
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen chemistry: magnetic dispersive solid phase extraction for simultaneous enrichment and determination of V, Ni, Ti and Ga in water samples by HR-CS ETAAS†
L.
Vázquez-Palomo‡
a,
P.
Montoro-Leal‡
a,
J. C.
García-Mesa‡
 a,
M. M.
López Guerrero‡
*ab and
E.
Vereda Alonso‡
*ab
a,
M. M.
López Guerrero‡
*ab and
E.
Vereda Alonso‡
*ab
aDepartment of Analytical Chemistry, Faculty of Sciences, University of Malaga, Campus de Teatinos, 29071, Malaga, Spain. E-mail: mmlopez@uma.es; eivereda@uma.es
bInstituto Universitario de Materiales y Nanotecnología, IMANA, University of Malaga, Campus de Teatinos, 29071, Málaga, Spain
First published on 14th November 2024
Abstract
This work presents a straightforward, highly sensitive, and cost-effective method for the simultaneous determination of V, Ti, Ni and Ga by high resolution-continuum source electrothermal atomic absorption spectrometer (HR-CS ETAAS) in aqueous environmental samples (tap and seawater samples). The system is based on retention of the analyte onto a novel magnetic nanomaterial (M@GO magnetic graphene oxide) functionalised with methylthiosalicilate (MTS). The formed complexes between the M@GO-MTS and the target analytes were broken, adding 1 mL of nitric acid (6%) and sonication for 5 min. The optimized method achieved detection limits of 0.71 μg L−1 for Ti, 0.20 μg L−1 for V, 0.04 μg L−1 for Ga, 0.66 μg L−1 for Ni. The accuracy of the proposed method was demonstrated by analysing two certified reference materials and by determining the analyte content in spiked environmental water samples. The results obtained using this method were in good agreement with the certified values of the standard reference materials, and the recoveries for the spiked tap water and seawater samples ranged from 94% to 120%.
1 Introduction
The toxicity of metals is governed by their physicochemical characteristics as ion size, geometry, and oxidation state, determining how metals interact in the bodies of animals, potentially affecting their overall homeostasis.1,2 Metals play crucial roles in biological processes and are often categorized into essential being required for metabolic functions, including growth, development, immunity, and reproduction, however, there are also non-essentials and borderline metals.3,4Vanadium, nickel, titanium, and gallium are elements widely found in the environment and used in various industrial and medical applications. Vanadium (V), found in products as catalysts and fertilizers,5–9 has been listed as a highly toxic substance according to the World Health Organization (WHO) classification and as the 13 priority pollutants mentioned in the American Environmental Protection Agency's Clean Water Act.10–12 The concentration of dissolved V in water should not exceed the long-term ecotoxicology limit of 1.2 μg L−1.13 Nickel (Ni), essential in trace amounts for biological processes,14 enters ecosystems through natural processes15,16 and human activities as mining.17,18 It is regulated due to its potential toxicity, common levels of nickel in uncontaminated freshwater sources can range from a few μg L−1 to around 20 μg L−1.18,19 However, higher levels may be found in areas with industrial discharges or pollution sources. Titanium (Ti), is typically found in low concentrations, generally below 10 μg L−1, depending on the geological and environmental characteristics of the area. Higher concentrations of Ti may be found in waters near industrial areas or due to anthropogenic contamination. The determination of Ti in environmental water can be significant, not so much due to its toxicity, but as an indicator of industrial pollution or for studying water treatment processes.20,21 Gallium (Ga), an emerging contaminant, is used in high-tech industries and medical procedures.22–25 Prolonged exposure can cause severe health issues include tachycardia and vertigo,26 but there are no established water quality guidelines for this metal in marine environments.22–24
Biologically active V, Ni, Ga and Ti compounds could pose a potential threat for marine biota when released into the environment. Marine invertebrates are known to accumulate high levels of metals in their tissues. Therefore, accurate risk assessment tools adapted to marine ecosystems based on relevant techniques are needed to identify the pollutants of interest. Such tools can assist in predict potential risks and the hazards associated with marine pollution. The monitoring and control of these trace elements in the environment sources require processing large numbers of samples to accurately characterize their abundance and to reach reliable conclusions.
Electrothermal atomic absorption spectrometry (ETAAS) has been extensively used for trace element analyses. The use of more efficient modifiers, advancements in controlling atomization temperatures, new designs of atomizers, and improvements in methods for correct background spectral interferences have allowed an enhancement in sensitivity, an increase in detection power, reduced sample handling and improved reproducibility of results. Furthermore, the current introduction of high resolution continuum source electrothermal atomic absorption spectrometry (HR-CS ETAAS) has facilitated direct solid sampling, reducing background noise and opening up the possibility of even faster quantification of certain elements and even the simultaneous determination of some elements.27 The combination with the higher radiation intensity of the Xe (only one lamp source is needed) makes it feasible to obtain better defined signals at lower analyte levels, resulting in improved limits of detections. The improved analytical performance of this technique can be seen in elsewhere.28–30 This technique shows a relatively cost-effective approach with low sample consumption, providing figures of merit that can be equivalent to those of inductively coupled plasma mass spectrometry (ICP-MS).30
To increase the sensitivity of the technique, solid phase extraction (SPE) is widely used as pretreatment method for the extraction and preconcentration of analytes. This approach is characterized by its easy to use, fast, low solvent consumption, and high enrichment factors. Thus, the selectivity, sensitivity and precision of the analytical method can be significantly enhanced.31,32 Graphene oxide (GO) and magnetic nanoparticles (MNPs) are materials which offer several advantages that make them attractive for analytical applications, including a large surface area and the presence of active sites on their surfaces. GO is an excellent sorbent; however, it is difficult to separate from the sample. The degradability, biocompatibility, chemical stability, low toxicity, and high magnetic response of iron oxide MNPs (Fe3O4) have been exploited as solid phase sorbents termed magnetic solid phase extraction (MSPE). To exploit the excellent characteristics of both sorbents and mitigate their drawbacks, e.g., to prevent the MNPs aggregates, and to facilitate the separation of GO from the sample, MNPs–GO coupling has been established as an excellent analytical tool for the development of MSPE pre-treatment methods.33
In this work, silica coated magnetic nanoparticles joined to graphene oxide (M@GO)34 modified with a chelating group methylthiosalicilate (MTS) were used as MSPE sorbent to increase the selectivity. The re-extraction of the analytes was reached with only 1 mL of eluent (HNO3 6%).
Considering the diverse sources and characteristics affecting water quality, it is imperative to analyze and understand the concentrations of specific elements, such as vanadium (V), nickel (Ni), gallium (Ga), and titanium (Ti), in aquatic ecosystems. The development of reliable analytical methods capable of the simultaneous determination of several elements (Ti, V, Ni and Ga) is challenging from an analytical point of view because considerably increasing sample throughput (by eliminating sequential determination) and reducing the risk of contamination and/or losses. Moreover, it is worth noting that this approach is particularly intriguing as it offers a cost-effective solution, given that simultaneous analysis of multiple elements is typically not feasible with this conventional equipment.
In this manuscript, the developed method has shown to be a highly useful tool for simultaneous quantitation of V, Ni, Ti and Ga in environmental samples. To the best of the authors' knowledge, this method represents the first instance allowing the simultaneous determination of the four target analytes (V, Ti, Ni and Ga) by ETAAS in water samples. For the quality control of the analytical performance and the validation of the developed method, the analysis of two certified samples, SPS-SW2 Batch 125 and TMDA 64.3, Fortified Lake Waters were addressed. The results obtained from the study exhibited an acceptable level of agreement with the certified values. The method has been applied to tap and seawater samples with goods results.
2 Experimental
2.1 Instrumentation
All experiments were carried out using a HR-CS ETAAS, ContrAA 700, (Analytik Jena AG, Jena, Germany), equipped with both a electrothermal atomizer and flame atomizers. High quality transversely heated pyrolytic graphite tubes with pyrolytic graphite platform (Analytik Jena AG, Jena, Germany) were used in this study. A description of this instrument can be found elsewhere.27The lines selected for the simultaneous absorbance measurements and their sensitivities were 294.318 nm (V), sensitivity 2.3%, 294.364 nm (Ga), sensitivity 100%, 294.199 nm (Ti), sensitivity 42%, and 294.391 nm (Ni), sensitivity 1.4%, respectively, and the type of background correction used was IBC (Iterative Baseline Correction). A similar spectral area was previously used by us for the direct analysis of Fe, V, Ga and Ni in solid fuel fly ash.35
The peak height selected absorbance was equivalent to three pixels (central pixel ± adjacent ones). The electrothermal atomizer heating program for the simultaneous determination of V, Ni, Ti and Ga is provided in Table 1.
| Stage | Temperature (°C) | Ramp (°C s−1) | Hold time (s) | Ar flow rate |
|---|---|---|---|---|
| Drying | 110 | 6 | 60 | Max |
| Pyrolysis | 1400 | 70 | 3 | Max |
| Gas adaptation | 1400 | 0 | 5 | Stop |
| Atomization | 2650 | 3000 | 5 | Stop |
| Cleaning | 2650 | 0 | 5 | Max |
Argon with a purity of 99.99% was used as purging and protective gas for the graphite atomizer.
A conductometer SensION™ + EC7 (HACH, Colorado, EEUU) was used for salinity measurements of the water samples.
2.2 Reagents and samples
High purity reagents were used in all experiments. All solutions were prepared using doubly de-ionized water (18 MΩ cm) obtained from a Milli-Q water system (Millipore, Bedford, MA, USA).For the synthesis of M@GO MTS graphite powder, sodium chloroacetate, ferrous chloride tetrahydrate (FeCl2·4H2O), ferric chloride hexahydrate (FeCl3·6H2O), 37% HCl (wt/wt), ammonium hydroxide 30% (wt/wt), 97% 3-aminopropyltriethoxysilane, ≥99% tetraethoxysilane (TEOS), methanol, sodium chloride, ≥99.5% ethylenediamine (EN) and 99% N,N′-dicyclohexylcarbodiimide (DCC) were used. 97% Methyl thiosalicylate (MTS) was used for functionalization. All reagents were supplied by Merck (Merck, Darmstadt, Germany).
V(V), Ti(IV), Ni(II), and Ga(III) stock standard solutions 1000 mg L−1 (Merck, Darmstadt, Germany) were used for preparing the calibration curve. Standards of 1000 mg L−1 Pd(II)/Mg(II), Ir(III), Nb(V), and W(VI) solutions (PerkinElmer pure, Darmstadt, Germany) were used to coat the graphite furnace platform.
A pH 5.5 buffer was prepared by mixing acetic acid (0.2 M, 14.8 mL) and sodium acetate (0.2 M, 35.2 mL) (Merck, Darmstadt, Germany) and diluting to 100 mL with de-ionized water.
The accuracy of the proposed procedure was determined by analysing the certified reference materials (CRMs) from National Research Council of Canada (NRCC) TMDA 64.3, Fortified Lake Waters and Standard reference material by National Institute of Standards and Technology (USA) SPS-SW2 Batch 125 Surface Water. To study the applicability of the proposed method, seawater and drinking water samples were also analysed.
2.3 Treatment of the graphite tube with a permanent modifier
Titanium and vanadium tend to form stable carbides in the graphite furnace which avoid their complete atomization. To solve this problem, permanent chemical modifiers were tested to cover the platform of the graphite furnace. The correct use of these modifiers has reduced interferences and improved the atomization process.35The pre-treatment of pyrolytically coated graphite tubes involved a specific procedure to ensure a permanent and high-quality Nb (niobium) coating. This process included the injection of a total of 20 aliquots of 20 μL of a 10 mg L−1 Nb modifier standard solution into the graphite tube, followed by running the device temperature program. The injections were dried slowly by heating the atomizer at 100 °C with a ramp rate and a hold time of 2 °C s−1 and 70 s, respectively. Subsequently, they were thermally treated at 1400 °C with a ramp rate and a hold time of 45 °C s−1 and 130 s, respectively. At the end of these 20 treatments, a reduction step at 1600 °C was applied for 15 s. A total mass of 0.004 mg of the modifier were deposited on the tube graphite wall.35–37 This coating enhances the performance of the tubes during subsequent analysis and extends its useful lifetime to around 350 firings.
On the other hand, the improvement of the vanadium atomization peak required further attention. Therefore, a study on non-permanent matrix modifiers was also conducted, including Pd/Mg (150 ng Pd/100 ng Mg), Ir/W (150 ng Ir/150 ng W), 50 ng and 100 ng Nb (5 μL Nb 5 and 10 mg L−1), as described in the literature.35–37
2.4 Analytical procedure
The synthesis of silica coating MNPs was described elsewhere38 and GO was prepared from natural graphite using the exfoliation process described by Diagboya et al.39 GO and MNPs were coupled using the patented synthesis of M@GO (Spain Patent with application for extension of European Patent ES2844942B2, EP4095097A1).40 After that, the organic group MTS was coupled to the material M@GO (M@GO-MTS) as described in ESI.† The ESI† (ESI) provides details on the synthesis route for the preparation of M@GO, the functionalization process, and the structure of the subsequent material (Fig. SM1†).For the extraction, aliquots of standard or samples were placed in 250 mL volumetric flasks, 25 mL buffer (pH = 5.5) and de-ionized water were added up to the mark. Then 20 mg M@GO-MTS was added. The M@GO-MTS and the sample were kept in contact for 5 min into an ultrasonic bath. After that, a strong Nd/Fe/B magnet was placed at the bottom of the tube and when the solution became clear, the supernatant was able to be easily decanted. The formed complexes between the M@GO-MTS and the target analytes were broken adding 1 mL of nitric acid (6%) and sonication for 5 min. Finally, the sorbent was separated using the Nd/Fe/B magnet and a volume of 20 μL of the supernatant was injected into the Nb coated graphite tube plus 5 μL Nb 10 mg L−1 and analysed by ETAAS.
The experimental conditions and the heating temperature program used were carefully optimized by a univariate form with standards of 15 μg L−1 V(V) and Ti(IV), 30 μg L−1 Ni(II) and 1.5 μg L−1 Ga(III). The optimized furnace temperature program is summarized in Table 1.
The absorbance was measured in quadruplicate (n = 4). All the experiments were carried out in graphite furnace with platforms, preliminary treated with Nb as permanent modifier. The temperature program for the graphite furnace treatment with Nb was described in Section 2.3.
2.5 Sample preparation
Two certified reference materials (CRMs), TMDA 64.3, Fortified Lake Waters and SPS-SW2 Batch 125 Surface Water were tested to validate the proposed procedure. The method was applied to the analysis of two type of water samples, sea and tap water, which were collected locally. These samples were collected in polypropylene bottles and stored at 4 °C as recommended by Method 3010B of the Environmental Protection Agency (USA), for 3 days. Samples were filtered through 0.45 μm pore size filters, acidified with HNO3 and stored until analysis.To analyse the CRMs, aliquots of 2.5 mL of sample were placed in 25 mL volumetric flasks, 2.5 mL of buffer pH 5.5 and de-ionized water were added up to the mark. For tap water analysis, 225 mL of tap water were place in 250 mL volumetric flasks, 25 mL of buffer pH 5.5, and for seawater, aliquots of 100 mL were placed in 250 mL volumetric flasks, 25 mL of buffer pH 5.5, and de-ionized water were added up to the mark.
Then, 2 mg of M@GO-MTS for CRMs and 20 mg for tap and sea water was added, following the procedure described in Section 2.4.
Four aliquots of each sample extract were injected for determination of target analytes under the optimal conditions of the proposed method. For seawater samples, the salinity of samples and standards were measured by conductimetry and adjusted at the same value with NaCl before the extraction. The salinity of each sample was analysed and found to be 35.5 g L−1 for seawater.
3 Results
3.1 Evaluation of analytical lines
Primarily, the appropriate wavelengths must be selected, and the sensitivity must be suitable for the analyte content. In the HR-CS ETAAS, the simultaneous determination requires atomic lines within the same spectral window. Thus, the instrument detector was centred at 294.318 nm, a secondary line of V, being for all the elements used secondary spectral lines, as well. Subsequently, the wavelength of 294.391 nm for Ni was chosen, 294.199 nm for Ti and 294.364 nm for Ga. The use of secondary lines is not a problem using a Xe short-arc lamp since its radiation intensity is high enough to provide good signal-to-noise ratios for all lines. The automatic correction for continuous background adsorption realized by 200 pixels on the CCD detector, gets even better signal-to-noise ratios. Thus, the spectral range between 294.134 nm and 294.499 nm was observed, Fig. 1.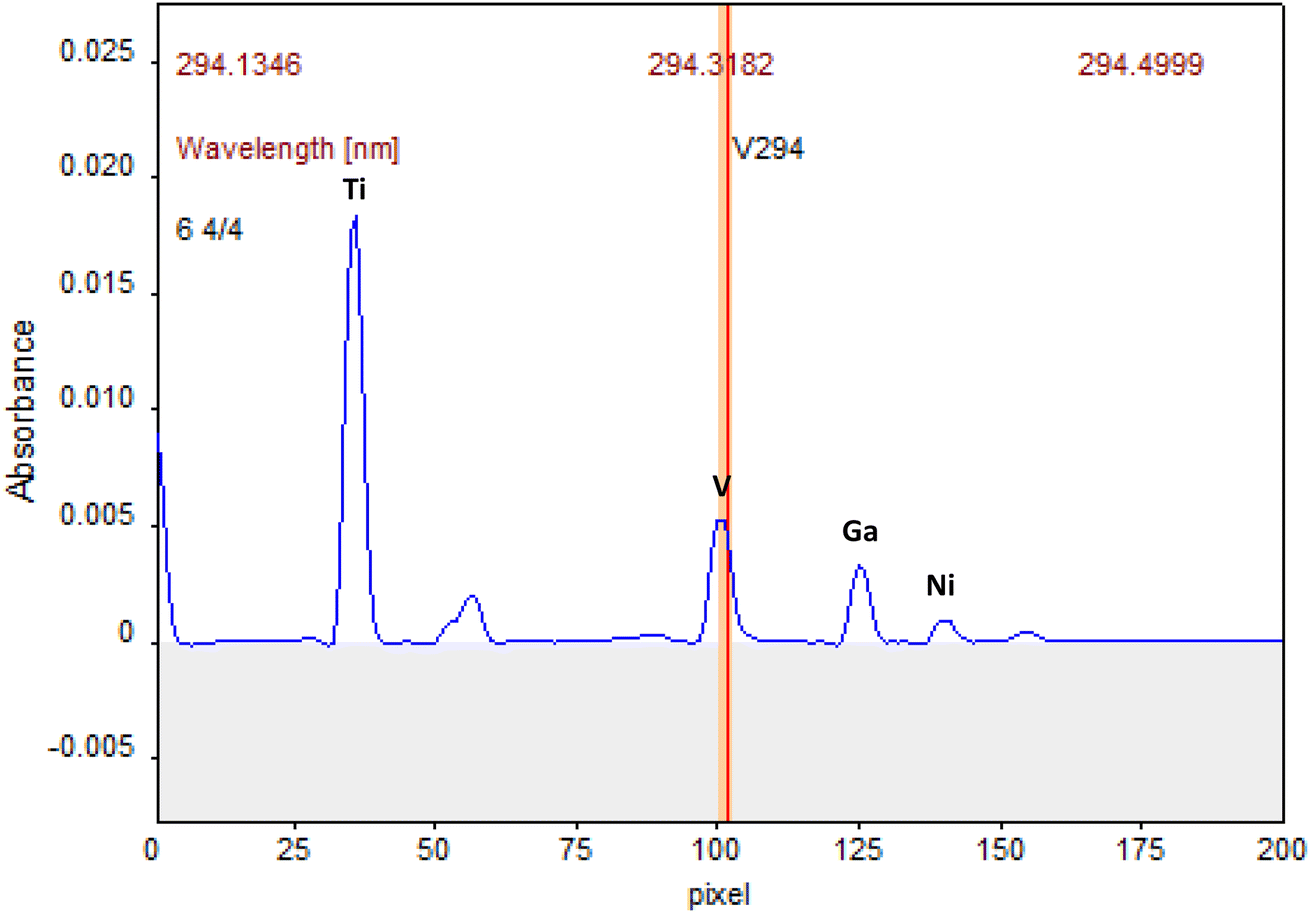 | ||
| Fig. 1 Atomic lines in the same spectral window for V, Ti, Ni and Ga. Atomization peaks obtained with a solution of 15 μg L−1 V, Ti, 30 μg L−1 Ni and 1.5 μg L−1 Ga. | ||
Additionally, the thermo-chemical behaviour of the target analytes in a graphite furnace requires to be considered. Thus, the furnace temperature program and modifiers were studied, as well.
The sensitivity was also studied varying the number of pixels for the determinations. In this case, the number of pixels was set to 3 (the central pixel plus the adjacent ones, CP ± 1). In this work, a further extension of the peak volume detection was not necessary because only noise would be integrated. The signals were measured in terms of both peak area and peak heights. The signals measured as peak height provided more reproducible results due to the tail of the vanadium and titanium atomization signals, which were reduced by the employ of modifiers, as will be discussed in the following sections. Peak height absorbance is probably less susceptible to interferences that can affect peak shape and, thus the accuracy of area integration.41
3.2 Optimization of method
The effect of different parameters on the accumulation and recovery of V, Ti, Ni and Ga were studied to optimize conditions for achieving the best results.The best signal-to-noise (S/N) ratios between a blank and a standard solution of 15 μg L−1 V, Ti, 30 μg L−1 Ni and 1.5 μg L−1 Ga were chosen as the optimization criteria. In the study, the analysis of each sample was repeated four times to ensure repeatability and reliability of the results.
Several parameters were optimized in the study to enhance the performance of the analytical method: (a) furnace program; (b) pH; (c) selection of the eluent; (d) extraction and elution times and amount of M@GO-MTS; and (e) volume of sample (preconcentration factor).
Pyrolysis and atomization steps are the most decisive steps to remove most of the matrix components without analyte loss, and to achieve well defined atomization peaks. Commonly, the pyrolysis temperature is established by considering the most volatile element present in the sample, and the atomization temperature is determined by considering the most refractory (least volatile) element present in the sample.42 The pyrolysis temperature was varied over the range 1200–1450 °C. When the pyrolysis temperature increased up to 1400 °C, the signal increased for three of all four, Fig. 2A. Although Ga showed a decrease in the signal, the sensitivity to the line is enough for the determinations. This method has shown the capability to determine concentrations of Ga up to ten times lower than those for the other elements. In comparison to nickel, gallium concentrations are twenty times lower. Consequently, temperature selection is based on maximizing the signal for nickel. The optimum pyrolysis temperature was determined to be 1400 °C. Slower pyrolysis temperature ramps help to eliminate matrix effect. Slower pyrolysis plays a crucial role in minimizing matrix effects during elemental analysis, allowing a better separation of analytes from interfering matrix components. This process enhances the accuracy and precision of measurements. Gradual heating helps the effective volatilization of matrix components without prematurely releasing the analytes, reducing the interference during the measurement phase.43
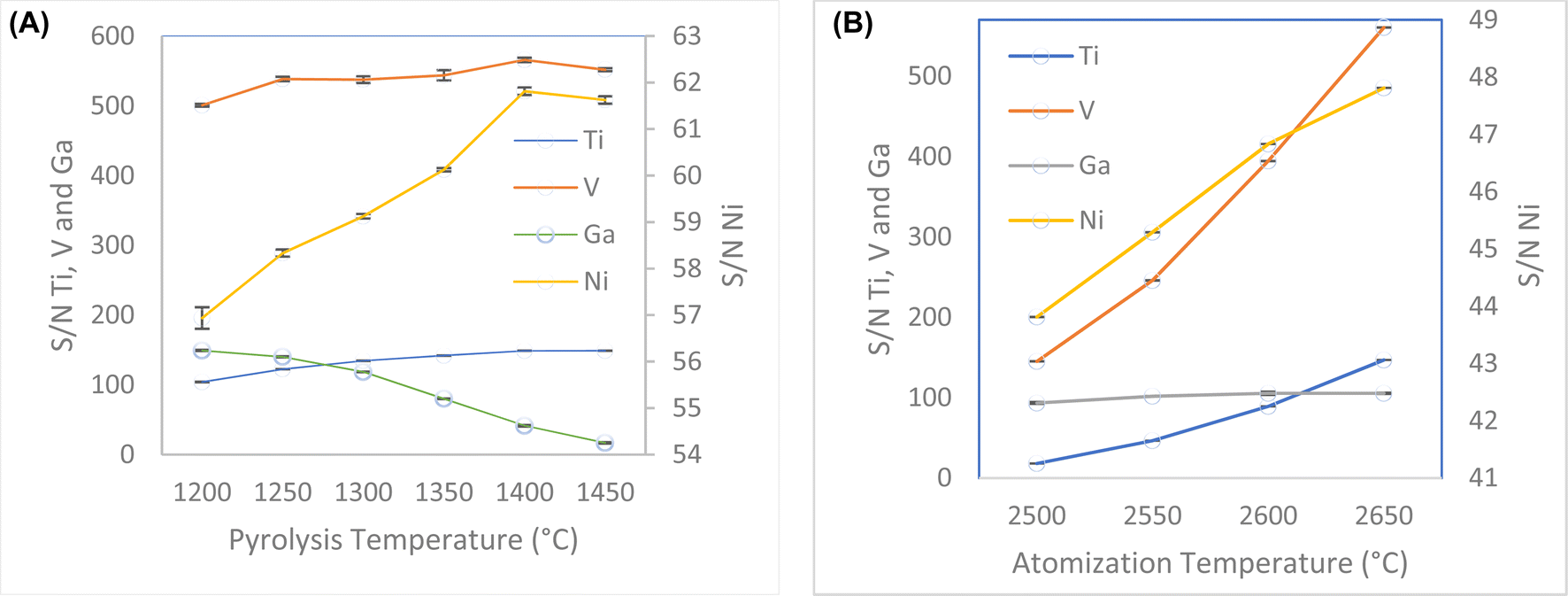 | ||
| Fig. 2 (A) Optimization of pyrolysis temperature between 1200–1450 °C. (B) Optimization of atomization temperature between 2500–2650 °C. | ||
The controlled temperature increase can lead to a more uniform particle size distribution, which aids in the consistent behaviour of the sample matrix, further mitigating matrix effects.43
Slower pyrolysis ramps can reduce the likelihood of signal suppression caused by matrix elements. This is particularly important when are used matrix modifiers, which can sometimes introduce additional variability in the results.44
The pyrolysis ramp was investigated between 30–110 °C s−1 and only a little influence on the analytical signal was found and was considered the optimal values 70 °C s−1. The pyrolysis hold time was varied between 3 and 10 s, only the signal of Ti was increased until 7 s, the signals of V, Ni and Ga started to decrease at times greater than 3 s. Thus, 3 s was selected as the optimum hold time.
Since most of the analytes are refractory, the atomization temperature was varied over the range 2500–2650 °C. When the atomization temperature increased, the signal also increased. The optimum atomization temperature was determined to be 2650 °C because at lower temperature, broader peaks were observed, Fig. 2B. The atomization ramp should be high enough to produce instantaneous atomization, thus a ramp time of 3000 °C s−1 was selected. The atomization hold time was studied between 3 to 10 s. The signals for all analytes increased until 5 s, after that the signals were constant or decreased. Thus, 5 s was selected as optimum hold time.
The high-atomization temperature obtained could potentially affect the thermal stability of the permanent chemical modifiers and lifetime of the graphite tube. However, the number of firings was also studied, and at least, 350 firings could be made without significant decrease in the target analyte signals. The optimal established furnace operation conditions for V, Ni, Ti and Ga based on permanent modifiers, have been listed in Table 1.
3.2.1.1 Use of chemical modifiers. In this study, several challenges were encountered, primarily related to the tail of the vanadium and titanium signals due to carbides formation, as documented by previous researchers.45,46 The formation of V, Ti carbides can interfere with the atomization process and affect the accuracy and precision of the analysis. To mitigate this issue, chemical modifiers that prevent carbide formation were explored and optimized. By adding Ir, W and Nb as permanent modifiers,46,47 the formation of carbides of V and Ti in the sample was reduced, improving the precision and accuracy of the analysis. Among permanent chemical modifiers, Nb showed beneficial for V and Ti determination, producing thinner V and Ti peaks, Fig. SM2A and SM2B.† The conditions of permanent modifiers deposition were optimized to achieve the best analytical signals.
The analysis of each sample was repeated four times, employing a one-at-a-time method to optimize the conditions. This optimization led to the selection of Nb as the most effective permanent modifier. In the case of the graphite surface modified Nb, the integrated absorbance for V is higher than for the graphite surface modified with other modifiers.
These results are supported by the study reported elsewhere36,37 where the niobium carbide formation on the graphite surface prevented the formation of vanadium and titanium carbides. Despite using a modifier and increased atomization temperatures, minor tailing in the atomization V peak still appeared on the spectra. To address this issue, several chemical matrix modifiers added along with the sample aliquot were also tested, including Ir/W, Nb, and Pd/Mg, Fig. 3. These modifiers have proven to be effective in thermally stabilizing metals, removing matrix constituents during vaporization and atomization, and extending the lifespan of the furnace.47 For analytical measurements, peak heights were used with better reproducibility to be independent of the peak tails.
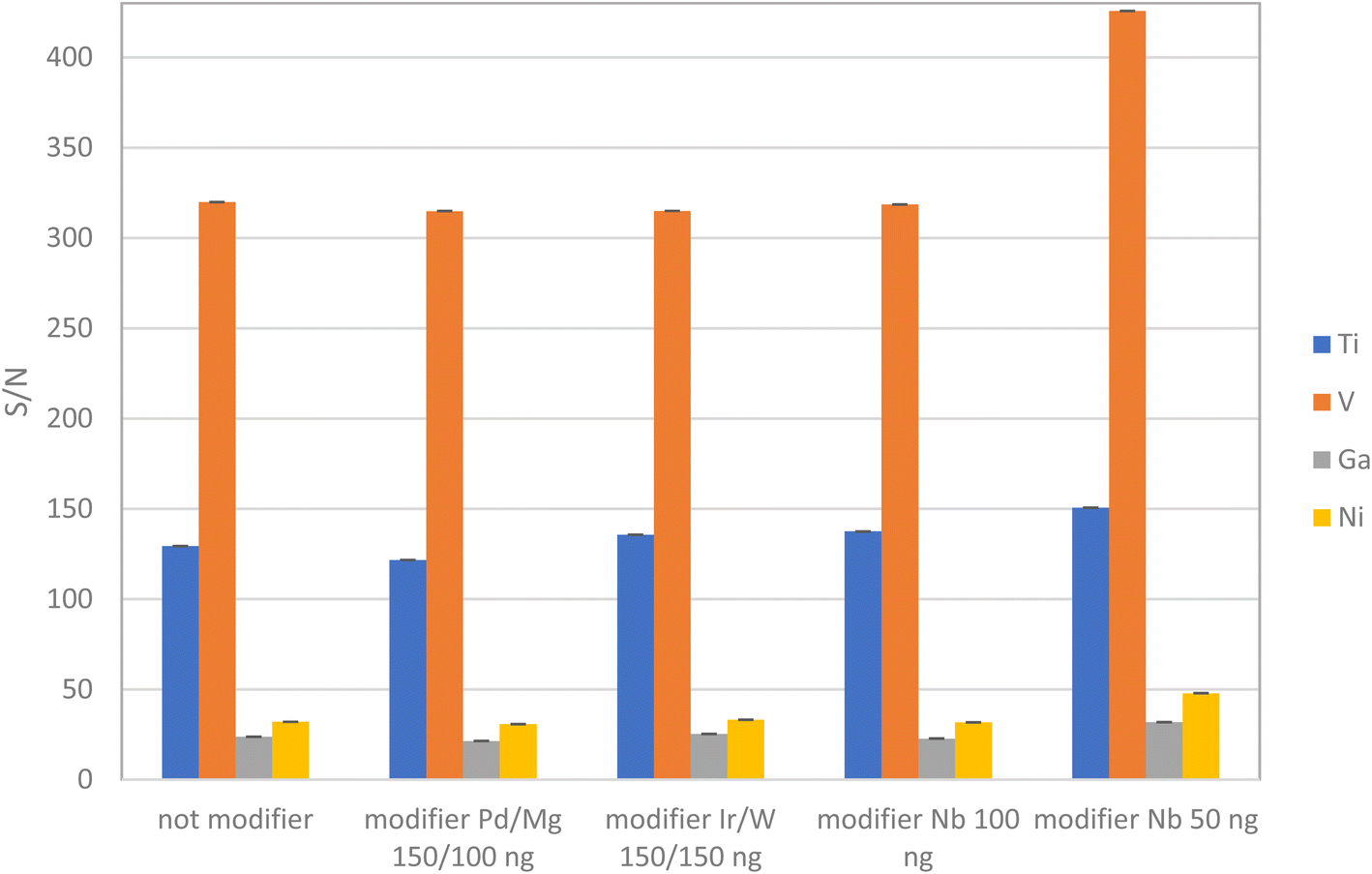 | ||
| Fig. 3 S/N ratio obtained using several chemical matrix modifiers and their mixtures. | ||
Thus, the quantities used for matrix modifiers were determined based on their effectiveness in facilitating the efficient thermal separation of the target analytes and concomitants during the pyrolysis step. Based on these studies, the optimal amount of Nb required was 0.004 mg as a permanent modifier plus 50 ng with each sample aliquot as a non-permanent modifier.
Thus, the pre-concentration of traces of the target analytes from buffered solutions at different pH values was studied. In this study, 25 mL of the standards buffered at different pHs were mixed with 2 mg M@GO-MTS during 5 min in the ultrasonic bath, after the magnetic decantation, 1 mL of 6% HNO3 was added and ultrasonicated 2 min. Finally, 20 μL of the decanted eluent was injected into the Nb coated graphite tube plus 5 μL Nb 10 mg L−1 and analysed by ETAAS. The pH of the buffered standards varied between 1.4 and 10.3. The pH from 1.4 to 5.0 was adjusted using glycine–HCl or sodium acetate–acetic acid buffer solution and from 5.0 to 9.9 using sodium hydroxide–boric acid buffer. The Fig. SM3† shows the absorbances versus pHs.
The optimum pre-concentration pH for Ti was from 4.0 to 6.0; for V was from 4.0 to 6.0, Ni showed optima pH interval, between pH 5.5–8.0, with a maximum to pH 5.5, and Ga showed a range between 4.0–7.0, being maximum at 5.0. Thus, to accomplish the simultaneous determination of the target elements, a pH value of 5.5 was chosen as overall optimum.
The sorption mechanism on the nanomaterial has been studied for other M@GO derivatives and analytes,48 revealing a complex competition among three mechanisms: chelation, hydrogen bonding, and π–electron interactions. In the case of Ni2+ and Ga3+, the predominant extraction mechanism probably involves the formation of chelates via thiol or diamine groups in M@GO-MTS.49,50 For Ti4+, the [TiO]2+ oxo-cation form in aqueous solutions, and its structure in a slightly acidic medium facilitates adsorption through chelation and hydrogen bonding, Fig. SM4A.†,51 As demonstrated in elsewhere,46 the orthovanadate and metavanadate anions possess double bonds, leading to a competition between chelation, hydrogen bonding, and π–electron interactions, similar to the sorption behaviour of arsenic species, Fig. SM4B.†48
The studied eluents were phosphoric acid at 1% plus EDTA 0.5%, nitric acid at 3% plus EDTA 0.5%, nitric acid 3%, nitric acid 6% and nitric acid 7%. As shown in Fig. 4, the best signals were obtained at 6% HNO3, being concluded that effective elution of target analytes was obtained using at eluent.
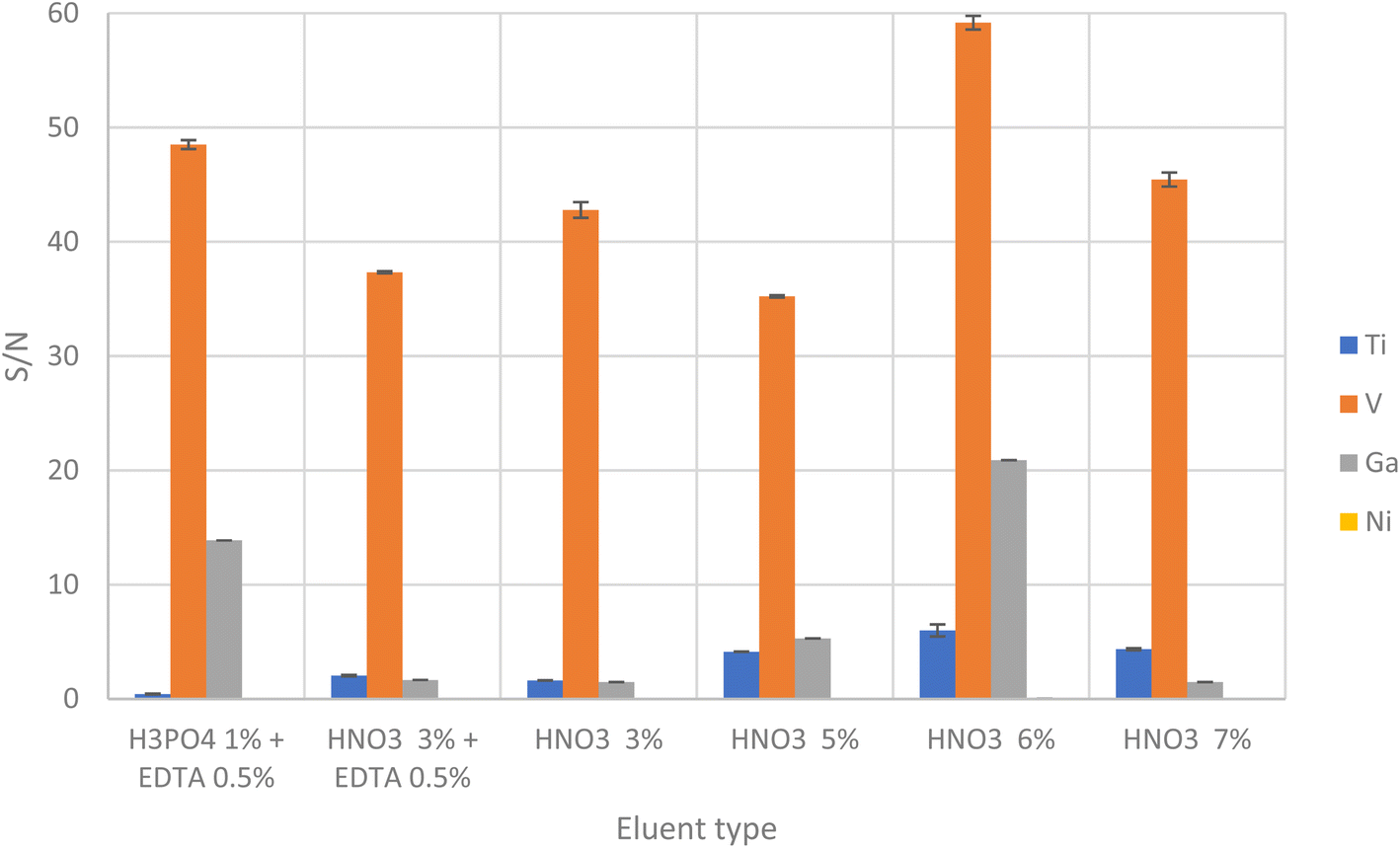 | ||
| Fig. 4 Study of different eluents, mixtures and their concentrations. | ||
The volume of eluent required for the quantitative desorption of target analytes was also investigated. The optimum eluent volume was chosen as the smallest possible volume to achieve maximum preconcentration. Quantitative elution was achieved using 1.0 mL of 6% HNO3.
With the optimized extraction parameters, a recovery evaluation study of target analytes was carried out by varying the amount of the sorbent across the range of 1–20 mg, maintaining the volume of sample in 250 mL.
The influence of the amount of M@GO-MTS on the extraction of V, Ni, Ti and Ga (60 μg L−1, 30 μg L−1, 30 μg L−1, 60 μg L−1 respectively) was assessed, Fig. SM6.† The extraction recovery increase with the increase of M@GO-MTS mass. On the other hand, the preconcentration was carried out with the least amount of material possible, allowing quantitative simultaneous recovery, and therefore, 20 mg was selected as optimal amount.
3.3 Preconcentration factor
With all the experimental parameters optimized, the metal ion sorption capacity was studied in the range of sample volume from 100 mL to 1000 mL maintaining the masses of the target analytes constant to 3.75 μg for V and Ti, 0.375 μg for Ga and 7.5 μg for Ni. Based on the results, the sorption capacity for all metal ions decreased with an increase of the volume above 250 mL, Fig. 5. Thus, the optimum volume selected was 250 mL.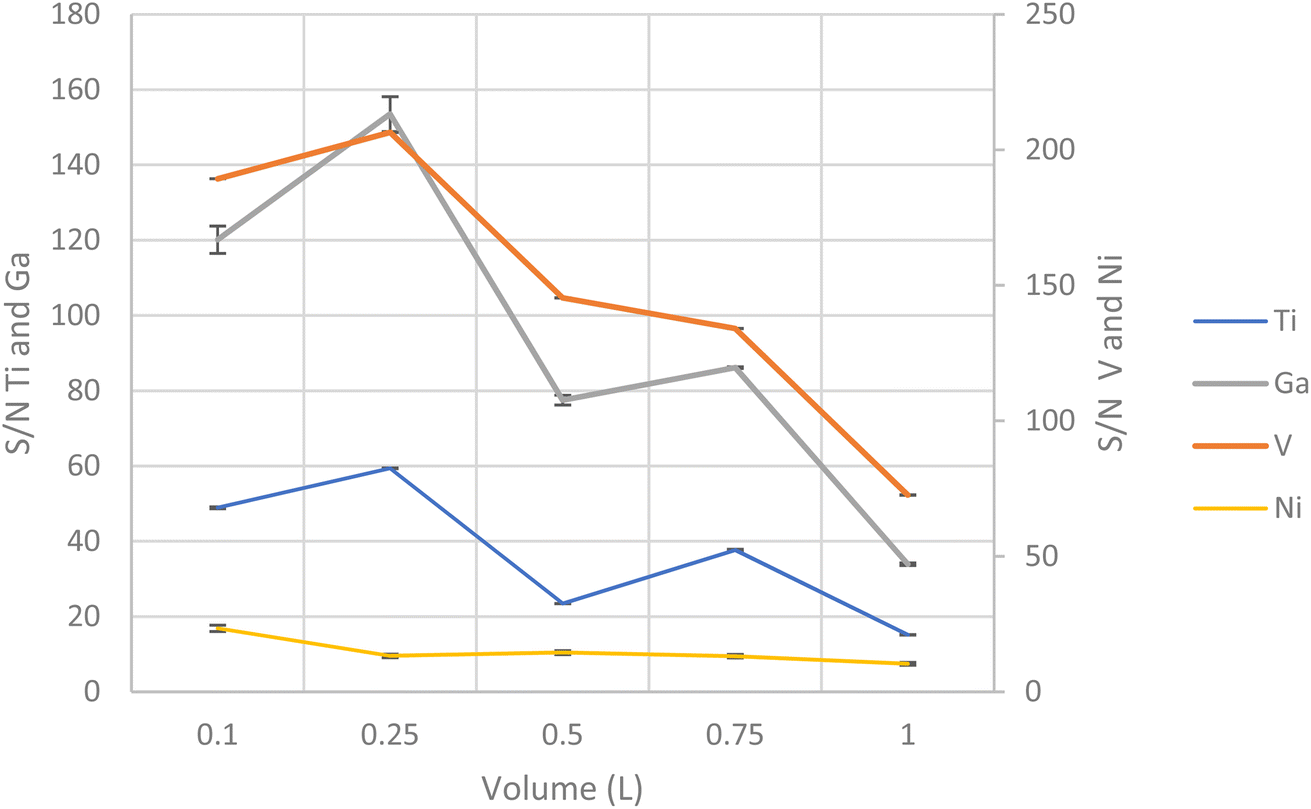 | ||
| Fig. 5 Study of maximum sample volume to a quantitative analysis. | ||
The preconcentration factor can be calculated as the sample to eluent volume ratio. A higher preconcentration factor can be provided by either increasing the sample volume or decreasing the eluent volume.
The recoveries were found to be stable until 250 mL and close to 100%; and the adsorbed V, Ti, Ni and Ga were able to elute with 1.0 mL of eluent. Thus, a preconcentration or enrichment factor (EF) of 250 can be obtained for the target analytes.
3.4 Figure of merit
The figures of merit for the determination of target analytes in environmental samples using proposed ETAAS procedure are summarised in Table 2.| Analyte | Calibration graph | Blank ± SD | LOD (pg) | LOQ (pg) | RSD intra-day (%) n = 7a | RSD intra-day (%) n = 7b | RSD Inter-day (%) n = 7c |
|---|---|---|---|---|---|---|---|
| a 5 μg L−1 Ti and V, 0.5 μg L−1 Ga, 10 μg L−1 Ni. b 10 μg L−1 Ti, and V, 1 μg L−1 Ga, 20 μg L−1 Ni. c 5 μg L−1 Ti, 2 μg L−1 V, 0.5 μg L−1 Ga, 10 μg L−1 Ni. | |||||||
| Ti(IV) | y = 0.0028x + 0.0032 | 0.0016 ± 0.0012 | 14.3 | 74.3 | 1.4 | 1.7 | 1.1 |
| V(V) | y = 0.0005x + 0.0003 | 0.0006 ± 0.0012 | 4.0 | 32.0 | 2.0 | 2.3 | 2.9 |
| Ni(II) | y = 1.22 × 10−5x + 0.00011 | 0.0001 ± 6 × 10−6 | 13.1 | 82.0 | 5.6 | 4.6 | 3.1 |
| Ga(III) | y = 0.025x + 0.00048 | 0.0007 ± 0.0003 | 0.9 | 2.6 | 4.6 | 4.3 | 5.6 |
Under the optima conditions described before, the linear ranges were evaluated using the optimised experimental and instrumental conditions. Four linear calibration graphs were obtained from 74.3 pg–0.28 ng for Ti; 32 pg–0.28 ng for V; 82 pg–0.6 ng for Ni, and 2.6 pg–0.1 ng for Ga, with correlation always better than 0.997.
The equations for calibration curves, blank signals, limits of detection and quantification (LODs and LOQs) and other analytical characteristics are presented in Table 2. The limits of detection (LOD) and quantification (LOQ) were calculated as the concentration of the target analyte giving signals equivalent to three and ten times (n = 11), respectively, the standard deviation of the blank plus the net blank intensity.
As can be seen in Table 2, LOD values, based on an EF of 250, ranged from 0.9 to 14.3 pg for the four elements. Considering 20 μL were injected, these LODs correspond with 0.71; 0.20; 0.66; and 0.04 μg L−1 for Ti, V, Ni, and Ga, respectively. The LODs obtained can be considered adequate for the pursued analytical purpose. Using the optimized conditions, this method enables the simultaneous determination of V, Ni, Ti and Ga using the external calibration technique. The precision, evaluated as the intra-day relative standard deviation (RSD) and inter-day values, for seven replicate measurements of solutions, are shown in Table 2. “Intraday RSD” assesses variability within a single day, and “Interday RSD” measures variability between three different days. A low RSD value in both categories indicates precise and consistent measurements. RSDs between 1.1 and 5.6% were obtained, indicating that the method is repeatable and reproducible, showing low variability among analyses performed on the same day and between days. Thus, the results indicated that the method for the simultaneous determination of V, Ti, Ga and Ni is sensible and precise.
For comparative purposes, the analytical performance data of analogous methods reported in the literature are registered in Table 3 (LODs has been shown with amount and concentration units for a better comparison). The direct comparison of the figures of merit is difficult due to the different experimental conditions. Nonetheless, the detection limits for the target elements and the precisions of the developed method were similar or superior to those reported in the bibliography.
3.5 Analytical applications
The accuracy of the proposed method was assessed by analysing certified reference materials, TMDA 64.3 Fortified Lake Waters and SPS-SW2 Batch 125 Surface Water for trace elements. Considering certified references materials include trace concentrations of other ions, it can be assumed that the method does not present any interferences at μg L−1 concentrations. The method was also applied to spiked and non-spiked seawater and tap water samples, allowing evaluate the accuracy of methods by recovery tests. The sample aliquots were spiked with metals of interest and analysed. The recoveries for the spiked samples were close to 100%. Three replicates of reference material and four replicates for real samples were analysed. The determined concentrations are shown in Tables 4 and 5.| Sample | Certificate value (μg L−1) | Found value (μg L−1) | Experimental t < 4.303 = tabulated t for 95% confidencea | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ti | V | Ni | Ga | Ti | V | Ni | Ga | Ti | V | Ni | Ga | |
| a t values obtained comparing certified and found value for 2 d.f. | ||||||||||||
| TMDA-64.3 | 124 ± 7 | 280 ± 18 | 251 ± 17 | 50.3 ± 3.4 | 125 ± 1 | 278 ± 2 | 267 ± 11 | 47 ± 2 | 1.73 | 1.73 | 2.52 | 2.85 |
| SPS-SW2 | — | 50.0 ± 0.3 | 50.0 ± 0.3 | — | — | 51.3 ± 0.8 | 52.5 ± 1.8 | — | — | 2.81 | 2.40 | — |
| Sample | Added (μg L−1) | Found (μg L−1) ± SDa | Recovery (%) ± SDa | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ti | V | Ni | Ga | Ti | V | Ni | Ga | Ti | V | Ni | Ga | |
| a The results are expressed as the average ± standard deviation for four separated determinations. | ||||||||||||
| Tap water | — | — | — | — | <LOD (0.71) | <LOD (0.20) | <LOD (0.66) | <LOD (0.04) | — | — | — | — |
| 5 | 5 | 20 | 3 | 5.9 ± 0.2 | 5.4 ± 0.7 | 22.4 ± 0.6 | 2.9 ± 0.3 | 118 ± 4 | 108 ± 14 | 112 ± 3 | 97 ± 10 | |
| 15 | 15 | 40 | 5 | 14.4 ± 0.2 | 15.5 ± 0.2 | 43 ± 5 | 4.8 ± 0.3 | 96 ± 1 | 103 ± 1 | 107.5 ± 12 | 96 ± 6 | |
| Seawater | — | — | — | — | <LOD (0.71) | <LOD (0.20) | 1.0 ± 0.1 | 0.44 ± 0.02 | — | — | — | — |
| 15 | 5 | 20 | 1 | 14 ± 1 | 6.0 ± 0.2 | 20 ± 1 | 1.4 ± 0.1 | 94 ± 6 | 120 ± 4 | 95 ± 5 | 96 ± 10 | |
| 30 | 15 | 40 | 3 | 29.9 ± 0.4 | 16.0 ± 0.2 | 41 ± 4 | 3.44 ± 0.02 | 100 ± 1 | 107 ± 1 | 100 ± 10 | 100 ± 1 | |
The obtained results highlight the good harmony between the certified and found values. The t-test applied to each individual result determined there is no significant difference between the experimental average and the population mean (μ) or “true value” = certified value for each result. No significant differences at 95% confidence level were found between the concentrations of V, Ni, Ti and Ga after M@GO-MTS enrichment and the certified concentrations, indicating the accuracy of the proposed method.
The t-test for paired samples was also used in the statistical analysis of data to demonstrate that there are no significant differences between certified and found concentrations of all results between the two samples. The tcalculated (1.85) value was lower than the critical value (tcritical = 2.57) at 95% confidence level, also indicating a good accuracy.
The validated method was applied to the analysis of two real samples: tap and sea water samples, Table 5. The recoveries for the spiked samples were not significantly different to 100%, indicating that the proposed method is useful to the simultaneous determination of the four analytes in samples with very complex matrix such as sea water with salinity about 35.5 g L−1.
3.6 Analytical greenness study
A novel analytical greenness metric, AGREEprep, has recently been proposed to evaluate the environmental sustainability of an analytical method.54 AGREEprep is founded upon ten evaluative steps that align with the ten principles of green sample preparation.55 The evaluations for each of the ten distinct components of AGREEprep are scaled from 0 to 1, with the extreme values representing the lowest and highest levels of achievement, respectively.A total score exceeding 0.5 is deemed indicative of a green analytical approach. The AGREEprep pictogram, Fig. 6, illustrates a favourable outcome, affirming that the established methodology aligns with contemporary green chemistry principles by eliminating toxic substances, facilitating a sustainability and renewability sorbent material, and minimizing energy usage, and also guaranteeing safe operational practices for the technician.
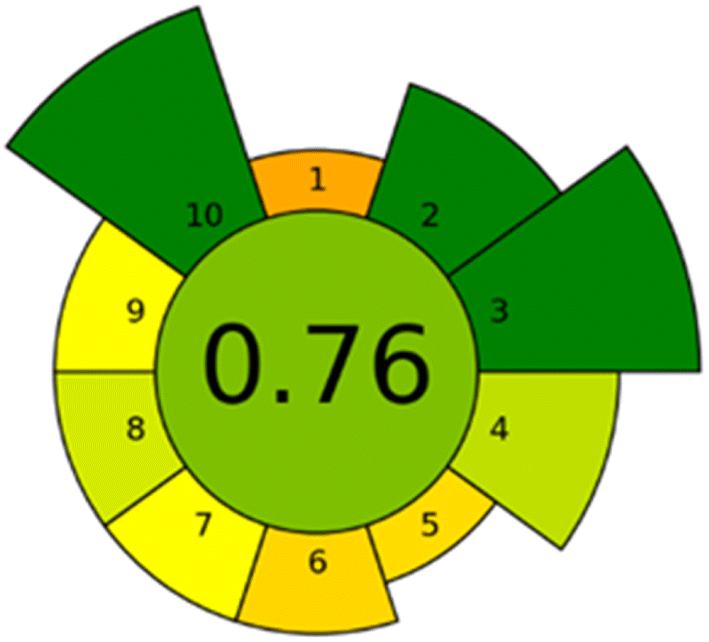 | ||
| Fig. 6 Pictogram representation of the analytical greenness about the proposed method. Each number indicate: (1) favor in situ sample preparation; (2) use safer solvents and reagents; (3) target sustainable, reusable, and renewable materials; (4) minimize waste; (5) minimize sample, chemical and material amounts; (6) maximize sample throughput; (7) integrate steps and promote automation; (8) minimize energy consumption; (9) choose the greenest possible post-sample preparation configuration for analysis; (10) ensure safe procedures for the operator. | ||
Nonetheless, this metric suggests that there exists a potential for enhancement, several parameters as sample preparation placement, waste generation, or the automation of the associated procedures.
4 Conclusions
In this work a novel M@GO functionalized with MTS as complexing agent has been successfully employed to the separation, preconcentration and simultaneous determination of V(V), Ti(IV), Ni(II) and Ga(III) ions from natural water samples, including seawater.The proposed method offers important advantages such as simplicity, reproducibility and economy. The sensitivity of the analytical technique is high, and no problems could be determined regarding the minimum content of the target elements. The proposed method was carried out using an external calibration since the standard addition method is time consuming demonstrating the absence of interferences. The sensitivity of different atomic lines was evaluated, and the LOQ and detection were shown to be adequate for the samples analysed. The accuracy of the method was checked by analysing certified reference waters and the results obtained confirmed a good concordance with the certified values.
Additionally, the proposed analytical protocol can be applied to real samples using a single extraction and a single heating program, enabling simultaneous determination, and providing precise and accurate data at low analysis costs. Furthermore, the proposed method adheres to green chemistry principles by avoiding the use of contaminating materials and requiring only a minimal amount of acid, rendering this method environmentally friendly. The greenness of the method has been quantitatively evaluated employing the AGREEprep metric, revealing that proposed method shows an acceptable green analytical method.
To conclude, magnetic dispersive solid phase extraction appears to be a promising approach for various applications, including the extraction of trace metals from environmental samples, the purification of pharmaceuticals, and the recovery of valuable elements from waste materials. Its versatility and efficiency in selectively isolating target analytes suggest that magnetic dispersive solid phase extraction could play a significant role in enhancing analytical methodologies across diverse fields, such as environmental monitoring, food safety, and materials recycling.
Data availability
The data supporting the findings of this study are available upon reasonable request from the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been partially supported by the University of Malaga (II Plan Propio (B1-2022_20, D5-2023_08 and B4-2023-19, Universidad de Málaga) and CEI MAR CEI-JD-17, FEDER funds and the Spanish Ministerio de Ciencia e Innovación (fellowship FPU18/05371) and Project PID2021-126794OB-100. Funding for open access charge: Universidad de Málaga/CBUA.References
- D. C. Crans, J. J. Smee, E. Gaidamauskas and L. Yang, The chemistry and biochemistry of vanadium and the biological activities exerted by vanadium compounds, Chem. Rev., 2004, 104, 849–902 CrossRef PubMed.
- R. J. Reeder, M. A. A. Schoonen and A. Lanzirotti, Metal speciation and its role in bioaccessibility and bioavailability, Rev. Mineral. Geochem., 2006, 64, 59–113 CrossRef.
- D. Tripathi, V. Mani and R. P. Pal, Biol. Trace Elem. Res., 2018, 186, 52–67 CrossRef PubMed.
- R. Chiarelli and M. C. Roccheri, Heavy metals and metalloids as autophagy inducing agents: focus on cadmium and arsenic, Cells, 2012, 1, 597–616 CrossRef PubMed.
- S. Hartung, N. Bucher, H. Y. Chen, R. Al-Oweini, S. Sreejith, P. Borah, Z. Yanli, U. Kortz, U. Stimming, H. E. Hoster and M. Srinivasan, Vanadium-based polyoxometalate as new material for sodium-ion battery anodes, J. Power Sources, 2015, 288, 270–277 CrossRef.
- IARC Monographs on the Evaluation of Carcinogenic Risks to Humans Volume, Lyon, France, 2006, pp. , pp. 227–292 Search PubMed.
- G. Caumette, C. P. Lienemann, I. Merdrignac, B. Bouyssiere and R. L. Lobinski, Element speciation analysis of petroleum and related materials, J. Anal. At. Spectrom., 2009, 24, 263–276 RSC.
- P. a Mello, J. S. F. Pereira, M. F. Merko, J. S. Barin and E. M. M. Flores, Sample preparation methods for subsequent determination of metals and non-metals in crude oil—A review, Anal. Chim. Acta, 2012, 746, 15–36 CrossRef.
- A. Desprez, B. Bouyssiere, C. Arnaudguilhem, G. Krier, L. Vernex-loset and P. Guisti, Study of the Size Distribution of Sulfur, Vanadium, and Nickel Compounds in Four Crude Oils and Their Distillation Cuts by Gel Permeation Chromatography Inductively Coupled Plasma High-Resolution Mass Spectrometry, Energy fuels, 2014, 28, 3730–3737 CrossRef.
- J. Liu, Y. Huang, H. Li and H. Duan, Recent advances in removal techniques of vanadium from water: A comprehensive review, Chemosphere, 2022, 287, 132021 CrossRef PubMed.
- L. A. Macdonald, J. A. Wiklund, M. C. Elmes, B. B. Wolfe and R. I. Hal, Paleolimnological assessment of riverine and atmospheric pathways and sources of metal deposition at a floodplain lake (Slave River Delta, Northwest Territories, Canada), Sci. Total Environ., 2016, 544, 811–823 CrossRef PubMed.
- Y. Zhou, L. Gao, D. Xu and B. Gao, An integrated exploration on health risk assessment quantification of potentially hazardous elements in soils from the perspective of sources, Sci. Total Environ., 2019, 660, 1338–1345 CrossRef PubMed.
- R. X. S. Tulcan, W. Ouyang, C. Lin, M. He and B. Wang, Vanadium pollution and health risks in marine ecosystems: Anthropogenic sources over natural contributions, Water Res., 2021, 207, 117838 CrossRef PubMed.
- M. Valko, H. Morris and M. Cronin, Metals, toxicity and oxidative stress, Curr. Med. Chem., 2005, 12, 1161–1208 CrossRef.
- Q. Y. Chen, J. Brocato, F. Laulicht and M. Costa, Mechanisms of nickel carcinogenesis, In Essential and Non-Essential metals. Molecular and Integrative Toxicology, ed. A. Mudipalli and J. T. Zelikoff, Springer International Publishing AG, New York, NY, USA, 2017, pp. 181–197 Search PubMed.
- M. Herrero, J. Rovira, M. Nadal and J. L. Domingo, Risk assessment due to dermal exposure of trace elements and indigo dye in jeans: Migration to artificial sweat, Environ. Res., 2019, 172, 310–318 CrossRef PubMed.
- M. Y. A. Khan and J. Wen, Evaluation of physicochemical and heavy metals characteristics in surface water under anthropogenic activities using multivariate statistical methods, Garra River, Ganges Basin, India, Environ. Eng. Res., 2020, 26(6), 200280 CrossRef.
- M. L. Alonso Castillo, I. Sánchez Trujillo, E. Vereda Alonso, A. García de Torres and J. M. Cano Pavón, Bioavailability of heavy metals in water and sediments from a typical Mediterranean Bay (Málaga Bay, Region of Andalucía, Southern Spain), Mar. Pollut. Bull., 2013, 73, 427–434 CrossRef PubMed.
- Directive, 2020/2184/EC of the European Parliament and the Council of 16.12.2020 A framework for community action in the field of water policy. Off. J. Eur, 2020, p. , p. 36 Search PubMed.
- G. Ravindiran, S. Rajamanickam, S. Sivarethinamohan, B. Karupaiya Sathaiah, G. Ravindran, S. Muniasamy and G. Hayder, A Review of the Status, Effects, Prevention, and Remediation of Groundwater Contamination for Sustainable Environment, Water, 2023, 15(20), 3662, DOI:10.3390/w15203662.
- U.S. Environmental Protection Agency (EPA), Introduction to In Situ Bioremediation of Groundwater. EPA's Technologies for Cleaning up Contaminated Sites, 2023 Search PubMed.
- ANZECC/ARMCANZ, Australian and New Zealand Guidelines for Fresh and Marine Water Quality, Australian and New Zealand Environmental and Conservation Council. Agriculture and Resource, 2000 Search PubMed.
- European Commission, Directive 2008/105/EC of the European Parliament and of the Council of 16 December 2008 on environmental quality standards in the field of water policy, amending and subsequently repealing Council Directives 82/176/EEC, 83/513/EEC, 84/156/EEC, 84/491/EEC, 86/280/EEC and amending 2000/60/EC of the European Parliament and of the Council, 2008, Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32008L0105 Search PubMed.
- USEPA, National Recommended Water Quality Criteria, 2017, Available from: https://www.epa.gov/wqc/aquatic-life-ambient-water-quality-criteria#guide. Accessed 1/12/2023 Search PubMed.
- F. Graham, M. Lord, D. Froment, H. Cardinal and G. Bollée, The use of gallium-67 scintigraphy in the diagnosis of acute interstitial nephritis, Clin. Kidney J., 2016, 1, 76–81 CrossRef PubMed.
- C. S. Ivanoff, A. E. Ivanoff and T. L. Hottel, Gallium poisoning: A rare case report, Food Chem. Toxicol., 2012, 50, 212–215 CrossRef PubMed.
- B. Welz, H. Becker-Ross, S. Florek and U. Heitmann, High Resolution Continuum Source AAS, The Better Way to Do Atomic Absorption Spectrometry, Wiley-VCH, 2005, pp. 60–74 Search PubMed.
- U. Heitmann, B. Welz, D. L. G. Borges and F. G. Lepri, Feasibility of peak volume, side pixel and multiple peak registration in high-resolution continuum source atomic absorption spectrometry, Spectrochim. Acta, Part B, 2007, 62, 1222–1230 CrossRef.
- J. Briceño, M. A. Belarra, K. A. C. De Schamphelaere, S. Vanblaere, C. Janssen, F. Vanhaecke and M. Resano, Direct determination of Zn in individual Daphnia magna specimens by means of solid sampling high-resolution continuum source graphite furnace atomic absorption spectrometry, J. Anal. At. Spectrom., 2010, 25, 503–510 RSC.
- J. C. García-Mesa, I. Morales-Benitez, P. Montoro-Leal, M. M. Lopez Guerrero and E. Vereda Alonso, Sp-ICP-MS and HR-CS-ETAAS as Useful Available Techniques for the Size Characterization and Speciation of Ionic and Nanoparticular Zinc in Cosmetic and Pharmaceutical Samples, Talanta, 2024, 268(1), 125360 CrossRef.
- D. Wang, X. Chen, J. Feng and M. Sun, Recent advances of ordered mesoporous silica materials for solid-phase extraction, J. Chromatogr. A, 2022, 1675, 463157 CrossRef PubMed.
- M. Mencin, M. Mikulic-Petkovsek, R. Veberič and P. Terpinc, Development and Optimisation of Solid-Phase Extraction of Extractable and Bound Phenolic Acids in Spelt (Triticum spelta L.) Seeds, Antioxidants, 2021, 10, 1085 CrossRef PubMed.
- I. Morales-Benítez, P. Montoro-Leal, J. C. García-Mesa, J. Verdeja-Galán and E. I. Vereda Alonso, Magnetic graphene oxide as a valuable material for the speciation of trace elements, TrAC, Trends Anal. Chem., 2022, 157, 116777 CrossRef.
- P. Montoro-Leal, J. C. García-Mesa, M. M. del López Guerrero and E. Vereda Alonso, Comparative Study of Synthesis Methods to Prepare New Functionalized Adsorbent Materials Based on MNPs–GO Coupling, Nanomaterials, 2020, 10, 304 CrossRef PubMed.
- A. Cárdenas Valdivia, E. Vereda Alonso, M. M. López Guerrero, J. Gonzalez-Rodriguez, J. M. Cano Pavón and A. García de Torres, Simultaneous determination of V, Ni and Fe in fuel fly ash using solid sampling high resolution continuum source graphite furnace atomic absorption spectrometry, Talanta, 2018, 179(1), 1–8 CrossRef.
- R. Dobrowolski, A. Adamczyk and M. Otto, Determination of vanadium in soils and sediments by the slurry sampling graphite furnace atomic absorption spectrometry using permanent modifiers, Talanta, 2013, 113, 19–25 CrossRef PubMed.
- R. Dobrowolski, A. Adamczyk and M. Otto, Comparison of action of mixed permanent chemical modifiers for cadmium and lead determination in sediments and soils by slurry sampling graphite furnace atomic absorption spectrometry, Talanta, 2010, 82, 1325–1331 CrossRef PubMed.
- A. González Moreno, M. M. López Guerrero, E. Vereda Alonso, A. García de Torres and J. M. Cano Pavón, Development of a new FT-IR method for the determination of iron oxide. Optimization of the synthesis of suitable magnetic nanoparticles as sorbent in magnetic solid phase extraction, New J. Chem., 2017, 41, 8804–8811 RSC.
- P. N. Diagboya, B. I. Olu-Owolabi, D. Zhou and B. H. Han, Graphene oxide–tripolyphosphate hybrid used as a potent sorbent for cationic dyes, Carbon, 2014, 79, 174–182 CrossRef.
- P. Montoro-Leal, J. C. Garcia-Mesa, M. M. del Lopez Guerrero and E. Vereda Alonso, ES 2 844 942 B2, EP 4 095 097 A1, 2021.
- M. Resano, M. Aramendía and M. A. Belarra, High-resolution continuum source graphite furnace atomic absorption spectrometry for direct analysis of solid samples and complex materials: a tutorial review, J. Anal. At. Spectrom., 2014, 29, 2229–2250 RSC.
- J. C. García-Mesa, P. Montoro-Leal, S. Maireles-Rivas, M. M. Lopez Guerrero and E. Vereda Alonso, Sensitive determination of mercury by magnetic dispersive solid-phase extraction combined with flow-injection-cold vapour-graphite furnace atomic absorption spectrometry, J. Anal. At. Spectrom., 2021, 36, 892 RSC.
- C. Bendicho and M. T. C. de Loos-Vollebregt, Solid sampling in electrothermal atomic absorption spectrometry using commercial atomizers. A review, J. Anal. At. Spectrom., 1991, 6, 353–374 RSC.
- A. D. Woolfson and G. M. Gracey, Matrix effects in the determination of aluminium in dialysis fluids by graphite furnace atomic absorption spectrometry, Analyst, 1987, 112, 1387–1389 RSC.
- E. C. Lima, J. L. Brasil and A. Santos, Evaluation of Rh, Ir, Ru, W-Rh, W-Ir and W-Ru as permanent modifiers for the determination of lead in ashes, coals, sediments, sludges, soils, and freshwaters by electrothermal atomic absorption spectrometry, Anal. Chim. Acta, 2003, 484(2), 233–242, DOI:10.1016/S0003-2670(03)00328-3.
- N. N. Meeravali and S. J. Kumar, The utility of a W–Ir permanent chemical modifier for the determination of Ni and V in emulsified fuel oils and naphtha by transverse heated electrothermal atomic absorption spectrometer, J. Anal. At. Spectrom., 2001, 16, 527–532 RSC.
- M. Resano, M. R. Flórez and E. García-Ruiz, High-resolution continuum source atomic absorption spectrometry for the simultaneous or sequential monitoring of multiple lines. A critical review of current possibilities, Spectrochim. Acta, Part B, 2013, 88, 85–97 CrossRef.
- P. Montoro-Leal, J. C. García-Mesa, I. Morales-Benitez, A. García de Torres and E. Vereda Alonso, Semiautomatic method for the ultra-trace arsenic speciation in environmental and biological samples via magnetic solid phase extraction prior to HPLC-ICP-MS determination, Talanta, 2021, 235, 122769 CrossRef PubMed.
- N. G Tsierkezos, D. Schröder and H. Schwarz, Complexation of nickel(II) by ethylenediamine investigated by means of electrospray ionization mass spectrometry, Int. J. Mass Spectrom., 2004, 235(1), 33–42 CrossRef.
- K. Kowolik, M. Shanmugam, T. W. Myers, C. D. Cates and L. A. Berben, A redox series of gallium(iii) complexes: ligand-based two-electron oxidation affords a gallium–thiolate complex, Dalton Trans., 2012, 41, 7969–7976 RSC.
- S. I. Uddin Ahmed, M. Shahid and S. Sankarasubramanian, Aqueous titanium redox flow batteries—State-of-the-art and future potential, Front. energy res., 2022, 10, 1–9 Search PubMed.
- E. Chevallier, R. Chekri, J. Zinck, T. Guerin and L. Noe, Simultaneous determination of 31 elements in foodstuffs by ICP-MS after closed-vessel microwave digestion: Method validation based on the accuracy profile, J. Food Compos. Anal., 2015, 41, 35–41 CrossRef.
- I. Lavilla, P. Vilas and C. Bendicho, Fast determination of arsenic, selenium, nickel and vanadium in fish and shellfish by electrothermal atomic absorption spectrometry following ultrasound-assisted extraction, Food Chem., 2008, 106(1), 403–409 CrossRef.
- F. Pena-Pereira, M. Tobiszewski, W. Wojnowski and E. Psillakis, A tutorial on AGREEprep an analytical greenness metric for sample preparation, Adv. Sample Prep., 2022, 1(3), 100025 CrossRef.
- A. I. Lopez-Lorente, F. Pena-Pereira, S. Pedersen-Bjergaard, V. G. Zuin, S. A. Ozkan and E. Psillakis, The ten principles of green sample preparation, TrAC, Trends Anal. Chem., 2022, 1(148), 116530 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ay01809e |
| ‡ Equally contribution. |
| This journal is © The Royal Society of Chemistry 2025 |