
Intermolecular interaction potential maps from energy decomposition for interpreting reactivity and intermolecular interactions†
Amin
Kiani‡
 ,
Wentong
Zhou‡
and
Lawrence M.
Wolf
*
,
Wentong
Zhou‡
and
Lawrence M.
Wolf
*
Department of Chemistry, University of Massachusetts Lowell, Lowell, MA 01854, USA. E-mail: lawrence_wolf@uml.edu
First published on 29th October 2024
Abstract
The electrostatic potential (ESP) has been widely used to visualize electrostatic interactions about a molecule. However, electrostatic effects are often insufficient for capturing the entirety of an interaction or a reaction of interest. In this investigation, intermolecular interaction potential maps (IMIPs), constructed from the potentials derived from energy decomposition analysis (EDA) using density functional theory, were developed and applied to provide unique insight into molecular interactions and reactivity. To this end, rather than constructing a potential map from probe point charge interactions, IMIPs were constructed from probe interactions with small molecular fragments, including CH3+, CH3−, benzene, and atomic probes including alkali metals, transition metals, and halides. The interaction potentials are further decomposed producing IMIPs for each interaction component using EDA (electrostatic, orbital, steric, etc.). The IMIPs are applied to the study of various interactions including cation–π and anion–π interactions, electrophilic and nucleophilic aromatic substitution, Lewis acid activation, π-stacking, endohedral fullerenes, and select organometallics which reveal fundamental insight into the positional preferences and physical origins of the interactions that otherwise would be difficult to uncover through other surface analyses.
Introduction
Molecular surfaces reflecting defined properties are often used to predict locations of greater interaction or reactivity within a molecule. The most applied surface properties for predicting interactions is the molecular electrostatic potential (MESP).1–4 MESP surfaces reflect the interaction of the system with a point charge at a designated constant electron density surrounding the molecule and have been applied for describing a wide range of interactions.3,4 While the MESP has proven to be a valuable property for analysis, which is also directly observable, it loses some of its utility in application to weaker dispersion type interactions and orbital based interactions, particularly with unoccupied orbitals, which are not represented by the MESP. Furthermore, the electrostatic interaction is only one component of the total interaction energy, and often is not the strongest. To address these limitations in part, extensions to the MESP that include polarization, from perturbation theory, and empirical dispersion effects have been developed5,6 and applied to anion–π interactions.6–8 It would be useful if molecular surfaces representing all components of the interaction, in addition to the electrostatic, were readily accessible at the QM level. The application and production of such surfaces that reflect all components of the interaction energy with atoms and small molecules of interest have been the focus of this investigation.The MESP belongs to a broader category of molecular surface property approaches (MSPA) that can be used to capture intermolecular interactions indirectly through rigorous manipulation of the wave function or electron density.9 These additional methods include the average local ionization energy (AIE)10,11 and the local electron attachment energy (LEAE).12,13 The AIE gives insight into electron rich sites in a molecule, particularly characterized as Lewis basic or nucleophilic. It accounts for the electron density contributions from all occupied orbitals, with greater weighting given to contributions from higher energy orbitals. In contrast, the LEAE gives a sense of electron deficient regions of the system, particularly those that can be characterized as Lewis acidic or electrophilic, with a similar expression to that of the AIE. These surfaces have been applied to non-covalent interactions,13,14 organic substitution and addition reactions,15–17 and nanoparticles and surfaces.18,19 Related approaches include Fukui index surfaces20 that reflect the most favorable sights within the HOMO and LUMO, orbital overlap distance,21 and through visualization of non-covalent interactions (NCIs) through various NCI analysis approaches22,23 including the electron localization function (ELF).24,25 While these approaches have been used to correlate with reactivity and intermolecular interactions, these correlations are indirect in part by excluding reacting or interacting components of interest, thereby missing some components of the total interaction, and many of these approaches are typically insufficient for capturing both weak and steric interactions in particular.
Conversely, intermolecular interactions can be directly captured through calculating probe interactions which can be mapped on a grid producing molecular interaction fields (MIFs). An MIF is a field of points of interaction around a target molecule with a probe.26 The interaction between the probe and molecule is typically evaluated classically through a Coulomb potential to capture electrostatic interactions and a Lennard-Jones potential to capture steric interactions. Recent efforts have been undertaken for the construction of DFT based MIFs with some success in describing protein–ligand interactions.27,28 MIFs have been used extensively as descriptors for the development of quantitative structure–activity relationship (QSAR) predictive models for drug design,29,30 as well as in asymmetric catalysis using machine learning.31 While MIFs have found success in predictive modeling, they have generally not been applied for interpretation or rationalization of intermolecular interactions nor for electronic structure analysis.
In this work, we sought to construct molecular surfaces that can be used for characterizing both weak and strong intermolecular interactions that expand the predictive capacity of MIFs and the MSPA by mapping relevant probe interactions on an isodensity surface (Fig. 1). This approach required judicious orientations for molecular probes about the isosurface. In particular, a planar CH3+ probe is positioned tangentially to the isodensity surface using its norm vector with planarity restricted (Fig. 1). These probe interactions are then further decomposed into contributions from electrostatic, orbital, steric, and dispersion interactions using energy decomposition analysis (EDA), which are mapped on an isodensity surface that can be used to uncover physical insight into the nature of the interactions and positional preferences. These intermolecular interaction potential maps (IMIPs) are then shown to capture interactions important in describing reactivity in electrophilic aromatic substitution, nucleophilic aromatic substitution, and organometallics as well as intermolecular interactions including cation–π, anion–π, and π–π stacking with appropriate probe selection. While this IMIP approach is computationally more costly, as each probe interaction represents an independent wave function determination, it can be used to better reflect these interactions, furnishing deeper insight into the nature of the interaction while also imparting information about the electronic structure.
 | ||
| Fig. 1 (a) MESP surface for benzene. (b) IMIP surfaces from probe interaction energies between benzene and a CH3+ probe, further decomposed using EDA. | ||
Computational methods
The IMIPs are generated using atomic or small molecule probes, from which interaction energies are calculated. EDA is used to further decompose these energies to generate EDA-IMIPs. EDA methods within Orca 5.1,32 Turbomole 7.7,33,34 and xTB 6.6.035 have been implemented in the code for molecular surface generation. The Python code for generating IMIP surfaces is hosted on GitHub.EDA methods
Multiple EDA methods are used for surface generation which include the LMO-EDA36 as implemented in Turbomole, Grimme Group's xTB-IFF,37 and the ETS-NOCV38 schemes as implemented in ORCA. All of the results discussed here are based on the LMO-EDA method for its available implementation in Turbomole and its applications in describing various non-covalent interactions, some considered here.39 The resulting surfaces are shown to be qualitatively similar surfaces constructed using the ETS-NOCV scheme (Fig. S1, ESI†). Details on the NOCV-ETS and xTB-IFF EDA methods and their respective surfaces are available in the ESI† (Fig. S1). The LMO-EDA method uses a supramolecular approach to obtain the interaction energies of two interacting fragments. The interaction energy (ΔEint) is decomposed into electrostatic (ΔEelec), exchange (ΔEex), repulsion (ΔErep), polarization (ΔEorb), dispersion (ΔEdisp), and correlation (ΔEcorr) energies (eqn (1)). Furthermore, the exchange and repulsion, and the dispersion and correlation terms are combined to construct terms for the repulsive (ΔEex–rep) and van der Waals like attractive (ΔEdisp–corr) interactions, respectively.| ΔEint = ΔEelec + ΔEex + ΔErep + ΔEorb + ΔEcorr + ΔEdisp | (1) |
Probe selection
The interpretation of the IMIFs is dependent on the probe structure. For the assessment of the varied interactions discussed here, many probe types are explored (Fig. S2, ESI†) including alkali cations, alkali earth cations, transition metals, halides, CH3+, CH3−, and C6H6. Where main-group metals, transition metals, and halide probes are suitable for studying systems where metal–ligand interactions, cation–π, or anion–π interactions are of interest, while C6H6 can be used to probe systems for regions where π-stacking interactions exist. The CH3+ probe is well-suited for capturing electron-rich regions susceptible to electrophilic attack, while CH3− is appropriate for exploring electron-deficient regions susceptible to nucleophilic attack. Finally, probes like CO and PR3 are ideal for studying unsaturated transition metal complexes. Additional probes can be readily implemented for IMIP construction.Surface generation
Molecular surfaces are constructed by generating a grid surrounding a target molecule. An EDA calculation is performed at each grid point between the probe and molecule. Grid generation, probe placement, positioning, orientation, and visualization are described in Fig. 2 (expanded in Fig. S4, ESI†). | ||
| Fig. 2 Overview for IMIP map generation (a), structure optimization (b), grid generation (c), isodensity selection and multiple filtration steps (d), probe positioning and orientation (e), EDA calculation and (f) surface generation (units: kcal mol−1). | ||
Grid generation method
Cubic (Fig. 2) or spherical (Fig. S3, ESI†) grid methods can be used for grid construction. The present study utilizes a cubic grid generation method. The surfaces generated are based on user-defined electron isodensity values. First, a sparse cubic grid is generated around the molecule (Fig. 2a and b). An isodensity range is then set to filter off points. Subsequently, a volume element is created for each remaining grid point, and within this volume, a finer grid is constructed and filtered. This refined grid selects grid points within a narrow electron density range of 0.01% (Fig. 2c and Fig. S4, ESI†). A more detailed illustration of this method is provided in the ESI† (Fig. S4).The cubic initial grid is typically dense; conducting EDA calculations for probes at each grid point would be computationally demanding, therefore grid filtering is necessary. A K-means clustering algorithm40 is used to filter grid points (Fig. 2c).
Probe positioning and orientation
After grid generation and filtration, probes are positioned at each grid point. Single-atom probes are positioned without ambiguity while the alignment of molecular probes is determined by vectors normal to the tangential plane of the molecular surface (Fig. 2d) based typically on the preferred orbital directionality of the probes. The normal vectors are computed using the Open3D Python library.41 Open3D estimates the normal vectors on the surface by statistical analysis of neighboring grid points.42 By combining this approach with a rigid rotor scan using GFN2-xTB (Fig. 2d), the lowest energy orientation of the probe is determined within reasonable computational cost. EDA calculations are then conducted on the grid points; their corresponding energy values are stored as extended XYZ files and are subsequently used to construct surfaces using Ovito Pro (Fig. 2e and f).43 Coupling the GFN2-xTB scan with DFT interaction energies produces surfaces that are qualitatively indistinguishable from surfaces produced from a full DFT scan coupled with DFT interaction energies (Fig. S6, ESI†), thus validating the use of the GFN2-xTB scan at substantially lower computational cost.Structure optimization and EDA
Quantum mechanical (QM) and semi-empirical methods were employed depending on the size of the system under investigation. For larger systems biomolecule or supramolecular systems, the GFN2-xTB44 method was utilized for geometry optimizations. For all other cases, DFT with the r2SCAN45 functional, def2-SVP46,47 basis set, D4 dispersion correction,48–50 and resolution of identity (RI) approximation51,52 was used with a full grid and radial grids sizes set to 7 and 5, respectively45,53,54 and SCF convergence was set to 10−6 for EDA calculations and 10−7 for geometry optimization. The r2SCAN functional combined with the D4 dispersion correction was chosen as a proven cost-effective all-purpose method capable of describing a broad range of chemistry54–56 determined to be suitable for the applications covered in this investigation. BSSE corrections were not employed here, although surfaces for select molecules were constructed with a larger basis set (def2-TZVPP) and were shown to be qualitatively very similar (Fig. S5, ESI†).Isodensity value selection
Isodensity selection for surface generation was based on two approaches (methods A and B). In method A, the isodensity is selected based on a restricted scan along a coordinate approximately normal to the surface, at a site of interest to the interaction, to locate the maximum interaction strength. This method was used for comparing the same molecule with different probes. In method B, isodensities were determined from exploratory work on weak and strong interactions. For weak interactions a smaller isodensity of 1.0 × 10−6 was used, and a larger isodensity of 6.0 × 10−5 was used for stronger interactions. Method B was applied to compare different structures with the same probe.Results and discussion
Cation–π interactions
The cation–π interaction is defined as occurring between a metal cation and an aromatic π-surface57,58 and influences the physical and chemical properties of various systems across fields including biology,58–60 organic chemistry,58,60–62 and materials chemistry.60 IMIP surfaces can be used to better understand the magnitude and positional preference for cation–π interactions across various systems. In this study, a series of aromatic hydrocarbons with the probes Li+, Na+, K+, Mg2+, Be2+, Ca2+, Ag+, and Zn2+ were investigated (Fig. 3a). This selection encompasses a broad spectrum of interactions within aromatic systems. | ||
| Fig. 3 ΔEint, ΔEelec, and ΔEorb (top to bottom) IMIP's of (a) benzene with Li+, Na+, K+, Mg2+, Be2+, Ca2+, Ag+, and Zn2+ probes (left to right). (b) Aniline, toluene, and benzonitrile (probe = Na+) (c) naphthalene, anthracene, phenanthrene, and tetracene (Ag+ probe). Isodensities (method A; benzene centroid): Be2+: 3.15 × 10−3, Mg2+: 5.49 × 10−4, Ca2+: 1.32 × 10−4, Li+: 7.52 × 10−4, Na+: 9.15 × 10−5, K+: 8.21 × 10−6, Zn2+: 5.49 × 10−4, Ag+: 1.93 × 10−4. | ||
ΔEint surfaces reveal that for all cations, the most favorable interactions occur over the center of the benzene (1) π-system. Divalent cations exhibit stronger total interaction energies with benzene than monovalent cations, manifested in both ΔEorb and ΔEelec. ΔEint decreases for probes along a periodic group. This reduction in ΔEint is predominantly attributed to decreased ΔEorb, as there is little variation in ΔEelec, consistent with poorer orbital overlap with the carbon π system (as probe size increases) resulting in reduced covalent character. This transition in bonding character is further highlighted by the ratio of ΔEelec to ΔEorb at the ring center. For Be2+, Mg2+ and Ca2+, the ratios are 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8.5, 1:2.4, 1:2, respectively, which suggests a more covalent bonding character. In contrast, Li+, Na+, and K+ have 1:1.4, 1:0.8, and 1:0.7 ratios, indicating a shift towards more ionic-like character. Other theoretical investigations have shown this shift in ionic to covalent character between the alkali and alkaline earth metals.63,64 The maximum in ΔEelec is centered over the π-system for all probes excluding Be2+, where the maximum is shifted slightly outward ∼10 kcal mol−1 relative to the center. Conversely, ΔEorb is strongest at the center for Be2+, with a ∼27 kcal mol−1 difference between these points and where the electrostatic interaction is maximal. It is the dominant ΔEorb that drives the ΔEint to the center, not the ΔEelec for Be2+, whereas for Mg2+ and Ca2+, ΔEelec is more important for central probe preference.
:8.5, 1:2.4, 1:2, respectively, which suggests a more covalent bonding character. In contrast, Li+, Na+, and K+ have 1:1.4, 1:0.8, and 1:0.7 ratios, indicating a shift towards more ionic-like character. Other theoretical investigations have shown this shift in ionic to covalent character between the alkali and alkaline earth metals.63,64 The maximum in ΔEelec is centered over the π-system for all probes excluding Be2+, where the maximum is shifted slightly outward ∼10 kcal mol−1 relative to the center. Conversely, ΔEorb is strongest at the center for Be2+, with a ∼27 kcal mol−1 difference between these points and where the electrostatic interaction is maximal. It is the dominant ΔEorb that drives the ΔEint to the center, not the ΔEelec for Be2+, whereas for Mg2+ and Ca2+, ΔEelec is more important for central probe preference.
The surfaces of mono-substituted benzenes (2–4) with a Na+ probe reveal interesting trends in substituent influence on the interaction (Fig. 3b; Fig. S51, ESI†). While the interaction is strongest with donating groups (NH2) and weakest with withdrawing groups (CN), the variation in ΔEint tracks with the variation in ΔEelec, while only a small variation is observed in ΔEorb (Table S3 and Fig. S52, ESI†). These trends indicate that the substituent influence on the cation–π interaction is primarily an electrostatic effect. Similar observations were made regarding substituent influence on cation–π interactions by Wheeler and Houk.65–67
The transition metals Zn2+ and Ag+ show a notable increase in ΔEint compared to metals with similar ionic radii (Mg2+ and K+, respectively; Fig. 3a). Like non-transition metal probes, ΔEint is most prominent at the center of the π-system. The increase in ΔEint stems from both ΔEelec and ΔEorb terms. The enhanced ΔEorb for the transition metals can be rationalized based on more favorable interaction with their higher energy occupied s and p orbitals with some back-bonding from their occupied d orbitals.
As polyaromatic cyclic hydrocarbon (PAH) size increases, ΔEint increases with increasing number of fused rings, driven largely by ΔEorb, translating to increased partial covalent character in the interactions (Fig. 3c; Fig. S7–S14, ESI†), which is in agreement with prior theoretical studies.68–70 Furthermore, ΔEorb reveals that alkali and alkaline earth metals mainly engage around the center of rings of the π-systems, whereas transition metals (eq. Ag+) are drawn to terminal rings, engaging in isolated π bonding. Notably, larger rings enhance orbital interactions at their peripheries, where the interacting probe can localize electron density most while maintaining delocalization and aromaticity in the unbound rings. This effect is more pronounced when contrasting anthracene and phenanthrene (Fig. 3c; Fig. S7 and S14, ESI†). This also clarifies why the outer rings of anthracene exhibit enhanced orbital interactions over its center. In contrast, carbons at ring junctions interact less favorably, where probe association results in reduced aromaticity. This behavior is most noticeable with transition metals. These effects are largely orbital based as reflected in the ΔEorb surfaces.
Anion–π interactions
Favorable interactions can occur between anions and electron deficient π systems called anion–π interactions.71 They are thought to operate in some biological and supramolecular systems, and more recently in small molecule catalysis,72,73 but are not as well understood as cation–π interactions. To give a better understanding of these interactions, IMIP surfaces are used to examine how different anionic probes engage with various electron-deficient π systems. Finding stable anion–π complexes is both experimentally and computationally challenging due to the propensity for Meisenheimer74,75 complex formation, therefore our focus is not necessarily on systems where anion–π interactions are experimentally verified, but rather on identifying regions in molecules where these interactions may be relevant.The molecules shown in Fig. 4 were selected for studying anion–π interactions as they have been extensively examined previously.76–80 The halide probes explored include F−, Cl−, Br− and I−. It is shown that ΔEint decreases as the halide probe size decreases, (Fig. 4; Fig. S52–S56, ESI†). For Cl−, Br−, and I− probes, the minimum ΔEint is over the hexafluorobenzene (9) center. However, with F−, the most favorable site shifts to between the π bonds. This shift for F− is attributed to preferences in ΔEorb which is likely reflecting favorable F−(2p) overlap with the π* orbitals resulting in an energetic preference of ∼2.3 kcal mol−1 compared to the center. Within the π system, steric repulsion is reduced with the probe nonconcentric. Conversely, ΔEelec is most favorable at the ring center, but ΔEorb and ΔEex–rep shift the ΔEint preference off-center. This trend is less pronounced with larger probes like Cl−, Br−, and I− and is ascribed to weaker ΔEorb than that with F−.
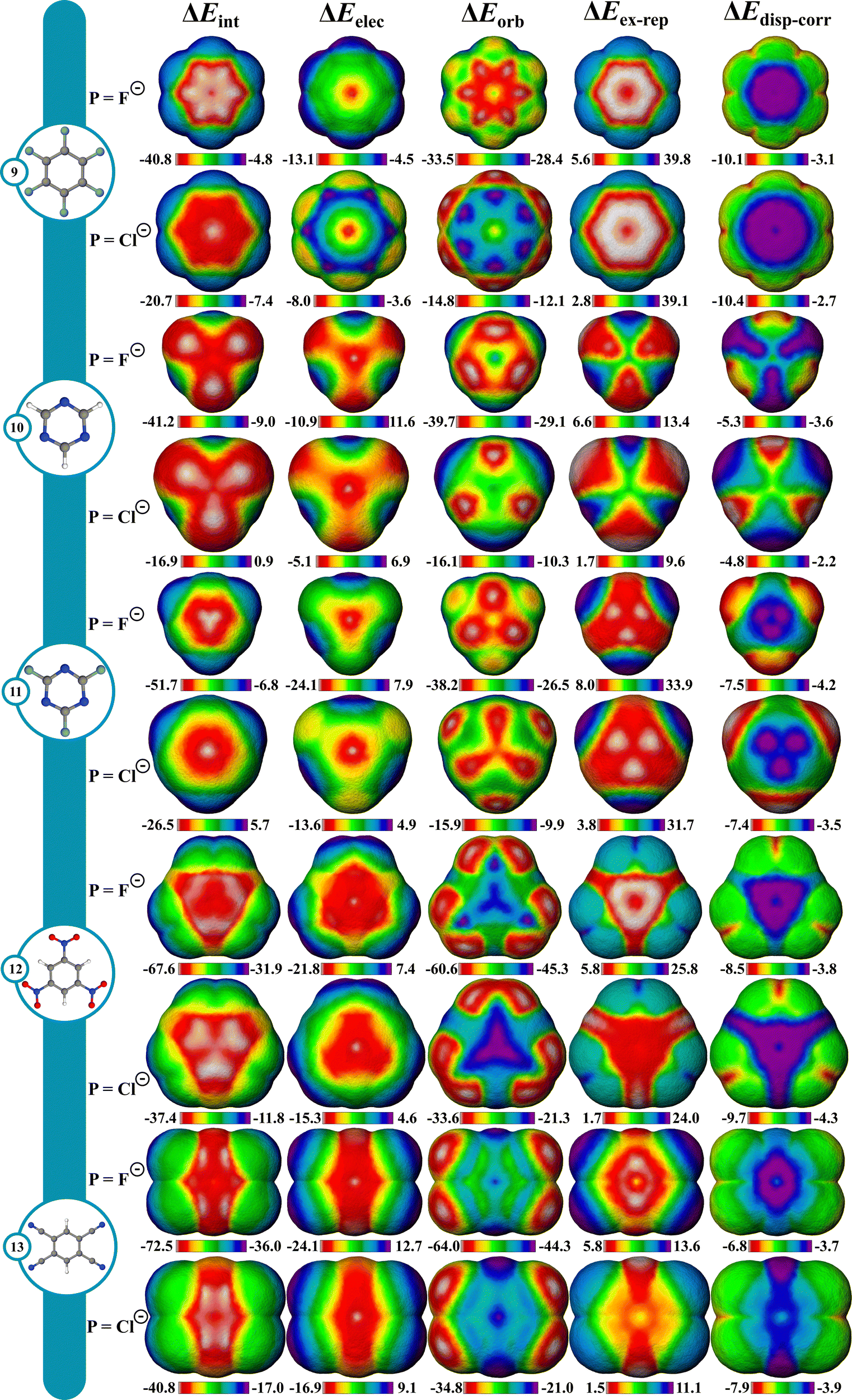 | ||
| Fig. 4 ΔEint, ΔEelec, ΔEorb, ΔEex–rep and ΔEdisp–corr IMIP's (left to right) of hexafluorobenzene, triazine, trifluoroazine, trinitrobenzene, and tetracyanobenzene (top to bottom), with F− and Cl− probes. Isodensities (method A; center of ring system): F− = 5.62 × 10−5 and Cl− = 1.22 × 10−6. | ||
With azines, trifluoro-triazine (11) interacts stronger with all halide probes than triazine (10) which is largely attributed to ΔEelec since ΔEorb is similar in magnitude between the two. Since the fluorine substituents are both inductively withdrawing and π-donating,81 these effects are partially cancelled out, leading to a small difference in ΔEorb. The heightened electrostatic interactions in 11 can be rationalized by considering the sum of the ion–dipole (C–F bond dipoles) interactions. This helps account for the observed decrease in ΔEelec as the probe size increases, since ion–dipole interactions are more pronounced with denser F− ion than with Cl−. In both azines, ΔEelec is most favorable in the center, with some reduced preference near the C–H bonds in triazine. Additionally, the ΔEex–rep surfaces reveal that the minimum repulsions are located around the C–H bonds for 10 whereas in 11, the maximum repulsions are around the C–F bonds. This combined steric and electrostatic effect results in different favorable energy landscapes between the molecules.
The most favorable interaction for compounds with strong π-withdrawing groups, trinitrobenzene (12) and tetracyano-benzene (13), is shifted off-center, driven by ΔEorb and ΔEex–rep with the center still being favored by ΔEelec. The shift off-center is a result of enhanced ΔEorb (where the LUMO is localized) with slight reinforcement from ΔEex–rep (Fig. 4 and Fig. S52–S56, ESI†). While both systems show similar ΔEint at their center, their most favorable sites differ by ∼4.8 kcal (F−) and ∼3.3 kcal (Cl−), with 13 being stronger given the extra π-withdrawing group.
From the surfaces provided, it is apparent that increasing the number of π-withdrawing substituents leads to increased overall interaction. Moreover, π-withdrawing groups enhance the anion–π interaction more effectively than σ-withdrawing groups (cf.9, 12, and 13). Lastly, while the probe interaction is not strongly influenced by ΔEdisp–corr, the proportion to ΔEint increases with increasing probe size.
EAS reactivity
Reactivity can often be interpreted through analysis of reactant structure, particularly through the use of FMO theory. In cases where the electronic structure of the reactant is useful for assessing reactivity, typically where substantial nuclear rearrangement has not occurred in the TS, the resulting IMIP surfaces may be useful.While ESP maps have been used to qualitatively assess relative rates of substituted aromatics in EAS, these surfaces are much less useful for understanding selectivity concepts pertaining to orbital overlap which influences chemical reactivity.21,82–84 IMIP surfaces can be particularly useful for revealing the electronic structure of π-surfaces with variable substitution, which strongly affects rates and selectivity in EAS reactions. The IMIP surfaces of various benzene derivatives were generated using the CH3+ probe, which can be used to pinpoint the most reactive sites for electrophilic addition. Benzene derivatives with substituents of –NH2, –OCH3, –CH3, –Cl, –H, –CHO, and –CF3 (14–20) have been compared. This list spans most of the electronic spectrum as supported by substituent constants including Hammett sigma values reflecting inductive and π-donating and withdrawing effects.85 Shown in Fig. 5a, the ΔEint surface clearly highlights enhanced interaction at the ortho and para positions, which is dominated by orbital interactions as illustrated in the ΔEorb surfaces. The most favorable ΔEorb within the π system correlates with relative reactivity patterns observed in EAS reactions.86 The reactivity order follows NH2 > OCH3 > CH3 > Cl > H > CHO > CF3 and correlates with Hammett σ+ constants which are known to reflect EAS reactivity (Fig. 6; Table S1, ESI†).87,88 This level of analysis is less readily extracted from ESP surfaces alone.
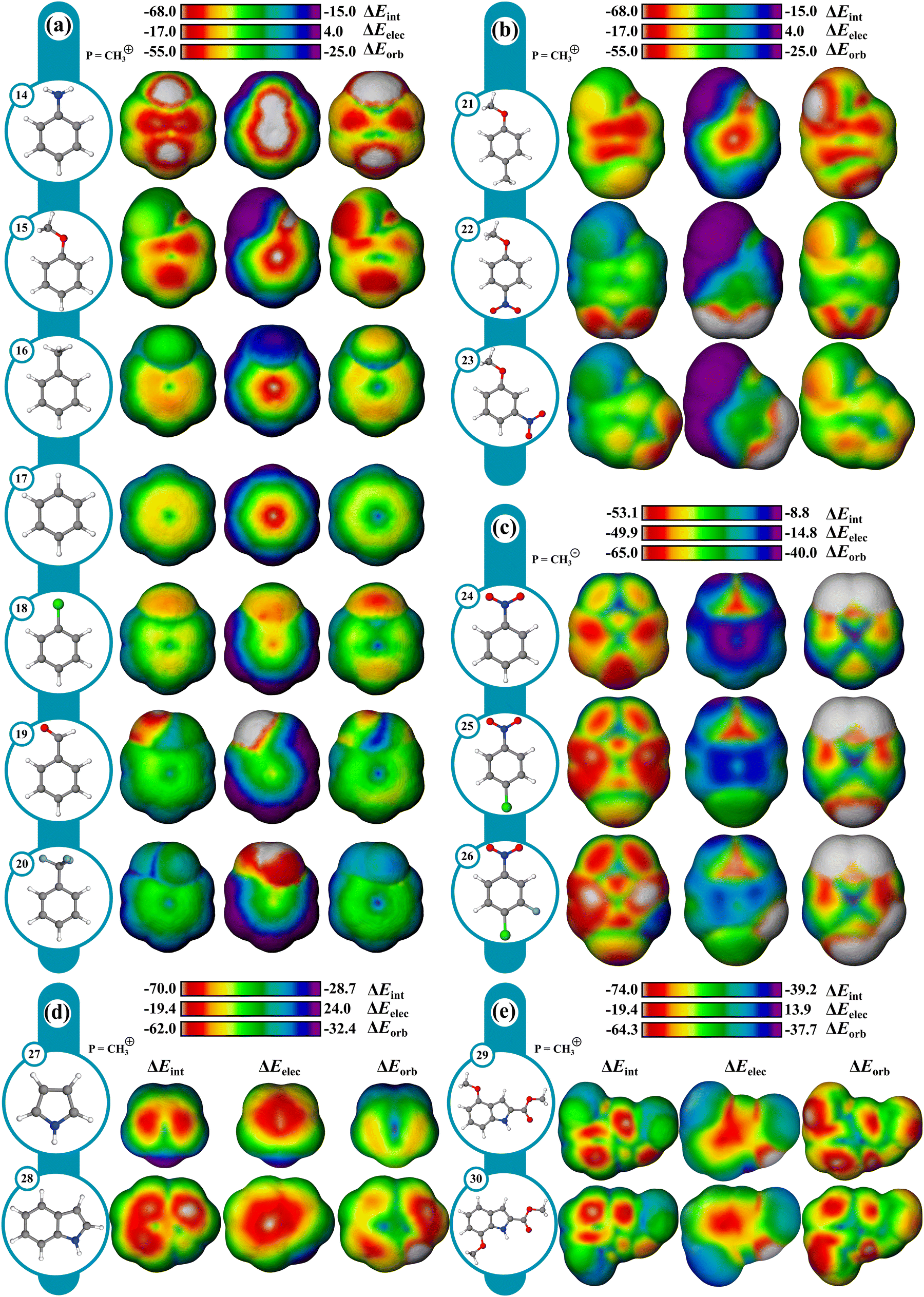 | ||
| Fig. 5 From left to right ΔEint, ΔEelec, and ΔEorb IMIP's of (a) mono-, (b) di-subsituted benzene derivatives (probe = CH3+), and (c) surfaces generated with CH3− probe for benzene derivatives. Surfaces for (d) pyrrole, indole, and (e) indole derivatives (probe = CH3+); isodensity: 6.00 × 10−5; method B. | ||
 | ||
| Fig. 6 Hammet plot of (a) ΔEintvs. σ+, (b) ΔEorbvs. σ+, and (c) ΔEelecvs. σ+. Energies are obtained at the para-position. | ||
The orbital surface for chloro-benzene (18) shows that the strongest probe interaction is directed to the ortho and para positions consistent with EAS selectivity with halogens, which is greater than the maximum orbital interaction in benzene (17). However, the orbital surface analysis does not capture the complete reactivity profile as 18 is deactivated relative to 17. The ΔEelec surface reveals that 18 is electrostatically deactivated relative to 17. The electrostatic deactivation in 18 offsets its orbital activation rendering it overall deactivating as reflected in the ΔEint surface. With meta-directing deactivating groups, like –CHO (19) and –CF3 (20), the ΔEint surfaces show significantly weaker interactions consistent with their reduced reactivity.88,89 Selectivity in poly-substituted benzenes (21–23; Fig. 5b) with meta and ortho–para directors (OMe vs. NO2) or competing ortho–para directors (OMe vs. Me) is readily visualized through the ΔEint and ΔEorb surfaces, and is in agreement with experiment.90,91
Moreover, useful reactivity patterns can be discerned with heteroaromatics in EAS (Fig. 5d). ΔEint and ΔEorb surfaces (Fig. 5d) for pyrrole (27) clearly show the 2-position to be the most reactive, while the ΔEelec surface shows little difference between the positions. For indole (28), the surfaces show the most reactive position to be closer to the 3-position than the 2-position, consistent with experimental patterns. Also, ΔEint between indole and pyrrole reveals indole to be more reactive, reflected mostly in ΔEorb, consistent with experimental nucleophilicity parameters.92,93 Regioselectivity in substituted indoles can also be assessed. For example, 29 shows an experimental preference for the 7 and 5 positions at an 85:15 ratio towards EAS. For 30, experiments also show the 4 and 6 positions to be reactive towards EAS at 80:20 ratio.94 The ΔEint and ΔEorb surfaces for both 29 and 30 are qualitatively consistent with the experimental site preferences.
SNAr reactivity
While EAS reactivity was probed using a CH3+ probe, nucleophilic aromatic substitution (NAS) can likewise be probed with an anionic CH3− probe. Using nitrobenzene (24) as a reference in the absence of a leaving group, the most reactive sites are the ortho and para positions with ∼2.1 kcal mol−1 difference between sites favoring the para as determined from ΔEint. para substitution shifts the preference to ortho. Computational and experimental findings revealed that nucleophiles add to unsubstituted ortho positions more rapidly than at the halogenated para positions.95 However, only addition at the halogenated para positions leads to the desired substitution product, while other additions result in transient adducts without productive outcomes, which the surfaces can readily highlight.To assess the influence of multiple leaving groups on reactivity, 1-chloro-4-nitrobenzene (25) and the 3-fluoro (26) analogue were compared. For both compounds the ortho positions are identified as the most reactive. Notably, 26 displays enhanced reactivity at both the ortho and para positions with energy differences of approximately 3–4 kcal mol−1 and 5–6 kcal mol−1, respectively, when compared to 25. This enhancement is driven largely by the ΔEelec component, which favors the ortho and para positions by 1 kcal mol−1 and 3 kcal mol−1 respectively. The ΔEorb component also contributes to the enhancement in 26, though to a lesser extent. The surfaces highlight the preference for nucleophilic attack at the p-chloro position over the m-fluoro position despite fluoro being more active in NAS.
π-stacking
The IMIP surfaces can be used to gain insight into weaker non-covalent interactions including π–π type interactions (Fig. 7a). Benzene can be used as a probe to generate surfaces revealing favorable π–π interactions which may include sandwich, T-shaped, and slip-stacking arrangements as well as cation–π, anion–π, and polar–π interactions. The ΔEint surface for stacked benzene (31) is unfavorable at the centroid (∼1.2 kcal mol−1), contrasted by favorable interaction encircling the centroid (∼−3.0 to −3.5 kcal mol−1). There is also favorable interaction at the ring periphery leading to T-shaped and edge-to-face interactions (∼−2.3 kcal mol−1). ΔEelec and ΔEorb surfaces mark the central region as having favorable interactions with each contributing ∼2.3 kcal mol−1 stabilization. ΔEdisp–corr provides ∼9 kcal mol−1 of stabilization in this region. ΔEex–rep surface reveals that maximal repulsions (∼15 kcal mol−1) occur when the rings are directly stacked, suggesting at close distances sterics drives the preference for a slipped arrangement (∼15 kcal mol−1vs. ∼4 kcal mol−1). The total energy surfaces for hexaflourobenzene (32) follow a similar trend, where the slip-stacked arrangement is still the most favored, as steric repulsion at the center is more unfavorable. The trends at sites of maximum interaction are summarized in Fig. 8.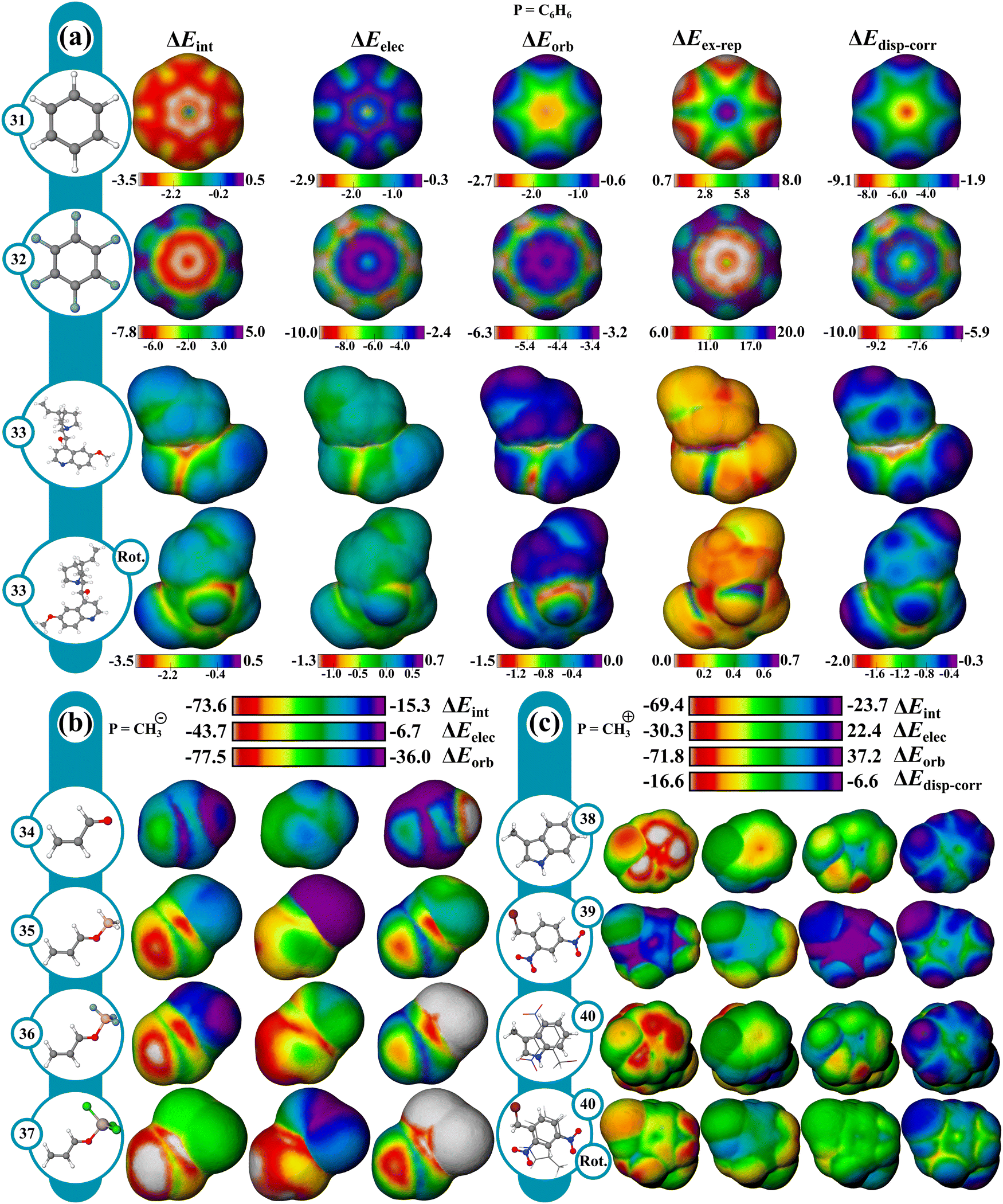 | ||
| Fig. 7 IMIPs: (a) benzene, hexafluorobenzene, cinchona alkaloid (probe = C6H6); (b) acrolein, –BH3, –BF3, and –AlCl3 coordinated acrolein (probe = CH3+); (c) methylindole, 2,4-dinitrobenzyl bromide, EDA complex (probe = CH3+). Isodensity: benzene, cinchona alkaloids, hexafluorobenzene (method B; 1.00 × 10−6), acrolein systems (method B; 6.00 × 10−5), and EDA complex (method B; 6.00 × 10−5). | ||
 | ||
| Fig. 8 Plots of (a) C6H6 (left) and (b) C6F6 (right) sites of maximum interaction, ΔEint (red), ΔEelec (orange), ΔEorb (yellow), ΔEdisp–corr (light green), and ΔEex–rep (dark green); probe = C6H6. | ||
The positional preference for the π–π interactions is controlled mostly by ΔEdisp–corr and ΔEex–rep consistent with the general understanding that dispersion interactions are important for describing π-stacking. Furthermore, despite the stabilizing roles of ΔEelec, ΔEorb, and ΔEdisp–corr, it is ΔEex–rep that often overrides these stabilizing effects, emphasizing its importance in determining molecular stacking arrangements. These results are in agreement with recent theoretical findings,96,97 which propose that at typical π-stacking and CH⋯π distances the traditional electrostatic model may not accurately capture the nature of these interactions.
Furthermore, complex π-type interactions can be isolated in larger molecules from these IMIPs. Using quinine (33) as an example, four types of favorable π interaction regions can be identified which include the interaction of the nitrogen lone pair on the quinoline ring (n → π*), the oxygen lone pair from the hydroxyl group (n → π*), the hydroxyl proton (polar–π), and the π-face interactions (π–π stacking).
Lewis acid-activated acrolein
The influence of Lewis acids on unsaturated carbonyls was explored using a nucleophilic CH3− probe, with focus on 1,2 vs. 1,4-addition to acrolein (Fig. 7b). Starting with unactivated acrolein (34), ΔEint surfaces show a favorable interaction with a probe at both the carbonyl carbon and β carbons, with the β carbon displaying a greater preference for nucleophilic attack. Under Lewis acid activation, the magnitude of ΔEint max increases, following the reactivity trend of unactivated (−46.2 kcal) < BH3 (−71.2 kcal, 35) < BF3 (−73.6 kcal, 36) < AlCl3 (−87.1 kcal, 37). The ΔEorb term more strongly influences this reactivity trend than ΔEelec. While the carbonyl carbon exhibits stronger ΔEelec, the ΔEorb term at the β carbon dominates. This orbital preference for the β-carbon is consistent with the known preference of soft nucleophiles favoring conjugate addition. Interestingly, BH3 increases the ΔEorb term at both sites more than BF3. BF3 offsets this disparity through increased ΔEelec. These surfaces uniquely reveal the enhanced activation of BF3 over BH3 which is explained as an electrostatic influence rather than an orbital influence and is in agreement with recent theoretical findings.98,99Electron donor–acceptor complexes
The electron donor–acceptor (EDA) complex (40) formed by 3-methylindole and 2,4-dinitrobenzyl bromide100 was investigated using a CH3+ probe to capture electron transfer effects (Fig. 7c). Comparison of the surfaces of the individual fragments (38 and 39) with the complex (40) shows a stronger total interaction near the acceptor and a weaker interaction near the donor, reflecting charge transfer from 38 to 39 manifested mostly in ΔEorb and to a lesser extent in ΔEelec. The probe interaction change is greater in 38, since the CH3+ interaction near 39 induces further electron transfer from 38, and probe interaction near 38 reduces charge transfer to 39.Organometallic complexes
Reactivity in select organometallic complexes is next shown in Fig. 9 using CH3− and CH3+ probes. The regioselectivity of nucleophilic attack on allyl–Pd complexes generally favors the less substituted position with standard phosphine ligands but can be reversed in the presence of strong cation stabilizing groups like OMe.101,102 Analysis of the corresponding reactant allyl–Pd complexes reproduces these trends (Fig. 9a and Fig. S58, ESI†). With methyl substitution, (42), the largest probe interaction favors the unsubstituted allyl carbon by 5.4 kcal mol−1 in ΔEint, which is favored by both ΔEorb and ΔEelec. Substitution of the methyl group for a methoxy group (43) reverses the most favored probe interaction site to the more substituted allyl carbon by 5.2 kcal mol−1 in ΔEint, driven primarily by a preference in ΔEelec (ca. 12.1 kcal mol−1). The preference for the methoxy position is enhanced further with methoxy and methyl substitution on each allyl carbon (44), (ΔΔEint = 23.4 kcal mol−1). The unsubstituted (41) reference system is also shown, where no such preference exists, with only slight differences based on ligand conformation. | ||
| Fig. 9 IMIPs: (a) substituted Pd(PPh3)3(allyl) complexes (probe = CH3−); (b) Cp*RuCl complex (CH3+ and CH3− probes); (c) fullerene and endohedral fullerenes@H2O, @Li+ (probe = CH3+). Isodensity: 6.00 × 10−5. | ||
CpRuX (X = halogen) complexes can be used to catalyze an array of transformations and are particularly useful for effecting trans-selective hydrogenation and hydrometalation.103 The surfaces for CpRuCl highlight its ambiphilic donating and accepting behavior (45). ΔEorb for the CH3+ and CH3− probes shows favorable orbital interactions at the metal, being −87 kcal mol−1 and ∼−60 kcal mol−1, respectively. ΔEelec for CH3+ demonstrates unfavorable interactions (∼7 kcal mol−1), stemming from the electropositive metal. CH3− interacts favorably at −27 kcal mol−1. Favorable ΔEorb and unfavorable ΔEelec indicate charge donation from the metal to the probe. Favorable ΔEorb and ΔEelec suggest donation from the probe to the metal indicating that the complex is a stronger donor than acceptor. These results are further corroborated by comparing surfaces constructed from NBO charge transfer values, which give charge transfers of ∼−0.8e with CH3+ and ∼+0.5e with CH3− (Fig. 9b).
Endohedral fullerenes & host guest effects
Endohedral fullerenes were also investigated to uncover the electronic influence of the guest on the C60 surface (Fig. 9c). With a CH3+ probe, the interaction is strongest at the 6-membered rings, comprising mostly ΔEorb. With an H2O guest (47), the C60 surface becomes polarized, which is mostly attributed to a change in ΔEelec induced by the H2O dipole moment. With a Li+ guest (48), the probe interaction is substantially weakened uniformly throughout. ΔEelec becomes highly repulsive with minor reduction in ΔEorb while the symmetry of the surface remains unchanged. The scale is adjusted for Li+@C60 (48) to provide contrast. The IMIP surfaces readily convey the effects of guests on the C60 electronic structure.Conclusions
In this investigation, the development and application of IMIP maps towards rationalizing reactivity and intermolecular interactions for a variety of systems in an intuitive way has been presented that is not readily achieved through other surface analyses. Within cation–π interactions, the surfaces demonstrate the ring center to be the site of maximum interaction, controlled by ΔEelec. They also reveal that for alkali metals, the reduction in the interaction along a periodic group is attributed to a decreasing orbital interaction. With transition metal probe examples including Zn2+ and Ag+, the interaction positional preference is shifted off-center on account of favorable π-bonding as indicated in ΔEorb.The IMIP maps revealed unique positional preferences in anion–π interactions with halide probes. In C6F6, the positional preference is for the π bonds as revealed by the ΔEorb surface using an F− probe, whereas larger halide probes exhibit preferential binding at the center on account of weaker π bonding interactions. The preference shifts for the triazines and stronger π-withdrawing groups.
EAS and NAS reactivity and selectivity were readily demonstrated from analysis of the surfaces with a CH3+ probe. The ortho/para selectivity was clearly highlighted from π-donating groups to halogens, which is reflected by ΔEorb, with ΔEelec modulating the level of activation from benzene. Selectivity for multiply substituted rings can be predicted from the inspection of the ΔEint and ΔEorb surfaces. Similar predictions can be made for more complex heteroaromatics including pyrrole and substituted indoles. Strong correlation between ΔEint and σ+ was demonstrated which is dominated by ΔEorb with lower correlation with ΔEelec.
The origins of positional preferences in weaker π–π type interactions were revealed through inspections of the IMIP surfaces using a C6H6 probe. The preference for the slipped-stack arrangement was shown to be determined by reduced ΔEexp–rep despite all favorable interactions favoring direct stacking, including the most favorable term, ΔEdisp–corr. The C6F6⋯C6H6 interaction still favored a slipped-stack arrangement, but shifted closer to direct stacking largely driven by reduced ΔEex–rep interactions. More complex π interactions were also probed in quinine.
Furthermore, Lewis acid activation on acrolein was inspected using a CH3− probe which revealed enhanced reactivity and enhanced site selectivity at the β carbon driven by ΔEorb. Interestingly, while it is shown that BF3 is more activating than BH3, the increased activation is electrostatic in origin as BH3 enhances ΔEorb more than BF3. Insight into interactions involved in select organometallics, endohedral fullerenes, and an EDA complex were also gained through the analysis of the IMIP surfaces.
The demonstrations of the utility of IMIP surfaces for the interactions discussed here are just a small subset of their potential applications. We predict these surfaces to have broader applicability and work into expanding their utility is ongoing.
Author contributions
A. K. and W. Z. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have approved the final version of the manuscript.Data availability
The Python code for generating IMIP surfaces has been made accessible at https://github.com/lwolflab/IMIP. All QM calculations were performed using Turbomole 7.7 (https://turbomole.org). Molecular probe scans and xTB-IFF EDA calculations were performed using the xTB package (https://github.com/grimme-lab/xtb). Multiwfn 3.8 (https://sobereva.com/multiwfn/) was used for extracting electron densities. All visualization was done using Ovito (https://ovito.org). Computational data including detailed methods, additional IMIP surfaces images, plots, tables, molecular Cartesian coordinates, and field files are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for partial financial support from the National Science Foundation (CHE-2305006). The authors thank the Massachusetts Green High-Performance Computing Center (MGHPCC) and the MIT SuperCloud and Lincoln Laboratory Supercomputing Center for providing computational resources.Notes and references
- M. Orozco and F. J. Luque, J. Comput. Chem., 1993, 14, 587–602 CrossRef CAS.
- E. B. Wilson, Jr., J. Chem. Phys., 1962, 36, 2232–2233 CrossRef.
- J. S. Murray and P. Politzer, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 153–163 CAS.
- C. H. Suresh, G. S. Remya and P. K. Anjalikrishna, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1601 CAS.
- F. J. Luque and M. Orozco, J. Comput. Chem., 1998, 19, 866–881 CrossRef CAS.
- D. Escudero, A. Frontera, D. Quiñonero, A. Costa, P. Ballester and P. M. Deyà, J. Chem. Theory Comput., 2007, 3, 2098–2107 CrossRef CAS PubMed.
- C. Estarellas, A. Frontera, D. Quiñonero and P. M. Deyà, J. Chem. Theory Comput., 2008, 4, 1981–1989 CrossRef CAS PubMed.
- C. Estarellas, A. Bauzá, A. Frontera, D. Quiñonero and P. M. Deyà, Phys. Chem. Chem. Phys., 2011, 13, 5696–5702 RSC.
- T. Brinck and J. H. Stenlid, Adv. Theory Simul., 2019, 2, 1800149 CrossRef.
- P. Sjoberg, J. S. Murray, T. Brinck and P. Politzer, Can. J. Chem., 1990, 68, 1440–1443 CrossRef CAS.
- F. A. Bulat, M. Levy and P. Politzer, J. Phys. Chem. A, 2009, 113, 1384–1389 CrossRef CAS.
- B. Ehresmann, B. Martin, A. H. C. Horn and T. Clark, J. Mol. Model., 2003, 9, 342–347 CrossRef CAS PubMed.
- T. Brinck, P. Carlqvist and J. H. Stenlid, J. Phys. Chem. A, 2016, 120, 10023–10032 CrossRef CAS PubMed.
- J. H. Stenlid, A. J. Johansson and T. Brinck, Crystals, 2017, 7, 222 CrossRef.
- J. J. Brown and S. L. Cockroft, Chem. Sci., 2013, 4, 1772–1780 RSC.
- M. Liljenberg, T. Brinck, B. Herschend, T. Rein, G. Rockwell and M. Svensson, J. Org. Chem., 2010, 75, 4696–4705 CrossRef CAS PubMed.
- J. H. Stenlid and T. Brinck, J. Org. Chem., 2017, 82, 3072–3083 CrossRef CAS PubMed.
- J. H. Stenlid and T. Brinck, J. Am. Chem. Soc., 2017, 139, 11012–11015 CrossRef CAS.
- J. Du, X. Cheng, X. Zhou, M. Shi, G. Jiang and D. Chen, J. Phys. Chem. A, 2024, 128, 9587–9595 CrossRef CAS PubMed.
- R. G. Parr and W. Yang, J. Am. Chem. Soc., 1984, 106, 4049–4050 CrossRef CAS.
- A. Mehmood, S. I. Jones, P. Tao and B. G. Janesko, J. Chem. Inf. Model., 2018, 58, 1836–1846 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- R. Laplaza, F. Peccati, R. A. Boto, C. Quan, A. Carbone, J.-P. Piquemal, Y. Maday and J. Contreras-García, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1497 CAS.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- A. Savin, R. Nesper, S. Wengert and T. F. Fässler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1808–1832 CrossRef CAS.
- P. J. Goodford, J. Med. Chem., 1985, 28, 849–857 CrossRef CAS PubMed.
- D. Hayakawa, N. Sawada, Y. Watanabe and H. Gouda, J. Mol. Graphics Modell., 2020, 96, 107515 CrossRef CAS PubMed.
- D. Hayakawa, Y. Watanabe and H. Gouda, J. Chem. Inf. Model., 2024, 64, 6003–6013 CrossRef CAS.
- R. D. Cramer, D. E. Patterson and J. D. Bunce, J. Am. Chem. Soc., 1988, 110, 5959–5967 CrossRef CAS PubMed.
- G. Klebe, U. Abraham and T. Mietzner, J. Med. Chem., 1994, 37, 4130–4146 CrossRef CAS PubMed.
- A. F. Zahrt, S. V. Athavale and S. E. Denmark, Chem. Rev., 2020, 120, 1620–1689 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding, C. Hättig, A. Hellweg, B. Helmich-Paris, C. Holzer, U. Huniar, M. Kaupp, A. Marefat Khah, S. Karbalaei Khani, T. Müller, F. Mack, B. D. Nguyen, S. M. Parker, E. Perlt, D. Rappoport, K. Reiter, S. Roy, M. Rückert, G. Schmitz, M. Sierka, E. Tapavicza, D. P. Tew, C. Van Wüllen, V. K. Voora, F. Weigend, A. Wodyński and J. M. Yu, J. Chem. Phys., 2020, 152, 184107 CrossRef CAS PubMed.
- Y. J. Franzke, C. Holzer, J. H. Andersen, T. Begušić, F. Bruder, S. Coriani, F. Della Sala, E. Fabiano, D. A. Fedotov, S. Fürst, S. Gillhuber, R. Grotjahn, M. Kaupp, M. Kehry, M. Krstić, F. Mack, S. Majumdar, B. D. Nguyen, S. M. Parker, F. Pauly, A. Pausch, E. Perlt, G. S. Phun, A. Rajabi, D. Rappoport, B. Samal, T. Schrader, M. Sharma, E. Tapavicza, R. S. Treß, V. Voora, A. Wodyński, J. M. Yu, B. Zerulla, F. Furche, C. Hättig, M. Sierka, D. P. Tew and F. Weigend, J. Chem. Theory Comput., 2023, 19, 6859–6890 CrossRef CAS.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1493 CAS.
- P. Su, Z. Tang and W. Wu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1460 CAS.
- S. Grimme, C. Bannwarth, E. Caldeweyher, J. Pisarek and A. Hansen, J. Chem. Phys., 2017, 147, 161708 CrossRef.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS.
- N. Kumar, S. Saha and G. N. Sastry, Phys. Chem. Chem. Phys., 2021, 23, 8478–8488 RSC.
- J. B. MacQueen, L. M. Le Cam and J. Neyman, Proceedings of the fifth berkeley symposium on mathematical statistics and probability, 1967 Search PubMed.
- Q.-Y. Zhou, J. Park and V. Koltun, arXiv, 2018, preprint DOI:10.48550/arXiv.1801.09847.
- M. Pauly, In Point primitives for interactive modeling and processing of 3D-geometry, 2003 DOI:10.3929/ethz-a-004612876.
- A. Stukowski, Modell. Simul. Mater. Sci. Eng., 2010, 18, 015012 CrossRef.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS.
- J. W. Furness, A. D. Kaplan, J. Ning, J. P. Perdew and J. Sun, J. Phys. Chem. Lett., 2020, 11, 8208–8215 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 15122 CrossRef PubMed.
- E. Caldeweyher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2017, 147, 034112 CrossRef PubMed.
- E. Caldeweyher, J.-M. Mewes, S. Ehlert and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 8499–8512 RSC.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–290 CrossRef CAS.
- G. Santra and J. M. L. Martin, Molecules, 2022, 27, 141 CrossRef CAS.
- S. Ehlert, U. Huniar, J. Ning, J. W. Furness, J. Sun, A. D. Kaplan, J. P. Perdew and J. G. Brandenburg, J. Chem. Phys., 2021, 154, 061101 CrossRef CAS PubMed.
- M. Bursch, J.-M. Mewes, A. Hansen and S. Grimme, Angew. Chem., Int. Ed., 2022, 61, e202205735 CrossRef CAS.
- S. Spicher, E. Caldeweyher, A. Hansen and S. Grimme, Phys. Chem. Chem. Phys., 2021, 23, 11635–11648 RSC.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS.
- D. A. Dougherty, Acc. Chem. Res., 2013, 46, 885–893 CrossRef CAS.
- J. P. Gallivan and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459–9464 CrossRef CAS PubMed.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2013, 113, 2100–2138 CrossRef CAS PubMed.
- R. R. Knowles, S. Lin and E. N. Jacobsen, J. Am. Chem. Soc., 2010, 132, 5030–5032 CrossRef CAS.
- C. R. Kennedy, S. Lin and E. N. Jacobsen, Angew. Chem., Int. Ed., 2016, 55, 12596–12624 CrossRef CAS.
- X. Jian Tan, W. Liang Zhu, M. Cui, X. Min Luo, J. De Gu, I. Silman, J. L. Sussman, H. Liang Jiang, R. Yun Ji and K. Xian Chen, Chem. Phys. Lett., 2001, 349, 113–122 CrossRef.
- W. Zhu, X. Tan, J. Shen, X. Luo, F. Cheng, P. C. Mok, R. Ji, K. Chen and H. Jiang, J. Phys. Chem. A, 2003, 107, 2296–2303 CrossRef CAS.
- S. E. Wheeler and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 3126–3127 CrossRef CAS.
- R. K. Raju, J. W. G. Bloom, Y. An and S. E. Wheeler, ChemPhysChem, 2011, 12, 3116–3130 CrossRef CAS PubMed.
- S. E. Wheeler, Acc. Chem. Res., 2013, 46, 1029–1038 CrossRef CAS PubMed.
- D. Vijay and G. N. Sastry, Phys. Chem. Chem. Phys., 2008, 10, 582–590 RSC.
- J. R. Premkumar, D. Vijay and G. N. Sastry, Dalton Trans., 2012, 41, 4965–4975 RSC.
- T. C. Dinadayalane, A. Hassan and J. Leszczynski, Theor. Chem. Acc., 2012, 131, 1131 Search PubMed.
- B. L. Schottel, H. T. Chifotides and K. R. Dunbar, Chem. Soc. Rev., 2008, 37, 68–83 RSC.
- Y. Zhao, Y. Cotelle, L. Liu, J. López-Andarias, A.-B. Bornhof, M. Akamatsu, N. Sakai and S. Matile, Acc. Chem. Res., 2018, 51, 2255–2263 CrossRef CAS PubMed.
- D.-X. Wang and M.-X. Wang, Acc. Chem. Res., 2020, 53, 1364–1380 CrossRef CAS PubMed.
- K. Hiraoka, S. Mizuse and S. Yamabe, J. Phys. Chem., 1987, 91, 5294–5297 CrossRef CAS.
- K. Hiraoka, S. Mizuse and S. Yamabe, J. Chem. Phys., 1987, 86, 4102–4105 CrossRef CAS.
- D. Quiñonero, C. Garau, C. Rotger, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Angew. Chem., Int. Ed., 2002, 41, 3389–3392 CrossRef.
- D. Quiñonero, C. Garau, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Chem. Phys. Lett., 2002, 359, 486–492 CrossRef.
- I. Alkorta, I. Rozas and J. Elguero, J. Am. Chem. Soc., 2002, 124, 8593–8598 CrossRef CAS PubMed.
- B. Han, J. Lu and J. K. Kochi, Cryst. Growth Des., 2008, 8, 1327–1334 CrossRef CAS.
- M. Mascal, A. Armstrong and M. D. Bartberger, J. Am. Chem. Soc., 2002, 124, 6274–6276 CrossRef CAS PubMed.
- T. X. Carroll, T. D. Thomas, H. Bergersen and K. J. Børve, J. Org. Chem., 2006, 71, 1961–1968 CrossRef CAS PubMed.
- K. Fukui, Science, 1982, 218, 747–754 CrossRef CAS.
- R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1982, 21, 711–724 CrossRef.
- G. Klopman, J. Am. Chem. Soc., 1968, 90, 223–234 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- B. Galabov, S. Ilieva, D. Cheshmedzhieva, V. Nikolova, V. A. Popov, B. Hadjieva and H. F. Schaefer, ACS Omega, 2022, 7, 8199–8208 CrossRef CAS.
- G. A. Olah, S. J. Kuhn and S. H. Flood, J. Am. Chem. Soc., 1961, 83, 4571–4580 CrossRef CAS.
- L. M. Stock and H. C. Brown, in Advances in Physical Organic Chemistry, ed. V. Gold, Academic Press, 1963, vol. 1, pp. 35–154 Search PubMed.
- P. W. Robertson, P. B. D. De La Mare and B. E. Swedlund, J. Chem. Soc., 1953, 782–788 RSC.
- L. M. Stock and H. C. Brown, J. Am. Chem. Soc., 1960, 82, 1942–1947 CrossRef CAS.
- J. G. Hoggett, Nitration and Aromatic Reactivity, Cambridge University Press, 1971 Search PubMed.
- B. Kempf, N. Hampel, A. R. Ofial and H. Mayr, Chem. – Eur. J., 2003, 9, 2209–2218 CrossRef CAS.
- S. Lakhdar, M. Westermaier, F. Terrier, R. Goumont, T. Boubaker, A. R. Ofial and H. Mayr, J. Org. Chem., 2006, 71, 9088–9095 CrossRef CAS.
- N. Roué, T. Delahaigue and R. R. Barret, Heterocycles, 1996, 43, 263 CrossRef.
- K. Błaziak, W. Danikiewicz and M. Mąkosza, J. Am. Chem. Soc., 2016, 138, 7276–7281 CrossRef.
- K. Carter-Fenk and J. M. Herbert, Chem. Sci., 2020, 11, 6758–6765 RSC.
- K. Carter-Fenk and J. M. Herbert, Phys. Chem. Chem. Phys., 2020, 22, 24870–24886 RSC.
- L. J. Duarte, W. E. Richter, R. E. Bruns and P. L. A. Popelier, J. Phys. Chem. A, 2021, 125, 8615–8625 CrossRef CAS.
- A. Thayyil and P. C. Parambil, ChemPhysChem, 2023, 24, e202200761 CrossRef CAS.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- F. Delbecq and C. Lapouge, Organometallics, 2000, 19, 2716–2723 CrossRef CAS.
- N. Vicart, B. Cazes and J. Goré, Tetrahedron Lett., 1995, 36, 535–538 CrossRef CAS.
- A. Fürstner, J. Am. Chem. Soc., 2019, 141, 11–24 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03237c |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2025 |