
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantification of dimethylamine in low concentration particulate matter by reducing the concentration of 9-fluorenylmethyl chloroformate†
Susana
García-Alonso
 *a,
Francisco Javier
Gómez-Moreno
b,
Elisabeth
Alonso-Blanco
b and
Rosa María
Pérez-Pastor
a
*a,
Francisco Javier
Gómez-Moreno
b,
Elisabeth
Alonso-Blanco
b and
Rosa María
Pérez-Pastor
a
aTechnology Department, Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas (CIEMAT), Madrid 28040, Spain. E-mail: susana.garcia@ciemat.es
bEnvironment Department, Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas (CIEMAT), Madrid 28040, Spain
First published on 22nd November 2024
Abstract
This study presents a refined method that uses liquid chromatography with a fluorescence detector (LC-FD) to quantify trace amounts of dimethylamine in particulate matter (PM). This method was optimized to prioritize simplicity, cost-effectiveness and practicality. To ensure accurate and reliable analysis, strict protocols and procedures were followed to minimize cross-contamination. Separate workspaces were designated for preparing control blanks and sample treatments in one area and standard solutions in another, thus mitigating the risk of cross-contamination. An evaluation was conducted on different concentrations of 9-fluorenylmethyl chloroformate to derivatize dimethylamine. The results showed that a concentration of 3 μg mL−1 was effective in derivatizing dimethylamine concentrations up to 300 ng mL−1. Increasing the concentration of the derivatization reagent from 2.9 to 7.3 μg mL−1 resulted in slightly elevated dimethylamine levels in blank measurements. Also, during the preparation of standards at low concentrations, high analytical coefficients of variation were observed. This highlights the importance of checking for potential sources of contamination. Method precision and quantification limits were evaluated through blank analysis, yielding values of approximately 20% and 20 ng mL−1, respectively, consistent with chromatographic determination for environmental analysis. The suitability of the method for environmental analysis was demonstrated by analyzing eight PM2.5 samples. The concentrations of methylamine and dimethylamine were found to range from 0.8 to 3 ng m−3 and 1.4 to 7.1 ng m−3, respectively, in accordance with the literature. Comparison with concurrent carbonyl measurements revealed similar concentration profiles. Both types of analyses can be performed using affordable methodologies that involve prior derivatization using a reduced concentration of the derivatization reagent.
Introduction
Atmospheric particles have been the subject of debate regarding their effects on human health and ecosystems. Reactive atmospheric gases play a key role in their formation and transformation, making it essential to know their implications in atmospheric processes. In this regard, recently, there has been increased attention to atmospheric amines and their impact on air quality and climate, especially since they have been linked to new particle formation.1,2 C1–C6 alkylamines are mainly emitted from various natural and anthropogenic sources, such as industry, vehicle emissions, biomass burning, and biological activity, particularly from marine environments.3,4 However, the absence of amine emission inventories contributes to high uncertainties in global and regional atmospheric models, limiting their ability to accurately simulate their distribution.3,5–7 Thus, further research and comprehensive observations are necessary to deepen the understanding of amine behavior and seasonal variations in aerosol concentrations.8,9Field observations are essential to gain a comprehensive understanding of organic behavior, including their temporal and spatial distribution. The growing interest in the role of amines in atmospheric processes is based on their reactive capacity. Amines contribute to the gas-to-particle conversion through processes such as direct dissolution, acid–base chemistry and heterogeneous reactions, serving as effective precursors of atmospheric new particles.3,5 They react with gaseous inorganic acids to form salt particles and with organic acids to form amides.3,4 They also have significant potential to react with atmospheric oxidants, contributing to secondary organic aerosol formation.3 Additionally, particle-phase amines may react further with carbonyl compounds or oxidants, leading to the formation of more oxidized compounds.5,6
Measuring short-chain amines in air is difficult, with the risk of loss during sampling due to their high polarity, water solubility, volatility, sticky nature and presence at trace levels.10,11 Gas-phase measurement has primarily relied on online technology,8 while offline methods such as diffusion denuders and impregnated filters encounter difficulties with sampling efficiency.9,12 Low flow rates (14–40 L min−1) and sampling times of several days or even weeks are often used, further complicating the process and data interpretation. However, some studies have used high-volume aerosol sampling conditions, in the order of hundreds of liters per minute, to measure short-chain aliphatic amines in particulate matter (PM).5,6,8,13 This method of sampling increases the sample size and facilitates chromatographic analysis, which requires high sensitivity due to the presence of trace levels.
Ion chromatography is the preferred method for direct analysis of low molecular weight aliphatic amines due to their polar nature.9,14–17 Analytical methods that use liquid chromatography and detectors such as ultraviolet/fluorescence for aliphatic amines require the transformation of these compounds into less polar derivatives to enable their detection.4 Various reagents have been used for derivatization,18 including 9-fluorenylmethyl chloroformate (Fmoc-Cl), a derivatizing reagent that can simultaneously derivatize primary and secondary amines19,20 and is mainly used in the biological field to determine amino acids or longer chain aliphatic amines.21
Jámbor et al.22 have already pointed out that there is some controversy in the literature regarding the most appropriate methodology depending on the suitable Fmoc concentration. As in any derivatization reaction, the concentration of the derivatizing agent plays a critical role in performance because it must sufficiently meet sensitivity requirements.20 Recommended concentrations range from milligrams per milliliter4,23,24 to hundreds of micrograms per milliliter.20,25,26 To achieve consistent and reliable chromatography, it is important to balance sensitivity and peak stability, while minimizing the contamination of the chromatographic system.11,19
Despite its utility, Fmoc-Cl exhibits limitations due to the possible generation of by-products. These by-products can interfere with chromatographic separation and reduce reaction efficiency, for instance in the presence of pH fluctuations. Some associated by-products include:
(1) Hydrolysis of Fmoc-OH in aqueous environments, leading to the formation of a polar compound. This results in chromatographic elution with short retention times.27
(2) Under acidic conditions, another by-product elutes with longer retention times, further decreasing the efficiency of derivatization due to the pH decrease.
(3) Gradual degradation occurs over time.
When preparing standards using amine reagents, some publications lack detailed descriptions or even provide incomplete or no information at all. The existing literature suggests two predominant methodologies: acquiring amine compounds in the form of solid hydrochloride salts4,6 or as a pure reagent solution dissolved in methanol25,28 or water.19
This study aims to adapt a simple and affordable LC-FD method to routinely quantify trace levels of dimethylamine in aerosols, included in the OASIS project (evolution of secondary organic aerosols: composition, sources and formation of new particles under ambient conditions), focused on characterizing particle nucleation processes. The main objectives are to reduce reagent consumption and ensure result reproducibility. In the evaluation of the analytical procedure, we focus on mitigating background levels, an inherent analytical challenge to the method. Finally, methodological reliability was assessed by analyzing eight aerosol samples obtained from a suburban area as a real environmental setting. The results obtained were compared with those obtained from the analysis of samples collected simultaneously to characterize carbonyl and BTEX compounds. These preliminary results contribute to the international database on the concentration levels of these compound families in the study area.
Experimental
Chemicals
The following products were purchased from Sigma-Aldrich (Deisenhofen, Germany): sodium hydroxide and Fmoc-Cl used as the derivatization reagent. Dimethylamine, methylamine, and diethylamine hydrochlorides (purity >98%) as a pure reagent solution dissolved in methanol (at a concentration of 2 M) obtained from Sigma-Aldrich (Deisenhofen, Germany) were used for the preparation of standards. High-purity water was sourced from a Milli-Q IQ7003 Water Purification System, while acetonitrile was obtained from Merck (Darmstadt, Germany).Boric acid used for buffer preparation was obtained from Honeywell (for analysis >99.8%, Fluka). Borate buffer (10 mL) was prepared from boric acid (0.55 M) and sodium hydroxide (0.5 M) for neutralization purposes. In detail, around 0.34 g of boric acid and 0.2 g of sodium hydroxide are dissolved in water and made up to a final volume of 10 mL, and was subsequently stored in a refrigerator (5 °C).
The PTFE syringe filter (13 mm, 0.45 μm) used in sample preparation was purchased from Membrane Solutions (Spring View Lane Plano, EEUU).
Chromatographic analysis
The liquid chromatograph used for the analysis was an Agilent 1100 series (Waldbronn, Germany). It was equipped with an Agilent column (Zorbax Eclipse XDB-C18, 5 mm, 250 × 4.6 mm) maintained at 36.7 °C and coupled with a fluorescence detector (excitation at 267 nm and emission at 314 nm). The injection volume used was 25 μL. The mobile phase, which consisted of a mixture of acetonitrile and water (55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :45 v/v), was delivered at a flow rate of 1.2 mL min−1 and programmed to reach 70% acetonitrile in 6 minutes and maintained for 3 minutes. An equilibration delay of 2 minutes was applied before the next injection, resulting in a total analysis time of 11 minutes.
:45 v/v), was delivered at a flow rate of 1.2 mL min−1 and programmed to reach 70% acetonitrile in 6 minutes and maintained for 3 minutes. An equilibration delay of 2 minutes was applied before the next injection, resulting in a total analysis time of 11 minutes.
Given the expected low levels of amines in PM aerosol, the calibration curve was constructed by assessing standards with concentrations below 100 ng mL−1 of the amine, with particular emphasis on those in the range of 30 to 60 ng mL−1. Final quantification was carried out using a response factor determined from a low-concentration standard solution, as described in the section of Results.
Derivatization reaction
The derivatized solution was prepared following the amino acid analysis procedure provided by Jámbor et al.22 Reagents for producing Fmoc-Cl derivatives must be freshly prepared daily. The addition order, which has been identified as a limiting factor through laboratory testing and by other authors,25 should be carried out in the specified order:(1) Borate buffer solution (82.5 mM) was prepared by diluting 150 μL of a primary borate solution (0.55 M) to 1 mL with Milli-Q grade water. The primary borate solution (0.55 M) was made by dissolving 0.35 g of boric acid and 0.22 g of sodium hydroxide in water to a total volume of 10 mL. This solution can be stored in a refrigerator (6 °C) for several weeks, but it is recommended to prepare fresh dilutions on a daily basis.
(2) The primary solution of Fmoc-Cl was prepared by weighing between 0.20 and 1.50 mg and dissolved it in 1 mL of acetonitrile at room temperature. This resulted in concentrations ranging from 200 to 1500 μg mL−1. Next, this solution was sequentially diluted to obtain a concentration of 10 μg mL−1, which was used as the working solution for the derivatization preparation.
(3) The solution volumes of buffer:Fmoc-Cl:amine in a ratio of 1:1:1 were used for analysis.22 The derivatization solution was prepared to a final volume of 300 μL, by adding 100 μL of Fmoc-Cl working solution to 100 μL of borate buffer (82.5 mM). Next, 100 μL of either standard or sample extract was added and vortexed. The vial was kept at room temperature throughout the process. No differences in the results were observed when the derivatized solutions were prepared using 150 μL of the three components (final volume of 450 μL). Therefore, for the minimization of the proposed method, the smallest volumes were considered.
PM aerosol: sampling and extraction
To validate the developed methodology, PM samples were collected at an urban background station in Madrid, Spain. The site is surrounded by two green areas: Casa de Campo, the largest peri-urban park in Madrid, and Dehesa de la Villa, an urban park. It is located approximately 7 km northwest of the city center. Eight PM2.5 samples were collected using a high-volume sampler (MCV CAV-A/mb sampler) at a flow rate of 30 m3 h−1 for 24 hours on preheated quartz fiber filters (400 °C for 24 h).DNPH-impregnated cartridges were employed to simultaneously sample carbonyl compounds in the PM + gas phase of ambient air within the designated area. A similar methodology was used for carbonyl measurements, utilizing reduced amounts of solvent and derivatization reagent (DNPH).29 The BTEX compounds were sampled on adsorbent tubes from the gas phase of ambient air and analyzed by thermal desorption with gas chromatography/mass spectrometry (refer to the ESI for the experimental conditions of analysis†).
Results and discussion
To offer valuable insights into quantifying short-chain aliphatic amines in ambient air, we conducted optimization of derivatization conditions, assessed method precision and detection limits, and demonstrated applicability for routine sample analysis. The results have been organized based on the following objectives:(1) Optimisation of derivatization conditions using solid hydrochloride salts to prepare standards.
(2) Assessment of method precision and detection limits. Comparison of results obtained from standards prepared using hydrochloride salts versus the pure reagent dissolved in methanol (2 M).
(3) Applicability of the method for sample analysis.
Optimization of derivatization conditions
Fig. 1 shows four representative chromatograms obtained from the analysis of a filter blank, a standard prepared at a very low concentration, and two PM extracts representing low and high concentrations measured during the method application study. The peaks assigned to methylamine and dimethylamine were included. Although the peak for diethylamine was also assigned in the chromatogram, this amine compound was either not detected in some aerosol extracts or, if detected, it appeared with interference at the retention time of the measurement. Therefore, diethylamine was not considered in this study. Youn et al.30 also reported interference at the retention time of the diethylamine measurement.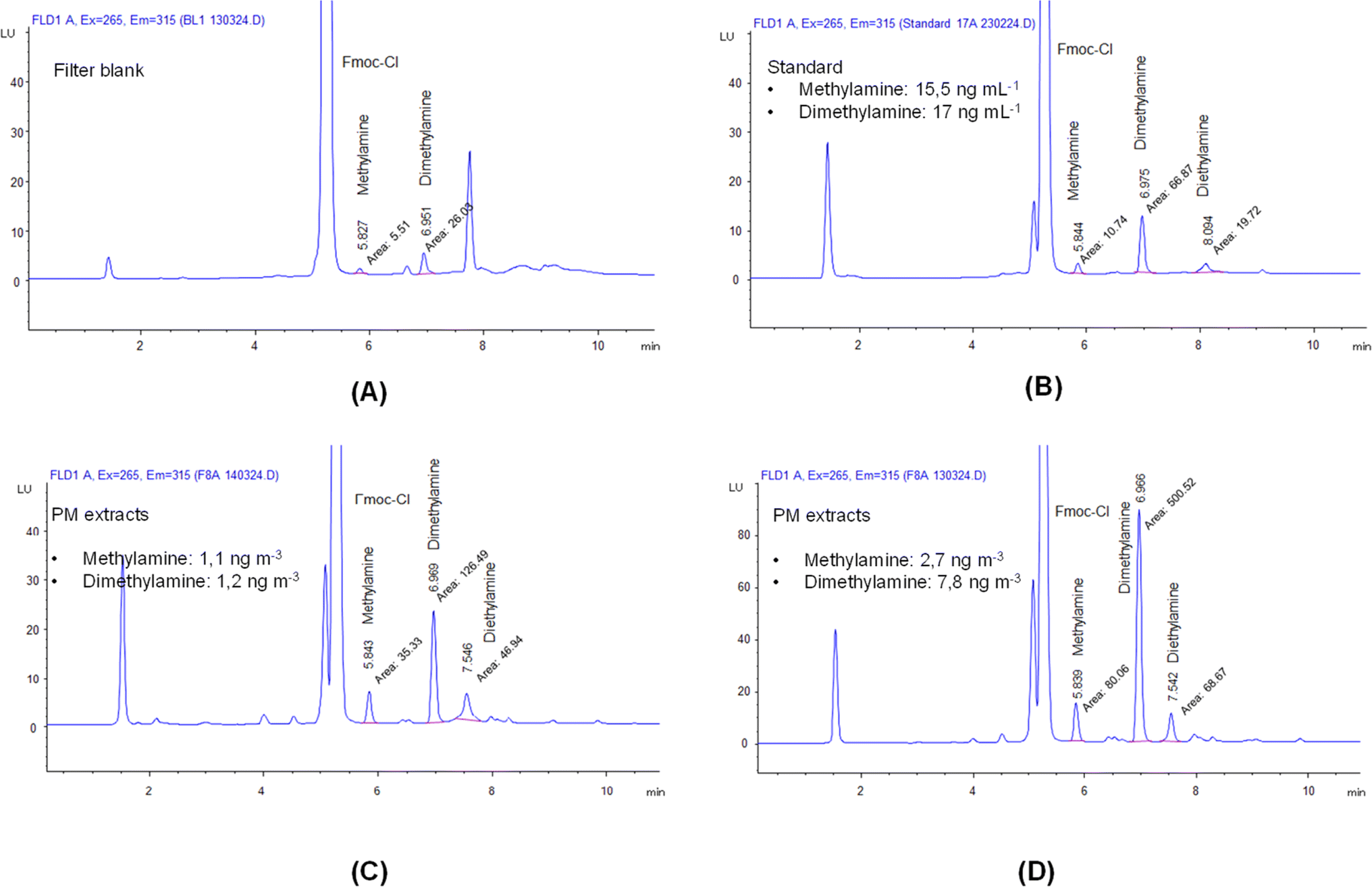 | ||
| Fig. 1 Representative chromatograms obtained from the analyses of three derivatized solutions: a blank (A), a standard with a very low concentration (B), and two aerosol sample extracts (C and D). | ||
Limiting background levels is a common challenge in analyses that require prior derivatization.29 This issue is particularly problematic when analysing dimethylamine due to its volatile, polar properties and sticky nature, which can cause rapid dispersion in the standard preparation environment. Following strict protocols and procedures was essential to reduce the background levels effectively. Given the adhesive nature of the compounds of interest, there is a high risk of contamination through surface adherence. Therefore, maintaining the cleanliness of vials, syringes, and reagents to prevent exposure to amine vapor from standard solutions was imperative. Implementing separate workspaces for preparing blanks/samples and standards further minimizes the risk of contamination.
The quality of solvent and pH play critical roles in influencing background levels during analysis. For example, raw acetonitrile is obtained as a by-product in the industrial production of acrylonitrile, which contains a wide range of impurities, such as aliphatic amines.19 Therefore, ensuring and maintaining quality solvent usage was essential. The production of hydrochloric acid during the derivatization process between the amine compounds and the Fmoc-Cl reagent is a key factor.19 Borate buffer solutions are often employed to maintain the desired pH level during derivatization, typically ranging from 20 (ref. 11 and 24) to 50 mM.25 Exceeding a pH of 10 or encountering excessive water can trigger hydrolysis reactions can occur in the Fmoc-Cl reagent, leading to the appearance of Fmoc-OH peaks in the chromatogram, thereby compromising result accuracy.22,27
Blanks and dimethylamine standards were prepared at different Fmoc-Cl concentrations. The objective was to ensure sufficient Fmoc-Cl for the amine compound reaction, while minimizing excess reagent that could interfere with the analysis or cause instrument deposits. Specifically, the minimum ratio of Fmoc-Cl:amine was 3:1. The concentration of Fmoc-Cl ranged from 2.9 to 29 μg mL−1 in a final volume of 300 μL. The tests were conducted three times on the same day. Fig. 2 shows the results obtained. As can be seen, slight elevations in dimethylamine levels were observed in blank measurements when the Fmoc-Cl concentration increased from 2.9 to 7.3 μg mL−1, remaining constant for higher concentrations (Fig. 2A). Furthermore, the results indicated that concentrations of 2.9 μg mL−1 of Fmoc-Cl were effective in derivatizing dimethylamine at concentrations of 184 ng mL−1, while at higher concentrations, above double that amount (586 ng mL−1), there was a significant decrease in the derivatization efficiency (Fig. 2B). These results are of interest in optimizing the final concentration of Fmoc-Cl for ensuring and producing the derivatization of dimethylamine concentrations in aerosol extracts, which are expected to be less than 180 ng mL−1.
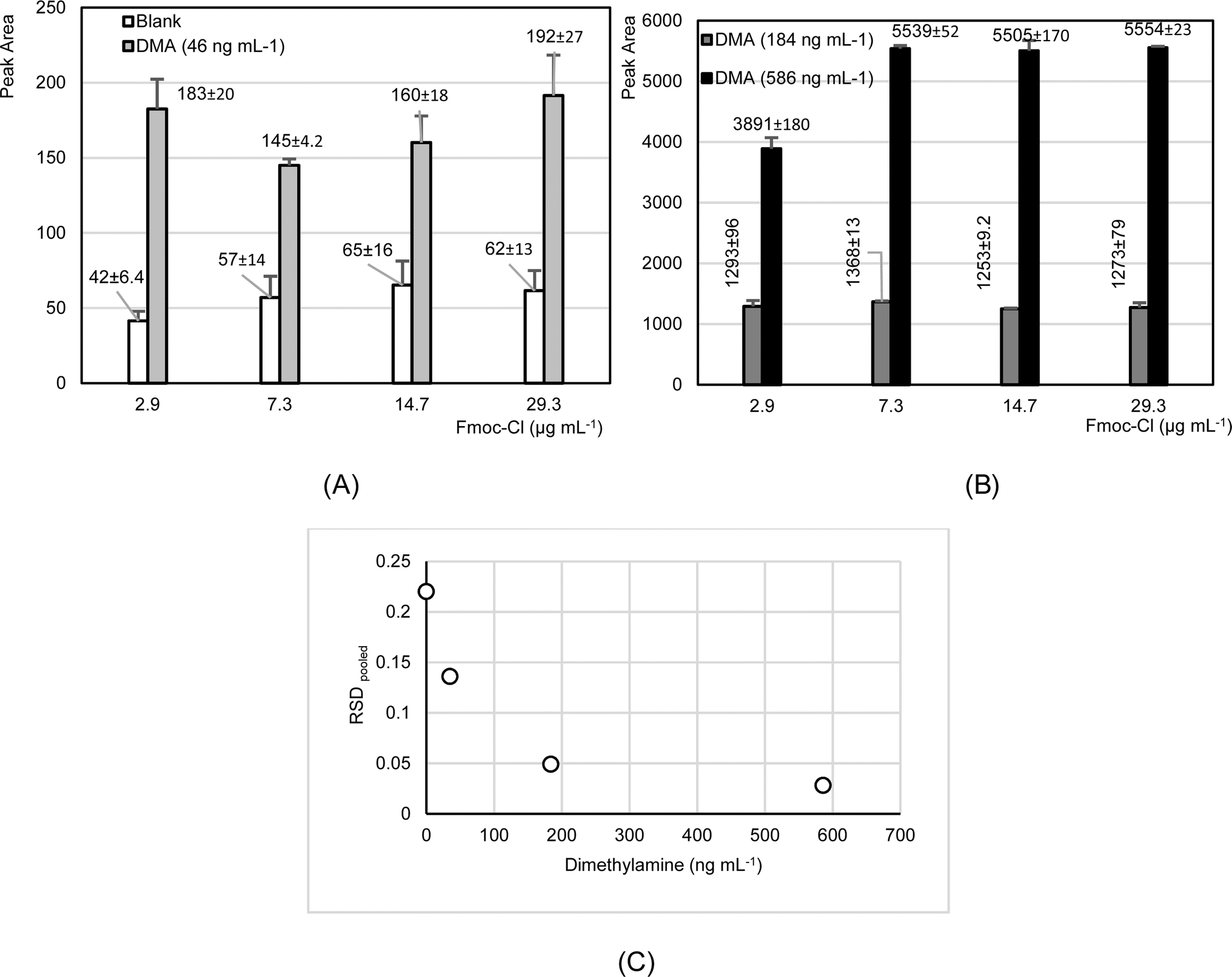 | ||
| Fig. 2 Variation in the analytical response of dimethylamine when analyzing blanks and solutions of amine at different concentrations (ng mL−1), relative to the Fmoc-Cl concentration (μg mL−1) (A and B). Furthermore, (C) presents the pooled relative standard deviation (RSDpooled) derived from measurements at each dimethylamine concentration investigated in the trials conducted on the same day. | ||
In order to provide an estimate of the intra-day variability of these measurements, intermediate precision was estimated for each of the studied dimethylamine concentration levels, which were influenced by the Fmoc-Cl concentrations added in the derivatization. In more detail, the following equation was applied:
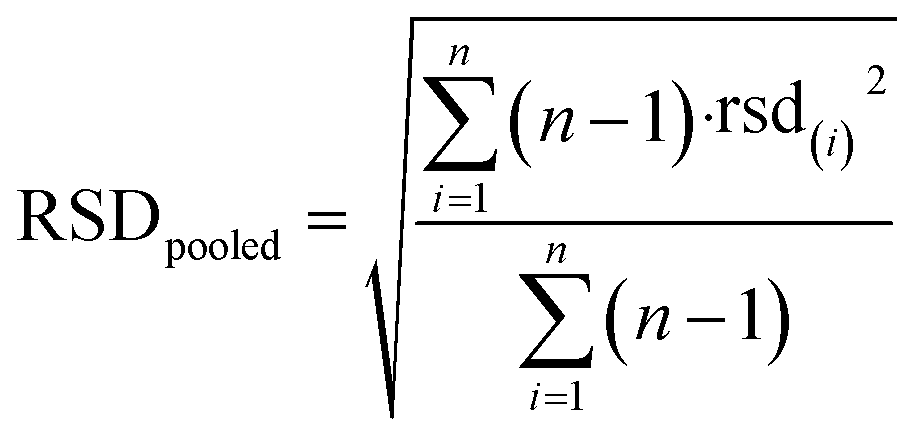 | (1) |
Based on the initial findings and considering the results from tests examining the impact of Fmoc-Cl concentration on chromatographic response across various dimethylamine concentrations, intermediate precision values were calculated for both measured blanks and each level of dimethylamine concentration. The results indicated a significant decrease in the measurement variability as the dimethylamine concentration increased, with the highest variability observed in the blank analyses (Fig. 2C). These preliminary findings highlight the considerable impact of analytical background variability on measurements at lower concentrations. Consequently, variations in background analyses have a significant effect on measurements at low concentrations.
Given the expected low levels of dimethylamine in PM samples, a concentration range of 3–7 μg mL−1 for the Fmoc-Cl reagent was chosen.
Analysis of standards: estimating basic quality parameters
Methodological variability significantly contributes to measurement uncertainty in the analysis of organic compounds.31 To assess result dispersion, an accuracy evaluation was conducted using tests on blanks, standard solutions, and particulate matter (PM) sample extracts. The outcomes from these blank and standard measurements are essential for evaluating fundamental quality parameters.To anticipate the most unfavourable outcome, we calculated the pooled relative standard deviation (RSDpooled) by measuring blanks and standard solutions across various sample batches and days.
The Fmoc-Cl derivatives of standards were prepared from hydrochloride salts and a commercial pure alkylamine solution (in a concentration of 2 M in methanol) for comparison. To prepare standard solutions, the first assays were done with 0.1 g of alkylamine primary standard as a hydrochloride salt, which was diluted with water to a final volume of 10 mL (concentration 10 mg mL−1). Alternatively, if methanol solution (2 M) was used as the stock solution, it was diluted accordingly. Then successive dilutions in acetonitrile were prepared to obtain solutions with concentrations of 100 and 200 ng mL−1. See Table 1S of the ESI for a detailed calculation example.†
Tables 2S and 3S (ESI)† present the results of experiments conducted on different days. Therefore, the intermediate precision derived can be used to assess insights into the day-to-day variation, which can impact analytical precision. Rigorous control over laboratory materials is crucial due to the necessity for high dilutions from concentrated stock solutions and the risk of cross-contamination.
To calculate the pooled standard deviation, the mean values and associated deviations of the areas measured at each of the concentration levels under investigation were obtained. Furthermore, the area values were corrected according to the blank measurements obtained in the same batch. The resulting pooled relative standard deviation reached values as high as 30%.
The results showed that methylamine solutions, especially those prepared by diluting the stock solution in methanol, were unstable within a few hours. On the other hand, for dimethylamine, a decrease in analytical variability was observed when the pure product was used in salt form. Therefore, the use of the pure product in salt form was preferred. By weighing approximately 0.01 and 0.02 g and dissolving it in 25 mL of water, the concentration of the stock solution in salt form should be between 400 and 800 μg mL−1. After the successive dilutions and application of a correction factor (Table 1S, ESI†), the Fmoc-amine derivative was prepared at a concentration of 200 ng mL−1. To minimize bias introduced by the preparation of derivatized standards at low concentration levels, the Fmoc-amine derivative (200 ng mL−1) was diluted to prepare lower calibration points. Analogously, intermediate precision values, derived from blank measurements performed in a standard-free work area, exhibited dispersions below 20% (Table 4S, ESI†). This approach involves assessing the consistency and reliability of chromatographic measurements specifically from blank samples, ensuring that potential interference from standards is minimized.
Subsequently, the limits of detection (LOD) and quantification (LOQ) were determined based on these results, as detailed in the ESI.† The LODs and LOQs estimated corresponded to 10 and 20 ng mL−1, respectively, based on the response factors derived from standard solutions around 30 ng mL−1. A summary of the variability of the results is presented in Fig. 3. As can be seen, the results indicated higher variability associated with methylamine, which could be attributed to its lower molecular weight and high volatility.
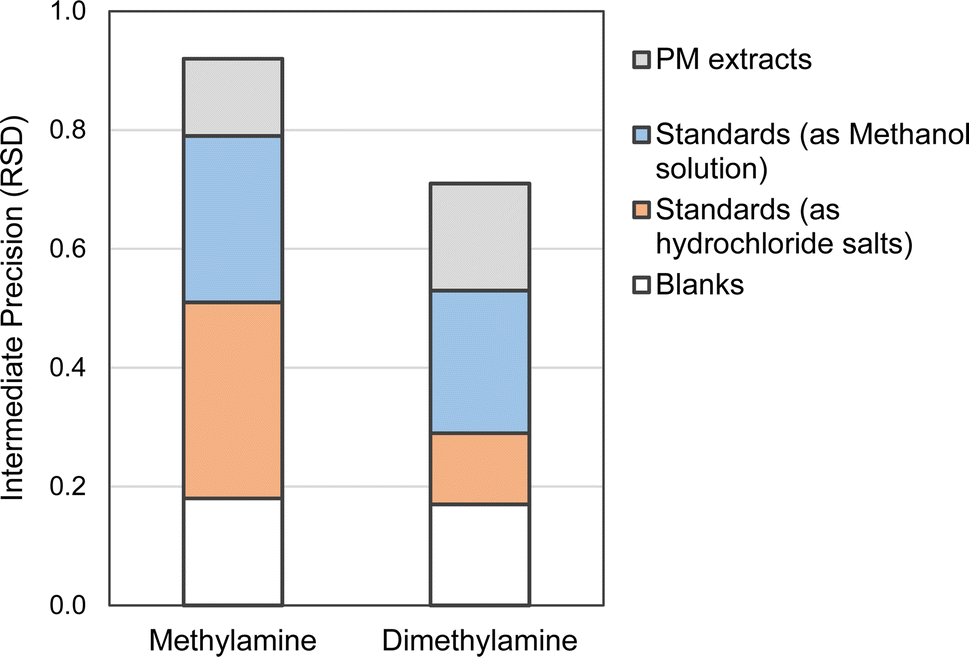 | ||
| Fig. 3 Intermediate precision deduced from the measurements of blank, standards at low concentration and extracts of PM samples. | ||
Certainly, for every sample batch analyzed, a minimum of two blanks and four sample extracts were prepared, along with two low-concentration standards (25–40 ng mL−1), with an additional blank included during standard preparation for quantification and contamination control purposes. Standards and extracts were meticulously prepared in separate rooms, as previously highlighted.
Aerosol sample analysis
The suitability of the method for determining methylamine and dimethylamine was evaluated by analyzing eight PM2.5 samples. In terms of analytical considerations, as mentioned in the section “PM aerosol: sampling and extraction”, the analyses were carried out in duplicate. Additionally, the area values were corrected to the blank measured in the same batch.Methylamine concentrations ranged from 0.8 to 3.0 ng m−3, while dimethylamine concentrations ranged from 1.4 to 7.1 ng m−3 (Table 5S, ESI†). The intermediate precision, assessed using the relative standard deviation (RSD), was found to be 0.13 for methylamine and 0.18 for dimethylamine, based on the analysis of the eight batches of extract measurements. As can be seen in Fig. 3, these RSD values were in general lower than those calculated from the standard tests, highlighting the importance of monitoring and mitigating the risk of cross-contamination.
Concerning the comparison among the concentrations of the studied amines, carbonyl, and BTEX, the concentration profiles of some of them (Table 5S, ESI†) were very similar, providing statistically significant correlation coefficients above 0.73 (Table 6S, ESI†). The highest correlations were observed between dimethylamine and aldehydes, especially long-chain aldehydes such as nonanal and decanal, and between methylamine and BTEX compounds. Fig. 4 shows the pattern for methylamine/BTEX and dimethylamine/aldehydes, reflecting the concordance between the measured organic families. Considering the diverse sampling and analysis methods used (varying sampling times, flow rates, derivatization techniques, and chromatographic methods), these correlation values obtained support the analytical validation of the developed methods.
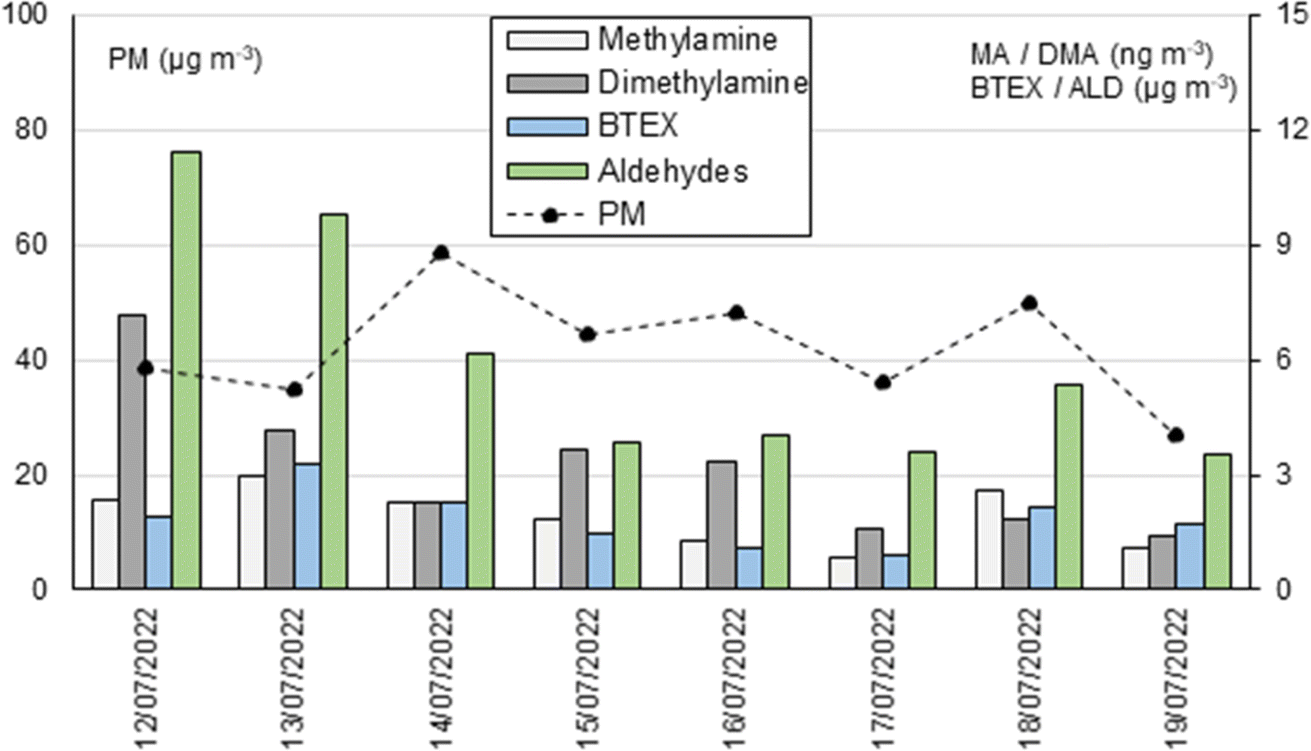 | ||
| Fig. 4 Concentration profiles for methylamine, dimethylamine, BTEX and aldehydes were obtained during one week in July 2022. | ||
From an interpretive standpoint, these findings may suggest a shared origin for these compounds. However, unfortunately, no studies have been found in the literature, apart from the work by Pusfitasari et al.,32 who noted a correlation between these nitrogen containing compounds with ethylbenzene and p-xylene. To gain deeper insights, a more comprehensive characterization study is required in the area, which would include seasonal variations, as well as other gaseous pollutants. This is especially of interest in simulating their atmospheric distribution.6
The amines in the particulate phase can react with carbonyl compounds, forming more oxidized compounds,5,6 such as imines. These imines are included as brown carbon species.33 According to the literature on the carbonyl-alkylamine reaction, organic matter in the particle phase produces small α-dicarbonyls, such as glyoxal and methylglyoxal. These compounds react irreversibly with ammonium salts and primary amines, resulting in the formation of imidazole and other nitrogen-containing and light-absorbing products.7,34 Furthermore, some studies suggest that reactions involving specific long-chain carbonyl compounds, particularly those facilitated by acids, also contribute to particle growth processes in urban areas.35 It is worth mentioning the possibility of a reaction between dimethylamine and nitrogen oxides leading to the formation of formaldehyde and acetaldehyde, as indicated by Saeki et al.,36 which would support the correlation results found in this work. To the best of our knowledge, this type of relationship has not been well documented in the field before.
Ultimately, to advance our understanding of precursors and the processes of aerosol growth, more field campaigns are needed to study the role of precursors and related compounds, as well as their interactions.37 This is one of the principal objectives of the ongoing OASIS project.
To conclude, Table 1 compiles recent literature data on dimethylamine concentration values measured in PM from various areas. No bibliographic references were found for measurements conducted in Spanish regions. As can be seen, the values measured in this study are consistent with those reported in the literature.
| Site (n) | Particle size, filter | Period | Sampling | Analysis (derivatization reagent) | Dimethylamine (ng m−3) | Ref. |
|---|---|---|---|---|---|---|
| Finland (8), forest | TSP | March–May 2011 | 4 L min−1 (5–10 days) | LC-MS/MS (dansyl) | 7.2 (<18) | 38 |
| China (unspecified), cruise seas | PM10 | Mar–Apr 2015 | 29.4 L min−1 (<10 h) | IC | 18 ± 17.3 | 39 |
| Apr 2015 | 13 ± 7.2 | |||||
| Aug 2015 | 42 ± 7.2 | |||||
| Aug–Sep 2015 | 20 ± 5.0 | |||||
| Nov 2012 | 13 ± 4.5 | |||||
| Nov 2012 | 19 ± 19.4 | |||||
| Nov 2013 | ||||||
| China (103), urban | PM2.5 | Nov 2015–Apr 2016 | 1.05 m3 min−1 (23 h) | LC-FD (FMOC) | 7.9 ± 5.4 | 5 |
| China (20), forest | PM2.5 | Oct 2016 | 1.05 m3 min−1 (23 h) | GC/MS | 2.37 ± 3.15 | 12 |
| May–Jun 2017 | 5.03 ± 2.23 | |||||
| China (131), urban | PM2.5 | March–Apr 2013 | 100 L min−1 (24 h) | IC | 6.4 ± 6.1 | 40 |
| Jul–Aug 2013 | 9.1 ± 15.2 | |||||
| Nov 2013 | 15.5 ± 13.4 | |||||
| Winter | 27.3 ± 29.0 | |||||
| China (128), urban | PM2.5 | Apr 2016–Nov 2016 | 1.05 m3 min−1 (22 h) | LC-FD (FMOC) | 3.6 (<19.2) | 6 |
| Seoul (117), urban | PM2.5 | Jan 2018–Dec 2018 | 1.02 m3 min−1 (22 h) | GC-MS/MS (ECF reagent) | 2.72 ± 1.49 | 13 |
| Japan (unspecified), forest | PM2, PM10 | Jan 2018–Jan 2019 | 20 L min−1 (14 days) | LC-UV | Methylamine | 41 |
| PM2 – 0.74 (2.54) | ||||||
| PM10 – 0.059 (0.18) | ||||||
| China (145), coastal/urban | PM2.5 | Winter 2018, 2019 | 100 L min−1 (11.5 h) | LC-MS/MS | 58.7 ± 25.8–86.3 ± 20.2 | 7 |
| Italy (106), urban | PM10 | Winter 2020 | 38 L min−1 (24 h) | LC-FD | 14.2 ± 5.6–24.9 ± 7.2 | 4 |
| China (71), industrial | PM2.5 | 2015–2016 | 1.05 m3 min−1 (23 h) | IC | 14.6 ± 8.2 | 3 |
| China (65); urban | PM2.5 | 2016–2017 | 1.13 m3 min−1 (24 h) | GC-MS (isobutyl chloroformate) | 20.81 ± 11.10 | 33 |
| Spain (8), urban background | PM2.5 | July, 2022 | 0.49 m3 min−1 (24 h) | LC-FD (FMOC) | 3.2 ± 1.9 | This study |
Conclusions
A simple and sensitive method using LC-FD with prior Fmoc-Cl derivatization is proposed to measure low ambient levels of dimethylamine and methylamine in atmospheric aerosol samples. Our approach emphasizes minimizing the quantities of derivatization reagent and solvent, which offers several advantages. The results provide valuable insights into understanding measurement variability and comparability among different concentration values. Moreover, this study contributes to the development of sustainable analytical methods for determining amine compounds by decreasing the generation of hazardous waste during sample treatment, resulting in time and cost savings.The effectiveness of the proposed method was evaluated by analyzing eight PM2.5 samples collected in a clean area of Madrid. Concentration results were consistent with the literature, being below ten ng m−3. The application study of the methodology demonstrated consistent results with two families of organic compounds, corresponding to different sampling and analysis methods. A correlation between dimethylamine and aldehydes, especially longer chain ones like nonanal and decanal, was observed. Similarly, methylamine was found to be correlated with ethylbenzene and o-xylene. These results can be used as a foundation for future environmental characterization of the compound families in question over extended periods. These efforts aim to advance our understanding of the dynamics of organic compounds in the atmosphere.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
S. García-Alonso: formal analysis, resources, investigation, validation and writing – original draft. F. J. Gómez-Moreno: funding acquisition, writing – review & editing and methodology. E. Alonso-Blanco: writing – review & editing and methodology. R. M. Pérez-Pastor: conceptualization, supervision, and writing – review & editing.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The authors are grateful for the financial support provided by the OASIS project (PID2021-127885OB-I00 funded by MCIN/AEI/10.13039/501100011033 and by ‘ERDF A way of making Europe’).References
- T. Kurtén, V. Loukonen, H. Vehkamäki and M. Kulmala, Atmos. Chem. Phys., 2008, 8, 4095–4103, DOI:10.5194/acp-8-4095-2008.
- M. Chen, M. Titcombe Lee, J. Jiang, C. Jen, C. Kuang, M. Fischer, F. Eisele, J. Siepmann, D. Hanson, J. Zhao and P. McMurry, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18713–18718, DOI:10.1073/pnas.1210285109.
- X.-Y. Yang, F. Cao, M.-Y. Fan, Y.-C. Lin, F. Xie and Y.-L. Zhang, Sci. Total Environ., 2023, 857, 159371, DOI:10.1016/j.scitotenv.2022.159371.
- D. Spolaor, L. Soldà, G. Formenton, M. Roverso, D. Badocco, S. Bogialli, F. A. Monikh and A. Tapparo, Aerosol Res., 2023, 1, 29–38, DOI:10.5194/ar-1-29-2023.
- W. Shen, L. Ren, Y. Zhao, L. Zhou, L. Dai, X. Ge, S. Kong, Q. Yan, H. Xu, Y. Jiang, J. He, M. Chen and H. Yu, Environ. Pollut., 2017, 230, 12–21, DOI:10.1016/j.envpol.2017.06.034.
- G. Cheng, Y. Hu, M. Sun, Y. Chen, Y. Chen, C. Zong, J. Chen and X. Ge, Atmos. Pollut. Res., 2020, 11, 296–302, DOI:10.1016/j.apr.2019.11.002.
- Z. Liu, M. Li, X. Wang, Y. Liang, Y. Jiang, J. Chen, J. Mu, Y. Zhu, H. Meng, L. Yang, K. Hou, Y. Wang and L. Xue, Sci. Total Environ., 2022, 839, 156281, DOI:10.1016/j.scitotenv.2022.156281.
- Y. Chen, Q. Lin, G. Li and T. An, J. Environ. Sci., 2022, 114, 401–411, DOI:10.1016/j.jes.2021.09.027.
- K. Matsumoto, T. Kuwabara and T. Nakano, Atmos. Environ., 2023, 309, 119885, DOI:10.1016/j.atmosenv.2023.119885.
- Y. Wang, G. Yang, Y. Lu, Y. Liu, J. Chen and L. Wang, Atmos. Environ., 2020, 243, 117875, DOI:10.1016/j.atmosenv.2020.117875.
- H. Xiongfeng, X. Qun and J. Rohrer, Thermo Scientific Application Note, 2016, vol. 1012, pp. 1–7 Search PubMed.
- F. Liu, X. Bi, G. Zhang, X. Lian, Y. Fu, Y. Yang, Q. Lin, F. Jiang, X. Wang, P. a. Peng and G. Sheng, Atmos. Environ., 2018, 195, 1–11, DOI:10.1016/j.atmosenv.2018.09.038.
- N. R. Choi, J. Y. Lee, Y. G. Ahn and Y. P. Kim, Chemosphere, 2020, 258, 127367, DOI:10.1016/j.chemosphere.2020.127367.
- M. Hemmilä, H. Hellén, A. Virkkula, U. Makkonen, A. P. Praplan, J. Kontkanen, L. Ahonen, M. Kulmala and H. Hakola, Atmos. Chem. Phys., 2018, 18, 6367–6380, DOI:10.5194/acp-18-6367-2018.
- M. van Pinxteren, K. W. Fomba, D. van Pinxteren, N. Triesch, E. H. Hoffmann, C. H. L. Cree, M. F. Fitzsimons, W. von Tümpling and H. Herrmann, Atmos. Environ., 2019, 203, 183–195, DOI:10.1016/j.atmosenv.2019.02.011.
- A. F. Corral, Y. Choi, B. L. Collister, E. Crosbie, H. Dadashazar, J. P. DiGangi, G. S. Diskin, M. Fenn, S. Kirschler, R. H. Moore, J. B. Nowak, M. A. Shook, C. T. Stahl, T. Shingler, K. L. Thornhill, C. Voigt, L. D. Ziemba and A. Sorooshian, Environ. Sci.: Atmos., 2022, 2, 1534–1550, 10.1039/D2EA00117A.
- D. Chen, X. Yao, C. K. Chan, X. Tian, Y. Chu, S. L. Clegg, Y. Shen, Y. Gao and H. Gao, Environ. Sci. Technol., 2022, 56, 5430–5439, DOI:10.1021/acs.est.1c08713.
- J. Xie, Y. Li, J. Zhang, L. Zeng, D. Lu, Y. Liu, Y. Yang and C. Sun, Anal. Methods, 2014, 6, 5140–5146, 10.1039/C4AY00509K.
- M. Asif Iqbal, J. E. Szulejko and K.-H. Kim, Anal. Methods, 2014, 6, 5697–5707, 10.1039/C4AY00740A.
- R. Herráez-Hernández, C. Cháfer-Pericás, J. Verdú-Andrés and P. Campíns-Falcó, J. Chromatogr. A, 2006, 1104, 40–46, DOI:10.1016/j.chroma.2005.11.121.
- X. Huang, Q. Zhang, X. Li, X. Ao and X. Wang, Chromatographia, 2021, 84, 463–471, DOI:10.1007/s10337-021-04020-3.
- A. Jámbor and I. Molnár-Perl, J. Chromatogr. A, 2009, 1216, 3064–3077, DOI:10.1016/j.chroma.2009.01.068.
- J. Verdú-Andrés, P. Campíns-Falcó and R. Herráez-Hernández, Chromatographia, 2002, 55, 129–134, DOI:10.1007/BF02492132.
- D. Shangguan, Y. Zhao, H. Han, R. Zhao and G. Liu, Anal. Chem., 2001, 73, 2054–2057, DOI:10.1021/ac001243c.
- M. C. Prieto-Blanco, C. Cháfer-Pericás, P. López-Mahía and P. Campíns-Falcó, J. Chromatogr. A, 2008, 1188, 118–123, DOI:10.1016/j.chroma.2008.02.056.
- I. Fradi, A.-C. Servais, C. Lamalle, M. Kallel, M. Abidi, J. Crommen and M. Fillet, J. Chromatogr. A, 2012, 1267, 121–126, DOI:10.1016/j.chroma.2012.05.098.
- J. E. Szulejko and K.-H. Kim, TrAC, Trends Anal. Chem., 2014, 57, 118–134, DOI:10.1016/j.trac.2014.02.010.
- X. Huang, C. Deng, G. Zhuang, J. Lin and M. Xiao, Environ. Sci.: Processes Impacts, 2016, 18, 796–801, 10.1039/C6EM00197A.
- S. García-Alonso, A. M. Bernal-Páez and R. M. Pérez-Pastor, Anal. Methods, 2021, 13, 1976–1985, 10.1039/d0ay02288h.
- J. S. Youn, E. Crosbie, L. C. Maudlin, Z. Wang and A. Sorooshian, Atmos. Environ., 2015, 122, 250–258, DOI:10.1016/j.atmosenv.2015.09.061.
- S. L. R. Ellison and A. Williams, 3rd edn, 2012, available from, https://www.eurachem.org/images/stories/Guides/pdf/QUAM2012_P1.pdf.
- E. D. Pusfitasari, J. Ruiz-Jimenez, A. Tiusanen, M. Suuronen, J. Haataja, Y. Wu, J. Kangasluoma, K. Luoma, T. Petäjä, M. Jussila, K. Hartonen and M. L. Riekkola, Atmos. Chem. Phys., 2023, 23, 5885–5904, DOI:10.5194/acp-23-5885-2023.
- C. W. Leung, X. Wang and D. Hu, J. Hazard. Mater., 2024, 469, 133899, DOI:10.1016/j.jhazmat.2024.133899.
- W. Marrero-Ortiz, M. Hu, Z. Du, Y. Ji, Y. Wang, S. Guo, Y. Lin, M. Gomez-Hermandez, J. Peng, Y. Li, J. Secrest, M. L. Zamora, Y. Wang, T. An and R. Zhang, Environ. Sci. Technol., 2019, 53, 117–126, DOI:10.1021/acs.est.8b03995.
- R. Zhang, G. Wang, S. Guo, M. L. Zamora, Q. Ying, Y. Lin, W. Wang, M. Hu and Y. Wang, Chem. Rev., 2015, 115, 3803–3855, DOI:10.1021/acs.chemrev.5b00067.
- K. Saeki, K. Ikari, S.-I. Ohira and K. Toda, Anal. Sci., 2024, 40, 1907–1918, DOI:10.1007/s44211-024-00626-3.
- F. J. Gómez-Moreno, E. Alonso-Blanco, E. Díaz, E. Coz, F. Molero, L. Núñez, M. Palacios, M. Barreiro, J. Fernández, P. Salvador, M. Piñeiro-Iglesias, P. López-Mahía, E. Borrás, T. Vera, A. Muñoz, T. Tritscher, S. H. Schmitt and B. Artíñano, Environ. Res. Commun., 2022, 4, 125010, DOI:10.1088/2515-7620/acacf0.
- J. Ruiz-Jiménez, S. Hautala, J. Parshintsev, T. Laitinen, K Hartonen, T. Petäjä, M. Kulmala and M. Riekkola, Talanta, 2012, 97, 55–62, DOI:10.1016/j.talanta.2012.03.062.
- P. Yu, Q. Hu, K. Li, Y. Zhu, X. Liu, H. Gao and X. Yao, Sci. Total Environ., 2016, 572, 813–824, DOI:10.1016/j.scitotenv.2016.07.114.
- S. Zhou, H. Li, T. Yang, Y. Chen, C. Deng, Y. Gao, C. Chen and J. Xu, Atmos. Chem. Phys., 2019, 19(16), 10447–10467, DOI:10.5194/acp-19-10447-2019.
- A. Hirai and K. Matsumoto, Tellus B, 2021, 73(1), 1875585, DOI:10.1080/16000889.2021.1875585.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ay00894d |
| This journal is © The Royal Society of Chemistry 2025 |