
Insights into the interface reaction between electrolyte and Li2MnO3 from ab initio molecular dynamics simulations†
Xiaotong
Yan
a,
Chunwei
Zhu
a,
Weijie
Huang
a and
Yu-Jun
Zhao
 *ab
*ab
aDepartment of Physics, South China University of Technology, Guangzhou 510640, PR China. E-mail: zhaoyj@scut.edu.cn
bKey Laboratory of Advanced Energy Storage Materials of Guangdong Province, South China University of Technology, Guangzhou 510641, China
First published on 5th August 2024
Abstract
The complex interface reaction plays a critical role in the Li2MnO3 cathode material with high energy density. Here, the interface reactions between the liquid electrolyte molecules and the typical surfaces of Li2MnO3 are systematically investigated by ab initio molecular dynamics (AIMD) simulation and first-principles calculation. We demonstrate that the decomposition of electrolyte molecules on the different surfaces of Li2MnO3 exhibits a high degree of similarity. The carbonyl carbon (CC) and ether oxygen (OE) of the electrolyte molecule that bind to the O and Mn of the Li2MnO3 surface, respectively, are the prerequisites for the decomposition of the electrolyte molecules. The redox reaction between the electrolyte molecule and the surface of Li2MnO3 considerably weakens the strength of the CC–OE bond. In particular, the surface of (001) is an inert surface, which does not react with electrolyte molecules through our AIMD simulations. This is due to the large electron occupation energy gap between the electrolyte molecule and the (001) surface. This study provides a theoretical insight into the interface reaction between lithium-rich cathode materials and liquid electrolytes.
1 Introduction
Lithium-rich cathode materials (LRCMs), with ultra-high energy densities exceeding 300 mA h g−1, are strong candidates for solving the range problem of electric vehicles and large-scale energy storage applications.1–3 The cathode material Li2MnO3 attracts much attention due to its high voltage (up to 4.5 V), high specific capacity (theoretical capacity of up to 458 mA h g−1), cost effectiveness and environmental friendliness.4,5 However, the complex surface reactions can negatively affect stability, safety, and energy density during the cycle.4,6–8In general, a multitude of side reactions occur at the interface between cathode materials and liquid electrolytes during the cycle. These reactions not only consume limited amounts of cathode material and electrolyte but also lead to the formation of inert cathode-electrolyte interphase (CEI) films and gases.1,6
Researchers have performed a significant amount of work to mitigate this issue. Yan et al.9 investigated the mechanism of structural evolution of Li2MnO3, and their results indicate that the structural evolution of Li2MnO3 proceeds from the surface to the interior. Li et al.10 reported LRCMs coated with Al2O3 using a combination of atomic layer deposition (ALD) and bulk phase modulation techniques. These modified cathode materials have demonstrated improved cycling stability and safety. Wei et al.11 utilized the solid acid Zr(HPO4)·H2O to modify the surface of manganese-based LRCMs, effectively inhibiting the release of oxygen and the occurrence of surface side reactions. Additionally, Chen et al.12 investigated the deposition of S on the surface of LRCMs based on the properties of polyanionic polymers. Their research demonstrated that the surface deposition of S can lead to the formation of a (SOn)m− polyanion layer, which interacts with the surrounding O ions, thereby enhancing the cycling stability of LRCMs. Si et al.13 utilized the piezoelectric material LiTaO3 to coat LRCMs, which not only enhances the structural stability, but also improves the diffusion properties of Li+ at the interface through its piezoelectric properties. Zhu et al.14 employed MoO3 as a surface reagent. This reagent reacts with LiO precipitates near the surface of LRCMs, thereby increasing the concentration of transition metals. The findings indicate that the valence of transition metal (TM) ions is reduced, and the oxidation and evolution of O ions are effectively mitigated.
To sum up, there are numerous reports focusing on improving the performance of LRCMs.15–23 However, the interfacial reaction mechanisms between Li-rich cathode materials and electrolytes have been rarely reported. This can be attributed to the complexity and dynamics of interface reactions, which pose significant challenges even when in situ characterization techniques are employed.24–26
The first-principles calculation and ab initio molecular dynamics (AIMD) have proven effective in addressing this issue.27–31 Through molecular dynamics simulations, Zhang et al.32 demonstrated that the defective CEI material LiF reacts with LiPF6 at the interface. Qin et al.33 conducted a theoretical study of the decomposition of ethylene carbonate (EC) molecules on the (110) surface of both LiCoO2 and LiNi1/3Co1/3Mn1/3O2. Their study indicates that the initial stage of this reaction involves a ring-opening reaction of CC–OE cleavage. Subsequently, the H from the electrolyte molecule adsorbs onto the cathode surface. In addition, theoretical research has identified an oxygen release phenomenon occurring with pure Li1.2Ni0.6Mn0.2O2 cathode materials across various Li ion concentrations. It has been shown that sulfur doping can significantly suppress oxygen release during the charging process.34 However, the interface reactions between Li2MnO3 and electrolyte have not been well studied, in either experimental or theoretical contexts.
In this study, we employ AIMD simulations to effectively model the interface reactions between liquid electrolytes and the typical surfaces of Li2MnO3, which are characterized in our previous research.35 We then carefully examine the interface stability, reaction barrier, electronic structure and reaction mechanism of electrolytes on these surfaces of Li2MnO3. Our finding reveals that the CC and OE atoms of electrolyte molecules engage in charge transfer (binding) with the O and Mn ions on the Li2MnO3 surfaces during the adsorption stage. Consequently, the bond strength of CC–OE is thus greatly weakened. Finally, we summarize the similarities of reactions of electrolyte molecules on different surfaces of Li2MnO3, and offer a theoretical explanation to elucidate the interfacial reaction between electrolyte molecules and surfaces of the Li2MnO3 cathode material.
2 Computational details
2.1 Computational methods
The AIMD simulations and first-principles calculation are performed using the projector augmented wave (PAW) scheme,36 as implemented in the Vienna Ab initio Simulation Package (VASP).37,38 The exchange-correlation energies are approximated with the generalized gradient approximation (GGA) of Perdew, Burke, and Ernzerhof (PBE).39 For AIMD simulations, the NVT ensemble is employed at an equilibrium temperature of 400 K for a total time of 10 ps with a time interval of 1 fs.31,32,34 The cutoff energy for the plane wave expansion is set to 400 eV, and the convergence criterion for the electronic self-consistent iteration is 1 × 10−4 eV. According to ref. 40–44 and our test results, the van der Waals (vdW) interaction is not considered in AIMD simulations, as it does not affect our main conclusions (see Sections S7 and S8 of the ESI†). For subsequent property studies based on first-principles calculations, the vdW interaction (DFT-D3 method of Becke–Johnson45) and the cutoff energy option of 650 eV are used to improve the accuracy of calculations. The ionic positions are relaxed until the forces on each ion converge to less than 0.01 eV Å−1. The climbing image nudged elastic band (CINEB) method is used to obtain energy barriers for electrolyte molecule decomposition.46 In addition, to account for the on-site Coulomb interactions for the localized 3d electrons of transition metals, an additional Hubbard parameter correction is applied according to ref. 47 and 48 Within the GGA + U scheme, a single effective parameter Ueff = U – J is used, and an Ueff value of 4.5 eV for Mn is determined by the deintercalation voltage and band gap tests, which are in good agreement with the experiment.4,49 The gamma point of the Brillouin zone is sampled without consideration of the symmetry of systems. Spin polarization is taken into all the calculations.2.2 Computational model and process
The primary focus of this work is on constructing an interface model and subsequently calculating and analyzing its properties. First, our AIMD calculations are based on an interface model that includes a liquid electrolyte and the typical surface of Li2MnO3. Here, the liquid electrolyte is composed of ethylene carbonate (EC), dimethyl carbonate (DMC), and lithium hexafluorophosphate (LiPF6), with a volume ratio of EC to DMC of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and a LiPF6 concentration of 1 mol L−1 in EC/DMC.32 To ensure the reliability of AIMD calculations, a stable electrolyte cluster is meticulously investigated and subsequently used. The typical surfaces for the interface model are selected from our previous work,35 including the (010), (10
:1 and a LiPF6 concentration of 1 mol L−1 in EC/DMC.32 To ensure the reliability of AIMD calculations, a stable electrolyte cluster is meticulously investigated and subsequently used. The typical surfaces for the interface model are selected from our previous work,35 including the (010), (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) , (131), and (001) planes. A slab model, consisting of atleast five atomic layers, is employed to simulate the surface structure, with one-third of the slab's thickness fixed to the bulk structure. The stability analysis of the electrolyte cluster, the selection of typical surfaces, and the process of creating the interface model are detailed in Section S1.† Ultimately, employing the computational method outlined in Section 2.1, we calculate and analyze the relevant properties of the interface. A schematic diagram of the research process is depicted in Fig. 1.
) , (131), and (001) planes. A slab model, consisting of atleast five atomic layers, is employed to simulate the surface structure, with one-third of the slab's thickness fixed to the bulk structure. The stability analysis of the electrolyte cluster, the selection of typical surfaces, and the process of creating the interface model are detailed in Section S1.† Ultimately, employing the computational method outlined in Section 2.1, we calculate and analyze the relevant properties of the interface. A schematic diagram of the research process is depicted in Fig. 1.
 | ||
| Fig. 1 The schematic diagram of the research process for the interface reaction of liquid electrolyte and the Li2MnO3 surface. | ||
3 Results and discussion
To clarify the interface reaction between the electrolyte molecules and the various surfaces of Li2MnO3, the AIMD simulations for each distinct interface are analyzed and discussed in this section. First, we described the decomposition process of electrolyte molecules during the AIMD simulations. Subsequently, we extracted the initial and final states of these decomposition structures and calculated the energy barriers for their decomposition (see Section S3.5† for detailed calculations of the energy barriers). Finally, we studied the properties exhibited during the decomposition of electrolyte molecules and explained these properties by analyzing the changes in electronic structure throughout the decomposition process. In the following figures, ISO stands for an isolated molecule or surface. The acronyms of IS, TS, and FS represent the initial, transitional, and final states, respectively, of different interfaces during the process of electrolyte molecule decomposition. In this study, the (001) surface is identified as inert. It exhibits no reaction with electrolyte molecules, regardless of the AIMD simulations conducted (further details are available in Sections S4 and S8†). A comprehensive explanation is provided in Section 3.4.3.1 The interface reaction of the (010) surface
The results of the interface reaction of the electrolyte molecule on the (010) surface show that the EC molecule is decomposed. The relevant data on the decomposition of the EC molecule are shown in Fig. 2. By examining Fig. 2(a) and (b), we observe the following sequence of events regarding the decomposition of the EC molecule on the (010) surface:(i) The OC of the EC molecule becomes adsorbed onto Mn1 of the (010) surface. (ii) Overtime, the distances between the CC and OE of the EC molecule and the O and Mn2 of the (010) surface decrease. (iii) At the time of 2.96 ps, the EC molecule is stably adsorbed on the (010) surface, marking the commencement of its decomposition reaction. (iv) Subsequently, the distance between CC and OE of the EC molecule increases progressively over time. (v) Commencing at 3.08 ps, the bond lengths of CC–O and OE–Mn2 remain constant, whereas the bond of CC–OE is broken. This observation suggests that the EC molecule undergoes a ring-opening reaction on the (010) surface.
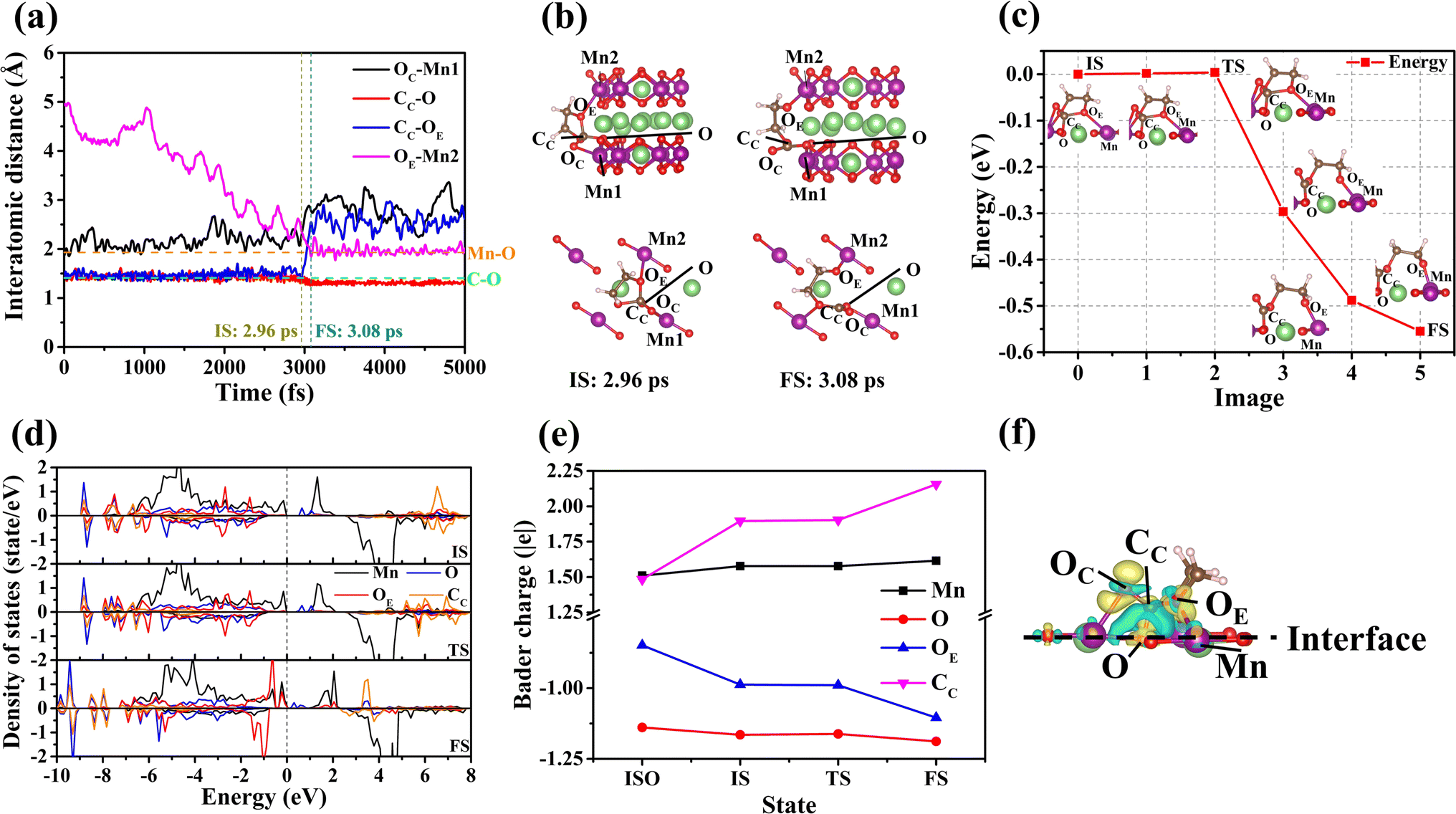 | ||
| Fig. 2 (a) The changes in atomic distance upon decomposition of EC on the (010) surface. (b) and (c) The schematic diagram and energy barrier of EC decomposition on the (010) surface. (d) and (e) The partial density of states (PDOS) and Bader charges of Mn, O, OE, and CC in different states. (f) The charge density difference between the EC and the (010) surface after adsorption (the iso-surface charge density is assumed to be 5 × 10−3 e bohr−3). | ||
To ascertain the difficulty of the ring-opening reaction for the EC molecule on the (010) surface, we calculated the energy barrier associated with the decomposition process, as depicted in Fig. 2(c). The calculated energy barrier for the decomposition of the EC molecule is found to be minimal, at only 4 meV (see Section S3.5†). This low value suggests that the decomposition of the EC molecules on the (010) surface is likely to occur spontaneously.
To address this issue, we have analyzed the changes in the electronic structure, specifically the partial density of states (PDOS) of the EC molecule as it decomposes on the (010) surface. As illustrated in Fig. 2(d), there are minimal changes in the electronic occupancy states for the Mn, O, OE, and CC (c.f.Fig. 2(c)) during the transition from the initial state (IS) to the transitional state (TS). This proves that no remarkable charge transfer occurs during this process. At the same time, the Bader charges for the Mn, O, OE, and CC have also been calculated across different states. Fig. 2(e) shows that the charges of CC and OE change significantly before and after adsorption. In contrast, the charge changes for Mn and O ions at the (010) surface are not obvious, which may be attributed to the redistribution of surface charge. However, the Bader charges for Mn, O, OE, and CC remain almost unchanged during the transition from the IS to the TS. This constancy, as depicted in Fig. 2(e), further supports the notion that no charge transfer occurs during the transition. The above analysis indicates that any charge transfer between the relevant ions during the adsorption process should influence the energy barrier. The charge density difference Δρ (see Fig. 2(f)) of EC and the (010) surface after adsorption is thus calculated according to formula (1).
| Δρ = ρads − ρsurf − ρmol, | (1) |
3.2 The interface reaction of the (10![[1 with combining macron]](https://www.rsc.org/images/entities/h3_char_0031_0304.gif) ) surface
) surface
The EC molecule decomposes at the interface between the liquid electrolyte and the (10) surface. The corresponding results of EC decomposition are shown in Fig. 3. Fig. 3(a) and (b) illustrate that the decomposition of the EC molecule occurs through the following steps:
 | ||
| Fig. 3 (a) The changes in atomic distance upon decomposition of EC on the (10) surface. (b) and (c) The schematic diagram and energy barrier of EC decomposition on the (10) surface. (d) and (e) The PDOS and Bader charges of Mn, O, OE, and CC in different states. (f) The charge density difference between EC and the (10) surface after adsorption (the iso-surface charge density is assumed to be 5 × 10−3 e bohr−3). | ||
(i) The Li1 of the surface adsorbs the OC of the EC molecule. (ii) The distances between CC–O and OE1–Mn1 decrease over time. (iii) The EC molecule becomes stably adsorbed on the surface and initiates dissociation at 1.00 ps. (iv) As time continues to increase, the distance between CC and OE1 of the EC molecule gradually increases. (v) From 1.30 ps onwards, the bond lengths of the CC-O and OE1–Mn1 bonds remain unchanged. However, the bond between CC and OE1 breaks, and ring-opening occurs within the EC molecule.
The energy barrier for the decomposition process of the EC molecule on the (10) surface has been calculated to ascertain the difficulty of the ring-opening reaction, as depicted in Fig. 3(c). According to Fig. 3(c), the energy barrier during the decomposition of the EC molecule is nearly zero, with a value of only 0.2 meV (see Section S3.5†). This suggests that the decomposition of EC molecules on the (10) surface is spontaneous. To further investigate this finding, the electronic structure of the EC molecule during decomposition on the (10) surface has been calculated (c.f.Fig. 3(d)). The electronic occupation states of Mn, O, OE, and CC (as seen in Fig. 3(c)) exhibit minimal changes from the IS to the TS, indicating that this process does not clearly involve a charge transfer. Additionally, the Bader charges of Mn, O, OE, and CC have been calculated across different states (c.f.Fig. 3(e)). Although the charges of CC and OE of the EC change significantly during adsorption, the Bader charges for Mn, O, OE, and CC remain constant throughout the transition from the IS to the TS. This consistency further supports the absence of charge transfer from the IS to the TS. Consequently, the energy barrier may be influenced by charge transfer between the relevant ions during the adsorption process. Fig. 3(f) illustrates the charge density difference before and after the adsorption of the EC molecule on the (10) surface. From Fig. 3(e) and (f), it is observed that the CC in the EC molecule has lost a significant number of electrons. These electrons are transferred to the O atom of the (10) surface as a result of the binding interaction. Additionally, due to the binding, the OE of the EC molecule receives electrons from the Mn of the (10) surface. In particular, there is a substantial loss of electrons surrounding CC in the CC–OE bond. This indicates that the bond strength of the CC–OE bond is greatly reduced, so that the energy barrier is nearly zero.
3.3 The interface reaction of the (131) surface
Interface reactions take place on the surface of (131). According to the AIMD simulation, the EC and DMC molecules decompose on this surface (DMC decomposition, see Section S5†). Fig. 4 shows the corresponding results of the EC molecule decomposition. As observed in Fig. 4(a) and (b), the decomposition of the EC molecule follows a similar decomposition process to that described previously:(i) The OC of the EC molecule is adsorbed onto the Mn1 of the (131) surface. (ii) The distances of CC–O and OE–Mn2 decrease over time. (iii) At the time point of 0.67 ps, the stably adsorbed EC molecule initiates the ring-opening reaction on the (131) surface. (iv) The distance between CC and OE continues to increase with time. (v) At 1.21 ps, the CC–OE bond is broken, while the bond lengths of CC–O and OE–Mn2 remain stable.
 | ||
| Fig. 4 (a) The changes in atomic distance upon decomposition of EC on the (131) surface. (b) and (c) The schematic diagram and energy barrier of EC decomposition on the (131) surface. (d) and (e) The PDOS and Bader charges of Mn, O, OE, and CC in different states. (f) The charge density difference between EC and the (131) surface after adsorption (the iso-surface charge density is assumed to be 7 × 10−3 e bohr−3). | ||
Similar to the previous results, the energy barrier of the ring-opening reaction of the EC molecule on the (131) surface has been calculated (see Fig. 4(c)). According to Fig. 4(c), there is nearly no energy barrier observed during the decomposition of the EC molecule, with a value of only 7 meV (see Section S3.5†). This minimal energy barrier suggests that the EC molecules decompose spontaneously on the (131) surface. To understand this phenomenon, the electronic structure and Bader charge of the EC molecule on the (131) surface have been calculated for various states (c.f.Fig. 4(d) and (e)). From Fig. 4(d) and (e), the electronic occupancies and Bader charges of Mn, O, OE, and CC (see Fig. 4(c)) are stable during the transition from the IS to the TS, indicating that no charge transfer occurs during this transition. However, significant changes in the charges of CC and OE of the EC are observed before and after adsorption on the (131) surface. According to the above analysis, the charge transfer between the relevant ions may affect the energy barrier in the adsorption process. The charge density difference of the adsorption of EC molecules on the (131) surface is shown in Fig. 4(f). When Fig. 4(e) and (f) are considered together, a similar conclusion can be drawn. The CC of the EC molecule and the O of the (131) surface display distinct electronic behaviors, with the CC releasing electrons and the O atom accepting them, due to their mutual binding. The OE of the EC molecule also accepts electrons from the Mn of the (131) surface, forming a bond. Consequently, the electrons surrounding the CC have significantly decreased in the CC–OE bond. The bond strength of the CC–OE bond is greatly reduced, so that the energy barrier for the EC ring-opening is nearly zero.
3.4 Discussion
Analysis of the above results reveals that the decomposition of the electrolyte molecules (EC/DMC) on the (010), (10), and (131) surfaces of Li2MnO3 shows an extremely high degree of similarity. This suggests that the decomposition of EC/DMC on the Li2MnO3 surfaces can be attributed to a combination of a special adsorption site and a stable electronic structure (redox reaction). The influence of the special adsorption site is illustrated in Fig. 5.
 | ||
| Fig. 5 Schematic diagram of decomposition of electrolyte molecules (EC/DMC) on the surfaces of Li2MnO3. Only atoms involved in the decomposition of electrolyte molecules are shown. | ||
According to the above analysis and as depicted in Fig. 5, the decomposition of the electrolyte molecule (EC/DMC) on the surfaces of Li2MnO3 can generally be summarized as follows:
(I) Localized adsorption: the OC of the electrolyte molecule adsorbs on to Li/Mn on the surface, which plays a role in positioning. Following this, the CC and OE of the electrolyte molecule are adsorbed onto the Li2MnO3 surface in the vicinity of this site.
(II) Specific adsorption leading to a decomposition reaction: this study shows that only the effective adsorption and binding of CC–O and OE–Mn can trigger the decomposition reaction of the electrolyte molecules. However, the adsorption and binding of CC–O and OE–Li do not lead to the decomposition reaction of electrolyte molecules (c.f. Sections S6 and S8†). This is mainly due to the strong redox reaction between electrolyte molecules and the Li2MnO3 surfaces (the detailed analysis shows the stable electronic structure). As a result, the stable chemical bonds CC–O and OE–Mn are formed and the strength of the CC–OE bond is greatly weakened. Therefore, the interaction between the electrolyte molecules and the Li2MnO3 surfaces is strengthened, while the internal strength of the electrolyte molecules is weakened.
(III) Distortion decomposition: in this study, the different bond lengths and angles between the atoms (ions) of electrolyte molecules and Li2MnO3 surfaces induce distortion. This distortion, in turn, leads to the decomposition of the electrolyte molecules, which fulfills the conditions established in step II.
In general, achieving a stable electronic structure is the starting point of a redox reaction. The above analysis shows that a large part of the charge transfer takes place mainly in the adsorption stage. The changes in the occupancy state of electrons before and after the adsorption of electrolyte molecules on Li2MnO3 surfaces are illustrated in Fig. 6, which takes into account the above PDOS, Bader charge, and charge density difference data.
 | ||
| Fig. 6 Schematic diagram of the electronic structure change of electrolyte molecules (EC/DMC) and surfaces of Li2MnO3. | ||
Fig. 6 shows that Mn–O and CC-OE are bound to the isolated surface and molecule, respectively (old bonds). However, this state is destroyed when the electrolyte molecules are adsorbed at specific sites on the Li2MnO3 surface. After adsorption, effective bonds (new bonds) are formed for the CC–O and OE–Mn of the electrolyte molecule and the Li2MnO3 surface. This is mainly due to the close occupation of the energy levels of the electrons between the binding atoms (ions), along with a subsequent effective reduction in the electronic energy levels after binding.
In addition, the (001) surface is considered inert in our research, as it does not react with the electrolyte molecules. This behavior can be well explained by referring to Fig. 6. The (001) surface is a stable surface that satisfies the stoichiometric ratio, with the outermost layer composed of Li ions (as detailed in Section S1.2 and ref. 35). However, Fig. 6 indicates that the electron occupation energy level of the Li ion is quite low (the large energy level gap with the electrolyte molecule). This makes it difficult to effectively bond or react (charge transfer) with the electrolyte molecules, which aligns with the findings of relevant experimental studies.50 Meanwhile, the (001) surface has the highest energy level of occupied electrons compared to other surfaces and electrolyte molecules. As a result, the (001) surface remains inert even under conditions of charging (c.f. Section S4†). Similarly, the large energy level gap between the Li ion and the electrolyte molecule also accounts for why the adsorption and binding of CC–O and OE–Li does not cause a decomposition reaction of the electrolyte molecules (see Section S6†). This indicates that the stable electronic structure leads to the specificity of the adsorption site.
It should be noted that only the decomposition of the DMC electrolyte molecule at the (131) surface has been observed, albeit due to the limited computational resources. However, the decomposition processes are very similar to the ring-opening process of EC molecules (see Section S5†). Therefore, we postulate that the findings from our discussion on the decomposition of electrolyte molecules on the Li2MnO3 surface are also applicable to the DMC molecule, although the primary basis for these conclusions is drawn from the ring-opening reaction of the EC molecule.
4 Conclusion
In summary, we have systematically investigated the interface reaction, reaction barrier, electronic structure, and reaction mechanism between the liquid electrolyte molecules and the typical surfaces of Li2MnO3 by the AIMD simulation and first-principles calculation. It is shown that the electrolyte molecules EC (or DMC) have decomposition reactions on the surfaces of (010), (10) , and (131), with the exception of the (001) surface, which remains inert. The energy barrier for the decomposition of the electrolyte molecule is nearly zero. This phenomenon is a critical redox reaction caused by the special adsorption of electrolyte molecules on the Li2MnO3 surfaces. On one hand, it weakens the internal bond strength (CC–OE) of the electrolyte molecule. On the other hand, it strengthens the interaction between the electrolyte molecule and the Li2MnO3 surface (CC–O and OE–Mn). Additionally, the different bond lengths and angles between the atoms (ions) of the electrolyte molecules and the Li2MnO3 surface influence the decomposition of the electrolyte molecules. The inertia of the (001) surface is due to the low electron occupation energy level of Li ions in the outermost layer of the (001) surface. This makes effective binding with the electrolyte molecule difficult. Although our computational methods and resources limit us from simulating the interface reactions over extended periods, the initial stages of these reactions have been effectively captured. This early insight is crucial for further elucidating the mechanisms of interface reactions and for enhancing the properties of cathode materials. Furthermore, given that cathode materials are subjected to frequent charge and discharge cycles, the study of the interface reactions of Li2MnO3 cathode materials during the charging process will be a focal point of our future research.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Xiaotong Yan: methodology, investigation, formal analysis, visualization, writing – original raft. Chunwei Zhu: methodology, writing – review & editing. Weijie Huang: methodology, writing – review & editing. Yu-Jun Zhao: conceptualization, supervision, resources, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 12074126).The computer time at the High Performance Computational center at the South China University of Technology is gratefully acknowledged.Notes and references
- W. He, W. Guo, H. Wu, L. Lin, Q. Liu, X. Han, Q. Xie, P. Liu, H. Zheng and L. Wang, et al. , Adv. Mater., 2021, 33, 2005937 CrossRef CAS PubMed.
- Y. Ding, Z. P. Cano, A. Yu, J. Lu and Z. Chen, Electrochem. Energy Rev., 2019, 2, 1–28 CrossRef CAS.
- W. Lee, S. Muhammad, C. Sergey, H. Lee, J. Yoon, Y.-M. Kang and W.-S. Yoon, Angew. Chem., Int. Ed., 2020, 59, 2578–2605 CrossRef CAS PubMed.
- N. Guerrini, L. Jin, J. G. Lozano, K. Luo, A. Sobkowiak, K. Tsuruta, F. Massel, L.-C. Duda, M. R. Roberts and P. G. Bruce, Chem. Mater., 2020, 32, 3733–3740 CrossRef CAS.
- J. Song, H. Wang, Y. Zuo, K. Zhang, T. Yang, Y. Yang, C. Gao, T. Chen, G. Feng and Z. Jiang, et al. , Electrochem. Energy Rev., 2023, 6, 20 CrossRef CAS.
- Y. Lei, J. Ni, Z. Hu, Z. Wang, F. Gui, B. Li, P. Ming, C. Zhang, Y. Elias and D. Aurbach, et al. , Adv. Energy Mater., 2020, 10, 2002506 CrossRef CAS.
- S. Sharifi-Asl, J. Lu, K. Amine and R. Shahbazian-Yassar, Adv. Energy Mater., 2019, 9, 1900551 CrossRef.
- E. Cho, K. Kim, C. Jung, S.-W. Seo, K. Min, H. S. Lee, G.-S. Park and J. Shin, J. Phys. Chem. C, 2017, 121, 21118–21127 CrossRef CAS.
- P. Yan, L. Xiao, J. Zheng, Y. Zhou, Y. He, X. Zu, S. X. Mao, J. Xiao, F. Gao and J.-G. Zhang, et al. , Chem. Mater., 2015, 27, 975–982 CrossRef CAS.
- Y. Li, Z. Shi, B. Qiu, J. Zhao, X. Li, Y. Zhang, T. Li, Q. Gu, J. Gao and Z. Liu, Adv. Funct. Mater., 2023, 33, 2302236 CrossRef CAS.
- H.-X. Wei, Y.-M. Liu, Y.-H. Luo, Y.-D. Huang, L.-B. Tang, Z.-Y. Wang, C. Yan, J. Mao, K.-H. Dai and Q. Wu, et al. , Adv. Funct. Mater., 2024, 34, 2307583 CrossRef CAS.
- Q. Chen, Y. Pei, H. Chen, Y. Song, L. Zhen, C.-Y. Xu, P. Xiao and G. Henkelman, Nat. Commun., 2020, 11, 3411 CrossRef CAS PubMed.
- M. Si, D. Wang, R. Zhao, D. Pan, C. Zhang, C. Yu, X. Lu, H. Zhao and Y. Bai, Advanced Science, 2020, 7, 1902538 CrossRef CAS PubMed.
- Z. Zhu, D. Yu, Y. Yang, C. Su, Y. Huang, Y. Dong, I. Waluyo, B. Wang, A. Hunt and X. Yao, et al. , Nat. Energy, 2019, 4, 1049–1058 CrossRef CAS.
- Z. Ye, B. Zhang, T. Chen, Z. Wu, D. Wang, W. Xiang, Y. Sun, Y. Liu, Y. Liu and J. Zhang, et al. , Angew. Chem., Int. Ed., 2021, 60, 23248–23255 CrossRef CAS PubMed.
- Y. Shin, W. H. Kan, M. Aykol, J. K. Papp, B. D. McCloskey, G. Chen and K. A. Persson, Nat. Commun., 2018, 9, 4597 CrossRef PubMed.
- P. Yan, J. Zheng, X. Zhang, R. Xu, K. Amine, J. Xiao, J.-G. Zhang and C.-M. Wang, Chem. Mater., 2016, 28, 857–863 CrossRef CAS.
- S. Zhao, B. Sun, K. Yan, J. Zhang, C. Wang and G. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 33260–33268 CrossRef CAS PubMed.
- S. Jeong, K. Choi, V.-C. Ho, J. Cho, J.-S. Bae, S. C. Nam, T. Yim and J. Mun, Chem. Eng. J., 2022, 434, 134577 CrossRef CAS.
- J. Liu, Z. Wu, M. Yu, H. Hu, Y. Zhang, K. Zhang, Z. Du, F. Cheng and J. Chen, Small, 2022, 18, 2106337 CrossRef CAS PubMed.
- W. He, P. Liu, B. Qu, Z. Zheng, H. Zheng, P. Deng, P. Li, S. Li, H. Huang and L. Wang, et al. , Advanced Science, 2019, 6, 1802114 CrossRef PubMed.
- W. Guo, Y. Zhang, L. Lin, Y. Liu, M. Fan, G. Gao, S. Wang, B. Sa, J. Lin and Q. Luo, et al. , Small, 2023, 19, 2300175 CrossRef CAS PubMed.
- D. Luo, H. Xie, F. Tan, X. Ding, J. Cui, X. Xie, C. Liu and Z. Lin, Angew. Chem., Int. Ed., 2022, 61, e202203698 CrossRef CAS PubMed.
- Y. Yan, S. Zhou, Y. Zheng, H. Zhang, J. Chen, G. Zeng, B. Zhang, Y. Tang, Q. Zheng and C. Wang, et al. , Adv. Funct. Mater., 2024, 34, 2310799 CrossRef CAS.
- J. F. Browning, L. Baggetto, K. L. Jungjohann, Y. Wang, W. E. Tenhaeff, J. K. Keum, D. L. Wood III and G. M. Veith, ACS Appl. Mater. Interfaces, 2014, 6, 18569–18576 CrossRef CAS PubMed.
- W. Lu, J. Zhang, J. Xu, X. Wu and L. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 19313–19318 CrossRef CAS PubMed.
- K. T. Butler, G. Sai Gautam and P. Canepa, npj Comput. Mater., 2019, 5, 19 CrossRef.
- S. Shi, J. Gao, Y. Liu, Y. Zhao, Q. Wu, W. Ju, C. Ouyang and R. Xiao, Chin. Phys. B, 2015, 25, 018212 CrossRef.
- K. Leung, J. Phys. Chem. C, 2012, 116, 9852–9861 CrossRef CAS.
- T. Tamura, M. Kohyama and S. Ogata, Phys. Rev. B, 2017, 96, 035107 CrossRef.
- J. L. Tebbe, T. F. Fuerst and C. B. Musgrave, ACS Appl. Mater. Interfaces, 2016, 8, 26664–26674 CrossRef CAS PubMed.
- B. Zhang, Z. Lin, H. Chen, L.-W. Wang and F. Pan, J. Mater. Chem. A, 2020, 8, 2613–2617 RSC.
- X. Qin, P. B. Balbuena and M. Shao, J. Phys. Chem. C, 2019, 123, 14449–14458 CrossRef CAS.
- K.-Y. Lin, S. Nachimuthu, H.-W. Huang and J.-C. Jiang, npj Comput. Mater., 2022, 8, 210 CrossRef CAS.
- X. Yan, X. Zhou, C. Zhu, W. Huang and Y.-J. Zhao, J. Mater. Chem. A, 2024, 12, 3722–3733 RSC.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- E. P. Kamphaus, S. Angarita-Gomez, X. Qin, M. Shao, M. Engelhard, K. T. Mueller, V. Murugesan and P. B. Balbuena, ACS Appl. Mater. Interfaces, 2019, 11, 31467–31476 CrossRef CAS PubMed.
- S.-H. Pan, K.-Y. Lin, W.-X. Wu, B. J. Hwang and J.-C. Jiang, ACS Appl. Energy Mater., 2023, 6, 3291–3300 CrossRef CAS.
- S. Angarita-Gomez and P. B. Balbuena, ACS Appl. Mater. Interfaces, 2022, 14, 56758–56766 CrossRef CAS PubMed.
- D. Kuai and P. B. Balbuena, ACS Appl. Mater. Interfaces, 2022, 14, 2817–2824 CrossRef CAS PubMed.
- J. Si and Y. Ma, J. Phys. Chem. C, 2023, 127, 23541–23550 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943 CrossRef CAS PubMed.
- F. Zhou, M. Cococcioni, C. A. Marianetti, D. Morgan and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 235121 CrossRef.
- M. Ranjeh, M. Masjedi-Arani, M. Salavati-Niasari and H. Moayedi, J. Mol. Liq., 2020, 300, 112292 CrossRef CAS.
- A. Quesne-Turin, D. Flahaut, G. S. Vallverdu, L. Croguennec, J. Allouche, F. Weill, M. Ménétrier and I. Baraille, Appl. Surf. Sci., 2021, 542, 148514 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04598j |
| This journal is © The Royal Society of Chemistry 2024 |