
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePharmacoNet: deep learning-guided pharmacophore modeling for ultra-large-scale virtual screening†
Seonghwan
Seo
 a and
Woo Youn
Kim
*abc
a and
Woo Youn
Kim
*abc
aDepartment of Chemistry, KAIST, 291 Daehak-ro, Yuseong-gu, Daejeon, 34141, Republic of Korea. E-mail: wooyoun@kaist.ac.kr
bGraduate School of Data Science, KAIST, 291 Daehak-ro, Yuseong-gu, Daejeon, 34141, Republic of Korea
cHITS Inc., 28 Teheran-ro 4-gil, Gangnam-gu, Seoul, 06234, Republic of Korea
First published on 4th November 2024
Abstract
As ultra-large-scale virtual screening becomes critical for early-stage drug discovery, highly efficient screening methods are gaining prominence. Deep-learning-based approaches which directly estimate binding affinities without binding conformation have attracted great attention as an alternative solution to molecular docking, but the generalization capability of existing methods in vast chemical space remains uncertain due to restricted training data. Here, we introduce PharmacoNet, the first deep-learning framework for pharmacophore modeling toward ultra-fast virtual screening. PharmacoNet offers fully automated protein-based pharmacophore modeling and evaluates the potency of ligands with a parameterized analytical scoring function, ensuring high generalization ability across unseen targets and ligands. Our benchmark study shows that PharmacoNet is extremely fast yet reasonably accurate compared to traditional docking methods and existing deep learning-based scoring models. We successfully identified selective inhibitors from 187 million compounds against cannabinoid receptors within 21 hours on a single CPU. This study uncovers the hitherto untapped potential of deep learning in pharmacophore modeling.
1 Introduction
Discovering new drug candidates often requires exploring a vast chemical space that can be accessible through ultra-large chemical libraries.1–4 The expansion of library sizes from millions to billions of molecules has significantly enhanced a hit rate, suggesting a paradigm shift towards ultra-large-scale virtual screening as a cornerstone of early drug discovery efforts.5 However, molecular docking, a fundamental evaluation strategy in virtual screening, takes seconds to minutes to evaluate each molecule.6,7 Consequently, the effective screening of such large volumes entails practical challenges due to the intense computational cost of molecular docking.8This bottleneck has spurred the development of innovative strategies to streamline the screening process, primarily focusing on efficient molecule exploration and pre-screening strategies. The former, including structured library searches, Bayesian searches, and active learning algorithms, concentrate on promising chemical subsets to boost screening efficiency.5,9 Meanwhile, the latter involves a tiered approach, whereby the whole library is preliminarily evaluated with a rapid assessment tool to prioritize promising candidates for an accurate and resource-intensive fine-screening assessment.10,11 This brute-force approach can uncover challenging inhibitors, such as selective inhibitors against multiple targets found in extremely sparse regions of chemical space. However, such pre-screening with low reliability is likely to overlook promising hits, highlighting the need for a method that balances accuracy and speed.
The pre-screening approach's overarching goal is to dramatically enhance computational efficiency, for example, thousands of fold speedups on standard computing setups, while preserving the accuracy necessary for meaningful virtual screening. To circumvent the prohibitive computational costs associated with molecular docking, recent endeavors have explored docking-free deep learning (DL) techniques that do not rely on protein–ligand binding conformations.11 These methods have demonstrated remarkable speed and even surpassed the performance of structure-based methodologies, which use binding conformations, in some benchmarking tests.12 Nevertheless, the limited diversity of experimental datasets, such as the PDBbind13 database comprising only 4200 unique ligand molecules apart from common biomolecules (e.g., ATP, GTP), constrains the generalization ability and scalability of these methods.14 They often memorize a structural bias in the training set rather than learn the desirable patterns of protein–ligand interaction (PLI), which impedes their reliability in evaluating vast chemical spaces.15,16
To address these limitations, the PLI prediction can be reformulated by abstracting the detailed topologies of protein–ligand binding to a pharmacophore level. By using the pharmacophore‡ information, one can focus on essential non-covalent interactions (NCIs) instead of numerous atom-pairwise interactions.18,19 This pharmacophore-level abstraction allows for rapid yet reliable evaluations that are difficult to achieve with traditional atomistic computational methods. However, traditional pharmacophore modeling methods often rely on the binding conformation of active molecules or manual processes by experts,20 which can be less adaptable to new targets or protein structures predicted by AlphaFold21 and RoseTTAFold.22,23 As a result, an automated protein-based pharmacophore modeling method that relies solely on protein structures is needed.
Here we present PharmacoNet, the first deep-learning framework for protein-based pharmacophore modeling, as schematically described in Fig. 1. PharmacoNet introduces instance segmentation DL modeling to automate the identification of critical protein functional groups (hotspots) and optimal locations of corresponding pharmacophore points to construct a pharmacophore model. PharmacoNet then incorporates a parameterized analytic function to evaluate the ligand by algorithmically calculating the compatibility with the pharmacophore at the NCI level. Our approach considerably reduces computational demands while preserving reasonable accuracy by shifting the focus from atomistic to pharmacophoric interaction. Furthermore, this coarse-grained evaluation method avoids the over-fitting inherent in deep learning models with excessive parameters, ensuring its reliability and generalization across diverse chemical spaces.
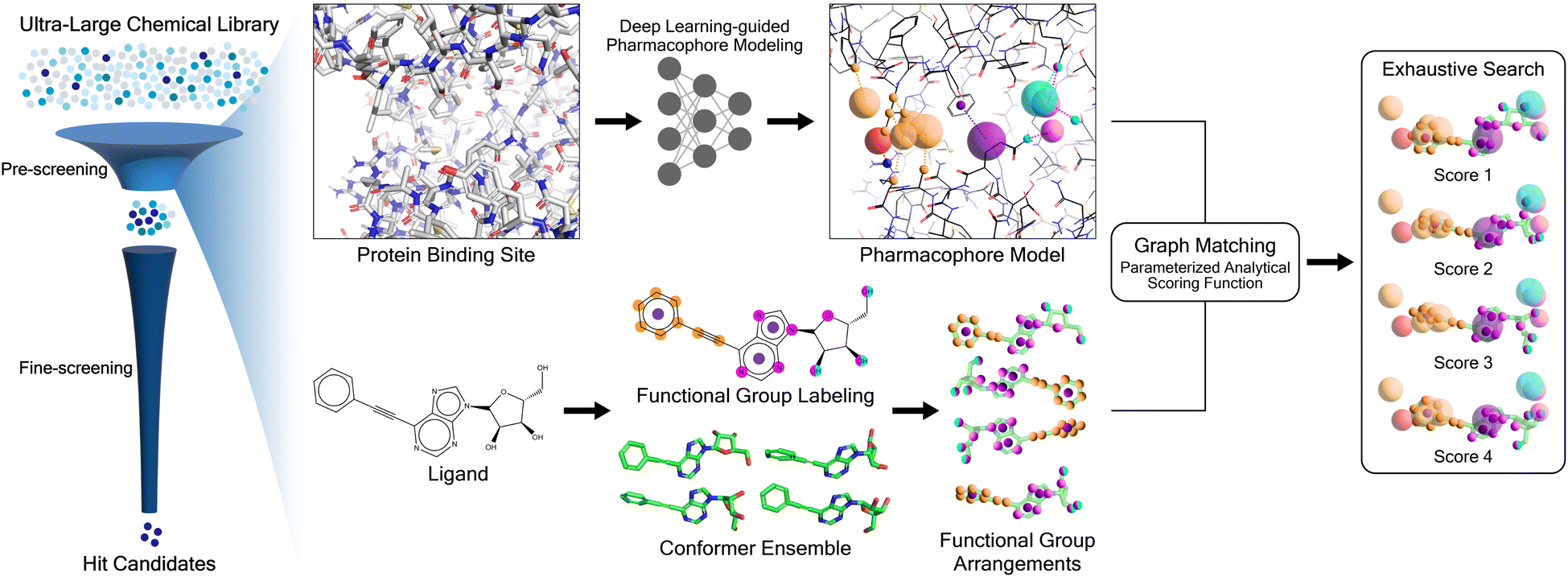 | ||
| Fig. 1 Overview of PharmacoNet. PharmacoNet comprises (1) a fully automated deep learning-based pharmacophore modeling from protein structure and (2) ligand evaluation for virtual screening. Pharmacophore modeling is performed only once before an actual virtual screening process. | ||
PharmacoNet is extremely fast yet reasonably accurate, achieving 3000-fold speedups while maintaining competitive performance against standard docking methods such as AutoDock Vina24 in virtual screening benchmarks. Furthermore, PharmacoNet evaluated 187 million molecules to discover the potential cannabinoid (CB) antagonist candidates with both potency and CB2/CB1 selectivity within 21 hours (11 years for AutoDock Vina) on a desktop computer with a single 32-core CPU. These results demonstrate the feasibility of PharmacoNet in accelerating drug discovery by enabling the rapid screening of vast chemical libraries. In addition, PharmacoNet provides a comprehensive graphical user interface (GUI) software, OpenPharmaco, designed to facilitate the use of protein-based pharmacophore modeling and high-throughput virtual screening by a broad range of users, including those without computational resources and expertise.
2 Results
2.1 PharmacoNet framework
PharmacoNet comprises three stages: (1) DL-based pharmacophore modeling, (2) coarse-grained graph matching, and (3) distance likelihood-based scoring, as illustrated in Fig. 1. First, the PharmacoNet constructs the pharmacophore model using only the structural information of a target protein binding site by determining the hotspots and optimal locations (pharmacophore point) for ligand functional groups to form stable NCIs with each hotspot. Then, the graph-matching algorithm effectively estimates the spatial relationship between ligands and the pharmacophore model. This pharmacophore-level prediction requires significantly less computation than the corresponding atomistic prediction. Finally, the scoring function gives the binding affinity of each pose with reasonable accuracy and high generalization ability thanks to the pharmacophore-level abstraction of PLIs.In PharmacoNet, deep learning is utilized to model the distribution of NCIs within a given protein binding site structure. Through instance segmentation modeling, the neural network first identifies protein interaction sites, known as protein hotspots. It then returns a spatial density map of ligand interaction sites corresponding to each identified protein hotspot. Consequently, our deep neural network constructs a protein-based pharmacophore model from the data distribution of the crystal structure dataset. In contrast, other protein-based pharmacophore modeling methods rely on biased methodologies. For example, Apo2ph4, proposed by Heider et al.,25 estimates key interactions based on molecular docking results of fragments rather than the crystal structure distribution. Similarly, PharmRL26 infers the spatial density map of ligand interaction sites from data distribution, but its hotspot detection process is trained using reinforcement learning to maximize the screening powers for the DUD-E benchmark.
2.2 Benchmark study for virtual screening
We performed a series of benchmark studies to validate our framework as an efficient pre-screening tool. We first adopted three widely used commercial molecular docking programs (GOLD,27 LeDock,28 and GLIDE SP29) and two popular open-source programs (AutoDock Vina24 and Smina30) as baselines to assess the accuracy and speed of virtual screening. In addition, we considered a DL-based docking method (KarmaDock6), sequence-based docking-free DL methods (TransformerCPI,31 PLAPT32) and structure-based docking-free DL methods (DeepBindGCN,11 and TANKBind12).We evaluated the screening power of each method on the standard virtual screening benchmark: Demanding Evaluation Kits for Objective In silico Screening 2.0 (DEKOIS2.0)33 We used the average top α% enrichment factor (EFα%), the average area under the receiver operating characteristic curve (AUROC), Boltzmann-enhanced discrimination of the receiver operating characteristic (BEDROC, α = 80.5), and the area under the precision–recall curve (PRAUC) as metrics to evaluate the screening power. For all metrics, higher is better. For the speed benchmark, we compared the average runtimes of PharmacoNet against those of the docking programs on both the PDBbind core set34 and the refined set when the initial conformers of each ligand were provided. For all benchmark tests, we considered 8 conformers for each ligand for a fair comparison with AutoDock Vina and Smina, which perform pose searches with a default exhaustiveness of 8.
However, widely used screening benchmark sets, such as DUD-E35 or DEKOIS2.0,33 may not reflect real-world screening scenarios since they are derived from decoys rather than experimentally confirmed inactive molecules. Therefore, we also employed the unbiased screening benchmark LIT-PCBA36 which mimics the experimental screening by constructing the true actives and inactives from PubChem bioassays37 and adjusting the active/inactive ratio. In particular, LIT-PCBA removes the structural bias of ligand libraries, allowing for more rigorous evaluation of ML methodologies. In this study, we included the state-of-the-art DL-based docking method (KarmaDock) and various conventional docking tools (GLIDE, Smina, AutoDock Vina). We also compared PharmacoNet to two automated protein-based pharmacophore modeling approaches, Apo2ph4-Pharmit§25,38 and PharmRL.26 Since both Apo2ph4-Pharmit and PharmRL perform classification rather than quantification, we reported EF instead of EFα%.
As shown in Fig. 2A and B, PharmacoNet was much faster than the conventional docking programs. Compared to AutoDock Vina, the fastest docking software in this benchmark study, PharmacoNet was 3956 and 3483 times faster on the core set and the refined set, respectively. Against GLIDE SP, the most accurate docking software in this benchmark study, PharmacoNet was 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 117 and 27731 times faster for the core set and the refined set, respectively. PharmacoNet's speed gain becomes more pronounced for larger molecules with over 60 heavy atoms in the refined set, where it outperformed AutoDock Vina and GLIDE SP by 7256 and 35474 times, respectively. Specifically, PharmacoNet evaluated the large molecules with 70 heavy atoms in an average of 5.15 (ms), whereas AutoDock Vina took 208 ms even for a simple benzene molecule (PDB ID 4w5z). This remarkable efficiency arises from PharmacoNet's unique strategic focus on evaluating NCIs, bypassing computationally intensive atom-pairwise interactions.
117 and 27731 times faster for the core set and the refined set, respectively. PharmacoNet's speed gain becomes more pronounced for larger molecules with over 60 heavy atoms in the refined set, where it outperformed AutoDock Vina and GLIDE SP by 7256 and 35474 times, respectively. Specifically, PharmacoNet evaluated the large molecules with 70 heavy atoms in an average of 5.15 (ms), whereas AutoDock Vina took 208 ms even for a simple benzene molecule (PDB ID 4w5z). This remarkable efficiency arises from PharmacoNet's unique strategic focus on evaluating NCIs, bypassing computationally intensive atom-pairwise interactions.
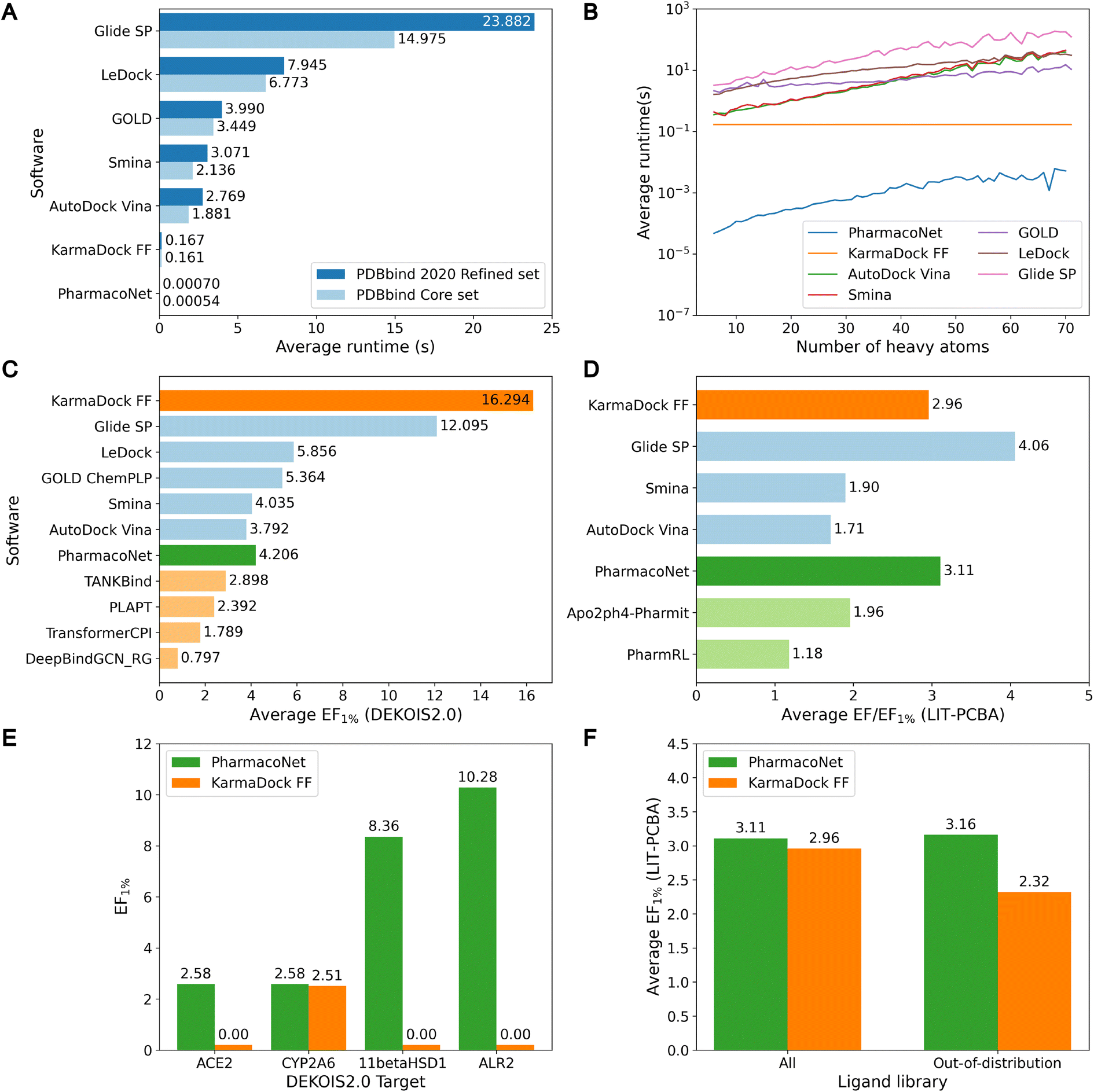 | ||
| Fig. 2 The speed and screening power of PharmacoNet and baseline models. (A) Average runtime of various ligand evaluation software on the PDBbind core set and the PDBbind 2020 refined set. The runtime of PharmacoNet, AutoDock Vina, and Smina is measured on a single 32-core Intel Xeon Gold 6326 CPU @ 2.90 GHz, that of GOLD, LeDock, and Glide SP is measured on a single 48-core Intel Xeon Gold 6240R CPUs @ 2.40 GHz, and that of KarmaDock is measured on a NVIDIA A4000. The runtime of KarmaDock is the sum of the data processing and GPU model runtimes. (B) Average runtime according to the number of heavy atoms on the PDBbind 2020 refined set. (C and D) Average screening powers (EF1%) for conventional docking softwares (blue), docking-free DL scoring methods (light orange), DL-based docking method (deep orange), pharmacophore modeling-based methods (light green), and PharmacoNet (deep green). (E) Screening powers on out-of-distribution targets in DEKOIS2.0. (F) Average screening powers on out-of-distribution ligands in LIT-PCBA. | ||
Despite PharmacoNet's ultrafast speed, its screening power was acceptable for virtual screening, as shown in Fig. 2C, D and S3.† It outperformed AutoDock Vina and closely competed with Smina on the DEKOIS2.0 and LIT-PCBA benchmarks. This desirable balance between accuracy and speed highlights the utility of PharmacoNet as a pre-screening tool in the high-throughput virtual screening of ultra-large chemical libraries to retain promising candidates for additional fine screening.
Notably, PharmacoNet surpassed existing protein-based pharmacophore modeling methods, Apo2ph4-Pharmit and PharmRL, on the LIT-PCBA benchmark. Apo2ph4 constructs pharmacophore models from the docking structures of numerous fragments instead of the crystal structure, restricting its performance to docking. PharmRL is trained on the DUD-E screening benchmark,35 but it is well-known that the inherent structural biases hinder the generalization.39 In contrast, PharmacoNet is free from these issues, as it is trained solely on crystal structures in PDBBind2020.13
Furthermore, PharmacoNet outperformed all the docking-free DL models including both structure-based and sequence-based approaches. It also showed better performance than the DL-based docking KarmaDock on LIT-PCBA which is the benchmark without the structural biases. It is known that the DL-based scoring methods show superior performance on PLI prediction on the PDBbind test set. However, they have been trained and evaluated only on drug molecules. Thus, they tend to show highly uncertain predictions for general molecules, making them vulnerable to the virtual screening of ultra-large chemical libraries designed to cover a huge chemical space.
PharmacoNet adopts a significantly different approach to evaluating binding affinities than existing DL methods. It estimates the binding affinities via an algorithmic process based on a pharmacophore-level scoring function. The scoring function contains only 7 parameters assigned to each pharmacophoric feature type, unlike DL-based scoring functions, which typically rely on millions of learnable parameters, which can cause overfitting unless a sufficient amount of data is provided. The following two factors explain why PharmacoNet was able to achieve such remarkable accuracy with the simple scoring function. First, our DL model enables us to construct accurate pharmacophore models for a given binding pocket. Second, the scoring function is designed to empirically evaluate the contribution of each NCI in the pharmacophore-level graph matching between target proteins and ligands. Consequently, PharmacoNet uses DL exclusively for protein-based pharmacophore modeling rather than for direct scoring. Compared to DL-based scoring, which is highly parameterized and the evaluation process is a black box, our analytical scoring is transparent and interpretable, reducing overfitting and improving generalization within the scope of the equation. This avoids overfitting problems even with a small training dataset and performs robustly across various chemicals and proteins, highlighting its effectiveness in ultra-large-scale virtual screening tasks.
To demonstrate the generalization ability of PharmacoNet, we evaluated its screening power in out-of-distribution settings. Specifically, we compared PharmacoNet with KarmaDock, a state-of-the-art deep learning-based docking tool, using the same train/test split. To assess generalization to unseen proteins, we measured the screening power on all out-of-distribution DEKOIS2.0 targets with sequence similarity below 0.5 to all training proteins.¶ For generalization to unseen ligands, we filtered out active and inactive molecules from LIT-PCBA with a Tanimoto similarity exceeding 0.7 to any training ligand.|| As shown in Fig. 2E and F, KarmaDock showed a significant degradation in screening power for unseen targets, whereas PharmacoNet maintained its performance. Similarly, for unseen ligands, PharmacoNet showed consistent performance (3.11 vs. 3.16), while KarmaDock's performance declined (2.94 vs. 2.32). These results suggest that focusing on coarse-grained NCIs provides better generalization in small drug datasets than detailed atom-wise interaction modeling. The similarity distributions of proteins and ligands between the training set and the test sets are illustrated in Fig. S4 and S5,† respectively.
2.3 Impact of the number of initial conformers
In the context of virtual screening, the utilization of a multi-conformer database is of paramount importance for he accurate modeling of protein–ligand interaction, given the pivotal role of ligand flexibility.1,5 PharmacoNet effectively addresses this necessity by providing the capacity to accommodate multiple conformers per ligand, analogous to exhaustive rigid-body docking methods.40Fig. 3A shows that increasing the number of initial conformers from 1 to 16 enhances the enrichment factor (EF1%) on the DEKOIS2.0 benchmark from 3.951 to 4.459.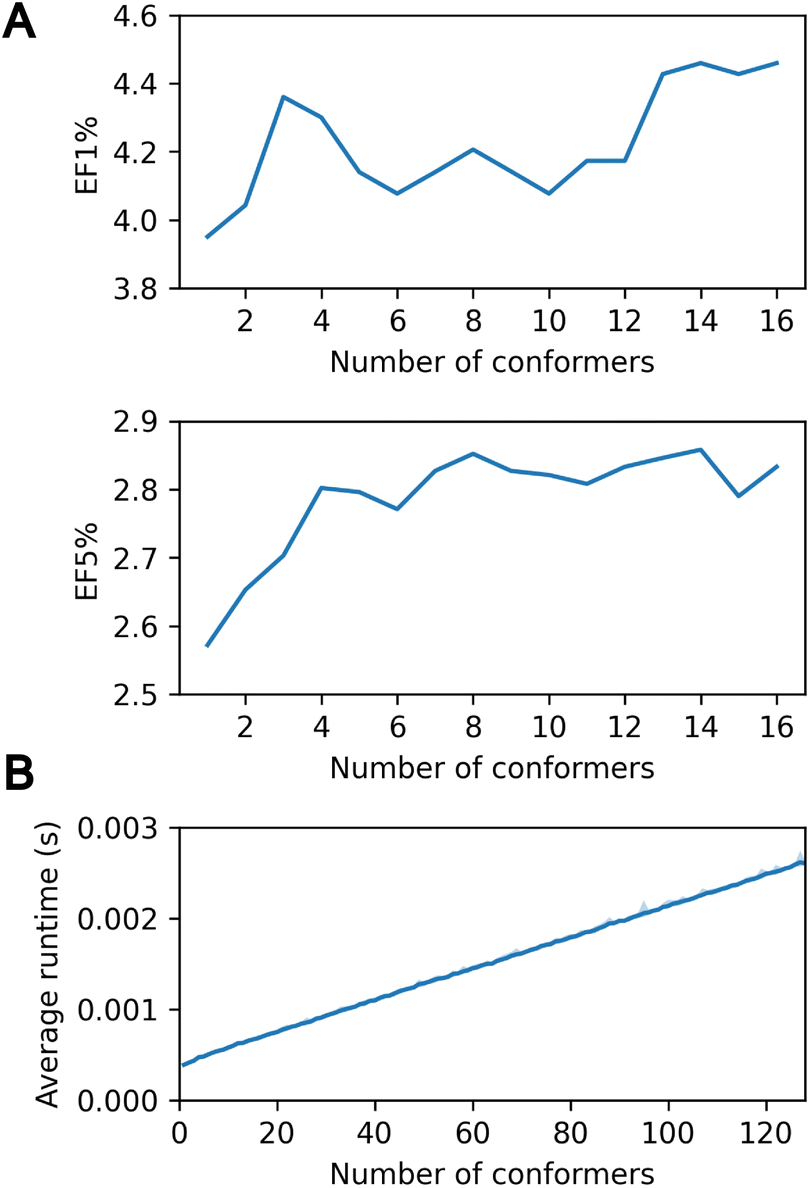 | ||
| Fig. 3 (A) Screening power EF1% and EF5% according to the number of RDKit ETKDG conformers. (B) Average runtime using up to 128 conformers for the PDBbind core set according to the number of RDKit ETKDG conformers. We measured the runtime 10 times for each number of conformers. All measurements were performed on a 32-core Intel Xeon Gold 6326 CPU @ 2.90 GHz. | ||
However, this accuracy improvement can come with a trade-off in computational speed due to the multiple-conformer calculation. Assessing up to 128 conformers using the PDBbind core set revealed minimal runtime increases: 0.39 ms for a single conformer versus 2.60 ms for 128 conformers (Fig. 3B). This efficiency surpasses standard docking programs such as AutoDock Vina or Smina, which show a linear runtime increase due to independent evaluations of each conformer. PharmacoNet uses internal coordinate systems of ligands to bypass the need to optimize absolute positions and orientations. Moreover, it first evaluates the core structure of the ligand conformer ensemble and then considers the remaining functional groups, enhancing computational efficiency. These findings highlight the high efficiency of PharmacoNet in integrating with multi-conformer databases in the virtual screening pipeline.
2.4 Performance as a pre-screening tool
The primary objective of pre-screening is to prioritize highly probable molecules that will be subjected to a fine-screening process to identify hit candidates. To assess the efficacy of pre-screening, we evaluated 1.6 million bioactive molecules sampled from the ChEMBL database.41 For the assessment, we selected two druggable targets not included in DEKOIS2.0 and LIT-PCBA: dihydrofolate reductase (DHFR, PDB ID 1dis) and epidermal growth factor receptor (EGFR) G719S/T790M double mutant (PDB ID 3ug2). In the pre-screening process, PharmacoNet utilized eight ETKDG conformers per molecule. For the fine-screening process, we used PIGNet2,42 a state-of-the-art structure-based scoring method in the virtual screening task.As illustrated in Fig. 4A, PharmacoNet was able to process the entire library in less than 10 minutes on a desktop with a single 32-core CPU, showing a notable reduction in time compared to the 35 days that would be required using traditional docking methods like AutoDock Vina or Smina. Fig. 4B shows that the average docking score of the remaining molecules increases as the filtration rate increases, indicating that PharmacoNet effectively screened out the low-potent molecules. Furthermore, PharmacoNet retained 40% and 70% of the top-10 hit candidates at a 95% filtration rate for DHFR and EGFR mutant, respectively (Fig. 4C). This result demonstrates that PharmacoNet is capable of achieving ultra-fast screening and maintaining sufficient accuracy as a pre-screening tool.
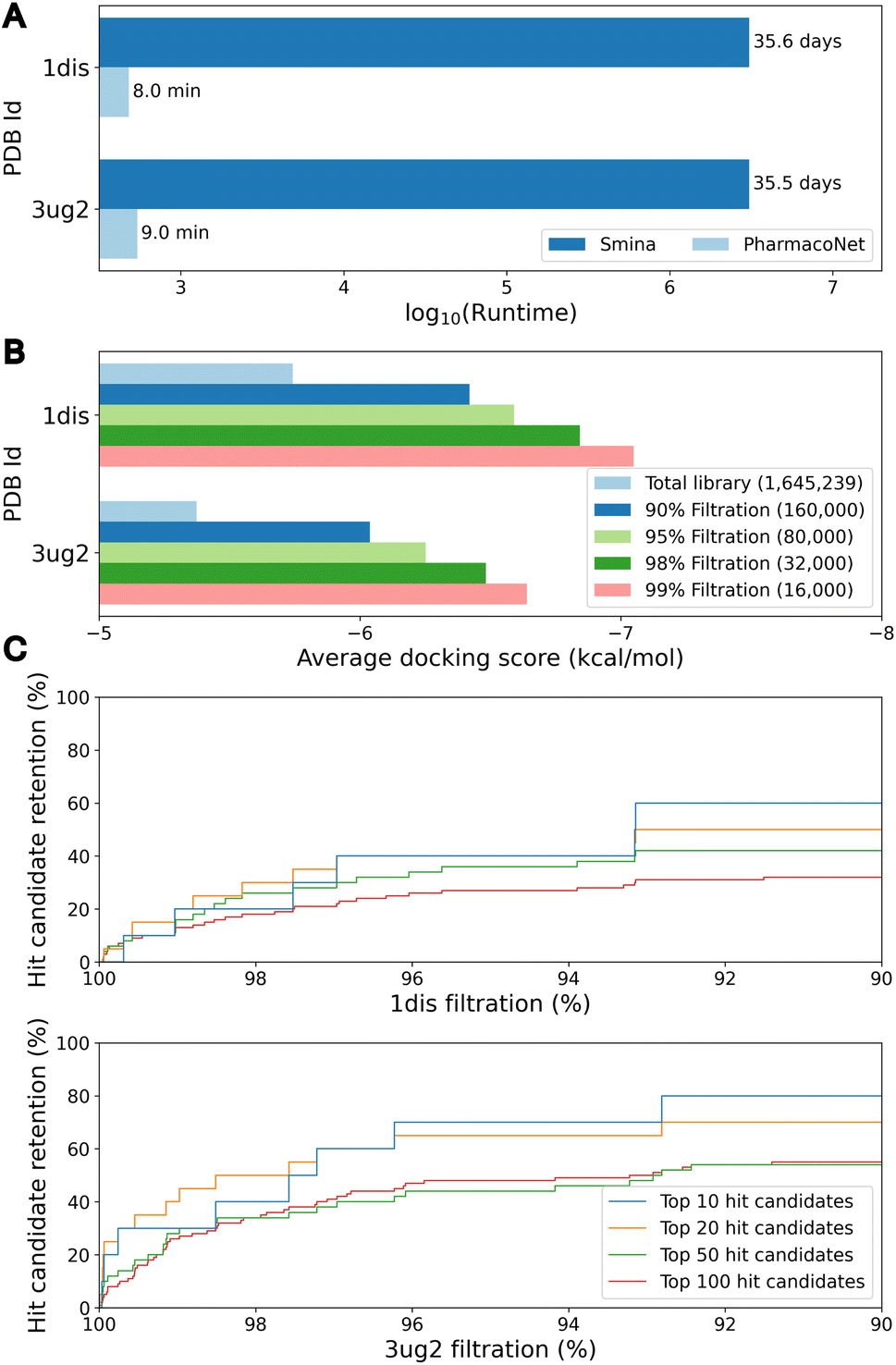 | ||
| Fig. 4 The speed and accuracy of pre-screening by PharmacoNet. (A) Total runtime to evaluate 1.6 million ChEMBL molecules for each target protein whose PDB ID is given on the y-axis. (B) Average docking score according to the filtration rate of pre-screening with PharmacoNet for each target protein whose PDB ID is given on the y-axis. (C) Retention rates of top N hit candidates according to the filtration rates for each target protein. | ||
Fig. 5A displays the generated pharmacophore model for the DHFR, and 5B shows its alignment with the X-ray crystal structure of brodimoprim-4,6-dicarboxylate, a known active ligand of DHFR. Notably, the PIGNet2 binding poses of CHEMBL1967896 (Fig. 5C) and CHEMBL3260001 (Fig. 5D), which received the highest scores from PharmacoNet, confirmed that the trifluoromethyl groups were aligned with the halogen pharmacophore points (yellow spheres) in our model. Intriguingly, these NCIs were absent in the complex structures of the active ligand, manifesting PharmacoNet's capability to identify novel pharmacophore points that may be overlooked by traditional complex-based pharmacophore modeling with known actives.
 | ||
| Fig. 5 Pharmacophore model and binders for the DHFR. Each color of the pharmacophore and protein hotspot indicates the following: orange for hydrophobic carbons, purple for aromatic rings, cyan for H-bond donors, and yellow for halogen atoms. (A) The generated pharmacophore model for the given binding site. (B) The crystal structure of the known active ligand (PDB ID BDM). (C and D) The PIGNet2 binding poses of CHEMBL1967896 (C) and CHEMBL3260001 (D), which are the ligands with the highest PharmacoNet score from the pre-screening. The circles denote the NCIs absent in the complex structure of the active ligand. | ||
2.5 Screening for potent and selective cannabinoid antagonists from an ultra-large chemical library
Developing potent and selective inhibitors poses significant challenges, particularly for targets with high structural similarity but distinct biological functions. In this scenario, employing a pharmacophore-focused scoring approach provides substantial benefits. Subtle differences at the binding site such as point mutations can alter specific NCIs, resulting in considerable changes to the pharmacophore model. These differences are pivotal for designing selective drugs as they allow the discrimination between closely related targets by emphasizing unique interaction opportunities. To demonstrate PharmacoNet's practicality in identifying potent and selective hit candidates, we performed an ultra-large-scale virtual screening targeting cannabinoid receptors (Fig. 6A).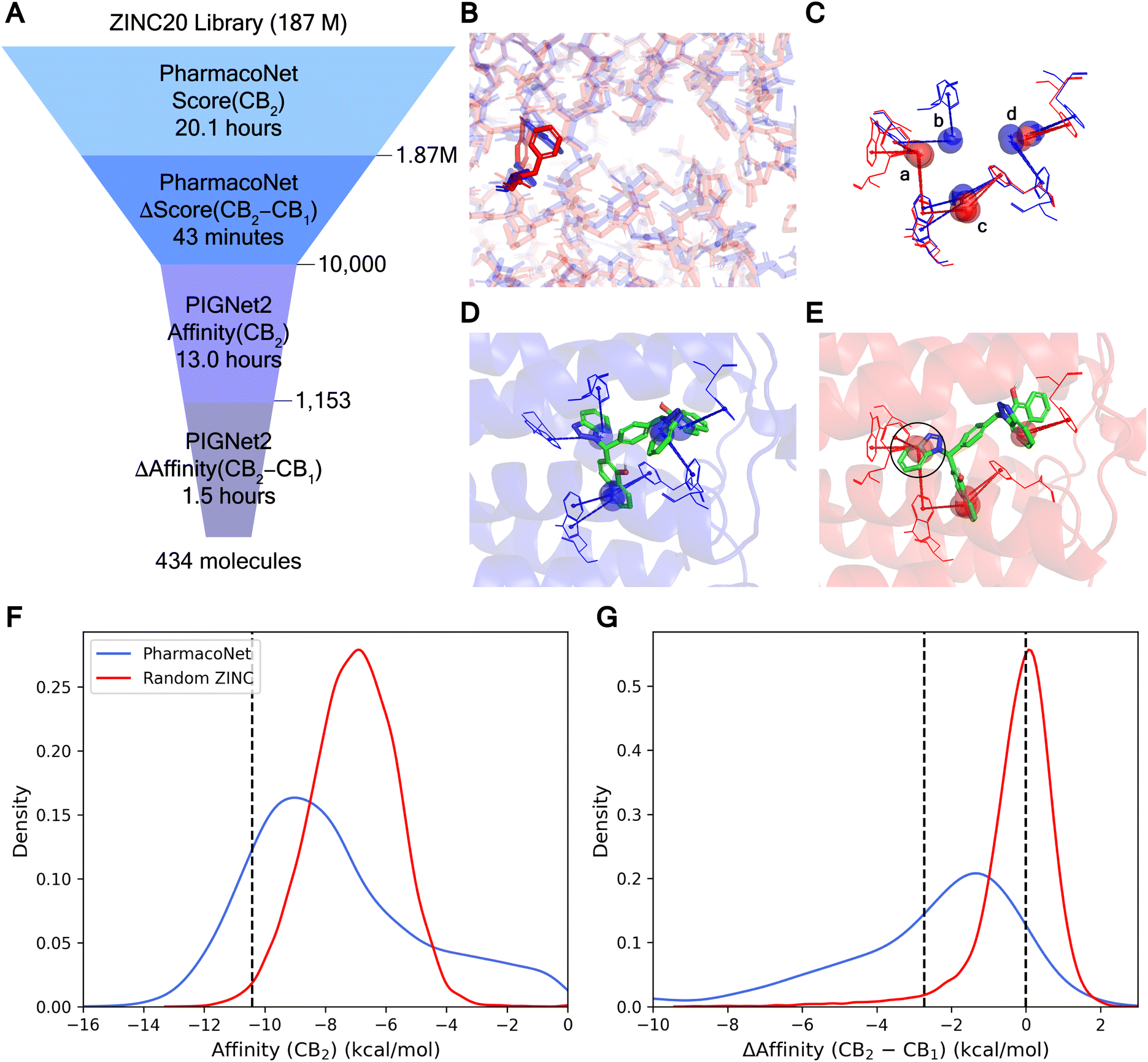 | ||
| Fig. 6 Virtual screening against cannabinoid receptors. (A) Virtual screening process with PharmacoNet on a desktop with a single 32-core CPU. (B) The aligned binding pocket structures of CB1 (blue) and CB2 (red). The unaligned residues are highlighted. (C) The pharmacophore models of CB1 and CB2. Only the pharmacophore points for the π–π stacking are visualized. (C) and (D) are overlapped, while (A) and (B) are not overlapped. (D and E) The PIGNet2 binding poses of ZINC100000809 against CB1 (D) and CB2 (E). (F) The distribution of docking scores against CB2 from the pre-screening result. The dashed line denotes −10.49 kcal mol−1 (top 1% affinity). (G) The distribution of docking score differences against CB1 and CB2 for potent molecules. The dashed lines represent 0 kcal mol−1 and −2.73 kcal mol−1 (100-fold selectivity), respectively. | ||
Cannabinoid receptors (CB), including CB1 and CB2, are components of the G protein-coupled receptor (GPCR) family, the key target of drug discovery. The high similarity between the binding sites of CB1 and CB2 complicates the development of potent and selective antagonists. As illustrated in Fig. 6B, the binding sites of CB1 (PDB ID 6kqi, blue) and CB2 (PDB ID 5zty, red) are nearly identical with subtle differences such as the placement of tryptophan and phenylalanine. These slight differences are clearly captured in the pharmacophore models (Fig. 6C). Since no hydrogen bonds or salt bridges were detected, only the π–π stacking was visualized for clarity. The alignment of pharmacophore points corresponding to overlapping residues appears similar (c, d), while those for non-overlapping residues are distinct (a, b), providing a key approach to achieving high selectivity.
Using these differential pharmacophore models, PharmacoNet can identify molecules whose aromatic groups are optimally positioned to form additional NCIs solely with a main target protein. For example, in the binding pose of ZINC100000809, the compound with the highest PharmacoNet score difference in screening, the benzotriazole group (black circle) formed three additional π–π stackings with CB2, which are absent in CB1 as shown in Fig. 6D and E. This selective interaction substantially influences the relative stability of the molecule for each target, yielding an energy difference of 7.50 kcal mol−1.
The high efficiency of our approach was verified with the computational time required for the virtual screening with selectivity, as summarized in Fig. 6A. Within about 20.1 hours, PharmacoNet completed the screening of 187 million ZINC20 (ref. 43) molecules against CB2 on a single 32-core Intel Xeon Gold 6326 CPU at 2.90 GHz. Then, the top 1% molecules were re-assessed against CB1 using PharmacoNet with an additional 43 minutes. The 10000 molecules with the largest score differences between CB2 and CB1 were selected for fine-screening with PIGNet2. The fine-screening first identified potent molecules against CB2 and then evaluated them against CB1 to estimate target selectivity. This two-step process took 14.5 hours.
Fig. 6F and G demonstrated that PharmacoNet efficiently identified molecules with both high potency and selectivity, respectively. A total of 1153 molecules among 10000 pre-screened molecules achieved a top 1% ranking in the entire library, and the average affinity difference against CB1 and CB2 of those potent molecules was 2.70 kcal mol−1. Ultimately, this screening process identified 434 molecules with over 100-fold selectivity and 278 molecules with over 1000-fold selectivity, outperforming the random selection by 65 and 67 times, respectively. These compelling results underscore PharmacoNet's ability to identify subtle yet critical differences between highly similar protein binding sites, enhancing selectivity in the screening process. In particular, its extremely high efficiency shows its potential as a transformative tool for navigating vast chemical spaces effectively in a time-sensitive and resource-constrained environment.
2.6 OpenPharmaco: graphical user interface software for PharmacoNet
OpenPharmaco promotes the accessibility of PharmacoNet with a user-friendly GUI for protein-based pharmacophore modeling and high-throughput virtual screening. This tool is particularly valuable for users without computational expertise, as illustrated in Fig. 7. OpenPharmaco allows for the import of common chemical file formats such as PDB, SDF, and MOL2, thus enabling to integrate it smoothly with various chemical informatics tools and workflows. The interface consists of modules dedicated to specific functions in PharmacoNet and the visualization with Open-Source PyMOL.44 | ||
| Fig. 7 OpenPharmaco workflow (A) the initial session of OpenPharmaco. (B) The structure of the KRAS-G12C mutant (PDB ID 6oim) imported from RCSB. (C) The protein-based pharmacophore modeling is performed automatically. (D) The generated pharmacophore model for KRAS-G12C mutant. (E) The comprehensive workspace for virtual screening. (F) The virtual screening result for example library. | ||
In OpenPharmaco, users can easily specify a target protein's binding site by importing it directly from the Research Collaboratory for Structural Bioinformatics (RCSB) with the corresponding PDB ID or loading a customized file. Once the binding site information is provided, PharmacoNet performs pharmacophore modeling based on it. Then, users need to import a chemical library for virtual screening on multiple CPU cores. If necessary, they can adjust the pre-optimized parameters in PharmacoNet's scoring function.
3 Methods
In the following sections, we describe the detailed framework of PharmacoNet, as illustrated in Fig. 8. Section 3.1 introduces the deep learning model for pharmacophore modeling (Fig. 8A). Section 3.2 explains the algorithm of the coarse-grained graph-matching process for protein–ligand spatial correlation estimation (Fig. 8B). Section 3.3 describes the parameterized analytical scoring function and its parameters (Fig. 8C).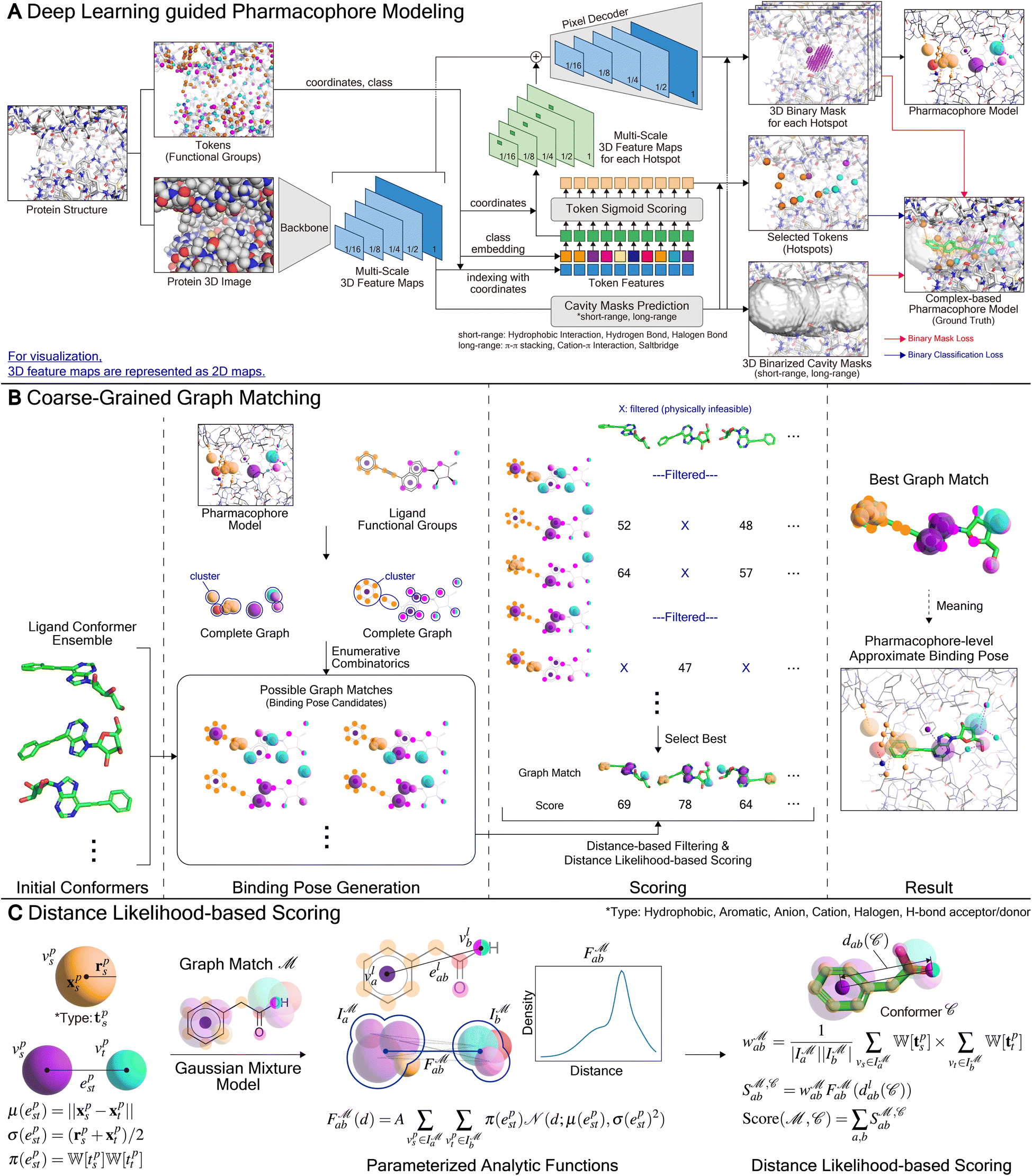 | ||
Fig. 8 Detailed architecture of PharmacoNet. (A) The architecture of deep learning model for fully automated protein-based pharmacophore modeling. For visualization, 3D feature maps are represented as 2D maps. For model training, the complex-based pharmacophore model is constructed from the crystal structure of the protein–ligand binding complex. (B) The graph-matching algorithm aligns the ligand and the pharmacophore model. All numbers in the figure are arbitrary values. (C) The distance likelihood-based scoring. For scoring, we use 7 pharmacophoric feature types: hydrophobic, aromatic ring, anion, cation, halogen, hydrogen bond (H-bond) acceptor, and donor. We use a set of weights 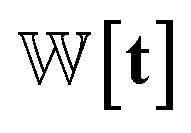 according to the pharmacophoric feature types as parameters for the scoring function. according to the pharmacophoric feature types as parameters for the scoring function. | ||
3.1 Deep learning-guided pharmacophore modeling
(1) They tend to identify excessive pharmacophoric features, making it difficult to find selective interaction patterns optimal to a specific ligand.50 Therefore, it is necessary to prioritize protein functional groups (FGs) for the selection of protein hotspots.
(2) Determining pharmacophore points is important for finding spatial information to ensure that a particular ligand forms optimal interactions with a given protein hotspot, but this is difficult to determine without protein–ligand binding information.
Due to these issues, protein-based methods heavily rely on expert-based manual processing and often require resource-intensive procedures like molecular dynamics, docking, or fragment crystallography.20 While a few automated protein-based approaches have been proposed,48,49 they are very slow due to energy-based optimization and thus used for fine-screening rather than pre-screening. As a result, there is no fully automated protein-based approach for pre-screening.
For the purpose of pharmacophore modeling, it is crucial to ascertain the nature of each pharmacophore point, including its coordinates, counterpart protein FGs, and an appropriate type of NCIs.17 To identify individual pharmacophore points and their natures, we frame pharmacophore modeling as an image instance segmentation problem, as illustrated in Fig. 8A. Image segmentation is the process of dividing an image into segments based on either categories or instance-level criteria.51,52 Semantic segmentation classifies pixels into categories, grouping different objects into a single segment. In contrast, instance segmentation achieves the recognition of both the segment and the category for each object by following a set of procedures: (1) detecting an object, (2) delineating the object's bounding box, (3) classifying its category, and (4) predicting a binary mask (segment). In the context of tailoring to the pharmacophore modeling, the key differences are:
(1) Instead of object detection in images, our deep learning model determines hotspots (instances) among protein FGs (tokens) in a given binding site. When a single FG can form multiple types of NCIs, it is treated as multiple tokens.
(2) Each instance already contains an NCI type (class). Moreover, a region (bounding box) to form a pharmacophore point can be obtained from prior knowledge of the maximum length of the given NCI type. Consequently, a deep learning model does not need to predict its class and bounding box.
(3) A single voxel may belong to multiple instances.
(4) The deep learning model also addresses an image inpainting problem, given that the space in the binding site is empty. Therefore, there is no definitive answer for the segmentation.
Similar to our approach, Skalic et al.53 developed LigVoxel, a 3D CNN-based deep learning model for inpainting the chemical functionality map from a binding pocket image. However, LigVoxel generates only one map for each of the three FG types (aromatic, H-bond donor, and H-bond acceptor), so it does not recognize individual pharmacophoric points or ensure NCIs with the binding site.
For the given protein binding site, the coordinates and atomic features of atoms are denoted as 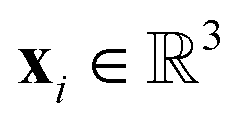 and
and 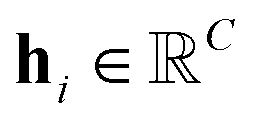 , respectively. Then, the 3D input image
, respectively. Then, the 3D input image 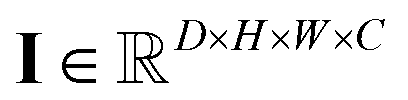 is represented as follows:
is represented as follows:
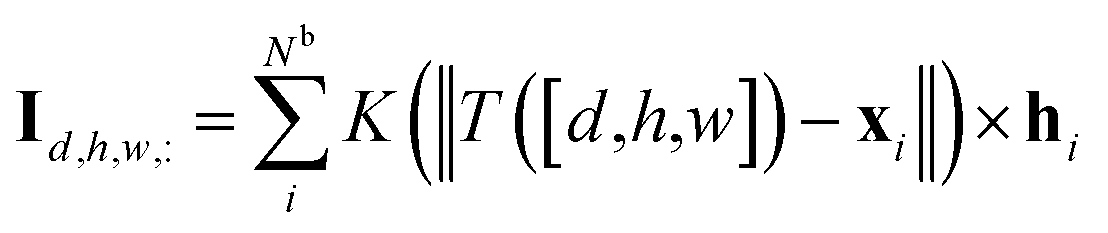 | (1) |
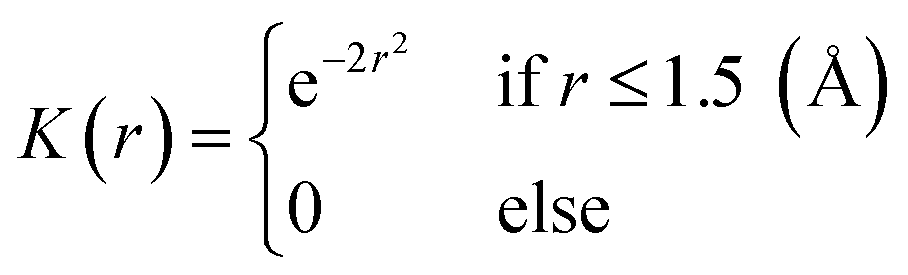 | (2) |
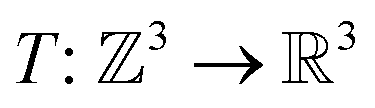 is the coordinate mapping function between the voxel indices and the real-world coordinates.
is the coordinate mapping function between the voxel indices and the real-world coordinates.
To extract features from the voxel image of a given protein binding site, our deep learning model (ϕ) uses the Feature Pyramid Network54 with a 3D extension of the Swin Transformer V2 encoder,55 which is typically used in object detection tasks. The feature pyramid network ϕbackbone obtains multi-scale 3D feature maps:
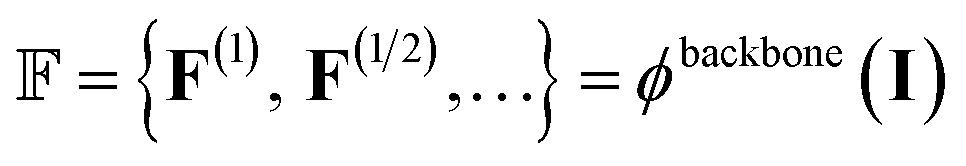 | (3) |
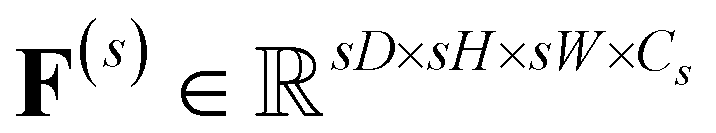 represents the feature map for each scale s with its corresponding hidden dimension Cs.
represents the feature map for each scale s with its corresponding hidden dimension Cs.
| Clong = sigmoid(ϕcavitylong(F(1))) | (4) |
| Cshort = sigmoid(ϕcavityshort(F(1))) | (5) |
In the cavity regions, PharmacoNet prioritizes the protein FGs to identify the protein hotspots. Each protein FG (token) contains grid indices [d,h,w] and its NCI type (class) ci. Some FGs can form multiple NCIs, so our deep learning model distinguishes them according to the NCI types. The model then calculates a sigmoid score for each token to determine whether it is a hotspot as follows:
| zi = ϕtoken(F(1)d,h,w,:,ci) | (6) |
| yi = sigmoid(ϕscore(zi)) | (7) |
To determine the score threshold for each NCI type, we utilize the score distribution of the tokens in the validation set. The score distribution and threshold for each NCI type are presented in Fig. S1.†
After the protein hotspots are determined, our model predicts the optimal spatial locations of the pharmacophore points for each protein hotspot in the instance segmentation manner:
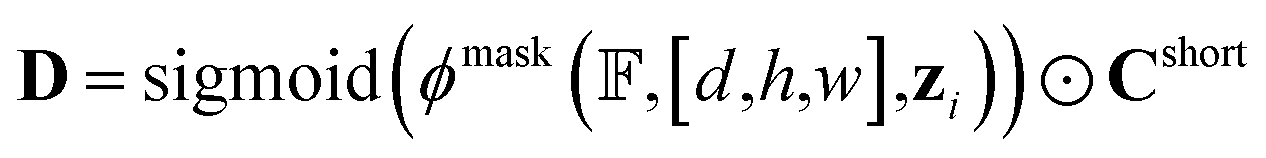 | (8) |
The spatial probability density of the pharmacophore points for each protein hotspot can be represented as the segments in the corresponding binary mask. When there are multiple segments in a single binary mask, each segment is considered a distinct pharmacophore point. Since the segment denotes the group of voxels, the center 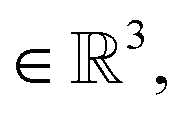 and the radius
and the radius 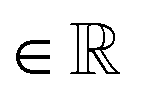 of the corresponding pharmacophore point are obtained from the density map D and the binary mask M as follows:
of the corresponding pharmacophore point are obtained from the density map D and the binary mask M as follows:
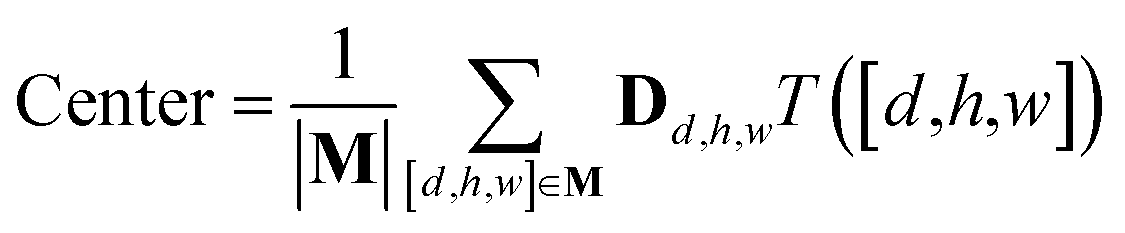 | (9) |
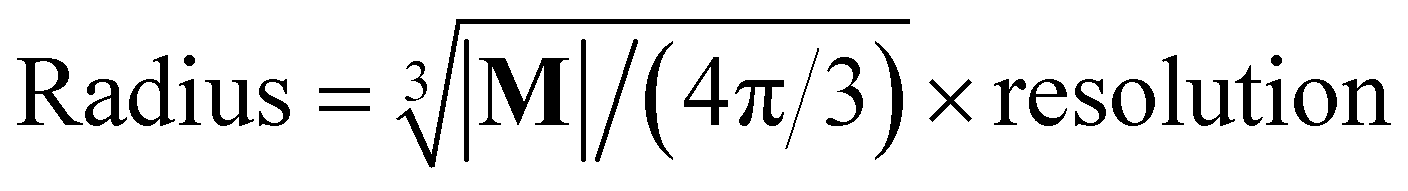 | (10) |
3.2 Coarse-grained graph matching
To predict the spatial relation between a target protein and a given ligand, PharmacoNet aligns the ligand to the pharmacophore model by a graph-matching algorithm. Both the pharmacophore model and the ligand FG arrangement can be represented as individual 3D complete graphs.The complete graph of the pharmacophore model GP = (VP,EP) uses pharmacophore points as nodes. Each node vps ∈ GP has a center position xps and a radius rps obtained from eqn (9) and (10) with a pharmacophoric feature type tps. For each edge epst ∈ Ep, the mean and standard deviation of its length are given by μ(epst) = ‖xps − xpt‖and σ(epst) = (rps + rpt)/2, respectively.
The complete graph of the ligand FG arrangement is denoted as GL = (VL,EL), where each node vla ∈ VL represents a specific FG and contains a set of its possible pharmacophoric feature types Tla. The edge is denoted as elab ∈ EL. For a ligand conformer 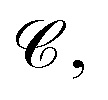 the position of the node vla is
the position of the node vla is 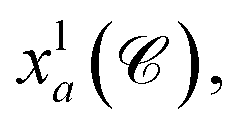 and the length of the edge elab is given by
and the length of the edge elab is given by 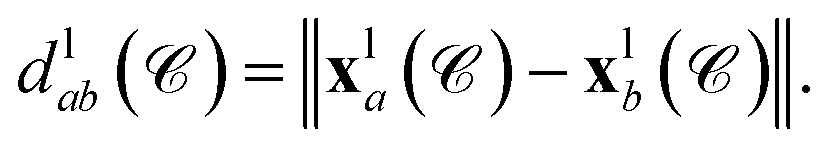
The FGs of ligand molecules often comprise numerous pharmacophoric features. For example, benzene contains 1 aromatic ring and 6 hydrophobic carbons. To improve the efficiency of the graph-matching process, we perform clustering for the same FGs. 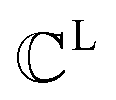 denotes a set of clusters, where each cluster
denotes a set of clusters, where each cluster 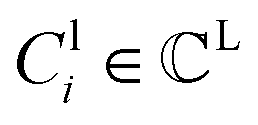 is a set of vl. In addition, since one ligand FG can form interactions with multiple protein hotspots, i.e., a single vl can be matched with multiple vp, we perform clustering for the pharmacophore model.
is a set of vl. In addition, since one ligand FG can form interactions with multiple protein hotspots, i.e., a single vl can be matched with multiple vp, we perform clustering for the pharmacophore model. 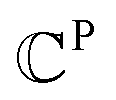 denotes a set of resulting clusters, where each cluster
denotes a set of resulting clusters, where each cluster 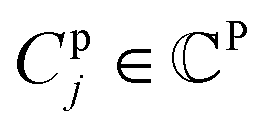 is a set of vp. Thanks to the clustering, the graph matching process can be done on a per-cluster basis, not a per-node basis, which helps accelerate the entire process.
is a set of vp. Thanks to the clustering, the graph matching process can be done on a per-cluster basis, not a per-node basis, which helps accelerate the entire process.
To formulate the graph-matching process, we define a matrix for a possible graph match (PGM), 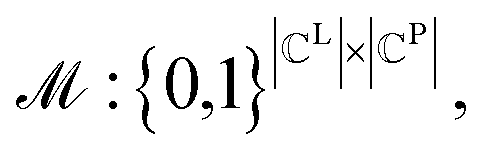 where
where 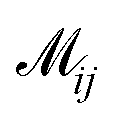 indicates the matching status between the ligand cluster Cli and pharmacophore model cluster Cpj. The constraints of our matching process are as follows: (1) a single pharmacophore model cluster Cpj can be matched to multiple ligand clusters Cli, which is expressed as
indicates the matching status between the ligand cluster Cli and pharmacophore model cluster Cpj. The constraints of our matching process are as follows: (1) a single pharmacophore model cluster Cpj can be matched to multiple ligand clusters Cli, which is expressed as 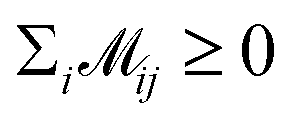 for all j (2) a single ligand cluster Cli can be matched with up to one pharmacophore model cluster Cpj, which is expressed as
for all j (2) a single ligand cluster Cli can be matched with up to one pharmacophore model cluster Cpj, which is expressed as 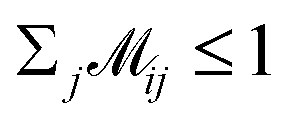 for all i (3) only clusters with the same pharmacophoric feature type can be matched. The time complexity of the graph matching is
for all i (3) only clusters with the same pharmacophoric feature type can be matched. The time complexity of the graph matching is 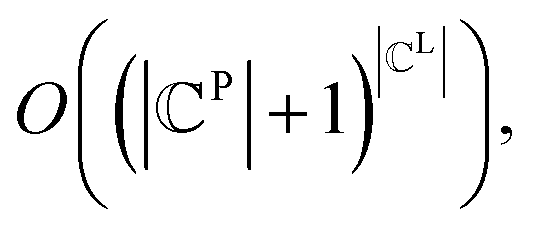 and the computational requirements increase exponentially with the complexity of the ligand or pharmacophore model. Therefore, the efficient graph-matching algorithm is mandatory.
and the computational requirements increase exponentially with the complexity of the ligand or pharmacophore model. Therefore, the efficient graph-matching algorithm is mandatory.
We note that most PGMs are physically infeasible. For example, it is not feasible for two ligand FGs with about 1 Å distance to match with two pharmacophore points separated by about 10 Å. To account for this, we use the following distance constraint between cluster pairs in for the conformer 
 | (11) |
Furthermore, an ensemble of ligand conformers shares a common core structure. Across various conformers, the feasible PGM patterns of the same core structure within the distance constraints show substantial similarities. As a result, the graph matching for numerous conformers can be performed simultaneously by identifying a unique PGM pattern for their core structure and then performing graph matching for the remaining part in a manner of depth-first search algorithm.
Finally, the graph-matching process can be formulated in terms of optimizing a scoring function. It is conceptually the same as the principle of conventional molecular docking, as illustrated in Fig. S3.† More details are in the ESI Section S5.†
3.3 Distance likelihood-based scoring function
Recently, Shen et al.58,59 reported the state-of-the-art scoring model based on the pairwise atom distance likelihood between proteins and ligands instead of scoring in the unit of energy. This has the advantage of allowing relative comparisons between ligands without additional efforts to map the structural information to binding affinities.16 Likewise, we introduce a distance likelihood-based scoring function to score and rank the PGMs.The Gaussian mixture model is used to express the probability density function  from the 3D arrangement of pharmacophore points GP. We define a scoring matrix,
from the 3D arrangement of pharmacophore points GP. We define a scoring matrix, 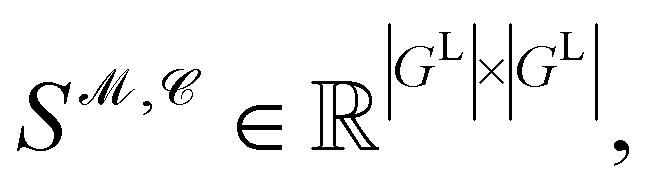 where
where 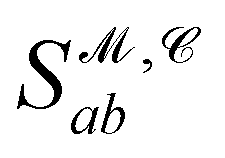 is the distance likelihood score vla and vlb with probability density function
is the distance likelihood score vla and vlb with probability density function 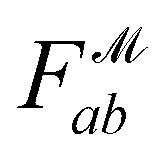 obtained from GP. When vla ∈ Cla′ and vlb ∈ Clb′ are matched to CpA and CpB in
obtained from GP. When vla ∈ Cla′ and vlb ∈ Clb′ are matched to CpA and CpB in 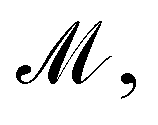 the Gaussian mixture model
the Gaussian mixture model 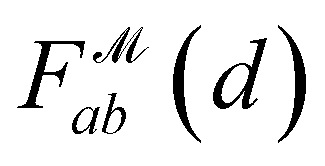 and the distance likelihood score
and the distance likelihood score 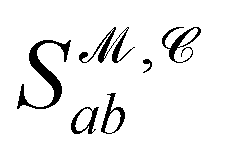 are follows:
are follows:
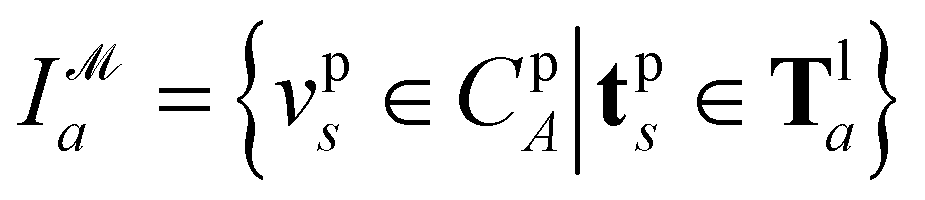 | (12) |
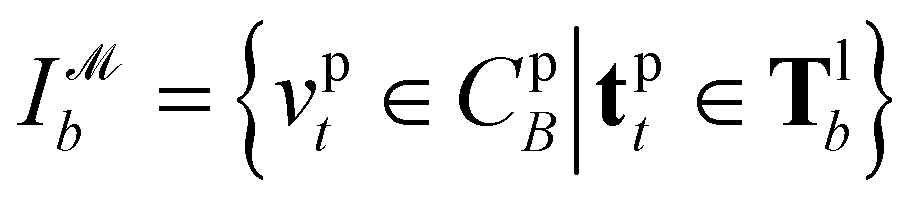 | (13) |
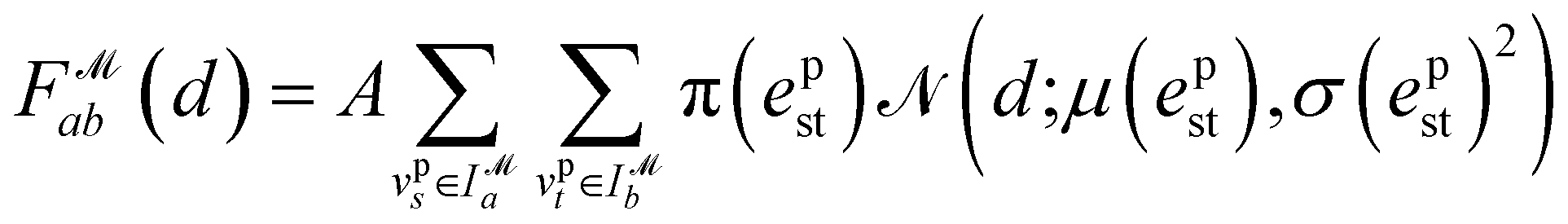 | (14) |
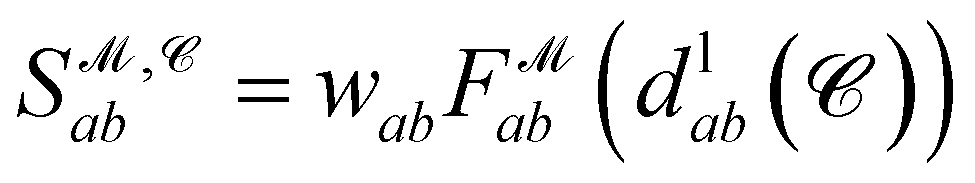 | (15) |
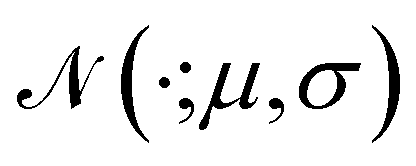 is the Gaussian function, A is the normalizing constant, and the coefficient of each Gaussian function is
is the Gaussian function, A is the normalizing constant, and the coefficient of each Gaussian function is 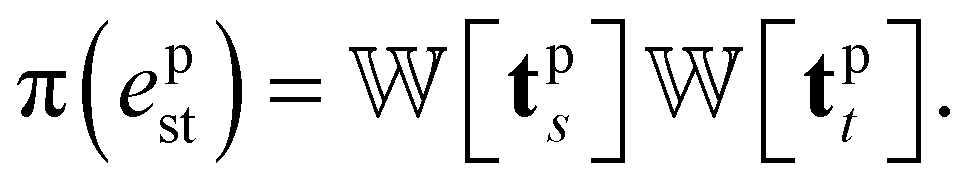 We introduce the weight for each ligand node pair as
We introduce the weight for each ligand node pair as 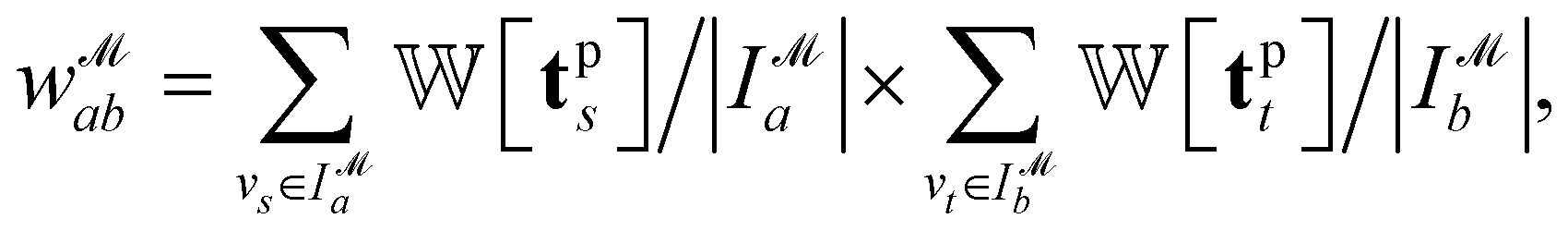 where
where 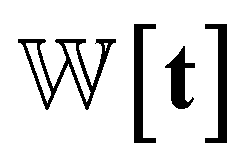 is the weights for each pharmacophoric feature type. We note that the distance likelihood score corresponding to the ligand node without any matched pharmacophore model points is 0.
is the weights for each pharmacophoric feature type. We note that the distance likelihood score corresponding to the ligand node without any matched pharmacophore model points is 0.
Finally, the score of PGM  for ligand conformer
for ligand conformer  can be represented as the sum of the scoring matrix:
can be represented as the sum of the scoring matrix:
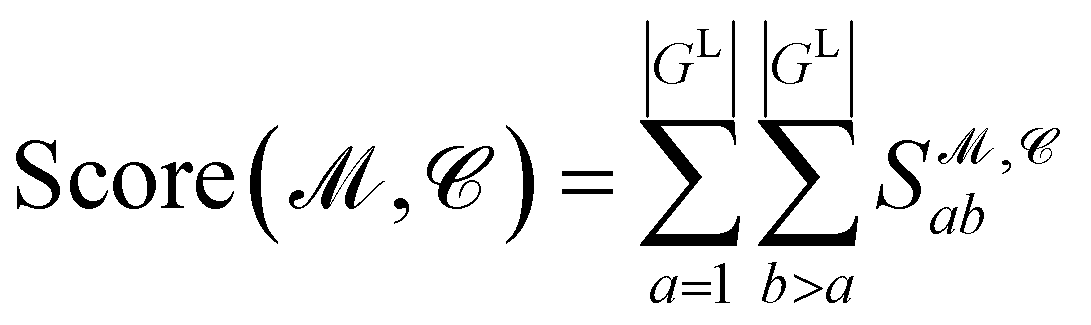 | (16) |
Our scoring function uses a set of weights,  assigned to each pharmacophoric feature type as parameters. In this study, these weights were determined based on prior knowledge of the relative contribution of NCIs to protein–ligand binding affinities. For example, hydrogen and halogen bonds were considered comparable in terms of their characteristics and strengths, with both significantly outweighing the influence of hydrophobic interactions. Salt bridges were considered stronger than both hydrogen and halogen bonds. On the other hand, π–π stacking was recognized as a predominant driving force contributing to complex stability, particularly in terms of entropy. Guided by this prior knowledge, we assigned 7 parameters as follows:
assigned to each pharmacophoric feature type as parameters. In this study, these weights were determined based on prior knowledge of the relative contribution of NCIs to protein–ligand binding affinities. For example, hydrogen and halogen bonds were considered comparable in terms of their characteristics and strengths, with both significantly outweighing the influence of hydrophobic interactions. Salt bridges were considered stronger than both hydrogen and halogen bonds. On the other hand, π–π stacking was recognized as a predominant driving force contributing to complex stability, particularly in terms of entropy. Guided by this prior knowledge, we assigned 7 parameters as follows:
• 1.0 for hydrophobic carbon.
• 4.0 for H-bond donor, H-bond acceptor, halogen atom, aromatic ring.
• 8.0 for cation, anion.
3.4 Training details for deep learning model
To train the deep learning model, we used the PDBbind v2020 dataset,13 a collection of high-resolution crystal structures, and measured binding affinities for 19443 protein–ligand complexes deposited in the Protein Data Bank (PDB).60 Following Shen et al.,58,59 we partitioned the dataset into 17658 training complexes and 1500 validation complexes, excluding 285 CASF-2016 (ref. 34) test complexes. We omitted 8 complexes with multiple ligands detected from the training set. The center of mass of active ligands was taken as the center of the binding site, and random translations and rotations were applied to augment the data.
To establish the ground truth for the protein-based pharmacophore modeling, we used the complex-based pharmacophore model obtained from the crystal structures in the PDBbind v2020 dataset. We considered the protein and ligand FG pairs that form NCIs as the pairs of protein hotspots and pharmacophore points. We first identified NCIs using the Protein–Ligand Interaction Profiler (PLIP)57 and then determined the protein hotspots and pharmacophore points from each NCI. For pharmacophore modeling, we used the following 6 NCI types: hydrophobic interaction, H-bond, halogen bond, π–π stacking, cation–π interaction, and salt bridge. Furthermore, we delineated two cavity regions based on the range of each interaction: one with 5.0 Å from the ligand atoms in a given crystal structure for short-range interactions (hydrophobic interaction, H-bond, and halogen bond) and another with 7.0 Å for long-range interactions (π–π stacking, cation–π interaction, and salt bridge).
As the ground truth, we used the hotspots in the complex-based pharmacophore model and the 3D binary mask with the dimension of (D,H,W) within 1.0 Å from each pharmacophore point. The cavities are also voxelized to 3D binary masks. For the instance segmentation modeling, we utilize two loss terms, which are pixel-wise binary mask loss and binary classification loss. Both loss terms employ the binary cross entropy loss function between the ground truth and deep learning model prediction obtained from eqn (4), (5), (7) and (8). More details are in the ESI Section S3.†
3.5 Benchmark test details
For commercial docking programs (GLIDE,29 GOLD27 and LeDock27), we reused the values reported by Zhang et al.,6 which were measured on 48-core Intel Xeon Gold 6240R CPUs @ 2.40 GHz. PharmacoNet and open-source docking programs (AutoDock Vina (version 1.2.5)24 and Smina30) were evaluated with a 32-core Intel Xeon Gold 6326 CPU @ 2.90 GHz. We used the default setting for AutoDock Vina with the search box size of (30 Å, 30 Å, 30 Å). For Smina, we also used the default setting with the auto box ligand configuration. To ensure the reproducibility of docking results, all data processing and docking protocols are explained in detail in ESI Section S4.†3.6 Virtual screening details
We used the ChEMBL library (version 33)41 and the ZINC20 library43 to demonstrate the applicability of PharmacoNet on diverse chemical libraries. We filtered the ChEMBL molecules with unidentified SMILES consisting of more than two molecules/ions, more than 40 heavy atoms, or failed ones with ETKDG,61,62 resulting in 1.64 million molecules out of 1.92 million molecules. For ZINC20, we randomly selected 10% (187 million) molecules from the whole library (1.87 billion).3.7 MolVoxel: molecular voxelization tool
We developed a new voxelization tool, MolVoxel, designed to enable on-the-fly voxelization in various machine-learning applications. Current voxelization tools often conflict with other ML packages. MolVoxel is implemented in Python with minimal dependencies (NumPy, SciPy), rendering it highly versatile and stable for various applications. Currently, it supports NumPy, Numba, and PyTorch (with CUDA support).4 Conclusions
In drug discovery, previous works on ultra-large-scale virtual screening have shown that expanding the search space significantly enhances the chances of identifying potent new molecules. However, it requires enormous computing resources due to the computational cost of molecular docking when dealing with billions of compounds. Our study addresses this problem by accelerating molecular docking through deep learning-based pharmacophore modeling.We developed PharmacoNet, the first deep-learning framework for pharmacophore modeling. It tackles key challenges in both pharmacophore modeling and large-scale virtual screening. A deep learning model trained on experimental binding structures performs automated pharmacophore modeling using only protein structures in a data-driven manner. By abstracting protein–ligand interactions to the pharmacophore level with an algorithmic scoring function, PharmacoNet alleviates generalization issues of deep learning approaches in a vast chemical space and simultaneously accelerates the computational speed of molecular docking. This allows for rapid and reliable scoring compared to conventional docking software. This result facilitates ultra-large-scale virtual screening with affordable costs, as demonstrated by successfully identifying potent and selective cannabinoid receptor antagonists from a library of 187 million molecules within 21 hours on a single CPU. Furthermore, we provide a user-friendly graphical user interface (GUI) for large-scale virtual screening, even on a desktop computer.
PharmacoNet's scoring function has only seven parameters, unlike deep learning-based scoring methods, which have numerous parameters. For instance, docking-free deep learning models rapidly predict binding affinities without resorting to the information of protein–ligand binding structures. However, limited training data often causes undesired biases in the models because molecules in ultra-large-scale chemical libraries will likely be out-of-distribution.x On the other hand, our framework can achieve high generalization ability in scoring by using a coarse-grained graph-matching algorithm with seven parameters.
Despite these advancements, there are areas for further enhancement. PharmacoNet relies on a scoring and graph-matching algorithm at the pharmacophore level, which provides rapid and reliable predictions. However, such pharmacophore-level abstraction comes with a trade-off. For example, since it does not capture atom-level features, it cannot discriminate variations in the strength of the same salt bridge, which is attributed to slight differences in charge and atom type. In addition, it cannot account for intramolecular energy changes. In this regard, the present approach is more suitable for pre-screening, followed by more accurate post-scoring. To overcome these limitations, atomistic features should be incorporated into the scoring function while maintaining a high generalization ability.
Consequently, PharmacoNet offers a new direction of deep learning approaches toward rapid and reliable ultra-large-scale virtual screening in drug discovery. We believe this approach will facilitate the practicalization of ultra-large-scale virtual screening in real-world applications.
Code availability
All the programs developed in this work are open-source. The source code, trained models, and GUI software are available at GitHub (https://github.com/SeonghwanSeo/PharmacoNet and https://github.com/SeonghwanSeo/OpenPharmaco) and Zenodo.63,64 The developed voxelization tool is available at PyPi https://pypi.org/project/molvoxel/.Data availability
The training data is from PDBbind v2020 and can be found at https://www.pdbbind.org.cn. The benchmark test sets are available at https://www.pharmchem.uni-tuebingen.de/dekois/, https://drugdesign.unistra.fr/LIT-PCBA/and https://www.pdbbind.org.cn/casf.php. The compound libraries for virtual screening are available at https://zinc20.docking.org and https://www.ebi.ac.uk/chembl/. The proteins for virtual screening are available at https://rcsb.org.Author contributions
S. S. was the main developer of PharmacoNet, OpenPharmaco, and MolVoxel. S. S. also performed data curation, formal analysis, investigation, methodology, and visualization. S. S. and W. Y. K. were the main contributors to the conceptualizing of this project and the writing & revising of the manuscript. W. Y. K. provided the supervision of this project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Basic Science Research Programs through the National Research Foundation of Korea (NRF), grant-funded by the Ministry of Science and ICT (NRF-2023R1A2C2004376, RS-2023-00257479).Notes and references
- J. Lyu, S. Wang, T. E. Balius, I. Singh, A. Levit, Y. S. Moroz, M. J. O'Meara, T. Che, E. Algaa and K. Tolmachova, et al. , Nature, 2019, 566, 224–229 CrossRef CAS PubMed.
- C. Gorgulla, A. Boeszoermenyi, Z.-F. Wang, P. D. Fischer, P. W. Coote, K. M. Padmanabha Das, Y. S. Malets, D. S. Radchenko, Y. S. Moroz and D. A. Scott, et al. , Nature, 2020, 580, 663–668 CrossRef CAS PubMed.
- R. M. Stein, H. J. Kang, J. D. McCorvy, G. C. Glatfelter, A. J. Jones, T. Che, S. Slocum, X.-P. Huang, O. Savych and Y. S. Moroz, et al. , Nature, 2020, 579, 609–614 CrossRef CAS PubMed.
- C. Gorgulla, A. Nigam, M. Koop, S. Selim Çınaroğlu, C. Secker, M. Haddadnia, A. Kumar, Y. Malets, A. Hasson and M. Li, et al., bioRxiv, 2023, preprint, DOI:10.1101/2023.04.25.537981.
- A. A. Sadybekov, A. V. Sadybekov, Y. Liu, C. Iliopoulos-Tsoutsouvas, X.-P. Huang, J. Pickett, B. Houser, N. Patel, N. K. Tran and F. Tong, et al. , Nature, 2022, 601, 452–459 CrossRef CAS PubMed.
- X. Zhang, O. Zhang, C. Shen, W. Qu, S. Chen, H. Cao, Y. Kang, Z. Wang, E. Wang and J. Zhang, et al. , Nat. Comput. Sci., 2023, 3, 789–804 CrossRef CAS PubMed.
- H. Stärk, O. Ganea, L. Pattanaik, R. Barzilay and T. Jaakkola, International conference on machine learning, 2022, pp. 20503–20521 Search PubMed.
- F. Gentile, J. C. Yaacoub, J. Gleave, M. Fernandez, A.-T. Ton, F. Ban, A. Stern and A. Cherkasov, Nat. Protoc., 2022, 17, 672–697 CrossRef CAS PubMed.
- D. E. Graff, E. I. Shakhnovich and C. W. Coley, Chem. Sci., 2021, 12, 7866–7881 RSC.
- L. Luo, A. Zhong, Q. Wang and T. Zheng, Mar. Drugs, 2021, 20, 29 CrossRef PubMed.
- H. Zhang, K. M. Saravanan and J. Z. Zhang, Molecules, 2023, 28, 4691 CrossRef CAS PubMed.
- W. Lu, Q. Wu, J. Zhang, J. Rao, C. Li and S. Zheng, Adv. Neural Inf. Process. Syst., 2022, 35, 7236–7249 Search PubMed.
- Z. Liu, Y. Li, L. Han, J. Li, J. Liu, Z. Zhao, W. Nie, Y. Liu and R. Wang, Bioinformatics, 2015, 31, 405–412 CrossRef CAS PubMed.
- A. S. Powers, H. H. Yu, P. Suriana, R. V. Koodli, T. Lu, J. M. Paggi and R. O. Dror, ACS Cent. Sci., 2023, 9, 2257–2267 CrossRef CAS PubMed.
- I. Wallach and A. Heifets, J. Chem. Inf. Model., 2018, 58, 916–932 CrossRef CAS PubMed.
- L. Chan, M. Verdonk and C. Poelking, arXiv, 2023, preprint, arXiv:2308.09086, DOI:10.48550/arXiv.2308.09086.
- C.-G. Wermuth, C. Ganellin, P. Lindberg and L. Mitscher, Pure Appl. Chem., 1998, 70, 1129–1143 CrossRef CAS.
- F. Imrie, T. E. Hadfield, A. R. Bradley and C. M. Deane, Chem. Sci., 2021, 12, 14577–14589 RSC.
- H. Zhu, R. Zhou, D. Cao, J. Tang and M. Li, Nat. Commun., 2023, 14, 6234 CrossRef CAS PubMed.
- S.-Y. Yang, Drug discovery today, 2010, 15, 444–450 CrossRef CAS PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard and J. Bambrick, et al. , Nature, 2024, 1–3 Search PubMed.
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch and R. D. Schaeffer, et al. , Science, 2021, 373, 871–876 CrossRef CAS PubMed.
- R. Krishna, J. Wang, W. Ahern, P. Sturmfels, P. Venkatesh, I. Kalvet, G. R. Lee, F. S. Morey-Burrows, I. Anishchenko and I. R. Humphreys, et al. , Science, 2024, 384, eadl2528 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- J. Heider, J. Kilian, A. Garifulina, S. Hering, T. Langer and T. Seidel, J. Chem. Inf. Model., 2022, 63, 101–110 CrossRef PubMed.
- R. Aggarwal and D. R. Koes, Research Square, 2024, preprint, DOI:10.21203/rs.3.rs-5033986/v1.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS PubMed.
- H. Zhao and A. Caflisch, Bioorg. Med. Chem. Lett., 2013, 23, 5721–5726 CrossRef CAS PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley and J. K. Perry, et al. , J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- D. R. Koes, M. P. Baumgartner and C. J. Camacho, J. Chem. Inf. Model., 2013, 53, 1893–1904 CrossRef CAS PubMed.
- L. Chen, X. Tan, D. Wang, F. Zhong, X. Liu, T. Yang, X. Luo, K. Chen, H. Jiang and M. Zheng, Bioinformatics, 2020, 36, 4406–4414 CrossRef CAS PubMed.
- T. Rose, N. Monti, N. Anand and T. Shen, bioRxiv, 2024, preprint, DOI:10.1101/2024.02.08.575577.
- M. R. Bauer, T. M. Ibrahim, S. M. Vogel and F. M. Boeckler, J. Chem. Inf. Model., 2013, 53, 1447–1462 CrossRef CAS PubMed.
- M. Su, Q. Yang, Y. Du, G. Feng, Z. Liu, Y. Li and R. Wang, J. Chem. Inf. Model., 2018, 59, 895–913 CrossRef PubMed.
- M. M. Mysinger, M. Carchia, J. J. Irwin and B. K. Shoichet, J. Med. Chem., 2012, 55, 6582–6594 CrossRef CAS PubMed.
- V.-K. Tran-Nguyen, C. Jacquemard and D. Rognan, J. Chem. Inf. Model., 2020, 60, 4263–4273 CrossRef CAS PubMed.
- Y. Wang, J. Xiao, T. O. Suzek, J. Zhang, J. Wang, Z. Zhou, L. Han, K. Karapetyan, S. Dracheva and B. A. Shoemaker, et al. , Nucleic Acids Res., 2012, 40, D400–D412 CrossRef CAS PubMed.
- J. Sunseri and D. R. Koes, Nucleic Acids Res., 2016, 44, W442–W448 CrossRef CAS PubMed.
- L. Chen, A. Cruz, S. Ramsey, C. J. Dickson, J. S. Duca, V. Hornak, D. R. Koes and T. Kurtzman, PLoS One, 2019, 14, e0220113 CrossRef CAS PubMed.
- G. P. Vigers and J. P. Rizzi, J. Med. Chem., 2004, 47, 80–89 CrossRef CAS PubMed.
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich and B. Al-Lazikani, et al. , Nucleic Acids Res., 2012, 40, D1100–D1107 CrossRef CAS PubMed.
- S. Moon, S.-Y. Hwang, J. Lim and W. Y. Kim, Digital Discovery, 2024, 3, 287–299 RSC.
- J. J. Irwin, K. G. Tang, J. Young, C. Dandarchuluun, B. R. Wong, M. Khurelbaatar, Y. S. Moroz, J. Mayfield and R. A. Sayle, J. Chem. Inf. Model., 2020, 60, 6065–6073 CrossRef CAS PubMed.
- L. L. C. Schrödinger, The PyMOL Molecular Graphics System, Version 1.8, 2015 Search PubMed.
- G. Jones, P. Willett and R. C. Glen, J. Comput.-Aided Mol. Des., 1995, 9, 532–549 CrossRef CAS PubMed.
- G. Wolber and T. Langer, J. Chem. Inf. Model., 2005, 45, 160–169 CrossRef CAS PubMed.
- P. D. Kirchhoff, R. Brown, S. Kahn, M. Waldman and C. Venkatachalam, J. Comput. Chem., 2001, 22, 993–1003 CrossRef CAS.
- V.-K. Tran-Nguyen, F. Da Silva, G. Bret and D. Rognan, J. Chem. Inf. Model., 2018, 59, 573–585 CrossRef PubMed.
- S. Jiang, M. Feher, C. Williams, B. Cole and D. E. Shaw, J. Chem. Inf. Model., 2020, 60, 4326–4338 CrossRef CAS PubMed.
- X. Qing, X. Yin Lee, J. De Raeymaeker, J. RH Tame, K. YJ Zhang, M. De Maeyer and A. RD Voet, J. Recept., Ligand Channel Res., 2014, 81–92 Search PubMed.
- J. Long, E. Shelhamer and T. Darrell, Proceedings of the IEEE conference on computer vision and pattern recognition, 2015, pp. 3431–3440 Search PubMed.
- K. He, G. Gkioxari, P. Dollár and R. Girshick, Proceedings of the IEEE international conference on computer vision, 2017, pp. 2961–2969 Search PubMed.
- M. Skalic, A. Varela-Rial, J. Jiménez, G. Martínez-Rosell and G. De Fabritiis, Bioinformatics, 2019, 35, 243–250 CrossRef CAS PubMed.
- T.-Y. Lin, P. Dollár, R. Girshick, K. He, B. Hariharan and S. Belongie, Proceedings of the IEEE conference on computer vision and pattern recognition, 2017, pp. 2117–2125 Search PubMed.
- Z. Liu, H. Hu, Y. Lin, Z. Yao, Z. Xie, Y. Wei, J. Ning, Y. Cao, Z. Zhang and L. Dong, et al., Proceedings of the IEEE/CVF conference on computer vision and pattern recognition, 2022, pp. 12009–12019 Search PubMed.
- D. Sehnal, S. Bittrich, M. Deshpande, R. Svobodová, K. Berka, V. Bazgier, S. Velankar, S. K. Burley, J. Koča and A. S. Rose, Nucleic Acids Res., 2021, 49, W431–W437 CrossRef CAS PubMed.
- S. Salentin, S. Schreiber, V. J. Haupt, M. F. Adasme and M. Schroeder, Nucleic Acids Res., 2015, 43, W443–W447 CrossRef CAS PubMed.
- C. Shen, X. Zhang, Y. Deng, J. Gao, D. Wang, L. Xu, P. Pan, T. Hou and Y. Kang, J. Med. Chem., 2022, 65, 10691–10706 CrossRef CAS PubMed.
- C. Shen, X. Zhang, C.-Y. Hsieh, Y. Deng, D. Wang, L. Xu, J. Wu, D. Li, Y. Kang and T. Hou, et al. , Chem. Sci., 2023, 14, 8129–8146 RSC.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- G. Landrum, et al., RDKit: Open-Source Cheminformatics, 2006 Search PubMed.
- S. Riniker and G. A. Landrum, J. Chem. Inf. Model., 2015, 55, 2562–2574 CrossRef CAS PubMed.
- S. Seo, PharmacoNet: The Second Release of PharmacoNet, Zenodo, 2024, DOI:10.5281/zenodo.12168475.
- S. Seo, OpenPharmaco: The First Release of OpenPharmaco, Zenodo, 2024, DOI:10.5281/zenodo.12168539.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04854g |
| ‡ IUPAC17 definition: “an ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response”. |
| § Aggarwal and Koes26 performed virtual screening with the pharmacophore models obtained from Apo2ph4 using Pharmit.38 |
| ¶ In DEKOIS2.0, there are 4 proteins with sequence similarity less than 0.5. In LIT-PCBA, there are only one out-of-distribution protein targets, so it is difficult to perform statistical analysis. |
| || Retention rates range from 90.1% to 99.7%, with an average of 97.0%. |
| This journal is © The Royal Society of Chemistry 2024 |