
The effect of a distorted MO6 octahedral unit on the activity and stability for the oxygen evolution reaction†
Haixiang
Yang‡
,
Xinran
Ning‡
,
Wenjun
Yan
,
Hua Gui
Yang
 and
Haiyang
Yuan
*
and
Haiyang
Yuan
*
Key Laboratory for Ultrafine Materials of Ministry of Education, Shanghai Engineering Research Center of Hierarchical Nanomaterials, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: hyyuan@ecust.edu.cn
First published on 22nd November 2024
Abstract
Metal oxides have garnered significant attention in the oxygen evolution reaction (OER), where a distorted MO6 octahedral unit has a marked impact on their performance. This study introduces the distortion coefficient to quantify the MO6 distortion composed of its volume and shape, and reports the effect of MO6 distortion on the activity and stability in rutile metal oxides for the OER. This provides a fundamental understanding for designing effective MO6 active units for the OER.
Hydrogen (H2) fuel has emerged as a potential solution to the prevailing energy conundrum.1 The H2 generation via water electrolysis is an exceptionally meritorious and efficient modality;2 however, its industrial application remains limited by the shortage of effective catalysts for the oxygen evolution reaction (OER).3 Metal oxides have garnered significant attention for the OER due to their modifiability and easy availability,4 such as nickel,5 manganese,6 cobalt,7 ruthenium and iridium oxides.8 Nonetheless, with the exception of precious iridium-based oxides, most of them are largely impeded in industry due to their undesirable activity or stability, particularly under harsh acidic conditions. It is strongly desired to develop efficient and inexpensive metal oxide catalysts for the OER.
Metal octahedral units (MO6) are typical active structures that are preferentially exposed on the surface of metal oxides.9 A slight structural change of the MO6 unit (i.e., octahedral distortion) has a significant effect on the OER activity and stability of the corresponding catalysts.10 Modifying the MO6 unit has become an effective strategy for designing high-performance metal oxide catalysts for the OER, for which various approaches have been proposed. For instance, the incorporation of Na and Ho into SrRuO3 and Y2RuO7 can weaken the octahedral distortion of the RuO6 unit, which in turn enhances the strength of the Ru–O bond, thus optimizing the adsorption energy of key oxygen-containing intermediates and facilitating the OER.11 The Ru incorporation into the SnO2 matrix introduces tensile strain in the RuO6 unit, which results in a weakened Ru–O bond, thereby significantly improving the long-term stability.12 Similarly, the distortions of CoO6 and IrO6 octahedrons have been reported for their effects on the OER.13 Obviously, the OER activity and stability of the MO6 active units in metal oxides are usually related to their distortion degree. Despite these findings, there is no comprehensive understanding of the relations between the distortion degree of the MO6 unit and the OER activity or stability. Therefore, one fundamental question is necessarily answered: How do different MO6 units distort to influence their OER activity and stability?
Aiming to address the above issue, we selected the representative non-iridium metal (M = Co, Ni, Ru) sites incorporated in different rutile metal oxides as the basic research objects, which are generally used as the active site models for the OER.13b,14 We introduced the distortion coefficient (D), which is composed of its volume and shape, to quantify MO6 distortion and help explore the effect of such distortion on the OER activity and stability of the MO6 unit. Accordingly, we uncovered the tuning role of the MO6 distortion in rutile metal oxides for the OER, and gave some suggestions for designing effective MO6-incorporated metal oxide catalysts for the OER. This work provides a fundamental insight into the structural dependence of MO6 active units for the OER, and may facilitate the design of metal oxide catalysts to some extent.
Before exploring the effect of the MO6 distortion on the OER, we constructed a doping model to delineate the varying degrees of the MO6 distortion, as shown in Fig. 1a, in which the metal active sites (M = Co, Ni, Ru) substitute a five-coordinated metal site on the (110) surface of some rutile metal oxides (CrO2, MnO2, RhO2, RuO2, IrO2). These metal oxides have different lattice sizes: MnO2 < CrO2 < RhO2 < IrO2 < RuO2, which can give rise to diverse modifications in the octahedral structure of the MO6 unit, involving either longitudinal contraction/extension or volumetric changes. The distortion of the MO6 octahedron typically manifests as changes in both volume and shape, as shown in Fig. 1b. To illustrate the distortion degree of the MO6 active unit, we have introduced a distortion coefficient (D, Fig. 1b), defined as D = V × (lp/le). Here, lp and le represent the average M–O bond lengths in the polar and equatorial directions, respectively. The ratio of lp and le (i.e., lp/le) represents the shape of the MO6 octahedron. lp/le < 1 (or > 1) is indicative of a flat (or leptosomic) type of MO6 octahedron. V is the volume of the MO6 octahedron, given by [(2le)2lp]/3, which represents the size of the MO6 unit.
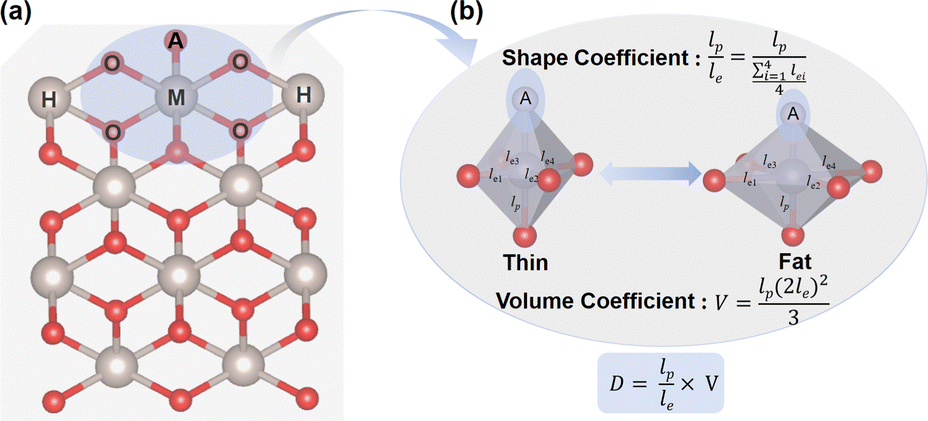 | ||
| Fig. 1 (a) Construction of the MO6 octahedron by doping M into different rutile metal oxide (110) surfaces. A, O, M and H represent the adsorbed oxygen-containing intermediates, oxygen atom, doped metal site and metal atoms in the host rutile metal oxides, respectively. (b) Scheme of the illustration of distortion coefficient (D). | ||
Based on the above models, we first explored the electrochemical stabilities of different MO6 units incorporated in different rutile metal oxides, by calculating the dissolution free energy (ΔGd) at the working conditions (pH = 0, U = 1.5 V vs. SHE, see the detail in Note S2, ESI†). A more positive ΔGd means better stability of the M site. Interestingly, after scaling ΔGd with the distortion coefficient (D) of the MO6 octahedron, there are very good linear relationships between D and ΔGd for these MO6 units (Fig. 2a). This suggests that D could be the effective indicator for describing the stability of MO6. Specifically, the incorporation of NiO6 and CoO6 octahedrons into different rutile metal oxides results in a significant variation in the value of D, which changes in a wide range of about 4.745 < D < 6.038 and 4.944 < D < 6.160 (see the blue and orange lines in Fig. 2a), respectively; only as D increases to a certain extent (> ∼6.0 for NiO6 and CoO6), ΔGd becomes negative, implying the instability of NiO6 and CoO6 octahedrons. This illustrates that the structural tunability of the NiO6 and CoO6 octahedrons for stability is considerable, which may be suitably modified to design more stable Ni-based and Co-based oxide catalysts for the acidic OER. For RuO6 in different rutile metal oxides, its relationship changes in a small range of about 5.136 < D < 5.521, implying its limited structural tunability; at the same time, ΔGd for the RuO6 unit is generally less than zero in this range (see the green line in Fig. 2a), implying the poor stability of the RuO6 active unit in the OER as reported in the experiment.15 Therefore, it is anticipated that the modification for the stability of NiO6 and CoO6 could be relatively easier compared to that of RuO6.
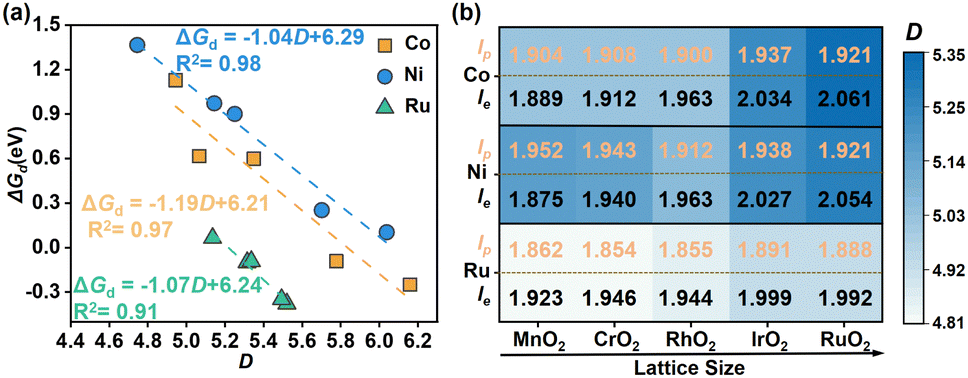 | ||
| Fig. 2 (a) Relationship of the dissolution free energy (ΔGd) of the metal site as a function of the distortion coefficient (D) of the MO6 octahedron (M = Co, Ni, Ru). (b) Variation trends of the polar (lp, orange) and equatorial (le, black) M–O bonds in different MO6 octahedrons. | ||
All these relationships in Fig. 2a have negative slopes, illustrating that ΔGd increases with the decrease of D, implying that the small D could contribute to the electrochemical stability of the MO6 unit. To further understand the origin of the better stabilized MO6 with small D, we performed structural and electronic analysis. Structurally, the reduction in D of the MO6 octahedron significantly extrudes the equatorial M–O bonds of MO6 (le, indicated by black values in Fig. 2b) and strengthens these equatorial M–O bonds, while the polar M–O bonds (lp, indicated by orange values in Fig. 2b) undergo relatively small changes. The shorter equatorial M–O bond indicates the stronger bond strength, resulting in better stability, as shown by the relationship between ΔGd and lp/le (Fig. S1, ESI†). Consequently, the central M site can be better stabilized by the equatorial O atoms as MO6 has a relatively small D, inhibiting its dissolution. Combining with the changes of le and the lattice parameter of different rutile metal oxide substrates, we can find that the small lattice parameter, which compresses the octahedral structure of MO6 in the equatorial direction, corresponds to the shorter le (Fig. 2b). Hence, for a given MO6, incorporating it into a rutile metal oxide with a relatively smaller lattice parameter can stabilize it in the OER. Furthermore, the charge density difference (CDD) analysis of CoO6, NiO6 and RuO6 units incorporated in different metal oxides was carried out. As shown in Fig. S2 (ESI†), it can be found that there are good linear relationships between ΔGd and electron transfer (Δq) from the metal center to the surrounding O atoms within the MO6 unit; their positive slopes show that with the increase of Δq, which strengthens the M–O bond in MO6, ΔGd becomes more positive, thus improving the stability of MO6 with relatively small D.
In addition to the stability of the MO6 unit, its catalytic activity stands as a pivotal determinant in its suitability in the OER. Here, we first calculated the adsorption free energies (Gi) of key oxygen-containing intermediates (*OH, *O and *OOH) involved in the OER by using the computational hydrogen electrode method16 (see the detailed calculation method in Note S3, ESI†), and examined the change trend of the theoretical overpotential η. As a consequence of the distortion of MO6 upon incorporation into different rutile metal oxides, we can find a deviation from the dependence with a smaller slope of 0.85 (G[*OOH] = 0.85G[*OH] + 3.06, R2 = 0.95) compared to the universal scaling relation of G[*OOH] = G[*OH] + (3.2 ± 0.2) observed in standard MO6 in rutile or perovskite oxides17 (Fig. 3a). This indicates that the scaling limitation for the standard MO6 octahedron on rutile metal oxides has been broken through. Based on this scaling relationship between G[*OH] and G[*OOH], a universal 3D volcanic map was constructed to describe the OER activity of these MO6 units incorporated in different rutile metal oxides (Fig. 3b).
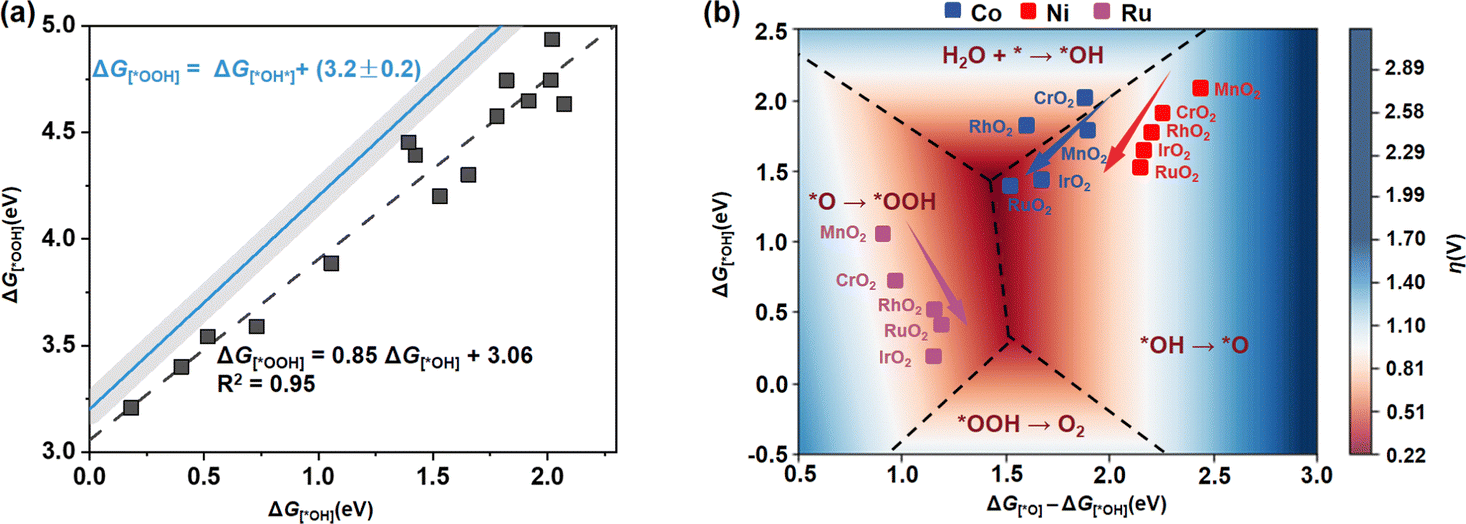 | ||
| Fig. 3 (a) Scaling relationship between G[*OOH] and G[*OH] on MO6 incorporated in different rutile metal oxides (black dashed line), and the universal relationship between G[*OOH] and G[*OH] observed in standard MO6 of rutile or perovskite metal oxides (blue line). (b) 3D volcanic map of theoretical overpotential η for MO6 in different rutile metal oxides as a function of G[*O]–G[*OH] and G[*OH], in which the blue, red and purple points represent CoO6, NiO6 and RuO6 units, respectively. The discussion for the boundary between the four rate-determining steps can be seen in Fig. S4 (ESI†), and the detailed overpotentials (η) are listed in Table S3 (ESI†). | ||
The 3D activity map in Fig. 3b shows that for each MO6 unit, η can gradually approach the optimum region in a certain pattern. To further understand the specific effect of the MO6 distortion on the OER activity, we proceeded to fit the relationship between the overpotential η and D. For all CoO6, NiO6 and RuO6 units, there are poor relationships between η and D (see Fig. S3a, ESI†). According to the definition of D = (lp/le) × V, we further scaled the relationships of η as a function of volume (V) and shape coefficient (lp/le), respectively. The distribution of η as a function of V remains relatively chaotic (Fig. S3b, ESI†), and the unique relationships between them have a relatively low R2. Interestingly, there are very good linear relationships between η and lp/le for CoO6, NiO6 and RuO6 units, as illustrated in Fig. 4a. This implies that in the MO6 distortion, the shape of MO6 (flat or leptosomic) could be the predominant factor influencing its OER activity.
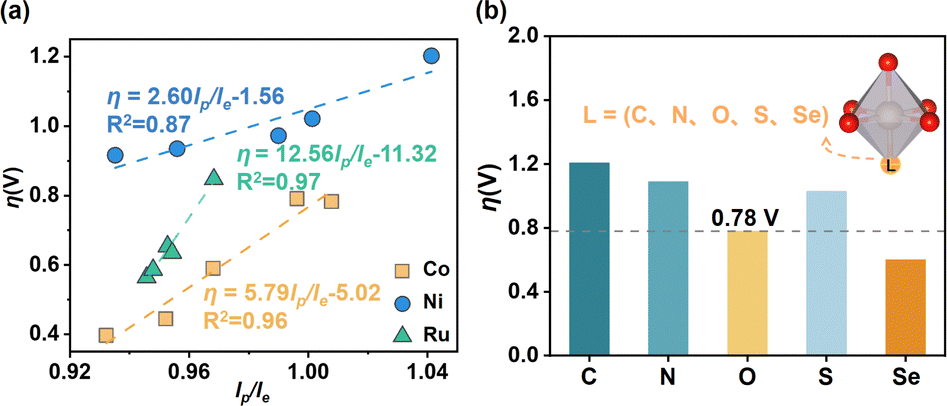 | ||
| Fig. 4 (a) Relationships of the overpotential η as a function of lp/le of the MO6 octahedron (M = Co, Ni, Ru). (b) Overpotential η for the OER of CoO6 with different ligand heteroatoms (C, N, S, Se) incorporated. | ||
The positive slopes of their relationships in Fig. 4a illustrate that η generally decreases as lp/le decreases, meaning that the small lp/le (i.e., the fat MO6 unit) could be more favourable for the OER. The slopes (α) of the relationships for the MO6 units decrease in the following order: RuO6(α = 12.56) > CoO6(α = 5.79) > NiO6(α = 2.60). A larger α means that the shape of the MO6 unit has a better sensitivity to the overpotential η (i.e., OER activity). The slopes of the relationships for RuO6 and CoO6 units are much larger than that for the NiO6 unit, implying their greater shape sensitivities to η compared with the NiO6 unit, especially the CoO6 unit. In addition, the intercepts (β) of the relationships for η, which illustrate the intrinsic activity of the MO6 unit for the OER, decrease with the order: NiO6(β = −1.56) > CoO6(β = −5.02) > RuO6(β = −11.32). A lower β means a better intrinsic activity. Thus, the RuO6 unit possesses the best intrinsic OER activity, and is widely demonstrated and used in experiments; the intrinsic OER activity for the NiO6 unit is the worst. Therefore, the activity regulatory effect of the RuO6 (or NiO6) unit for the OER is the greatest (or worst).
Specifically, as demonstrated by the activity map in Fig. 3b, NiO6 units incorporated into different rutile metal oxides are distant from the optimal range, indicating the relatively poor activity in these rutile metal oxides. For RuO6, although the RuO6 unit has the optimal activity sensitivity (i.e., larger α) and the best intrinsic activity (i.e., lowest β), its incorporation into these rutile metal oxides has a relatively small distortion degree compared with NiO6 or CoO6 units, thus leading to the relatively small change of the overpotential η for the OER. Hence, rutile metal oxides may not be suitable as a substrate for modifying RuO6, unless metal oxides that differ greatly from RuO2 lattice size are used. The CoO6 unit can locate near the optimum range (Fig. 3b), and CoO6 cooperated in IrO2 or RuO2 can reach an overpotential η as low as 0.44 V and 0.39 V, respectively. This illustrates the excellent structural tunability of CoO6 for activity, which could be a more viable option for the OER.
Scaling the adsorption free energies of *OH, *O and *OOH on these MO6 units incorporated in different rutile metal oxides with lp/le, as shown in Fig. S5 (ESI†), we can find that the decrease of lp/le leads to the strengthened adsorption of the intermediates *O, *OH and *OOH to different extents. For CoO6 and NiO6, the promotion of *O adsorption is greater, contributing to the rate-determining *OH → *O step and thus facilitating the OER. For RuO6, decreasing lp/le changes *OOH more visibly compared to *O, thereby promoting the rate-determining *O → *OOH step and facilitating the OER. In general, the OER activity of the MO6 unit incorporated in rutile metal oxide is inversely proportional to its shape (i.e., lp/le). Decreasing the M–O bond lengths in the polar direction (lp) or increasing the M–O bond lengths in the equatorial direction (le) can effectively improve the OER activity of the corresponding MO6 unit in rutile metal oxide.
In terms of structure, the polar M–O bond (lp) of the MO6 unit is directly related to the interaction of the O-containing intermediate on the M site, and the shorter lp means stronger adsorption strength of the intermediate in the polar direction. In addition, according to the bond order conservation principles, the longer le (i.e., the weaker equatorial M–O bond) can also strengthen the adsorption in the polar direction on the M site.18 Therefore, decreasing lp/le can enhance the adsorption of the O-containing intermediate, facilitating the OER on the MO6 unit incorporated in rutile metal oxide.
Based on the above analysis of the distortion of MO6 in terms of stability and OER activity, we can obtain the tuning rule of the MO6 active unit incorporated in rutile metal oxides for the OER. The M–O bond length (i.e., strength) in MO6 plays a key role in its stability and OER activity. A reduction in the M–O bond strength of MO6, especially the equatorial one, results in enhanced stability. However, for the OER activity, the fat MO6 (i.e., a lower lp/le) could be favourable, in which a longer le or shorter lp is required. In general, controlling the MO6 active unit with a relatively short le (i.e., strong equatorial M–O bond) and low lp/le value at the same time could be effective for designing a MO6 unit in rutile metal oxide for the OER, which can simultaneously maintain the high activity and stability. The above analysis of the change trends of M–O bonds in MO6 shows that MO6 incorporated in different rutile metal oxides primarily mainly affects the changes in the equatorial M–O bonds, with a comparatively minor impact from changes in the polar M–O bond. The change trends of the equatorial M–O bond strength and lp/le of MO6 in rutile metal oxides could be consistent. In other words, to achieve the good stability of MO6, the short le, i.e., strong equatorial M–O bond, is required, but this may mean a large lp/le value, which is detrimental to the OER activity. Therefore, breaking this inherent distortion limitation of MO6 for stability and activity is necessary for designing rutile metal oxide catalysts for the OER.
According to the above discussion, for the stable MO6 in rutile metal oxides, which has a strong equatorial M–O bond, the directional enhancement of the polar M–O bond could be effective in improving its OER activity. The CoO6 unit has excellent structural tunability for OER activity and good stability as discussed above, and MnO2 as a substrate has been widely used in the OER due to its superior stability and low cost.19 Accordingly, the CoO6 unit incorporated into MnO2, which has superior stability in the OER (ΔGd = 2.29 eV), was chosen as the basic object to verify this idea. Here, the oriented doping engineering was adopted to directly modify the polar Co–O bond strength in CoO6, in which several ligand heteroatoms (e.g., C, N, S, Se) were introduced into the polar O position (Fig. 4b). One can see that the Se incorporation into CoO6 units in MnO2 can achieve a low overpotential η of 0.60 V compared with the original η = 0.78 V of the pure CoO6 in MnO2. Compared with the O and S atoms, the introduction of the Se atom results in more overlap between the Co 3d of the Co site and O 2p orbitals of O-containing species adsorbed on the Co site, significantly promoting the key *O adsorption and thereby facilitating the OER (see the detailed discussion in Fig. S6 and S7, ESI†). This result verifies the feasibility of our strategy, which may be worth experimenting with in the future.
In summary, this work investigated the effect of the distortion of MO6 octahedrons in rutile metal oxide, which generally involves the change in volume and shape of the MO6 unit, on the activity and stability for the OER through the introduction of a distortion coefficient (D). The results show that the M–O bond length (i.e., strength) and shape of MO6 are the key factors in the stability and OER activity. The shorter M–O bond of MO6, especially the equatorial one, promotes its stability due to more electron transfer between the M center and the adjacent O atoms, which strengthens the M–O bond and thus stabilizes the M center. The MO6 unit with a fat shape (i.e., small lp/le value) has a higher OER activity. However, the short le of MO6 with superior stability generally results in a large lp/le value, which is not conducive to the OER activity. It is therefore evident that breaking this cooperative limitation resulting from the structural distortion of the MO6 unit in rutile metal oxides on the stability and activity is necessary. Accordingly, the oriented doping engineering was proposed to modify the MO6 unit, directing the proposal of a cost-effective Se-doped CoO6 unit incorporated into MnO2 for the OER. This work provides a fundamental understanding of the effect of the MO6 octahedral distortion on the activity and stability in the OER, promoting the rational design of metal oxide catalysts.
This work was financially supported by the National Natural Science Foundation of China (22472053 and 22239001), Science and Technology Commission of Shanghai Municipality (23ZR1416800 and 22ZR1416400), and Innovation Program of Shanghai Municipal Education Commission (E00014).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- J. A. Turner, Science, 2004, 305, 972 CrossRef CAS PubMed.
- (a) X. Wang, S. Xi, P. Huang, Y. Du, H. Zhong, Q. Wang, A. Borgna, Y.-W. Zhang, Z. Wang, H. Wang, Z. G. Yu, W. S. V. Lee and J. Xue, Nature, 2022, 611, 702 CrossRef CAS PubMed; (b) X. X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148 RSC.
- N. Suen, S. Hung, Q. Quan, N. Zhang, Y. Xu and H. Chen, Chem. Soc. Rev., 2017, 46, 337 RSC.
- (a) D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef; (b) S. Lee, Y. J. Lee, G. Lee and A. Soon, Nat. Commun., 2022, 13, 3171 CrossRef CAS PubMed.
- N. Zhang, X. Feng, D. Rao, X. Deng, L. Cai, B. Qiu, R. Long, Y. Xiong, Y. Lu and Y. Chai, Nat. Commun., 2020, 11, 4066 CrossRef CAS.
- M. Huynh, D. K. Bediako and D. G. Nocera, J. Am. Chem. Soc., 2014, 136, 6002 CrossRef CAS PubMed.
- H. Jia, N. Yao, C. Yu, H. Cong and W. Luo, Angew. Chem., Int. Ed., 2023, 62, 202313886 CrossRef PubMed.
- (a) P. Ping, R. J. Nielsen and W. A. GoddardIII, J. Am. Chem. Soc., 2017, 139, 149 CrossRef PubMed; (b) Y. H. Fang and Z. P. Liu, J. Am. Chem. Soc., 2010, 132, 18214 CrossRef CAS.
- (a) J. Druce, H. Téllez, M. Burriel, M. D. Sharp, L. Fawcett, S. N. Cook, D. Mcphail, T. Ishihara, H. H. Brongersma and J. A. Kilner, Energy Environ. Sci., 2014, 7, 3593 RSC; (b) D. K. Bediako, B. Lassalle-Kaiser, Y. Surendranath, J. Yano, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6801 CrossRef CAS.
- (a) B. Kim, A. Oh, M. K. Kabiraz, Y. Hong, J. Joo, H. Baik, S. Choi and K. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 10115 CrossRef CAS; (b) J. Yan, J. Zhu, D. Chen, S. Liu, X. Zhang, S. Yu, Z. Zeng, L. Jiang and F. Du, J. Mater. Chem. A, 2022, 10, 9419 RSC.
- (a) N. Zhang, C. Wang, J. Chen, C. Hu, J. Ma, X. Deng, B. Qiu, L. Cai, Y. Xiong and Y. Chai, ACS Nano, 2021, 15, 8537 CrossRef CAS PubMed; (b) Y. Xu, Z. Mao, J. Zhang, J. Ji, Y. Zou, M. Dong, B. Fu, M. Hu, K. Zhang, Z. Chen, S. Chen, H. Yin, P. Liu and H. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202316029 CrossRef CAS.
- F. Yang, J. Xie, D. Rao, X. Liu, J. Jiang and X. Lu, Nano Energy, 2021, 85, 106020 CrossRef CAS.
- (a) W. Sun, Y. Song, X. Gong, L. Cao and J. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 820 CrossRef CAS PubMed; (b) L. Sun, Z. Dai, L. Zhong, Y. Zhao, Y. Cheng, S. Chong, C. Guanjun, C. Yan, X. Zhang, H. Tan, L. Zhang, K. N. Dinh, S. Li, F. Ma and Q. Yan, Appl. Catal., B, 2021, 297, 120477 CrossRef CAS.
- (a) Z. Wu, F. Chen, B. Li, S. Yu, Y. Z. Finfrock, D. M. Meira, Q. Yan, P. Zhu, M. Chen, T. Song, Z. Yin, H. Liang, S. Zhang, G. Wang and H. Wang, Nat. Mater., 2023, 22, 100 CrossRef CAS; (b) J. Wang, Y. Ji, R. Yin, Y. Li, Q. Shao and X. Huang, J. Mater. Chem. A, 2019, 7, 6411 RSC.
- J. Chen, Y. Ma, T. Huang, T. Jiang, S. Park, J. Xu, X. Wang, Q. Peng, S. Liu, G. Wang and W. Chen, Adv. Mater., 2024, 36, e2312369 CrossRef.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311 RSC.
- M. Bajdich, M. García-Mota, A. Vojvodić, J. K. Nørskov and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 13521 CrossRef CAS.
- L. Zhang, H. Yuan, L. Wang, H. Zhang, Y. Zang, Y. Tian, Y. Wen, F. Ni, H. Song, H. Wang, B. Zhang and H. Peng, Sci. China Mater., 2020, 63, 2509 CrossRef CAS.
- C. Lin, J.-L. Li, X. Li, S. Yang, W. Luo, Y. Zhang, S.-H. Kim, D.-H. Kim, S. S. Shinde, Y.-F. Li, Z.-P. Liu, Z. Jiang and J.-H. Lee, Nat. Catal., 2021, 4, 1012 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04992f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |