
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural and mechanistic insights into KslB, a bacterial Pictet–Spenglerase in kitasetaline biosynthesis†
Wantae
Kim‡
a,
Ziyang
Zheng‡
 b,
Kangsan
Kim
c,
Yu-Hsuan
Lee
d,
Hung-wen
Liu
*bd and
Y. Jessie
Zhang
*c
b,
Kangsan
Kim
c,
Yu-Hsuan
Lee
d,
Hung-wen
Liu
*bd and
Y. Jessie
Zhang
*c
aMcKetta Department of Chemical Engineering, University of Texas at Austin, Austin, Texas 78712, USA
bDepartment of Chemistry, University of Texas at Austin, Austin, Texas 78712, USA. E-mail: h.w.liu@mail.utexas.edu
cDepartment of Molecular Biosciences, University of Texas at Austin, Austin, Texas 78712, USA. E-mail: jzhang@cm.utexas.edu
dDivision of Chemical Biology and Medicinal Chemistry, College of Pharmacy, University of Texas at Austin, Austin, Texas 78712, USA
First published on 16th April 2025
Abstract
KslB is one of the few bacterial Pictet–Spenglerases recently identified in the biosynthesis of the β-carboline compound kitasetaline. While previous in vitro studies established that KslB catalyzes the condensation between L-tryptophan and α-ketoglutarate, the reaction mechanism, particularly its stereochemistry, remains poorly understood. This study presents five crystal structures of KslB, capturing key stages of reaction, shedding light on its catalytic dynamics. Among these, alternative binding poses of substrate and reaction product highlighted two significant features: (1) an additional pocket that accommodates L-tryptophan, and (2) two positively charged residues, Lys264 and Arg256, which form salt bridges with the product C1′ and C5′ carboxylate groups derived from α-ketoglutarate, ensuring a stereoselective process. These structural insights elucidate how KslB governs the stereochemistry of the cyclization process. Accordingly, we propose the configurations for the cyclized intermediate that align with the reaction's stereochemical outcome. Together, these findings offer valuable structural and mechanistic insights into KslB, paving the way for its potential engineering as a Pictet–Spengler biocatalyst.
Introduction
The Pictet–Spengler reaction is a widely used reaction to synthesize a diverse array of heterocyclic compounds.1 It is an acid catalyzed reaction involving the condensation between a β-arylethylamine and an aldehyde or a ketone followed by intramolecular cyclization to form a fused ring adduct. When the Pictet–Spengler reaction is performed with an aldehyde other than formaldehyde, a new chiral center will be generated in the product. Several substrate- or auxiliary-controlled diastereoselective Pictet–Spengler reactions have been developed for chemical synthesis of natural products.1–5 This seemingly complicated reaction can also be catalyzed by enzymes and has been found operative in the biosynthesis of many heterocyclic alkaloids in nature.1,6 Enzymes that evolve to catalyze this type of reaction have thus been coined as Pictet–Spenglerases.6Majorities of Pictet–Spenglerases are discovered in plants; however, a few derived from bacteria have also been reported in recent years, including mikimopine synthase and cucumopine synthase from Agrobacterium rhizogenes,7,8 McbB from Marinactinospora thermotolerans,9 NscbB from Nocardiopsis synnemataformans,10 StnK2 from Streptomyces flocculus,11 and SfmC from Streptomyces lavendulae.12 The best studied example among them is McbB that catalyzes a Pictet–Spengler reaction between L-tryptophan (L-Trp) and oxaloacetaldehyde (1) to form 2, which is believed to undergo rapid nonenzymatic oxidation and decarboxylation to yield final products 3 and 4 (Fig. 1A). Interestingly, McbB has low sequence (ca. 15% sequence identity) and structural similarity when compared to Pictet–Spenglerases isolated from plants. For example, X-ray crystallographic analysis of McbB showed that it adopts a completely different folding pattern from that of strictosidine synthase, a plant Pictet–Spenglerase from Rauvolfia serpentina which catalyzes the first committed step in indole alkaloid biosynthesis by coupling tryptamine to secologanin to form strictosidine (see Fig. 1B).13,14 Since McbB is the only bacterial Pictet–Spenglerase whose structure has been determined, biochemical and structural investigations of other bacterial Pictet–Spenglerases deem necessary to gain more insights into their catalytic properties which may offer opportunity to explore their potential as useful biocatalysts.
 | ||
| Fig. 1 (A) Reaction catalyzed by McbB. (B) Reaction catalyzed by strictosidine synthase. (C) Reaction of KslB with L-Trp and α-KG. (D) Reaction of KslB with L-Trp and succinic semialdehyde (7). (E) Structure of N-oxalylglycine (NOG). | ||
Our recent studies led to the discovery of a new bacterial Pictet–Spenglerase referred as KslB from Kitasatospora setae, which catalyzes the formation of 5 through condensation between L-Trp and α-ketoglutarate (α-KG) in the biosynthesis of the β-carboline kitasetaline (6) (Fig. 1C).15 KslB shows low sequence homology to both McbB (9% identity) and strictosidine synthase (2% identity). In addition to the ketone substrate α-KG, KslB also accepts succinic semialdehyde (7) to generate 8 with L-Trp (Fig. 1D). The stereochemistry of the C2′ carbon in 8 was determined by NMR analysis to have the S-configuration.15 The C2′ carbon in 5 was thus inferred to be S-configured as well albeit with no direct evidence. Importantly, how the cyclization step is stereochemically controlled during KslB catalysis remains poorly understood. While this work was in progress, a preprint on the crystal structures of KslB complexed with its substrate (L-Trp) and native product (5) was released.16 However, the mechanistic interpretations of the binding between α-KG and KslB were unavailable in that report leaving the above question unaddressed.
The appearance of the above article prompted us to publish our study of KslB aiming to better understand its mode of substrate recognition and mechanism of stereoselectivity using X-ray crystallography. In this work, we observed two different substrate binding poses: one as reported in the recent release, but also an alternative binding pose of L-Trp that occupies an unexplored hydrophobic cavity. Moreover, we have obtained two additional structures of KslB complexed with its native product (5) and another product (8). Importantly, our KslB·5 complex obtained through co-crystallization exhibits a catalytically relevant configuration, revealing how the enzyme governs the stereochemical course to form the product. This study captured different snapshots to complete the full profile of KslB active site interactions, which paves the path for enzyme engineering and development of biocatalysts for Pictet–Spengler reactions.
Results and discussion
Structures of apo KslB (structure I)
To gain insights into KslB catalysis, we sought to capture its crystal structures at different stages of its catalytic reaction. The N-terminal His6-tagged KslB construct was expressed in Escherichia coli and purified using Ni-NTA affinity chromatography followed by size exclusion chromatography (SEC). The purified KslB was crystallized and diffracted to a resolution of 2.95 Å in space group P32, with six molecules per asymmetric unit. The crystal structure shows that KslB forms a homodimer (structure I, Fig. 2A) with a dimer interface area of 3251.3 Å2, which is 21.1% of the surface area of a KslB monomer.17 The dimer formation of KslB observed in the crystal structure is consistent with the molecular weight measured from SEC (Fig. S2, ESI†). The overall structure and dimerization of KslB are very similar to those previously reported for the bacterial Pictet–Spenglerase, McbB, despite low sequence conservation of 9% (Fig. 2B and Fig. S3, ESI†). | ||
| Fig. 2 Crystal structure of KslB and comparison with McbB structure. (A) Overall homodimer structure of KslB shown in green and white cartoons. (B) Relative position of each domain in KslB/McbB illustrated in amino acid sequence notations (top) and crystal structures of the apo-enzymes (bottom). Structure of McbB is taken from its previously reported structure in RCSB PDB (3X27). | ||
Both enzymes share a two-domain architecture comprising an α-helix bundle domain and a β-barrel domain. However, in KslB, the order of these two domains is swapped when compared to McbB (Fig. 2B). Consequently, the disordered loop linking two domains in KslB and McbB adopts different connecting positions (Fig. 2B). Despite the low sequence identity (9%) and the domain order swap, the overall homodimeric structures of KslB and McbB are highly similar. This structural similarity implies a conserved catalytic mechanism for Pictet–Spenglerase activity. However, the key amino acid residues in substrate binding and chemical transformation may be different and are not apparent from primary sequence alignment of these two enzymes.
Structure of KslB complexed with substrate L-Trp (structure II)
To confirm the binding pose of L-Trp within the active site of KslB, we co-crystallized KslB in complex with L-Trp. The KslB·L-Trp complex was prepared by running the final SEC purification step using a buffer supplemented with 1 mM L-Trp. The addition of L-Trp significantly stabilized KslB, as revealed by differential scanning fluorimetry (DSF). Compared to the apo enzyme, the KslB·L-Trp complex exhibited a substantial increase in protein melting temperature (Tm) from 40 °C to 56.5 °C (Fig. S4, ESI†), indicating significantly enhanced stability upon substrate binding.The binary complex was crystallized, and X-ray diffraction data were collected to a resolution of 3.1 Å. A strong positive electron density was observed consistently in each of the six molecules per asymmetric unit at the position corresponding to the proposed active site of KslB (Fig. S5A, ESI†). The electron density in each chain was individually fitted with L-Trp and refined (structure II, Fig. 3A), resulting in highly consistent binding poses across all six molecules within the crystal asymmetric unit (Fig. S6A, ESI†). This high degree of conservation in ligand binding, despite the unique crystal packing environment for each molecule, strengthens confidence in the ligand-binding model and minimizes potential biases from packing artifacts.
 | ||
| Fig. 3 Two different binding poses of L-Trp observed within the active site of KslB. (A) Electron density map shown around L-Trp within the active site of KslB·L-Trp binary complex structure (structure II). (B) Active site interaction between KslB and L-Trp. (C) HPLC analysis of the reactions of KslB mutants with L-Trp and α-KG. AU indicates absorbance units at 276 nm. (D) Electron density map shown around L-Trp in an alternative binding mode (structure III). (E) Active site interaction between KslB and L-Trp in a different binding mode. (F) Active site cavity that accommodates an alternative binding pose of L-Trp is shown as a gray surface. 2Fo–Fc map (gray mesh, contoured at 1.0σ) covers the stick model of L-Trp (yellow in carbon atoms). Hydrogen bonding interactions are shown in yellow dashed lines. The cartoon and stick models in white and green represent two KslB monomers within a dimer. | ||
Upon L-Trp binding, the active site of KslB remains largely unchanged, except for a 1 Å shift in the phenyl side chain of Phe89, which may arise from KslB recognizing the aromatic indole moiety of L-Trp through π–π stacking interaction with Phe89 (Fig. 3B). Additionally, the α-amino group of L-Trp and the nitrogen atom within the indole moiety are both stabilized by negatively charged Glu274 (Fig. 3B). The carboxyl group of L-Trp is anchored within an oxyanion hole through hydrogen bonding with amide backbones of Val223, Val224, and Ser225 (Fig. 3B). Furthermore, the hydroxyl group of the Tyr230 side chain is also positioned within the hydrogen bond distance to the α-carboxylate of L-Trp, providing additional stabilization of substrate binding (Fig. 3B). The resulting binding pose of L-Trp in the active site of KslB closely resembles that observed in McbB·L-Trp complex structure (Fig. S7A, ESI†), further supporting the catalytic similarity between KslB and McbB.
To locate the binding site of the co-substrate α-KG or succinic semialdehyde (7), we tried to obtain the complex of KslB with these co-substrates in the absence of L-Trp. In the co-crystallization experiment, KslB was incubated with α-KG or succinic semialdehyde (7) prior to crystallization. Alternatively, a soaking approach was also employed in which these compounds were introduced into preformed KslB apo crystals. However, neither method revealed distinctive positive electron density in the active site, indicating the absence of stable co-substrate binding under these conditions. Given the strong stabilizing effect of L-Trp on KslB, these results suggest that binding of co-substrates such as α-KG and succinic semialdehyde (7) likely requires prior formation of the KslB·L-Trp complex.
Residues critical for L-Trp recognition and alternative binding mode (structure III)
In the KslB·L-Trp complex structure, Phe89, Glu274, and Tyr230 are within the distance to form favorable interaction with the substrate. To assess the functional significance of these interactions in the Pictet–Spengler reaction, we performed site-directed mutagenesis, in which Phe89, Glu274, and Tyr230 were mutated to alanine (F89A), glutamine (E274Q), and phenylalanine (Y230F), respectively. Each variant was expressed and purified with a similar protocol as the wild-type enzyme (Fig. S1, ESI†). In vitro assays were conducted involving 1 mM L-Trp, 1 mM α-KG, and 5 μM of either wild-type KslB or its mutants (F89A, E274Q or Y230F) in 50 mM HEPES buffer (pH 7.5) at room temperature for 16 h. The results in Fig. 3C showed that the F89A mutant exhibited only a slight reduction in activity compared with the wild-type enzyme, while the Y230F mutant is indistinguishable from the wild-type KslB. These suggest that π–π interaction between the indole ring and Phe89 or the hydrogen bonding between the α-carboxylate and Tyr230 is not essential for the reaction. In contrast, the E274Q mutation completely abrogated enzyme activity, indicating Glu274 is indispensable for the catalysis. Notably, sequence and structural alignments between the C-terminus of KslB and the N-terminus of McbB revealed that Glu274 in KslB aligns with the proposed catalytic Glu97 in McbB (Fig. S7B, ESI†). This strongly supports that Glu274 is the catalytic residue critical for KslB enzymatic function.To investigate the binding of L-Trp together with the co-substrate α-KG or succinic semialdehyde (7) in KslB, we attempted to capture the ternary complex using the non-reactive α-KG analogue, N-oxalylglycine (NOG, see Fig. 1E), to prevent turnover. However, despite extensive co-crystallization and soaking efforts, we only observed small patches of density close to Arg256, but not enough to build NOG structure in any of the six protein molecules within the asymmetric unit. Unexpectedly, in the 2.68 Å resolution structure obtained under these conditions, we observed a strong electron density that can be attributed to the side chain of L-Trp in an alternative conformation (structure III, Fig. 3D and Fig. S5B, ESI†). This alternative conformation was again consistent among all six KslB molecules in the asymmetric unit (Fig. S6B, ESI†). Compared to L-Trp in structure II, despite the same binding pose at the amino acid end, the indole ring of L-Trp in structure III underwent a ∼60° rotation and could no longer engage in the π–π stacking with Phe89 (Fig. 3E). Instead, the indole ring gets stabilized inside a pocket formed by Phe46, Trp232, Lys264, Tyr230, and His134 (Fig. 3E and F). The observed flexibility of the binding pose of the indole ring seen in structure II and III is consistent with its π–π stacking with Phe89 being not critical for the catalysis, providing a structural explanation for the mild reduction in enzymatic activity of the F89A mutant.
Structure of KslB·L-Trp·7 reaction product (KslB·8) (product-trapping complex, structure IV)
Next, we examined the binding of succinic semialdehyde (7) within the active site cavity of KslB. To obtain the KslB·L-Trp·7 ternary complex structure, we incubated KslB·L-Trp with 5 mM 7 for 3 h, successfully yielding crystals diffracting up to 2.87 Å. Unexpectedly, we observed a strong positive electron density that is substantially bulkier than L-Trp in the L-Trp binding cavity (Fig. S5C, ESI†). Attempts to fit this density with 7 and L-Trp revealed that the density was consistent with the trapping of the reaction product (8) (structure IV, Fig. 4A). Notably, despite being independently fitted, the binding poses of 8 were practically the same across all six KslB molecules in the asymmetric unit (Fig. S6C, ESI†). The stereochemistry of the C2′ carbon in compound 8 is all refined to be S-configuration, consistent with that determined by NMR.15 The tryptophan moiety in 8 occupied the same binding pocket as L-Trp did in structure II, maintaining key anchoring interactions (Fig. 4B). The newly formed six-member heterocycle induced a conformational change in the hydroxyl side chain of Ser225, allowing it to form a hydrogen bond with the α-nitrogen atom of 8 (Fig. 4B). Additionally, the carboxylate group from succinic semialdehyde (7) extends outward, displacing the loop containing the catalytic Glu274 by 3 Å (Fig. 4C). Simultaneously, Thr273 within the loop loses its hydrogen bonding interaction with Tyr92 from the dimer assembly partner upon trapping of the reaction product (8). The loss of the Thr273–Tyr92 interaction causes Tyr92 to swing 120° outward, which provides ample space to accommodate the side chain carboxylate group in 8 (Fig. 4C).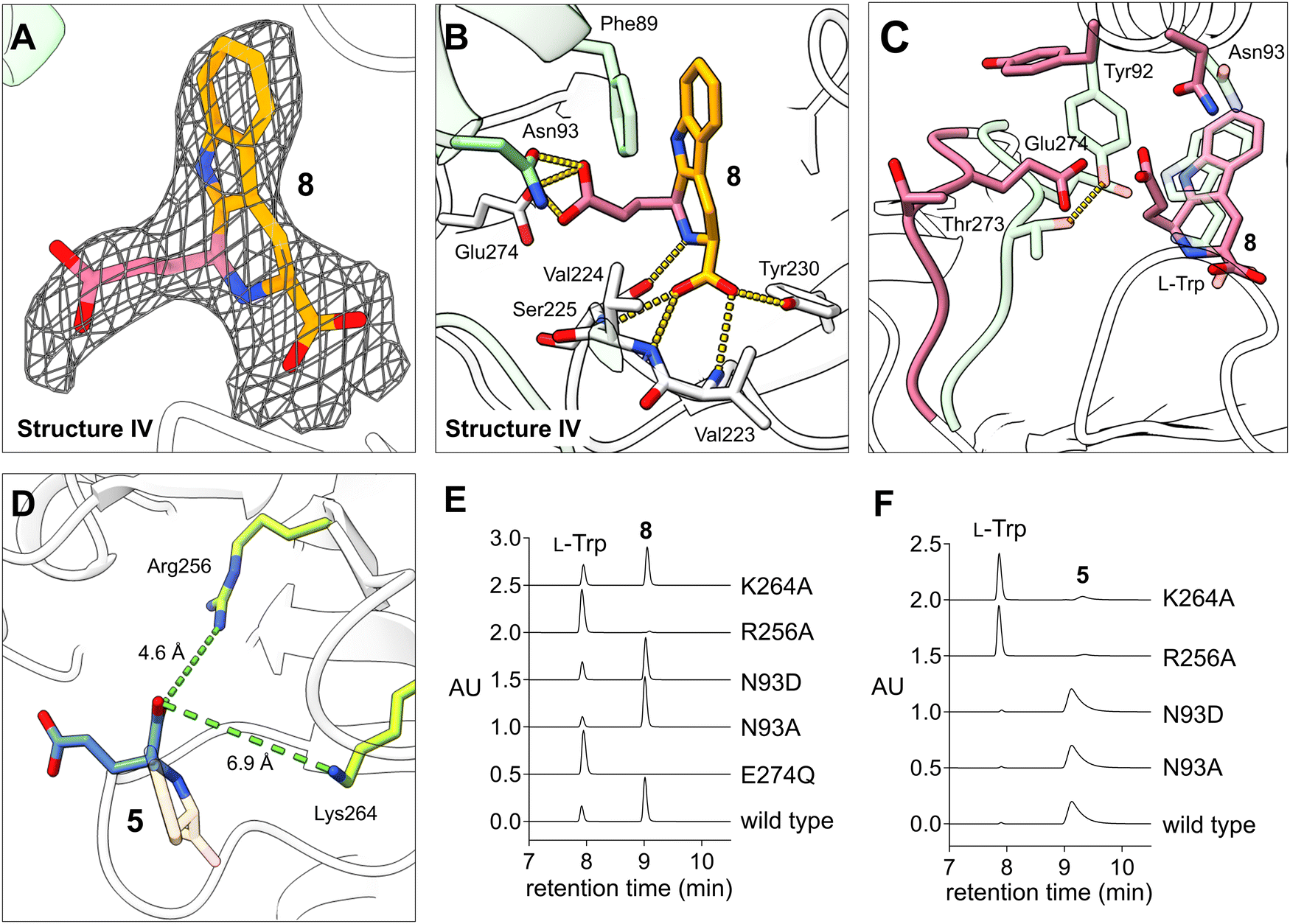 | ||
| Fig. 4 Structure of KslB·8 binary complex. (A) Electron density map shown around 8 within the active site of KslB. 2Fo–Fc map (gray mesh, contoured at 1.0σ) covers the stick model of 8. (B) Active site interactions observed between KslB and 8. Fragments of 8 derived from L-Trp and 7 are respectively colored yellow and pink. Hydrogen bonding interactions are shown in yellow dashed lines. The cartoon and stick models in white and green represent two KslB monomers within a dimer. (C) Superposed structures of KslB·8 (pink, structure IV) and KslB·L-Trp (pale green, structure II). The interaction between Tyr92 and Thr273 is shown in yellow dashed lines. (D) The modeled KslB·5 structure based on our KslB·8 structure (structure IV), which aligns with the soaked structure of KslB·5 reported by Renata and coworkers.16 The distances of Arg256 and Lys264 from the product in the product-trapping mode are shown. (E) and (F) HPLC analysis of the reactions of KslB mutants with L-Trp and (E) succinic semialdehyde (7) forming 8, or (F) α-KG forming 5. AU indicates absorbance units at 276 nm. | ||
As aforementioned, while we were preparing this manuscript, a paper deposited in ChemRxiv reported the crystal structure of KslB associated with 5 from the reaction with α-KG using crystal soaking.16 Their product-bound structure KslB·5 highly resembles our product-trapped structure of KslB·8 (see structure IV, Fig. 4A and B) where the loop region that contains Glu274 undergoes a ∼3 Å conformational shift in both cases and the C5′ carboxyl group derived from α-KG or 7 forms hydrogen bonds with Glu274 and Asn93 (Fig. 4B). Since the trapping of these two products involves an identical set of active site residues, we referred this mode of complex as the “product-trapping” mode.
Residues contributing to Pictet–Spengler reaction
Based on the product-trapping structures with either compound 516 or 8 (represented by structure IV), we investigated the catalytic significance of Glu274 and Asn93. Previous mutation studies identified Glu274 as a critical catalytic residue (Fig. 3C). Consistently, the E274Q variant failed to produce any product (8) when L-Trp and 7 were reacted (Fig. 4E). Surprisingly, mutation of Asn93 to alanine or even to the negatively charged aspartate had no effect on the reactions of L-Trp with either 7 or α-KG (Fig. 4E and F), suggesting that Asn93 is unlikely to play a catalytic role, despite the interactions found in product-trapping structures.Since the active site seems to be reasonably flexible judging from the observation of an alternative binding mode for substrate L-Trp (structures II and III, Fig. 3D), we suspected that the captured crystal structures of enzyme-product complex may simply represent a product-trapping state stabilized by the strongest interactions with residues which are not necessarily catalytically relevant. To identify functionally important residues for the Pictet–Spengler reaction, we opted to focus on residues near the active site that might undergo conformational changes upon substrate binding to interact with the reactants. Toward this goal, Arg256 and Lys264, two positively charged residues near the active site that may function as Lewis acid/base or act as additional binding ligands were selected (Fig. 4D). Notably, neither residue showed interactions in the product-trapping structures of KslB with 516 or 8 (see structure IV) (Fig. 4B and D). To assess their functional roles, Arg256 and Lys264 variants were generated and their enzymatic activities tested in vitro. Our results demonstrated that substituting Arg256 with alanine essentially abolished KslB activity to make either 5 or 8 (Fig. 4E and F). In contrast, the K264A variant significantly diminished production of 5, whereas its effect on the reaction with 7 was minimal (Fig. 4E and F). These results indicate that both residues are critical for KslB reaction.
Alternative structure of KslB·5 complex (structure V)
To further probe the roles of key residues Arg256 and Lys264, we next tried to capture an active binding mode of the KslB·L-Trp·α-KG complex by allowing for residue flexibility during structure analysis. We performed co-crystallization at room temperature by incubating the KslB·L-Trp complex with 5 mM α-KG for 3 h before setting up crystallization trays. The resulting crystals yielded X-ray diffraction data at 3.30 Å resolution. Despite the moderate resolution, the electron density at the active site was well-defined and consistent with the molecular shape of the reaction product (5) formed via the Pictet–Spengler reaction between L-Trp and α-KG (structure V, Fig. 5A and Fig. S5D, ESI†). Given the resolution limitation, we took a rigorous approach to interrogate the reliability of our structural model. We evaluated the electron density map quality and selected only four out of six molecules with strongest positive density at the active site for ligand modeling. We then independently built and refined the ligand models for each of these molecules. Several lines of evidence supported the reliability of our final KslB·5 co-crystallization model: First, the four independently modeled ligands adopted an identical conformation (Fig. S6D, ESI†). Second, the configuration of the C2′ stereocenter in product 5 was unambiguously determined by fitting and refining both S- and R-configured products into the omit map individually. Fitting of the R-configured product resulted in significant positive and negative difference densities (Fig. S8B, ESI†), whereas the S-configured model showed high consistency with the experimental map (Fig. S8A, ESI†). Third, we fitted the density using the product-trapping model (structure IV), which resembles soaked KslB·5 structure,16 and the calculated map exhibited significant deviation with both positive and negative difference density near 5 (Fig. S8C, ESI†), indicating a different configuration of the ligand in our structure (structure V, Fig. 5D). Additionally, the conformational shift of Glu274 observed upon product binding in the soaked structure16 (Fig. 4C) was absent in our co-crystallized KslB·5 structure, further highlighting the discrepancy of the structures obtained by co-crystallization versus those obtained using the soaking method (Fig. 5D and F). | ||
| Fig. 5 Structure of KslB·5 binary complex obtained from co-crystallization. (A) Electron density map shown around 5 and two interacting residues Arg256 and Lys264 within the active site of KslB. 2Fo–Fc map (gray mesh, contoured at 1.0σ) covers the stick model of 5, Arg256 and Lys264. (B) Superposed structures of the cocrystal of KslB·5 (blue, structure V) and KslB·L-Trp (pale green, structure II). (C) Active site interactions observed between KslB and 5 in the catalytic mode. Fragments of 5 derived from L-Trp and α-KG are respectively colored yellow and blue. Hydrogen bonding interactions are shown in yellow dashed lines. The cartoon and stick models in white and green represent two KslB monomers within a dimer. (D) Superimposition of 5 in the modeled KslB·5 structure (transparent sticks) and that in structure V (opaque sticks). (E) Salt bridge interactions between 5 and two positively charged residues, Arg256 and Lys264 (green sticks) in structure V. (F) Superimposition structures of KslB·5 from experimental observations in the catalytic mode (structure V, blue) and that from the modeled KslB·5 structure (pink). | ||
A closer examination of this new KslB·5 structure (structure V) revealed that the fragment derived from L-Trp in 5 remained in the same position as those observed in both structures II and IV (Fig. 3B, 4B and 5B). With most interactions preserved for the co-crystallized or soaked KslB·5 structures, the carboxylate groups derived from α-KG, however, are positioned rather differently in the two structures (Fig. 5D). Most notably, strong salt bridge interactions are formed between the C1′ and C5′ carboxylate moieties of 5 and two positively charged residues, Lys264 and Arg256, respectively in our co-crystallized KslB·5 structure (Fig. 5E). Such positioning of the carboxylate groups in α-KG nicely explains the biochemical assay results of the mutants (Fig. 4E and F), which was unaccounted for using the product-trapping structures (soaked structure KslB·516 and our KslB·8 complex). As shown in our co-crystallization KslB·5 structure (Fig. 5E), Arg256 forms salt bridge with the C5′ carboxylate group of 5 and very likely with the C5′ carboxylate group of 8. Consistently, the R256A mutation abolished all enzymatic activity for using both α-KG and succinic semialdehyde (7) in the reaction, because its elimination prevents the binding of both co-substrates. In contrast, Lys264 specifically interacts with the second carboxylate group unique to α-KG in this active configuration (Fig. 5E) but lacking in 7. Consequently, the K264A mutation impaired the reaction with α-KG but had no effect when 7 was used as the co-substrate. It should be noted that Arg256 and Lys264 in KslB are not conserved in McbB (Fig. S7B, ESI†). The absence of this conservation in McbB is consistent with its use of oxaloacetaldehyde (1) as the substrate, which differs structurally from α-KG. It is therefore plausible that McbB utilizes a different set of residues for carbonyl substrate recognition.
Besides, in this active configuration identified under co-crystallization conditions, Asn93 is positioned ∼12 Å far from the C5′ carboxylate group of 5, again consistent with the biochemical assay results that it does not participate in catalysis. Instead, the Tyr92-Thr273 hydrogen bond is restored compared to product-trapping mode (Fig. 5F). Overall, the binding mode observed with our new KslB·5 structure is more likely catalytically relevant since it is consistent with the biochemical findings. Hence, we denoted it as the “catalytic” mode.
Implication of mechanism
Based on the aforementioned five KslB structures captured under different conditions, we tried to understand the reaction mechanism using AutoDock Vina18,19 to computationally dock the imine intermediate (9) formed between L-Trp and α-KG in the active site. In the docking trials, the conformation of the substrate L-Trp is not restraint so that all possible binding modes will be explored. The resulting docked structure of 9 exhibits high confidence with the molecule occupying the active site cavity of KslB (Fig. 6A). Significantly, the binding pose of the indole ring in the imine intermediate (9) is consistent with that in the alternative substrate binding mode observed in structure III (Fig. 3D). In contrast, adaptation of the tryptophan binding mode in structure II or the substrate binding complex reported by Renata and coworkers16 leads to steric clashes between indole and the imine moiety (Fig. S9, ESI†). | ||
| Fig. 6 Mechanistic investigation of KslB-catalyzed Pictet–Spengler reaction. (A) Computationally docked structure of the imine intermediate (9). Fragments of 5 derived from L-Trp and α-KG are respectively colored yellow and blue. Hydrogen bonding interactions are shown in yellow dashed lines. White stick models represent the KslB residues that interact with 9. (B) Proposed nucleophilic attack from the si face of the indole ring to the si face of the imine carbon (C2′). (C) Proposed binding orientation of the carbocation intermediate (10). Distances between the catalytic Glu274 and the C2 hydrogen are shown as green dashed lines. (D) Proposed mechanism of the KslB-catalyzed Pictet–Spengler reaction. | ||
The docked model is consistent with a mechanism where the tight anchoring of the C1′ and C5′ carboxylate groups of 9 by Lys264 and Arg256 allows the C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C3 double bond of the indole ring to attack C2′ only from its si face, yielding the S-configuration at C2′ (Fig. 6B). In contrast, attack from the re face at C2′ is prohibited due to the steric clash between Lys264 and the C5′ carboxylate group (Fig. S10A, ESI†). These stereochemical restrictions are consistent with the stereochemistry of 5 observed in structure V (Fig. 5C and E) and the stereochemical characterization recently reported by Renata and coworkers.16 The KslB reaction of L-Trp with 7 likely proceeds via the same stereochemical course to generate only the S-configured stereocenter in product 8 as observed in the case of α-KG (Fig. S10B, ESI†).
C3 double bond of the indole ring to attack C2′ only from its si face, yielding the S-configuration at C2′ (Fig. 6B). In contrast, attack from the re face at C2′ is prohibited due to the steric clash between Lys264 and the C5′ carboxylate group (Fig. S10A, ESI†). These stereochemical restrictions are consistent with the stereochemistry of 5 observed in structure V (Fig. 5C and E) and the stereochemical characterization recently reported by Renata and coworkers.16 The KslB reaction of L-Trp with 7 likely proceeds via the same stereochemical course to generate only the S-configured stereocenter in product 8 as observed in the case of α-KG (Fig. S10B, ESI†).
Likewise, the nucleophilic attack initiated from the C2C3 double bond of 9 should proceed from the si face of the indole ring as the attack from its re face requires the indole ring to rotate resulting in clashes with Phe89 (Fig. S11, ESI†). Consequently, the C2 and C2′ of the cyclized product (10) have the R- and S-stereoconfiguration, respectively (state III, Fig. 6D). The catalytic Glu274 can then act as a general base to remove the C2 proton in 10 given their proximity to afford 5 (Fig. 6C and state III to state IV in 6D).
Conclusion
In summary, we solved five crystal structures of the bacterial Pictet–Spenglerase KslB, including the apo structure and four complex structures comprising two different conformations of KslB·L-Trp complexes, a KslB·8 complex, and a KslB·5 complex. Analysis of the active site revealed a putative catalytic glutamate residue Glu274, which completely deactivates KslB upon mutation. Moreover, upon co-crystallization with NOG, we identified an additional hydrophobic pocket that accommodates the indole moiety in an alternative binding mode.Importantly, our KslB·5 structure reveals a different binding pose of the product from that in our KslB·8 structure which is similar to the recently reported KslB·5 structure obtained by crystal soaking method.16 We propose that the two binding poses represent different stages in the catalytic cycle: one pose right after the reaction (KslB·5 co-crystal, i.e., structure V), and the other when the product exits the active site (KslB·8 co-crystal structure, i.e., structure IV and the KslB·5-soaked structure16). When L-Trp is introduced to the active site or when 5 is generated inside the active site cavity, KslB is in its ‘closed’ conformation governed by the interaction between Thr273 and Tyr92 in the dimer assembly partner, which resembles the catalytically active state of the enzyme (Fig. 5F). In contrast, KslB adopts an ‘open’ conformation characterized by a shift in Glu274 and loss of the Thr273–Tyr92 interaction when the reaction product (5) is artificially introduced into the enzyme by crystal soaking16 or when 8 is generated and exits the active site (Fig. 4C).
Investigation of the KslB·5 co-crystal structure in a closed enzyme conformation revealed two catalytically essential residues, Arg256 and Lys264 that specifically recognize the carboxylate groups derived from α-KG. Combined with the docking study, it is suggested that the α-KG moiety in 9 is tightly anchored by two salt bridge interactions mediated by Arg256 and Lys264, which position the si face of C2′ iminium carbon ready for cyclization with the indole moiety from its si face (state II, Fig. 6D). Hence, a carbocation intermediate (10) with two defined stereocenters (R-configuration in C2 carbon and S-configuration in C2′ iminium carbon) is produced (state III, Fig. 6D). The Glu274 is in position to deprotonate the C2 hydrogen generating the final product. Collectively, this work provides substantial insights into the mechanisms of bacterial Pictet–Spenglerases and paves the way for future enzyme engineering to develop biocatalysts for Pictet–Spengler reactions.
Data availability
The crystallographic data supporting this study have been deposited in the Protein Data Bank (PDB) under the following accession codes: KslB apo structure (structure I): PDB 9NS6, available at https://www.rcsb.org/structure/9NS6. KslB·L-Trp complex (structure II): PDB 9NSC, available at https://www.rcsb.org/structure/9NSC. KslB·L-Trp complex with an alternative binding pose (structure III): PDB 9NSS, available at https://www.rcsb.org/structure/9NSS. KslB·8 complex (structure IV): PDB 9NSU, available at https://www.rcsb.org/structure/9NSU. KslB·5 complex (structure V): PDB 9NST, available at https://www.rcsb.org/structure/9NST.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank funding for NIH (R35GM148356 for Y. J. Z. and R35GM153203 for H.-w. L.). Crystallographic data collections were conducted at Advanced Light Source (BCSB - BL 8.2.2), and Advanced Photon Source (BL23-ID-B and BL24-ID-E), Department of Energy scientific user facility at Lawrence Berkeley National Laboratory and Argonne National Laboratory.References
- J. Stockigt, A. P. Antonchick, F. Wu and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 8538–8564 CrossRef CAS PubMed.
- K. M. Czerwinski, L. Deng and J. M. Cook, Tetrahedron Lett., 1992, 33, 4721–4724 CrossRef CAS.
- J. Li, T. Wang, P. Yu, A. Peterson, R. Weber, D. Soerens, D. Grubisha, D. Bennett and J. M. Cook, J. Am. Chem. Soc., 1999, 121, 6998–7010 CrossRef CAS.
- M. S. Reddy and J. M. Cook, Tetrahedron Lett., 1994, 35, 5413–5416 CrossRef CAS.
- C. Gremmen, B. Willemse, M. J. Wanner and G. J. Koomen, Org. Lett., 2000, 2, 1955–1958 CrossRef CAS PubMed.
- R. Roddan, J. M. Ward, N. H. Keep and H. C. Hailes, Curr. Opin. Chem. Biol., 2020, 55, 69–76 CrossRef CAS PubMed.
- J. Brevet, D. Borowski and J. Tempé, Mol. Plant-Microbe Interact., 1988, 1, 75–79 CrossRef.
- K. Suzuki, N. Tanaka, H. Kamada and I. Yamashita, Gene, 2001, 263, 49–58 CrossRef CAS PubMed.
- Q. Chen, C. Ji, Y. Song, H. Huang, J. Ma, X. Tian and J. Ju, Angew. Chem., Int. Ed., 2013, 52, 9980–9984 CrossRef CAS PubMed.
- Q. Chen, S. Zhang and Y. Xie, J. Biotechnol., 2018, 281, 137–143 CrossRef CAS PubMed.
- X. Wang, D. Kong, T. Huang, Z. Deng and S. Lin, Org. Biomol. Chem., 2018, 16, 9124–9128 RSC.
- K. Koketsu, K. Watanabe, H. Suda, H. Oguri and H. Oikawa, Nat. Chem. Biol., 2010, 6, 408–410 CrossRef CAS PubMed.
- T. Mori, S. Hoshino, S. Sahashi, T. Wakimoto, T. Matsui, H. Morita and I. Abe, Chem. Biol., 2015, 22, 898–906 CrossRef CAS PubMed.
- X. Ma, S. Panjikar, J. Koepke, E. Loris and J. Stöckigt, Plant Cell, 2006, 18, 907–920 CrossRef CAS PubMed.
- Z. Zheng, H. Choi and H.-W. Liu, J. Am. Chem. Soc., 2024, 146, 28553–28560 CAS.
- R. Jiang, N. Wasfy, T. Mori, M. Hoang, I. Abe and H. Renata, ChemRxiv, 2025, preprint DOI:10.26434/chemrxiv-2025-fvct3.
- E. Krissinel and K. Henrick, J. Mol. Biol., 2007, 372, 774–797 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cb00070j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |