
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Searching the chemical space of hetero-atom bridged norbornadienes†
Nils
Oberhof
*a,
Andreas Erbs
Hillers-Bendtsen
 b,
Oscar Berlin
Obel
b,
Karoline
Schjelde
b,
Kurt V.
Mikkelsen
b and
Andreas
Dreuw
*a
b,
Oscar Berlin
Obel
b,
Karoline
Schjelde
b,
Kurt V.
Mikkelsen
b and
Andreas
Dreuw
*a
aInterdisciplinary Center For Scientific Computing, Heidelberg University, Im Neuenheimer Feld 205, Heidelberg, Germany. E-mail: dreuw@uni-heidelberg.de; nils.oberhof@iwr.uni-heidelberg.de
bDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen, Denmark
First published on 2nd December 2024
Abstract
The efficient utilization of solar energy as renewable source is a central pillar of societal future energy production. So-called molecular solar thermal energy storage (MOST) systems have attracted considerable attention as storage solution and heat release on demand. Substituted norbornadiene/quadricyclane (NBD/QC) derivatives have been shown to be well suited for this task, in particular when substituted with electron donating and accepting functional groups. The introduction of a hetero-atom in the main structural framework, however, has not been investigated thoroughly, yet. In this study, a previously established high-throughput screening procedure is used to investigate carbon, nitrogen and oxygen-bridged norbornadiene derivatives for their potential as MOST system employing their theoretical solar power conversion efficiency as scoring metric. Therefore, we explore a large chemical space considering also plausible synthetic availability and propose a set of 5 molecules per bridge head as best candidates for further experimental evaluation.
1 Introduction
With a projected global temperature rise surpassing the 1.5° mark1 in the near future, it is becoming even more pressing to equip the world with adequate renewable energy sources. So far, the renewable energy production stems mostly from water, wind and solar energy. These systems, however, are particularly susceptible for daily and seasonal variations in production, which have to be levelled out, and therefore energy storage is of particular interest.2 With the sun providing a great amount of energy to the Earth's surface, it is an efficient energy source even in remote regions.1One approach for energy storage is represented by so-called molecular solar thermal (MOST) storage systems. These incorporate photoswitches for the absorption of sun light, which undergo a photochemical switching process (Fig. 1) forming a meta-stable high-energy state. The stored energy can be released either spontaneously or on demand through a controlled external stimulus such as heat, a (heterogeneous) catalyst,3,4 light source5,6 or an applied electric potential.7,8 Several photoswitches with their stable and meta-stable states, such as (E/Z)-azobenzene ((E/Z)-AB),5,6,9,10 1,2-azaborine and its dewar isomer (BNB/BND),11 the dihydroazulene/vinylheptafulvene (DHA/VHF) couple12–14 and especially the norbornadiene/quadricyclane (NBD/QC) system,15–17 are promising MOST candidates.
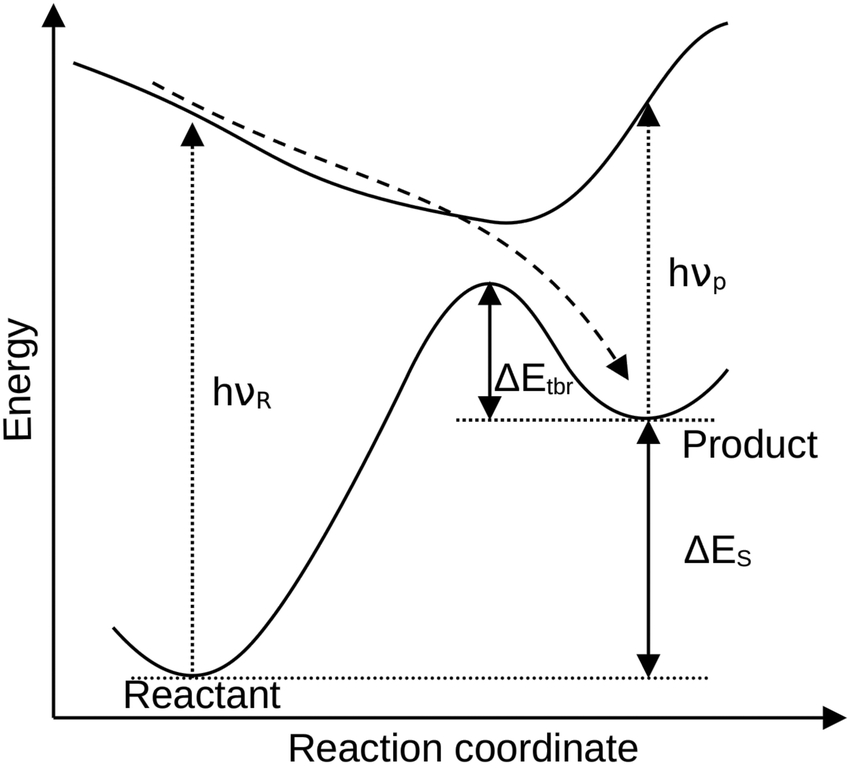 | ||
| Fig. 1 Schematic potential energy surface of a molecular solar thermal system highlighting the quantities computed for the calculation of the solar conversion efficiency (storage energy ΔES, thermal back reaction barrier ΔEtbr and the absorption wavelength of reactant νR and product νP). | ||
The applicability of these switches is, however, strongly dependent on several properties that can – and must be – tuned through chemical modification. The most important properties of efficient MOST systems are: (I) the storage energy, i.e. how much energy can be accumulated in the meta-stable state for downstream heat release (II) the thermal back-reaction barrier, determining the lifetime of the storage state, and (III) the absorption spectrum, since the overlap with the solar spectrum must be high to efficiently capture the sun's energy.18
For a direct comparison between systems, these interacting parameters can be combined within one principal descriptor, the so-called solar power conversion efficiency (SCE).18,19 It has been employed previously in experimental as well as quantum chemical studies.20–23 The SCE is given by
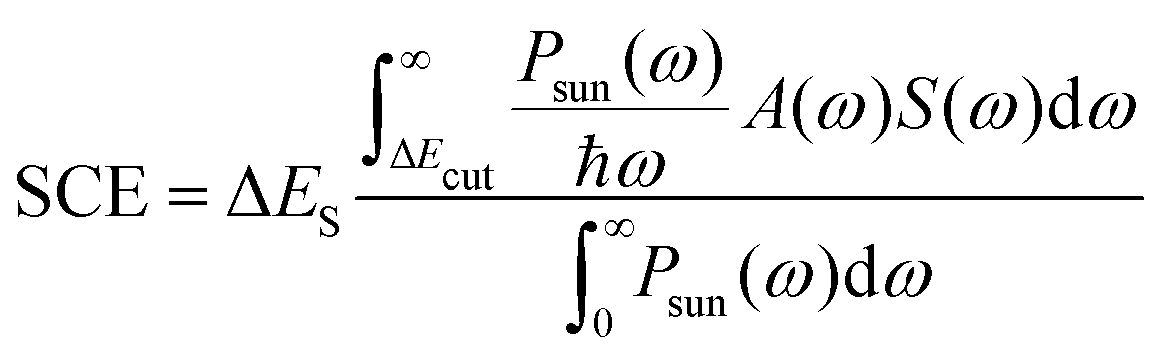 | (1) |
| A(ω) = [1 − 10(εR(ω)cR+εP(ω)cPL)] | (2) |
The fraction of sunlight absorbed by the reactant (S(ω)) is
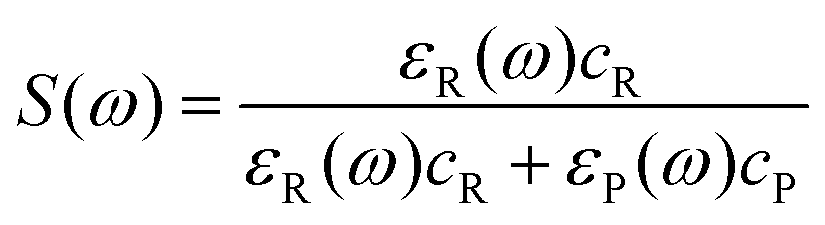 | (3) |
Common photoswitches are most often chemically modified via chemical substitutions at the main framework. Some desired changes of the (photochemical) properties can, however, also be achieved through direct alteration of the main framework, which has, for example, already been achieved for hetero-cyclic AB derivatives.25 NBDs with different substitution patterns, have been previously screened for MOST applications,23,26–28 however, only very little is still known about the properties of their hetero-atom bridged derivatives. Studying these systems provides a chance for a deeper understanding of the NBD/QC interconversion and also to possibly further improve properties relevant for MOST applications.
To explore this, we have screened a chemical space of 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 662 possible hetero-atom bridged NBD/QC derivatives in a (partially) automated fashion with respect to their MOST properties, scoring them by their SCE. In the following, we show that the combination of a screening procedure at the xTB/sTDA-xTB level of theory with a consecutive reevaluation at the DFT/TDDFT level of theory is an efficient and sufficiently accurate approach for hetero-atom bridged NBD/QC systems. Based on these findings some novel derivatives are proposed for future synthesis and spectroscopic studies.
662 possible hetero-atom bridged NBD/QC derivatives in a (partially) automated fashion with respect to their MOST properties, scoring them by their SCE. In the following, we show that the combination of a screening procedure at the xTB/sTDA-xTB level of theory with a consecutive reevaluation at the DFT/TDDFT level of theory is an efficient and sufficiently accurate approach for hetero-atom bridged NBD/QC systems. Based on these findings some novel derivatives are proposed for future synthesis and spectroscopic studies.
2 Methods
2.1 Chemical space
The three considered (hetero-atom) bridge heads are methylene (–CH2–), methyl–carbamate (–N(C(O)OMe)–) and an oxygen (–O–), which in the following are called carbon (C), nitrogen (N) and oxygen (O) bridge, respectively. They are analogous to the simplest previously synthesized stable bridges,29–31 are thus expected to also lead to novel synthetically accessible derivatives. As a counter example, the simple –NH– bridged NBD would be a purely theoretical toy model, since it is known to have a strong tendency to decompose quickly in experiment and is thus not of interest for actual MOST applications.32 Therefore, it is excluded from this study.Since especially push–pull NBDs have proven to be promising MOST compounds, electron-donating groups (EDG) and electron-withdrawing groups (EWG) constitute the biggest part of the chosen substituent space of our study (see Fig. 2). Additionally, some other promising substituents, such as the dicyanovinyl group27 and some heterocycles – previously not explored – were included. Considering these substituents 2 data sets per bridging unit were realized.
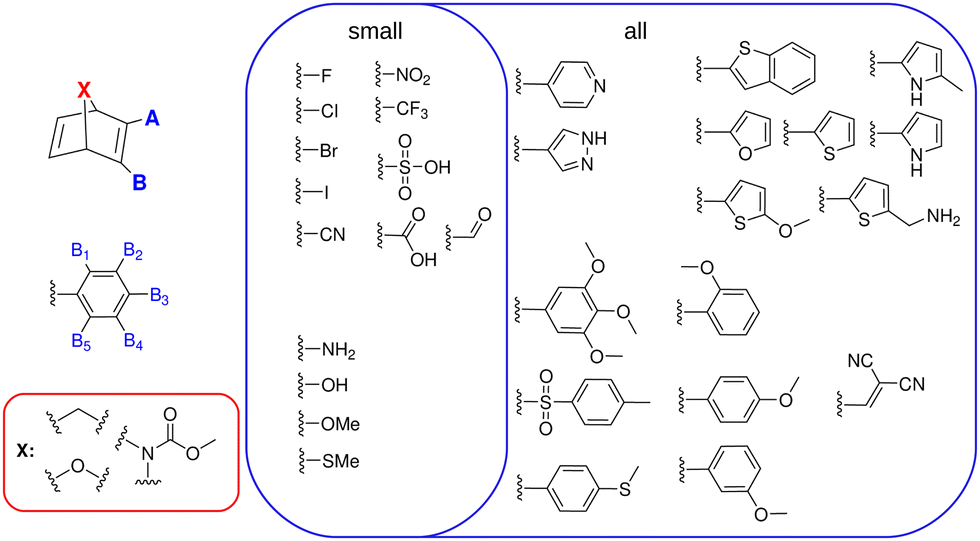 | ||
| Fig. 2 Substitution patterns at positions A & B (dataset 1) and A & B1–5 (dataset 2) in the top left. The employed substituents are shown in the blue boxes and the employed bridge heads (X) in the red box. | ||
The first data set incorporates all substituents with up to 2 substituents at A and B directly (see Fig. 2). With 30 substituents and –H as the “standard” substituent, this yields (31 × 30)/2 = 465 unique systems per bridge head, corrected for double counting due to the symmetry of the system.
The second, larger data set combines all small EDG and EWG groups (14 overall) in the A and B1–5 positions (see Fig. 2) with up to 3 substituents present at once. This gives 12089 unique molecules per bridge head, when correcting for double counting due to the symmetry of the phenyl ring (see also ESI†). The complete data set of possible derivatives then amounts to 37662 compounds, which is a large chemical space, given the incorporated synthetic plausibility emphasized in this study.
2.2 Computational details
The resulting 37662 compounds are investigated following a previously developed and benchmarked screening procedure. A quick overview of the screening method is given in the following (for further details see ESI† or ref. 23 and 27).
The differently substituted NBD and QC systems are generated as SMILES strings and subsequently their minimum energy conformers identified through a systematic conformer search using the ETKDG method33 within the RDKit module.34 The transition state between both minima is estimated by a constrained potential energy surface scan along the symmetrical opening of the C–C bonds broken in the thermal back-reaction, between 1.5 and 2.2 Å. The storage energy as well as the thermal back-reaction barrier are then evaluated at the GFN2-xTB35 level of theory. The photochemical properties of the reactant and photo-product are subsequently investigated with sTDA-xTB36 and consequently the collected properties are compiled into the solar power conversion efficiency metric for each system according to eqn (1). Whenever the SCE would be negative the value is set to 0. From this, a scoring of the molecules is performed and evaluated.
The geometries of the 20 highest (40 for C bridge) and lowest scoring systems for each bridging unit are subjected to ground state geometry optimizations and transition state searches at the DFT/M06-2X37/def2-SVPD38 level of theory with consecutive TDDFT calculations of the first 10 singlet excited states with the same combination of functional and basis set, done in the Orca software package.39 With these results, the solar power conversion efficiency of each compound is reevaluated and scored. This methodology has been validated in previous work, showing that the sTDA-xTB excitation energies are underestimated compared to the TDDFT/M06-2X/def2-SVPD energies.23 At the same time excitation energies calculated at the TDDFT/M06-2X level are expected to be higher than in reality. The underestimation of excitation energies of charge-transfer transitions, however, is still possible, since a functional is employed which does not incorporate full asymptotic exchange.40–42 This approach will, in general, lead to an overestimation of the theoretical SCE within the xTB framework relative to the (TD)DFT values, which in turn underestimates the real SCE. Therefore, one can expect the screening method to yield no “false negatives” and only, if at all, “false positives” for promising MOST compound candidates.
3 Results
3.1 General trends
For a comprehensive understanding of the interplay between molecular properties and the solar power conversion efficiency, scatter plots of each relation are presented in Fig. 3. Therefore, the storage energy (ΔES), the back-reaction barrier (ΔEtbr), the absorption wavelength and the corresponding oscillator strength of the lowest-lying excited state selected for the evaluation for both the NBD and the QC derivatives, are evaluated. This includes the whole data set of all three bridges (for each individual hetero-atom bridge see ESI†). The resulting storage energy for many compounds is negative, which is in stark contrast to the expectations of MOST systems storing energy in their meta-stable state. A possible explanation is a deficiency of xTB, underestimating the storage energy of NBDs and consequently its derivatives within this study. Previously this underestimation was shown to be systematic.23 A high number of low/negative storage energies appear in the O and N bridge head systems (for further calrification see ESI†). Since these negative storage energies result in very low SCEs anyways, this issue is of low concern for our search for good MOST molecules.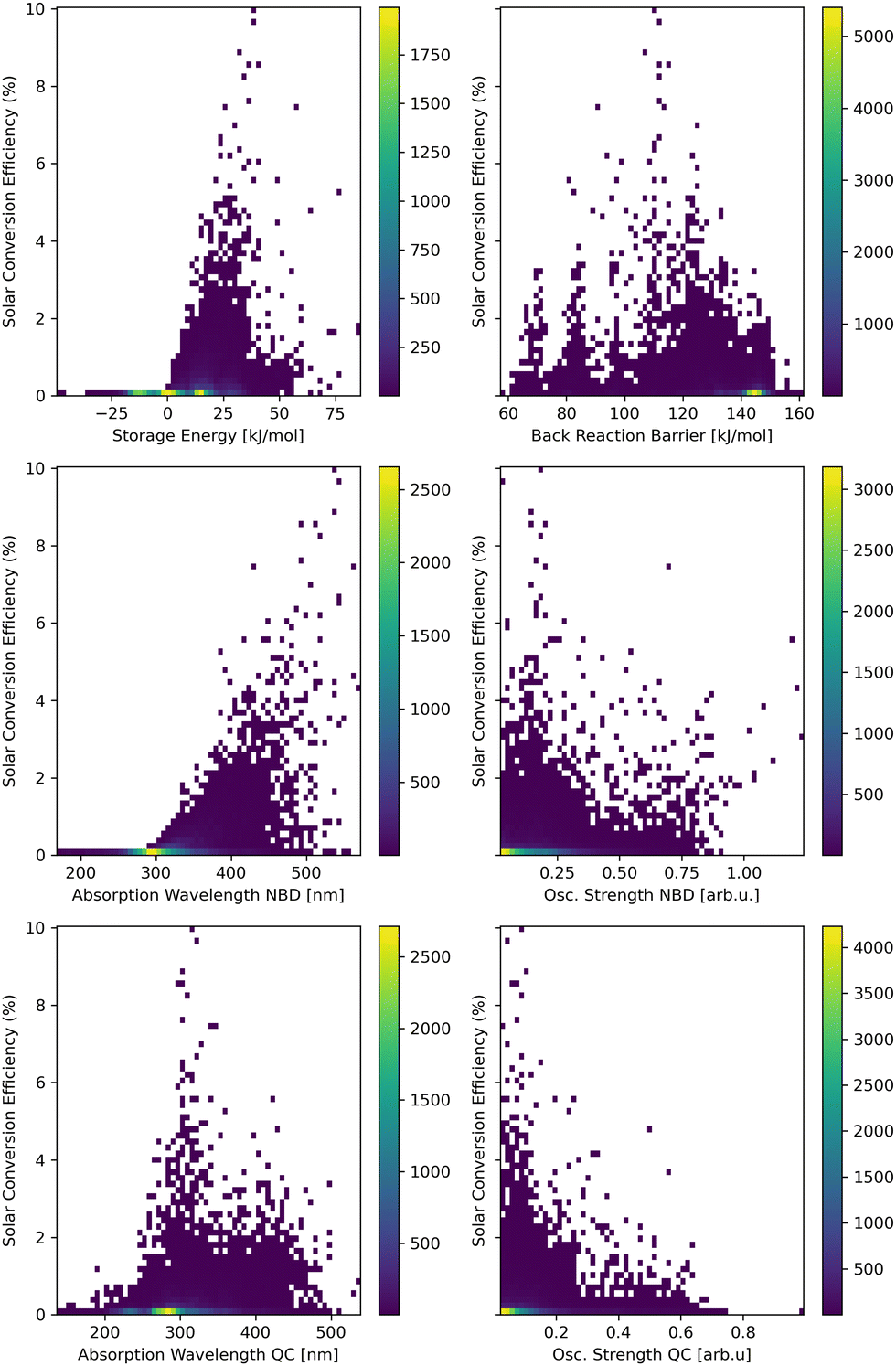 | ||
| Fig. 3 Scatter plots of the SCE calculated according to eqn (1) for the full data set. The contributing properties are analyzed: storage energy (ΔES, top left), thermal back-reaction barrier (ΔEtbr, top right), the absorption wavelengths and the oscillator strengths of the lowest-lying singlet excited state with an oscillator strength above 0.01 of the NBD (middle) and QC (bottom) isomers. | ||
In addition, a general tendency for higher SCE with higher storage energy can be observed, since the storage energy is a direct multiplicative factor for the calculated SCE (see eqn (1)). This is, however, partially counteracted by the storage energy contributing to the cut-off energy (Ecut) within the integration. This ensures the excitation to provide enough energy for the photo-reaction and the storage cycle to be performed. Thus, the highest SCE does not (necessarily) correspond to the highest storage energy. This interplay between storage energy and back-reaction barrier balances the distribution of high SCEs to storage energies around 40 kJ mol−1 and back-reaction barriers of around 110 kJ mol−1. Though in general that distribution is found to be relatively broad.
The strongest correlation is seen between longer absorption wavelengths of the NBD and high SCEs. Since the intensity of sunlight increases sharply above 400 nm, absorption wavelengths above this threshold significantly contribute to higher SCEs due to the larger overlap with the solar spectrum allowing for more photons to be absorbed and thus more energy to be converted.
The absorption wavelength of QC shows a more disperse relationship to the SCE, where initially a lower energy of the QC absorption correlates to higher SCEs, as this also correlates with longer NBD absorption wavelenghts. With even longer absorption wavelengths of the QC, however, this contributes to a reduced SCE via competition with the reactant NBD for solar power, giving an unfavorable photostationary state.
The oscillator strengths of the excited states of NBD and QC do not show a clear trend with respect to the SCE. Systems with high SCE possess a broad distribution with a tendency towards lower oscillator strengths. This might correlate to the favorably a red-shifted absorption, which leads to lower oscillator strengths.
This investigation aims not only to provide the best candidates for MOST applications of C, N and O bridged NBD systems, but also to shine light on their respective MOST properties in relation to each other. The averages of the respective parameters evaluated within the SCE expression are given in Table 1. The maximum SCEs from GFN2-xTB calculations for the bridges point towards a tendency that C-bridged NBDs exhibit the highest potential. Here, the N-bridged systems are slightly worse but still better than the O-bridged versions. The respective best systems have a maximum solar power conversion efficiency of 10.0%, 7.5% and 4.7% respectively. This is also reflected in the average SCE values of 0.32%, 0.14% and 0.03% being in the same order.
| Bridging unit | ΔES (kJ mol−1) | ΔEtbr (kJ mol−1) | λ NBD (nm) | f NBD | λ QC (nm) | f QC | SCE |
|---|---|---|---|---|---|---|---|
| –(CH2)– | 19.88 | 134.35 | 317.52 | 0.16 | 295.18 | 0.11 | 0.32 |
| –(NC(O)OMe)– | 6.32 | 136.97 | 320.79 | 0.16 | 296.81 | 0.13 | 0.14 |
| –(O)– | −6.42 | 140.45 | 318.90 | 0.17 | 294.98 | 0.12 | 0.03 |
The average storage energies for the data sets shift in a similar fashion. For the C-bridges the majority of systems can be found around 15–20 kJ mol−1, whereas for the N-bridges they accumulate around 0 kJ mol−1 and for the O-bridges this clustering even shifts to negative energies of approximately −15 kJ mol−1 (see Fig. 3 or ESI† for individual plots). This implies a generally reduced stability and reduced storage energy for N- and O-bridged NBD compounds. This suggests that a synthetic investigation and application in devices of these hetero-atom bridged NBDs is less promising.
Interestingly, the thermal back-reaction barrier is barely affected by changing bridge atoms. The majority of the investigated molecules cluster slightly above 140 kJ mol−1 for all three modifications (see Fig. 3 and individual plots in ESI†). This indicates some independence of the thermal back-reaction of the ring opening from the bridge. The distribution of absorption wavelengths, for NBD as well as QC, again is very similar, with a majority of systems absorbing around and slightly below 300 nm, giving an average of around 320 nm (3.87 eV) for all three designs.
At the same time the oscillator strengths for all three bridges also show very similar distributions and extremely similar averages. These findings indicate the absorption behavior of these NBD derivatives is only marginally influenced by the substitution of the bridge atom. Thus, the GFN2-xTB calculations suggest that the largest difference within the general behavior of these systems is the change in storage energy dependent on the bridge (hetero-)atom. This is also reflected in the close resemblance in the substitution patterns of the best compounds identified for the differing bridge heads (see below and ESI†).
3.2 Identification of best candidates
The 20 best compounds for each bridging unit are analyzed for common properties (for the full list of 60 compounds from xTB predictions see ESI†). The majority of compounds include strongly electron withdrawing substituents directly substituted to the NBD core, with additionally two electron donating substituents on the phenyl ring. The nitro group as core substituent appears to be a preferred motive in these systems with its very strong electron withdrawing characteristic. Most systems have similar storage energies of approximately 30–40 kJ mol−1, 20–30 kJ mol−1 and 10–20 kJ mol−1 for the C,N and O bridges, respectively. For the respective data of the two best systems from xTB predictions see Table 2. The thermal back-reaction barriers amount to approx. 110 kJ mol−1 for the majority of molecules. In addition, the first absorption lies above 500 nm, and thus have a significant overlap with the solar spectrum, allowing for a high theoretical solar power conversion efficiency.| Property | C1 | C2 | N1 | N2 | O1 | O2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| xTB | DFT | xTB | DFT | xTB | DFT | xTB | DFT | xTB | DFT | xTB | DFT | |
| ΔES (kJ mol−1) | 37.8 | 48.9 | 38.6 | 62.0 | 25.4 | 35.3 | 23.7 | 40.2 | 15.2 | 46.4 | 15.3 | 61.2 |
| ΔEtbr (kJ mol−1) | 110.1 | 185.7 | 112.0 | 148.7 | 113.9 | 202.5 | 111.2 | 178.9 | 103.4 | 195.3 | 126.5 | 158.4 |
| λ NBD (nm) | 535.2 | 407.0 | 542.6 | 383.0 | 561.3 | 428.2 | 540.3 | 406.2 | 560.7 | 425.8 | 571.7 | 406.2 |
| f NBD | 0.18 | 0.14 | 0.03 | 0.02 | 0.19 | 0.17 | 0.19 | 0.15 | 0.20 | 0.16 | 0.04 | 0.03 |
| λ QC (nm) | 313.3 | 273.1 | 319.1 | 246.5 | 349.3 | 313.3 | 319.4 | 277.4 | 350.4 | 324.1 | 285.7 | 246.5 |
| f QC | 0.09 | 0.09 | 0.04 | 0.03 | 0.03 | 0.02 | 0.08 | 0.06 | 0.02 | 0.02 | 0.07 | 0.03 |
| SCE | 10.0 | 3.15 | 9.6 | 3.24 | 7.5 | 2.05 | 6.7 | 3.43 | 4.7 | 2.40 | 4.3 | 4.67 |
Following the initial screening with GFN2-xTB the 20 best scoring systems for each bridge head were again subjected to geometry optimization and subsequent excited state calculations at the (TD)DFT/M06-2X/def2-SVPD level of theory. This leads to some changes in their predicted SCE, which are discussed in the following. Most importantly, the SCE is generally reduced, which has also been observed in previous works.23,27 This, however, originates from different property changes dependent on the specific system.
In general the storage energies increase at DFT level. This again is consistent with literature23 for the –CH2– bridge head, but also holds true for the hetero-atom bridges additionally investigated here. This should lead to an increased SCE, but this effect is usually overcompensated by other property changes at DFT level.
The change in thermal reaction barrier is less predictable in magnitude and direction. In most cases, the reaction barrier is increased significantly, which contributes to a reduced SCE indirectly via an increased cut-off energy. In some cases however the reaction barrier is lowered, due to the simplistic TS search algorithm employed due to limited resources. In other cases where the back-reaction barrier is reduced, the straight forward approach of a symmetric stretch does not represent the actual transiton state well, which is less symmetric. A reduced thermal back-reaction barrier then contributes to a higher predicted SCE.
The most dramatic change, however, is the significant shift of the absorption maxima to higher energies upon TDDFT calculation. This contributes drastically to the observed reduced SCEs, at DFT level, as the overlap with the solar spectrum is substantially reduced. This is the most important effect in the reduction of SCE, when going from (sTDA-)xTB screening to DFT.
The change in absolute value for the solar power conversion efficiencies can therefore be related to previously expected deficiencies within the (sTDA-)xTB methods. The simplified approach for the prediction of the transition state and the underestimation of the excitation energy are the major contributions. The general tendency of the best identified molecules to also be scored highly within the DFT framework remains, however, and verifies the approach taken via our screening method.
The five best candidated for efficient MOST systems from each subset after the combined screening procedure are shown in Fig. 4. Interestingly, the best scoring derivatives of the C-, N- or O-bridged NBD systems possess an identical substitution pattern, being substituted with a methoxy-thiophenyl and a dicyano-vinyl substituent. The combination of strong electron-donating and electron-withdrawing effects obviously provide an optimal interplay of the different properties, leading to high SCEs for these systems.
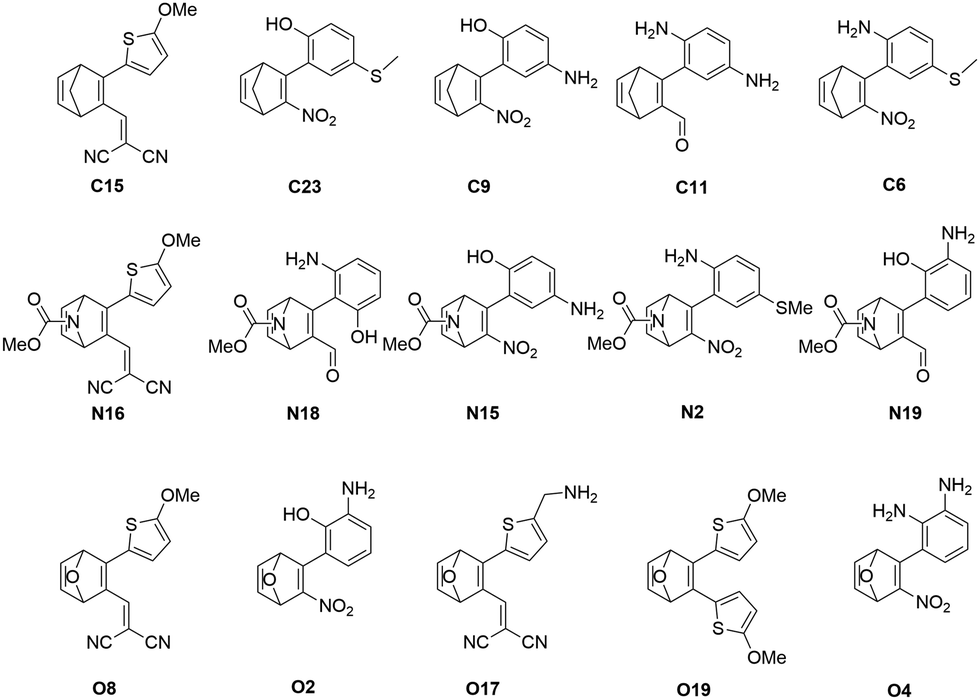 | ||
| Fig. 4 The predicted five best MOST systems from each bridge head subset. The names indicate the scoring position after xTB scoring. | ||
For completeness and as a consistency check, the re-investigation of the worst molecules of the xTB screening via (TD)DFT does not change their evaluation as inappropriate MOST compounds. The higher thermal back-reaction barrier as well as the even worse overlap with the solar spectrum through higher excitation energies, still lead to negligible SCEs.
4 Conclusion
We have investigated carbon, nitrogen and oxygen-bridged NBD compounds and screened them with a high-throughput GFN2-xTB procedure for their expected solar power conversion efficiency. The established database spans a large chemical space for exploratory investigation of this kind of NBD derivatives with potential synthetic availability. We show the classical carbon bridge to be the most promising variant for further MOST investigations. The 20 best scoring candidates from the xTB pipeline are reevaluated with (TD)DFT and scored again. From this, we propose a set of five substituted derivatives per bridge head as best MOST candidates, which have not been investigated previously. They are typically characterized by one electron donating and one electron withdrawing substituent resulting in an overall pronounced push–pull character of the NBD derivatives. In the future these compounds should be considered for further synthesis and subsequent investigation in MOST devices.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgesellschaft (DFG) within the Research Unit 5499 “Molecular Solar Energy Management – Chemistry of MOST Systems”, the Young Researchers Fund of the University of Heidelberg and a mobility grant issued by the 4EU+ alliance. Financial support is acknowledged from the European Commission (Grant No. 765739), and the Danish Council for Independent Research, DFF-0136-00081B.References
- I. P. C. C., Global Warming of 1.5 °C, 2022, 49–92 Search PubMed.
- J. Rugolo and M. J. Aziz, Energy Environ. Sci., 2012, 5, 7151 RSC.
- R. Eschenbacher, F. Hemauer, E. Franz, A. Leng, V. Schwaab, N. J. Waleska-Wellnhofer, E. M. Freiberger, L. Fromm, T. Xu, A. Görling, A. Hirsch, H. Steinrück, C. Papp, O. Brummel and J. Libuda, ChemPhotoChem, 2024, 8, e202300155 CrossRef CAS.
- G. L. Hallett-Tapley, C. D'Alfonso, N. L. Pacioni, C. D. McTiernan, M. González-Béjar, O. Lanzalunga, E. I. Alarcon and J. C. Scaiano, Chem. Commun., 2013, 49, 10073 RSC.
- Z. Wang, R. Losantos, D. Sampedro, M. Aki Morikawa, K. Börjesson, N. Kimizuka and K. Moth-Poulsen, J. Mater. Chem. A, 2019, 7, 15042–15047 RSC.
- L. Dong, Y. Feng, L. Wang and W. Feng, Chem. Soc. Rev., 2018, 47, 7339–7368 RSC.
- E. Franz, A. Kunz, N. Oberhof, A. H. Heindl, M. Bertram, L. Fusek, N. Taccardi, P. Wasserscheid, A. Dreuw, H. A. Wegner, O. Brummel and J. Libuda, ChemSusChem, 2022, 15, e202200958 CrossRef CAS.
- A. Goulet-Hanssens, M. Utecht, D. Mutruc, E. Titov, J. Schwarz, L. Grubert, D. Bléger, P. Saalfrank and S. Hecht, J. Am. Chem. Soc., 2017, 139, 335–341 CrossRef CAS.
- A. M. Kolpak and J. C. Grossman, Nano Lett., 2011, 11, 3156–3162 CrossRef CAS.
- K. Ishiba, M. Morikawa, C. Chikara, T. Yamada, K. Iwase, M. Kawakita and N. Kimizuka, Angew. Chem., Int. Ed., 2015, 54, 1532–1536 CrossRef CAS PubMed.
- K. Edel, X. Yang, J. S. A. Ishibashi, A. N. Lamm, C. Maichle-Mössmer, Z. X. Giustra, S. Liu and H. F. Bettinger, Angew. Chem., Int. Ed., 2018, 57, 5296–5300 CrossRef CAS PubMed.
- M. B. Nielsen, N. Ree, K. V. Mikkelsen and M. Cacciarini, Russ. Chem. Rev., 2020, 89, 573–586 CrossRef CAS.
- J. Daub, T. Knöchel and A. Mannschreck, Angew. Chem., 1984, 96, 980–981 CrossRef CAS.
- H. Goerner, C. Fischer, S. Gierisch and J. Daub, J. Phys. Chem., 1993, 97, 4110–4117 CrossRef CAS.
- M. Quant, A. Lennartson, A. Dreos, M. Kuisma, P. Erhart, K. Börjesson and K. Moth-Poulsen, Chem. – Eur. J., 2016, 22, 13265–13274 CrossRef CAS PubMed.
- J. Orrego-Hernández, A. Dreos and K. Moth-Poulsen, Acc. Chem. Res., 2020, 53, 1478–1487 CrossRef.
- V. Gray, A. Lennartson, P. Ratanalert, K. Börjesson and K. Moth-Poulsen, Chem. Commun., 2014, 50, 5330–5332 RSC.
- K. Börjesson, A. Lennartson and K. Moth-Poulsen, ACS Sustainable Chem. Eng., 2013, 1, 585–590 CrossRef.
- D. A. Strubbe and J. C. Grossman, J. Phys.: Condens. Matter, 2019, 31, 034002 CrossRef PubMed.
- M. Kuisma, A. Lundin, K. Moth-Poulsen, P. Hyldgaard and P. Erhart, ChemSusChem, 2016, 9, 1786–1794 CrossRef CAS.
- W. Sun, Z. Shangguan, X. Zhang, T. Dang, Z. Zhang and T. Li, ChemSusChem, 2023, 16, e202300582 CrossRef CAS.
- Z. Wang, H. Moïse, M. Cacciarini, M. B. Nielsen, M. Morikawa, N. Kimizuka and K. Moth-Poulsen, Adv. Sci., 2021, 8, 2013060 Search PubMed.
- J. L. Elholm, A. E. Hillers-Bendtsen, H. Hölzel, K. Moth-Poulsen and K. V. Mikkelsen, Phys. Chem. Chem. Phys., 2022, 24, 28956–28964 RSC.
- A. T. Young, Appl. Opt., 1994, 33, 1108–1110 CrossRef CAS PubMed.
- J. Calbo, C. E. Weston, A. J. P. White, H. S. Rzepa, J. Contreras-García and M. J. Fuchter, J. Am. Chem. Soc., 2017, 139, 1261–1274 CrossRef CAS.
- N. Ree, M. Koerstz, K. V. Mikkelsen and J. H. Jensen, J. Chem. Phys., 2021, 155, 184105 CrossRef CAS.
- A. E. Hillers-Bendtsen, J. L. Elholm, O. B. Obel, H. Hölzel, K. Moth-Poulsen and K. V. Mikkelsen, Angew. Chem., Int. Ed., 2023, 62, e202309543 CrossRef CAS PubMed.
- A. E. Hillers-Bendtsen, P. G. Iuel Lunøe Dünweber, L. H. Olsen and K. V. Mikkelsen, J. Phys. Chem. A, 2022, 126, 2670–2676 CrossRef CAS PubMed.
- E. Moreno-Clavijo, A. J. Moreno-Vargas, R. Kieffer, T. Sigstam, A. T. Carmona and I. Robina, Org. Lett., 2011, 13, 6244–6247 CrossRef CAS.
- R. A. Valiulin, T. M. Arisco and A. G. Kutateladze, J. Org. Chem., 2013, 78, 2012–2025 CrossRef CAS.
- C. Zhang, C. J. B. Ii and M. L. Trudell, J. Chem. Soc., Perkin Trans. 1, 1999, 675–676 RSC.
- H. Prinzbach, G. Kaupp, R. Fuchs, M. Joyeux, R. Kitzing and J. Markert, Chem. Ber., 1973, 106, 3824–3849 CrossRef CAS.
- S. Riniker and G. A. Landrum, J. Chem. Inf. Model., 2015, 55, 2562–2574 CrossRef CAS PubMed.
- RDKit: Open-source cheminformatics. https://www.rdkit.org Search PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- S. Grimme and C. Bannwarth, J. Chem. Phys., 2016, 145, 054103 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- C. Katan, P. Savel, B. M. Wong, T. Roisnel, V. Dorcet, J.-L. Fillaut and D. Jacquemin, Phys. Chem. Chem. Phys., 2014, 16, 9064–9073 RSC.
- Y. Shao, Y. Mei, D. Sundholm and V. R. I. Kaila, J. Chem. Theory Comput., 2020, 16, 587–600 CrossRef PubMed.
- A. E. Raeber and B. M. Wong, J. Chem. Theory Comput., 2015, 11, 2199–2209 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04179h |
| This journal is © the Owner Societies 2025 |