
Colorimetric aptasensors for sensitive low-density lipoprotein detection based on reduced oxide graphene@molybdenum disulfide-ferrocene nanosheets with peroxidase-like activity†
Guiyin
Li
 a,
Tingting
Yu
ab,
Haimei
Li
b,
Bingbing
Wan
ab,
Xiaohong
Tan
a,
Xueqing
Zhou
*c,
Jintao
Liang
*b and
Zhide
Zhou
*b
a,
Tingting
Yu
ab,
Haimei
Li
b,
Bingbing
Wan
ab,
Xiaohong
Tan
a,
Xueqing
Zhou
*c,
Jintao
Liang
*b and
Zhide
Zhou
*b
aCollege of Chemistry, Guangdong University of Petrochemical Technology, Guandu Road, Maoming, Guangdong 525000, People's Republic of China
bSchool of Life and Environmental Sciences, Guilin University of Electronic Technology, Guilin, Guangxi 541004, People's Republic of China
cClinical Laboratory, the 924th Hospital of Chinese People's Liberation Army Joint Logistic Support Force, Guilin, Guangxi 541004, People's Republic of China
First published on 22nd November 2024
Abstract
Low-density lipoprotein (LDL) is a key biomarker for cardiovascular disease (CVD) risk assessment. Monitoring LDL for the early diagnosis of CVD and its complications is an important clinical analysis tool. In this work, a novel colorimetric aptasensor for LDL detection was constructed via reduced graphene oxide@molybdenum disulfide-ferrocene-carboxylic nanosheets (rGO@MoS2-Fc) with excellent peroxidase-like activity. On this basis, the LDL aptamer (LDLapt) immobilized on the surface of rGO@MoS2-Fc served as a signal probe (rGO@MoS2-Fc/LDLapt), while the unmodified LDLapt served as a capture probe. When LDL was present, it was recognized by the LDLapt and rGO@MoS2-Fc/LDLapt to form an rGO@MoS2-Fc/LDLapt/LDL/LDLapt sandwich-type conjugate with excellent enzymatic catalytic properties that can catalyze the generation of hydroxyl radicals (·OH) from hydrogen peroxide (H2O2), which in turn oxidized the colorless substrate o-phenylenediamine (OPD) to the yellow compound 2,3-diamino phenothiazine (DAP). In addition, the catalytic mechanism of the reaction was confirmed to be induced by ·OH through free radical experiments. The aptasensor had a linear range of 15.0 to 200.0 μg mL−1, and a limit of detection (LOD) of 2.199 μg mL−1. Overall, the assay has high selectivity, sensitivity and operability, showing broad application prospects in the clinical diagnosis of CVD.
1 Introduction
According to a 2022 report, cardiovascular disease (CVD) was the leading cause of death in urban and rural areas, accounting for 48.00% and 45.86% of deaths in urban and rural areas, respectively. Therefore, it is very important to assist patients in the early diagnosis and monitoring of CVD.Levels of low-density lipoprotein (LDL) affect the incidence of CVD, and as LDL levels increase, the risk of CVD also increases.1,2 On the basis of data from statin trials, a correlation between CVD risk and LDL levels can be observed.3,4 The risk of CVD increases in individuals at very high risk with LDL ≥ 70 mg dL−1 and in those at moderate risk with LDL ≥ 130 mg dL−1 in adults.5 Thus, LDL becomes a key biomarker involved in CVD risk assessment.6 Clinically, the biologically relevant concentration of LDL in the serum of healthy people is 114–218 mg dL−1,7 and the LDL concentration is divided into three concentration ranges:8 ideal (<120 mg dL−1), moderate-risk (120–159 mg dL−1), and high risk (≥160 mg dL−1). Therefore, the determination of LDL concentration in human serum is of great significance for monitoring CVD.
So far, various methods have been used to detect LDL including the Friedewald method,9 beta-quantification,10 precipitation methods,11 homogeneous methods,12 and sensor immunoassays.13 Except for the homogeneous method and sensor immunoassay, other methods require additional protein concentrations to calculate LDL concentrations. While the homogenous method requires large instruments, and the immunoassay uses antibodies as recognition molecules, which are all expensive and easily inactivated, limiting the application of biosensors in the early diagnosis of CVD.
Compared with antibodies, aptamers, obtained through SELEX (systematic evolution of ligands by exponential enrichment) technology, have the advantages of high chemical stability and low synthesis cost. Therefore, aptamers have been widely used in biosensing, bioimaging, therapy, and the theragnostic field.14 Fortunately, Klapak et al.15 selected an aptamer that has a 1.6 pM dissociation constant (KD) to LDL-protein using the SELEX process. In recent years, research on biosensors based on aptamers (i.e., aptasensor) has emerged, offering advantages of simple operation, strong specificity and reliability, and flexible design by utilizing aptamers with high specificity and affinity.16 Our group17 has constructed a light-addressable potentiometric sensor (LAPS) to detect LDL using an aptamer of LDL as the recognition molecule. The sensor demonstrated good linearity from 1.0 to 100.0 μg mL−1 with a LOD of 0.79 μg mL−1, showing good precision.
Colorimetric biosensors achieve quantitative detection of objects that cause color changes through visual recognition or spectrophotometer monitoring. Thus, colorimetric biosensors have been widely applied because of their low cost, simple application, high sensitivity and real-time monitoring capability. Using natural enzymes or nanoenzymes with catalytic activity to catalyze substrate color development is one of the main strategies to realize colorimetric adaptive sensing.19 At present, nano-enzymes are characterized by low cost, batch production and easy modification. They can be used to replace natural enzymes for catalysis and analysis in the field of colorimetry.20–22 For example, Tan et al.18 constructed a simple colorimetric aptasensor for profenofos detection using aptamer-AuPtRh as the capture probe with outstanding peroxidase catalytic activity. The linear range was 1–300 ng L−1 and the limit of detection (LOD) was 0.725 ng L−1. Wang et al.23 prepared Fe3O4@SiO2@NiCo2S4 nanocomposites that exhibited good peroxidase-like activity, and a colorimetric assay for sarcosine was carried out with a linear range of 1.25–350 μM and a detection limit of 0.42 μM. Layered molybdenum disulfide (MoS2) has good anisotropy, catalytic performance and stability. It is used in electronic devices, biomedicine, catalysis and degradation.24,25 Also, MoS2 has peroxidase activity and can catalyze the decomposition of hydrogen peroxide (H2O2), which in turn catalyzes the oxidation of colorless 3,3,5,5-tetramethylbenzidine (TMB) to blue.26 For instance, Ran et al.27 synthesised Au/Cu2S/MoS2 composites and utilised their peroxidase activity to develop a smartphone-assisted portable detection platform with dual modes of photo-thermal and colorimetric detection for the detection of cortisol in human saliva. However, MoS2 tends to aggregate due to the interaction of the Johannes Diderik van der Waals force and the surface force. To improve MoS2 performance, reduced graphene oxide (rGO) can be introduced as a good support to prevent aggregation. Meanwhile, the preparation of rGO@MoS2 can greatly improve the structural characteristics because both MoS2 and rGO show a layered structure and similar morphology.28
In this design, we attempted to construct a colorimetric aptasensor for the detection of LDL based on reduced graphene oxide@molybdenum disulfide-ferrocene nanosheets (rGO@MoS2-Fc). The prepared rGO@MoS2-Fc has strong peroxidase-like activity and excellent stability. Then, a “sandwich-type” colorimetric aptasensor was proposed using the LDL aptamer (LDLapt) as the capture probe and LDLapt-labelled rGO@MoS2-Fc as the recognition probe, respectively. Under optimal experimental conditions, the proposed aptasensor was able to detect LDL with ultra-high sensitivity and good selectivity over a wide range. The aptasensor was also used for the detection of LDL in human serum samples and showed a small relative error.
2 Experimental
2.1 Materials and reagents
Materials and reagents are provided in the ESI.†2.2 Apparatus
The information of the apparatus is listed in the ESI.†2.3 Synthesis of rGO@MoS2-Fc nanosheets
RGO was prepared based on a previous study with some changes.29 Briefly, 10.0 mL of 1.0 mg mL−1 GO solution was crushed using an ultrasonic crusher for 1 h; then 60.0 mg of AA was added and stirred at room temperature for 6 h to obtain the rGO solution. Afterward, 20.0 mg of MoS2 was added to 20.0 mL of DMF solution to form a homogeneous suspension, which was then crushed through an ultrasonic crusher for 1 h. 20.0 mg of β-ME was then added to the abovementioned well-crushed MoS2 solution under magnetic stirring at 25 °C for 12 h, and then the above mixture was centrifuged at 8000 rpm to obtain the aminated MoS2 (NH2-MoS2). Following that, 60.0 mg of Fc was added to a 1.0 mL DMF solution to form a homogeneous suspension through ultrasonication for 30 min; then, the above NH2-MoS2 solution was mixed with Fc solution, while adding 20.0 mL of EDAC/NHS (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). After stirring for 6 h, MoS2-Fc solution was obtained by centrifugation. Finally, 10.0 mL of 2.0 mg mL−1 MoS2-Fc nanosheet solution and 10.0 mL of 1.0 mg mL−1 rGO solution were mixed and stirred at 25 °C for 24 h, then centrifuged at 8000 pm to obtain rGO@MoS2-Fc nanosheets.
:1). After stirring for 6 h, MoS2-Fc solution was obtained by centrifugation. Finally, 10.0 mL of 2.0 mg mL−1 MoS2-Fc nanosheet solution and 10.0 mL of 1.0 mg mL−1 rGO solution were mixed and stirred at 25 °C for 24 h, then centrifuged at 8000 pm to obtain rGO@MoS2-Fc nanosheets.
2.4 Steady-state kinetics of rGO@MoS2-Fc nanosheets
The peroxidase-like activity of rGO@MoS2-Fc was assessed in the presence of H2O2 using TMB and o-phenylenediamine (OPD) as peroxidase substrates, respectively. The OPD–H2O2 color system was as follows: 20.0 μL of 1 mg mL−1 of rGO@MoS2-Fc, 10.0 μL of 50 mM OPD, 10.0 mL of 100 mM H2O2 and 60.0 μL of 125 mM phosphate buffer (PBS) at pH = 5.5 were added to a test tube and incubated for 60 min at room temperature. The TMB–H2O2 chromogenic system was prepared by the same method and incubated at room temperature for 10 min. The absorbance of OPD–H2O2 and TMB–H2O2 at 425 nm and 652 nm was measured using a UV-vis photometer, respectively.In addition, using OPD as the chromogenic agent, a steady-state kinetic assay was performed to investigate the peroxidase-like activity of rGO@MoS2-Fc by varying the concentration of one substrate (OPD or H2O2) while keeping the other fixed. The steady-state kinetic parameters were calculated by using the Lineweaver–Burk plot:30
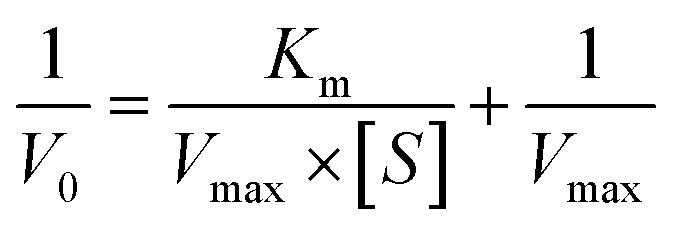
2.5 Verification of the catalytic mechanism of rGO@MoS2-Fc nanosheets
A fluorescence photometer was used to verify the reaction mechanism in the experiments. Terephthalic acid (PTA) was used as a fluorescence probe, and the fluorescence was measured in a test tube by adding 100.0 μL of 5 mM PTA, 200.0 μL of 125 mM PBS at pH = 4.0, 100.0 μL of 50 mM H2O2, and 100.0 μL of 1.0 mg mL−1 rGO@MoS2-Fc after incubation for 12 h at room temperature and protected from light. The excitation and emission wavelengths of PTA were 315 nm and 425 nm, respectively.332.6 Preparation of the rGO@MoS2-Fc/LDLapt signal probe
2.0 mg of rGO@MoS2-Fc and 500.0 μL of 0.25% GA solution were mixed by sonication for 1 h, and then centrifuged and washed three times with pure water; rGO@MoS2-Fc was re-dispersed in 490.0 μL of pure water, and 10.0 μL of 100 μM LDLapt was added to the above solution and incubated in the dark at 4 °C for 12 h. Then, 250.0 μL of the supernatant was removed by centrifugation and mixed with 550.0 μL of pure water. Through the cross-linking amide reaction between the amino and carboxyl groups, rGO@MoS2-Fc/LDLapt was finally synthesized.2.7 Construction of the rGO@MoS2-Fc-based colorimetric aptasensor
First, 20.0 μL of 1 μM LDLapt was added dropwise into a centrifuge tube and incubated at 4 °C for 60 min. Second, 20.0 μL of 0.5% bovine serum albumin (BSA) was added and incubated at 25 °C for 30 min. Third, 20.0 μL of LDL (or serum) was added and incubated at 25 °C for 60 min. Fourth, 20.0 μL of 1.0 mg mL−1 rGO@MoS2-Fc/LDLapt was added and incubated at 25 °C for 60 min. After each step, the excess waste liquid was removed with a pipette. Finally, 10.0 μL of 50 mM OPD, 10.0 μL of 100 mM H2O2, and 60.0 μL of HAc–NaAc buffer (125 mM) were added and incubated in the dark for 25 min. After that, the absorbance was measured. All experiments were conducted in triplicate or more to ensure reproducibility and reliability.2.8 Detection of LDL in real human serum samples using the assay
To verify the clinical usability of the designed sensors, actual human serum samples were tested under optimal experimental conditions. All experiments were performed in compliance with the relevant laws and institutional guidelines (the Ethics Committee of 924th Hospital of Chinese people's Liberation Army (No. 2023-52, Guilin, China)), and informed consent was obtained for any experimentation with human subjects.20.0 μL of 1 mg mL−1 rGO@MoS2-Fc/LDLapt recognition probe was taken, and the actual human serum samples were mixed with a known concentration of LDL by the homogenisation method and then incubated for 1 h at 25 °C. The OPD–H2O2 chromogenic substrate and buffer were added, and finally the absorbance change at 450 nm was determined by UV-vis spectrophotometric analysis.
3 Results and discussion
3.1 Principle of a colorimetric aptasensor for the determination of LDL
The principle of the rGO@MoS2-Fc based colorimetric aptasensor for the detection of LDL is illustrated in Fig. 1A. First, the rGO@MoS2-Fc/LDLapt recognition probe was prepared, and LDL was added after immobilisation of LDLapt in a test tube. When LDL was present, LDLapt specifically bound to LDL. The specificity between LDLapt and LDL was verified using fluorescence spectra (Fig. S1†) and circular dichroism analysis (Fig. S2†), which confirmed the formation of the rGO@MoS2-Fc/LDLapt/LDL/LDLapt sandwich-type complex. At this point, the peroxidase activity of the rGO@MoS2-Fc/LDLapt probe was enhanced. And when the peroxidase substrate OPD was added in the presence of H2O2, a yellow product was formed, turning the colourless solution yellow; then the concentration of LDL could be detected by measuring the change in absorbance.32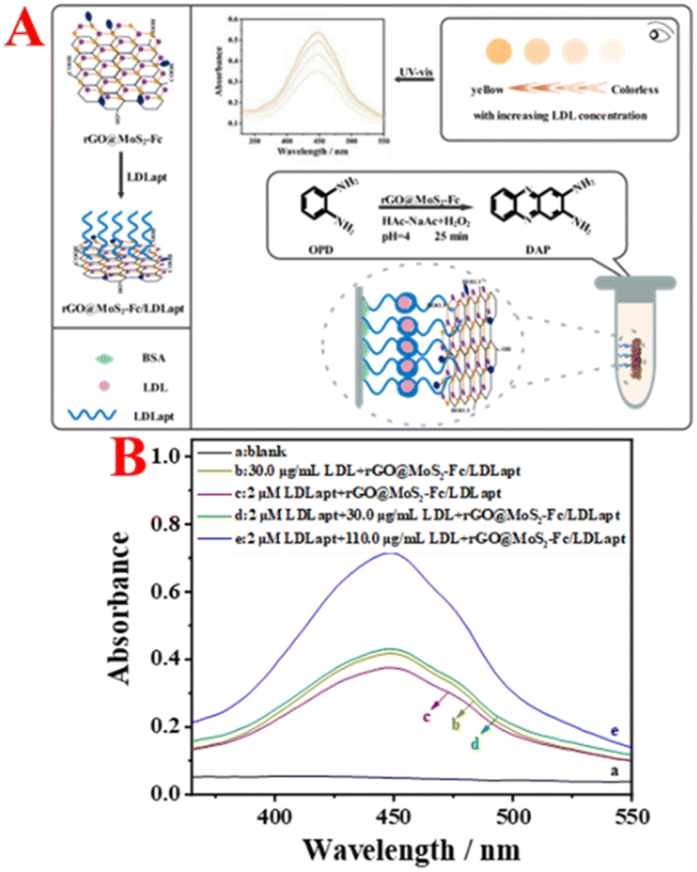 | ||
| Fig. 1 (A) Schematic illustration of rGO@MoS2-Fc as a peroxidase mimetic for colorimetric detection of LDL. (B) Feasibility of colorimetric detection of LDL. | ||
Fig. 1B shows the feasibility analysis of the colorimetric aptasensor for LDL detection. As shown in Fig. 1B, no absorption peak is generated in the blank group (curve a). When LDL is incubated with rGO@MoS2-Fc/LDLapt (curve b), an absorption peak is observed, because rGO@MoS2-Fc/LDLapt specifically binds to LDL and remains in the test tube, resulting in an absorption peak. When LDLapt is incubated with rGO@MoS2-Fc/LDLapt (curve c), the absorption peak is weaker. However when LDL and LDLapt are incubated with rGO@MoS2-Fc/LDLapt (curve d), a strong absorption peak appears due to the formation of the stable sandwich-type rGO@MoS2-Fc/LDLapt/LDL/LDLapt complex. Moreover, the absorption peak increases with the increase in LDL concentration (from curve d to curve e). The results show that the prepared colorimetric aptasensor is effective and can be used for the detection of LDL.
3.2 Characterization of rGO@MoS2-Fc nanosheets
Fig. 2A–C show the transmission electron microscope (TEM) images of rGO@MoS2-Fc. As shown in Fig. 2A, MoS2 has a sheet structure with a multi-layer stacking arrangement. When MoS2 is combined with Fc, the surface of MoS2-Fc (Fig. 2B) displays many round or oval particles, indicating that MoS2-Fc has been successfully prepared. When rGO is combined with MoS2-Fc, the resulting rGO@MoS2-Fc (Fig. 2C) exhibits a membrane structure of rGO, and MoS2-Fc are evenly distributed on the surface of rGO without obvious aggregation, which is attributed to the dispersion of MoS2-Fc caused by the combination of rGO and MoS2-Fc. This proves the successful preparation of rGO@MoS2-Fc.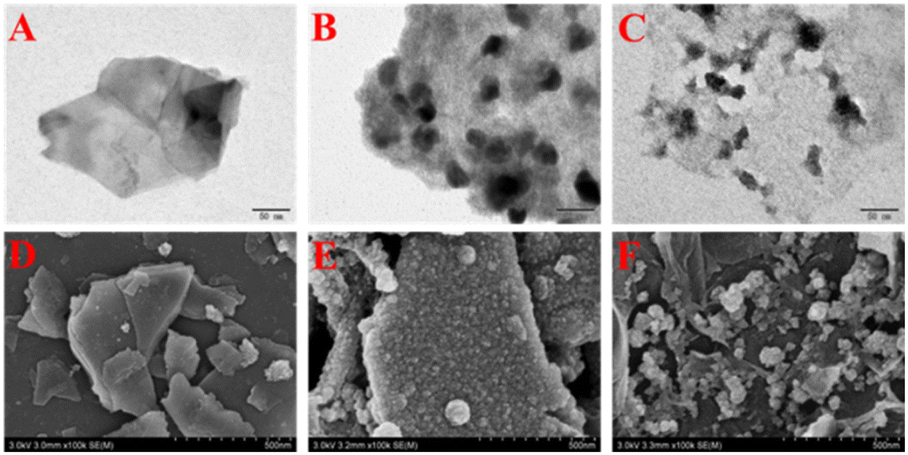 | ||
| Fig. 2 TEM images of MoS2 (A), MoS2-Fc (B), and rGO@MoS2-Fc (C). SEM images of MoS2 (D), MoS2-Fc (E), and rGO@MoS2-Fc (F). | ||
Fig. 2D–F show the scanning electron microscopy (SEM) images of rGO@MoS2-Fc. As shown in Fig. 2D, MoS2 shows a smooth, multilayered and sheet-like structure. MoS2-Fc (Fig. 2E) has a rough surface with many granular substances attached to the surface. As shown in Fig. 2F, MoS2-Fc is dispersed on the surface of rGO or coated with rGO, indicating the successful preparation of rGO@MoS2-Fc.
The FT-IR spectra, X-ray diffraction (XRD), particle size potential analysis, are used to further characterize the rGO@MoS2-Fc nanosheets. As shown in Fig. 3A, the Fc spectrum (curve a) exhibits strong vibrational absorption peaks of the C–O bond, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, and CO bond at 1282 cm−1, 1469 cm−1, and 1650 cm−1, respectively, because of the presence of its own carboxyl group. The MoS2-Fc spectrum (curve b) showed M-type peaks at 3500–4000 cm−1, which were different from those observed in the Fc spectrum, and the intensity of the vibrational absorption peaks of the C–O bond, CC bond, and CO bond diminished, demonstrating the successful binding of MoS2-Fc. The continued diminution of the intensity of the vibrational absorption peaks of the C–O bond, CC bond, and CO bond in the rGO@MoS2-Fc spectrum (curve c) is caused by the successful binding of rGO to MoS2-Fc.
C bond, and CO bond at 1282 cm−1, 1469 cm−1, and 1650 cm−1, respectively, because of the presence of its own carboxyl group. The MoS2-Fc spectrum (curve b) showed M-type peaks at 3500–4000 cm−1, which were different from those observed in the Fc spectrum, and the intensity of the vibrational absorption peaks of the C–O bond, CC bond, and CO bond diminished, demonstrating the successful binding of MoS2-Fc. The continued diminution of the intensity of the vibrational absorption peaks of the C–O bond, CC bond, and CO bond in the rGO@MoS2-Fc spectrum (curve c) is caused by the successful binding of rGO to MoS2-Fc.
 | ||
| Fig. 3 (A) FT-IR spectra, (B) XRD spectra and (C) XPS analysis of rGO@MoS2-Fc. (D) XPS spectra of S 2p, (E) XPS spectra of Mo 3d and (F) XPS spectra of Fe 2p of rGO@MoS2-Fc. (G) UV-vis spectrum of the rGO@MoS2-Fc/LDLapt probe. (H) Size distribution and (I) zeta potential of rGO, Fc, MoS2, MoS2-Fc, rGO@MoS2-Fc, and rGO@MoS2-Fc/LDLapt probes, respectively. | ||
The XRD spectrum of the as-synthesized rGO@MoS2-Fc is shown in Fig. 3B. The MoS2-Fc spectrum (curve b), compared with the Fc spectrum (curve a), not only shows the characteristic peaks of the Fc spectrum, but also has other characteristic peaks that do not belong to the Fc spectrum, which are unique to MoS2 when compared with the MoS2 standard card (JCPDS: 24-0513),34 indicating the successful preparation of MoS2-Fc. In the rGO@MoS2-Fc spectrum (curve c), the intensity of both the MoS2 characteristic peak and the Fc characteristic peak were weakened, which may be due to the binding process of rGO to MoS2-Fc, proving the successful preparation of rGO@MoS2-Fc.
XPS is used to analyze the elements and composition of rGO@MoS2-Fc as shown in Fig. 3(C–F). Fig. 3C shows the elemental distribution of rGO@MoS2-Fc, where the Fe element is from Fc, and C and O elements are from both Fc and rGO. Mo, S and N elements are from amino-modified MoS2. The inset of Fig. 3C shows that the C element accounts for 59.49%, the O element accounts for 30.50%, the Fe element accounts for 3.43%, the Mo element accounts for 0.72%, the S element accounts for 2.80%, and the N element accounts for 3.06%. Fig. 3D shows the XPS energy spectrum analysis of S 2p, where S 2p3/2 at 161.5 eV and S 2p1/2 at 162.7 eV correspond to the divalent sulfide ion (S2−) and the S–C bond of MoS2, respectively, while the peak at 167.6 eV corresponds to the S–O bond, indicating the binding of MoS2-Fc to rGO.35Fig. 3E shows the XPS analysis of Mo 3d;36 the peak at 226.1 eV corresponds to S 2s, indicating S–Mo bonding; the peaks at 228.7 eV, 231.9 eV, 231.1 eV, and 233.9 eV correspond to (Mo4+)Mo 3d5/2, (Mo4+)Mo 3d3/2, (Mo6+)Mo 3d5/2, and (Mo6+)Mo 3d3/2, respectively, indicating the presence of MoS2 in the prepared rGO@MoS2-Fc and the presence of both the tetravalent and hexavalent state of MoS2. Fig. 3F shows the XPS spectra of Fe 2p with the peaks at 710.0 eV, 711.4 eV, 715.0 eV, 719.0 eV, 723.6 eV, 725.5 eV, 729.0 eV, and 732.8 eV corresponding to (FeO)Fe 2p3/2, (Fe2O3)Fe 2p3/2, Fe(II)Fe 2p3/2, Fe(III)Fe 2p3/2, (FeO)Fe 2p1/2, (Fe2O3)Fe 2p1/2, Fe(II)Fe 2p1/2, and Fe(III)Fe 2p1/2, respectively, demonstrating the presence of Fc in the prepared rGO@MoS2-Fc. In addition, XPS analysis of C1s, N1s, and O1s in rGO@MoS2-Fc is presented in Fig. S3.†
As shown in Fig. 3H (curves b, d, e, f, and g), the particle sizes of rGO (curve d), Fc (curve e), MoS2 (curve f), MoS2-Fc (curve g), rGO@MoS2-Fc (curve b) are 124.9 nm, 165.9 nm, 135.2 nm, 401.3 nm, and 255 nm, respectively. The combination of MoS2 with Fc increases the particle size, and when combined with rGO, the particle size of rGO@MoS2-Fc decreases, indicating that rGO helps disperse MoS2-Fc, which proves the successful preparation of rGO@MoS2-Fc. As shown in Fig. 3I (curves b, d, e, f, and g), the potentials of rGO (curve d), Fc (curve e), MoS2 (curve f), MoS2-Fc (curve g), and rGO@MoS2-Fc (curve b) are −2.14 mV, 13.4 mV, −19.1 mV, −22.8 mV, and −0.151 mV, respectively. The small value for rGO indicates that it has certain instability, which is caused by the easy redox properties of rGO. Since MoS2 has a negative charge and Fc has a positive charge, the combination of MoS2-Fc is feasible due to electrostatic adsorption, and the high values of MoS2 and Fc lead to the high value of MoS2-Fc, thus resulting in a certain degree of stability. Due to the combination with unstable rGO, the value of rGO@MoS2-Fc is relatively low, thus resulting in a certain degree of instability.
3.3 Characterization of the rGO@MoS2-Fc/LDLapt signal probe
Fig. 3G shows the UV-Vis characterization of rGO@MoS2-Fc/LDLapt. LDLapt (curve a) exhibits a strong absorbance at 260 nm, representing the characteristic absorption peak of single-stranded DNA. The supernatant of rGO@MoS2-Fc/LDLapt (curve b) shows no characteristic absorption peak at 260 nm, indicating that LDLapt is bound to rGO@MoS2-Fc through chemical bonds or π–π bonds. rGO@MoS2-Fc/LDLapt (curve c) exhibits a weak characteristic absorption peak at 260 nm, which may be the characteristic absorption peak of rGO or LDLapt on rGO@MoS2-Fc/LDLapt, indicating the successful preparation of rGO@MoS2-Fc/LDLapt.Fig. 3H (curves a, b, and c) shows the particle size diagram of rGO@MoS2-Fc/LDLapt. The particle sizes of LDLapt (curve a), rGO@MoS2-Fc (curve b), and rGO@MoS2-Fc/LDLapt (curve c) are 434.5 nm, 255 nm, and 600.9 nm, respectively. It can be seen that the particle size of rGO@MoS2-Fc/LDLapt increases due to the combination of rGO@MoS2-Fc and LDLapt, indicating the successful preparation of the rGO@MoS2-Fc/LDLapt signal probe.
Fig. 3I (curves a, b, and c) shows the potential characterization diagram of rGO@MoS2-Fc/LDLapt. The potentials of LDLapt (curve a), rGO@MoS2-Fc (curve b), and rGO@MoS2-Fc/LDLapt (curve c) are −3.84 eV, −0.151 eV, and −14.9 eV, respectively. rGO@MoS2-Fc/LDLapt has a more negative potential, which may be due to the negatively charged phosphate skeleton of the aptamer,37 indicating the successful preparation of rGO@MoS2-Fc/LDLapt.
3.4 Enzymatic kinetics and catalytic studies of rGO@MoS2-Fc
As shown in Fig. 4A, no adsorption peak but straight lines are observed for OPD + H2O2, OPD + rGO@MoS2-Fc, and H2O2 + rGO@MoS2-Fc (curves 1, 2, and 3 in Fig. 4A). Besides, the OPD + H2O2, OPD + rGO@MoS2-Fc, and H2O2 + rGO@MoS2-Fc show no color change (tubes 1, 2, and 3 in Fig. 4A). With the addition of OPD, H2O2, and rGO@MoS2-Fc, a color change can be observed (tube 4 in Fig. 4A) and a big adsorption peak appeared at 425 nm (curve 4 in Fig. 4A), indicating that rGO@MoS2-Fc catalyzed the oxidation of H2O2 and OPD to DAP. As shown in Fig. 4B, no adsorption peaks are observed for TMB + H2O2, TMB + rGO@MoS2-Fc, and H2O2 + rGO@MoS2-Fc (curves 1, 2, and 3 in Fig. 4B). Besides, the TMB + H2O2, TMB + rGO@MoS2-Fc, and H2O2 + rGO@MoS2-Fc show no color change (tubes 1, 2, and 3 in Fig. 4B). With the addition of TMB, H2O2, and rGO@MoS2-Fc, a color change can be observed (tube 4 in Fig. 4B) and a big adsorption peak appeared at 650 nm (curve 4 in Fig. 4B). This is because rGO@MoS2-Fc catalyzed the oxidation of TMB in the presence of H2O2, which proved that rGO@MoS2-Fc catalyzed the oxidation of H2O2 and caused the oxidation of TMB to oxTMB, changing from a colorless liquid to a blue liquid. Therefore, it can be concluded that rGO@MoS2-Fc has the ability to catalyze H2O2 and thus cause the color change of the chromogenic substrate.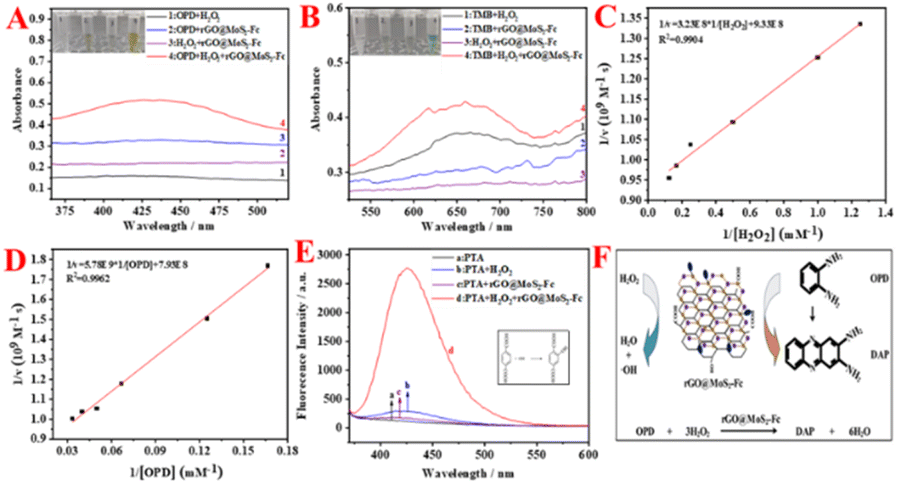 | ||
| Fig. 4 UV-vis absorption spectra obtained using (A) OPD–H2O2 solution and (B) TMB–H2O2 solution. Double reciprocal plots of different concentrations of (C) H2O2 and (D) OPD. (E) Fluorescence experimental diagram of the reaction of PTA with rGO@MoS2-Fc and (F) Schematic illustration of the catalytic mechanism of rGO@MoS2-Fc. | ||
To analyse the steady-state kinetics of rGO@MoS2-Fc catalysis, the concentrations of OPD and H2O2 were varied, and the Michaelis–Menten constants (Km) and the reaction rate (Vmax) were calculated. The lower the value of Km, the higher the enzyme's affinity for the substrate. Fig. 4C & D show the double reciprocal plots obtained by varying the concentrations of H2O2 and OPD, respectively. The Vmax and Km of rGO@MoS2-Fc with H2O2 are 1.07 × 10−9 (M s−1) and 0.346 mM, respectively. Similarly, the Vmax and Km values for rGO@MoS2-Fc with OPD as the substrate are 1.26 × 10−9 (M s−1) and 7.29 mM, respectively. Compared to HRP with a Km of 13.247 mM (H2O2) and 18.657 mM (OPD),38 the Km (H2O2) and Km (OPD) of rGO@MoS2-Fc are smaller, indicating that rGO@MoS2-Fc has stronger binding ability to the OPD chromogenic substrate. Furthermore, the pH stability and temperature stability of rGO@MoS2-Fc were measured along with those of HRP, and the two were compared as shown in Fig. S4.†
Fig. 4E shows the fluorescence diagram of the reaction of PTA with rGO@MoS2-Fc, and the inset shows the reaction equation of PTA with ·OH. PTA alone (curve a) and PTA + rGO@MoS2-Fc (curve c) do not fluoresce. When PTA + H2O2 (curve b) is observed, a weak fluorescence peak change is visible, which is because H2O2 can be catalytically decomposed to produce ·OH, and PTA reacts with free ·OH, thus generating fluorescence. When PTA + H2O2 + rGO@MoS2-Fc (curve d) is observed, a clear fluorescence peak change is seen, which proves that rGO@MoS2-Fc can catalyze the decomposition of H2O2 to generate ·OH, indicating that rGO@MoS2-Fc has peroxidase-like properties. Thus, the schematic illustration of the catalytic mechanism rGO@MoS2-Fc is depicted in Fig. 4F. H2O2 is catalyzed in the presence of rGO@MoS2-Fc to produce H2O and ·OH, the latter of which could promote the oxidation of OPD substrates to form DAP, thus turning the substrate yellow. Furthermore, the optimal enzymatic catalytic activity of rGO@MoS2-Fc was observed at 25 min in HAc–NaAc buffer (in Fig. S5†).
3.5 Performance analyses of the LDL colorimetric aptasensor
Under the optimized experimental conditions (concentration of LDLapt: 1.0 μM, incubation time for LDL: 60 min, incubation temperature for LDL: 25 °C, and pH value of HAc–NaAc buffer solution: 4.0, as shown in Fig. S6†), the concentration of LDL was varied and the absorbance was detected by UV-vis. Fig. 5A shows the UV curves of the LDL colorimetric aptasensor at different LDL concentrations (0, 15.0, 30.0, 50.0, 70.0, 90.0, 110.0, 130.0, 160.0, 170.0, 180.0, 190.0, and 200.0 μg mL−1). The absorbance values increase with the increase in LDL concentration. The color of the corresponding experimental phenomenon (inset of Fig. 5B) changed from light to dark with the increase in LDL concentration. This is because with the increase in LDL concentration, the number of binding sites for rGO@MoS2-Fc/LDLapt increased, which led to an increase in the number of catalytic sites and increased catalytic H2O2 decomposition to generate more ·OH, resulting in the deepening of the color of the OPD chromogenic substrate. Fig. 5B shows the calibration curve between the absorbance values and different LDL concentrations. In the range of 15.0–200.0 μg mL−1, there is a linear relationship between absorbance of the color change and the LDL concentration. The linear equation is Y = 0.00386 × CLDL + 0.2912 (R2 = 0.9946). And the limit of detection (LOD) is calculated to be 2.199 μg mL−1 from the triple signal-to-noise ratio (S/N = 3).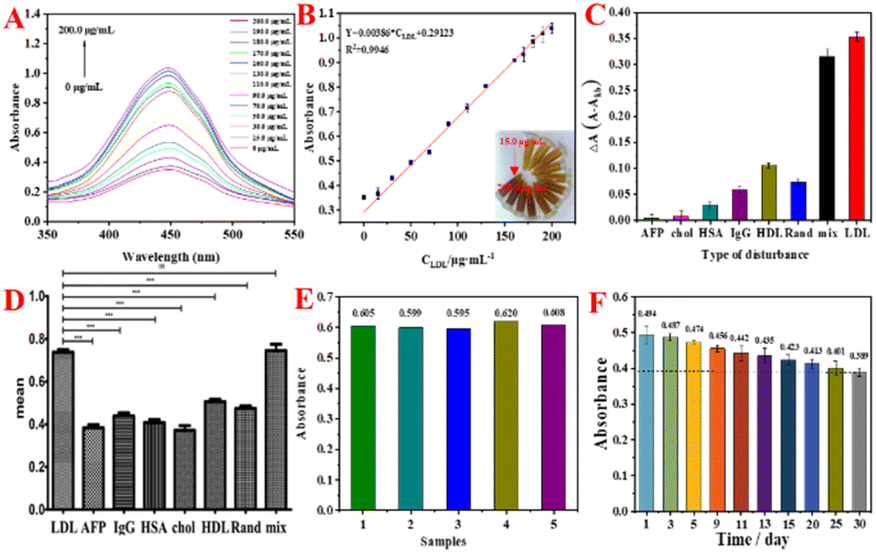 | ||
| Fig. 5 (A) UV-vis spectra of the colorimetric detection of LDL. (B) The linear calibration curve for LDL detection. (C and D) Specificity analysis of the colorimetric aptasensor. (E) Reproducibility and (F) stability of the colorimetric aptasensor. | ||
Moreover, the constructed colorimetric aptasensor was compared with other LDL assays, and the results are shown in Table 1. Compared with other methods, the LOD is slightly higher than that of other methods. The constructed colorimetric aptasensor using the OPD–H2O2 colorimetric system has a wider detection range that meets the clinical requirements.
| Materials | Method | Detected range | LOD | Ref. |
|---|---|---|---|---|
| LDL/BSA/AAB/Gly/N-CDs/Ni-MnFe-LDHs | Colorimetric (TMB) | 0.0625–0.75 mg dL−1 | 0.051 mg dL−1 | 39 |
| Ab-Fc/4-ATP | Electrochemical detection | 0.01–1.0 ng mL−1 | 0.53 ng mL−1 | 40 |
| N-CDs/Ni-MnFe-LDHs | Immunoassay | 625.0–7500.0 ng mL−1 | 51.0 ng mL−1 | 41 |
| Functionalised gold electrodes | Non-faraday impedance | 0.05–500.0 ng mL−1 | 0.12 ng mL−1 | 3 |
| MoS2-Fc-Au | Electrochemical detection | 0.001–100.0 μg mL−1 | 0.42 ng mL−1 | 42 |
| rGO@MoS2-Fc/LDLapt/LDL/LDLapt | Colorimetric (OPD) | 15.0–200.0 μg mL−1 | 2.199 μg mL−1 | This work |
In the actual detection of serum, the designed sensor has a shorter pretreatment time compared with the enzyme linked immunosorbent assay (ELISA) method; the pretreatment time for the sensor was 90 min, and the pretreatment time for ELISA was 5 h. After adding protein, the detection time is similar to that of ELISA, taking about 2 hours,44 but it can roughly display the results immediately; the operation mode during detection is more convenient, and the detection time is shorter than that of clinical detection method, which takes 3 h to 6 h.
To study the specificity of the colorimetric aptasensor for LDL, different interfering proteins were used in the experiments such as AFP, chol, HSA, IgG, HDL, Rand, and a mixture of these proteins. Fig. 5C shows the histogram for specificity of the colorimetric aptasensor. The vertical coordinate is ΔA obtained by subtracting the absorbance value of the blank group from the absorbance value of different substances. The ΔA value of each interfering protein is much lower than one-third of the value for LDL. And the ΔA for the mixture is close to that of LDL, indicating that the constructed colorimetric sensor has good specificity and selectivity for the target LDL. Meanwhile, the colorimetric sensor modified with Rand shows a lower value, indicating that Rand does not have the specificity to recognize LDL, proving that LDLapt has selective specificity and high affinity for LDL. In addition, the specificity data were analyzed using Graphpad prism statistical analysis software43 to verify the specificity of the LDL colorimetric aptasensor. The data were processed using one-way ANOVA for the comparison of data from the same colorimetric sensor detecting different proteins, and the results are shown in Fig. 5D. There is a significant difference between LDL protein and other interfering proteins (the differences between different interfering proteins has been indicated by ‘*’) due to P < 0.001, which is statistically significant. But there is no significant difference between LDL and the mix. The results show that the constructed colorimetric sensor has good selectivity and specificity.
Fig. 5E shows the reproducibility graph of the colorimetric aptasensor. As shown in Fig. 5E, the RSD between the absorbance values is small (RSD = 0.016), indicating that the prepared colorimetric sensor has good reproducibility. In addition, Fig. 5F shows the stability graph of the colorimetric aptasensor. The absorbance value gradually decreased with the increase in storage days. Compared with the first day, the absorbance value on the 30th day was still 79.3% of the original value, which showed that the prepared colorimetric sensor had good stability.
3.6 Detecting LDL in human serum samples using the colorimetric aptasensor
The constructed colorimetric sensor was used to directly detect a 10-fold dilution human serum sample and was compared with the clinically known serum LDL concentration. The results are shown in Table 2. The relative error between the constructed colorimetric sensor and the LDL concentration in the actual serum samples ranged from 1.70% to 4.71%, and the relative standard deviation (RSD) ranged from 0.12% to 2.45%. A paired t test with clinical data showed that the t value was very small (0.372); the P value was 0.721 (P > 0.05). There is no difference between the paired t-test and P value, which proves that the detection results of human serum are not significantly different from the clinical data. Hence, the constructed colorimetric sensor has good potential for the determination of LDL concentration in human serum samples.| Real serum samples | Clinically LDL concentration (μg mL−1) | Detected LDL concentration of (μg mL−1) | Relative error (%) | RSD (%) | Absolute value of t | P value |
|---|---|---|---|---|---|---|
| a Samples 1, 2, and 3: healthy person, samples 4, 5, and 6: patients with moderate coronary artery disease, and samples 7 and 8: patients with severe coronary artery disease. | ||||||
| Sample 1 | 38.2833 | 37.09 | 3.22 | 0.90 | 0.372 | 0.721 |
| Sample 2 | 48.7242 | 49.63 | 1.83 | 0.41 | ||
| Sample 3 | 56.0715 | 57.09 | 1.78 | 1.35 | ||
| Sample 4 | 133.7982 | 130.96 | 2.17 | 1.01 | ||
| Sample 5 | 143.8524 | 150.96 | 4.71 | 0.12 | ||
| Sample 6 | 152.7465 | 148.29 | 3.01 | 0.41 | ||
| Sample 7 | 171.6948 | 168.83 | 1.70 | 2.45 | ||
| Sample 8 | 177.882 | 184.83 | 3.76 | 2.15 | ||
4 Conclusions
In summary, rGO@MoS2-Fc was prepared with strong peroxidase catalytic activity that can catalyze H2O2, which in turn oxidized the colorless substrate OPD to the yellow color DAP. The rGO@MoS2-Fc/LDLapt recognition probe was synthesized by linking it to LDLapt, which enabled signal amplification and specific recognition of LDL. Based on this, a colorimetric aptamer sensor for detecting LDL was constructed using rGO@MoS2-Fc and LDLapt. The aptasensor exhibited a linear range of 15.0–200.0 μg mL−1 with a LOD of 2.199 μg mL−1. Overall, the assay has high selectivity, sensitivity and operability and can be applied for detecting LDL in actual human serum samples.However, the sensor has not been used for intracellular detection due to its higher LOD. In future research, the peroxidase catalytic properties of the materials will be further explored to obtain a lower LOD. And more in-depth research will be conducted on the cytotoxicity and biocompatibility of the materials. In brief, the designed colorimetric sensor will be improved to enhance its clinical applicability in the diagnosis of CVD.
Ethical approval
The human serum samples used in this study were approved by the 924th Hospital of Chinese People's Liberation Army Joint Logistic Support Force in Guilin, China.Data availability
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.Author contributions
Guiyin Li: methodology, funding acquisition, conceptualization, writing – review & editing. Tingting Yu: methodology, writing – original draft, formal analysis. Haimei Li: methodology, writing – original draft, data curation. Bingbing Wan: investigation, data curation. Xiaohong Tan: data curation, validation. Xueqing Zhou: conceptualization, writing – review & editing. Jintao Liang: project administration, funding acquisition, conceptualization, validation. Zhide Zhou: project administration, writing – review & editing, supervision.Conflicts of interest
The authors declare that they have no conflict of interest or personal relationship that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 62161009), the Special Projects in Key Areas of Ordinary Universities in Guangdong Province (No. 2023ZDZX2040), the projects of Talents Recruitment of GDUPT in Guangdong Province (No. RC-XJ2022000401), and the Guangxi Science and Technology Major Program (No. AA24011005).Notes and references
- J. Yu, X. Liu, S. Chen, Y. Liu, H. Liu, H. Zheng, N. Yang, S. Wu and Y. Li, Endocrine, 2021, 75, 418–426, DOI:10.1007/s12020-021-02870-3.
- H. Yao, C. Hou, W. Liu, J. Yi, W. Su and Q. Hou, BMC Cardiovasc. Disord., 2020, 20, 426, DOI:10.1186/s12872-020-01696-7.
- A. K. Assaifan, F. A. Alqahtani, S. Alnamlah, R. Almutairi and H. I. Alkhammash, BioChip J., 2022, 16, 197–206, DOI:10.1007/s13206-022-00058-z.
- Y. Qiao, Y. Zou and S. Guo, Front. Physiol., 2022, 13, 931, DOI:10.3389/fphys.2022.931931.
- J. Noh, M. Moon, E. Rhee, S. Park, H. Kim, B. Kim, H. Kim, S. Choi, J. Na, Y. Hyun, B. Kim, K. Han and I. Jeo, Diabetes Metab. J., 2023, 47, 59–71, DOI:10.4093/dmj.2021.0320.
- X. Sun, W. Chen, A. C. Razavi, M. Shi, Y. Pan, C. Li, M. Argos, B. T. Layden, M. L. Daviglus, J. He, O. T. Carmichael, L. A. Bazzano and T. N. Kelly, J. Am. Coll. Cardiol., 2024, 9, 577–590, DOI:10.1016/j.jacbts.2024.01.018.
- J. Cao, P. Lv, Y. Shu and J. Wang, Biosens. Bioelectron., 2022, 196, 113743, DOI:10.1016/j.bios.2021.113743.
- M. Tohidi, S. Asgari, A. Chary, F. Azizi and F. Hadaegh, Clin. Biochem., 2022, 109, 28–36, DOI:10.1016/j.clinbiochem.2022.08.007.
- A. Wolska and A. T. Remaley, Clin. Chem., 2019, 65, 1067–1069, DOI:10.1373/clinchem.2019.307678.
- M. Oztug, B. Vatansever, G. Altin, M. Akgoz and S. Z. Can, J. Mass Spectrom. Adv. Clin. Lab, 2024, 31, 40–48, DOI:10.1016/j.jmsacl.2024.01.002.
- G. Feng, W. Xie, F. Jiang, C. Shao, J. Yu, Q. Wu, Y. Jin, Q. Yang, W. Jin, J. Liu and T. Xu, Colloid Interface Sci. Commun., 2023, 57, 100748, DOI:10.1016/j.colcom.2023.100748.
- K. Shimura, S. Yoshida, H. Oikawa and T. Fujitani, Microporous Mesoporous Mater., 2022, 338, 111955, DOI:10.1016/j.micromeso.2022.111955.
- K. Xue, B. Cai, Y. Yang, A. He, Z. Chen and C. Zhang, Anal. Chim. Acta, 2024, 1309, 342646, DOI:10.1016/j.aca.2024.342646.
- C. Fan, S. Wang, K. Schanze and L. E. Fernandez, ACS Appl. Mater. Interfaces, 2021, 13, 9289–9290, DOI:10.1021/acsami.1c02475.
- D. Klapak, S. Broadfoot, G. Penner, A. Singh and E. Inapuri, PLoS One, 2018, 13, 1–16, DOI:10.1371/journal.pone.0205460.
- J. Wang, Q. Wang, Y. Zhong, D. Wu and N. Gan, Microchem. J., 2020, 158, 105288, DOI:10.1016/j.microc.2020.105288.
- G. Li, S. Li, Q. Huang, X. Li, Z. Zhang, J. Liang and Z. Zhou, Microchem. J., 2023, 194, 109314, DOI:10.1016/j.microc.2023.109314.
- X. Tan, W. Xie, Q. Jia, F. Zhao, W. Wu, Q. Yang and X. Hou, Analyst, 2022, 147, 4105–4115, 10.1039/d2an00668e.
- B. Liu, J. Zhuang and G. Wei, Environ. Sci.: Nano, 2020, 7, 2195–2213, 10.1039/d0en00449a.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076, 10.1039/c8cs00457a.
- H. Wang, K. Wan and X. Shi, Adv. Mater., 2019, 31, 1805368, DOI:10.1002/adma.201805368.
- Z. Chi, Q. Wang and J. Gu, Analyst, 2023, 148, 487–506, 10.1039/d2an01850k.
- X. Wang, M. Chen and L. Zhao, Chem. Eng. J., 2023, 468, 143612, DOI:10.1016/j.cej.2023.143612.
- L. Gong, L. Feng, Y. Zheng, Y. Luo, D. Zhu, J. Chao, S. Su and L. Wang, Biosensors, 2022, 12, 87, DOI:10.3390/bios12020087.
- J. Li, Y. Wang, D. Liu, K. He and L. Lu, Powder Metall. Technol., 2021, 39, 471–478, DOI:10.19591/j.cnki.cn11-1974/tf.2021020008.
- W. Sheng, M. Sun, D. Bai, L. Ren, T. Ya, Z. Jin, S. Wang, Z. Wang and X. Tang, Microchem. J., 2024, 201, 110637, DOI:10.1016/j.microc.2024.110637.
- J. Ran, D. Yang, K. Luo and B. Liu, Microchem J., 2024, 196, 109657, DOI:10.1016/j.microc.2023.109657.
- K. Ahmad, M. A. Shinde and H. Kim, Microchem. J., 2021, 169, 106583, DOI:10.1016/j.microc.2021.106583.
- G. Li, S. Li, Z. Wang, Y. Xue, C. Dong, J. Zeng, Y. Huang, J. Liang and Z. Zhou, Anal. Biochem., 2018, 547, 37–44, DOI:10.1016/j.ab.2018.02.012.
- R. Dadigala, R. Bandi, S. Han, G. Kwon and S. Lee, Int. J. Biol. Macromol., 2023, 234, 123657, DOI:10.1016/j.ijbiomac.2023.123657.
- C. Yuan, X. Qin, Y. Xu, R. Shi, S. Cheng and Y. Wang, Spectrochim. Acta, Part A, 2021, 254, 119678, DOI:10.1016/j.saa.2021.119678.
- G. Li, H. Li, C. Wang, X. Li, J. Chen, J. Liang and Z. Zhou, IEEE Sens. J., 2023, 23, 111–118, DOI:10.1109/jsen.2022.3224554.
- L. Zhao, J. Wang, D. Su, Y. Zhang, H. Lu, X. Yan, J. Bai, Y. Gao and G. Lu, Nanoscale, 2020, 12, 19420–19428, 10.1039/d0nr05649a.
- Z. Cui, D. Li, W. Yang, K. Fan, H. Liu, F. Wen, L. Li, L. Dong, G. Wang and W. Wu, Anal. Methods, 2022, 14, 1956–1962, 10.1039/d2ay00467d.
- N. H. A. Rosli, K. S. Lau, T. Winie, S. X. Chin, S. Zakaria and C. H. Chia, J. Energy Storage, 2022, 52, 104991, DOI:10.1016/j.est.2022.104991.
- J. S. Arya Nair, S. Saisree and K. Y. Sandhya, Analyst, 2022, 147, 2966–2979, 10.1039/D2AN00531J.
- Y. Zeng, M. Wang, Z. Sun, L. Sha, J. Yang and G. Li, J. Mater. Chem. B, 2022, 10, 450–455, 10.1039/d1tb02192c.
- H. Cheng, Z. Wang, H. Sun, B. Chen, J. Huang, R. Jia, X. He and K. Wang, Chem. Commun., 2022, 58, 13487–13490, 10.1039/d2cc05578c.
- G. Chen, T. Chai, J. Wang and F. Yang, J. Pharm. Biomed. Anal., 2023, 236, 115695, DOI:10.1016/j.jpba.2023.115695.
- A. Prakobkij, P. Jarujamrus, S. Chunta, R. Chawengkirttikul, T. Keawin, N. Malahom, S. Tamuang, M. Amatathongchai and D. Citterio, Microchim. Acta, 2022, 189, 72, DOI:10.1007/s00604-022-05173-0.
- D. Rudewicz-Kowalczyk and I. Grabowska, Molecules, 2022, 27, 5492, DOI:10.3390/molecules27175492.
- A. Prakobkij, P. Jarujamrus, S. Chunta, R. Chawengkirttikul, T. Keawin, N. Malahom, S. Tamuang, M. Amatathongchai and D. Citterio, Mikrochim. Acta, 2022, 189, 72, DOI:10.1007/s00604-022-05173-0.
- G. Li, H. Li, X. Li, H. Huang, H. Bian, J. Liang and Z. Zhou, Microchem. J., 2023, 193, 109068, DOI:10.1016/j.microc.2023.109068.
- R. J. Bevan, M. F. Durand, P. T. Hickenbotham, G. D. Kitas, P. R. Patel, I. D. Podmore, H. R. Griffiths, H. L. Waller and J. Lunec, Free Radical Biol. Med., 2003, 35, 517–527, DOI:10.1016/s0891-5849(03)00359-9.
Footnote |
| † Electronic supplementary information (ESI) available: Reagents and instrumentation, stability of rGO@MoS2-Fc, XPS, validation of experimental mechanisms, aptamer studies, and optimisation of experimental conditions. See DOI: https://doi.org/10.1039/d4ay01648c |
| This journal is © The Royal Society of Chemistry 2025 |