
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Taylor dispersion analysis as a tool for size measurement of PAMAM dendrimers: the effect of generation, functionality and pH†
Vikesh
Chhabria‡
 *,
Robert
Forbes
and
Zhengyuan
Zhou
*,
Robert
Forbes
and
Zhengyuan
Zhou
School of Pharmacy and Biomedical Sciences, University of Central Lancashire, Maudland Building, Preston, Lancashire PR1 2HE, UK. E-mail: ChhabriaV2@cardiff.ac.uk; vikeshc@gmail.com
First published on 14th November 2024
Abstract
Taylor Dispersion Analysis (TDA) is explored to measure the hydrodynamic sizes of full and half generation PAMAM dendrimers up to generation 4.5 in various buffer solutions. A method was used to minimize the interaction between the capillary and cationic dendrimers. The effects of generation, surface functionality, pH and ionic strength on the hydrodynamic radii of PAMAM dendrimers were investigated. Our results show that TDA can accurately measure the sizes of PAMAM dendrimers with a relatively low standard deviation especially for half generations. It was found that the ionisation of functional groups at various pH values led to a conformational change due to electrostatic repulsion or back-folding of the branches. Furthermore, adding salt to a half-generation dendrimer (G4.5) can lead to a profound size change that is dependent on ionic strength. A 17% increase in the size of the G4.5 dendrimer was observed in a 1 M NaCl solution compared to that in a 0.1 M solution. Compared to dynamic light scattering, TDA is more reliable and tolerant to large particles in the solutions. The findings of this study indicate that TDA could serve as a viable alternative technique for assessing dendrimer size and conformation, as well as studying their binding behavior.
1. Introduction
Pharmaceutical companies are investing heavily in nanotherapeutics to address challenges related to drug solubility, bioavailability, biodistribution, and mimicry. Recent years have seen the transition from bench to market of formulations such as Doxil®, DaunoXome®, Oncaspar®, Feridex®, and NanoTherm®.1 Dendrimers are a class of nanotherapeutics that have been shown to overcome certain barriers to drug development such as improving drug solubility and the ability to carry the drug across cell membranes and to mimic the in vivo environment.2 Dendrimers have attracted considerable interest owing to their unique structural and physicochemical properties.3 These macromolecules possess a globular shape with internal cavities, exhibit narrow molecular weight distribution, and offer controllable peripheral surface functionality. Such characteristics facilitate drug incorporation through either encapsulation within the internal cavities or surface binding to the functional groups on the periphery, thereby enhancing their utility in drug delivery applications.4 Now that dendrimer-drug conjugates are entering clinical trials, regulators and industrialists are keen to identify critical quality attributes and have robust methods of their measurement. Recently, the challenges of meeting the prescient needs for in-depth characterisation of drug-dendrimer conjugates have been comprehensively addressed in the literature.5Despite the synthesis of numerous dendrimers with diverse structures over the years, PAMAM dendrimers, as the pioneering commercially available dendrimer family, continue to be the most extensively studied, attributed to their outstanding attributes in pharmaceutical applications.3 PAMAM dendrimers have shown potential for e.g. cancer diagnosis and treatment,6 delivery of immunosuppressives,7 prophylaxis of eye infections,8 and transdermal delivery of ketoprofen.9 Size measurement of PAMAM dendrimers is essential for understanding their structural properties and optimising their performance in various applications. Although techniques such as molecular dynamics simulations, small angle X-ray scattering (SAXS) and small angle neutron scattering (SANS)10–12etc. have been employed to characterize PAMAM dendrimers and provide useful insight into the structure and conformation of dendrimers, challenges remain in accurately measuring the size of PAMAM dendrimers, including the difficulty in characterising small dendrimer sizes, the influence of solvent and environmental conditions on dendrimer conformation, and the need for improved data analysis algorithms and standards for comparison between techniques. These analytical techniques face limitations in sizing dendrimers,13–18 often due to their specialized requirements and complexities, demanding strict method development and sample preparation. Moreover, they often entail lengthy analysis times and may not fully replicate real experimental conditions, potentially resulting in discrepancies with measurements in actual solutions.
Recently, Taylor Dispersion Analysis (TDA) has been applied by several research groups to measure the size of nanomaterials, e.g. inorganic gold and super-paramagnetic nanoparticles19–21 and lipid-nanoparticle formulations for mRNA vaccines.22 Cottet and coworkers were the first to explore the use of TDA to measure the diffusion coefficients and hydrodynamic radii of poly-L-lysine dendrigrafts.23 They also used TDA to monitor the functionalization of dendrigraft polylysine via click chemistry 24 and successfully used TDA to monitor the impact of buffer compositions and ionic strength (e.g. phosphate ions) on the size of linear polylysines.25 A comparison was made to the data obtained from dynamic light scattering and size exclusion chromatography. Higher generations showed notable differences in size measurements, mainly due to aggregate contributions to light scattering intensity. TDA was concluded to be the most appropriate technique for evaluating hyperbranched macromolecules. Full generation L-lysine dendrimers up to G6 with ammonium trifluoroacetate and Boc-protected surfaces were prepared and characterized by TDA and size exclusion chromatography to accurately measure various properties including hydrodynamic radii.26 These experimental techniques were combined with molecular dynamics simulations to gain insight into the dendrimer structure.
Although Taylor Dispersion Analysis (TDA) has been shown to offer structural insights for several dendrimers in previous studies,5,26 there is a lack of extensive reports utilising TDA to measure the sizes of PAMAM dendrimers with diverse surface functionalities. Moreover, investigations into the effects of pH and ionic strength on the conformational changes of dendrimer structures in real solutions are limited. Such studies could enhance our understanding of dendrimer interactions with guest molecules, such as drugs or proteins, and inform the design of dendrimer-based formulations with optimized binding and release behaviour. To address this, we investigated the characterisation of low-generation PAMAM dendrimers (full or half) with various surface functionalities using TDA in conjunction with dynamic light scattering (DLS). The size and conformation changes of dendrimers in various buffer solutions upon ionisation of functional groups were monitored and the effect of counterions and ionic strength on dendrimer size was also investigated.
2. Materials and methods
2.1. Materials
PAMAM dendrimers (G1.5, G3.5, and G4.5, half generation) were purchased from Dendritech Ltd. Full generation PAMAM dendrimers (G3 and G4), Hellmanex III, L-tryptophan, caffeine, DEAE-dextran hydrochloride, sodium acetate trihydrate, acetic acid, sodium hydroxide, 0.22 μm PVDF membrane filter, 1 M hydrochloric acid, carbonate buffer, sodium phosphate dibasic heptahydrate and sodium phosphate monobasic monohydrate were purchased from Sigma-Aldrich Ltd (Poole, UK).2.2. Taylor dispersion analysis
A Viscosizer 200 (VS200) was used (Malvern Instruments, Malvern, UK) with a Malvern fused silica capillary. The dimensions were 75 μm i.d. and 200 μm o.d. and a total length of 130 cm. It is also possible to determine the capillary i.d. optically.27 The length to window 1 is 45 cm and to window 2 is 85 cm, respectively. The instrument was set to a constant oven and tray temperature of 25.0 °C. Each series of runs was carried out using an optical filter at 214 nm. The instrument was calibrated by stray light corrections using an appropriate stray light test solution of 10 mg cm−3L-tryptophan dissolved in water.2.3. Hydrodynamic radius measurements of PAMAM dendrimers
Measurements of the hydrodynamic radius (Rh) were conducted using a well-established method that was optimized for full generation dendrimers, so that the Rh values of PAMAM dendrimers were analysed accurately, with minimal interference from the capillary walls.28 The samples were run in triplicate according to the schedule shown in Table 1. Prior to running the PAMAM dendrimer samples, a 1 mg cm−3 caffeine standard solution was analysed for Rh (Fig. S1‡) to assess the accuracy and repeatability of the instrument. The analytical instrument utilizes UV imaging and TDA for determining the diffusion coefficients and hydrodynamic radii of dendrimers in solution. The UV detector monitors band broadening of the dendrimer solution injected into a stream of buffer solution, driven by a pump through a fused-silica capillary. The dendrimers are imaged at two windows. This band broadening phenomenon is calculated from absorbance versus time data, using the peak times at the first (t1) and second window (t2), and standard deviations τ1 and τ2, using the equation shown below:where Rh is the hydrodynamic radius; kβ is the Boltzmann constant; η is the viscosity of run buffer; r is the radius of the capillary; T is the temperature; t1 and t2 correspond to peak centre times at the first window and second window, and the corresponding standard deviations, τ1 and τ2 (band broadening).

| Injection | Sequence | Pressure (mbar) | Time (min) |
|---|---|---|---|
| 1 | Rinse (run buffer) | 2000 | 1.00 |
| 2 | Fill (run buffer) | 2000 | 1.00 |
| 3 | Reset baseline | 140 | 1.00 |
| 4 | Load (sample) | 140 | 0.20 |
| 5 | Dip (run buffer) | 0 | 0.15 |
| 6 | Run (run buffer) | 140 | Auto |
PAMAM dendrimers samples (G1.5, G3, G3.5, G4 and G4.5) were prepared at 1% w/v in buffers at various pH values and then filtered once before TDA measurement. The samples were measured using the corresponding buffer solutions as the run buffer: 0.067 M phosphate (pH 7.4, ionic strength 0.168 M), 0.085 M hydrochloric acid (pH 1.2, ionic strength 0.135 M) and 0.05 M carbonate buffer (pH 10.6, ionic strength 0.132 M), which allowed investigating the effect of pH on the size, conformation and stability of the dendrimers under stressful environments. G1.5, 3.5, and 4.5 dendrimer samples were measured using an uncoated fused silica capillary, which carried an overall negative charge. The half-generation dendrimers carry an overall negative charge and thus have no interaction with the wall of the capillary due to a repulsion effect. In addition, to assess the effect of ionic strength of half-generation dendrimers, 1% w/v dendrimer solutions were prepared by dissolving G4.5 dendrimers in PBS buffer with 0.1 and 1 M sodium chloride solutions. The size was measured by the method above. The viscosities of the run buffers were determined by TDA and used for the calculation of the hydrodynamic radius of the dendrimers under various conditions. The viscosities of the three buffer solutions are similar to that of water (0.890 cP) due to their low concentrations while the viscosities with 0.1 and 1 M sodium chloride are 0.899 and 0.972 cP, respectively. However, the full-generation dendrimers (G3 and G4) carry an overall positive charge and interact with the walls of the capillary creating a tailing Taylor dispersion profile (Scheme 1), which leads to unreliable results. To avoid this, we employed a semi-permanent dextran coating to immobilize the charges on the capillary walls.
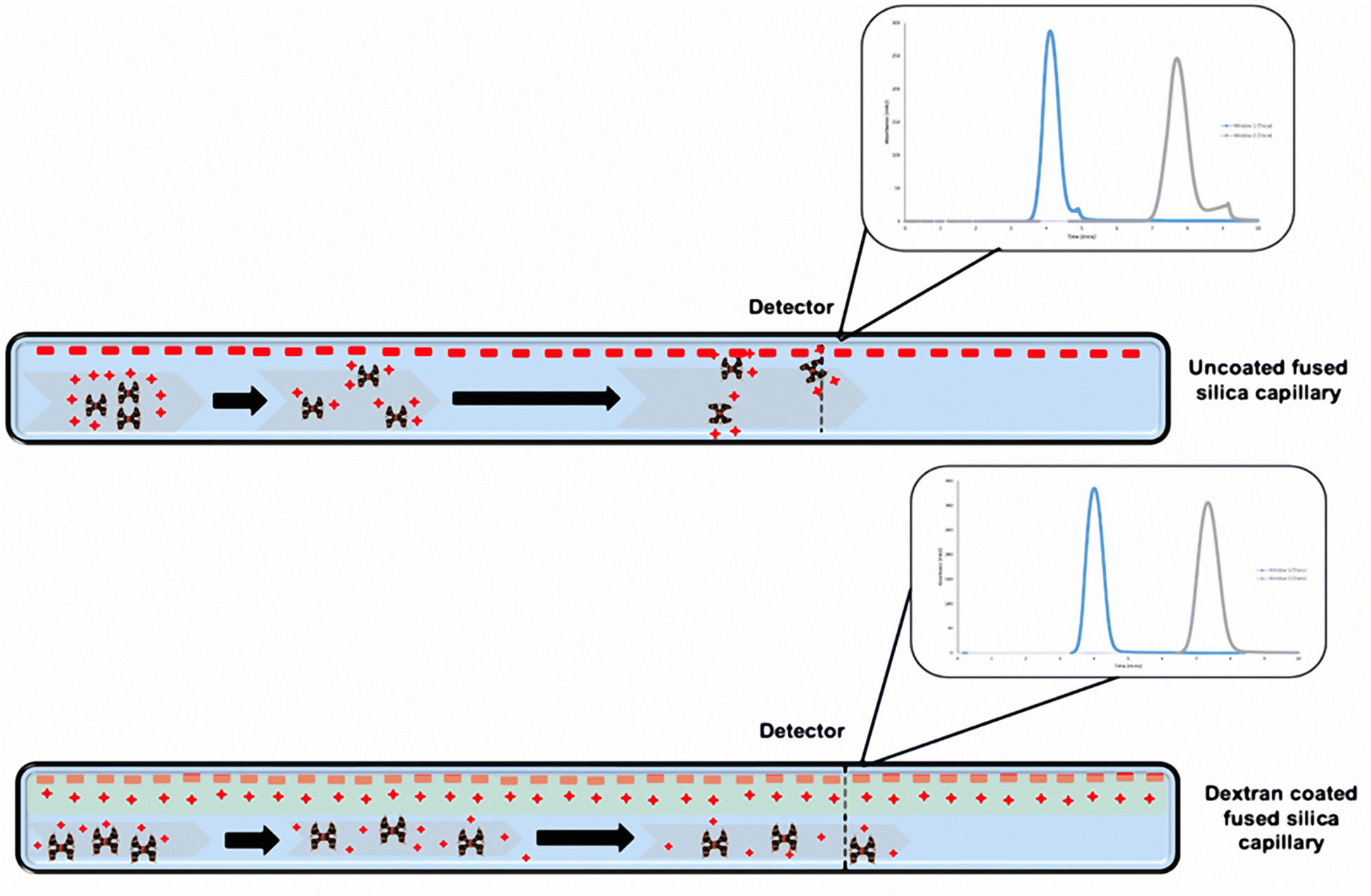 | ||
| Scheme 1 Schematic representation of the charges on the capillary walls before and after dextran coating and its effect on full generation PAMAM dendrimers with respect to the Taylorgram. | ||
2.4. Capillary pre-coating with dextran
In order to coat the capillary wall with dextran, a 25 mM sodium acetate buffer was prepared and adjusted to pH 5.5 using 1 M sodium hydroxide, to a final volume of 200 mL. A 2% w/v DEAE dextran solution was prepared in 10 mL sodium acetate buffer. This mixture was filtered through a 0.45 μm nylon membrane filter. The capillary coating was obtained by washing the capillary with 1 M sodium hydroxide, which activates the capillary surface. The 2% w/v DEAE dextran solution was then applied to the walls of the capillary. The coating was applied by the method shown in Table 2.| Injection | Sequence | Pressure (mbar) | Time (min) |
|---|---|---|---|
| Schedule 1 | |||
| 1 | Water | 3000 | 5.00 |
| 2 | 1 M NaOH | 3000 | 5.00 |
| 3 | Water | 3000 | 5.00 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Schedule 2 | |||
| 1 | Sodium acetate | 3000 | 1.00 |
| 2 | 2% (w/v) dextran | 3000 | 10.00 |
| 3 | Sodium acetate | 0 | 10.00 |
| 4 | Sodium acetate | 3000 | 2.00 |
2.5. Capillary washing procedure
The fused silica capillary was washed according to a well published study.29 A surfactant solution of Hellmanex III (2% v/v) was flushed for 10 minutes at 2000 mbar to wash the capillary after each run, followed by a wash with water. At times, the capillary was also flushed with nitrogen gas to remove any blockages.2.6. Dynamic light scattering
Size analysis of PAMAM dendrimers was conducted using dynamic light scattering (Zetasizer Nano, Malvern Instruments, UK). The same batch of dendrimer samples for TDA measurement was analysed by DLS without further filtration to compare the results of these two techniques. The dendrimer solutions (1% w/v) in phosphate buffer (0.067 M, pH 7.4), hydrochloric acid buffer (0.085 M, pH 1.2), and carbonate buffer (0.05 M, pH 10.6) were filtered through a PVDF filter (Millipore, 0.22 μm pore size) into a clean scattering cell. The measurement was conducted in triplicate.3. Results and discussion
3.1. Measurement of hydrodynamic radii of half and full generation dendrimers
In this work we explored the size measurement of PAMAM dendrimers using TDA. To ensure accurate and reliable measurement of samples, a 1 mg cm−3 caffeine standard was run multiple times to assess the state of the VS200 TDA analyser. As compared to studies by the same group,29–31 the Rh of caffeine shown in Fig. S1‡ was within the range of 0.40–0.42 nm. The sizes of PAMAM dendrimers measured by TDA were assessed by the repeatability of the samples and in reference to the values reported by other techniques.32–34The divergent synthesis of PAMAM dendrimers involves a two-step reaction: Michael addition of methyl acrylate to amino groups, followed by amidation of the esters with ethylene diamine. An amine-terminated full generation dendrimer is produced for one complete synthesis cycle. However, if the amidation step is omitted, a half generation dendrimer terminated with carboxyl groups will be formed. A range of lower generation PAMAM dendrimers (full and half generations) were selected for TDA measurements and the effect of pH on the conformation was investigated in different buffer solutions. The pKa values of full generation PAMAM dendrimers are 7–9 for primary amine groups on the surface and 3–6 for interior tertiary amine groups.31 For half generation dendrimers, the pKa for surface carboxyl groups is estimated to be around 3, which is a common value for carboxylic acids. Compared to higher generation dendrimers, lower generation dendrimers have relatively open structures and less densely packed surfaces.3
Since the wall of the TDA fused silica capillary is negatively charged, the half generation G4.5 PAMAM dendrimer was initially measured by TDA. The G4.5 PAMAM dendrimer carries negative charges under neutral or basic conditions due to carboxyl surface groups. The repulsion between the G4.5 dendrimer and the walls of the capillary was preferred and therefore the dendrimer did not interact with the capillary walls. Fig. 1a shows a standard TDA profile of a 1% w/v G4.5 dendrimer with a hydrodynamic radius of 2.67 nm (±0.06). As shown in Fig. 1b, the same sample was run in triplicate, and each Taylorgram fit the previous run perfectly, displaying the repeatability of the technique. The positively charged full generation PAMAM dendrimers had minor interactions with the capillary wall and showed a tailing effect in Fig. S2,‡ which obviously results in a relatively higher variation in the measurement. However, this interaction was reduced by the presence of a dextran coating that neutralized the surface of the capillary wall.
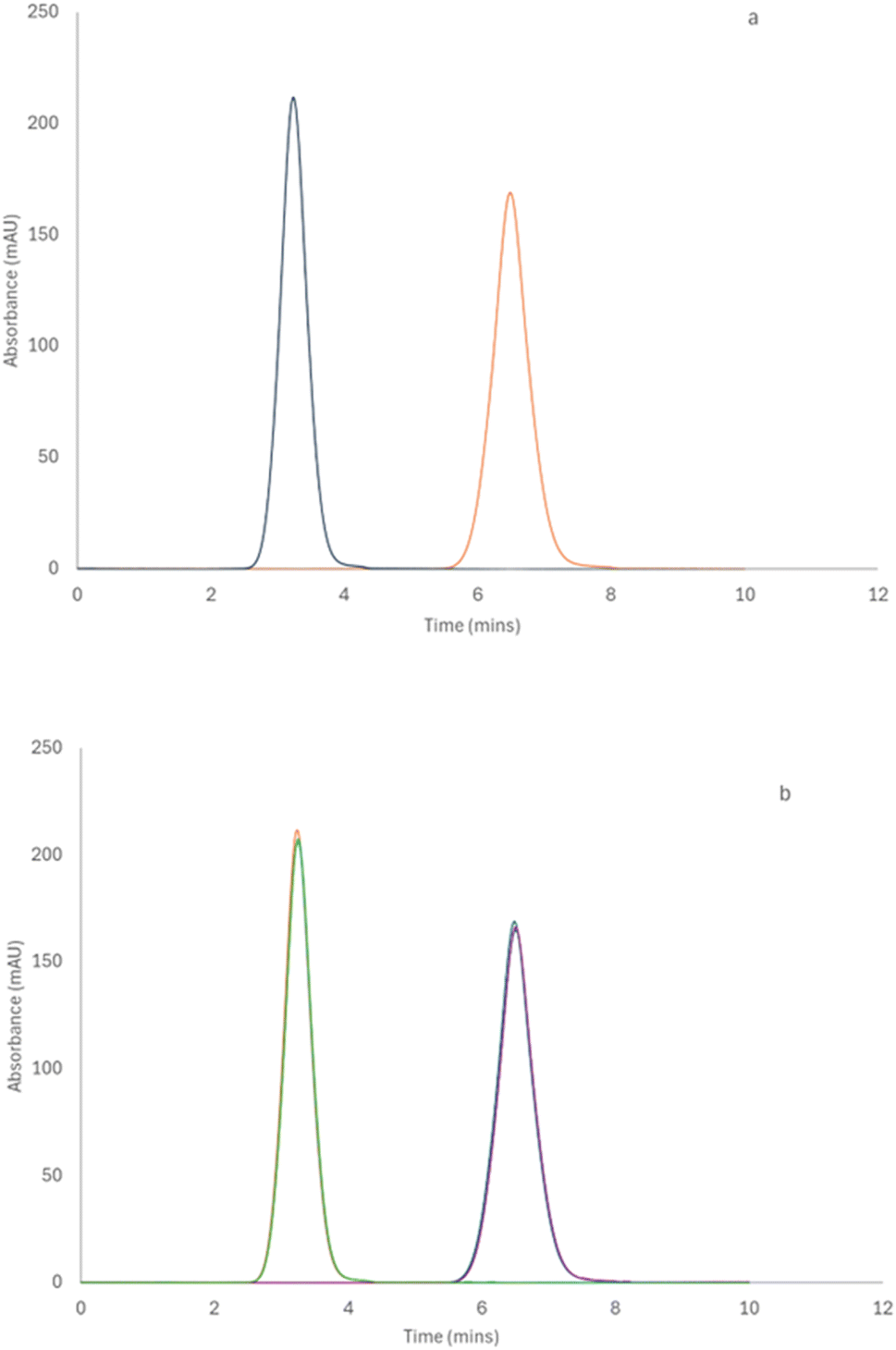 | ||
| Fig. 1 (a) TDA profile for a 1% w/v G4.5 PAMAM dendrimer solution in phosphate buffer (pH 7.4) recorded at 214 nm, showing the first and second passes at windows 1 and 2. (b) TDA profile showing an overlay of three runs for a 1% w/v G4.5 PAMAM dendrimer solution in PBS (n = 3). | ||
As shown in Table 3, a range of PAMAM dendrimers G1.5, G3.5, G4.5, G3 and G4 were analysed by TDA in various buffer solutions: phosphate (pH 7.4), carbonate (pH 10.6) and HCl (pH 1.2). The measurement of Rh was conducted in three separate runs and showed minute differences with relatively small standard deviations. Various pH values were employed to facilitate monitoring the effect of ionisation of the functional groups on the conformation of dendrimers. Our data show that at the same pH, the Rh increases with the generation of dendrimers, e.g. the Rh values of G1.5, G3.5 and G4.5 dendrimers at pH 7.4 are 1.36, 2.32, and 2.67 nm, respectively. This correlation is in agreement with other studies using SAXS, SANS and molecular dynamics.13,15,34,35 This increment in size is mainly attributed to an increase of the focal points within the branches, leading to additional layers of the dendrimer.
| Sample (1% w/v) | pH | R h (nm) | Average Rh (nm) | ||
|---|---|---|---|---|---|
| 1 | 2 | 3 | |||
| G1.5 | 7.4 | 1.31 | 1.36 | 1.41 | 1.36 (±0.05) |
| 1.2 | 1.06 | 1.07 | 1.10 | 1.08 (±0.02) | |
| G3.5 | 7.4 | 2.26 | 2.32 | 2.37 | 2.32 (±0.06) |
| 1.2 | 1.61 | 1.55 | 1.64 | 1.60 (±0.04) | |
| G4.5 | 7.4 | 2.73 | 2.70 | 2.61 | 2.67 (±0.06) |
| 1.2 | 1.85 | 1.89 | 1.87 | 1.87 (±0.02) | |
| G3 | 7.4 | 2.23 | 2.20 | 2.10 | 2.18 (±0.07) |
| 10.6 | 2.21 | 2.27 | 2.57 | 2.35 (±0.19) | |
| 1.2 | 1.98 | 2.57 | 2.12 | 2.23 (±0.31) | |
| G4 | 7.4 | 2.40 | 2.64 | 2.77 | 2.60 (±0.19) |
| 10.6 | 2.24 | 2.82 | 2.45 | 2.50 (±0.30) | |
| 1.2 | 2.62 | 2.74 | 2.63 | 2.66 (±0.07) | |
Compared to pH 7.4, there is a significant decrease in the average Rh of the half generation PAMAM dendrimers at pH 1.2 (Fig. 2). However, the degree of reduction in Rh varies across generations. Despite efforts by various research groups,35 half-generation PAMAM dendrimers remain inadequately characterized. The structures of charged half-generation dendrimers are expected to be more expanded due to coulombic repulsion on the surface of the dendrimers and also to an increased solvent cage effect, in which the charged groups were surrounded and stabilized by the solvents while the movement of branches was restricted. Hence under acidic conditions (pH 1.2), the surface carboxyl moieties of half generation dendrimers become neutralized while the interior tertiary amines are protonated. This reduces the repulsion of the dendrimer surface and leads to possible back folding of the dendrimer branches, especially for lower generation dendrimers having an open structure. It was observed that TDA enabled a repeatable size measurement of the G1.5 dendrimer at pH 1.2, yielding a size of approximately 1.08 nm. This measurement is particularly noteworthy as it approaches the lower detection limits of conventional techniques, such as light scattering, which often struggle to accurately measure such small particle sizes due to their inherent sensitivity and resolution constraints. This highlights the potential of TDA in providing precise size determinations for very small dendrimer molecules under challenging conditions.
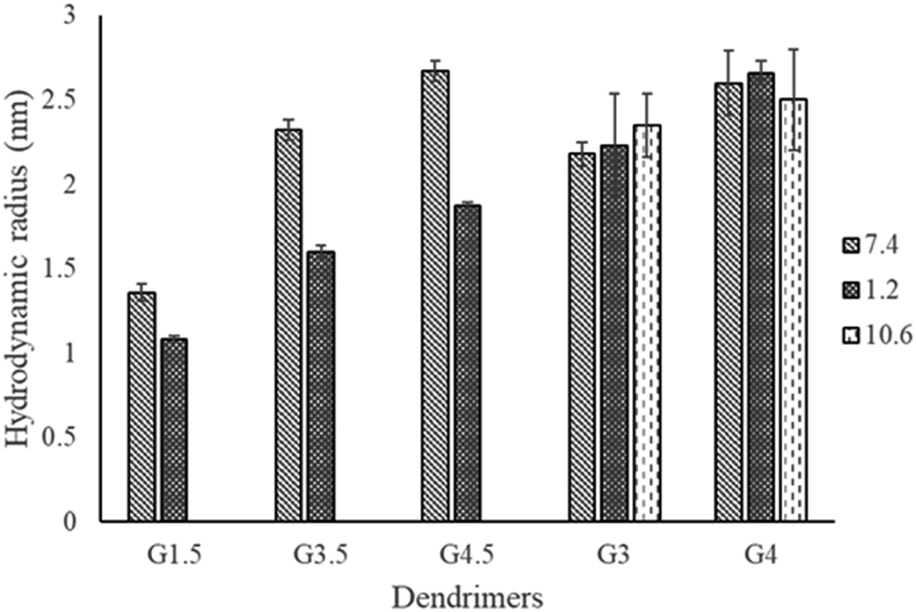 | ||
| Fig. 2 Comparison of the hydrodynamic radii of PAMAM dendrimers at various pH values measured by TDA. | ||
The effect of pH on the conformation of full generation PAMAM dendrimers is ambiguous. The conformational behaviour of full generation PAMAM dendrimers was investigated using molecular dynamics simulation,13 SAXS and SANS.10,36 It was found that the radius of gyration of the dendrimers was pH dependent due to electrostatic repulsion between protonated dendritic branches or back-folding of the neutralized surface. In contrast, several studies also reported that the dendrimer size is essentially insensitive to the pH change of the solvent by measuring G4 and G8 PAMAM dendrimers at various pH values using SANS.11,37 Further simulation work with an optimized method suggested pH-induced conformational changes for G4 dendrimers from a ‘dense core’ at high pH to a ‘dense shell’ at low pH without significant size swelling.18 In our work, as shown in Fig. 2, no significant difference in Rh was found under various pH conditions for either G3 or G4 dendrimers. G4 dendrimers exhibit a slightly expanded structure at pH 1.2, which may be due to the repulsion effect of the protonated surface and tertiary amine groups. Under basic conditions, the surface amine moieties are mainly neutralized and less electrostatic repulsion was expected, which leads to a relatively compact structure and back-folding of surface branches. For G3 dendrimers, the hydrodynamic radii are very similar in different buffer solutions. This is probably due to the smaller size and cavity structure of G3 that leads to a less apparent size change with pH.
3.2. Effect of ionic strength on the size of half-generation dendrimers
PAMAM dendrimers are polyelectrolytes due to their multi-functional surface. The conformational behaviour of dendrimers is considered to be influenced by counterions and ionic strength in solutions. Current literature mainly focuses on full generation dendrimers with protonated amino moieties.38,39 These studies suggest that solutions of monovalent salts with low concentrations, e.g. ≤1 M NaCl solutions, show no change in the size of PAMAM dendrimers. In this study, we investigated the effect of ionic strength on the size of half generation PAMAM dendrimers by dissolving G4.5 dendrimers in phosphate buffers with 0.1 and 1 M NaCl (ionic strength of 0.268 M and 1.168 M respectively). As shown in Fig. 3, the hydrodynamic radius of G4.5 in phosphate buffer with 0.1 M NaCl was decreased compared to that in phosphate buffer alone. G4.5 dendrimers are polyelectrolytes with carboxylate ions, which are efficiently solvated by water. Like conventional polyelectrolytes e.g., polyacrylic acids,40 added sodium ions can surround the negative charges on the dendrimer surface, weaken the electrostatic repulsion between the dendrimer branches because of electrostatic shielding, which leads to a relatively compact structure. When the salt concentration increases further, it will push the counterions in the diffusion layer of the dendrimer into the absorb layer and thus compress the electric double layer of the surface hydration shell of the dendrimer. This results in an increase in the unneutralized surface charge and repulsion between the branches. Hence, the hydrodynamic radius in phosphate buffer with 1 M NaCl is larger than that in buffers with lower salt concentrations.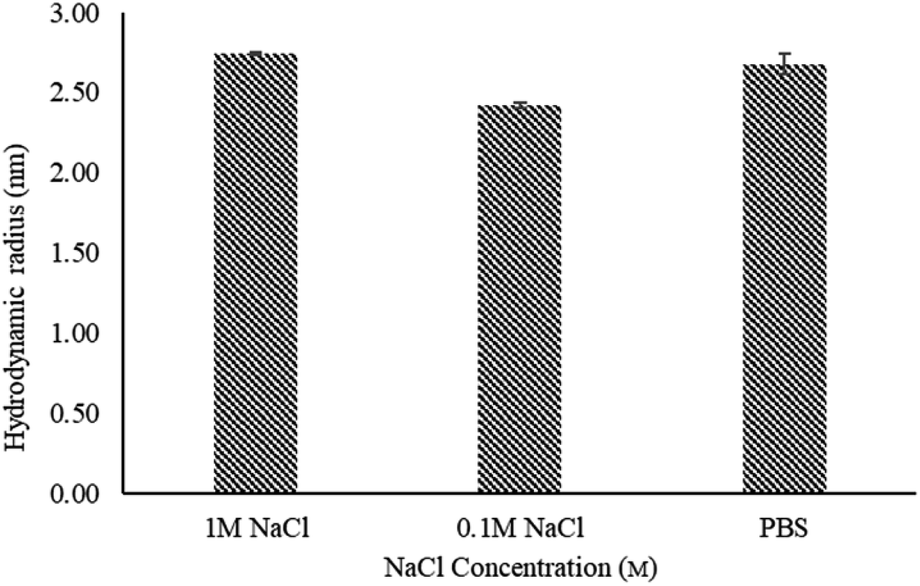 | ||
| Fig. 3 Hydrodynamic radius of G4.5 PAMAM dendrimers in phosphate buffer (pH 7.4) with varying NaCl concentrations, measured by TDA at 25 °C. Error bars represent STDEV (n = 3). | ||
3.3. TDA vs. DLS
Dynamic light scattering has been widely employed to measure the hydrodynamic size of nanoparticles and investigate the interaction with the surrounding solvent. DLS is one of the most cost-effective sizing techniques with respect to time and volume.29 However, it is limited by its quality of light scattering and requires model fitting to properly analyse the raw data. Furthermore, DLS has very strict requirements for sample preparation and struggles with analysing nanoparticles in complex environments.21 We analysed the Rh of G4 and G4.5 PAMAM dendrimers using DLS to compare with TDA. The same batch of samples was analysed by both techniques and the results are shown in Table 4. The results indicate that TDA is a superior technique compared to DLS for measuring PAMAM dendrimers, which allows for the easy acquisition of undisturbed data. The DLS technique is intensity-weighted, yielding a harmonic z-average hydrodynamic radius and is highly sensitive to the presence of larger particles in a sample. In contrast, TDA employs a UV detector sensitive to mass concentration, resulting in a weight-average hydrodynamic radius.41 The DLS results have shown the presence of 2 peaks (Intensity PSD), leading to an increased z-average radius. This is probably due to the guest particles or aggregates in the samples, despite careful filtration. The presence of larger particles observed in the DLS measurements likely represents a small number of loosely associated clusters of monomeric PAMAM molecules, a common occurrence in DLS due to the stationary and small sample volume. However, despite using identical samples, these larger particles were absent in the TDA measurements, which offers greater tolerance for sample preparation. Under TDA flow conditions, the loose aggregates of PAMAM dendrimers likely dissociate due to flow dynamics, rendering them undetectable. Moreover, when only considering the smaller peaks from DLS, both techniques yielded hydrodynamic radii within a comparable range.| Sample (1% w/v) | pH | DLS | TDA | ||
|---|---|---|---|---|---|
| Intensity mean peak 1 (nm) | Intensity mean peak 2 (nm) | Intensity z-average (nm) | Average Rh (nm) | ||
| G4 | 7.4 | 2.97 (±0.02) | 126.1 (±2.10) | 8.82 (±0.14) | 2.60 (±0.19) |
| 10.6 | 2.86 (±0.02) | 114.5 (±11.63) | 7.31 (±0.13) | 2.50 (±0.30) | |
| 1.2 | 2.72 (±0.11) | 121.5 (±11.27) | 3.31 (±0.46) | 2.66 (±0.07) | |
| G4.5 | 7.4 | 2.94 (±0.02) | 89.98 (±7.08) | 4.94 (±0.10) | 2.67 (±0.06) |
| 1.2 | 4.68 (±0.06) | 1867 (±16.64) | 4.17 (±0.02) | 1.87 (±0.02) | |
3.4. TDA vs. conventional sizing techniques
Apart from DLS, PAMAM dendrimers have been investigated using other well-established techniques such as SAXS,10 SANS,18 molecular dynamics simulation,5,36 gel permeation chromatography (GPC)41 and differential mobility analyser.17Table 5 shows the dendrimer sizes under neutral conditions reported by the aforementioned techniques in comparison to the data obtained by TDA. The dendrimer sizes from TDA are relatively larger due to the solvation of the dendrimers in buffer solution. It must be borne in mind that the types of size information obtained from various techniques differ. SANS and SAXS measure the radius of gyration (Rg), while MD simulations provide dynamic sizes depending on the simulation conditions. The differential mobility analyser measures the mobility size in the gas state instead of in solvent. GPC with viscosity or light scattering detectors can provide information on the dendrimer size and structure in aqueous and organic solvents. SANS and SAXS are the preferred methods to experimentally measure the dendrimers size in an appropriate medium.18,36,42 However, these techniques are normally expensive and time-consuming and also require deuterated solvents for enhanced contrast and lower incoherent scattering. Molecular dynamics simulations are capable of estimating the hydrodynamic radius by taking into account the interactions with the solvent and ions, but it is not experimentally measured in real solution. Nevertheless, in this study, TDA has demonstrated its efficacy as a fast, easily prepared, and cost-efficient method. This suggests its potential as a reliable alternative technique for determining the sizes of dendrimers or hyperbranched macromolecules. Notably, it was able to characterize half-generation PAMAM moieties.| Sample | Literature values | |||||
|---|---|---|---|---|---|---|
| SAXS (Å)a | SANS (Å)a | Molecular dynamics (Å)a | GPC (nm) | DMA (nm) | TDA (nm) | |
| R g | R g | R g | R h | R p | R h | |
| a 1 angstrom = 0.1 nanometers. | ||||||
| G3 neutral pH (∼7) | 15.8 (ref. 10) | — | 17.2 (ref. 15) | 1.78 (ref. 2) | 1.54 (ref. 17) | 2.18 |
| G4 neutral pH (∼7) | 17.1 (ref. 10) | 21.58 (ref. 18) | 21.7 (ref. 15) | 2.24 (ref. 42) | 1.86 (ref. 17) | 2.60 |
4. Conclusions
In summary, this study has demonstrated the efficiency of TDA in sizing various PAMAM dendrimers across diverse buffer solutions. The precision of the results has facilitated the determination of structural insights and revealed the sensitivities of dendrimers to their solution environment. Our findings indicate the utility of TDA as a promising orthogonal sizing method for hyperbranched macromolecules like PAMAM dendrimers. This could be valuable for developing dendrimers as effective carriers for drugs or proteins with customized binding capabilities.Data availability
The data that support the findings of this study are available in the ESI Section of the paper.‡Author contributions
Zhengyuan Zhou: data curation, formal analysis, investigation, writing – original draft, resources, review and editing. Robert Forbes: conceptualization, investigation, methodology, project administration, supervision, writing – original draft, review and editing. Vikesh Chhabria: data curation, formal analysis, investigation, writing – original draft, validating, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the University of Central Lancashire under a Science Research Program.Notes and references
- A. Narain, S. Asawa, V. Chhabria and Y. Patil-Sen, Nanomedicine, 2017, 11, 2677 CrossRef PubMed.
- D. P. Otto and M. M. de Villiers, J. Pharm. Sci., 2018, 107, 75 CrossRef PubMed.
- R. Esfand and D. A. Tomalia, Drug Discovery Today, 2001, 6, 427 CrossRef CAS PubMed.
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J., 1985, 17, 117 CrossRef CAS.
- S. Sonzini, F. Caputo, D. Mehn, L. Calzolai, S. E. Borgos, A. Hyldbakk, K. Treacher, W. Li, M. Jackman, N. Mahmoudi, M. J. Lawrence, C. Patterson, D. Owen, M. Ashford and N. Akhtar, Int. J. Pharm., 2023, 637, 122905 CrossRef CAS PubMed.
- B. William, R. Shukla and J. Baker, US Pat., US11/503742, 2007 Search PubMed.
- W. M. Saltzman, T. Fahmy and P. Fong, US Pat., US7534449, 2009 Search PubMed.
- J. K. Fairley, C. P. Barrett and J. R. Paull, US Pat., US14/427970, 2015 Search PubMed.
- J. Manikkath, A. Manikkath, G. V. Shavi, K. Bhat and S. Mutalik, J. Drug Delivery Sci. Technol., 2017, 1, 334 CrossRef.
- P. K. Maiti, T. Çaǧın, G. Wang and W. A. Goddard, Macromolecules, 2004, 37, 6236 CrossRef.
- W. R. Chen, L. Porcar, Y. Liu, P. D. Butler and L. J. Magid, Macromolecules, 2007, 40, 5887 CrossRef.
- T. J. Prosa, B. J. Bauer and E. J. Amis, Macromolecules, 2001, 34, 4897 CrossRef.
- B. Wang, Y. Sun, T. P. Davis, P. C. Ke, Y. Wu and F. Ding, ACS Sustain. Chem. Eng., 2018, 6, 11704 CrossRef PubMed.
- Y. Liu, L. Porcar, K. L. Hong, C. Y. Shew, X. Li, E. Liu, P. D. Butler, K. W. Herwig, G. S. Smith and W. R. Chen, J. Chem. Phys., 2010, 132, 124901 CrossRef PubMed.
- P. K. Maiti and B. Bagchi, J. Chem. Phys., 2009, 131, 214901 CrossRef PubMed.
- H. Lee, J. S. Choi and R. G. Larson, Macromolecules, 2011, 44, 8681 CrossRef.
- M. Imanaka, Y. Okada, K. Ehara and K. Takeuchi, J. Aerosol Sci., 2006, 37, 1643 CrossRef.
- Y. Liu, V. S. Bryantsev, M. S. Diallo and W. A. Goddard, J. Appl. Chem. Sci., 2009, 131, 2798 Search PubMed.
- J. Gouyon, A. Boudier, F. Barakat, A. Pallotta and I. Clarot, Electrophoresis, 2002, 43, 2377 CrossRef PubMed.
- P. Lemal, A. Petri-Fink and S. Balog, Anal. Chem., 2018, 17, 1217 Search PubMed.
- D. A. Urban, A. M. Milosevic, D. Bossert, F. Crippa, T. L. Moore, C. Geers, S. Balog, B. Rothen-Rutishauser and A. Petri-Fink, Colloids Interface Sci. Commun., 2018, 1, 29 CrossRef.
- C. Malburet, L. Leclercq, J. F. Cottet, J. Thiebaud, E. Bazin, M. Garinot and H. Cottet, Gene Ther., 2022, 1, 1 Search PubMed.
- H. Cottet, M. Martin, A. Papillaud, E. Souaïd, H. Collet and A. Commeyras, Biomacromolecules, 2007, 8, 3235 CrossRef PubMed.
- T. Liu, F. Oukacine, H. Collet, A. Commeyras, L. Vial and H. Cottet, J. Chromatogr. A, 2013, 1273, 111–116 CrossRef.
- X. Jin, L. Leclercq, N. Sisavath and H. Cottet, Macromolecules, 2014, 47, 5320–5327 CrossRef.
- R. M. England, S. Sonzini, D. Buttar, K. E. Treacher and M. B. Ashford, Polym. Chem., 2022, 13, 2626 RSC.
- S. Roca, L. Leclercq, C. Somnin, L. Dhellemmes, J. Chamieh and H. Cottet, Electrophoresis, 2024 DOI:10.1002/elps.202400156.
- H. Zaman, A. G. Bright, K. Adams, D. M. Goodall and R. T. Forbes, Int. J. Pharm., 2017, 522, 98 CrossRef.
- W. L. Hulse and R. T. Forbes, Int. J. Pharm., 2011, 411, 64 CrossRef PubMed.
- W. L. Hulse and R. T. Forbes, Int. J. Pharm., 2011, 416, 394 CrossRef PubMed.
- W. L. Hulse, J. Gray and R. T. Forbes, Int. J. Pharm., 2013, 453, 351 CrossRef.
- X. Wang, L. Guerrand, B. Wu, X. Li, L. Boldon, W. R. Chen and L. Liu, Polymers, 2012, 4, 600 CrossRef.
- D. A. Tomalia, A. M. Naylor and W. A. Goddard III, Angew. Chem., Int. Ed., 1990, 29, 138 CrossRef.
- T. J. Prosa, B. J. Bauer, E. J. Amis, D. A. Tomalia and R. Scherrenberg, J. Polym. Sci., Part B: Polym. Phys., 1997, 35, 2913 CrossRef.
- P. M. Paulo, J. N. Lopes and S. M. Costa, J. Phys. Chem. B, 2007, 111, 10651 CrossRef PubMed.
- P. K. Maiti, T. Çaǧın, S. T. Lin and W. A. Goddard, Macromolecules, 2005, 38, 979 CrossRef.
- G. Nisato, R. Ivkov and E. J. Amis, Macromolecules, 2000, 33, 4172, DOI:10.1021/ma991474p.
- Y. Q. Ma, Soft Matter, 2010, 6, 1308, 10.1039/B923960J.
- H. Lee, Simulations, 2012, 38, 589 CrossRef.
- H. Markovitz and G. E. Kimball, J. Colloid Interface Sci., 1950, 5, 115 CrossRef.
- H. Cottet, J.-P. Biron and M. Martin, Anal. Chem., 2007, 79, 9066–9073 CrossRef PubMed.
- S. Uppuluri, D. A. Tomalia and P. R. Dvornic, PMSE Prepr., 1997, 77, 116 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ay01769b |
| ‡ Current address: School of Biosciences, Cardiff University, Sir Martin Evans Building, Cardiff, CF10 3AX, UK |
| This journal is © The Royal Society of Chemistry 2025 |