
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembled tetracyanoquinodimethane derivatives: differential fluorescent responses on sensing copper and mercury ions in an aqueous medium†
Anuradha Sureshrao
Mohitkar
,
Nilanjan
Dey
and
Subbalakshmi
Jayanty
 *
*
Department of Chemistry, Birla Institute of Technology and Science, Pilani-Hyderabad Campus, Jawahar Nagar, Kapra Mandal, Medchal Malkajgiri District, Hyderabad 500078, Telangana, India. E-mail: jslakshmi@hyderabad.bits-pilani.ac.in
First published on 10th October 2024
Abstract
Copper and mercury metal ions are known to cause serious harm to biological and environmental systems, and therefore the development of new efficient sensory systems for the detection of copper and mercury ions in aqueous media is always encouraged and in need. Herein, we present the employment of di-substituted tetracyanoquinodimethane (TCNQ) derivatives for metal ion recognition. Owing to their fluorescent properties, single-step synthesis and easy purification, small molecules, namely 7,7-bis(1-(2-aminoethyl)pyrrolidino)-8,8-dicyanoquinodimethane (AEPRDQ) [1], 7,7-bis(1-(2-aminoethyl)piperidino)-8,8-dicyanoquinodimethane (AEPIDQ) [2] and 7,7-bis(N,N-diethylethylenediamino)-8,8-dicyanoquinodimethane (BDEDDQ) [3], have been utilized for Cu2+ and Hg2+ sensing applications. [1]–[3] considered in this study showed the formation of self-assembled nanoaggregates in aqueous media. A thorough investigation on the effect of the microenvironment, such as temperature, pH, polarity, etc., on the extent of self-agglomeration was done spectroscopically. Interestingly, in an acetonitrile–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) solvent mixture, the compounds showed diverse responses to Cu2+ and Hg2+ ions. [1] manifested fluorescence quenching with Cu2+ ions, while enhanced emission was noted with Hg2+. Hg2+ induced fluorescence quenching with [2] while no prominent quenching is seen on the addition of Cu2+. Adding Cu2+ and Hg2+ resulted in a turn-off fluorescence signal in [3]. Hence, alteration by simply varying the terminal functional groups among the said [1]–[3] resulted in varied metal ion sensing responses.
:9) solvent mixture, the compounds showed diverse responses to Cu2+ and Hg2+ ions. [1] manifested fluorescence quenching with Cu2+ ions, while enhanced emission was noted with Hg2+. Hg2+ induced fluorescence quenching with [2] while no prominent quenching is seen on the addition of Cu2+. Adding Cu2+ and Hg2+ resulted in a turn-off fluorescence signal in [3]. Hence, alteration by simply varying the terminal functional groups among the said [1]–[3] resulted in varied metal ion sensing responses.
Sustainability spotlightToxic heavy metals like Hg and Cu can easily accumulate in aquatic ecosystems, ultimately affecting human health. Thus, accurate and sensitive detection of Hg2+ and Cu2+ in water is essential for environmental monitoring and public health safety. Therefore, TCNQ derivative fluorophores have been utilized originally for Hg2+ and Cu2+ recognition, particularly in aqueous medium. A thorough investigation of the effect of microenvironment such as temperature, pH, polarity, etc. on the extent of self-agglomeration was executed; furthermore, a detailed analysis of photophysical properties, metal-ion sensing, mechanistic investigations and morphological studies have been conducted and are presented in this article. Interestingly, the presence of different amino functionalities at the terminal position of the ethylene spacer affected the metal ion sensing property with varied fluorescence response towards Hg2+ and Cu2+ in aqueous medium. Current work is consistent with the WHO's and UN's sustainable development goals of good health/well-being (SDG 3), clean water and sanitation (SDG 6), and responsible production (SDG 12). |
1 Introduction
Tetracyanoquinodimethane (TCNQ) is a strong electron acceptor molecule and promising member exhibiting organic electronic applications.1 When reacted with primary or secondary amines in corresponding quantities, TCNQ tends to form diaminodicyanoquinodimethanes (DADQs) through nucleophilic aromatic substitution. Further, DADQs have been employed for diverse applications like nonlinear optical properties,2 optoelectronic devices,3,4 and bioimaging.5 Nevertheless, TCNQ-based compounds utilized for sensing are limited.6 For instance, tetrathiafulvalene (TTF)-TCNQ complex is sensitive to many gas species such as alkylamines (butylamine, hexylamine etc.), aromatic amines (aniline, p-toluidine etc.), NO2 and O2.7,8 Liu et al. developed TTF-TCNQ/HKUST-1 composite structures arranged as nanosheet arrays for NO2 detection with a detection limit of 10 parts per billion (ppb) and exclusive selectivity of NO2 in the presence of 10 times the concentration of other interfering gases.9 Wang et al. have demonstrated sensing of bovine serum albumin (BSA), DNA and hydroxyl radical generated from Fenton's reagent when TCNQ was combined with tetraphenylethene (TPE) derivatives.10 Increased surface-enhanced Raman scattering (SERS) activity was exhibited by a low-cost, facilely prepared, and highly stable F4TCNQ/molybdenum disulfide (MoS2) nanocomposite platform. It showed a 10−10 M limit of detection (LOD) against methylene blue, and thus was utilized for trace molecule detection.11 A non-covalent charge transfer (CT) complex of di(hydroxymethyl)-di-(2-pyrrolyl)methane and TCNQ has shown selective detection of sulfite ion in a neutral buffered aqueous system in the presence of other inorganic anions.12 Apart from these, TCNQ readily formed metal organic complexes13,14 Cu-TCNQ and Ag-TCNQ; especially with copper (Cu) and silver (Ag). Several 2D Cu-TCNQ-based frameworks have been developed for semiconducting properties,15 sensing,9 as photodetectors16etc. Nonetheless, to the best of our knowledge, TCNQ derivatives have not been utilized for metal ion sensing application primarily in solutions.Hg2+ and Cu2+ are considered as toxic heavy metal ions when they exceed their permissible limits, i.e., 2 ppb and 1.3 ppm, respectively, in drinking water as per the Environmental Protection Agency (EPA) guidelines.17,18 They tend to accumulate in living organisms and are non-biodegradable, leading to severe health issues like kidney, brain and developing fetus damage.19 Fluorescent sensors such as carbon nanoparticles,20o-phthalimide-based AIEgen of 2,3-diphenylquinoxaline-6,7-dicarboximide,21 thioacetal-modified phenothiazine22etc. have been widely used for Hg2+ detection. For Cu2+ detection, fluorescent sensors like polymer nanodots,23 graphitic carbon nitride nanosheets,24 2,2′-((1E,1′E)-((4-chloro-1,2-phenylene)bis(azaneylylidene))bis(methane-ylylidene))bis(4-bromophenol),25 bispicolyl appended oxidized bisindole derivative,26etc. have been reported. TCNQ-based fluorescent sensors have been employed for metal ion detection; for example, colorimetric sensing of ferrous (Fe2+), Cu2+ and Ag2+ by polystyrene TCNQ adducts has been reported, wherein Fe2+and Cu2+ displayed a decrease in CT band intensity in the visible-near infrared (IR) region whereas, for Ag2+, the CT band showed a bathochromic shift.27 Singh et al. have reported a CT complex of TCNQ with N-methylpyrrole, as a selective Hg2+ chemodosimeter in liquid and solid state.28 Appreciating the diversity of TCNQ-based compounds, their sensing applicability, and the scarcity of TCNQ-derived small-molecule chromophores motivated us to utilize DADQs for metal ion recognition.
The beauty of the diversified application of DADQs is based on subtle changes in amines, leading to varied photophysical and molecular material properties. This enables the design of DADQs for a given application by the appropriate selection of amine. DADQ with 4-aminophenylmorpholinone has shown enhanced fluorescence in solution and potassium (K+) ion recognition exclusively in the solid state.29 The nonlinear optical property of 7,7-bis(1-(2-aminoethyl)pyrrolidino)-8,8-dicyanoquinodimethane (AEPRDQ) [1]30 and 7,7-bis(1-(2-aminoethyl)piperidino)-8,8-dicyanoquinodimethane (AEPIDQ) [2]30 has been previously studied by us, wherein AEPIDQ solid manifested second harmonic generation (SHG) of about six times that of urea. The physical and chemical stimuli response of AEPIDQ was reported by Sudhakar et al.31 Further, intense solid-state emission in 7,7-bis(N,N-diethylethylenediamino)-8,8-dicyanoquinodimethane (BDEDDQ) [3]32 was reported by us recently, owing to the role of supramolecular self-assembly in the BDEDDQ crystal. However, though these DADQs, i.e.[1], [2], and [3], exhibited fluorescence in solutions as well as in their solid forms, as of now, neither [1]–[3] nor any other DADQs have been utilized for metal ion sensing applications in solutions. Small-molecule fluorescent sensors are in higher demand owing to their facile synthesis, better repeatability, ease of purification, and higher quantum yields; DADQs have proved themselves as fluorescent small-molecule probes in solids and solutions, facilitating sensing applications. Further, single-step, quick synthesis and easy purification have attracted attention towards [1]–[3] as small-molecule fluorophores. Table S1† reflects the robustness of these DADQs for Cu2+and Hg2+sensing.
On this basis, herein, we have considered and explored the three DADQs [1]–[3], initially for sensing various metal ions such as cobalt (Co2+), copper (Cu2+), lead (Pb2+), magnesium (Mg2+), manganese (Mn2+), mercury (Hg2+), nickel (Ni2+), zinc (Zn2+) and silver (Ag2+) and we further studied in detail their selectively in recognizing metal ions Hg2+ and Cu2+ in water medium.
All three DADQs [1]–[3] showed the formation of self-assembled nanoaggregates in aqueous medium. The effect of the microenvironment (temperature, pH, polarity, etc.) on the extent of self-agglomeration has been thoroughly investigated. Interestingly, in acetonitrile–water (1:9) solvent mixture, the compounds showed varied responses to Cu2+ and Hg2+ ions. For [1], fluorescence quenching was observed with Cu2+ ions, while Hg2+ induced enhanced emission. Hg2+ induced fluorescence quenching in [2] with no prominent quenching on the addition of Cu2+. A turn-off fluorescence signal in the case of [3] was noted on adding Cu2+ and Hg2+.
2 Experimental
2.1 Materials and methods
[1]–[3] were synthesized (Scheme 1) following reported procedures, i.e.[1], [2],30 and [3].32 To a hot solution of TCNQ (1 equiv.) under N2 atmosphere, 2 equiv. of primary amines (i.e. 1-(2-aminoethyl)pyrrolidine, (a) in Scheme 1 for [1]; 1-(2-aminoethyl)piperidine, (b) in Scheme 1 for [2]; and N,N-diethylethylenediamine, (c) in Scheme 1 for [3]) was added and allowed to stir for three hours to obtain [1]–[3] as yellow-colored microcrystalline products (caution: ferrous sulfate solution dipped scrubber was used to capture HCN liberated during the process); the obtained products were further recrystallized and utilized for the metal ion sensing studies. | ||
| Scheme 1 Compounds [1]–[3] considered in this study. | ||
2.2 Solvatochromism, aggregation, temperature dependence and pH studies
:THF in different proportions maintaining a constant total volume of 2 mL solution. Also, the change in emission intensity was determined on diluting [1]–[3] (50 μM) solution (in MeCN:H2O (1:9) solvent mixture) to 0.75 μM.
:H2O (1:9) solvent mixture was subjected to varying temperatures ranging from 20 to 80 °C with 10 °C increments.
:H2O (1:9) solvent mixture was studied at acidic pH (2) and basic pH (9) by adding a 10 μL saturated solution of hydrochloric acid (HCl) and sodium hydroxide (NaOH) respectively.
:H2O (1:1) solvent mixture was considered and gradually 100 μL of glycerol was added in the same solution until it attained saturation.
2.3 Studies with metal ions in aqueous environment
[1]–[3] (10 μM) solution in MeCN:H2O (1:9) solvent and 250 μM of metal ion solution from a 20 mM stock in H2O was examined for metal ion (Co2+, Cu2+, Pb2+, Mg2+, Mn2+, Hg2+, Ni2+, Zn2+, Ag2+) interaction. [1]–[3] (10 μM) solution in MeCN:H2O (1:9) solvent was gradually titrated against Cu2+/Hg2+ (10 μM) until it reached saturation. Furthermore, quenching was studied using Stern–Volmer plots33 by plotting Io/I vs. [Mn+] (Cu2+/Hg2+), where I and Io are the emission intensities in the presence and absence of the metal ion respectively and [Mn+] is the concentration of metal ion. LOD was calculated from a plot of log of the metal ion concentration vs. Imax–I/Imax–Imin. In the case of enhancement of emission intensity, LOD was calculated from a plot of log of the metal ion concentration vs. I–Imin/Imax–Imin.34 Here, Imax is the maximum and Imin is the minimum emission intensity within the selected range of concentrations, and I is the intensity calculated at particular concentrations. The quenching constant (Ksv) is obtained from the equation Io/I = 1 + Ksv [Mn+], where Ksv is the slope of the plot of (Io − I)/I vs. [Mn+].
:H2O (1:9) solvent was gradually titrated against Cu2+/Hg2+ (10 μM) until it reached saturation. Herein, real water (tap water/pond water) samples were utilized for the analysis.
2.4 Morphological studies
An Oxford X-maxN LEICA EM ACE200 scanning electron microscope (SEM) was used to examine the morphology of drop-cast films. Scanning electron microscopy was carried out to study the morphology of drop-cast films for [1]–[3] and [1]–[3]:metal ion (Cu2+/Hg2+) (1:1) of 10 μM concentration on a silicon wafer using gold sputtering. The silicon wafers were thoroughly cleaned with acetone and water and dried in an oven at 100 °C prior to use. Energy dispersive X-ray analysis (EDAX) and mapping was also executed. The morphology of the aggregates was observed using 10 kV and 1 μm magnification.
2.5 UV-visible, IR, fluorescence, NMR studies
A JASCO V-650 spectrophotometer was used for absorption studies of [1]–[3]. Emission investigations were conducted with a Fluorolog–Horiba. 1H and 13C NMR spectra were obtained with a Bruker Advance DRX (400–500 MHz) using deuterated dimethyl sulphoxide (DMSO-d6) solvent and tetramethylsilane (TMS) as the internal standard. NMR spectra for [1]–[3] of 20 mM concentration and [1]–[3]:metal ion (Cu2+/Hg2+) (1:1) in DMSO-d6 were recorded. FT-IR spectra were recorded using a PerkinElmer FT-IR Spectrum BX system. FT-IR spectra were obtained for [1]–[3] of 20 mM concentration and [1]–[3]:metal ion (Cu2+/Hg2+) (1:1) in MeCN:H2O (1:9) solvent. All the samples were sonicated to obtain clear spectra. Since clear peaks were not visible at lower concentrations, 20 mM concentration was optimized.
3 Results and discussion
3.1 Photophysical studies
Absorption (λmax,abs) and emission (λmax,emi) spectra of compounds [1]–[3] were investigated with concentrations as described in Section 2.2. λmax,abs and λmax,emi in different solvents were recorded to deduce the solvatochromic behaviour. The λmax,abs of [1] in various solvents indicated a blueshift (∼64 nm) with increasing solvent polarity (Fig. 1a), indicating the highly polar nature of the ground state, a common occurrence reported in several DADQs.4,35 This blueshift also leads to the formation of H-aggregates.36λmax,abs at 414 nm for toluene followed by 401 nm (DCM), 385 nm (THF, MeOH), 382 nm (MeCN, DMF), 379 nm (EtOH), and 350 nm (H2O) highlight this observation. On the contrary, [1] manifested a redshift (∼27 nm) with quenching behaviour in the emission spectra with increasing solvent polarity (Fig. 1b) owing to the vibrational relaxation of the excited states.29 Significant emission peaks at 455 nm, 472 nm, 476 nm and 485 nm for THF, MeOH, DMF and H2O respectively highlight the redshift behaviour, i.e. λmax,emi emission shifting to longer wavelengths with an increase in solvent polarity attributed to the internal charge transfer (ICT), owing to the presence of both donor and acceptor moieties in the same molecule. A shift of charge from donor to acceptor moiety in the excited state occurs, which results in a large dipole moment in the excited state. The interaction of polar solvent molecules and larger dipole moment in the excited state reduces the excited state energy, and hence longer wavelengths are noticed with an increase in polarity of the solvent.37 The increased ICT effect in the [1]–[3] fragment is responsible for this redshift.37[1]–[3], being zwitterionic in nature and polar, are not soluble in hexane; therefore, no significant peak was observed when the solutions as described in Section 2.2 were subjected to the solvatochromism study with hexane. On the other hand, in the emission study, fine spectra with sharp vibronic bands were observed in toluene and DCM (Fig. 1b) due to the presence of more than one excited vibrational energy level.37,38 The fluorescence emission spectra of [1] showed broad feature, as the addition of water fraction (fw) increased from 0 to 90% with a gradual redshift in the emission maxima from 433 nm to 482 nm (Fig. 1c). The fluorescence intensity enhanced until 20% (v/v) water fraction (fw) in the mixture, followed by systematic quenching. Approximately 5-fold enhancement in emission intensity was observed with decreasing fw, i.e. from 100% fw to 20% fw. However, enhancement in intensity was observed with increased viscosity (Fig. S1†). This study further elucidated that [1] showed similar behaviour for lower fw as manifested with an increase in viscosity. This is possibly owing to the twisted intramolecular charge transfer (TICT), with restricted rotation due to weaker [1]–[3] molecular dipole–dipole interaction and water-induced aggregation; and hydrogen bonding could lead to local rigidification, consequently, nonradiative pathways are reduced thereby leading to enhanced emission. Whereas, with an increase in fw after 20%, ACQ is being followed.37 The decreased emission intensity was witnessed at elevated temperatures (Fig. 1d, and S1c†), once again supporting aggregate formation.37 The formation of aggregated structures in aqueous medium was also confirmed by the dilution experiment. As shown in Fig. S2a,† the fluorescence intensity of [1] gradually decreased with dilution in aqueous medium. Broadening of peaks is observed on dilution due to aggregation. The pH-variation studies indicated that [1] remained comparably fluorescent across the wide range of pH (2–9) conditions (Fig. S2b†). With an increase in pH, the fluorescence intensity increased by ∼2.5-fold. Lower emission intensity at lower pH suggests protonation of N–H moieties, thereby reducing the electrostatic repulsion, and facilitating aggregate formation, whereas in basic conditions, deprotonation of N–H will lead to enhancement in the intensity. No significant change in wavelength is observed with variation in pH. Aggregate formation is exhibited for the drop-cast DMSO solution of [1] (Fig. S2c†).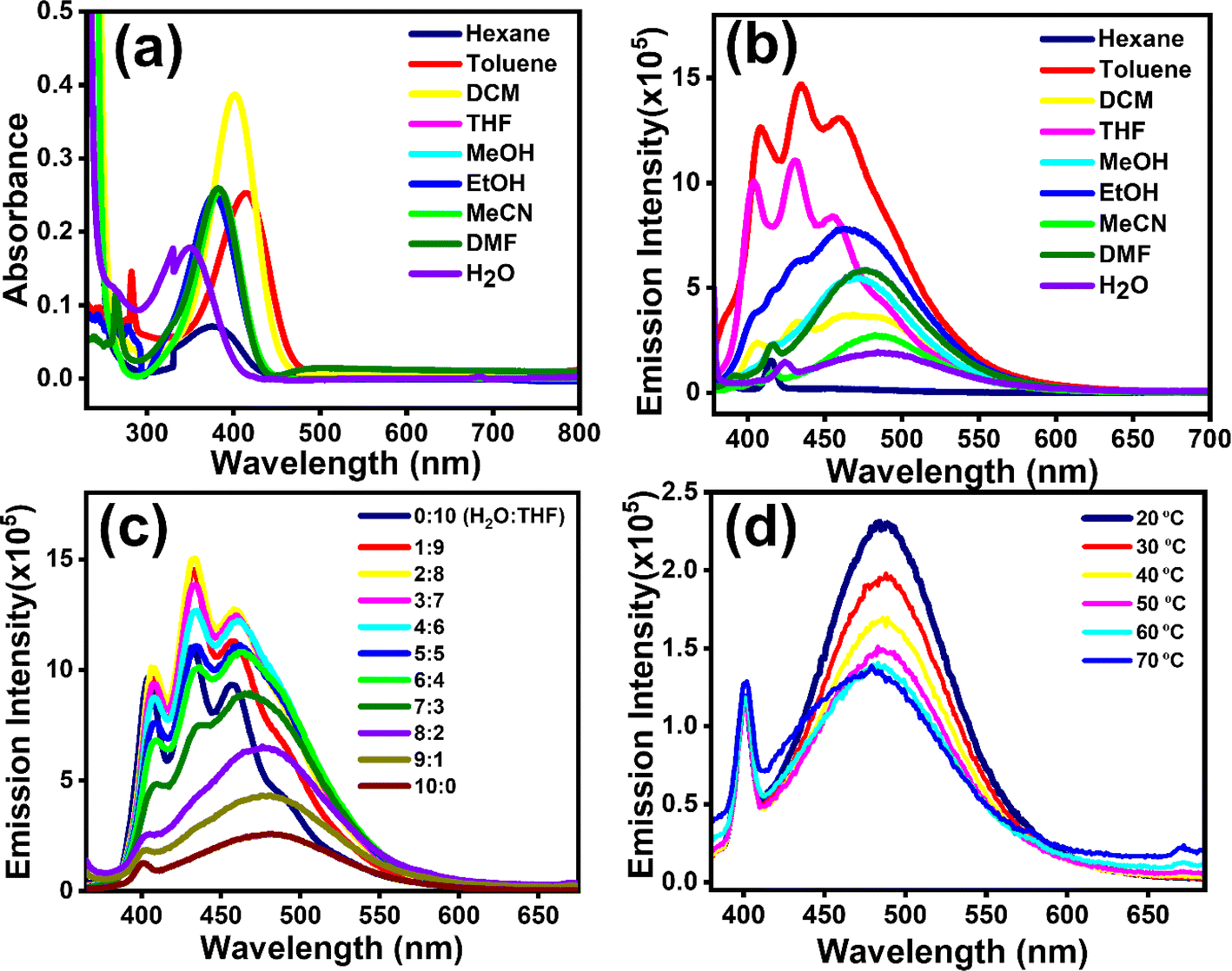 | ||
| Fig. 1 Results for [1]. (a) Shift in absorption wavelength and (b) emission spectra (λexc ∼358 nm) in solvents with different polarity. (c) Quenching of emission intensity due to aggregation in varying proportions of H2O:THF solvent mixture. (d) Lowering of emission intensity with increase in temperature. | ||
For [2], ∼69 nm blueshift in absorption spectra was observed with an increase in polarity with λmax,abs at 417 nm (toluene, THF); 405 nm (DCM); 385 nm (DMF, MeCN); 379 nm (EtOH); 372 nm (MeOH); and 348 nm (H2O) revealing the formation of H-aggregates36 (Fig. S3a†). Emission spectra for [2] exhibited a redshift (∼27 nm): 455 nm, 472 nm, 476 nm and 485 nm peaks for THF, MeOH, DMF and H2O similar to [1] (Fig. S3b†). Though the ACQ effect is noticed in [2] (Fig. S3c†) like in [1], an about 1.3-fold increase in emission intensity of [1] was observed when compared to [2] (Fig. S3d†). [3] also follows a similar blueshift of ∼62 nm with increasing polarity with λmax,abs peaks at 414 nm (toluene, THF); 402 nm (DCM); 401 nm (DMF); 384 nm (MeCN); 381 nm (EtOH); 369 nm (MeOH); and 348 nm (H2O) (Fig. S4a†) revealing H-aggregates.36 A slightly larger redshift (∼40 nm) trend is evident for [3] with significant λmax,emi peaks at 456 nm, 470 nm, 476 nm and 487 nm for THF, MeOH, DMF and H2O (Fig. S4b†). An ACQ effect was perceived for [3] as well (Fig. S4c†) and a 1.2-fold increase in emission intensity of [1] was observed in comparison to [3] (Fig. S4d†). The possibility of an overlap of S1/T1 vibronic modes with S0 may lead to a decrease in emission intensity in [2] and [3] compared with [1].29 Hence, [2] and [3] exhibited comparable observations among the absorption and emission properties which also could be understood with a similar rationale to that discussed above for [1].
Fig. 2a shows emission intensity with respect to solvent polarity for all three compounds [1]–[3]. Fig. 2b indicates initial enhanced intensity until 60% fw with respect to [2] and [3], followed by quenching, suggesting aggregate formation. Higher fluorescence intensity, ∼2.5-fold increase, was noted in [1] (for all the fw considered) compared to [2] and [3]. Further, the effect of temperature on aggregate formation was investigated. The fluorescence intensity at the aggregate emission band decreased with increasing temperature, irrespective of the substituent amine present (Fig. 2c). We believe that at lower temperatures, the solvent reorientation would be prolonged which would slow down the solvent relaxation and thereby facilitating aggregate formation accompanied with enhanced emission.37Fig. 2d illustrates ∼1.6-fold enhanced fluorescence intensity of [1] for all the concentrations when compared to [2] and [3]. Similar emission intensities were observed for [2] and [3] for all the concentrations considered in the study. [2] shows ∼28 nm blueshift (Fig. S5a†) with increasing temperature. Similar trend to that for [1] is seen for [2] with respect to pH studies, i.e. lowering of emission intensity with lower pH (Fig. S5b†). The formation of aggregates is clearly exhibited in [2] (Fig. S5c†). [3] shows ∼17 nm blueshift (Fig. S6a†) with increasing temperature. A trend similar to that for [1] and [2] is followed for [3] with varying pH (Fig. S6b†).37 Similar to [1] and [2], aggregates are revealed in [3] (Fig. S6c†) as well. [3] shows a 1.38-fold higher emission intensity compared to [1] and [2] with varying temperature, perhaps because the presence of the ethyl moiety at the end in [3] facilitates effective H-bonding and interchain interactions giving rigidity due to restricted intramolecular interactions when compared to pyrrole and piperidine substituents in [1] and [2] respectively (Fig. 2c).32
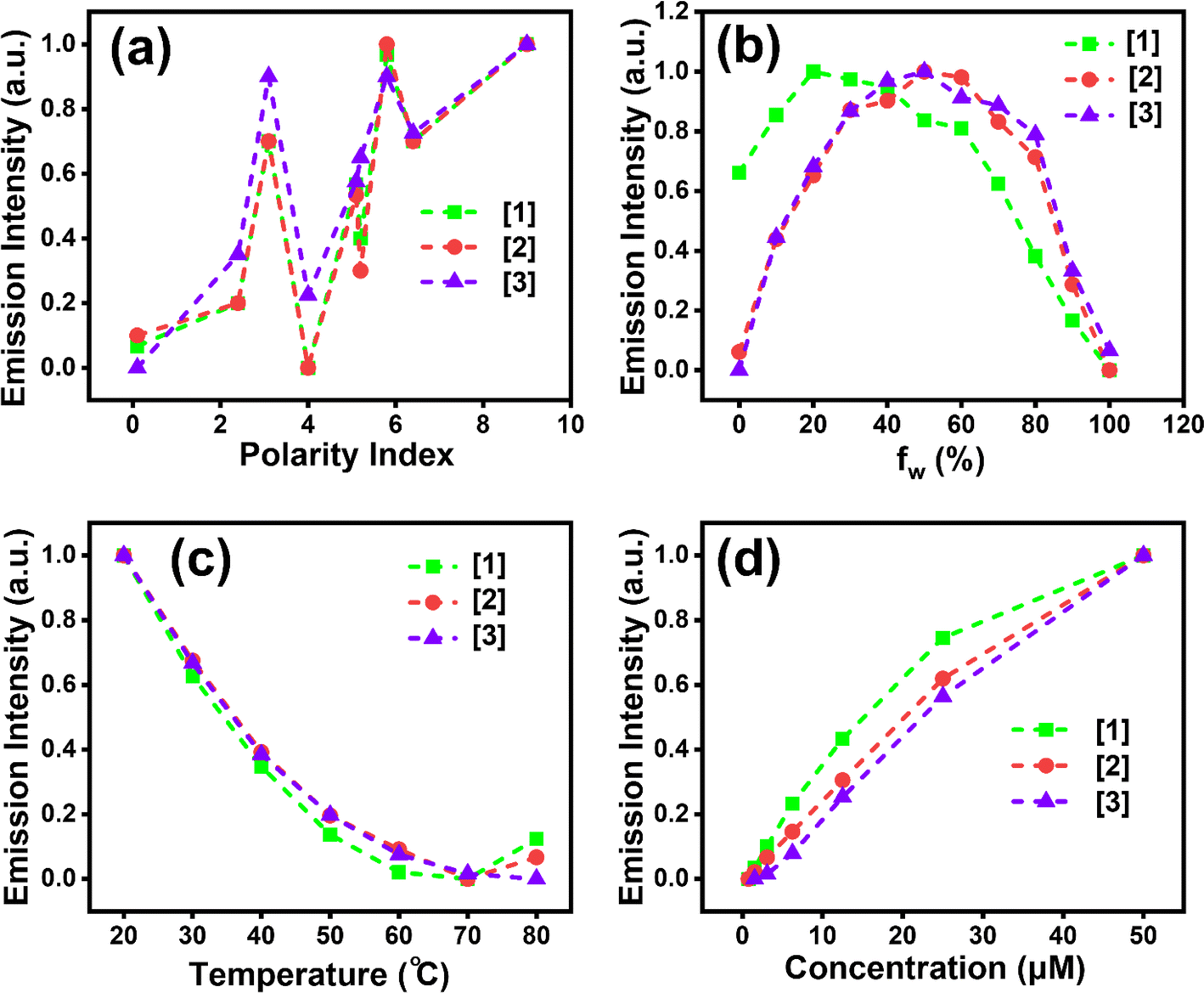 | ||
| Fig. 2 For [1]–[3]: (a) solvent effect on fluorescence maxima with increasing polarity, (b) fluorescence intensity change with respect to increasing water fraction, (c) relation of emission intensity with increasing temperature and (d) effect on emission intensity of dilution. | ||
3.2 Metal ion sensing
Structurally the selected [1], [2] and [3] TCNQ derivatives possessed varied amines with ethylene spacers, manifested by fluorescence in solutions besides in their solid state. Solid [1]–[3] exhibited supramolecular ensembles mediated by short contacts and strong hydrogen bonds, established from crystal structural analysis.30,32 The presence of heteroatoms in fluorophores leads to susceptibility to changes in properties when there is contact between the heteroatom and metal ions.22,34,39–41 On the supposition of directional noncovalent interactions, supramolecular assemblies in solutions show stimuli-responsive features that have drawn attention for application in sensing and detection. Thus, for understanding the structural properties of [1]–[3] and learning from their photophysical properties, we were intrigued to investigate their interaction with transition metal ions (Co2+, Cu2+, Pb2+, Mg2+, Mn2+, Hg2+, Ni2+, Zn2+, Ag2+). [1]–[3] all showed selective response for Cu2+ and Hg2+ ions compared to other transition metal ions. Here, it is important to note that toxic heavy metals, such as Cu and Hg, can easily accumulate in aquatic ecosystems, leading to bioaccumulation and biomagnification in the food chain, which ultimately affects human health. Elevated levels of copper can cause gastrointestinal distress and liver and kidney damage, while mercury exposure is associated with various neurological and developmental ailments. Additionally, the presence of these metals can disrupt aquatic life, affecting both biodiversity and stability of ecosystems. Therefore, accurate and sensitive detection of Cu and Hg in water is essential for environmental monitoring and public health safety.For [1], the addition of Cu2+ led to quenching of fluorescence intensity, whereas the presence of Hg2+ resulted in 3.6-fold enhancement in intensity with a redshift from 469 nm to 499 nm (Fig. 3a). [2] and [3] experienced fluorescence quenching (Fig. 3b and c) in the presence of both Cu2+ and Hg2+ ions in aqueous medium, but no shift in emission maxima was observed. Interestingly, for [2], the extent of quenching was more prominent with Hg2+ ion than with Cu2+ (Fig. S7(a)–(d)†). On the contrary, both metal ions caused a similar extent of quenching for [3] (Fig. S8(a)–(d)†).
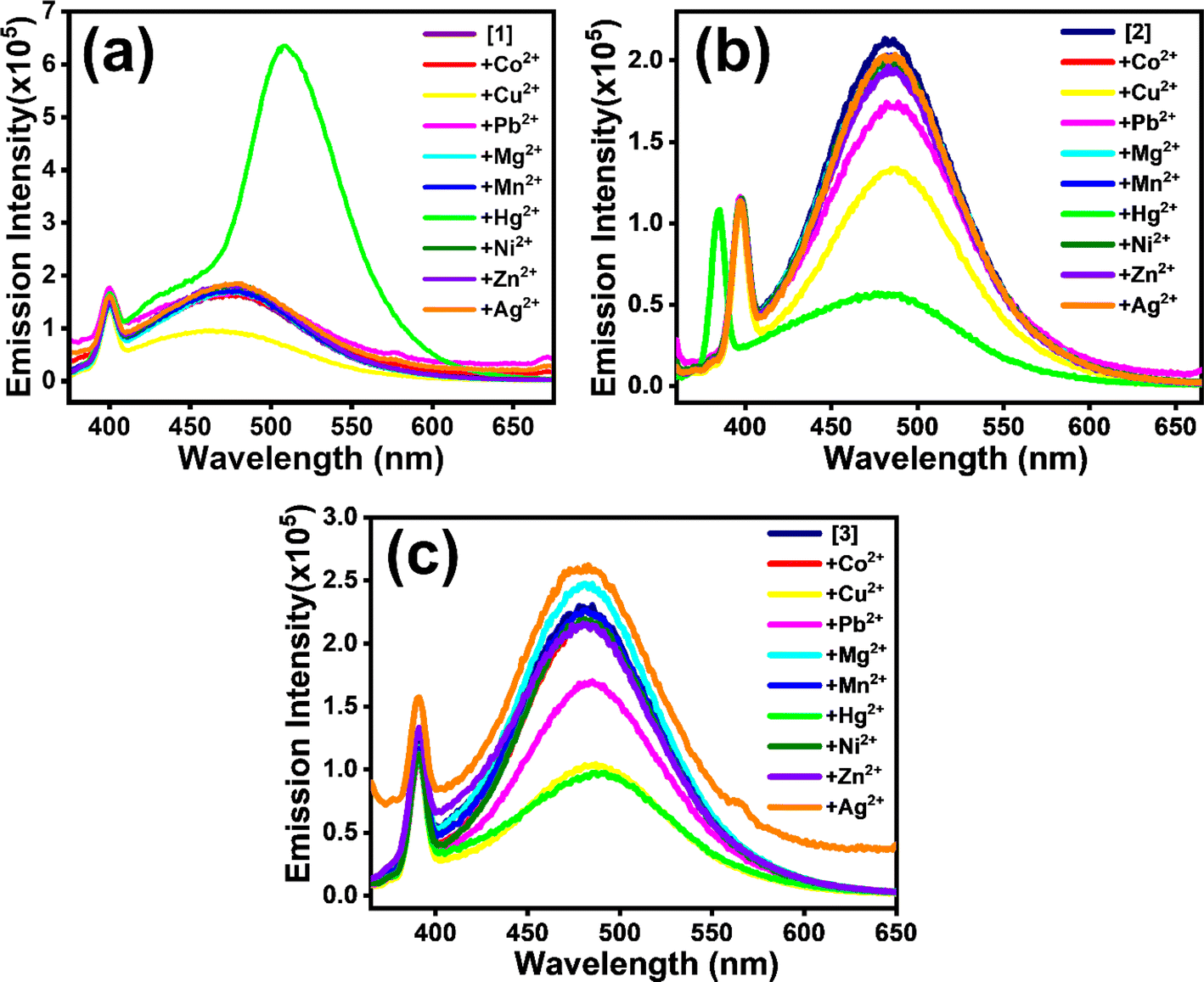 | ||
| Fig. 3 Emission spectra (λexc ∼358 nm) showing reactivity towards different metal ions for (a) [1], (b) [2] and (c) [3] considered in the study. | ||
Further, absorption and fluorescence titrations of [1]–[3] were performed against Cu2+ and Hg2+ ions under similar conditions. In the case of [1], both absorption at 352 nm and fluorescence intensity at 474 nm were quenched upon the addition of Cu2+ (Fig. 4a, and c). A blueshift (∼25 nm) is observed with an increase in Cu2+ ions in emission spectra indicating H-aggregate formation (Fig. 4a).36 On the contrary, the fluorescence titration with Hg2+ showed a gradual increase in the emission intensity with systematic redshift (∼40 nm) in the emission maxima forming J-aggregates (Fig. 4b).36 Though the addition of Hg2+ ions also triggered quenching of absorption, the extent was less prominent than that observed with Cu2+ (Fig. 4d). Consequently, the yellow-colored solution of [1] turned colorless with Cu2+ (Fig. 4e(i)). Both [2] and [3] showed decreased absorption along with emission intensities when titrated with Cu2+ and Hg2+ ions (Fig. S7, and S8†). The phenomenon of quenching in [1]–[3] was elaborated with the help of Stern–Volmer plots and Ksv was calculated with a standard reported procedure (Fig. S9–S11†).37,42 For [2], the UV-visible titrations with both Cu2+ and Hg2+ showed hypochromic shifts of similar magnitude. However, in the case of fluorometric analysis, the extent of response with Hg2+ ion was found to be larger (∼3.7-fold) than that noted with Cu2+ ions (Fig. 5). In [3], a completely different scenario was witnessed; fluorescence titration with Cu2+ and Hg2+ resulted in comparable amount of quenching (Fig. 5). However, the UV-visible titration studies showed that the hypochromic shift is more prominent with Cu2+ than with Hg2+ ions (see Fig. 4 for [1], S7, and S8† for [2], [3]). Based on the spin–orbit coupling phenomenon, Hg2+ ions are known to quench fluorescence, whereas Cu2+ induces a photoinduced electron transfer that turns off the fluorescence response.26 Also, the UV-visible titrations with metal ions sometimes resulted in substantial tailing at longer wavelength regions.37 This suggested metal ion-mediated, large aggregated structure formation, in solution. Fig. 4e shows variation in emission intensity with the addition of Cu2+ and Hg2+ ions in [1]–[3] in MeCN:H2O (1:9) solvent in short-wavelength UV (254 nm) light.
 | ||
| Fig. 4 Variation in emission intensity (λexc ∼358 nm) of [1] with an increase in metal ion concentration. (a) Quenching effect for Cu2+ and (b) enhancement with respect to Hg2+. Lowering of absorbance with increased concentration of (c) Cu2+ and (d) Hg2+ for [1]. (e) From left to right: (i) [1], [1]:Cu2+, [1]:Hg2+; (ii) [2], [2]:Cu2+, [2]:Hg2+; (iii) [3], [3]:Cu2+, [3]:Hg2+ in MeCN:H2O (1:9) solvent in short-wavelength UV (254 nm) light. | ||
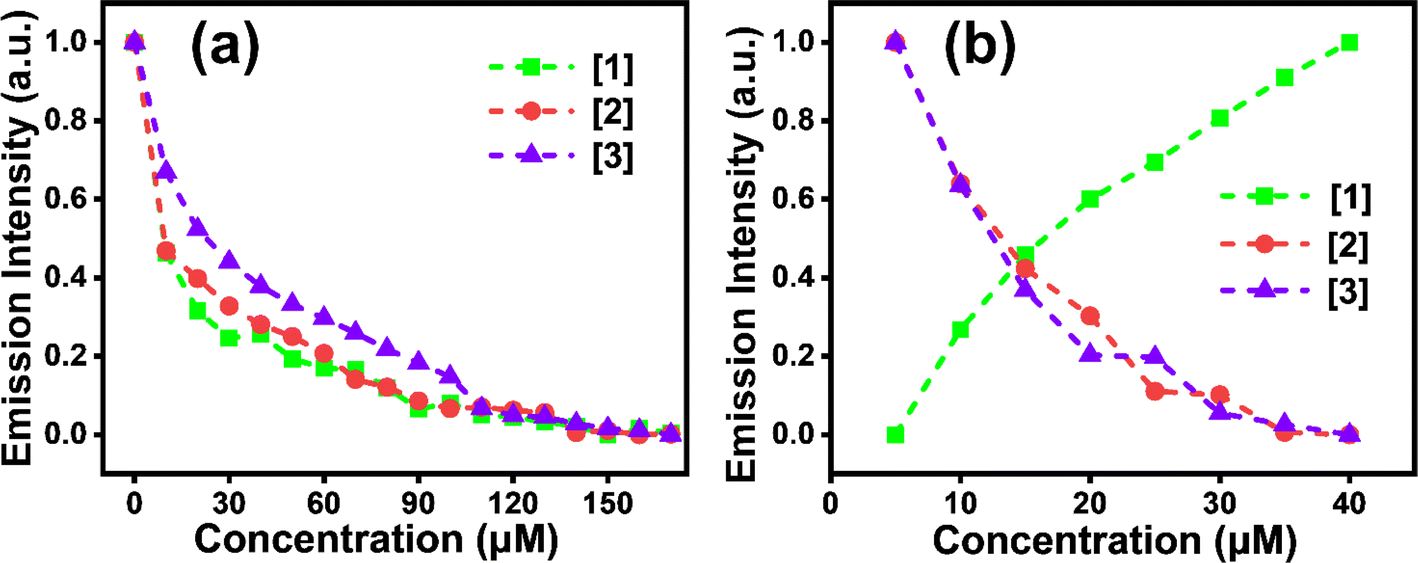 | ||
| Fig. 5 Variation in emission intensity (λexc ∼358 nm) with increase in (a) Cu2+ and (b) Hg2+ for [1]–[3]. Quenching is observed with Cu2+ for [1]–[3] while quenching is seen with Hg2+ for only [2] and [3]. | ||
The combination of static and dynamic quenching is observed for the considered [1]–[3]; the linear nature of the plot at initial concentration indicates static quenching.37 All [1]–[3] have shown a higher quenching effect with Cu2+ ions than Hg2+ (Table 1). Higher Ksv explains a stronger quenching effect.37 The sensitivity of [1] and [2] is more towards Cu2+ compared to [3] as a stronger quenching effect (higher Ksv) is witnessed in a lower concentration range (∼1–15 μM). [1] has a higher sensitivity for Cu2+ than [2] and [3]; [1] has almost 1.5 times higher Ksv than [2] and ∼3 times higher than [3]. In response to Hg2+, [3] has higher Ksv than [2], i.e., almost 1.9 times in the concentration range of (∼5–40 μM). LOD values from Table 1 depict the least LOD for [2] for Cu2+, i.e. 0.75 μM (Fig. S9c, S10e, f, S11e, and f†). Consequently, dynamic quenching ascribed to collisions with regard to metal ions and [1], [2] and [3] with an increase in metal ion concentration leads to aggregation quenching.37 Nonetheless, interestingly only [1] led to fluorescence enhancement with Hg2+. Notably the solid form of [1] formed supramolecular assemblies with one of the amino groups in a gauche conformation so as to fold backward and create a strong intramolecular hydrogen bond with another amino ethylene group.30 Further, the average dihedral angle of ∼57° among the amine and the quinone moiety in [1] and [2] leads to the molecular twist in solids. However, in solution, especially for [1], there is a possibility of attaining rigidity on coming into contact with Hg2+, as a result, restricting rotation/twisting of the molecule, leading to Hg-induced emission,21 likely due to the smaller ring size of pyrrolidine in [1] when compared to the piperidine substituent in [2]. Moreover, the smaller five-membered pyrrole ring possibly facilitates CT interaction between [1] and metal ions (Cu2+/Hg2+), generating higher sensitivity. Though pyrrolidine and piperidine nitrogen ends have almost similar pKa values (pKa ∼11.1), pyrrolidine could bind more strongly with Cu2+ than piperidine, probably due to its smaller ring size, less steric hindrance, and higher ring strain. These aspects make the pyrrolidine nitrogen's lone pair more accessible for coordination. Conversely, piperidine, owing to its larger and more flexible six-membered ring, introduces more conformational freedom and steric bulk, making the metal binding relatively weak.43 Further, in the case of ethylenediamine residue, due to its acyclic nature and two nitrogen atoms with a flexible carbon chain, its effective binding to the metal ion is hindered, and the binding process is weaker because both nitrogen atoms must coordinate with the metal ion.44
| DADQ | K sv (Cu2+) (μM−1) | Concentration range (μM) | K sv (Hg2+) (μM−1) | Concentration range (μM) | LOD Cu2+ (μM) | LOD Hg2+ (μM) |
|---|---|---|---|---|---|---|
| [1] | 0.03905 | 1–4 | — | — | 0.7906 | 4.1873 |
| [2] | 0.03512 | 1.5–5 | 0.00448 | 5–25 | 0.7510 | 3.1937 |
| [3] | 0.00288 | 5–25 | 0.00287 | 5–20 | 3.9810 | 3.7505 |
Cu2+, being smaller, less polarizable, and favouring a square planar or tetrahedral geometry, binds to the nitrogen in pyrrolidine in a more rigid, localized, and faster manner. Hg2+, being larger and more polarizable, binds more flexibly and diffusely, often adopting different coordination geometries and involving softer, less directional interactions with the nitrogen.45
3.3 Mechanistic investigations
After the above-discussed photophysical and metal ion sensing studies, the mechanism of interaction between [1]–[3] and the corresponding metal ions was examined. As previously stated, [1] and [2] contain heterocyclic pyrrolidine and piperidine rings respectively, whereas [3] comprises a simple aliphatic amine. Resuming the solid-state interactions, molecular packing in the crystal structures of [1]–[3] revealed supramolecular assemblies of the molecular dipoles with antiparallel orientations with C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N of one DADQ molecule aligned opposite to the CN of the adjacent DADQ (gauche conformation).30,32 Moreover, Crozier et al. reported the attachment of one Cu+ to four nitrogens, toward CN of four different TCNQ molecules.16 Furthermore, the H and J aggregates observed in the absorption spectra and the supramolecular assemblies of molecular dipoles [1]–[3] lead to an efficient sensing of several metal ions, with selectivity towards Hg2+ and Cu2+, under very low concentrations (μM mM−1). Also absorption λmax of solid [1]–[3] matches with the absorption λmax of solutions [1]–[3] (in polar solvents), probably with similar coulombic intermolecular interactions among molecular dipoles. Accordingly, both 1H-NMR and FT-IR spectra were recorded and analyzed for [1], [2] and [3] in solutions with and without the presence of the concerned metal ions (Hg2+ and Cu2+) (Fig. 6, S12 and S13†) to understand the interaction of Hg2+ and Cu2+ with [1]–[3]. Though the spectroscopic studies were performed in CH3CN–H2O (1:9) mixture medium, the 1H-NMR studies could not be executed in CD3CN–D2O (1:9) mixture medium due to limited solubility. Due to indefinite peaks under 10 μM concentration, a 20 mM concentration was optimised for 1H-NMR and FT-IR studies. To a 20 mM solution of [1]–[3] equal amounts, i.e. 20 mM, metal ion (Hg2+ and Cu2+) solution were added for the 1H-NMR titration study (Fig. 6, S12 and S13†). Notably, the DMSO-d6 solutions of 20 mM of all [1]–[3] turned intense yellow on adding 20 mM of Hg2+, whereas they immediately became brown-black on the addition of 20 mM Cu2+. According to reports, 1H-NMR studies in the case of Cu2+ sensing are undivulged, owing to the difficulty in detecting the exact peak positions, while the broadening of peaks is seen due to the paramagnetic quenching exhibited by Cu2+ (3d[9] electronic configuration).26 The plausible interaction of Cu2+ and Hg2+ with the aromatic as well as aliphatic moieties of [1]–[3] is clearly visible with a significant change in the nature of peaks. However, the change is more prominent for Cu2+ with charge transfer characteristics (Fig. 6a, S12a, and S13a†). Fig. 7 illustrates the possible binding of Cu2+ and Hg2+ to δ+ carbon attached moieties and δ− carbon connected CN nitrogen. Consequently, the change in shape and shift of aliphatic protons is visible for [1]–[3]. Thus, the binding of metal ions (Hg2+ and Cu2+) to N (CN) of TCNQ is prominent. Correspondingly, the aromatic protons (f, and g) belonging to the benzenoid ring are further split into two doublets along with a slight downfield shift for Hg2+; in contrast, a broadening of peaks occurs, losing the doublet nature for Cu2+.
N of one DADQ molecule aligned opposite to the CN of the adjacent DADQ (gauche conformation).30,32 Moreover, Crozier et al. reported the attachment of one Cu+ to four nitrogens, toward CN of four different TCNQ molecules.16 Furthermore, the H and J aggregates observed in the absorption spectra and the supramolecular assemblies of molecular dipoles [1]–[3] lead to an efficient sensing of several metal ions, with selectivity towards Hg2+ and Cu2+, under very low concentrations (μM mM−1). Also absorption λmax of solid [1]–[3] matches with the absorption λmax of solutions [1]–[3] (in polar solvents), probably with similar coulombic intermolecular interactions among molecular dipoles. Accordingly, both 1H-NMR and FT-IR spectra were recorded and analyzed for [1], [2] and [3] in solutions with and without the presence of the concerned metal ions (Hg2+ and Cu2+) (Fig. 6, S12 and S13†) to understand the interaction of Hg2+ and Cu2+ with [1]–[3]. Though the spectroscopic studies were performed in CH3CN–H2O (1:9) mixture medium, the 1H-NMR studies could not be executed in CD3CN–D2O (1:9) mixture medium due to limited solubility. Due to indefinite peaks under 10 μM concentration, a 20 mM concentration was optimised for 1H-NMR and FT-IR studies. To a 20 mM solution of [1]–[3] equal amounts, i.e. 20 mM, metal ion (Hg2+ and Cu2+) solution were added for the 1H-NMR titration study (Fig. 6, S12 and S13†). Notably, the DMSO-d6 solutions of 20 mM of all [1]–[3] turned intense yellow on adding 20 mM of Hg2+, whereas they immediately became brown-black on the addition of 20 mM Cu2+. According to reports, 1H-NMR studies in the case of Cu2+ sensing are undivulged, owing to the difficulty in detecting the exact peak positions, while the broadening of peaks is seen due to the paramagnetic quenching exhibited by Cu2+ (3d[9] electronic configuration).26 The plausible interaction of Cu2+ and Hg2+ with the aromatic as well as aliphatic moieties of [1]–[3] is clearly visible with a significant change in the nature of peaks. However, the change is more prominent for Cu2+ with charge transfer characteristics (Fig. 6a, S12a, and S13a†). Fig. 7 illustrates the possible binding of Cu2+ and Hg2+ to δ+ carbon attached moieties and δ− carbon connected CN nitrogen. Consequently, the change in shape and shift of aliphatic protons is visible for [1]–[3]. Thus, the binding of metal ions (Hg2+ and Cu2+) to N (CN) of TCNQ is prominent. Correspondingly, the aromatic protons (f, and g) belonging to the benzenoid ring are further split into two doublets along with a slight downfield shift for Hg2+; in contrast, a broadening of peaks occurs, losing the doublet nature for Cu2+.
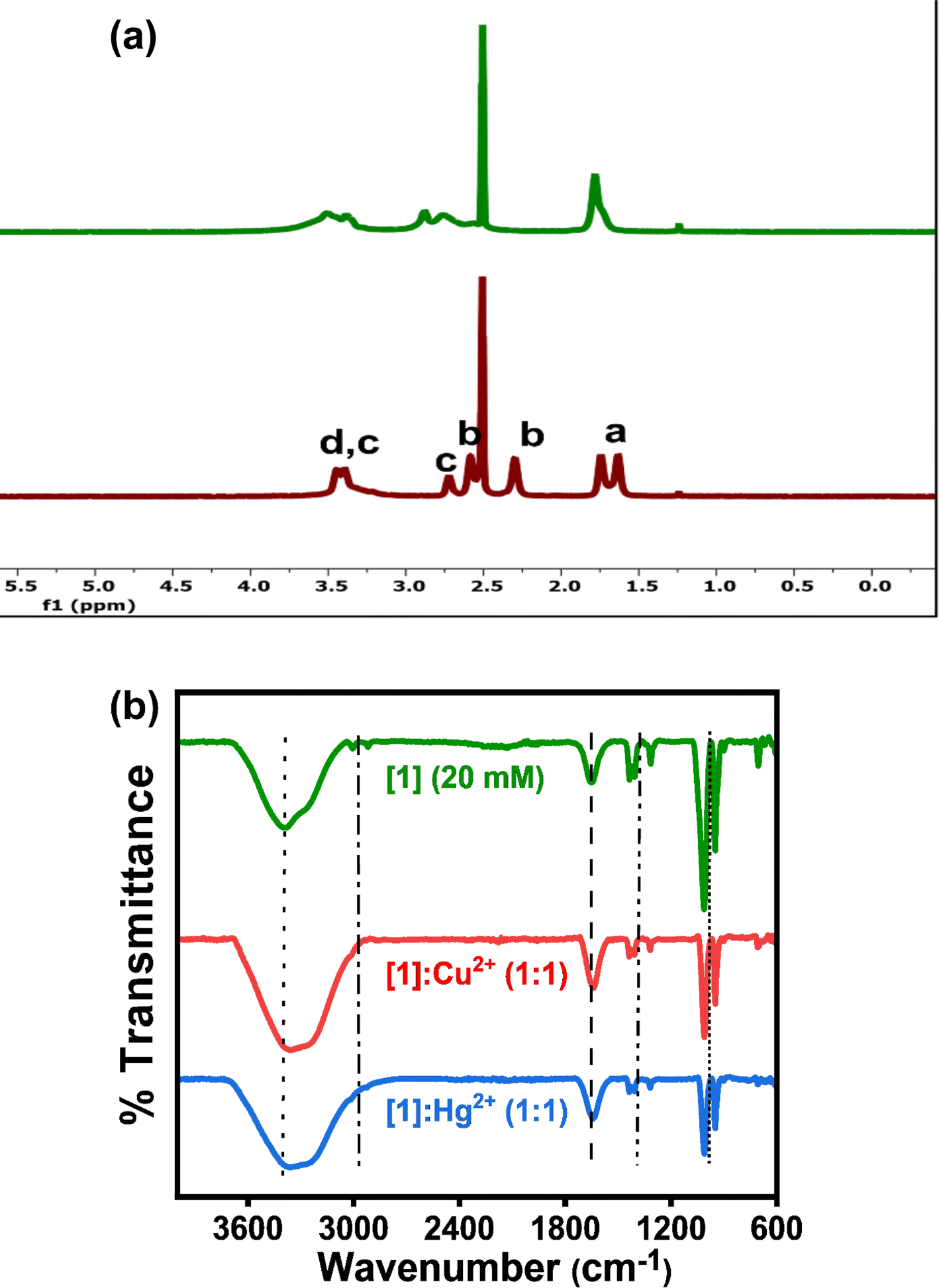 | ||
| Fig. 6 (a) 1H NMR and (b) FT-IR spectra of [1] (20 mM); [1]:Cu2+ (1:1) and [1]:Hg2+ (1:1) to understand the possible interaction of [1] with Hg2+ and Cu2+. | ||
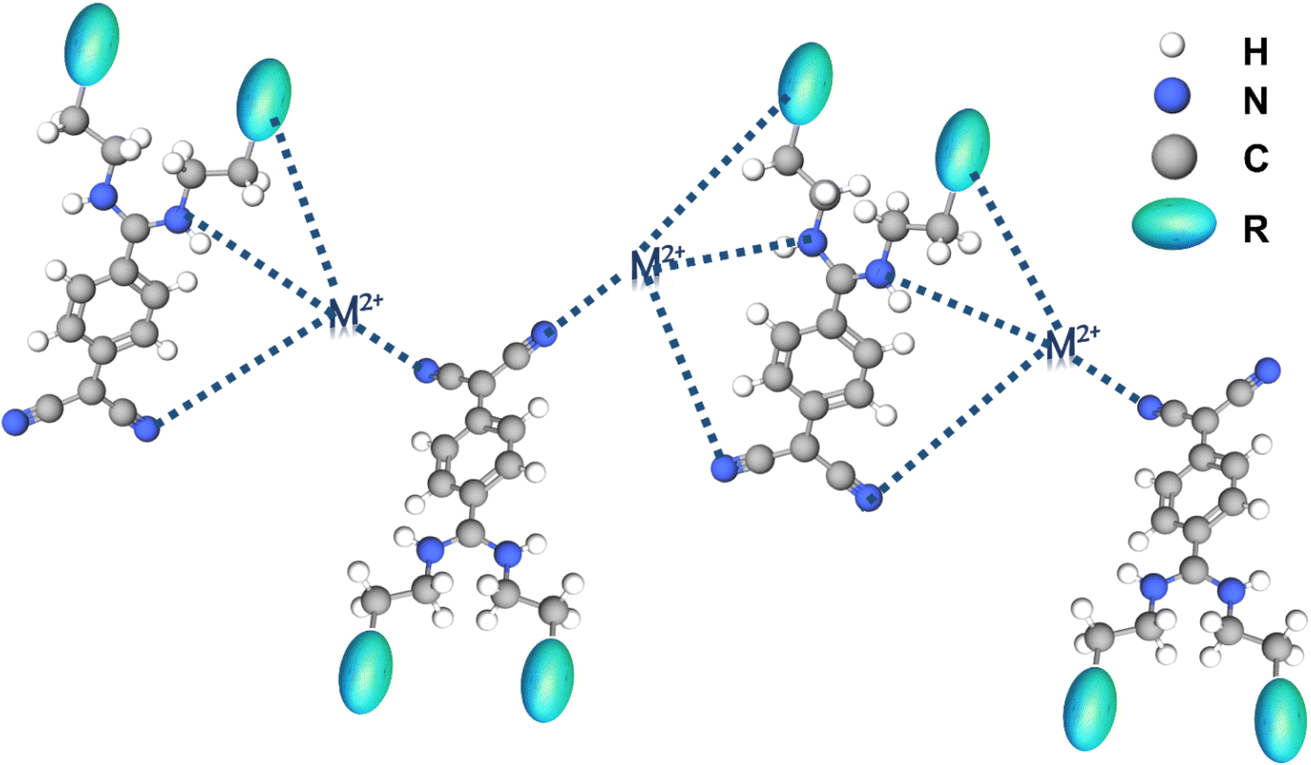 | ||
| Fig. 7 Plausible mechanism for Hg2+ and Cu2+ metal ion sensing by [1]–[3]. | ||
A new peak at ∼7.5 ppm (Fig. 6a, S12a, and S13a†) can be likely attributed to the shift of e protons (9.3 ppm) towards the upfield region due to the interaction of electron-rich N (NH) with electron-deficient Hg2+. The broadening, lower resolution and downfield shift of c, and b protons by 1 ppm ([1]), subtle 0.5 ppm ([2]) and 2 ppm ([3]) with respect to Hg2+ are perceived. Whereas the disappearance of c and b protons or their merging in the DMSO-d6 peak (2.5 ppm) is observed on the addition of Cu2+ (Fig. 6a, S12a, and S13a†). These observations highlight the interaction of metal ions (Hg2+ and Cu2+) with the N between c and b protons. Further, modest downfield shift is seen for a protons in [1] and broadening of a protons is noticed in [2] and [3] with no major shift of peaks. FT-IR studies also support the binding of Cu2+/Hg2+ towards the aromatic region along with the aliphatic region in [1]–[3] (peak at ∼1660 cm−1 represents the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch). Broadening of N–H stretch band (3399 cm−1) of secondary amine and absence of C–H stretch bands (3005, 2921 cm−1) from ethylene moiety on introducing metal ions (Hg2+ and Cu2+) are observed. Less intense peaks of C–N bending (1434, 1321 cm−1) and intense peaks of C–N stretch (1007, 947 cm−1) confirm the engagement of metal ions (Hg2+ and Cu2+) with N atoms of [1]–[3], this further influencing the e, f, and g proton chemical shifts. Combining the observations from the detailed NMR and FT-IR studies, a plausible mechanism is depicted in Fig. 7.
C stretch). Broadening of N–H stretch band (3399 cm−1) of secondary amine and absence of C–H stretch bands (3005, 2921 cm−1) from ethylene moiety on introducing metal ions (Hg2+ and Cu2+) are observed. Less intense peaks of C–N bending (1434, 1321 cm−1) and intense peaks of C–N stretch (1007, 947 cm−1) confirm the engagement of metal ions (Hg2+ and Cu2+) with N atoms of [1]–[3], this further influencing the e, f, and g proton chemical shifts. Combining the observations from the detailed NMR and FT-IR studies, a plausible mechanism is depicted in Fig. 7.
3.4 Morphological investigation
Scanning electron microscopy study of the drop-cast solutions confirms aggregate formation leading to quenching in [1]–[3] (Fig. 8). Interaction of [1]–[3] with Cu2+ and Hg2+ was confirmed further with EDAX measurements and mapping (Fig. S14–S16†) revealing the elemental composition. Larger aggregate formation with Cu2+ thus leading to faster quenching is clearly observed, when compared to Hg2+. Fig. S14b, S15b, and S16b† illustrate the presence of Cu2+ and Hg2+. The elemental composition of Cu2+ was obtained as 3.20 wt% and Hg2+ as 27.51 wt% in [1]:Cu2+(1:1) and [1]:Hg2+(1:1). 2 wt% and 17.56 wt% were seen for Cu2+ and Hg2+, respectively, in [2]:Cu2+ (1:1) and [2]:Hg2+ (1:1) besides their obvious absence in pristine [1] and [2]. 4.14 wt% and 22.87 wt% compositions of Cu2+ and Hg2+ marked their presence in [3]:Cu2+ and [3]:Hg2+ respectively. A 2.5 μm magnification area was used in mapping all the samples except for [2]:Hg2+ (1:1); for clarity, the magnification region was 5 μm.
 | ||
| Fig. 8 SEM images (at 1 μm magnification) showcasing aggregate formation in [1]–[3] on the addition of equimolar 1:1 ratio of Cu2+ and Hg2+ metal ions. From left to right: (a) (i) only [1] at 10 μM concentration, (ii) [1]:Cu2+ (1:1 molar ratio), (iii) [1]:Hg2+; (b) (i) only [2], (ii) [2]:Cu2+, (iii) [2]:Hg2+; (c) (i) only [3], (ii) [3]:Cu2+, (iii) [3]:Hg2+. | ||
3.5 Sensing of metal ions in real water samples
To understand the applicability of the proposed DADQs for real water samples, water samples were collected from a lotus pond and a tap. The pond water sample showed higher emission intensities without any DADQ added, which may be due to microorganisms and suspended particulate matter present in it. Since [1] has shown varied responses to Cu2+ and Hg2+ ions, [1] was chosen for real water sample analysis. Metal ion spiking was done in the real water samples as mentioned in Section 2.2. Fluorescence quenching behavior remained the same for Cu2+ in both samples (Fig. S17†). There was a 1.3-fold reduction in emission intensity for pond water (Fig. S17a†) whereas emission intensity was lowered by 1.7 times for the tap water sample (Fig. S17b†). Enhancement in intensity against Hg2+ was not seen for [1] in both pond and tap water samples. Therefore, [1] was found to be sensitive to Cu2+ and retain its properties in real water samples following quenching behaviour at lower concentrations.4 Conclusions
In summary, we synthesized [1]–[3] following the reported procedure and initially applied [1]–[3] solutions for effective metal ion sensing of Cu2+ and Hg2+. [1]–[3] exhibited solvatochromism. We thoroughly investigated the effects of the microenvironment such as temperature, pH, polarity, etc. on the aggregation. Furthermore, the metal ion sensing ability of [1]–[3] was investigated in an acetonitrile–water (1:9) medium. Interestingly, the presence of different amino functional groups at the terminal position of the ethylene spacer affected the metal ion sensing property in [1]–[3]. [1], with a pyrrolidine ring, showed fluorescence quenching with Cu2+ and fluorescence enhancement with Hg2+ ions. [2] with a piperidine substituent resulted in fluorescence quenching selectively with Hg2+ and not remarkably with Cu2+. Conversely, the addition of both Cu2+ and Hg2+ to [3], with N,N-diethylamine substituent, resulted in a turn-off fluorescence signal. Higher sensitivity for Cu2+ ions is observed in real water samples. Interestingly, diverse responses with Cu2+ and Hg2+, merely by varying the amine substituents on [1]–[3], could assist in engineering new sensory systems with better efficiency going forward.
Data availability
All the data collected, studied and analysed in this work are presented in the main manuscript and ESI.†Author contributions
Anuradha Mohitkar: writing – initial draft data curation, investigation, formal analysis, methodology. Nilanjan Dey: methodology, investigation, formal analysis, validation. Subbalakshmi Jayanty: conceptualization, resources, supervision, investigation, validation, methodology, administration and organization, writing – reviewing, and editing the original draft.Conflicts of interest
Authors declare no conflicts of interest.Acknowledgements
SJ thanks BITS-Pilani Hyderabad campus for financial assistance to AM. The authors thank the BITS-Pilani Hyderabad campus for the Central Analytical Laboratory (CAL) facility and the DST-FIST grant facility sanctioned to the Department of Chemistry, BITS-Pilani Hyderabad Campus. Also, the HRMS facility, sponsored by DST-FIST (grant number: SR/FST/CS-I/2020/158), at BITS Pilani, Hyderabad Campus, is acknowledged.References
- T. H. Kim, J. H. Kim and K. Kang, Jpn. J. Appl. Phys., 2023, 62(SE0803), 1–15 Search PubMed.
- T. P. Radhakrishnan, Acc. Chem. Res., 2008, 41, 367–376 CrossRef CAS PubMed.
- A. Mohitkar, H. Renuka, S. Goel and S. Jayanty, ACS Omega, 2023, 8, 40836–40847 CrossRef CAS PubMed.
- A. Syed, H. Battula, P. K. R. Boppidi, S. Kundu, C. Chakraborty and S. Jayanty, Org. Electron., 2020, 76, 105457 CrossRef.
- N. Senthilnathan, C. G. Chandaluri and T. P. Radhakrishnan, Sci. Rep., 2017, 7, 10583 CrossRef CAS PubMed.
- G. Kamarchuk, O. Pospelov, V. Vakula and E. Faulques, Sens. Actuators, B, 2024, 402, 135064 CrossRef CAS.
- C. Wang, N. Wu, D. L. Jacobs, M. Xu, X. Yang and L. Zang, Chem. Commun., 2017, 53, 1132–1135 RSC.
- J. Y. Liao and K. C. Ho, Sens. Actuators, B, 2008, 130, 343–350 CrossRef CAS.
- C. Lin, K. Kim, Z. Wang, Z. Yan, Z. Tang and Y. Liu, Nano Res., 2023, 16(12), 13366–13374 CrossRef CAS.
- I. Yamaguchi, K. Ikawa, N. Takimiya and A. Wang, Molecules, 2023, 28, 5663 CrossRef CAS PubMed.
- M. Liu, W. Liu, W. Zhang, P. Duan, M. Shafi, C. Zhang, X. Hu, G. Wang and W. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 56975–56985 CrossRef CAS PubMed.
- J. Xu, K. Liu, D. Di, S. Shao and Y. Guo, Inorg. Chem. Commun., 2007, 10, 681–684 CrossRef CAS.
- R. A. Heintz, H. Zhao, X. Ouyang, G. Grandinetti, J. Cowen and K. R. Dunbar, Inorg. Chem., 1999, 38, 144–156 CrossRef CAS.
- B. Mukherjee, M. Mukherjee, J. Park and S. Pyo, J. Phys. Chem. C, 2010, 114, 567–571 CrossRef CAS.
- A. L. Sutton, B. F. Abrahams, C. J. Commons, A. D. Dharma, L. Goerigk, S. G. Hardin, T. A. Hudson and R. Robson, J. Mater. Chem. C, 2023, 11, 15030 RSC.
- S. Balendhran, Z. Hussain, V. R. Shrestha, J. Cadusch, M. Ye, N. S. Azar, H. Kim, R. Ramanathan, J. Bullock, A. Javey, V. Bansal and K. B. Crozier, ACS Appl. Mater. Interfaces, 2021, 13(32), 38544–38552 CrossRef CAS PubMed.
- Y. S. Wu, A. I. Osman, M. Hosny, A. M. Elgarahy, A. S. Eltaweil, D. W. Rooney, Z. Chen, N. S. Rahim, M. Sekar, S. C. B. Gopinath, N. N. I. Mat Rani, K. Batumalaie and P. S. Yap, ACS Omega, 2024, 9, 5100–5126 CrossRef CAS PubMed.
- H. Patton, E. Sarver, L. Lehmann, J. Parks and L. A. Krometis, Environ. Adv., 2024, 16, 100543 CrossRef CAS.
- Y. Zohrabi, Mater. Sci. Eng., B, 2024, 299, 117024 CrossRef CAS.
- W. Lu, X. Qin, S. Liu, G. Chang, Y. Zhang, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Anal. Chem., 2012, 84, 5351–5357 CrossRef CAS PubMed.
- Y. He, Y. Li, H. Su, Y. Si, Y. Liu, Q. Peng, J. He, H. Hou and K. Li, Mater. Chem. Front., 2019, 3, 50–56 RSC.
- F. Lafzi, A. S. Hussein, H. Kilic and S. Bayindir, J. Photochem. Photobiol., A, 2023, 444, 114958 CrossRef CAS.
- S. Liu, J. Tian, L. Wang, Y. Zhang, X. Qin, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Adv. Mater., 2012, 24, 2037–2041 CrossRef CAS PubMed.
- J. Tian, Q. Liu, A. M. Asiri, A. O. Al-Youbi and X. Sun, Anal. Chem., 2013, 85, 5595–5599 CrossRef CAS PubMed.
- N. R. Divyashree, H. D. Revanasiddappa, N. R. Bhavya, M. Mahendra, B. Jayalakshmi, C. Shivamallu and S. P. Kollurf, Spectrochim. Acta, Part A, 2023, 292, 122435 CrossRef CAS PubMed.
- R. S. Fernandes and N. Dey, Mater. Chem. Phys., 2023, 302, 127637 CrossRef CAS.
- Y. Li, M. Ashizawa, S. Uchida and T. Michinobu, Polym. Chem., 2012, 3, 1996–2005 RSC.
- P. Kaur, S. Kaur, Y. Kasetti, P. V. Bharatam and K. Singh, Talanta, 2010, 83, 644–650 CrossRef CAS PubMed.
- A. Syed, H. Battula, S. Mishra and S. Jayanty, ACS Omega, 2021, 6, 3090–3105 CrossRef CAS PubMed.
- P. Raghavaiah, R. Kuladeep, D. Narayana Rao, A. Jyothi Lakshmi, P. Srujana and J. Subbalakshmi, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 709–715 CrossRef CAS PubMed.
- P. Sudhakar and T. P. Radhakrishnan, Chem.–Asian J., 2019, 14, 4754–4759 CrossRef CAS PubMed.
- A. Syed, S. Mishra and S. Jayanty, J. Fluoresc., 2022, 32, 115–124 CrossRef CAS PubMed.
- S. Kaja, D. P. Damera and A. Nag, Anal. Chim. Acta, 2020, 1129, 12–23 CrossRef CAS PubMed.
- H. Battula, S. Muduli, S. Priyanka Bandi, S. Kapoor, S. Mishra, H. Aggarwal, V. V. Venuganti and S. Jayanty, J. Photochem. Photobiol., A, 2022, 426, 113748 CrossRef CAS.
- S. Jayanty and T. P. Radhakrishnan, Chem.–Eur. J., 2004, 10, 791–797 CrossRef CAS PubMed.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2006 Search PubMed.
- A. SD, A. Mohitkar and S. Jayanty, New J. Chem., 2022, 46, 8069–8078 RSC.
- B. Yu, R. Yuan, T. He, L. Liang and K. Huang, J. Fluoresc., 2022, 32, 2077–2086 CrossRef CAS PubMed.
- N. Kaewnok, N. Chailek, W. Muansrichai, S. Wangngae, A. Petdum, W. Panchan, A. Kamkaew, J. Sirirak, P. Swanglap, T. Sooksimuang and N. Wanichacheva, J. Photochem. Photobiol., A, 2023, 444, 114968 CrossRef CAS.
- J. Pan, J. Ma, L. Liu, D. Li, Y. Huo and H. Liu, J. Photochem. Photobiol., A, 2021, 416, 113322 CrossRef CAS.
- N. Savita, A. Nandikolla, A. K. Shukla, K. V. G. Chandra Sekhar and A. Bhattacharya, Dalton Trans., 2023, 52, 4103–4111 RSC.
- G. L. Petri, M. V. Raimondi, V. Spanò, R. Holl, P. Barraja and A. Montalbano, Top. Curr. Chem., 2021, 379, 34 CrossRef PubMed.
- P. Paoletti, Pure Appl. Chem., 1984, 56(4), 491–522 CrossRef.
- L. Rulíšek and J. Vondrášek, J. Inorg. Biochem., 1998, 71, 115–127 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Absorption, emission spectra, solvatochromism, SEM images, emission intensity changes on addition of Cu2+ and Hg2+ of [2], [3]; Stern–Volmer plots, LOD, Ksv for [1]–[3];1HNMR, FT-IR of [2], [3] and EDAX for [1]–[3] are detailed in the supporting information. See DOI: https://doi.org/10.1039/d4su00445k |
| This journal is © The Royal Society of Chemistry 2024 |