
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and catalytic testing of the first hydrophilic derivative of Shvo's catalyst†
Justus
Diekamp
 a,
Annika
Schmidt
b,
Julian J.
Holstein
b,
Carsten
Strohmann
b and
Thomas
Seidensticker
*a
a,
Annika
Schmidt
b,
Julian J.
Holstein
b,
Carsten
Strohmann
b and
Thomas
Seidensticker
*a
aTU Dortmund University, Department for Biochemical and Chemical Engineering, Laboratory of Industrial Chemistry, Emil-Figge-Straße 66, 44227 Dortmund, Germany. E-mail: thomas.seidensticker@tu-dortmund.de
bTU Dortmund University, Inorganic Chemistry, Otto-Hahn Str. 6, 44227 Dortmund, Germany
First published on 27th November 2024
Abstract
Although commonly applied in various reactions, Shvo's catalyst has not been modified towards solubility in highly polar solvents until now. Here, we report the straightforward synthesis of a disulfonate derivative of the complex, which allows to (transfer) (de-)hydrogenate aldehydes and ketones in aqueous solutions. A proof of principle for the recycling of the catalyst is also provided.
The late-stage introduction of sulfonate groups into ligands to gain hydrophilic complex catalysts is a commonly applied strategy in the research of recyclable homogeneous catalysts.1–5 Usually, sulfonation is achieved with highly acidic and oxidising reagents (H2SO4, Oleum, and chloro sulphuric acid), but not all ligands are stable enough to survive such conditions. This is most likely the reason why there has not been a sulfonated derivative of Shvo's catalyst,6,7 a commonly applied and versatile complex catalyst,8–10 until now.
In 2015, Holdcroft and coworkers disclosed a mild sulfonation protocol for tetraphenylcyclopentadienone (TPCP) furnishing the 4,4′-disulfonic acid.11 According to their procedure, we sulfonated the commercially available and inexpensive TPCP (1), the main ligand of Shvo's catalyst, with trimethylsilyl chlorosulfonate (3). The resulting disulfonic acid (4) was dissolved in EtOH. 2.5 equivalents of NaOH dissolved in EtOH were added to the solution of the acid (Scheme 1). The disodium sulfonate precipitated instantly, was filtered off and dried in a vacuum oven at 100 °C, yielding a grey-violet solid. Additional attempts to access a tetrasulfonated species of the ligand with sulfuric acid and chlorosulfuric acid were unsuccessful and resulted in the decomposition of TPCP (1). In order to obtain the isolated, sulfonated derivative of Shvo's catalyst, ligand 5 was refluxed with trirutheniumdodecacarbonyl (Ru3(CO)12) in MeOH for 60 h (Scheme 1). This synthesis route is based on the simplified complex synthesis established by Casey's12 and Williams’ groups8 for the original complex 2 and a tolyl substituted derivative. While at the beginning of the reaction, the almost insoluble Ru3(CO)12 was suspended in a dark violet solution of the ligand, the reaction mixture turned into a clear orange solution after 60 h. The solution was concentrated in vacuo, resulting in rapid crystallisation of an orange solid. The remaining solvent was also removed in vacuo, and the residual orange solid was washed with diethyl ether. The product was dried under a high vacuum. Slow recrystallisation of the solid from MeOH yielded yellow crystals suitable for analysis via X-ray diffraction. The complex’ (7) molecular structure (Fig. 1 and 2) matches the known dimeric structures of the derivatives of Shvo's complex.7,13,14
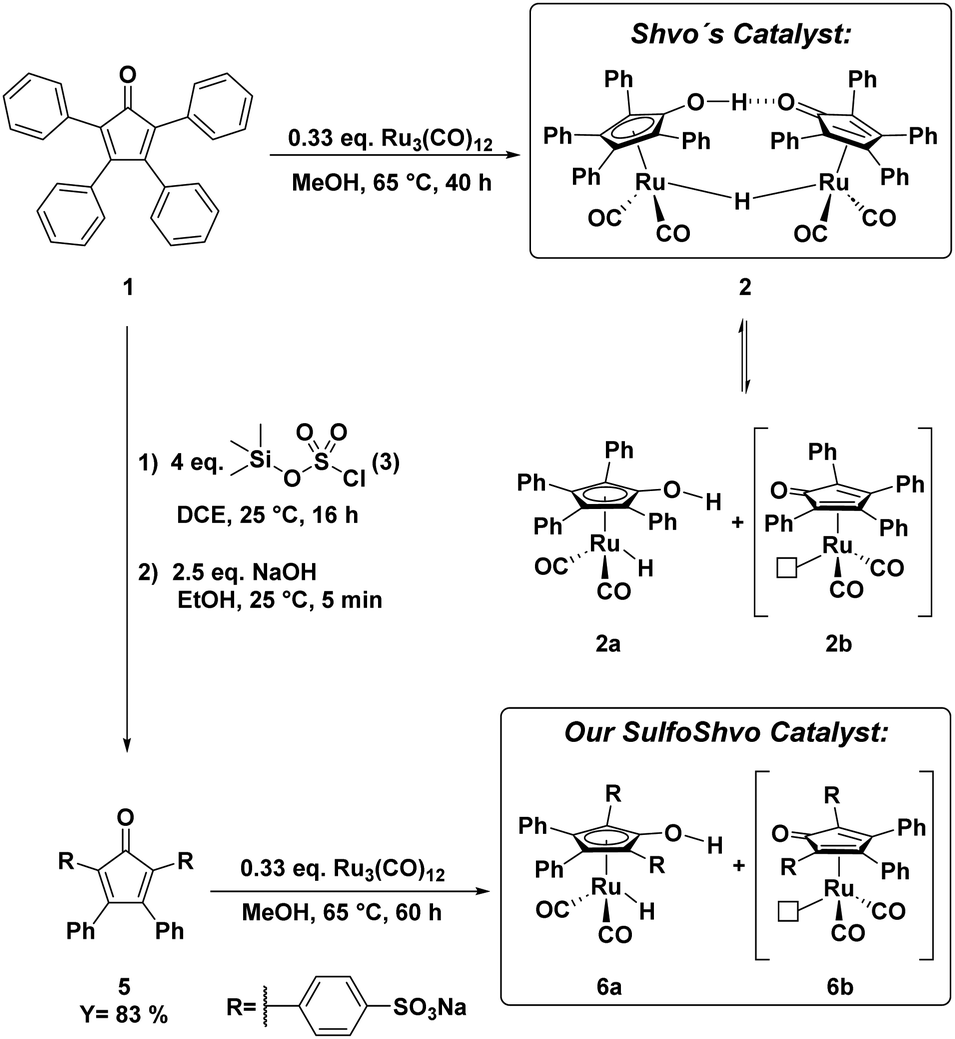 | ||
| Scheme 1 Synthesis and structure of Shvo's catalyst (2) and our novel sulfoShvo catalyst (6a + 6b).8,11,12 | ||
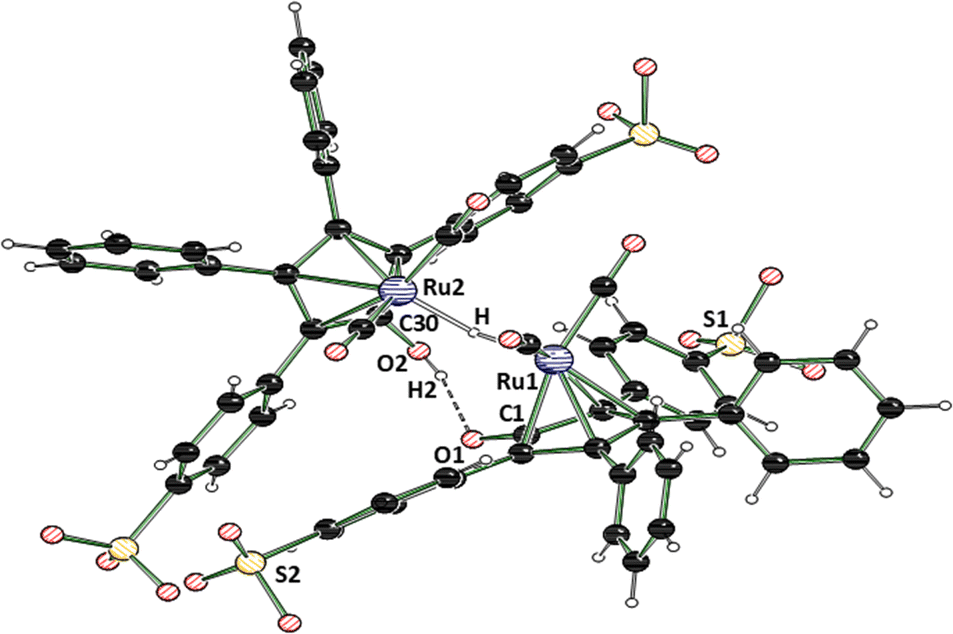 | ||
| Fig. 1 Molecular structure of the crystallised dimeric hydride species 7. To improve clarity, only one Ru dimer of the asymmetric unit is shown. Sodium cations and MeOH molecules are omitted for clarity. For more detailed data, please see the ESI.† CCDC deposition number 2336908.† Selected distances [Å] and angles [°]: Ru1–H 1.72(7) Å, O1–C1 1.287(4) Å, O2–C30 1.300(4) Å, Ru1–Ru2 3.1758(4) Å, Ru1–H–Ru2 140(4)°.15 | ||
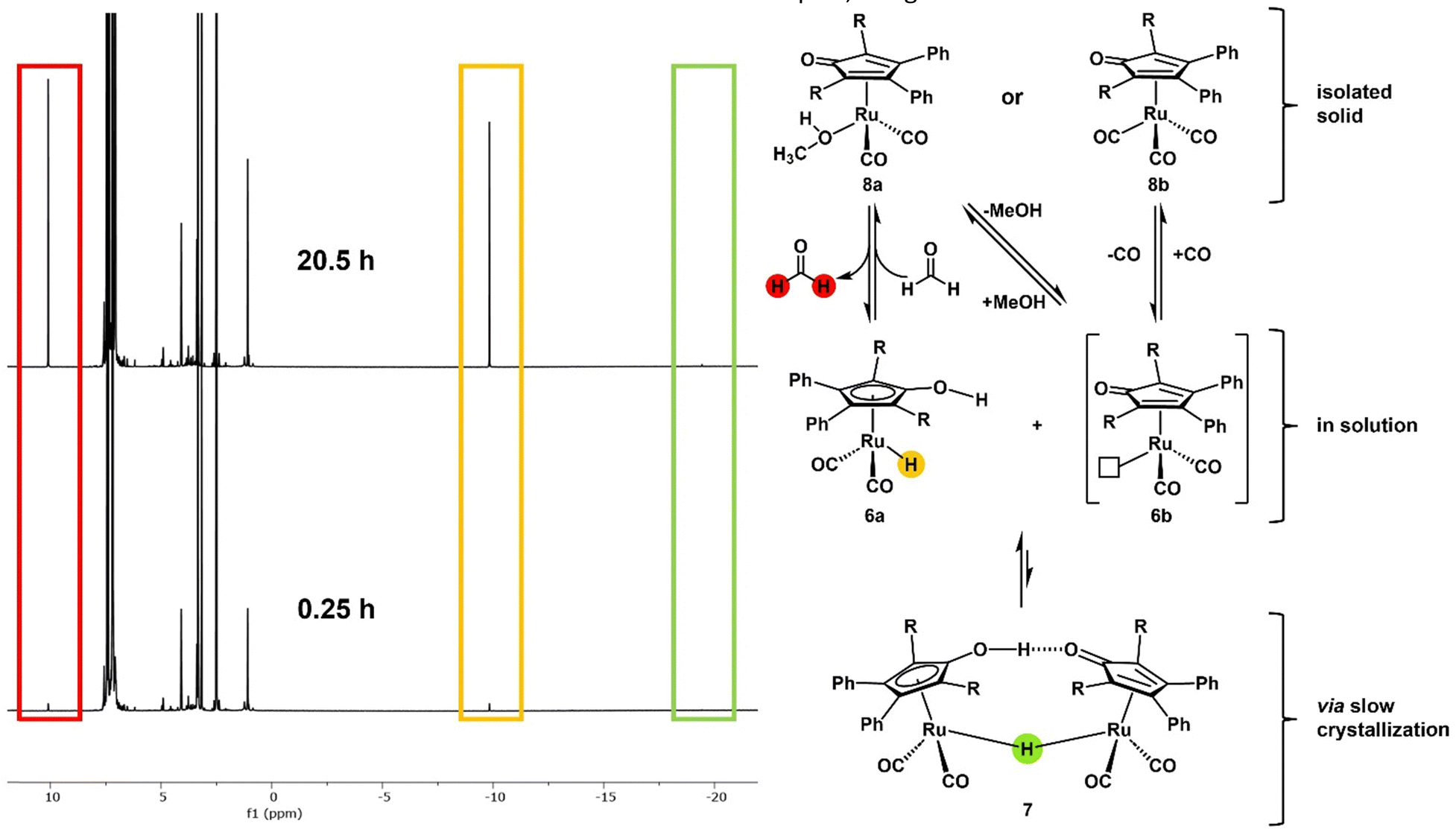 | ||
| Fig. 2 Proposed formation of the catalytically active monomeric hydride species 6avia the dehydrogenation of coordinated and/or residual methanol from the crystals in DMSO-d6 at 25 °C. The 1H NMR spectra show the formation of two singlets for formaldehyde (red) and the monomeric hydride 6a (yellow) over 20.5 h. The spectra show minimal to no formation of the dimeric hydride 7 (green). R = sodium p-phenylsulfonate. | ||
Although the crystal structure shows the existence of the dimeric complex, NMR and IR studies of the gained solid cannot support this finding. Fábos et al. have shown the formation of the catalytically active monomeric hydride species by NMR studies. While the bridging hydride appears at −18.14 ppm in the 1H NMR spectrum, the monomeric hydride complex (1a) displays a signal at −9.73 ppm (in toluene-d8).16 To avoid dissociation and subsequent hydride formation through dehydrogenation of the solvent, we performed NMR experiments on the obtained complex in DMSO-d6. Complex 6a showed a signal at −9.83 ppm in the 1H NMR spectrum that indicates the formation of the monomeric hydride complex. A minimal signal at −19.44 ppm was detected (see ESI†), which can be attributed to the dimeric hydride complex 7. The repeated measurement of the NMR spectrum in DMSO-d6 over 20.5 h showed that the monohydride complex 6a is formed over time (Fig. 2) (see ESI† for detailed NMR data). Since DMSO cannot provide hydrogen to the complex and no relevant amount of the dimeric hydride species can be found at any time, the monomeric hydride must be traced back to the dehydrogenation of residual methanol (which was found to co-crystallise in the crystalline solid state) or coordinated methanol (Fig. 2, complex 8a). This hypothesis was supported by the 1H-NMR spectrum that shows the formation of a formaldehyde signal (1H δ = 10.10 ppm) (Fig. 2). Furthermore, the solid-state IR spectrum shows two distinct bands at 2088 cm−1 and 2020 cm−1, a more similar pattern to the IR bands of the literature-known monomeric derivatives rather than the dimeric species as shown in an in situ IR study of the complex dissociation.16 HRMS of the orange solid in MeOH provided only the mass of the monomeric hydride species (6a) (calcd [M-2Na]2− = 350.9805, found [M-2Na]2− = 350.9782). It is important to note that while the monohydride species of our complex is analogue to the monohydride of Shvo's catalyst it already forms at 25 °C while Shvo's complex requires higher temperatures (>70 °C)16,17 to dissociate into the monomeric species. The reasons for the increased presence of the monomeric species in solution could be the electron-withdrawing effect of the sulfonate moieties or the increased steric demand of the sulfonated phenyl rings facing the centre of the potential dimeric species 7. Although the various analytical methods yield different outcomes and interpretations of the molecular structure of our obtained complex, the general structural motifs of Shvo's complex can be found in our sulfonated derivative. Possible alternative molecular structures for the isolated solid could be a tricarbonyl piano stool complex (8b) or an analogous species in which methanol substitutes a carbonyl ligand in coordination with the ruthenium (8a) (Fig. 2). Since we cannot determine the structure of our orange complex with certainty and most likely have residual MeOH co-crystallised in the solid state, for subsequent experiments and catalyst weighing, we proceeded to measure the ruthenium content in our solid via ICP-OES, based on the assumption that every possible complex species will form the active monomeric hydride complex when presented with a hydrogen source at reaction temperature. The potential of polar-tagged homogeneous catalysts lies in various applications. First, they allow the conversion of highly polar substrates in environmentally benign solvents such as water or ethanol. Second, catalyst separation (and potentially recycling) via extraction becomes an option. Finally, it opens a way towards tandem catalysis with catalysts which require polar solvents. The (transfer) hydrogenation of levulinic acid (LA) (9) to 4-hydroxy valeric acid (10) with subsequent formation of γ-valero lactone (11) (GVL) has already been published with Shvo's complex as a catalyst and different hydrogen sources.16,18 Levulinic acid possesses a high solubility in water and is, therefore, an ideal substrate for first monophasic reactions in an aqueous phase. A comparison with the original complex showed sulfoShvo's (transfer) hydrogenation capability in general but also reduced catalytic activity compared to Shvo's catalyst (Table S1 and ESI†). The reason for this is most likely the electron-withdrawing effect of the sulfonate groups which results in an electron poorer Ru centre. Since Mazzoni and coworkers reported the rapid microwave-assisted synthesis of Shvo-type complexes,14 we tried to generate our catalyst system in situ in a microwave synthesizer. The obvious change of colour of the solution from dark violet to orange and the results of the subsequent transfer hydrogenation (Table 1, entries 4 and 5) indicate a successful formation of our catalyst. Apart from the first “accidental” reactions with Shvo's complex6 and our publication with a tandem catalytic set-up,19 we are not aware of the use of in situ-generated Shvo-type catalysts. It is important to note, that an in situ generated catalyst system can circumvent the difficulties regarding the Ru-equivalents in the not fully characterised, isolated orange solid. Sulfonated ligands are often specially designed for liquid–liquid biphasic reactions where they retain the catalyst complex in a polar liquid phase. To assess our catalyst's capability to perform catalysis in such a system, the hydrogenation of undecanal (12) to undecanol (13) – both nearly insoluble in water – with an aqueous catalyst phase was chosen. A first run showed the necessity of mediation by randomly methylated (RAME) β-cyclodextrins (CD) (see ESI†). Based on this, undecanal was successfully hydrogenated followed by recycling of the catalyst phase and a second successful undecanal hydrogenation (Scheme 2).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | H2-source | Cat. | Solvent | Reaction set-up | t [h] | T [°C] | Y GVL [%] |
| a GC yield. b In situ catalyst formation in microwave (MW) (110 °C, 30 min). | |||||||
| 1 | H2 (30 bar) | SulfoShvo | H2O | Pressure autoclave | 37 | 100 | 98 |
| 2 | H2 (30 bar) | Shvo | Toluene | Pressure autoclave | 4 | 100 | 97 |
| 3 | FA | SulfoShvo | H2O | Reflux | 43 | 100 | 90 |
| 4 | IPA | SulfoShvo | H2O | MWb | 2 | 110 | 21 |
| 5 | IPA | Shvo | Toluene | MWb | 2 | 110 | 41 |

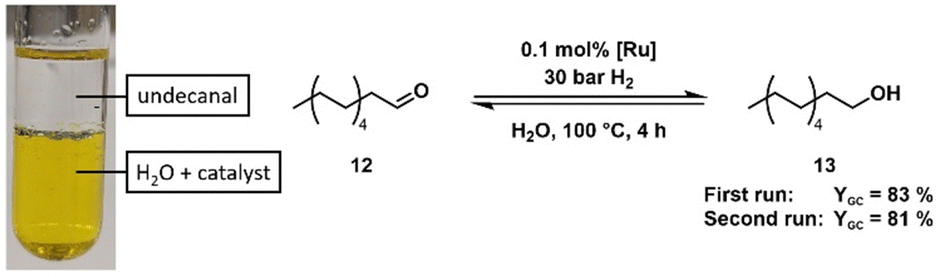 | ||
| Scheme 2 Hydrogenation of undecanal (12) catalysed by the sulfoShvo catalyst and mediated by RAME-β-cyclodextrins (CD)in two consecutive runs. | ||
Shvo's catalyst has also been successfully applied in the dynamic kinetic resolution of racemic mixtures of alcohols20 and amines.21,22 Its capability to convert an alcohol into a ketone and back again without a selectivity towards one enantiomer is a great asset when combined with an enantioselective enzyme catalyst like a lipase.23 While the lipase's affinity for non-polar solvents is advantageous when one is working with non-polar substrates and Shvo's catalyst, many enzymes require/prefer water as a solvent. This again favours our novel hydrophilic variant. Before such an orthogonal system can be developed, we want to show here that the catalyst is able to racemise a model substrate in an aqueous biphasic system. To prove this, sulfoShvo was successfully applied in the racemisation of (R)-1-phenylethanol ((R)-14) in a biphasic reaction, which consisted of a solution of 1 mol% catalyst in water and a pure substrate/product phase (Scheme 3).
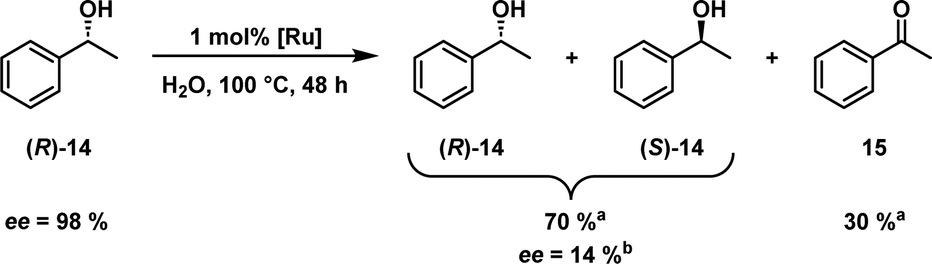 | ||
Scheme 3 Biphasic aqueous racemisation of (R)-1-phenyethanol ((R)-14) catalysed by the sulfoShvo catalyst in water at 100 °C. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) GC-FID area percent. bBased on chiral HPLC area percent. GC-FID area percent. bBased on chiral HPLC area percent. | ||
After 48 h the ee decreased to 14% while simultaneously a considerable amount of 30% acetophenone (15) was found. The remaining excess can be attributed to the mass transport limitations in the biphasic reaction system. The loss of H2 between the dehydrogenation and hydrogenation steps is most likely a result of the open reaction set-up with a reflux condenser.
In conclusion, we developed a simple synthesis route based on inexpensive synthons to gain the first sulfonate derivative of Shvo's complex, readily soluble in highly polar solvents. X-ray diffraction, NMR, IR, and HRMS studies confirmed the complex's general structural motif. The catalyst was successfully applied in the monophasic (transfer) hydrogenation of levulinic acid (pressure autoclave, classic reflux, and microwave assisted), the biphasic, cyclodextrin-mediated hydrogenation of undecanal including catalyst recycling, and the racemisation of (R)-1-phenylethanol. Additionally, we established its use in an in situ-generated catalyst system. Further studies investigating the long-term stability of the complex and a viable catalyst recycling are on the way. To fully examine the catalyst's potential, we will happily provide other research groups with the ligand and/or the complex if they are interested in working with this new system.
The authors thank the network program ‘Sustainable Chemical Synthesis 2.0’ (SusChemSys 2.0) for the support and fruitful discussions between the disciplines. Finally, we would also like to thank the German Federal Ministry of Food and Agriculture (Bundesministerium für Ernährung und Landwirtschaft), represented by the FNR (Fachagentur Nachwachsende Rohstoffe) for the financial support of the junior research group ‘Renewlysis’ (project number 2219NR355). Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 421173804.
Data availability
Crystallographic data for compound 7 has been deposited at the Cambridge Crystallographic Data Centre (CCDC) with the accession number 2336908,† which can be accessed at https://www.ccdc.cam.ac.uk/. Materials, instrumentation details, synthesis procedures, and additional data are provided in the ESI,† accompanying this article.Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Diekamp and T. Seidensticker, Angew. Chem., Int. Ed., 2023, e202304223 CAS.
- W. A. Herrmann and C. W. Kohlpaintner, Angew. Chem., Int. Ed. Engl., 1993, 32, 1524–1544 CrossRef.
- T. Schlatzer and R. Breinbauer, Adv. Synth. Catal., 2021, 363, 668–687 CrossRef CAS.
- K. H. Shaughnessy, Chem. Rev., 2009, 109, 643–710 CrossRef CAS PubMed.
- F. Lin and S. Mecking, Angew. Chem., Int. Ed., 2022, 61, e202203923 CrossRef CAS PubMed.
- Y. Blum and Y. Shvo, Isr. J. Chem., 1984, 24, 144–148 CrossRef CAS.
- Y. Shvo, D. Czarkie, Y. Rahamim and D. F. Chodosh, J. Am. Chem. Soc., 1986, 108, 7400–7402 CrossRef CAS.
- B. L. Conley, M. K. Pennington-Boggio, E. Boz and T. J. Williams, Chem. Rev., 2010, 110, 2294–2312 CrossRef CAS.
- A. M. Afanasenko, X. Wu, A. de Santi, W. A. M. Elgaher, A. M. Kany, R. Shafiei, M.-S. Schulze, T. F. Schulz, J. Haupenthal, A. K. H. Hirsch and K. Barta, Angew. Chem., Int. Ed., 2024, 63, e202308131 CrossRef CAS.
- N. Menashe, E. Salant and Y. Shvo, J. Organomet. Chem., 1996, 514, 97–102 CrossRef CAS.
- T. J. G. Skalski, B. Britton, T. J. Peckham and S. Holdcroft, J. Am. Chem. Soc., 2015, 137, 12223–12226 CrossRef CAS.
- C. P. Casey, S. W. Singer, D. R. Powell, R. K. Hayashi and M. Kavana, J. Am. Chem. Soc., 2001, 123, 1090–1100 CrossRef CAS PubMed.
- C. P. Casey, J. B. Johnson, S. W. Singer and Q. Cui, J. Am. Chem. Soc., 2005, 127, 3100–3109 CrossRef CAS PubMed.
- C. Cesari, L. Sambri, S. Zacchini, V. Zanotti and R. Mazzoni, Organometallics, 2014, 33, 2814–2819 CrossRef CAS.
- CCDC Deposition number 2336908† (7).
- V. Fábos, L. T. Mika and I. T. Horváth, Organometallics, 2014, 33, 181–187 CrossRef.
- O. Verho and J.-E. Bäckvall, J. Am. Chem. Soc., 2015, 137, 3996–4009 CrossRef CAS PubMed.
- C. A. M. R. van Slagmaat, M. A. F. Delgove, J. Stouten, L. Morick, Y. van der Meer, K. V. Bernaerts and S. M. A. de Wildeman, Green Chem., 2020, 22, 2443–2458 RSC.
- M. R. L. Furst, V. Korkmaz, T. Gaide, T. Seidensticker, A. Behr and A. J. Vorholt, ChemCatChem, 2017, 9, 4319–4323 CrossRef CAS.
- A. L. E. Larsson, B. A. Persson and J.-E. Bäckvall, Angew. Chem., Int. Ed. Engl., 1997, 36, 1211–1212 CrossRef CAS.
- O. Pàmies, A. H. Éll, J. S. Samec, N. Hermanns and J.-E. Bäckvall, Tetrahedron Lett., 2002, 43, 4699–4702 CrossRef.
- J. Paetzold and J. E. Bäckvall, J. Am. Chem. Soc., 2005, 127, 17620–17621 CrossRef CAS PubMed.
- F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12–22 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2336908. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc04390a |
| This journal is © The Royal Society of Chemistry 2025 |