponding 1H and UV-visible spectra were acquired and data was processed using multiparameter fitting with HypSpec and HypNMR programs to determine pH dependent complex speciation, and complex pKa, logKML and pM values (Fig. 8, Tables S10–S11†). Our data indicates that the difference in ligand charge lowers the pKa of formation of the [M(OH)(L111)] when compared with [MOH(L201)]–. Fig. 9 compares the speciation of the H2L111 complex series with the previously characterized H3L201 complexes and identifies the following trends: formation of Sc complexes occurs at lower pH in both instances, whereas the formation of the MOH(Lxxx) species requires lower OH− concentrations for Lu complexes. This correlates inversely with the reported ionic hardness values for the individual M3+ ions.29
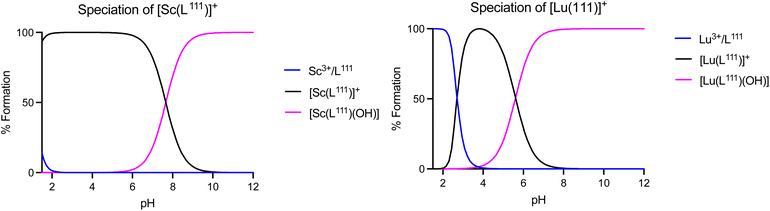 |
| | Fig. 8 Speciation plots of [Sc(L111)]+ (top), [Lu(L111)]+ (bottom left). | |
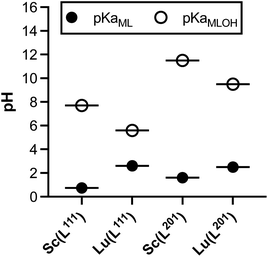 |
| | Fig. 9 Summary of pKa values obtained for the M(L111) series in direct comparison with the M(L201) complexes, M = Sc, Lu. | |
The acquired speciation data helps rationalize biodistribution and stability study results: specifically, the speciation data indicates likely prevalence of [M(OH)(L111)], and not the aqua ion [MOH2(L111)]+ (for M = Sc, Lu) under physiologically relevant conditions. For both metals, the OH− complex is more prevalent at physiological pH when complexed with H2L111 in comparison to H3L201. This contrasts with the [M(L201)] complexes, where the hydroxide species remains negligible at physiological pH and indicates that OH− complex species are biocompatible and can remain inert in vivo.
Conclusions
Here, we have employed a variety of characterization methods to explore the coordinative and chemical reactivity landscape of ten triaza-macrocycle chelates in the context of the 18F–Sc/44Sc/177Lu triad. In this study density functional theory, convergent chemical synthesis, heteronuclear NMR spectroscopy, radiochemical screening, vivo biodistribution, metabolite analysis, and pH-dependent speciation were utilized. In accordance with our hypothesis, 7-coordinate chelators are ideally suited to form F–Sc/Sc/Lu coordination complexes. Solution coordination number can be affirmed by 45Sc NMR spectroscopy, with corresponding complexes exhibiting a characteristic chemical shift of 65–75 ppm in MeOH. Charge equilibration is paramount, to stabilize all three complex-types; as such, complexes outside of the −1 to +1 charge range were not sufficiently inert in solution. All complexes form Δ- and Λ-enantiomers, with H2L111forming additional M- and P- forms, producing 4 distinct isomers consisting of two diastereomeric pairs of Δ- and Λ-enantiomers. While radiochemical labelling was feasible (and in some instances high-yielding) with 6- and 7-coordinate chelators, 6-coordinative complexes exhibited limited in vivo stability as evidenced by enhanced blood retention of the 44Sc complexes and bone-deposition of 18F. In vivo analysis identifies complexes of H3L201 and H2L111 as most ideally suited for the future construction of bifunctional derivatives; pH dependent speciation indicates that both ligands form neutral, charge-balanced [M(L201)] and [M(OH)(L111)] species respectively between pH 6–8. This work serves as a blueprint for the design of bifunctional chelators compatible with 18F/44Sc/177Lu isotopes, providing clear spectroscopic, radiochemical labeling and biological performance benchmarks.
Data availability
Crystallographic data for [Sc(L003)]·3H2O been deposited at the CCDC under 2369298. The datasets supporting this article have been uploaded as part of the ESI† specifically, we provide processed NMR spectroscopic information, chromatographic analysis spectra, UV-Vis spectra and tabulated biodistribution and stability analysis data as an average of single cohorts of animals as indicated. Additional, raw data is available upon request to the corresponding author.
Author contributions
CAAK, OMG and EB designed all experimental approaches and wrote the manuscript. CAAK, OMG, JNW conducted radiochemical labeling and animal experimentation. KMS and IAG conducted crystallographic data acquisition and analysis. TEB, EAS, JCM, JWE conducted isotope production and separation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Dr Heike Hofstetter is acknowledged for her support in establishing various heteronuclear NMR pulse programs and acquisition methods. E. B. and J. W. E. acknowledge the National Institutes of Bioimaging and Bioengineering (R01EB032349) and the Department of Energy (0000262368) for funding support of this work. The following instrumentation in the Paul Bender Chemical Instrumentation Center at the University of Wisconsin-Madison was supported by: NSF CHE-2017891 (Bruker Avance Neo 500); by a generous gift from Paul J. and Margaret M. Bender (Bruker Quazar APEX2 and Bruker 500); NIH S10 OD012245 (Bruker Avance-600). The Bruker D8 VENTURE Photon III X-ray diffractometer was partially funded by NSF Award #CHE-1919350 to the UW-Madison Department of Chemistry and the Electrospray Ionization-Quadrupole-Ion Trap Mass Spectrometer was funded by NIH 1S10 OD020022.
References
- A. C. Civelek, Reawakening of Nuclear Medicine through Molecular Imaging: Quantitative Theranostics and PSMA PET, Radiology, 2023, 307, e230627 CrossRef PubMed.
- N. Drude, L. Tienken and F. M. Mottaghy, Theranostic and nanotheranostic probes in nuclear medicine, Methods, 2017, 130, 14–22 CrossRef CAS PubMed.
- X. Deng, J. Rong, L. Wang, N. Vasdev, L. Zhang, L. Josephson and S. H. Liang, Chemistry for positron emission tomography: recent advances in 11C-, 18F-, 13N-, and 15O-labeling reactions, Angew. Chem., Int. Ed., 2019, 58, 2580–2605 CrossRef CAS PubMed.
- S. Richter, M. Wuest, C. N. Bergman, J. D. Way, S. Krieger, B. E. Rogers and F. Wuest, Rerouting the metabolic pathway of 18F-labeled peptides: the influence of prosthetic groups, Bioconjugate Chem., 2015, 26, 201–212 CrossRef CAS PubMed.
- V. Bernard-Gauthier, J. J. Bailey, Z. Liu, B. r. Wängler, C. Wängler, K. Jurkschat, D. M. Perrin and R. Schirrmacher, From unorthodox to established: The current status of 18F-trifluoroborate-and 18F-SiFA-based radiopharmaceuticals in PET nuclear imaging, Bioconjugate Chem., 2016, 27, 267–279 CrossRef CAS PubMed.
- M. L. Lepage, H. T. Kuo, Á. Roxin, S. Huh, Z. Zhang, R. Kandasamy, H. Merkens, J. O. Kumlin, A. Limoges, S. K. Zeisler, K. S. Lin, F. Bénard and D. M. Perrin, Toward 18 F-Labeled Theranostics: A Single Agent that Can Be Labeled with 18F, 64Cu, or 177Lu, ChemBioChem, 2019, 20, cbic.201900632 Search PubMed.
- W. J. McBride, R. M. Sharkey and D. M. Goldenberg, Radiofluorination using aluminum-fluoride (Al18F), EJNMMI Res., 2013, 3, 1–11 CrossRef PubMed.
- C. Fersing, A. Bouhlel, C. Cantelli, P. Garrigue, V. Lisowski and B. Guillet, A comprehensive review of non-covalent radiofluorination approaches using aluminum [18F] fluoride: Will [18F] AlF replace 68Ga for metal chelate labeling?, Molecules, 2019, 24, 2866 CrossRef CAS PubMed.
- E. W. Price and C. Orvig, Matching chelators to radiometals for radiopharmaceuticals, Chem. Soc. Rev., 2014, 43, 260–290 RSC.
- N. P. van der Meulen, M. Bunka, K. A. Domnanich, C. Muller, S. Haller, C. Vermeulen, A. Turler and R. Schibli, Cyclotron production of 44Sc: From bench to bedside, Nucl. Med. Biol., 2015, 42, 745–751 CrossRef CAS PubMed.
- C. Muller, M. Bunka, S. Haller, U. Koester, V. Groehn, P. Bernhardt, N. van der Meulen, A. Tuerler and R. Schibli, Promising prospects for 44Sc-/47Sc-based theragnostics: application of 47Sc for radionuclide tumor therapy in mice, J. Nucl. Med., 2014, 55, 1658–1664 CrossRef CAS PubMed.
- K. A. Domnanich, C. Müller, M. Benešová, R. Dressler, S. Haller, U. Köster, B. Ponsard, R. Schibli, A. Türler and N. P. van der Meulen,
47Sc as useful β–-emitter for the radiotheragnostic paradigm: a comparative study of feasible production routes, EJNMMI Radiopharm. Chem., 2017, 2, 5 CrossRef PubMed.
- J. N. Whetter, B. A. Vaughn, A. J. Koller and E. Boros, An Unusual Pair: Facile Formation and In Vivo Validation of Robust Sc–18F Ternary Complexes for Molecular Imaging, Angew. Chem., Int. Ed., 2022, 61, e202114203 CrossRef CAS PubMed.
- G. Nagy, D. Szikra, G. Trencsényi, A. Fekete, I. Garai, A. M. Giani, R. Negri, N. Masciocchi, A. Maiocchi, F. Uggeri, I. Tóth, S. Aime, G. Giovenzana and Z. Baranyai, AAZTA: an ideal chelating agent for the development of 44Sc PET imaging agents, Angew. Chem., Int. Ed., 2017, 56, 2118–2122 CrossRef CAS PubMed.
- C. Gateau, M. Mazzanti, J. Pécaut, F. A. Dunand and L. Helm, Solid-state and solution properties of the lanthanide complexes of a new nonadentate tripodal ligand derived from 1, 4, 7-triazacyclononane, Dalton Trans., 2003, 2428–2433 RSC.
- G. N. Nocton, A. Nonat, G. Christelle and M. Marinella, Water Stability and Luminescence of Lanthanide Complexes of Tripodal Ligands Derived from 1,4,7-Triazacyclononane: Pyridinecarboxamide versus Pyridinecarboxylate Donors, Helv. Chim. Acta, 2009, 92, 2257–2273 CrossRef CAS.
- K. Mason, A. C. Harnden, C. W. Patrick, A. W. J. Poh, A. S. Batsanov, E. A. Suturina, M. Vonci, E. J. L. McInnes, N. F. Chilton and D. Parker, Exquisite sensitivity of the ligand field to solvation and donor polarisability in coordinatively saturated lanthanide complexes, Chem. Commun., 2018, 54, 8486–8489 RSC.
- R. Bhalla, W. Levason, S. K. Luthra, G. McRobbie, G. Sanderson and G. Reid, Radiofluorination of a pre-formed gallium (III) aza-macrocyclic complex: towards next-generation positron emission tomography (PET) imaging agents, Chem.–Eur. J., 2015, 21, 4688 CrossRef CAS PubMed.
- N. Kim, C.-H. Hsieh and J. F. Stebbins, Scandium Coordination in Solid Oxides and Stabilized Zirconia: 45Sc NMR, Chem. Mater., 2006, 18, 3855–3859 CrossRef CAS.
- A. J. Rossini and R. W. Schurko, Experimental and Theoretical Studies of 45Sc NMR Interactions in Solids, J. Am. Chem. Soc., 2006, 128, 10391–10402 CrossRef CAS PubMed.
- A. Jaworski, T. Charpentier, B. Stevensson and M. Edén, Scandium and Yttrium Environments in Aluminosilicate Glasses Unveiled by 45Sc/89Y NMR Spectroscopy and DFT Calculations: What Structural Factors Dictate the Chemical Shifts?, J. Phys. Chem. C, 2017, 121, 18815–18829 CrossRef CAS.
- E. Curnock, W. Levason, M. E. Light, S. K. Luthra, G. McRobbie, F. M. Monzittu, G. Reid and R. N. Williams, Group 3 metal trihalide complexes with neutral N-donor ligands – exploring their affinity towards fluoride, Dalton Trans., 2018, 47, 6059–6068 RSC.
- A. Hu, M. E. Simms, V. Kertesz, J. J. Wilson and N. A. Thiele, Chelating rare-earth metals (Ln3+) and 225Ac3+ with the dual-size-selective macrocyclic ligand Py2-macrodipa, Inorg. Chem., 2022, 61, 12847–12855 CrossRef CAS PubMed.
- A. Hu, V. Brown, S. N. MacMillan, V. Radchenko, H. Yang, L. Wharton, C. F. Ramogida and J. J. Wilson, Chelating the alpha therapy radionuclides 225Ac3+ and 213Bi3+ with 18-membered macrocyclic ligands macrodipa and Py-macrodipa, Inorg. Chem., 2021, 61, 801–806 CrossRef PubMed.
- S. Aime, M. Botta, M. Fasano, M. P. M. Marques, C. F. Geraldes, D. Pubanz and A. E. Merbach, Conformational and coordination equilibria on DOTA complexes of lanthanide metal ions in aqueous solution studied by 1H-NMR spectroscopy, Inorg. Chem., 1997, 36, 2059–2068 CrossRef CAS PubMed.
- S. F. Yuk, I. Sargin, N. Meyer, J. T. Krogel, S. P. Beckman and V. R. Cooper, Putting error bars on density functional theory, Sci. Rep., 2024, 14, 20219 CrossRef CAS PubMed.
- N. Mardirossian and M. Head-Gordon, Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals, Mol. Phys., 2017, 115, 2315–2372 CrossRef CAS.
- A. Marlin, A. Koller, E. Madarasi, M. Cordier, D. Esteban-Gómez, C. Platas-Iglesias, G. Tircsó, E. Boros, V. Patinec and R. Tripier, H3 nota Derivatives Possessing Picolyl and Picolinate Pendants for Ga3+ Coordination and 67Ga3+ Radiolabeling, Inorg. Chem., 2023, 62, 20634–20645 CrossRef CAS PubMed.
- R. G. Parr and R. G. Pearson, Absolute hardness: companion parameter to absolute electronegativity, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Owen M.
Glaser
ab,
Jennifer N.
Whetter
ab,
Eduardo
Aluicio-Sarduy
c,
Jason C.
Mixdorf
c,
Kyana M.
Sanders
ab,
Owen M.
Glaser
ab,
Jennifer N.
Whetter
ab,
Eduardo
Aluicio-Sarduy
c,
Jason C.
Mixdorf
c,
Kyana M.
Sanders