
Reactivity in Friedel–Crafts aromatic benzylation: the role of the electrophilic reactant†
Diana
Cheshmedzhieva
 *a,
Ivan
Atanasov
a,
Sonia
Ilieva
a,
Boris
Galabov
*a and
Henry F.
Schaefer III
*b
*a,
Ivan
Atanasov
a,
Sonia
Ilieva
a,
Boris
Galabov
*a and
Henry F.
Schaefer III
*b
aDepartment of Chemistry and Pharmacy, University of Sofia, Sofia 1164, Bulgaria. E-mail: dvalentinova@chem.uni-sofia.bg; galabov@chem.uni-sofia.bg
bCenter for Computational Quantum Chemistry, University of Georgia, Athens, GA 30602, USA. E-mail: ccq@uga.edu
First published on 23rd October 2024
Abstract
Density functional theory is employed in understanding the reactivity in the TiCl4 catalyzed Friedel–Crafts benzylation of benzene with substituted benzyl chlorides in nitromethane solvent. A series of ten substituted (in the aromatic ring) benzyl chlorides are characterized by theoretical reactivity indices. The theoretical parameters are juxtaposed to experimental relative rates of benzylation. It is established that the carbon–chlorine ionic bond dissociation energy and the Hirshfeld charge at the chlorine atom for the benzyl chlorides reactants – describe quite satisfactorily the reactivity trends. These results provide further insights into the factors governing reactivity in EAS reactions, which so far have been mostly focused on rate variations induced by changes in the structure of the aromatic substrate. The EAS benzylation investigated is quite unusual since, in contrast to most EAS reactions, the latest experimental kinetic results suggest that the aromatic substrate does not participate in the kinetic equation of the process. To shed more light on this unexpected result, we also conducted a theoretical study on the mechanistic pathway by applying M06-2X density functional computations combined with several basis sets: 6-311+G(d,p), 6-311+G(2df,2p), and def2-TZVPP. Because of the well-known difficulties in evaluating realistic free energy barriers for organic reactions, we tested two solvent models in determining the barrier for the TiCl4-catalyzed Friedel–Crafts benzylation of benzene by benzyl chloride. Since all methods employed did not provide satisfactory results for the free energy barriers, we used a combination of theoretically estimated enthalpy barriers and the available (from kinetic experiments) entropy contribution. This approach enabled us to verify that indeed the rate of this EAS reaction does not depend on the nature of the aromatic substrate. The computations revealed the structure and relative energies of the critical structures along the mechanistic pathway. Four intermediates were established along the reaction route.
Introduction
The Friedel–Crafts electrophilic substitution of aromatic compounds provides effective routes for the synthesis of various alkylated or acylated systems, intermediates for further transformations, as well as numerous industrial products.1–6 The kinetic and mechanistic studies of the Friedel–Crafts alkylation reactions have a long history.5–12 In the present research we characterize the reactivity associated with a particular reaction from this class electrophilic aromatic substitution (EAS) reactions: the TiCl4-catalyzed benzylation of benzene by benzyl chlorides in nitromethane solvent (eqn (1)). The kinetic results of Olah et al.7 provide a rare opportunity to investigate the role of the changing structure of the electrophilic reactant on the rate of an electrophilic aromatic substitution (EAS) reaction. | (1) |
Early kinetic results7–10 on the Friedel–Crafts alkylation revealed that depending on the type of reactants, catalyst, and solvent, alternative mechanistic pathways may be followed. The benzylation of arenes with substituted benzyl chlorides catalyzed by AlCl3 in nitrobenzene suggested a displacement mechanism involving nucleophilic attack of the arene on the benzyl chloride-AlCl3 initially formed complex.9 A third order kinetic equation was suggested:
| Rate = k3[RCl][AlCl3][arene] | (2) |
| Rate = k3[RBr][GaBr3][arene] | (3) |
The rate equation for the TiCl4-catalyzed benzylation of benzene with benzyl chloride in nitromethane, reported by DeHaan et al.,11,12 has the form:
| Rate = k3[PhCH2Cl][TiCl4]2 | (4) |
| Rate = k4[ArCH2Cl][TiCl4]2[arene] | (5) |
Theoretical modeling of the mechanisms of EAS acylation and alkylation reactions have been also conducted.23–27 Kim et al.23 investigated the role of the Lewis acid interaction with the aromatic ring in the Friedel–Crafts reactions using the MP2/6-311++G(d,p) method. Yamabe et al.25 applied B3LYP/6-31+(d,p) density functional theory computations in modeling the benzene acylation with acetyl chloride and the alkylation with 2-chloropropane. Catalysis by the Al2Cl6 dimer in simulated dichloromethane solvent was considered. For both reactions the formation of an arenium ion intermediate was shown to be an essential step. Gandon et al.26 conducted M06-2X/6-311+G(2d,2p) computations in studying the GaCl3-catalyzed methylation of benzene. These authors showed that a Ga2Cl6 adduct is a more active catalyst compared to the monomeric GaCl3. Three alternative possible mechanistic pathways were considered. The results indicated that the reaction follows a concerted mechanism, which does not involve the formation of an arenium ion intermediate. Nogeira and Pliego27 investigated the AlCl3-catalyzed alkylation of benzene by isopropyl chloride in simulated benzene solution. The reaction pathways considering both AlCl3 and Al2Cl6 catalysts were theoretically modeled using X3LYP/def2-SVP (ma-def2-SVP) computations. These authors showed that the energetically favored route involves catalysis by the Al2Cl6 adduct. The reaction pathway includes the formation of a CH3CHCH3+⋯Al2Cl7− ion pair, which then reacts with benzene to form a Wheland (arenium ion) intermediate in the rate-controlling step of the process.
The chemical reactivity associated with electrophilic aromatic substitution (EAS) processes has been of prime interest over the long history of application of these reactions.14–21 The progress on the topic has been presented in recent studies and reviews.17–21,28–36 Notably, theoretical parameters describing aromatic reactivity are frequently verified by comparisons with Hammett type substituent constants.13,17,18,21,28 Machine learning methods employing theoretically evaluated proton affinities that aimed at providing the experimental chemists with effective tools for the prediction of both relative reactivity and positional selectivity were also proposed.34–36
As mentioned, we apply in the present research theoretically derived quantities characterizing the role of the electrophilic reactant on the rates of benzylation of benzene with substituted in the ring benzyl chlorides. The detailed experimental study of Olah et al.7 on the rates of the Lewis acid catalyzed benzylation of benzene and toluene with substituted benzyl chlorides provides an excellent platform to test the performance of theoretically derived quantities in describing reactivity. In Table 16 of ref. 7 these authors present results on the relative rates for the competitive benzylation of benzene by benzyl chloride and substituted in the aromatic ring derivatives under catalysis of TiCl4 in nitromethane solvent. These kinetic results are employed in the present study as a basis for assessment of the applicability of theoretically derived parameters in describing the reactivity trends for these reactions. The interpretation of reactivity patterns is strongly associated with the mechanism of the reaction of Friedel–Crafts benzylation considered. We, therefore, report first the mechanistic features of this reaction.
Computational methods
The computational results discussed in most detail in the present research were conducted using the M06-2X density functional37 combined with the 6-311+G(d,p) basis set.38,39 This level of theory has been shown to provide reliable information on the energetics and mechanistic features of organic reactions.40–44 To assess the influence of size and type of the basis set, we applied also the 6-311+G(2df,2p)38,39 and def2-TZPVPP45 basis sets with the M06-2X functional in evaluating the critical structures along the reaction paths. To assess the potential additional influences of dispersion effects on the computed reaction barriers we also performed M06-2X(D3)/6-311+G(d,p) computations using the zero-damping D3 correction.46 Harmonic vibrational frequencies were calculated to verify that the optimized structures are true minima or saddle points on the potential energy surfaces. The effects of the nitromethane solvent were modeled using the CPCM method.47 In view of the difficulties in evaluating reliable estimates of the entropy contributions to free energies,48 we also tested the SMD solvent model of Marenich, Cramer, and Truhlar.49 The computations employed ultrafine (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 590) integration grids and tight SCF convergence. The intrinsic reaction coordinate (IRC) method50 was applied in connecting the critical structures along the reaction path for the TiCl4 catalyzed Friedel–Crafts benzylation of benzene in nitromethane. The IRC computations employed the M06-2X/6-311+G(d,p) theoretical method. All computations were conducted with the Gaussian16 suite of programs.51
590) integration grids and tight SCF convergence. The intrinsic reaction coordinate (IRC) method50 was applied in connecting the critical structures along the reaction path for the TiCl4 catalyzed Friedel–Crafts benzylation of benzene in nitromethane. The IRC computations employed the M06-2X/6-311+G(d,p) theoretical method. All computations were conducted with the Gaussian16 suite of programs.51
Results and discussion
Mechanism of benzylation
As discussed above, the kinetic equations for the TiCl4 catalyzed benzylation of arenes in nitromethane derived by DeHaan et al.11,12 differ substantially from earlier kinetic results referring to Friedel–Crafts benzylation employing other Lewis acids, such as AlCl3, GaCl3, and FeCl3.2,7–10 We conducted transition state theory computations on the potential energy landscapes of two possible reaction paths: (a) catalysis by two TiCl4 molecules; (b) catalysis by a single TiCl4 molecule. As mentioned, the basic approach in modeling the potential energy surfaces employed M06-2X/6-311+G(d,p) computations. To obtain more accurate energetics we also conducted M06-2X/6-311+G(2df,2p) and M06-2X/def2-TZVPP computations on the potential energy surface. Considering the paramount difficulties in the theoretical evaluation of the entropy contributions to free energies,48,52,53 we present in Tables 1 and 2 the obtained theoretical estimates of the enthalpy shifts along the potential energy surfaces. The theoretical predictions of the free energy changes along the reaction pathway for all methods employed turned out highly overestimated (see Table S1 in the ESI†) compared to the experimental value reported by DeHaan et al.12 To avoid uncertainties associated with the theoretical evaluation of the entropy contribution to free energy barriers,48 we adopted a somewhat unusual approach. We discuss in the text the variations of the calculated ΔH‡ values accompanied by discussion of the potential contributions to the free energies of the experimentally determined entropy shift ΔS‡ for the reaction considered, ΔS‡ = 78 J (deg mol)−1, as reported by DeHaan et al.12 The contribution from the TΔS‡ term to the free energies for standard conditions is then −5.6 kcal mol−1. This approach allows us to obtain more realistic comparisons of the barriers associated with the two alternative reaction paths (involving one or two TiCl4 catalyst) considered. Similarly, the discussion of the relative heights of the sequential transition states along the same reaction route is based also on more solid grounds.| Theoretical approach | ΔH | ||
|---|---|---|---|
| TS1 | TS2 | P | |
| a Using zero-damping D3 dispersion correction (ref. 46). b From ref. 12. | |||
| CPCM M06-2X/6-311+G(d,p) | 7.5 | −0.8 | −7.9 |
| CPCM M06-2X(D3)/6-311+G(d,p)a | 6.5 | −2.7 | −8.2 |
| SMD M06-2X/6-311+G(d,p) | −2.1 | −11.7 | −7.7 |
| CPCM M06-2X/6-311+G(2df,2p) | 7.9 | 0.5 | −7.5 |
| CPCM M06-2X/def2-TZVPP | 9.9 | 3.8 | −7.8 |
| Exp. valueb | 12.7 | — | — |
| Structure | ΔH |
a |
b |
|---|---|---|---|
| a In this research, ΔG(est.) for TS1 and TS2 are obtained by summing the theoretical enthalpy shifts and the experimental TΔS contribution to the free energy shifts. The experimental value for ΔS of 78 J (deg mol)−1 determined by DeHaan et al. (ref. 12) is employed. b From ref. 12. | |||
| R | 0.0 | 0.0 | |
| Pre-reaction complex | −4.1 | — | |
| TS1 | 7.5 | 13.1 | 18.3 |
| I2 | −1.6 | — | |
| TS2 | −0.8 | 4.8 | |
| I3 σ-complex | −3.8 | — | |
| P | −7.9 | — | |

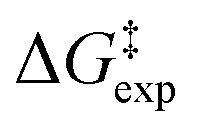
DeHaan et al.12 discussed two distinct steps of the TiCl4-catalyzed benzylation of benzene with benzyl chloride in nitromethane. Dissociation of the benzyl chloride and formation of a carbenium ion take place at the first step. These authors consider that this is the rate-controlling step of the reaction. It involves the electrophile and two molecules of the catalyst. At the second step, the formed ion interacts with the benzene substrate. DeHaan et al. assumed that this step results in the formation of a σ-complex intermediate. Removal of a proton leads to the final products. Still, there are several questions regarding the mechanism of the reaction: (a) How is the process initiated? (b) Does the rate-controlling step involves one or two TiCl4 molecules? (c) What is the structure of potential intermediates? (d) Does the reaction route involves a Sigma complex intermediate?
It is of considerable interest to compare how the different levels of theory employed perform in predicting the barrier heights of the reaction investigated. Table 1 presents the enthalpy shifts associated with the two found transition states, theoretically estimated from the different methods applied. According to DeHaan et al.12 the enthalpy shift associated with the dissociation of the C–Cl bond is 12.7 kcal mol−1.12Table 1 shows that all three methods employing the CPCM simulation of the nitromethane solvent produce results for the highest energy transition state that are in reasonable agreement with the experimental enthalpy barrier. The M06-2X employing the largest basis set (def2-TZVPP) produces an enthalpy barrier (9.9 kcal mol−1) that is in close correlation with the experimental ΔH‡ value. In contrast, the application of the SMD solvation model47 resulted in an enthalpy shift in poorer agreement with the experiment (Table 1). The application of the Grimme46 zero-damping D3 dispersion correction reduces the TS1 enthalpy barrier by 1 kcal mol−1 (Table 1). For the TS2 transition state, the dispersion corrections lead to a ΔH‡ value, which is nearly 2 kcal mol−1 lower. The enthalpy shifts associated with TS2 do not compare well with the experimental value. In brief, the theoretical computations conducted support the conclusions of DeHaan et al.12 regarding the rate-controlling step of the reaction. Since all IRC computations were performed using the M06-2X/6-311+G(d,p) computations, in the subsequent section we discuss the found critical structures – intermediates and transition states – as obtained at this level of theory.
 | ||
| Fig. 1 Critical structures and enthalpy variations (in kcal mol−1) along the reaction pathway for benzylation catalyzed by two TiCl4 molecules in simulated nitromethane solvent. All from M06 2X/6-311+G(d,p) computations. Energies are relative to reactants (benzene and benzyl chloride) and two TiCl4 molecules. | ||
The enthalpy shifts accompanying the different steps of the reaction are provided in Table 2 and illustrated in Fig. 1. The transition state for the last step of the reaction – the removal of a proton – could not be located. Similar problems have been encountered in attempts to locate analogous transition states for other EAS reactions.20 Liljenberg et al.20 emphasized that the proton transfer is usually a low energy step. The results obtained in the present research clearly show that the rate-controlling step of the reaction is the dissociation of the carbon–chlorine bond in the benzyl chloride and formation of a carbenium ion. The interpretation considering the estimated free energy barrier 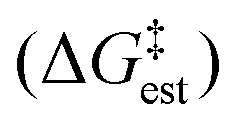 confirms this finding. The theoretical enthalpy changes combined with the experimental TΔS contribution results in a ΔG‡ barrier value of 13.1 kcal mol−1 (Table 2). A more accurate value for this barrier comes from the M06-2X/def2-TZVPP computations (Table 1). The ΔG‡ value for the reaction becomes 15.6 kcal mol−1, in good accord with the experimentally determined barrier of 18.3 kcal mol−1.12 The fully theoretical computed ΔG‡ value for TS1 from M06-2X/6-311+G(d,p) computations is 29.7 kcal mol−1 (see Table S1 in ESI†). The contribution from the TΔS‡ term is significantly overestimated when compared to the experimentally determined value. Overall, the results obtained clearly indicate that the TS1 barrier, associated with the dissociation of the C–Cl bond in the electrophilic reactant, governs the rate of the reaction. The reaction pathway includes four intermediates. In contrast to previous considerations, the initial attack does not involve a TiCl4 dimer but a single catalyst TiCl4 molecule.
confirms this finding. The theoretical enthalpy changes combined with the experimental TΔS contribution results in a ΔG‡ barrier value of 13.1 kcal mol−1 (Table 2). A more accurate value for this barrier comes from the M06-2X/def2-TZVPP computations (Table 1). The ΔG‡ value for the reaction becomes 15.6 kcal mol−1, in good accord with the experimentally determined barrier of 18.3 kcal mol−1.12 The fully theoretical computed ΔG‡ value for TS1 from M06-2X/6-311+G(d,p) computations is 29.7 kcal mol−1 (see Table S1 in ESI†). The contribution from the TΔS‡ term is significantly overestimated when compared to the experimentally determined value. Overall, the results obtained clearly indicate that the TS1 barrier, associated with the dissociation of the C–Cl bond in the electrophilic reactant, governs the rate of the reaction. The reaction pathway includes four intermediates. In contrast to previous considerations, the initial attack does not involve a TiCl4 dimer but a single catalyst TiCl4 molecule.
Reactivity trends
C–Cl dissociation energy (Ediss). The kinetic data of DeHaan et al.11,12 for the TiCl4 catalyzed benzylation of benzene in nitromethane show that the rate-controlling step of the reactions involves dissociation of the C–Cl bond in the electrophilic reactant. Our theoretical modeling confirmed this result. It appears, therefore, that the ionic C–Cl bond dissociation energy (Ediss) in the series of benzyl chlorides may well describe the experimentally determined kinetic trends. We estimated theoretically the dissociation energies for the series of the ten substituted in the aromatic ring benzyl chlorides considered (Table 3). These quantities were then juxtaposed to the experimental relative rates.
| RC6H4CH2Cl | k R/kHa | logkR/kH |
E diss (kcal mol−1) | q(Cl)d (e) |
|---|---|---|---|---|
| a From ref. 7. b Theoretical parameters from M06-2X/6-311+G(2df,2p) computations. c Ionic dissociation energies for substituted benzyl chlorides. d Hirshfeld charge at the chlorine atom in ArCH2Cl. e Absolute values of correlation coefficients. | ||||
| o-F | 0.15 | −0.8239 | 44.83 | −0.1338 |
| m-F | 0.08 | −1.0969 | 47.38 | −0.1330 |
| p-F | 1.29 | 0.1106 | 41.28 | −0.1407 |
| o-Cl | 0.07 | −1.1549 | 46.49 | −0.1343 |
| m-Cl | 0.06 | −1.2219 | 47.56 | −0.1321 |
| p-Cl | 0.22 | −0.6576 | 43.14 | −0.1360 |
| H | 1 | 0 | 42.94 | −0.1412 |
| o-CH3 | 3.23 | 0.5092 | 40.80 | −0.1319 |
| m-CH3 | 1.29 | 0.1106 | 42.10 | −0.1435 |
| p-CH3 | 12.4 | 1.09342 | 38.12 | −0.1463 |
| Correlation coef.e with logkR/kH |
0.968 | 0.971 | ||
Hirshfeld charges. Hirshfeld charges55 at the reactive carbon centers in aromatic substrates were shown to correspond very well to theoretically estimated reaction barriers for the BF3 catalyzed electrophilic chlorination of an extended series of substituted benzenes.56 It was also demonstrated that Hirshfeld charges at the carbon atoms correlate strongly with the Hammett constants for substituted benzenes.57 This type of population analysis has been successfully applied in rationalizing properties of various molecular systems (see, e.g. ref. 58 and 59). In the present research, the aromatic substrate is just the benzene molecule. Nonetheless, it was of interest to test whether this type of atomic charges would provide insights into the reactivity trends for systems, where the changes of structures are in the electrophile.
Table 3 presents the experimental relative rates for the benzylation of benzene with benzyl chloride and nine ring substituted derivatives as determined by Olah et al.7 The reactivity indexes are evaluated using DFT computations at the M06-2X/6-311+G(2df,2p) level, including CPCM modeling of the nitromethane solvent. As may be expected, in accordance with the rate equation (eqn (3)), the changes in the structure of the electrophile will lead to substantial differences in the relative rates of benzylation compared to the rate for the parent electrophile (benzyl chloride). The theoretically evaluated reactivity parameters are shown in Table 3. In most cases, the electron withdrawing chlorine and fluorine aromatic substituents cause strengthening of the C–Cl bond in the CH2Cl moiety. The stronger C–Cl bonds in these compounds lead to higher bond dissociation energies (Ediss), resulting in slower reaction rates. Somewhat surprizingly, the p-F substituent increases the reactivity (Table 3). This influence is well reflected in the computed C–Cl dissociation energy (Ediss = 41.3 kcal mol−1), which is lower than in the unsubstituted benzyl chloride (Ediss = 42.9 kcal mol−1). The strong hyperconjugation effect of p-F group evidently overwhelms the reverse inductive (and through-space) influences of the same substituent. As expected, the hyperconjugation effects of the methyl group in the methyl substituted derivatives, in particular the para-methylbenzyl chloride, cause lowering of the C–Cl bond dissociation energies and, respectively, higher reaction rates. Altogether, the shifts in dissociation energies are in quite good accord with the experimental trends of relative rates (Table 3).7 The dependence between log kR/kH and Ediss is illustrated in Fig. 2.
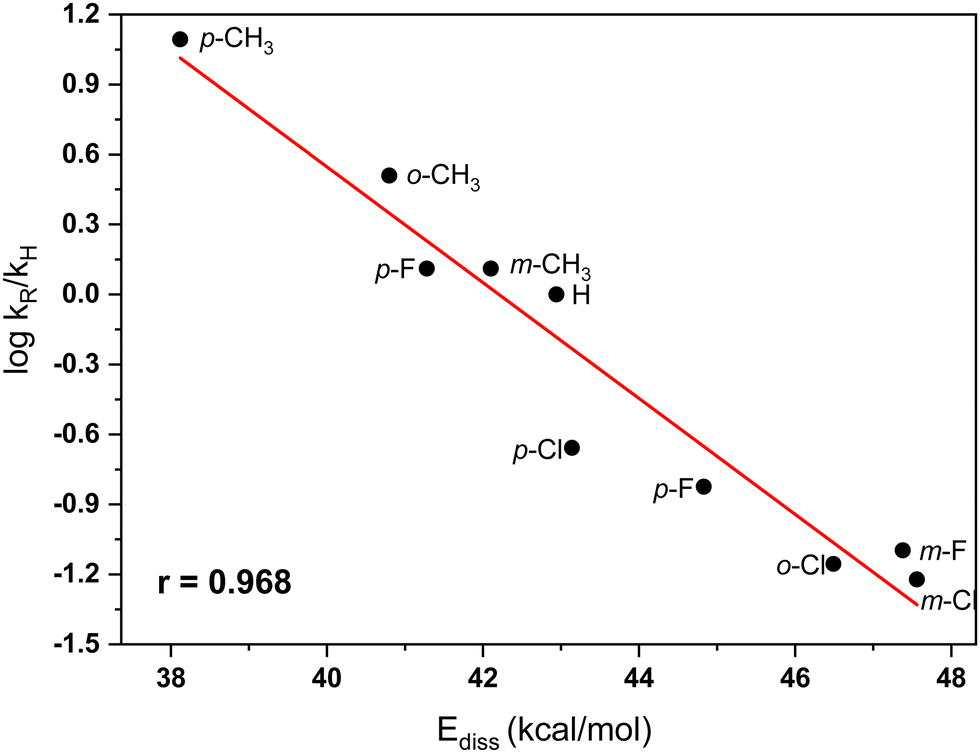 | ||
| Fig. 2 Plot of the dependence between C–Cl ionic bond dissociation energies in substituted benzyl chlorides and relative rates (ref. 7) of the TiCl4-catalyzed Friedel–Crafts benzylation of benzene in nitromethane solvent. | ||
The partial charge at the chlorine in the ArCH2Cl molecules is expected to influence the interaction between the TiCl4 catalyst and the electrophilic reactants. A good correlation between relative rates and Hirshfeld charges at the chlorine atom of the CH2Cl group was found (r = 0.971). The relationship established shows again that Hirshfeld charges provide a useful theoretical approach in rationalizing molecular properties (see, e.g. ref. 56–59). Fig. 3 shows the plot between charges at the chlorine center and the rates of benzylation. An outlier for this correlation is the charge at the chlorine atom in the o-CH3 derivative. A possible hydrogen bonding interaction between the chlorine and a methyl C–H bond may explain the deviation.
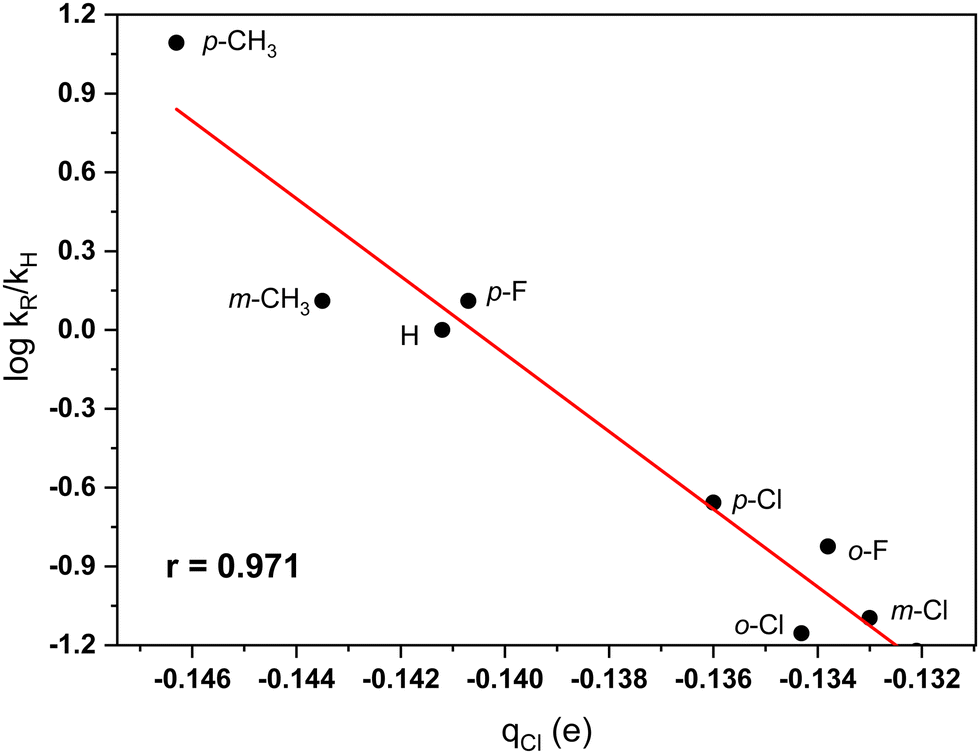 | ||
| Fig. 3 Plot of the dependence between Hirshfeld charges at the chlorine of the CH2Cl group in substituted benzyl chlorides and relative rates (ref. 7) of the TiCl4-catalyzed Friedel–Crafts benzylation of benzene in nitromethane solvent. | ||
An interesting case for this type of Friedel–Crafts benzylation is the reaction involving p-nitrobenzyl chloride. The computed (at the same level of theory) C–Cl ionic dissociation energy for this derivative is 54.6 kcal mol−1, higher than all values shown in Table 3. The strong electron-withdrawing effects of the nitro group invokes strengthening of the C–Cl bond. Based on this result, one might expect that the rate of the p-nitrobenzylation may also be determined by the C–Cl dissociation energy. However, the form of the kinetic equation (eqn (5)) suggests a very different mechanistic pathway for this reaction, with participation of the arene in the rate-controlling step. The Hirshfeld charge on the CH2Cl chlorine in the p-nitrobenzyl chloride is −0.12 e. This value for the partial charge is less favorable for the initial interaction between the p-nitrobenzyl chloride and the catalyst.
The application of bond dissociation energy as a reactivity index for the Friedel–Crafts benzylation reactions investigated proved quite appropriate considering the mechanistic features of the reaction. The good correlations between relative rates and the theoretical reactivity parameters indicate that the ten TiCl4-catalyzed benzylation reactions considered follow the same mechanistic route.
Conclusions
The experimental relative rates7 of the TiCl4-catalyzed Friedel–Crafts benzylation of benzene by benzyl chloride and a series of ring substituted derivatives are investigated based on results from theoretical modeling of the mechanism of the reaction. Density functional theory computations employing the M06-2X functional combined with several types of basis sets and two alternative solvent models reveal the potential energy landscape for the basic reaction from the series. The usual difficulties in evaluation free energy barriers in solution are avoided by applying theoretically evaluated enthalpy shifts combined with experimental12 entropy contributions to arrive at physically plausible potential energy surface. Our comprehensive mechanistic DFT computations reveal full details of the reaction pathway, including the structures of transition states as well as of four intermediates. Contrary to previous assumptions, the present results show that while the rate-controlling transition state involves two catalyst molecules, the initial interaction is between the electrophile and a single TiCl4 molecule.On the basis of the revealed mechanistic features, the reactivity trends for the series of reactions investigated are rationalized with the aid of reactivity descriptors. Most significantly, the computed C–Cl ionic dissociation energies for the benzyl chlorides correlate strongly with the relative rates of benzylation, thus confirming that the kinetics is governed by this process. Quite satisfactory correlations also exist between the experimental relative rates of benzylation7 and theoretically evaluated Hirshfeld charges at the reacting chlorine atom.
Data availability
The data supporting this article have been included in the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The research in Sofia is financed by the European Union-NextGenerationEU, through the National Recovery and Resilience Plan of the Republic of Bulgaria, project SUMMIT BG-RRP-2.004-0008-C01; the research in Georgia was supported by Department of Energy, Computational and Theoretical Chemistry Program, Contract DE-SC0018412.References
- (a) C. Friedel and J. M. Crafts, Sur Une Nouvelle Méthode Générale de Synthèse d’Hydrocarbures, d’Acétones, etc., Compt. Rend., 1877, 84, 1392–1395 Search PubMed; (b) C. Friedel and J. M. Crafts, Sur Une Nouvelle Méthode Générale de Synthèse d’Hydrocarbures, d’Acétones, etc., Compt. Rend., 1877, 84, 1450–1454 Search PubMed.
- G. A. Olah, Friedel–Crafts and Related Reactions, Wiley, New York, 1963 Search PubMed.
- R. M. Roberts and A. A. Khalaf, Friedel–Crafts Alkylation Chemistry, Marcel Dekker, New York, 1984 Search PubMed.
- M. Rueping and B. J. Nachtsheim, A Review of New Developments in the Friedel–Crafts Alkylation – From Green Chemistry to Asymmetric Catalysis, Beilstein J. Org. Chem., 2010, 6, 6 CrossRef PubMed.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, New York, 2007 Search PubMed.
- F. A. Carrey and R. J. Sandberg, Advanced Organic Chemistry. Part A: Structure and Mechanisms, Springer, New York, 5th edn, 2006 Search PubMed.
- G. A. Olah, S. Kobayashi and M. Tashiro, Aromatic Substitution. XXX. Friedel–Crafts Benzylation of Benzene and Toluene with Benzyl and Substituted Benzyl Halides, J. Am. Chem. Soc., 1972, 94, 7448–7461 CrossRef CAS.
- H. Jungk, C. R. Smoot and H. C. Brown, Kinetics of Methylation and Ethylation of Benzene and Toluene in 1,2,4-Trichlorobenzene under the Influence of Aluminum Bromide: Mechanism of the Alkylation Reaction, J. Am. Chem. Soc., 1956, 78, 2185–2190 CrossRef CAS.
- H. C. Brown and M. Grayson, Kinetics of the Reaction of Representative Benzyl Halides with Aromatic Compounds: Evidence for a Displacement Mechanism in the Friedel–Crafts Reaction of Primary Halides, J. Am. Chem. Soc., 1953, 75, 6285–6292 CrossRef CAS.
- S. U. Choi and H. C. Brown, Kinetic and Relative Rates of the Gallium Bromide-Catalyzed Reaction of Alkyl Bromides with Aromatic Compounds in 1,2,4-Trichlorobenzene Solution, J. Am. Chem. Soc., 1963, 85, 2596–2600 CrossRef CAS.
- F. P. DeHaan, G. L. Delker, W. D. Covey, J. Ahn, M. S. Anisman, E. C. Brehm, J. Chang, R. M. Chicz, R. L. Cowan, D. M. Ferrara, C. H. Fong, J. D. Harper, C. D. Irani, J. Y. Kim, R. W. Meinhold, K. D. Miller, M. P. Roberts, E. M. Stoler, Y. J. Suh, M. Tang and E. L. Williams, Electrophilic Aromatic Substitution. 8. A Kinetic Study of the Friedel–Crafts Benzylation Reaction in Nitromethane, Nitrobenzene, and Sulfolane. Substituent Effects in Friedel–Crafts Benzylation, J. Am. Chem. Soc., 1984, 106, 7038–7046 CrossRef CAS.
- F. P. DeHaan, W. H. Chan, J. Chang, T. B. Cheng, D. A. Chiriboga, M. M. Irving, C. R. Kaufman, G. Y. Kim, A. Kumar, J. Na, T. T. Nguyen, D. T. Nguyen, B. R. Patel, N. P. Sarin and J. H. Tidwell, Kinetic Studies of the TicCl4 Catalyzed Reactions of Benzyl Chloride, p-Xylyl Chloride, and p-Nitrobenzyl Chloride with Benzene and Anisole in Nitromethane and Dichloromethane, J. Am. Chem. Soc., 1990, 112, 356–363 CrossRef CAS.
- L. P. Hammett, Some Relations Between Reaction Rates and Equilibrium Constants, Chem. Rev., 1935, 17, 125–136 CrossRef CAS.
- H. H. Jaffé, A Reexamination of the Hammett Equation, Chem. Rev., 1953, 53, 191–261 CrossRef.
- C. Hansch, A. Leo and R. W. Taft, A Survey of Hammett Substituent Constants and Resonance and Field Parameters, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- K. Fukui, T. Yonezawa and H. Shingu, A Molecular Orbital Theory of Reactivity in Aromatic Hydrocarbons, J. Chem. Phys., 1952, 20, 722–725 CrossRef CAS.
- J. J. Brown and S. L. Cockroft, Aromatic Reactivity Revealed: Beyond Resonance Theory and Frontier Orbitals, Chem. Sci., 2013, 4, 1772–1780 RSC.
- B. Galabov, S. Ilieva, G. Koleva, W. D. Allen, H. F. Schaefer and P. V. R. Schleyer, Structure-Reactivity Relationships for Aromatic Molecules: Application of the Electrostatic Potential at Nuclei and Electrophile Affinity Electronic Indices, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 37–55 CAS.
- T. Brinck and M. Liljenberg, in Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds, ed. J. Mortier, Wiley, Hoboken, NJ, 2016, ch. 4 Search PubMed.
- M. Liljenberg, H. J. Stenlid and T. Brinck, Theoretical Investigation into Rate-Determining Factors in Electrophilic Aromatic Halogenation, J. Phys. Chem. A, 2018, 122, 3270–3279 CrossRef CAS PubMed.
- B. Galabov, S. Ilieva, D. Cheshmedzhieva, V. Nikolova, V. A. Popov, B. Hadjieva and H. F. Schaefer III, Mini-review on Structure-Reactivity Relationship for Aromatic Molecules: Recent Advances, ACS Omega, 2022, 7, 8199–8208 CrossRef CAS.
- (a) P. R. Rablen and A. Yett, The Relative Favorability of Placing Substituents Ortho or Para in the Cationic Intermediate for Electrophilic Aromatic Substitution, J. Phys. Org. Chem., 2022, e4457 Search PubMed; (b) A. Yett and P. R. Rablen, A G4 Approach to Computing the Hammett Substituent Constants σp, σm, σ−, σ+, and σ+m, J. Phys. Org. Chem., 2022, e4436 Search PubMed.
- P. Tarakeshwar, J. Y. Lee and K. S. Kim, Role of Lewis Acid (AlCl3) – Aromatic Ring Interactions in Friedel–Crafts Reactions; An Ab Initio Study, J. Phys. Chem. A, 1998, 102, 2253–2258 CrossRef CAS.
- S. Csihony, H. Mehdi, Z. Homonnay, A. Vertes, O. Farkas and I. T. Horvath, In Situ Spectroscopic Studies Related to the Mechanism of the Friedel–Crafts Acetylation of Benzene in Ionic Liquids Using AlCl3 and FeCl3, J. Chem. Soc., Dalton Trans., 2002, 5, 680–685 RSC.
- S. Yamabe and S. Yamazaki, A Remarkable Difference in the Deprotonation Steps of the Friedel–Crafts Acylation and Alkylation Reactions, J. Phys. Org. Chem., 2009, 22, 1094–1103 CrossRef CAS.
- S. Yang, C. Bour and V. Gandon, Superelectrophilic Gallium(III) Homodimers in Gallium Chloride Mediated Methylation of Benzene: a Theoretical Study, ACS Catal., 2020, 10, 3027–3033 CrossRef CAS.
- I. C. Nogueira and J. R. Pliego Jr., Friedel–Crafts Reaction: Theoretical Study of the Mechanism of Benzene Alkylation with Isopropyl Chloride Catalyzed by AlCl3 and Al2Cl6, J. Braz. Chem. Soc., 2023, 34, 194–200 CAS.
- (a) T. M. Krygowski and B. T. Stepien, Sigma- and pi-electron delocalization: focus on substituent effects, Chem. Rev., 2005, 105, 3482–3512 CrossRef CAS PubMed; (b) H. Szatylowicz, T. Siodla, O. A. Stasyuk and T. M. Krygowski, Towards Physical Interpretation of Substituent Effects: The Case of meta- and para- substituted Anilines, Phys. Chem. Chem. Phys., 2016, 18, 1711–1721 RSC.
- B. Galabov, G. Koleva, P. V. R. Schleyer and H. F. Schaefer III, Electrophile Affinity: Quantifying Reactivity for the Bromination of Arenes, J. Org. Chem., 2010, 75, 2813–2819 CrossRef CAS PubMed.
- M. Liljenberg, J. H. Stenlid and T. Brinck, Mechanism and Regioselectivity of Electrophilic Aromatic Nitration in Solution: The Validity of the Transition State Approach, J. Mol. Model., 2017, 24, 15 CrossRef PubMed.
- (a) S. R. Gadre and C. H. Suresh, Electronic Perturbations of the Aromatic Nucleus: Hammett Constants and Electrostatic Potential Topography, J. Org. Chem., 1997, 62, 2625–2627 CrossRef CAS; (b) S. R. Gadre, C. H. Suresh and N. Mohan, Electrostatic Potential Topology for Probing Molecular Structure, Bonding and Reactivity, Molecules, 2021, 26, 3289 CrossRef CAS.
- A. V. Salnikov, A. M. Genaev, G. E. Salnikov, H. S. Rzepa and V. G. Shubin, J. Comput. Chem., 2016, 37, 210–225 CrossRef PubMed.
- X. He, M. Li, C. Rong, D. Zhao, W. Liu, P. W. Ayers and S. Liu, Some Recent Advances in Density-Based Reactivity Theory, J. Phys. Chem. A, 2024, 128, 1183–1196 CrossRef CAS PubMed.
- J. M. Kromann, J. H. Jensen, M. Kruszyk, M. Jessing and M. Jorgensen, Fast and Accurate Prediction of the Regioselectivity of Electrophilic Aromatic Substitution Reactions, Chem. Sci., 2018, 9, 660–665 RSC.
- A. Tomberg, M. J. Johansson and P.-O. Norrby, A Predictive Tool for Electrophilic Aromatic Substitutions Using Machine Learning, J. Org. Chem., 2019, 84, 4695–4703 CrossRef CAS.
- K. Jorner, T. Brinck, P.-O. Norrby and D. Buttar, Machine Learning Meets Mechanistic Modelling for Accurate Prediction of Experimental Activation Energies, Chem. Sci., 2021, 12, 1163–1175 RSC.
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 other Functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3-21+G Basis Set for First-Row Elements, Li–F, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- F. M. Bickelhaupt and K. N. Houk, Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed.
- Y. H. Lam, M. N. Grayson, M. C. Holland, A. Simon and K. N. Houk, Theory and Modeling of Asymmetric Catalytic Reactions, Acc. Chem. Res., 2016, 49(4), 750–762 CrossRef CAS PubMed.
- T. A. Hamlin, B. J. Levandowski, A. K. Narsaria, K. N. Houk and F. M. Bickelhaupt, Structural Distortion of Cycloalkynes Influences Cycloaddition Rates Both by Strain and Interaction Energies, Chem. – Eur. J., 2019, 25, 6342–6348 CrossRef CAS PubMed.
- M. Walker, A. J. A. Harvey, A. Sen and C. E. H. Dessent, Performance of M06, M06-2X, and M06-HF Density Functionals for Conformationally Flexible Anionic Clusters: M06 Functionals Perform Better than B3LYP for a Model System with Dispersion and Ionic Hydrogen-Bonding Interactions, J. Phys. Chem. A, 2013, 117, 12590–12600 CrossRef CAS.
- K. Yang, J. L. Zheng, Y. Zhao and D. G. Truhlar, Tests of the RPBE, revPBE, τ-HCTHhyb, ω-B97X-D, and MOHLYP Density Functional Approximations and 29 Others Against Representative Database for Diverse Bond Energies and Barrier Heights in Catalysis, J. Chem. Phys., 2010, 132, 164117 CrossRef.
- F. Weigend and R. Ahlrichs, Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- (a) V. Barone and M. Cossi, Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS; (b) M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- E. Plata and D. A. Singleton, A Case Study of the Mechanism of Alcohol-Mediated Morita Baylis–Hillman Reactions. The Importance of Experimental Observations, J. Am. Chem. Soc., 2015, 137, 3811–3826 CrossRef.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Co6stant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- K. Fukui, The Path of Chemical Reactions – the IRC Approach, Acc. Chem. Res., 1981, 141, 363–368 CrossRef.
- M. J. Frisch, et al., Gaussian 16, Revision 16. B.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- J. N. Sanders, H. J. Jun, R. A. Yu, J. L. Gleason and K. N. Houk, Mechanism of an Organocatalytic Cope Rearrangement Involving Iminium Intermediates: Elucidating the Role of Catalyst Ring Size, J. Am. Chem. Soc., 2020, 142, 16877–16886 CrossRef CAS.
- D. Cheshnedzhieva, S. Ilieva, B. Hadjieva and B. Galabov, The Mechanism of Alkaline Hydrolysis of Amides: A Comparative Computational and Experimental Study of the Hydrolysis of N-Methylacetamide, N-Methylbenzamide, and Acetanilide, J. Phys. Org. Chem., 2009, 22, 619–631 CrossRef.
- E. Ritter and S. Kvisle, Spectral Evidence for Dimerization of Titanium Tetrachloride at Cryogenic Temperatures, Inorg. Chem., 1985, 24, 640–642 CrossRef.
- (a) F. L. Hirshfeld, Bonded-Atom Fragments for Describing Molecular Charge Densities, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS; (b) J. P. Ritchie, Electron Density Distribution Analysis for Nitromethane, Nitromethide, and Nitramide, J. Am. Chem. Soc., 1985, 107, 1829–1837 CrossRef CAS; (c) J. P. Ritchie and S. M. Bachrach, Some Methods and Applications of Electron Density Distribution Analysis, J. Comput. Chem., 1987, 8, 499–509 CrossRef CAS.
- S. Liu, Quantifying Reactivity for Electrophilic Aromatic Substitution Reactions with Hirshfeld Charge, J. Phys. Chem. A, 2015, 119, 3107–3111 CrossRef CAS.
- V. Nikolova, D. Cheshmedzhieva, S. Ilieva and B. Galabov, Atomic Charges in Describing Properties of Aromatic Molecules, J. Org. Chem., 2019, 84, 1908–1915 CrossRef CAS PubMed.
- K. B. Wiberg and P. R. Rablen, Atomic Charges, J. Org. Chem., 2018, 83, 15463–15469 CrossRef CAS.
- P. G. Seybold, Quantum Chemical-QSPR Estimation of the Acidities and Basicities of Organic Compounds, Adv. Quant. Chem., 2012, 64, 83–104 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03181d |
| This journal is © the Owner Societies 2025 |