
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural and computational analysis of H-bond mediated anion⋯anion interactions in new salts of fumaric and maleic acids†
Mahdi
Jemai
 ab,
Miquel
Barceló-Oliver
c,
Houda
Marouani
b,
Thierry
Roisnel
d,
Antonio
Frontera
*c and
Rafel
Prohens
*a
ab,
Miquel
Barceló-Oliver
c,
Houda
Marouani
b,
Thierry
Roisnel
d,
Antonio
Frontera
*c and
Rafel
Prohens
*a
aLaboratory of Organic Chemistry, Faculty of Pharmacy and Food Sciences, University of Barcelona, Avda. Joan XXIII, 08028 Barcelona, Spain. E-mail: rafel_prohens@ub.edu
bLaboratory of Material Chemistry, LR13ES08, Faculty of Sciences of Bizerte, University of Carthage, 7021 Bizerte, Tunisia
cDepartment of Chemistry, University of Balearic Islands, 07122 Palma de Mallorca, Spain
dUniversité de Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes), UMR 6226, F-35000 Rennes, France
First published on 28th May 2025
Abstract
We report the synthesis and the combined crystallographic/computational analysis of a series of ammonium salts of fumaric and maleic acids. In the solid state, the structures form a variety of non-covalent interactions including N–H⋯O, O–H⋯O and C–H⋯O H-bonds, supported additionally by other aromatic interactions such as π⋯π and C–H⋯π. They have been investigated through quantum chemical calculations, such as molecular electrostatic potential (MEP) surface analysis, quantum theory of atoms in molecules (QTAIM), and noncovalent interaction plot (NCIPlot) methodologies, specially focused on the formation of anion⋯anion interactions facilitated by hydrogen bonds. Our findings show that specific hydrogen bonds, can establish attractive forces between like-charged anionic units in the presence of counterions or within high-dielectric environments such as water, highlighting that cooperative noncovalent assemblies can overcome coulombic repulsion driving the formation of organized anionic networks in the solid state.
1. Introduction
Fumaric acid (FA), whose name is inspired by Fumaria officinalis, the medicinal plant from which the acid was originally obtained, is an organic acid comprising two carboxylic groups double-bonded in the trans position.1,2 This valuable compound has a wide range of applications in different fields. It is widely used in food and beverage production as an acidifying agent over many years, since 1946.3 FA is also frequently present in a wide variety of pharmaceutical products, serving as an important compound used in the production of therapeutic drugs and cosmetics.4–6 It is not possible to introduce FA without mentioning its important cis isomer, maleic acid (MA), which also has a wide range of commercial applications. It is used in the production of polyester resin, plastics, paint, copolymers and agrochemicals.7 Both FA8,9 and MA10,11 have been extensively used as coformers for the synthesis of pharmaceutical salts and co-crystals,12 or as a drug compound in the case of FA.13–15 Salts and co-crystals are made up by the formation of non-covalent interactions (NCIs) between MA and FA with the coformers of interest, being the formation of hydrogen bonds the principal mode of the supramolecular assembly framework, followed by the mutual development of weaker NCIs such as π⋯π, C–H⋯π and N–H⋯π interactions.16In recent years, computational chemistry has played an increasingly central role in the analysis of noncovalent interactions within molecular crystals. Techniques such as quantum theory of atoms in molecules (QTAIM), noncovalent interaction plot (NCIPlot) analysis, and energy decomposition schemes have been successfully used to identify and characterize the subtle interplay of forces that govern crystal packing.16–19 These methods enable quantitative and visual insight into directional interactions, which may not be fully captured by crystallographic data alone. Several recent studies have demonstrated the power of these tools in unravelling noncovalent patterns in complex organic systems and guiding rational supramolecular design strategies.17–19
Scientists now recognize noncovalent anion⋯anion interactions as important structural and functional motifs in supramolecular chemistry despite their initial perception as counterintuitive because of the electrostatic repulsion between same-charged species.20 When hydrogen bonding participates in these interactions between like-charged anions they break away from standard coulombic predictions by demonstrating complex directional forces working alongside solvation effects and geometric limitations.21–24 When anions are incorporated into structured networks or stabilized by hydrogen-bond donors like protonated amines, hydroxyls, or acidic heterocycles the formation of anion⋯anion contacts becomes possible through the creation of cooperative bonding networks.25–27 The H-bond functions as a bridge between anions through direct interaction or it maintains anionic clusters that promote energetically favourable anion⋯anion closeness. This effect reaches beyond crystallographic interest to influence multiple domains such as crystal engineering and molecular recognition as well as ion transport and catalysis.28
To gain comparative insights into how the maleic acid (MA) and fumaric acid (FA) isomers interact in the solid state and to explore their potential to engage in anion⋯anion interactions, we conducted a salt screening and carried out a supramolecular analysis of the resulting crystal structures through computational methods. The formation of ammonium salts of carboxylic acids is a well-established phenomenon, which allows the study of a great diversity of intermolecular interaction in the solid-state.29,30 Thus, in this paper, we report the synthesis and X-ray crystal structures of a series of ammonium salts of fumaric and maleic acids (salts I–VI), as outlined in Scheme 1. Notably, we have identified and analysed in details several intriguing anion⋯anion binding motifs involving the hydrogen maleate anions (salts I, III, and V), fumarate (IV), and hydrogen fumarates (II and VI).
 | ||
| Scheme 1 Salts reported in this manuscript and motifs studied computationally. | ||
2. Materials and methods
2.1 Crystallization
Dissolution of stoichiometric amounts (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) of MA:NBPEA; FA:NBPEA; MA:1,4-DAD; FA:4-DAD; MA:6-MQ and FA:MXDA in ethanol and water followed by slow evaporation at room temperature for a few days produced high-quality I–VI single crystals suitable for SCXRD.
:1) of MA:NBPEA; FA:NBPEA; MA:1,4-DAD; FA:4-DAD; MA:6-MQ and FA:MXDA in ethanol and water followed by slow evaporation at room temperature for a few days produced high-quality I–VI single crystals suitable for SCXRD.
2.2 Single X-ray crystallographic analysis
The crystallographic data of the compounds I, II, III, V and VI have been determined using a D8 VENTURE Bruker AXS diffractometer equipped with a CCD detector (Cu Kα radiation, λ = 1.54178 Å) and Incoatec-Helios-MX multilayer optics. Compound IV has been determined using a D8-VENTURE-Bruker-AXS diffractometer with the CCD detector: Mo Kα radiation, λ = 0.71073 Å.The reduction of the data and the refinement of the cells were carried out using the Bruker-APEX5 program.31 Using the SADABS-2016/2 program,31 a multi-scan method was then employed to correct the data collected. Applying the Olex2-1.5 suite,32 the crystal structures were then solved by intrinsic phasing with SHELXT-2018/2 and further refined by the full matrix least squares technique with SHELXL-2019/3,33 and the WINGX program was used to solve the structure of compound IV directly.34
Compounds I, II and V were shown to be twinned. For compound I, racemic twinning was found and was refined using the instruction TWIN −1 0 0 0 −1 0 0 0 −1 2, and the BASF scale factors refined to 0.01(3). For compound II, using Olex2-1.5 the TWIN law was found to be −1 0.0393 −0.0352 0 −0.0502 −0.9483 0 −1.0502 0.0518, and an HKLF5 file was prepared for the subsequent refinement of both twinned fractions, with a BASF scale factor of 0.476(12). For compound V, the APEX-5 suite was used to select the reflections from the major component (99.6%), and the structure was solved and refined as a non-twinned crystal. Due to the quality of crystals, the fraction of measured diffractions was just of 95.3% in the best of the crystals tested.
Refining of all non-hydrogen atoms (in compounds I–VI) was performed with anisotropic thermal parameters by full-matrix least-squares calculations on F2. The hydrogen atoms were included in the refinement at idealized geometry and refined “riding” on the corresponding parent atoms. Hydrogen atoms bound to carbon were then added to the calculated positions and refined as circumscription atoms, with Uiso(H) = 1.2Ueq(C). The structures were verified for higher symmetry using the PLATON program.35Table 1 summarizes the crystallographic data of all the compounds we have reported.
| Compound | I | II | III | IV | V | VI |
|---|---|---|---|---|---|---|
| Empirical formula | C19H21NO4 | C19H21NO4 | C11H17NO6 | C9H15NO4 | C14H13NO4 | C16H26N2O11 |
| Formula weight | 327.37 | 327.37 | 259.25 | 201.22 | 259.25 | 422.39 |
| Temperature (K) | 100 | 100 | 100 | 150 | 100 | 100 |
| Crystal system | Orthorhombic | Triclinic | Monoclinic | Triclinic | Triclinic | Monoclinic |
| Space group | P212121 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P |
P |
P21/n |
| a (Å) | 5.6443(5) | 7.9935(7) | 8.8182(5) | 5.6190(5) | 5.6275(3) | 8.0431(7) |
| b (Å) | 17.2728(14) | 10.9224(10) | 5.5773(3) | 8.2905(7) | 9.6420(5) | 14.8328(12) |
| c (Å) | 17.5092(15) | 11.3553(10) | 25.6130(14) | 11.0972(9) | 11.8288(6) | 16.9537(14) |
| α (°) | 90 | 76.150(5) | 90 | 99.262(3) | 100.639(2) | 90 |
| β (°) | 90 | 75.668(5) | 97.959(2) | 99.099(3) | 90.079(2) | 98.675(4) |
| γ (°) | 90 | 72.689(5) | 90 | 102.841(3) | 102.129(2) | 90 |
| V (Å3) | 1707.0(3) | 902.21(14) | 1247.56(12) | 487.25(7) | 616.18(6) | 1999.5(3) |
| Z | 4 | 2 | 4 | 2 | 2 | 4 |
| Density (calc. Mg m−3) | 1.274 | 1.205 | 1.380 | 1.372 | 1.397 | 1.403 |
| Final R indices [I > 2σ(I)] | R 1 = 0.044 | R 1 = 0.091 | R 1 = 0.055 | R 1 = 0.039 | R 1 = 0.042 | R 1 = 0.060 |
| wR2 = 0.110 | wR2 = 0.285 | wR2 = 0.150 | wR2 = 0.109 | wR2 = 0.110 | R 2 = 0.160 | |
| CCDC | 2440160 | 2440159 | 2440156 | 2440152 | 2440157 | 2440161 |
Crystal explorer software was used to perform the Hirshfeld surface analysis36,37 and estimate the normalized contact distance (dnorm), and the shape index mode was carried out with a rescale surface property ranging from −0.5 to 1.5 and from −1 to 1 Å, respectively; the 2D-fingerprint plots were plotted over the range 0.4 Å −2.6 Å in each of di and de distances.
2.3 Theoretical methods
For the DFT calculations of the supramolecular assemblies, the PBE0-D3/def2-TZVP level of theory was employed using the Gaussian 16 software package.38–41 The binding energies were determined as the difference between the total energy of the assembly and the sum of the energies of the isolated monomers, with corrections applied for the basis set superposition error (BSSE).42 The molecular electrostatic potential (MEP) surfaces were calculated using the 0.001 a.u. isosurface to approximate the van der Waals envelope. The anion⋯anion dimers were fully optimized without symmetry constraints and they correspond to true minima on the potential energy surface. The optimization was performed in water using the PCM continuum model.43To analyze the interactions within the assemblies, quantum theory of atoms in molecules (QTAIM)44 and noncovalent interaction plot (NCIPlot)45 methods were applied at the same level of theory using the AIMAll software.46 The NCIPlot method is particularly effective for visualizing noncovalent interactions in real space. It employs reduced density gradient (RDG)47 isosurfaces and a color-coded scheme based on the sign of the second eigenvalue of the electron density Hessian (λ2) to differentiate between attractive and repulsive interactions. For this study, the settings used were RDG = 0.5, density cut-off = 0.04 a.u., and a color scale ranging from −0.04 a.u. ≤ signλ2(ρ) ≤ 0.04 a.u. Strongly attractive interactions are represented in blue, while moderately attractive interactions are shown in green.
3. Results and discussion
Employing the same synthesis conditions, the combination of the two isomeric carboxylic acids FA and MA with amine coformers such as N-benzylphenethylamine (NBPEA), 1,4-dioxa-8-azaspiro(4.5)decane (1,4-DAD), 6-methylquinoline (6-MQ) in the case of MA and m-xylylenediamine (MXDA) for FA has produced the following six new salt compounds: NBPEA/MA (I); NBPEA/FA (II); 1,4-DAD/MA (III); 1,4-DAD/FA (IV); 6-MQ/MA (V) and MXDA/FA-trihydrate (VI).3.1 Structural description and supramolecular details
 | ||
Fig. 1 Ortep representation of compound I (ellipsoids are drawn at the 45% probability level) (a), projection along the (![[b with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0062_20d1.gif) , , ![[c with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0063_20d1.gif) ) plane (b). ) plane (b). | ||
This H-bonding distribution allows the formation of well-organized layers of anions distributed in Z = 1/4 and Z = 3/4 directions as shown in Fig. 1(b). The distribution of anions and cations in the structure held by N–H⋯O and C–H⋯O bonds produces a combination of supramolecular synthons of the R22(8), R24(10), R23(8), R21(4) and R23(10) types,48 which make the anion⋯cation network extremely ramified. Concerning the hydrogen maleate anion, in addition to the C–H⋯O intramolecular interaction giving rise to an R23(7) interaction motif, intramolecular H-bonds are established to form supramolecular synthons of the S(7) type (Fig. 2(a)).49 Moreover, the N-benzylphenethylammonium cation contributes to structural stability through C–H⋯π intramolecular interactions established between the cations distributed throughout the cell with hydrogen to centroid distances equal to 3.09, 3.77 and 3.88 Å, as shown in Fig. 2(b).
 | ||
| Fig. 2 Intra- and intermolecular interaction motifs formed in salt I (a) and C–H⋯π interaction distances (b). | ||
In order to evaluate the most influential intermolecular interactions in the structures, we have carried out a Hirshfeld surface (HS) analysis and generated the related fingerprint plots using Crystal Explorer software.
HS analysis highlights the strong intermolecular H⋯O/O⋯H contacts (24.2%) depicted as red areas in the 3D dnorm picture (Fig. 3(a)) and sharp peaks in the associated 2D fingerprint plots (Fig. 3(c)). The dnorm mode also shows the existence of a white zone around the NBPEA rings, usually corresponding to weak interactions that can be attributed to the C–H⋯π contacts established between the cations; this is confirmed by the significant presence of C⋯H/H⋯C contacts (26.8%) on the one hand, and the appearance of a large red zone spanning the acceptor–electron system in the shape index surface and the blue zone appearing on the C⋯H donor (Fig. 3(b)).50–53 The contribution of all weaker interactions are also marked on the fingerprint plots by giving the percentage involved of each contact.
 | ||
| Fig. 3 Hirshfeld surface analysis shown in dnorm (a) and shape index (b) modes for compound I and computed fingerprint plots (c). | ||
and with an N-benzylphenethylammonium cation (NBPEA+) and two hydrogen fumarate half-molecules in the asymmetric unit. One of NBPEA+ rings shows a disorder with an occupancy probability of 74.6% for the C11–C12–C13–C14–C15–C16 ring and 25.4% for the C11–C12′–C13′–C14′–C15′–C16′ ring. In the crystal lattice, the hydrogen fumarate anions form strong carboxylate–carboxylic self-complementary ribbons through two charge-assisted H-bonds (d(O1A–H1A⋯H1A) = 1.221(2)Å; O1A–H1A⋯O1A = 180.0(2)°) and (d(O5A–H5A⋯H5A) = 1.230(3) Å; O5A–H5A⋯O5A = 180.0(3)°) as shown in Fig. 4(a). Regarding the cationic part, and as in compound I, besides the C–H⋯ π interaction that participates in the intermolecular connection between the cations, NBPEA+ rings are stacked in a way that allows the formation of π⋯π interactions as shown in Fig. 4(b). The anion⋯cation connection is made up by N8–H8A⋯O3A and N8–H8B⋯O7A as shown in Fig. 5(a). The projection of the structure along the (, ) plane (Fig. 5(b)) shows how the N-benzylphenethylamine molecules are ordered in straight lines intersected by zigzag lines formed by the anions at nodes made of nitrogen atoms, which makes the molecules inside the cell form a network resembling a “trawling net”.
 | ||
| Fig. 4 Intramolecular H-bonding interactions formed between anions (a) and presentation of C–H⋯π and π⋯π stacking between N-benzylphenethylaminium cations (b). | ||
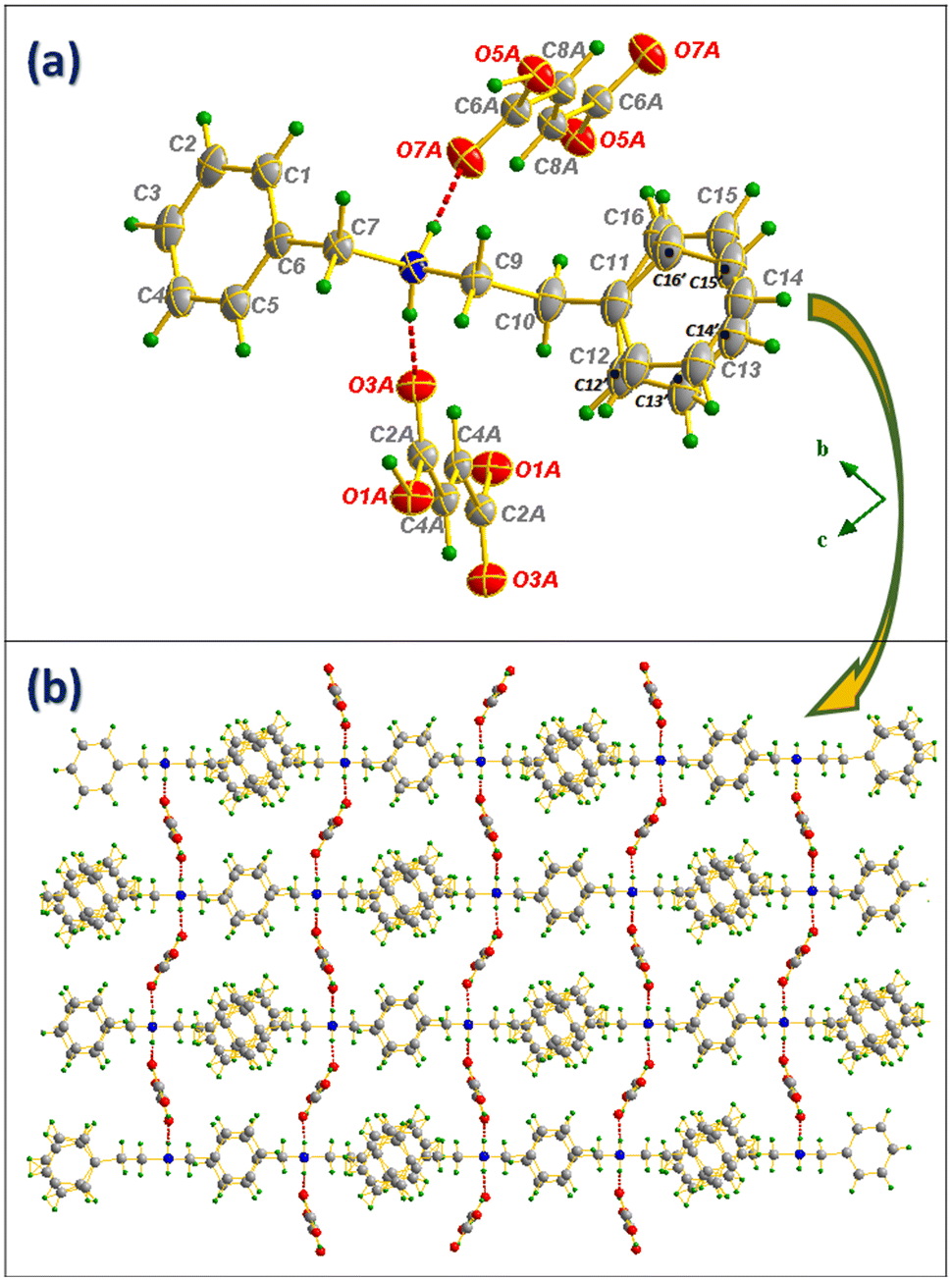 | ||
| Fig. 5 Ortep representation of N-benzylphenethylamine/fumaric acid salt (a) and projection of the compound II structure along the (, ) plane (b). | ||
A Hirshfeld surface analysis with this structure has not been included since the asymmetric unit contains two independent half-molecules of fumaric acid and the resulting fingerprint plot showed an unrealistically short contact between the hydrogen atoms of the carboxylic acid.
![[a with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0061_20d1.gif) , ) plane, the clearest atomic arrangement view and as shown in Fig. 6(b); the molecular packing is consolidated via N(C)–H⋯O H-bonds, leading to the formation of supramolecular synthons of types R23(8) and R21(4) (Fig. 6(c)), while the MA anions interact intra- and intermolecularly by C(O)–H⋯O H-bonds giving rise to the S(7) and R23(7) supramolecular synthons (Fig. 6(d)).
, ) plane, the clearest atomic arrangement view and as shown in Fig. 6(b); the molecular packing is consolidated via N(C)–H⋯O H-bonds, leading to the formation of supramolecular synthons of types R23(8) and R21(4) (Fig. 6(c)), while the MA anions interact intra- and intermolecularly by C(O)–H⋯O H-bonds giving rise to the S(7) and R23(7) supramolecular synthons (Fig. 6(d)).
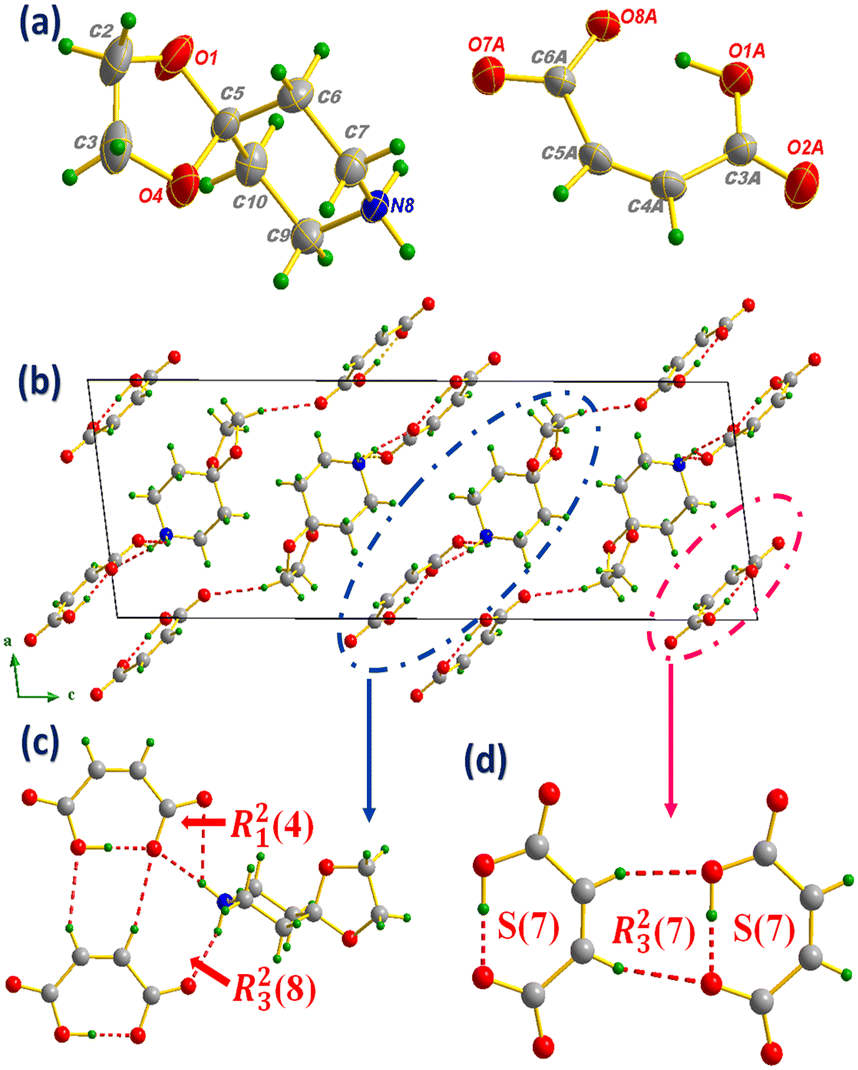 | ||
| Fig. 6 Asymmetric unit of compound III (a), projection of the structure along the (, ) plane (b) marking the formation of R23(8) and R21(4) supramolecular synthons between anions and cations (c), intra- and intermolecular S(7) and R23(7) interaction motifs formed between MAs (d). | ||
The HS analysis depicted in Fig. 7 highlights the H⋯O/O⋯H contacts by displaying the dnorm mode associated with the fingerprint plots of the 1,4-DAD/MA complex and the individual anion and cation. The high percentage contribution of the O⋯H contact (51%) on the anion and H⋯O (31%) on the cation clearly indicates the reciprocal donor/acceptor H-bonding roles between MA and 1,4-DAD responsible for the counterion connection.
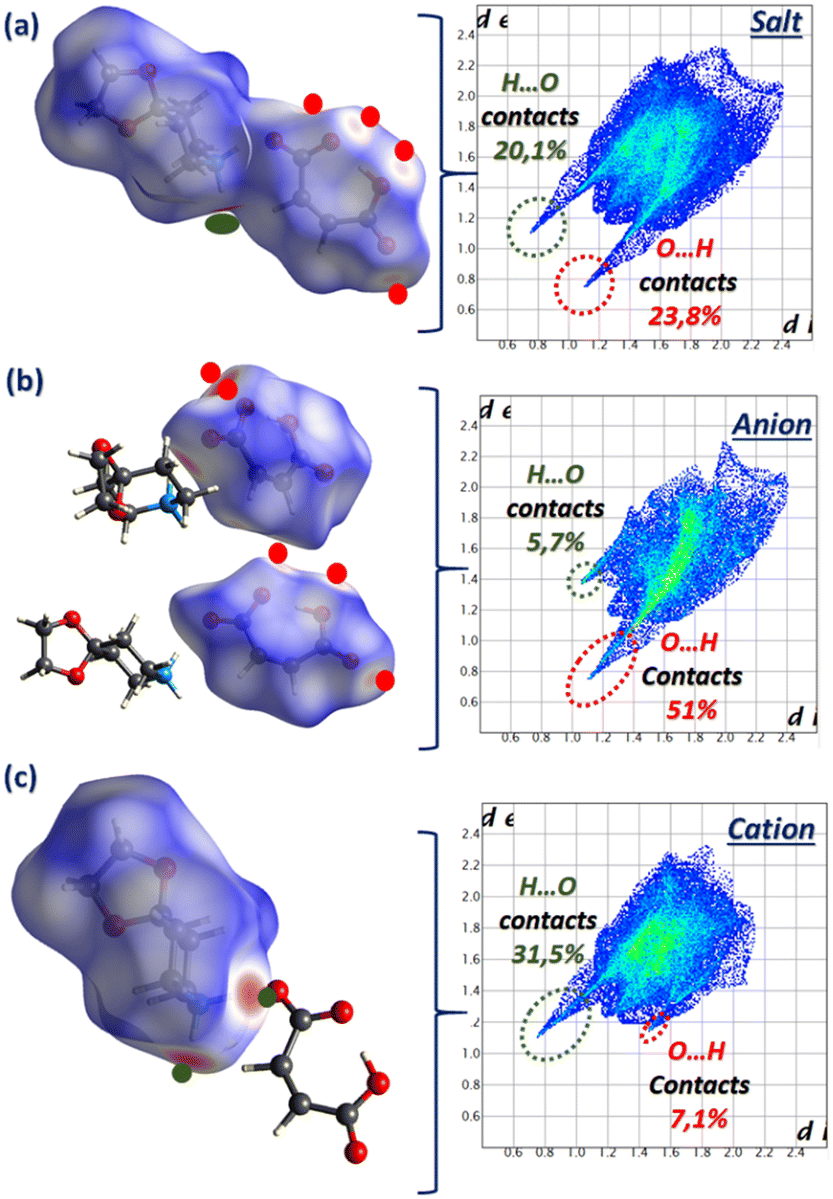 | ||
| Fig. 7 Hirshfeld surface mapped over the dnorm mode with the associated finger print plots of (a) 1,4-DAD/MA salt and its (b) anion and (c) cation. | ||
and with a 1,4-dioxa-8-azaspiro(4.5)decane cation and half molecule of the fumarate anion in the asymmetric unit. As in 1,4-DAD/MA, intermolecular contacts are also observed between FA anions by means of C–H⋯O H-bonds which have given rise to the R22(10) interaction motif shown in Fig. 9(b), as well as being linked to 1,4-DAD via N(C)–H⋯O H-bonds (Fig. 9(a)), allowing all components inside the cell to interconnect and therefore participate in the formation of a massive supramolecular synthon of type R44(26) formed with the support of two additional smaller intramolecular interaction motifs of the R22(8) type.
 | ||
| Fig. 8 Ortep representation of 1,4-dioxa-8-azaspiro(4.5)decane/fumaric acid salt. | ||
 | ||
| Fig. 9 Formation of supramolecular synthons R44(26) and R22(8) between anions and cations (a) and R22(10) between anions separately (b). | ||
The Hirshfeld surface analysis of compound IV presented in Fig. 10 shows a comparative account of the contribution of intermolecular H-bonds contacts (H⋯O/O⋯H).The participation of each part of this supramolecular unit is displayed by the dnorm mode with the associated fingerprint plots as follows: the salt on the one hand, and the anion and cation separately on the other hand. The results of the analysis show that compound IV shares similar characteristics to its related previous compound in terms of H-bond contribution.
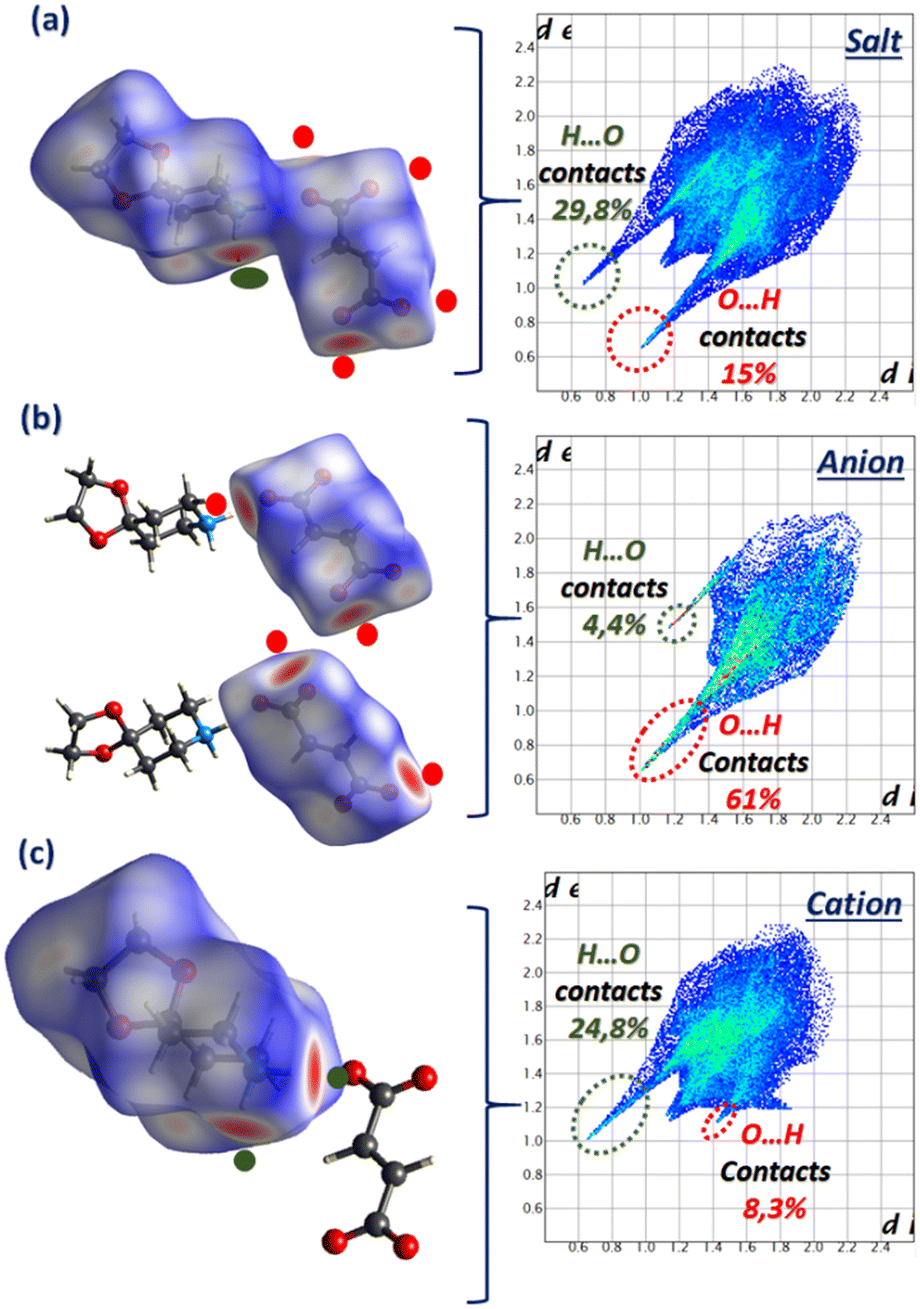 | ||
| Fig. 10 Hirshfeld surfaces of compound IV (a) and its anion (b) and cation (c) mapped in the dnorm mode with their 2-D fingerprint plots. | ||
and with a 6-methylquinoline cation and one hydrogen maleate anion. In the cell, anions and cations are linked via N–H⋯O and C–H⋯O H-bonds forming layers parallel to the (, ) plane located at y = 1/4 and y = 3/4 (Fig. 12(a)) and supported by the intramolecular interaction motif of the R22(7) type (Fig. 12(b)). Both inter- and intramolecular hydrogen bonds are also present between MA, forming R23(7) and S(7) supramolecular motifs. The cation⋯cation intermolecular connection is supported by the π-stacking between the benzene and pyridine rings of 6-MQ with a distance between centroids equal to 4.09 Å (Fig. 13(a)). Fig. 13(b) shows that the connection between anions and cations via H-bonds gives rise to layers arranged with the shape of arrowheads in the opposite sequence, and π⋯π interactions interconnecting them and allowing the appearance of a chair like conformation. This weak intermolecular interaction has been primarily detected by the Hirshfeld surface analysis shown in Fig. 14, particularly with the appearance of a blue-red bow-tie located on the 6-MQ ring shape index surface. The contributions of each contact are shown in detail in Fig. 14(c), where the lowest contributors are shaded out from the figure (H⋯N/N⋯H: 1.5%; C⋯N/N⋯C: 0.5%; N⋯O/O⋯N: 0.4%).
 | ||
| Fig. 11 Asymmetric unit representation of 6-methylquinoline/maleic acid salt. | ||
 | ||
| Fig. 12 Projection of the compound V structure along the (, ) plane (a) and intra- and intermolecular interaction motifs formed in compound V (b). | ||
 | ||
| Fig. 13 Representation of 6-methylquinoline π⋯π arrangement (a) and combination of hydrogen bonding and π-stacking interactions (b). | ||
 | ||
| Fig. 14 d norm mode (a), shape index (b), and calculated fingerprint plots of 6-methylquinoline/maleic acid salt (c). | ||
, ) plane, located at y = 0 and y = 1/2. Water molecules are present between layers, allowing them to interconnect via C–H⋯O and OW–HW⋯O bonds. As for the anions, cations are also linked to water molecules to form wavy chains by means of N–H⋯O H-bonds, which are in turn interlocked by the appearance of the CH⋯π interaction with H⋯centroid distances equal to 3.57 Å, as shown in Fig. 16(b), and further identified by the HS analysis through the shape index surface featured in Fig. 18.
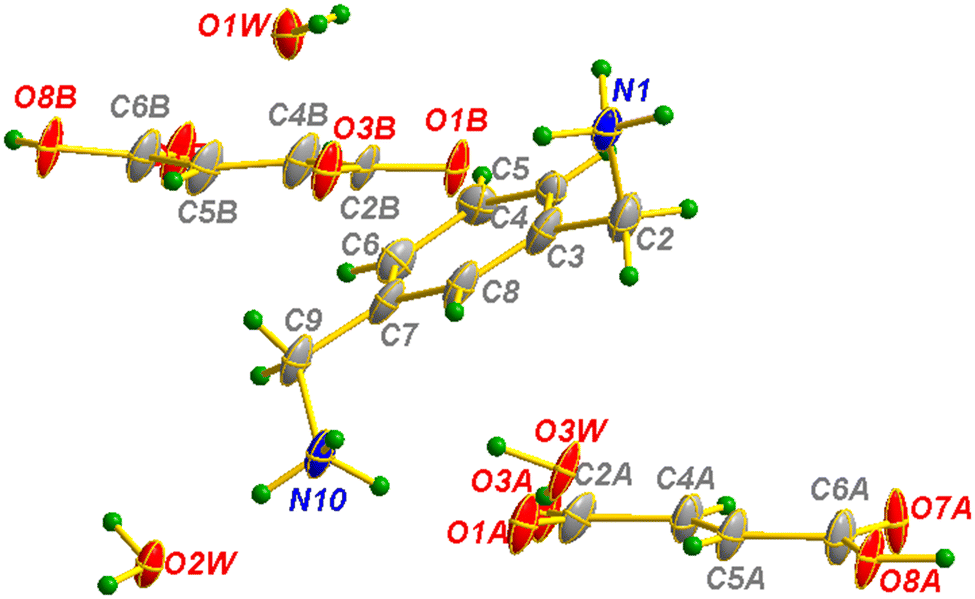 | ||
| Fig. 15 Asymmetric unit of m-xylylenediamine/fumaric acid-trihydrate salt (VI) with the atom-labeling scheme. | ||
 | ||
| Fig. 16 Molecular packing of anions (a) and cations (b) separately in m-xylylenediamine/fumaric acid-trihydrate salt (VI). | ||
The connection between all the constituents of salt VI: MXDA, FA and water molecules is provided by a combination of N–H⋯O, C–H⋯O, O–H⋯O and OW–HW⋯O which leads to a super-branched structure; the m-xylylenediaminium cations and hydrogen fumarate anions are represented in the molecular packing along the (, ) plane as wavy chains intersecting at nodes formed by nitrogen atoms as depicted in Fig. 17.
 | ||
| Fig. 17 Molecular packing of m-xylylenediamine/fumaric acid-trihydrate salt (VI) along the (, ) plane. | ||
HS analysis (Fig. 18) in the dnorm mode shows the distribution of the red zones over the whole surface, supported by the high contribution of the O⋯H/H⋯O contact equal to 59.9%, the highest value compared to all previous compounds, highlighting the rich network of hydrogen bonds in the structure.
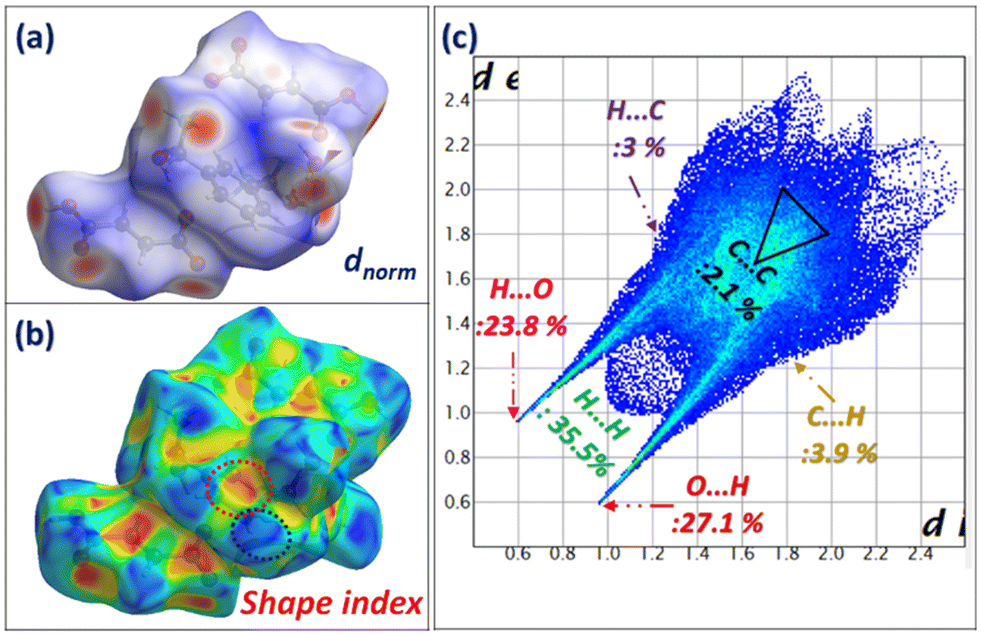 | ||
| Fig. 18 Hirshfeld surface analysis in dnorm (a) and shape index modes (b) and the calculated fingerprint plots (c) of m-xylylenediamine/fumaric acid-trihydrate salt (IV). | ||
The identification of the supramolecular synthons formed in the dense molecular framework of compound VI shows the generation of different types, which are distributed as follows: R22(7) and R45(15) resulting from the FA⋯water interaction (Fig. 19); R12(7), R22(14) and R23(8) resulting from the FA⋯MXDA interaction (Fig. 20) and finally R45(12), R24(8), R35(10) and R46(16) appearing via a FA⋯MXDA⋯water inter-connection (Fig. 21).
 | ||
| Fig. 19 Supramolecular synthons formed via the FA⋯water inter-molecular interaction in m-xylylenediamine/fumaric acid-trihydrate salt (IV). | ||
 | ||
| Fig. 20 FA⋯MXDA intermolecular interaction motifs in m-xylylenediamine/fumaric acid-trihydrate salt (IV). | ||
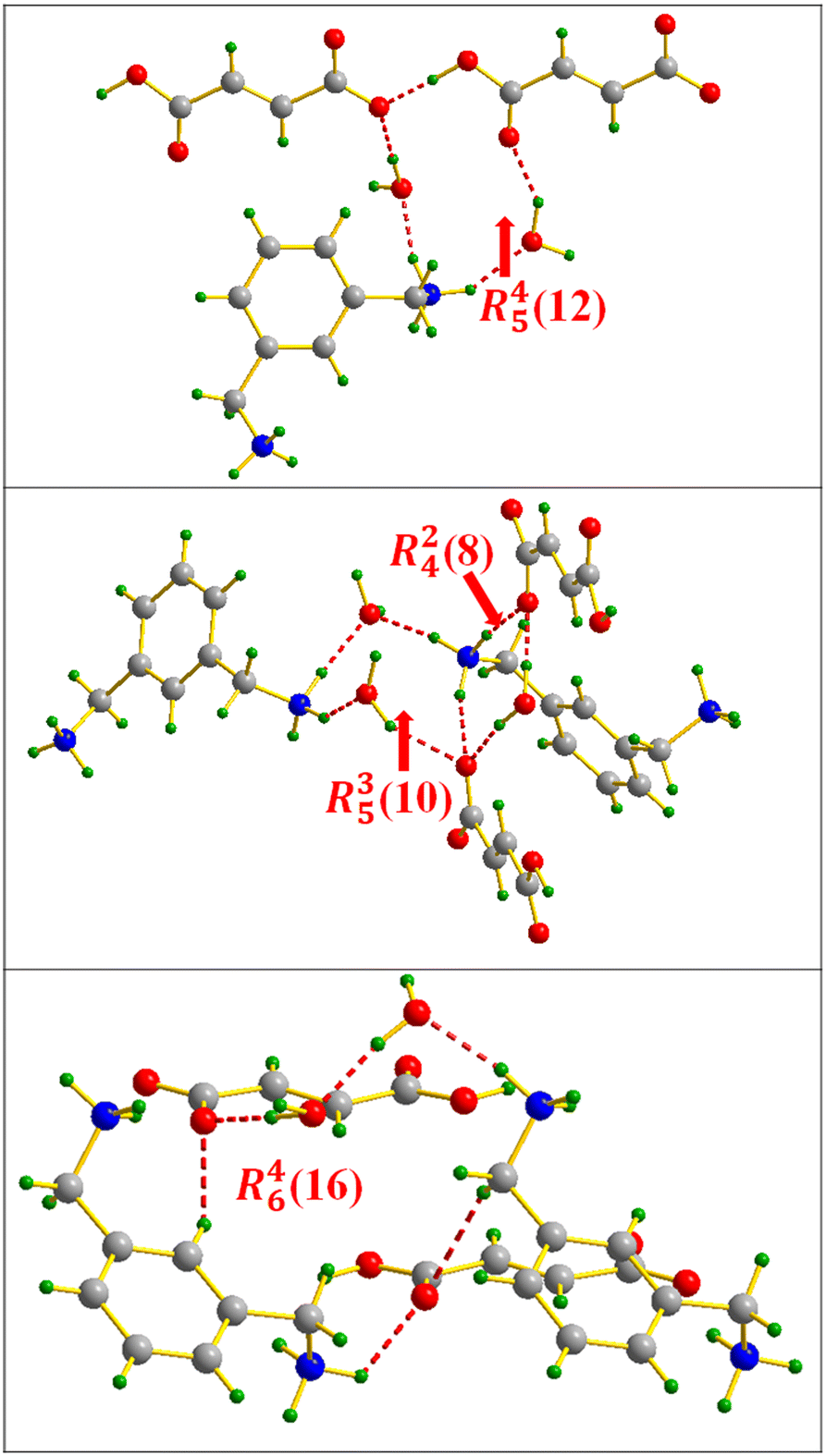 | ||
| Fig. 21 Supramolecular synthons resulting from the FA⋯MXDA⋯water interaction in m-xylylenediamine/fumaric acid-trihydrate salt (IV). | ||
3.2 DFT study
The DFT study focuses on analyzing the hydrogen bonds observed between anions in compounds III, IV, and VI, which serve as representative models of the three binding motifs highlighted in Scheme 1. As a first step, we computed the molecular electrostatic potential (MEP) surfaces of the salts, shown in Fig. 22. | ||
| Fig. 22 MEP surfaces of the salts of compounds III, IV and VI. Energies at selected points are indicated. Isosurface 0.001 a.u. | ||
The molecular electrostatic potential (MEP) maps reveal that the MEP maxima are located on the ammonium groups, ranging from 57.7 kcal mol−1 in salt IV to 84.1 kcal mol−1 in salt VI. The MEP minima are found on the oxygen atoms of the carboxylic or carboxylate groups of FA and MA, with values spanning from −60.9 kcal mol−1 in salts III and VI to −84.7 kcal mol−1 in salt IV. Particularly noteworthy are the MEP values at the hydrogen atoms in the monoanionic forms of MA and FA.
In MA, the hydrogens on the double bond exhibit slightly positive potentials (2.5 kcal mol−1), while in FA, the carboxylic hydrogen atoms show significantly higher positive values of 43.9 and 56.5 kcal mol−1. These elevated MEP values underscore the strong hydrogen-bonding capability of the carboxylic hydrogens, even in the anionic form, as a result of the stabilizing effect of the counter-cation. In the case of the dianionic form of FA (Fig. 22b), the MEP at the hydrogen atom of the double bond becomes negative (−15.7 kcal mol−1), reflecting the excess negative charge on the FA2− unit that is not fully compensated by the counter-cations.
Fig. 23 presents the QTAIM/NCIplot analysis of several assemblies derived from compound III. We first examined the isolated anion⋯anion dimer observed in the solid state, which forms an R23(7) synthon. QTAIM analysis confirms the presence of two CH⋯O hydrogen bonds, where both hydrogen atoms of the double bond in one anion interact with the oxygen atoms involved in the intramolecular OH⋯O hydrogen bond of the adjacent anion. Each hydrogen bond is characterized by a bond critical point (BCP, indicated by a small pink sphere) and a bond path connecting the hydrogen and oxygen atoms. In the graphical representation, noncovalent hydrogen bonds are shown as dashed lines, while those with significant covalent character are depicted as solid lines. Notably, only the CH⋯O interactions exhibit RDG isosurfaces (in green), indicating attractive noncovalent interactions. Despite this, the overall interaction energy of the dimer in the gas phase is strongly repulsive due to dominant coulombic repulsion between the anions. Indeed, gas-phase optimization of the dimer leads to complete dissociation of the monomers. Nevertheless, the individual CH⋯O hydrogen bonds are attractive in nature, as supported by the green RDG isosurfaces and QTAIM-derived interaction energies, which amount to −2.4 kcal mol−1 for the pair of CH⋯O contacts.
 | ||
| Fig. 23 QTAIM (BCPs in pink and bond path as dashed bonds) and NCIplot (RDG = 0.5, ρ cut-off = 0.04 a.u., color scale −0.035 a.u. ≤ (signλ2)ρ ≤ 0.035 a.u.) for the H-bonded anion⋯anion dimer in the X-ray (a) and optimized (b) geometries. (c) QTAIM analysis of a tetrameric fragment of III. The energies of the assemblies and those derived from potential energy densities (in red) are indicated in kcal mol−1. | ||
Fig. 23b shows the optimized structure of the anion⋯anion dimer in the presence of solvent effects (water). Unlike the gas-phase scenario, where the dimer dissociates completely, the inclusion of solvation leads to a stable hydrogen-bonded minimum, with a dimerization energy of −2.8 kcal mol−1 in water. This result suggests that the anion⋯anion dimer can exist in aqueous environments. While the dielectric environment in the crystal structure is unknown, it is likely that the stabilizing effect of the surrounding counterions is even greater than that provided by water. Interestingly, the optimized geometry differs from that observed in the X-ray structure. Instead of two CH⋯O interactions, the QTAIM/NCIplot analysis of the solvated structure reveals the formation of three CH⋯O hydrogen bonds. Notably, the total hydrogen-bonding energy estimated from QTAIM parameters—based on the potential energy density (V) at the bond critical points—is also −2.8 kcal mol−1, in excellent agreement with the computed dimerization energy. This correlation lends further credibility to the use of QTAIM-derived energy estimates for such interactions.
Finally, Fig. 23c illustrates a neutral tetrameric assembly, analysed to highlight the crucial role of counterions in the solid-state structure. The total formation energy is significantly large (−189.8 kcal mol−1), primarily due to strong coulombic attraction between oppositely charged species. To isolate the contribution of directional hydrogen bonds, we turned to QTAIM analysis. This reveals a network of interactions combining strong NH⋯O hydrogen bonds—characterized by blue RDG isosurfaces—and weaker CH⋯O interactions between cations and anions, alongside the R23(7) synthon. The total energy contribution of these directional hydrogen bonds amounts to −23.8 kcal mol−1, underscoring their essential role in shaping the solid-state architecture of compound III, even in the presence of much stronger but non-directional electrostatic forces.
A similar analysis was carried out for compound IV, where fumaric acid (FA) is in its dianionic form (see Fig. 24). The dimerization energy of the X-ray dimer (the R22(10) synthon) is highly repulsive at +185.8 kcal mol−1, owing to the strong coulombic repulsion between the two dianions. However, the CH⋯O hydrogen bonds, characterized by the presence of bond critical points (BCPs), bond paths, and green RDG isosurfaces, exhibit an attractive interaction energy of −1.2 kcal mol−1. Upon optimization in water, the dimer adopts a very similar geometry, with slightly shorter hydrogen bond distances (2.742 Å). The dimerization energy in this solvated environment is slightly positive at +1.4 kcal mol−1, representing a significant stabilization compared to the gas phase. The QTAIM-derived energy for the CH⋯O interactions in this optimized structure is −1.4 kcal mol−1, confirming their attractive nature, although still insufficient to fully overcome the electrostatic repulsion between the dianions, even in water.
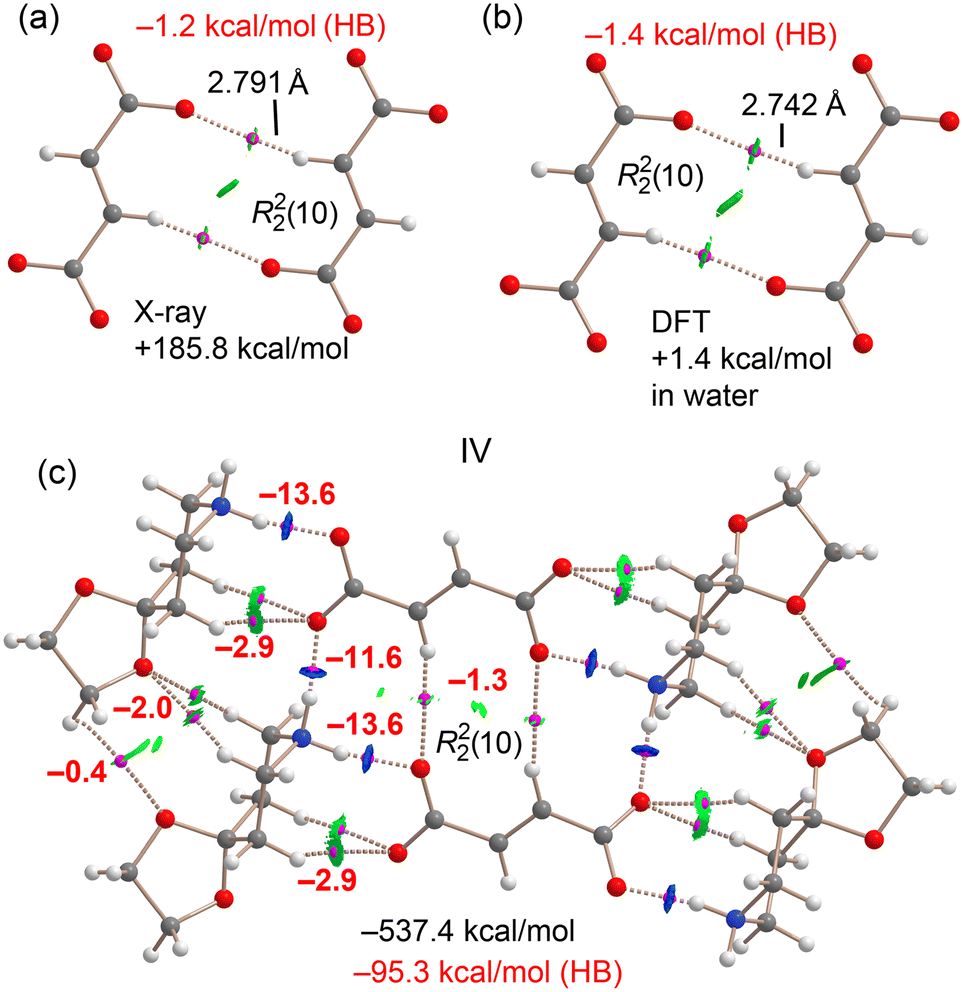 | ||
| Fig. 24 QTAIM (BCPs in pink and bond path as dashed bonds) and NCIplot (RDG = 0.5, ρ cut-off = 0.04 a.u., color scale −0.035 a.u. ≤ (signλ2)ρ ≤ 0.035 a.u.) for the H-bonded anion⋯anion dimer in the X-ray (a) and optimized (b) geometries. (c) QTAIM analysis of a hexameric fragment of IV. The energies of the assemblies and those derived from potential energy densities (in red) are indicated in kcal mol−1. | ||
For the larger assembly (Fig. 24c), which includes two dianions and four counterions, the total formation energy is substantially favorable at −539.4 kcal mol−1, driven largely by electrostatic attraction. Some NH⋯O hydrogen bonds are particularly strong, with interaction energies of −11.6 and −13.6 kcal mol−1, consistent with their short H⋯O distances (see the previous section) and the high MEP values. The QTAIM/NCIplot analysis again reveals a complex network of NH⋯O and CH⋯O interactions. The total energy contribution from these directional hydrogen bonds is −95.3 kcal mol−1, which, while smaller than the overall electrostatic stabilization, plays a decisive role in dictating the relative orientation of anions and cations in the solid-state packing of compound IV.
Finally, a similar analysis was performed for compound VI, as shown in Fig. 25. The hydrogen-bonded anion⋯anion dimer extracted from the X-ray structure exhibits a repulsive interaction energy of +20.1 kcal mol−1, consistent with coulombic repulsion.
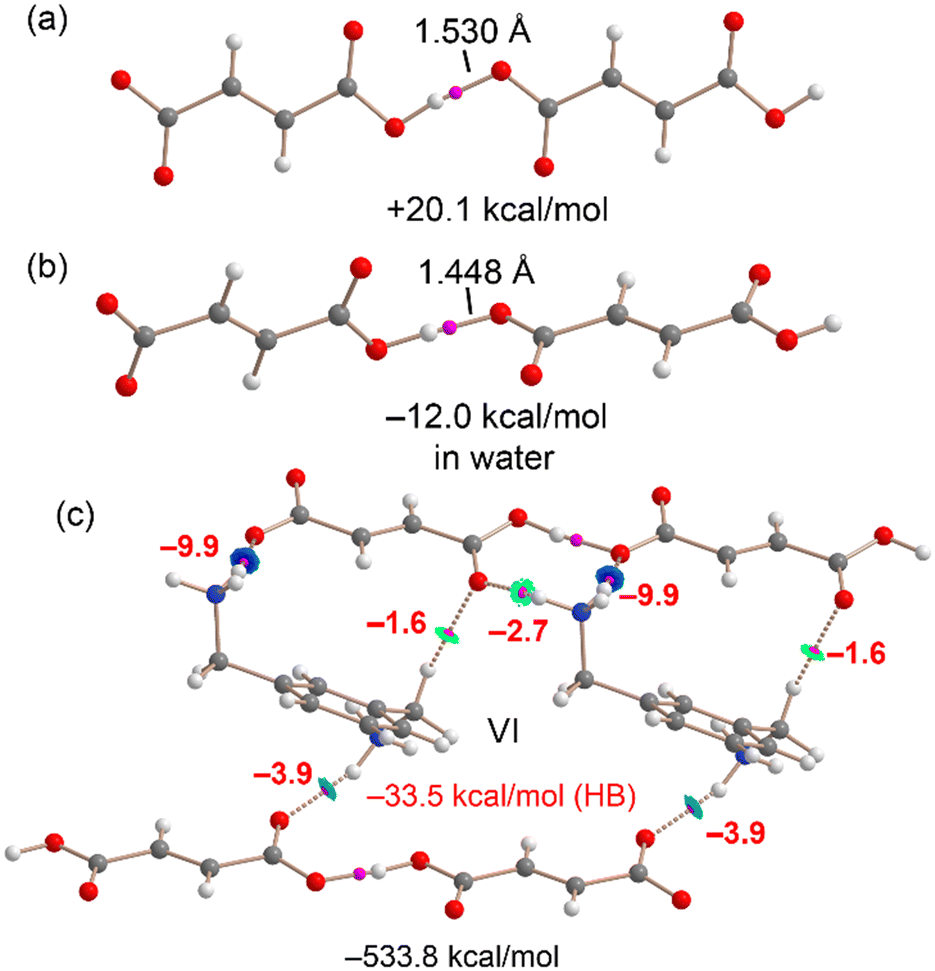 | ||
| Fig. 25 QTAIM (BCPs in pink and bond path as dashed bonds) and NCIplot (RDG = 0.5, ρ cut-off = 0.04 a.u., color scale −0.035 a.u. ≤ (signλ2)ρ ≤ 0.035 a.u.) for the H-bonded anion⋯anion dimer in the X-ray (a) and optimized (b) geometries. (c) QTAIM analysis of a hexameric fragment of VI. The energies of the assemblies and those derived from potential energy densities (in red) are indicated in kcal mol−1. | ||
However, QTAIM analysis reveals a short O⋯H hydrogen bond with significant covalent character, as indicated by the condition |V| > G at the bond critical point and the absence of an RDG isosurface (ρ cut-off = 0.04 a.u.). Upon optimization in water, the O⋯H distance shortens from 1.53 Å to 1.448 Å, and the dimerization energy becomes attractive (−12.0 kcal mol−1), suggesting that such anion⋯anion dimers are likely to form in aqueous environments.
In the larger assembly composed of two cations and four anions (Fig. 25c), the total formation energy is highly favorable at −533.8 kcal mol−1, primarily due to coulombic attraction. QTAIM analysis identifies several strong NH⋯O hydrogen bonds, with interaction energies of −9.9 kcal mol−1, as well as moderately strong ones at −3.9 and −2.7 kcal mol−1, depending on whether the acceptor is a carboxylate or a carboxylic acid group. Additionally, multiple CH⋯O interactions are observed, particularly involving the methylene groups. The total contribution from noncovalent hydrogen bonds is −33.5 kcal mol−1, while those with significant covalent character contribute −24.0 kcal mol−1, giving a combined stabilization energy of −57.5 kcal mol−1 in the tetrameric assembly. These results further underscore the structure-directing role of both anion⋯anion and anion⋯cation hydrogen bonds in the solid-state organization of compound VI.
4. Conclusions
In this work, we have synthesized and structurally characterized a series of six new salts based on fumaric acid (FA) and maleic acid (MA) combined with various ammonium-based coformers. The comparative crystallographic study revealed a rich landscape of supramolecular architectures, largely governed by classical and non-classical hydrogen bonding interactions. Notably, the observed structures demonstrate the ability of both isomers to participate in the formation of anion⋯anion interactions, a feature traditionally considered electrostatically unfavorable.Through comprehensive computational studies including MEP surface analysis, QTAIM, and NCIPlot methodologies, we have gained deeper insight into the energetic and directional nature of these anion⋯anion contacts. The results highlight how specific hydrogen bonds, including CH⋯O interactions, can mediate attractive forces between like-charged anionic units, especially in the presence of counterions or within high-dielectric environments such as water. The energetics derived from QTAIM correlate well with the computed interaction energies, supporting the robustness of these theoretical descriptors.
In larger assemblies that include counterions, strong NH⋯O and moderate CH⋯O interactions contribute significantly to the overall supramolecular stabilization. These findings underscore the structural and energetic importance of directional hydrogen bonds—both between anions and between anions and cations—in determining the final packing arrangement. Furthermore, our study provides compelling evidence that cooperative noncovalent networks can overcome coulombic repulsion and guide the formation of organized anionic assemblies in the solid state.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data have been deposited at the CCDC under CCDC numbers 2440152, 2440156, 2440157 and 2440159–2440161 and can be obtained from https://www.ccdc.cam.ac.uk.Author contributions
This manuscript was collaboratively written by all authors, who have approved the final version. Rafel Prohens contributed to the study's conception, experimental work, and manuscript writing. Antonio Frontera contributed to the study's conception, computational work and manuscript writing. Mahdi Jemai conducted experiments on crystal form screening and characterization techniques and manuscript writing. Houda Marouani contributed to manuscript writing. Miquel Barceló-Oliver and Thierry Roisnel analysed data on X-ray crystal structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the MICIU/AEI of Spain (projects PID2020-115637GB-I00, PID2023-148453NB-I00 and PID2023-146632OB-I00, FEDER funds) for funding. We thank the CTI (UIB) for computational facilities.References
- L. Georgieva, I. Ivanov, A. Marchev, I. Aneva, P. Denev, V. Georgiev and A. Pavlov, Appl. Biochem. Biotechnol., 2015, 176, 287–300 CrossRef CAS PubMed.
- R. A. Ilica, L. Kloetzer, A. I. Galaction and D. Cascaval, Biotechnol. Lett., 2019, 41, 47–57 CrossRef CAS PubMed.
- M. Mohmad, N. Agnihotri and V. Kumar, Phys. Sci. Rev., 2022, 9, 143–168 Search PubMed.
- F. Guo, M. Wu, Z. Dai, S. Zhang, W. Zhang, W. Dong, J. Zhou, M. Jiang and F. Xin, Biochem. Eng. J., 2020, 153, 107397 CrossRef CAS.
- Q. Xu, S. Li, H. Huang and J. Wen, Biotechnol. Adv., 2012, 30, 1685–1696 CrossRef CAS PubMed.
- M. Jemai, N. Issaoui, T. Roisnel, A. S. Kazachenko, H. Marouani and O. M. Al-Dossary, Z. fur Phys. Chem., 2023, 238, 509–529 CrossRef.
- R. Wojcieszak, F. Santarelli, S. Paul, F. Dumeignil, F. Cavani and R. V. Gonçalves, Sustain. Chem. Process., 2015, 3, 9 CrossRef.
- L. Kotte, V. Pendota, B. Sreedhar and J. B. Nanubolu, CrystEngComm, 2023, 25, 2662–2678 RSC.
- K. Nakai, K. Miki, T.i. Kikuchi and M. Yamano, Cryst. Growth Des., 2025, 25(1), 129–135 CrossRef CAS.
- A. Srivastava, P. Prajapati, D. P. Kale, P. Tandon and A. K. Bansal, J. Mol. Struct., 2025, 1319, 139471 CrossRef CAS.
- Y. Shen, M. Aucamp, H. E. Abdelhakim, X. Li, Y. Ghazali and K. Edkins, RSC Pharm, 2024, 1, 132–140 RSC.
- J. Wouters and L. Quéré, Pharmaceutical Salts and Co-Crystals, The Royal Society of Chemistry, Cambridge, 2012 Search PubMed.
- R. K. Das, S. K. Brar and M. Verma, Pharmacol. Rep., 2016, 68, 404–414 CrossRef CAS PubMed.
- A. B. Gottlieb, Nat. Rev. Drug Discovery, 2005, 4, 19–34 CrossRef CAS PubMed.
- R. Gold, R. A. Linker and M. Stangel, Clin. Immunol., 2012, 142, 44–48 CrossRef CAS PubMed.
- K. F. Bowes, G. Ferguson, A. J. Lough and C. Glidewell, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2003, 59, 100–117 CrossRef PubMed.
- M. J. Percino, M. Udayakumar, M. Cerón, E. Pérez-Gutiérrez, P. Venkatesan and S. Thamotharan, Front. Chem., 2023, 11, 1209428 CrossRef CAS PubMed.
- S. Suda, A. Tateno, D. Nakane and T. Akitsu, Int. J. Org. Chem., 2023, 13, 57–85 CrossRef CAS.
- R. Soria-Martínez and S. García-Granda, J. Mol. Struct., 2025, 1322, 140667 CrossRef.
- F. Weinhold and R. A. Klein, Angew. Chem., Int. Ed., 2014, 53, 11214–11217 CrossRef CAS PubMed.
- R. Beccaria, A. Dhaka, M. Calabrese, A. Pizzi, A. Frontera and G. Resnati, Chem. – Eur. J., 2023, 30, e202303641 CrossRef PubMed.
- A. P. Novikov, K. E. German, A. V. Safonov and M. S. Grigoriev, ChemistrySelect, 2022, 7, e202202814 CrossRef CAS.
- I. Mata, I. Alkorta, E. Molins and E. Espinosa, Chem. Phys. Lett., 2013, 555, 106–109 CrossRef CAS.
- I. Alkorta, I. Mata, E. Molins and E. Espinosa, Chem. – Eur. J., 2016, 22, 9226–9234 CrossRef CAS PubMed.
- D. Mungalpara, H. Kelm, A. Valkonen, K. Rissanen, S. Keller and S. Kubik, Org. Biomol. Chem., 2017, 15, 102–113 RSC.
- D. Mungalpara, A. Valkonen, K. Rissanen and S. Kubik, Chem. Sci., 2017, 8, 6005–6013 RSC.
- L. González, F. Zapata, A. Caballero, P. Molina, C. Ramírez de Arellano and I. Alkorta, Chem. Eur. J, 2016, 22, 7533–7544 CrossRef PubMed.
- W. Zhao, A. H. Flood and N. G. White, Chem. Soc. Rev., 2020, 49, 7893–7906 RSC.
- J. A. Odendal, J. C. Bruce, K. R. Kocha and D. A. Haynes, CrystEngComm, 2010, 12, 2398–2408 RSC.
- A. Lemmerer, S. Govindraju, M. Johnston, X. Motloung and K. L. Savig, CrystEngComm, 2015, 17, 3591–3595 RSC.
- Bruker, Bruker AXS Inc., Madison, Wisconsin, USA, 2023.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–11 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. A. Spackman and J. J. Mckinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110(13), 6158–6170 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- R. F. W. Bader, J. Phys. Chem. A, 1998, 102, 7314–7323 CrossRef CAS.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- T. A. Keith, AIMAll (Version 13.05.06), TK Gristmill Software, Overland Park, KS, 2013 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1997, 74, 1004–1010 Search PubMed.
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1990, 46, 256–262 CrossRef PubMed.
- M. N. Ahmed, M. A. Bashir, M. Akhtar, H. Andleeb, M. A. A. Ibrahim, I. Arshad, R. M. Gomila, P. A. Sidhom, M. N. Tahir, D. M. Gil and A. Frontera, CrystEngComm, 2023, 25, 6710–6718 RSC.
- N. Karaush-Karmazin, G. Baryshnikov and B. Minaev, MATEC Web Conf., 2022, 355, 01020 CrossRef CAS.
- M. Z. Shaikh, R. Alabada, Y. Ajaj, A. Abduldayeva, Z. M. Almarhoon, R. J. Butcher and M. A. Bhat, J. Mol. Struct., 2024, 1308, 138032 CrossRef CAS.
- S. L. Beko, M. U. Schmidt and A. D. Bond, CrystEngComm, 2012, 14, 1967–1971 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2440152, 2440156, 2440157 and 2440159–2440161. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ce00380f |
| This journal is © The Royal Society of Chemistry 2025 |