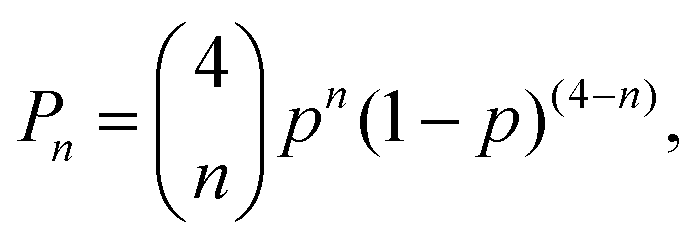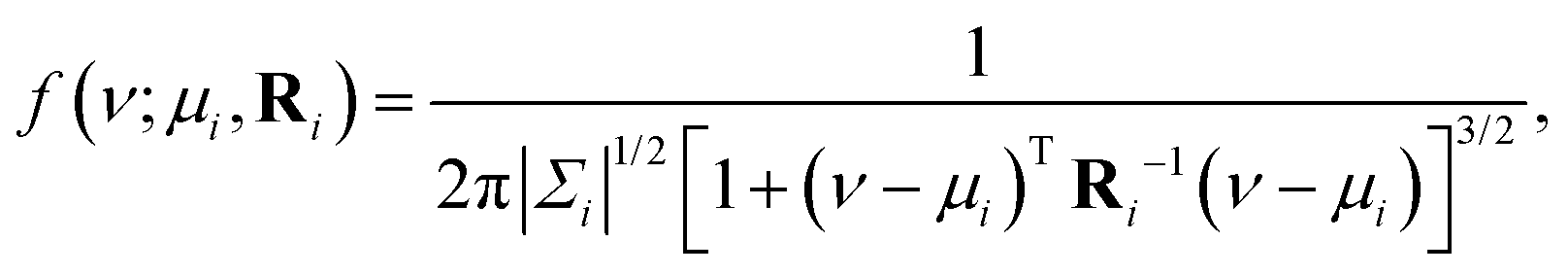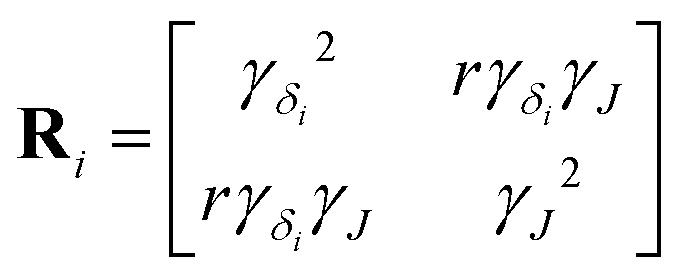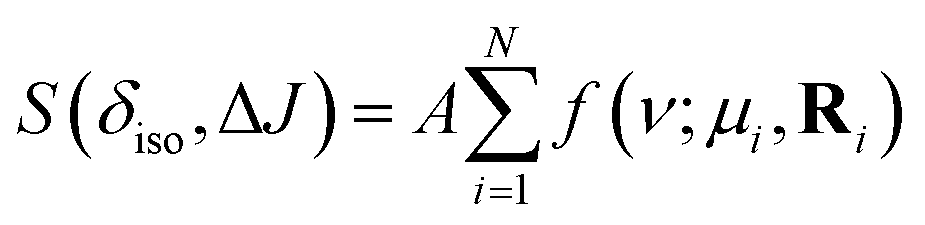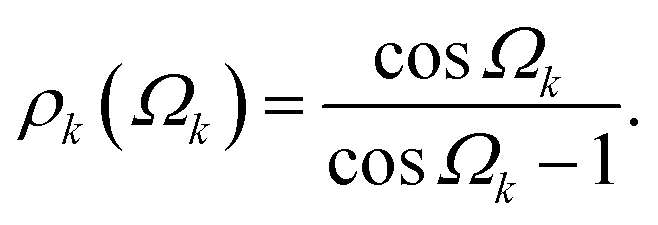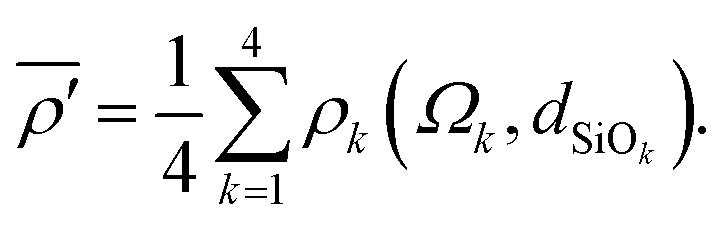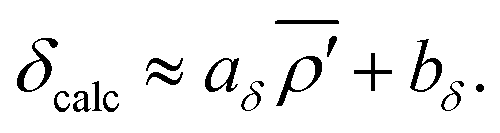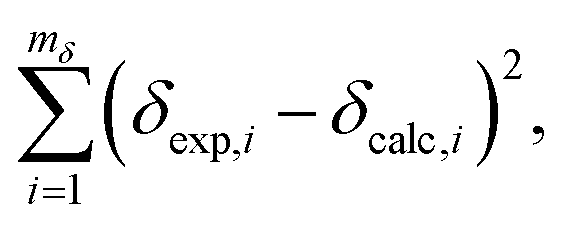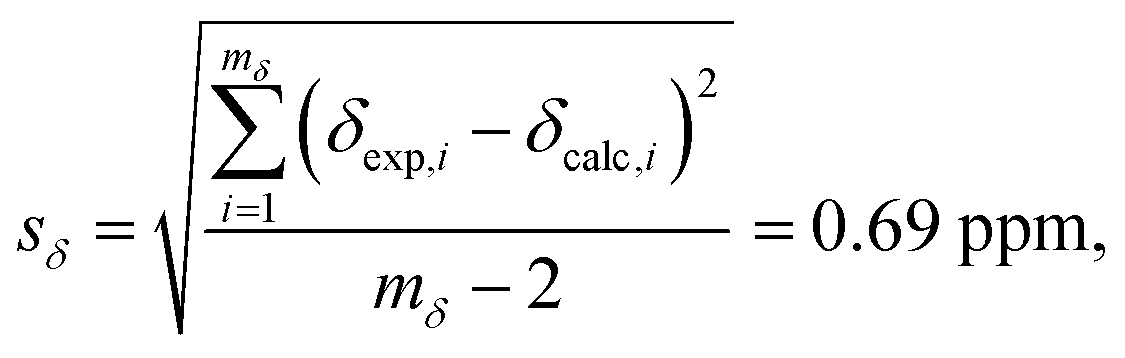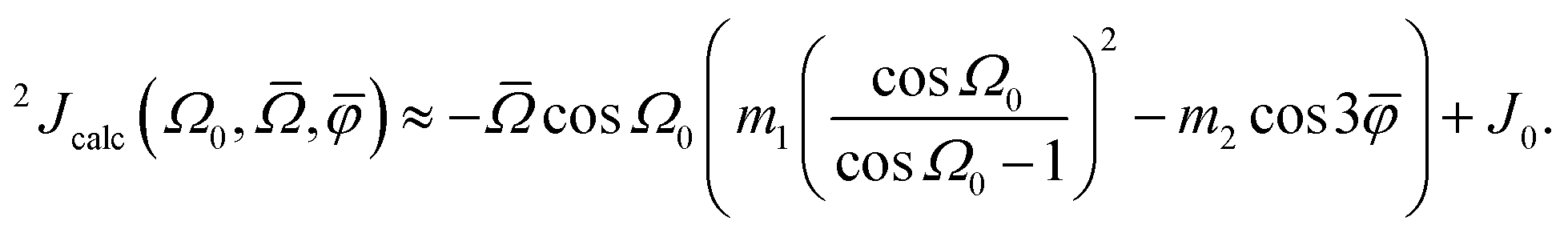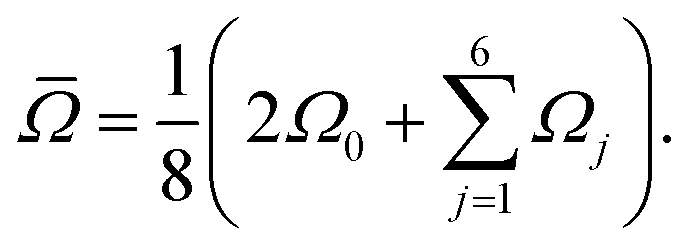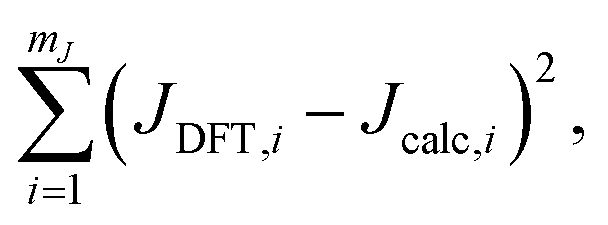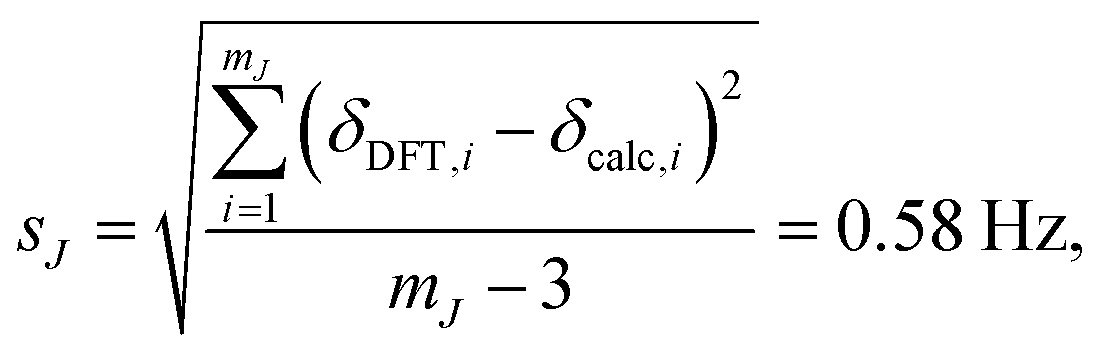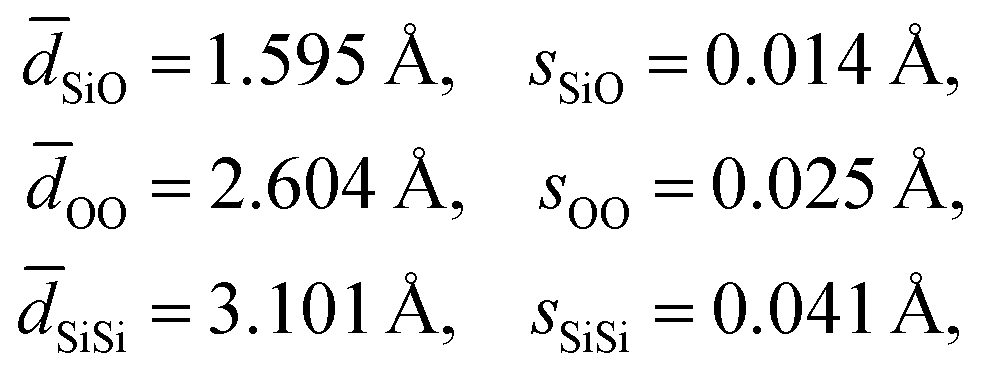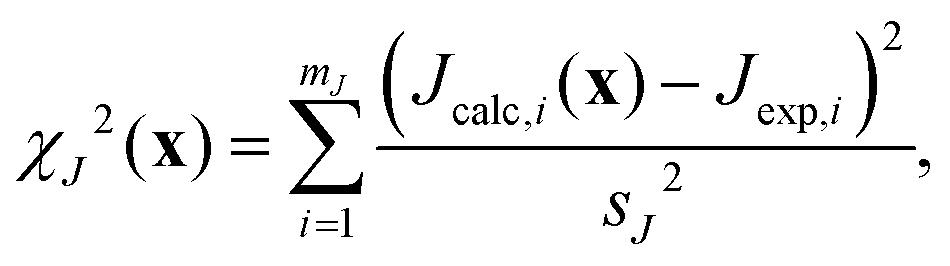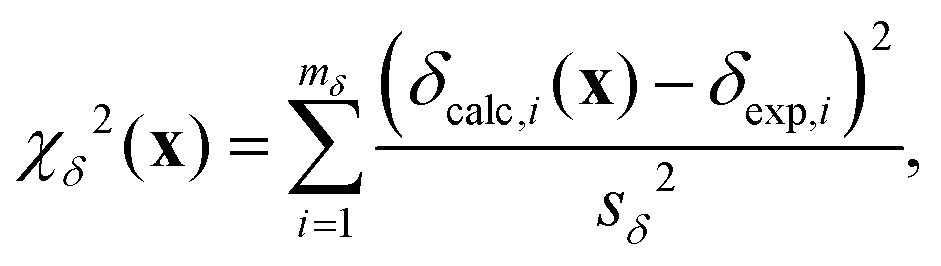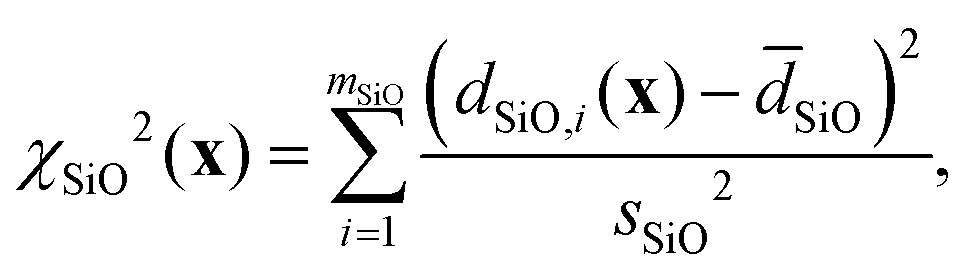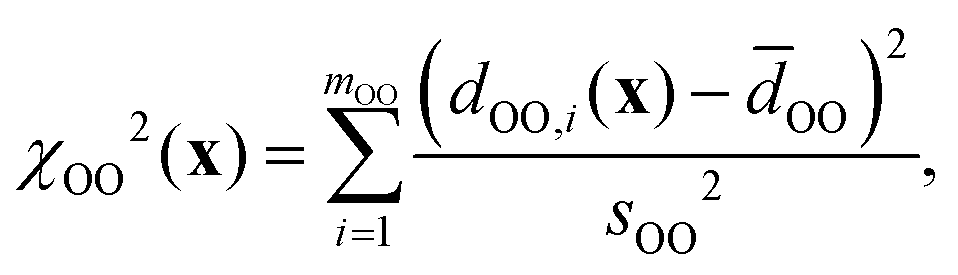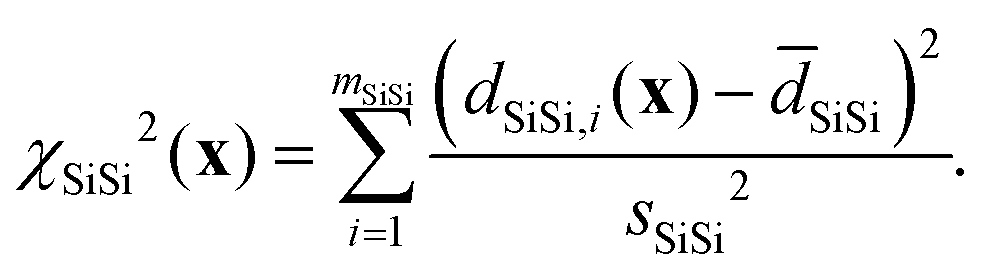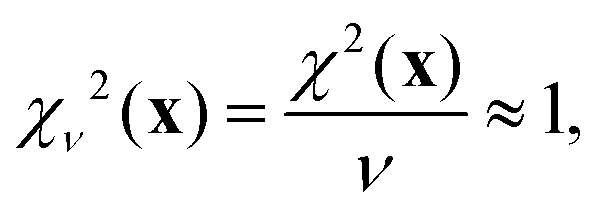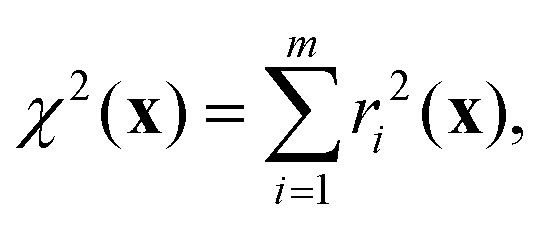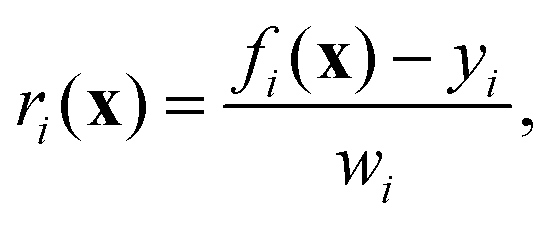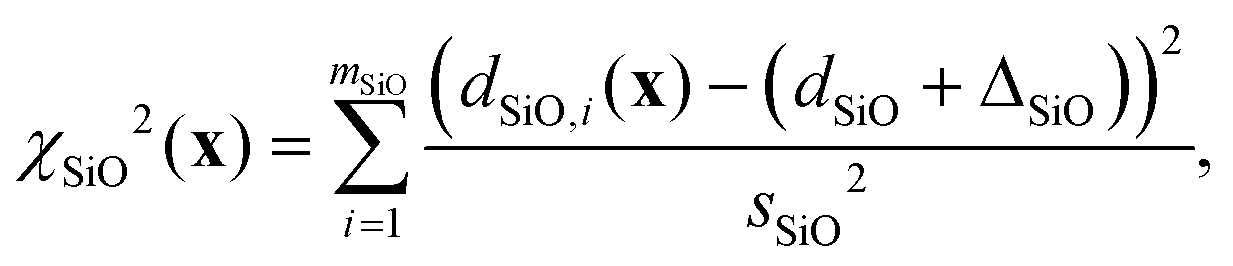Refining siliceous zeolite framework structures with 29Si 2D J-resolved NMR spectroscopy†
Received
11th September 2024
, Accepted 27th November 2024
First published on 27th November 2024
Abstract
A modified shifted-echo PIETA pulse sequence is developed to acquire natural abundance 29Si 2D J-resolved spectra in crystalline silicates. The sequence is applied to the highly siliceous zeolites Sigma-2 and ZSM-12. The 2D J-resolved spectra are used to develop a silicate framework structure refinement strategy based on Si–O, O–O, and Si–Si distance restraints and analytical relationships between local structure and 29Si chemical shifts and geminal 2JSi–O–Si couplings. The refinement of the Sigma-2 structure showed that the Si–O and O–O distances were in excellent agreement with the single-crystal X-ray diffraction (SCXRD) data. The refinement of the ZSM-12 structure, initially determined from synchrotron powder XRD data, highlighted significant improvements in Si–O and O–O distances, and better agreement between calculated and experimental chemical shifts and J-couplings.
1 Introduction
Zeolites display diverse structures resulting from the interaction between their framework's chemical composition and topology. These materials possess unique and desirable chemical and physical properties, making them highly valuable for many applications in sustainable chemistry,1 such as catalysis and separations. Pure and high-silica zeolites are especially interesting due to their superior thermal and hydrothermal stability compared to their aluminosilicate counterparts. They are highly effective as molecular sieves for removing organic micro-pollutants, making them useful in various industrial applications, including water and wastewater treatment.2
A complete knowledge of the zeolite framework structure is of the utmost importance for improving performance in these applications. However, since synthetic zeolites are, more often than not, polycrystalline, single-crystal diffraction measurements of zeolites are rare as it is difficult to grow large enough crystals. As the quality of structure refinements from powder diffraction data is lower than those obtained from single-crystal measurements, other characterization techniques become essential in their structure refinement.
Solid-state nuclear magnetic resonance (NMR) spectroscopy has long played an important role in characterizing zeolite structures. In particular, the 29Si isotropic chemical shift, δiso, has been a primary structural probe in zeolites.3–7 In pure-silica zeolites, the high tetrahedral symmetry of the 4QSi sites, with little deviation from silicon sp3 hybridization, leads to the 29Si isotropic chemical shift of 4QSi sites being dominated by variations in the four inter-tetrahedral Si–O–Si angles of its second-coordination sphere.
Beyond the 29Si isotropic chemical shifts, other nuclear spin interactions such as dipolar couplings,8–10 nuclear-shielding anisotropies,10–12 quadrupolar couplings,13 and J couplings14 also have great potential to serve as additional structural constraints in zeolites. The two most direct NMR probes of a Si–O–Si bond angle are the 17O quadrupolar coupling tensor, i.e., in Si–17O–Si,15,16 and the geminal 2JSi–O–Si coupling across a 29Si–O–29Si linkage.14,17,18 Although the natural abundance of 29Si at 4.683% puts the abundance of 29Si–O–29Si linkages at 0.76%, this is still an order of magnitude higher than the natural abundances of 17O at 0.038%. Even with 17O enrichment, determining the 17O quadrupolar coupling tensor parameters from multiple-quantum magic-angle spinning spectra19 can be complicated by low sensitivity and distorted anisotropic lineshapes. Thus, although less commonly used, the geminal 2JSi–O–Si coupling with 29Si NMR offers a promising alternative to 17O NMR.
More recently, Srivastava et al.18 showed that the geminal 2JSi–O–Si coupling across a QSi4–QSi4 linkage depends primarily upon the s-character of the valence hybrid-type orbitals associated with the Si(i)–O and Si(j)–O bonds across the Si(i)–O–Si(j) linkage. Based on systematic DFT investigations, they proposed an approximate analytical relationship relating the geminal 2JSi–O–Si coupling to the bridging oxygen linkage angle with a more subtle variation due to the double mean Si–O–Si angle of the two coupled 29Si nuclei, 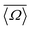 , and the dihedral angle of the QSi4–QSi4 cluster, φ.
, and the dihedral angle of the QSi4–QSi4 cluster, φ.
In an attempt to exploit both the 29Si isotropic chemical shift and correlated 2JSi–O–Si coupling as structural constraints in refining pure-silica zeolite structures, we have developed the interleaved-shifted-echo phase-incremented echo train acquisition (PIETA) NMR pulse sequence as a rapid means of measuring the natural abundance 29Si two-dimensional (2D) J spectrum of a crystalline silicate. This method provides a direct measurement of the correlation of 29Si isotropic chemical shifts to geminal 2JSi–O–Si couplings across a QSi4–QSi4 linkage. Using this approach, we have obtained the 2D J spectrum of the pure-silica zeolites, Sigma-220 and ZSM-12.21 This new NMR method not only reveals the connectivity of the siliceous zeolite framework but also provides the most precise measurements of geminal 2JSi–O–Si couplings in a pure-silica zeolites to date. In this work, we further develop a modified distance least-squares method22–26 to incorporate these NMR priors in the crystal structure refinement of both materials.
2 NMR spectroscopy
Two-dimensional J-resolved spectroscopy is an NMR method focusing primarily on resolving the J-couplings along the indirect dimension. The characteristic feature of this experiment is the J-modulation of the spin echo27 where the chemical shift evolution refocuses into an echo in the direct dimension (t2) and the evolution of the echoes along the indirect (t1) dimension, provides the desired J-modulation. Like incredible natural abundance DoublE QUAntum transfer experiment (INADEQUATE),28–34 this method reveals the atom-connectivity. In J-resolved spectroscopy, the connectivity information is in the correlation of the chemical shift to the J-resolved frequencies. Additionally, the J-coupling can be measured directly from the splittings along the J-resolved dimension, where the removal of inhomogeneous line broadenings leads to the sub-hertz site resolution along the J-resolved dimension and reveals the connectivity information. In the INADEQUATE method, the connectivity information is in the correlation of the single- to double-quantum transition frequencies. A comparison of the two methods is schematically shown in Fig. 1. In contrast to J-resolved spectroscopy, the J-couplings need to be known in advance to optimize the double-quantum build-up times in the INADEQUATE method. Furthermore, if there is a large variation of J couplings across the different sites, INADEQUATE only observes the peaks with J couplings captured by the double quantum build-up times chosen. Additionally, the refocused version of INADEQUATE32 is needed to avoid anti-phase doublets that cancel out for broad peaks common in solids. Thus, the J-resolved method, which does not require double quantum excitation, is more quantitative. A disadvantage of 2D J-resolved spectroscopy is the presence of uncoupled spin resonances in the 2D J-resolved spectrum at 0 Hz, schematically represented with open red circles in Fig. 1C. Furthermore, when a coupling deviates from the weak coupling limit, its 2D J-resolved spectrum can exhibit four additional “intermediate-coupling” resonances that are weaker in amplitude and appear at a frequency that is midway between the shifts of the two sites in the chemical shift dimension of the 2D J spectrum.35 The effect starts to become pronounced at |νA − νB|/JAB < 2. Fortunately, the frequencies of the “weak-coupling” resonances along the J-resolved dimension, i.e., those illustrated in Fig. 1, are not affected by these intermediate coupling effects, although their amplitudes are progressively reduced to zero as the coupling approaches the strong limit.
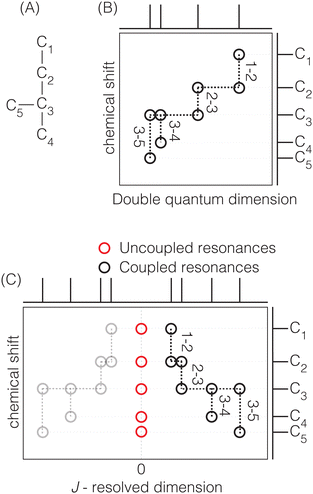 |
| | Fig. 1 (A) Model structure used to compare the network connectivity information in 2D J-resolved and 2D INADEQUATE experiments. A schematic representation of the network connectivity elucidation using (B) 2D INADEQUATE and (C) 2D J-resolved spectroscopy. The pairs along the vertical lines represent the connected atoms. In this example with naturally occurring (∼0.01%) 13C pairs, there are four pairs corresponding to C1–C2, C2–C3, C3–C4, and C3–C5. The structural connectivity is highlighted with dotted lines. While both experiments can provide the same connectivity information, the 2D J-resolved spectrum has quantitative intensities and can provide J couplings. | |
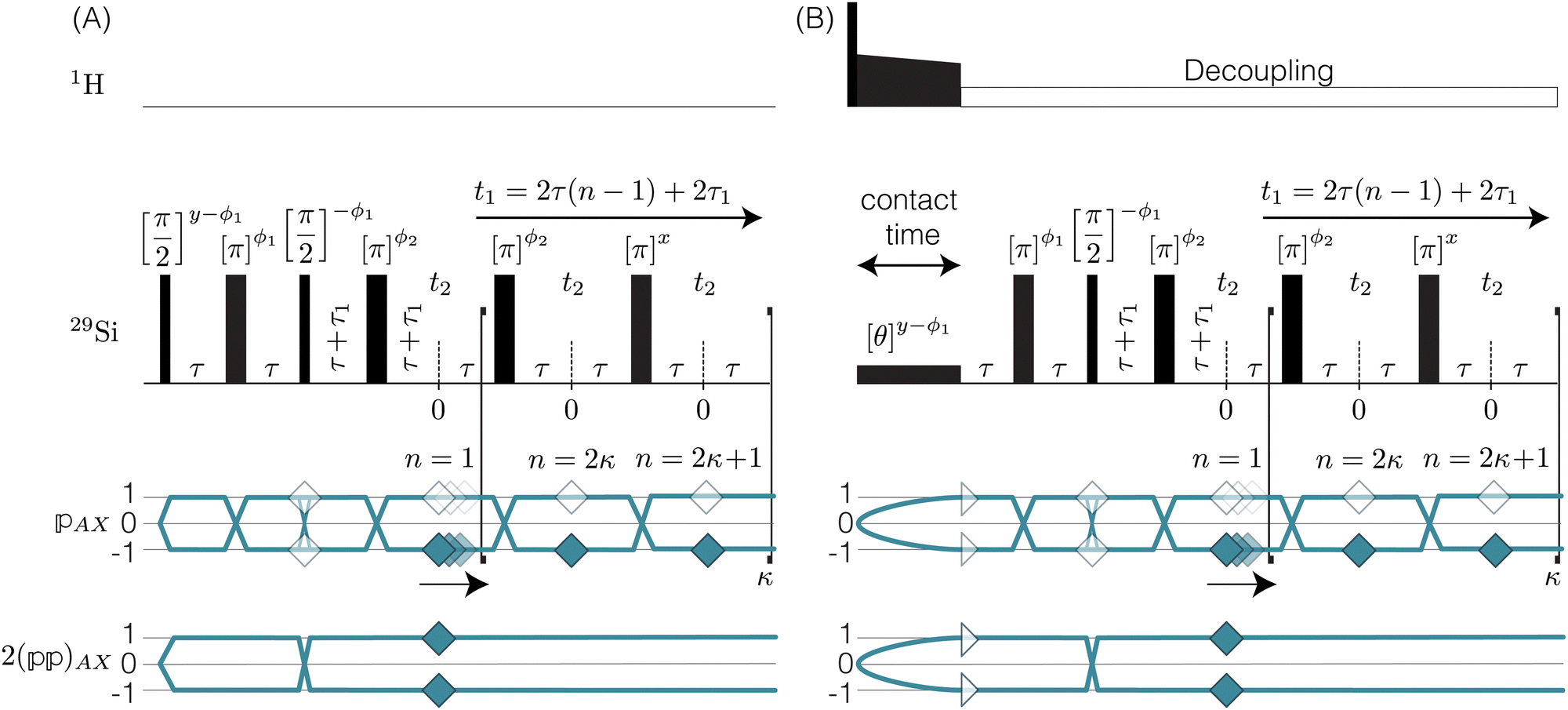
2.1 Shifted-echo PIETA with t1 interleaving scheme
Acquiring a 2D J-resolved 29Si spectrum where every t1 point requires waiting for longitudinal magnetization recovery can be prohibitively long, particularly in the case of natural abundance (4.67%) 29Si NMR in solids, where the T1 relaxation times can be on the order of hours. Fortunately, the PIETA sequence36 provides a faster method than the traditional 2D J-resolved pulse sequences.17,37,38 The PIETA sequence acquires a 2D spectrum in a “pseudo-single-scan” experiment.36 We write “single-scan” because the entire 2D-time domain signal is obtained in a single acquisition and “pseudo” because the separate “single-scan” signals must also be acquired along a radio frequency (RF) pulse phase dimension. However, sampling in the RF pulse phase dimension need not increase the total experiment time, for it is implemented in place of conventional phase cycling and signal averaging.
The recently developed shifted-echo PIETA sequence39 eliminates a signal artifact when using the original PIETA experiment for 2D J-resolved spectroscopy, which arises from an inability to acquire a full echo for the “t1 = 0” (n = 1) cross-section. A remaining limitation of the shifted-echo PIETA method is that the spectral resolution in the isotropic chemical shift dimension can only be improved at the expense of the spectral width along the J-resolved dimension and vice versa. In the case of 2JSi–O–Si coupling18 across the Si–O–Si linkage, the coupling is of the order of 0–25 Hz. Assuming a maximum resolvable 2J-coupling of 25 Hz, the Nyquist theorem invokes a maximum limit for the inter-echo spacing interval, i.e., Δt1, of 40 ms, giving, in turn, a total t2 acquisition time, of 40 ms for each echo in the echo train. This is usually not an issue in applications to amorphous silicates where the full echo (total t2) acquisition times are 4–20 ms. However, a full echo can last an order of magnitude longer in polycrystalline silicates. To resolve this issue and ensure adequate resolution and spectral width along the chemical shift and J-resolved dimensions, respectively, we introduce the interleaved-shifted-echo PIETA pulse sequence with t1 interleaving scheme as shown in Fig. 2. This sequence is similar to the shifted-echo PIETA pulse sequence except for an additional τ1 dimension with the definition
| | 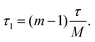 | (1) |
Here,
m ∈ [1, 2,…
M] is an integer counter, and
M is the total number of
t1 interleaving points. This slight modification decouples the resolution of the MAS dimension from the
J-resolved dimension by allowing subsampling of the
t1 dimension along the
τ1 dimension. The integer
M is chosen such that the spectral width along the
J-resolved dimension, given as
M/
τ, is sufficiently large to resolve the
J-couplings. This approach, however, comes at an added price of increased experiment time,
i.e., the experiment time increases by a factor of
M compared to the shifted-echo PIETA sequence. To allow rotor synchronization of the echo train, the echo spacing,
τ, is set to an integer multiple of
Mτr, where
τr is the rotation period.
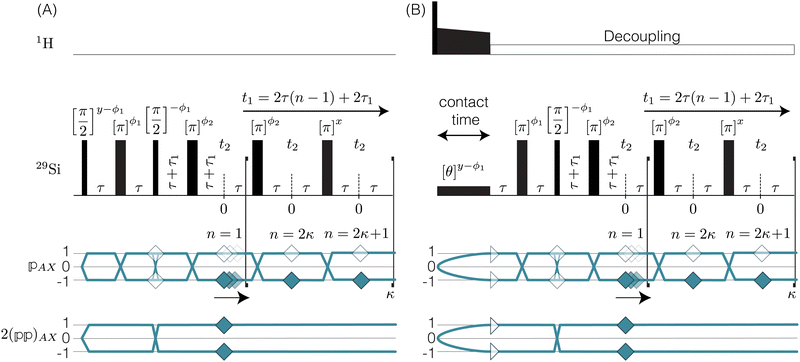 |
| | Fig. 2 (A) A graphical representation of J-resolved shifted-echo PIETA sequence with t1 interleaving and relevant symmetry pathways. Here κ = 1…N and n = 1…2N is the echo counter with 2N number of echoes per t1 interleaving point, m. Here, the τ1 increment provides the t1 interleaving dimension. (B) The shifted-echo PIETA pulse sequence with t1 interleaving is similar to the sequence in (A) with an initial 1H to 29Si cross-polarization and constant low power 1H decoupling. | |
The interleaved-shifted-echo PIETA sequence follows the same set of spin transitions and transition symmetry pathways as the shifted-echo PIETA sequence, which are described in detail elsewhere.39,40 As before, the RF phases of the first three pulses are defined in terms of ϕ1 and every other π pulse phase set to ϕ2 in the echo train acquisition, as shown in Fig. 2. Because the τ1 increment is added only after the second π/2 pulse, and because  values of transitions remain invariant under the π pulse, there will always be an echo of
values of transitions remain invariant under the π pulse, there will always be an echo of 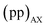 symmetry at the end of 4τ period regardless of the duration of τ1. On the contrary, the echoes from the
symmetry at the end of 4τ period regardless of the duration of τ1. On the contrary, the echoes from the 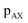 symmetries will move forward in time by the duration of 2τ1. Note, for m = 1 or τ1 = 0, the pulse sequence in Fig. 2 is the same as the shifted-echo PIETA sequence.39
symmetries will move forward in time by the duration of 2τ1. Note, for m = 1 or τ1 = 0, the pulse sequence in Fig. 2 is the same as the shifted-echo PIETA sequence.39
The Bruker code for interleaved-shifted-echo PIETA pulse sequence using ϕ1 and ϕ2 phase dimensions is provided in the ESI.†
2.2 Signal processing
As acquired, the shifted-echo PIETA with t1 interleaving scheme is a 5D signal, s(ϕ2,m,ϕ1,n,t2), where ϕ1 and ϕ2 are the two phase dimensions, n is the echo count index dimension encoding the J-modulated t1 evolution in the echo train, m is the interleaved index encoding the additional J-modulated t1 evolution, and t2 is the direct time acquisition dimension, occurring in between the π pulses. Fourier transforms with respect to the ϕ1 and ϕ2 dimensions transform the 5D signal into s(Δp2,m,Δp1,n,t2) where Δp1 is the accumulated change in coherence order through the first three pulses, and Δp2 is the accumulated change in coherence order through the echo train acquisition. Based on the desired transition pathways,39 the desired signals at the nth echo appear at the coordinate| | 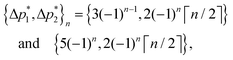 | (2) |
where ⌈·⌉ is the ceiling function. The Δp1 values for all desired odd and even echoes are fixed to 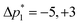 and
and 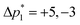 , respectively. After zeroing the signal in the undesired (Δp1, Δp2) coordinate, the 5D signal, s(Δp2,m,Δp1,n,t2), is projected down to the 3D signal s(m,n,t2), followed by a Fourier transform with respect to t2 to obtain s(m,n,ω2). An interactive zeroth and first-order phase correction is applied along the ω2 dimension of s(m,n,ω2) to obtain a pure absorption mode spectrum of the first “t1 = 0” echo, i.e., s(m,n = 1,ω2). As the J-resolved dimension is symmetric about zero, there should be little to no signal remaining in the imaginary part of the signal. However, an additional zeroth-order phase correction may need to be applied only to the even echoes to reduce the signal in the imaginary part if intermediate couplings are present.
, respectively. After zeroing the signal in the undesired (Δp1, Δp2) coordinate, the 5D signal, s(Δp2,m,Δp1,n,t2), is projected down to the 3D signal s(m,n,t2), followed by a Fourier transform with respect to t2 to obtain s(m,n,ω2). An interactive zeroth and first-order phase correction is applied along the ω2 dimension of s(m,n,ω2) to obtain a pure absorption mode spectrum of the first “t1 = 0” echo, i.e., s(m,n = 1,ω2). As the J-resolved dimension is symmetric about zero, there should be little to no signal remaining in the imaginary part of the signal. However, an additional zeroth-order phase correction may need to be applied only to the even echoes to reduce the signal in the imaginary part if intermediate couplings are present.
Once the signal in the imaginary part is minimized, the imaginary part of the signal is set to zero to improve sensitivity and symmetrize the J-resolved dimension about zero. Finally, using t1 = 2τ(n − 1) + 2τ1, the 3D signal is interleaved and reduced to the 2D signal s(t1,ω2), followed by a Fourier transform to obtain the 2D J-resolved spectrum, s(ω1,ω2). A step-by-step guide illustrating this signal processing in the RMN software41 is given in the ESI.†
Since acquiring the datasets presented in this paper, we have developed a version of the interleaved-shifted-echo PIETA pulse sequence that replaces the ϕ1 phase dimension with a phase cycle. The dataset obtained with this version of the pulse sequence requires fewer signal processing steps, and the Bruker code for this version is also provided in the ESI.†
2.3 NMR measurements
The J-couplings were measured for Sigma-2 and ZSM-12 samples using the interleaved-shifted-echo PIETA sequence. The two samples are those previously used by Brouwer and Enright.11 The experiments were performed on a Bruker Avance III HD 400 MHz NMR spectrometer operating at 9.4 T with 29Si and 1H Larmor frequency of 79.56935 MHz and 400.5989344 MHz, respectively, using a 4 mm Bruker MAS probe. A precise setting of the magic angle, within ±0.01°, is required for this experiment to avoid residual dipolar coupling in the J-resolved spectra. Thus, the magic angle was calibrated using a STMAS42 measurement on Na2SO4. This method better calibrates the magic angle within 0.001° than the traditional KBr spinning sidebands method. The sample spinning speed was set for both measurements at 12.5 ± 0.001 kHz. The 29Si chemical shift was referenced with respect to TMS at 0 ppm.
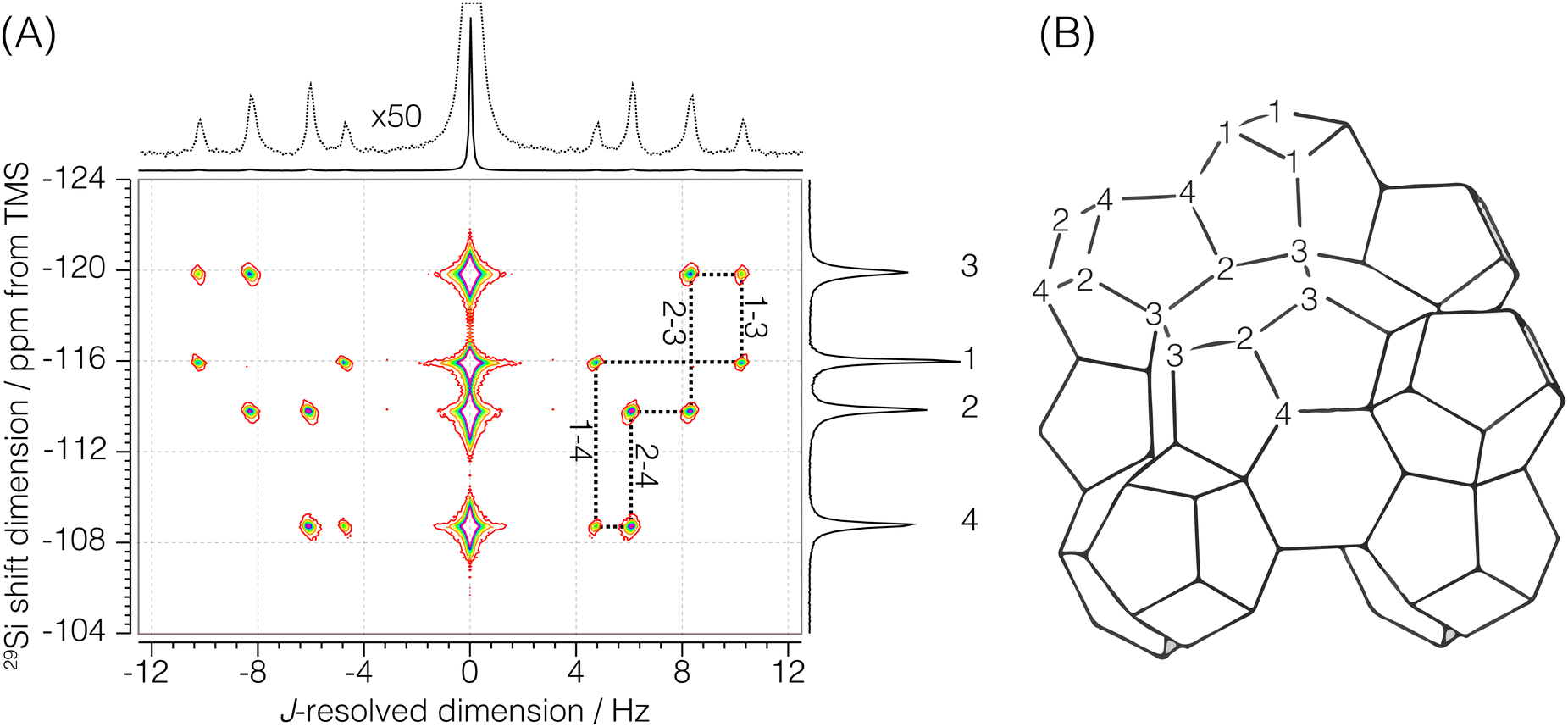
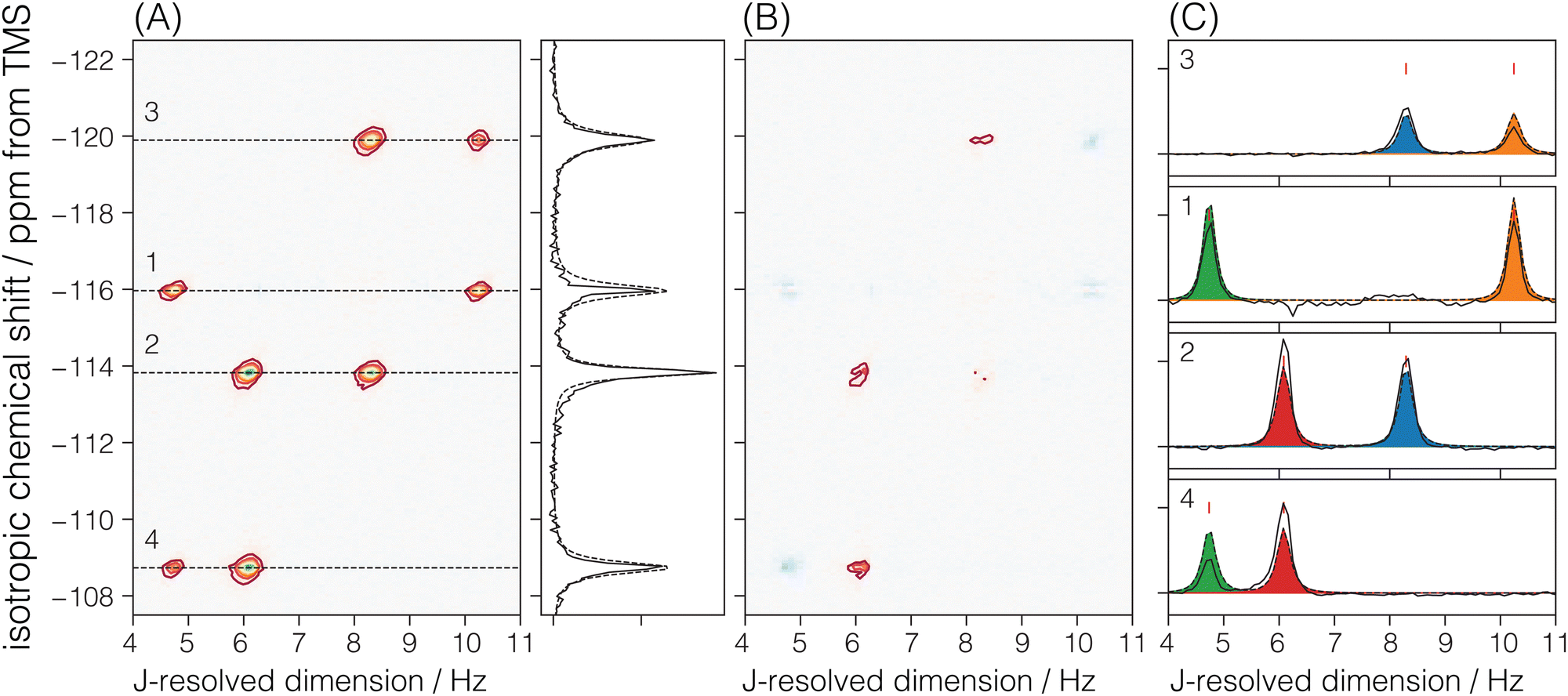
Sigma-2: for this sample, a 1H to 29Si cross-polarization step was prepended to the shifted-echo PIETA sequence, as shown in Fig. 2B. The 1H polarization was excited using an RF pulse of field strength 125 kHz and a τ90° of 2 μs. This polarization was then transferred to 29Si using the simultaneous spin lock RF pulses on each nucleus for a contact period of 9 ms. During the contact period, the RF field strength for 29Si was set to 8.7 kHz, while the RF field strength for 1H was amplitude modulated using a linear 100–90% ramp with the maximum RF field strength of 19.14 kHz. During the echo train acquisition, the RF field strength on 29Si was set to 117.65 kHz with a τ90° and τ180° of 2.125 μs and 4.25 μs, respectively. A low power (∼0.051 W, RF field strength ∼2.5 kHz) 1H–29Si constant decoupling was applied during the measurement using the SPINAL-16 decoupling scheme. The sample temperature was maintained at 25 °C, and the recycle delay was set to 5 s. The 1H relaxation time, T1, was measured to be 2.08 ± 0.05 s. The echoes along the t2 dimension were acquired for 159.36 ms at a sampling rate of 311.25 μs per point for 512 complex points. A total of 288 echoes were collected with four (M = 4) t1 interleaving steps of 72 echoes each. The inter-echo period, 2τ, was set to 160 ms, giving a single signal acquisition time of 11.48 s. This gives a resolution of 160/4 = 40 ms along the t1 dimension, corresponding to a spectral width of 25 Hz along the J-resolved dimension. The phase increments on the first, ϕ1, and the second, ϕ2, phase dimensions were set to π/6 and π/40 with 12 and 80 phase points, respectively. The total experiment time with a single shifted-echo PIETA measurement was approximately 17.92 h. The 29Si 2D J-resolved spectrum for Sigma-2 is shown in Fig. 3.
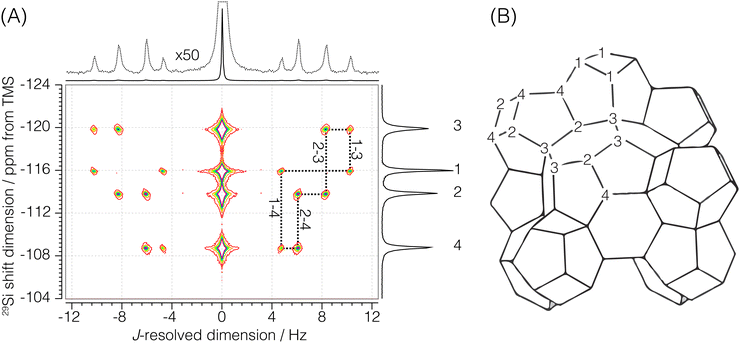 |
| | Fig. 3 (A) Experimental 2D J-resolved 29Si NMR spectrum of siliceous zeolite Sigma-2 showing contributions from both coupled and uncoupled 29Si resonances. Numbered isotropic chemical shift peaks are assigned to the corresponding numbered sites in the Sigma-2 framework structure shown in (B). The structural connectivity is highlighted with dotted lines between the coupled resonance peaks. | |
ZSM-12: the 29Si relaxation time, T1, was measured to be 12 ± 2 s; however, only 20 s of recovery time was allowed per scan. For this reason, four dummy scans were added before the start of the experiment to reach a steady non-equilibrium density state. The RF field strength on 29Si was set to 96.15 kHz with a τ90° and τ180° of 2.6 μs and 5.2 μs, respectively. A total of 288 echoes were acquired with three (M = 3) t1 interleaving steps of 96 echoes each.
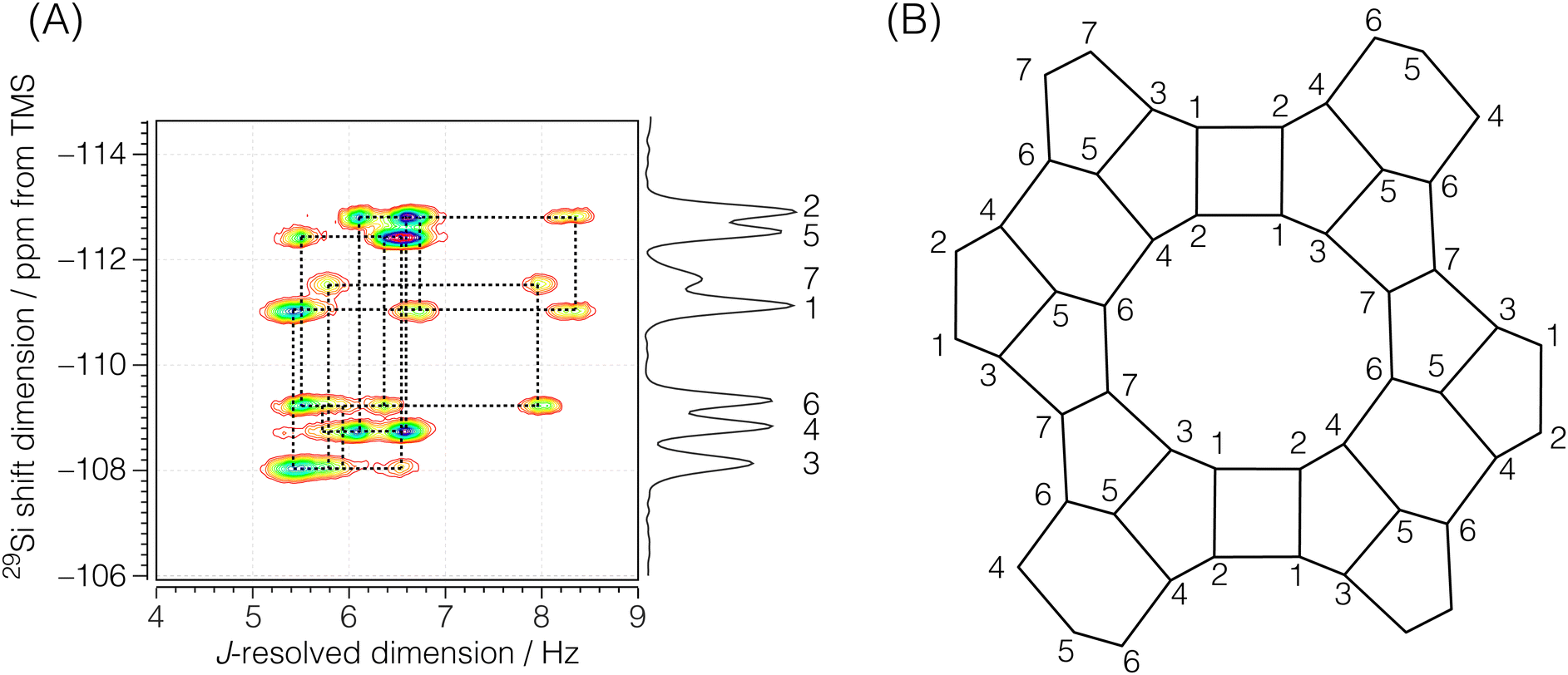
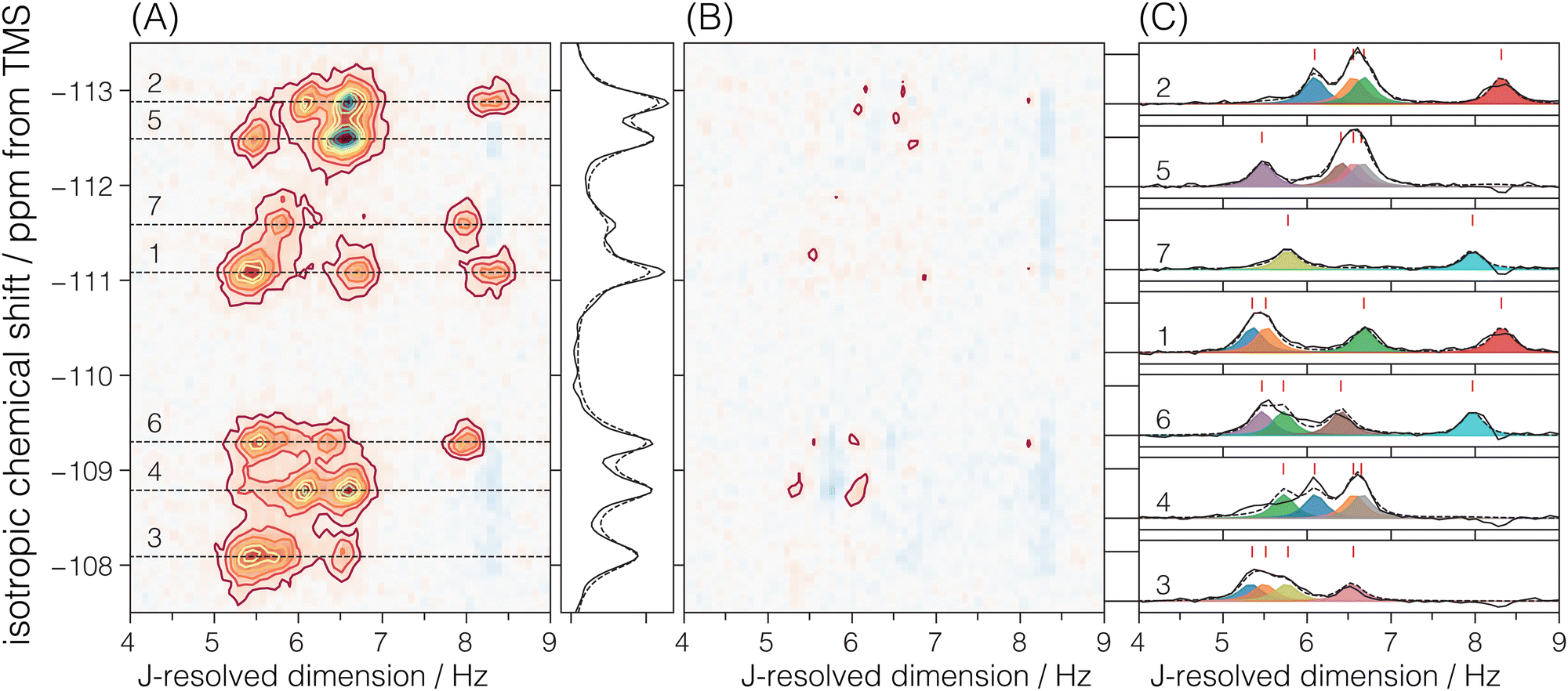
Each echo was acquired with 384 complex data points sampled at every 311.25 μs with an echo acquisition window of 119.52 ms along the t2 dimension. The inter-echo period, 2τ, was set to 120 ms, resulting in a single signal acquisition time of 11.48 s. The phase increments on the first, ϕ1, and the second, ϕ2, phase dimensions were set to π/6 and π/50 with 12 and 100 phase points, respectively. With this setup, a single shifted-echo PIETA measurement was approximately 31.8 h. The signal was averaged over 15 shifted-echo PIETA measurements spanning about 20 days. The sample temperature was maintained at 29 °C throughout the experiment. The resolution and sensitivity of the processed ZSM-12 spectrum were further enhanced by first apodizing the corresponding signal in the t1 − t2 domain with the function exp(|t2|/Ta)exp(−t22/2σa2) where Ta = 16 ms and σa = 18 ms. The apodized signal was Fourier transformed back into the frequency domain and subjected to a noise reduction filter using singular value decomposition (SVD), retaining only the first ten singular values. The full spectrum before and after this resolution and denoising step is shown in Fig. S6 of the ESI.† Additionally, a Python Jupyter notebook performing this operation is provided in the ESI.† The region of the 2D J spectrum of ZSM-12 showing the positive J splittings of the coupled resonances is shown in Fig. 4. The measured J-resolved spectrum and the framework connectivity for ZSM-12 are shown in Fig. 4.
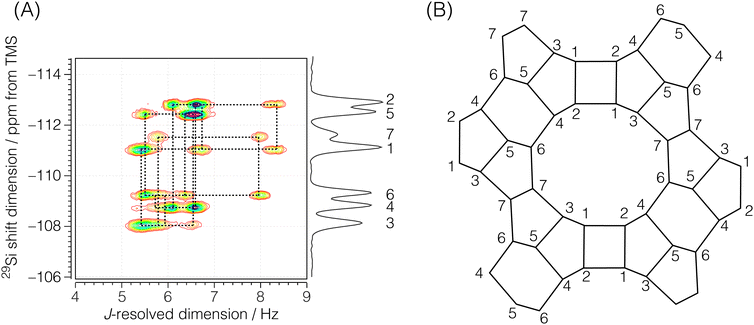 |
| | Fig. 4 (A) Region of the experimental 2D J-resolved 29Si NMR spectrum of siliceous zeolite ZSM-12 showing positive J splittings of the coupled 29Si resonances. Numbered isotropic chemical shift peaks are assigned to the corresponding numbered sites in the ZSM-12 framework structure shown in (B). The structural connectivity is highlighted with dotted lines between the coupled resonance peaks. | |
2.4 NMR spectral analysis
At 29Si natural abundance, the relative probability of 2JSi–O–Si couplings in a 29Si NMR spectrum is given by| | 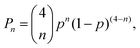 | (3) |
where n is the number of 29Si sites connected to a 29SiO4 tetrahedron, and p = 0.04683 is the natural abundance of 29Si. This expression predicts that the relative intensity of J multiplets decrease rapidly with increasing n, with P0 = 0.8254 for the uncoupled resonance, P1 = 0.1622 for the two spin multiplet, P2 = 0.01195 for the three spin multiplet, P3 = 0.0003916 for the four spin multiplet, and P4 = 4.809 × 10−6 for the five spin multiplet. While the 29Si NMR spectrum contains contributions from all five cases, we can confidently take the observed intensity as arising entirely from the uncoupled and two-spin doublet resonances. Thus, in a weak coupling limit, we expect to see up to four 2JSi–O–Si couplings for a given silicon site at 29Si natural abundance.
We model each (δiso, ΔJ) cross-peak with a bivariate Lorentzian distribution, given by
| | 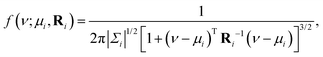 | (4) |
where Δ
Jk =
Jk/2,
ν = [
δiso,Δ
Jk] are the coordinates of a point on a
δiso − Δ
J 2D grid,
μi = [
μδ,
μJ] is the mode, and
| | 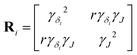 | (5) |
is a matrix of the
ith (
δiso, Δ
J) cross-peak. Here,
γδi is the half-width at half maximum (HWHM) for the isotropic chemical shift distribution of the
ith Si site, and
γJ is the HWHM for all the
J-splitting distributions. Note,
Ri is not a covariance matrix, as the moments of the Lorentzian distribution are undefined.
A non-linear least-squared analysis of the 2D J spectrum, modeled by
| | 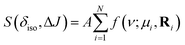 | (6) |
where
A is the peak amplitude, and the summation runs over the
N observable cross-peaks. We only report the mode and HWHM for each observable peak in the discussion below.
2.4.1 Sigma-2.
There are four crystallographic Si sites in Sigma-2, where each site is tetrahedrally connected to four other Si sites. We should observe this connectivity in the 2D J spectrum as four J-coupled doublets at a given 29Si isotropic chemical shift. A total of 8 cross-peaks, (δiso,ΔJ), are observed in Fig. 5A. Here, we only observe two J-splittings for each isotropic chemical shift. This is because each Si is connected to two other Si sites with the same crystallographic site, resulting in unobservable strongly coupled Si–O–Si pairs. Thus, the model for the Sigma-2 spectrum is taken as eqn (6) with N = 8. Additionally, the constraint ΔJij = ΔJji across a given Sii–O–Sij linkage is invoked during the least-squares analysis of the spectrum. A total of four unique J-couplings are found. Table 1 gives the optimized parameters from a non-linear least-squares fit. Also included in Table 1 are the chemical shift differences between coupled sites given in hertz. These values show that all couplings with an observed splitting in Sigma-2 are well within the weak coupling limit. Fig. 5C shows the J-splitting contributions from the individual Si–O–Si pairs. Table 3 lists all optimized parameter values and constraints in the least-squares analysis.
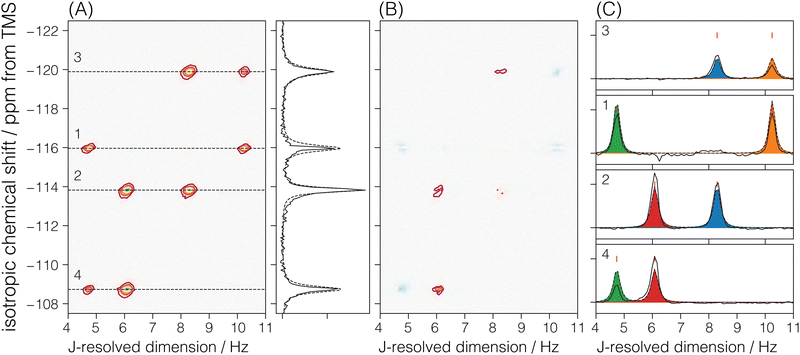 |
| | Fig. 5 The figure in (A) depicts the 2JSi–O–Si coupling and isotropic chemical shift correlation spectrum of Sigma-2 along with the isotropic projections, shown to the side. Here, the solid black line is the projection from the measurement, while the dotted line represents the projection from the model. In (B) are the residuals. The figure in (C) shows the J-coupling contribution from individual isotropic chemical shifts. The black solid line is the measurement, the black dotted line is the fit from the model, and the colored fill in-between plots represent couplings from the individual Si–O–Si pairs. The J-couplings from the coupled Sii–O–Sij pairs are similarly colored across their respective 29Si isotropic chemical shifts. The contours in (A) and (B) are drawn at 10%. | |
Table 1 The experimentally determined 29Si nuclear spin interaction parameters in Sigma-2. The isotropic chemical shifts, δiso, and geminal couplings, 2JSi–O–Si, are obtained from the 2D J spectrum shown in Fig. 3A. The ΔΩ values are isotropic shift differences at 9.4 T
| Site |
δ
iso/ppm |
|
Sii–Ok–Sij |
2
J
Si–O–Si/Hz |
ΔΩ/Hz |
| Si1 |
−115.97 ± 0.18 |
|
Si4–O2–Si2 |
12.16 ± 0.30 |
405.1 |
| Si2 |
−113.82 ± 0.13 |
|
Si1–O3–Si3 |
20.50 ± 0.30 |
312.4 |
| Si3 |
−119.98 ± 0.22 |
|
Si4–O4–Si1 |
9.48 ± 0.30 |
575.7 |
| Si4 |
−108.73 ± 0.18 |
|
Si2–O6–Si3 |
16.60 ± 0.30 |
482.9 |
Cadars et al.14 observed resolved J splittings in cross-sections of the 2D refocused-INADEQUATE 29Si{29Si} spectrum of Sigma-2. By fitting these cross-sections to a doublet of (tilted) Lorentzian peaks centered about a fixed isotropic chemical shift, they obtained 2JSi–O–Si values of 10.0 Hz, 23.5 Hz, 6.3 Hz, 16.5 Hz, across the bridging oxygen O2, O3, O4, and O6, respectively, Even though, they estimate the uncertainty as ±1 Hz, the disagreement with the values in Table 1 is as large as 3 Hz. This is likely a result of the poor resolution of the J splitting in the INADEQUATE cross-sections, where they report line widths with full-width at half maximum (FWHM) of 8 to 16 Hz. In contrast, the FWHM of peaks along the J-resolved dimension of the SE-PIETA spectrum in Fig. 3 of Sigma-2 are on the order of 0.3 Hz. The FWHM of peaks along the J-resolved dimension in SE-PIETA is inversely proportional to the observed echo-train coherence lifetime under MAS of each site, which are provided in the ESI.†
2.4.2 ZSM-12.
There are seven crystallographic silicon sites in ZSM-12. Of these seven sites, six are connected across all four Si–O–Si linkages to magnetically inequivalent sites with observable J splittings, i.e., none of these J couplings are in the strong limit. Thus, six of the seven resonances along the isotropic chemical shift dimension correlate to four cross-peaks along the J-resolved dimension, as seen in Fig. 6. In the case of crystallographic site 7, connected across the Si–O–Si linkages to two magnetically equivalent and two magnetically inequivalent sites, only two cross-peaks are observed along the J-resolved dimension. Thus, the model for the ZSM-12 spectrum is taken as eqn (6) with N = 26. Again, the constraint ΔJij = ΔJji across a given Sii–O–Sij linkage is invoked during the least-squares analysis of the spectrum. A total of 13 unique J couplings were determined. Table 2 gives the optimized parameters from a non-linear least-squares fit. From the chemical shift differences in Table 2, given in hertz, we find that all the couplings are within the weak-coupling limit, although the smaller shift difference between sites 4 and 6 suggests that it may show signs of intermediate coupling effects, i.e., slightly weaker amplitude than the other splittings. Fig. 6C shows the J-coupling contributions from the individual Si–O–Si pairs. Table 4 lists all optimized parameter values and constraints in the least-squares analysis. While we can tentatively assign these splittings based on the expected increase in 2JSi–O–Si with ∠Si–O–Si, these assignments remain the same after the structure refinement.
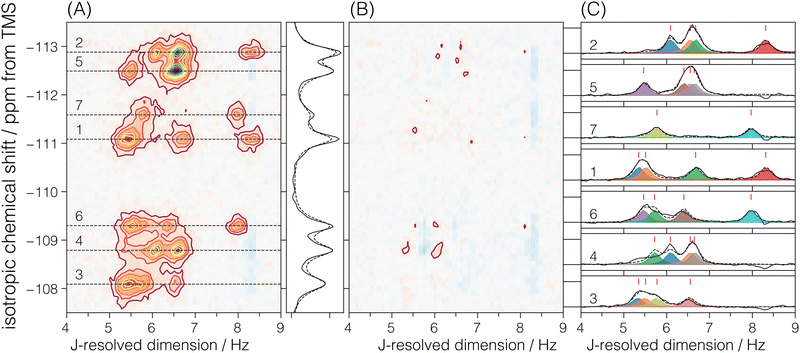 |
| | Fig. 6 The figure in (A) depicts the 2JSi–O–Si coupling and isotropic chemical shift correlation spectrum of ZSM-12 along with the isotropic projections, shown to the side. Here, the solid black line is the projection from the measurement, while the dotted line represents the projection from the model. In (B) are the residuals. The figure in (C) shows the J-coupling contribution from individual isotropic chemical shifts. The black solid line is the measurement, the black dotted line is the fit from the model, and the colored fill in-between plots represent couplings from individual Si–O–Si pairs. The J-couplings from the coupled Sii–O–Sij pairs are similarly colored across their respective 29Si isotropic chemical shifts. The contours in (A) and (B) are drawn at 10%. | |
Table 2 Experimental 29Si isotropic chemical shifts (δiso) and geminal couplings (2JSi–O–Si) in ZSM-12 from Fig. 4A. The ΔΩ values are isotropic shift differences at 9.4 T
| Site |
δ
iso/ppm |
| Si1 |
−111.10 ± 0.16 |
| Si2 |
−112.89 ± 0.15 |
| Si3 |
−108.10 ± 0.19 |
| Si4 |
−108.80 ± 0.17 |
| Si5 |
−112.50 ± 0.17 |
| Si6 |
−109.31 ± 0.18 |
| Si7 |
−111.60 ± 0.22 |
| Sii–Ok–Sij |
2
J
Si–O–Si/Hz |
ΔΩ/Hz |
| Si1–O1–Si2 |
16.62 ± 0.38 |
143.0 |
| Si1–O2–Si2 |
13.36 ± 0.38 |
143.0 |
| Si1–O3–Si3 |
10.70 ± 0.38 |
238.3 |
| Si3–O4–Si5 |
13.10 ± 0.38 |
350.3 |
| Si2–O5–Si4 |
13.10 ± 0.38 |
325.8 |
| Si3–O6–Si7 |
11.54 ± 0.38 |
278.3 |
| Si6–O8–Si5 |
12.80 ± 0.38 |
254.0 |
| Si4–O9–Si6 |
11.44 ± 0.38 |
40.7 |
| Si4–O10–Si5 |
13.30 ± 0.38 |
294.7 |
| Si7–O11–Si6 |
15.94 ± 0.38 |
182.0 |
| Si1–O12–Si3 |
11.02 ± 0.38 |
238.3 |
| Si4–O13–Si2 |
12.18 ± 0.38 |
325.8 |
| Si5–O14–Si6 |
10.92 ± 0.38 |
254.0 |
Table 3 Fit parameters and the corresponding optimum values from the Sigma-2 model
| Parameter |
Optimized values |
Constraints |
| Site 1 |
|
δ
iso,1
|
−115.967 ± 0.001 ppm |
|
| ΔJ14 |
4.735 ± 0.001 Hz |
|
| ΔJ13 |
10.245 ± 0.001 Hz |
ΔJ13 = ΔJ31 |
|
γ
δ,1
|
0.183 ± 0.002 ppm |
|
|
|
| Site 2 |
|
δ
iso,2
|
−113.824 ± 0.001 ppm |
|
| ΔJ24 |
6.084 ± 0.001 Hz |
|
| ΔJ23 |
8.295 ± 0.001 Hz |
ΔJ23 = ΔJ32 |
|
γ
δ,2
|
0.130 ± 0.001 ppm |
|
|
|
| Site 3 |
|
δ
iso,3
|
−119.893 ± 0.002 ppm |
|
| ΔJ32 |
8.295 ± 0.001 Hz |
|
| ΔJ31 |
10.245 ± 0.001 Hz |
|
|
γ
δ,3
|
0.218 ± 0.002 ppm |
|
|
|
| Site 4 |
|
δ
iso,4
|
−108.733 ± 0.001 ppm |
|
| ΔJ43 |
4.735 ± 0.001 Hz |
ΔJ43 = ΔJ34 |
| ΔJ42 |
6.084 ± 0.001 Hz |
ΔJ42 = ΔJ24 |
|
γ
δ,2
|
0.177 ± 0.002 ppm |
|
|
|
|
γ
J
|
0.148 ± 0.001 Hz |
|
|
A
|
0.00328 ± 2 × 10−5 |
|
|
r
|
−0.285 ± 0.006 |
|
Table 4 Fit parameters and the corresponding optimum values from the ZSM-12 model
| Parameter |
Optimized values |
Constraints |
| Site 1 |
|
δ
iso,1
|
−111.097 ± 0.001 ppm |
|
| ΔJ13a |
5.347 ± 0.002 Hz |
|
| ΔJ13b |
5.508 ± 0.002 Hz |
|
| ΔJ12a |
6.676 ± 0.002 Hz |
ΔJ12a = ΔJ21a |
| ΔJ12b |
8.312 ± 0.002 Hz |
ΔJ12b = ΔJ21b |
|
γ
δ,1
|
0.161 ± 0.001 ppm |
|
|
|
| Site 2 |
|
δ
iso,2
|
−112.894 ± 0.001 ppm |
|
| ΔJ24a |
6.090 ± 0.002 Hz |
|
| ΔJ24b |
6.551 ± 0.003 Hz |
|
| ΔJ21a |
6.676 ± 0.002 Hz |
|
| ΔJ21b |
8.312 ± 0.002 Hz |
|
|
γ
δ,2
|
0.153 ± 0.001 ppm |
|
|
|
| Site 3 |
|
δ
iso,4
|
−108.102 ± 0.001 ppm |
|
| ΔJ31a |
5.347 ± 0.002 Hz |
ΔJ31a = ΔJ13a |
| ΔJ31b |
5.508 ± 0.002 Hz |
ΔJ31b = ΔJ13b |
| ΔJ37 |
5.774 ± 0.003 Hz |
ΔJ37 = ΔJ73 |
| ΔJ35 |
6.550 ± 0.003 Hz |
ΔJ35 = ΔJ53 |
|
γ
δ,3
|
0.190 ± 0.002 ppm |
|
|
|
| Site 4 |
|
δ
iso,4
|
−108.800 ± 0.001 ppm |
|
| ΔJ46 |
5.718 ± 0.002 Hz |
ΔJ46 = ΔJ64 |
| ΔJ42a |
6.090 ± 0.002 Hz |
ΔJ42a = ΔJ24a |
| ΔJ42b |
6.551 ± 0.003 Hz |
ΔJ42b = ΔJ24b |
| ΔJ45 |
6.645 ± 0.003 Hz |
ΔJ45 = ΔJ54 |
|
γ
δ,4
|
0.168 ± 0.001 ppm |
|
|
|
| Site 5 |
|
δ
iso,5
|
−112.504 ± 0.001 ppm |
|
| ΔJ56a |
5.462 ± 0.002 Hz |
|
| ΔJ56b |
6.402 ± 0.002 Hz |
|
| ΔJ53a |
6.550 ± 0.003 Hz |
|
| ΔJ54b |
6.645 ± 0.003 Hz |
|
|
γ
δ,5
|
0.166 ± 0.001 ppm |
|
|
|
| Site 6 |
|
δ
iso,6
|
−109.312 ± 0.001 ppm |
|
| ΔJ65a |
5.462 ± 0.002 Hz |
ΔJ65a = ΔJ56a |
| ΔJ64 |
5.718 ± 0.002 Hz |
|
| ΔJ65b |
6.402 ± 0.002 Hz |
ΔJ65b = ΔJ56b |
| ΔJ67 |
7.970 ± 0.002 Hz |
ΔJ67 = ΔJ76 |
|
γ
δ,6
|
0.175 ± 0.002 ppm |
|
|
|
| Site 7 |
|
δ
iso,5
|
−111.599 ± 0.003 ppm |
|
| ΔJ73 |
5.774 ± 0.003 Hz |
|
| ΔJ76 |
7.970 ± 0.002 Hz |
|
|
γ
δ,7
|
0.221 ± 0.004 ppm |
|
|
|
|
γ
J
|
0.190 ± 0.001 Hz |
|
|
A
|
2145 ± 6 |
|
|
r
|
−0.242 ± 0.007 |
|
In the case of ZSM-12, Cadars et al.14 only observed eight out of the thirteen 2JSi–O–Si couplings. Similar to Sigma-2, the reported values of Cadars et al.14 for ZSM-12 agree within 3 Hz of the values reported here. They report FWHM of ∼8 Hz in the refocused-INADEQUATE cross-sections, i.e., significantly larger than the FWHM of 0.4 Hz observed along the J-resolved dimension of the SE-PIETA spectrum of ZSM-12 in Fig. 4. Note, the FWHM of J-coupling peaks in SE-PIETA spectra of ZSM-12 is slightly broader compared to SE-PIETA spectra of Sigma-2. This is likely from the relatively shorter 29Si echo-train coherence lifetimes in ZSM-12 compared to Sigma-2. The 29Si site-specific lifetimes for ZSM-12 are provided in the ESI.†
3 Structure refinement
The local structural geometry around the 29Si affects the chemical shifts and couplings of 29Si nuclei. These NMR parameters can be used to refine zeolite framework structures, similar to how powder X-ray diffraction (PXRD) data is refined using Rietveld refinement. The optimized zeolite structure would be the one that best matches the experimentally measured NMR parameters. To achieve this, we need a way to calculate the 29Si chemical shifts and J-couplings for a given structure.
Regarding accuracy, the best way to calculate NMR parameters for a given structure is through ab initio calculations using density functional theory (DFT) methods. This can be done with clusters of sufficient size extracted from the zeolite network structure or the full crystal structure using periodic plane-wave DFT methods. These methods have been proven successful in calculating 29Si chemical shifts11,13,43–45 and 29Si J-couplings14,18 of pure silica network structures. In fact, a method for optimizing zeolite structures based on ab initio calculations of 29Si chemical shift tensors has been demonstrated.10,12 However, the main drawback of these ab initio calculations is the lengthy computation time (usually several hours) required to calculate NMR parameters for just one structure. This high computation cost makes it impractical for an iterative structure optimization routine.
To efficiently optimize zeolite structures, it is necessary to employ faster approaches for calculating 29Si chemical shifts and J-couplings without compromising accuracy. In this study, we utilized simple functions based on geometrical parameters such as bond lengths, bond angles, and torsion angles of Si-centered and O-centered clusters extracted from the zeolite framework (as shown in Fig. 7). Once these functions are parameterized, they can rapidly calculate the 29Si chemical shifts and J-couplings for a given structure with a reasonable level of accuracy. These calculations can be incorporated into an iterative structure optimization routine to identify the optimal structure that best matches the experimental NMR parameters.
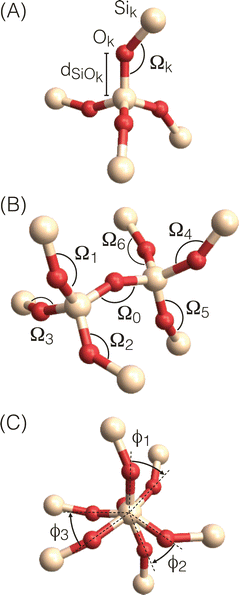 |
| | Fig. 7 Local geometry parameters for (A) Si-centered clusters used in calculations of isotropic 29Si chemical shifts (B) and (C) O-centered clusters used in calculations of 29Si–O–29Si J-couplings with (B) view perpendicular to central Si–O–Si linkage showing Si–O–Si bond angles and (C) view along central Si–O–Si linkage showing O–Si–Si–O torsion angles. | |
3.1 Rapid calculation of 29Si chemical shifts
The relationship between the isotropic 29Si chemical shift and the four Si–O–Si bond angles in Si-centered clusters has been recognized for some time.3,46 However, efforts to train model functions have been hampered by the scarcity of large datasets and the absence of high-quality single-crystal X-ray diffraction (SCXRD) structures of zeolites. Recently, Brouwer et al.47 augmented the International Zeolite Association's Database of Zeolite Structures48 with solid-state NMR data. They conducted an exhaustive review of the zeolite literature, gathering high-resolution 29Si solid-state NMR spectra and corresponding SCXRD-determined crystal structures of pure silica zeolites. This comprehensive dataset, the largest of its kind, established a robust relationship, described below, between the isotropic 29Si chemical shift and the local geometry of Si-centered clusters.
Fig. 7A shows a Si-centered cluster with the relevant Si–O distances and Si–O–Si bond angles labeled. The oxygen s-character for a given Si–Ok–Si linkage with angle Ωk is approximated as
| | 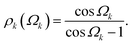 | (7) |
To account for the effect of bond length on the chemical shift, each oxygen s-character is modified by multiplying by
dSiOk,
i.e., the SiO
k bond length,
49| | | ρk(Ωk,dSiOk) = ρk(Ωk)dSiOk. | (8) |
The mean of the modified s-character of the four oxygens that surround Si is then calculated according to
| | 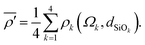 | (9) |
Using only the highest quality SCXRD structures of pure silica zeolites, Brouwer
et al.47 showed there is a strong linear correlation between
29Si chemical shifts and the average modified s-character parameter:
| | 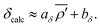 | (10) |
A linear least-squares analysis of
mδ = 142 unique Si sites from 22 SCXRD pure silica network structures (see ESI
† for raw data) determined that values of
aδ = −208.33 ppm Å
−1 and
bδ = 42.04 ppm minimized the sum of the squares of the residuals,
| | 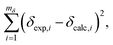 | (11) |
where
δexp,i are the experimentally measured
29Si chemical shifts.
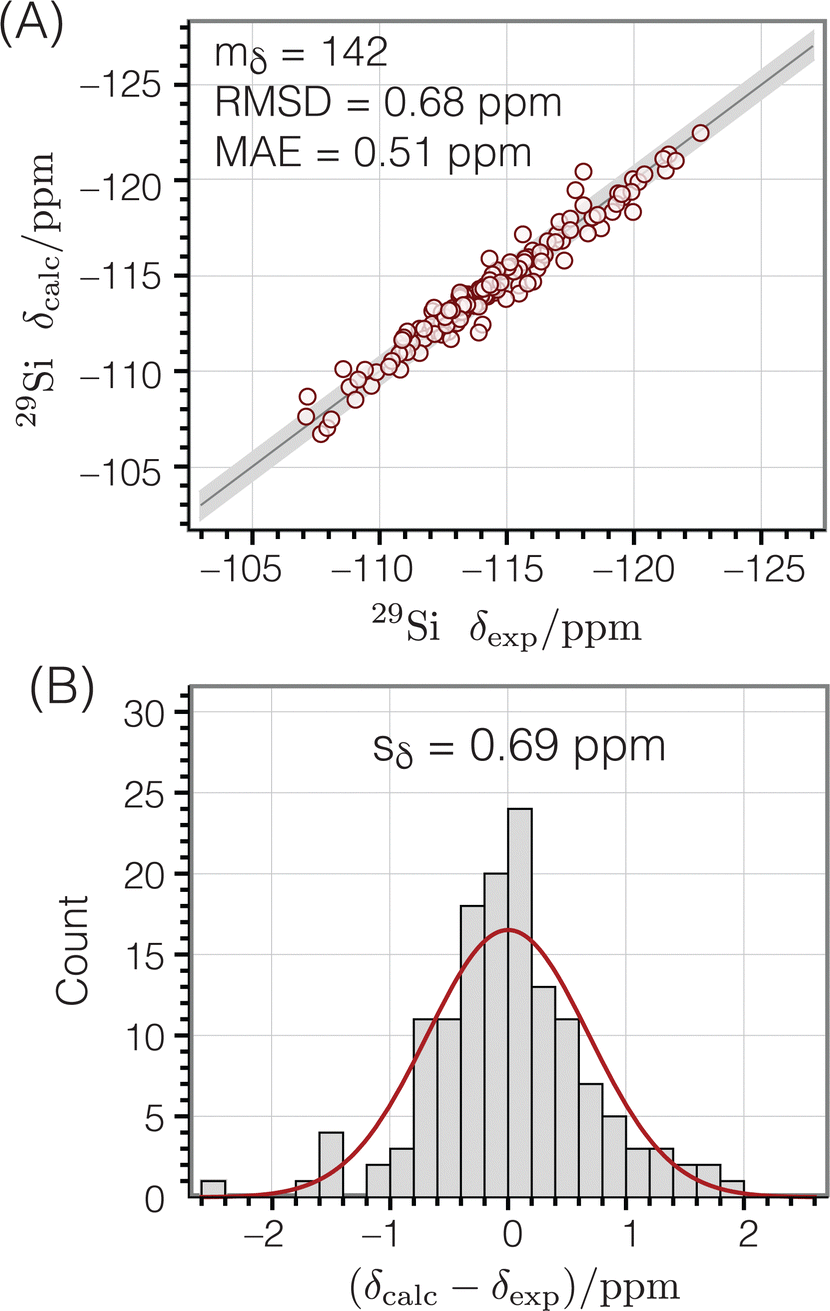
Fig. 8A shows the correlation between calculated and experimental chemical shifts, while
Fig. 8B displays a histogram of the residuals. The standard deviation of the residuals between calculated and experimental chemical shifts was calculated according to
| | 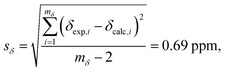 | (12) |
with
mδ − 2 degrees of freedom to account for the two parameters (
aδ and
bδ) used to parameterize
eqn (10). This standard deviation of 0.69 ppm is approximately 4% of the range of
29Si chemical shift values (−107 to −123 ppm).
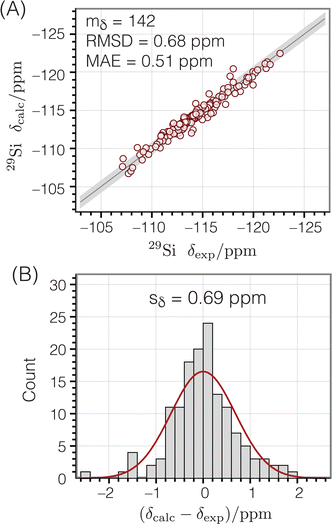 |
| | Fig. 8
29Si isotropic chemical shifts calculated from the geometries of Si-centered clusters using eqn (10) compared to corresponding experimental chemical shifts:47 (A) correlation plot (B) residual histogram plot. The root mean square deviation (RMSD) and mean absolute error (MAE) are also reported. The shaded bands show the calculated standard deviations between experimental and predicted isotropic chemical shifts. | |
3.2 Rapid calculation of 29Si–O–29Si J-couplings
It is known that the geminal 2JSi–O–Si couplings in silicate network materials are related in some manner to their corresponding Si–O–Si bond angles.14,18 Unfortunately, there does not exist a collection of experimental J-couplings that could be used to develop a model for rapidly calculating J-couplings from local geometry in the manner described above for 29Si isotropic chemical shifts. However, the relationship between 2JSi–O–Si-couplings and the local geometry of O-centered clusters has been investigated in detail by Srivastava et al.18 with ab initio DFT calculations. Through an extensive set of calculations of 2JSi–O–Si in O-centered clusters with varying geometries, they developed a robust model for calculating 2JSi–O–Si given by| | 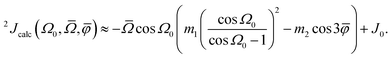 | (13) |
where Ω0 is the central Si–O–Si bond angle, ![[small phi variant, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d7.gif) is the mean of the three O–Si–Si–O torsional angles, φ1, φ2, and φ3, and
is the mean of the three O–Si–Si–O torsional angles, φ1, φ2, and φ3, and ![[capital Omega, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c0.gif) is the double mean of Si–O–Si angles around each Si atom in the cluster, given by
is the double mean of Si–O–Si angles around each Si atom in the cluster, given by| | 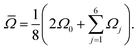 | (14) |
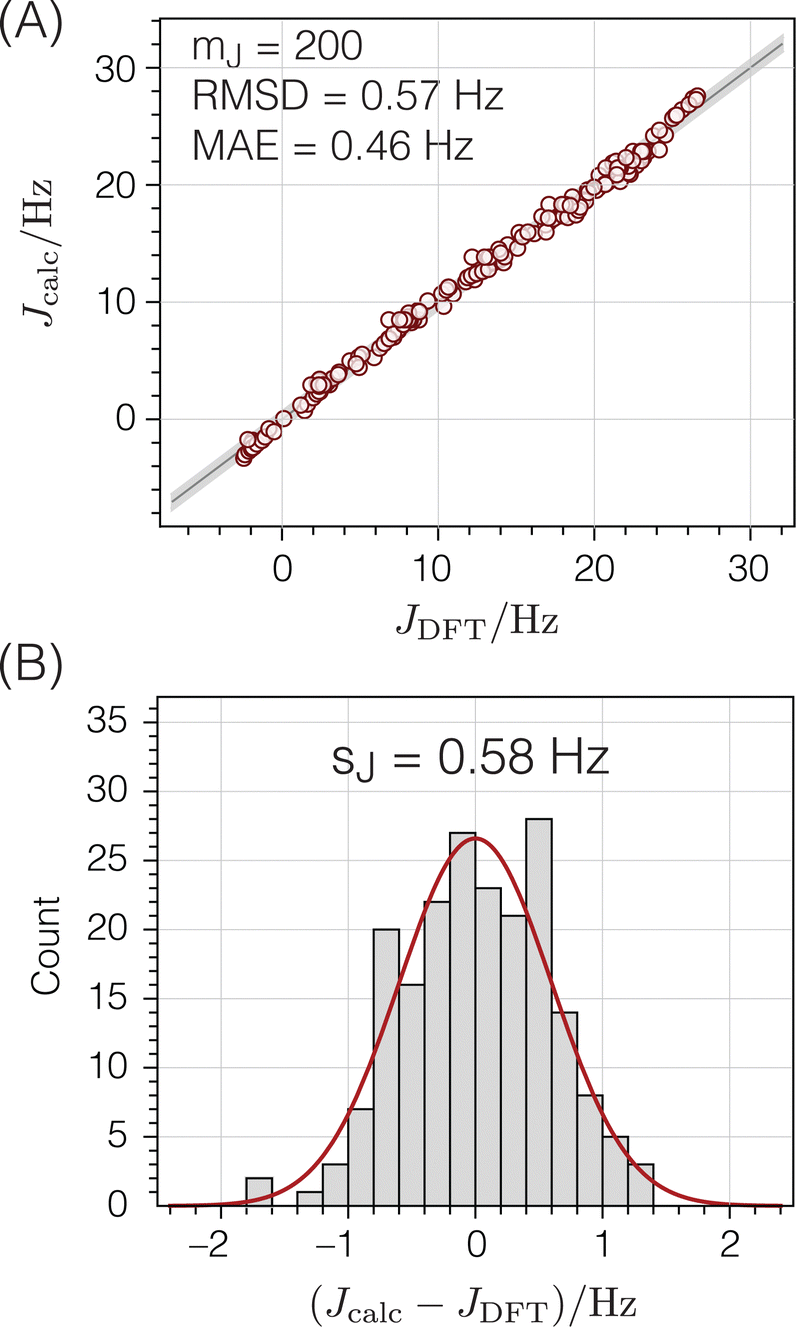
Fig. 7B and C show the relevant geometric parameters of an O-centered cluster extracted from a silicate network structure. The parameters m1, m2, and J0 are determined by fitting the calculated J-couplings to the ab initio DFT-calculated values. A linear least-squares analysis of the data set reported by Srivastava et al.18 with mJ = 200 unique O-centered clusters on which ab initio DFT-calculations had been performed (see ESI† for raw data) determined that values of m1 = 0.778 Hz/°, m2 = 0.00577 Hz/°, and J0 = −8.25 Hz minimized the sum of the squares of the residuals,| | 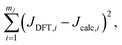 | (15) |
where JDFT,i are the ab initio DFT-calculated 29Si–O–29Si geminal J-couplings and Jcalc,i are the J-couplings calculated according to geometric model in eqn (13). Fig. 9A shows the correlation between the J-couplings calculated with the geometric model and ab initio DFT-calculated values, while Fig. 9B displays a histogram of the residuals. The standard deviation of the residuals between geometric and DFT calculated J-couplings was calculated according to| | 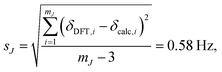 | (16) |
with mJ − 3 degrees of freedom to account for the three parameters (m1, m2, and J0) used to parameterize eqn (13). This standard deviation of 0.58 Hz is approximately 2% of the range of 29Si–O–29Si J-coupling values (−2.5 to 26.6 Hz).
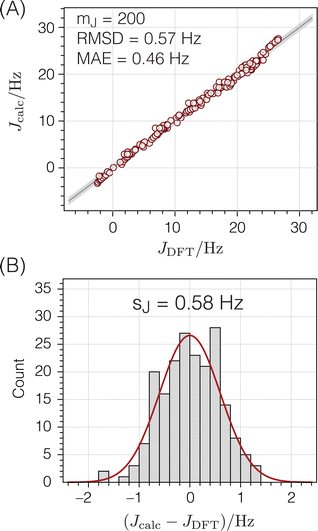 |
| | Fig. 9
2
J
Si–O–Si couplings calculated from the geometries of O-centered clusters using eqn (13) compared to corresponding ab initio DFT-calculated values:18 (A) correlation plot (B) residual histogram plot. The root mean square deviation (RMSD) and mean absolute error (MAE) are also reported. The shaded bands show the standard deviations between DFT and eqn (13) predicted 2JSi–O–Si couplings. | |
3.3 Expected geometry
The optimization method presented here aims to improve structures to achieve the best match between calculated and experimental NMR parameters (isotropic chemical shifts and J-couplings). It also takes into account the assumption that the optimized structures should be consistent with expected bond lengths and angles compared to other known structures for pure silica zeolites. Including this geometric information as a constraint ensures that the optimization process results in structures with realistic geometries.
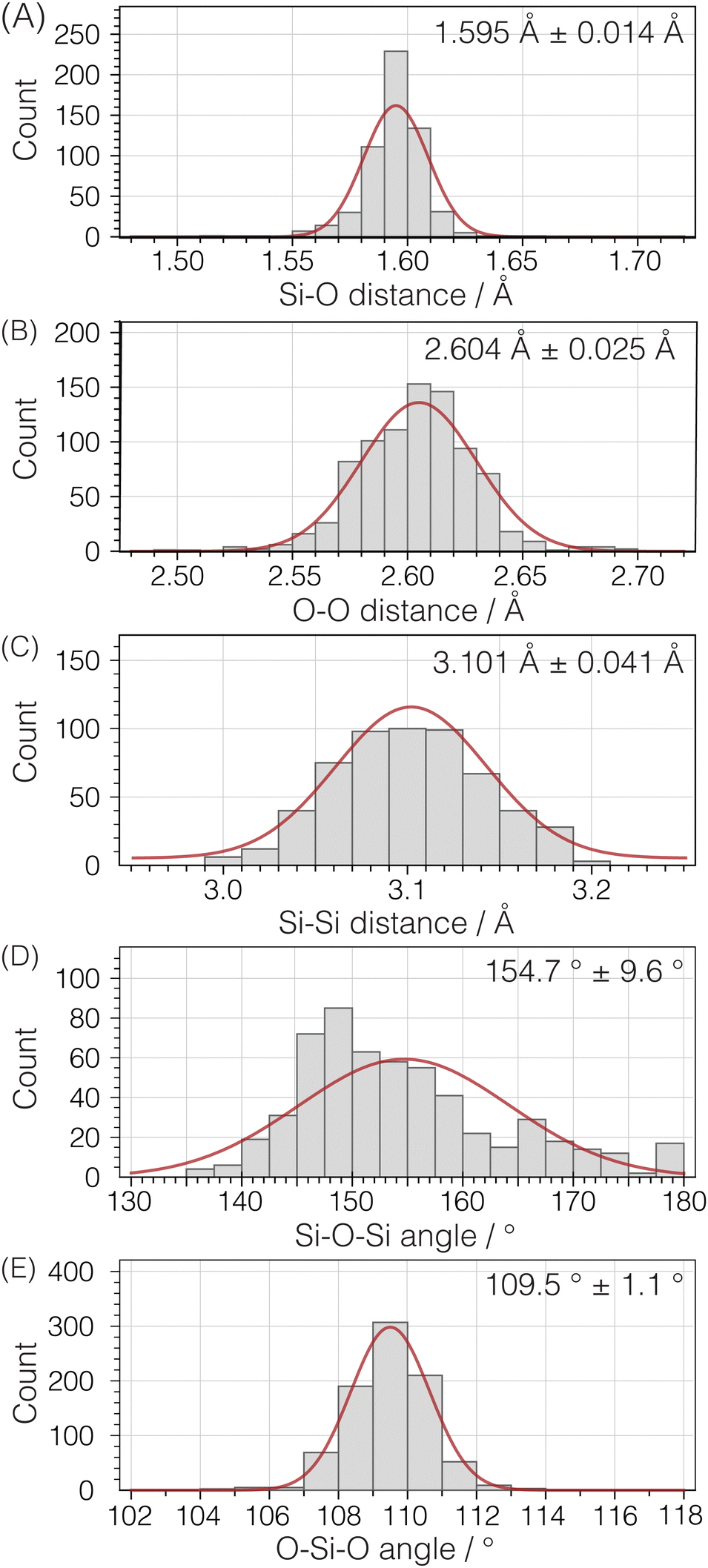
To parameterize the range of expected geometries, we examined the same set of pure silica structures used for calculating 29Si isotropic chemical shifts. This group consists of 142 unique Si atoms in 22 pure silica structures, encompassing zeolites and dense silica phases, with each structure determined by single crystal X-ray diffraction. Fig. 10 illustrates histograms for Si–O distances, O–O distances within a Si tetrahedron, and Si–Si distances between Si tetrahedra. Additionally, the distributions of inter-tetrahedral Si–O–Si angles and intra-tetrahedral O–Si–O bond angles are displayed despite not being directly integrated into the structure optimization.
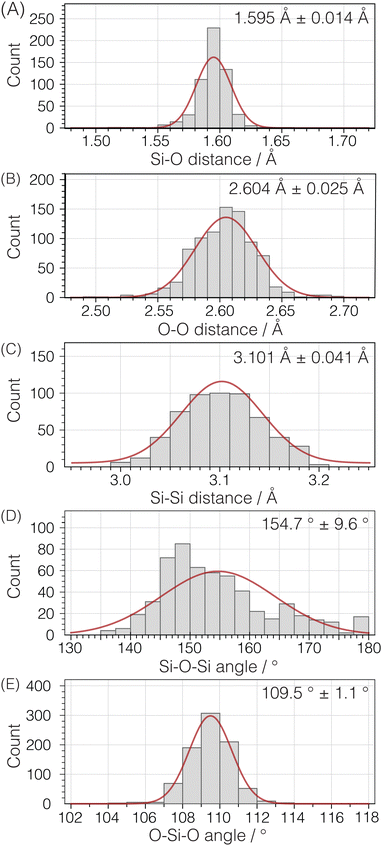 |
| | Fig. 10 Histogram plots and normal distribution curves for (A) Si–O, (B) intra-tetrahedral O–O, and (C) inter-tetrahedral Si–Si distances as well as (D) inter-tetrahedral Si–O–Si and (E) intra-tetrahedral O–Si–O bond angles. Analysis based on 142 unique Si sites in 22 crystal structures (see ESI† for raw data). | |
An analysis of the three distance histograms reveals that they each closely follow normal distributions. This analysis provides the target Si–O, O–O, and Si–Si distances, ![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) and associated standard deviations, s, for the expected geometries in pure silica network structures:
and associated standard deviations, s, for the expected geometries in pure silica network structures:
| | 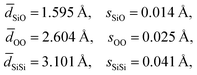 | (17) |
It should be noted that an optimization of the structure against just these target distances can be considered a “distances least squares” (DLS) refinement of a structure,
22,50 although the target values and associated standard deviations used here have been updated based on a large set of SCXRD structures compared to what is typically employed in a DLS optimization.
48
3.4 Cost function
To refine the structure, we must establish a cost function that quantifies the agreement with the relevant NMR parameters and expected distances for a given structure. This function is a weighted sum of the squares of the differences between target and calculated values for a given structure across the various types of information available, i.e.,| | | χ2(x) = χJ2(x) + χδ2(x) + χd2(x), | (18) |
where x = {x1, x2,…,xn} is the set of n crystallographically unique and adjustable Si and O fractional atomic coordinates being optimized. Here χJ2(x) quantifies how well the calculated J couplings agree with the set of mJ experimental J couplings,| | 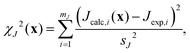 | (19) |
χδ2(x) quantifies how well the calculated chemical shifts agree with the set of mδ experimental chemical shifts,| | 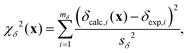 | (20) |
and χd2(x) quantifies how well the intra- and inter-tetrahedral distances agree with expected distances based on single-crystal XRD structures of silica materials, i.e.,| | | χd2(x) = χSiO2(x) + χOO2(x) + χSiSi2(x), | (21) |
where| | 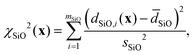 | (22) |
| | 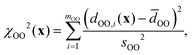 | (23) |
| | 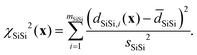 | (24) |
Here, SiO, OO, and SiSi are the target distances, and sSiO, sOO, and sSiSi are the associated standard deviations based on the analysis of single crystal XRD structures (see Fig. 10). For each distance type, mSiO, mOO, and mSiSi are the number of crystallography unique distances of that type in the structure. Good estimates of the standard deviations are essential to ensure that each observable or restraint is weighted appropriately and to prevent overfitting. Ideally, the reduced chi-square (χν2) value, which adjusts for degrees of freedom, should approximate 1, indicating a well-fitted model:| | 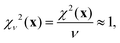 | (25) |
where ν = m − n is the degrees of freedom and m = mJ + mδ + mSiO + mOO + mSiSi is the number of observations.
3.5 Optimization method
In this study, structure optimization was carried out using the Gauss–Newton non-linear least-squares method,51 a technique detailed by Brouwer12 for refining zeolite frameworks based on 29Si chemical shift tensor components. This method was adopted here, with the primary modification being the cost function. Additionally, instead of relying on computationally demanding DFT calculations for chemical shielding tensors, this work utilized rapidly evaluated parameterized equations to calculate chemical shifts (eqn (10)) and J-couplings (eqn (13)), streamlining the optimization process.
The cost function in eqn (18) can be re-expressed as a sum of squared weighted residuals
| | 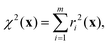 | (26) |
where
r = {
r1,
r2,…,
rm} is the set of
m weighted residuals
| | 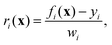 | (27) |
between observations
y = {
y1,
y2,…,
ym} (a concatenated list of
J-couplings, isotropic shifts, and target distances) and their corresponding calculated values
f(
x) = {
f1(
x),
f2(
x),…,
fm(
x)} for a given set of coordinates, with weights
w = {
w1,
w2,…,
wm} set to the standard deviations associated with each value.
At each iteration k of the optimization, a new set of atomic coordinates xk+1 is generated from the current values xk and a small adjustment Δx that leads to a decrease in the cost function:
The adjustment Δ
x =
αp is calculated from a step direction vector
p and a scalar value
α describing how far to step in this direction. The step direction vector
p is calculated from the Jacobian matrix
J according to
| | | p = (JTJ)−1·JT·r(xk), | (29) |
where
J is an
m ×
n matrix of partial derivatives
Jij = ∂
ri/∂
xj of the weighted residuals with respect to each atomic coordinate parameter. These partial derivatives were estimated numerically by calculating the change in the residuals for a small change in the value of the parameters
| | 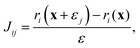 | (30) |
where
εj is a vector of zeros except for the
jth element which is a small amount
ε (set to 10
−6) such that
| | | x + εj = {x1,…,xj + ε,…,xn}. | (31) |
Once the step direction vector
p is determined, a line search is performed along this direction to determine the step length parameter
α, leading to a sufficient decrease in
χ2(
x). Details of the line search process can be found in ref.
12 and
51.
These refinement steps were iterated until the changes in all fractional atomic coordinates were less than 10−6, and the change in the cost function was less than 10−5. Convergence was achieved in about ten iterations (approximately 0.5 seconds per iteration) for Sigma-2 and 35 iterations (approximately 2 seconds per iteration) for ZSM-12.
3.6 Estimation of uncertainties
The uncertainties of the optimized fractional atomic coordinates were estimated using a Monte Carlo approach. The optimizations were repeated numerous times, and different random values selected from normal distributions governed by the associated standard deviations were added to the cost function calculation. For example, the geometric model for calculating J-couplings was parameterized with an associated uncertainty of sJ = 0.57 Hz. To incorporate this uncertainty into the optimization, the χJ2(x) contribution to the overall cost function was modified to be| | 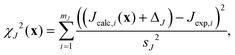 | (32) |
where ΔJ is a random number selected from a normal distribution with zero mean and standard deviation sJ. A similar modification was made for the χδ2(x) chemical shift contribution to the cost function, with a random number Δδ selected from a normal distribution with standard deviation sδ.
The contributions to the cost function from distances were modified similarly. For example, the Si–O distances have an associated standard deviation of sSiO = 0.014 Å. To incorporate this distribution of expected distances into the optimization, the χSiO2(x) contribution to the cost function was modified to be
| | 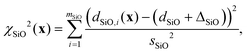 | (33) |
where Δ
SiO is a random number selected from a normal distribution with zero mean and standard deviation
sSiO. Similar modifications were made for the
χOO2(
x) and
χSiSi2(
x) contributions to the cost function.
The optimization of each zeolite framework was repeated 100 times with different values of ΔJ, Δδ, ΔSiO, ΔOO, and ΔSiSi values, leading to 100 optimized structures with distributions of fractional atomic coordinates. For each coordinate, the standard deviation of these 100 values was calculated and used to estimate its uncertainty.
4 Results and discussion
4.1 Sigma-2
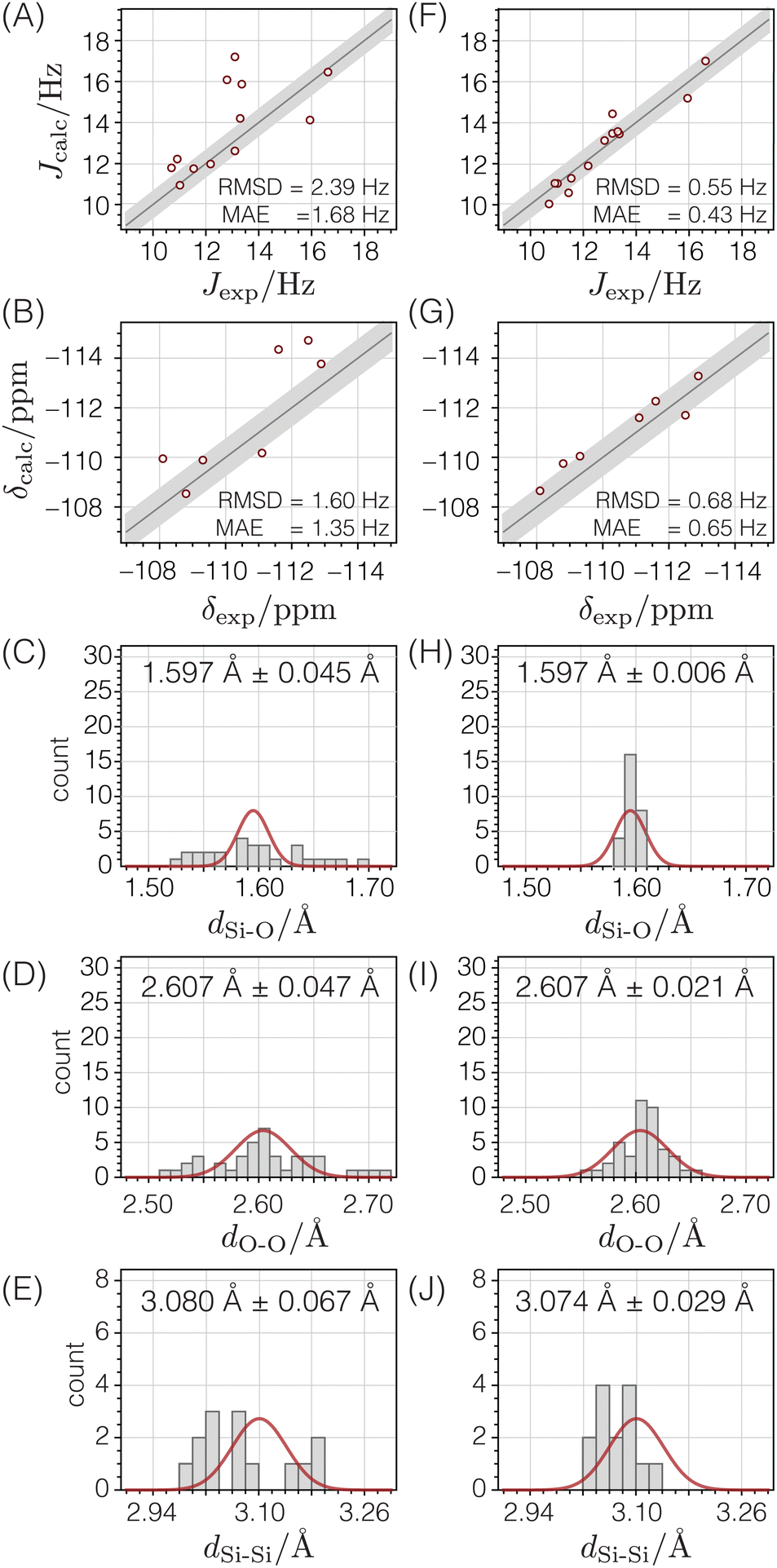
We first carried out a structure optimization of the zeolite Sigma-2 to test the validity of this structure optimization approach. The existence of a high-quality SCXRD structure for Sigma-211 provides a reference against which the accuracy of the optimized structure can be evaluated. In the spirit of demonstrating how this structure optimization strategy fits within a broader NMR crystallography approach, our starting structure here was the set of Si atomic coordinates for Sigma-2 that were solved from 29Si double-quantum dipolar recoupling NMR experiments,8,10 following the structure-solution strategy outlined in ref. 9. Oxygen atoms were added midway between Si atoms known to be connected through Si–O–Si linkages and an optimization of only the oxygen coordinates against target Si–O, O–O, and Si–Si distances was first performed to ensure that the local environments around the Si atoms were approximately tetrahedral.
The first column in Fig. 11 shows that the quality of agreement between calculated and experimental isotropic chemical shifts and J couplings for this initial structure derived from the 29Si dipolar recoupling experiments is poor. Additionally, while the Si–O and Si–Si distances are quite reasonable, there are several O–O distances that are out of the expected range.
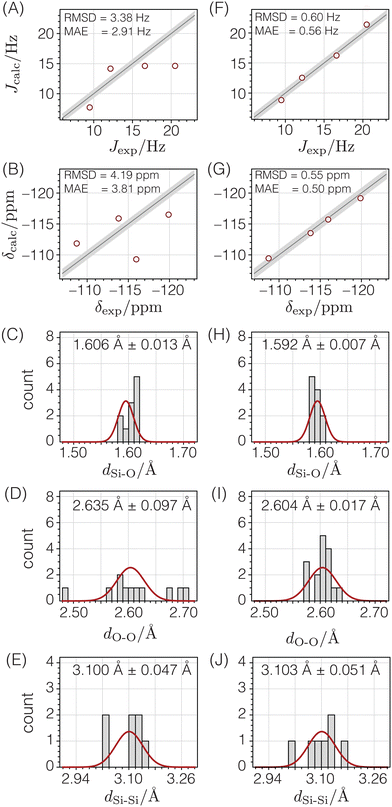 |
| | Fig. 11 Comparison of NMR and geometry parameters for Sigma-2 before (A)–(E) and after (F)–(J) structure optimization. The quality of agreement between calculated and experimental J-couplings (A) and (F) and isotropic chemical shifts (B) and (G) are shown as correlation plots in which the straight lines represent perfect agreement, and the shaded bands (±sJ or ±sδ) show the expected standard deviations. The quality of agreement between expected distributions (solid red lines) of Si–O distances (C) and (H), O–O distances (D) and (I), and Si–Si distances (E) and (J) are shown as histograms. | |
To improve this structure, all atomic coordinates were optimized against distances, then against distances and chemical shifts, and finally against distances, chemical shifts, and J couplings. The ESI† reports additional details of these optimizations, as well as the final optimized atomic coordinates and their estimated uncertainties.
After optimizing the structure against distances, chemical shifts, and J couplings (see second column in Fig. 11), the distances become consistent with expected distances, and the agreement between calculated and experimental chemical shifts and J couplings is dramatically improved. From a statistical perspective, it is satisfying that the resulting RMSD values are similar to the standard deviations determined in the parameterization stage with a goodness of fit parameter of χν2 = 1.19.
The accuracy of this NMR-solved and NMR-optimized structure was evaluated by comparing it to the reported SCXRD structure for Sigma-2.11Fig. 12A displays the deviations between each unique atom in the optimized structure compared to the SCXRD structure. The average displacement of the silicon atoms in the optimized structure from the SCXRD structure is only 0.012 Å, while the oxygen atoms differ by an average of only 0.020 Å. Fig. 12B displays the magnitudes of the differences of the x, y, and z components of the atomic coordinates between the two structures relative to the associated uncertainties for the optimized structure. Most of the optimized atomic coordinates are within one multiple of their uncertainty in comparison to the SCXRD structure, except for the O4 coordinates and one of the O6 components. This suggests that, for the most part, the optimized Sigma-2 structure is not statistically different from the SCXRD structure, perhaps except for the O4 atomic coordinates.
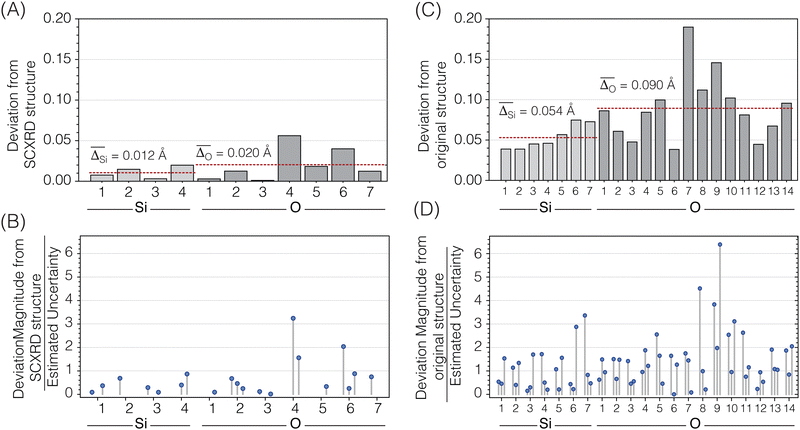 |
| | Fig. 12 (A) Atom-by-atom distance differences between the atomic coordinates in the optimized Sigma-2 structure and the single-crystal XRD structure of Sigma-2. (B) Magnitudes of the differences between the x, y, and z fractional atomic coordinates in the optimized structure and the SCXRD structure of Sigma-2 divided by the estimated uncertainty of the optimized structure coordinates. (C) Atom-by-atom distance differences between the atomic coordinates in the optimized ZSM-12 structure and the original powder XRD structure of ZSM-12. (D) Magnitudes of the differences between the x, y, and z fractional atomic coordinates in the optimized structure and the original PXRD structure of ZSM-12 divided by the estimated uncertainty of the optimized structure coordinates. The coordinate components fixed by symmetry are not displayed. | |
4.2 ZSM-12
We optimized the structure of ZSM-12, starting from a structure determined from synchrotron powder XRD data.52 As the first column in Fig. 13 shows, the agreement between calculated and experimental chemical shifts and J-couplings was poor for this initial structure. The structure also has several Si–O and O–O distances outside the expected range, suggesting that the reported PXRD structure of ZSM-12 is not all that accurate and would benefit from the improvement that an optimization against NMR data potentially provides.
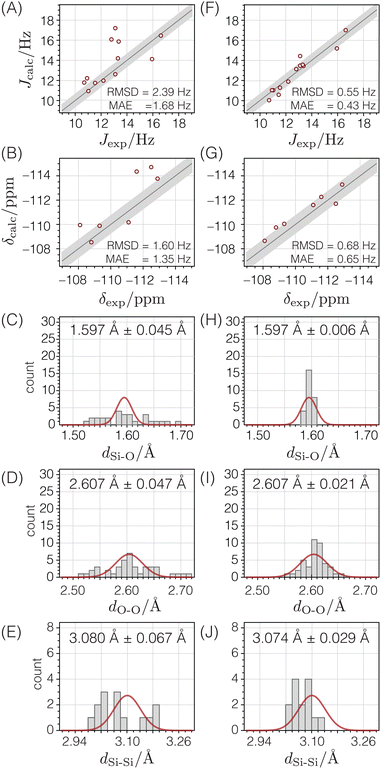 |
| | Fig. 13 Comparison of NMR and geometry parameters for ZSM-12 before (A)–(E) and after (F)–(J) structure optimization. The quality of agreement between calculated and experimental J-couplings (A) and (F) and isotropic chemical shifts (B) and (G) are shown as correlation plots in which the straight lines represent perfect agreement. The shaded bands (±sJ or ±sδ) show the expected standard deviations. The quality of agreement between expected distributions (solid red lines) of Si–O distances (C) and (H), O–O distances (D) and (I), and Si–Si distances (E) and (J) are shown as histograms. | |
Starting from the reported PXRD structure,52 the structure underwent optimization based on distances, followed by optimization involving distances and chemical shifts, and ultimately, optimization involving distances, chemical shifts, and J-couplings. The ESI† provides additional details of these optimizations, as well as the final optimized atomic coordinates and their estimated uncertainties.
After optimizing the structure based on distances, chemical shifts, and J-couplings, there is improved agreement between the calculated and experimental chemical shifts and J-couplings, as shown in the second column of Fig. 13. The Si–O and O–O distances are now within their expected ranges. The RMSD values are similar to the standard deviations determined in the parameterization stage, and the goodness of fit parameter χν2 = 1.61.
Fig. 12C displays how far each atom moved before and after the structure optimization. The average displacement of silicon atoms was 0.054 Å and the oxygen atoms was 0.090 Å. Fig. 12D displays how much the x, y, or z components of the atomic coordinates changed relative to their associated uncertainties. In this case, only about half of the atomic coordinates are within one multiple of their standard deviation, and many are greater than a factor two times their standard deviation, with one coordinate six times its standard deviation. This suggests that the optimized ZSM-12 structure significantly differs from the original powder XRD structure. Based on the improvements shown in Fig. 13, we are confident that this optimization against chemical shifts, J-couplings, and distances has led to a more accurate crystal structure for ZSM-12.
5. Conclusions
This study represents a significant advance in the structural refinement of highly siliceous zeolites using 29Si 2D J-resolved NMR spectroscopy. We have developed a modified shifted-echo PIETA pulse sequence using a t1 interleaving scheme to acquire high-resolution 2D J spectra in crystalline materials. This approach eliminates an additional artifact associated with short inter-echo periods, 2τ, on the J modulation frequency during the echo train acquisition.53 In this work, we demonstrate its application of both shifted-echo PIETA and shifted-echo PIETA with the t1 interleaving scheme in measuring the 2JSi–O–Si couplings in two highly siliceous zeolites, Sigma-2 and ZSM-12, at 29Si natural abundance where the 29Si–O–29Si linkage abundance is 0.756%.
The applicability of this approach requires site resolution along the chemical shift dimension, which can become more challenging as the number of crystallographic sites in the zeolite framework increases. Additionally, this approach requires all observable J splitting to be near the weak coupling limit. In the case of Sigma-2 and ZSM-12, no significant issues with deviations from the weak coupling limit were encountered at the modest magnetic field strength of 9.4 T used in this study. However, for other samples, particularly those with smaller chemical shift differences and a greater number of crystallographic sites, higher magnetic field strengths may be necessary to obtain better site resolution in the chemical shift dimension and to ensure that all J-couplings are near the weak-coupling limit.
The acquired spectra facilitated the introduction of a new silicate framework structure refinement strategy. This strategy integrates Si–O, O–O, and Si–Si distance restraints with analytical relationships derived from 29Si chemical shifts and geminal 2JSi–O–Si couplings. The application of this method to the ZSM-12 zeolite resulted in a refined structure that showed substantial improvements in the accuracy of Si–O and O–O distances, and a better fit between calculated and experimental chemical shifts and J-couplings. These refinements underscore the potential of 29Si 2D J-resolved NMR spectroscopy as a powerful tool for elucidating the detailed structures of siliceous zeolites.
While J-based refinements currently suffer from a lack of available data, it is anticipated that the SE-PIETA method and the ease at which refinements can be performed will increase available data, which may further strengthen the models used for refinement.
Data availability
The experimental raw Bruker NMR datasets and CSDM-compliant54 datasets, the Bruker pulse sequences, the Python notebook for denoising the 2D J-resolved spectrum of ZSM-12, the Mathematica notebook for performing the optimization of the structure of Sigma-2 and ZSM-12, the raw data used in the optimizations, and the CIF files with the optimized atomic coordinates are openly available in Zenodo at https://doi.org/10.5281/zenodo.10053064, ref. 55.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This material is based upon work supported by the Chemical Measurement and Imaging program in the National Science Foundation Division of Chemistry under Grant No. CHE-2107636 (with partial co-funding from the Ceramics program in the Division of Materials Research) as well as support from the Natural Sciences and Engineering Research Council (NSERC) of Canada in the form of a Discovery Grant (funding reference number 03836).
Notes and references
- Y. Li, L. Li and J. Yu, Chem, 2017, 3, 928–949 CAS.
- N. Jiang, R. Shang, S. G. Heijman and L. C. Rietveld, Water Res., 2018, 144, 145–161 CrossRef CAS.
- G. Engelhardt and R. Radeglia, Chem. Phys. Lett., 1984, 108, 271–274 CrossRef CAS.
- N. Janes and E. Oldfield, J. Am. Chem. Soc., 1985, 107, 6769 CrossRef CAS.
-
G. Engelhardt and D. Michel, High-resolution solid-state NMR of silicates and zeolites, John Wiley & Sons, Chichester, 1987 Search PubMed.
-
C. A. Fyfe, Solid-State NMR for Chemists, C.F.C. Press, Guelph, 1983 Search PubMed.
-
K. J. D. Mackenzie and M. E. Smith, Multinuclear Solid-State NMR of Inorganic Materials, Pergamon, 2002 Search PubMed.
- D. H. Brouwer, P. E. Kristiansen, C. A. Fyfe and M. H. Levitt, J. Am. Chem. Soc., 2005, 127, 542–543 CrossRef CAS.
- D. H. Brouwer, R. J. Darton, R. E. Morris and M. H. Levitt, J. Am. Chem. Soc., 2005, 127, 10365–10370 CrossRef CAS.
- D. H. Brouwer, J. Am. Chem. Soc., 2008, 130, 6306–6307 CrossRef CAS.
- D. H. Brouwer and G. D. Enright, J. Am. Chem. Soc., 2008, 130, 3095–3105 CrossRef CAS.
- D. H. Brouwer, J. Magn. Reson., 2008, 194, 136–146 CrossRef CAS PubMed.
- L. M. Bull, B. Bussemer, T. Anupold, A. Reinhold, A. Samoson, J. Sauer, A. K. Cheetham and R. Dupree, J. Am. Chem. Soc., 2000, 122, 4948–4958 CrossRef CAS.
- S. Cadars, D. H. Brouwer and B. F. Chmelka, Phys. Chem. Chem. Phys., 2009, 11, 1825–1837 RSC.
- P. J. Grandinetti, J. H. Baltisberger, U. Werner, A. Pines, I. Farnan and J. F. Stebbins, J. Phys. Chem., 1995, 99, 12341–12348 CrossRef CAS.
- T. M. Clark and P. J. Grandinetti, J. Phys.: Condens. Matter, 2003, 15, S2387–S2395 CrossRef CAS.
- P. Florian, F. Fayon and D. Massiot, J. Phys. Chem. C, 2009, 113, 2562–2572 CrossRef CAS.
- D. J. Srivastava, P. Florian, J. H. Baltisberger and P. J. Grandinetti, Phys. Chem. Chem. Phys., 2018, 20, 562–571 RSC.
- L. Frydman and J. S. Harwood, J. Am. Chem. Soc., 1995, 117, 5367–5369 CrossRef CAS.
- L. McCusker, J. Appl. Crystallogr., 1988, 21, 305–310 CrossRef CAS.
- R. LaPierre, A. Rohrman, J. Schlenker, J. Wood, M. Rubin and W. Rohrbaugh, Zeolites, 1985, 5, 346–348 CrossRef CAS.
- W. M. Meier and H. Villiger, Z. Kristallogr., 1969, 129, 411–423 CrossRef CAS.
- W. H. Baur, Phys. Chem. Miner., 1977, 2, 3–20 CrossRef CAS.
- D. H. Brouwer and K. P. Langendoen, CrystEngComm, 2013, 15, 8748–8762 RSC.
- M. H. D. H. Brouwer, Solid State Nucl. Magn. Reson., 2015, 65, 89–98 CrossRef PubMed.
- D. Brouwer and J. V. Huizen, Magn. Reson. Chem., 2019, 57, 167–175 CrossRef CAS PubMed.
- E. L. Hahn, Phys. Rev., 1950, 80, 580–594 CrossRef.
- A. Bax, R. Freeman and T. A. Frenkiel, J. Am. Chem. Soc., 1981, 103, 2102–2104 CrossRef CAS.
- R. Freeman, T. Frenkiel and M. B. Rubin, J. Am. Chem. Soc., 1982, 104, 5545–5547 CrossRef CAS.
- A. Bax, R. Freeman and S. P. Kempsell, J. Am. Chem. Soc., 1980, 102, 4849–4851 CrossRef CAS.
- C. A. Fyfe, H. Gies, Y. Feng and G. T. Kokotailo, Nature, 1989, 341, 223–225 CrossRef CAS.
- A. Lesage, M. Bardet and L. Emsley, J. Am. Chem. Soc., 1999, 121, 10987–10993 CrossRef CAS.
- D. H. Brouwer, J. Magn. Reson., 2003, 164, 10–18 CrossRef CAS PubMed.
- E. Kupče and R. Freeman, J. Am. Chem. Soc., 2008, 130, 10788–10792 CrossRef.
- G. Bodenhausen, R. Freeman, G. A. Morris and D. L. Turner, J. Magn. Reson., 1978, 31, 75–95 CAS.
- J. H. Baltisberger, B. J. Walder, E. G. Keeler, D. C. Kaseman, K. J. Sanders and P. J. Grandinetti, J. Chem. Phys., 2012, 136, 211104 CrossRef PubMed.
- S. P. Brown, M. Pérez-Torralba, D. Sanz, R. M. Claramunt and L. Emsley, Chem. Commun., 2002, 1852–1853 RSC.
- L. Duma, W. C. Lai, M. Carravetta, L. Emsley, S. P. Brown and M. H. Levitt, ChemPhysChem, 2004, 5, 815–833 CrossRef CAS PubMed.
- D. Srivastava, J. Baltisberger, P. Florian, F. Fayon, R. Shakhovoy, M. Deschamps, N. Sadiki and P. Grandinetti, Phys. Rev. B, 2018, 98, 134202 CrossRef CAS.
-
D. Srivastava, PhD thesis, The Ohio State University, 2018.
- PhySy Ltd., RMN 2.0, 2019, https://www.physyapps.com/rmn.
- Z. H. Gan, J. Chem. Phys., 2001, 114, 10845–10853 CrossRef CAS.
- X. Xue and M. Kanzaki, Solid State Nucl. Magn. Reson., 2000, 16, 245–259 CrossRef CAS.
- D. H. Brouwer, I. L. Moudrakovski, R. J. Darton and R. E. Morris, Magn. Reson. Chem., 2010, 48, S113–S121 CrossRef CAS.
- D. M. Dawson, R. F. Moran and S. E. Ashbrook, J. Phys. Chem. C, 2017, 121, 15198–15210 CrossRef CAS.
- J. Thomas, J. Klinowski, S. Ramdas, B. Hunter and D. Tennakoon, Chem. Phys. Lett., 1983, 102, 158–162 CrossRef CAS.
- D. H. Brouwer, C. C. Brouwer, S. Mesa, C. A. Semelhago, E. E. Steckley, M. P. Sun, J. G. Mikolajewski and C. Baerlocher, Microporous Mesoporous Mater., 2020, 297, 110000 CrossRef CAS.
-
C. Baerlocher and L. McCusker, Database of Zeolite Structures, 2017 Search PubMed.
- J. R. Lewis, C. C. Freyhardt and M. E. Davis, J. Phys. Chem., 1996, 100, 5039–5049 CrossRef CAS.
-
C. Baerlocher, A. Hepp and W. M. Meier, DLS-76, a program for the simulation of crystal structures by geometric refinement, Zürich, 1978 Search PubMed.
-
S. J. Wright and J. Nocedal, Numerical Optimization, Springer, New York, 2006, p. 664 Search PubMed.
- C. A. Fyfe, H. Gies, G. T. Kokotailo, B. Marler and D. E. Cox, J. Phys. Chem., 1990, 94, 3718–3721 CrossRef CAS.
- A. Allerhand, J. Chem. Phys., 1966, 44, 1 CrossRef CAS.
- D. J. Srivastava, T. Vosegaard, D. Massiot and P. J. Grandinetti, PLoS One, 2020, 15, e0225953 CrossRef CAS PubMed.
-
D. Srivastava, M. C. Venetos, L. McCarthy-Carney, J. H. Baltisberger, P. J. Grandinetti and D. Brouwer, Refining siliceous zeolite framework structures with 29Si 2D J-resolved NMR Spectroscopy, 2024 DOI:10.5281/zenodo.10053064.
Footnote |
| † Electronic supplementary information (ESI) available: Additional details on the design of the shifted-echo PIETA pulse sequence and its implementation on the Bruker spectrometer, the processing of the experimental spectra with the RMN software, the Python notebook for denoising the 2D J-resolved spectrum of ZSM-12, the Mathematica notebook for performing the optimization of the structure of Sigma-2 and ZSM-12, and the results of the optimizations. The raw data used in the optimizations, the optimized atomic coordinates, and the estimated uncertainties of the atomic coordinates. See DOI: https://doi.org/10.1039/d4cp03530e |
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Maxwell C.
Venetos
a,
Maxwell C.
Venetos
 , and the dihedral angle of the QSi4–QSi4 cluster, φ.
, and the dihedral angle of the QSi4–QSi4 cluster, φ.
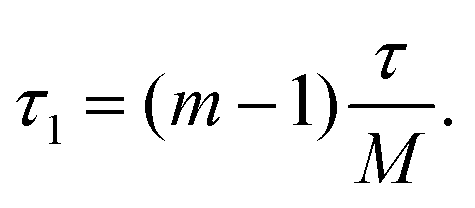
 values of transitions remain invariant under the π pulse, there will always be an echo of
values of transitions remain invariant under the π pulse, there will always be an echo of 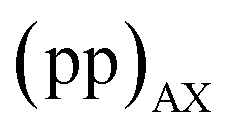 symmetry at the end of 4τ period regardless of the duration of τ1. On the contrary, the echoes from the
symmetry at the end of 4τ period regardless of the duration of τ1. On the contrary, the echoes from the 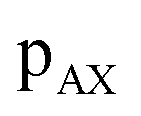 symmetries will move forward in time by the duration of 2τ1. Note, for m = 1 or τ1 = 0, the pulse sequence in Fig. 2 is the same as the shifted-echo PIETA sequence.39
symmetries will move forward in time by the duration of 2τ1. Note, for m = 1 or τ1 = 0, the pulse sequence in Fig. 2 is the same as the shifted-echo PIETA sequence.39
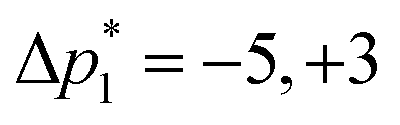 and
and 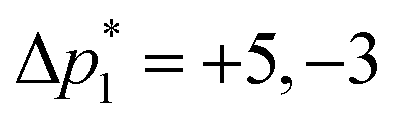 , respectively. After zeroing the signal in the undesired (Δp1, Δp2) coordinate, the 5D signal, s(Δp2,m,Δp1,n,t2), is projected down to the 3D signal s(m,n,t2), followed by a Fourier transform with respect to t2 to obtain s(m,n,ω2). An interactive zeroth and first-order phase correction is applied along the ω2 dimension of s(m,n,ω2) to obtain a pure absorption mode spectrum of the first “t1 = 0” echo, i.e., s(m,n = 1,ω2). As the J-resolved dimension is symmetric about zero, there should be little to no signal remaining in the imaginary part of the signal. However, an additional zeroth-order phase correction may need to be applied only to the even echoes to reduce the signal in the imaginary part if intermediate couplings are present.
, respectively. After zeroing the signal in the undesired (Δp1, Δp2) coordinate, the 5D signal, s(Δp2,m,Δp1,n,t2), is projected down to the 3D signal s(m,n,t2), followed by a Fourier transform with respect to t2 to obtain s(m,n,ω2). An interactive zeroth and first-order phase correction is applied along the ω2 dimension of s(m,n,ω2) to obtain a pure absorption mode spectrum of the first “t1 = 0” echo, i.e., s(m,n = 1,ω2). As the J-resolved dimension is symmetric about zero, there should be little to no signal remaining in the imaginary part of the signal. However, an additional zeroth-order phase correction may need to be applied only to the even echoes to reduce the signal in the imaginary part if intermediate couplings are present.