
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrategic design of GalNAc-helical peptide ligands for efficient liver targeting†
Takahito
Ito
 ab,
Nobumichi
Ohoka
c,
Michihiko
Aoyama
d,
Takashi
Nishikaze
e,
Takashi
Misawa
a,
Takao
Inoue
c,
Akiko
Ishii-Watabe
d and
Yosuke
Demizu
*abf
ab,
Nobumichi
Ohoka
c,
Michihiko
Aoyama
d,
Takashi
Nishikaze
e,
Takashi
Misawa
a,
Takao
Inoue
c,
Akiko
Ishii-Watabe
d and
Yosuke
Demizu
*abf
aDivision of Organic Chemistry, National Institute of Health Sciences, 3-25-26 Tonomachi, Kawasaki, Kanagawa 210-9501, Japan. E-mail: demizu@nihs.go.jp
bGraduate School of Medical Life Science, Yokohama City University, 1-7-29 Yokohama, Kanagawa 230-0045, Japan
cDivision of Molecular Target and Gene Therapy Products, National Institute of Health Sciences, Kanagawa, Japan
dDivision of Biological Chemistry and Biologicals, National Institute of Health Sciences, 3-25-26 Tonomachi Kawasaki-ku, Kawasaki, Kanagawa 210-9501, Japan
eSolutions COE, Analytical & Measuring Instruments Division, Shimadzu Corporation, 1 Nishinokyo Kuwabara-cho, Nakagyo-ku, Kyoto, 604-8511, Japan
fGraduate School of Medicine, Dentistry and Pharmaceutical Sciences, Division of Pharmaceutical Science of Okayama University, 1-1-1 Tsushimanaka, Kita 700-8530, Japan
First published on 21st October 2024
Abstract
There is a growing need for liver-selective drug delivery systems (DDS) in the treatment and diagnosis of liver diseases. The asialoglycoprotein receptor, a trimeric protein specifically expressed in the liver, is a key target for DDS. We hypothesized that peptides with reduced main-chain flexibility and strategically positioned N-acetylgalactosamine (GalNAc) moieties could enhance liver selectivity and uptake efficiency. The helical peptides designed in this study demonstrated superior uptake efficiency and liver selectivity compared with the conventional triantennary GalNAc DDS. These peptides also showed potential in protein delivery. Furthermore, we explored their application in lysosome-targeting chimeras (LYTACs), gaining valuable insights into the requirements for effective LYTAC functionality. This study not only highlights the potential of helical peptides as liver-selective DDS ligands, but also opens avenues for their use in various therapeutic and diagnostic applications, making significant strides in the targeted treatment of liver diseases.
Introduction
The liver is involved in many biological functions and contributes to the maintenance of homeostasis. Therefore, liver diseases can be life-threatening.1 In recent years, the number of reported cases of non-alcoholic steatohepatitis/non-alcoholic fatty liver disease has increased.2 Additionally, hepatitis B infections remain prevalent in Asia and Africa, and a lack of effective treatments poses a potential threat in the development of liver disease,3 as these infections often progress to liver cirrhosis and/or liver cancer. Estimates from the International Agency for Research on Cancer for 2022 indicate that approximately 870![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 individuals are diagnosed with liver cancer annually, of which 760000 succumb to the disease worldwide.4 This alarming mortality rate highlights the severity of liver cancer as a global health issue, emphasizing the pressing need for effective preventive measures, early diagnostic techniques, and innovative treatment strategies to combat this devastating malignancy. Consequently, there is a high demand for liver-targeted therapies.
000 individuals are diagnosed with liver cancer annually, of which 760000 succumb to the disease worldwide.4 This alarming mortality rate highlights the severity of liver cancer as a global health issue, emphasizing the pressing need for effective preventive measures, early diagnostic techniques, and innovative treatment strategies to combat this devastating malignancy. Consequently, there is a high demand for liver-targeted therapies.
Among recent advances in therapeutic modalities, medium-sized molecules have attracted significant attention. Some of these molecules target liver diseases, but often require combination with drug delivery systems (DDS) because of their relatively large molecular weights compared with traditional small molecule drugs. The asialoglycoprotein receptor (ASGPR), which is specifically expressed in the liver, recognizes galactose and N-acetylgalactosamine (GalNAc) and internalizes these via endocytosis.5 GalNAc is therefore used in liver-targeted DDS. ASGPR typically forms a trimer comprising two H1 units and one H2 unit.6 Thus, ligands with three GalNAc moieties are considered effective liver-selective carriers.7–9 Triantennary (Tri)-GalNAc, a representative liver-selective carrier with three GalNAc moieties, has been reported to enhance the antisense activity of loaded antisense oligonucleotides more than ten-fold.10 It has also been applied to diagnostic agents through conjugation with radioactive nuclides.11 The structure–activity relationships of Tri-GalNAc have been studied in terms of linker length and structure, revealing that all versions exhibit highly flexible structures. In silico simulations suggest that ASGPR is arranged at each vertex of a triangle, although highly flexible ligand structures are thought to be thermodynamically unfavorable to such conformations.12 Therefore, we hypothesized that more efficient DDS ligands could be developed by attaching GalNAc on molecules that can define their conformations (Fig. 1a).
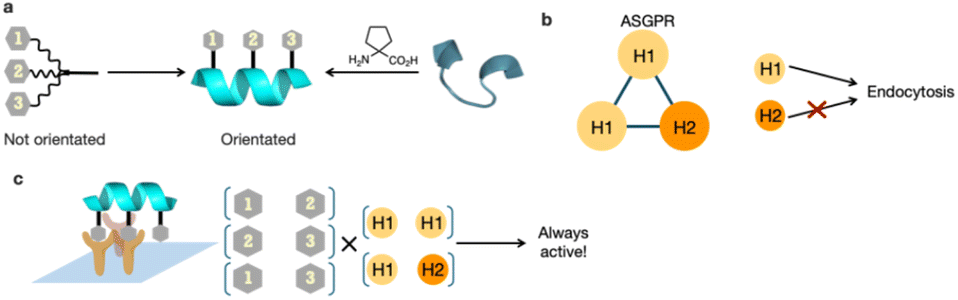 | ||
| Fig. 1 (a) Strategic design of N-acetylgalactosamine (GalNAc)-helical peptide ligands. (b) Geometry of each asialoglycoprotein receptor (ASGPR) unit and its function. (c) The hypothesis that controlling GalNAc orientation may lead to efficient endocytosis. | ||
Short-chain peptides, although easily synthesized, generally struggle to form helical structures. However, it is well-established that α,α-disubstituted amino acids13,14 and side-chain stapling15,16 stabilize helical conformations. Their incorporation into short-chain peptides enables the formation of helical structures even in typically recalcitrant sequences.14,17 Such helical peptides have been included in various functional peptides, including protein–protein interaction inhibitors,18–21 DDS,22–25 and antimicrobial peptides.26–28 These applications rely on the consistent orientation of specific residues to function effectively.
We hypothesized that the orientation of GalNAc could be controlled by using helical peptides as scaffolds and attaching GalNAc moieties to their side chains, while simultaneously reducing the molecule's degrees of freedom. We posited that this approach could lead to the development of potent ligands for ASGPR. The ASGPR subunits H1 and H2 form a trimer at a 2:1 ratio (H1:H2)5 and it is well established that H1 internalizes efficiently, whereas H2 does not (Fig. 1b).29 Considering their linear nature, helical peptides can engage only two GalNAc moieties simultaneously during binding. However, we hypothesized that efficient endocytosis could still be induced, because at least one of the bound receptors would be H1. Furthermore, we postulated that incorporating three GalNAc moieties would create three potential GalNAc pairs, thereby potentially enhancing binding (Fig. 1c). This study therefore presents a ligand design strategy based on secondary structure control for the development of hepatocyte-selective carriers.
Results and discussion
We first designed template peptides capable of forming helical structures. To determine the optimal position for introducing GalNAc, we designed the template peptides with three repeats of a seven-residue unit. In the center (4th residue) of each unit, the α,α-disubstituted amino acid Ac5c was introduced. Three GalNAc moieties were introduced into the peptide, all designed to face the same side of the helix (Fig. 2a). GalNAc was modified with an alkyne via propargylation of the anomeric hydroxy group and introduced into the peptide through a click reaction (copper-catalyzed azide–alkyne cycloaddition [CuAAC]) to face the same side of the helix (Fig. 2b). In later studies, we found that the peptides exhibited high hydrophobicity. Therefore, we designed peptides to form salt bridges, thereby reducing hydrophobicity and further stabilizing the helix.30 To investigate the importance of the helical structure in cellular uptake, we also designed peptide LD, based on 5S, incorporating alternating L- and D-amino acids to disrupt the secondary structure (Fig. 2). Tri-GalNAc was synthesized according to methods previously reported10 and was used as the control.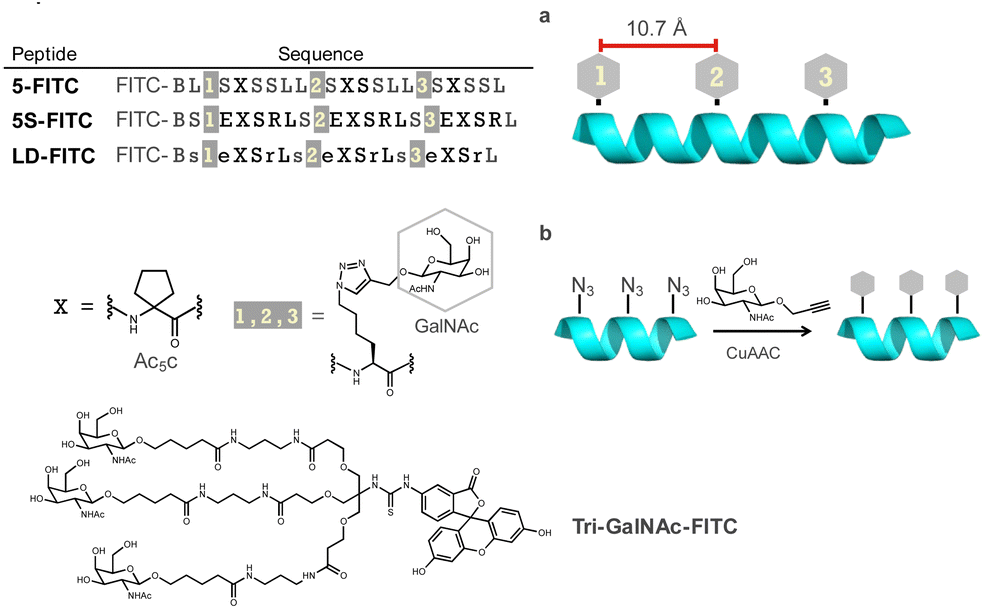 | ||
| Fig. 2 (a) Peptides designed showing the positional relationship of N-acetylgalactosamine (GalNAc) on the peptide. (b) The method used to introduce GalNAc molecules on the peptide. B, β-alanine; FITC, fluorescein isothiocyanate. | ||
The stability of the helical structures of peptides 5 and 5S was verified using computer-based molecular modeling. The energy of the most stable conformation was negative for both peptides, suggesting that the helical structure was stable. In peptide 5S, hydrogen bonding was observed between the side chains of glutamic acid and arginine. Considering that the energy of the most stable conformation was lower than that of peptide 5, it was suggested that the Glu–Arg salt bridge contributed to the stability of the helix (Fig. 3a). Circular dichroism spectrum measurements revealed that peptide 5S had a larger maximum than peptide 5, indicating it formed a more stable helical structure (Fig. 3b). The helix content also showed a two-fold difference. In contrast, peptide LD did not form a defined secondary structure (Fig. 3c).
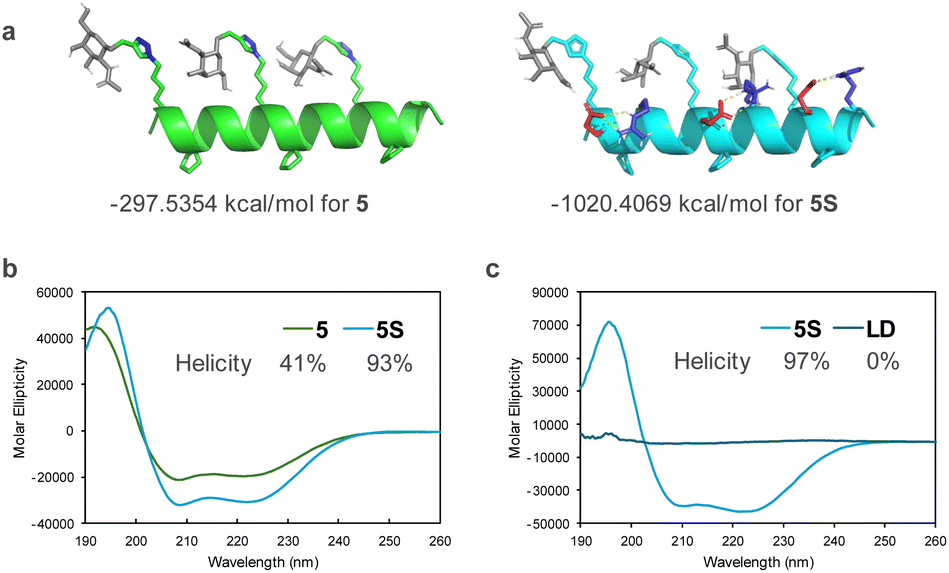 | ||
| Fig. 3 (a) Calculated secondary structures of peptides 5 and 5S. (b) and (c) Circular dichroism spectra of peptides <100 μM in (b) 50% MeCN aqueous and (c) 20 mM phosphate buffer (pH 7.2). | ||
The cellular uptake of fluorescently labelled peptides into HepG2 cells, which are derived from the human liver and express ASGPR, was evaluated by flow cytometry. Tri-GalNAc showed cellular uptake similar to that previously reported,31 but the helix-forming peptides 5-FITC and 5S-FITC exhibited more efficient uptake. In contrast, the non-helix-forming peptide LD-FITC showed lower uptake than peptides 5-FITC and 5S-FITC, similar to that of Tri-GalNAc-FITC (Fig. 4a). Therefore, it was suggested that the helical structure contributes to cellular uptake. In a later study, we confirmed that GalNAc competitively inhibited the cellular uptake of peptide 5S-FITC (Fig. 6). We investigated whether this uptake was mediated by ASGPR by comparing ASGPR positive and negative cell lines. The uptake evaluation was performed in HeLa cells, which do not express ASGPR. No significant uptake of cellular uptake of Tri-GalNAc or peptide 5S-FITC was observed in HeLa cells. In contrast, peptide 5-FITC showed uptake in HeLa cells at lower levels than in HepG2 cells (Fig. 4b) suggesting peptide 5-FITC was also internalized via an ASGPR-independent route. Tri-GalNAc-FITC and peptide 5S-FITC exhibit high hydrophilicity, whereas peptide 5-FITC is comparatively hydrophobic. Therefore, the high hydrophobicity of peptide 5-FITC is thought to affect its uptake in HeLa cells. Based on these results, liver cell selectivity was calculated as the uptake in HepG2 cells divided by the uptake in HeLa cells. Peptide 5S-FITC demonstrated approximately twice the selectivity of Tri-GalNAc, whereas peptide 5-FITC exhibited lower selectivity.
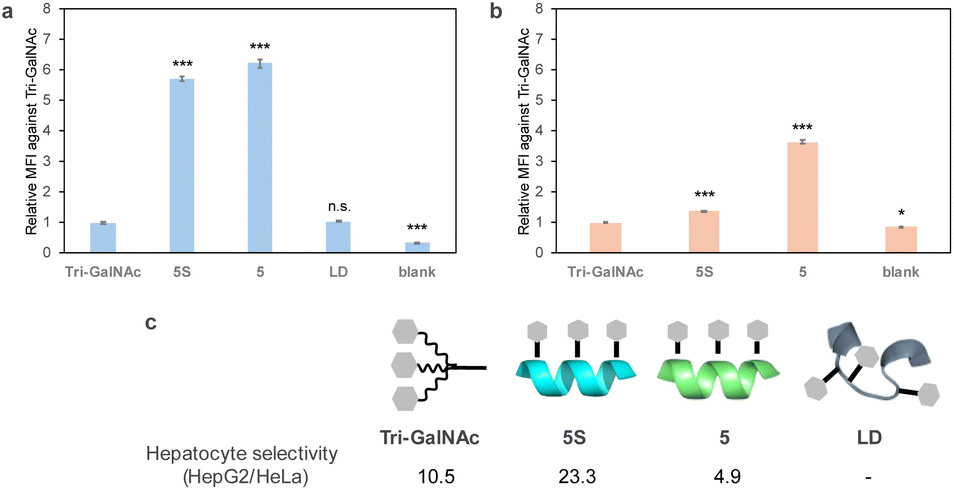 | ||
| Fig. 4 Cellular uptake of ligands against (a) HepG2 and (b) HeLa cells. HepG2 or HeLa cells were treated with 5 μM of each compound for 2 h. Cellular uptake activities are indicated as the mean fluorescence intensity (MFI) of three samples recorded using a flow cytometer. The relative MFI was defined as the MFI of triantennary (Tri)-N-acetylgalactosamine (GalNAc) as one. (c) Hepatocyte selectivity was calculated from the absolute MFI value of HepG2 divided by that of HeLa. Both MFI values were subtracted from the background value. Significant differences were calculated by the Dunnett's test for Tri-GalNAc-FITC. *P < 0.05, ***P < 0.001 n.s., not significant. Error bars represent the standard error of three samples. | ||
 | ||
| Fig. 5 The effect of the number and position of N-acetylgalactosamine (GalNAc) moieties in a molecule on cellular uptake. (a) Cellular uptake of peptides from 5S-FITC to 5S-2′′-FITC. Cellular uptake activities are indicated as the mean fluorescence intensity (MFI) of three samples recorded using a flow cytometer. The relative MFI was defined as the MFI of 5S-FITC as one. Significant differences were calculated by the Dunnett's test for 5S-FITC. ***P < 0.001 n.s., not significant. (b) The side-chain flexibility of GalNAc-mounted residues calculated using molecular dynamics simulation. The flexibility is expressed as root mean square deviation. | ||
 | ||
| Fig. 6 Cell-based binding study. HepG2 cells were treated with 5 μM compound in the presence of 0.03–100 mM GalNAc for 2 h. The mean fluorescence intensity (MFI) of three samples was recorded using a flow cytometer. The concentration of N-acetylgalactosamine (GalNAc) and MFI were plotted on the X- and Y-axis, respectively. Sigmoidal curves were fitted to plots to calculate the 50% inhibitory concentration (IC50) of GalNAc against the compounds tested. The dissociation constant (Kd) was calculated by substituting the inhibition constant (Ki; 150 μM) of GalNAc and the IC50 into the Cheng–Prusoff equation. | ||
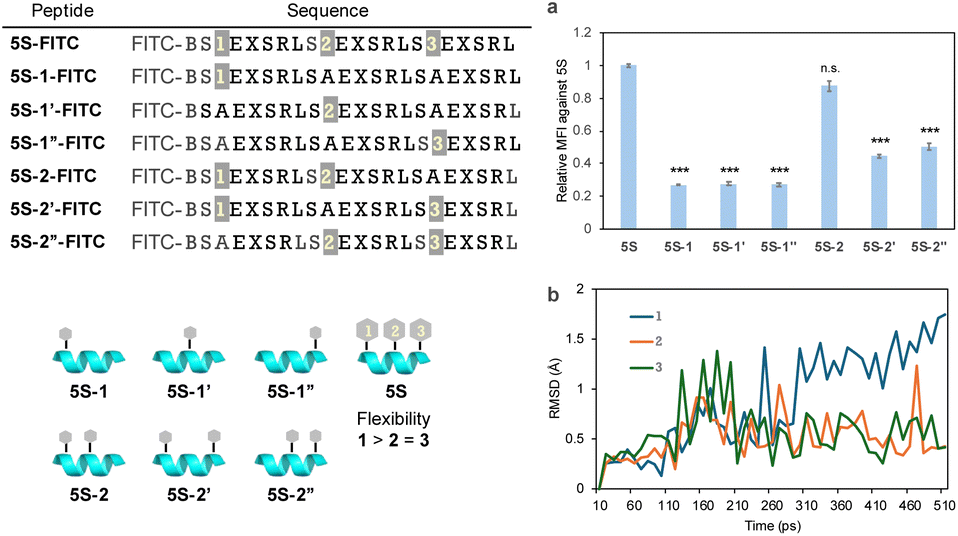
ASGPRs are thought to be arranged at each vertex of a triangle. It may be challenging for all three GalNAc moieties in peptide 5S-FITC to bind simultaneously as these are arranged in a straight line. Therefore, to identify which GalNAc units contribute to ASGPR binding, we designed peptides 5S-1-FITC to 5S-2′′-FITC, which contain fewer GalNAc moieties than peptide 5S-FITC (Fig. 5). The uptake of peptides 5S-1-FITC to 5S-1′′-FITC, each containing only one GalNAc moiety, was significantly reduced compared with that of 5S-FITC (Fig. 5a). The binding affinity of a single GalNAc moiety to ASGPR has been reported to be 40 μM,32 indicating that the inclusion of a single GalNAc moiety is ineffective. Among the peptides containing two GalNAc moieties, only 5S-2-FITC showed significant uptake, while 5S-2′-FITC and 5S-2′′-FITC exhibited greatly reduced uptake (Fig. 5a). This result was contrary to our expectations. Since there was no difference in the secondary structure among these peptides (Fig. S1†) and no clear difference in the relative positions of the GalNAc moieties between 5S-2-FITC and 5S-2′′-FITC. We hypothesized that the GalNAc moiety at position 3 would exhibit a stronger orientation than the moiety at position 1, as it is closer to the center of the sequence and thus subject to more restricted side-chain freedom. Because there was no difference in the uptake of molecules containing a single GalNAc moiety, we concluded that the ease of binding at each position does not differ significantly. Molecular dynamics simulations of peptide 5S-FITC revealed that the root mean square deviation values (RMSD) of the alkyl side chains of GalNAc at position 1 were larger than those of side chains at other positions, suggesting higher flexibility (Fig. 5b). This observation indicates that the GalNAc moiety at the N-terminus of the peptide may possess greater conformational freedom, potentially enabling it to adopt a more favorable orientation for binding to ASGPR. This suggests that the binding of GalNAc to ASGPR occurs in a stepwise manner rather than simultaneously. Previous reports have indicated that the length of the GalNAc linker should be neither too short nor too long, suggesting that appropriate flexibility of the ligand is necessary.33
The protein knockdown activity of various GalNAc ligand-small interfering RNA (siRNA) conjugates is reportedly related to the binding activity of the GalNAc ligands to ASGPR.9 Therefore, we evaluated the binding activity of the ligands to ASGPR. Although surface plasmon resonance and isothermal titration calorimetry have been used for binding activity evaluation previously, these may not mimic the geometric environment of trimeric ASGPR in a physiological state. Thus, we decided to perform a cell-based evaluation using flow cytometry. By co-administering GalNAc to competitively inhibit uptake and by varying its concentration, we were able to calculate the 50% inhibitory concentration (IC50) of the ligands. The dissociation constant (Kd) of the ligands was calculated by substituting the known inhibition constant (Ki) of GalNAc and the calculated IC50 into the Cheng–Prusoff equation.34–36 The results suggested that the binding activity of peptide 5S-FITC to HepG2 was approximately three times higher than that of peptide LD-FITC and Tri-GalNAc-FITC (Fig. 6). The difference between LD-FITC and Tri-GalNAc did not appear, which may be related to the fact that both are highly flexible molecules.
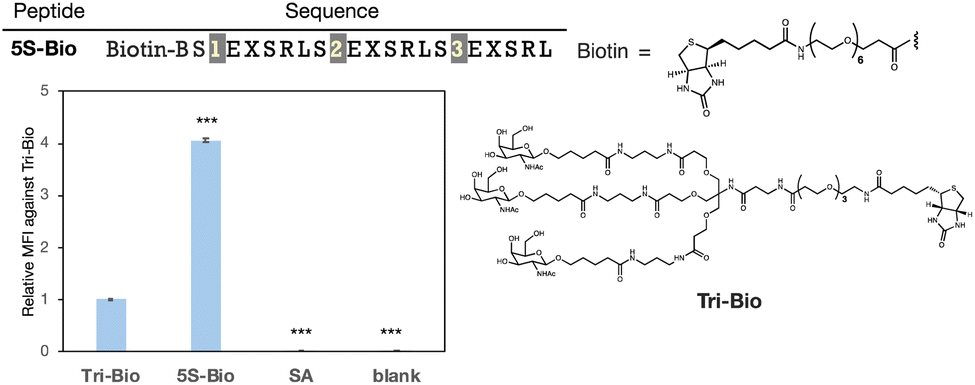
Next, we evaluated the molecules' delivery efficiency. The peptide was biotinylated and the uptake of streptavidin (54 kDa), which binds to the biotinylated ligands, was evaluated. We showed that peptide 5S-Bio could transport approximately four times more streptavidin than Tri-GalNAc and significantly than streptavidin itself (SA) suggesting its potential to deliver large proteins that cannot pass through the cell membrane on their own (Fig. 7). Although, the uptake trends between peptide 5S-Bio and Tri-Bio were similar to the result of FITC-labelled ligands, their differences became slightly smaller. Streptavidin forms a tetramer with biotin, which is able to bind to each streptavidin subunit. Therefore, there are four binding sites and the apparent binding activity of Tri-GalNAc may have increased due to this multivalent effect.36
 | ||
| Fig. 7 Cellular uptake of streptavidin. Each compound (1 μM) was preincubated with 250 nM Alexa Fluor™ 647-conjugated streptavidin. HepG2 cells were then incubated with the streptavidin complex for 2 h. Cellular uptake activities are reported as the mean fluorescence intensity (MFI) of three samples recorded using a flow cytometer. The relative MFI was defined as the MFI of Tri-GalNAc (Tri-Bio) as one. Significant differences were calculated by the Dunnett's test for Tri-Bio. ***P < 0.001. SA: Alexa Fluor647 labelled streptavidin without carriers. | ||
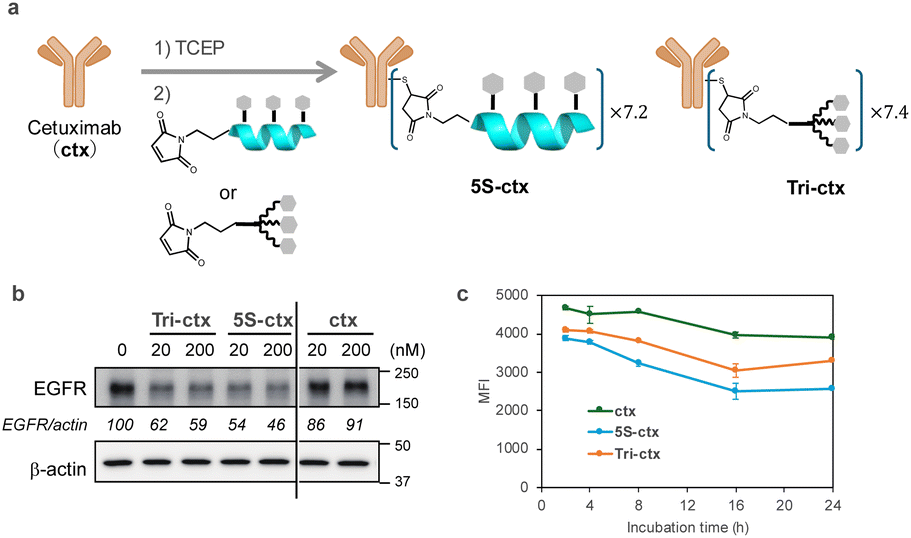
Ligands comprising GalNAc or its analogs have been used as extracellular protein degraders (e.g., lysosome-targeting chimeras [LYTACs] or ASGPR-targeting chimeras).37–39 LYTACs, which comprise a ligand that triggers endocytosis and a ligand that binds to the target protein, have recently attracted attention. Therefore, we attempted to use peptide 5S as an endocytosis ligand for LYTACs. A conjugate compound comprising ligands and an anti-epidermal growth factor receptor (EGFR) antibody (cetuximab) was synthesized with an intended drug-to-antibody ratio of 8.0 (Fig. 8a). This ratio was subsequently confirmed to be 7.2 for 5S and 7.4 for Tri-GalNAc via MALDI mass spectrometry. The EGFR degradation activity in HepG2 cells was evaluated using western blotting. Both peptide 5S (5S-ctx) and Tri-GalNAc (Tri-ctx) conjugates showed significant degradation activity at 20 and 200 nM (Fig. 8b). Next, we evaluated the clearance of EGFR from membrane by flow cytometry. The experiment was conducted using 1 nM of antibody-conjugates on HepG2 cells. 5S-ctx showed significant EGFR clearance at 8 h in HepG2 compared to Tri-ctx, and this clearance reached a plateau at 16 h (Fig. 8c). Although this experiment does not evaluate the degradation of EGFR, when combined with the western blot results, these findings suggest that 5S-ctx may have a higher degree of EGFR degradation than Tri-ctx.
 | ||
| Fig. 8 Application to LYTACs for membrane protein degradation. (a) Synthesis of antibody-conjugates by reaction between maleimide and cysteine side-chain. (b) HepG2 cells were incubated with compounds for 24 h. Epidermal growth factor receptor (EGFR) degradation was calculated relative to the expression of β-actin, which was used as the internal standard. (c) Evaluation of the EGFR clearance activity of ligand–cetuximab conjugates. Ligand–cetuximab conjugates were incubated with HepG2 cells for several hours. Cells were washed and incubated with Alexa Fluor 488 labeled F(ab′)2 Fragment Goat Anti-Human IgG, and the fluorescence intensity was measured using a flow cytometer. The EGFR internalization of ctx, Tri-ctx, and 5S-ctx is indicated as the mean fluorescence intensity (MFI) of four samples. Error bars represent the standard error of the mean of four samples. ctx, cetuximab. | ||
Conclusions
In this study, we successfully developed peptide-based hepatocyte-targeting DDS ligands using helical structures to control the orientation of GalNAc moieties. Our key peptide, 5S, demonstrated greater uptake efficiency and hepatocyte selectivity than the conventional Tri-GalNAc ligand. These findings underscore the effectiveness of our approach in enhancing ligand binding to ASGPR through geometric control. Our comprehensive analysis revealed that peptide 5S could facilitate the endocytosis of large proteins, such as streptavidin, more efficiently than Tri-GalNAc. This indicates its potential applicability in delivering macromolecules that typically cannot penetrate cellular membranes by themselves. Furthermore, the use of peptide 5S as an endocytosis ligand for LYTACs shows promise, despite the need for further optimization to enhance protein degradation efficiency. The flexibility of the Tri-GalNAc linker emerged as a critical factor in binding efficiency, highlighting the necessity for balanced structural rigidity and adaptability. Our molecular dynamics simulations and binding studies provided a deeper understanding of how the positional and structural attributes of GalNAc influence ASGPR engagement. Optimizing these ligands for in vivo applications remains essential. The inherent versatility of peptides, coupled with their ability to incorporate functional sequences, offers a promising avenue for overcoming current limitations.40 Overall, our approach involving the use of helical peptides to control ligand orientation represents a significant step forward in designing effective liver-targeted therapies. This methodology not only enhances the specificity and efficiency of DDS ligands but also holds potential for broader applications in targeting multivalent receptors, such as those involved in various viral infections.41,42 Our ongoing efforts aim to further refine these systems, potentially addressing membrane permeability challenges in drug discovery modalities that cannot penetrate membranes or target specific tissues themselves.43–47Data availability
Additional experimental data supporting this article are included in the ESI.† Reasonable requests for additional information can be made to the corresponding authors.Author contributions
Y. D. designed the research, and Takahito Ito and Y. D. wrote the paper. Takahito Ito, N. O., M. A., and T. N. performed the experiments and analyzed the results. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported in part by grants from the Japan Agency for Medical Research and Development (AMED; grant numbers 24mk0121286, 24ama221127, 24ak0101185, all to Y. D., 23K06043 to T. M., 24ak0101186 to N. O. and T. I., 24fk0310504 to N. O., and 24ae0121013 to M. A. and A. I.). The study also received support from the Japan Society for the Promotion of Science (KAKENHI; grant numbers 21K05320, 23H04926, both to Y. D., JP21K06490 and JP24K08645 to N. O.), the Japan Science and Technology Agency (JST), the establishment of university fellowships towards the creation of science technology innovation (grant number JPMJFS2140, to Takahito Ito), and the Sasakawa Scientific Research Grant from The Japan Science Society (to Takahito Ito). We thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.Notes and references
- H. Devarbhavi, S. K. Asrani, J. P. Arab, Y. A. Nartey, E. Pose and P. S. Kamath, Global burden of liver disease: 2023 update, J. Hepatol., 2023, 79, 516–537 CrossRef PubMed.
- Z. M. Younossi, P. Golabi, J. M. Paik, A. Henry, C. Van Dongen and L. Henry, The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): a systematic review, Hepatology, 2023, 77, 1335–1347 CrossRef PubMed.
- A. Schweitzer, J. Horn, R. T. Mikolajczyk, G. Krause and J. J. Ott, Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013, Lancet, 2015, 386, 1546–1555 CrossRef PubMed.
- J. Ferlay, M. Ervik, F. Lam, M. Lavresanne, M. Colonbet, L. Mery, M. Piñeros, A. Znaor, I. Soerjomataram and F. Bray, Global Cancer Observatory: Cancer Today, https://gco.iarc.who.int/today, accessed May 2024.
- J. U. Baenziger and D. Fiete, Galactose and N-Acetylgalactosamine-Specific Endocytosis of Glycopeptides by Isolated Rat Hepatocytes, Cell, 1980, 221, 611–620 CrossRef PubMed.
- R. J. Stockert, The Asialoglycoprotein Receptor: Relationships between Structure, Function, and Expression, Physiol. Rev., 1995, 75, 591–609 CrossRef CAS.
- E. A. Biessen, D. M. Beuting, H. C. P. F. Roelen, G. A. Marel, J. H. Boom and T. J. C. Berkel, Synthesis of Cluster Galactosides with High Affinity for the Hepatic Asialoglycoprotein Receptor, J. Med. Chem., 1995, 38, 1538–1546 CrossRef CAS PubMed.
- T. P. Prakash, J. Yu, M. T. Migawa, G. A. Kinberger, W. B. Wan, M. E. Ostergaard, R. L. Carty, G. Vasquez, A. Low, A. Chappell, K. Schmidt, M. Aghajan, J. Crosby, H. M. Murray, S. L. Booten, J. Hsiao, A. Soriano, T. Machemer, P. Cauntay, S. A. Burel, S. F. Murray, H. Gaus, M. J. Graham, E. E. Swayze and P. P. Seth, Comprehensive Structure-Activity Relationship of Triantennary N-Acetylgalactosamine Conjugated Antisense Oligonucleotides for Targeted Delivery to Hepatocytes, J. Med. Chem., 2016, 59, 2718–2733 CrossRef CAS PubMed.
- P. Kandasamy, S. Mori, S. Matsuda, N. Erande, D. Datta, J. L. S. Willoughby, N. Taneja, J. O'Shea, A. Bisbe, R. M. Manoharan, K. Yucius, T. Nguyen, R. Indrakanti, S. Gupta, J. A. Gilbert, T. Racie, A. Chan, J. Liu, R. Hutabarat, J. K. Nair, K. Charisse, M. A. Maier, K. G. Rajeev, M. Egli and M. Manoharan, Metabolically Stable Anomeric Linkages Containing GalNAc-siRNA Conjugates: An Interplay among ASGPR, Glycosidase, and RISC Pathways, J. Med. Chem., 2023, 66, 2506–2523 CrossRef CAS PubMed.
- T. P. Prakash, M. J. Graham, J. Yu, R. Carty, A. Low, A. Chappell, K. Schmidt, C. Zhao, M. Aghajan, H. F. Murray, S. Riney, S. L. Booten, S. F. Murray, H. Gaus, J. Crosby, W. F. Lima, S. Guo, B. P. Monia, E. E. Swayze and P. P. Seth, Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice, Nucleic Acids Res., 2014, 42, 8796–8807 CrossRef CAS PubMed.
- A. Mishra, T. R. Castaneda, E. Bader, B. Elshorst, S. Cummings, P. Scherer, D. S. Bangari, C. Loewe, H. Schreuder, C. Poverlein, M. Helms, S. Jones, G. Zech, T. Licher, M. Wagner, M. Schudok, M. de Hoop, A. T. Plowright, J. Atzrodt, A. Kannt, I. Laitinen and V. Derdau, Triantennary GalNAc Molecular Imaging Probes for Monitoring Hepatocyte Function in a Rat Model of Nonalcoholic Steatohepatitis, Adv. Sci., 2020, 7, 2002997 CrossRef CAS PubMed.
- S. K. Ramadugu, Y. H. Chung, E. J. Fuentes, K. G. Rice and C. J. Margulis, In silico Prediction of the 3D Structure of Trimeric Asialoglycoprotein Receptor Bound to Triantennary Oligosaccharide, J. Am. Chem. Soc., 2010, 132, 9087–9095 CrossRef CAS PubMed.
- M. Crisma, M. De Zotti, F. Formaggio, C. Peggion, A. Moretto and C. Toniolo, Handedness preference and switching of peptide helices. Part II: Helices based on noncoded α-amino acids, J. Pept. Sci., 2015, 21, 148–177 CrossRef CAS PubMed.
- M. Crisma and C. Toniolo, Helical screw-sense preferences of peptides based on chiral, Cα-tetrasubstituted α-amino acids, Biopolymers, 2015, 104, 46–64 CrossRef CAS.
- J. Zhang and S. Dong, In-Bridge Stereochemistry: A Determinant of Stapled Peptide Conformation and Activity, ChemBioChem, 2024, 25, e202300747 CrossRef CAS PubMed.
- M. Moiola, M. G. Memeo and P. Quadrelli, Stapled Peptides—A Useful Improvement for Peptide-Based Drugs, Molecules, 2019, 24, 3654 CrossRef CAS PubMed.
- M. Tanaka, Design and Synthesis of Chiral α,α-Disubstituted Amino Acids and Conformational Study of Their Oligopeptides, Chem. Pharm. Bull., 2007, 55, 349–358 CrossRef CAS PubMed.
- R. Banerjee, G. Basu, P. Chene and S. Roy, Aib-based peptide backbone as scaffolds for helical peptide mimics, J. Pept. Res., 2002, 60, 88–94 CrossRef CAS PubMed.
- Y. Demizu, S. Nagoya, M. Shirakawa, M. Kawamura, N. Yamagata, Y. Sato, M. Doi and M. Kurihara, Development of stapled short helical peptides capable of inhibiting vitamin D receptor (VDR)-coactivator interactions, Bioorg. Med. Chem. Lett., 2013, 23, 4292–4296 CrossRef CAS PubMed.
- A. Dhar, S. Mallick, P. Ghosh, A. Maiti, I. Ahmed, S. Bhattacharya, T. Mandal, A. Manna, K. Roy, S. Singh, D. K. Nayak, P. T. Wilder, J. Markowitz, D. Weber, M. K. Ghosh, S. Chattopadhyay, R. Guha, A. Konar, S. Bandyopadhyay and S. Roy, Simultaneous inhibition of key growth pathways in melanoma cells and tumor regression by a designed bidentate constrained helical peptide, Biopolymers, 2014, 102, 344–358 CrossRef CAS PubMed.
- J. Morimoto, Y. Hosono and S. Sando, Isolation of a peptide containing d-amino acid residues that inhibits the alpha-helix-mediated p53-MDM2 interaction from a one-bead one-compound library, Bioorg. Med. Chem. Lett., 2018, 28, 231–234 CrossRef CAS PubMed.
- S. Wada, Y. Hashimoto, Y. Kawai, K. Miyata, H. Tsuda, O. Nakagawa and H. Urata, Effect of Ala replacement with Aib in amphipathic cell-penetrating peptide on oligonucleotide delivery into cells, Bioorg. Med. Chem., 2013, 21, 7669–7673 CrossRef CAS PubMed.
- H. Yamashita, T. Misawa, M. Oba, M. Tanaka, M. Naito, M. Kurihara and Y. Demizu, Development of helix-stabilized cell-penetrating peptides containing cationic alpha,alpha-disubstituted amino acids as helical promoters, Bioorg. Med. Chem., 2017, 25, 1846–1851 CrossRef CAS PubMed.
- T. Misawa, N. Ohoka, M. Oba, H. Yamashita, M. Tanaka, M. Naito and Y. Demizu, Development of 2-aminoisobutyric acid (Aib)-rich cell-penetrating foldamers for efficient siRNA delivery, Chem. Commun., 2019, 55, 7792–7795 RSC.
- M. Oba, Y. Nagano, T. Kato and M. Tanaka, Secondary structures and cell-penetrating abilities of arginine-rich peptide foldamers, Sci. Rep., 2019, 9, 1349 CrossRef PubMed.
- M. Hirano, C. Saito, C. Goto, H. Yokoo, R. Kawano, T. Misawa and Y. Demizu, Rational Design of Helix-Stabilized Antimicrobial Peptide Foldamers Containing alpha,alpha-Disubstituted Amino Acids or Side-Chain Stapling, ChemPlusChem, 2020, 85, 2731–2736 CrossRef CAS PubMed.
- T. Ito, W. Hashimoto, N. Ohoka, T. Misawa, T. Inoue, R. Kawano and Y. Demizu, Structure-Activity Relationship Study of Helix-Stabilized Antimicrobial Peptides Containing Nonproteinogenic Amino Acids, ACS Biomater. Sci. Eng., 2023, 9, 4654–4661 CrossRef CAS PubMed.
- T. Ito, N. Matsunaga, M. Kurashima, Y. Demizu and T. Misawa, Enhancing Chemical Stability through Structural Modification of Antimicrobial Peptides with Non-Proteinogenic Amino Acids, Antibiotics, 2023, 12, 1326 CrossRef CAS PubMed.
- C. Fuhrer, I. Geffen, K. Huggel and M. Spiess, The two subunits of the asialoglycoprotein receptor contain different sorting information, J. Biol. Chem., 1994, 269, 3277–3282 CrossRef CAS.
- M. Wolny, M. Batchelor, G. J. Bartlett, E. G. Baker, M. Kurzawa, P. J. Knight, L. Dougan, D. N. Woolfson, E. Paci and M. Peckham, Characterization of long and stable de novo single alpha-helix domains provides novel insight into their stability, Sci. Rep., 2017, 7, 44341 CrossRef CAS PubMed.
- O. Khorev, D. Stockmaier, O. Schwardt, B. Cutting and B. Ernst, Trivalent, Gal/GalNAc-containing ligands designed for the asialoglycoprotein receptor, Bioorg. Med. Chem., 2008, 16, 5216–5231 CrossRef CAS PubMed.
- S. K. Mamidyala, S. Dutta, B. A. Chrunyk, C. Preville, H. Wang, J. M. Withka, A. McColl, T. A. Subashi, S. J. Hawrylik, M. C. Griffor, S. Kim, J. A. Pfefferkorn, D. A. Price, E. Menhaji-Klotz, V. Mascitti and M. G. Finn, Glycomimetic ligands for the human asialoglycoprotein receptor, J. Am. Chem. Soc., 2012, 134, 1978–1981 CrossRef CAS PubMed.
- E. Zacco, J. Hutter, J. L. Heier, J. Mortier, P. H. Seeberger, B. Lepenies and B. Koksch, Tailored Presentation of Carbohydrates on a Coiled Coil-Based Scaffold for Asialoglycoprotein Receptor Targeting, ACS Chem. Biol., 2015, 10, 2065–2072 CrossRef CAS PubMed.
- D. Stokmaier, O. Khorev, B. Cutting, R. Born, D. Ricklin, T. O. Ernst, F. Boni, K. Schwingruber, M. Gentner, M. Wittwer, M. Spreafico, A. Vedani, S. Rabbani, O. Schwardt and B. Ernst, Design, synthesis and evaluation of monovalent ligands for the asialoglycoprotein receptor (ASGP-R), Bioorg. Med. Chem., 2009, 17, 7254–7264 CrossRef CAS PubMed.
- M. Monestier, P. Charbonnier, C. Gateau, M. Cuillel, F. Robert, C. Lebrun, E. Mintz, O. Renaudet and P. Delangle, ASGPR-Mediated Uptake of Multivalent Glycoconjugates for Drug Delivery in Hepatocytes, ChemBioChem, 2016, 17, 590–594 CrossRef CAS PubMed.
- Y. C. Cheng and W. H. Prusoff, Relationship between the Inhibition Constant (Ki) and the Concentration of Inhibitor which Cause 50 per cent Inhibition (I50) of an Enzymatic Reaction, Biochem. Pharmacol., 1973, 22, 3099–3108 CrossRef CAS PubMed.
- S. Cecioni, A. Imberty and S. Vidal, Glycomimetics versus multivalent glycoconjugates for the design of high affinity lectin ligands, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed.
- G. Ahn, S. M. Banik, C. L. Miller, N. M. Riley, J. R. Cochran and C. R. Bertozzi, LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation, Nat. Chem. Biol., 2021, 17, 937–946 CrossRef CAS PubMed.
- D. F. Caianiello, M. Zhang, J. D. Ray, R. A. Howell, J. C. Swartzel, E. M. J. Branham, E. Chirkin, V. R. Sabbasani, A. Z. Gong, D. M. McDonald, V. Muthusamy and D. A. Spiegel, Bifunctional small molecules that mediate the degradation of extracellular proteins, Nat. Chem. Biol., 2021, 17, 947–953 CrossRef CAS PubMed.
- Y. Zhou, P. Teng, N. T. Montgomery, X. Li and W. Tang, Development of Triantennary N-Acetylgalactosamine Conjugates as Degraders for Extracellular Proteins, ACS Cent. Sci., 2021, 7, 499–506 CrossRef CAS PubMed.
- T. Braulke and J. S. Bonifacino, Sorting of lysosomal proteins, Biochim. Biophys. Acta, 2009, 1793, 605–614 CrossRef CAS PubMed.
- D. S. Kwon, G. Gregorio, N. Bitton, W. A. Hendrickson and D. R. Littman, DC-SIGN-Mediated Internalization of HIV Is Required for Trans-Enhancement of T Cell Infection, Immunity, 2002, 16, 135–144 CrossRef CAS PubMed.
- B. Tassaneetrithep, T. H. Burgess, A. Granelli-Piperno, C. Trumpfheller, J. Finke, W. Sun, M. A. Eller, K. Pattanapanyasat, S. Sarasombath, D. L. Birx, R. M. Steinman, S. Schlesinger and M. A. Marovich, DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells, J. Exp. Med., 2003, 197, 823–829 CrossRef CAS PubMed.
- H. Osawa, T. Kurohara, T. Ito, N. Shibata and Y. Demizu, CRBN ligand expansion for hematopoietic prostaglandin D(2) synthase (H-PGDS) targeting PROTAC design and their in vitro ADME profiles, Bioorg. Med. Chem., 2023, 84, 117259 CrossRef CAS PubMed.
- C. A. Foley, F. Potjewyd, K. N. Lamb, L. I. James and S. V. Frye, Assessing the Cell Permeability of Bivalent Chemical Degraders Using the Chloroalkane Penetration Assay, ACS Chem. Biol., 2020, 15, 290–295 CrossRef CAS PubMed.
- H. Yokoo, M. Naito and Y. Demizu, Investigating the cell permeability of proteolysis-targeting chimeras (PROTACs), Expert Opin. Drug Discovery, 2023, 18, 357–361 CrossRef PubMed.
- Y. Asami, K. Yoshioka, K. Nishina, T. Nagata and T. Yokota, Drug delivery system of therapeutic oligonucleotides, Drug Discovery Ther., 2016, 19, 256–262 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05606j |
| This journal is © The Royal Society of Chemistry 2024 |