
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Challenges in aptamer-based sensor development using carbon nanotube networks†
Laura
Ferrer Pascual
 a,
Eero
Gustafsson
a,
Juha
Siitonen
b,
Vasuki
Durairaj
c and
Tomi
Laurila
*ab
a,
Eero
Gustafsson
a,
Juha
Siitonen
b,
Vasuki
Durairaj
c and
Tomi
Laurila
*ab
aDepartment of Electrical Engineering and Automation, School of Electrical Engineering, Aalto University, PO Box 13500, 00076 Aalto, Finland. E-mail: tomi.laurila@aalto.fi
bDepartment of Chemistry and Materials Science, School of Chemical Engineering, Aalto University, PO Box 16200, 00076 Aalto, Finland
cSustainable Products and Materials, VTT Technical Research Centre of Finland, P.O. Box 1000, FI-02044 Espoo, Finland
First published on 2nd October 2024
Abstract
Electrochemical aptamer-based (EAB) sensors represent a promising biosensing platform, leveraging the selectivity of aptamers and the advantages of electrochemical methods. These sensors offer high sensitivity, rapid response, low limits of detection, cost-effectiveness, and miniaturization potential. While gold electrodes have been predominantly used in EAB sensors, alternatives such as carbon nanotubes (CNTs) are gaining attention. CNTs offer advantages like large surface area and conductivity but pose challenges due to their reactivity and 3D network structure. In this study, we explore the development of EAB sensors using single-wall carbon nanotube (SWCNT) networks, emphasizing on the challenges and electroanalytical insights. Three key electrochemical parameters are proposed for assessing EAB sensor performance: (i) variations in peak current, (ii) shifts in peak position, and (iii) the restoration of the background current. Focusing solely on peak current changes can be misleading, as factors like aptamer surface depletion can influence it. Additionally, both partial and integrated currents should be monitored in square wave voltammetry (SWV) analysis, considering both ON and OFF behaviours across frequencies. This comprehensive approach provides a preliminary assessment of successful binding and surface passivation in EAB sensors when combined with surface analytical techniques such as surface plasmon resonance (SPR) measurements.
1. Introduction
Electrochemical aptamer-based (EAB) sensors offer significant promise in the field of biosensing. EAB sensors combine the high specificity of aptamers with the high sensitivity, rapid response, low limits of detection, low cost, and miniaturization of electrochemical methods.1,2 In addition, EAB sensors are promising for multi-drug screening because their aptamer sequence can be customised for different target molecules and their signal arises from the binding of the target instead of its chemical reactivity.3Aptamers are short nucleic acid strands with high selectivity towards their target arising from systematic evolution. The aptamer refolds when binding to the target, changing its three-dimensional conformation. The folding of the strand changes the distance between a redox reporter attached to the aptamer and the electrode surface, producing a change in the electron transfer kinetics (Fig. 1). The signal is classified as “signal on” or “signal off” depending on whether the binding-induced conformational changes moves the redox label closer to or further away from the electrode surface, respectively.4
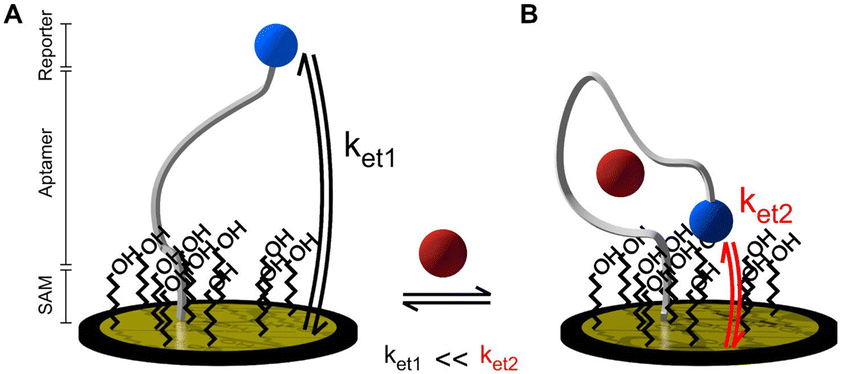 | ||
| Fig. 1 Working principle of an ideal electrochemical aptamer-based sensor in A) the absence and B) presence of target.5 | ||
As of date, most electrochemical aptamer-based studies are focused on the use of gold electrodes as sensing material due to the widely known gold-alkanethiol chemistry.6,7 Despite their potential, gold-based systems face a limitation in their operational lifespan due to the inherent instability of the weak thiol-bond. This instability hinders the continuous application of voltage during measurements, as it can lead to the desorption of the aptamer-SAM monolayer.8 In addition, gold presents a high cost compared to other available sensing materials such as carbon nanotubes (CNTs). There have been a few studies investigating alternative materials such as glassy carbon,1,9,10 silicon,11 or graphene12 for EAB sensor development. Carbon nanotubes are often considered promising electrode materials for electrochemical sensing due to their large surface area, high conductivity, good sensitivity, wide potential windows, and fast response times.13 While the elevated reactive surface area of CNTs is typically advantageous, it also results in increased non-specific interactions, which are unfavourable for aptamer binding. Moreover, the 3D structure of CNTs promotes volume interactions over surface interactions, hindering the uniform orientation of aptamers during immobilization. Lastly, the typically advantageous flexibility in the functionalisation of carbon surfaces proves disadvantageous for EAB sensors, as it leads to surfaces with large heterogeneity.14,15
Recently, there has been increasing scrutiny regarding the practical functionality of electrochemical aptamer sensors, even when deployed on traditional gold (Au) surfaces. In particular, the oversimplified depiction of their operation, as illustrated in Fig. 1, has faced criticism.16 As mentioned earlier, substituting carbon-based materials for Au in these sensors amplifies the contribution of non-specific interactions, thereby adding complexity to the underlying challenges. In this context, we present the findings of our recent investigations aimed at developing electrochemical aptamer-based sensors integrated with single-wall carbon nanotube (SWCNT) networks.
Our system under investigation involves SWCNT network electrodes on a glass support, onto which aptamers specific to vancomycin have been immobilized using four distinct strategies. The first strategy is centred on the direct immobilization of aptamers through non-specific interactions with the SWCNTs. The second strategy involves the covalent binding of the amine terminal group of the aptamers to the carboxylic groups of SWCNTs via EDC/NHS coupling. The last strategy employs a linker molecule (1-pyrenebutyric acid N-hydroxysuccinimide ester, Pyr-NHS) with two different protocols. In one protocol, termed the ‘layer-by-layer (LbL)’ approach, the linker is first deposited, followed by the drop-casting of the aptamer. The alternative protocol involves pre-mixing the aptamers and the linker molecule before drop-casting the solution onto the SWCNTs. In summary, these four protocols are denoted as (i) direct non-covalent, (ii) direct covalent, (iii) linker LbL, and (iv) linker pre-mixed, respectively (Fig. 2). Detailed descriptions of these immobilization protocols, along with the materials and methods used, are provided in the experimental section. Vancomycin (VA) is selected as the target molecule because it is classified as one of the critical antibiotics for human medicine by the World Health Organization (WHO). It serves as the last resort in treating resistant MRSA and holds the top position on the US Center for Disease Control's list of antibiotics associated with high costs in treating antibiotic-resistant infections.17 Our goal is to shed light on the challenges that arise when transitioning from flat Au electrodes to the three-dimensional world of SWCNT networks. Furthermore, we aim to provide readers with valuable electroanalytical insights into the system, aiding in the assessment of successful aptamer binding to the electrode. We argue that to evaluate the electroanalytical performance of an EAB sensor, it is crucial to consider three key parameters:
(i) Variations in the measured peak current,
(ii) Shifts in the peak position, and
(iii) The restoration of the original background current and peak position after the target measurement.
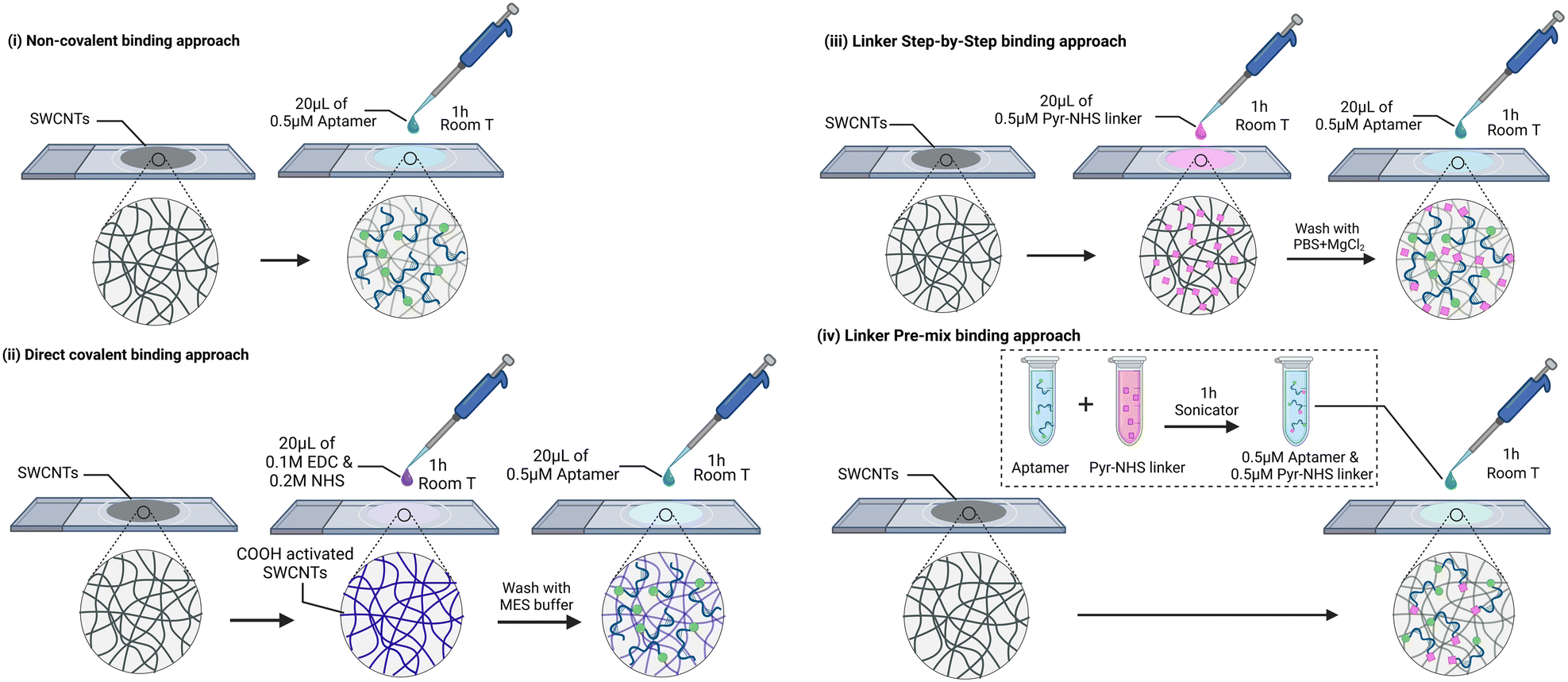 | ||
| Fig. 2 Aptamer binding approaches of this work: (i) non-covalent immobilization through aptamer adsorption to SWCNTs, (ii) direct-covalent immobilization through amide bond formation, (iii) indirect immobilization through linker LbL method, and (iv) indirect immobilization through linker pre-mix method. | ||
At present, the assessment of EAB sensors relies exclusively on changes in peak current—a criterion that may be prone to misinterpretation, given that alterations in peak current can stem from factors such as the gradual depletion of aptamers from the electrode surface. Furthermore, when employing the commonly used Square-Wave Voltammetry (SWV) technique, it is crucial to monitor the three key parameters defined above using both partial and integrated currents. Focusing solely on the latter can obscure vital information regarding peak position and peak current. Additionally, it is imperative to examine both the ON and OFF behaviours as a function of frequency. This comprehensive analysis is essential to confidently affirm the successful binding and surface passivation of the sensor. Alongside electrochemical investigations, physicochemical characterizations are essential to substantiate our arguments. However, it is crucial to recognize the limitations of commonly used techniques like Infrared spectroscopy, Surface Plasmon Resonance (SPR), and atomic force microscopy, especially when applied to physically and chemically complex 3D electrode structures, as demonstrated in this manuscript. In this study, SPR measurements are conducted to comprehend adsorption interactions at the SWCNT networks and elucidate the efficacy of our different aptamer binding protocols. Nonetheless, deriving quantitative conclusions about the efficacy of aptamer binding to the electrode surface or their target response requires consideration of the three aforementioned electrochemical response parameters, as discussed in the following manuscript.
To clarify the primary focus of our investigation, it is important to delineate the scope of this study. This manuscript focuses on the fundamental challenges encountered in the development of aptamer-based sensors, emphasizing conditions relevant to physiological environments, with all measurements conducted in phosphate-buffered saline at pH 7.4. The inherent selectivity of aptamers, combined with the passivation of the sensor's surface with the linker molecules, is anticipated to reduce interference from electrochemically active inner-sphere probes (ISR). While studies on pH variations and interferents are beyond the scope of this work, these will be explored in a forthcoming study focused on sensor performance optimization.
2. Experimental
Sulfo-NHS (N-hydroxysulfosuccinimide), EDC (N-(3-dimethylamino-propyl)-N′-ethylcarbodiimide hydrochloride), 0.1 M MES buffer (2-(N-morpholino)ethanesulfonic acid, 0.9% NaCl, pH 4), and vancomycin hydrochloride were purchased from Thermo Fisher. Magnesium chloride (MgCl2), 1× phosphate buffered saline (PBS), Atto MB2 methylene blue derivative, Pyr-NHS linker (1-pyrenebutyric acid N-hydroxysuccinimide ester) was purchased from Sigma Aldrich. Potassium chloride (KCl) was purchased from Merck Suprapur. The modified aptamer was purchased from Integrated DNA Technologies. The aptamer sequence: 5′-CG AGG GTA CCG CAA TAG TAC TTA TTG TTC GCC TAT TGT GGG TCG G-3′ with modification on the 5′ with an amino group and the redox reporter methylene blue (ATTO-MB2) on the 3′ end. The cartoon model (made by M-Fold) is shown in the ESI† (Fig. SI1).2.1 Electrode fabrication
The single-wall carbon nanotubes (SWCNTs) were provided by Canatu Oy. Briefly, the SWCNT networks were fabricated in a laminar flow reactor by chemical vapor deposition and were collected using a membrane filter. The SWCNTS fabrication protocol is described in detail in.18 The electrode fabrication consisted of press-transferring the SWCNTs onto a glass substrate where the network was then densified by adding 99.7% ethanol and letting it evaporate in air. Electrical contact was made by adding conductive silver paste and letting it dry for a minimum of 30 min. Then a conductive copper tape was used to contact the silver paste to the copper slide. PTFE-tape was used to insulate the electrode from other regions except a 3 mm hole.2.2 Aptamer binding methods
Four different EAB sensors were fabricated based on the aptamer binding method (Fig. 2): (i) in the non-covalent binding approach, the aptamer was attached to the CNT surface due to the adsorption of the DNA strand and methylene blue. To do so, 20 μL of 0.5 μM aptamer solution in PBS + 2 mM MgCl2 was drop-casted onto the electrodes for one hour at room temperature and then cleaned with PBS + 2 mM MgCl2. (ii) In the direct covalent binding approach, 20 μL of a solution of 0.1 M EDC and 0.2 M NHS in MES buffer was drop-casted to the electrode for 1 h at room temperature. Then, the electrode was cleaned with MES buffer and immediately 20 μL of 0.5 μM aptamer solution in PBS + 2 mM MgCl2 was drop-casted for another hour at room temperature. Finally, the electrode was cleaned with PBS + 2 mM MgCl2. The EDC/NHS concentration and ratio was optimised based on the largest peak shift upon VA binding (Table SI1†). For the linker approach, two different protocols were used: a so-called “layer-by-layer (LbL)” functionalization and a “pre-mix” functionalization. (iii) The “LbL” functionalization consisted in first drop-casting 20 μL of 0.5 μM Pyr-NHS linker to the electrode for 1 h at room temperature. Then, the electrode was cleaned with PBS + 2 mM MgCl2 and 20 μL of 0.5 μM aptamer solution in PBS + 2 mM MgCl2 was drop-casted for 1 h at room temperature. Finally, the electrode was cleaned with PBS + 2 mM MgCl2. (iv) The “pre-mix” functionalization consisted of bath sonication for 1 h a solution containing 0.5 μM of aptamer and 0.5 μM of linker. The temperature of the bath increased to 44 °C. Then, 20 μL of the solution was drop-casted to the electrodes for 1 h and then cleaned with PBS + 2 mM MgCl2.2.3 Washing methods
After the electrode functionalization, the electrodes underwent washing using two different protocols to remove most of the loosely attached aptamers. One method involved immersing the electrodes in a 500 μM vancomycin bath solution in PBS with 2 mM MgCl2 for 30 minutes. The second method consisted of repetitive washings in a 6 M GuHCl solution: 40 μL of GuHCl solution was drop-casted onto each electrode for 15 minutes, followed by immersion in PBS with 2 mM MgCl2. This procedure was repeated two additional times. Following the washing protocols, the electrodes were placed in PBS with 2 mM MgCl2 before the SWV measurements.2.4 Surface plasmon resonance
The SWCNT networks were press-transferred onto pre-cleaned (UV-ozone treated) Au-coated SPR sensors (BioNavis Ltd.) and densified with 99.7% ethanol, followed by air drying. The SPR measurements were carried out using a multi parametric surface plasmon resonance instrument (MP-SPR model Navi 200, BioNavis Ltd.). All CNT modified Au sensors were first stabilised in the measurement buffer for 2 hours following which the adsorption studies were carried out from different test solutions at 10 μl min−1 flow rate. Following each adsorption step, the sensors were rinsed with the measurement buffer solution at 10 μl min−1 flow rate for 25 min.2.5 Electrochemical techniques
Electrochemical investigations were carried out using both conventional Cyclic Voltammetry (CV) and Square-Wave Voltammetry (SWV). Experiments were conducted with a Gamry Reference 600 + potentiostat. A three-electrode cell was used with an Ag/AgCl reference electrode (Radiometer Analytical) and a Pt wire (Goodfellow) counter electrode. The solutions were purged with N2 for 30 min before the experiments. All the measurements were carried out at room temperature in a Faraday cage.2.6 Measurement protocols
All measurements were performed using four different sensor types, with three nominally identical electrodes per type (N = 3). For each sensor type, the electrodes measured the same solution, representing technical repeats. This setup allowed us to assess the precision and consistency of the biosensor system across replicates.2.7 Data analysis
The partial currents in the SWVs have been potential corrected by the addition of 50 mV to forward currents and subtraction of 50 mV to reverse currents. The “current decrease” and “signal change” are calculated from eqn (1):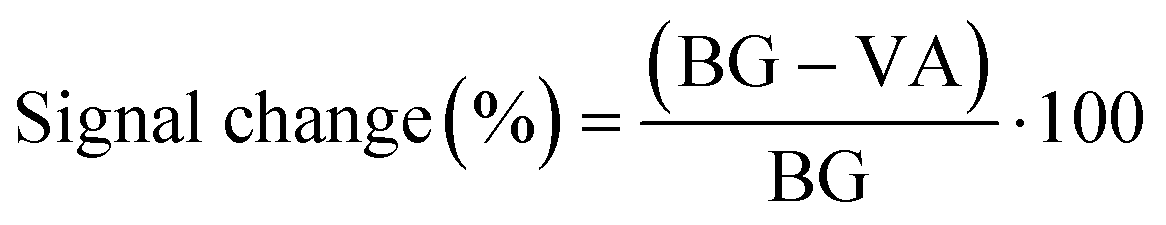 | (1) |
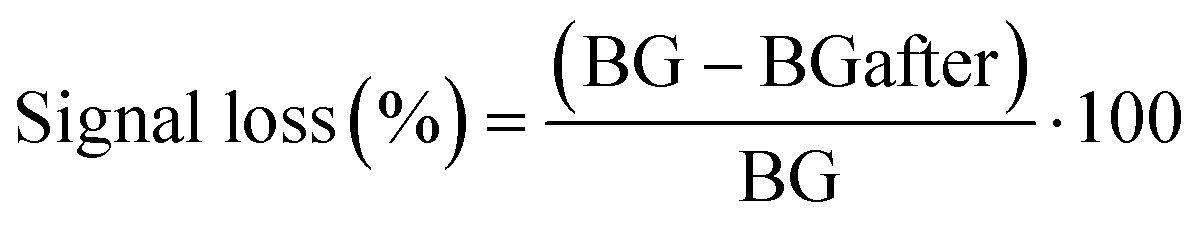 | (2) |
Limit of detection (LOD) is calculated with eqn (3):
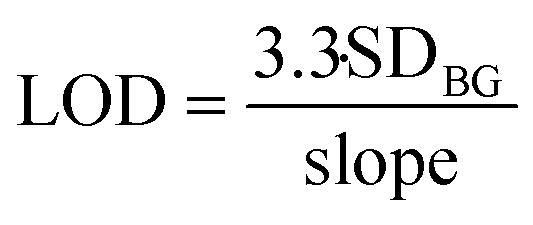 | (3) |
3. Results
3.1 Surface plasmon resonance
Au coated SPR sensors were first pre-cleaned through UV-ozone treatment, followed by rinsing with MilliQ water and air drying. SWCNT network was then press-transferred on top of the Au sensor surface and densified by adding a drop of 99.7% ethanol and letting it evaporate in air. Note that this sample preparation was done solely for the purpose of SPR measurements. SPR sensograms obtained using a 670 nm laser wavelength for the different adsorption studies carried out in this work are shown in Fig. 2. The angle at which the SPR effect occurs for a particular wavelength is highly sensitive to the refractive index (measured in refractive index unit – RIU) of the medium that is in contact with the metal surface of the SPR sensor.19 Adsorption events occurring at the sensor surface thus result in a change in the SPR angle, corresponding to the refractive index change due to the adsorbed layer. The thickness of the adsorbed layer can be determined using the eqn (4):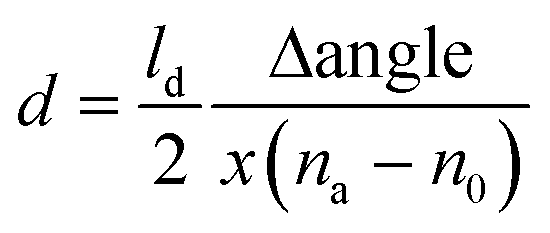 | (4) |
| Δm = d·ρ | (5) |
| Aptamer binding | Material adsorbed | Refractive index (RI) | Density (g cm−3) | Thickness (nm) | Mass (mg m−2) |
|---|---|---|---|---|---|
| a Average values of aptamer and linker refractive index and density have been used here to estimate the adsorption parameters from this equimolar mixture. | |||||
| — | Vancomycin21 | 1.735 | 1.7 | 6.0 | 10.3 |
| — | ATTO-MB2 (ref. 22) | 1.676 | 1.2 | 7.3 | 8.7 |
| Direct non-covalent | Aptamer23–25 | 1.520 | 1.3 | 14.2 | 18.5 |
| Vancomycin | 1.735 | 1.7 | 3.2 | 5.4 | |
| Direct covalent | EDC/NHS26 | 1.528 | 1.6 | 45.7 | 36.5 |
| Aptamer | 1.520 | 1.3 | 1.2 | 1.5 | |
| Vancomycin | 1.735 | 1.7 | 0.3 | 0.5 | |
| Linker LbL | Linker27 | 1.852 | 1.2 | 4.8 | 5.7 |
| Aptamer | 1.676 | 1.2 | 2.0 | 2.7 | |
| Vancomycin | 1.520 | 1.3 | 0.2 | 0.4 | |
| Linker pre-mixed | Linker + aptamera | 1.686a | 1.3a | 9.2 | 11.4 |
| Vancomycin | 1.735 | 1.7 | 2.2 | 3.8 | |
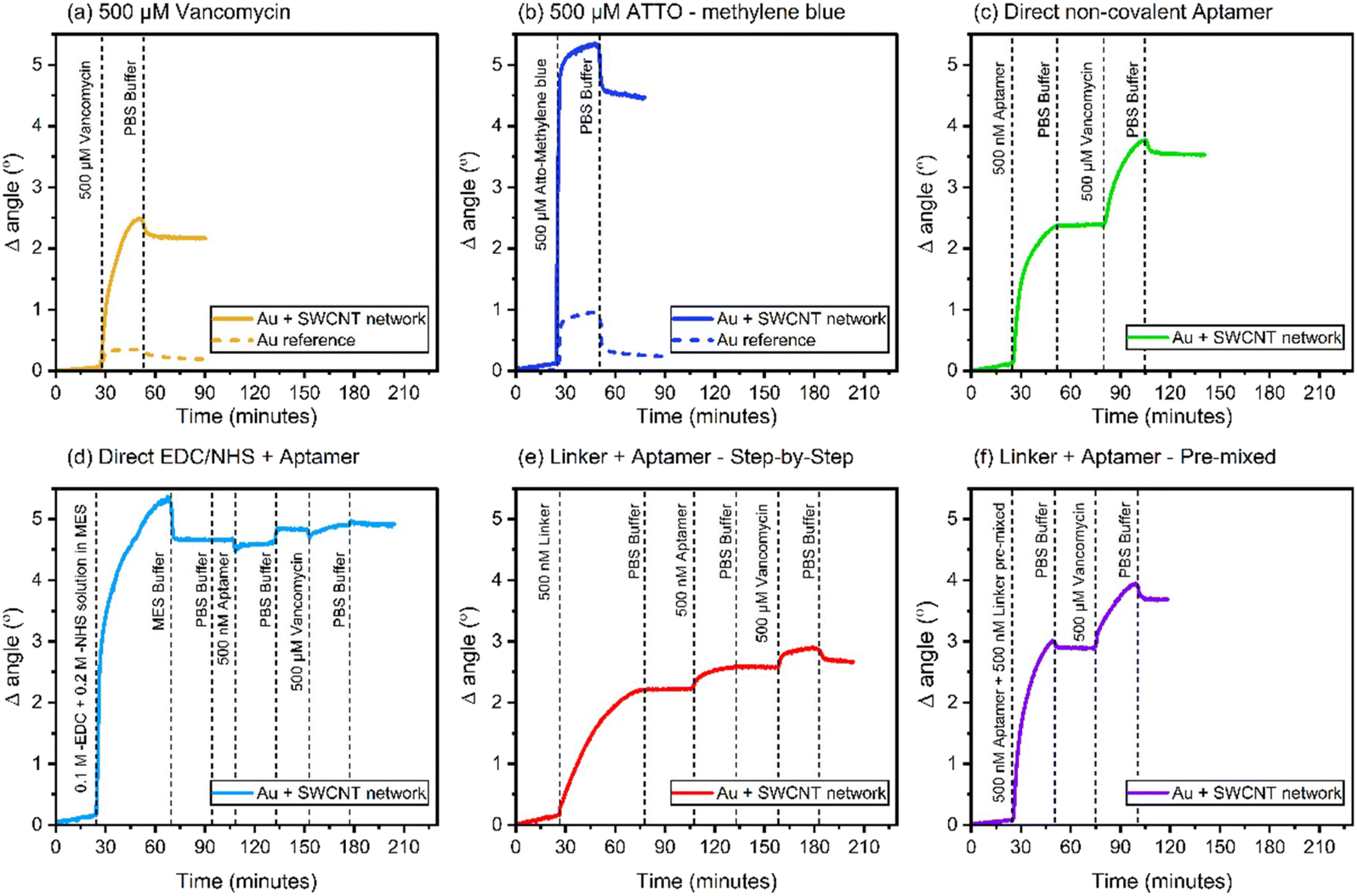 | ||
| Fig. 3 SPR sensograms showing the adsorption of (a) 500 μM vancomycin, (b) 500 μM ATTO-MB2, (c) 500 nM aptamer via direct non-covalent binding, (d) direct covalent activation followed by 500 nM aptamer, (e) 500 nM linker followed by 500 nM aptamer (LbL approach) and (f) 500 nM linker + 500 nM aptamer – pre-mixed solution. The responses of adsorbed aptamers to buffer rinse and 500 μM vancomycin injection were also followed for each aptamer linking protocol (c–f). | ||
According this SPR results, the direct covalent and linker LbL systems exhibit similar behaviour in terms of their aptamer adsorption characteristics, hinting at the presence of ordered aptamers on their surfaces. It is crucial to note that these observed adsorption patterns sharply contrast with the other two system types. Both the direct covalent and linker LbL systems display relatively modest levels of both aptamer and vancomycin adsorption, when compared to direct non-covalent and linker pre-mixed approaches which demonstrate approximately ten times greater adsorption of both aptamers and vancomycin. These results suggest that the direct covalent and the linker LbL systems yield more controlled aptamer responses to vancomycin, possibly indicating a degree of ordering in the arrangement of aptamers on their surfaces. Conversely, the alternative systems result in less organized and more random responses. Further, the SPR results suggest that there is some degree of coupling of the aptamers to the SWCNT network especially in the case of direct covalent linking. However, due to the limited number of carboxylic groups on the SWCNT network materials (as documented in ref. 28 and 29), it is likely that most of the applied EDC/NHS reacts further, leading to the formation of by-products,30 which then partially foul the surface (Fig. 3d).
3.2 Electrochemical characterizations
In this section, we present a comprehensive analysis of the electrochemical performance of our designed electrochemical aptamer-based sensors. Our investigation focuses on four different aptamer binding methods to evaluate their efficacy and stability. Additionally, we examine the optimal cleaning methods for removing non-specifically bound aptamers, comparing no cleaning, guanidinium hydrochloride (GuHCl) cleaning, and vancomycin cleaning.The electrochemical characterizations were performed using square wave voltammetry (SWV). In SWV, the current is measured at the end of each potential pulse. This technique allows us to study the current's behaviour based on the relationship between the rate of the electron transfer reaction and the frequency of the square wave pulse. Specifically, the current shows a quasireversible maximum at a critical frequency, which is related to the surface standard rate constant (ksur).31 By studying the current change at different SWV frequencies, we can approximate the rates of the aptamer's bound and unbound states to a target. This facilitates the identification of regions exhibiting signal ON and OFF behaviours in EAB sensors.32,33 In addition, the electrochemical reversibility and thus the peak location of a surface bound redox probe in SWV measurements is mainly controlled by the dimensionless kinetic parameter ω = ksur/f, where f is the measurement frequency.34–36 When a target is measured with an EAB electrode at a specific frequency, all changes in ω can be taken to arise from the changes in ksur. If ksur decreases and thus ω decreases, there will be an anodic shift in the peak position for the oxidation process, while opposite is true in the case of increase in ksur. Therefore, with SWV we can also evaluate the nature of the signal changes with EAB sensors.
Through our analysis, we investigate the frequency response of the sensors and evaluate the impact of different linker chemistries on their signal output. We also examine the sensors' stability and repeatability, which are crucial for reliable long-term measurements. Furthermore, we assess the sensors' response to varying concentrations of VA to establish their sensitivity and detection limits. Lastly, we explore strategies for passivating the sensor surface to minimize non-specific binding of aptamers and methylene blue, thereby enhancing the specificity and accuracy of the sensors.
Each aspect of this investigation provides detailed insights into the mechanisms underlying sensor performance and offers guidelines for optimizing EAB sensor design.
All sensors exhibited a maximum current at a frequency of 170 Hz, irrespective of the presence of the target (Fig. 4A–D), corresponding to a surface rate constant of approximately 170 s−1 for both bound and unbound states. Despite the similar rate constant for both binding states, the signal change plot reveals frequency-dependent behaviour. Specifically, a signal-ON region is observed from 10 to 30 Hz, while a signal-OFF region is evident from 40 to 2000 Hz. However, this frequency dependent behaviour cannot be directly associated with conformational changes since unbound and bound states present the same ksur. The observed signal-OFF behaviour could be attributed to both a detachment of non-covalently bound aptamers from the electrode surface and the conformational change of the aptamers upon target binding (Fig. 5A and B). However, the former will not produce a consistent peak shift. In our case, all sensor systems exhibited an anodic shift of the forward (oxidation) peak position upon VA addition (Fig. SI4†), indicating that the reaction becomes kinetically less feasible. In addition, the magnitude of peak position changes depended on the applied SWV frequency. At lower frequencies (40 Hz), the sensors showed larger peak shifts and higher peak position restoration than at medium frequencies (160 Hz).
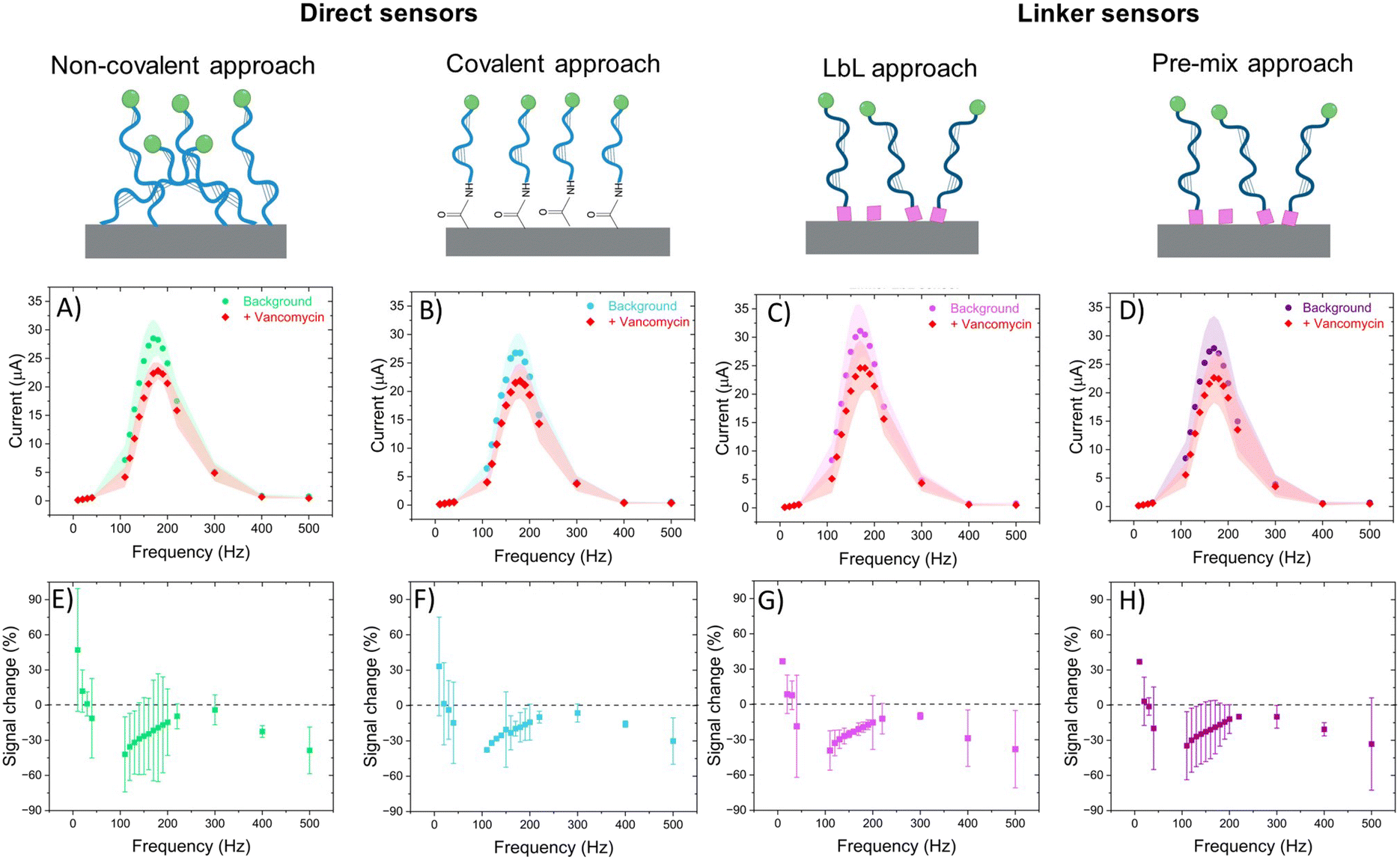 | ||
| Fig. 4 Frequency behaviour study of EAB sensors with different aptamer immobilization methods. Direct immobilization was performed with two approaches: non-covalent (A and E) and covalent binding (B and F). The indirect immobilization by the use of linker was performed by two approaches: layer-by-layer (C and G) and premix (D and H) methods. The frequency behaviour was studied with current vs. frequency plots (A–D) in absence (background) and presence (+vancomycin) of target as well as with signal change vs. frequency plots (E–H) (N = 3). | ||
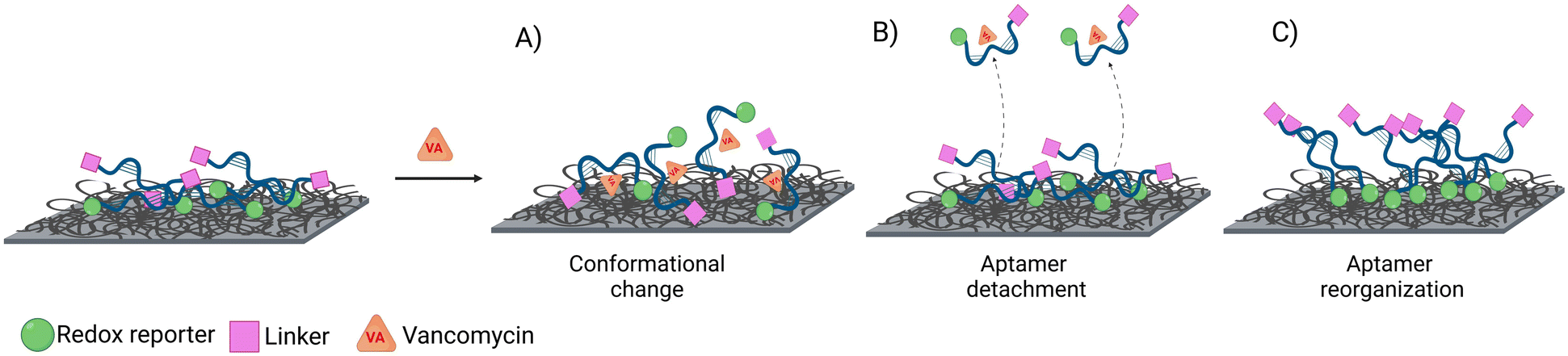 | ||
| Fig. 5 Scheme of the three possible scenarios producing a signal change upon target addition: A) conformational change of the aptamer, B) detachment of the non-covalently bound aptamers, and/or C) reorganization of the non-covalently attached aptamers. | ||
Our hypothesis for the observed signal-ON behaviour is a possible reorganization of non-covalently bound aptamers upon vancomycin binding (Fig. 5C). This binding event displaces some aptamers from the electrode surface, creating space for others to adsorb to the carbon nanotubes via their methylene blue (MB) end. The adsorption of MB induces two effects: an anodic peak shift and a slight increase in background peak current. The shift results from the higher overpotential required to drive the reaction due to the adsorption of the reduced form of MB (the reactant) on the SWCNT surface. However, this peak shift will be of much smaller magnitude than that produce by a conformational change. Simultaneously, the small current increase is attributed to the higher concentration of redox reporters on the electrode surface within the effective electron transfer distance. The SPR studies (Fig. 3) corroborate the pronounced adsorption tendency of the redox probe on the CNT networks, providing further support for our observations. Therefore, it is vital to also monitor the restoration of the background current to its original peak position (prior to vancomycin binding), considering that a comparable anodic shift may result from two vastly distinct physical mechanisms. In the context of conformational changes, a return is anticipated since the aptamer will get reoriented upon vancomycin removal; yet such a return is not observed in the case of the strong adsorption of MB. Among all sensor types, the linker LbL sensor presented the most samples with both anodic peak shift and peak position restoration at 160 Hz.
The signal loss was defined as the change in background peak current (in PBS + MgCl2) before and after vancomycin addition. This signal loss was similar across all four sensor types (Table SI2†) and is likely primarily due to the detachment of aptamers from the electrode surface. Additionally, the similar signal loss observed in the direct non-covalent sensor compared to the other sensors again suggests strong adsorption of the aptamers to the CNT network, an observation supported by the SPR results.
First, we studied the effect of 6 M GuHCl solution on non-covalent aptamer sensors and a control group (no cleaning). We used SWV at both low (40 Hz) and high (2000 Hz) frequencies during repetitive measurements of saturating concentrations of VA, monitoring signal loss after each VA measurement. The guanidinium ion (Gu+) possesses flat hydrophobic faces that can participate in hydrophobic interactions and directional H-bonding via its three NH2 groups. Therefore, Gu+ has been shown to denaturate protein secondary structure by competing for H-bonds and is additionally capable of stacking with nonpolar side chains, particularly those containing planar aromatic groups.37,38 In a similar fashion, aptamers can readily bind to guanidinium ions, potentially releasing any bound species as they do so.39,40 Our result showed that GuHCl cleaning reduced the signal loss at low frequencies compared to non-cleaned electrodes, suggesting removal by Gu+ ions of non-specifically bound aptamers from the outer layers of the electrode surface (Fig. 6A and B). Additionally, the non-cleaned electrodes exhibited higher standard deviations and total signal loss at low frequencies (Table SI3†). Conversely, at high frequencies, the signal loss was similar regardless of the cleaning method, indicating that aptamers close to the electrode surface were strongly bound and unaffected by Gu+ ions.
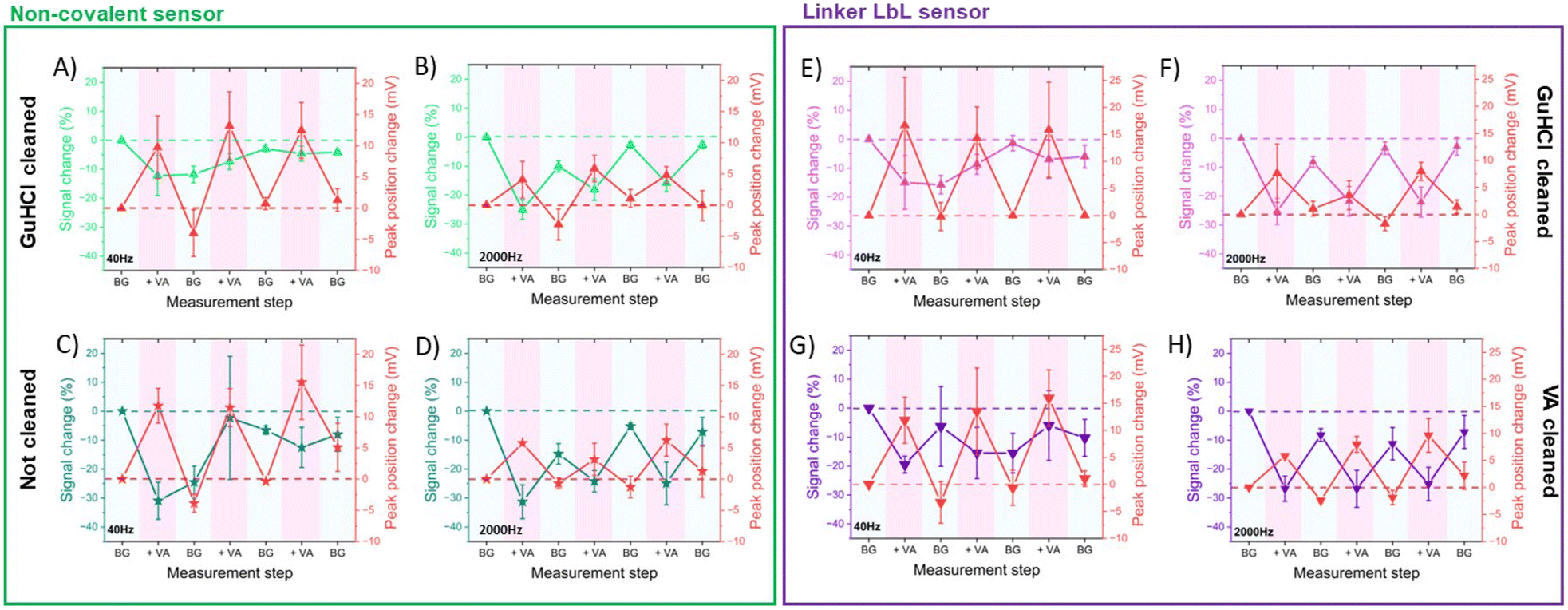 | ||
| Fig. 6 Stability upon repetitive measurements of vancomycin at 40 and 2000 Hz of non-covalent sensors previously cleaned with GuHCl (A and B) and without a previous cleaning (C and D). Additionally, the stability upon repetitive measurements of vancomycin at 40 and 2000 Hz of linker LbL sensors previously cleaned with GuHCl (E and F) and with VA cleaning (G and H) is presented. The stability was assessed in terms of signal and peak position changes (N = 3). | ||
Observing significant signal loss after the initial VA measurement in non-covalent non-cleaned sensors (Fig. 6C and D), we hypothesized that VA may indirectly detach non-specifically bound aptamers from the electrode surface. This detachment could occur as the aptamers preferentially bind to vancomycin rather than to the electrode surface. To test this hypothesis, we evaluated the effectiveness of GuHCl and VA cleaning methods in linker LbL sensors (Fig. 6E–H). Both methods exhibited similar signal loss, albeit with slightly higher variability in the VA cleaning method (Table SI3†). This suggests that both proposed cleaning methods effectively removed unbound aptamers and prepared the aptamers to be in their unbound state.
In terms of sensor stability during repetitive measurements of saturating concentrations of VA, both direct non-covalent and linker LbL sensors exhibited changes in signal and peak position upon exposure to VA at high frequencies, which were nearly restored during background measurements following VA measurement. Therefore, the sensors demonstrated good stability and repeatability when measuring saturating concentrations of VA at high frequencies. On the other hand, the signal change at low frequencies upon VA or BG measurement was inconsistent, likely due to the presence of more randomly oriented and loosely bound aptamer at the outer layers of the electrode surface.
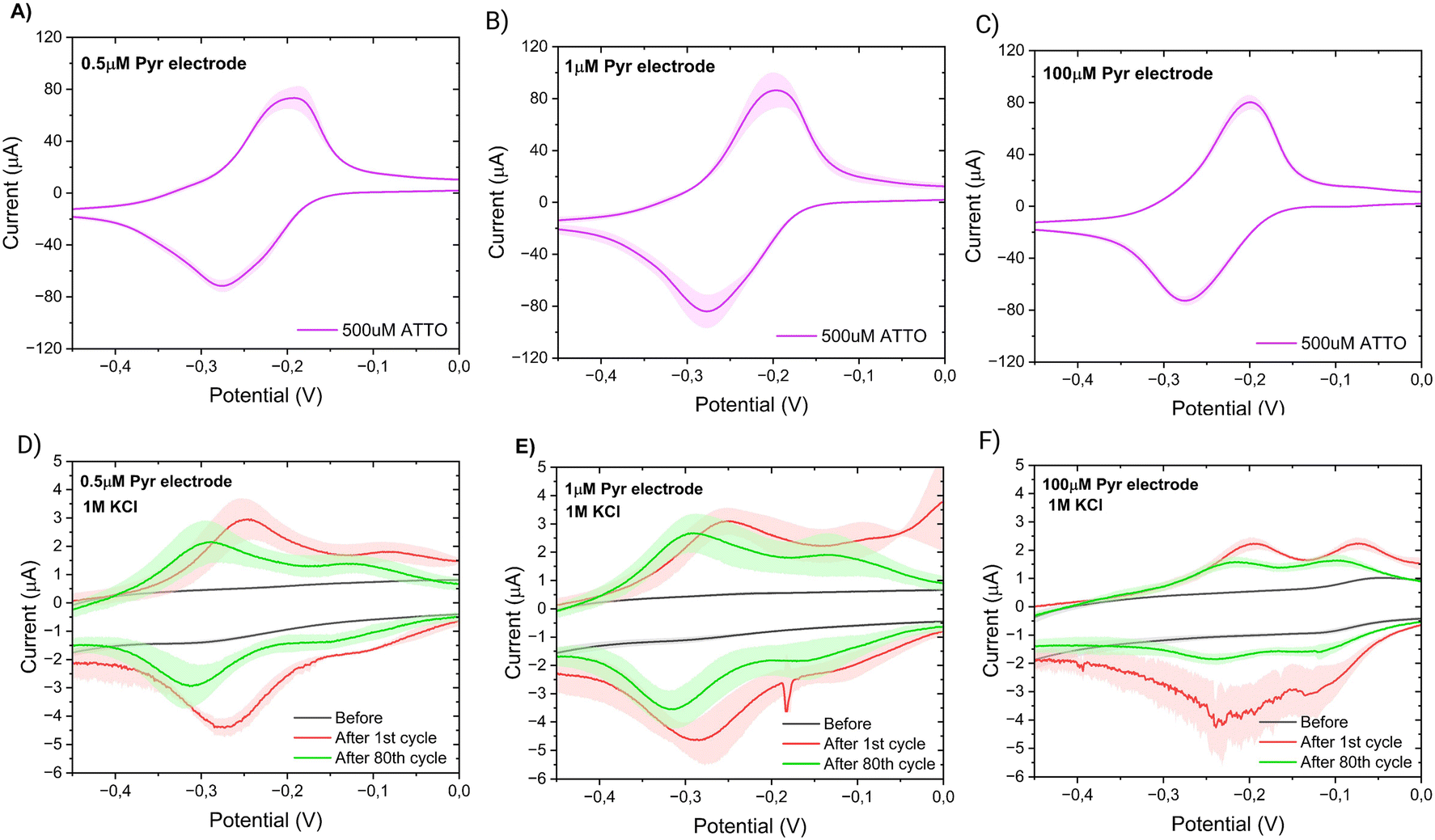 | ||
| Fig. 7 Passivation against methylene blue of a SWCNTs electrode with different linker concentrations. CV response of passivated electrodes to 500 μM of methylene blue (ATTO) (A–C) (N = 3). CV response of passivated electrodes in background (KCl) before and after the exposure of the electrodes to methylene blue (D–F) (N = 3). | ||
| Plain electrode | Passivated electrode | ||
|---|---|---|---|
| 0.5 μM linker | 1 μM linker | 100 μM linker | |
| 31% | 33 ± 7% | 8 ± 14% | 45 ± 10% |
| [VA] (μM) | Signal change (%) | |||
|---|---|---|---|---|
| Direct non-covalent | Direct covalent | Linker LbL | Linker pre-mixed | |
| 1 | 0.5 ± 0.3 | 0.4 ± 0.5 | 0.3 ± 1.0 | 2.0 ± 2.0 |
| 10 | 1.2 ± 1.6 | 1.1 ± 0.0 | 1.5 ± 1.0 | 3.5 ± 1.0 |
| 25 | 3.5 ± 0.8 | 2.3 ± 0.6 | 2.9 ± 1.2 | 4.7 ± 1.1 |
| 50 | 5.9 ± 0.7 | 3.9 ± 1.4 | 5.5 ± 1.4 | 5.5 ± 1.1 |
| 100 | 9.8 ± 2.7 | 7.0 ± 0.6 | 8.7 ± 1.4 | 8.3 ± 0.8 |
| 250 | 17.4 ± 5.7 | 14.8 ± 0.8 | 15.5 ± 0.6 | 14.4 ± 1.1 |
| Signal loss (%) | 11.3 ± 6.8 | 6.9 ± 0.5 | 8.5 ± 0.2 | 9.9 ± 1.1 |
Regarding peak shifts upon VA addition, considerable variability in sensor performance was observed within the same sample type. Some sensors exhibited an anodic shift, while others showed a cathodic shift upon VA addition (Table SI4†). This supports the hypothesis that there is a mix of differently attached aptamers on the surface exhibiting a variety of local reaction constants. Additionally, none of these sensors showed restoration of the peak position when tested in PBS + MgCl2 after the VA concentration series measurements.
Based on the aforementioned results, a calibration curve could be obtained to detect different concentrations of vancomycin in the physiological range (1–50 μM) (Fig. 8) for all the sensor types. The calibration curves revealed that direct non-covalent sensors exhibit the highest sensitivity and LOD (Table 4), which might misleadingly suggest enhanced performance compared to the other sensors. However, this sensor fails to meet our predefined criteria for proper operation. Furthermore, the signal generated by this sensor does not originate from a target-induced conformational change. One can apply the same reasoning to the other EAB sensor found in the literature (Table 4), where although it presents a current change with the addition of VA, there is no data on peak shift, thus it cannot be confirmed that the sensor signal arises from a conformational change of the aptamer. Consequently, when employing these commonly used calibration curves, it is crucial to carefully consider the underlying physical signal generation mechanism.
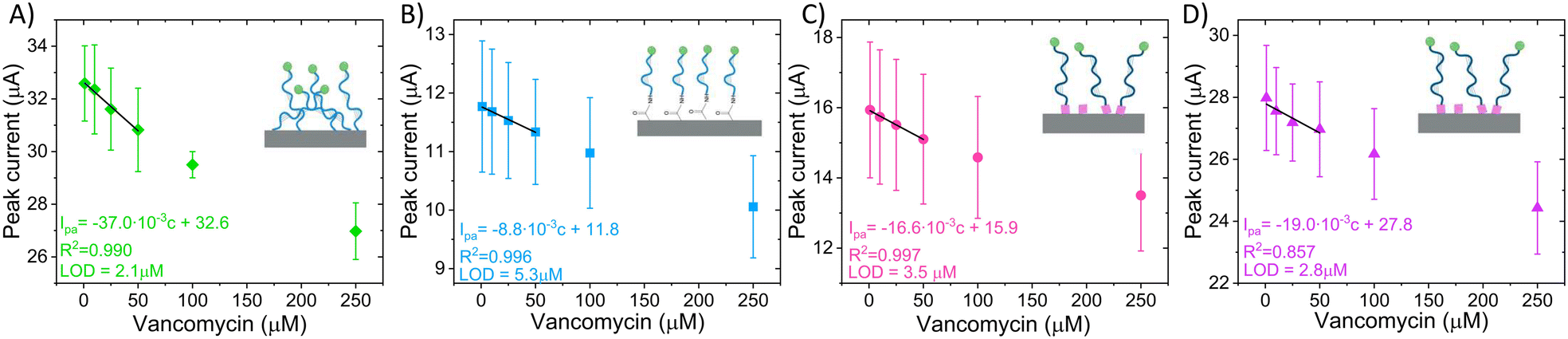 | ||
| Fig. 8 Calibration curves and linear fit from 1–50 μM VA for A) direct noncovalent, B) direct covalent, C) linker LbL, and D) linker premix electrodes (Ipa = forward partial peak current, c = vancomycin concentration, LOD = limit of detection, N = 3). | ||
| Material | LOD (μM) | Sensitivity (μA μM−1) | Ref. |
|---|---|---|---|
| a Approximated from Fig. 1C (1–10 μM) from ref. 44. | |||
| rGO-modified glassy carbon | 0.2 | 0.810 | 44 |
| Au-SAM EAB | NA | 0.006a | 45 |
| Hybrid graphene-Au nanocomposite | 0.3 | 0.768 | 46 |
| Direct noncovalent EAB (N = 3) | 2.1 | 0.037 | This work |
| Direct covalent EAB (N = 3) | 5.3 | 0.009 | |
| Linker premix EAB (N = 3) | 2.8 | 0.019 | |
| Linker LbL EAB (N = 3) | 3.5 | 0.016 | |
To conclude, for a proper sensor operation, it is imperative to address the two central challenges: (i) efficient aptamer binding and (ii) effective surface passivation. While the former affects the stability of the sensor the latter causes a substantial amount of background noise originating from the randomly oriented non-specifically bound aptamers. One potential approach to solve these challenges could involve transitioning from a 3D to a 2D carbonaceous material, allowing for better control over the orientation of aptamers and simultaneously reducing the adsorption volume. In addition, we are currently working on a bio-derived linker which is displaying extremely promising results with respect to surface passivation, together with the known ability for it to be used for direct linking of biomolecules using EDC/NHS chemistry.
4. Conclusions
While the literature contains numerous aptamer-based electrochemical sensors, their operational depiction is oversimplified, preventing their optimization, and understanding of the phenomena taking place on the electrode surface. Moreover, it is paramount to understand how the aptamers' conformational changes and factors such as redox reporter adsorption are shown in the SWV signal, as this understanding is pivotal for advancing the field. Here we have proposed three electrochemical parameters to determine if a conformational change of the aptamer occurs in an EAB sensor: peak shift, current amplitude change, and the restoration of the original background current and peak position after the target measurement. These parameters, supported by the SPR measurements, provide us with insight about the plausible interfacial structure of the EAB sensors in different cases. Hereby we have monitored the behaviour of four types of SWCNTs EAB sensors using different aptamer binding methods: direct (covalent and non-covalent) and indirect using a linker molecule. At this stage, the sensors could only be used as single-use sensors if an appropriate calibration curve is performed. In the future, it will be necessary to optimize binding and passivation, in order to use these EAB sensors in continuous and accurate measurements.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.† Raw data that supports the findings of this study are available from the corresponding author upon request.Author contributions
LFP: investigation, methodology, writing – original draft, writing – review & editing, formal analysis, visualization. EG: investigation & methodology. JS: conceptualization, writing – original draft, writing – review & editing. VD: investigation, methodology, writing – original draft, writing – review & editing, formal analysis, visualization. TL: conceptualization, methodology, writing – original draft, writing – review & editing, resources, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Canatu Oy for providing the SWCNT networks and Elli Leppänen for her help during the investigation process.References
- M. A. Pellitero and N. Arroyo-Currás, Anal. Bioanal. Chem., 2022, 414, 5627–5641 CrossRef CAS.
- E. Verrinder, N. Wester, E. Leppänen, T. Lilius, E. Kalso, B. R. Mikladal, I. Varjos, J. Koskinen and T. Laurila, ACS Omega, 2021, 6(17), 11563–11569 CrossRef CAS.
- P. Dauphin-Ducharme, K. Yang, N. Arroyo-Currás, K. L. Ploense, Y. Zhang, J. Gerson, M. Kurnik, T. E. Kippin, M. N. Stojanovic and K. W. Plaxco, ACS Sens., 2019, 4(10), 2832–2837 CrossRef CAS PubMed.
- A. A. Lubin and K. W. Plaxco, Acc. Chem. Res., 2010, 43(4), 496–505 CrossRef CAS PubMed.
- M. A. Pellitero, S. D. Curtis and N. Arroyo-Currás, ACS Sens., 2021, 6(3), 1199–1207 CrossRef CAS PubMed.
- C. Vericat, M. E. Vela, G. Benitez, P. Carro and R. C. Salvarezza, Chem. Soc. Rev., 2010, 39(5), 1805 RSC.
- F. V. Oberhaus, D. Frense and D. Beckmann, Biosensors, 2020, 10, 45 CrossRef CAS PubMed.
- A. Shaver and N. Arroyo-Currás, Curr. Opin. Electrochem., 2022, 32, 100902 CrossRef CAS.
- F. Hamdi, M. Roushani, M. Nasibipour and J. S. Hoseini, Anal. Chim. Acta, 2024, 1291, 342235 CrossRef CAS PubMed.
- M. Z. Karazan, M. Roushani, J. S. Hoseini and M. Nasibipour, Microchem. J., 2024, 204, 110975 CrossRef.
- P. Michaels, M. T. Alam, S. Ciampi, W. Rouesnel, S. G. Parker, M. H. Choudhury and J. J. Gooding, Chem. Commun., 2014, 50(58), 7878–7880 RSC.
- K. Xu, X. Meshik, B. M. Nichols, E. Zakar, M. Dutta and M. A. Stroscio, Nanotechnology, 2014, 25(20), 205501 CrossRef.
- T. Laurila, S. Sainio and M. A. Caro, Prog. Mater. Sci., 2017, 88, 499–594 CrossRef CAS.
- M. Caro, A. Aarva, V. Deringer, G. Csányi and T. Laurila, Chem. Mater., 2018, 30, 7446–7455 CrossRef CAS.
- V. Deringer, M. Caro, R. Jana, A. Aarva, S. Elliott, T. Laurila, G. Csányi and L. Pastewka, Chem. Mater., 2018, 30, 7438–7445 CrossRef CAS.
- A. Nano, A. L. Furst, M. G. Hill and J. K. Barton, J. Am. Chem. Soc., 2021, 143(30), 11631–11640 CrossRef CAS PubMed.
- Centers for Disease Control and Prevention, https://www.cdc.gov/drugresistance/solutions-initiative/stories/partnership-estimates-healthcare-cost.html (accessed 16:39, Oct 4, 2023).
- E. Leppänen, J. Etula, P. Engelhardt, S. Sainio, H. Jiang, B. Mikladal and T. Laurila, Electroanal. Chem., 2021, 896, 115255 CrossRef.
- L. S. Jung, C. T. Campbell, T. M. Chinowsky, M. N. Mar and S. S. Yee, Langmuir, 1998, 14(19), 5636–5648 CrossRef CAS.
- Bionavis, https://www.bionavis.com/en/news/warning-about-edc/ (accessed 16:39, Oct 4, 2023).
- ChemSpider, https://www.chemspider.com/Chemical-Structure.14253.html (accessed 13:01, Oct 4, 2023).
- ChemSpider, https://www.chemspider.com/Chemical-Structure.144378.html (accessed 14:32, Oct 4, 2023), https://www.chemspider.com/Chemical-Structure.144378.html?rid=b99ec56a-d12f-4b71-b350-d07b006f0e37&page_num=0.
- K. A. Peterlinz, R. M. Georgiadis, T. M. Herne and M. J. Tarlov, J. Am. Chem. Soc., 1997, 119(14), 3401–3402 CrossRef CAS.
- X. Campbell and G. Kim, Biomaterials, 2007, 28(15), 2380–2392, DOI:10.1016/j.biomaterials.2007.01.047.
- L. S. Jung, C. T. Campbell, T. M. Chinowsky, M. N. Mar and S. S. Yee, Langmuir, 1998, 14(19), 5636–5648, DOI:10.1021/la971228b.
- ChemSpider, https://www.chemspider.com/Chemical-Structure.7703.html (accessed 14:33, Oct 4, 2023), https://www.chemspider.com/Chemical-Structure.7703.html?rid=5e3f1d68-19a5-4e42-a622-608230802c57.
- ChemSpider, https://www.chemspider.com/Chemical-Structure.29153.html (accessed 14:34, Oct 4, 2023), https://www.chemspider.com/Chemical-Structure.29153.html?rid=4a5add03-2345-4c02-b4a5-a264d47962f5&page_num=0.
- S. Sainio, N. Wester, A. Aarva, C. Titus, D. Nordlund, E. Kauppinen, E. Leppänen, T. Palomäki, J. Koehne, O. Pitkänen, K. Kordas, M. Kim, H. Lipsanen, M. Mozetič, M. Caro, M. Meyyappan, J. Koskinen and T. Laurila, J. Phys. Chem. C, 2021, 125, 973–988 CrossRef CAS.
- E. Leppänen, E. Gustafsson, N. Wester, I. Varjos, S. Sainio and T. Laurila, Electrochim. Acta, 2023, 466, 143059 CrossRef.
- S. Sam, L. Touahir, J. Salvador Andresa, P. Allongue, J.-N. Chazalviel, A. C. Gouget-Laemmel and S. Djebbar, Langmuir, 2010, 26(2), 809–814 CrossRef CAS.
- V. Mirčeski, Š. Komorsky-Lovrić and M. Lovrić, Electrode Mechanisms, in Square-Wave Voltammetry, Monographs in Electrochemistry, Springer, Berlin, Heidelberg, 2008 Search PubMed.
- H. Li, P. Dauphin-Ducharme, G. Ortega and K. W. Plaxco, J. Am. Chem. Soc., 2017, 139(32), 11207–11213 CrossRef CAS.
- R. J. White and K. W. Plaxco, Anal. Chem., 2010, 82(1), 73–76 CrossRef CAS PubMed.
- V. Mirceski, S. Komorsky-Lovric and M. Lovric, Square-wave voltammetry: theory and application, Springer Science & Business Media, 2007 Search PubMed.
- Š. Komorsky-Lovrić and M. Lovrić, Anal. Chim. Acta, 1995, 305(1–3), 248–255 CrossRef.
- V. Mirceski, R. Gulaboski, M. Lovric, I. Bogeski, R. Kappl and M. Hoth, Electroanalysis, 2013, 25(11), 2411–2422 CrossRef CAS.
- M. Hanke, N. Hansen, E. Tomm, G. Grundmeier and A. Keller, Int. J. Mol. Sci., 2022, 23, 8547 CrossRef CAS PubMed.
- M. Kurnik, G. Ortega, P. Dauphin-Ducharme, H. Li, A. Caceres and K. Plaxco, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8352–8357 CrossRef CAS.
- L. N. Cella, P. Sanchez, W. Zhong, N. V. Myung, W. Chen and A. Mulchandani, Anal. Chem., 2010, 82(5), 2042–2047 CrossRef CAS PubMed.
- Y. Xiao, A. A. Lubin, A. J. Heeger and K. W. Plaxco, Angew. Chem., Int. Ed., 2005, 44(34), 5456–5459 CrossRef CAS PubMed.
- V. Sanz, E. Borowiak, P. Lukanov, A. M. Galibert, E. Flahaut, H. M. Coley and J. McFadden, Carbon, 2011, 49(5), 1775–1781 CrossRef CAS.
- Y. Yan, M. Zhang, K. Gong, L. Su, Z. Guo and L. Mao, Chem. Mater., 2005, 17(13), 3457–3463 CrossRef CAS.
- V. V. Chagovets, M. V. Kosevich, S. G. Stepanian, O. A. Boryak, V. S. Shelkovsky, V. V. Orlov and V. A. Karachevtsev, J. Phys. Chem. C, 2012, 116(38), 20579–20590 CrossRef CAS.
- M. Hadi and T. Mollaei, Electroanalysis, 2018, 31(7), 1224–1228 CrossRef.
- A. Shaver, J. D. Mahlum, K. Scida, M. L. Johnston, M. A. Pellitero, Y. Wu, G. V. Carr and N.l. Arroyo-Currás, ACS Sens., 2022, 7(12), 3895–3905 CrossRef CAS PubMed.
- A. Blidar, B. Feier, A. Pusta, A.-M. Drăgan and C. Cristea, Coatings, 2019, 9(10), 652 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sd00250d |
| This journal is © The Royal Society of Chemistry 2024 |