
Ultrafast structural dynamics of UV photoexcited cis,cis-1,3-cyclooctadiene observed with time-resolved electron diffraction†
Sri Bhavya
Muvva
 *a,
Yusong
Liu
bc,
Pratip
Chakraborty
de,
Joao Pedro Figueira
Nunes‡
a,
Andrew R.
Attar
b,
Surjendu
Bhattacharyya
f,
Kurtis
Borne
f,
Elio G
Champenois
b,
Nathan
Goff
g,
Kareem
Hegazy
bh,
Matthias C
Hoffmann
b,
Fuhao
Ji
b,
Ming-Fu
Lin
b,
Duan
Luo
b,
Lingyu
Ma
g,
Asami
Odate
g,
Shashank
Pathak
f,
Daniel
Rolles
f,
Artem
Rudenko
f,
Sajib Kumar
Saha
a,
Xiaozhe
Shen
b,
Xijie
Wang§
b,
Matthew R
Ware
b,
Stephen
Weathersby
b,
Peter M
Weber
g,
Kyle J
Wilkin
a,
Thomas J. A.
Wolf
b,
Yanwei
Xiong
a,
Xuan
Xu
g,
Jie
Yang
b,
Spiridoula
Matsika
d,
Thomas
Weinacht
c and
Martin
Centurion
*a
*a,
Yusong
Liu
bc,
Pratip
Chakraborty
de,
Joao Pedro Figueira
Nunes‡
a,
Andrew R.
Attar
b,
Surjendu
Bhattacharyya
f,
Kurtis
Borne
f,
Elio G
Champenois
b,
Nathan
Goff
g,
Kareem
Hegazy
bh,
Matthias C
Hoffmann
b,
Fuhao
Ji
b,
Ming-Fu
Lin
b,
Duan
Luo
b,
Lingyu
Ma
g,
Asami
Odate
g,
Shashank
Pathak
f,
Daniel
Rolles
f,
Artem
Rudenko
f,
Sajib Kumar
Saha
a,
Xiaozhe
Shen
b,
Xijie
Wang§
b,
Matthew R
Ware
b,
Stephen
Weathersby
b,
Peter M
Weber
g,
Kyle J
Wilkin
a,
Thomas J. A.
Wolf
b,
Yanwei
Xiong
a,
Xuan
Xu
g,
Jie
Yang
b,
Spiridoula
Matsika
d,
Thomas
Weinacht
c and
Martin
Centurion
*a
aDepartment of Physics and Astronomy, University of Nebraska–Lincoln, Lincoln, Nebraska, USA. E-mail: bhavya.m@huskers.unl.edu; martin.centurion@unl.edu
bSLAC National Accelerator Laboratory, Menlo Park, California, USA
cDepartment of Physics and Astronomy, Stony Brook University, Stony Brook, New York, USA
dDepartment of Chemistry, Temple University, Philadelphia, Pennsylvania, USA
eSchool of Chemistry, Pharmacy and Pharmacology, University of East Anglia, Norwich NR4 7TJ, UK
fJ.R. Macdonald Laboratory, Department of Physics, Kansas State University, Manhattan, Kansas, USA
gDepartment of Chemistry, Brown University, Providence, Rhode Island, USA
hDepartment of Physics, Stanford University, Stanford, USA
First published on 3rd December 2024
Abstract
Conjugated diene molecules are highly reactive upon photoexcitation and can relax through multiple reaction channels that depend on the position of the double bonds and the degree of molecular rigidity. Understanding the photoinduced dynamics of these molecules is crucial for establishing general rules governing the relaxation and product formation. Here, we investigate the femtosecond time-resolved photoinduced excited-state structural dynamics of cis,cis-1,3-cyclooctadiene, a large-flexible cyclic conjugated diene molecule, upon excitation with 200 nm using mega-electron-volt ultrafast electron diffraction and trajectory surface hopping dynamics simulations. We tracked the photoinduced structural changes from the Franck–Condon region through the conical intersection seam to the ground state. Our findings revealed a novel primary reaction coordinate involving ring distortion, where the ring stretches along one axis and compresses along the perpendicular axis. The nuclear wavepacket remains compact along this reaction coordinate until it reaches the conical intersection seam, and it rapidly spreads as it approaches the ground state, where multiple products are formed.
1 Introduction
Investigating ultrafast dynamics of photoexcited molecules, such as tracking transient states,1 mapping reaction pathways,2 and probing quantum effects,3 yields insights into fundamental natural processes3–7 and paves the way for controlling photochemical reactivity.8 Photoexcitation of polyatomic molecules can lead to the breakdown of the Born–Oppenheimer approximation, i.e., non-adiabatic coupling of electronic and nuclear motions, facilitating radiation-less transitions between electronic states through conical intersections (CIs).9–11 Tracking the wavepacket dynamics following photoexcitation along the reaction path to the CIs provides insights into the underlying mechanisms determining the reaction's outcome.12–15 Conjugated dienes, molecules having two double bonds separated by a single bond, exhibit photoinduced dynamics leading to various reactions via CIs, such as isomerization16,17 and ring transformations.11,18–20 The reaction outcomes are influenced by the dienes’ double bonds and the molecular rigidity they impose.21–23Many time-resolved studies investigated the photoinduced dynamics of small diene systems such as 1,3-cyclohexadiene (CHD)11,18,24–26 and butadiene (BD).16,17,27,28 CHD has been extensively studied as a prototypical example of electrocyclic ring transformation reactions,11 the general reactivity of which is described by the Woodward–Hoffmann rules.29 Upon UV excitation, CHD relaxes to the ground state through a CI seam and undergoes electrocyclic ring-opening to form 1,3,5-hexatriene.11,18,24–26 In contrast, UV excitation of BD leads to competition between various processes as the molecule relaxes to the ground state through CIs. These processes include cis–trans isomerization, electrocyclic ring closure to cyclobutene, and the formation of other products.16,17,27,28 The excited-state dynamics of these molecules have been investigated by considering the motions of the double bonds as the primary reaction coordinate. Although the initial photoexcitation is delocalized over both double bonds for both molecules, large amplitude motion allows the localization of dynamics on one of the double bonds in BD,17 unlike CHD.18,26 Studies on larger and more flexible systems are required to develop the general rules governing the dynamics of the diene systems.
So far there have been few studies on larger ring systems, and none with diffraction methods that give direct structural information. A few experimental and theoretical studies have been carried out on cis,cis-1,3-cyclooctadiene (cc-COD),30–35 which is a large flexible cyclic molecule with an eight-membered ring and conjugated double bonds (as shown in Fig. 1). Fuß et al.,30 using femtosecond mass spectrometry on gas phase cc-COD, reported the photoinduced products to be cis,trans-1,3-cyclooctadiene (ct-COD) through isomerization and bicyclo [4.2.0] oct-7-ene (BCO) through electrocyclic ring closure. A prior study reported the formation of BCO using selective cyclization of cc-COD in the solution phase by nanosecond pulsed UV irradiation.31 An earlier study on irradiation of solution of cc-COD with added sensitizer reported isomerization of cc-COD to ct-COD, which then converts to BCO.32 Recently, the excited-state time-resolved dynamics of cc-COD after photoexcitation to the S1 state was investigated theoretically using trajectory surface hopping (TSH) simulations33 and experimentally using time-resolved photoelectron spectroscopy (TRPES).34 The theory showcased the internal conversion from the S1 to the S0 state occurring over an extended region of the CI seam involving distortions around the double bonds, with the photoproducts formed in the ground state after traversing the CI seam.33 The TRPES experiment employed a 267 nm pump laser pulse to excite the cc-COD molecules to the S1 state, and its measurements showed a qualitative agreement with the TSH simulations, suggesting that the calculations could capture the main features of the dynamics observed experimentally.34 However, the measurements revealed an excited state lifetime of about 50 fs,34 faster than the 100 fs lifetime predicted by theory. An analysis of the calculations reported in ref. 34 indicated that the discrepancy between the experiment and theory could not be easily explained in terms of systematic issues with the experiment or the calculations but was likely a more subtle issue specific to the molecule. Therefore, this earlier work motivates further development of the calculations and comparison with other measurement approaches. Moreover, these previous spectroscopic measurements on cc-COD were not sensitive to structural changes and, therefore, could not trace the wavepacket during the nuclear motions from the Franck–Condon (FC) region to the ground state through a CI seam and the formation of multiple products.
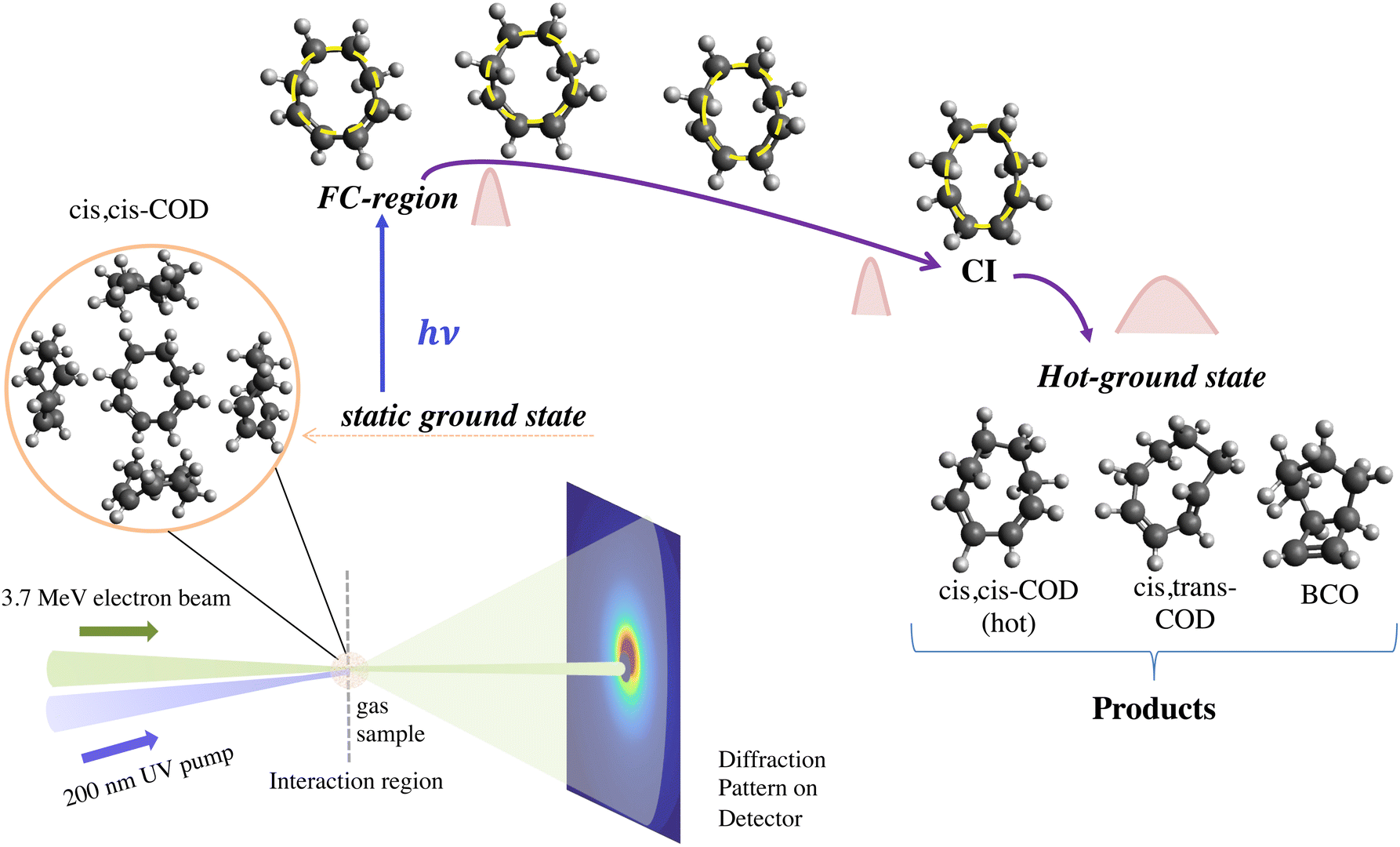 | ||
| Fig. 1 Schematic illustrating the UED experimental set-up and the photoinduced structural dynamics of cc-COD. The cc-COD molecules in the gas phase are introduced in the interaction region via a flow cell. The inset shows the randomly oriented cc-COD molecules. A 200 nm UV pump pulse is used to excite the cc-COD molecules from the static ground state, and 3.7 MeV electron pulses with a variable time delay are used to probe the molecules. The scattered diffraction patterns are recorded on the detector. The structures of the molecule in the static ground state/FC-region (t = 0 fs), in the excited-state at t = 20 fs, t = 40 fs, and at a conical intersection seam (t = 74 fs) are shown. The qualitative depiction of ring distortion motion (highlighted in yellow) in the molecule, as it approaches the CI seam, is shown along with the width of the nuclear wavepacket (in light red), which remains compact until the CI seam is reached and spreads out rapidly after passing through the CI seam and into the ground state where multiple products are formed. Representative structures of the photoproducts predicted from our TSH theoretical simulations at a pump–probe delay of 350 fs are shown. The products are cc-COD, ct-COD, and BCO, and their calculated yields (at t ∼ 400 fs) from TSH simulations are 63%, 32%, and 5%, respectively. | ||
We have carried out a time-resolved study of photoexcited cc-COD using mega-electron-volt ultrafast electron diffraction (MeV-UED),10,11,19,20,36–40 supported by TSH41 dynamics simulations. The TSH simulations, which were previously published in ref. 34, are used to calculate the UED observables and compared to the experimental measurements. The direct sensitivity of UED to changes in the nuclear geometry allows us to follow the structural dynamics and provides a direct experimental comparison to theoretically predicted dynamics.10,11,19,20,36–40,42–47 In this work, we tracked the molecular structure of cc-COD as the wavepacket moves from the FC region through the CI seam and to the electronic ground state. Our investigation unveiled a previously unrecognized primary reaction coordinate, predominantly characterized as a ring distortion, where the ring stretches along one axis and compresses along the perpendicular axis. The distortion was predicted by the TSH simulations and experimentally verified by comparing the measured diffraction signal with the diffraction signal predicted by the simulations. The simulations show that the nuclear wavepacket formed in the FC region remains compact as it moves along this ring distortion reaction coordinate until it reaches the CI seam, from where it rapidly spreads out as multiple photoproducts are formed in the ground state (illustrated in Fig. 1).
A schematic representation of the experimental setup is shown in Fig. 1. In brief, we used a 200 nm pump laser pulse to excite the cc-COD molecules in the gas phase and 3.7 MeV electrons as a probe. The time delay between pump and probe pulses was varied, and the diffraction pattern at each time delay was recorded. The instrument response time is 150 fs.36,37 Standard UED data analysis methodologies were employed in the retrieval of real space information, i.e., atomic pair distribution functions (PDFs) from reciprocal space (diffraction) data. In this approach, structural changes are reflected in the difference PDF (ΔPDF), which are extracted by subtracting the static signal from the time-dependent signal. The experimental results were interpreted with the aid of TSH dynamics simulations carried out at the extended multi-state second-order perturbation theory (XMS-CASPT2) level.48–50 TSH trajectories were propagated for 400 fs for excitation to the bright S1 state. The 200 nm pump used in our experiment is estimated to primarily populate the S1 state, as the S2 state is a dark state, as described in a prior publication.33 Detailed descriptions of the TSH simulations and UED data analysis are presented in the methods section and ESI.†
2. Methods
2.1. Experiment details
We performed our experiment at the Mega-electronvolt ultrafast electron diffraction (MeV-UED) facility at SLAC National Accelerator Laboratory.36 The experimental setup involved the use of a pump laser to excite the gas molecules and a MeV-electron beam as a probe to track the structural changes. The details of the experimental setup are described elsewhere.36,37 In summary, the 800 nm laser output of a Ti: sapphire laser system was separated into two beam paths. One path was used for generating the probe pulse of electrons, and the other path was for generating the pump laser beam to excite the gas sample. In the probe path, laser pulses were frequency tripled and then directed onto the photocathode of a radiofrequency (RF) gun to generate electron bunches containing ∼104 electrons. These electron bunches were accelerated to a kinetic energy of 3.7 MeV by an RF cavity and focused to a spot size of about 200 μm full width at half-maximum at the interaction region of the experimental chamber. The pulses in the pump path were converted to 200 nm pulses. These pump pulses, at an energy of 15 μJ, were focused at the interaction region of the experimental chamber to a spot size of about 260 × 200 μm full width at half-maximum and were nearly collinearly overlapped with the electron pulses. The sample cis,cis-1,3-cyclooctadiene (purity 98%) was purchased from Sigma-Aldrich and used without further purification. The sample reservoir was maintained at room temperature. We used a 4 mm flow cell (∼500 μm orifices, heated to 55 °C) to deliver the sample in the interaction region. The experimental response function of the set-up is estimated to be 150 fs,36,37 and we estimate that about 3.25% of the molecules were excited based on the relative signal levels of the experiment and theory (for details, see ESI†).The electrons scattered from the gas sample were detected using a combination of a phosphor screen and an electron-multiplying charge-coupled device (EMCCD) camera. The time delay between the pump and electron probe pulses was varied by adjusting the delay stage in the pump path. Time-dependent diffraction patterns were recorded for delay time points between −1 and 1 ps with separation between the time delay points as 62.5 fs, and a few other early and later delay points were also recorded. Diffraction patterns were recorded with 5-second exposure. A total of approximately 22 minutes of data was collected at each pump–probe time delay. Delay points were visited in random order to avoid systematic errors. Time-dependent diffraction data was integrated for the total data collected per delay point. The signal range was limited in the momentum space range of 0.6 Å−1 to 8 Å−1 due to a hole at the center of the detector and the limited signal-to-noise-ratio beyond 8 Å−1, respectively. We calculated the 1-D difference-modified scattering intensity signals in momentum space and ΔPDFs for each time delay. The data processing details are described in the ESI.† We used a bootstrapping algorithm to calculate the uncertainties in the experimental data (details are in Section S7 of ESI†).
2.2 Trajectory simulation details
We carried out TSH dynamics simulations of cc-COD at the extended multi-state second-order perturbation theory (XMS-CASPT2) level.48–50 The trajectories were propagated for 400 fs using the Newton-X package,51,52 after excitation to the bright S1 state. On-the-fly electronic structure calculations (energies, gradients, non-adiabatic couplings) were performed using the XMS-CASPT2 method along with three-state averaging and an active space of 4 electrons in 4 orbitals (SA3-CAS(4,4)) with cc-pVDZ53 basis set, using BAGEL package.54,55 A total of 200 trajectories were started and 116 reached the ground (S0) state and formed products. The remaining trajectories failed within 200 fs, before or soon after reaching the ground state. These 116 trajectories were used to generate a pool of sorted structures to compare with the end-product signal observed experimentally. The simulated time-dependent signal is based on the subset of trajectories (95) that propagated for more than 350 fs. Further details are described in the ESI.†3. Results and discussion
3.1. Comparison of experiment and theoretical diffraction signals
Fig. 2a shows the excellent agreement between the experimental and simulated static (ground state) PDF(r), where r is the internuclear distance. The distributions of different carbon–carbon distances at time zero from all simulated trajectories are shown in Fig. 2(a), which reflects the initial distribution of ground state structures. The distance between two, three, four, and five consecutive carbon atoms in the molecule's ring is denoted by R2C, R3C, R4C, and R5C, respectively.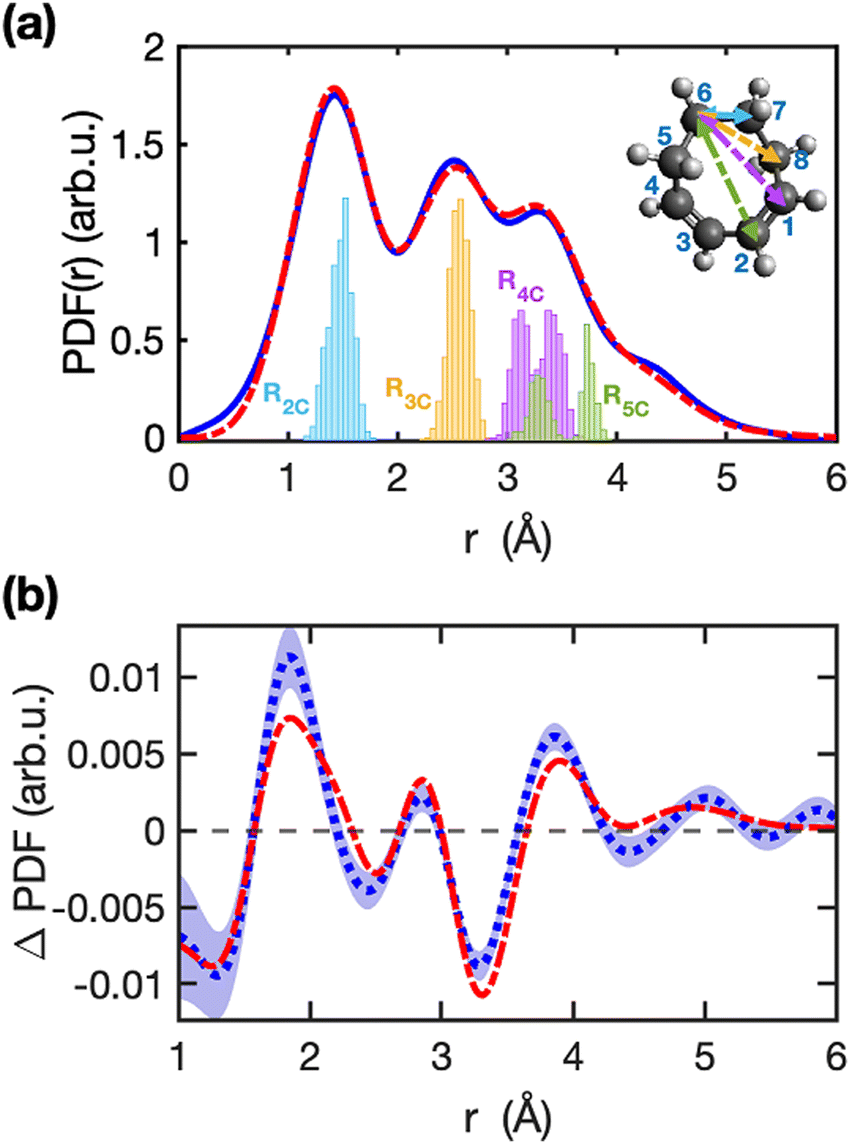 | ||
| Fig. 2 Comparison of experimental and simulated diffraction signals in real space. Panel (a) shows the experimental (blue-solid line) and simulated (red dashed line) stationary ground-state PDF(r) of COD. The blue, yellow, purple, and green histograms in panel (a) depict the distribution of carbon–carbon distances of the geometries in the TSH simulations at time-zero, for the ground-state Wigner ensemble. The inset illustrates the averaged stationary-ground state molecular structure (at T = 0 fs) with different carbon–carbon distances marked in color code for one of the carbon atoms in the molecule (R2C, R3C, R4C, and R5C distances are marked in blue, yellow, purple, and green arrows, respectively). Panel (b) shows the experimental (dotted blue line) and simulated (dashed red line) ΔPDF averaged over delays between 250 fs to 400 fs. The shaded blue area in panel (b) represents the statistical one-standard deviation error of the experiment signals calculated from a bootstrap analysis. | ||
The simulations show that after 250 fs, the population is back in the ground state, multiple hot structures are formed and there is no interconversion between these structures across the delays from 250 fs to 400 fs. The products predicted from simulations are classified into three categories: hot cc-COD, ct-COD, and BCO (structures shown in Fig. 1), and the details of the end-products formation are discussed in the sub-section: ‘Products formed’. The photoproducts of cc-COD, as identified in the TSH simulations here, have also been predicted in a separate theoretical study.35Fig. 2b shows the experimental and simulated difference diffraction signals in real space (ΔPDF(r)), averaged for time delays between 250 fs and 400 fs. The difference diffraction signal calculated from the structures in the simulation is in close agreement with the experimental measurement. The main information here is in the position of the positive signals which indicates new distances, and the valleys indicating lost distances. Small discrepancies in the amplitude of the peaks are likely due to artifacts introduced when transforming from momentum to real space (more details are in ESI† Sections S7 and S8). Based on the amplitude of the difference diffraction signal in the experiment, we extracted an excitation percentage of 3.3%. The real space signal allows us to directly identify the main interatomic distances that are changing after photoexcitation. As new structures are formed, we see a depletion in the region around 3.3 Å (R4C and R5C) and an increase in both shorter and longer distances around this value.
Fig. 3(a) shows the experimental ΔPDF(r, t), and Fig. 3(b) and (c) show the simulated ΔPDF. In Fig. 3(b) the predicted diffraction signal is convoluted in time with a Gaussian function of 150 fs full width at half maximum (FWHM) to match the temporal resolution of the measurement and provide a quantitative comparison. The red (positive) regions correspond to an increase in the signal intensity at those distances, while blue (negative) corresponds to a decrease. The trajectories extend up to 400 fs, so we use this time range for comparing simulations with experimental data. The same features are observed in experimental and simulated signals, with some fast oscillations in the simulated signal (Fig. 3(c)) not captured experimentally due to the finite temporal resolution. Three positive signals appear in the ΔPDF in the regions α (1.6 < r (Å) < 2.25), β (2.6 < r (Å) < 3), and γ (3.6 < r (Å) < 4.3), as seen in Fig. 3. Interestingly, we observe that the signal in region β, marked in Fig. 3, shows a transient increase in amplitude at early times. Fig. 4 shows lineouts of the experimental and theoretical ΔPDF at four different interatomic distances, which shows very good agreement between experiment and theory across multiple distances and timescales.
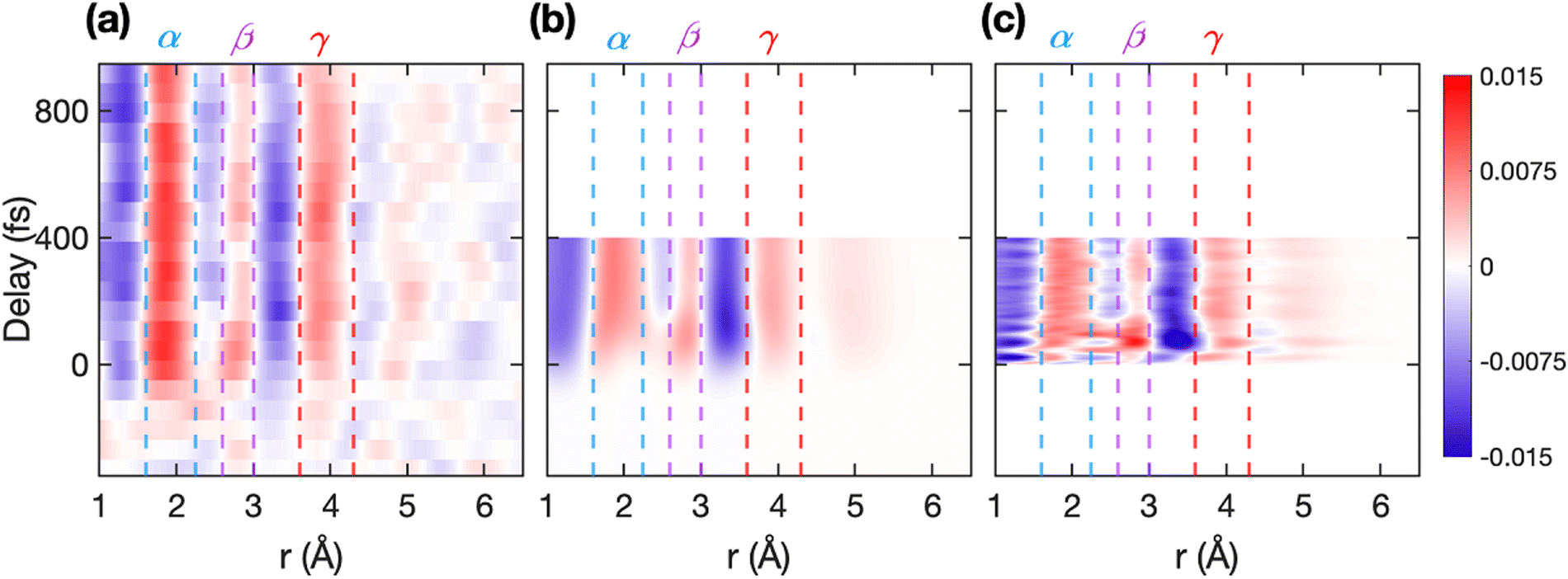 | ||
| Fig. 3 Experimental and simulated ΔPDFs. The false color plots show the difference diffraction signal (ΔPDF (r, t)) in real space as the function of pump–probe delay for (a) experiment, (b) (scaled) simulation convolved in time with a Gaussian of 150 fs full width at half maximum (FWHM), and (c) (scaled) simulation without any convolution in time. The regions α (1.6 < r (Å) < 2.25), β (2.6 < r (Å) < 3), and γ (3.6 < r (Å) < 4.3) are marked in all three panels with blue, purple, and red broken lines, respectively. The ΔPDF in region β (2.6 < r (Å) < 3) has a modulation in intensity with the delay. | ||
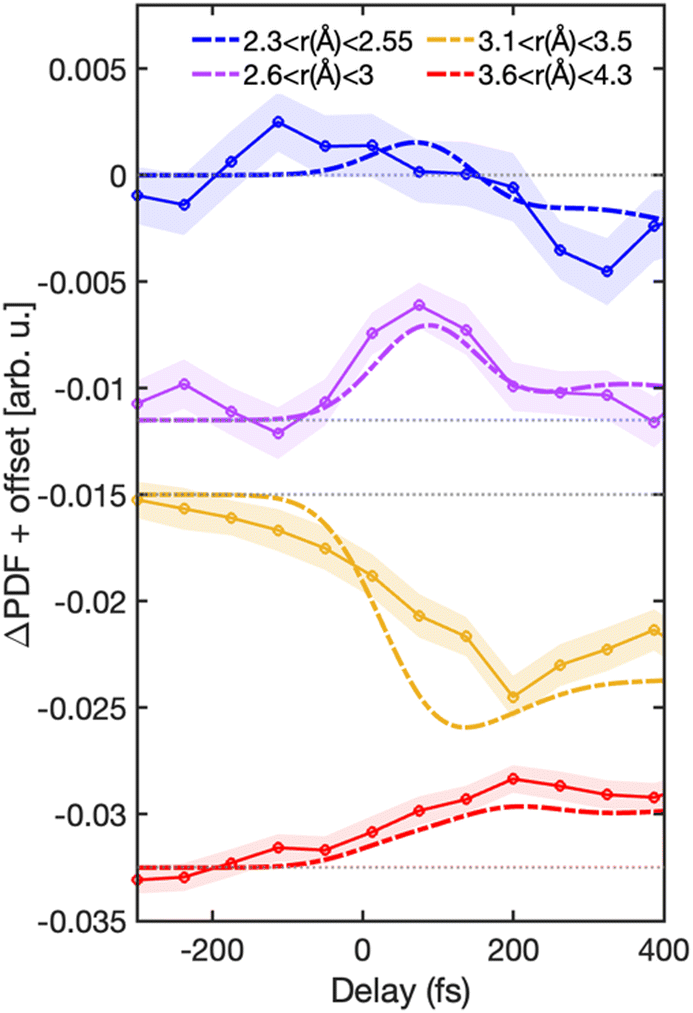 | ||
| Fig. 4 Comparison between experimental and simulated ΔPDF signals at different regions. The lineouts in blue, purple, yellow, and red colors represent the summed ΔPDF signal in regions of 2.3 < r (Å) < 2.55, 2.6 < r (Å) < 3, 3.1 < r (Å) < 3.5, and 3.6 < r (Å) < 4.3, respectively. The experimental signals are depicted by solid lines with circle markers, and the simulated signals are illustrated with broken lines. The shaded area in all panels represents the statistical (1-σ) error of the experimental signals for the corresponding region, calculated from a bootstrap analysis. An offset in the y-axis is added to the lineouts to displace them vertically for visualization. Each feature is plotted relative to the offset added. The offset is marked in grey dotted lines. | ||
3.2 Trajectory simulations
The early time dynamics, up to 150 fs, map the motion of the wavepacket from the excited-state, through nonadiabatic transitions back to the ground state. We found that the structural changes taking place from the FC region down to the CI seam can be captured by a new reaction coordinate: a distortion of the ring in which it simultaneously stretches along one axis while compressing along the perpendicular axis. This motion can be qualitatively visualized as a stretching of the ring, although it includes both in-plane and out-of-plane motions. In particular, the direction of the ring stretching results in a reduction in the separation between the double bonds. This reduction occurs after an initial increase of this separation within the first 20 fs of excitation, during which the ring expands (refer to Fig. S9 of ESI†). The molecule's ring is asymmetric in the ground state, as seen from the histograms of R5C and R4C distances displayed in Fig. 2(a). After photoexcitation, the ring stretches in a direction such that the asymmetry increases. This ring distortion motion creates a distinctive feature in the region β of the ΔPDF as multiple distances contribute positively to the signal in this narrow region (2.6 < r (Å) < 3) when the ring stretches. During the stretching motion, the shorter R5C and R4C distances compress, generating a positive signal in this region, while the longer R3C distances stretch leading also to an increase in the signal in the same region (shown in ESI† Fig. S8). The corresponding motion does not exhibit clear intensity signal modulations in ΔPDF at large distances (r (Å) < 3.6), i.e., in γ region, as observed in Fig. 3. This is because while the longer R4C distances stretch into the region, the longer R5C distances simultaneously stretch out from that region. The positive signal observed in region α of the ΔPDF is primarily due to the increased C–C bond distances in the ring and some of the shorter R5C distances compressing to this region. Furthermore, the depletion in the ΔPDF around 2.4 Å and 3.3 Å is caused by the R3C, R4C, and R5C distances moving out of these respective regions. Additional details describing the changes in carbon–carbon distances can be found in the ESI† Sections S11 and S12.The ring distortion can be parameterized by the difference between the long and the short R5C distances. For this, we introduce the parameter ε(t), which is the difference between these largest and shortest R5C distances at each pump–probe delay:
| ε(t) = [R5C]max(t) − [R5C]min(t) |
While this parameter cannot be directly observed experimentally because multiple distances contribute to the ΔPDF signal, we use it as a straightforward metric of the ring distortion and show that it is directly correlated with the signal in the region β of the ΔPDF.
Fig. 5(a) shows, for each trajectory, the largest R5C distance ([R5C]max, marked in red) and shortest R5C distance ([R5C]min, marked in blue) as a function of time. After an initial expansion of both distances over the first 20 fs, [R5C]max increases while [R5C]min decreases simultaneously, reaching a maximum difference (stretching) at a time around 70 fs (TS). This motion corresponds to a stretching of the ring with the initial longer distances becoming longer and the shorter distances becoming shorter. This motion occurs approximately simultaneously in all trajectories, which can be interpreted as a coherent wavepacket motion along this reaction coordinate. Subsequently, the ring returns towards the initial configuration as seen by the distances reverting towards their original values. Following this, the oscillations persist in all trajectories, albeit with different phases. This dephasing in the trajectories corresponds to a broadening of the wavepacket after reaching the maximum stretching of the ring.
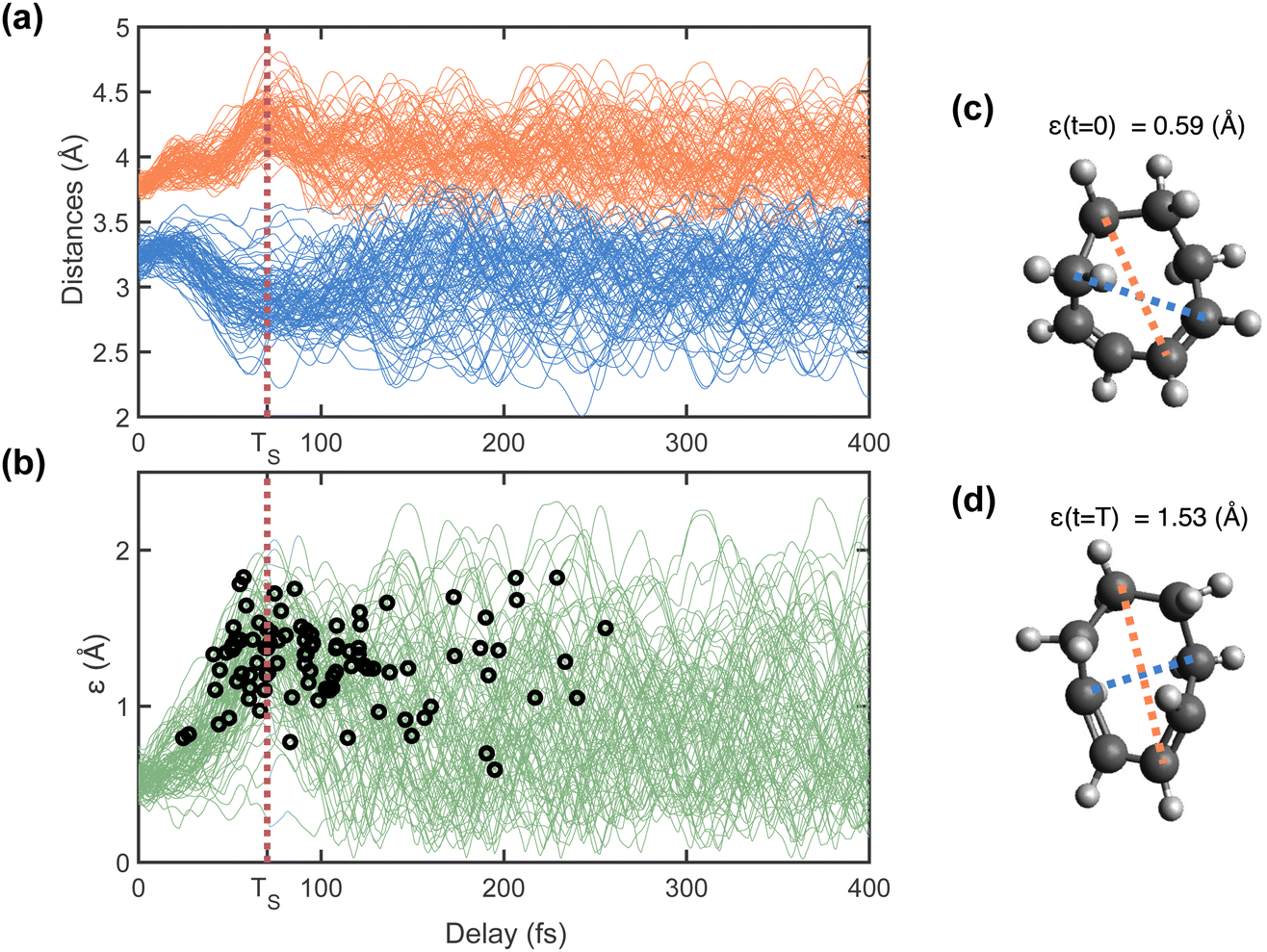 | ||
| Fig. 5 Ring distortion motion. Panel (a) shows the changes in largest ([R5C]max) and shortest ([R5C]min) R5C distances of all the 95 trajectories used in calculation of simulated signal, in orange and blue, respectively, as a function of delay for the first 400 fs. Panel (b) shows the evolution of the ring distortion motion parameter (ε(t)), which is equal to the difference of [R5C]max, and [R5C]min(t). The ε(t) values of all the 95 trajectories used for calculating simulated signal are shown in this plot. The black circles represent the S1 → S0 hop times of the trajectories. The vertical dotted line (in maroon) in panels (a) and (b) corresponds to the time TS (t = 70 fs) at which maximum ring stretching motion is observed over all the 95 trajectories. Panels (c) and (d) show representative structures from a trajectory simulation at t = 0 and TS (t = 70 fs) respectively, as an example to show the amount of ring stretch present. The largest and shortest R5C distances are marked with orange and blue lines, respectively, and the corresponding ε values are noted. | ||
Fig. 5(b) shows ε(t), overlaid with the time at which each trajectory hops from the excited-state to the ground state (S1 to S0), shown as the black open circles. The hop times are clearly clustered around time TS (t = 70 fs). This indicates that the wavepacket moves compactly along this coordinate until it reaches the CI seam, then quickly disperses as it reaches the ground state. The spreading is likely caused by the excess vibrational energy and by splitting into multiple products when the wavepacket reaches the ground state. Fig. 5(c) and (d) show the structures from a representative trajectory at the FC point (t = 0 fs) and at time TS (70 fs) to highlight the significant changes in the ring shape.
Fig. 6(a) shows the value ε(t) averaged over all trajectories. Fig. 6(b) shows the histogram of the probability distribution of hop times from S1 to S0 state for all trajectories, which has a peak at 74 fs. This time matches the maximum of εavg(t), which implies that the ring distortion motion described here is necessary for reaching the CI seam. The shaded area in Fig. 6(a) represents the standard deviation of ε(t), which clearly shows how the wavepacket spreads significantly and rapidly after traversing through the CI seam. The rapid spreading can be attributed to the excess vibrational energy as the wavepacket reaches the ground state and multiple products are formed. In the following section, we show that the temporal profile of the ring distortion motion signature identified in the simulation matches the temporal evolution of the signal in the β region of the experimental ΔPDF.
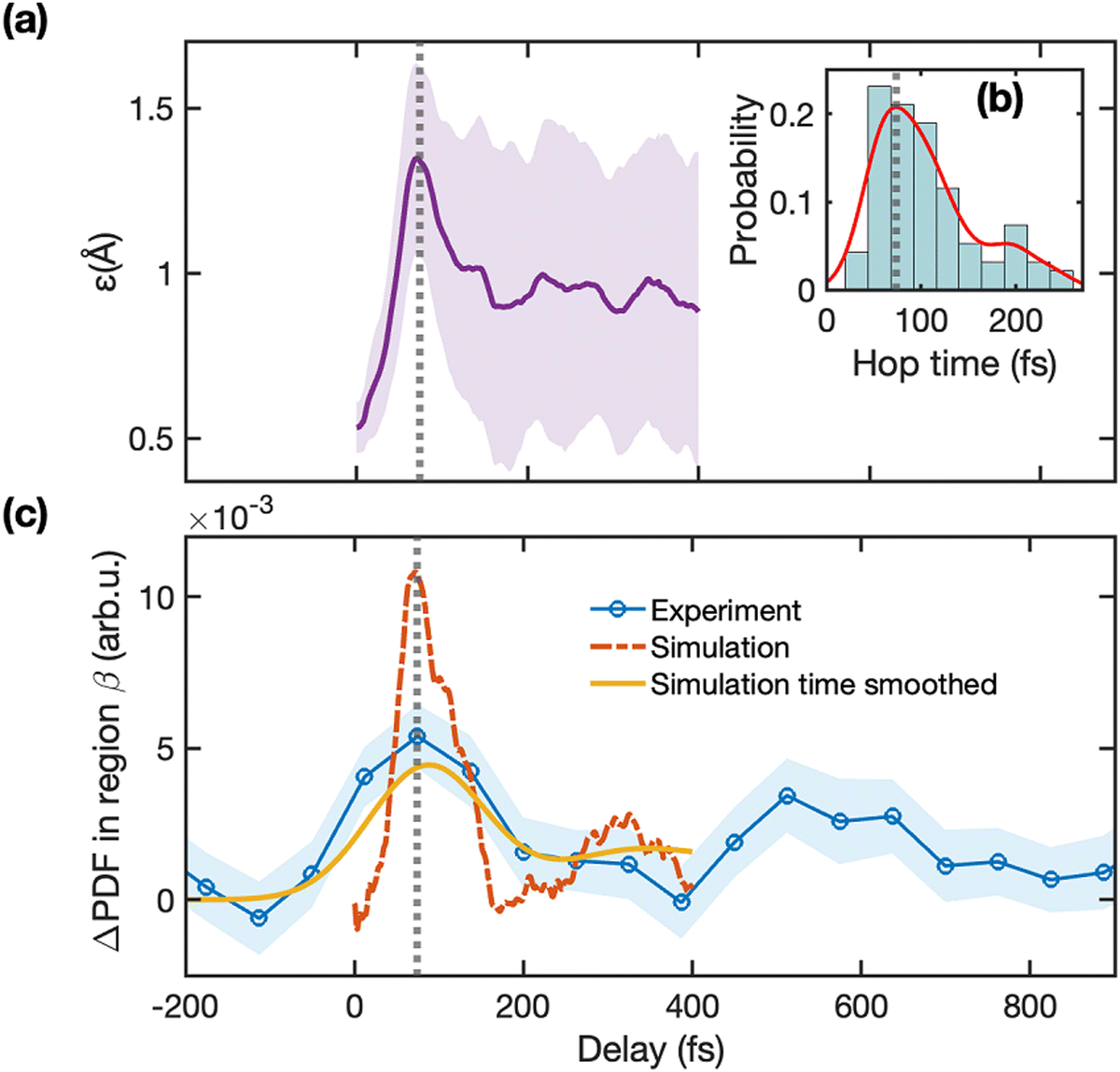 | ||
| Fig. 6 Ring stretching motion dynamics reflected in the experimental and simulated ΔPDFs. Panel (a) shows the amount of ring stretching averaged over the simulated trajectories as a function of time delay (εavg(t)) along with the standard deviation from the mean. The inset (b) shows the probability distribution of hop times from S1 to S0 (excited-state to ground-state) for all trajectories. Panel (c) shows the sum of ΔPDF intensity in the region β (2.6 < r (Å) < 3) for the experiment (blue) along with the one-standard-deviation error (shaded region), the corresponding signal from the simulation without (red) and with (yellow) 150 fs Gaussian time smoothing applied. The vertical grey dotted line in Panels (a), (b), and (c) marks the most likely time (74 fs) for the wavepacket to reach the CI seam. | ||
3.3. Experimental evidence of the ring distortion motion
Fig. 2, 3, and 4 have highlighted the strong overall agreement between experiment and theory, here we focus on the specific signature of the ring stretching in the diffraction signal. As discussed earlier, the ring distortion motion has a prominent signature in the region β of the ΔPDF. Fig. 6(c) shows the integrated intensity of the ΔPDF in the region β (2.6 < r (Å) < 3), where we observed a modulation of the intensity in both experiment and simulations (refer to Fig. 3). The dashed red line in Fig. 6(c) shows the intensity in the region β of the simulated diffraction signal generated from the trajectories. This signal is strongly correlated with ε(t). The solid yellow line shows the theoretical signal after convolution to match the temporal resolution of the experiment. This predicted signal (yellow line) quantitatively matches the measurement (blue line), further validating the interpretation of the trajectories. The longer time window of our UED data allows us to catch the revival of this modulation, with a lower amplitude, approximately 400 fs after the first peak. It is not clear if this second, weaker modulation is due to periodic stretching of the ring or due to the interaction of other vibrational modes, but it suggests that some vibrational coherence persists in the ground state. With the aid of the TSH trajectories, we have identified that the ring distortion motion in the ground state persists only in the hot cc-COD product (more details are in the ESI† Section S13), indicating that after the large structural changes needed to form the ct-COD and BCO products the energy in this mode quickly spreads to other vibrational modes.3.4 Reaction coordinate
While in previous experiments with CHD and BD,17,18,26 and in general in photophysics and photochemistry of molecules with double bonds, the motion involving double bonds stretching and twisting is considered to be a dominant reaction coordinate, here we have found a new reaction coordinate of ring distortion (ε) that produces a compact motion through a CI seam. In the simulations, we also observed oscillations in the bond length alternation (BLA) coordinate with a period of approximately 25 fs and decreasing amplitude (refer to ESI† Section S14). The BLA coordinate, which involves the stretching motion of double bonds, was coherent across the trajectories over the time the wavepacket takes to reach the CI seam (approximately 70 fs), after which this coherence behavior is lost. We observed the stretching of the ring during the first 70 fs in 98% of the trajectories. Additionally, we note that the ring stretching is slower as it involves a larger structural distortion, and the maximum distortion coincides with the wavepacket reaching the CI seam. The advantage of this slower motion is that it can be easier to observe experimentally, while the very fast double bond stretching would require a much shorter time resolution. Thus, both coordinates of ε and BLA are signatures of the CI, and the main difference is the frequency, which makes the ring distortion coordinate (ε) easier to capture experimentally. In addition, we also compared the ring distortion parameter reaction coordinate with torsional and pyramidalization motions. From our comparisons, we found that while all these motions of double bond stretching, torsional angles twisting, and pyramidalization angles changing take place after photoexcitation of cc-COD with 200 nm to S1 state, none of these parameters strongly correlate with the ring distortion reaction coordinate. These other motions do not oscillate on the same time scale as the ring distortion, and they undergo multiple oscillation periods before the molecule reaches the CI. The ring distortion, on the other hand, changes monotonically between excitation and reaching the CI, which makes it preferable as a reaction coordinate. The detailed comparison is in the Section S14 of ESI.†While all three molecules-BD, CHD, and COD, have the same two double bonds connected by a single bond, the photochemistry of CHD is quite different from the dynamics of COD and BD. The photochemistry of CHD leads to opening of the ring, while COD and BD behave similarly, as discussed in Chakraborty et al.33 In both cases, the majority of the dynamics involves cis–trans isomerization of the double bonds, with a single bond twisting being more prevalent than both double bonds twisting concurrently. The difference between BD and COD, however, is that COD has additional restrictions because of its ring structure. This ring structure leads to the ring distortion observed in this work, which is indeed a consequence of the torsional and bond stretching distortions.
3.5. Products formed
We classified the end-products broadly into three categories: hot cc-COD, ct-COD, and BCO (shown in Fig. 1). Within this broad classification, the simulations also predict an additional four end-products in which hydrogen migration takes place across the ring. Refer to ESI† Section S2 for details on the product classification. The hydrogen migration does not significantly change the geometry, and these end products are included within the three main groups. Our diffraction measurements are not sufficiently sensitive to distinguish the hydrogen migration, but the three main categories are clearly distinguishable. The product yields predicted from the TSH simulations are 63% cc-COD, 32% ct-COD, and 5% BCO. We performed a fitting analysis on the experimental data to determine the photoproduct yields. The signal from each photoproduct from the simulations is taken as the fitting basis (refer to ESI† Section S15). The best fit to the experiment is for product yields of 87.4 ± 8.8% cc-COD, 12.3 ± 7.1% ct-COD, and 0.3 ± 1% BCO for a time-averaged signal between 250 fs and 400 fs. Our experiment results indicate that hot cc-COD is the dominant product, followed by ct-COD as the subsequent significant product, which is in qualitative agreement with the theoretical prediction. We did not observe a significant change over the product distribution for up to the measured time of 900 fs. We captured the product yield after the wavepacket returns to the ground state, however, the product yield may change over longer time scales of nanoseconds and longer. Thus, the observed distribution of products may not match the final distribution of products of this reaction.4. Conclusions
Through a combination of experiment and theory, we followed the structural dynamics of photoexcited cc-COD from the excited-state to the ground state through the CI seam. We have identified a primary reaction coordinate of the nuclear motion from the FC region to the CI seam to be a ring distortion motion. This ring distortion motion produces a clear signature in the ΔPDF that was identified in the experiment in good agreement with the theoretical predictions. This ring distortion motion provides a new, alternative, signature of the distortions needed to reach CIs with the ground state in cyclic dienes, besides the well-established coordinates of double bond stretching and carbon pyramidalization. The TSH simulations show that the nuclear wavepacket formed in the FC region evolves in a compact manner towards the CI seam along this coordinate, and upon returning to the ground state, it rapidly splits into multiple photoproduct isomers: hot cis,cis-COD, cis,trans-COD, and BCO (with and without hydrogen migration). Our experiment results indicate that the dominant photoproduct is hot cis,cis-COD, followed by cis,trans-COD as the next significant photoproduct, qualitatively matching the theoretical predictions.Author contributions
MC and TW conceived the experiment. YL, JPFN, ARA, SB, KB, EGC, NG, KH, MCH, FJ, MFL, DL, LM, AO, SP, DR, AR, SKS, XS, XW, MRW, SW, PMW, KJW, TJAW, YX, XX, JY, TW, and MC participated in the data acquisition at the SLAC ultrafast electron diffraction facility. PC and SM performed the trajectory surface hopping simulations, and SBM calculated the theoretical scattering signal from the trajectory surface hopping simulated trajectories. SBM and YL analyzed the data with inputs from PC, JPFN, SM, TW, and MC. SBM and MC wrote the manuscript with inputs from all the authors.Data availability
The data supporting this article is available at Zenodo at the following link: https://doi.org/10.5281/zenodo.12720149.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The SLAC MeV UED facility is supported in part by the US Department of Energy, Office of Basic Energy Sciences, SUF Division Accelerator & Detector R&D program, the Linac Coherent Light Source Facility, and SLAC under contract no. DE-AC02-05-CH11231 and DE-AC02-76SF00515. SBM, JPFN, SB, SKS, KJW, YX, AR, DR, and MC were funded through the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, Office of Science, US Department of Energy under DOE-EPSCoR grant no. DE-SC0020276. SP and KB were funded by the US Department of Energy under grant no. DE-FG02-86ER1349. NG, AO, and PMW were funded by the US Department of Energy under grant no. DE-SC0017995. SM and PC acknowledge support from DOE, Award no. DE-FG02-08ER15983. TW and YL acknowledge support from DOE, Award No. DE-FG02-08ER15984. The computational work was performed using the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant no. ACI-1548562.Notes and references
- A. H. Zewail, J. Phys. Chem. A, 2000, 104, 5660–5694 CrossRef CAS.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- Y. C. Cheng and G. R. Fleming, Annu. Rev. Phys. Chem., 2009, 60, 241–262 CrossRef CAS PubMed.
- C. T. Middleton, K. de La Harpe, C. Su, Y. K. Law, C. E. Crespo-Hernández and B. Kohler, Annu. Rev. Phys. Chem., 2009, 60, 217–239 CrossRef CAS PubMed.
- C. E. Crespo-Hernández, B. Cohen, P. M. Hare and B. Kohler, Chem. Rev., 2004, 104, 1977–2020 CrossRef PubMed.
- G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. Van Grondelle, Nat. Chem., 2011, 3, 763–774 CrossRef CAS PubMed.
- Q. Wang, R. W. Schoenlein, L. A. Peteanu, R. A. Mathies and C. V. Shank, Science, 1994, 266, 422–424 CrossRef CAS.
- J. S. Lim and S. K. Kim, Nat. Chem., 2010, 2, 627–632 CrossRef CAS.
- D. Polli, P. Altoè, O. Weingart, K. M. Spillane, C. Manzoni, D. Brida, G. Tomasello, G. Orlandi, P. Kukura, R. A. Mathies and M. Garavelli, Nature, 2010, 467, 440–443 CrossRef CAS PubMed.
- J. Yang, X. Zhu, T. J. A. Wolf, Z. Li, J. P. F. Nunes, R. Coffee, J. P. Cryan, M. Gühr, K. Hegazy, T. F. Heinz, K. Jobe, R. Li, X. Shen, T. Veccione, S. Weathersby, K. J. Wilkin, C. Yoneda, Q. Zheng, T. J. Martinez, M. Centurion and X. Wang, Science, 2018, 361, 64–67 CrossRef CAS PubMed.
- T. J. A. Wolf, D. M. Sanchez, J. Yang, R. M. Parrish, J. P. F. Nunes, M. Centurion, R. Coffee, J. P. Cryan, M. Gühr, K. Hegazy, A. Kirrander, R. K. Li, J. Ruddock, X. Shen, T. Vecchione, S. P. Weathersby, P. M. Weber, K. Wilkin, H. Yong, Q. Zheng, X. J. Wang, M. P. Minitti and T. J. Martínez, Nat. Chem., 2019, 11, 504–509 CrossRef CAS.
- ed. W. Domcke, D. Yarkony and H. Köppel, Conical intersections: theory, computation and experiment, World Scientific Publishing, Singapore, Hackensack NJ and London, Vol. 17, 2011 Search PubMed.
- S. Matsika and P. Krause, Annu. Rev. Phys. Chem., 2011, 62, 621–643 CrossRef CAS.
- B. G. Levine and T. J. Martínez, Annu. Rev. Phys. Chem., 2007, 58, 613–634 CrossRef CAS PubMed.
- M. S. Schuurman and A. Stolow, Annu. Rev. Phys. Chem., 2018, 69, 427–450 CrossRef CAS PubMed.
- W. J. Glover, T. Mori, M. S. Schuurman, A. E. Boguslavskiy, O. Schalk, A. Stolow and T. J. Martínez, J. Chem. Phys., 2018, 148, 164303 CrossRef PubMed.
- B. G. Levine and T. J. Martínez, J. Phys. Chem. A, 2009, 113, 12815–12824 CrossRef CAS.
- S. Deb and P. M. Weber, Annu. Rev. Phys. Chem., 2011, 62, 19–39 CrossRef CAS.
- Y. Liu, D. M. Sanchez, M. R. Ware, E. G. Champenois, J. Yang, J. P. F. Nunes, A. Attar, M. Centurion, J. P. Cryan, R. Forbes, K. Hegazy, M. C. Hoffmann, F. Ji, M. F. Lin, D. Luo, S. K. Saha, X. Shen, X. J. Wang, T. J. Martínez and T. J. A. Wolf, Nat. Commun., 2023, 14, 2795 CrossRef CAS PubMed.
- E. G. Champenois, D. M. Sanchez, J. Yang, J. P. Figueira Nunes, A. Attar, M. Centurion, R. Forbes, M. Gühr, K. Hegazy, F. Ji, S. K. Saha, Y. Liu, M. F. Lin, D. Luo, B. Moore, X. Shen, M. R. Ware, X. J. Wang, T. J. Martínez and T. J. A. Wolf, Science, 2021, 374, 178–182 CrossRef CAS.
- P. Chakraborty, Y. Liu, S. McClung, T. Weinacht and S. Matsika, J. Phys. Chem. A, 2022, 126, 6021–6031 CrossRef CAS.
- O. Schalk, A. E. Boguslavskiy, A. Stolow and M. S. Schuurman, J. Am. Chem. Soc., 2011, 133, 16451–16458 CrossRef CAS PubMed.
- S. L. Horton, Y. Liu, P. Chakraborty, S. Matsika and T. Weinacht, Phys. Rev. A, 2017, 95, 063413 CrossRef.
- A. R. Attar, A. Bhattacherjee, C. D. Pemmaraju, K. Schnorr, K. D. Closser, D. Prendergast and S. R. Leone, Science, 2017, 356, 54–59 CrossRef CAS PubMed.
- M. P. Minitti, J. M. Budarz, A. Kirrander, J. S. Robinson, D. Ratner, T. J. Lane, D. Zhu, J. M. Glownia, M. Kozina, H. T. Lemke, M. Sikorski, Y. Feng, S. Nelson, K. Saita, B. Stankus, T. Northey, J. B. Hastings and P. M. Weber, Phys. Rev. Lett., 2015, 114, 255501 CrossRef CAS PubMed.
- I. Polyak, L. Hutton, R. Crespo-Otero, M. Barbatti and P. J. Knowles, J. Chem. Theory Comput., 2019, 15, 3929–3940 CrossRef CAS.
- Y. Dou, B. R. Torralva and R. E. Allen, J. Phys. Chem. A, 2003, 107, 8817–8824 CrossRef CAS.
- M. Olivucci, F. Bernardi, I. N. Ragazos and M. A. Robb, J. Am. Chem. Soc., 1993, 115, 3710–3721 CrossRef CAS.
- R. B. Woodward and R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1969, 8, 781–853 CrossRef CAS.
- W. Fuß, S. Panja, W. E. Schmid and S. A. Trushin, Mol. Phy., 2006, 104, 1133–1143 CrossRef.
- K. Komori-Orisaku, Y. Hirose and I. Iwakura, Photochem. Photobiol. Sci., 2017, 16, 146–150 CrossRef CAS.
- R. S. H. Liu, J. Am. Chem. Soc., 1967, 89, 112–114 CrossRef CAS.
- P. Chakraborty, Y. Liu, T. Weinacht and S. Matsika, J. Chem. Phys., 2020, 152, 174302 CrossRef CAS.
- Y. Liu, P. Chakraborty, S. Matsika and T. Weinacht, J. Chem. Phys., 2020, 153, 074301 CrossRef CAS.
- C. Cisneros, Master Thesis, California State University, Long Beach, 2022 Search PubMed.
- S. P. Weathersby, G. Brown, M. Centurion, T. F. Chase, R. Coffee, J. Corbett, J. P. Eichner, J. C. Frisch, A. R. Fry, M. Gühr, N. Hartmann, C. Hast, R. Hettel, R. K. Jobe, E. N. Jongewaard, J. R. Lewandowski, R. K. Li, A. M. Lindenberg, I. Makasyuk, J. E. May, D. McCormick, M. N. Nguyen, A. H. Reid, X. Shen, K. Sokolowski-Tinten, T. Vecchione, S. L. Vetter, J. Wu, J. Yang, H. A. Dürr and X. J. Wang, Rev. Sci. Instrum., 2015, 86, 073702 CrossRef CAS PubMed.
- X. Shen, J. P. F. Nunes, J. Yang, R. K. Jobe, R. K. Li, M. F. Lin, B. Moore, M. Niebuhr, S. P. Weathersby, T. J. A. Wolf, C. Yoneda, M. Guehr, M. Centurion and X. J. Wang, Struct. Dyn., 2019, 6, 054305 CrossRef CAS PubMed.
- J. Yang, M. Guehr, T. Vecchione, M. S. Robinson, R. Li, N. Hartmann, X. Shen, R. Coffee, J. Corbett, A. Fry, K. Gaffney, T. Gorkhover, C. Hast, K. Jobe, I. Makasyuk, A. Reid, J. Robinson, S. Vetter, F. Wang, S. Weathersby, C. Yoneda, M. Centurion and X. Wang, Nat. Commun., 2016, 7, 11232 CrossRef CAS PubMed.
- J. Yang, X. Zhu, J. P. F. Nunes, J. K. Yu, R. M. Parrish, T. J. A. Wolf, M. Centurion, M. Gühr, R. Li, Y. Liu, B. Moore, M. Niebuhr, S. Park, X. Shen, S. Weathersby, T. Weinacht, T. J. Martinez and X. Wang, Science, 2020, 368, 885–889 CrossRef CAS.
- J. P. Figueira Nunes, L. M. Ibele, S. Pathak, A. R. Attar, S. Bhattacharyya, R. Boll, K. Borne, M. Centurion, B. Erk, M. F. Lin, R. J. G. Forbes, N. Goff, C. S. Hansen, M. Hoffmann, D. M. P. Holland, R. A. Ingle, D. Luo, S. B. Muvva, A. H. Reid, A. Rouzée, A. Rudenko, S. K. Saha, X. Shen, A. S. Venkatachalam, X. Wang, M. R. Ware, S. P. Weathersby, K. Wilkin, T. J. A. Wolf, Y. Xiong, J. Yang, M. N. R. Ashfold, D. Rolles and B. F. E. Curchod, J. Am. Chem. Soc., 2024, 146, 4134–4143 CrossRef CAS.
- M. Barbatti, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 620–633 CAS.
- M. Centurion, J. Phys. B: At., Mol. Opt. Phys., 2016, 49, 062002 CrossRef.
- K. J. Wilkin, Y. Xiong, H. Zhao, S. B. Muvva, S. K. Saha and M. Centurion, Struct. Dyn., 2022, 9, 054303 CrossRef CAS PubMed.
- Y. Xiong, K. J. Wilkin, S. K. Saha, S. B. Muvva, H. Zhao and M. Centurion, Phys. Rev. A, 2022, 106, 033109 CrossRef CAS.
- Y. Liu, S. L. Horton, J. Yang, J. P. F. Nunes, X. Shen, T. J. A. Wolf, R. Forbes, C. Cheng, B. Moore, M. Centurion, K. Hegazy, R. Li, M. F. Lin, A. Stolow, P. Hockett, T. Rozgonyi, P. Marquetand, X. Wang and T. Weinacht, Phys. Rev. X, 2020, 10, 021016 CAS.
- J. P. F. Nunes, M. Williams, J. Yang, T. J. A. Wolf, C. D. Rankine, R. Parrish, B. Moore, K. Wilkin, X. Shen, M.-F. Lin, K. Hegazy, R. Li, S. Weathersby, T. J. Martinez, X. J. Wang and M. Centurion, Phys. Chem. Chem. Phys., 2024, 26, 17991–17998 RSC.
- K. Hegazy, V. Makhija, P. Bucksbaum, J. Corbett, J. Cryan, N. Hartmann, M. Ilchen, K. Jobe, R. Li, I. Makasyuk, X. Shen, X. Wang, S. Weathersby, J. Yang and R. Coffee, Commun. Phys., 2023, 6, 325 CrossRef CAS.
- J. Finley, P. Å. Malmqvist, B. O. Roos and L. Serrano-Andrés, Chem. Phys. Lett., 1998, 288, 299–306 CrossRef CAS.
- A. A. Granovsky, J. Chem. Phys., 2011, 134, 214113 CrossRef PubMed.
- T. Shiozaki, W. Gyroffy, P. Celani and H. J. Werner, J. Chem. Phys., 2011, 135, 081106 CrossRef PubMed.
- M. Barbatti, M. Ruckenbauer, F. Plasser, J. Pittner, G. Granucci, M. Persico and H. Lischka, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 26–33 CAS.
- M. Barbatti; G. Granucci; M. Ruckenbauer; F. Plasser; R. Crespo-Otero; J. Pittner; M. Persico and H. LischkaNEWTON-X: A package for Newtonian dynamics close to the crossing seam, version 2, 2016, https://www.newtonx.org Search PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- T. Shiozaki, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1331 Search PubMed.
- BAGEL, Brilliantly Advanced General Electronic-structure Library. https://www.nubakery.org under the GNU General Public License, ( 2017) Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02785j |
| ‡ Present address: Diamond Light Source Ltd, Didcot, UK. |
| § Present addresses: Department of Physics, University of Duisburg-Essen 47052 Duisburg, Germany; Department of Physics, University Dortmund, 44221 Dortmund, Germany; Research Center Chemical Sciences and Sustainability, Research Alliance Ruhr, 44780 Bochum, Germany. |
| This journal is © the Owner Societies 2025 |