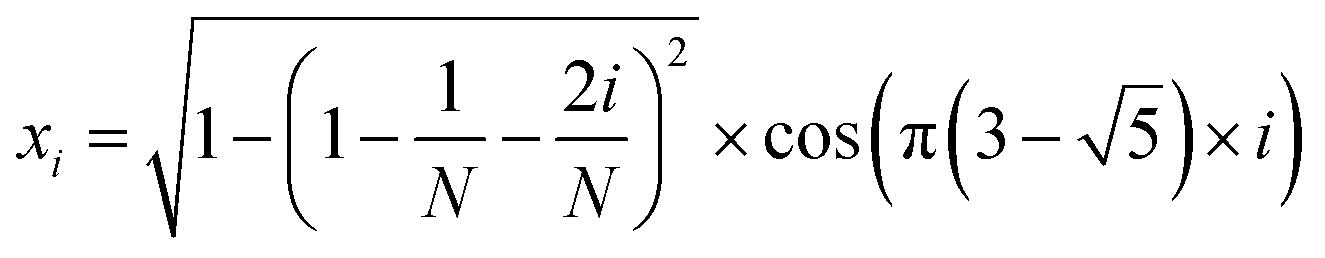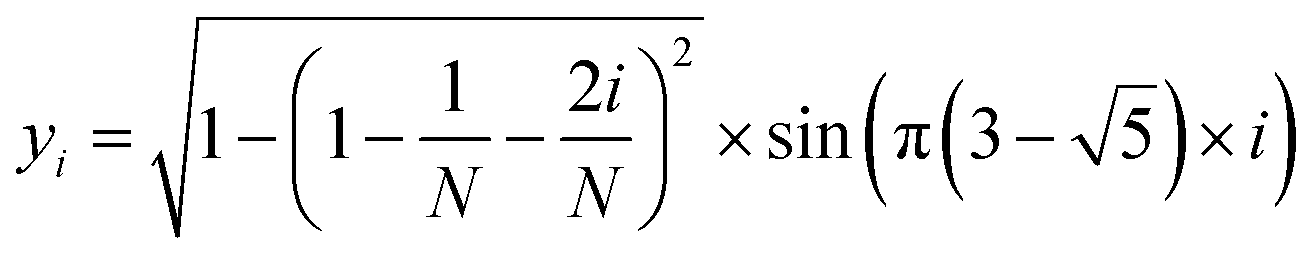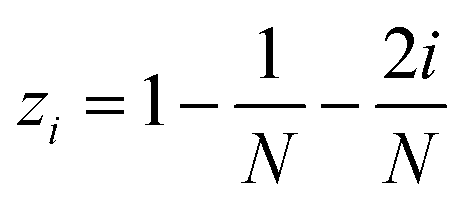Design of bisamide inhibitors of the TASK-1 potassium channel in silico†
Received
10th September 2024
, Accepted 23rd November 2024
First published on 9th December 2024
Abstract
TWIK-related acid-sensitive potassium channel 1 (TASK-1) is expressed ubiquitously across various tissues and plays a significant role in neural activity and anesthetic modulation, making it a crucial target for pharmaceutical research. The high conservation of binding site residues within the TASK family, particularly between TASK-1 and TASK-3, necessitates the development of selective inhibitors for TASK-1. In this study, we utilized a combination of structure-based drug design (SBDD) and ligand-based drug design (LBDD) approaches. Initially, several bisamide-centered molecules were designed using the program MolAICal, which is recognized for its ability to generate selective inhibitors containing bisamide segments, and conducted preliminary screening via molecular docking. Subsequently, 3D-QSAR models were developed for 56 bisamide derivatives targeting TASK-1 and TASK-3, with the models exhibiting robust predictive capabilities (TASK-1: Q2 = 0.61, R2pred = 0.84; TASK-3: Q2 = 0.60, R2pred = 0.71). Using these models, the candidate molecules were subjected to activity prediction and subsequent filtering. Ultimately, molecular dynamics simulations, coupled with free energy calculations, pinpointed two bisamide-core molecules with favorable ADMET properties as potential selective inhibitors for TASK-1. Furthermore, molecular dynamics simulations revealed the critical role of the key residue Leu122 in conferring selectivity to bisamide compounds for TASK-1 channel proteins.
Introduction
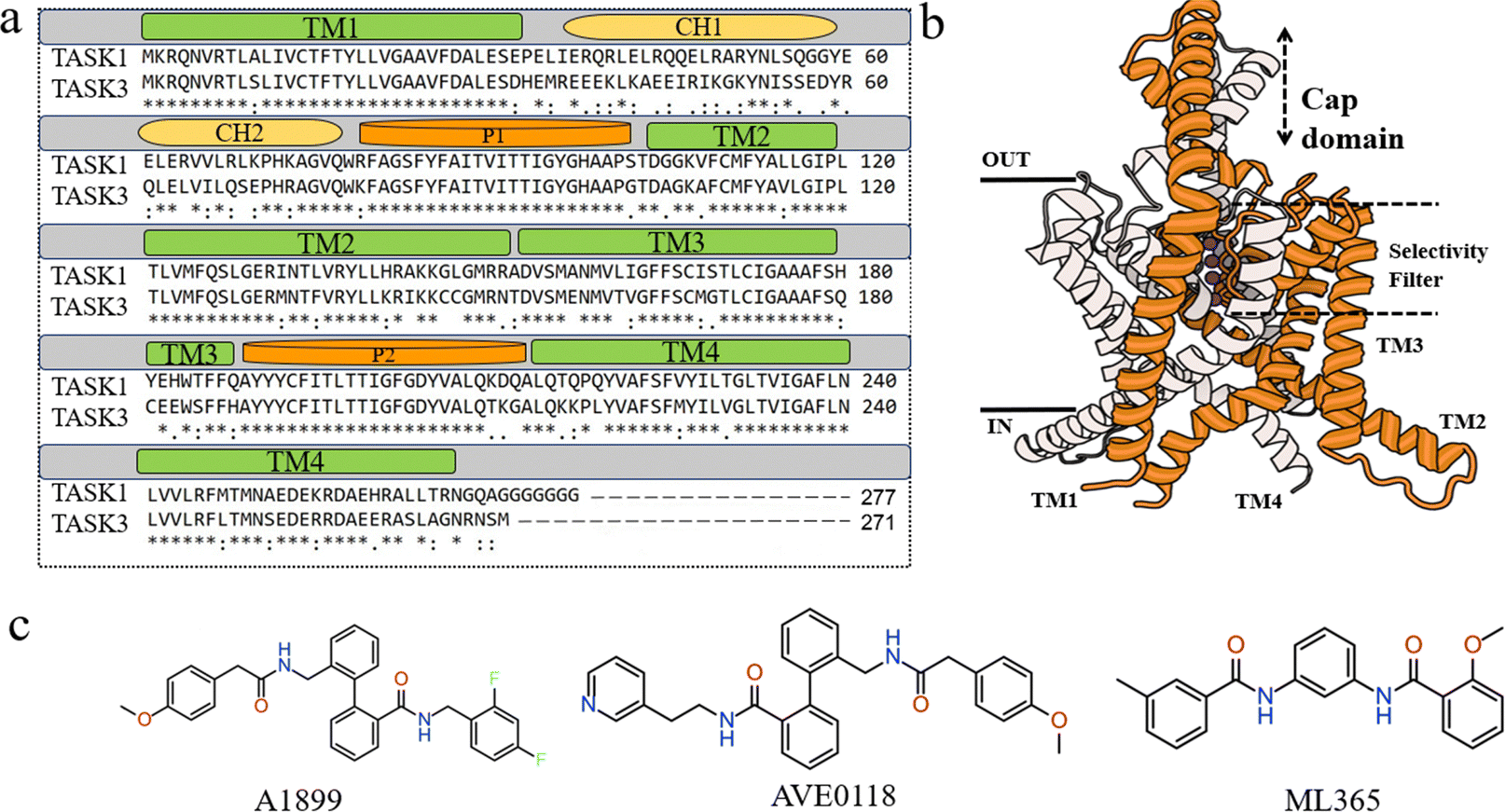
Two-pore-domain potassium (K2P) channels are broadly distributed across diverse cell types and are pivotal in the release of neurotransmitters.1,2 The first mammalian K2P channel identified was named TWIK-1, which stands for tandem of pore domains in a weak inward rectifying K+ channel. The TASK channel (TWIK-related acid-sensitive potassium channel 1) is a member of the K2P channel family, found in neuronal cells, cardiomyocytes, and the central nervous system, where it serves as a key regulator of various physiological processes.1,3 Crystallized structures of K2P channels have shown that the cap consists of two helices per monomer, termed the inner and outer helices (Fig. 1a). In contrast, the outer helix of the cap in other crystallized K2P channels interacts with the inner helix of the adjacent monomer, forming the domain-swapped conformation.4 The K2P family primarily includes TASK-1, TASK-3, and TASK-5. TASK-1 and TASK-3 channel proteins are targeted by various neurotransmitters and modulators, including norepinephrine, serotonin, hydrogen ions, and zinc ions.5 Their activity is modulated by oxygen, volatile anesthetics, hormones, and neurotransmitters. In contrast, TASK-5 channel protein is predominantly involved in muscle contraction.6–9 Within the TASK family, TASK-1 shares the highest amino acid sequence homology with TASK-3, reaching up to 58.9% (Fig. 1a),3,10,11 with 51.4% homology to TASK-5 and 55.1% between TASK-3 and TASK-5. Amino acid sequence similarity exceeding 50% can indicate similar co-expression patterns and potential heterodimerization; however, no functional expression of TASK-5 was observed in the recombinant system. Consequently, the selectivity between TASK-1 and TASK-3 poses a significant challenge in the study of TASK-1 channel proteins.8,12–14
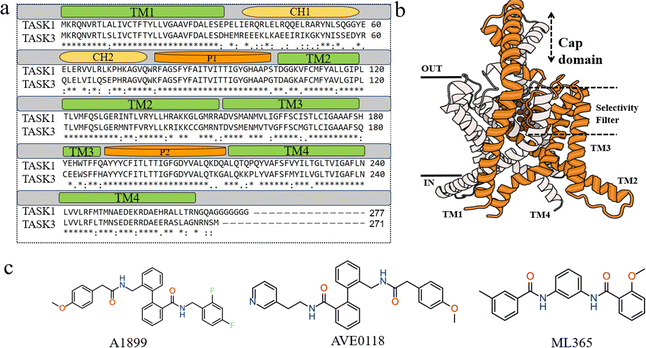 |
| | Fig. 1 (a) Amino acid sequence alignment of TASK-1 and TASK-3 in humans. (b) Three-dimensional structure of TASK-1, featuring two subunits depicted in gray and orange cartoon representations, with potassium ions shown in purple. (c) Two-dimensional structures of A1899, AVE0118, and ML365. | |
Overactivation of TASK-1 can lead to changes in the intracellular membrane potential, resulting in arrhythmias, atrial fibrillation, inflammation, and sleep apnea, among other complications, making TASK-1 an attractive therapeutic target in recent years.15,16 TASK channel proteins exhibit a distinctive double-pore structure, differing from other potassium channels. They form a homodimer, consisting of a large extracellular “cap” domain, four transmembrane helices (TM1–TM4), and two pore-forming domains (P1–P2) (Fig. 1b).17 Within the TASK family, homology is relatively low between the C- and N-termini, with higher homology observed in the central cavity and transmembrane regions of the channel protein.18 TASK channels have emerged as promising drug targets for a range of conditions, such as atrial fibrillation, sleep apnea, diabetes, pulmonary hypertension, and cardiac conduction disorders. There is a significant medical need for selective TASK channel blockers or activators. Compared to other potassium channels, the K2P channel blockers identified to date preferentially bind to a conserved site within the central cavity. Consequently, compounds A189917 and A29374, which inhibit TASK-1, are classified as classical open channel blockers due to their binding to the central cavity. Currently, a few TASK-1 inhibitors, including Doxapram and Anandamide, have been approved by the US Food and Drug Administration (FDA), with several others in various stages of clinical trials. Consequently, the development of a selective inhibitor for TASK-1 is of critical importance. To date, highly selective inhibitors for TASK-1 are scarce. Nonetheless, several TASK-1 selective inhibitors have been identified, including A1899, AVE0118, and ML365, which contain aromatic rings, hydrogen-bond acceptors, hydrophobic groups, and bisamide cores.15,19–22 High-throughput screening has revealed bisamide derivatives as the most promising candidates for TASK-1 selective inhibitors.23,24 Subsequently, Flaherty et al. reported the discovery of bisamide derivatives as novel selective central TASK channel blockers.25 They synthesized a series of derivatives based on the bisamide scaffold, with most exhibiting selectivity for TASK-1. The most effective and selective compound exhibited an IC50 value for TASK-1 that was 62-fold higher than that for TASK-3 in QPatch automated electrophysiology tests. We observed that many selective central blockers for TASK-1 contain bisamide cores and demonstrate relatively superior inhibitory activity and selectivity.19 Consequently, we posit that focusing on compounds with bisamide cores could significantly enhance the likelihood of identifying novel selective TASK-1 channel inhibitors.
Computer-aided drug design (CADD) is an effective technique for drug design and discovery, encompassing two primary categories: structure-based drug design (SBDD) and ligand-based drug design (LBDD).26 Typically, only one of these methods is employed in the drug design process.27,28 To enhance the quality of candidate inhibitors, we integrated both SBDD and LBDD methods to identify high-quality inhibitor molecules. Traditional molecular design relies on manual processes, demanding experienced researchers with extensive knowledge and adherence to explicit rules.29–31 With the rapid advancement of artificial intelligence, deep learning has become a crucial branch of AI applied to drug design. MolAICal is a 3D drug design software that integrates the strengths of genetic algorithms and classical algorithms within deep learning to generate 3D ligands within protein binding pockets.32 This process eschews reliance on the researcher's experience, instead producing highly efficient and diverse molecules by modifying the scaffold and functional groups of known ligands based on the receptor protein. Conventional approaches in ligand-based drug design encompass quantitative structure–activity relationships (QSAR), a fundamental chemometric tool in computational drug design, which is further categorized into 2D-QSAR and 3D-QSAR. 3D-QSAR incorporates the three-dimensional structure of drug molecules into QSAR studies, indirectly revealing the non-covalent interaction characteristics between molecules and macromolecules during their interaction.26 This method offers a more precise physical interpretation and richer information content than 2D-QSAR. Moreover, 3D-QSAR is a widely employed method in drug design, guiding the design of specific drug fragments and predicting and evaluating the activity of designed molecules.
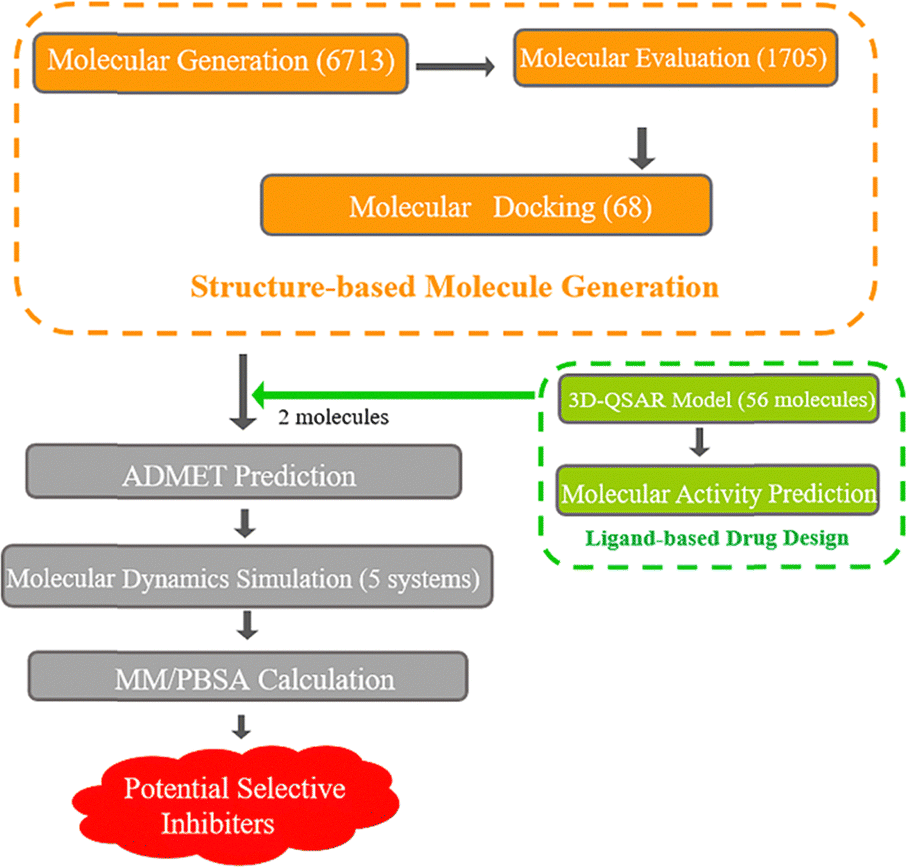
Building upon the aforementioned research background, the objective of this study is to design selective inhibitors for the TASK-1 channel using a combined SBDD and LBDD approach, and to further elucidate the binding characteristics of molecules to TASK-1 and TASK-3, as well as the rationale for the selective inhibition of bisamide compounds through dynamic simulations. Initially, we utilized MolAICal to automatically generate three-dimensional ligand molecules based on the protein pocket, with bisamide fragments serving as the initial building blocks. Potential molecules were then selected through molecular docking. Subsequently, 3D-QSAR models were constructed using 56 bisamide derivatives as the research basis, and the molecules were predicted. We focused on molecules with high selectivity for TASK-1, performing molecular dynamics simulations in conjunction with free energy calculations. Ultimately, we identified two potential selective TASK-1 inhibitors (Fig. 2). This methodological approach provides new insights into the design of selective inhibitor drugs.
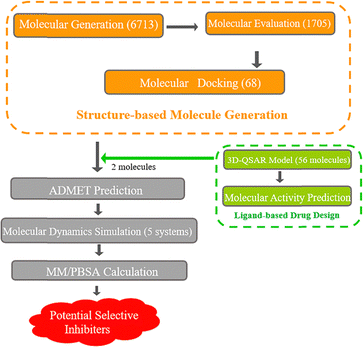 |
| | Fig. 2 Workflow diagram. | |
Methods
Data collection
A dataset comprising 56 reported bisamide derivatives was utilized, detailing their structures and inhibitory properties against TASK-1 and TASK-3 channel proteins in Table S1 (ESI†)25 Given that the IC50 values (half-maximal inhibitory concentration values) of the compounds are discrete and unsuitable for model development, these values were converted to pIC50 values.
Molecule generation
The MolAICal software package generates three-dimensional ligand molecules based on receptor proteins.32 The generation model, AIGenMols, within the software, integrates genetic algorithms and classical methods to address the challenge of three-dimensional ligand design based on protein pockets. Using bisamide fragments as the initial growth units and a box size of 15 Å × 15 Å × 15 Å, the subsequent fragment is generated based on the preceding molecular fragment through a perturbation search employing the Fibonacci algorithm. The Fibonacci algorithm utilizes the golden angle 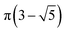 to distribute points on a sphere. The points on the spherical surface are calculated according to the following equations:
to distribute points on a sphere. The points on the spherical surface are calculated according to the following equations:| | 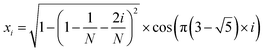 | (1) |
| | 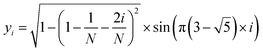 | (2) |
| | 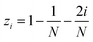 | (3) |
The coordinates (xi, yi, and zi) represent the positions of the spherical points, and (N) denotes the total number of generated points. The subsequent fragment searches for the optimal anchor pose by perturbing the generated points around the growth atom. Once the ligand has expanded sufficiently for testing, the genetic algorithm further optimizes the growing ligand within the receptor pocket. The evolving ligand is treated as a combination of rigid fragments and rotatable bonds. The selection of the genetic algorithm (GA) is determined based on the binding scores between the ligands and receptors. MolAICal employs the Vinardo score to assess the affinity between ligands and receptors. The Vinardo score is trained using experimental affinity data and high-resolution complex crystal structures from the PDBbind v2018 database.33 Consequently, this score can identify elite ligands for the subsequent genetic algorithm (GA) evolutionary process. To obtain a more optimal selection of elite ligands, we established cutoff values for molecular weight, hydrogen bond acceptors, and rotatable bonds at 1000, 12, and 14, respectively, to generate structurally reasonable molecules. The well-scored generated molecules were selected and filtered according to MolAICal's application of Lipinski's rule of five (Ro5) and synthetic accessibility (SA) custom rules to eliminate molecules that did not conform to these criteria.
Molecular docking
We utilized AutoDock Vina for batch docking of the generated molecules.34,35 Referring to the crystal structures of TASK-1 and TASK-3 (PDB IDs: 6RV3 and 8K1Q),17,36 the docking box was positioned in the central cavity of the co-crystal ligand, with box dimensions set to 25 Å × 25 Å × 25 Å and a grid spacing of 0.375 Å to effectively encapsulate the central cavity. Prior to docking, any missing residues and atoms in the protein were completed using Jackal software. AutoDock tools were employed to prepare the ligand, maintaining the number of rotatable bonds to fewer than six. The Lamarckian genetic algorithm (LGA) was then utilized to identify the optimal binding state between the ligand and the receptor, generating ten docking conformations for each docked molecule, of which the lowest energy conformation was selected.
Establishment of 3D-QSAR model
We utilized the TASK-1/TASK-3 inhibitor dataset, characterized by the bisamide fragment as a common core. The modeling tool Cloud 3D-QSAR (https://chemyang.ccnu.edu.cn/ccb/server/cloud3dQSAR/) was employed for the 3D-QSAR modeling of TASK-1 and TASK-3 separately.37 The biological activity IC50 values were converted to pIC50 values, which served as the dependent variable for the 3D-QSAR model. We developed 3D-QSAR models centered on bisamide scaffolds utilizing the comparative molecular field analysis (CoMFA) method.
ADMET prediction
Absorption, distribution, metabolism, excretion, and toxicity (ADMET) are critical components of contemporary drug design and screening. ADMET prediction serves as a fundamental criterion for evaluating drug-like properties. We utilized two online prediction tools, SwissADME (https://www.swissadme.ch/)38 and ADMETlab 2.0 (https://admetmesh.scbdd.com/),39 to assess the ADMET characteristics of the generated molecules.
Molecular dynamics simulation
GROMACS 2020 software was utilized for molecular dynamics simulations of the protein–ligand complex systems. Prior to simulation, it was necessary to prepare the structural parameter file for the small molecule. First, Gaussian was employed to generate restrained electrostatic potential (RESP) charges. Subsequently, the ACPYPE program was used to create the Amber force field parameter file for the molecule, replacing the original charges with those calculated by Gaussian in the generated file.40–42 Additionally, we utilized CHARMM-GUI (https://charmm-gui.org/) to generate the required POPC membrane.43
After preparation, five complex systems were constructed, with the generated molecules individually bound to TASK-1 and TASK-3 channel proteins, as well as to the co-crystal complexes of TASK-1, for dynamic simulation. The AMBER14 SB force field and TIP3P water model44 were used, with Na+ and Cl− ions added to neutralize the system and simulate the human physiological environment. Energy minimization was achieved using the steepest descent method to obtain the lowest energy conformation.45 A 200 picosecond isothermal–isochoric (NVT) simulation was conducted at 310 K, employing the V-rescale coupling algorithm, followed by a 200 picosecond isothermal–isobaric (NPT) simulation at the same temperature and pressure, using the Parrinello–Rahman coupling algorithm. A 200 nanosecond molecular dynamics simulation was then performed. The cutoff value was set at 1.2 Å, and the time step was 2 fs. During the simulation, the LINCS algorithm was utilized to constrain all bonds, and the particle mesh Ewald (PME) method was applied to calculate long-range electrostatic forces.46
Binding free energy calculation
The molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) approach was employed for the calculation of binding free energy (Gbind).47 This approach encompasses three energy terms: vacuum potential energy (EMM), polar solvation energy (GGB), and nonpolar solvation energy (GSA). EMM includes bonded interactions (bonds, angles, and dihedrals) and non-bonded interactions (electrostatic interactions Eele and van der Waals interactions EvdW), which are calculated using molecular mechanics parameters. Similarly, GGB and GSA are computed using the Poisson–Boltzmann equation and solvent accessible surface area (SASA), respectively.48,49 The equation for complex systems in a solvent is as follows:| | | Gbind = ΔEMM + ΔGGB + ΔGSA + −TΔS | (4) |
| | | = ΔEvdW + ΔEele + ΔGGB + ΔGSA − TΔS | (5) |
TΔS represents the entropy contribution at temperature (T).
Results and discussion
Structure-based molecule generation
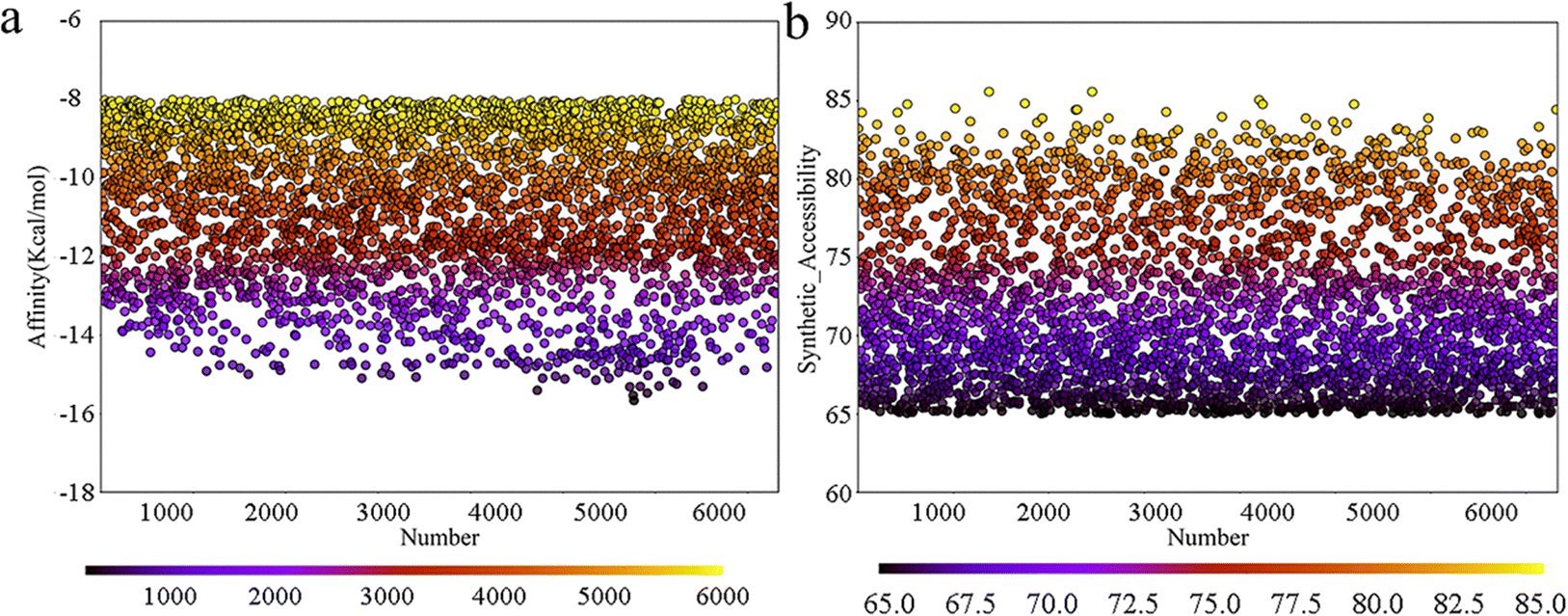
Deep learning models are typically employed for generating one-dimensional sequences or two-dimensional structures; however, rational drug design necessitates the generation of three-dimensional molecular structures. MolAICal software integrates genetic algorithms within deep learning with classical algorithms in new drug design (by obtaining output results from input data and designed algorithm rules) to tackle the challenge of 3D ligand design based on protein pockets. To achieve molecules with diverse three-dimensional chemical structures, we employed MolAICal software to facilitate ligand growth within the receptor pocket. Based on the biological activity of the inhibitors we collected, the bisamide fragment demonstrated exceptional performance in the selectivity of inhibitory abilities for TASK-1 and TASK-3 channels. Therefore, the bisamide fragment was selected as the starting fragment for molecular growth. Subsequent to the initial molecular fragment, the next stage of molecular fragment growth was executed through perturbation search utilizing the Fibonacci algorithm, resulting in the generation of a total of 6713 molecules. To ascertain the reliability of the generated molecules, the Vinardo score was employed to assess the affinity of the molecules within the protein pocket, and the synthetic ability (SA) of the molecules was further evaluated. The prediction of SA can be utilized to forecast the complexity of compound synthesis. As depicted in Fig. 3, the affinities of the generated molecules were all below −8 kcal mol−1, suggesting that the molecules could effectively bind to the protein pocket. Concurrently, their SA scores were all above 65, with values approaching 100 indicating simpler synthesis; thus, all the molecules exhibited ease of synthesizability. Given that the molecules must adhere to Lipinski's rule of five (RO5) custom rules and synthetic accessibility (SA) custom rules, a total of 1705 molecules were retained after screening. In summary, the molecules generated by MolAICal can serve as an exclusive database for the screening of potential selective inhibitors in the subsequent step.
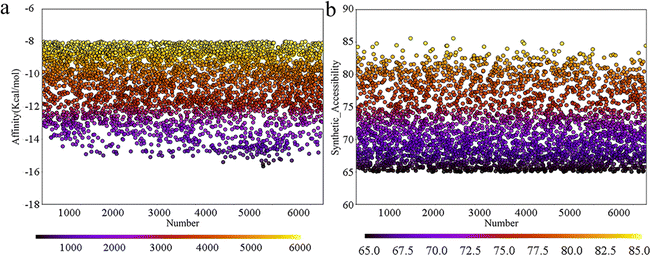 |
| | Fig. 3 (a) Affinity energy scatter plot; (b) SA score scatter plot. | |
Molecular docking
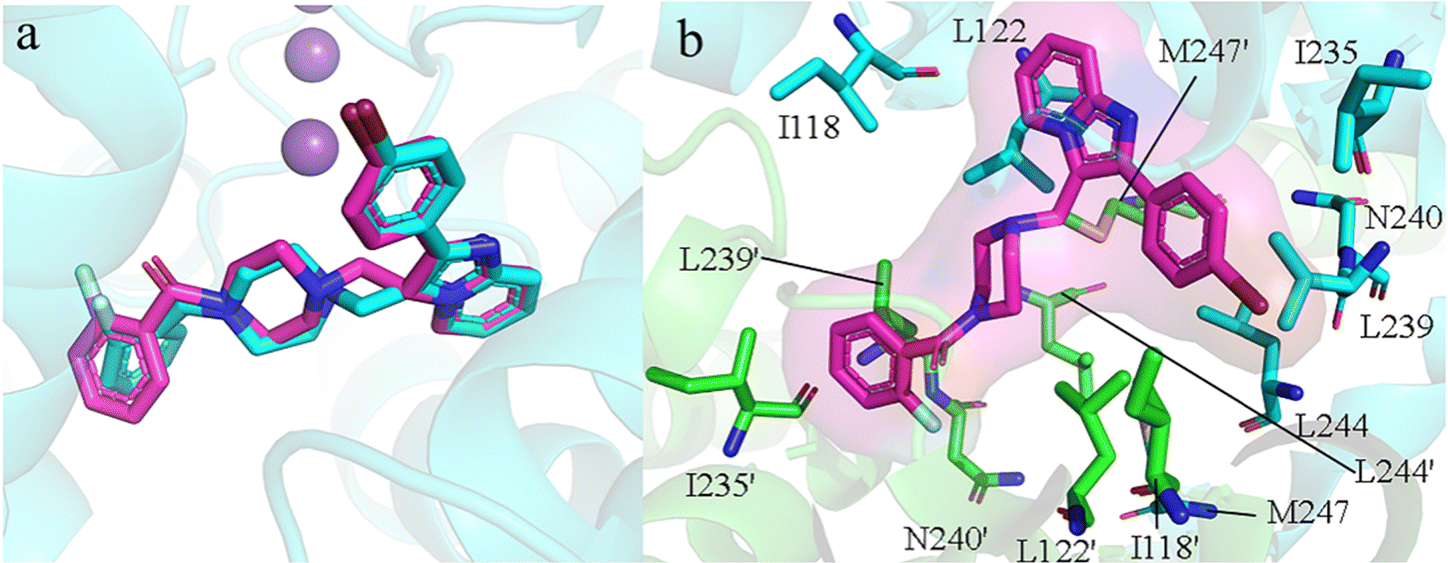
To identify elite ligands, we employed Autodock Vina for batch molecular docking of the generated molecules. The docking protocol included re-docking the co-crystal ligand (PDB ID: 6RV3) into the central pocket of TASK-1, which served to evaluate the reliability of our molecular docking. The re-docked ligand was largely consistent with the original co-crystal ligand in terms of spatial orientation and conformation (Fig. 4a), with a root mean square deviation (RMSD) value of 1.10 Å and a docking affinity of −10.0 kcal mol−1, suggesting that our docking results were reliable. We docked the 1705 screened generated molecules against both TASK-1 and TASK-3 channel proteins individually. We then selected molecules that exhibited good binding energy following docking with both channels. The active site of TASK-1 within the protein pocket is composed of Thr93, Ile118, Ile118′, Leu122, Leu122′, Ile235, Ile235′, Leu239, Leu239′, Asn240, Asn240′, Leu244, Leu244′, Met247, and Met247′ (Fig. 4b). Based on mutation experiments, Thr93, Leu122, Thr199, G236, Leu239, and Asn240 are identified as key active site residues. Initially, we excluded molecules whose binding sites were not within the active pocket. Given that the re-docking affinity of the co-crystal complex was −10.0 kcal mol−1, we selected elite ligands with docking affinities below −10 kcal mol−1 and RMSD values below 1.10 Å. Following screening, we identified 68 candidate molecules for further analysis.
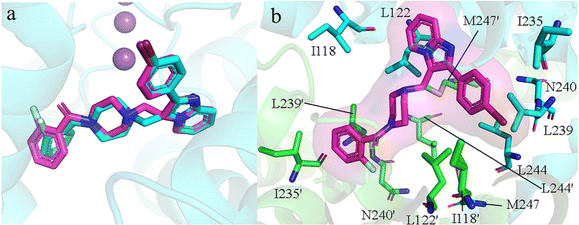 |
| | Fig. 4 (a) Co-crystal ligand re-docking comparison: cyan represents the original ligand, and magenta represents the re-docked ligand. (b) Green and cyan rod structures represent amino acids on the two protein subunits, and the magenta rod structure represents the co-crystal ligand molecule. | |
Ligand-based 3D-QSAR modeling
Based on the collection of 56 bisamide derivatives with biological activity against TASK-1 and TASK-3 channel proteins, we developed two 3D-QSAR models centered on bisamide scaffolds. These models were established based on the different pIC50 values of the molecules against TASK-1 and TASK-3, with the aim of predicting the activity values of the generated molecules. Molecular superimposition is an essential prerequisite for achieving a robust 3D-QSAR model. Before proceeding with the modeling, we first superimposed the active molecules that we had collected. As depicted in Fig. S1 (ESI†), the active molecules of both models exhibited good overlap in the bisamide fragment, which was consistent with our expectations and indicated that the modeling standards were met. The specific results of the two models are presented in Table 1. Generally, models with Q2 > 0.5 are considered to have good internal validation capabilities, and models with R2pred > 0.6 are deemed to have good external validation capabilities. As can be seen from Table 1, both models that we constructed have Q2 values exceeding 0.5 and R2pred values above 0.6, suggesting that our models possess good internal validation and external predictive capabilities and are suitable for predicting molecular activity values.
Table 1 Model evaluation indicators
| Model |
Q
2
|
R
2
|
R
2
pred
|
| TASK-1 |
0.61 |
0.92 |
0.84 |
| TASK-3 |
0.60 |
0.95 |
0.71 |
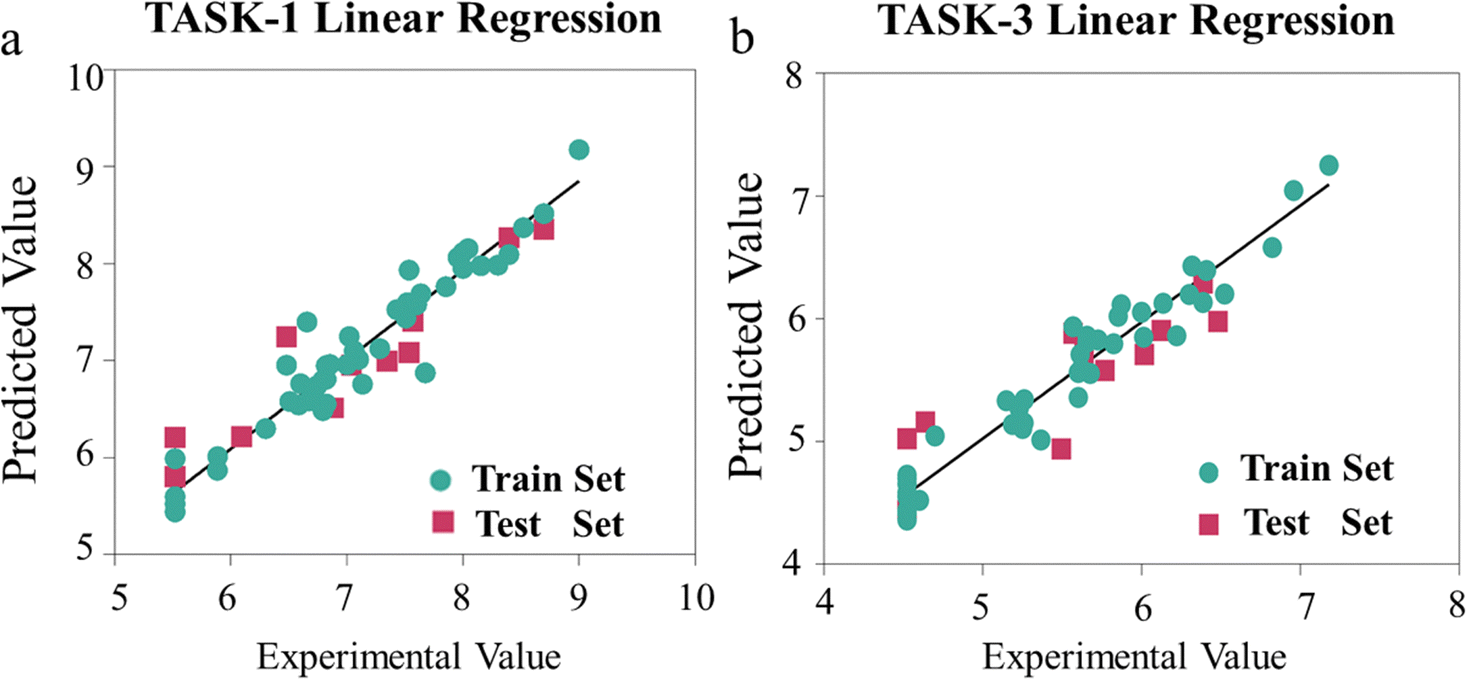
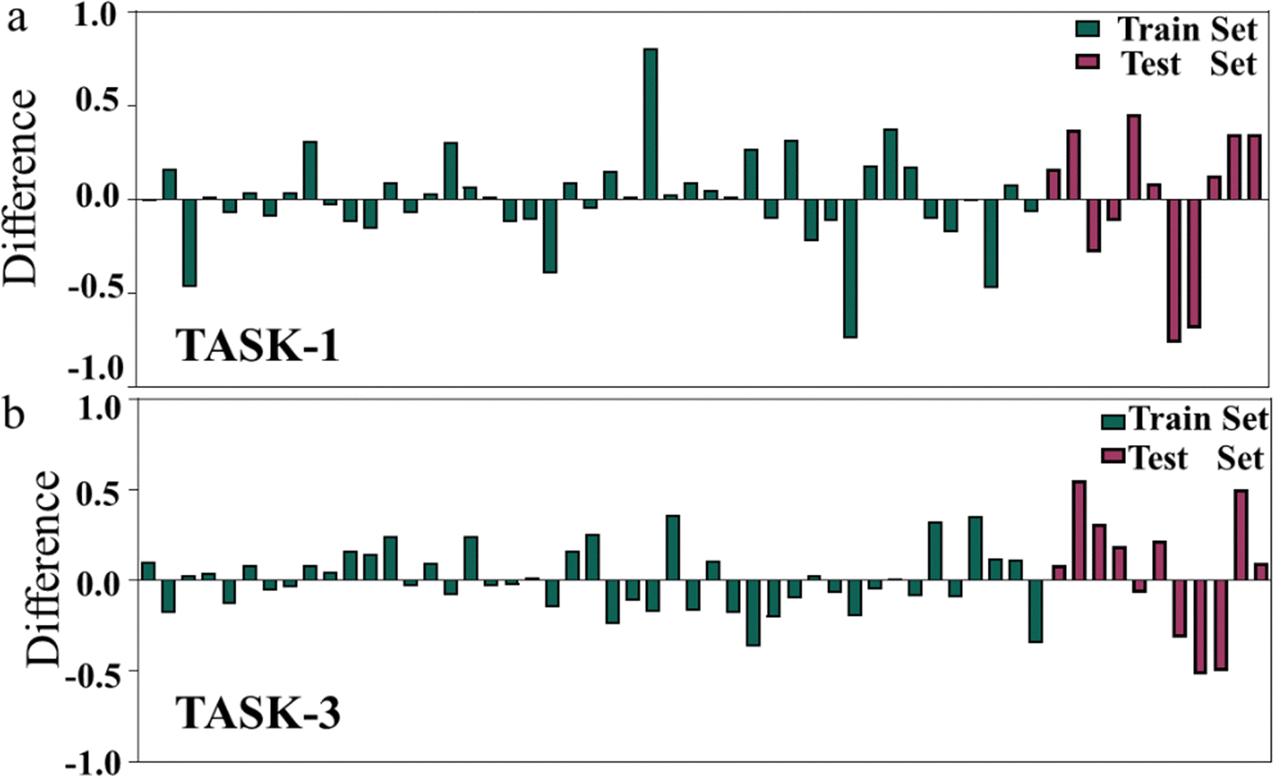
To visually depict the predictive capabilities of the two models, we plotted linear regression model scatter plots for the training and test sets. From Fig. 5, we can intuitively observe the relationship between the experimental values and the model-predicted values in both sets, with the x-axis representing the experimental values (pIC50 values) and the y-axis representing the predicted values (pIC50 values). It is evident that the points in the training and test sets of the two models are predominantly distributed close to the fitted line, suggesting a good agreement between the predicted pIC50 values and the experimental values (Fig. 5). The linear equations of the fitted lines are (y = 0.9208x + 0.564) and (y = 0.9475x + 0.2916), respectively, which also reflect the accuracy of the predictive capabilities of the two models. To gain a deeper insight into the predictive capabilities of the models, we represented the test errors of the training and test sets in the form of histograms. As depicted in Fig. 6, we observed that the maximum error in the test set relative to the experimental values does not exceed 0.8, which further confirms the reliability and accuracy of our model predictions. In summary, our 3D-QSAR models are suitable for predicting molecular activity values. The predicted results are listed in Table S2 (ESI†).
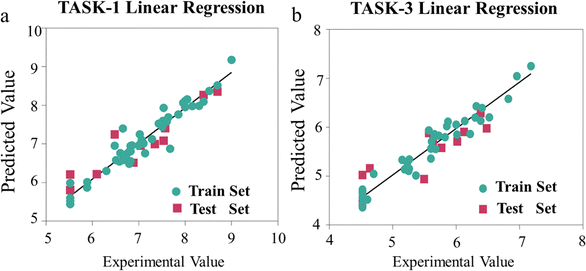 |
| | Fig. 5 (a) Linear regression scatter plot of TASK-1 model; (b) linear regression scatter plot of TASK-3 model. | |
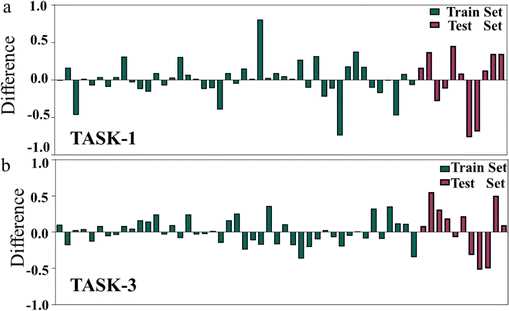 |
| | Fig. 6 (a) Histogram of errors between experimental and predicted values in TASK-1 model; (b) histogram of errors between experimental and predicted values in TASK-3 model. | |
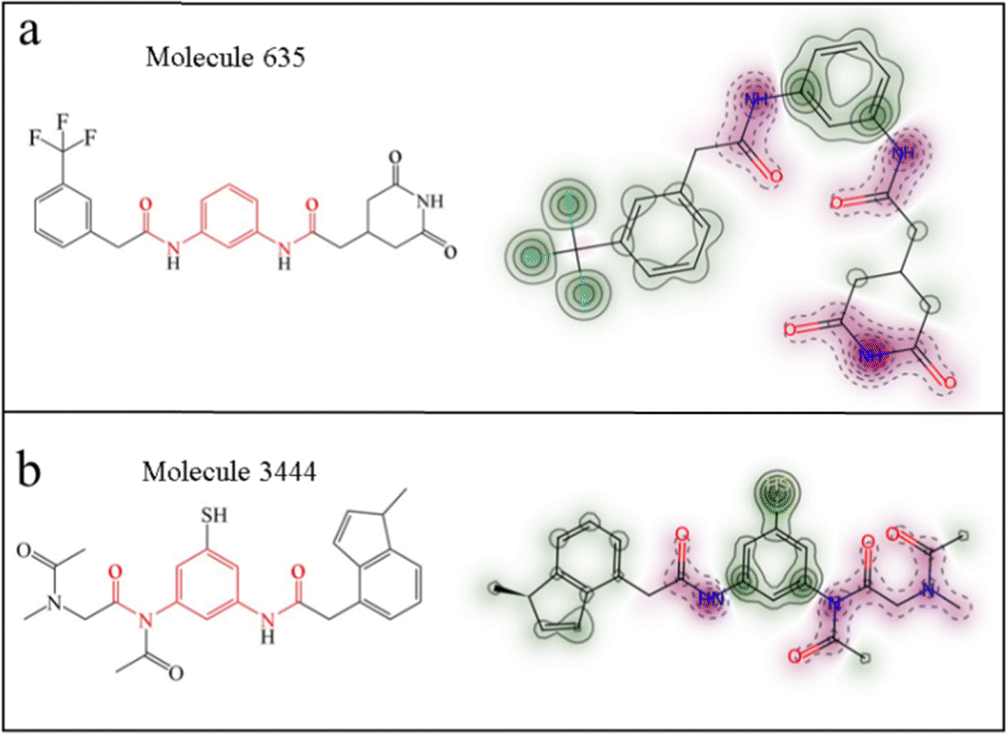
Through the established 3D-QSAR models, we proposed an efficient compound screening strategy. The 68 candidate molecules were subjected to screening through the 3D-QSAR model for prediction. To maximize the selection of selective molecules, we chose two molecules with large predicted pIC50 value differences: molecule 635 and molecule 3444 (the generated molecules). The predicted pIC50 values are listed in Table 2. From the lipophilicity contour maps of these two molecules, it can be observed that the heads of both molecule 635 and molecule 3444 exhibit strong lipophilicity (hydrophobicity) (Fig. 7a and b). Therefore, we believe that both molecules are likely to enter the central cavity of the protein and interact with the receptor protein.
Table 2 Predicted molecular pIC50 values
| Generated molecules |
pIC50 (TASK-1) |
pIC50 (TASK-3) |
| 635 |
9.199 |
5.144 |
| 3444 |
8.718 |
4.690 |
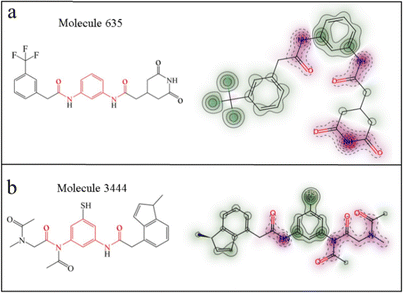 |
| | Fig. 7 (a) and (b) Depict the two-dimensional structures (red highlighted bisamide fragment structure) and lipophilicity contour maps of molecule 635 and molecule 3444, respectively. The deeper the green color, the higher the lipophilicity. | |
ADMET prediction
We employed ADMET property prediction methods to evaluate molecules 635 and 3444, and the results are presented in Table 3. All molecules meet the Lipinski drug-likeness standard and exhibit good synthetic feasibility (SA values less than 3; the closer the value is to 1, the easier the synthesis), suggesting that our selected molecules are readily synthesizable. From the perspective of the physicochemical properties of the selected molecules: lipophilicity (Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) values are within the range of 0.7–6.0, indicating that all the molecules are hydrophobic; solubility (LogS) values are less than 6 (insoluble < −10 < less soluble < −6 < soluble), suggesting that the selected molecules are soluble. Regarding human intestinal absorption (HIA), all molecules have a high probability of being absorbed by the intestine, and they are less likely to permeate the skin (the more negative LogKp, the fewer skin-permeating molecules). For the toxicity parameter AMES, the highest probability of molecules being positive is 18.30%, indicating that we can consider these molecules to be non-AMES toxic. The ADMET prediction results suggest that our selected molecules possess favorable ADMET properties.
P) values are within the range of 0.7–6.0, indicating that all the molecules are hydrophobic; solubility (LogS) values are less than 6 (insoluble < −10 < less soluble < −6 < soluble), suggesting that the selected molecules are soluble. Regarding human intestinal absorption (HIA), all molecules have a high probability of being absorbed by the intestine, and they are less likely to permeate the skin (the more negative LogKp, the fewer skin-permeating molecules). For the toxicity parameter AMES, the highest probability of molecules being positive is 18.30%, indicating that we can consider these molecules to be non-AMES toxic. The ADMET prediction results suggest that our selected molecules possess favorable ADMET properties.
Table 3 ADMET property prediction of molecules
| Generated-compound |
Physicochemical properties |
Pharmacokinetics |
Toxicity |
Druglikeness |
SA |
| LogP |
LogS |
HIA (%) |
LogKp (cm s−1) |
AMES (%) |
Lipinski rule |
| 635 |
−4.51 |
−4.51 |
79.90 |
−7.84 |
81.70 |
Accepted |
2.37 |
| 3344 |
2.71 |
−3.87 |
79.30 |
−7.27 |
91.20 |
Accepted |
3.75 |
Stability of the complex systems
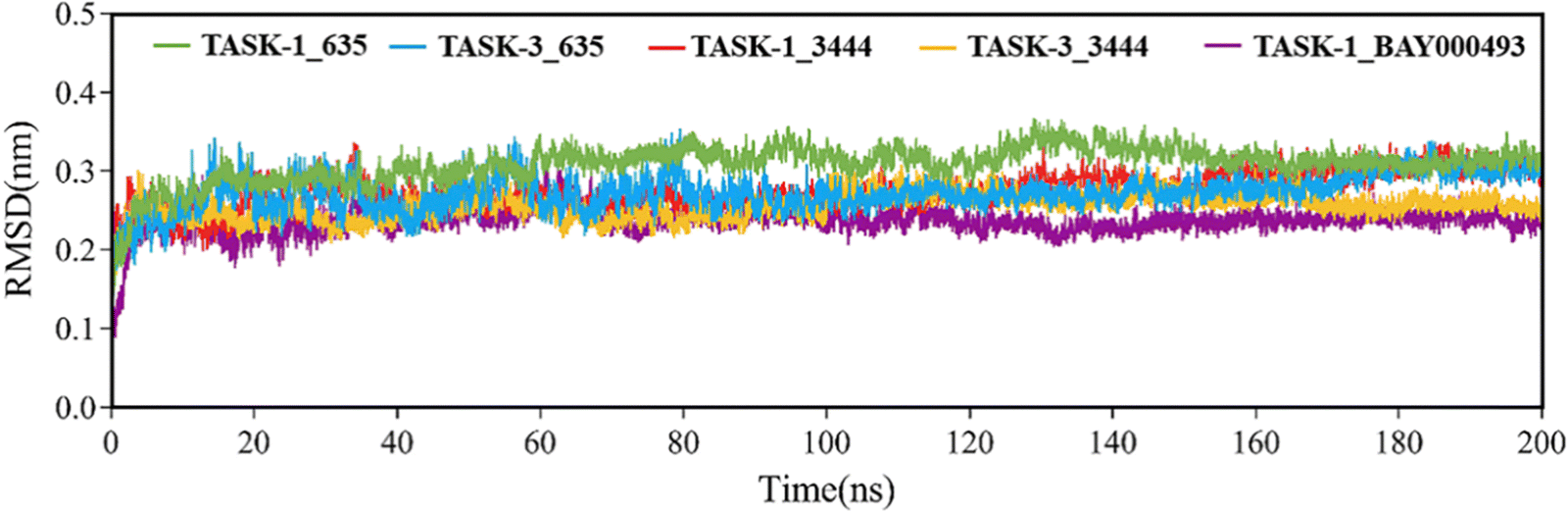
Molecular dynamics simulations were performed on five complex systems. The root mean square deviation (RMSD) was employed to analyze the 200-nanosecond simulation trajectory, thereby assessing the stability of protein–ligand complexes. The results are presented in Fig. 8. RMSD values for all systems were approximately 0.3 nanometers, suggesting that the systems had achieved equilibrium and were stably associated. Fig. 8 illustrates that, with the exception of the TASK-3_635 complex, the RMSD for the remaining complexes stabilized at 20 nanoseconds. This suggests that these three systems reached a stable state following 20 nanoseconds. Conversely, the TASK-3_635 complex displayed minor fluctuations between 0 and 80 nanoseconds before stabilizing at 80 nanoseconds. This indicates that the system became stable after 80 nanoseconds, although conformational alterations might have occurred throughout the simulation.
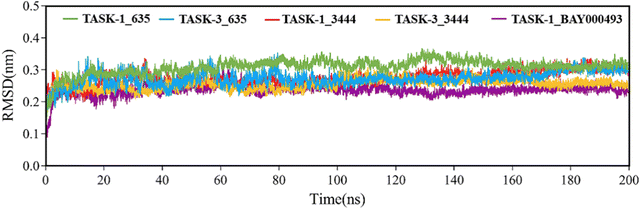 |
| | Fig. 8 RMSD values of the five complex systems. | |
Binding free energy analysis
The calculation of the binding free energy for ligand–protein complexes is pivotal for elucidating ligand binding affinities. To assess the binding capabilities of the two molecules with TASK-1 and TASK-3 channel proteins, we employed the Molecular Mechanics–Poisson Boltzmann Surface Area (MM/PBSA) method to determine binding free energies, as detailed in Table 4. MM/PBSA is a robust method for estimating the binding free energy between compounds and proteins, with lower free energies indicative of stronger binding. Table 4 reveals that the differences in binding free energies between the TASK-1 and TASK-3 complex systems with the two molecules are all approximately 20 kcal mol−1, with the complexes containing molecule 635 exhibiting particularly significant differences. The substantial energy difference indicates that the selected molecules are viable candidates for selective inhibitors of TASK-1, aligning with the predicted trend in pIC50 values from the 3D-QSAR model. van der Waals interactions are the predominant contributors to the energy landscape. To validate the accuracy of the computed free energies, we conducted molecular dynamics simulations and MM/PBSA calculations on the co-crystallized ligand–protein complex. The binding free energy of −38.73 kcal mol−1 for the co-crystallized ligand, a selective inhibitor for the TASK-1 protein, is marginally higher than that for TASK-1_635 and TASK-1_3444 but lower than that for TASK-3_635 and TASK-3_3444. This suggests that the calculated free energies are reliable, and molecules 635 and 3444 are suitable candidates for selective inhibitors of TASK-1.
Table 4 Binding free energies of the four complex systems
| Binding free energy (kcal mol−1) |
TASK-1 |
TASK-3 |
| BAY1000493 |
635 |
3444 |
635 |
3444 |
| ΔEele |
−12.27 |
−10.29 |
−8.19 |
−7.69 |
−3.38 |
| ΔEvdW |
−50.12 |
−54.18 |
−60.85 |
−46.73 |
−59.51 |
| ΔGPB |
26.52 |
21.04 |
30.17 |
33.16 |
36.86 |
| ΔGNP |
−8.00 |
−6.52 |
−7.23 |
−6.65 |
−7.15 |
| −TΔS |
5.11 |
4.34 |
3.82 |
5.01 |
6.51 |
| ΔGbind |
−38.73 |
−45.60 |
−42.82 |
−22.89 |
−26.67 |
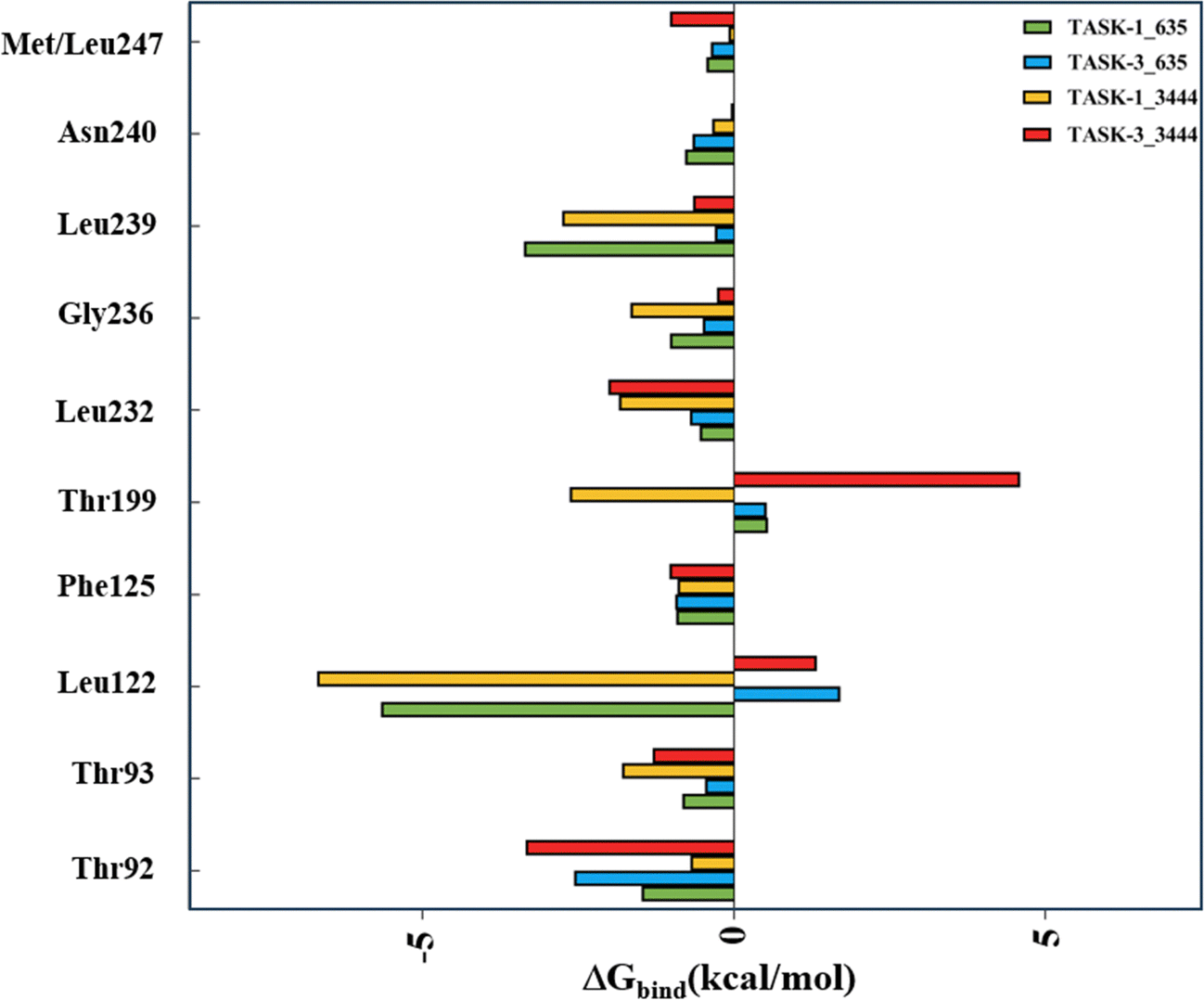
Moreover, we utilized energy decomposition to delineate the contributions of individual residues throughout the complex binding process. Fig. 9 illustrates that residues Leu122 and Thr199 display distinct trends and substantial disparities. Leu122 is a pivotal amino acid for the binding of molecules to TASK-1/3 proteins, with its energy contribution values being significantly more favorable for TASK-1 than for TASK-3. Consequently, we propose that residue Leu122 is pivotal in mediating the selectivity between TASK-1 and TASK-3. In contrast, residue Thr199 exhibits favorable contributions only within the TASK-1_3444 complex system, which we conjecture is associated with the specific binding mode of molecule 3444 to TASK-3.
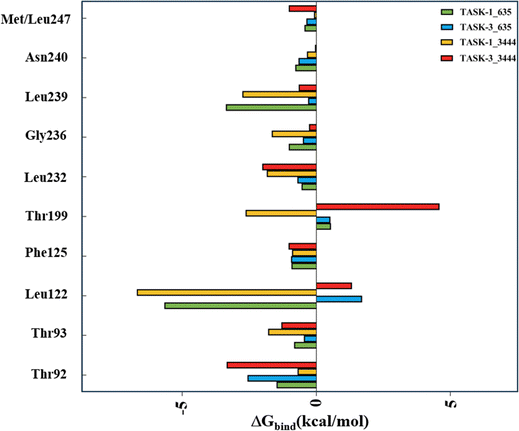 |
| | Fig. 9 Energy contributions of key residues in the four systems. | |
Interactions of molecules with TASK-1 and TASK-3
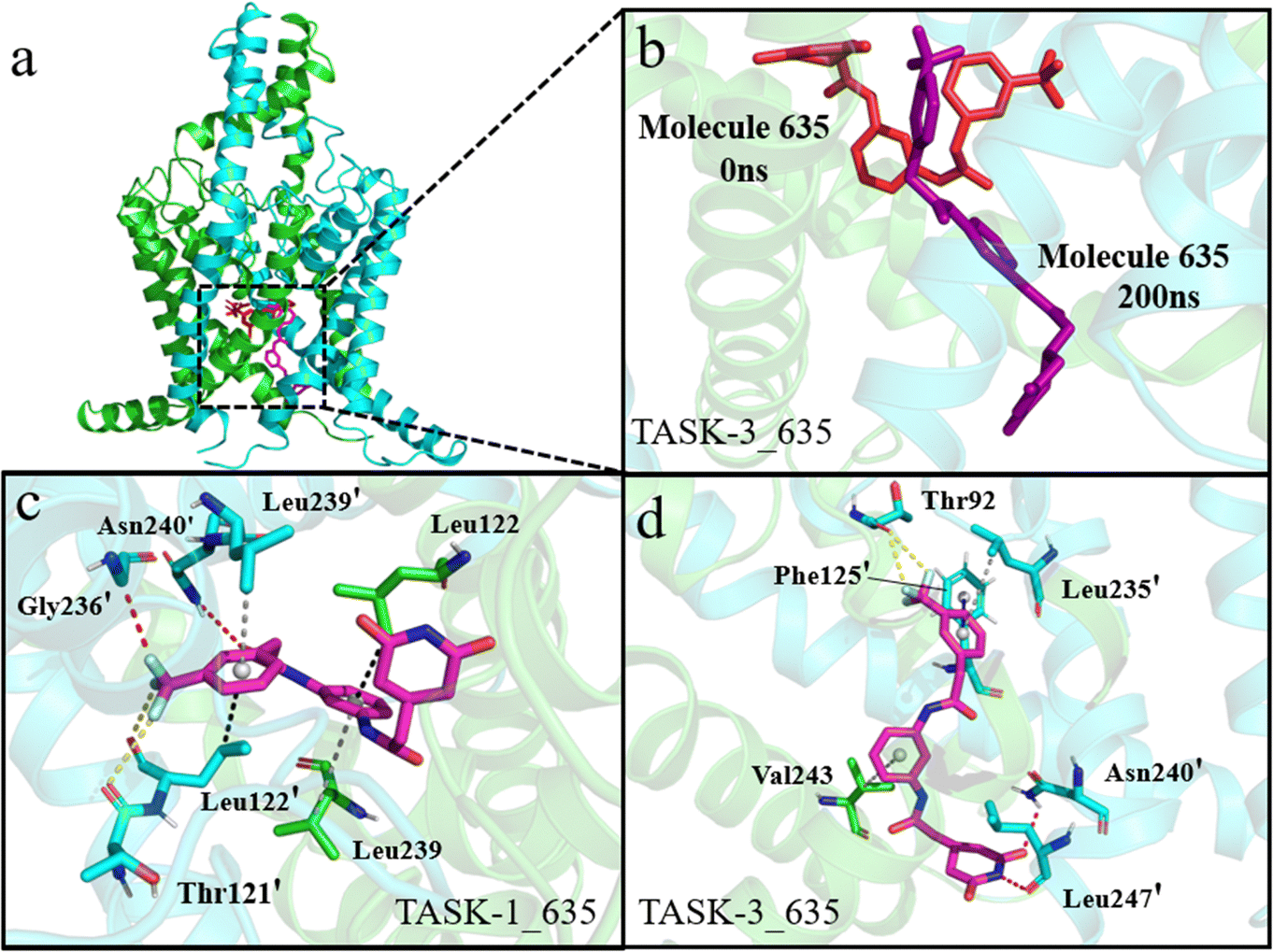
To gain a comprehensive understanding of the protein–ligand binding, we identified the most stable conformations with the lowest relative energy from the free energy landscape (FEL) to evaluate protein–ligand interactions. Initially, we examined the interactions within the TASK-1_635 and TASK-3_635 complex systems. In TASK-1, residues that interact with molecule 635 are Thr121′, Leu122, Leu122′, Gly236′, Leu239, Asn239′, and Asn240; in TASK-3, these residues are Thr92′, Phe125′, Leu235′, Asn240′, Val243, and Leu247′ (Fig. 10a, c and d). Mutations in Leu122, Leu122′, Gly236, and Gly236′ markedly diminish the inhibitory activity of inhibitors against TASK-1 and TASK-3 proteins, with Leu122 being particularly critical for the binding of pore blockers. A mutation at Leu122 results in an EC50 value for the inhibitor that is more than 100-fold lower than that of the wild type.50,51 As depicted in Fig. 11, the bisamide moiety of molecule 635 engages in a π–σ interaction with the pivotal residue Leu122 in the TASK-1 protein, whereas it does not interact with the corresponding Leu122 residue in the TASK-3 protein. We hypothesize that this discrepancy may account for the selectivity of bisamide scaffolds towards TASK-1 and TASK-3 proteins. Notably, we observed that molecule 635 experiences a conformational shift from a horizontal to a vertical arrangement during the simulation with TASK-3, as illustrated in Fig. 10b. This observation is consistent with our RMSD analysis, which indicates conformational alterations within the complex system. We propose that this conformational change is likely attributed to the fluorine atom in molecule 635 forming a halogen bond with residue Thr92. Experimental evidence indicates that the mutation of Thr92 does not compromise the inhibitory efficacy of inhibitors against TASK-1 and TASK-3. Consequently, we attribute the significantly lower free energy of the TASK-1_635 complex compared to the TASK-3_635 complex system to the role of residue Leu122.
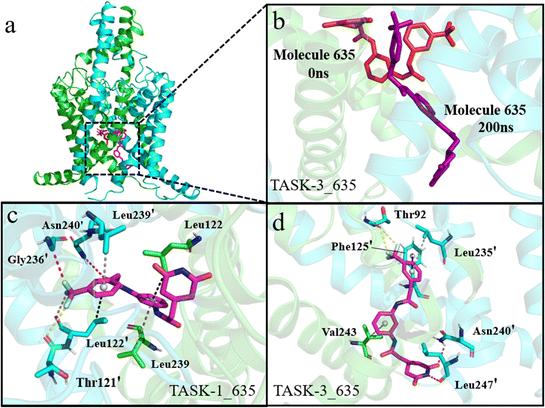 |
| | Fig. 10 Interactions of small molecules with TASK-1 and TASK-3. (a) and (b) Green and blue cartoon representations depict the two subunits of the TASK-1 protein. Red and purple stick models illustrate molecule 635 at 0 ns and 200 ns in the simulation, respectively. (c) and (d) Green and blue stick models show residues involved in interactions with the molecule, magenta stick models represent the generated molecule, red dashed lines denote hydrogen bonds, yellow dashed lines indicate halogen bonds, blue dashed lines signify π–π stacking interactions, gray dashed lines denote π–alkyl interactions, and black dashed lines represent π–sigma interactions. | |
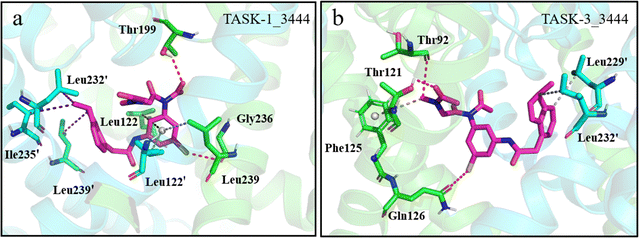 |
| | Fig. 11 (a) and (b) Green and blue stick models depict residues involved in interactions with the molecule, magenta stick models illustrate the generated molecule, red dashed lines denote hydrogen bonds, purple dashed lines indicate hydrophobic interactions, black dashed lines represent π–σ interactions, and gray dashed lines denote π–alkyl interactions. | |
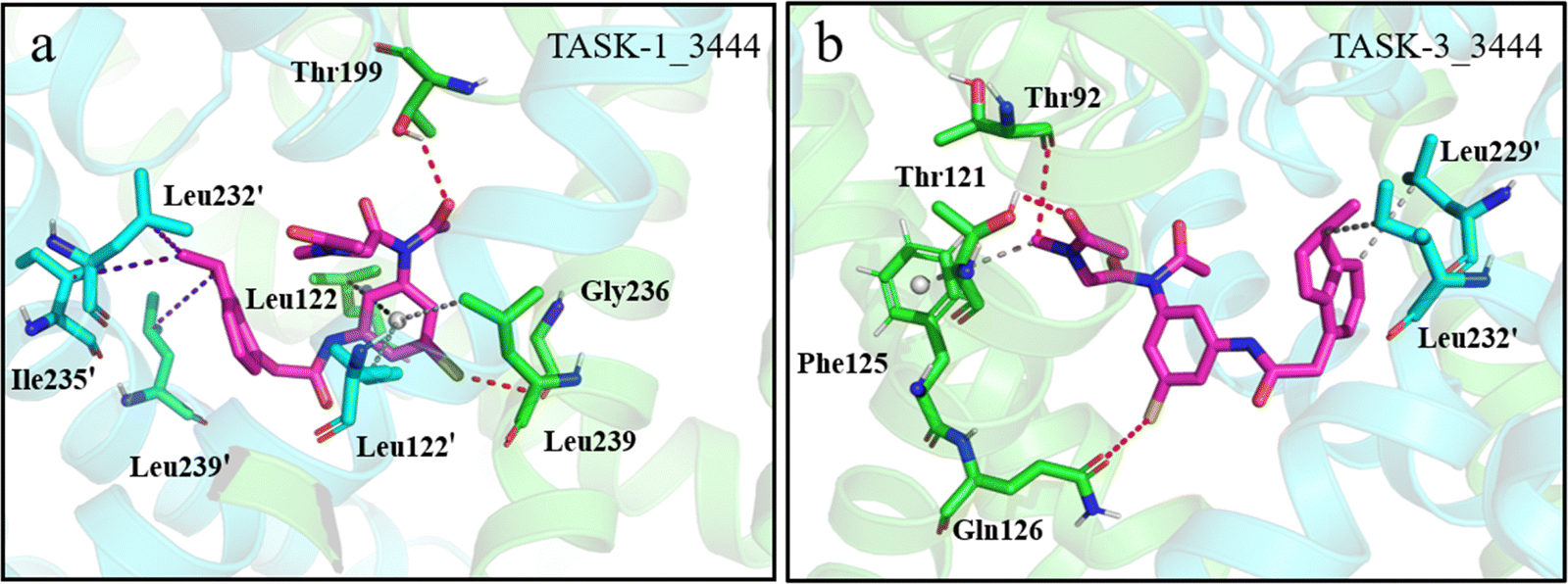
Next, we investigated the interactions within the TASK-1_3444 and TASK-3_3444 complex systems. In TASK-1, residues interacting with molecule 3444 are Leu122, Leu122′, Thr199, Leu232′, Ile235′, Gly236, Leu239, and Leu239′; in TASK-3, these residues are Thr92, Thr121, Phe125, Gln126, Leu229′, and Leu232′ (Fig. 11a and b). Similar to the TASK-1_635 complex system, the bisamide fragment of molecule 3444 engages in π–σ interactions with residues Leu122 and Leu122′ in the TASK-1 protein within the TASK-1_3444 complex. We posit that the formation of these interactions could be the rationale behind the distinct energy contribution trends observed for residue Leu122 in the energy contributions. In contrast, the phenyl ring of the bisamide fragment participates in π–alkyl interactions with residue Gly236. The oxygen atom on the bisamide fragment of molecule 3444 forms a hydrogen bond with residue Thr199. The formation of these interactions may account for the distinct energy contribution trends observed for residue Thr199 in the energy contributions. Simultaneously, the large-volume indole group on the side chain of molecule 3444 engages in hydrophobic interactions with residue Leu239′ in TASK-1, while the negatively charged thiol group forms a hydrogen bond with residue Leu239. All these mentioned residues are considered key amino acids. However, these specific interactions were not observed in the TASK-3_3444 complex system. Therefore, we posit that the interactions formed between molecule 3444 and key amino acids in TASK-1 are the rationale behind the significantly lower free energy of the TASK-1_3444 complex compared to the TASK-3_3444 complex system. To confirm the persistence of the aforementioned interactions, we conducted hydrogen bond occupancy statistics across the entire simulation trajectory. As indicated in Table 5, all hydrogen bonds identified in the interactions occupy more than 30% of the simulation trajectory. This suggests that the observed interactions between molecules and proteins are stable and significant. In summary, the interactions within the complex systems validate the reliability of our calculated binding free energies and demonstrate that residue Leu122 is exclusively present in TASK-1 complex systems, confirming that these two molecules are viable candidates for selective inhibitors of TASK-1.
Table 5 Hydrogen bond occupancy in ligand–protein interactions
|
|
Molecule |
Residues |
Occupancy (%) |
| TASK-1 |
635 |
Gly263 |
80.7 |
| Asn240 |
36.1 |
| 3444 |
Thr199 |
60.3 |
| Gly236 |
80.5 |
| TASK-3 |
635 |
Asn240 |
30.2 |
| Leu247 |
50.8 |
| 3444 |
Thr92 |
50.8 |
| Gln126 |
60.8 |
Conclusions
TASK-1 channel proteins are essential for maintaining the resting membrane potential in cells. Given the high sequence homology between TASK-1 and TASK-3, the development of a selective inhibitor for TASK-1 is of considerable importance. In this study, we integrated structure-based and ligand-based drug design approaches. Leveraging the high selectivity of bisamide derivatives for TASK-1 channel proteins, we initially employed the bisamide fragment as a starting point to generate 6713 molecules using MolAICal software. Following filtering, 1705 molecules were retained. To refine the selection, we conducted batch molecular docking of the remaining molecules against TASK-1 and TASK-3 channel proteins, identifying 68 molecules with binding affinities below −10.0 kcal mol−1. Subsequently, we constructed two robust 3D-QSAR models with strong predictive capabilities, utilizing the diverse activity profiles of 56 bisamide derivatives as inhibitors against TASK-1 and TASK-3 proteins. Through the prediction of the 68 shortlisted molecules, we identified two compounds exhibiting the greatest activity value disparity between the models. These compounds also possessed favorable ADMET (absorption, distribution, metabolism, excretion, and toxicity) profiles. Subsequently, molecular dynamics simulations and MM/PBSA calculations provided further evidence supporting the hypothesis that these two molecules could exhibit selectivity towards TASK-1. We also identified that the interaction with residue Leu122 in TASK-1 is pivotal for the selectivity of the bisamide fragment. In summary, we utilized a combination of structure-based drug design (SBDD) and ligand-based drug design (LBDD) methods to efficiently screen candidate molecules as selective inhibitors of TASK-1 channel proteins. Nonetheless, it is essential to recognize the limitations of this study. As the work focused on designing potential selective inhibitors for TASK-1, the sample size of TASK-1 inhibitors was limited, which may have influenced the accuracy of the findings. Furthermore, the accuracy of the free energy calculation method was subject to limitations, thereby affecting the precision of our results. Consequently, a future study with a larger sample size is required to construct robust models. In our subsequent work, we aim to enhance the accuracy of our results by employing more precise energy calculation methods for comparative analyses. We anticipate that our research will offer a methodology for the design and investigation of novel selective inhibitors for TASK-1.
Author contributions
Conceptualization: Lu Liu and Xi Zhao; methodology: Lu Liu, and Jixiang Liu; software: Lu Liu, Liang Chen, and Jixiang Liu; visualization: Risong Na and Xiaoping Liu; formal analysis: Lu Liu, and Lianjuan Yang; investigation: Risong Na and Xiaoping Liu; resources: Xi Zhao; data curation: Lu Liu and Liang Chen; writing – original draft preparation: Lu Liu and Xi Zhao; writing – review and editing: Lu Liu and Xi Zhao; funding acquisition: Xi Zhao, Lianjuan Yang and Risong Na. All authors have read and agreed to the published version of the manuscript.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article or its ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC), grant number 82173429 and 32472613; Excellent Youth Foundation of He’nan Scientific Committee, grant number 232300421008; Henan Natural Fund general project, grant number 232300420009; Shanghai Municipal Health Commission, grant number 202140336.
References
- F. Lesage and M. Lazdunski, Molecular and functional properties of two-pore-domain potassium channels, Am. J. Physiol., 2000, 279(5), F793–F801 CAS.
- N. Comes, A. Serrano-Albarrás, J. Capera, C. Serrano-Novillo, E. Condom and S. R. Y. Cajal,
et al., Involvement of potassium channels in the progression of cancer to a more malignant phenotype, Biochim. Biophys. Acta, Biomembr., 2015, 1848(10), 2477–2492 CrossRef CAS.
- E. M. Talley, Q. B. Lei, J. E. Sirois and D. A. Bayliss, TASK-1, a two-pore domain K channel, is modulated by multiple neurotransmitters in motoneurons, Neuron, 2000, 25(2), 399–410 CrossRef CAS.
- C. Navarro-Retamal and J. Caballero, Energetic differences between non-domain-swapped and domain-swapped chain connectivities in the K2P potassium channel TRAAK, RSC Adv., 2018, 8, 26610–26618 RSC.
- J. F. Cotten, TASK-1 (KCNK3) and TASK-3 (KCNK9) Tandem Pore Potassium Channel Antagonists Stimulate Breathing in Isoflurane-Anesthetized Rats, Anesth. Analg., 2013, 116(4), 810–816 CrossRef CAS.
- S. Trapp, M. I. Aller, W. Wisden and A. V. Gourine, A role for TASK-1 (KCNK3) channels in the chemosensory control of breathing, J. Neurosci., 2008, 28(35), 8844–8850 CrossRef CAS.
- P. Ortega-Sáenz, K. L. Levitsky, M. T. Marcos-Almaraz, V. Bonilla-Henao, A. Pascual and J. López-Barneo, Carotid body chemosensory responses in mice deficient of TASK channels, J. Gen. Physiol., 2010, 135(4), 379–392 CrossRef PubMed.
- Y. Kim, H. Bang and D. Kim, TASK-3, a new member of the tandem pore K(+) channel family, J. Biol. Chem., 2000, 275(13), 9340–9347 CrossRef CAS PubMed.
- D. Kim and C. Gnatenco, TASK-5, a new member of the tandem-pore K channel family, Biochem. Biophys. Res. Commun., 2001, 284(4), 923–930 CrossRef CAS PubMed.
- S. Rajan, E. Wischmeyer, G. X. Liu, R. P. Müller, J. Daut and A. Karschin,
et al., TASK-3, a novel tandem pore domain acid-sensitive K channel -: An extracellular histidine as pH sensor, J. Biol. Chem., 2000, 275(22), 16650–16657 CrossRef CAS.
- S. G. Brohawn, J. del Mármol and R. MacKinnon, Crystal Structure of the Human K2P TRAAK, a Lipid- and Mechano-Sensitive K Ion Channel, Science, 2012, 335(6067), 436–441 CrossRef CAS PubMed.
- F. Duprat, F. Lesage, M. Fink, R. Reyes, C. Heurteaux and M. Lazdunski, TASK, a human background K+ channel to sense external pH variations near physiological pH, EMBO J., 1997, 16(17), 5464–5471 CrossRef CAS PubMed.
- A. Mathie, Neuronal two-pore-domain potassium channels and their regulation by G protein-coupled receptors, J. Physiol., 2007, 578(Pt 2), 377–385 CrossRef CAS.
- S. Rajan, E. Wischmeyer, G. Xin Liu, R. Preisig-Müller, J. Daut and A. Karschin,
et al., TASK-3, a novel tandem pore domain acid-sensitive K+ channel. An extracellular histiding as pH sensor, J. Biol. Chem., 2000, 275(22), 16650–16657 CrossRef CAS PubMed.
- X. G. Wang, N. X. Yuan, X. P. Li and F. F. Chen, TASK-1 induces gefitinib resistance by promoting cancer initiating cell formation and epithelial-mesenchymal transition in lung cancer, Exp. Ther. Med., 2018, 15(1), 365–370 CrossRef CAS.
- L. Ma, D. Roman-Campos, E. D. Austin, M. Eyries, K. S. Sampson and F. Soubrier,
et al., A Novel Channelopathy in Pulmonary Arterial Hypertension, N. Engl. J. Med., 2013, 369(4), 351–361 CrossRef CAS.
- K. E. J. Rödström, A. K. Kiper, W. Zhang, S. Rinné, A. C. W. Pike and M. Goldstein,
et al., A lower X-gate in TASK channels traps inhibitors within the vestibule, Nature, 2020, 582(7812), 443–447 CrossRef PubMed.
- G. Czirják and P. Enyedi, Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits, J. Biol. Chem., 2002, 277(7), 5426–5432 CrossRef.
- M. Bedoya, S. Rinné, A. K. Kiper, N. Decher, W. González and D. Ramírez, TASK Channels Pharmacology: New Challenges in Drug Design, J. Med. Chem., 2019, 62(22), 10044–10058 CrossRef CAS PubMed.
- A. K. Streit, M. F. Netter, F. Kempf, M. Walecki, S. Rinné and M. K. Bollepalli,
et al., A Specific Two-pore Domain Potassium Channel Blocker Defines the Structure of the TASK-1 Open Pore, J. Biol. Chem., 2011, 286(16), 13977–13984 CrossRef CAS PubMed.
- A. K. Kiper, S. Rinné, C. Rolfes, D. Ramírez, G. Seebohm and M. F. Netter,
et al., Kv1.5 blockers preferentially inhibit TASK-1 channels: TASK-1 as a target against atrial fibrillation and obstructive sleep apnea?, Pflugers Arch., 2015, 467(5), 1081–1090 CrossRef CAS PubMed.
-
B. Zou, D. P. Flaherty, D. S. Simpson, B. E. Maki, M. R. Miller and J. Shi, et al. ML365: Development of Bis-Amides as Selective Inhibitors of the KCNK3/TASK1 Two Pore Potassium Channel. Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD): National Center for Biotechnology Information (US); 2010.
- C. D. Weaver, D. Harden, S. I. Dworetzky, B. Robertson and R. J. Knox, A thallium-sensitive, fluorescence-based assay for detecting and characterizing potassium channel modulators in mammalian cells, J. Biomol. Screening, 2004, 9(8), 671–677 CrossRef CAS.
- J. Kutchinsky, S. Friis, M. Asmild, R. Taboryski, S. Pedersen and R. K. Vestergaard,
et al., Characterization of potassium channel modulators with QPatch automated patch-clamp technology: system characteristics and performance, Assay Drug Dev. Technol., 2003, 1(5), 685–693 CrossRef CAS PubMed.
- D. P. Flaherty, D. S. Simpson, M. Miller, B. E. Maki, B. Y. Zou and J. Shi,
et al., Potent and selective inhibitors of the TASK-1 potassium channel through chemical optimization of a bis-amide scaffold, Bioorg. Med. Chem. Lett., 2014, 24(16), 3968–3973 CrossRef CAS PubMed.
- M. Rudrapal and D. D. Chetia, Virtual Screening, Molecular Docking and QSAR Studies in Drug Discovery and Development Programme, J. Drug Delivery Ther., 2020, 10, 225–233 CrossRef CAS.
- W. L. Zhang, J. F. Pei and L. H. Lai, Computational Multitarget Drug Design, J. Chem. Inf. Model., 2017, 57(3), 403–412 CrossRef CAS PubMed.
- J. Achenbach, F. M. Klingler, R. Blöcher, D. Moser, A. K. Häfner and C. B. Rödl,
et al., Exploring the Chemical Space of Multitarget Ligands Using Aligned Self-Organizing Maps, ACS Med. Chem. Lett., 2013, 4(12), 1169–1172 CrossRef CAS PubMed.
- G. Schneider and U. Fechner, Computer-based de novo design of drug-like molecules, Nat. Rev. Drug Discovery, 2005, 4(8), 649–663 CrossRef CAS PubMed.
- M. Segall, Advances in multiparameter optimization methods for de novo drug design, Expert Opin. Drug Discovery, 2014, 9(7), 803–817 CrossRef PubMed.
- M. Hartenfeller, H. Zettl, M. Walter, M. Rupp, F. Reisen and E. Proschak,
et al., DOGS: reaction-driven de novo design of bioactive compounds, PLoS Comput. Biol., 2012, 8(2), e1002380 CrossRef CAS.
- Q. Bai, S. Tan, T. Xu, H. Liu, J. Huang and X. Yao, MolAICal: a soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm, Briefings Bioinf., 2021, 22(3), bbaa161 CrossRef.
- R. X. Wang, X. L. Fang, Y. P. Lu and S. M. Wang, The PDBbind database: Collection of binding affinities for protein-ligand complexes with known three-dimensional structures, J. Med. Chem., 2004, 47(12), 2977–2980 CrossRef CAS PubMed.
- R. Huey, G. M. Morris, A. J. Olson and D. S. Goodsell, A semiempirical free energy force field with charge-based desolvation, J. Comput. Chem., 2007, 28(6), 1145–1152 CrossRef CAS.
-
Y. Chabchoub and C. Fricker, Classification of the Velib Stations Using Kmeans, Dynamic Time Wraping and Dba Averaging Method. 2014 International Workshop on Computational Intelligence for Multimedia Understanding (Iwcim). 2014.
- H. Lin, J. Li, Q. Zhang, H. Yang and S. Chen, C-type inactivation and proton modulation mechanisms of the TASK3 channel, Proc. Natl. Acad. Sci. U. S. A., 2024, 121(17), e2320345121 CrossRef CAS.
- Y. L. Wang, F. Wang, X. X. Shi, C. Y. Jia, F. X. Wu and G. F. Hao,
et al., Cloud 3D-QSAR: a web tool for the development of quantitative structure-activity relationship models in drug discovery, Briefings Bioinf., 2021, 22(4), bbaa276 CrossRef.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717 CrossRef.
- J. Dong, N. N. Wang, Z. J. Yao, L. Zhang, Y. Cheng and D. F. Ouyang,
et al., ADMETlab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database, J. Cheminf., 2018, 10, 29 Search PubMed.
- J. Tirado-Rives and W. L. Jorgensen, Performance of B3LYP density functional methods for a large set of organic molecules, J. Chem. Theory Comput., 2008, 4(2), 297–306 CrossRef CAS PubMed.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo and K. M. Merz,
et al., The Amber biomolecular simulation programs, J. Comput. Chem., 2005, 26(16), 1668–1688 CrossRef CAS.
- T. Lu and F. W. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS PubMed.
- J. Lee, X. Cheng, S. Jo, A. D. MacKerell, J. B. Klauda and W. Im, CHARMM-GUI Input Generator for NAMD, Gromacs, Amber, Openmm, and CHARMM/OpenMM Simulations using the CHARMM36 Additive Force Field, Biophys. J., 2016, 110(3), 641a-a CrossRef.
- C. J. Dickson, B. D. Madej, A. A. Skjevik, R. M. Betz, K. Teigen and I. R. Gould,
et al., Lipid14: The Amber Lipid Force Field, J. Chem. Theory Comput., 2014, 10(2), 865–879 CrossRef CAS PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Comparison of multiple amber force fields and development of improved protein backbone parameters, Proteins, 2006, 65(3), 712–725 CrossRef CAS PubMed.
- K. Kholmurodov, W. Smith, K. Yasuoka, T. Darden and T. Ebisuzaki, A smooth-particle mesh Ewald method for DL_POLY molecular dynamics simulation package on the Fujitsu VPP700, J. Comput. Chem., 2000, 21(13), 1187–1191 CrossRef CAS.
- R. Kumari, R. Kumar and A. Lynn, g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations, J. Chem. Inf. Model., 2014, 54(7), 1951–1962 CrossRef CAS PubMed.
- H. Gohlke and D. A. Case, Insights into protein-protein binding by binding free energy calculation and free energy decomposition using a generalized born model, Abstr. Pap. Am. Chem. Soc., 2003, 225, U791-U Search PubMed.
- S. Keretsu, S. P. Bhujbal and S. J. Cho, Computational study of paroxetine-like inhibitors reveals new molecular insight to inhibit GRK2 with selectivity over ROCK1, Sci. Rep., 2019, 9, 13053 CrossRef PubMed.
- S. Rinné, A. K. Kiper, K. S. Vowinkel, D. Ramirez, M. Schewe and M. Bedoya,
et al., The molecular basis for an allosteric inhibition of K plus -flux gating in TASK channels, Acta Physiol., 2019, 227 Search PubMed.
- D. Ramírez, M. Bedoya, A. K. Kiper, S. Rinné, S. Morales-Navarro and E. W. Hernández-Rodríguez,
et al., Structure/Activity Analysis of TASK-3 Channel Antagonists Based on a 5,6,7,8 tetrahydropyrido[4,3-d]pyrimidine, Int. J. Mol. Sci., 2019, 20(9), 2252 CrossRef.
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Jixiang
Liu
a,
Jixiang
Liu
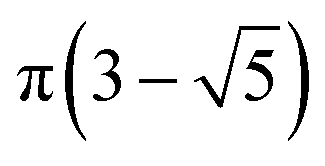 to distribute points on a sphere. The points on the spherical surface are calculated according to the following equations:
to distribute points on a sphere. The points on the spherical surface are calculated according to the following equations: