
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient single-component nickel catalysts with tetradentate aminopyridine ligands for cycloaddition reactions of CO2 and epoxides under mild conditions†
Congcong
Zhang‡
a,
Minghui
Shi‡
a,
Ning
Yu
a,
Bowen
Zhang
*ab,
Feng
Han
 *a and
Chengxia
Miao
*a
*a and
Chengxia
Miao
*a
aKey Laboratory of Low-Carbon and Green Agriculture Chemistry in Universities of Shandong, College of Chemistry and Material Science, Shandong Agricultural University, Tai'an, Shandong 271018, P. R. China. E-mail: fenghan@sdau.edu.cn; chxmiao@sdau.edu.cn
bDepartment of Chemistry, Texas A&M University, College Station, Texas 77843, USA
First published on 28th October 2024
Abstract
A series of single-component nickel catalysts (L1-NiBr2/L2-NiBr2/L3-NiBr2) with tetradentate aminopyridine ligands are presented, which exhibit excellent capabilities and selectivity for the synthesis of cyclic carbonates from epoxides and carbon dioxide. A green crystal of L1-NiBr2 was obtained in CH3CN, and the ligand adopted a cis-α conformation in the complex. The conversion of styrene oxide could reach 98%, providing 100% selectivity at 90 °C, 1 MPa CO2 pressure and 5 mol% of L1-NiBr2 under solvent-free conditions, while the yield and selectivity values were still as high as 92% and 99%, respectively, under 1 atm CO2 and 0.5 mol% of the catalyst at the same temperature. The catalysts also exerted efficient catalytic coupling reactions of terminal epoxides (90–98% yields of cyclic carbonates), except those bearing a long aliphatic chain under mild conditions. The catalyst exhibited excellent recyclability and stability, which were further proved through ICP to test the catalyst leaching, TGA and IR spectra. Moreover, the catalytic cycloaddition reaction mechanism was investigated using density functional theory (DFT) calculations.
Sustainability spotlightThe rapid increase in CO2 emission has had an adverse impact on the environment; therefore, developing sustainable chemical processes to transform CO2 into value-added chemicals is crucial. This study focuses on the production of efficient single-component nickel catalysts with tetradentate aminopyridine ligands and their application for transforming CO2 into cyclic carbonates. The investigation demonstrated that the developed nickel catalysts presented excellent bifunctional catalytic performance, stability and substrate scope under mild conditions, even in low catalyst amounts and atmospheric pressure of CO2 in the absence of solvent, which is industrially attractive for future applications. Our technologies align with the United Nations Sustainable Development Goals, particularly Goals 9, 12 and 13. |
1. Introduction
As a potent greenhouse gas, carbon dioxide (CO2) emissions have increased worldwide on a massive scale due to the generation of energy by the combustion of fossil fuels, adversely impacting the environment.1 Thus, reducing the level of atmospheric CO2 is of utmost importance in the coming decade. In contrast, CO2 is a very significant C1 feedstock in organic synthesis due to its non-flammable, non-toxic, sustainable and inexpensive characteristics.2 Therefore, the transformation of CO2 into industrially attractive chemicals by chemical strategies has attracted increasing attention.3 Among them, the production of cyclic carbonates from epoxides and CO2, as an atom-economical reaction, is regarded a considerate method.4 Moreover, cyclic carbonates have a vibrant area of application in academic and industrial arenas, such as polar aprotic solvents, electrolytes in lithium-ion batteries, and intermediates of biodegradable polymers.5A crucial challenge in the reaction between epoxides and CO2 is the development of suitable catalysts due to the high thermodynamic stability of CO2.6 To date, numerous catalytic systems, which usually contain both a Lewis acidic center to activate the C–O bond of the epoxide and a Lewis basic site that can be used as a nucleophilic reagent to open the epoxide ring, have been successfully synthesized, including organo-catalysts,7 metal complexes catalysts,8 organic frameworks9 and others, to catalyze the cycloaddition reaction.10 Among these active catalytic systems, metal-containing catalysts have attracted increasing attention due to their higher intrinsic Lewis acidity, lower catalyst loadings and lower reaction temperatures.11 However, most of them are binary catalytic systems, and Lewis bases are usually added as co-catalysts, such as DMAP (4-dimethylaminopyridine) or onium salts.8c,12 Consequently, considerable efforts to develop efficient one-component bifunctional metal catalyst systems have been proposed to promote cyclic carbonate synthesis without co-catalysts.
As a part of the metal complex, ligands play a pivotal role in developing bifunctional metal catalysts with high catalytic reactivity, and tetradentate nitrogen ligands have been proven efficient for various oxidation reactions, as well as the conversion of CO2.13 In view of this, a series of tetradentate aminopyridine-containing metal catalysts have been designed for the transformation of CO2.14 For example, various kinds of bifunctional zinc coordination compounds with tetradentate aminopyridine (N4) ligands have been proven efficient catalysts for the coupling reaction between CO2 and epoxides with a broad substrate scope under solvent-free conditions by Sun et al.14a Despite of this, the development of novel N4 bifunctional catalysts under milder conditions is still needed.
In order to achieve the goal, a more active metal center is of utmost importance to obtain high catalytic activity.15 Nickel (Ni) is inexpensive and earth-abundant, and nickel complexes usually exhibit distinct properties in small molecule activation chemistry and redox chemistry, such as functionalization of alkene and CO2.16 Moreover, nickel complexes have been rarely reported for direct coupling of CO2 with epoxides.17
Encouraged by this, metal complexes composed of nickel and N4 were synthesized, and their catalytic activity for the coupling of epoxides and CO2 to produce cyclic carbonates in the absence of co-catalysts have been evaluated (Scheme 1) and found to be efficient bifunctional catalysts without a co-catalyst. Moreover, a plausible mechanism for the coupling reaction was investigated by density functional theory (DFT).
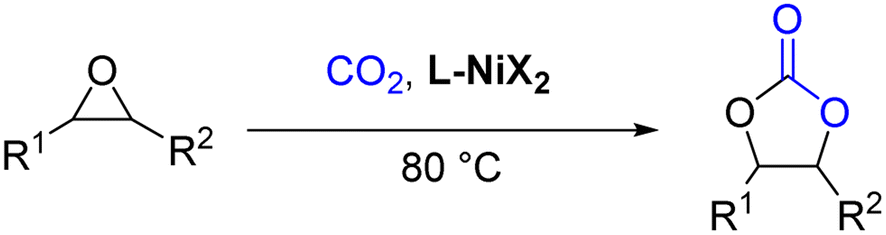 | ||
| Scheme 1 The cycloaddition reaction of CO2 and epoxides catalyzed by the Ni complex. | ||
2. Experimental
2.1 General information
Solvents and chemicals obtained from various companies, such as Aladdin and Macklin, were used without further purification unless stated otherwise. Analytical thin-layer chromatography (TLC) was carried out using 0.25 mm commercial silica gel plates. The purification of product was carried out by flash chromatography using silica gel (200–300 mesh). NMR spectra were performed at 298 K on a Bruker 400. Chemical shift values for 1H and 13C are reported as δ values (ppm) relative to the deuterated solvent and coupling constants (J) in Hz. Single-crystal X-ray diffraction measurements were carried out on a Bruker Smart APEX II CCD diffractometer at 100 (2) K using Cu Kα radiation (λ = 1.54184 Å). High-resolution mass spectroscopy experiments were carried out with a Bruker Daltonics microTOF-QII mass spectrometer. The gas chromatography (GC) was recorded by a Shimadzu GC-2010, SE-54 column using diphenyl as the internal standard. Infrared spectra were recorded with a Thermo Scientific Nicolet IS50-IR spectrometer at room temperature.2.2 Synthesis and characterization of nickel complexes
The ligands L1–L3 were prepared using previously reported methods.18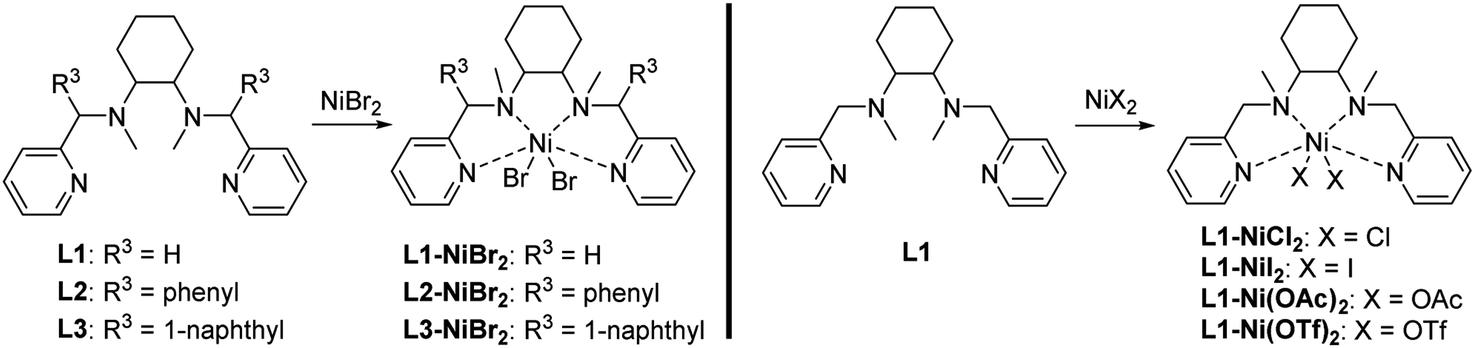 | ||
| Scheme 2 Synthesis of nickel complexes. | ||
2.2.1.1 L1-NiBr2 . Green solid (92%, yield), m. p. 112–114 °C, HRMS (ESI-MS) m/z: calcd for C20H28BrN4Ni [M–Br]+: 461.0851, found: 461.0845.
2.2.1.2 L2-NiBr2 . Green solid (90%, yield), m. p. 120–122 °C, HRMS (ESI-MS) m/z: calcd for C32H36BrN4Ni [M–Br]+: 613.1477, found: 613.1473.
2.2.1.3 L3-NiBr2 . Green solid (90%, yield), m. p. 130–132 °C, HRMS (ESI-MS) m/z: calcd for C40H40BrN4Ni [M–Br]+: 713.1790, found: 713.1786.
2.2.1.4 L1-NiCl2 . Green solid (93%, yield), m. p. 110–112 °C, HRMS (ESI-MS) m/z: calcd for C20H28ClN4Ni [M–Cl]+: 417.1356, found: 417.1350.
2.2.1.5 L1-NiI2 . Green solid (92%, yield), m. p. 110–112 °C, HRMS (ESI-MS) m/z: calcd for C20H28IN4Ni [M–I]+: 509.0712, found: 509.0707.
2.2.1.6 L1-Ni(OTf)2 . Blue solid (90%, yield), m. p. 60–62 °C, HRMS (ESI-MS) m/z: calcd for C21H28F3O3SN4Ni [M-OTf]+: 531.1188, found: 531.1182.
2.2.1.7 L1-Ni(OAc)2 . Blue solid (91%, yield), m. p. 60–62 °C, HRMS (ESI-MS) m/z: calcd for C22H31O2N4Ni [M–OAc]+: 441.1800, found: 441.1795.
2.3 General procedure for the synthesis of cyclic carbonates
Epoxides (1 mmol) and a specified amount of nickel complexes are added to a 25 mL high-pressure vessel along with magnetic particles in the presence of CH3CN (2 mL) or solvent-free. The internal gas is then replaced with CO2 three times. The reaction mixture is stirred at the desired temperature for the required time, and the remaining CO2 is slowly released after cooling the high-pressure vessel to room temperature. The conversion rate of epoxides and the yield of cyclic carbonates are determined by analyzing the crude product with gas chromatography when biphenyl is used as the internal standard. The calculation formula of conversion, yield, selectivity, and TON are given in the ESI.†3. Results and discussion
The L1-NiBr2 powder was dissolved in CH3CN solution and recrystallized at room temperature for a few days, and then a green crystal was obtained. Its structure was characterized by X-ray diffraction analysis, and the Oak Ridge thermal ellipsoid plot (ORTEP) diagram described in Fig. 1 shows that the ligand coordinated with nickel bromide using four N atoms in a cis-α topology. The data collection and structure refinement are shown in Table S1,† and selected bond lengths and bond angles are summarized in Table S2.†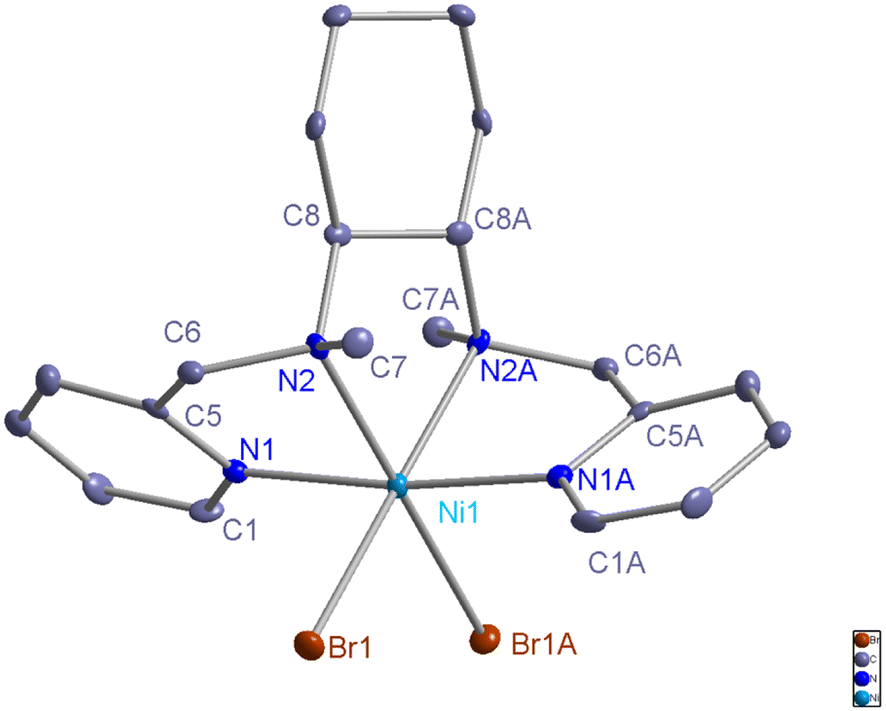 | ||
| Fig. 1 ORTEP diagram of the nickel complex L1-NiBr2 (CCDC: 2307070). The hydrogen atoms are omitted for clarity in the metal complex. | ||
3.1 Optimization of the reaction conditions
Originally, L1-NiBr2 was used to investigate the bifunctional catalytic performance for the coupling reaction of CO2 and styrene oxide (1a) in the presence of 2 mL CH3CN under the conditions of 2.5 MPa CO2 and 80 °C for 8 h. It could be seen that the conversion of 1a and the yield of styrene carbonate (2a) were 95% and 94% with the selectivity of 99% when 5 mol% of L1-NiBr2 was added alone and no co-catalyst was used, which indicated that L1-NiBr2 was an efficient bifunctional catalyst (Table 1, entry 1). Under the same conditions, the effects of common solvents such as CH3CN, DCE (dichloroethane), toluene, CH3OH, DMC (dimethyl carbonate), DMF (N,N-dimethylformamide), THF (tetrahydrofuran) and solvent-free for the catalytic performance were investigated (Tables S3† and 1, entry 2). The result showed that the catalytic system performed the highest conversion and yield with 100% selectivity in the absence of solvent (Table 1, entry 2). Consequently, the catalytic performance of the cycloaddition between CO2 and 1a could be optimized without any co-catalyst under solvent-free conditions in the next test.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | t (h) | P (MPa) | Conv.b (%) | Yieldb (%) | Sel.b (%) |
| a Reaction conditions: 1 mmol styrene oxide (1a), catalyzed by 5 mol% of catalyst at 80 °C with solvent-free. b Determined by gas chromatography using biphenyl as the internal standard. c In CH3CN (2 mL). d 1 mol% of L1-NiBr2. e 3 mol% of L1-NiBr2. f 7 mol% of L1-NiBr2. | ||||||
| 1c | L1-NiBr2 | 8 | 2.5 | 95 | 94 | 99 |
| 2 | L1-NiBr2 | 8 | 2.5 | 98 | 98 | 100 |
| 3 | L1-NiBr2 | 4 | 2.5 | 99 | 98 | 99 |
| 4 | L1-NiBr2 | 2 | 2.5 | 99 | 98 | 99 |
| 5 | L1-NiBr2 | 1 | 2.5 | 98 | 98 | 100 |
| 6 | L1-NiBr2 | 0.5 | 2.5 | 86 | 78 | 91 |
| 7 | L1-NiBr2 | 1 | 1.5 | 98 | 98 | 100 |
| 8 | L1-NiBr2 | 1 | 1.0 | 98 | 98 | 100 |
| 9 | L1-NiBr2 | 1 | 0.5 | 92 | 88 | 96 |
| 10 | L1-NiBr2 | 1 | 0.1 | 59 | 57 | 97 |
| 11d | L1-NiBr2 | 1 | 1.0 | 51 | 50 | 99 |
| 12e | L1-NiBr2 | 1 | 1.0 | 96 | 94 | 98 |
| 13f | L1-NiBr2 | 1 | 1.0 | 98 | 97 | 99 |
| 14 | L1-NiCl2 | 1 | 1.0 | 85 | 79 | 93 |
| 15 | L1-NiI2 | 1 | 1.0 | 98 | 96 | 98 |
| 16 | L1-Ni(OAc)2 | 1 | 1.0 | 31 | — | — |
| 17 | L1-Ni(OTf)2 | 1 | 1.0 | 30 | — | — |
| 18 | L2-NiBr2 | 1 | 1.0 | 95 | 95 | 100 |
| 19 | L3-NiBr2 | 1 | 1.0 | 97 | 96 | 99 |
| 20 | L1 | 1 | 1.0 | 8 | — | — |
| 21 | NiBr2 | 1 | 1.0 | 22 | — | — |
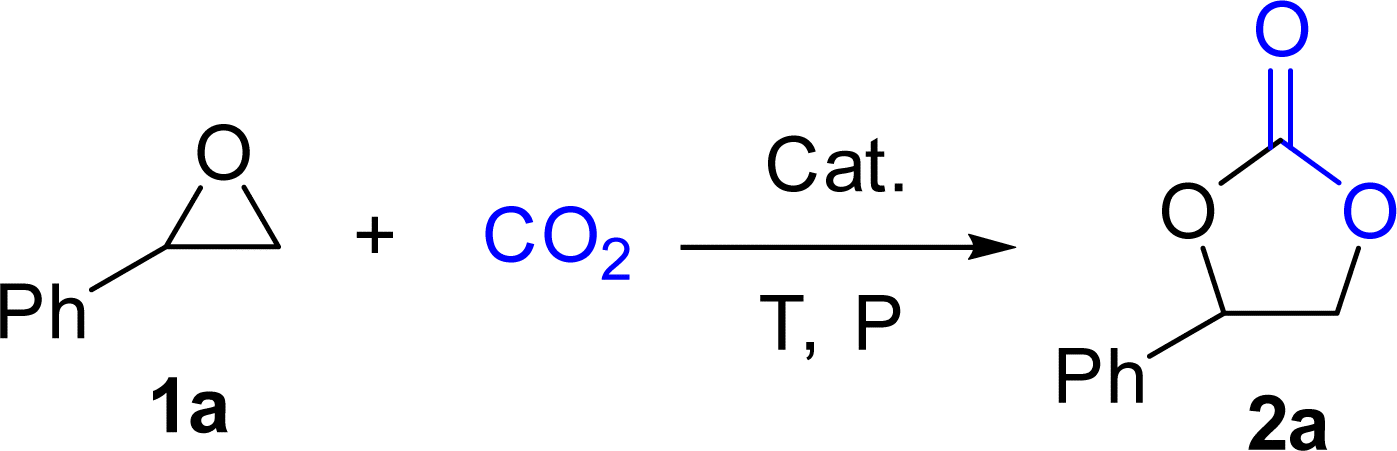
When the reaction temperature decreased from 80 °C to 60 °C, the conversion of 1a and the yield of 2a reduced to 59% and 56%, respectively (Table S4,† entries 1–3). At the same time, the selectivity of the coupling reaction decreased from 100% and 95%, respectively. Increasing the reaction temperature from 80 °C to 90 °C, the conversion and the yield would not decrease, presenting the stability of this catalytic system at higher temperatures (Table S4,† entries 3 and 4). The impact of reaction time on the catalytic performance was next studied with 5 mol% of L1-NiBr2 as the catalyst at 80 °C under 2.5 MPa CO2 pressure (Table 1, entries 3–6). Gradually decreasing the reaction time from 8 to 1 h, all of the reactions gave complete conversion from 1a to 2a. The conversion and selectivity would be turned to 86% and 91%, respectively, when the reaction time is reduced to 0.5 h. Therefore, 1 h is a more appropriate reaction time for the coupling reaction.
To further investigate more economical reaction conditions, the effect of CO2 pressure was examined, and good activity could be observed (98% conversion of 1a and 98% yield of 2a) under 1 MPa CO2 (Table 1, entries 7 and 8). However, the conversions of 1a were 59% and 92%, accompanied by 57% and 88% yields of 2a, when the CO2 pressures were 0.1 and 0.5 MPa, respectively (Table 1, entries 9 and 10). Despite the lower conversions of 1a to 2a, both presented a satisfactory selectivity (97% at a CO2 pressure of 0.1 MPa and 96% at a CO2 pressure of 0.5 MPa) (Table 1, entries 9 and 10). As described in Table 1 (entries 8 and 11–13), the amounts of catalyst were investigated under the conditions of 1 MPa CO2 and 80 °C for 1 h with L1-NiBr2 as the catalyst. It could be found that 5 mol% of L1-NiBr2 was enough to catalyze the coupling reactions of CO2 with epoxides (Table 1, entry 8).
Then, the activities of several nickel complexes were assessed. Compared to L1-NiBr2, L1-NiCl2 was a little sluggish, and 2a was isolated in 79% yield, owing to a lower nucleophilicity of Cl− (Table 1, entry 14). Although the nucleophilicity of I− is stronger than Br−, L1-NiBr2 showed similar transformation and yield, which may be attributed to the steric hindrance effect of I− (Table 1, entry 15).19 However, 2a was not tested, providing 31% or 30% conversion of 1a by employing L1-Ni(OAc)2 or L1-Ni(OTf)2 as the catalyst, due to the bulky steric hindrance and weak nucleophilicity of OAc− and OTf− (Table 1, entries 16 and 17).20 Besides, 5 mol% of L2-NiBr2 or L3-NiBr2 was added into this coupling reaction system to evaluate the effect of the ligand on the catalytic performance under the conditions of 1 MPa CO2 and 80 °C for 1 h, which also presented satisfactory catalytic ability due to their analogous scaffold (Table 1, entries 18 and 19). The activity of L-NiBr2 would not be affected by the steric hindrance and electronic property of the ligands. That is to say, anion, as a nucleophilic reagent to open the epoxide ring, played an important role in the reaction. Considering L1 as a much cheaper ligand than the others, L1-NiBr2 was the optimized catalyst for the cycloaddition reaction between epoxides and CO2. When ligand L1 (5 mol%) or NiBr2 (5 mol%) was added separately for the cyclization reaction, the conversion of 1a was just 8% or 22% with no 2a tested in the same conditions (Table 1, entries 20 and 21). The results indicated that there were synergistic catalytic effects between L1 and NiBr2. After all, one of the optimized conditions is 1 MPa CO2 pressure and 80 °C for 1 h catalyzed by 5 mol% L1-NiBr2 under solvent-free.
In order to investigate milder and more commercial reaction conditions, the pressure of CO2 and the amount of catalyst have been explored. Based on the above-investigated conditions (Table 1, entry 8), the conversion of 1a was 95%, and the selectivity was about 100% after stirring at 80 °C for 8 h under 1 atm CO2 (Table 2, entry 1). Consequently, 92% conversion and 92% yield were still obtained when the amount of catalyst decreased to 1 mol% (Table 2, entry 2). With the extension of reaction time, there was no obvious assistance to improve the yield of 2a (Table 2, entry 3). In contrast, the conversion of 1a/yield of 2a could be increased from 65%/64% to 92%/91% accompanied by prolonging the reaction time from 8 h to 14 h when the amount of catalyst was 0.5 mol% (Table 2, entries 4 and 5). However, the conversion of 1a and the yield of 2a were still less than 80%, even at 80 °C for 24 h at 1 atm CO2 pressure, when the amount of catalyst reduced to 0.2 mol% (Table 2, entries 6 and 7). In conclusion, the investigated milder reaction conditions were 1 atm CO2 and 80 °C for 14 h with 0.5 mol% L1-NiBr2 as the catalyst under solvent-free conditions. It was worth noting that the catalytic system also exhibited great catalytic activity, providing 88% conversion and 85% yield by extending the amount of styrene oxide (1a) to 10 mmol (1.20 g) under the optimized conditions (Table 2, entry 8).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Amount of catalyst (mol%) | t (h) | Conv.b (%) | Yieldb (%) | Sel.b (%) | TON |
| a Reaction conditions: 1a (1 mmol), 80 °C, 1 atm CO2, solvent-free. b Gas chromatography is used to determine the conversion rate and yield of cyclic carbonates, with biphenyl as the internal standard. c 10 mmol styrene oxide (1a). | ||||||
| 1 | 5 | 8 | 95 | 95 | 100 | 19 |
| 2 | 1 | 8 | 92 | 92 | 100 | 92 |
| 3 | 1 | 12 | 96 | 94 | 98 | 94 |
| 4 | 0.5 | 8 | 65 | 64 | 99 | 128 |
| 5 | 0.5 | 14 | 92 | 91 | 99 | 182 |
| 6 | 0.2 | 12 | 46 | 46 | 100 | 230 |
| 7 | 0.2 | 24 | 76 | 74 | 98 | 370 |
| 8c | 0.5 | 14 | 88 | 85 | 97 | 170 |
The stability of catalyst L1-NiBr2 was investigated by recycling experiments using 1a (1 mmol) as the model substrate under 1 atm CO2 pressure at 80 °C for 14 h with no solvent added. After the first 14 h period, the yield of 2a was determined by gas spectroscopy, and the reaction mixture was then washed with anhydrous diethyl ether (5.0 mL) to remove 2a and 1a and dried. Subsequently, the system was directly added to another 1 mmol 1a to complete the next circulation. As listed in Fig. 2, this procedure could be repeated five times with almost no loss of its catalytic activity (the yields of 2a were 92%, 92%, 90%, 91%, and 91%, respectively). The ICP test recorded a 0.76% catalyst leaching in every recycling. The recycling experiments further proved that the catalytic system of L1-NiBr2 is stable and efficient for the coupling of CO2 and epoxides under mild conditions. It is worth noting that the stability of L1-NiBr2 could also be verified by the TGA (thermogravimetric analysis) and IR spectra, which exhibited almost no changes between before and after the cyclic experiment (Fig. S1 and S2†).
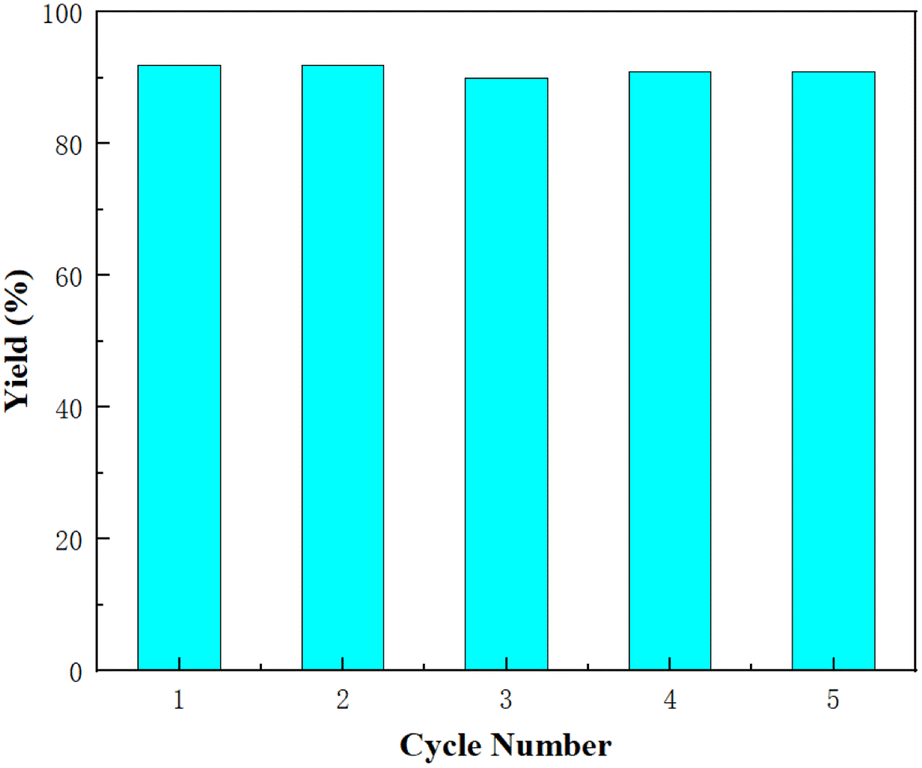 | ||
| Fig. 2 Recyclability of catalyst L1-NiBr2 for the coupling of CO2 and 1a (1 mmol). | ||
3.2 Extend the epoxide substrates
In addition, to investigate the potential and general applicability of the bifunctional catalyst L1-NiBr2, the coupling reactions between CO2 and a variety of epoxides were investigated under the above two optimal reaction conditions. It should be noted that L1-NiBr2 exhibited a good catalytic performance with more than 90% of yield for the transformation of substituted epoxides 1a–1f bearing various aliphatic and aromatic groups, such as phenyl, methyl, chloromethyl, alkenyl and ether groups under both optimal conditions (Table 3, entries 1–6). However, when the substituted group was a long aliphatic chain, the yields of 2g decreased to 42% and 35% under the two optimal conditions due to its weak dissolvability for the catalyst (Table 3, entry 7). Fortunately, under the same conditions, increasing the reaction time from 1 h to 6 h, the yield of 2g would be increased from 42% to 95%. However, the internal epoxide 1h gave the lowest yield under different reaction conditions due to its steric hindrance (Table 3, entry 8).|
|
||||
|---|---|---|---|---|
| Entry | Epoxide | Product | Yielda,b (%) | Yielda,c (%) |
| a Isolated yield. b Reaction conditions: 1 mmol epoxide, 1 MPa CO2, 80 °C, 1 h, 5 mol% (relative to the epoxide) L1-NiBr2, solvent-free. c Reaction conditions: 1 mmol epoxide, 1 atm CO2, 80 °C, 14 h, 0.5 mol% (relative to the epoxide) L1-NiBr2, solvent-free. d 6 h. | ||||
| 1 |
|
|
98 | 91 |
| 2 |
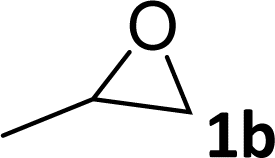
|
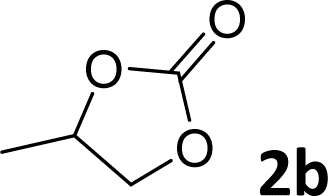
|
95 | 92 |
| 3 |
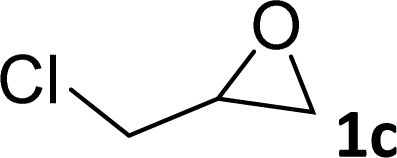
|
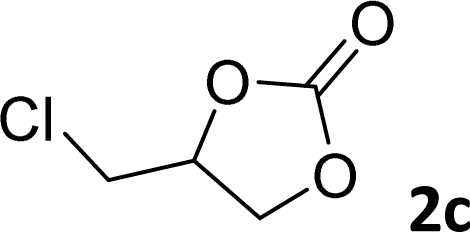
|
97 | 90 |
| 4 |
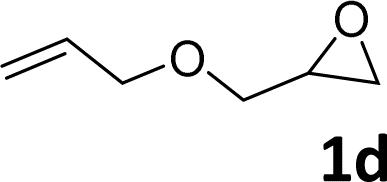
|
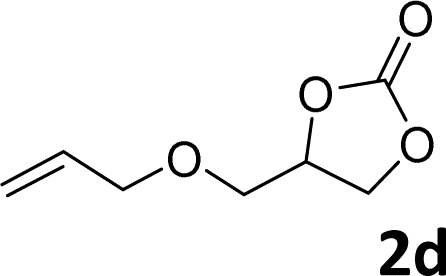
|
91 | 95 |
| 5 |
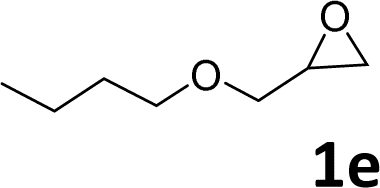
|
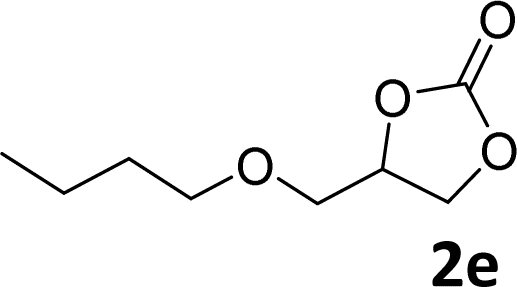
|
92 | 95 |
| 6 |
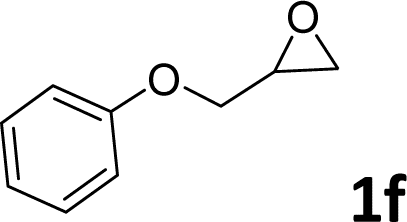
|
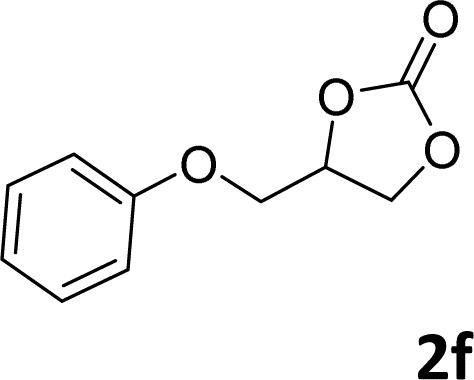
|
92 | 95 |
| 7 |
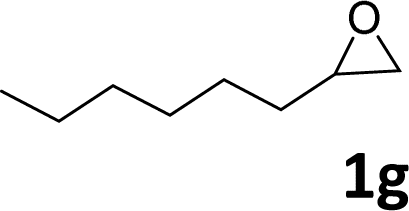
|
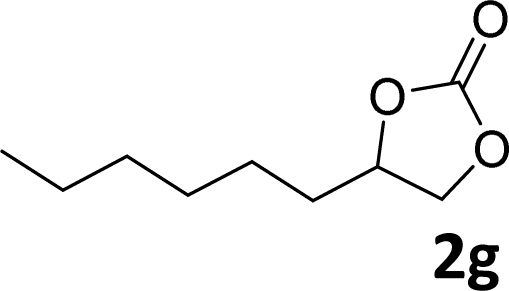
|
42(95d) | 35 |
| 8 |
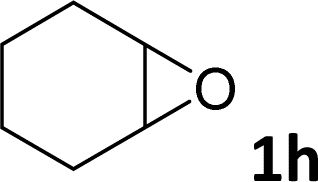
|
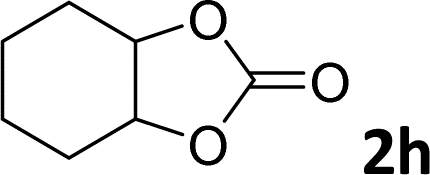
|
10 | 6 |
3.3 Unravelling the reaction mechanism with DFT calculations
The catalytic mechanism of the coupling reaction of epoxide and CO2 was explored with DFT calculations. Initiation of the catalytic cycle involves a ligand substitution process in which one Br− leaves the Ni coordination sphere in exchange for an epoxide. Then, the Br− rebinds to form IM3 before a nucleophilic attack occurs on the epoxide ring, yielding IM4 (Fig. 3). A 4.9 kcal mol−1 Gibbs free energy barrier was estimated for this ring-opening process. Next, the addition of CO2 occurs, in which the C atom of CO2 weakly binds the O atom originally from the epoxide and currently coordinating to Ni, giving IM5 (Fig. 4). The insertion of CO2 into the Ni-coordinated bromoethanolate is computed to have a 4.4 kcal mol−1 Gibbs free energy barrier. The final step is the ring closure of the bromoethyl carbonate ligand in IM6, which is calculated to be the rate-determining step with a Gibbs free energy barrier of 18.7 kcal mol−1 (Fig. 4). Releasing the ethyl carbonate molecule closes the catalytic cycle with a net ΔH of −21.5 kcal mol−1. A summary of the proposed reaction mechanism, as per previous reports, is shown in Scheme 3.21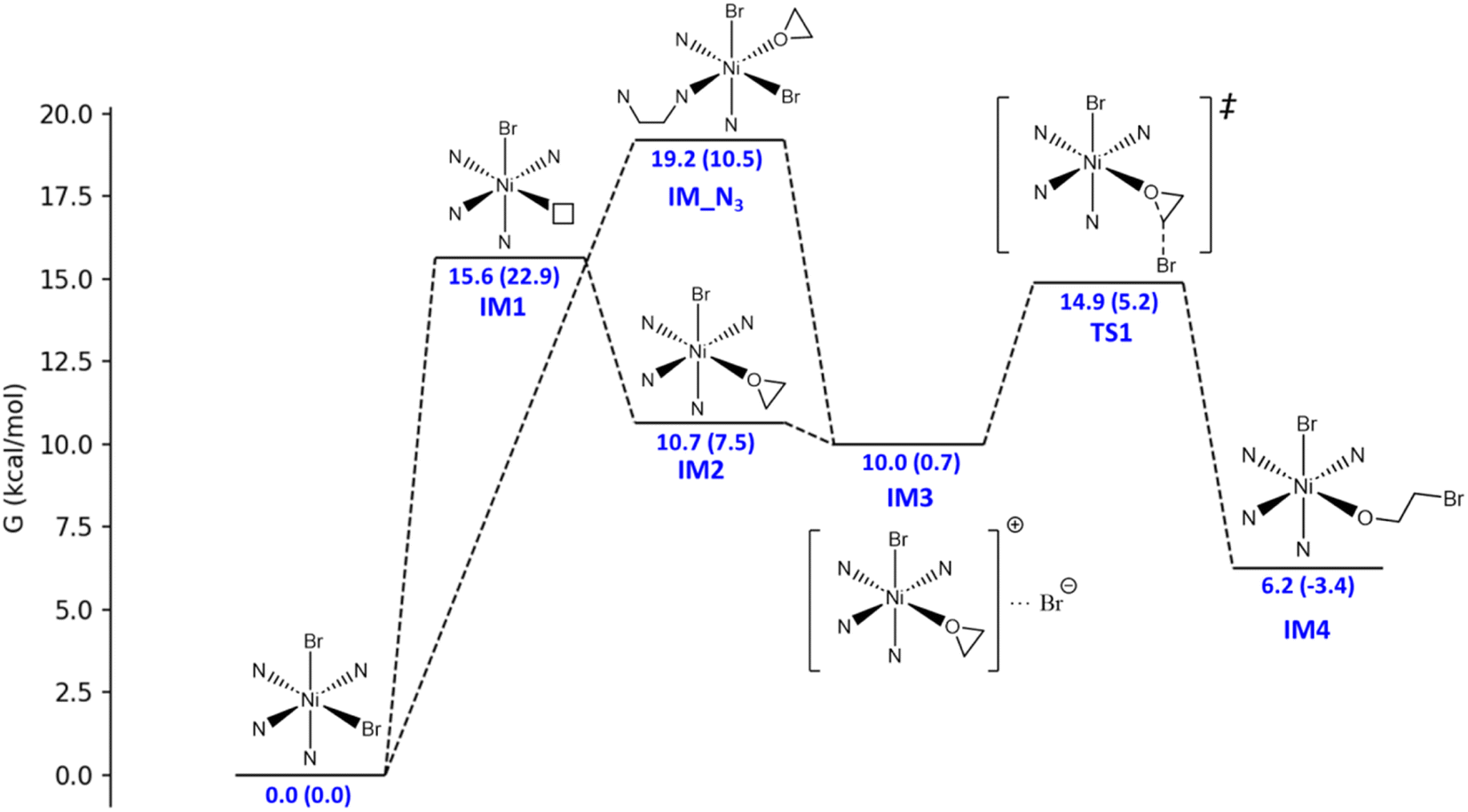 | ||
| Fig. 3 Energy profile for the binding of the L1-NiBr2 catalyst with the ethylene oxide substrate, followed by the nucleophilic ring opening process. The relative Gibbs free energies and the relative electronic energies (in parentheses) in kcal mol−1 are labelled. | ||
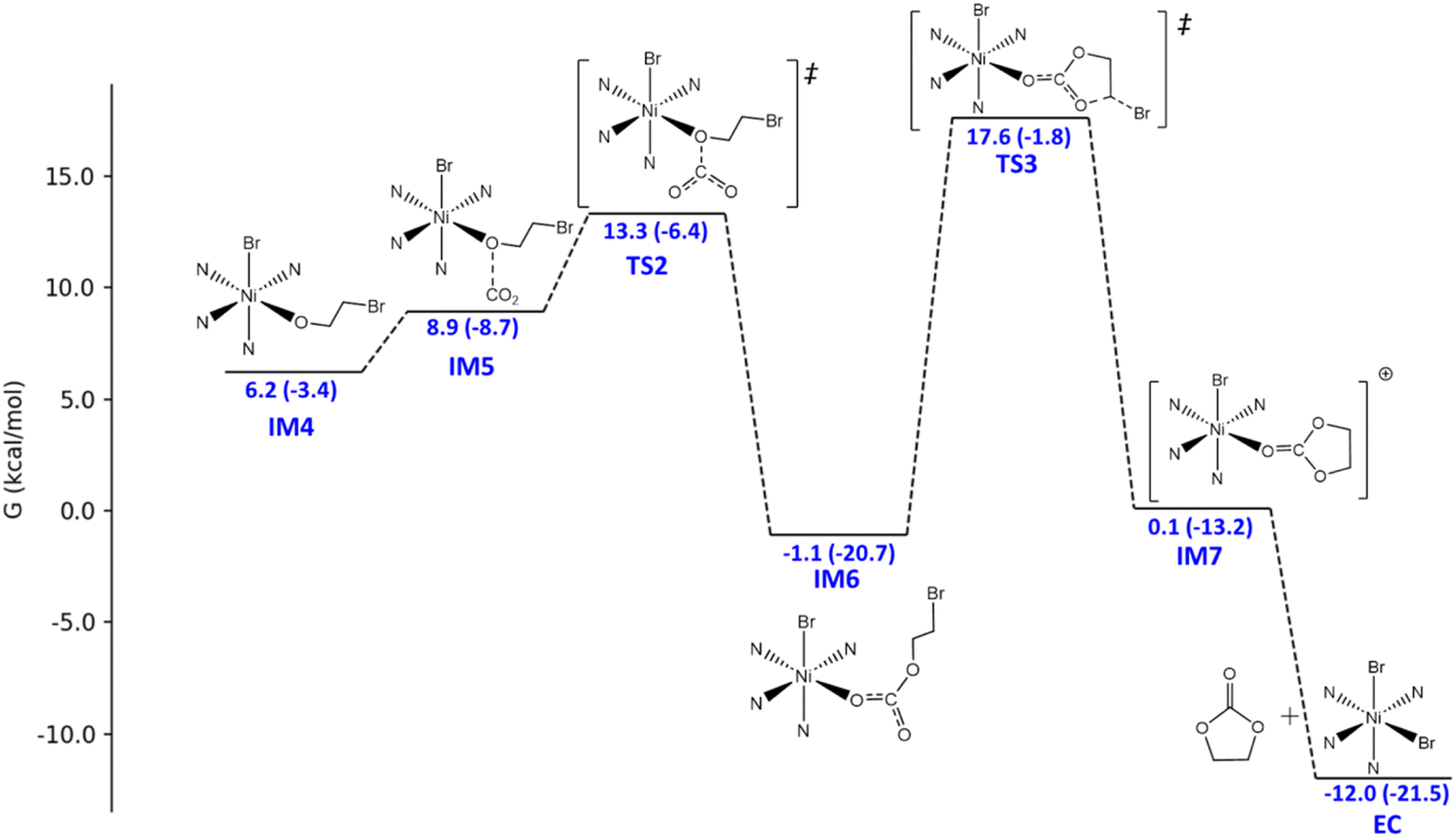 | ||
| Fig. 4 Energy profile for the conversion from the CoN4BrOCH2CH2Br intermediate (IM4) to the final product, ethylene carbonate (EC). The original L1-NiBr2 catalyst was selected to be the zero-energy scale to stay consistent with Fig. 3. The relative Gibbs free energies and the relative electronic energies (in parentheses) in kcal mol−1 are labelled. | ||
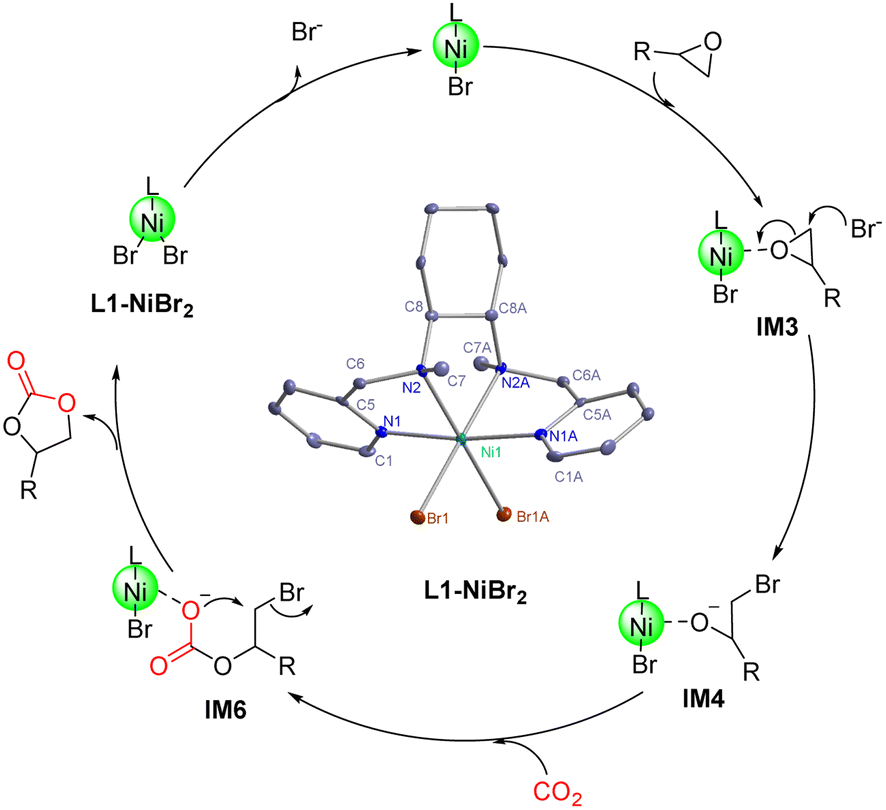 | ||
| Scheme 3 A proposed mechanism for the cycloaddition reaction catalyzed by L1-NiBr2. | ||
4. Conclusion
In summary, novel bifunctional single-component nickel catalysts with tetradentate aminopyridine ligands have been developed for cycloaddition reactions of CO2 and epoxides. The results indicated that L1-NiBr2, a promising candidate for commercial applications, showed excellent catalytic activity and substrate compatibility under co-catalyst-free and solvent-free conditions. As compared to the disubstituted one, most of the cyclic carbonates achieved high yields (>90%) and selectivity even at 80 °C under 1 atm CO2 and 0.5 mol% of the catalyst, except the long aliphatic chain substituted one, suggesting that the large epoxide substrates restricted diffusion into the confined spaces of the catalyst. Besides, the developed catalyst has good recyclability and stability, which were further proved through ICP to test the catalyst leaching, TGA and IR spectra. Furthermore, mechanistic investigations followed by DFT suggested that Ni acted as a Lewis acid and its anion Br− acted as a nucleophile to facilitate the ring opening.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We acknowledge the financial support of this work from the National Natural Science Foundation of China (22272094 and 21972079), Natural Science Foundation of Shandong Province (ZR2020QB034, ZR2019MB002 and ZR2023QB285) and Incubation Program of Youth Innovation in Shandong Province. Computer time was provided by the TAMU High-Performance Research Computing Facility, and software was provided by the Laboratory for Molecular Simulation.Notes and references
- (a) J. Rogelj, D. Huppmann, V. Krey, K. Riahi, L. Clarke, M. Gidden, Z. Nicholls and M. Meinshausen, Nature, 2019, 573, 357–363 CrossRef CAS PubMed; (b) A. Modak, P. Bhanja, S. Dutta, B. Chowdhury and A. Bhaumik, Green Chem., 2020, 22, 4002–4033 RSC; (c) M. Thangamuthu, Q. Ruan, P. O. Ohemeng, B. Luo, D. Jing, R. Godin and J. Tang, Chem. Rev., 2022, 122, 11778–11829 CrossRef CAS PubMed.
- (a) F. Han, F. F. Xie, M. Y. Yin, L. H. Jing and P. Han, Org. Biomol. Chem., 2024, 22, 5724–5728 RSC; (b) M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2022, 134, e202112835 CrossRef; (c) Z. Q. Wang, C. H. Deng, X. Liu and W. M. Wang, Dalton Trans., 2023, 52, 11163–11167 RSC.
- (a) A. Goeppert, M. Czaun, J.-P. Jones, G. K. S. Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC; (b) Z. C. Zhang, A. Q. Guan, J. Y. Yu, X. D. Jiang, S. Han, Z. Y. Wen, B. Y. Du and B. Y. Song, New J. Chem., 2024, 48, 13245–13250 RSC; (c) Q. Zou, G. Long, T. Zhao and X. Hu, Green Chem., 2020, 22, 1134–1138 RSC.
- (a) Z. C. Yang, J. L Ni, C. Y. Duan, Y. Feng and J. F. Yao, J. Mater. Chem., 2024, 12, 21279–21287 RSC; (b) Y. Yao, H. Zhang, C. Lu, H. Shang and Y. Tian, Eur. J. Org Chem., 2023, 26, e202300111 CrossRef CAS.
- (a) B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed; (b) W. Yu, E. Maynard, V. Chiaradia, M. C. Arno and A. P. Dove, Chem. Rev., 2021, 121, 10865–10907 CrossRef CAS PubMed; (c) A. Centeno-Pedrazo, J. Perez-Arce, Z. Freixa, P. Ortiz and E. J. Garcia-Suarez, Ind. Eng. Chem. Res., 2023, 62, 3428–3443 CrossRef CAS.
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 RSC.
- (a) Y. A. Alassmy, Z. A. Pour and P. P. Pescarmona, ACS Sustainable Chem. Eng., 2020, 8, 7993–8003 CrossRef CAS; (b) R. Jin, H. Xu, J. Easa, A. Chapero-Planell and C. P. O'Brien, J. Phys. Chem., 2023, 127, 1441–1454 CAS.
- (a) B.-H. Xu, J.-Q. Wang, J. Sun, Y. Huang, J.-P. Zhang, X.-P. Zhang and S.-J. Zhang, Green Chem., 2015, 17, 108–122 RSC; (b) N. H. Kim, E. Y. Seong, J. H. Kim, S. H. Lee, K.-H. Ahn and E. J. Kang, J. CO2 Util., 2019, 34, 516–521 CrossRef CAS; (c) T. Chuasaard, M. Sinchow, N. Chiangraeng, P. Nimmanpipug and A. Rujiwatra, J. CO2 Util., 2024, 80, 102686 CrossRef CAS; (d) A. Rehman, F. Saleem, F. Javed, A. Ikhlaq, S. W. Ahmad and A. Harvey, J. Environ. Chem. Eng., 2021, 9, 105113–105140 CrossRef CAS; (e) M. Navarro, A. Garcés, L. F. Sánchez-Barba, D. González-Lizana and A. Lara-Sánchez, Dalton Trans., 2023, 52, 6105–6116 RSC.
- (a) E. M. Maya, M. González-Lucas and M. Iglesias, ChemistrySelect, 2017, 2, 9516–9522 CrossRef CAS; (b) Z. Gao, L. Liang, X. Zhang, P. Xu and J. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 61334–61345 CrossRef CAS PubMed.
- (a) Y. Liu, S. Wang, Z. Dai and Y. Xiong, Ind. Eng. Chem. Res., 2021, 60, 18218–18229 CrossRef CAS; (b) Y. L. Hu, X. B. Liu and Q. Rong, Green Chem. Lett. Rev., 2023, 16, 2163192 CrossRef; (c) K. Guo, N. Ji, F. han, Q. Yang, N. Wang and C. Miao, RSC Sustainability, 2024, 2, 1074–1080 RSC.
- (a) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS; (b) C. K. Karan and M. Bhattacharjee, Inorg. Chem., 2018, 57, 4649–4656 CrossRef CAS PubMed; (c) J. H. Kim, S. H. Lee, N. H. Kim and E. J. Kang, J. CO2 Util., 2021, 50, 101595–101601 CrossRef CAS; (d) M. Dolai, S. Biswas, E. Moreno-Pineda, W. Wernsdorfer, M. Ali, R. A. Alshgari, S. M. Wabaidur and A. Ghosh, Cryst. Growth Des., 2023, 23, 801–810 CrossRef CAS.
- M. H. Chisholm and Z. P. Zhou, J. Am. Chem. Soc., 2004, 126, 11030–11039 CrossRef CAS PubMed.
- (a) H. M. Hüppe, L. I. Mühlhaus, J. Heck, M. Eilers, H. Gildenast, S. Schönfeld, A. Dürrmann, A. Hoffmann, B. Weber, U. P. Apfel and S. H. Pawlis, Inorg. Chem., 2023, 62, 4435–4455 CrossRef; (b) M. Kourgiantaki, G. G. Bagkavou, H. I. Stathakis and A. L. Zografos, Org. Chem. Front., 2023, 10, 2095–2114 RSC; (c) B. Wang, X. Cao, L. Wang, X. Meng, Y. Wang and W. Sun, Inorg. Chem., 2024, 63, 9156–9163 CrossRef CAS.
- (a) B. Wang, L. Wang, J. Lin, C. Xia and W. Sun, ACS Catal., 2023, 13, 10386–10393 CrossRef CAS; (b) J. E. Dengler, M. W. Lehenmeier, S. Klaus, C. E. Anderson, E. Herdtweck and B. Rieger, Eur. J. Inorg. Chem., 2011, 3, 336–343 CrossRef.
- (a) M. Q. Xing, N. Liu, C. N. Dai and B. H Chen, Catalysts, 2024, 14, 370 CrossRef CAS; (b) W. Moschkowitsch, S. Gonen, K. Dhaka, N. Zion, H. Honig, Y. Tsur, M. C. Toroker and L. Elbaz, Nanoscale, 2021, 13, 4576–4584 RSC.
- (a) I. Zilbermann, E. Maimon, H. Cohen and D. Meyerstein, Chem. Rev., 2005, 105, 2609–2625 CrossRef CAS; (b) J. Diccianni, Q. Lin and T. N. Diao, Acc. Chem. Res., 2020, 53, 906–919 CrossRef CAS PubMed; (c) S. K. Kariofillis and A. G. Doyle, Acc. Chem. Res., 2021, 54, 988–1000 CrossRef CAS; (d) B. H. Teng, Z. P. Bao, Y. Y. Zhao and X. F. Wu, Org. Lett., 2024, 26, 4779–4783 CrossRef CAS.
- (a) K. Takaishi, B. D. Nath, Y. Yamada, H. Kosugi and T. Ema, Angew. Chem., Int. Ed., 2019, 58, 9984–9988 CrossRef CAS PubMed; (b) T. Nouri and M. Khorasani, J. Phys. Chem., 2024, 128, 166–176 CAS.
- (a) M. Wu, B. Wang, S. Wang, C. Xia and W. Sun, Org. Lett., 2009, 11, 3622–3625 CrossRef CAS PubMed; (b) S. Yu, C. Miao, D. Wang, S. Wang, C. Xia and W. Sun, J. Mol. Catal. A: Chem., 2012, 353–354, 185–191 CrossRef CAS.
- Y. A. Rulev, V. A. Larionov, A. V. Lokutova, M. A. Moskalenko, O. L. Lependina, V. I. Maleev, M. North and Y. N. Belokon, ChemSusChem, 2016, 9, 216–222 CrossRef CAS PubMed.
- Z. Alaji, E. Safaei and A. Wojtczak, New J. Chem., 2017, 41, 10121–10131 RSC.
- (a) F. Chen, Q. Zhang, D. Wei, Q. Bu, B. Dai and N. Liu, J. Org. Chem., 2019, 84, 11407–11416 CrossRef CAS PubMed; (b) R. Das and C. M. Nagaraja, Inorg. Chem., 2020, 59, 9765–9773 CrossRef CAS; (c) S. Saltarini, N. V. Escobar, J. Martinez, C. G. Daniliuc, R. A. Matute, L. H. Gade and R. S. Rojas, Inorg. Chem., 2021, 60, 1172–1182 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystal information of L1-NiBr2, part screening reaction conditions, NMR data and spectra of ligands and products and computational methods. CCDC 2307070. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4su00556b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |