
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymatic polymerization of furan-based polymers in biobased solvents†
Fitrilia
Silvianti
 a,
Dina
Maniar
a,
Tijn C.
de Leeuw
b,
Jur
van Dijken
a and
Katja
Loos
*a
a,
Dina
Maniar
a,
Tijn C.
de Leeuw
b,
Jur
van Dijken
a and
Katja
Loos
*a
aMacromolecular Chemistry & New Polymeric Materials, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: k.u.loos@rug.nl
bCarbExplore Research B.V., 9747 AA Groningen, The Netherlands
First published on 13th September 2024
Abstract
The demand for biobased polymers is on the rise, driven by increasing environmental awareness and the imperative for sustainability. Biobased materials, which offer renewability, have emerged as a solution to the depletion of petroleum-based resources. Among biobased raw materials, 2,5-furandicarboxylic acid (2,5-FDCA) has gained prominence as an extensively studied monomer in the last decade. Polyesters based on 2,5-FDCA have shown compatibility and potential as biobased alternatives to polyethylene terephthalate (PET) for packaging applications. Besides FDCA, 2,5-bis(hydroxymethyl)furan (2,5-BHMF), a furan hetero-aromatic diol derivable from carbohydrates, has been identified as a versatile building block, presenting interesting properties for polymeric materials. In adherence to sustainability principles, the choice of catalyst for biobased polymer production is crucial. Biocatalysts, such as enzymes, not only provide renewability but also offer advantages such as mild reaction conditions, aligning with sustainable practices. However, many enzymatic polymerizations are reported in organic solvents, that are not environmentally friendly and/or non-renewable. To address this issue, this study explored the use of biobased solvents—namely, p-cymene, pinacolone, and D-limonene—for the enzymatic polymerization of dimethyl 2,5-furan dicarboxylate (2,5-FDCA-based) polyesters and copolyesters with 2,5-BHMF. By employing Candida antarctica lipase B (CALB), the enzymatic polymerization of this enzyme, particularly with p-cymene, has demonstrated high performance, resulting in high-molecular-weight polyester and copolyester products up to 7000 and 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 g mol−1, respectively. This study examined the thermal properties and crystallinity of the obtained products by analyzing their structure–property relationships. This research contributes to the advancement of sustainable polymer synthesis by considering biobased raw materials, environmentally friendly catalysts, and biobased solvents.
800 g mol−1, respectively. This study examined the thermal properties and crystallinity of the obtained products by analyzing their structure–property relationships. This research contributes to the advancement of sustainable polymer synthesis by considering biobased raw materials, environmentally friendly catalysts, and biobased solvents.
Sustainability spotlightThe traditional production of polymers heavily relies on fossil-based resources, posing significant environmental challenges. This study addresses these issues by exploring enzymatic polymerization techniques for biobased furanic–aliphatic (co)-polyesters. Utilizing renewable biobased solvents, such as citrus-derived solvents and pinacolone, represents a pivotal innovation. This approach offers a sustainable alternative, reducing dependence on non-renewable resources and supporting environmentally friendly manufacturing processes. Aligned with the United Nations Sustainable Development Goals (SDGs) 9 (Industry, Innovation, and Infrastructure), 12 (Responsible Consumption and Production), and 13 (Climate Action), this research promotes sustainable industrial practices, fosters responsible production methods, and contributes to climate change mitigation. By advancing polymer science towards the use of sustainable and renewable resources, this study underscores a critical shift in industrial applications. |
Introduction
In light of growing environmental concerns, polymer science increasingly aligns with a sustainable paradigm, demonstrating a dedication to sustainability principles. Consequently, in recent years, considerable attention has been directed towards producing biobased polymers to replace those derived from fossil-based resources with innovative, sustainable macromolecular materials. A wide range of biobased polymers has been produced using various renewable resources, contributing to the diversity of bio-derived polymers available in the current market. Notable examples include polylactic acid (PLA), poly(butylene succinate) (PBS), poly(ε-caprolactone) (PCL), poly(butylene adipate-co-butylene terephthalate) (PBAT), biopoly(ethylene terephthalate), biopoly(trimethylene terephthalate) (Bio-PTT), biopolyamide (Bio-PA), and poly(ethylene furanoate) (PEF).1–3 Among these, PEF has gained increased recognition due to its superior properties compared to polyethylene terephthalate (PET), particularly in packaging materials.2,4,5 This interest aligns with PET's established status as one of the most widely produced polyesters. Consequently, the increasing popularity of PEF is coupled with a proportional increase in the utilization of 2,5-furandicarboxylic acid (2,5-FDCA) derived from 5-hydroxymethylfurfural (HMF) from carbohydrates, a biobased building block employed in synthesizing PEF. Besides 2,5-FDCA, 2,5-bishydroxymethylfuran (2,5-BHMF) also derivable from HMF is a recognized versatile hetero-aromatic diol used as a polymer building block.6 2,5-BHMF imparts rigidity or stiffness to the resulting polymer chains, thereby affecting the material characteristics, including on 2,5-FDCA-based polymers. Therefore, developing polymeric materials utilizing 2,5-BHMF is also attractive following 2,5-FDCA popularity.In the last decade, research has explored various methods to produce 2,5-FDCA-based polymers by varying monomers, catalysts, and synthesis methods. Examples include two-step melt polycondensations using titanium catalysts,7 solid-state postcondensation for high-molecular-weight polymers,8 and hetero-phase step-addition for FDCA-based epoxy vitrimers with self-healing properties.9 Enzymatic catalysis, a green approach, has also been effective in producing 2,5-FDCA-based copolyesters, incorporating 2,5-BHMF using Candida antarctica lipase B (CALB).10,11 The use of enzymes as biocatalysts offers a more sustainable method due to their renewability, nontoxic nature, and mild reaction conditions.1,5,12 Moreover, enzymes have high selectivity, enabling the polymerization to be more effective toward specific substrates, and frequently do not require protecting groups, thereby reducing energy and waste disposal expenses.1,12–14
The enzymatic synthesis of FDCA-based polyesters has been extensively reported in recent years.15–19 Optimization of polymerization conditions to achieve high molecular weight has been reported, such as applying variations in vacuum or temperature conditions in one-step or two-step methods, as well as varying monomers and comonomers.1,10,12,15,17,20 The solvent used as the reaction medium could affect the enzyme catalytic performance, thus influencing the rendered results.1,15,16,18 Lipase, an active catalyst for synthesizing FDCA-based polyesters, typically works best in nonpolar organic solvents, which are hydrophobic and have higher logP (>4.0), the solvent distribution coefficient between water and octanol.21–24 However, traditional organic solvents, often derived from fossil fuels, pose sustainability issues due to their volatility and environmental impact.25 Thus, a shift towards biobased solvents is imperative.
Biobased solvents, derived from renewable resources and exhibiting low toxicity, have emerged as environmentally friendly alternatives.26,27 These solvents ensure process safety and environmental acceptability and have expanded to enzymatic catalysis. Studies have shown that enzymes are active in biobased solvents such as citrus-derived alkenes (D-limonene and p-cymene), 3,3-dimethyl-2-butanone (pinacolone), 2-methyltetrahydrofuran (2-MeTHF), γ-valerolactone (GVL), and Cyrene.27–32 Among these biobased solvents, citrus-derived solvents exhibited remarkable efficiency in the esterification of 2-phenylpropionic acid using immobilized CALB, reaching an 86% conversion rate.27 These citrus-based solvents performed better than several other organic solvents, including hexane, toluene, chloroform, and THF.27 In another report, pinacolone, regarded as an alternative to toluene, was reported to demonstrate a noteworthy conversion rate of more than 80% when employed in the enzymatic polymerization of poly(1,4-butylene adipate) and poly(1,8-octylene adipate) and more than 50% for poly(1,4-butylene 2,5-furandicarboxylate) using CALB.29,33 Pinacolone can be produced from renewable resources. It has been reported that isobutene, which is the precursor of pivalic acid that produces pinacolone, can be generated by sugar fermentation.33
The novelty of this study lies in the use of biobased solvents, specifically citrus-derived solvents and pinacolone, for the enzymatic polymerization of furanic–aliphatic polyesters or copolyesters utilizing dimethyl 2,5-furandicarboxylate (2,5-DMFDCA). To the best of our knowledge, there has been no prior research focusing on this aspect. This study investigates the influence of these solvents on CALB-catalyzed synthesis of these polymers. In the first part, we studied enzymatic polymerization involving 2,5-DMFDCA with two variations of aliphatic diols, i.e., 1,8-octanediol and 1,12-dodecanediol, in both p-cymene or pinacolone. In the second part, we presented the enzymatic copolymerization of 2,5-DMFDCA with 2,5-bis(hydroxymethyl)furan (2,5-BHMF) as a rigid diol and 1,10-decanediol as an aliphatic diol in p-cymene or D-limonene. We assessed the influence of solvent on enzymatic catalysis and investigated the correlation between the structure and product properties.
Our strategy focuses on fostering sustainability through enzymatic polymerization, incorporating conditions that minimize environmental impact. Specifically, we utilize enzymes as sustainable catalysts, facilitating reactions at significantly lower temperatures (below 100 °C), representing up to a 100% reduction compared to the higher temperatures typically required in conventional approaches employing metal-based catalysts. Additionally, biobased solvents are employed for the enzymatic polymerization process, replacing the non-sustainable organic solvents traditionally used in previous systems, which presented a limitation for more eco-friendly enzymatic polymerization methods.34–36 Furthermore, this study addresses the coloration issues frequently encountered in furan-based (co)-polyester products synthesized via conventional metal-catalyzed processes at elevated temperatures. By conducting the reactions under milder conditions using biobased solvents and sustainable catalysts, we successfully produced colorless products, thereby overcoming a common challenge in the production of furan-based polymers.
Materials and methods
Materials
Novozym 435 (N435, Candida antarctica lipase B (CALB)) immobilized on acrylic resin (5000+ U g−1), 1,8-octanediol (1,8-ODO, 98%), 1,10-decanediol (1,10-DDO, 98%), 1,12-dodecanediol (1,12-DODO, 99%), chloroform (CHCl3, Chromasolv HPLC, ≥99.8%, amylene stabilized), deuterated chloroform (CDCl3, 99.8 atom% D), potassium trifluoroacetate (KTFA, 98%), 2,5-bis(hydroxymethyl)furan (2,5-BHMF, 97%), 3,3-dimethyl-2-butanone (pinacolone, 97%), and p-cymene (99%) were purchased from Sigma-Aldrich. Toluene (anhydrous, 99.8%) was purchased from Alfa Aesar. Dimethyl 2,5-furandicarboxylate (2,5-DMFDCA, 97%) was purchased from Fluorochem UK. Absolute methanol (MeOH, AR grade) was obtained from Biosolve Chemicals. 1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP, ≥99%) and D-limonene (95%) were acquired from TCI Europe. Dithranol (≥98%) was purchased from Fluka.N435 was predried as reported previously.37 The molecular sieves (4 Å) were preactivated at 200 °C. All the other chemicals were used as received.
One-step approach for iCALB-catalyzed polycondensations of polyesters
A one-step enzymatic polymerization method was employed to synthesize FDCA-based polyesters (FPEs) based on our previous research by combining the monomer 2,5-DMFDCA at a 1:1 mol ratio with diols, predried N435, and a preactivated molecular sieve (15% and 150% of the monomer, respectively). For example, we placed 2,5-DMFDCA (0.2787 grams, 1.51 mmol), 1,8-ODO (0.2212 grams, 1.51 mmol), a molecular sieve (1.5 grams, 150%), N435 (0.15 grams, 15%), and 2.5 mL of either p-cymene or pinacolone as a solvent in a 25 mL round bottom flask. Then, we heated the flask to 90 °C in a nitrogen atmosphere and agitated it magnetically for 72 hours. Afterward, we cooled and terminated the reaction, added 20 mL of chloroform to dissolve the products, filtered N435 and the molecular sieve, washed them with 30 mL of chloroform, mixed all the obtained solutions, and concentrated them using a rotary evaporator at 40 °C and reduced pressure (400–480 mbar). We solidified the concentrated solution by adding excess cold methanol, collected the precipitated product through centrifugation (5 minutes, 4500 rpm, 4 °C) using a Thermo/Heraeus Labofuge 400 R, and dried it under vacuum at 40 °C for three days before conducting our analysis. The resulting products were stored at room temperature.
Procedure of the one-step approach for iCALB-catalyzed polycondensations of copolyesters
Based on previous research, enzymatic polymerization was performed to produce furan-based copolyesters (co-FPEs). An experimental copolymerization of 2,5-DMFDCA, 2,5-BHMF, and 1,10-DDO as aliphatic diols was performed with a 2,5-DMFDCA/2,5-BHMF/1,10-DDO ratio of 50:12.5:37.5 in the following manner: initially, we introduced predried N435 and preactivated molecular sieves in amounts equivalent to 15% and 150%, respectively, of the monomer into a 25 mL round bottle in a nitrogen environment. Next, we added 2,5-DMFDCA (265.4 mg, 1.44 mmol), 2,5-BHMF (46 mg, 0.36 mmol), 1,10-DDO (188.4 mg, 1.08 mmol), and 2.5 mL of either anhydrous toluene, p-cymene or D-limonene to the flask. The procedure for experimental polymerization and the subsequent purification of the product closely followed the one-step method previously described for polyester synthesis.
Two-step iCALB-catalyzed polycondensations for copolyesters
Two-stage enzymatic polymerizations of co-FPEs were carried out under two distinct vacuum conditions. Following a two-hour reaction at 90 °C with continuous magnetic stirring under a nitrogen atmosphere, the second step was initiated as follows:(1) In the first method, a vacuum at 600 mmHg was applied for 70 hours.
(2) In the second method, the vacuum pressure is slowly reduced to 60 mmHg over 70 hours for the reaction in p-cymene and D-limonene.
The experimental polymerization procedure and the composition of the monomers used are similar to those in the one-step method for preparing co-FPEs, as previously described.
Furan-based polyesters
ATR-FTIR (ν, cm−1): 3057–3200 (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H stretching vibration of the furan ring); 2780–3021 (C–H stretching vibrations); 1720–1724 (CO stretching vibration of ester); 1573–1576 (aromatic CC bending vibration); 1491–1493, 1468–1475 (C–H deformation and wagging vibration); 1392 (C–H rocking vibration); 1142–1144, 1270–1284 (asymmetrical and symmetrical stretching vibrations of the ester C–O–C group); 1230–1234, 1010–1016 (C–O–C ring vibration, furan ring); 966–977, 820–822, 764 (C–H out-of-plane deformation vibration, furan ring).
C–H stretching vibration of the furan ring); 2780–3021 (C–H stretching vibrations); 1720–1724 (CO stretching vibration of ester); 1573–1576 (aromatic CC bending vibration); 1491–1493, 1468–1475 (C–H deformation and wagging vibration); 1392 (C–H rocking vibration); 1142–1144, 1270–1284 (asymmetrical and symmetrical stretching vibrations of the ester C–O–C group); 1230–1234, 1010–1016 (C–O–C ring vibration, furan ring); 966–977, 820–822, 764 (C–H out-of-plane deformation vibration, furan ring).
Poly(octamethylene-2,5-furanoate)
1H NMR (600 MHz, CDCl3, δ, ppm): 7.16 (2H, s, –CH, 2,5-DMFDCA), 4.30 (4H, m, –CO–O–CH2–, from 1,8-ODO), 1.74 (4H, m, –CO–O–CH2–CH2–, from 1,8-ODO), 1.36 (4H, m, –CH2–, from 1,8-ODO), 3.90 (s, –O–CH3, end group from 2,5-DMFDCA), 3.62 (t, –CH2–OH, end group from 1,8-ODO).
Poly(dodecamethylene-2,5-furanoate)
1H NMR (600 MHz, CDCl3, δ, ppm): 7.17 (2H, s, –CH, 2,5-DMFDCA), 4.23 (4H, m, –CO–O–CH2–, from 1,12-DODO), 1.69 (4H, m, –CO–O–CH2–CH2–, from 1,12-DODO), 1.28 (4H, m, –CH2–, from 1,12-DODO), 3.84 (s, –O–CH3, end group from 2,5-DMFDCA), 3.62 (t, –CH2–OH, end group from 1,12-DODO).
Furan-based copolyesters
ATR-FTIR (ν, cm−1): 3118–3137 (C–H stretching vibrations of the furan ring); 2914–2954, 2848–2869 (asymmetric and symmetric C–H stretching vibrations); 1710–1729 (CO stretching vibrations); 1573–1583, 1506–1511 (aromatic CC bending vibrations); 1434–1471, 1371–1392 (C–H deformation and wagging vibrations); 1329 (C–H rocking vibrations); 1122–1151, 1268–1276 (asymmetric and symmetric stretching vibrations of the ester C–O–C groups); 1203–1228, 1004–1031 (C–O–C ring vibrations, furan ring); 948–979, 798–835, 763–771 (C–H out-of-plane deformation vibrations, furan ring).
Poly(2,5-furandimethylene furanoate-co-decamethylene-2,5-furanoate)
1H NMR (600 MHz, CDCl3, δ, ppm): 7.18 (2 H, m, –CH, 2,5-DMFDCA), 6.48 (2 H, m, –CH, 2,5-BHMF), 5.29 (4 H, s, –CO–O–CH2–, 2,5-BHMF), 4.31 (4 H, m, –CO–O–CH2–, from 1,10-DDO), 1.74 (4 H, m, –CO–O–CH2–CH2–, from 1,10-DDO), 1.36 (4 H, m, –CH2–, from 1,10-DDO), 1.29 (8 H, m, –CH2–, from 1,10-DDO), 4.61 (s, –CH2OH, end group from 2,5-BHMF), 3.92 (s, –O–CH3, end group from 2,5-DMFDCA), 3.63 ppm (t, –CH2–OH, end group from 1,10-DDO).
Analytics
Proton nuclear magnetic resonance (1H-NMR; 600 MHz) spectra were recorded on a Bruker Ascend™ NMR600 spectrometer using CDCl3 as the solvent.Attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectra were recorded on a Bruker VERTEX 70 spectrometer equipped with an ATR diamond single reflection accessory. The measurement resolution was 4 cm−1, and the spectra were collected in the range of 4000–400 cm−1, with 16 scans for each sample. Atmospheric compensation and baseline correction were applied to the collected spectra using OPUS spectroscopy software (v7.0) (Bruker Optics).
The molecular weights (number-average, 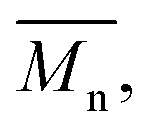 and weight-average,
and weight-average, 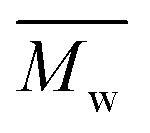 ) of the FPEs were determined via a size exclusion chromatography (SEC) instrument equipped with a triple detector, consisting of a Malvern dual detector and Schambeck RI2012, a refractive index detector. Separation was carried out by utilizing two PLgel 5 μm MIXED-C 300 mm columns from Agilent Technologies at 35 °C. HPLC grade chloroform was used as the eluent, with a flow rate of 0.5 mL min−1. Data acquisition and calculations were performed using Viscotek OmniSec software version 5.0. Molecular weights were determined based on a conventional calibration curve generated from narrow dispersity polystyrene standards (Agilent and Polymer Laboratories,
) of the FPEs were determined via a size exclusion chromatography (SEC) instrument equipped with a triple detector, consisting of a Malvern dual detector and Schambeck RI2012, a refractive index detector. Separation was carried out by utilizing two PLgel 5 μm MIXED-C 300 mm columns from Agilent Technologies at 35 °C. HPLC grade chloroform was used as the eluent, with a flow rate of 0.5 mL min−1. Data acquisition and calculations were performed using Viscotek OmniSec software version 5.0. Molecular weights were determined based on a conventional calibration curve generated from narrow dispersity polystyrene standards (Agilent and Polymer Laboratories, 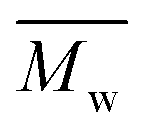 = 645–3001000 g mol−1). The samples were filtered through a 0.2 μm PTFE filter prior to injection.
= 645–3001000 g mol−1). The samples were filtered through a 0.2 μm PTFE filter prior to injection.
Matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-ToF MS) was performed using a 4800 Plus MALDI TOF/TOF Analyzer (Applied Biosystems) in reflector positive mode. The matrix, cationization agent, and solvent used were dithranol, KTFA, and HFIP, respectively. First, dithranol (20 mg mL−1), KTFA (5 mg mL−1) and a polymer sample (1–2 mg mL−1) were premixed at a ratio of 5:1:5. The mixture was subsequently hand-spotted on a stainless steel plate and left to dry. The presence of polyesters and copolyesters with different end groups was determined by using eqn (1) and (2), respectively:
| MP = MEG + (n × MRU) + Mcation+ | (1) |
| MP = MEG + (r1 × MRU1 + r2 × MRU2) + Mcation+ | (2) |
The analysis of the thermal properties was performed on a TA-Instruments Q1000 DSC instrument, which was calibrated with indium as the standard. The heating rate was 10 °C min−1 under nitrogen flow. The product melting points (Tm) were measured by a second heating scan. The glass transition temperature (Tg) was measured by a second heating scan or temperature-modulated differential scanning calorimetry (TMDSC) at 2 °C min−1 with a temperature modulation of ±0.50 °C every 60 seconds.
The thermal stability and degradation temperature were analyzed via thermogravimetric analysis (TGA) on a TA-Instruments Discovery TGA 5500 instrument at a heating rate of 10 °C min−1 in a nitrogen environment.
Wide-angle X-ray diffraction (WAXD) patterns were recorded on a Bruker D8 Endeavor diffractometer with Cu Kα radiation (λ = 0.1542 nm) in the angular range of 5–50° (2θ) at room temperature. The percentage of crystallinity (χc) was determined using the WAXD spectra through the following formula: χc (%) = 100 × k × Ic/(Ic + Ia).38,39 Here, k represents the relative scattering factor between a polymer's crystalline and amorphous parts (typically assumed to be k = 1 due to the difficulty of determination). The integration intensities of the crystal peaks (Ic) and the integration intensity of the amorphous halo (Ia) were obtained using the Peak Analyzer tool available in OriginPro 9.1 software from OriginLab Corporation. The d-spacings were calculated from the diffraction peak maxima through Bragg's equation.
Results and discussion
Synthesis and structural characterization of furan-based polyesters
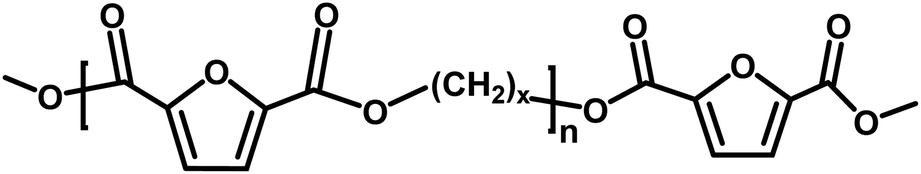
In the first part of this manuscript, we produced FPEs via the CALB-catalyzed polymerization of 2,5-DMFDCA with two different aliphatic linear diols, as illustrated in Scheme 1. These diols contained either eight or twelve methylene units (x), as listed in Table 1. To promote a more green polymerization system, we introduced two biobased solvents, p-cymene and pinacolone, as media and evaluated their influence on the enzymatic synthesis.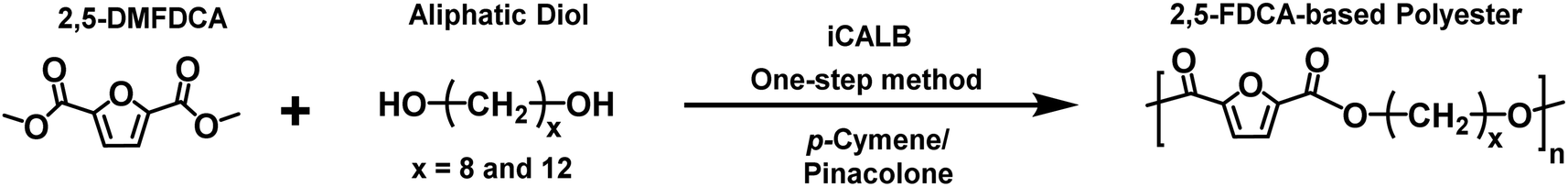 | ||
| Scheme 1 Enzymatic synthesis of furan-based polyesters using biobased solvents. | ||
| x | Polyesters | Abbreviation |
|---|---|---|
| a The number of methylene units in the aliphatic linear diol. | ||
| 8 | Poly(octamethylene-2,5-furanoate) | 2,5-POF |
| 12 | Poly(dodecamethylene-2,5-furanoate) | 2,5-PDOF |
The successful enzymatic synthesis of the FPEs listed in Table 1 was confirmed by analyzing 1H-NMR and ATR-FTIR spectra, as depicted in Fig. 1. The signal at 4.3 ppm, corresponding to –CO–O–CH2–, provided evidence of polyester formation. Furthermore, the ATR-FTIR spectra supported ester linkage formation, as evidenced by the sharp band at approximately 1720 cm−1, corresponding to the CO stretching vibration of the ester groups. A detailed assignment of the 1H-NMR and ATR-FTIR peaks can be found in the Materials and methods section.
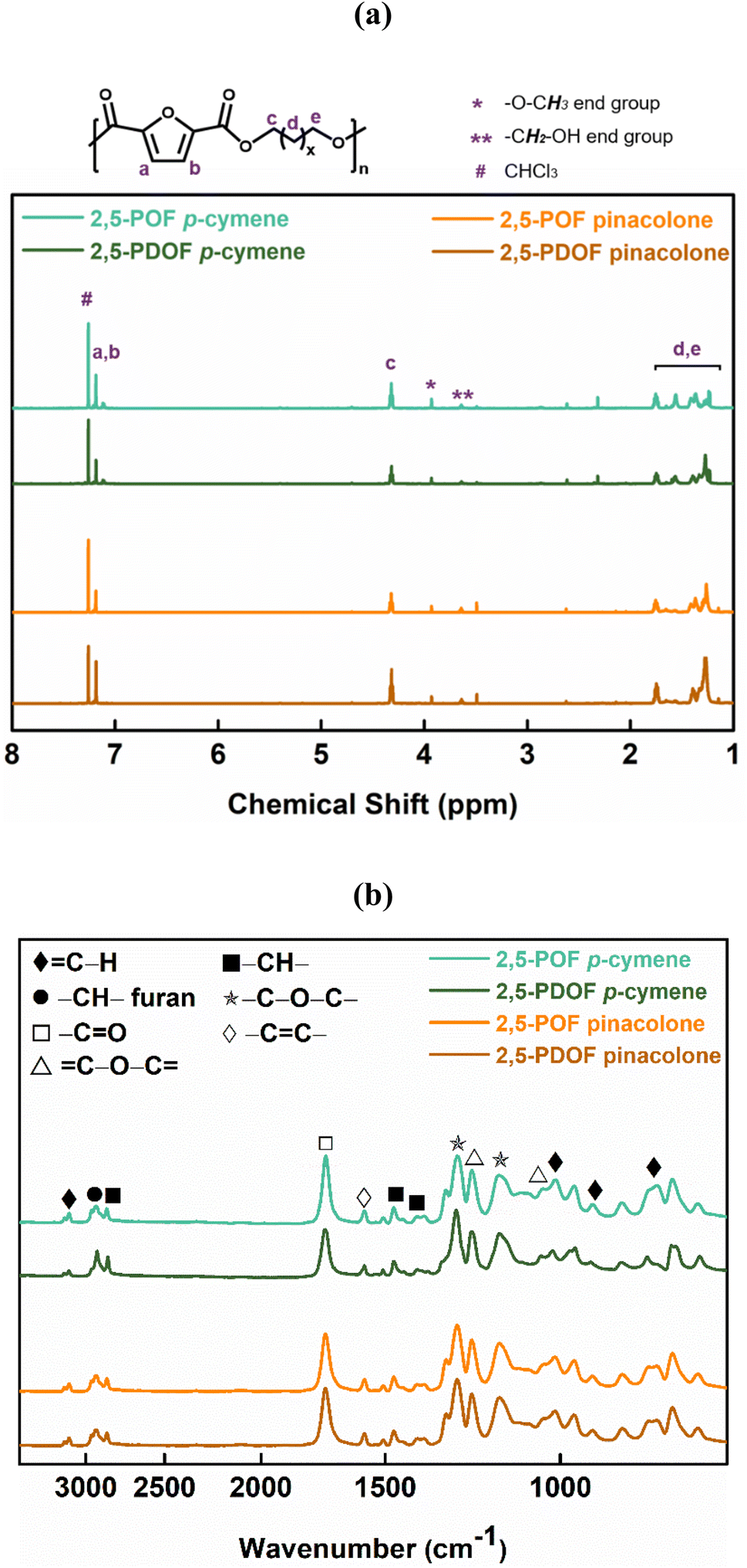 | ||
| Fig. 1 (a) 1H-NMR and (b) ATR-FTIR spectra of POF and PDOF obtained from the utilized biobased solvents. | ||
Matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-ToF MS) was employed to further investigate the microstructures of the FPEs and their end groups. The representative mass spectra of the FPEs are shown in Fig. 2, covering the m/z range from 500 to 3500. Distinct peak separations (m/z) were observed, corresponding to 2,5-POF and 2,5-PDOF, with aliphatic repeating unit molecular weights of 266.11 g mol−1 and 322.17 g mol−1, respectively, confirming the presence of both 2,5-POF and 2,5-PDOF as the products. For all the tested FPEs, a similar pattern was observed in the MALDI-ToF MS spectra, revealing the presence of eight species based on the end group provided in Table 2. Fig. 2 shows that generally, the FPEs are linear products of species A and G with ester/–OH and ester/ester end groups. The MALDI-ToF result also confirmed the presence of cyclic species (B) in the product, which usually occurs during CALB catalytic polymerization.40–42 This finding agrees with previous work on furan-based polyesters, which utilize diphenyl ether as the enzymatic reaction medium.39
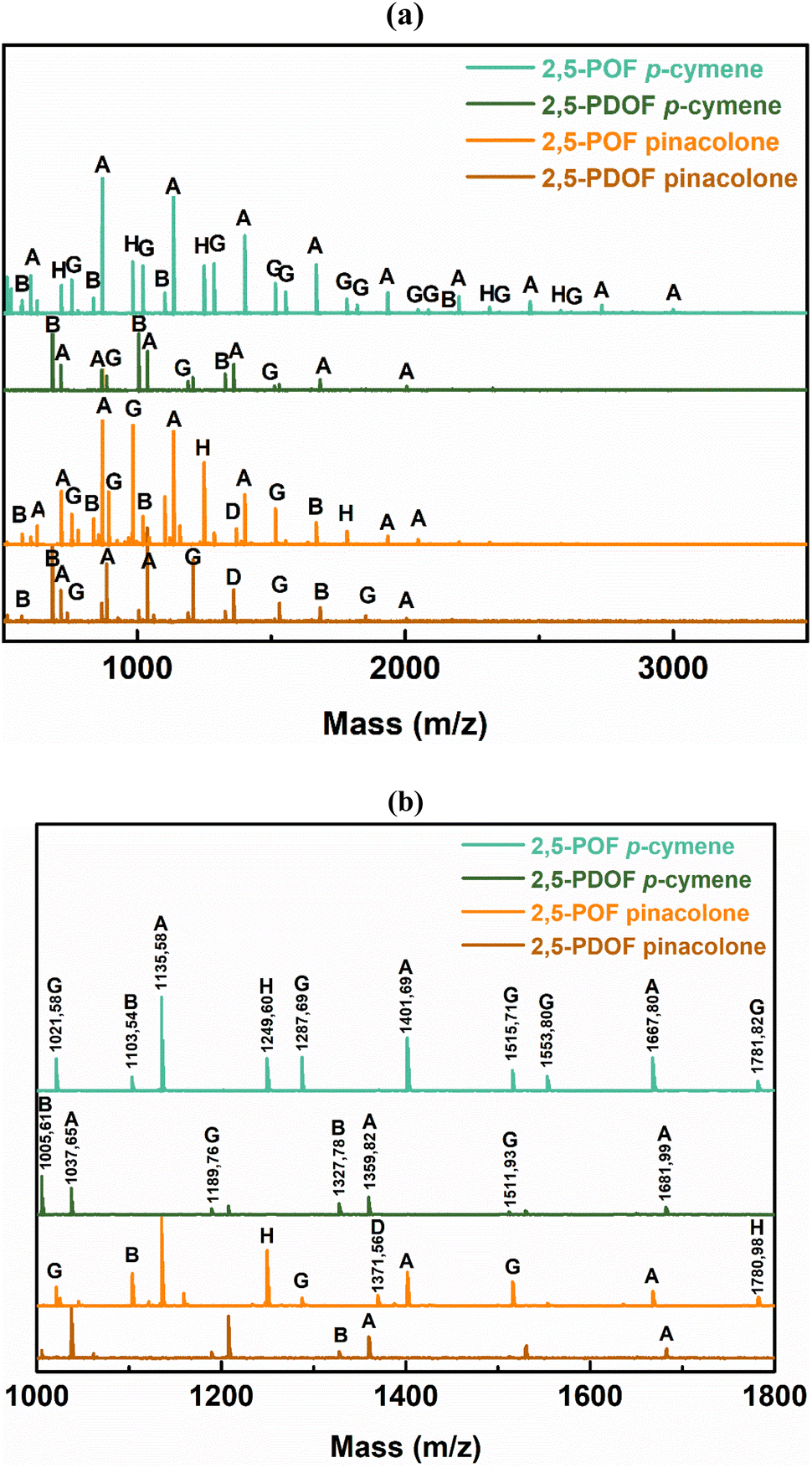 | ||
| Fig. 2 (a) MALDI-ToF MS spectra of 2,5-POF and PDOF obtained from different biobased solvents and the (b) magnified version. | ||
| Entry | Polyester species | End groups | Remaining massa (amu) |
|---|---|---|---|
| a Remaining mass refers to the mass of the residual fragment after fragmentation in mass spectrometry, expressed in atomic mass units (amu), where 1 amu is defined as one-twelfth of the mass of a carbon-12 atom. | |||
| A |
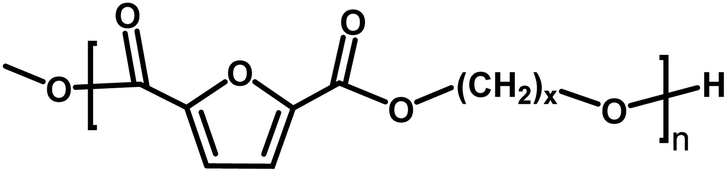
|
Ester/–OH | 32.04 |
| B |
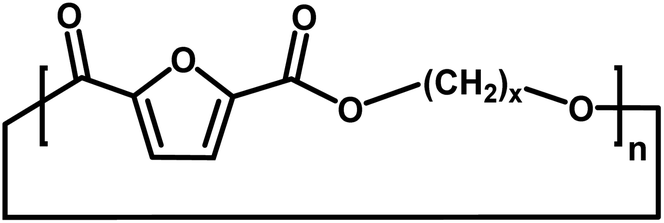
|
Cyclic | 0 |
| C |
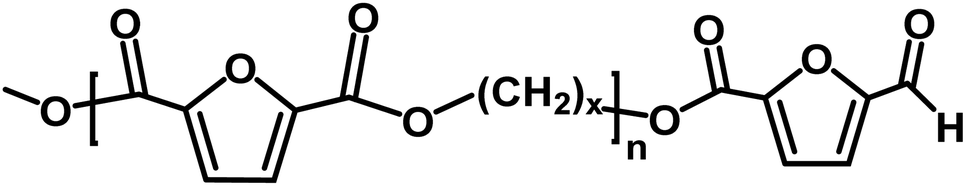
|
Ester/aldehyde | 154.03 |
| D |
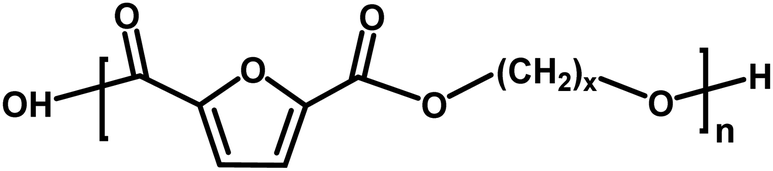
|
Acid/alcohol | 18.02 |
| E |
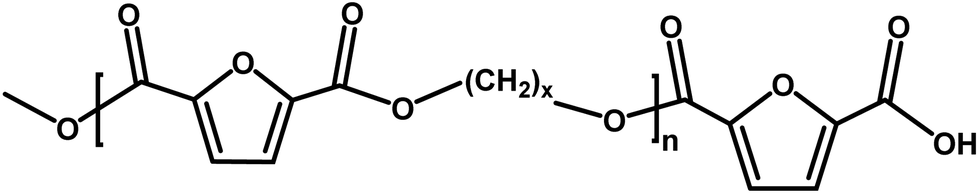
|
Ester/acid | 170.03 |
| F |
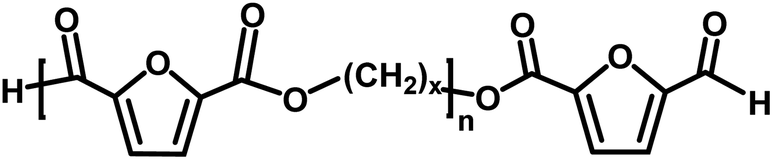
|
Aldehyde/aldehyde | 124.1 |
| G |
|
Ester/ester | 184.15 |
| H |
|
Alcohol/alcohol | 118.18 (x = 6) |
| 145.22 (x = 8) | |||
| 174.28 (x = 10) | |||
| 202.34 (x = 12) | |||
Influence of biobased solvents on the enzymatic synthesis of furan-based polyesters
In the realm of enzymatic polymerization, prior studies extensively documented the synthesis of FDCA-based polymers utilizing lipase in conventional organic solvents such as diphenyl ether and toluene.10,19,38,43 Notably, a two-stage method in diphenyl ether yielded a high molecular weight 2,5-PDF polymer of 23700 g mol−1 which is seven times higher than the molecular weight of 2,5-POF obtained with the same method, due to the longer aliphatic diol enhancing the molecular weight.39 The one-stage method in toluene resulted in 2,5-PDOF and poly(octamethylene furanamide) (PA8F) with molecular weights of 13300 and 54000 g mol−1, respectively, as reported previously.34,43 However, the environmental implications associated with diphenyl ether and toluene necessitated a shift towards enzymatic polymerization utilizing potentially greener alternatives, specifically biobased solvents. Consequently, this study focused on employing two bioderived solvents, p-cymene and pinacolone, for polymerizing 2,5-POF and 2,5-PDOF. These solvents were chosen based on the reported higher activity of CALB in these solvents.27,29
Fig. 3 and Table S1† reveal that using p-cymene resulted in a higher average molecular weight 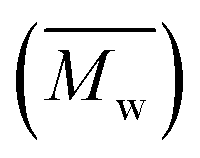 for 2,5-PDOF and 2,5-POF, achieving
for 2,5-PDOF and 2,5-POF, achieving 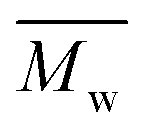 up to 7000 and 5000 g mol−1, respectively. In contrast, polymerizations using pinacolone yielded
up to 7000 and 5000 g mol−1, respectively. In contrast, polymerizations using pinacolone yielded 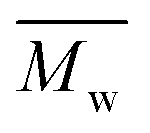 6200 and 4100 g mol−1, respectively. The SEC traces of the polyesters exhibit slight tailing, with the polyesters synthesized in p-cymene more distinctly revealing multiple populations (Fig. S8†). The higher
6200 and 4100 g mol−1, respectively. The SEC traces of the polyesters exhibit slight tailing, with the polyesters synthesized in p-cymene more distinctly revealing multiple populations (Fig. S8†). The higher 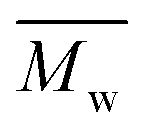 in p-cymene is attributed to the higher logP of p-cymene (3.47) compared to pinacolone (1.21), a critical factor in enzymatic catalysis.33,44 It is known that a solvent with a logP higher than 4 is favored for enzymatic catalysis because it tends to be less deactivating to the enzyme.21,23
in p-cymene is attributed to the higher logP of p-cymene (3.47) compared to pinacolone (1.21), a critical factor in enzymatic catalysis.33,44 It is known that a solvent with a logP higher than 4 is favored for enzymatic catalysis because it tends to be less deactivating to the enzyme.21,23
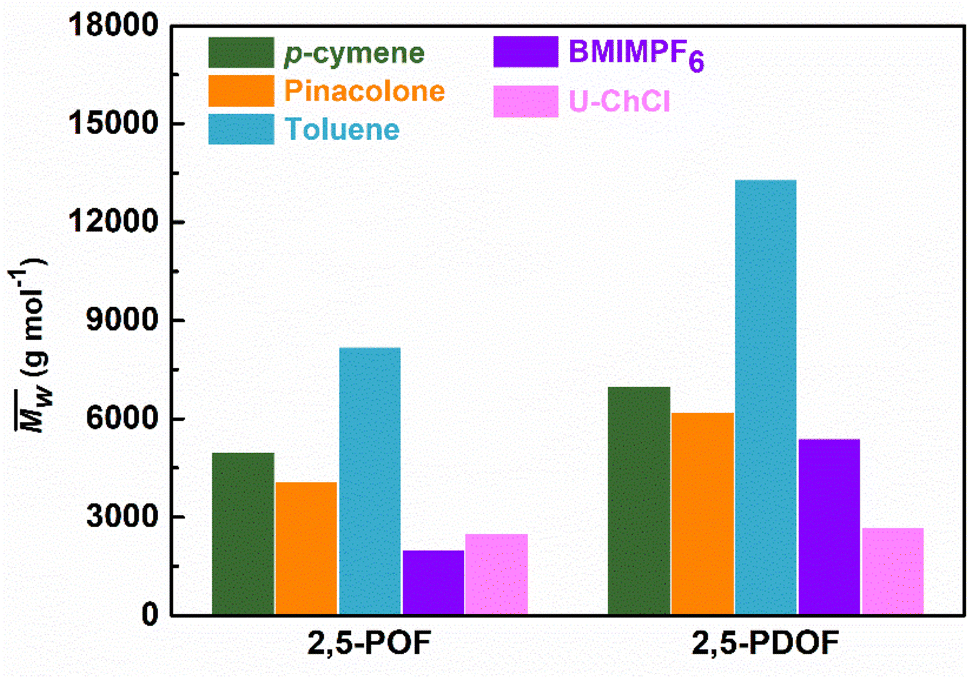 | ||
Fig. 3 Comparison of weight-average molecular weights 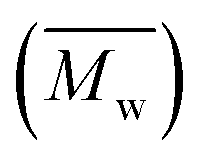 of furanic–aliphatic polyesters synthesized in different solvents via enzymatic polymerizations using CALB. The data on BMIMPF6 and U–ChCl from a previous study,16 and toluene from a previous study.34 of furanic–aliphatic polyesters synthesized in different solvents via enzymatic polymerizations using CALB. The data on BMIMPF6 and U–ChCl from a previous study,16 and toluene from a previous study.34 | ||
Considering additional solvent parameters from Table S3,† such as hydrogen bond accepting ability (β) and molar volume (Vm), which were previously reported to affect the enzymatic reaction rate, further reinforces p-cymene's efficacy.26,27p-Cymene, with a lower β (0.13) than pinacolone (0.58), signifies reduced interference at the solvent–enzyme catalytic site, facilitating favorable substrate–enzyme interactions.26,27,45 Moreover, higher Vm of p-cymene (156.058 mL mol−1) suggests a more conducive environment for catalytic reactions than pinacolone (125.2 mL mol−1). As previously reported, a higher Vm signifies a bulkier structure, leading to less solvent interaction with the enzyme and substrate, thereby promoting the enzymatic catalytic reaction of the substrates.26,27 In summary, these solvent parameters, namely logP, β, and Vm, support the suitability of p-cymene as the reaction medium over pinacolone in this study.
Furthermore, as previously reported,46,47p-cymene has excellent dissolution capability for conjugated polymers, allowing the polymerization to proceed without any interference from precipitation of oligomers or polymers during the reaction. Favorable polymer or oligomer interactions with the solvent during polymer chain growth affect the availability of reactive groups for inducing polymerization.18,48 This might provide additional insight into the high percentage of enzymatic polycondensations achieved with p-cymene. There is an opportunity to improve the results for better comparability with toluene. Previous research by Pellis et al.49 suggests that p-cymene yields higher molecular weight at lower temperatures, known to promote the formation of linear polymer products in CALB-catalyzed polymerization.50 Further investigation at lower temperatures could clarify this hypothesis.
Expanding on the results obtained from the investigation of MALDI-ToF MS peak intensities, the study confirms that altering the aliphatic diol chain length from eight to twelve influences linear and cyclic production during enzymatic catalysis using p-cymene or pinacolone. Despite the caution on the potential impact of end groups on ionization efficiency,51–55 MALDI-ToF MS offers direct identification of polymer species. The linear product of species A and G generated using p-cymene or pinacolone indicates ester/OH and ester/ester end groups. Additionally, cyclic product percentages increased to 86% for p-cymene and 48% for pinacolone with the extension of the aliphatic diol chain from eight (2,5-POF) to twelve (2,5-PDOF). This observation aligns with prior findings using toluene, which is attributed to the greater flexibility of longer aliphatic diol chain length molecules.34 However, in contrast to the previous findings that linear products were infrequent when utilizing toluene,34 we observe that CALB-catalyzed polymerization with p-cymene or pinacolone generates more linear products than cyclic FPE products. This underscores the versatility of p-cymene and pinacolone in producing a linear structure rather than a cyclic one, attributed to their higher Vm than toluene as a previously reported small size solvent was preferred for cyclization.56
Comparative analyses with enzymatic syntheses in ionic liquids or deep eutectic solvents, namely 1-butyl-3-methylimidazolium hexafluorophosphate (BMIMPF6) and urea–choline chloride (U–ChCl) from a previously reported study,16 highlighted the superior results of 2,5-POF and 2,5-PDOF obtained using p-cymene or pinacolone in this study (see Fig. 3). The  values demonstrated that the enzymatic synthesis using these biobased solvents was up to 61% more effective for p-cymene and 55% for pinacolone compared to using BMIMPF4 or U–ChCl. Furthermore, the yield increased to 35% for 2,5-PDOF using p-cymene and 14% for 2,5-PDOF using pinacolone. Although the molecular weights were generally lower than those achieved with toluene, comparable yields hinted at the potential for more environmentally friendly enzymatic polymerization methods with biobased solvents, particularly for 2,5-FDCA-based polyesters.
values demonstrated that the enzymatic synthesis using these biobased solvents was up to 61% more effective for p-cymene and 55% for pinacolone compared to using BMIMPF4 or U–ChCl. Furthermore, the yield increased to 35% for 2,5-PDOF using p-cymene and 14% for 2,5-PDOF using pinacolone. Although the molecular weights were generally lower than those achieved with toluene, comparable yields hinted at the potential for more environmentally friendly enzymatic polymerization methods with biobased solvents, particularly for 2,5-FDCA-based polyesters.
In addressing the common issue of discoloration in FDCA-based polymers, p-cymene has emerged as a promising solution, resulting in a white-powder product without discoloration, similar to that obtained with toluene from previous work.34 In contrast, pinacolone yielded a slightly yellowish product, requiring additional purification steps to enhance its appearance. The presence of a pronounced methylene proton peak at 3.49 ppm suggests that the yellow appearance may be attributed to the presence of ketal side products, as previously observed in CALB-catalyzed galactarate-based polymers using Cyrene, which also contains ketone functional groups.57–59 The discoloration issue associated with pinacolone could potentially be addressed through a two-step method, which would facilitate more effective removal of methanol side products, likely contributing to increased ketal formation due to the shorter chain alcohol in the system.60 Further research is needed to draw conclusive insights into this discoloration phenomenon (Fig. 4).
 | ||
| Fig. 4 From left to right, the products of poly(dodecamethylene-2,5-furanoate) synthesized with toluene from previous work,34p-cymene, and pinacolone. | ||
Synthesis and structural characterization of furan-based copolyesters
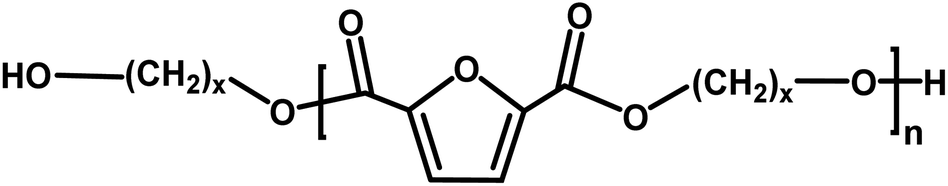
This study further explores utilizing biobased solvents applied to the enzymatic synthesis of furan-based copolyesters by incorporating 2,5-BHMF. It is known that the rigid structure is frequently affected by the enzymatic polymerization with CALB, as well as material properties.10,61 Therefore, exploring the copolymerization with 2,5-BHMF in a greener solvent, such as biobased solvents, is fascinating. In light of the results from the enzymatic synthesis of polyesters, in which we discovered the discoloration issue with pinacolone, in the second scheme of this study, besides utilizing p-cymene, we chose to use another variation of a citrus-based solvent, D-limonene, which was previously also reported to perform well in CALB catalysis.27 In addition, we performed copolymerization in toluene as the comparison system with the traditional organic solvent. As presented in Scheme 2, the copolymerization of 2,5-DMFDCA with 2,5-BHMF as a rigid diol and 1,10-decanediol as the linear aliphatic monomer was performed utilizing CALB as a catalyst. The main focus is to investigate the viability of the two citrus-based solvents, considering their sustainability, in the synthesis of furan-based copolyesters and to assess their impact on enzymatic catalysis.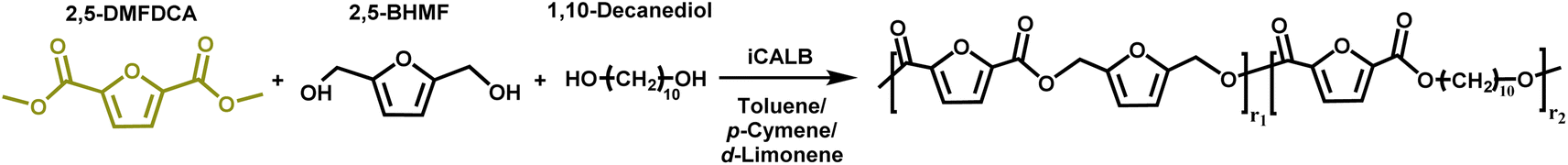 | ||
| Scheme 2 Enzymatic synthesis of P(2,5-FMF-co-2,5-DF). | ||
1H-NMR signals in Fig. 5a at approximately 4.31 and 5.29 ppm, prove the presence of –CO–O–CH2– in the ester formed by the combination of 2,5-DMFDCA with aliphatic 1,10-decanediol, as well as in the ester formed by 2,5-DMFDCA and 2,5-BHMF. These signals confirmed the formation of copolyesters, consistent with prior research on furan-based copolyesters.10 In the ATR-FTIR spectra shown in Fig. 5b, a prominent band at 1721 cm−1, representing the stretching vibration of the CO bond in the ester groups, offers additional evidence of ester linkage formation. Furthermore, specific bands related to the furan structure were observed, including a weak band corresponding to the stretching vibration of the C–H bonds in the furan ring at 3118–3137 cm−1 and bands at 1573–1583 cm−1 and 1506–1511 cm−1 corresponding to the bending vibrations of the aromatic CC bonds. These findings confirm the successful synthesis and structural attributes of P(2,5-FMF-co-2,5-DF) in this study. For precise 1H-NMR and ATR-FTIR peak assignments, please refer to the Materials and methods section. To investigate the microstructures of P(2,5-FMF-co-2,5-DF) obtained from three different solvents and their end groups, MALDI-ToF MS analysis was employed. Fig. 6 shows their mass spectra, ranging from m/z 700 to 2500. Clear distinctions in peak separations (m/z) were evident in those co-FPEs with ten aliphatic unit chain lengths, confirming the presence of this copolyester with aliphatic and hetero-aromatic segments with molecular weights of 294.35 g mol−1 and 263.23 g mol−1, respectively. Across all tested co-FPEs, a consistent profile appeared in the MALDI-ToF MS results for approximately eleven species, as outlined in Table 3. These spectra displayed similar peaks for the three co-FPEs, highlighting the prevalence of linear species A, D, H, and F and cyclic species B. These findings align with prior observations on furanic copolyesters,35 confirming the occurrence of typical cyclic and linear products in enzyme-catalyzed polymerization.40–42
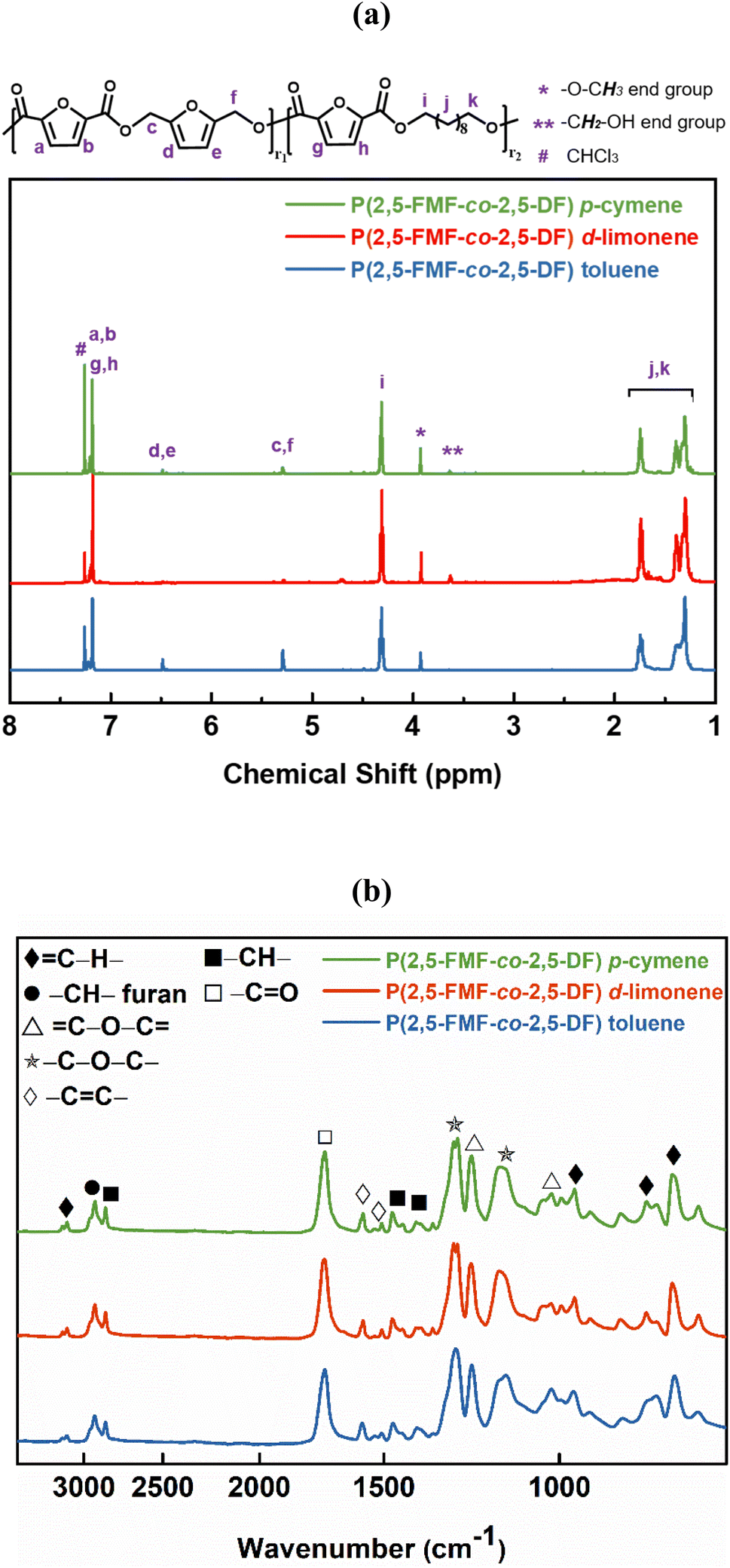 | ||
| Fig. 5 (a) 1H-NMR and (b) ATR-FTIR spectra of P(2,5-FMF-co-2,5-DF) resulting from the enzymatic polymerization of 2,5-DMFDCA, 2,5-BHMF and 1,10-decanediol in different solvents. | ||
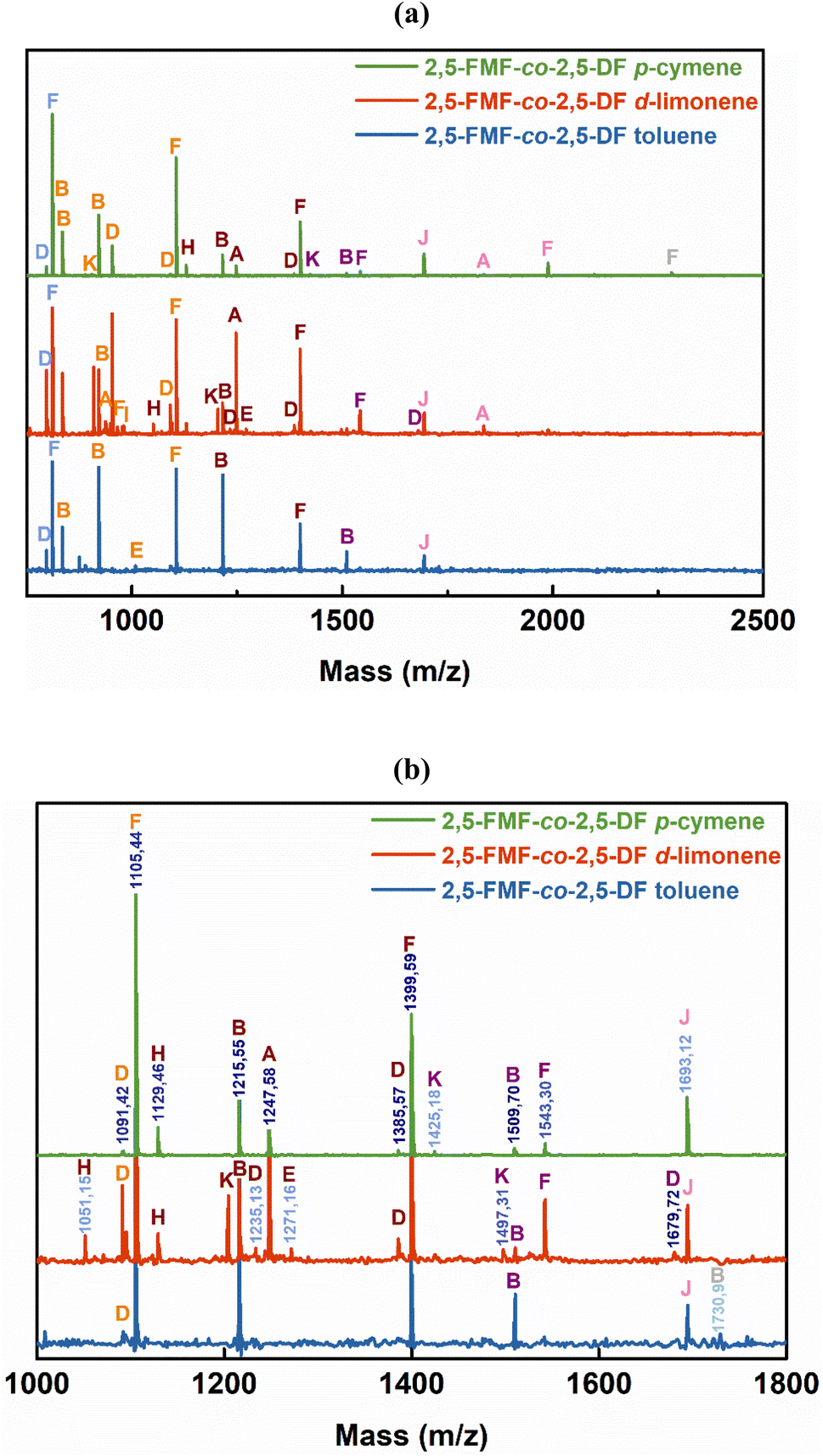 | ||
| Fig. 6 (a) MALDI-ToF MS spectra of P(2,5-FMF-co-2,5-DF) obtained from different solvents from the two-step method under 600 mmHg vacuum conditions and (b) the magnified version. | ||
| Entry | Copolyester species | End groups | Remaining massa (amu) |
|---|---|---|---|
| a Remaining mass refers to the mass of the residual fragment after fragmentation in mass spectrometry, expressed in atomic mass units (amu), where 1 amu is defined as one-twelfth of the mass of a carbon-12 atom. | |||
| A |
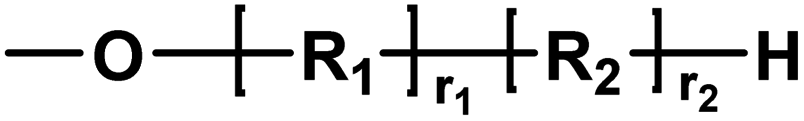
|
Ester/alcohol | 32.04 |
| B |
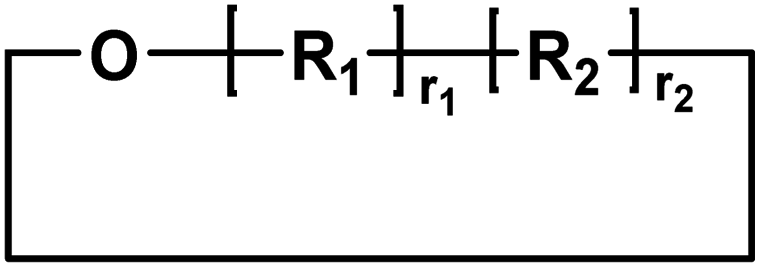
|
Cyclic | 0 |
| C |
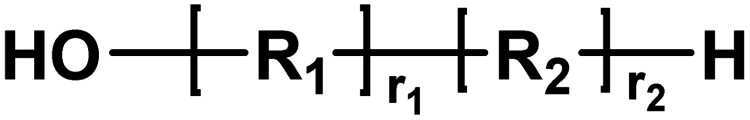
|
Acid/alcohol | 18.02 |
| D |
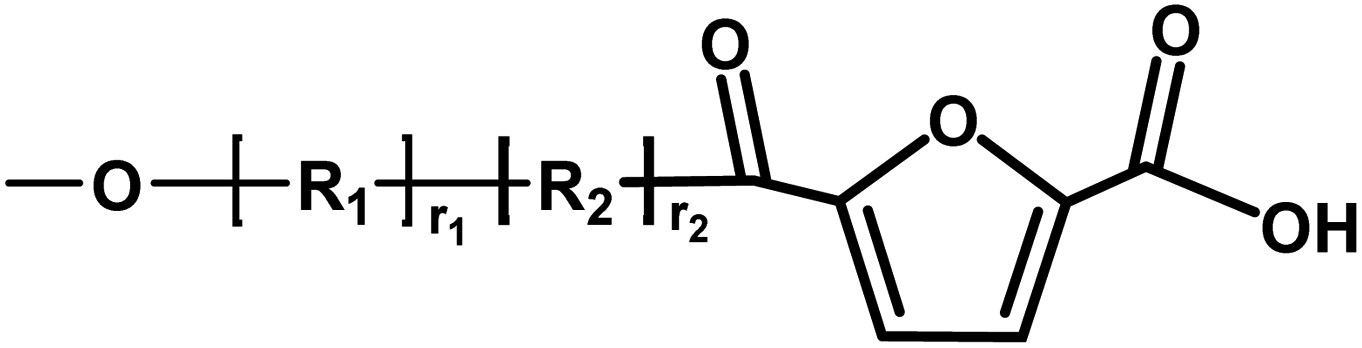
|
Ester/acid | 170.03 |
| E |

|
Aldehyde/aldehyde | 124.1 |
| F |
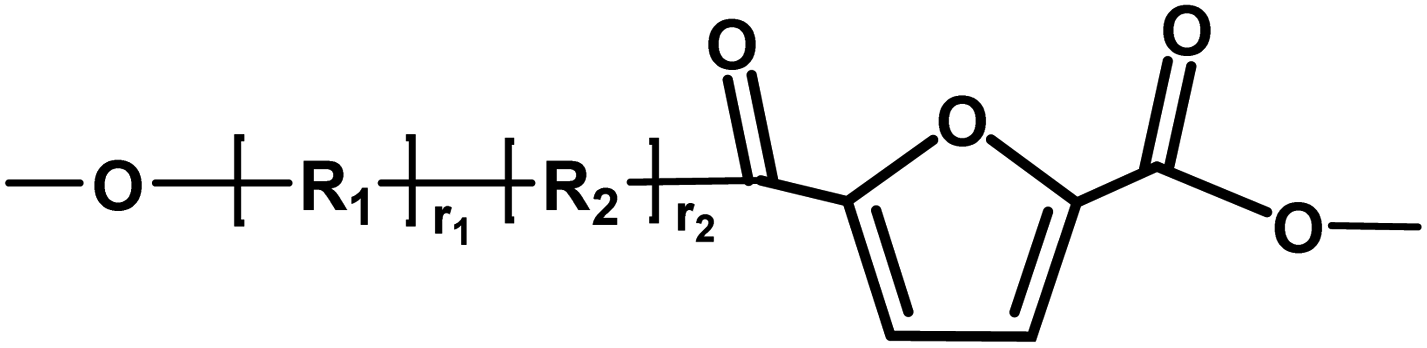
|
Ester/ester | 184.15 |
| G |
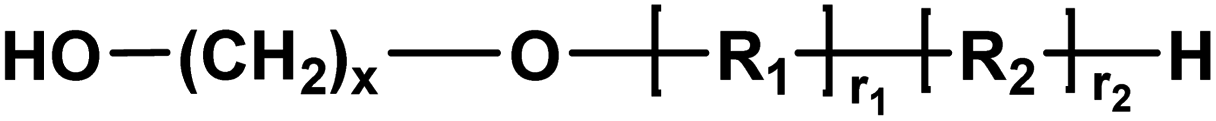
|
Alcohol/alcohol | 128.05 (2,5-BHMF) |
| 118.18 (x = 6) | |||
| 146.23 (x = 8) | |||
| 174.28 (x = 10) | |||
| 202.34 (x = 12) | |||
| H |
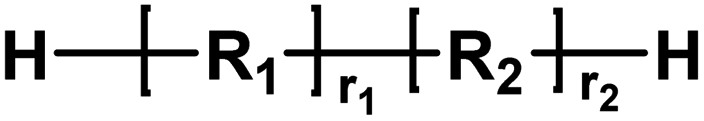
|
Aldehyde/alcohol | 2.02 |
| I |
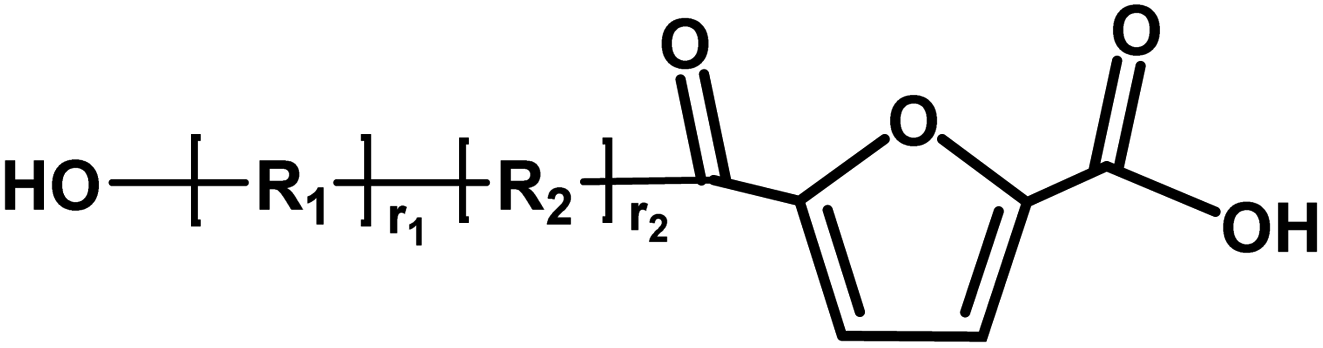
|
Acid/acid | 156.01 |
| J |
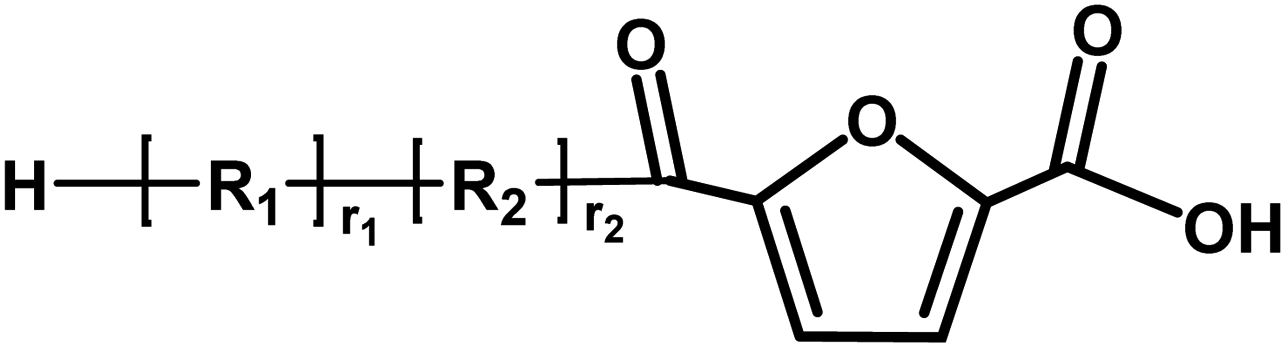
|
Acid/aldehyde | 140.01 |
| K |
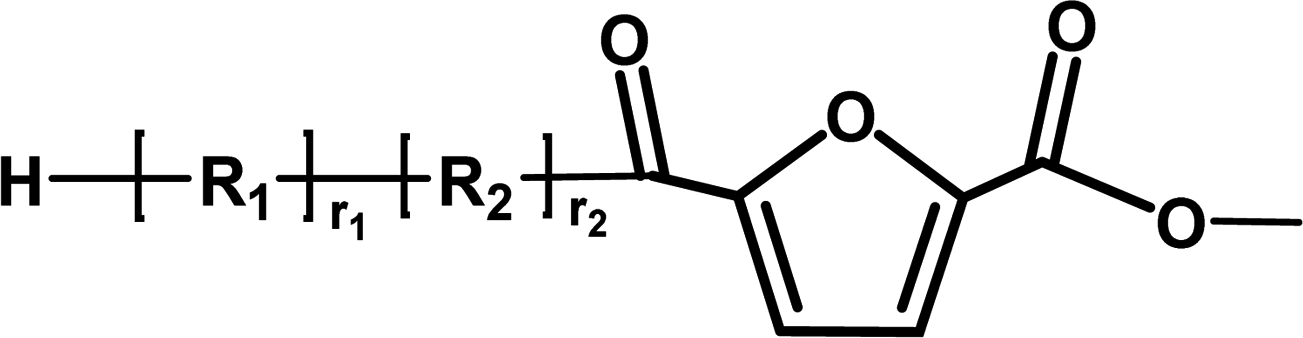
|
Ester/aldehyde | 154.03 |
| Copolyesters unit and segment | |||
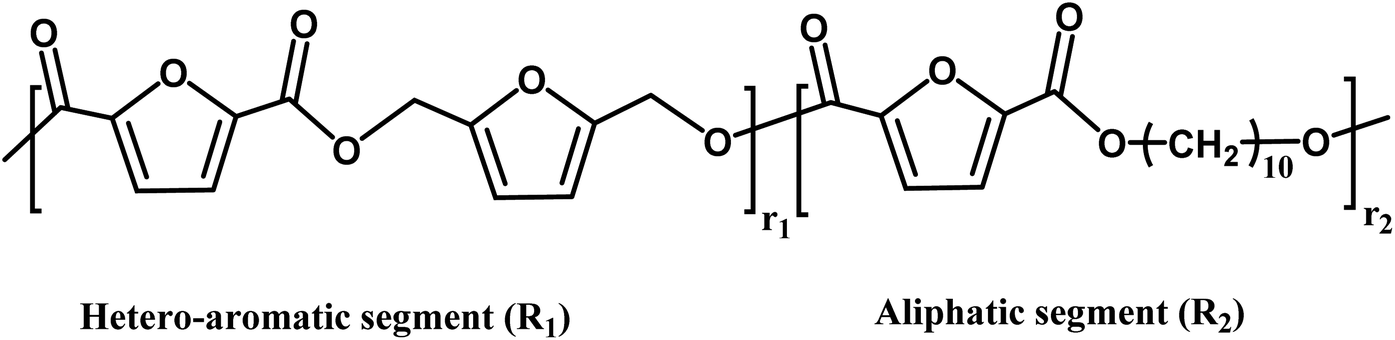
|
|||
| n = r1 + r2 | |||
Another insight from MALDI-ToF MS and the estimated m/z values based on the variations in repeating units revealed that co-FPEs exposed random polymer structures. As evident in Fig. 6a, species with similar repeating unit numbers (n) are identified with the same color. In addition, the gradual increase in the number of aliphatic segments (r1) is visually depicted by the blue color gradation for each m/z peak in Fig. 6b, inversely corresponding to the decrease in the number of hetero-aromatic segments (r2). In agreement with the previous work on furanic copolyesters,35 this random polymer structure often appeared due to transesterification polymerization involving the three monomers.62,63
Influence of citrus-based solvents on the enzymatic synthesis of furan-based copolyesters
The use of two citrus-based solvents led to the successful synthesis of P(2,5-FMF-co-2,5-DF), with results comparable to those obtained with the organic solvent toluene. As seen in Fig. 7 and Table S2,† we observed that from the one-step method, toluene possesses 30 and 44% higher compared to results with p-cymene and D-limonene, respectively. However, when we applied the two-step method with p-cymene, the obtained polymer had a molecular weight comparable to that obtained with toluene. P(2,5-FMF-co-2,5-DF) possesses
compared to results with p-cymene and D-limonene, respectively. However, when we applied the two-step method with p-cymene, the obtained polymer had a molecular weight comparable to that obtained with toluene. P(2,5-FMF-co-2,5-DF) possesses 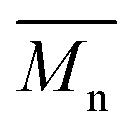 5600 and
5600 and 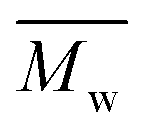 12800 with p-cymene when vacuum is applied to 60 mmHg, which is more or less similar to the results from the two-step method in toluene with
12800 with p-cymene when vacuum is applied to 60 mmHg, which is more or less similar to the results from the two-step method in toluene with 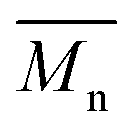 5800 and
5800 and  11400. Moreover, polymerization utilizing D-limonene did not substantially differ between
11400. Moreover, polymerization utilizing D-limonene did not substantially differ between 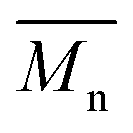 and
and 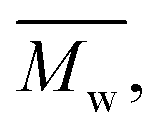 regardless of the applied method. The values are consistent for each method, with
regardless of the applied method. The values are consistent for each method, with 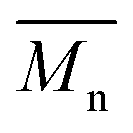 at 2400, 2300, and 3000 g mol−1 and
at 2400, 2300, and 3000 g mol−1 and 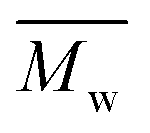 at 3500, 3400, and 4500 g mol−1. As previously mentioned, solvents with higher logP values (>4.0) are preferable for enzymatic reactions.24 However, the logP preference did not apply to the higher p-cymene results since the p-cymene logP (3.47) is lower than the D-limonene logP (4.57).
at 3500, 3400, and 4500 g mol−1. As previously mentioned, solvents with higher logP values (>4.0) are preferable for enzymatic reactions.24 However, the logP preference did not apply to the higher p-cymene results since the p-cymene logP (3.47) is lower than the D-limonene logP (4.57).
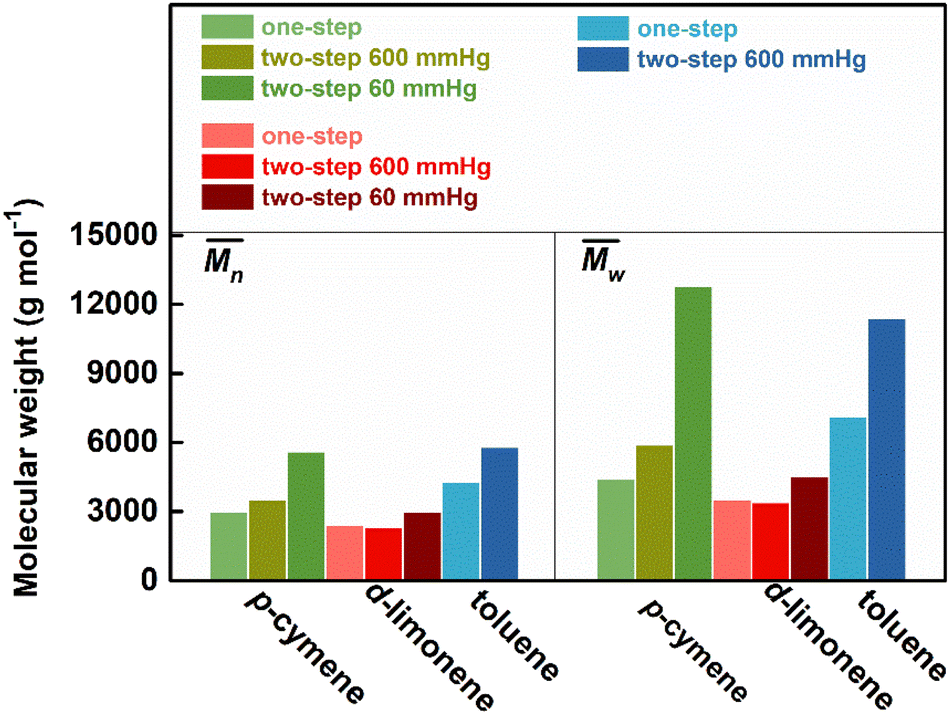 | ||
Fig. 7 Number-average molecular weight 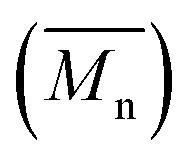 and weight-average molecular weight and weight-average molecular weight 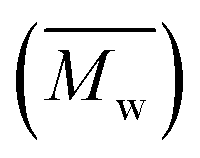 of P(2,5-FMF-co-2,5-DF) synthesized in different solvents and using different methods. of P(2,5-FMF-co-2,5-DF) synthesized in different solvents and using different methods. | ||
p-Cymene and toluene have demonstrated superior performance compared to D-limonene in both 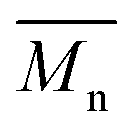 (Fig. 7) and the % mol-fraction of 2,5-BHMF in the copolyester (Fig. S3†). This trend closely aligns with the solvent β values outlined in Table S3† (0.13, 0.12 > 0.00), as discussed previously for polyesters.26,27 Furthermore, this alignment extends to the aromaticity trends of the solvents: toluene and p-cymene are aromatic, whereas D-limonene lacks aromatic properties. Additionally,
(Fig. 7) and the % mol-fraction of 2,5-BHMF in the copolyester (Fig. S3†). This trend closely aligns with the solvent β values outlined in Table S3† (0.13, 0.12 > 0.00), as discussed previously for polyesters.26,27 Furthermore, this alignment extends to the aromaticity trends of the solvents: toluene and p-cymene are aromatic, whereas D-limonene lacks aromatic properties. Additionally,  and % mol-fraction of hetero-aromatic diol 2,5-BHMF are linearly correlated (r2 = 89.3%), indicating that aromatic solvents yield a longer product enriched in hetero-aromatic monomer units. Aromatic solvent π–π stacking with furan is favored by approximately −4 kcal mol−1 per interaction; this solvation needs to be broken to access the CALB active site.64,65 We hypothesize that aromatic solvent shielding of 2,5-BHMF repeat units throughout the copolyester worsens product–CALB interaction. At high conversion, such a cleavage-resistant copolyester better retains molecular weight. Furthermore, D-limonene degrades by approximately 17% at 90 °C,66,67 suggesting that it may not be inert under the temperature conditions employed in this work. This degradation could account for the low conversion observed with D-limonene. However, identifying the specific peak corresponding to D-limonene degradation derivatives, such as in the 1H-NMR spectrum, was challenging due to the presence of numerous low-intensity overlapping peaks. In summary, the ability to accept hydrogen bonds (β) and π–π stacking plays a role in generating stronger catalytic activity of CALB in this copolymerization. These results suggest that p-cymene is a superior biobased solvent alternative for furan-based polymers, and optimizing the removal of side products with a two-step method considerably enhances the comparability of results with toluene.
and % mol-fraction of hetero-aromatic diol 2,5-BHMF are linearly correlated (r2 = 89.3%), indicating that aromatic solvents yield a longer product enriched in hetero-aromatic monomer units. Aromatic solvent π–π stacking with furan is favored by approximately −4 kcal mol−1 per interaction; this solvation needs to be broken to access the CALB active site.64,65 We hypothesize that aromatic solvent shielding of 2,5-BHMF repeat units throughout the copolyester worsens product–CALB interaction. At high conversion, such a cleavage-resistant copolyester better retains molecular weight. Furthermore, D-limonene degrades by approximately 17% at 90 °C,66,67 suggesting that it may not be inert under the temperature conditions employed in this work. This degradation could account for the low conversion observed with D-limonene. However, identifying the specific peak corresponding to D-limonene degradation derivatives, such as in the 1H-NMR spectrum, was challenging due to the presence of numerous low-intensity overlapping peaks. In summary, the ability to accept hydrogen bonds (β) and π–π stacking plays a role in generating stronger catalytic activity of CALB in this copolymerization. These results suggest that p-cymene is a superior biobased solvent alternative for furan-based polymers, and optimizing the removal of side products with a two-step method considerably enhances the comparability of results with toluene.
Despite the caution previously mentioned in investigating the MALDI-ToF MS peak height intensities, as illustrated in Fig. S2,† similar trends in the prevalence of cyclic species were generally indicated for the products synthesized with p-cymene and toluene. In contrast, when D-limonene was used, the dominant species consisted primarily of linear products. Using the one-step method with p-cymene, D-limonene, and toluene provided additional insights. The results indicated fairly consistent trends, with predominantly cyclic species B occurring at 24%, 25%, and 45%, respectively. Additionally, linear products of species F were observed at 32%, 25%, and 20%. Applying 600 mmHg vacuum conditions to the two-step method revealed remarkable trends. Notably, cyclic species were prevalent in both products when p-cymene and toluene were used. The percentage of cyclic species doubled to 54% with p-cymene and increased to 52% with toluene. This shift correlated with an increase in species J with the hydroxy group, which supports intramolecular backbiting to form cyclic conformations during lipase-catalyzed polymerization.40–42 In addition, consistent with previous reports on the preference of CALB for generating cyclic species with symmetrical substrates such as 2,5-DMFDCA, attributed to the free rotation of hydroxy groups,41 as discussed in previous work on furanic copolyesters, these changes may be attributed to more effective cyclization through backbiting.35 The two-step method likely facilitated fewer interruptions from side products in the system. Subsequently, a higher vacuum of 60 mmHg was applied to improve the removal of side products or water from the system. This adjustment revealed no substantial difference in results, specifically maintaining the cyclic species at 51% when using p-cymene.
In contrast to the results obtained with p-cymene and toluene, the application of vacuum at both 600 mmHg and 60 mmHg during enzymatic polymerization using D-limonene resulted in comparable decreases in the proportions of cyclic species, reaching 15% and 14%, respectively. The reversal of trends with D-limonene compared to p-cymene is consistent with the molecular weight trends discussed earlier. Hypothetically, the explanation related to p-cymene’s excellent dissolution capability might apply. As previously mentioned in the discussion section for FPEs, p-cymene, known for its excellent dissolution capability for conjugated polymers,46,47 is likely to contribute to facile and mild polymerization, including cyclization, without any interference from solid traces of oligomers or polymers during the reaction. The SEC traces of the copolyesters exhibit significant tailing for those synthesized in toluene and p-cymene under strong vacuum conditions. In contrast, the protocols using D-limonene and other p-cymene methods produced more uniform peaks, characterized by only minimal tailing (Fig. S9†).
Consistent with the earlier discussion on the FPE product, P(2,5-FMF-co-2,5-DF) from enzymatic polymerization with p-cymene also appeared as a white powder similar to that obtained with toluene (see Fig. 8). Moreover, the product from the enzymatic reaction with D-limonene was a dark yellow powder, and multistep purification was required for a better appearance. The dark yellow color is likely attributed to the presence of trace amounts of D-limonene degradation products as discussed previously; besides D-limonene undergoes autoxidation in the presence of light and air, resulting in the formation of various oxygenated monocyclic terpenes.67–70 These conditions could subsequently lead to the formation of side products. This discoloration issue could be effectively addressed by reducing the reaction temperature to at least 70 °C, considering that D-limonene degrades less than 10% around this temperature.66,67 Further research is necessary to conclusively validate this hypothesis. In summary, these findings underscore the superiority of p-cymene as a citrus-based solvent for furan-based polymerization compared to D-limonene.
 | ||
| Fig. 8 From left to right: product appearance of P(2,5-FMF-co-2,5-DF) synthesized with toluene, p-cymene, and D-limonene. | ||
Thermal properties and crystallinity of the obtained furan-based polyesters
The thermal properties of the FPEs obtained in this work were evaluated via DSC and TGA analyses, as presented in Fig. S4 and Table S4.† All the obtained polyesters exhibited a one-step degradation profile and decompose at temperatures ranging from 390 to 396 °C, which agrees with the previous work on 2,5-FDCA-based polyesters.16,34 They possess relatively high thermal stability, making them suitable for diverse potential applications. The decomposition temperature of PDOF was slightly greater than that of POF; this difference could be attributed to the greater molecular weight of PDOF.The thermal properties of polyesters are notably influenced by factors such as the length of the aliphatic methylene chain, the degree of crystallinity or the presence of an amorphous phase, and the molecular weight. As observed from the DSC results, the obtained 2,5-PDOF with twelve aliphatic chain lengths showed lower Tg than 2,5-POF with eight aliphatic chain lengths, especially for the product from p-cymene. This could be attributed to the longer aliphatic chain, which makes the materials more flexible than the shorter chain, and agrees with previous work on furanic polyesters.16,18,34 The melting temperature (Tm) shows the same trend as the Tg; the Tm of 2,5-PDOF was lower than that of 2,5-POF. This behavior represents the regularity of the molecular structure of the products and is related to their crystal packing, as a higher Tm generally indicates a greater degree of crystallinity (χc). This agrees with the trends presented in Table S4;† a higher Tm was associated with a greater χc. As we observed, double Tm peaks consistently appeared after the first and second heating cycles for all the sequences of the FPE products, which aligns with the findings of previous studies on furanic polyesters.16 This multiple Tm profile suggested that the FPEs crystallize as metastable lamellae during the heating scan.71–74 According to the melt-recrystallization model, the heating process involves a competitive interplay between the melting of crystallites and their subsequent recrystallization. The melting peaks at both low and higher temperatures are attributed to the melting of a specific amount of the initial crystals and the melting of crystals formed through the melt-recrystallization process during a heating scan.72,75–77 However, further investigations, such as additional variations in the predetermined crystallization temperature during the cooling step and heating rate, are needed to support this hypothesis.
In accordance with the DSC findings, the wide-angle X-ray diffraction (WAXD) patterns affirm that the furanic–aliphatic polyesters obtained possess a semicrystalline structure with a similar diffraction pattern, which would support the multiple Tm mentioned above due to the melt-recrystallization process of metastable lamellae.78 As depicted in Fig. S6,† 2,5-POF from both p-cymene and pinacolone exhibited similar profiles with prominent diffraction peaks at 24.23° and 16.70° and broad signals across 18.25 to 22.93° and 11.84° to 14.27°. Additionally, 2,5-PDOF from the two solvents showed comparable profiles, with one prominent peak at 24.01° and a broad signal across 15.49° to 19.59° and 6.21° to 11.08°. However, in alignment with previous studies on furan–aliphatic polyesters synthesized in various solvents, such as toluene, diphenyl ether, ionic liquids, and deep eutectic solvents, the aliphatic chain apparently affects the crystallite size since 2,5-POF has a more intense peak at 16.70° than 2,5-PDOF at 15.49°.16,34,39 As previously reported for poly(butylene terephthalate), sharper diffraction peaks correspond to diffraction from larger crystallites, which also aligns with the higher crystallinity of 2,5-POF products (see Table S4†).79 Furthermore, the diffraction peak (2θ) shifted to lower values with increasing diol chain length, transitioning from 16.70° (2,5-POF) to 15.49° (2,5-PDOF). This corresponds to an increase in the spacing between atomic lattice planes (d-spacing) from 2.68 Å to 2.88 Å, which could be attributed to the extended alkylene chain of diol units requiring additional space to construct identical crystal lattices.39
Thermal properties and crystallinity of the obtained furan-based copolyesters
The thermal degradation properties of the obtained copolyesters follow two-step degradation profiles, as presented in Fig. S5 and Table S5.† The first-step maximum decomposition rate (Td-max-1) occurs between 227 and 243 °C, and the second-step maximum decomposition rate (Td-max-2) occurs at higher temperatures between 391 and 395 °C. This finding clearly supports the existence of two segments within the copolyester structure, i.e., aliphatic and hetero-aromatic segments. Considering that the monomer feed contains a lower proportion of hetero-aromatic diols than aliphatic diols, as the initial step results in a relatively minor weight loss of approximately 10%, we deduce that the Td-max-1 occurrences are associated with the hetero-aromatic segment of the copolyester. Moreover, Td-max-2 is due to the degradation of the aliphatic segment. This profile aligns with prior research on (co)-polyesters based on 2,5-BHMF,10,38,61 which also exhibit degradation in the same temperature range within the appropriate frame of aliphatic and hetero-aromatic segments.DSC analysis revealed the thermal transition of the copolyesters with a Tg below 0 °C, ranging from −1 to −5 °C. This could be attributed to the length of the ten aliphatic chains, which enhances the flexibility of the material. In addition, a low molecular weight would also increase the mobility and flexibility of the polymer molecular chains, which agrees with the lower Tg exhibited by the one-step method, as these polymers have lower molecular weights than the products from the two-step method. In general, all the obtained P(2,5-FMF-co-2,5-DF) show multiple Tm values, which could be explained similarly to the previous discussion on the multiple Tm values of the FPE products, considering the similar WAXD profiles of P(2,5-FMF-co-2,5-DF) in this work presented in Fig. S7† to those in a previous report on the same furanic-copolyester synthesized in diphenyl ether.10
As shown in Table S5,† variations in crystallization profiles were observed, with the one-step method showing a crystallization peak from the cooling step for all the sequences, while the two-step method showed cold crystallization. This difference could be attributed to the larger spherulites for P(2,5-FMF-co-2,5-DF) for the one-step products than for the two-step products; as previously reported for poly(L-lactic acid), larger spherulites were generated from melt crystallization than from cold crystallization.80 From the WAXD spectra presented in Fig. S7,† we confirmed the semicrystalline properties of all the obtained P(2,5-FMF-co-2,5-DF). We observed a strong 2θ peak at 24.03–24.25° in addition to two broad peaks at 16.73–16.98° and 7.57–7.77°; these findings agree with previous work on furanic–aliphatic copolyesters synthesized in diphenyl ether.10 All the products display similar WAXD profiles; this implies that despite the varied crystallization temperatures and spherulites, the samples shared a uniform crystal structure.
Conclusions
This study successfully investigated CALB-catalyzed polymerization utilizing biobased solvents. The first scheme successfully synthesized 2,5-POF and 2,5-PDOF with eight and twelve aliphatic methylene chains utilizing p-cymene or pinacolone. A higher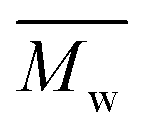 was achieved with p-cymene than with pinacolone;
was achieved with p-cymene than with pinacolone; 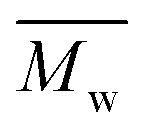 5000 g mol−1 2,5-POF and 7000 g mol−1 2,5-PDOF were detected. This is attributed to the higher logP of p-cymene than that of pinacolone, which results in less deactivating enzyme activity. In addition, compared to pinacolone, p-cymene has a lower β value, higher Vm, and excellent dissolution capability for solid traces of the oligomer, which support a more effective catalytic process of the enzyme toward the substrate. Moreover, we found that linear products were more apparent than cyclic products for 2,5-POF compared to 2,5-PDOF when utilizing p-cymene or pinacolone.
5000 g mol−1 2,5-POF and 7000 g mol−1 2,5-PDOF were detected. This is attributed to the higher logP of p-cymene than that of pinacolone, which results in less deactivating enzyme activity. In addition, compared to pinacolone, p-cymene has a lower β value, higher Vm, and excellent dissolution capability for solid traces of the oligomer, which support a more effective catalytic process of the enzyme toward the substrate. Moreover, we found that linear products were more apparent than cyclic products for 2,5-POF compared to 2,5-PDOF when utilizing p-cymene or pinacolone.
In the second scheme, P(2,5-FMF-co-2,5-DF) was successfully produced with two citrus-based solvents, i.e., p-cymene and D-limonene, and with toluene for comparison to non-biobased solvents. A higher molecular weight, 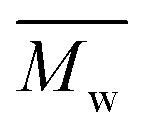 12800 g mol−1, was achieved when utilizing p-cymene than when utilizing D-limonene (
12800 g mol−1, was achieved when utilizing p-cymene than when utilizing D-limonene (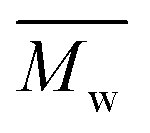 4500 g mol−1), and the result was comparable to that obtained when utilizing toluene (
4500 g mol−1), and the result was comparable to that obtained when utilizing toluene (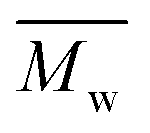 11400 g mol−1). Unlike what was previously mentioned for FPE enzymatic polymerization, logP did not apply to the superior catalytic activity of p-cymene. The β value and π–π stacking may play additional roles in preserving the effective enzyme catalytic activity during the polymerization of P(2,5-FMF-co-2,5-DF). In addition, it has been discovered that the abundance of cyclic products is greater when p-cymene and toluene are used, and this trend becomes more apparent when the two-step method is applied due to less disturbance from the side products to form cyclic products via intramolecular backbiting.
11400 g mol−1). Unlike what was previously mentioned for FPE enzymatic polymerization, logP did not apply to the superior catalytic activity of p-cymene. The β value and π–π stacking may play additional roles in preserving the effective enzyme catalytic activity during the polymerization of P(2,5-FMF-co-2,5-DF). In addition, it has been discovered that the abundance of cyclic products is greater when p-cymene and toluene are used, and this trend becomes more apparent when the two-step method is applied due to less disturbance from the side products to form cyclic products via intramolecular backbiting.
Comprehensive material analysis revealed that all the FPE products degrade in one step at temperatures ranging from 390 to 396 °C. Typical two-step decomposition was observed for the obtained P(2,5-FMF-co-2,5-DF), representing the occurrence of aliphatic segment decomposition between 391 and 395 °C and the hetero-aromatic segment decomposing between 227 and 243 °C. All the products obtained are semicrystalline with multiple Tm profiles attributed to the presence of unstable lamella. The aliphatic chain length of the obtained FPEs affects the crystallite size, which agrees with the findings of previous reports on furan-based polyesters. Among the P(2,5-FMF-co-2,5-DF) products, diverse crystallizations were observed despite their uniform WAXD diffractogram profiles, which could be due to the diversity of their crystal spherulites.
Overall, p-cymene offers a valuable option as a biobased solvent for enzymatic catalysis of furan-based (co)-polyesters without discoloration of the obtained products. This study highlights the applicability of bioderived solvents in enzymatic polymerization to enhance the sustainability of biocatalysis systems. Future research could improve the molecular weight by varying the polymerization temperature since this parameter likely affects enzymatic catalysis, as previously reported in some studies. To enhance the sustainability of the entire system, the product purification of furan-based polymers using greener methods should be considered in future investigations.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Fitrilia Silvianti: conceptualization, investigation, data collection, analysis, writing – original draft manuscript. Dina Maniar: conceptualization, supervision, writing – review and editing. Tijn C. de Leeuw: analysis, writing – review and editing. Jur van Dijken: data collection. Katja Loos: supervision, funding acquisition, critical revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are deeply grateful to Albert J. J. Woortman for his support with the SEC analysis. F. S. gratefully acknowledges the financial support from the Industrial Human Resource Development Agency of the Ministry of Industry of the Republic of Indonesia (BPSDMI, Kemenperin).References
- F. Silvianti, D. Maniar, L. Boetje and K. Loos, ACS Symp. Ser., 2020, 1373, 3 CrossRef CAS.
- K. Loos, R. Zhang, I. Pereira, B. Agostinho, H. Hu, D. Maniar, N. Sbirrazzuoli, A. J. D. Silvestre, N. Guigo and A. F. Sousa, Front. Chem., 2020, 8, 585 CrossRef CAS PubMed.
- A. Vasileiadis Vasileiou, S. T. Korfia, M. Sarigiannidou, D. Maniar and K. Loos, Polymer, 2024, 127652 CrossRef.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G. J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961 RSC.
- L. Lalanne, G. S. Nyanhongo, G. M. Guebitz and A. Pellis, Biotechnol. Adv., 2021, 48, 107707 CrossRef CAS PubMed.
- C. Post, D. Maniar, V. S. D. Voet, R. Folkersma and K. Loos, ACS Omega, 2023, 8, 8991 CrossRef CAS PubMed.
- G. Guidotti, M. Soccio, M. C. García-Gutiérrez, T. Ezquerra, V. Siracusa, E. Gutiérrez-Fernández, A. Munari and N. Lotti, ACS Sustain. Chem. Eng., 2020, 8, 9558 CrossRef CAS PubMed.
- J. Wu, P. Eduard, S. Thiyagarajan, B. A. J. Noordover, D. S. van Es and C. E. Koning, ChemSusChem, 2015, 8, 67 CrossRef CAS PubMed.
- E. Manarin, F. Da Via, B. Rigatelli, S. Turri and G. Griffini, ACS Appl. Polym. Mater., 2023, 5, 828 CrossRef CAS PubMed.
- D. Maniar, Y. Jiang, A. J. J. Woortman, J. van Dijken and K. Loos, ChemSusChem, 2019, 12, 990 CrossRef CAS PubMed.
- F. Silvianti, D. Maniar, B. Agostinho, T. C. de Leeuw, X. Lan, A. J. J. Woortman, J. van Dijken, S. Thiyagarajan, A. F. Sousa and K. Loos, Polymer, 2024, 309, 127441 CrossRef CAS.
- Y. Jiang and K. Loos, Polymers, 2016, 8, 243 CrossRef PubMed.
- J. L. Adrio and A. L. Demain, Biomolecules, 2014, 4, 117 CrossRef PubMed.
- Enzymatic Polymerization towards Green Polymer Chemistry, ed. S. Kobayashi, H. Uyama and J. Kadokawa, Springer, Singapore, 2019 Search PubMed.
- D. Maniar, F. Silvianti, V. M. Ospina, A. J. J. Woortman, J. van Dijken and K. Loos, Polymers, 2020, 205, 122662 CrossRef CAS.
- F. Silvianti, D. Maniar, L. Boetje, A. J. J. Woortman, J. van Dijken and K. Loos, ACS Polym. Au, 2022, 3(1), 82–95 CrossRef.
- P. Skoczinski, M. K. E. Cangahuala, D. Maniar, R. W. Albach, N. Bittner and K. Loos, ACS Sustain. Chem. Eng., 2019, 8, 1068 CrossRef.
- A. Bazin, L. Avérous and E. Pollet, Eur. Polym. J., 2021, 159, 110717 CrossRef CAS.
- A. Pellis, J. W. Comerford, S. Weinberger, G. M. Guebitz, J. H. Clark and T. J. Farmer, Nat. Commun., 2019, 10, 1 CrossRef CAS PubMed.
- A. Pellis, J. W. Comerford, S. Weinberger, G. M. Guebitz, J. H. Clark and T. J. Farmer, Nat. Commun., 2019, 10, 1762 CrossRef PubMed.
- C. Laane, S. Boeren, K. Vos and C. Veeger, Biotechnol. Bioeng., 1987, 30, 81 CrossRef CAS PubMed.
- C. Li, T. Tan, H. Zhang and W. Feng, J. Biol. Chem., 2010, 285, 28434 CrossRef CAS PubMed.
- Yu. L. Khmelnitsky, A. V Levashov, N. L. Klyachko and K. Martinek, Enzyme Microb. Technol., 1988, 10, 710 CrossRef.
- J. Seo, M. Shin, J. Lee, T. Lee, J.-M. Oh and C. Park, J. Ind. Eng. Chem., 2021, 93, 430 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288 RSC.
- G. Paggiola, A. J. Hunt, C. R. McElroy, J. Sherwood and J. H. Clark, Green Chem., 2014, 16, 2107 RSC.
- A. Iemhoff, J. Sherwood, C. R. Mcelroy and A. J. Hunt, Green Chem., 2018, 20, 136–140 RSC.
- A. G. Lanctôt, T. M. Attard, J. Sherwood, C. R. McElroy and A. J. Hunt, RSC Adv., 2016, 6, 48753 RSC.
- A. Pellis, F. P. Byrne, J. Sherwood, M. Vastano, J. W. Comerford and T. J. Farmer, Green Chem., 2019, 21, 1686 RSC.
- C. M. Warne, S. Fadlallah, A. C. Whitwood, J. Sherwood, L. M. M. Mouterde, F. Allais, G. M. Guebitz, C. R. McElroy and A. Pellis, Green Chem. Lett. Rev., 2023, 16, 2154573 CrossRef.
- N. Guajardo and P. Domínguez de María, Mol. Catal., 2020, 485, 110813 CrossRef CAS.
- G. de Gonzalo, Biocatal. Biotransform., 2022, 40, 252 CrossRef CAS.
- F. P. Byrne, B. Forier, G. Bossaert, C. Hoebers, T. J. Farmer and A. J. Hunt, Green Chem., 2018, 20, 4003 RSC.
- F. Silvianti, D. Maniar, B. Agostinho, T. C. de Leeuw, T. Pelras, L. Dijkstra, A. J. J. Woortman, J. van Dijken, S. Thiyagarajan, A. F. Sousa and K. Loos, Adv. Sustainable Syst., 2024, 2300542 CrossRef CAS.
- F. Silvianti, D. Maniar, B. Agostinho, T. C. de Leeuw, A. J. J. Woortman, J. van Dijken, S. Thiyagarajan, A. F. Sousa and K. Loos, Biomacromolecules, 2024, 25(5), 2792–2802 CrossRef CAS PubMed.
- A. Pellis, S. Weinberger, M. Gigli, G. M. Guebitz and T. J. Farmer, Eur. Polym. J., 2020, 130, 109680 CrossRef CAS.
- Y. Jiang, G. O. R. A. van Ekenstein, A. J. J. Woortman and K. Loos, Macromol. Chem. Phys., 2014, 215, 2185 CrossRef CAS.
- Y. Jiang, A. J. J. Woortman, G. O. R. Alberda Van Ekenstein, D. M. Petrović and K. Loos, Biomacromolecules, 2014, 15, 2482 CrossRef CAS PubMed.
- Y. Jiang, A. J. J. Woortman, G. O. R. Alberda Van Ekenstein and K. Loos, Polym. Chem., 2015, 6, 5198 RSC.
- S. Kobayashi, Macromol. Rapid Commun., 2009, 30, 237 CrossRef CAS PubMed.
- I. Hilker, A. E. J. Schaafsma, R. A. H. Peters, A. Heise and A. J. Nijenhuis, Eur. Polym. J., 2008, 44, 1441 CrossRef CAS.
- C. Berkane, G. Mezoul, T. Lalot, M. Brigodiot and E. Maréchal, Macromolecules, 1997, 30, 7729 CrossRef CAS.
- Y. Jiang, D. Maniar, A. J. J. Woortman, G. O. R. Alberda Van Ekenstein and K. Loos, Biomacromolecules, 2015, 16, 3674 CrossRef CAS PubMed.
- J. H. Clark, D. J. Macquarrie and J. Sherwood, Green Chem., 2012, 14, 90 RSC.
- G. B. Perin and M. I. Felisberti, Biomacromolecules, 2022, 23, 2968 CrossRef CAS PubMed.
- L. Ye and B. C. Thompson, ACS Macro Lett., 2021, 10, 714 CrossRef CAS PubMed.
- M. T. García, I. Gracia, G. Duque, A. de Lucas and J. F. Rodríguez, Waste Manage., 2009, 29, 1814 CrossRef PubMed.
- G. B. Perin and M. I. Felisberti, Macromolecules, 2020, 53, 7925 CrossRef CAS PubMed.
- A. Pellis, F. P. Byrne, J. Sherwood, M. Vastano, J. W. Comerford and T. J. Farmer, Green Chem., 2019, 21, 1686 RSC.
- S.-H. Pyo, K. Nuszkiewicz, P. Persson, S. Lundmark and R. Hatti-Kaul, J. Mol. Catal. B: Enzym., 2011, 73, 67 CrossRef CAS.
- C. Puglisi, F. Samperi, R. Alicata and G. Montaudo, Macromolecules, 2002, 35, 3000 CrossRef CAS.
- R. Alicata, G. Montaudo, C. Puglisi and F. Samperi, Rapid Commun. Mass Spectrom., 2002, 16, 248 CrossRef CAS PubMed.
- M. Florczak, A. Michalski, A. Kacprzak, M. Brzeziński, T. Biedroń, A. Pająk, P. Kubisa and T. Biela, React. Funct. Polym., 2016, 104, 71 CrossRef CAS.
- H. R. Kricheldorf, S. Böhme, G. Schwarz and C.-L. Schultz, Macromol. Rapid Commun., 2002, 23, 803 CrossRef CAS.
- H. R. Kricheldorf, G. Schwarz, S. Böhme, C.-L. Schultz and R. Wehrmann, Macromol. Chem. Phys., 2003, 204, 1398 CrossRef CAS.
- M. V Goryaeva, Y. V Burgart and V. I. Saloutin, J. Fluorine Chem., 2013, 147, 15 CrossRef.
- S. Kandula, L. Stolp, M. Grass, B. Woldt and D. Kodali, J. Am. Oil Chem. Soc., 2014, 91, 1967 CrossRef CAS.
- M. Vastano, A. Pellis, C. Botelho Machado, R. Simister, S. J. McQueen-Mason, T. J. Farmer and L. D. Gomez, Macromol. Rapid Commun., 2019, 40, 1900361 CrossRef CAS PubMed.
- I. Gavrila, P. Raffa and F. Picchioni, Polymers, 2018, 10(3), 248 CrossRef PubMed.
- N. B. Lorette, W. L. Howard and J. H. Brown Jr, J. Org. Chem., 1959, 24, 1731 CrossRef CAS.
- A. Pellis, S. Weinberger, M. Gigli, G. M. Guebitz and T. J. Farmer, Eur. Polym. J., 2020, 130, 109680 CrossRef CAS.
- Y. Dong, J. Wang, Y. Yang, Q. Wang, X. Zhang, H. Hu and J. Zhu, Polym. Degrad. Stab., 2022, 202, 110010 CrossRef CAS.
- L. Urpí, A. Alla and A. Martínez de Ilarduya, Polymers, 2023, 266, 125624 CrossRef.
- M. do R. G. Dias, G. P. C. da Silva, A. de Pauloveloso, N. Krieger and C. Pilissão, Chirality, 2022, 34, 1008 CrossRef CAS PubMed.
- R. G. Huber, M. A. Margreiter, J. E. Fuchs, S. von Grafenstein, C. S. Tautermann, K. R. Liedl and T. Fox, J. Chem. Inf. Model., 2014, 54, 1371 CrossRef CAS PubMed.
- S. Levi, V. Rac, V. Manojlovi, V. Raki, B. Bugarski, T. Flock, K. E. Krzyczmonik and V. Nedovi, Procedia Food Sci., 2011, 1, 1816 CrossRef.
- A. Hazra, D. Dollimore and K. Alexander, Thermochim. Acta, 2002, 392–393, 221 CrossRef CAS.
- M. Kamphoff, T. Thiele and B. Kunz, J. AOAC Int., 2007, 90, 1623 CrossRef CAS PubMed.
- A.-T. Karlberg and A. Dooms-Goossens, Contact Dermatitis, 1997, 36, 201 CrossRef CAS PubMed.
- U. Nilsson, K. Magnusson, O. Karlberg and A.-T. Karlberg, Contact Dermatitis, 1999, 40, 127 CrossRef CAS PubMed.
- F. Li, S. Luo, C. Ma, J. Yu and A. Cao, J. Appl. Polym. Sci., 2010, 118, 623 CrossRef CAS.
- M. Yasuniwa and T. Satou, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 2411 CrossRef CAS.
- E. S. Yoo and S. S. Im, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 1357 CrossRef CAS.
- K. Jamshidi, S.-H. Hyon and Y. Ikada, Polymers, 1988, 29, 2229 CrossRef CAS.
- H. G. Kim and R. E. Robertson, J. Polym. Sci., Part B: Polym. Phys., 1998, 36, 1757 CrossRef CAS.
- X. Ling and J. E. Spruiell, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 3200 CrossRef CAS.
- M. Yasuniwa, S. Tsubakihara, Y. Sugimoto and C. Nakafuku, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 25 CrossRef CAS.
- Z. Jie, L. Fa-xue and Y. Jiang-yong, J. Therm. Anal. Calorim., 2013, 111, 711 CrossRef.
- M. Yasuniwa, S. Tsubakihara, K. Ohoshita and S. Tokudome, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 2005 CrossRef CAS.
- Y. Wang, S. S. Funari and J. F. Mano, Macromol. Chem. Phys., 2006, 207, 1262 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00358f |
| This journal is © The Royal Society of Chemistry 2024 |