
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFully conjugated tetraborylethylene: selenium mediated C–C double bond formation from diborylcarbenoid†
Yuki
Shibutani
 a,
Shuhei
Kusumoto
*b and
Kyoko
Nozaki
a
a,
Shuhei
Kusumoto
*b and
Kyoko
Nozaki
a
aDepartment of Chemistry and Biotechnology, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, Japan
bDepartment of Chemistry, Graduate School of Science, Tokyo Metropolitan University, 1-1 Minamiosawa Hachioji, Tokyo, Japan. E-mail: kusumoto@tmu.ac.jp
First published on 2nd October 2024
Abstract
Heteroatom-substituted ethylenes have long been studied owing to their potential application to electronic devices. In contrast to well-studied π-donor substituted ethylene, the π-acceptor substituted one has only been limitedly reported. While boron can be a candidate of π-acceptors, there has still been no example of fully conjugated tetraborylethylene (TBE). Herein, we synthesized the first fully conjugated TBE 2 by selenium-mediated C–C double bond formation from diborylcarbenoid 1, a synthetic equivalent of diborylcarbene (DBC). An intermediate of bis(diborylmethylene)-λ4-selane 3Se, wherein two DBC fragments were bound to one selenium atom, was confirmed. TBE 2 has a longer C–C bond length of 1.368(2) Å than typical C–C double bonds (1.34 Å) owing to π-electron deficiency. By density functional theory calculations, the LUMO was found to be low-lying at −1.75 eV by the contribution of vacant p-orbitals on the boron atoms adjacent to the C–C double bond.
Ethylene H2C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 has a planar geometry with all atoms placed on the same plane in order to maximize the overlap of the p-orbitals on the carbon atoms.1 By substituting the hydrogen atoms on the carbon atoms, the molecular structure and electronic properties of ethylene can be drastically diversified.2,3 Substituted ethylenes have been well-studied owing to their potential application to electronic devices.4 Heteroatoms adjacent to the carbon atoms have significant effects on the molecular/electronic structure of ethylene.2,3,5 Elements of group 15 or 16 bound to the CC structure can work as π-donors by the unshared p-type lone pair to make the C–C double bond electron-rich (Fig. 1a).2 Contrastively, while introduction of π-acceptors onto the central carbon atoms would cause electron deficiency to the ethylene π-orbital, only limited examples have been reported.3,5 The silyl group (–SiR3) has been known to behave as a π-acceptor when substituted onto ethylene because
CH2 has a planar geometry with all atoms placed on the same plane in order to maximize the overlap of the p-orbitals on the carbon atoms.1 By substituting the hydrogen atoms on the carbon atoms, the molecular structure and electronic properties of ethylene can be drastically diversified.2,3 Substituted ethylenes have been well-studied owing to their potential application to electronic devices.4 Heteroatoms adjacent to the carbon atoms have significant effects on the molecular/electronic structure of ethylene.2,3,5 Elements of group 15 or 16 bound to the CC structure can work as π-donors by the unshared p-type lone pair to make the C–C double bond electron-rich (Fig. 1a).2 Contrastively, while introduction of π-acceptors onto the central carbon atoms would cause electron deficiency to the ethylene π-orbital, only limited examples have been reported.3,5 The silyl group (–SiR3) has been known to behave as a π-acceptor when substituted onto ethylene because 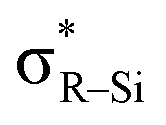 can interact with the πCC to pull the π-electrons from the CC moiety (Fig. 1b).3 Boron, which has a vacant p-orbital, would also show π-accepting ability when connected to CC structure. However, compared to the silicon-substituted ethylene, there are much fewer reports on tetraborylethylene (TBE).5 Reported TBEs have all been synthesized by the addition of diborane to diborylacetylene with a platinum catalyst, and their structures were discussed based on theoretical methods (Fig. 1c-i).5a–c Recently, Masarwa et al. achieved the first crystallographic analysis of TBE, tetrakis(pinacolatoboryl)ethylene I, which was first synthesized by Stang and Zhao et al. in 2020.5c,d In the obtained X-ray structure, only two of the four vacant orbitals on boron atoms are aligned parallel to the π-orbital of the CC moiety to participate in the π-conjugation system. So far, this is the sole example for structural analysis of TBE and there has still been no example of TBE wherein all the vacant p-orbitals on the boron atoms have full conjugation with the π-orbital of the CC unit. In recent years, not only in the field of cross-coupling reactions, polyborylated compounds have been featured as precursors of polyborylated reactive intermediates, which can be utilized for kinds of transformations.6 For example, Masarwa et al. recently demonstrated that the tetraborylated biradical generated from compound I was active for [2 + 2]-cycloaddition with olefins.6i In the reports, it is discussed that the stability of such transient borylated species is derived from the conjugation of the p-orbitals on carbon and boron atoms.6a,7 In this context, development of the synthetic method for fully conjugated TBE, which can serve a wide π-conjugation system over the B2CCB2 moiety, has been demanded.
can interact with the πCC to pull the π-electrons from the CC moiety (Fig. 1b).3 Boron, which has a vacant p-orbital, would also show π-accepting ability when connected to CC structure. However, compared to the silicon-substituted ethylene, there are much fewer reports on tetraborylethylene (TBE).5 Reported TBEs have all been synthesized by the addition of diborane to diborylacetylene with a platinum catalyst, and their structures were discussed based on theoretical methods (Fig. 1c-i).5a–c Recently, Masarwa et al. achieved the first crystallographic analysis of TBE, tetrakis(pinacolatoboryl)ethylene I, which was first synthesized by Stang and Zhao et al. in 2020.5c,d In the obtained X-ray structure, only two of the four vacant orbitals on boron atoms are aligned parallel to the π-orbital of the CC moiety to participate in the π-conjugation system. So far, this is the sole example for structural analysis of TBE and there has still been no example of TBE wherein all the vacant p-orbitals on the boron atoms have full conjugation with the π-orbital of the CC unit. In recent years, not only in the field of cross-coupling reactions, polyborylated compounds have been featured as precursors of polyborylated reactive intermediates, which can be utilized for kinds of transformations.6 For example, Masarwa et al. recently demonstrated that the tetraborylated biradical generated from compound I was active for [2 + 2]-cycloaddition with olefins.6i In the reports, it is discussed that the stability of such transient borylated species is derived from the conjugation of the p-orbitals on carbon and boron atoms.6a,7 In this context, development of the synthetic method for fully conjugated TBE, which can serve a wide π-conjugation system over the B2CCB2 moiety, has been demanded.
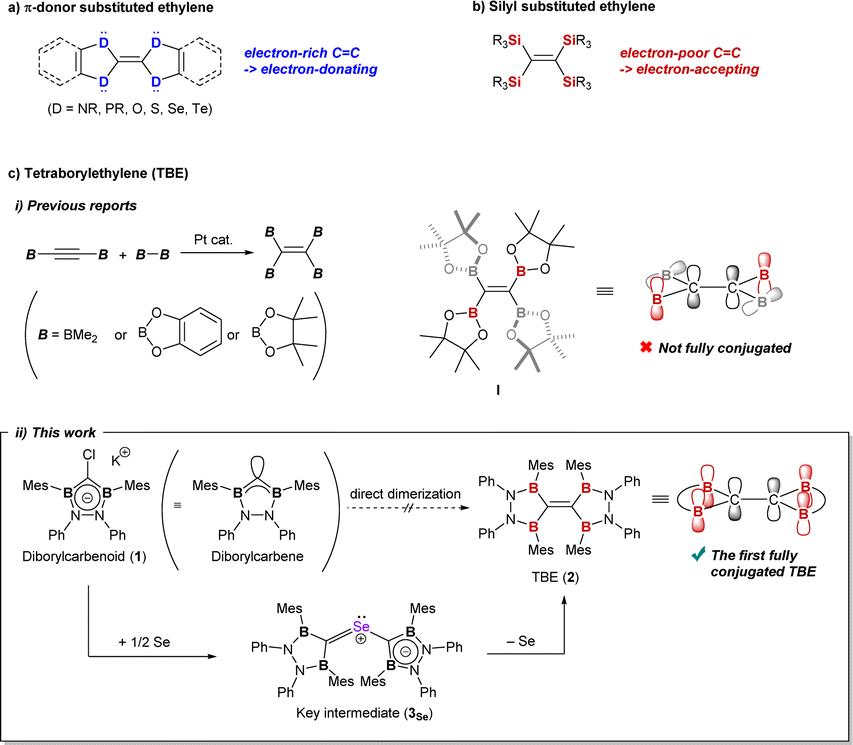 | ||
| Fig. 1 (a) General description of π-donor substituted ethylenes. (b) General description of ethylenes bearing silyl groups. (c-i) Reported synthetic method for TBEs and p-orbital interaction in compound I. (c-ii) Selenium-mediated TBE 2 formation described in the Edge article and p-orbital interaction in compound 2. | ||
Dimerization of diborylcarbene (DBC) would be an alternative approach to TBE. Some carbenes are reported to be in an equilibrium between monomeric (=free carbene) and dimeric (=olefin) states known as Wanzlick equilibrium.2c,8 Our calculation suggested that two DBC molecules readily dimerize into TBE; TBE was much more stable than two DBC molecules by 137 kcal mol−1 (see the ESI† for details). Previously, we synthesized diborylcarbenoid 1 as a DBC analogue but no distinct evidence for dimerization was obtained when DBC was generated from diborylcarbenoid.9 Here, in this report, we developed a novel synthetic method to approach TBE from diborylcarbenoid by employing selenium-mediated dimerization of DBC (Fig. 1c-ii). Two DBC moieties can get close to each other by forming bis(diborylmethylene)-λ4-selane (3Se), and this enabled the selective formation of TBE 2. This is analogous to the Barton–Kellogg reaction wherein elimination of the sulfur atom from thiirane by a phosphine molecule affords the corresponding olefin.10 Compound 2 is the first fully conjugated TBE; fixed-conformation by the CB2N2 cyclic structure made all p-orbitals on the central carbon and the adjacent boron atoms aligned parallel to form a π-conjugation system over six atoms. Contribution of the boron atom as a π-acceptor to the conjugation system was shown by electrochemical measurement and theoretical calculations.
The reaction of K/Cl-diborylcarbenoid 1 with a half equivalent of selenium powder in a benzene solution at 60 °C for 2 days afforded TBE 2 in a quantitative yield (Scheme 1-(i)). The 1H NMR spectrum of compound 2 in benzene-d6 indicated that 2 has a symmetric structure in the solution state; the four phenyl groups on nitrogen atoms and the two methyl groups at 2- and 6-positions on each Mes substituent on boron atoms showed equivalent signals to each other. The sp2-hybridized carbon between two boron atoms showed a broad signal at 196 ppm on the 13C NMR spectrum, which is significantly lower-field shifted than the reported TBE (165 ppm for ((C6H4O2)B)2CC(B(O2C6H4))2).5b Red crystals of compound 2 suitable for single crystal X-ray diffraction (scXRD) analysis were obtained by slowly concentrating a hexane solution at room temperature and the X-ray structure of 2 was unambiguously determined as shown in Fig. 2. The dihedral angles of B1–C1–C1′–B1′ and B2–C1–C1′–B2′ were 27° and 17° respectively, showing the slightly twisted geometry of TBE 2. This distorted structure would be due to the steric repulsion between facing mesityl groups (vide infra). The C–C double bond length of 2 was 1.368(2) Å, which was longer than the typical C–C double bond length (1.34 Å) and also than that in compound I (1.348(3) Å).5d This elongation of the C–C double bond in 2 can be attributed to the electron deficiency owing to the conjugation of the CC π-orbital with four boron p-orbitals. Our theoretical investigation of BH2-substituted ethylene ((H2B)2CC(BH2)2), the simplest TBE, showed that the introduction of boryl groups conjugated with the CC π-orbital leads to low-electron density of the CC moiety (see the ESI† for the detailed discussion). When diborylcarbenoid 1 reacted with a half equivalent of selenium powder at 0 °C for 5 days, formation of bis(diborylmethylene)-λ4-selane 3Se was confirmed by NMR measurement and scXRD analysis (Scheme 1-(ii) and Fig. 2b left, and also see the ESI† for details). As the heating of the reaction mixture gave compound 2 as the major product, it is suggested that compound 3Se is the key intermediate for the formation of TBE 2.10–12 When the stoichiometry of selenium was increased to more than one equivalent to carbenoid 1, formation of some selenium-containing compounds was suggested by 77Se NMR. The existence of compounds 4 and 5 was confirmed by mass spectroscopy and scXRD analyses, although compounds 4 and 5 could not be isolated, which retarded the complete characterization (Scheme 1-(iii) and Fig. 2c, also see the ESI† for details).13
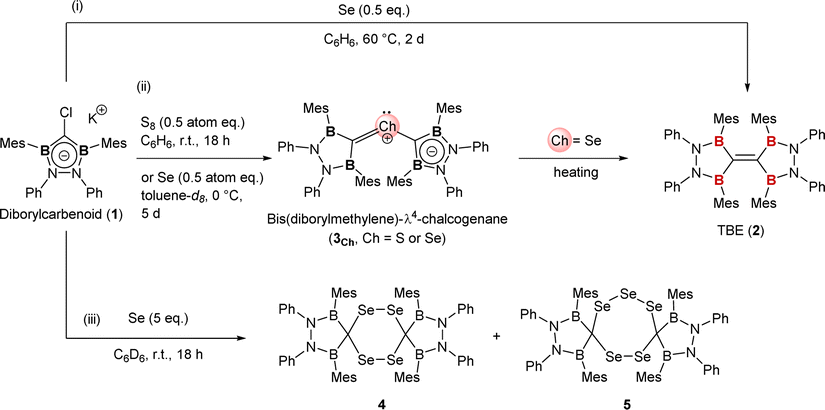 | ||
| Scheme 1 Reactions of diborylcarbenoid 1 with elemental chalcogens (sulfur or selenium). | ||
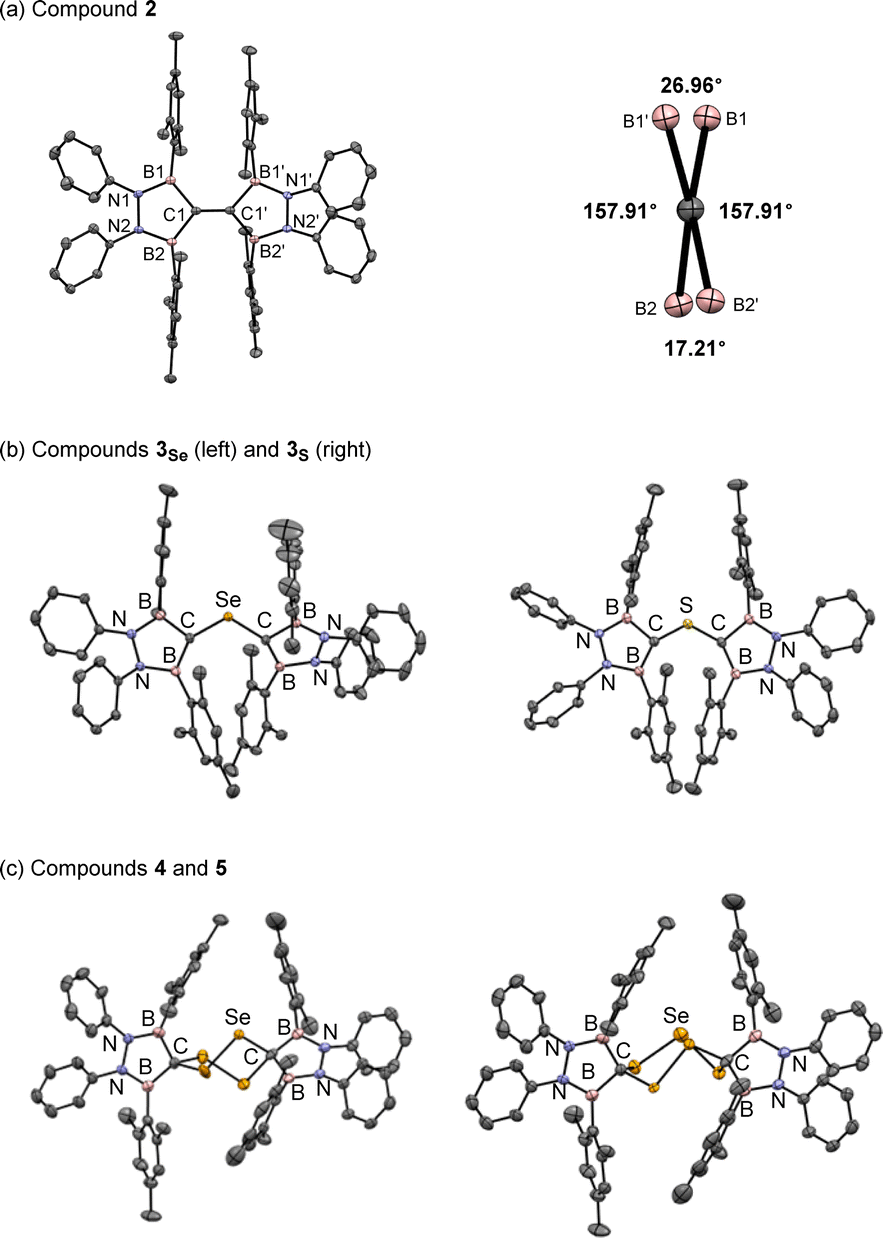 | ||
| Fig. 2 (a) X-ray structure of TBE 2. C1–C1′: 1.368(2) Å, C1–B1: 1.6004(15) Å, C1–B2: 1.5957(15) Å, and B1–C1–B2: 102.79(8)°. (b) X-ray structures of compounds 3Se and 3S. (c) X-ray structures of compounds 4 and 5. Thermal ellipsoids are drawn with a 50% probability. Hydrogen atoms are omitted for clarity. | ||
It should be noted that the reaction of compound 1 with a half-atom equivalent of elemental sulfur (S8) instead of selenium afforded bis(diborylmethylene)-λ4-sulfane 3S, a sulfur analogue of compound 3Se (Scheme 1-(ii) and Fig. 2b right, see the ESI† for details).14 In sharp contrast to the formation of TBE 2 from 3Se, compound 3S showed no conversion in a C6D6 solution even when heated at 50 °C (see the ESI† for details about the synthesis and VT-NMR measurement of compound 3S). Although the detailed reaction mechanism is still unclear, our theoretical investigation revealed that only in the case of selenium, the elimination of the chalcogen atom from bis(diborylmethylene)-λ4-chalcogenane (3Se or 3S) to yield TBE 2 was thermodynamically favorable (see the ESI† for the details).
To obtain more insight into the molecular and electronic structures of TBE 2, density functional theory (DFT) calculations at the M06-2X/6-311G(d,p) level of theory were conducted. The optimized structure of 2 well reproduced the X-ray structure with 1.360 Å of C–C double bond length and 15.4° and 22.6° of torsion angles. To observe the effect of bulky substituents on the boron atoms, simplified model structure 2′ wherein all mesityl and phenyl groups of 2 were replaced by hydrogen atoms was also considered at the same level of theory. The optimized structure of 2′ showed a completely planar structure, which suggested that the twisted geometry of compound 2 was driven by the steric repulsion at the bulky substituents on the boron atoms. On the other hand, the C–C double bond length of 2′ was 1.353 Å, which was just slightly shorter than that in 2 (1.360 Å) but still much longer than that of unsubstituted ethylene (1.324 Å). This suggested that the effect of bulky substituents on the C–C double bond length was quite small, and conjugation with the vacant p-orbitals on boron atoms was significant. When 2′ was compared with fulvalene, an isoelectronic compound, the lengths of C–C double bonds were almost comparable (1.353 Å for 2′ and 1.352 Å for fulvalene).
Fig. 3 shows the energy diagram of the frontier and π-orbitals of compound 2 and related molecules, ethylene, fulvalene, tetrathiafulvalene, and compound I. The bond lengths given in Fig. 3 are the ones for the optimized structures at the M06-2X/6-311G(d,p) level of theory. π-Donor substitution on the C–C double bond results in increasing the energy level of the π-bonding orbital (−9.08 eV for ethylene and −5.81 eV for tetrathiafulvalene), while having a small effect on the π-antibonding orbital (+1.35 eV for ethylene and +1.05 eV for tetrathiafulvalene). On the other hand, π-acceptor substitution results in lowering the energy level of the π-antibonding orbital. For compound I, the LUMO is located at −0.17 eV, 1.52 eV lower than that of ethylene. The effect of full conjugation was clearly exhibited by comparing compounds I and 2; there was a distinct contribution of the four vacant p-orbitals on the boron atoms of TBE 2 to the LUMO (−1.75 eV), which lies 1.58 eV lower than the LUMO of partly conjugated TBE I. The energy level of compound 2 was quite similar to that of fulvalene whose LUMO is delocalized to p-orbitals on the methine carbons adjacent to the central C–C double bond (see the ESI† for the description of molecular orbitals).
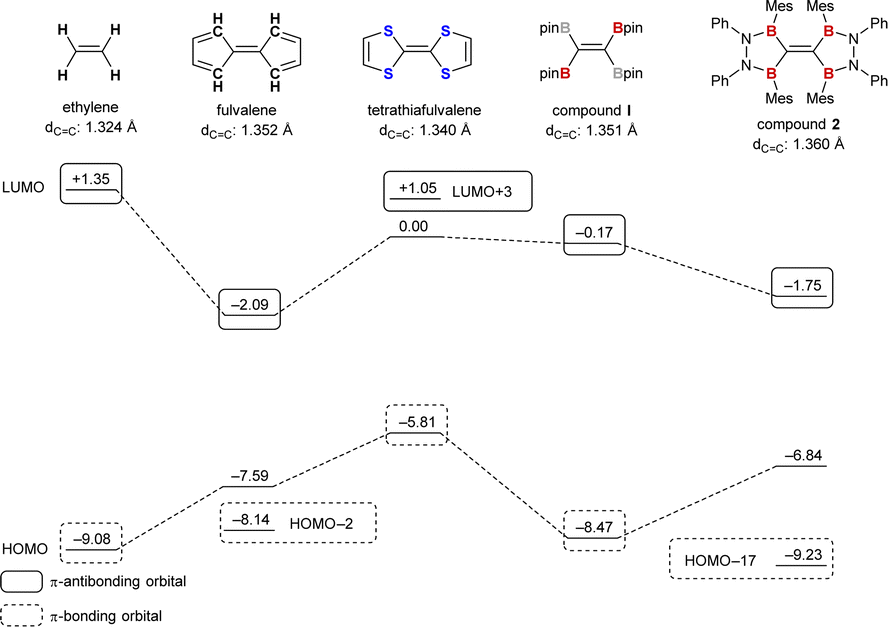 | ||
| Fig. 3 Frontier orbitals and π-orbitals of ethylene, fulvalene, tetrathiafulvalene, partly conjugated TBE I and fully conjugated TBE 2 calculated at the M06-2X/6-311G(d,p) level of theory. Calculated C–C bond length for the optimized structure is given below each structure. | ||
Natural bond orbital (NBO) analysis of compound 2 was also conducted. A π-bonding orbital over central carbon atoms was described with a small electron occupancy of 1.77 e. Second-order perturbation analysis clearly revealed π-electron delocalization of the CC moiety to the vacant p-orbitals on the four boron atoms with an electron donation of 81.4 kcal mol−1 in total (19.06–21.65 kcal mol−1 for each). The existence of the π-conjugation system on the central six atoms (B2CCB2) is clear; however, π-interaction between B and N atoms (73.69–77.65 kcal mol−1 for each) is much stronger than the donation from the CC π-bonding orbital. This suggests that B–N bonds in compound 2 behave like C–C double bonds in the backbone of fulvalene which resulted in a minimal difference in the electronic situation between these two molecules as described above.
Fig. 4 depicts the UV-Vis spectrum of compound 2 in a benzene solution. Two absorption bands were found at around 320 nm and 400–550 nm. TD-DFT calculations of compound 2 at the M06-2X/6-311G(d,p) level of theory were conducted and exhibited the same trend, relatively strong absorption at 304.84 nm (fcalc = 0.0287) and weak absorptions at around 400–500 nm (λmax = 391.72/395.73/482.21 nm, fcalc = 0.0073/0.0053/0.0019). According to the TD-DFT calculations, absorption at 320 nm is derived from πCC to 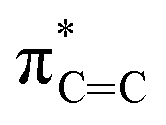 and absorption at 450 is from πBN to the
and absorption at 450 is from πBN to the 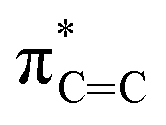 transition, respectively.
transition, respectively.
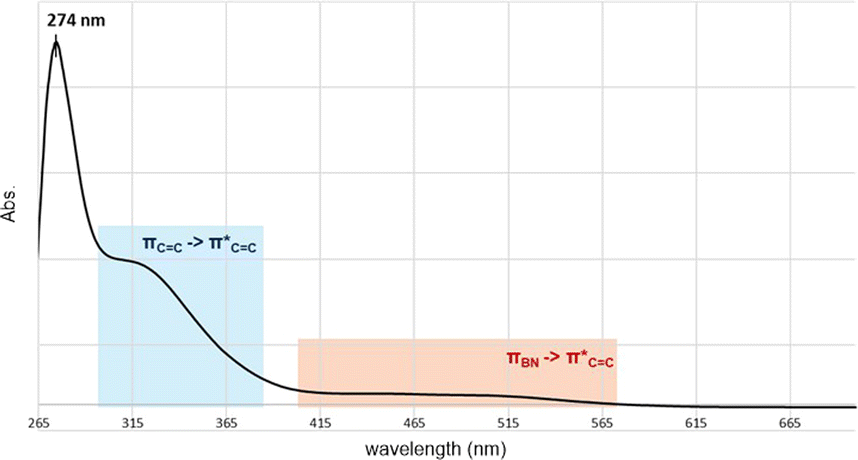 | ||
| Fig. 4 UV-Vis spectrum of compound 2 (1 × 10−5 M in C6H6). | ||
The cyclic voltammogram of TBE 2 in a THF solution at ambient temperature is shown in Fig. 5. When the scan rate was 0.1 V s−1, one oxidation step at +0.49 V and one reduction step at −1.92 V, both of which were irreversible, were found. These shifted to +0.32 V and −1.73 V when the scan rate was decreased to 0.02 V s−1. This suggests that one electron oxidation/reduction to afford a radical cation (2+)/anion (2−) can occur at the given voltage, but the generated species was not stable enough to be re-reduced/oxidized. The observed reduction potential for 2 was much lower than the redox potential of representative electron-accepting organic molecules such as 7,7,8,8-tetracyanoquinodimethane (TCNQ, +0.115 V in water, +0.21 V in CH3CN, vs. SCE)15a and the first to third reduction potentials of fullerene (−0.97, −1.34, −1.78 V in CH3CN/toluene at −10 °C, vs. Fc/Fc+),15b which meant that the electron-accepting nature of compound 2 was lower than those of highly electrophilic organic compounds.
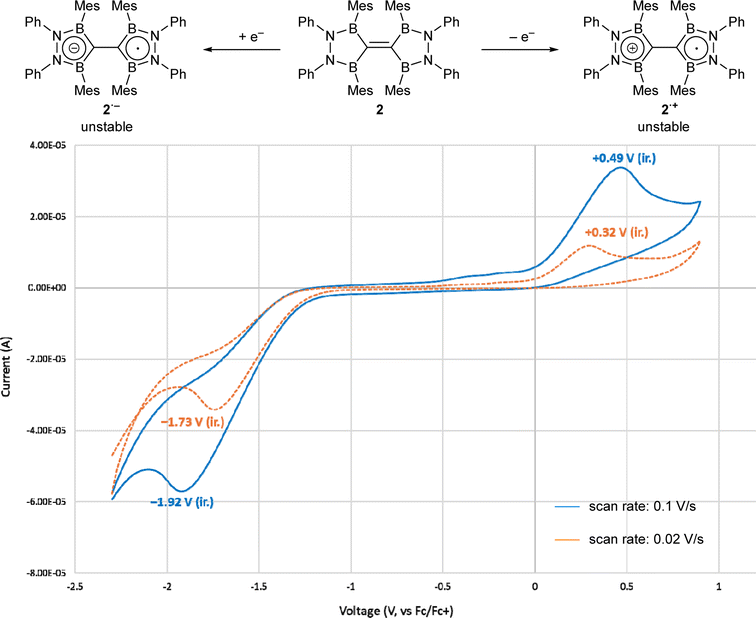 | ||
| Fig. 5 Cyclic voltammogram of compound 2 (0.1 M in THF with [NBu4][PF6] as a supporting electrolyte). | ||
TBE 2 was found to be applicable to the energy transfer (EnT) process by the Stern–Volmer luminescence quenching experiment with fac-Ir(ppy)3 as a photosensitizer (Fig. 6a). A linear relationship between the relative luminescence intensity and the concentration of TBE 2 was confirmed. From the linear approximation, the Stern–Volmer constant (KSV) for TBE 2 was calculated to be 12.6 mM−1 (Fig. 6b). It was recently reported that Bpin-substituted ethylenes including partly conjugated TBE I were active toward EnT from fac-Ir(ppy)3 (T1), and the KSV value increased from 0.095 mM−1 for geminal bis-Bpin-substituted ethylene to 0.1428 mM−1 for TBE I.6i The increase of the KSV value for borylethylenes along with the number of boryl substituents was explained by the stabilization of the borylethylene biradical, which was generated by the EnT process; unpaired electrons of the biradical species could be effectively delocalized with the increased number of boryl substituents.6i Although it must be noted that the solvent used here for the quenching experiment (THF) was different from that in the literature (CH2Cl2) and the KSV values of TBEs 2 and I cannot be compared directly, the large KSV value for TBE 2 can be understood as the result of full-conjugation.
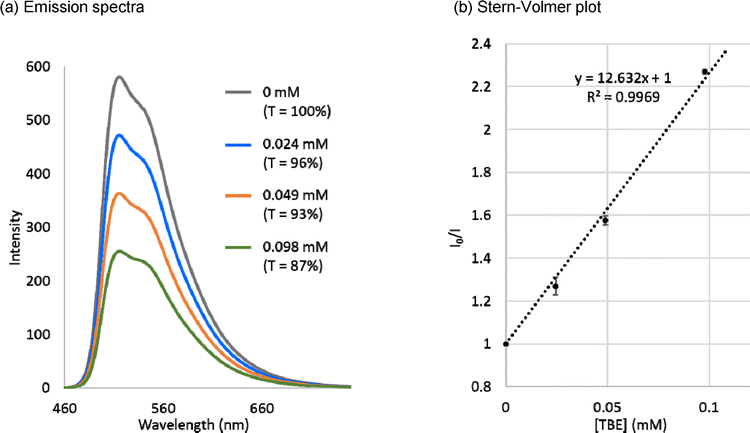 | ||
| Fig. 6 (a) Emission spectra of fac-Ir(ppy)3 (0.11 mM) in THF under the coexistence of TBE 2 as a luminescence quencher. T means the transmittance of the excitation light through TBE 2 calculated from the absorption at 1 × 10−5 M. (b) Stern–Volmer plot for TBE 2. To make the plot, intensity at 510 nm was employed. | ||
Conclusions
In summary, we developed a novel synthetic method for TBE employing diborylcarbenoid and elemental selenium to synthesize the first fully conjugated TBE. TBE 2 exhibited an overcrowded-ethylene-like crystal structure with a slightly twisted conformation and a long C–C double bond due to the π-electron withdrawal by the boron atoms. DFT calculations on 2 revealed the distinct contribution of vacant p-orbitals on the boron atoms and the high electron-accepting nature of compound 2.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 2, 3Se, 3S and co-crystals of 4 and 5 have been deposited at the CCDC under 2357519, 2357520, 2381240 and 2381489 and can be obtained from https://www.ccdc.cam.ac.uk/.Author contributions
S. K. and K. N. conceived and designed the study. Y. S. performed all the experiments and analyzed the data. All the authors discussed the results and co-wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The computations in this work were performed at the Research Center for Computational Science, Okazaki, Japan (Project: 24-IMS-C015). A part of this work was supported by the JST FOREST Program (Grant Number JPMJFR223K), JSPS KAKENHI, and Grant-in-Aid for Challenging Research (Pioneering) JP22K19021. This work was supported by “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Grant Number JPMXP1224UT0051.Notes and references
- (a) E. San-Fabián and F. Moscardó, J. Comput. Chem., 2014, 35, 1356–1363 CrossRef PubMed; (b) A. I. Krylov, C. D. Sherrill, E. F. C. Byrd and M. Head-Gordon, J. Chem. Phys., 1998, 109, 10669–10678 CrossRef CAS.
- For synthesis and properties of π-donor substituted ethylenes, see the following: (a) A. E.-W. A. O. Sarhan, Tetrahedron, 2005, 61, 3889–3932 CrossRef; (b) E. Çetinkaya, P. B. Hitchcock, H. Küçükbay, M. F. Lappert and S. Al-Juaid, J. Organomet. Chem., 1994, 481, 89–95 CrossRef; (c) F. E. Hahn, L. Wittenbecher, D. L. Van and R. Fröhlich, Angew. Chem., Int. Ed., 2000, 39, 541–544 CrossRef CAS; (d) E. Cetinkaya, P. B. Hitchcock, H. A. Jasim, M. F. Lappert and K. Spyropoulos, J. Chem. Soc., Perkin Trans. 1, 1992, 561–567 RSC; (e) T. A. Taton and P. Chen, Angew. Chem., Int. Ed., 1996, 35, 1011–1013 CrossRef CAS; (f) U. S. D. Paul and U. Radius, Chem.–Eur. J., 2017, 23, 3993–4009 CrossRef CAS PubMed; (g) M. K. Nayak, S. Suhr, N. Chrysochos, H. Rawat, C. Schulzke, V. Chandrasekhar, B. Sarkar and A. Jana, Chem. Commun., 2021, 57, 1210–1213 RSC; (h) D. M. Anderson, P. B. Hitchcock and M. F. Lappert, J. Organomet. Chem., 1989, 363, C7–C11 CrossRef CAS; (i) P. I. Jolly, S. Zhou, D. W. Thomson, J. Garnier, J. A. Parkinson, T. Tuttlea and J. A. Murphy, Chem. Sci., 2012, 3, 1675–1679 RSC; (j) N. Maigrot, L. Ricard, C. Charrier and F. Mathey, Angew. Chem., Int. Ed., 1988, 27, 950–951 CrossRef; (k) N. Maigrot, L. Ricard, C. Charrier and F. Mathey, Angew. Chem., Int. Ed., 1992, 31, 1031–1032 CrossRef.
- (a) H. Sakurai, Y. Nakadaira, M. Kira and H. Tobita, Tetrahedron Lett., 1980, 21, 3077–3080 CrossRef CAS; (b) H. Sakurai, H. Tobita, M. Kira and Y. Nakadaira, Angew. Chem., Int. Ed., 1980, 19, 620 CrossRef; (c) H. Sakurai, Y. Nakadaira, H. Tobita, T. Ito, K. Toriumi and H. Ito, J. Am. Chem. Soc., 1982, 104, 300–302 CrossRef CAS; (d) H. Sakurai, H. Tobita and Y. Nakadaira, Chem. Lett., 1982, 1251–1254 CrossRef CAS; (e) H. Sakurai, K. Ebata, K. Sakamoto, Y. Nakadaira and C. Kabuto, Chem. Lett., 1988, 965–968 CrossRef CAS; (f) H. Sakurai, K. Ebata, C. Kabuto and Y. Nakadaira, Chem. Lett., 1987, 301–304 CrossRef CAS; (g) A. Sekiguchi, M. Ichinohe, C. Kabuto and H. Sakurai, Bull. Chem. Soc. Jpn., 1995, 68, 2981–2988 CrossRef CAS; (h) A. Sekiguchi, K. Ebata, C. Kabuto and H. Sakurai, Chem. Lett., 1990, 539–542 CrossRef CAS; (i) H. G. V. Schnering, E. Krahé and G. Fritz, Z. Anorg. Allg. Chem., 1969, 365, 113–118 CrossRef; (j) A. Sekiguchi, M. Ichinohe, M. Takahashi, C. Kabuto and H. Sakurai, Angew. Chem., Int. Ed., 1997, 36, 1533–1534 CrossRef CAS; (k) A. Sekiguchi, T. Nakanishi, C. Kabuto and H. Sakurai, J. Am. Chem. Soc., 1989, 111, 3748–3750 CrossRef CAS; (l) A. Sekiguchi, M. Ichinohe, C. Kabuto and H. Sakurai, Organometallics, 1995, 14, 1092–1094 CrossRef CAS; (m) A. Sekiguchi, M. Ichinohe, C. Kabuto and H. Sakurai, Bull. Chem. Soc. Jpn., 1995, 68, 2981–2988 CrossRef CAS.
- (a) M. Mas-Torrent and C. Rovira, Chem. Soc. Rev., 2008, 37, 827–838 RSC; (b) W. Wu, Y. Liu and D. Zhu, Chem. Soc. Rev., 2010, 39, 1489–1502 RSC.
- (a) A. Maderna, H. Pritzkow and W. Siebert, Angew. Chem., Int. Ed., 1996, 35, 1501–1503 CrossRef CAS; (b) M. Bluhm, A. Maderna, H. Pritzkow, S. Bethke, R. Gleiter and W. Siebert, Eur. J. Inorg. Chem., 1999, 1693–1700 CrossRef CAS; (c) M. Zhang, Y. Yao, P. J. Stang and W. Zhao, Angew. Chem., Int. Ed., 2020, 59, 20090–20098 CrossRef CAS PubMed; (d) N. Eghbarieh, N. Hanania and A. Masarwa, Nat. Commun., 2023, 14, 2022 CrossRef CAS PubMed.
- (a) A. Marotta, C. E. Adams and J. J. Molloy, Angew. Chem., Int. Ed., 2022, 61, e202207067 CrossRef CAS PubMed; (b) N. Eghbarieh, N. Hanania, A. Zamir, M. Nassir, T. Stein and A. Masarwa, J. Am. Chem. Soc., 2021, 143, 6211–6220 CrossRef CAS PubMed; (c) K. N. Babu, F. Massarwe, R. R. Reddy, N. Eghbarieh, M. Jakob and A. Masarwa, Molecules, 2020, 25, 959 CrossRef CAS PubMed; (d) M. H. Zhang, Y. S. Yao, P. J. Stang and W. X. Zhao, Angew. Chem., Int. Ed., 2020, 59, 20090–20098 CrossRef CAS PubMed; (e) O. Salvado and E. Fernandez, Molecules, 2020, 25, 1758 CrossRef CAS PubMed; (f) J. Royes, A. B. Cuenca and E. Fernández, Eur. J. Org Chem., 2018, 2728–2739 CrossRef CAS; (g) X. J. Wang, Y. Wang, W. Huang, C. G. Xia and L. P. Wu, ACS Catal., 2021, 11, 1–18 CrossRef CAS; (h) X. C. Liu, W. B. Ming, A. Friedrich, F. Kerner and T. B. Marder, Angew. Chem., Int. Ed., 2020, 59, 304–309 CrossRef CAS PubMed; (i) N. Hanania, N. Eghbarieh and A. Masarwa, Angew. Chem., Int. Ed., 2024, 63, e202405898 CrossRef CAS PubMed.
- (a) A. Marotta, H. Fang, C. E. Adams, K. S. Marcus, C. G. Daniliuc and J. J. Molloy, Angew. Chem., Int. Ed., 2023, 135, e202307540 CrossRef; (b) C. Hwang, Y. Lee, M. Kim, Y. Seo and S. H. Cho, Angew. Chem., Int. Ed., 2022, 61, e202209079 CrossRef CAS PubMed.
- (a) H.-W. Wanzlick and E. Schikora, Chem. Ber., 1961, 94, 2389–2393 CrossRef CAS; (b) P. Bazinet, T.-G. Ong, J. S. O'Brien, N. Lavoie, E. Bell, G. P. A. Yap, I. Korobkov and D. S. Richeson, Organometallics, 2007, 26, 2885–2895 CrossRef CAS.
- Y. Shibutani, S. Kusumoto and K. Nozaki, J. Am. Chem. Soc., 2023, 145, 16186–16192 CrossRef CAS PubMed.
- (a) D. H. R. Barton and B. J. Willis, J. Chem. Soc. D, 1970, 1225–1226 RSC; (b) R. M. Kellogg and S. Wassenaar, Tetrahedron Lett., 1970, 11, 1987–1990 CrossRef; (c) R. M. Kellogg, Tetrahedron, 1976, 32, 2165–2184 CrossRef CAS.
- For bis(methylene)-λ4-selane or selenocarbonyl ylide, see the following: (a) K. Sugamata, Y. Urao and M. Minoura, Chem. Commun., 2019, 55, 8254–8257 RSC; (b) R. Allmann, A. F.-J. Kaiser, M. Krestel and G. Seitz, Angew. Chem., Int. Ed., 1986, 25, 183–184 CrossRef.
- For generation of a C–C double bond from episulfide/selenide, an isomer of bis(methylene)-λ4-sulfane/selane, see the following: (a) W. Ando, Y. Kumamoto and N. Tokitoh, Tetrahedron Lett., 1987, 28, 2867–2870 CrossRef CAS; (b) A. Schönberg and E. Frese, Chem. Ber., 1968, 101, 701–715 CrossRef; (c) M. Kamata, K. Murayama and T. Miyashi, Tetrahedron Lett., 1989, 30, 4129–4132 CrossRef CAS; (d) A. N. Bell, R. Fields, R. N. Haszeldine and D. Moran, J. Chem. Soc., Perkin Trans. 1, 1980, 487–489 RSC; (e) R. J. Bushby and M. D. Pollard, Tetrahedron Lett., 1977, 18, 3671–3672 CrossRef; (f) B. F. Bonini, L. Grossi, L. Lunazzi and D. Macciantelli, J. Org. Chem., 1986, 51, 517–522 CrossRef CAS; (g) A. Krebs, W. Rüger, B. Ziegenhagen, M. Hebold, I. Hardtke, R. Müller, M. Schütz, M. Wietzke and M. Wilke, Chem. Ber., 1984, 117, 277–309 CrossRef CAS; (h) A. Krebs and W. Rüger, Tetrahedron Lett., 1979, 20, 1305–1308 CrossRef; (i) A. Krebs, B. Kaletta, W.-U. Nickel, W. Rüger and L. Tikwe, Tetrahedron, 1986, 42, 1693–1702 CrossRef CAS; (j) A. Krebs, W. Rüger and W.-U. Nickel, Tetrahedron Lett., 1981, 22, 4937–4940 CrossRef CAS; (k) R. J. Bushby, M. D. Pollard and W. S. McDonald, Tetrahedron Lett., 1978, 19, 3851–3854 CrossRef.
- (a) T. M. Klapötke, B. Krumm, K. Polborn and M. Scherr, Eur. J. Inorg. Chem., 2006, 2006, 2937–2941 CrossRef; (b) K. Sugamata, T. Asakawa and M. Minoura, Eur. J. Inorg. Chem., 2023, 26, e202200780 CrossRef CAS.
- K. Sugamata, D. Hashizume, Y. Suzuki, T. Sasamori and S. Ishii, Chem.–Eur. J., 2018, 24, 6922–6926 CrossRef CAS PubMed.
- (a) A. R. Harris, A. Nafady, A. P. O'Mullan and A. M. Bond, Chem. Mater., 2007, 19, 5499–5509 CrossRef CAS; (b) Q. Xie, E. Perez-Cordero and L. Echegoyen, J. Am. Chem. Soc., 1992, 114, 3978–3980 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 235751923575202381240 and 2381489. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05928j |
| This journal is © The Royal Society of Chemistry 2024 |