
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Task-specific boronium ionic liquids as ashless lubricant additives†
Novina
Malviya
a,
Farah Fazlina M.
Yasin
ab,
Maria Teresa
Sateriale
c,
Fergal
Coleman
a,
H. Q. Nimal
Gunaratne
 a,
Andrea
Dolfi
c,
Geetha
Srinivasan
b and
Małgorzata
Swadźba-Kwaśny
*a
a,
Andrea
Dolfi
c,
Geetha
Srinivasan
b and
Małgorzata
Swadźba-Kwaśny
*a
aThe QUILL Research Centre, School of Chemistry and Chemical Engineering, Queen's University Belfast, UK. E-mail: m.swadzba-kwasny@qub.ac.uk
bPETRONAS Research SDN BHD (PRSB), Malaysia
cPETRONAS Global Research & Technology Centre, Santena, Turin, Italy
First published on 12th September 2024
Abstract
Modern engines are designed for very close contact between shearing planes, which requires high-performance boundary lubrication, delivered by lubricant base oils formulated with an array of additives. Commercial additive packages typically contain metals, sulfur, and phosphorus, which act as poisons to catalytic converters (thereby increasing emissions), increase wear and contribute to corrosion (which lowers the lifespan of engines). Ionic liquids (ILs), which are low-melting organic salts, have been extensively studied as lubricant additives; although some commercially available ionic liquids perform well as friction modifiers, they suffer from low solubility in the oil matrix and may cause corrosion due to residual chloride content. Here, we report nine new, task-specific ionic liquids, designed to act as ashless lubricant additives, comprising boron-containing cations for enhanced wear reduction, carboxylic acid anions to reduce friction, and modified alkyl chains to enhance solubility in the base oil. All ILs were inherently free from metals, sulfur, and phosphorus, and synthesised through a halide-free route. Their speciation was studied through multinuclear NMR and Raman spectroscopies, followed by studies of solubility in Group III+ base oil. Their performance as lubricant additives was assessed in terms of friction reduction and wear scar reduction, benchmarked against glycerol mono-oleate (GMO), a commercially availabe lubricant additive.
Sustainability spotlightAbout 23% of world's energy consumption originates from tribological contacts: 20% is required to overcome friction and 3% is used to refabricate worn parts and address wear-related failures (Friction, 2017, 5, 263). This accounts for 119 exajoules, generated from 2287 million tonnes of oil equivalent, generating 7000 metric tons of CO2 equivalent. Effective tribological solutions may potentially reduce global, annual CO2 emissions by 1460 MtCO2 short term, and 3140 MtCO2 long term (over 15 years), directly addressing SDG 13 – Climate Action. Whilst improvements of materials, surfaces and designs are effective, advanced customised lubricant formulations are amongst the most cost-effective approaches for decreasing frictional loss and prolonging the service life of machines (Sci. China Technol. Sci., 2013, 56, 2888). |
Introduction
Modern automotive oils are complex formulations of mineral and/or synthetic oils fortified with additives: antioxidants, antiwear agents, corrosion inhibitors, detergents, dispersants and friction modifiers.1 Lubricant additives make up 10 to 30% of the oil formulation and are particularly important in high-performance lubricant oils, designed to work in a boundary lubrication regime (in which two moving surfaces are very close and the main lubricant action is taken over by a tribofilm formed on the surface of moving elements).2,3 Antiwear and friction-reducing additives must have an appropriate chemical structure to be soluble in the base oil, but attracted to the metal surface, as well as a suitable elemental composition, to form an efficient tribofilm under shear.4,5Commercial antiwear and friction-modifying additives contain sulphur, phosphorus and heavy metals, exemplified by molybdenum dialkyl dithiophosphate (MoDTP) and zinc dialkyl dithiophosphate (ZDDP).6,7 As these additives degrade under tribological conditions, compounds of sulfur, phosphorus and metals get carried over to catalytic converters, where they act as catalyst poisons. This, in turn, results in increased emissions of toxic exhaust gases such as carbon monoxide (CO), sulphur oxides (SOx), nitrogen oxides (NOx), as well as ash particles.8,9 This drives the search for sulphur, phosphorus and metal-free lubricant additives, leaving only a few triboactive elements (oxygen, nitrogen, boron) to choose from as building blocks of organic friction modifiers: esters and epoxidised esters, amines, amides, and imides, and finally, borate esters and their derivatives.
Ionic liquids (ILs), organic salts that are liquid under ambient conditions, have been extensively studied in the context of lubricant applications as lubricants,10 and more recently as lubricant additives.11 Their advantages as lubricant additives are that they are polar ions promoting surface adsorption, and have extremely low volatility and therefore nonflammability, high thermal stability, and robust rheological behaviour.12 The main challenge is their miscibility with nonpolar base oils, but since 2012, reasonably oil-soluble (up to several per cent by weight) and triboactive ionic liquids have been reported.13–16 The vast majority of ILs tested as lubricant additives were based on commercially available cations (ammonium, phosphonium and dialkylimidazolium), combined with lipophilic anions, either common in ionic liquids (alkyl sulfonates, alkyl sulphates) or borrowed from the surfactant industry, e.g. bis(2,4,4-trimethyl pentyl) phosphinate (BTMPP), di(2-ethylhexyl) phosphate (DEHP) and dioctyl sulfosuccinate (DOSS) (Fig. S1†).11
In this work, we have taken a bottom-up “designer liquid” approach17 to synthesise task-specific, lipophilic and triboactive ionic liquids, in which both the cation and anion have a tribological function, ideally addressing simultaneously two tasks: wear reduction and friction reduction. Furthermore, they contain only benign main group elements: C, H, O, N and B (Fig. 1).
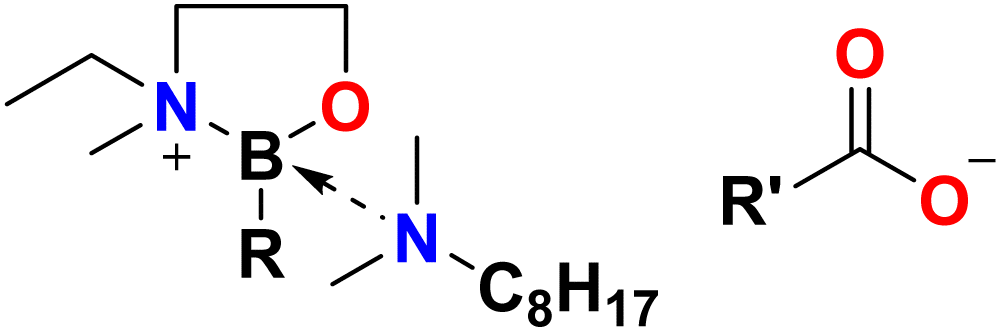 | ||
| Fig. 1 The general formula of task-specific triboactive ionic liquids (R,R′ – alkyl chains). | ||
Nine new ILs have been synthesised and characterised in terms of cationic speciation, thermal stability and solubility in Group III+ base oil. Finally, their performance as friction modifying additives and wear reducing additives has been benchmarked against glycerol mono-oleate (GMO), a commercial organic friction modifying additive.
Experimental
Materials and methods
n-Butylboronic acid, sec-butylboronic acid, 2-methylpropylboronic acid, iso-butylboronic acid, 2-(methylamino)ethanol (C2mea), ethyl methane sulfonate, N,N-dimethyl octylamine (N8111) and glycerol mono-oleate (GMO) were purchased from Sigma-Aldrich or Fluorochem, and used as received. All solvents were of analytical HPLC grade, dried by storage over activated 3 Å molecular sieves under a nitrogen atmosphere and kept stored in a glovebox. Sodium salts of 2-ethyl hexanoate, Na[EH], methacrylate, Na[ME] and oleate, Na[Ol], were purchased from Sigma Aldrich and used as received. Group III+ base oil (64742-54-7) was supplied by PETRONAS. Cobalt internal standard (5000 ppm in oil), organometallate standard (50 ppm in oil) and Premisolv as solvent for ICP analysis were purchased from Conostan.Samples for NMR spectroscopy of boronium ILs were prepared inside a glovebox (MBraun LabMaster dp, <0.3 ppm O2 and H2O, argon atmosphere). Neat ILs were loaded into NMR tubes (5 mm, borosilicate glass) containing sealed capillaries with acetonitrile-d3 (external lock). The tubes were closed with a standard cap, sealed with parafilm and taken out from the glovebox immediately before measurement.
Raman spectra were recorded using a Thermo Scientific DXR3 SmartRaman Spectrometer (Thermofisher Scientific) using a 785 nm laser. Data were collected in the range of 50–3300 cm−1 at an estimated resolution of 5.5–8.3 cm−1.
Differential scanning calorimetry (DSC) measurements were carried out using a TA Instruments Q2000 DSC with an RCS90 cooling system. All samples were prepared in the glovebox using Tzero aluminium pans and hermetic lids, with a typical sample mass of 10 mg. All scans were recorded at 2 °C min−1, between −90 and up to 110 °C (accounting for thermal stability limits), in triplicate. Isothermal steps (5 min) were introduced between each heating and cooling scan, allowing for equilibration. Melting points were read from peak maxima.
Synthesis
[B(C2mea)(n-Bu)(N811)][OMs]
A neutral boron precursor B(C2mea)(n-Bu) (39 mmol, 5.5 g) was mixed with ethyl methane sulfonate (39 mmol, 4.9 g). The reaction mixture was stirred for 10 min at room temperature under an inert atmosphere (glovebox). N,N-Dimethyl octylamine (39 mmol, 6.14 g) was added to the reaction mixture. The resultant solution was stirred overnight under an inert atmosphere (glovebox). A dark yellow colour liquid of [B(C2mea)(n-Bu)(N811)][OMs] is obtained, which crystallised when stored overnight in a fridge (5 °C) under an Ar atmosphere. 1H NMR (600 MHz, MeOH-d4) δ 3.71 (2H,t), 2.89 (2H,t), 2.54 (3H,s), 1.83 (1H,m), 0.91 (6H,m), 0.69 (2H,m); 1H NMR (600 MHz, MeOH-d4) δ 3.33 (4H,m), 2.90 (6H,s), 2.31 (6H,m), 1.45 (3H,m), 1.02 (19H,m), 0.57 (7H,m), 0.36 (2H,m); 13C NMR (600 MHz, MeOH-d4) δ 62.41, 58.23, 49.15, 42.86, 39.22, 31.24, 28.62, 26.47, 25.98, 25.02, 21.92, 13.34, 7.69; 11B NMR (193 MHz, MeOH-d4) δ 3.38, 0.36 (tetracoordinate boron)IL1 – [B(C2mea)(n-Bu)(N811)][EH]
[B(C2mea)(n-Bu)(N811)][OMs] (7.2 mmol, 3.042 g) was mixed with sodium 2-ethyl hexanoate, Na[EH] (7.2 mmol, 1.2085 g). The resultant reaction mixture was stirred overnight under an inert atmosphere (glovebox). The reaction mixture was then dissolved in 10 ml of dry acetonitrile which resulted in precipitation of sodium methanesulfonate, Na[OMs], which was filtered out. Subsequently, to promote further Na[OMs] precipitation, the acetonitrile solution of the product was shaken 3 times with 5 ml of dry hexane, followed by filtration and decantation of hexane after each step. The acetonitrile was removed under vacuum giving a light-yellow clear liquid. Yield: 3.1 g, 91.5%. 1H NMR (600 MHz, acetonitrile-d3) δ 3.30 (2H,dd), 3.16 (2H,dd), 2.95 (6H,s), 2.48 (3H,m), 1.74 (2H,m), 1.47 (2H,m), 1.28 (24H,m), 0.85 (16H,m), 0.56 (2H,t); 13C NMR (600 MHz, acetonitrile-d3) δ 182.99, 64.93, 60.91, 51.46, 51.32, 51.29, 40.36, 34.11, 32.86, 31.48, 30.14, 28.29, 27.31, 26.22, 24.15, 23.75, 14.91, 14.81, 13.26, 8.95, 8.72; 11B NMR (193 MHz, MeOH-d4) δ 33.41, 4.92, 0.54, −21.66 ppm; MS (TOF) of [B(C2mea)(n-Bu)(N811)][EH]: m/z = 327.3550 (in positive mode), 143.1072 (in negative mode).IL2 – [B(C2mea)(n-Bu)(N811)][MA]
[B(C2mea)(n-Bu)(N811)][OMs] (4.73 mmol, 2 g) was mixed with sodium methacrylate, Na[MA] (4.74 mmol, 0.52 g). The resultant reaction mixture was stirred overnight under an inert atmosphere (glovebox). The reaction mixture was then dissolved in 10 ml of dry acetonitrile which resulted in precipitation of sodium methanesulfonate, Na[OMs]. The precipitate was filtered out. Subsequently, to promote further Na[OMs] precipitation, the acetonitrile solution of the product was shaken 3 times with 5 ml of dry hexane, followed by filtration and decantation of hexane after each step. The acetonitrile was removed under vacuum giving a yellow clear liquid. Yield: 1.9 g, 97.2%. 1H NMR (600 MHz, acetonitrile-d3) δ 5.83 (1H,m), 5.27 (1H,m), 3.44 (2H,dd), 3.32 (2H,dd), 3.08 (9H,s), 2.56 (3H,s), 2.08 (2H,m), 1.76 (3H,t), 1.39 (20H,m), 0.99 (6H,m); 13C NMR (600 MHz, acetonitrile-d3) δ 172.72, 143.67, 63.47, 59.43, 49.89, 39.13, 31.53, 28.82, 27.13, 26.01, 22.41, 22.18, 19.14, 13.50, 7.65; 11B NMR (193 MHz, MeOH-d4) δ 31.90, 5.08, 0.45, −21.86 ppm; MS (TOF) of [B(C2mea)(n-Bu)(N811)][MA]: m/z = 327.3550 (in positive mode), 85.0290 (in negative mode).IL3 – [B(C2mea)(n-Bu)(N811)][Ol]
[B(C2mea)(n-Bu)(N811)][OMs] (11.04 mmol, 4.664 g) was mixed with sodium oleate, Na[Ol] (11.05 mmol, 3.4 g). The resultant reaction mixture was stirred overnight under an inert atmosphere (glovebox). The reaction mixture was then dissolved in 10 ml of dry acetonitrile which resulted in precipitation of sodium methanesulfonate, Na[OMs]. The precipitate was filtered out. Subsequently, to promote further Na[OMs] precipitation, the acetonitrile solution of the product was shaken 3 times with 5 ml of dry hexane, followed by filtration and decantation of hexane after each step. The acetonitrile was removed under vacuum giving a clear yellow liquid. Yield: 6.2 g, 92.26%. 1H NMR (600 MHz, acetonitrile-d3) δ 6.13 (1H,s), 5.34 (1H,s), 3.33 (8H,m), 2.97 (10H,m), 2.48 (6H,s), 2.25 (2H,m), 1.67 (2H,m), 1.27 (32H,m), 0.88 (12H,m), 0.57 (3H,m); 13C NMR (600 MHz, acetonitrile-d3) δ 163.06, 131.10, 64.70, 60.68, 51.15, 40.30, 32.96, 32.77, 31.89, 31.11, 30.05, 28.70, 27.22, 26.13, 23.66, 23.40, 14.73, 8.87, 8.64; 11B NMR (193 MHz, MeOH-d4) δ 33.35, 5.01, 1.06, −21.41 ppm; MS (TOF) of [B(C2mea)(n-Bu)(N811)][Ol]: m/z = 327.3550 (in positive mode), 281.2480 (in negative mode).[B(C2mea)(sec-Bu)(N811)][OMs]
A neutral boron precursor B(C2mea)(sec-Bu) (39 mmol, 5.5 g) was mixed with ethyl methane sulfonate (39 mmol, 4.9 g). The reaction mixture was stirred for 10 min at room temperature under an inert atmosphere (glovebox). N,N-Dimethyl octylamine (39 mmol, 6.14 g) was added to the reaction mixture. The resultant solution was stirred overnight under an inert atmosphere (glovebox). A light brown colour liquid of [B(C2mea)(sec-Bu)(N811)][OMs] is obtained, which crystallised when stored overnight in a fridge (5 °C) under an Ar atmosphere. 1H NMR (600 MHz, MeOH-d4) δ 3.18 (6H,m), 2.83 (6H,s), 2.18 (6H,s), 0.94 (19H,m), 0.51 (10H,m);13C NMR (600 MHz, MeOH-d4) δ 66.68, 62.15, 58.11, 43.30, 39.21, 36.55, 32.58, 31.34, 28.74, 25.95, 22.11, 15.05, 13.45, 13.03, 9.71, 7.72; 11B NMR (193 MHz, MeOH-d4) δ 3.51, 0.54 (tetracoordinate boron).IL-4 – [B(C2mea)(sec-Bu)(N811)][EH]
[B(C2mea)(sec-Bu)(N811)][OMs] (7.2 mmol, 3.042 g) was mixed with sodium 2-ethyl hexanoate, Na[EH] (7.2 mmol, 1.2085 g). The resultant reaction mixture was stirred overnight under an inert atmosphere (glovebox). The reaction mixture was then dissolved in 10 ml of dry acetonitrile which resulted in precipitation of sodium methanesulfonate, Na[OMs]. The precipitate was filtered out. Subsequently, to promote further Na[OMs] precipitation, the acetonitrile solution of the product was shaken 3 times with 5 ml of dry hexane, followed by filtration and decantation of hexane after each step. The acetonitrile was removed under vacuum giving a clear dark yellow liquid. Yield: 3.2 g, 94.4%. 1H NMR (600 MHz, acetonitrile-d3) δ 3.32 (2H,dd), 3.19 (2H,dd), 2.97 (6H,s), 2.49 (3H,m), 1.97 (3H,m), 1.32 (28H,m), 0.89 (15H,m); 13C NMR (600 MHz, acetonitrile-d3) δ 182.17, 64.89, 60.89, 51.30, 50.86, 40.29, 33.91, 32.82, 31.34, 30.11, 30.08, 27.32, 24.06, 23.72, 14.82, 14.76, 13.10, 8.66; 11B NMR (193 MHz, MeOH-d4) δ 32.56, 5.18, 1.32, −21.64 ppm; MS (TOF) of [B(C2mea)(sec-Bu)(N811)][EH]: m/z = 327.3550 (in positive mode), 143.21 (in negative mode).IL5 – [B(C2mea)(sec-Bu)(N811)][MA]
[B(C2mea)(sec-Bu)(N811)][OMs] (4.73 mmol, 2 g) was mixed with sodium methacrylate, Na[MA] (4.74 mmol, 0.52 g). The resultant reaction mixture was stirred overnight under an inert atmosphere (glovebox). The reaction mixture was then dissolved in 10 ml of dry acetonitrile which resulted in precipitation of sodium methanesulfonate, Na[OMs]. The precipitate was filtered out. Subsequently, to promote further Na[OMs] precipitation, the acetonitrile solution of the product was shaken 3 times with 5 ml of dry hexane, followed by filtration and decantation of hexane after each step. The acetonitrile was removed under vacuum giving a clear brown liquid. Yield: 1.88 g, 96.26%.1H NMR (600 MHz, acetonitrile-d3) δ 5.74 (1H,m), 5.20 (1H,m), 3.71 (2H,dd), 3.31 (2H,dd), 2.94 (9H,s), 2.48 (3H,m), 2.30 (1H,s), 1.84 (5H,m), 1.66 (3H,m), 1.27 (16H,m), 0.89 (6H,m); 13C NMR (600 MHz, acetonitrile-d3) δ 172.86, 143.31, 63.59, 59.56, 49.96, 49.94, 39.03, 31.53, 28.79, 25.96, 22.42, 21.89, 19.04, 13.48, 7.61; 11B NMR (193 MHz, MeOH-d4) δ 33.55, 5.44, 1.36, −21.22 ppm; MS (TOF) of [B(C2mea)(sec-Bu)(N811)][MA]: m/z = 327.3550 (in positive mode), 85.0290 (in negative mode).IL6 – [B(C2mea)(sec-Bu)(N811)][Ol]
[B(C2mea)(sec-Bu)(N811)][OMs] (3.6 mmol, 1.5 g) was mixed with sodium oleate, Na[Ol] (3.6 mmol, 1.107 g). The resultant reaction mixture was stirred overnight under an inert atmosphere (glovebox). The reaction mixture was then dissolved in 10 ml of dry acetonitrile which resulted in precipitation of sodium methanesulfonate, Na[OMs]. The precipitate was filtered out. Subsequently, to promote further Na[OMs] precipitation, the acetonitrile solution of the product was shaken 3 times with 5 ml of dry hexane, followed by filtration and decantation of hexane after each step. The acetonitrile was removed under vacuum giving a clear yellow liquid. Yield: 2 g, 92.5%. 1H NMR (600 MHz, acetonitrile-d3) δ 5.34 (1H,s), 5.13 (1H,s), 3.32 (8H,m), 2.95 (9H,m), 2.48 (6H,m), 2.12 (3H,m), 1.67 (3H,m), 1.29 (31H,m), 0.89 (14H,m), 0.71 (1H,m); 13C NMR (600 MHz, acetonitrile-d3) δ 163.52, 130.89, 64.57, 60.56, 51.01, 50.97, 39.99, 32.71, 32.51, 30.31, 29.79, 29.77, 27.14, 26.95, 23.40, 23.13, 14.44, 14.07, 8.58; 11B NMR (193 MHz, MeOH-d4) δ 32.42, 5.07, 1.18, −21.83 ppm; MS (TOF) of [B(C2mea)(sec-Bu)(N811)][Ol]: m/z = 327.3550 (in positive mode), 281.2480 (in negative mode).[B(C2mea)(iso-Bu)(N811)][OMs]
A neutral boron precursor B(C2mea)(iso-Bu) (39 mmol, 5.5 g) was mixed with ethyl methane sulfonate (39 mmol, 4.9 g). The reaction mixture was stirred for 10 min at room temperature under an inert atmosphere (glovebox). N,N-Dimethyl octylamine (39 mmol, 6.14 g) was added to the reaction mixture. The resultant solution was stirred overnight under an inert atmosphere (glovebox). A light-yellow colour liquid of [B(C2mea)(iso-Bu)(N811)][OMs] is obtained, which crystallised when stored overnight in a fridge (5 °C) under an Ar atmosphere. 1H NMR (600 MHz, MeOH-d4) δ 3.96, 3.42 (2H,m), 3.20 (4H,m), 2.84 (6H,s), 2.15 (6H,m), 1.39 (3H,m), 0.96 (14H,m), 0.53 (9H,m), 0.27 (2H,m); 13C NMR (600 MHz, MeOH-d4) δ 63.78, 59.63, 50.56, 44.65, 40.69, 32.65, 30.18, 30.04, 27.85, 27.41, 26.26, 25.47, 23.43, 14.82, 9.15; 11B NMR (193 MHz, MeOH-d4) δ 3.06, 0.40 (tetracoordinate boron).IL7 – [B(C2mea)(iso-Bu)(N811)][EH]
[B(C2mea)(iso-Bu)(N811)][OMs] (3.6 mmol, 1.5 g) mixed with sodium 2-ethyl hexanoate, Na[EH] (3.6 mmol, 0.604 g). The resultant reaction mixture was stirred overnight under an inert atmosphere (glovebox). The reaction mixture was then dissolved in 10 ml of dry acetonitrile which resulted in precipitation of sodium methanesulfonate, Na[OMs]. The precipitate was filtered out. Subsequently, to promote further Na[OMs] precipitation, the acetonitrile solution of the product was shaken 3 times with 5 ml of dry hexane, followed by filtration and decantation of hexane after each step. The acetonitrile was removed under vacuum giving a clear yellow liquid. Yield: 1.51 g, 90.37%.1H NMR (600 MHz, acetonitrile-d3) δ 3.29 (2H,dd), 3.17 (2H,dd), 2.94 (6H,s), 2.95 (3H,s), 1.77 (4H,m), 1.28 (24H,m), 0.89 (16H,m), 0.53 (2H,m);13C NMR (600 MHz, acetonitrile-d3) δ 182.47, 64.96, 60.97, 51.34, 51.15, 40.35, 34.03, 32.85, 31.68, 30.11, 27.11, 26.34, 24.23, 23.74, 23.48, 14.78, 13.19, 8.92; 11B NMR (193 MHz, MeOH-d4) δ 31.32, 4.87, 0.45, −21.78 ppm; MS (TOF) of [B(C2mea)(iso-Bu)(N811)][EH]: m/z = 327.3550 (in positive mode), 143.1072 (in negative mode).IL8 – [B(C2mea)(iso-Bu)(N811)][MA]
[B(C2mea)(iso-Bu)(N811)][OMs] (3.6 mmol, 1.5 g) was mixed with sodium methacrylate, Na[MA] (3.6 mmol, 0.39 g). The resultant reaction mixture was stirred overnight under an inert atmosphere (glovebox). The reaction mixture was then dissolved in 10 ml of dry acetonitrile which resulted in precipitation of sodium methanesulfonate, Na[OMs]. The precipitate was filtered out. Subsequently, to promote further Na[OMs] precipitation, the acetonitrile solution of the product was shaken 3 times with 5 ml of dry hexane, followed by filtration and decantation of hexane after each step. The acetonitrile was removed under vacuum giving a clear dark yellow liquid. Yield: 1.40 g, 95.6%. 1H NMR (600 MHz, acetonitrile-d3) δ 5.75 (1H,m), 5.20 (1H,m), 3.28 (2H,dd), 3.16 (2H,dd), 2.93 (9H,s), 2.48 (3H,s), 1.84 (6H,m), 1.28 (14H,m), 0.90 (10H,m), 0.54 (2H,m); 13C NMR (600 MHz, acetonitrile-d3) δ 173.10, 143.05, 118.01, 63.64, 59.64, 50.04, 50.02, 38.97, 31.53, 28.80, 25.01, 24.92, 22.42, 22.13, 18.97, 13.46, 7.58; 11B NMR (193 MHz, MeOH-d4) δ 31.57, 5.07, 0.50, −21.26 ppm; MS (TOF) of B(C2mea)(iso-Bu): MS (TOF) of [B(C2mea)(iso-Bu)(N811)][MA]: m/z = 327.3550 (in positive mode), 85.0290 (in negative mode).IL9 – [B(C2mea)(iso-Bu)(N811)][Ol]
[B(C2mea)(iso-Bu)(N811)][OMs] (2.4 mmol, 1 g) was mixed with sodium oleate, Na[Ol] (2.4 mmol, 0.73 g). The resultant reaction mixture was stirred overnight under an inert atmosphere (glovebox). The reaction mixture was then dissolved in 10 ml of dry acetonitrile which resulted in precipitation of sodium methanesulfonate, Na[OMs]. The precipitate was filtered out. Subsequently, to promote further Na[OMs] precipitation, the acetonitrile solution of the product was shaken 3 times with 5 ml of dry hexane, followed by filtration and decantation of hexane after each step. The acetonitrile was removed under vacuum giving a clear yellow liquid. Yield: 1.35 g, 93.75%. 1H NMR (600 MHz, acetonitrile-d3) δ 5.58 (1H,s), 5.34 (1H,s), 3.37 (8H,m), 2.95 (11H,m), 2.50 (7H,s), 2.12 (2H,m), 1.67 (5H,m), 1.27 (25H,m), 0.89 (14H,m), 0.53 (3H,m); 13C NMR (600 MHz, acetonitrile-d3) δ 163.74, 131.07, 64.79, 60.79, 51.23, 40.29, 33.00, 32.81, 31.79, 30.10, 30.07, 28.65, 27.24, 25.56, 23.69, 14.79, 14.75, 8.89, 8.65; 11B NMR (193 MHz, MeOH-d4) δ 31.61, 4.86, 0.45, −21.84 ppm; MS (TOF) of [B(C2mea)(iso-Bu)(N811)][Ol]: m/z = 327.3550 (in positive mode), 281.2480 (in negative mode).Formulations of ionic liquids in base oil
Formulations containing 1 wt% of boron additive in Group III+ mineral base oil were prepared by the dissolution of nine boronium ILs (0.1 g) in neat base oil (9.99 g), respectively. The benchmark additive glycerol mono-oleate (GMO) was also prepared by the same procedure. The mixtures were stirred (60 °C, 1 h) and then left to equilibrate at room temperature prior to tribo tests.The actual miscibility of the IL candidates was determined based on their boron content in the earlier prepared mixtures, by first subjecting them to centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min. The supernatant layer of each sample was then collected, and the boron content was analysed using Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES), using Dual View 5100 ICP-OES model equipment from Agilent. The analysis was conducted in accordance with the ASTM D4951 method by first preparing the solutions, diluting 10 wt% of the supernatant into a hydrocarbon solvent, and spiking with 40 ppm cobalt (Co) salt as the internal standard. Concentration values were determined with reference to boron standard calibration curves prepared at 3 different concentration points. The blank and all standards were prepared in the same way as the measured samples.
000 rpm for 15 min. The supernatant layer of each sample was then collected, and the boron content was analysed using Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES), using Dual View 5100 ICP-OES model equipment from Agilent. The analysis was conducted in accordance with the ASTM D4951 method by first preparing the solutions, diluting 10 wt% of the supernatant into a hydrocarbon solvent, and spiking with 40 ppm cobalt (Co) salt as the internal standard. Concentration values were determined with reference to boron standard calibration curves prepared at 3 different concentration points. The blank and all standards were prepared in the same way as the measured samples.
Solubility of ionic liquids in base oil
The solubility of ionic liquids in base oil was determined from their boron content.In a typical experiment, an ionic liquid (0.10 g, 1 wt%) was added to Group III+ base oil (9.99 g, 99 wt%), placed in a vial equipped with a magnetic stirrer. The mixture was stirred (500 rpm, 60 °C, 1 h) and then left to equilibrate (ambient temperature, 1 h). Subsequently, the mixture was centrifuged (10000 rpm, 15 min), the supernatant layer was collected and its boron content analysed by inductively coupled plasma - optical emission spectrometry (ICP-OES), using a Dual View 5100 ICP-OES (Agilent) (Fig. S2†).
Boron content analysis was carried out following the ASTM D4951 method. The supernatant was diluted to 10 wt% in Premisolv as the solvent and spiked with 40 ppm cobalt internal standard. Concentration values were determined with reference to boron standard calibration curves, prepared from the 50 ppm organometallate standard solutions at three different concentrations. The blank and all standards were prepared in the same way as the measured samples.
Friction and wear test
In a typical experiment, an ionic liquid (0.10 g, 1 wt%) was added to Group III+ base oil (9.99 g, 99 wt%), placed in a vial equipped with a magnetic stirrer. The mixture was stirred (500 rpm, 60 °C, 1 h) and then left to equilibrate (ambient temperature, 1 h).The friction and wear characteristics were evaluated using a high frequency reciprocating rig (HFRR), model PCS-002817 (PCS Instruments). The upper ball specimen was a 6 mm diameter ball that was loaded into the upper specimen holder, specified to grade 28 (ANSI B3.12), ANSI E-52100 steel, with a Rockwell hardness “C” scale (HRC) number of 58–66 (ISO 6508), and a surface finish of less than 0.05 μm Ra. The lower specimen (10 mm disc) was loaded into the lower specimen holder, specified to AISI E-52100 steel machined from an annealed rod, with Vickers hardness 800 Hv polished to a surface finish of less than 0.02 μm Ra. The tests (Table 1) were repeated until two reproducible curves were recorded. Upon the completion, the wear scar diameters of the ball specimens were measured, and images were captured using a PCS Instruments metallurgical microscope model PCS-005311 (100 times magnification).
| Test parameters | Test conditions |
|---|---|
| T (°C) | 120 |
| Frequency (Hz) | 20 |
| Stroke (mm) | 2 |
| Speed (mm s−1) | 125 (boundary) |
| Load (N) | 4 |
| Pressure (GPa) | 1 |
| Type of motion | Pure sliding |
| Test duration (min) | 60 |
Results and discussion
Design
Ionic liquids reported in this work were designed to comprise tetracoordinate boronium cations and carboxylate anions, each with a built-in tribological function.The presence of the B–O bond in the form of IL anions has been reported to give a favourable effect on friction and wear reduction, particularly chelated orthoborate anions.18 As borocations, the presence of both B–O and B–N bonds was yet to be investigated and was expected to benefit tribological performance as well; borate esters, particularly when combined with N-donors (amines), have been reported to enhance both friction and wear, enhancing the formation of iron nitride and iron boride.19–22 This design principle was incorporated into the design of the cations. Furthermore, bidentate coordination will limit ligand scrambling and the B–N coordination has been envisaged to increase the hydrolytic stability of the boron centre.23–25 Three different butyl isomers, R, were used to probe the influence of branching on melting point, oil miscibility and tribological properties.
Compared to tetraalkylammonium, tetraalkylphosphonium and 1,3-dialkylimidazolium cations, borocations are not extensively used in the design of ionic liquids. This work is preceded by boronium ILs developed by Davis and co-workers (Fig. 2a and b)26–29 and borenium ILs developed in our group (Fig. 2c and d),30–32 but introduces to the realm of ionic liquids a new design motif based on the 2-(methylamino)butylborane, B(C2mea)R, protected with an amine (Fig. 2e), in which boron is coordinated exclusively by oxygen, nitrogen and carbon.
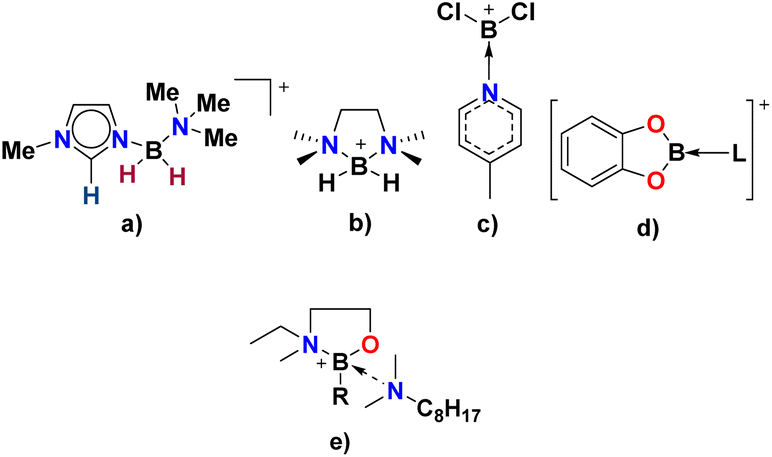 | ||
| Fig. 2 Examples of borocations used in the design of ionic liquids by Davis (a and b),26 Swadżba-Kwaśny (c and d),31,32 and reported here (e). L – phosphine, phosphine oxide, amine; R – n-Bu, iso-Bu, or sec-Bu. | ||
Carboxylate anions have been widely used in the design of ILs, being biodegradable, potentially bio-based, and in some cases – offering excellent performance as lubricants and lubricant additives.33–35
Several carboxylate anions were selected in this work to understand the effect of branching, unsaturated bond and long chain anions (Fig. 3): oleate, [Ol]−, methacrylate, [MA]− and 2-ethyl hexanoate, [EH]−, were used to encourage miscibility in the oil medium and adhesion of the IL onto the metal surface.
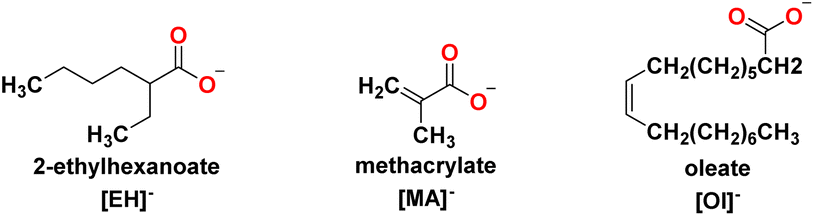 | ||
| Fig. 3 Carboxylate anions used in this work. | ||
All anions were commercially available in the form of inexpensive sodium salts and have been previously used in the design of ionic liquids. The presence of either unsaturated bonds or branching within the carboxylate anions was selected to aid miscibility in non-polar oil medium.
Chloride corrosion is a known problem in ionic liquids used as lubricants/lubricant additives, even if it only originates from traces of chloride from one of the precursors.36 To avoid this issue, all syntheses were chloride-free.
Synthesis of boronium ionic liquids
Boronium ionic liquids were synthesised in three steps. In the first step, butylboronic acids (n-Bu, iso-Bu, or sec-Bu) were reacted with 2-(methylamino)ethanol in the presence of dry toluene to form their respective cyclic boranes, B(C2mea)R (Fig. 4a). These dehydration reactions were carried out as reactive distillations in Dean–Stark apparatus.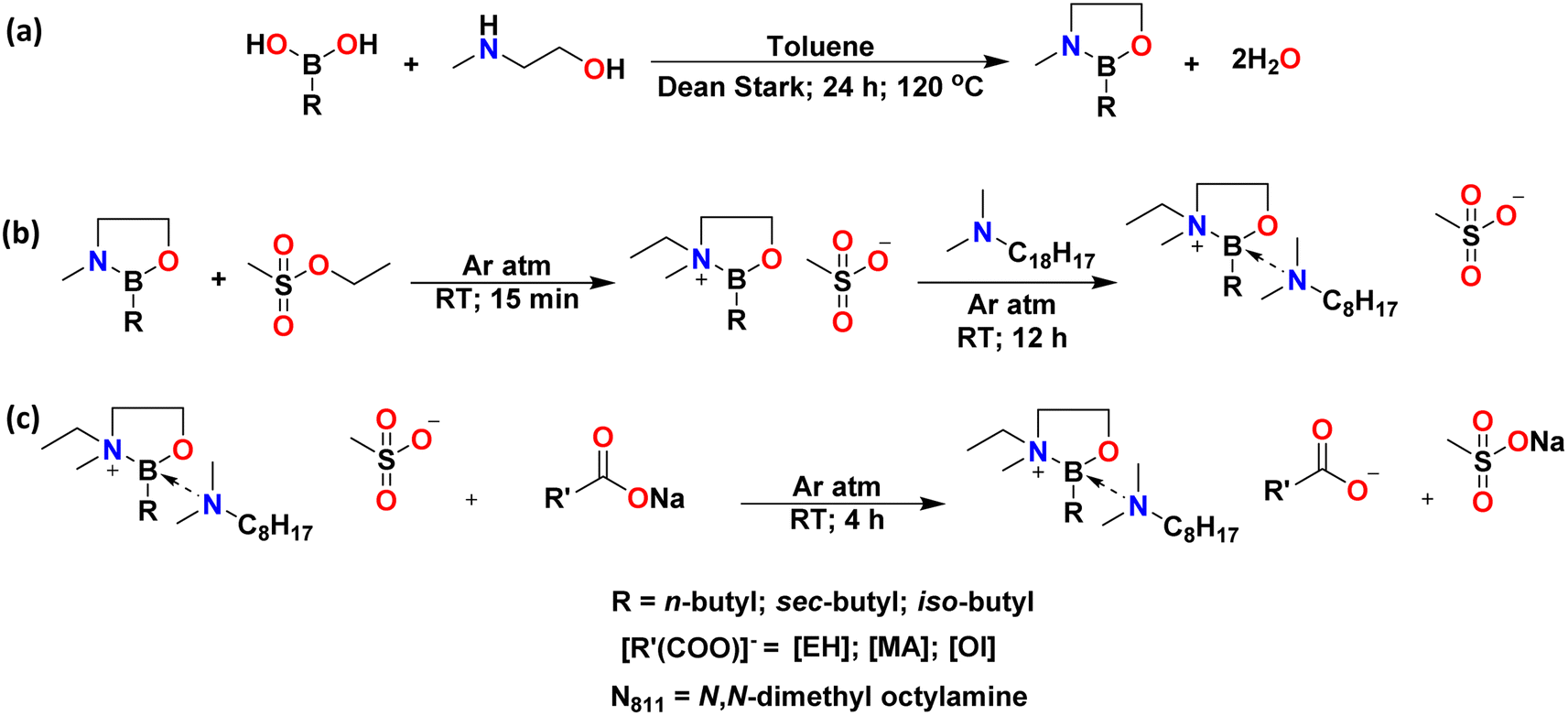 | ||
| Fig. 4 Three-step synthetic scheme for the synthesis of boronium ionic liquids: dehydration (a), alkylation (b) and ion exchange (c). | ||
All products were obtained in quantitative yields and dried under high vacuum; B(C2mea)(n-Bu) and B(C2mea)(sec-Bu) were pale yellow powders, and B(C2mea)(iso-Bu) was a colourless powder (Fig. 5).
 | ||
| Fig. 5 Photographs of 2-(methylamino)butylboranes with three isomers of the Bu chain. | ||
In the second step, cyclic boranes were alkylated with ethyl methanesulfonate, followed by the addition of N,N-dimethyloctylamine (Fig. 4b). All reactions were carried out solventless, at ambient temperature, under an inert argon atmosphere. The products were yellow viscous ionic liquids with methanesulfonic (mesylate) anions, [OMs]−: [B(C2mea)(n-Bu)][OMs], [B(C2mea)(sec-Bu)][OMs], and [B(C2mea)(iso-Bu)][OMs] (Fig. 6).
 | ||
| Fig. 6 Photographs of mesylate ionic liquids. | ||
Mesylate ionic liquids were not the target compounds in this work, as they contained a sulfur-based anion. An initial screening has shown that they were insoluble in nonpolar base oil used in this work (Group III+) due to the hydrophilic nature of mesylate. However, they were convenient halide-free precursors to more lipophilic carboxylate ILs, which cannot be accessed by direct alkylation (esters of carboxylic acids are very poor alkylating agents).
The third step was an anion exchange. Mesylate ILs were stirred with sodium salts: Na[EH], Na[MA], or Na[Ol] (Fig. 4c). All reactions were carried out solventless, at room temperature, under an argon atmosphere. Subsequently, addition of a mixture of dry acetonitrile and hexane resulted in precipitation of a white powder (sodium methanesulfonate), whereas ionic liquids remained in solution and were easily separated by solvent removal under reduced pressure. The structures and photographs of the lipophilic ionic liquids synthesised in this work (IL-1 to IL-9) are listed in Fig. 7 and S3.†
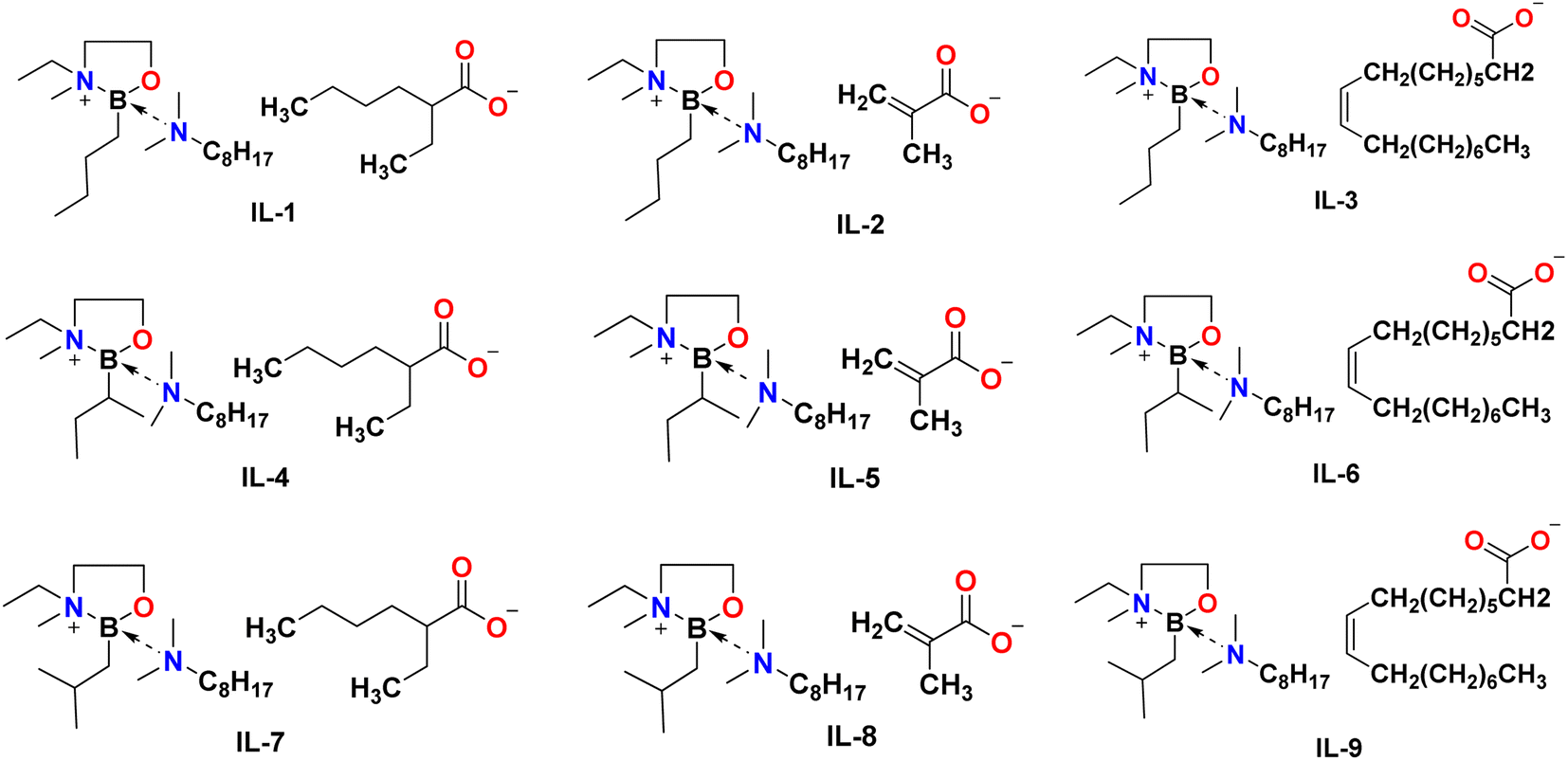 | ||
| Fig. 7 Structures of lipophilic boronium ionic liquids developed in this study. | ||
Speciation of boronium ionic liquids
Fig. 7 depicts idealised structures of ionic liquids, assuming that there is no ligand scrambling at the boron centre. Scrambling of the bidentate alcohol amine ligand was unlikely, also the B–C bond is not expected to be labile37 (boron–carbon bond enthalpy EB–C = 350 ± 10 kJ mol−1).38,39 However, it was possible that carboxylate anions would compete with N811 for the 4th coordination position, and/or that tricoordinate borenium ion would be liberated, resulting in speciation different from that in, including the potential for several boron species existing in a dynamic equilibrium (Fig. 8).40–42 The existence of several equilibrated species will have had an impact on the physical–chemical properties and performance of boronium ionic liquids, therefore it is crucial to have a good understanding of their speciation.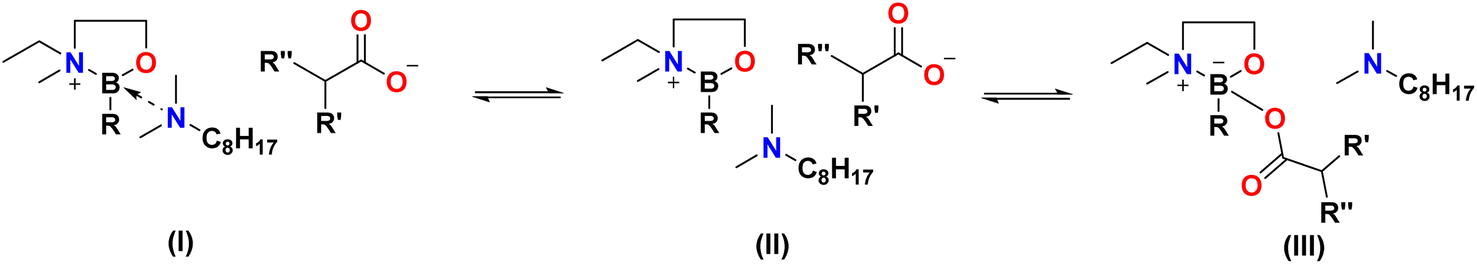 | ||
| Fig. 8 Plausible equilibria in hydrophobic boronium ILs. | ||
All boronium ILs were studied using multinuclear (1H, 13C, 11B) NMR and Raman spectroscopies, as well as mass spectrometry. Complete NMR and mass spectrometry analysis recorded for all 3 mesylate ILs, all ILs 1–9, as well as their precursors, are provided in the ESI† (Fig. S4–S60†).
The coordination environment of boron species was probed using 11B NMR spectroscopy.37,43–4511B NMR spectra recorded for B(C2mea)(n-Bu) and all derived ILs are shown in Fig. 9. 11B NMR chemical shifts and proposed species are listed in Table 2.
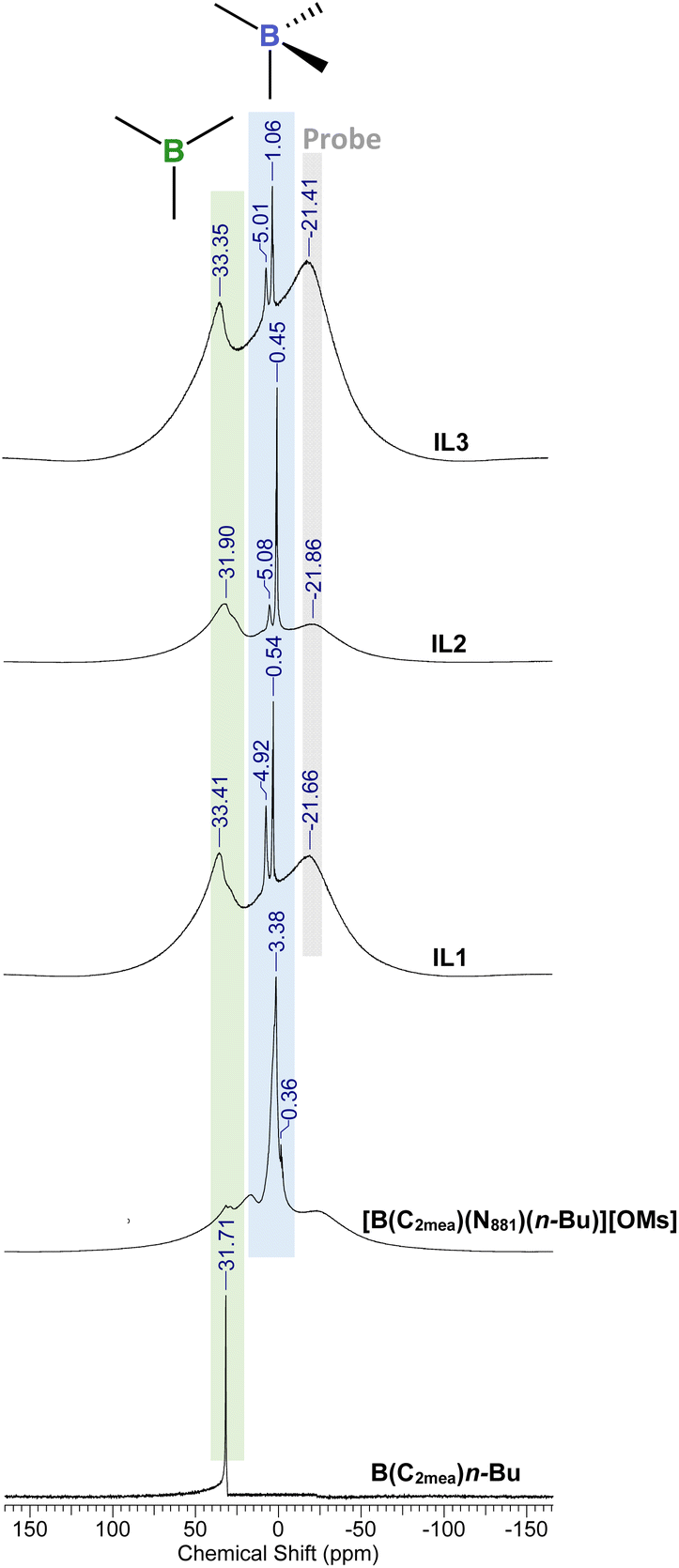 | ||
| Fig. 9 11B NMR spectra of B(C2mea)(n-Bu) and derived ionic liquids: [B(C2mea)(n-Bu)(N811)][OMs], IL-1, IL-2 and IL-3. The green-shaded area designates the chemical shift regions of tricoordinate boron compounds while the blue area designates tetracoordinate species. | ||
| pKa | 11B chemical shift (ppm) and boron species assignment | |||
|---|---|---|---|---|
| Tricoordinate | 4-Coordinate (I) | 4-Coordinate (III) | ||
| [OMs]− | −2.0 | 31.7 | 3.38 | 0.36 |
| [EH]− | 4.82 | 33.41 | 4.92 | 0.54 |
| [MA]− | 4.70 | 31.90 | 5.08 | 0.45 |
| [Ol]− | 9.85 | 33.35 | 5.18 | 1.32 |
The B(C2mea)(n-Bu) spectrum featured one sharp signal at 31.71 ppm, corresponding to a single tricoordinate boron species.37,46–48 The spectrum of [B(C2mea)(n-Bu)(N811)][OMs] was more complex: a sharp signal in the tetracoordinate region (3.38 ppm) corresponds to the [B(C2mea)(n-Bu)(N811)]+ cation – viz. species (I) in Fig. 8, whereas the minute signal at 0.36 ppm can be attributed to a charge-neutral zwitterionic compound [B(C2mea)(n-Bu)(OMs)] – species (III) in Fig. 8. There are also two low-intensity, broad signals in the tricoordinate region, at ca. 18 and 33 ppm, possibly overemphasised because of the overlap with the broad signal at ca. −21 ppm, originating from the NMR tube.
In carboxylate analogues, speciation is significantly different: each spectrum features a prominent signal in the tricoordinate region, corresponding to the borenium cation, species (II) in Fig. 8. The tetracoordinate region has two sharp signals: at ca. 5 ppm, assigned to species (I), and a more shielded signal corresponding to species (III).
11B chemical shifts corresponding to the signal originating from species (III), indicating tetracoordinate boron with coordinated anions, can be related to pKa values of the corresponding parent acids of these anions (Table 2).49–52 The conjugate base of the strong HOM acids (pKa = −2) is weak, and does not compete with the amine (N811, pKa = 9.86)53 for the boron centre, therefore the signal for species (III) is minute and shielded. Carboxylates are stronger bases; stronger the base, more deshielded the tetracoordinate signals. Similar observations were made for the analogous series derived from B(C2mea)(iso-Bu) and B(C2mea)(sec-Bu) – see Fig. S61 and S62,† respectively.
A complete set of 13C NMR spectra of the cyclic boranes and their boronium IL derivatives are shown in Fig. S5, S8, S11, S17, S20, S23, S26, S29, S32, S35, S38, S41, S44, S47 and S50.†13C NMR signals corresponding to the methylsulphonate anion were found at 42.86 ppm for [B(C2mea)(n-Bu)][OMs], 43.30 ppm for [B(C2mea)(sec-Bu)][OMs], and at 44.65 ppm for [B(C2mea)(iso-Bu)][OMs], which is in agreement with the literature data for free [OMs]−.54 The 13C NMR signals originating from carboxylate groups of ILs 1–9 are shown in Fig. 10. The [EH]− anion gave signals at 182.99 ppm for IL-1, 182.17 ppm for IL-4, and 182.47 ppm for IL-7, matching literature data for free 2-ethylhexanoate.55 Peaks for the [MA]− anion were found at 172.72 ppm for IL-2, 172.86 ppm for IL-5, and 173.10 ppm for IL-8, again matching the literature data for free methacrylate (it is more shielded due to the conjugate double bond).56,57 In contrast, signals for [Ol]− were recorded at 163.06 ppm (IL-3), 163.52 ppm (IL-6), and 163.74 ppm (IL-9), which is in the ester region, significantly shielded with respect to the free oleate anion (183.8 ppm).57,58
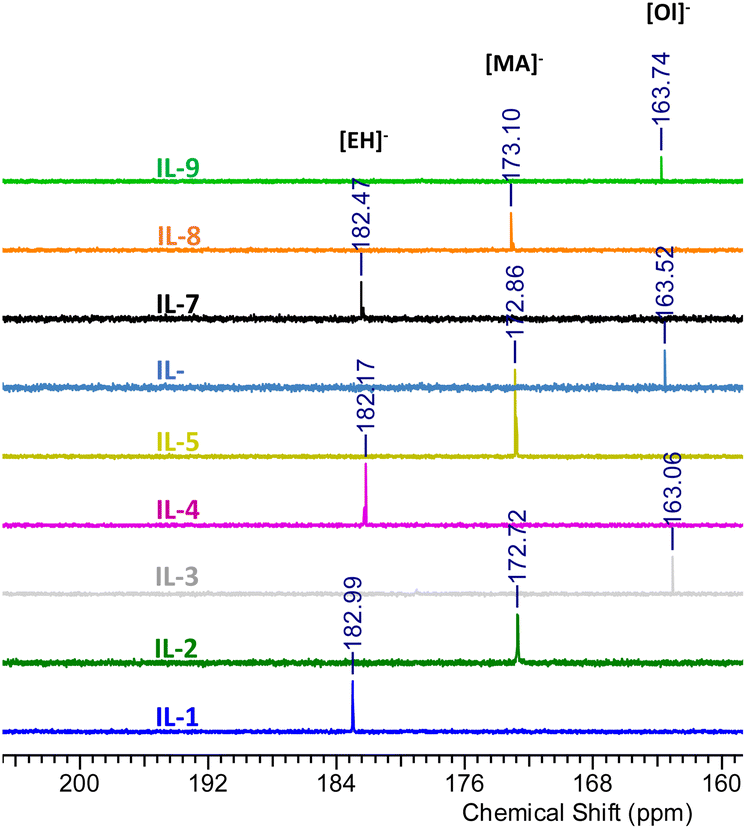 | ||
| Fig. 10 Fragments of 13C NMR spectra of carboxylate anions containing boronium ILs, showing signals originating from the carboxylate groups. | ||
Speciation suggested by 13C NMR spectroscopy is simplified compared to that from 11B NMR spectroscopy, showing the averaged coordination of each anion. However, it is consistent with conclusions drawn from 11B NMR spectroscopy; anions with lower basicity (lower pKa of the conjugate acid) are present mainly in their free form, favouring species (I) and/or (II). The more basic oleate is coordinated to the boron centre, favouring species (III).
Raman spectra recorded for B(C2mea)(n-Bu) and all derived ILs are shown in Fig. 11 and S63.† The B(C2mea)(n-Bu) spectrum features B–O symmetric stretch at νB–O s = 1455 cm−1, B–O asymmetric stretch at νB–O as = 1053 cm−1, and a group of weak signals in the B–N symmetric stretch area at νB–N s = 890, 869 and 786 cm−1.59–62 In all four IL derivatives, the [B(C2mea)(n-Bu)(N811)]+ cation retained the νB–O s = 1450 cm−1 peak in all ILs, although the asymmetric B–O signal has been blue-shifted to νB–O as = 1040 cm−1, indicating elongation of the bond. The weak band at νB–N s = 890 cm−1 appears to remain, a strong band at νB–N s = 776 cm−1 appears, likely due to boron coordinating to N811via a longer, dative bond.49–52
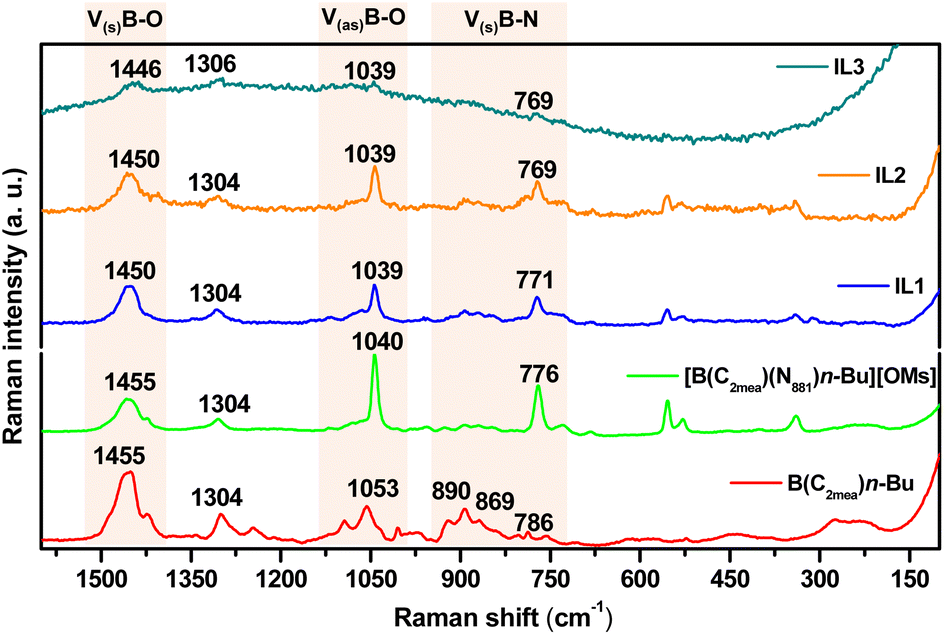 | ||
| Fig. 11 Fragments of Raman spectra of B(C2mea)(n-Bu) and the four derived ionic liquids: [B(C2mea)(n-Bu)(N811)][OMs], IL-1, IL-2 and IL-3, showing B–O and B–N stretching frequencies. | ||
Analogous conclusions were drawn for the compound series derived from B(C2mea)(iso-Bu) and B(C2mea)(sec-Bu) – see Fig. S64 and S65,† respectively.
Ligand scrambling that leads to multiple equilibrated boron species is characteristic of borocation chemistry in solution,53 and in borocation-based ionic liquids.30 Beyond boron, dynamic equilibria between several species are found in archetypal chloroaluminate ionic liquids that contain [AlCl4]− equilibrated with [Al2Cl7]− or Cl−, depending on their composition,54 and in a plethora of other halometallate systems, including both ionic liquids55,56 and liquid coordination complexes.57,58 This can be expanded even further, to “constitutionally dynamic” metal-free ionic liquids with equilibrated cations, where several cationic species exist in equilibria that involve addition/elimination of a small molecule, such as water.59,60 In this context, the complicated speciation of ILs reported in this work is their inherent feature and could be considered as an avenue to fine-tune their properties.
Thermal analysis
Thermal stability (low volatility) of lubricant additives is important in the context of their sustained performance under operative conditions. Thermal stability of the cyclic boron precursors, B(C2mea)R, was generally low, with Td onset around 71 °C (Fig. 12). Although it was expected that quaternisation would significantly improve the thermal stability, two out of three mesylate ILs, [B(C2mea)(n-Bu)(N811)][OMs] and [B(C2mea)(iso-Bu)(N811)][OMs] retained low thermal stability, with the onset of decomposition below 100 °C. Upon conversion to carboxylates, thermal stability improved, with Td values oscillating between 120 and 150 °C (Fig. 12).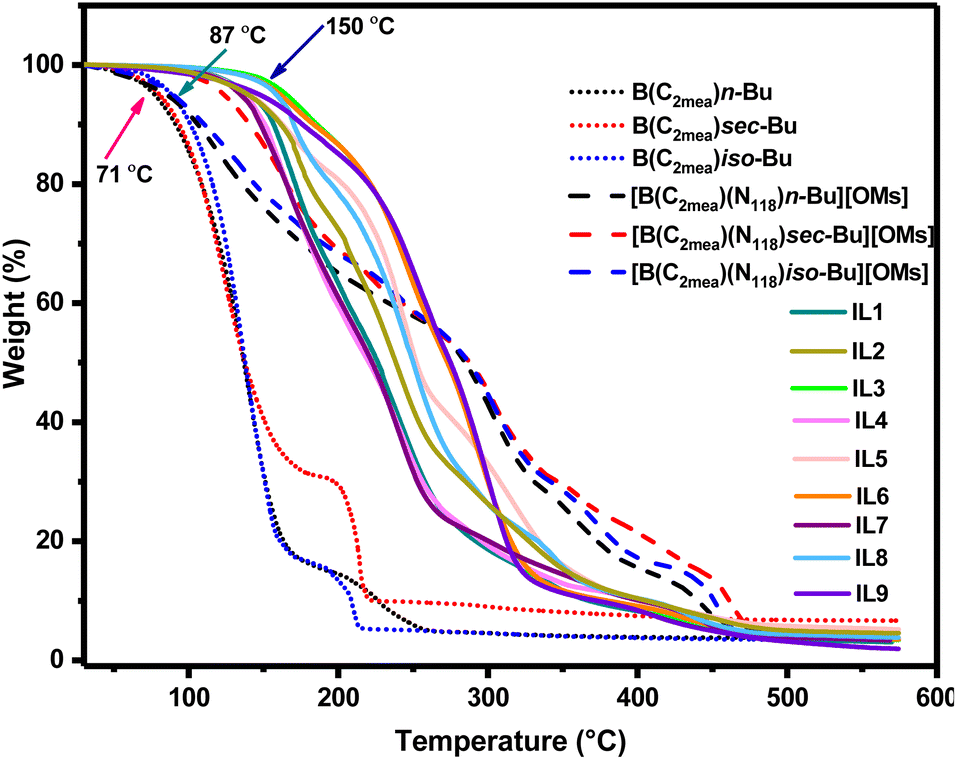 | ||
| Fig. 12 Thermogravimetric analysis of cyclic boranes and derived ionic liquids (ramp 5 °C min−1). Arrows indicate 5% mass loss for selected curves. | ||
DSC analysis (Fig. S66–68†) resulted in very complex measured curves, which is expected, given the complicated speciation of the boronium ILs. All endothermic events that could be classified as melting occurred below 25 °C, classifying IL-1 to IL-9 as room-temperature ionic liquids. In all samples the crystallisation was evidently hindered, which manifested itself either by samples undergoing only a glass transition, or by prominent cold crystallisation events (exothermic crystallisation peaks occurring upon heating).
Solubility of boronium ionic liquids in base oil
The miscibility of ILs 1–9 with non-polar group III+ base oil was measured by the addition of 1 wt% of an ionic liquid into the oil sample, vigorous stirring, followed by centrifugation, and the quantification of boron content in the supernatant by ICP (Fig. S69† and Table 3). The measured values were also compared against the theoretical values that assumed full miscibility of the ILs at 1 wt% concentration.| IL | Theoretical boron content at 1 wt% (mg kg−1) | Measured boron content (mg kg−1) | Calculated IL loading (wt%) |
|---|---|---|---|
| IL-1 | 230 | 59 | 0.26 |
| IL-2 | 262 | 68 | 0.26 |
| IL-3 | 178 | 114 | 0.64 |
| IL-4 | 230 | 45 | 0.20 |
| IL-5 | 262 | 67 | 0.26 |
| IL-6 | 178 | 128 | 0.72 |
| IL-7 | 230 | 43 | 0.19 |
| IL-8 | 262 | 77 | 0.29 |
| IL-9 | 178 | 136 | 0.76 |
Oleate ionic liquids (IL-3, IL-6 and IL-9) were found to be the most soluble, reaching the highest boron concentrations, 114–136 mg kg−1 (ca. 0.7 wt% IL loading). This can be partially attributed to structural features of the oleate anion: the long carbon chain is compatible with the non-polar base oil, and cis-unsaturated double bond results in a kink of the alkyl chain, which prevents localisation of electron density or structured packing, thus disrupting crystallisation and decreasing melting points, which in turn enhances dispersion in the oil.16,61,62 However, the deciding factor is probably speciation: from 11B and 13C NMR spectra it was evident that oleate ILs exist mainly as zwitterionic, charge-neutral species (III) – viz. Fig. 8 – with charges much more shielded than in other ionic liquids reported in this work. Considering cations of the three oleate ionic liquids, IL-9, with an iso-Bu chain (Fig. 4), was the most soluble. This is in agreement with the literature; it is known that branching aids miscibility possibly with oils due to the higher free volume and lower tendency of closed packing.62,63
Methacrylate ionic liquids (IL-2, IL-5 and IL-8) have shown consistent levels of miscibility in oil, between 67 and 77 mg kg−1, which amounts to ca. 0.25 wt% of IL. Despite branching and the presence of double bonds, the short alkyl chain and the greater ionic character of these ILs contributed to low solubility, irrespective of structural variation on the cation.
The overall oil miscibility of 2-ethylhexanoate ionic liquids (IL-1, IL-4 and IL-7) was the lowest of all samples, between 19 and 26%, despite features potentially aiding miscibility in oil: longer carbon chain than [MA]− and the presence of ethyl group branching. As shown in Fig. 10, 13C NMR signals from [EH]− anions appeared at the most de-shielded position, possibly suggesting the most localised charge and therefore higher polarity gap against the base oil. The position of the ethyl branching adjacent to the carboxylate group could be the main contributing factor, due to the weaker inductive effect from the tertiary carbon.
Tribological behaviour of boronium ionic liquids
The boronium64 ionic liquids were designed for both the cation and the anion to be triboactive, and to contain only benign main group elements: C, H, O, N and B. Their ability in reducing wear and friction was compared against a conventional organic commercial friction modifier that fulfils similar criteria, glycerol mono-oleate (GMO) – full results measured for GMO are presented in Fig. S70–S73.†The tribological behaviour of the IL additives in oil was studied using the high-frequency reciprocating rig (HFRR) method, which simulates the frictional behaviour of lubricated test specimens under a boundary lubrication regime, as a result of reciprocating sliding motion between an upper metal ball specimen against a flat lower specimen surface. This method provides two key indications of tribological behaviour: coefficient of friction (CoF), measured during the test, and the wear scar diameter (WSD), measured after each test from the ball specimens.
A load of 4 N was selected to attain Hertzian of ca. 1 GPa, which approximates the average contact pressures observed in key types of contacting surfaces within automotive systems, for the given geometry of the tribo-specimens. The temperature of 120 °C was selected because the benchmark (GMO) is known to effectively form tribofilm at higher temperatures (100–120 °C), elevated temperature enables direct comparison between the ILs and the benchmark.
The CoF values, recorded over 1 h, are shown in Fig. 13. The mean CoF values, averaged over the plateau part of each plot, are shown in Fig. 14 (see Fig. S70† for HFRR data).
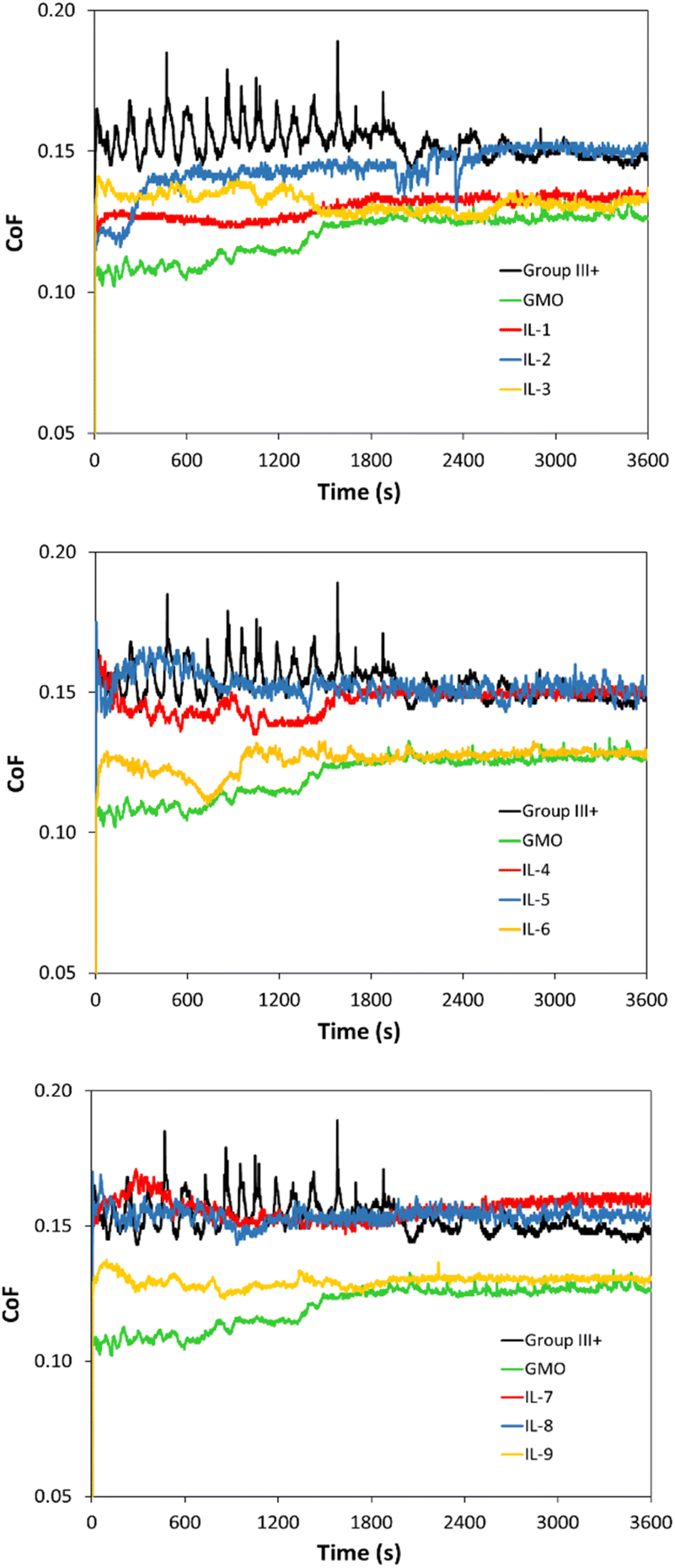 | ||
| Fig. 13 CoF profiles of saturated solutions of boronium ILs in Group III+ base oil, compared to neat Group III+. Top: [B(C2mea)(n-Bu)]+ ILs, middle: [B(C2mea)(sec-Bu)]+ ILs and bottom: [B(C2mea)(iso-Bu)]+ ILs. Anions: 2-ethylhexanoate (IL-1, 4 and 7), methacrylate (IL-2, 5 and 8) and oleate (IL-3, 5 and 9). | ||
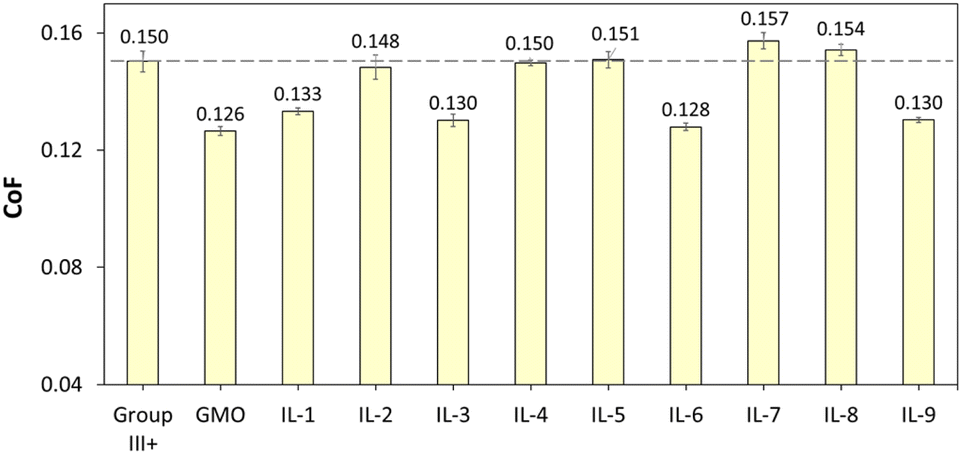 | ||
| Fig. 14 CoF values of Group III+ base oil, and its formulations containing 1 wt% of GMO or saturated solutions of boronium ILs; data recorded between 1800 and 3600 s. | ||
All ionic liquids (Fig. 13) had a pronounced influence on smooth sliding behaviour, from the beginning of the test and across the test duration, contrasting with the CoF profile for neat Group III+, which showed severe fluctuations due to “stick-and-slip” phenomena. In the absence of polar additives, Group III+ formed an unstable, disoriented and desorbed tribofilm. On the other hand, initial decrease in CoF induced by the ILs was in all cases lower than that of GMO, which is known for high metal surface affinity and ability for near-instantaneous lowering of friction upon rubbing.65–67
Although the smooth sliding effect was similar to that of GMO, CoF reduction throughout the experiments was either poorer than, or on par with that of GMO. GMO was fully miscible in oil at 1 wt%, and therefore had higher amounts of surface-active moieties from the ester group on the same level of additive concentration, sufficient to form a strong tribo-film.
Oleate-based ILs (IL-3, 6 and 9 – yellow plots in Fig. 13) were at loadings of ca. 0.70 wt%, but after a stable tribofilm was established (18000 s onwards), averaged CoFs were on par with GMO at 1 wt%. This performance could be attributed to the high solubility of oleate ILs compared to other tested ILs on one hand, and higher content of triboactive elements compared to GMO on the other.
With respect to cations, the [B(C2mea)(n-Bu)]+ cation appeared to have a positive influence on tribofilm formation. [B(C2mea)(n-Bu)][EH] established a good quality tribofilm (IL-1 – red plot in Fig. 13, top), despite its limited solubility of 0.26 wt%. Also [B(C2mea)(n-Bu)][MA], present at 0.26 wt% (IL-2 – blue plot in Fig. 13, top), demonstrated some CoF reduction within the first half of the test, which however deteriorated over time. This could imply low durability of the film that could have been formed earlier on the metal surface; however, due to the shorter alkyl chain and presence of the double bond within the alkyl group, formation of both stable and ordered packing of tribofilm to protect the metal surface from further friction may have been inhibited.68 Branched sec-Bu and iso-Bu groups have been introduced to increase the solubility of ILs in oil; apparently, not only has this not been successful, but also it is plausible that steric hindrance introduced by these groups has been detrimental to early tribofilm formation.
When comparing averaged CoF data recorded for stable tribofilms (Fig. 14), it was apparent that oleate ILs (IL-3, 6 and 9) have shown consistent propensity towards lowering friction (by 13.3–14.8% with respect to neat Group III+), showing performance similar to that of GMO, albeit at lower loadings per weight (0.7 wt% vs. 1 wt%). This could be attributed to their higher solubility in oil, which in turn could be related to their speciation and is consistent with the literature, where increased fatty acid concentration resulted in lower CoFs. Furthermore, the [B(C2mea)(n-Bu)]+ cation has shown transient ability to support tribofilm formation, but a sustained positive effect, other than that arising from oleate, was only seen for [B(C2mea)(n-Bu)][EH], which seems to necessitate the presence of carboxylate with a long alkyl chain.
To evaluate reduction in wear, wear scar diameters from the spent HFRR metal ball specimens were imaged (Fig. 15) and measured (Fig. 16). All ILs have shown wear scar reduction when compared to neat Group III+, the trends in performance were largely contrasting with those found for CoF, and dependent (within error bars) entirely on the anion.
 | ||
| Fig. 15 Wear scar microscopic images of metal ball specimens obtained after HFRR tests, from neat Group III+ and its formulations with 1 wt% of GMO or saturated solutions of boronium ILs. | ||
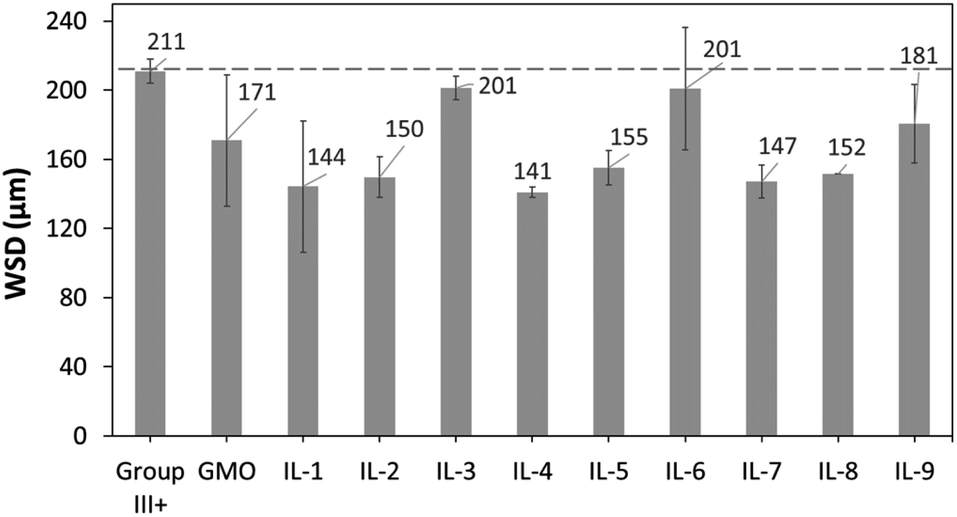 | ||
| Fig. 16 WSD values of metal ball specimens obtained after HFRR tests, from neat Group III+ and its formulations with 1 wt% of GMO or saturated solutions of boronium ILs. | ||
In contrast to CoF experiments, oleate ILs consistently performed the worst, with minimal wear scar reduction (201 μm, 5%; 197 μm, 7%; and 181 μm, 14%), slightly worse than that of GMO – with a caveat that in all these cases, there was a large error associated with the measurements.
Despite poor solubility in the base oil (0.19–0.26 wt%), ethylhexanoate ionic liquids (IL-1, 4 and 7) performed very well in reducing wear scar. IL-4 gave the lowest wear scar diameter in absolute numbers (143 μm, 32% better than Group III+ base oil, and 16% better than GMO), but all three ILs performed the same within the error of the measurement. Methacrylate ILs gave slightly higher wear (all ca. 151 μm and the same within error of measurement), also outperforming GMO.
Very surprisingly, the results were inversely proportional to boron content (Table 3); smaller quantities of boron gave the best performance, while the highest gave the worst. It is unclear whether boron really had a detrimental effect on WSD in this case, or the less soluble ionic liquids, with more polar ethylhexanoate and methacrylate anions, were better in reducing wear, and the lower content of boron was just a side effect of their lower solubility.
Conclusions
Boronium ionic liquids can be synthesised from abundant starting materials, through a halide-free route, and used as multi-functional additives in base oil. Speciation of such ILs is complicated, due to the ligand scrambling on the boron centre, and will affect their performance as additives.There is no one-size-fits-all solution to the design of these ILs. In terms of friction modification, oleate ILs perform on par with GMO, at slightly lower loadings. However, due to several synthetic steps required to synthesise them, for friction modification alone, GMO is likely the more viable option. ILs with ethylhexanoate and methacrylate anions were better in reducing wear, while largely underperforming in terms of friction reduction.
Within the matrix of nine studied ILs, the best performing was IL-1, [B(C2mea)(n-Bu)][MA], which reduced CoF by 11.3% (compared to 15.8% by GMO) and reduced wear scar by 30% (compared to 16% by GMO). From these preliminary results, it is difficult to explain the synergistic effect of this cation and anion combination, but identification of this well-performing system within the matrix of just nine samples gives hope for future structural improvement, especially using computation modelling to explain surface interactions.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
NM designed, synthesised and characterised all neutral cyclic boron & their ILs, with advice from HQNG. Preliminary syntheses were carried out by FC. FFMY performed miscibility tests, interpretation of NMR characterisation and tribological data; tribology tests were conducted by MTS. AD provided advice from the lubricant perspective while GS from the IL perspective for industrial applications. GSK supervised the findings of this work. All authors provided critical feedback and helped shape the research, analysis and manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
PETRONAS is acknowledged for funding this research.Notes and references
- T. Rüther, T. D. Huynh, J. Huang, A. F. Hollenkamp, E. Alan Salter, A. Wierzbicki, K. Mattson, A. Lewis and J. H. Davis, Chem. Mater., 2010, 22, 1038–1045 CrossRef.
- G. W. Stachowiak and A. W. Batchelor, Engineering Tribology, Butterworth-Heinemann, 2013 Search PubMed.
- M. I. De Barros'Bouchet, J. M. Martin, T. Le-Mogne and B. Vacher, Tribol. Int., 2005, 38, 257–264 CrossRef.
- H. Spikes, Lubr. Sci., 2008, 20, 103–136 CrossRef CAS.
- M. I. De Barros, J. Bouchet, I. Raoult, T. Le Mogne, J. M. Martin, M. Kasrai and Y. Yamada, Wear, 2003, 254, 863–870 CrossRef CAS.
- A. Morina, A. Neville, M. Priest and J. H. Green, Tribol. Int., 2006, 39, 1545–1557 CrossRef CAS.
- C. Grossiord, K. Varlot, J. Martin, T. Le Mogne and C. Esnouf, Tribol. Int., 1999, 31, 737–743 CrossRef.
- D. N. Khaemba, A. Neville and A. Morina, RSC Adv., 2016, 6, 38637–38646 RSC.
- A. M. Barnes, K. D. Bartle and V. R. A. Thibon, Tribol. Int., 2001, 34, 389–395 CrossRef CAS.
- I. Minami, Molecules, 2009, 14, 2286–2305 CrossRef CAS PubMed.
- Y. Zhou and J. Qu, ACS Appl. Mater. Interfaces, 2017, 9, 3209–3222 CrossRef CAS PubMed.
- M. Cai, Q. Yu, W. Liu and F. Zhou, Chem. Soc. Rev., 2020, 49, 7753–7818 RSC.
- G. Huang, Q. Yu, Z. Ma and M. Cai, Tribol. Int., 2017, 107, 152–162 CrossRef CAS.
- Q. Yu, C. Zhang, R. Dong, Y. Shi, Y. Wang, Y. Bai, J. Zhang, M. Cai and F. Zhou, Tribol. Int., 2019, 132, 118–129 CrossRef CAS.
- R. Ma, Q. Zhao, E. Zhang, D. Zheng, W. Li and X. Wang, Tribol. Int., 2020, 151, 106446 CrossRef CAS.
- B. Yu, D. G. Bansal, J. Qu, X. Sun, H. Luo, S. Dai, P. J. Blau, B. G. Bunting, G. Mordukhovich and D. J. Smolenski, Wear, 2012, 289, 58–64 CrossRef CAS.
- M. Freemantle, Chem. Eng. News, 1998, 76, 12 Search PubMed.
- F. U. Shah, S. Glavatskih, D. R. MacFarlane, A. Somers, M. Forsyth and O. N. Antzutkin, Phys. Chem. Chem. Phys., 2011, 13, 12865–12873 RSC.
- Z. Zheng, G. Shen, Y. Wan, L. Cao, X. Xu, Q. Yue and T. Sun, Wear, 1998, 222, 135–144 CrossRef CAS.
- J. Yan, X. Zeng, E. van der Heide and T. Ren, Tribol. Int., 2014, 71, 149–157 CrossRef CAS.
- J. Li, Z. Li, T. Ren, X. Zeng and E. van der Heide, Tribol. Int., 2014, 73, 101–107 CrossRef CAS.
- J. Yao and J. Dong, Tribol. Int., 1996, 29, 429–432 CrossRef CAS.
- W. E. Piers, S. C. Bourke and K. D. Conroy, Angew. Chem., Int. Ed., 2005, 44, 5016–5036 CrossRef CAS PubMed.
- T. S. De Vries, A. Prokofjevs and E. Vedejs, Chem. Rev., 2012, 112, 4246–4282 CrossRef CAS PubMed.
- X. Zhang, S. Wang, Z. Jiang, Y. Li and X. Jing, J. Am. Chem. Soc., 2020, 142, 21852–21860 CrossRef CAS PubMed.
- T. Rüther, T. D. Huynh, J. Huang, A. F. Hollenkamp, E. Alan Salter, A. Wierzbicki, K. Mattson, A. Lewis and J. H. Davis, Chem. Mater., 2010, 22, 1038–1045 CrossRef.
- M. Soltani, T. J. Ravine and J. H. Davis, Bioorg. Med. Chem. Lett., 2021, 36, 127808 CrossRef CAS PubMed.
- M. D. Soutullo, C. I. Odom, A. B. Smith, D. R. McCreary, R. E. Sykora, E. A. Salter, A. Wierzbicki and J. H. Davis, Inorg. Chim. Acta, 2007, 360, 3099–3102 CrossRef CAS.
- P. A. Fox, S. T. Griffin, W. M. Reichert, E. A. Salter, A. B. Smith, M. D. Tickell, B. F. Wicker, E. A. Cioffi, J. H. Davis, R. D. Rogers and A. Wierzbicki, Chem. Commun., 2005, 3679–3681 RSC.
- S. Coffie, J. M. Hogg, L. Cailler, A. Ferrer-Ugalde, R. W. Murphy, J. D. Holbrey, F. Coleman and M. Swadźba-Kwaśny, Angew. Chem., Int. Ed., 2015, 54, 14970–14973 CrossRef CAS PubMed.
- K. Matuszek, S. Coffie, A. Chrobok and M. Swadźba-Kwaśny, Catal. Sci. Technol., 2017, 7, 1045–1049 RSC.
- J. M. Hogg, A. Ferrer-Ugalde, F. Coleman and M. Swadźba-Kwaśny, ACS Sustain. Chem. Eng., 2019, 7, 15044–15052 CrossRef CAS.
- G. M. J. Al Kaisy, M. I. Abdul Mutalib and T. V. V. L. N. Rao, J. Mol. Liq., 2017, 242, 349–356 CrossRef CAS.
- M. R. Ortega Vega, J. Ercolani, S. Mattedi, C. Aguzzoli, C. A. Ferreira, A. S. Rocha and C. F. Malfatti, Ind. Eng. Chem. Res., 2018, 57, 12386–12396 CrossRef CAS.
- T. Myrdek, M. Stapels and W. Kunz, Proc. Inst. Mech. Eng. J., 2022, 236, 1409–1419 CrossRef CAS.
- S. A. Iqbal, J. Pahl, K. Yuan and M. J. Ingleson, Chem. Soc. Rev., 2020, 49, 4564–4591 RSC.
- M. Devillard, S. Mallet-Ladeira, G. Bouhadir and D. Bourissou, Chem. Commun., 2016, 52, 8877–8880 RSC.
- H. Y. Sun and D. G. Hall, At the Forefront of the Suzuki–Miyaura Reaction: Advances in Stereoselective Cross-Couplings, in Synthesis and Applications of Organoboron Compounds, ed. E. Fernandez and A. Whiting, Springer, 2015, pp. 221–242 Search PubMed.
- H. Nöth, S. Weber, B. Rasthofer, C. Narula and A. Konstantinov, Pure Appl. Chem., 1983, 55, 1453–1461 CrossRef.
- D. D. Perrin, B. Dempsey and E. P. Serjeant, pKa Prediction for Organic Acids and Bases, Springer, 1981, vol. 1 Search PubMed.
- A. E. Martell and R. M. Smith, Critical Stability Constants, Springer, 1974, vol. 1 Search PubMed.
- A. Martell, Critical Stability Constants: Volume 2: Amines, Springer Science & Business Media, 2012 Search PubMed.
- S. H. Yalkowsky, Y. He and P. Jain, Handbook of Aqueous Solubility Data, CRC press, 2016 Search PubMed.
- Y. Kosugi and T. Takeuchi, Org. Magn. Reson., 1979, 12, 435–437 CrossRef CAS.
- L. F. Johnson and W. C. Jankowski, Carbon-13 NMR Spectra: A Collection of Assigned, Coded and Indexed Spectra, Wiley-Interscience, 1972 Search PubMed.
- J. C. Elkaïm, S. Pace and J. G. Riess, J. Phys. Chem., 1980, 84, 354–360 CrossRef.
- T. Hjertberg, T. Hargitai and P. Reinholdsson, Macromol., 1990, 23, 3080–3087 CrossRef CAS.
- A. P. Tulloch and M. Mazurek, Lipids, 1976, 11, 228–234 CrossRef CAS.
- F. S. Freitas, A. S. Gonçalves, A. De Morais, J. E. Benedetti and A. F. Nogueira, J. Sustain. Energy, 2012, 11002–11003 Search PubMed.
- S. Ayyappan, N. Sundaraganesan, M. Kurt, T. R. Sertbakan and M. Özduran, J. Raman Spectrosc., 2010, 41, 1379–1387 CrossRef CAS.
- J. Andrieux, C. Goutaudier, L. Laversenne, E. Jeanneau and P. Miele, Inorg. Chem., 2010, 49, 4830–4835 CrossRef CAS PubMed.
- Y. Liao, Z. Chen, J. W. Connell, C. C. Fay, C. Park, J. W. Kim and Y. Lin, Adv. Funct. Mater., 2014, 24, 4497–4506 CrossRef CAS.
- S. A. Solomon, A. Del Grosso, E. R. Clark, V. Bagutski, J. J. W. McDouall and M. J. Ingleson, Organomet., 2012, 31, 1908–1916 CrossRef CAS.
- S. Takahashi, M. L. Saboungi, R. J. Klingler, M. J. Chen and J. W. Rathke, J. Chem. Soc., Faraday Trans., 1993, 89, 3591–3595 RSC.
- J. Estager, A. A. Oliferenko, K. R. Seddon and M. Swadźba-Kwaśny, Dalton Trans., 2010, 39, 11375–11382 RSC.
- J. Estager, J. D. Holbrey and M. Swadźba-Kwaśny, Chem. Soc. Rev., 2014, 43, 847–886 RSC.
- F. Coleman, G. Srinivasan and M. Swadźba-Kwaśny, Angew. Chem., Int. Ed., 2013, 52, 12582–12586 CrossRef CAS PubMed.
- K. Matuszek, A. Chrobok, J. M. Hogg, F. Coleman and M. Swadźba-Kwaśny, Green Chem., 2015, 17, 4255–4262 RSC.
- M. D. Soutullo, R. A. O'Brien, K. E. Gaines and J. H. Davis, Chem. Commun., 2009, 2529–2531 RSC.
- K. Kuroda, Y. Shimada and K. Takahashi, New J. Chem., 2018, 42, 15528–15532 RSC.
- J. Qu, D. G. Bansal, B. Yu, J. Y. Howe, H. Luo, S. Dai, H. Li, P. J. Blau, B. G. Bunting, G. Mordukhovich and D. J. Smolenski, ACS Appl. Mater. Interfaces, 2012, 4, 997–1002 CrossRef CAS PubMed.
- C. L. Camphen, PhD thesis, Imperial Colledge London, 2012.
- K. R. Wormuth and S. Zushma, Langmuir, 1991, 7, 2048–2053 CrossRef CAS.
- B. W. Pfennig, Principles of Inorganic Chemistry, Wiley, 2015th edn, 2015 Search PubMed.
- F. P. Bowden and L. Leben, Natural, 1938, 141, 691–692 CrossRef.
- R. Gusain, A. Khan and O. P. Khatri, J. Mol. Liq., 2020, 301, 112322 CrossRef CAS.
- E. Nyberg, J. Mouzon, M. Grahn and I. Minami, Appl. Sci., 2017, 7, 433 CrossRef.
- A. C. F. Mendonça, P. Malfreyt and A. A. H. Pádua, J. Chem. Theory Comput., 2012, 8, 3348–3355 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00451e |
| This journal is © The Royal Society of Chemistry 2024 |