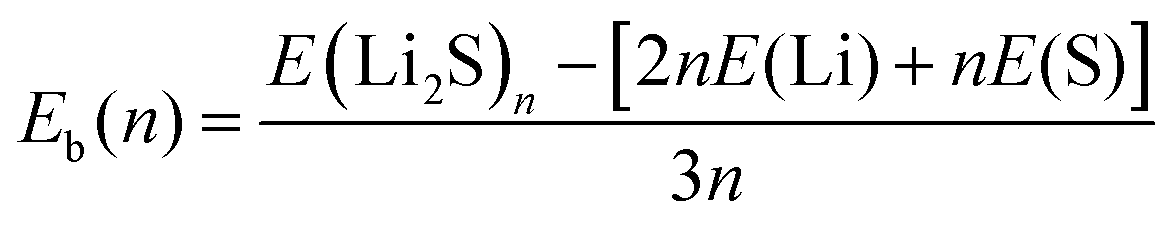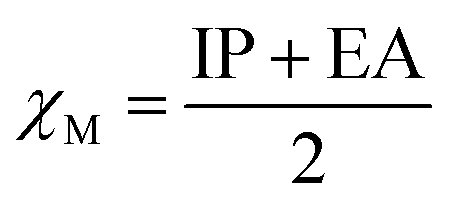Structure and property exploration of two-dimensional, bulk, and cluster lithium sulfide using the IM2ODE method†
Received
16th September 2024
, Accepted 28th November 2024
First published on 29th November 2024
Abstract
Lithium sulfide (Li2S) plays an important role in fields such as energy, environment and semiconductors. Exploration of the microstructure of Li2S has significant implications for developing new materials and optimizing related material properties. In this work, the inverse design of materials by the multi-objective differential evolution (IM2ODE) method combined with density functional theory (DFT) calculations was used to predict the two-dimensional (2D), three-dimensional (3D), and cluster structures of Li2S. Their structural stabilities and electronic properties were further investigated. Novel monolayer and double-layer hexagonal structures of 2D Li2S are predicted. The double-layer structure has better thermal stability and a wider band gap of 3.5 eV than the single-layer structure. Various novel structures of 3D Li2S are predicted. Some structures are similar to 1T-MoS2 and the double-layer hexagonal structure of 2D Li2S. With increasing number of atoms, the (Li2S)n clusters converge into a cage-like structure and their average binding energies decrease. The second-order energy differences of (Li2S)n clusters show an odd–even oscillation rule. The ionization potentials, electron affinities, electronegativities, and chemical hardnesses also decrease. These findings should improve theoretical understanding of the properties and behavior of new 2D, 3D, and cluster functional materials.
1. Introduction
As a typical alkali metal sulfide with a wide band gap, lithium sulfide (Li2S) has received much attention in recent years.1–3 It has been widely used in ceramics, semiconductors and energy materials.4–6 Li2S can serve as a cathode material with a theoretical capacity of 1166 mA h g−1 for lithium–sulfur (Li–S) batteries.7 However, certain deficiencies, such as low conductivity, a shuttle effect, and volume expansion, limit its performance and practical applications.8–13 Nanoscale Li2S has unique advantages and has facilitated many advances.14,15 Since the discovery of graphene, remarkable progress has been made in the field of two-dimensional (2D) materials, which has led to a wide range of applications in several fields.16–21 The structures of (Li2S)n clusters have also been reported.22 These can be used as electrolyte and cathode materials to achieve high energy density and long life for Li-ion or Li–S batteries.23–25
Focusing on the prediction of lithium sulfide material structures, this study aims to explore and reveal the effectiveness and feasibility of these new structures in enhancing the overall performance of lithium–sulfur batteries. Through a combination of the inverse design of materials by the multi-objective differential evolution (IM2ODE) method and density functional theory (DFT) calculations, novel two-dimensional (2D), three-dimensional (3D), and cluster structures of Li2S were predicted. Their stabilities and electronic properties were investigated. The discovery of new Li2S structures may deepen our understanding of the essential properties and behavior of new functional materials.
2. Computational details
IM2ODE is a structural search method based on a multi-objective differential evolutionary algorithm, which is capable of searching for structures with specific properties.26 To date, IM2ODE software has demonstrated powerful search and optimization capabilities, being applied to Aun and TiO2.27,28 Here, IM2ODE was used to perform structural searches for 2D, 3D, and clusters of Li2S composed of Li and S in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry. For the structure search of 2D Li2S, the thickness of the vacuum layer was set to 15.0 Å and the projected area was set to 20.0 Å2. For the structural search of 3D Li2S, the box volume and the number of atoms were correspondingly increased. For the cluster search, the ball mode was used. With increasing number of (Li2S)n cluster atoms, both the initial box volume and the cluster radius were increased appropriately.
:1 stoichiometry. For the structure search of 2D Li2S, the thickness of the vacuum layer was set to 15.0 Å and the projected area was set to 20.0 Å2. For the structural search of 3D Li2S, the box volume and the number of atoms were correspondingly increased. For the cluster search, the ball mode was used. With increasing number of (Li2S)n cluster atoms, both the initial box volume and the cluster radius were increased appropriately.
Structure optimization and electronic property calculations for all predicted Li2S structures were further performed using the Vienna ab initio simulation package (VASP) and DS-PAW, which are based on DFT.29,30 The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional was adopted.31 The DFT-D3 method was used for dispersion correction.32,33 The convergence criteria of the total energy and final force on all relaxed atoms were 10−6 eV and 0.01 eV Å−1, respectively. For 2D and 3D Li2S structures, default values were used for K points. For the Li2S clusters, the gamma point was used for K points.
3. Results and discussion
3.1. Structures and properties of 2D Li2S
3.1.1. Double- and single-layer hexagonal 2D Li2S.
The stability of 2D Li2S structures can be assessed by the average binding energy (Eb), which is defined according to eqn (1):| | 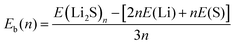 | (1) |
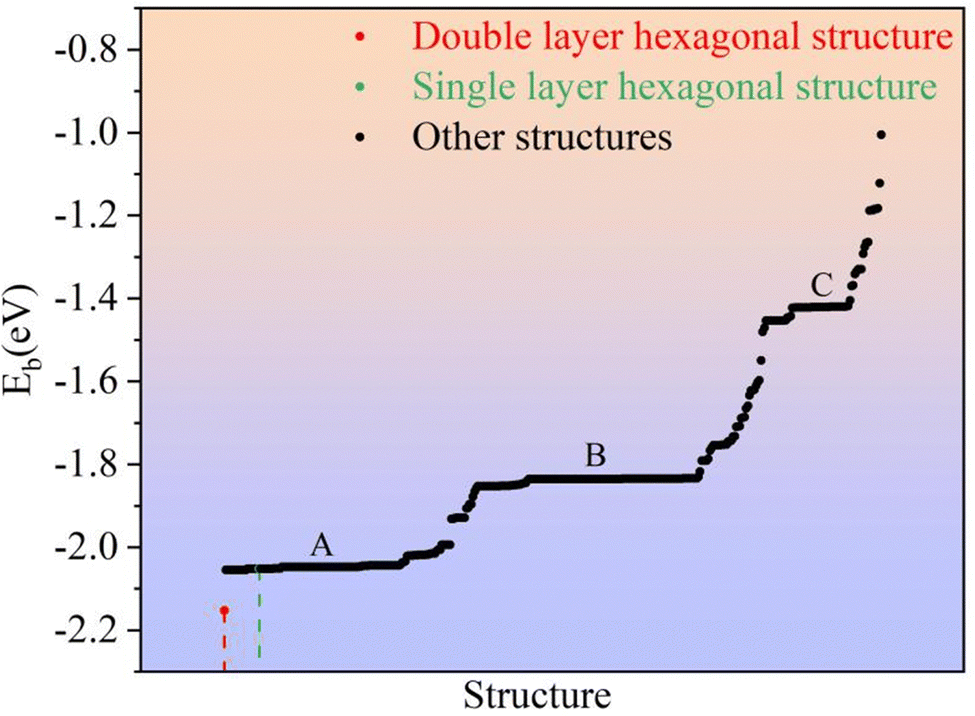
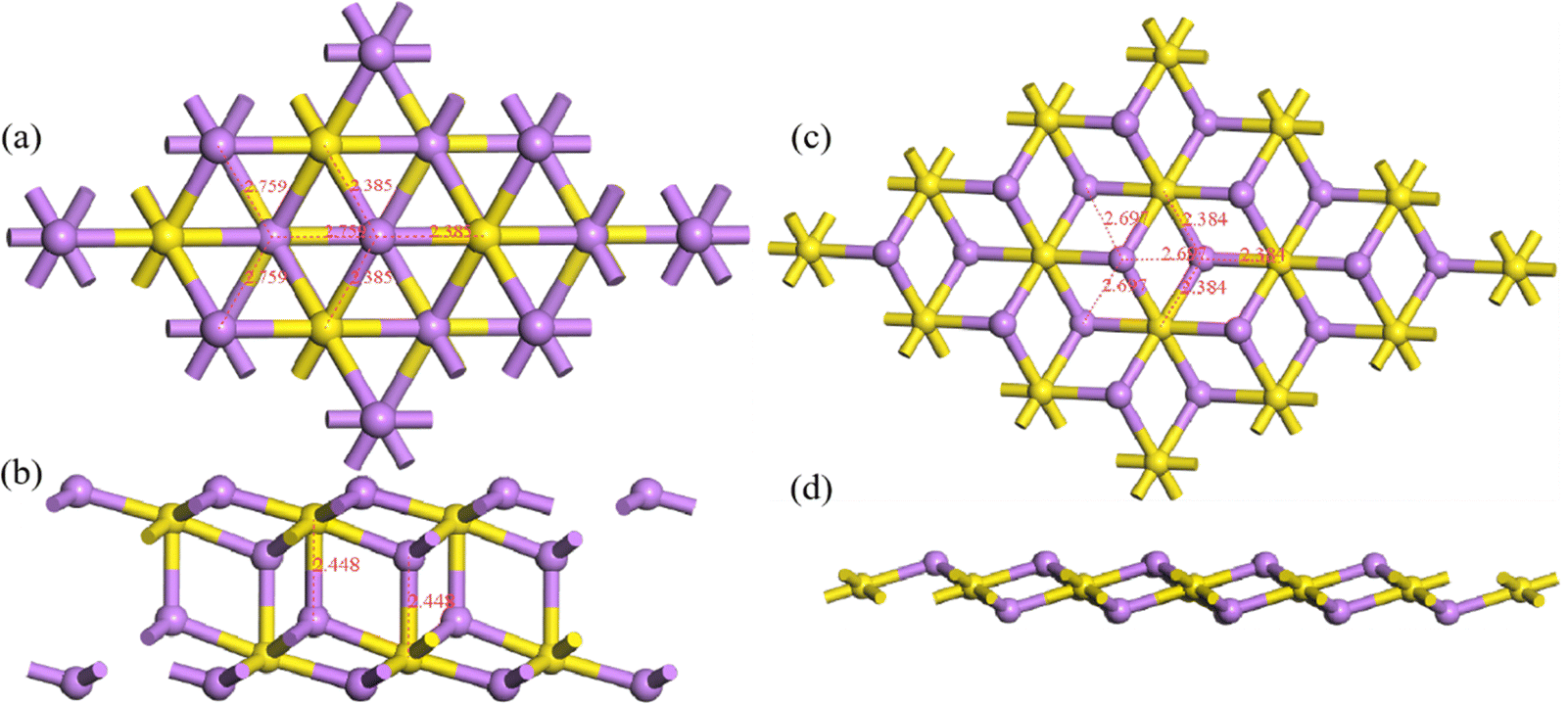
where E(Li2S)n is the energy of the Li2S structure, and E(Li) and E(S) are the energies of the Li and S atoms, respectively. Fig. 1 shows the Eb ranking of all structures found in the IM2ODE search. There are three energy plateaus. The first plateau is located between −2.05 and −1.99 eV. Among these structures, there are some similar structures with different space groups. The second plateau is located between −1.93 and −1.82 eV and demonstrates relatively stable structures slightly higher in energy. The third plateau is between −1.42 and −1.40 eV, in which the structures of higher energy are unstable. Here, a novel double-layered hexagonal structure of 2D Li2S with the lowest Eb of −2.15 eV was found, as shown in Fig. 2a and b. During extensive exploration, a single-layer hexagonal structure of 2D Li2S was also investigated, as shown in Fig. 2c and d.
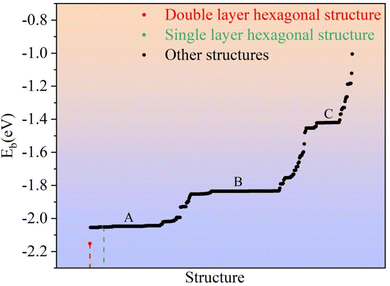 |
| | Fig. 1 The average binding energies of all predicted 2D structures. | |
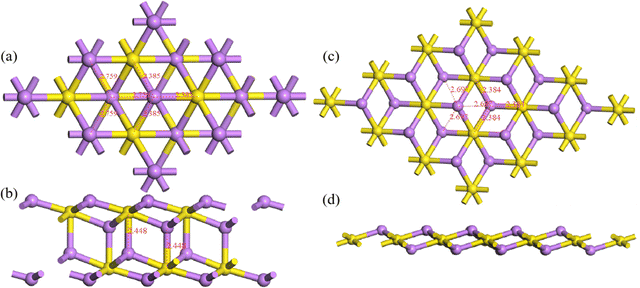 |
| | Fig. 2 Top (a and c) and side (b and d) views of double- and single-layer hexagonal 2D structures. The yellow and purple balls represent S and Li atoms, respectively. | |
The single-layer hexagonal structure of 2D Li2S has space group 164 (P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1). It is a sandwich structure, with the lithium atoms in the outer layers and the sulfur atoms in the inner layer, similar to that of 1T-MoS2.34,35 The distances between Li and three S atoms are 2.384 Å. The linear distances between Li and three neighboring Li atoms are 2.697 Å. In the novel double-layer hexagonal structure of 2D Li2S with space group 164 (Pm1), the upper and lower layers are similar to the single-layer hexagonal structure. These two single-layer structures are alternately arranged, connected through Li–S chemical bonds. In the double-layer structure, the distances between Li and three S atoms are 2.385 Å. The distances between Li and three neighboring Li atoms are 2.759 Å, respectively. The lengths of the S–Li bonds between the double layers are 2.448 Å. Their geometrical parameters, Eb and band gap (Eg) of double- and single-layer hexagonal structures are shown in Table 1.
m1). It is a sandwich structure, with the lithium atoms in the outer layers and the sulfur atoms in the inner layer, similar to that of 1T-MoS2.34,35 The distances between Li and three S atoms are 2.384 Å. The linear distances between Li and three neighboring Li atoms are 2.697 Å. In the novel double-layer hexagonal structure of 2D Li2S with space group 164 (Pm1), the upper and lower layers are similar to the single-layer hexagonal structure. These two single-layer structures are alternately arranged, connected through Li–S chemical bonds. In the double-layer structure, the distances between Li and three S atoms are 2.385 Å. The distances between Li and three neighboring Li atoms are 2.759 Å, respectively. The lengths of the S–Li bonds between the double layers are 2.448 Å. Their geometrical parameters, Eb and band gap (Eg) of double- and single-layer hexagonal structures are shown in Table 1.
Table 1 The parameters of double- and single-layer hexagonal structures of 2D Li2S
| Structure |
Space group |
Lattice parameter a, b and c (Å) |
Thickness (Å) |
E
b (eV) |
E
g (eV) |
| Double-layer |
Pm1 |
3.976, 3.976, 24.264 |
4.633 |
−2.153 |
3.50 |
| Single-layer |
Pm1 |
3.932, 3.932, 11.953 |
1.457 |
−2.052 |
3.14 |
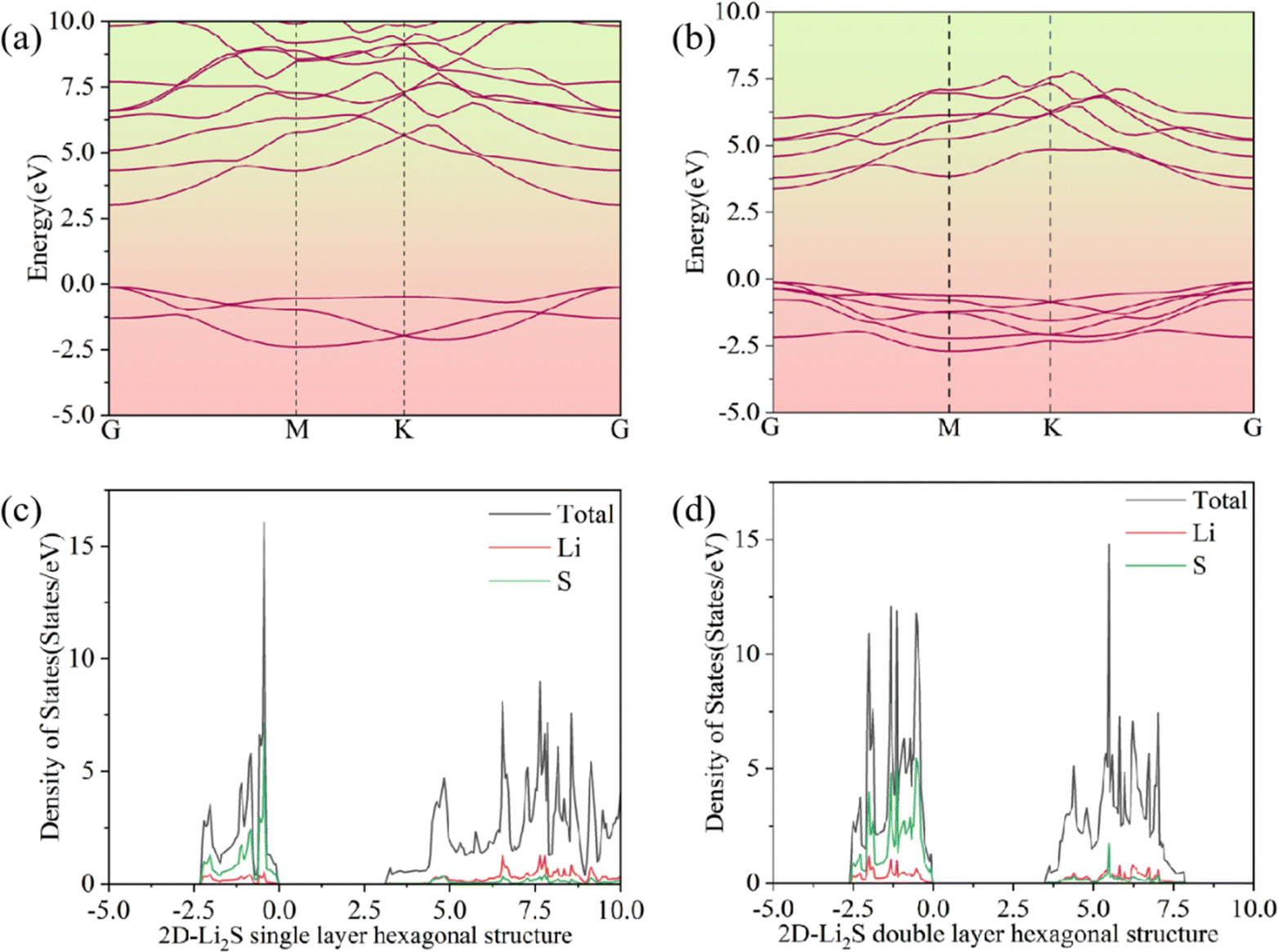
As shown in Fig. 3a and b, the double-layer hexagonal structure of 2D Li2S has a wider band gap than the monolayer hexagonal structure. The single-layer hexagonal structure has a direct band gap of 3.14 eV and the double-layer hexagonal structure has a direct band gap of 3.50 eV. The spin-up and spin-down densities of states (DOSs) are uniformly distributed. The spin-up DOSs of double-layer and monolayer 2D Li2S are shown in Fig. 3c and d. In the DOS on the left of the Fermi energy, the Li 2s and S 3p orbitals of 2D Li2S make the main contributions. On the right, the Li 2p and S 4s orbitals of 2D Li2S make the main contributions. Due to strong chemical bonds between the two monolayers, the gap between the valence and conduction bands further broadens.
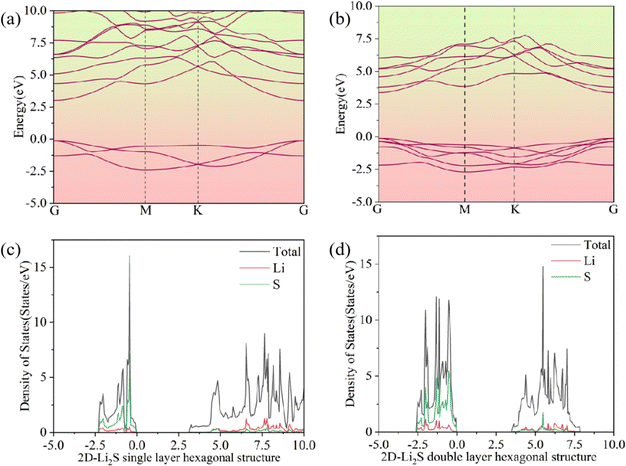 |
| | Fig. 3 Band structures (a and b) and densities of states (c and d) of double- and single-layer hexagonal structures of 2D Li2S. | |
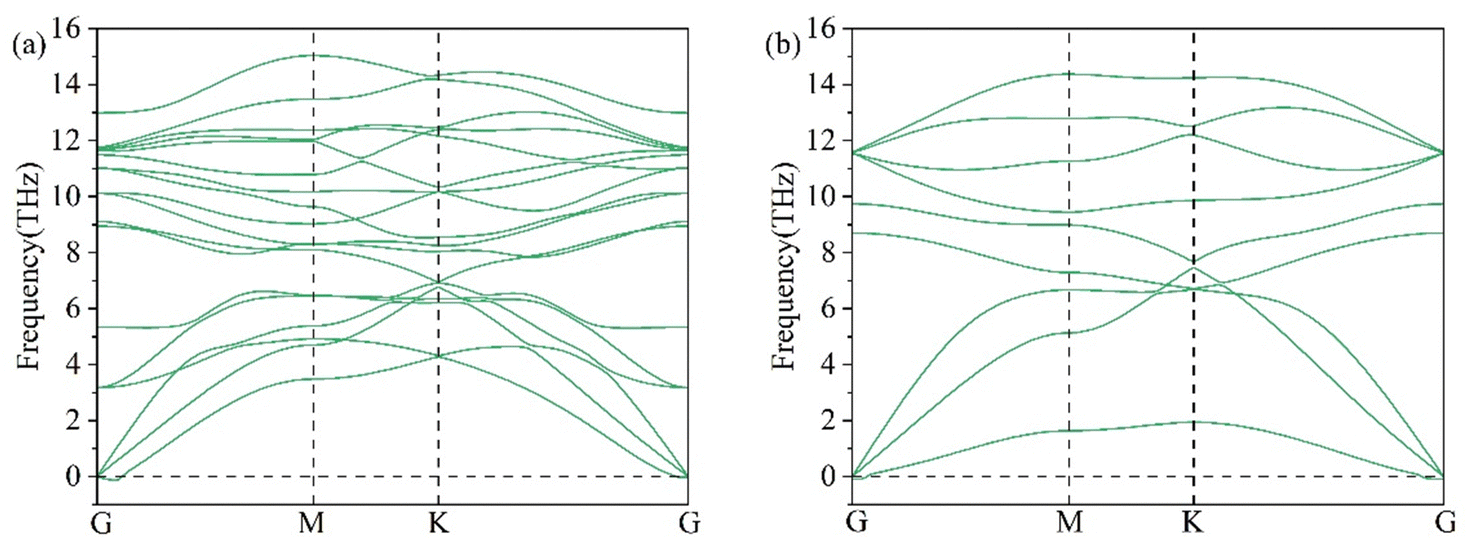
Phonons are quantized vibrations of a crystal lattice, which have a discrete frequency spectrum.36 Phonons are important in the thermodynamic properties of solids, as in heat and sound propagation. The phonon spectrum of 2D Li2S was predicted by density functional perturbation theory.37 The phonon spectra of the single- and double-layer hexagonal structures of 2D Li2S are shown in Fig. 4. For example, the single-layer hexagonal structure has 9 phonon modes. There are no imaginary frequencies throughout the Brillouin zone, which means that these two structures are stable.
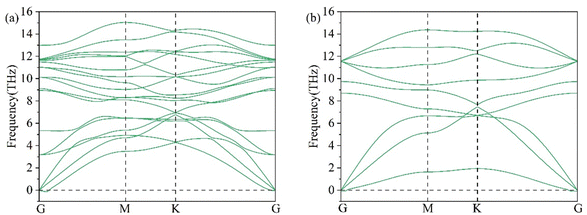 |
| | Fig. 4 Phonon spectra of double- (a) and single-layer (b) 2D Li2S. | |
3.1.2. Other monolayer 2D Li2S.
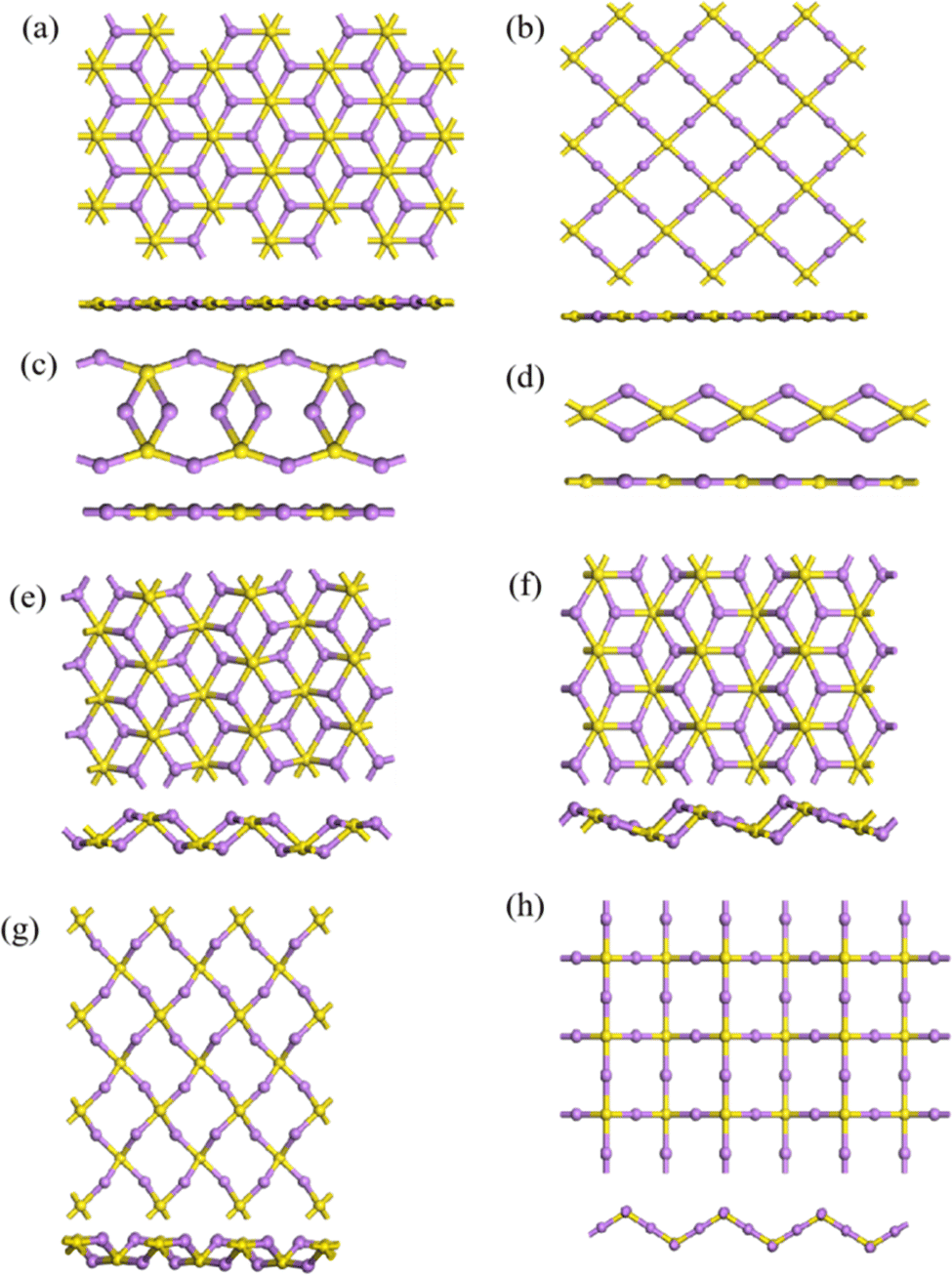
Besides the single- and double-layer hexagonal structures mentioned above, a number of other 2D structures were predicted by IM2ODE. Fig. 5 shows some predicted 2D structures of Li2S. The first four are flat monolayer structures of 2D Li2S. Structure 2D-a is a flat, single-layer hexagonal array with S atoms occupying the center of the hexagon. Structure 2D-b is a flat monolayer rectangular array formed by four S atoms and four Li atoms. Structure 2D-c is a flat, single-layer octagonal array. Structure 2D-d is a flat, single-layer chain structure. The latter four structures are twisted monolayer hexagonal structures of 2D Li2S. Both 2D-e and 2D-f are twisted hexagonal structures. The difference between them is that the atoms are not twisted in the same position. Structure 2D-g has a twisted rectangular form, while structure 2D-h shows a regular wavy form.
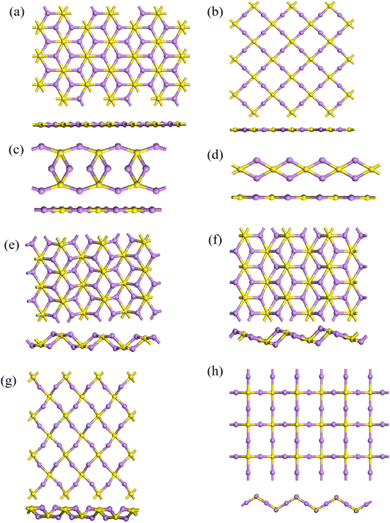 |
| | Fig. 5 Other flat 2D-a–2D-d (a)–(d) and twisted 2D-e–2D-h (e)–(h) monolayer structures of 2D Li2S. | |
For the predicted flat and twisted monolayer structures of 2D Li2S, the geometrical parameters, Eb and Eg, are shown in Table 2. According to Table 2, the Eb of 2D Li2S is inversely proportional to its band gap. The maximum band gap is 3.94 eV and the minimum band gap is 1.57 eV. Among the twisted monolayer structures, the hexagonal structure (Fig. 5e) with the band gap of 3.30 eV is the most stable. Another hexagonal structure (Fig. 5f) has a band gap of 3.69 eV.
Table 2 The parameters of other 2D Li2S
| Structure |
Space group |
Lattice parameter/Å (a, b, c) |
Thickness (Å) |
E
b (eV) |
E
g (eV) |
| 2D-a |
P6/mmm |
4.162, 4.162, 10.937 |
Planar |
−1.930 |
3.94 |
| 2D-b |
P4/mmm |
4.508, 4.508, 8.367 |
Planar |
−1.845 |
2.69 |
| 2D-c |
P2/m |
14.105, 4.405, 5.477 |
Planar |
−1.602 |
1.83 |
| 2D-d |
Pmmm
|
3.745, 6.297, 7.032 |
Planar |
−1.665 |
1.57 |
| 2D-e |
P21/m |
13.592, 6.359, 3.951 |
1.98 |
−2.055 |
3.30 |
| 2D-f |
P21/m |
6.532, 4.097, 12.824 |
2.84 |
−1.994 |
3.69 |
| 2D-g |
P21212 |
5.336, 6.300, 10.114 |
1.42 |
−1.852 |
3.16 |
| 2D-h |
Pmma
|
8.132, 4.633, 8.935 |
2.75 |
−1.851 |
3.01 |
3.2. Structure and properties of 3D Li2S
3.2.1. Typical 3D Li2S.
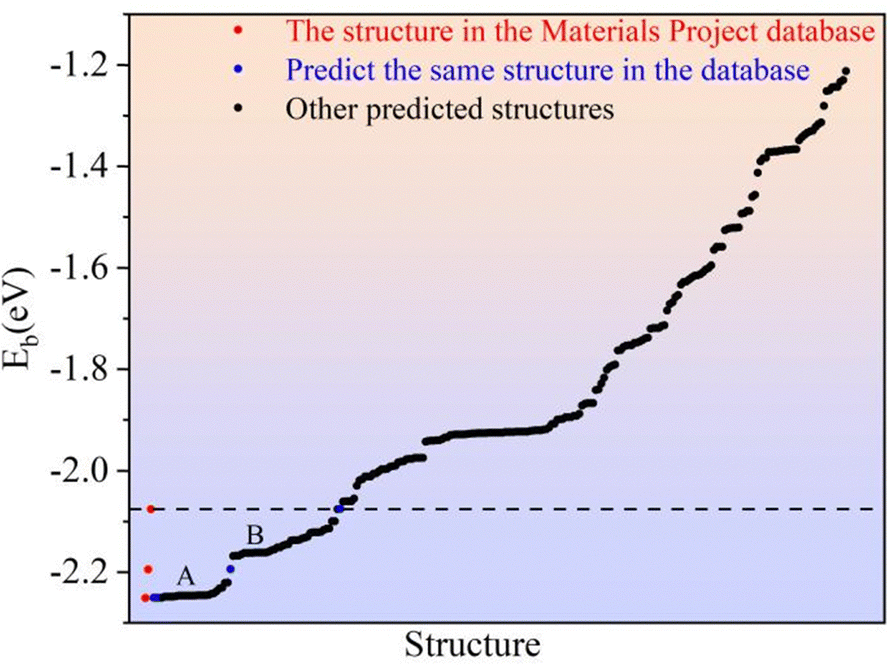
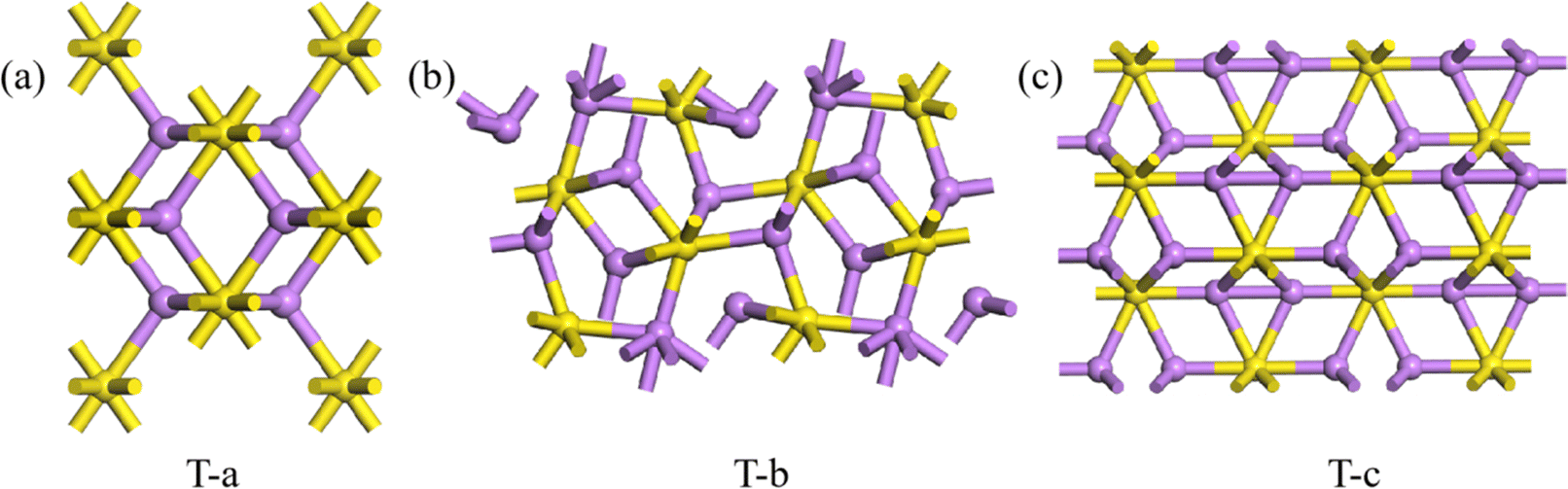
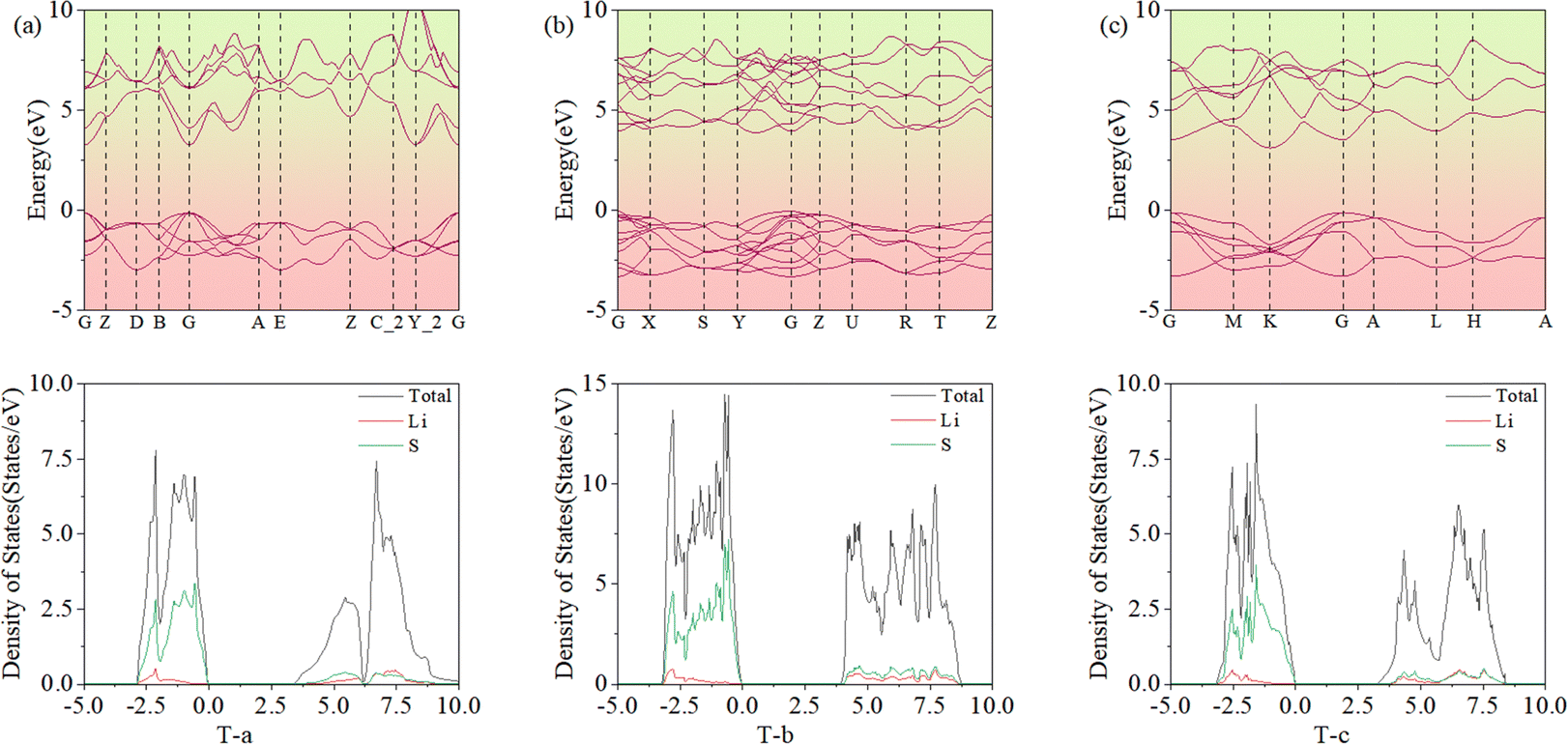
As described for 2D Li2S, the Eb of all predicted structures of 3D Li2S were ranked. In Fig. 6, the red dots indicate the Eb of the structures in the Materials Project (MP) database. The blue dots indicate the Eb of the structures identified in the IM2ODE search that match those in the MP database. The black dots indicate the Eb of novel structures predicted by IM2ODE. There are three distinct plateaus, which are located at −2.25 to −2.19 eV, −2.17 to −2.07 eV, and −1.94 to −1.87 eV. In Fig. 6, three typical structures of 3D Li2S predicted by IM2ODE match the MP database. Their structures are shown in Fig. 7. Their lattice parameters, space groups, and band gaps are listed in Table 3.
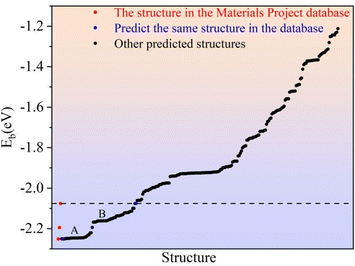 |
| | Fig. 6 The average binding energies of the predicted 3D structures. | |
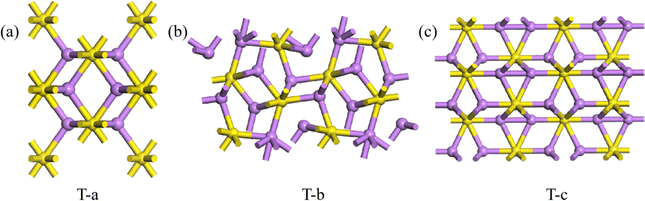 |
| | Fig. 7 Three typical structures, T-a(a), T-b (b) and T-c of 3D Li2S. | |
Table 3 Predicted lattice parameters, space groups, and band gaps of 3D Li2S structures that match the MP database
| Structure |
Lattice parameter a, b and c (Å) |
Cell volume (Å3) |
Space group |
|
T-a
|
5.716, 5.716 5.716 |
186.771 |
Fmm |
|
T-b
|
6.297, 3.811, 7.235 |
173.615 |
Pnma
|
|
T-c
|
3.941, 3.941, 7.086 |
95.296 |
P63/mmc |
Fig. 8 presents the band structures and DOS for specific space group structures. Their band gaps are 3.40, 3.94, and 3.20 eV, which are consistent with the MP values of 3.40, 3.93, and 3.17 eV.38 From the DOS, it can be seen that the Li 2s and S 3p orbitals of 3D Li2S make the main contributions.
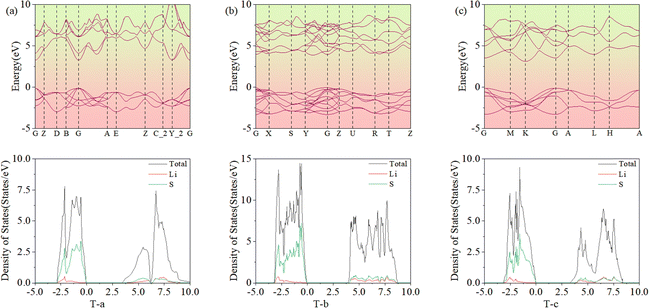 |
| | Fig. 8 Band structures and densities of states for three typical structures, T-a(a), T-b (b) and T-c (c). | |
3.2.2. Other 3D Li2S.
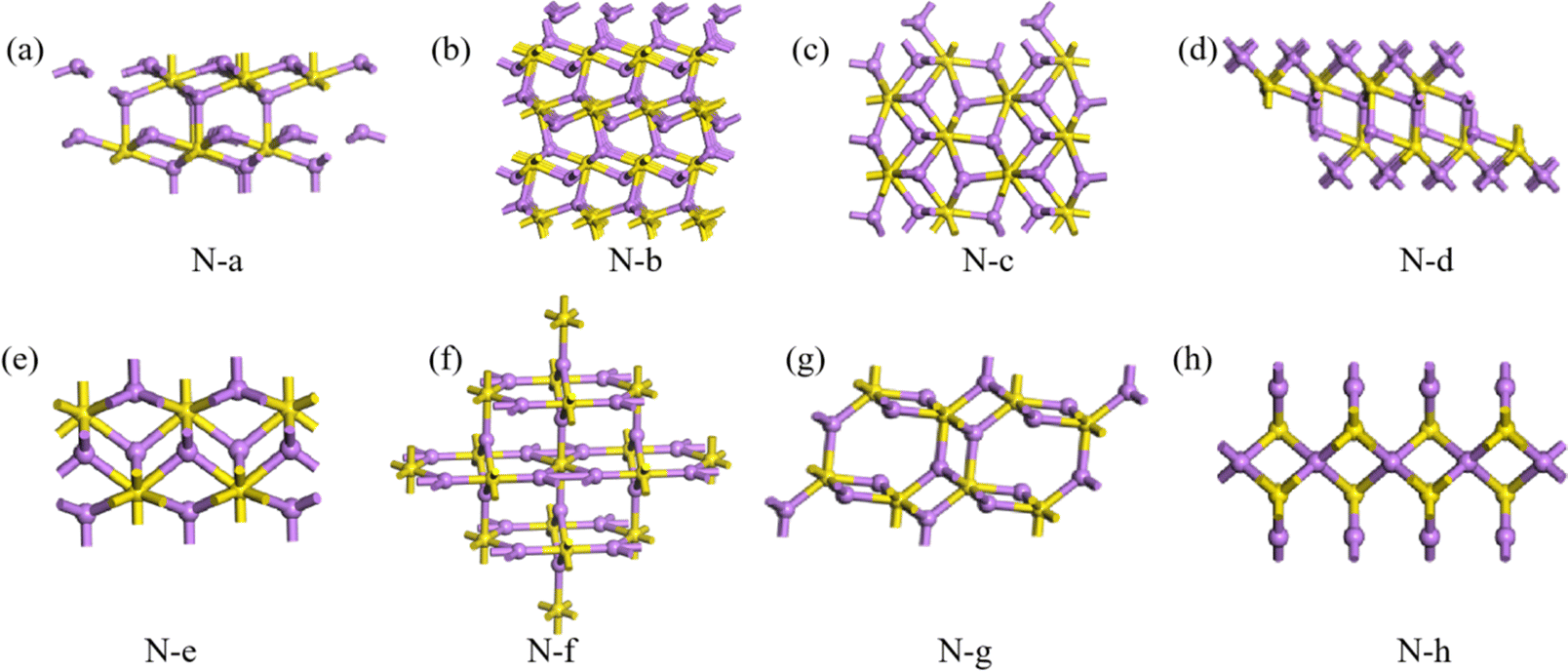
Here, the highest Eb of the three structures of 3D Li2S in the MP database was used as a cut-off criterion. The structures below this energy were further investigated and divided into two parts, A and B, as shown in Fig. 6. On plateau A, the structures are essentially the same as those in the library with the space group Fmm. Most of the novel structures are found on plateau B. Excluding similar structures, the structures on plateau B can be categorized into eight types, as shown in Fig. 9. The geometrical parameters of the structures are shown in Table 4. The crystal systems can be categorized in terms of space groups. Structure N-a has a hexagonal crystal form. Structures N-b and N-h have orthorhombic crystal forms. Structures N-c and N-g have monoclinic crystals. Structures N-d, N-e, and N-f have tetragonal crystal forms. An interesting phenomenon is that a similar sandwich structure to that of 1T-MoS2 also exists in the 3D structures, as shown in Fig. 9a and b.
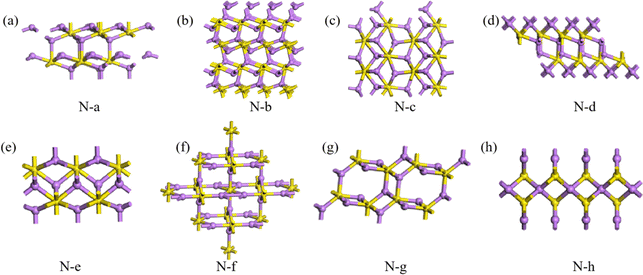 |
| | Fig. 9 Eight structures (a–h) of 3D Li2S. | |
Table 4 Geometrical parameters of the predicted 3D Li2S structures
| Structure |
Lattice parameter a, b and c (Å) |
Cell volume (Å3) |
Space group |
|
N-a
|
3.982, 3.982, 6.787 |
93.216 |
P63mc |
|
N-b
|
4.291, 6.464, 6.493 |
180.081 |
Cmc21 |
|
N-c
|
6.089, 5.297, 5.662 |
182.619 |
Abm2 |
|
N-d
|
5.518, 5.545, 7.985 |
186.289 |
C2 |
|
N-e
|
3.829, 3.829, 6.120 |
89.712 |
P4/nnm |
|
N-f
|
5.304,5.304, 3.915 |
110.151 |
P42/mnm |
|
N-g
|
4.429, 4.146, 6.481 |
107.801 |
P21/m |
|
N-h
|
6.067, 5.978, 5.915 |
214.482 |
Cmma
|
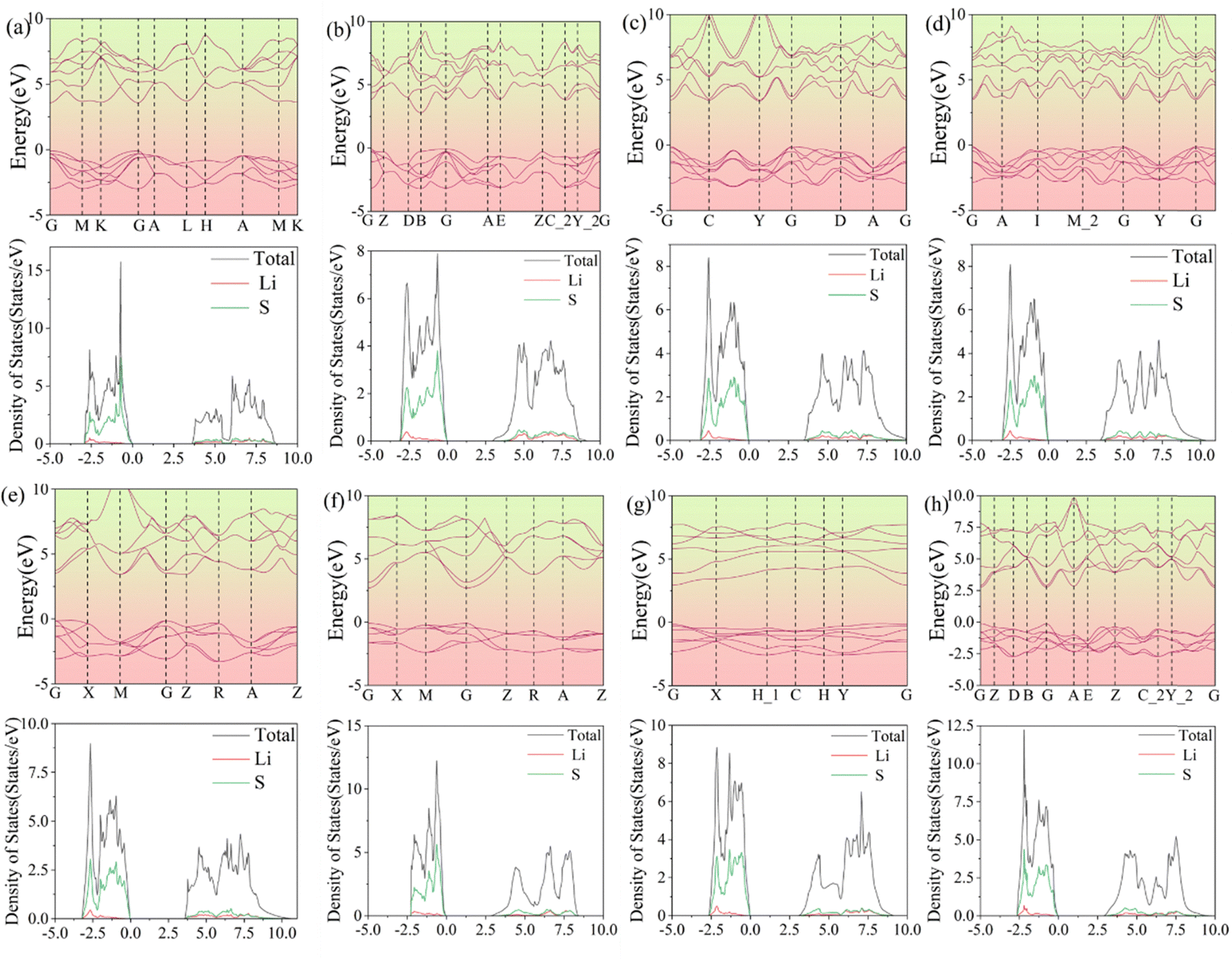
The band structures and DOS are shown in Fig. 10. Among these 3D structures, the direct band gaps of structures N-a, N-f, and N-h are 3.68, 2.80, and 2.89 eV, respectively. The indirect band gaps of structures N-b, N-c, N-d, N-e, and N-g are 2.87, 3.48, 3.38, 3.55, and 3.04 eV, respectively. As regards the DOS, both the valence and conduction bands are mainly contributed by the Li 2s, S 3s, and S 3p orbitals. In Fig. 10, the widths of the DOS at the Fermi level are in accordance with the gaps in the band structures.
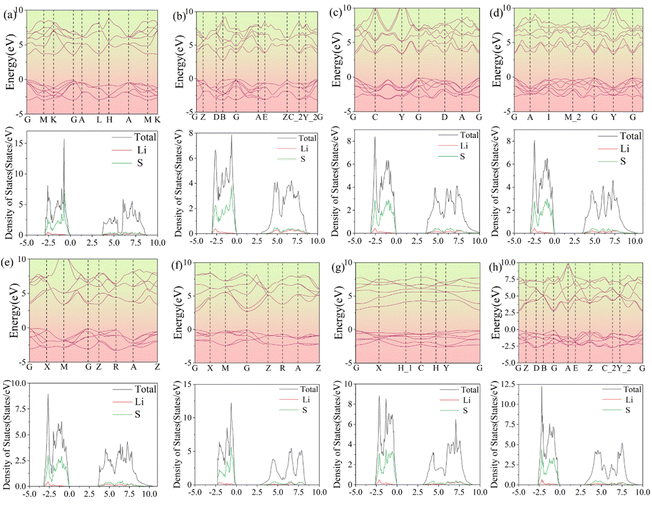 |
| | Fig. 10 Band structures and densities of states of the other eight structures (a–h) of 3D Li2S. | |
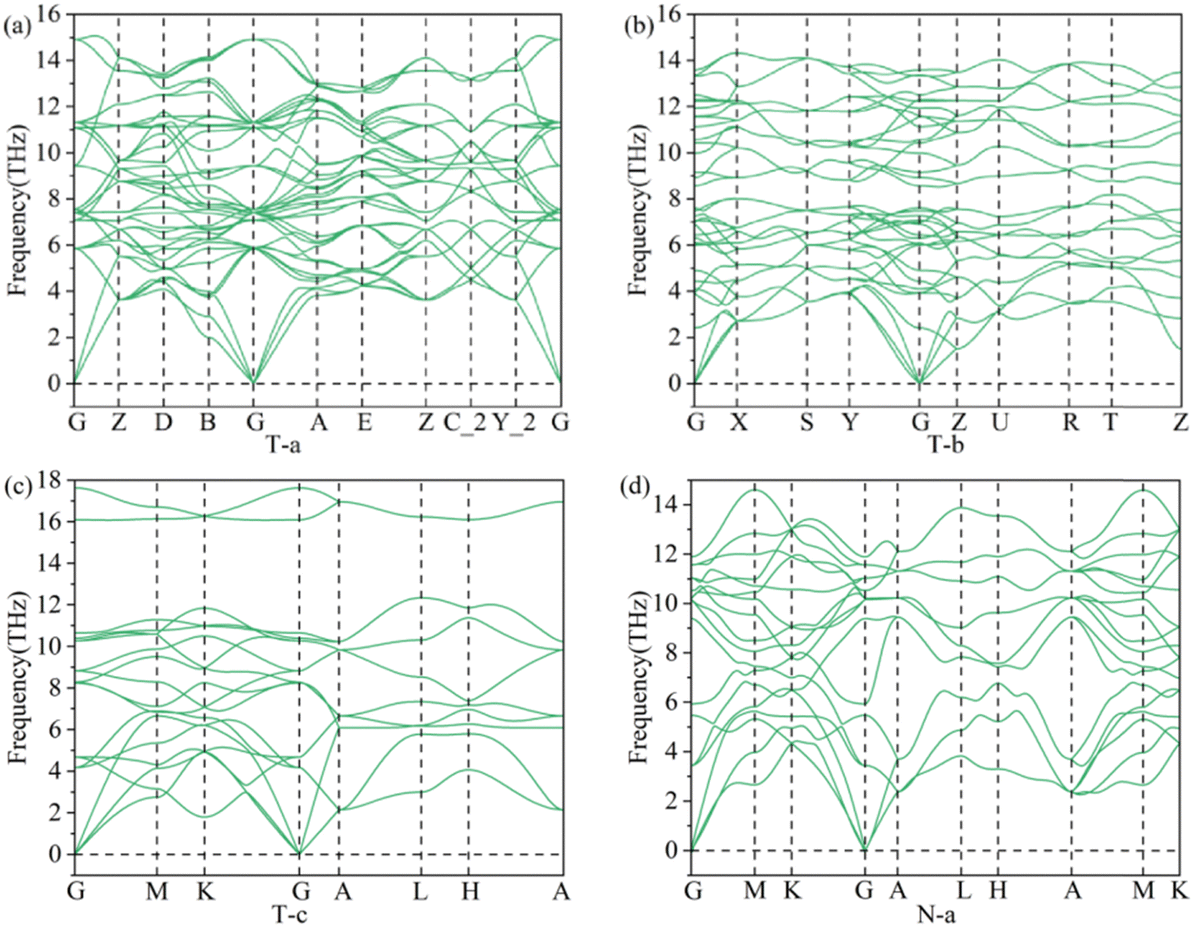
The phonon spectra of all these structures of 3D Li2S were obtained. According to the calculation results, only the phonon spectrum of structure N-a and three typical structures (T-a, T-b and T-c) do not show imaginary frequencies, as shown in Fig. 11. These indicate that the typical structures T-a, T-b and T-c and the new N-a structures are stable.
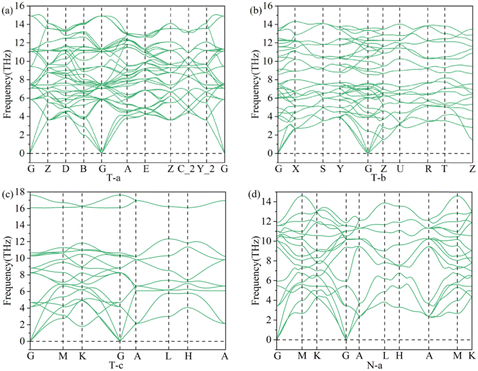 |
| | Fig. 11 Phonon spectrum of the structures, T-a (a), T-b (b), T-c (c) and N-a (d). | |
3.3. Structures and properties of (Li2S)n clusters
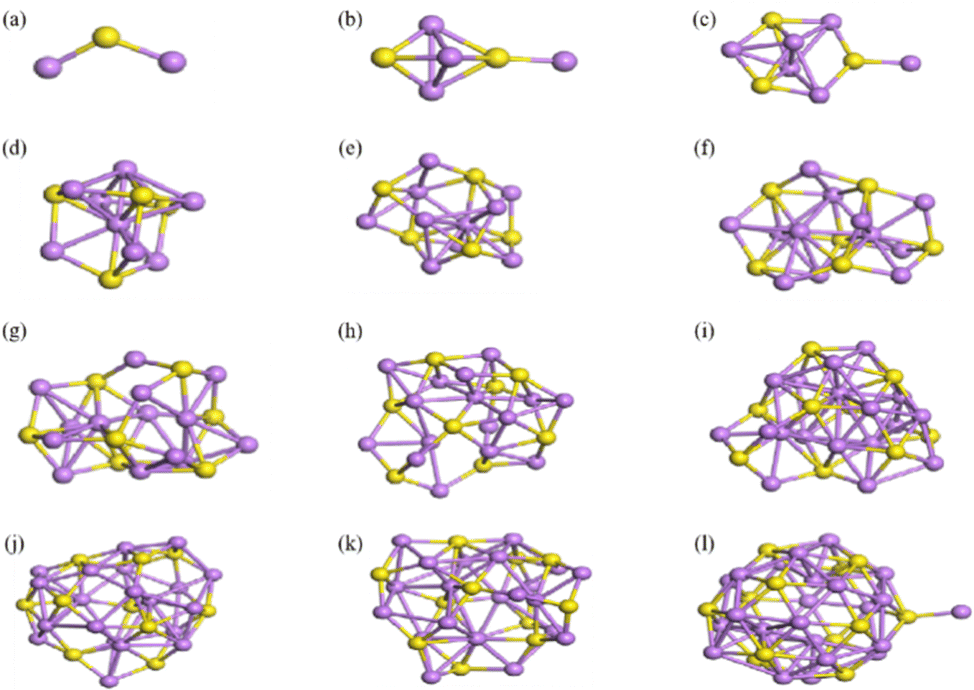
The structures of (Li2S)n (n = 1–12) clusters were systematically searched by means of IM2ODE. This process aimed to identify the most stable structure with the lowest energy at different sizes, as shown in Fig. 12. It may be noted that the (Li2S)n (n = 1–12) clusters gradually converge to a cage-like structure as the number of atoms is increased. The stabilities and electronic properties of these cluster structures were analyzed in detail.
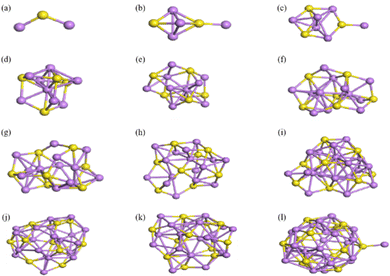 |
| | Fig. 12 Lowest energy structures of (Li2S)n clusters: (a)–(l) correspond to n = 1–12. The yellow and purple balls represent S and Li atoms, respectively. | |
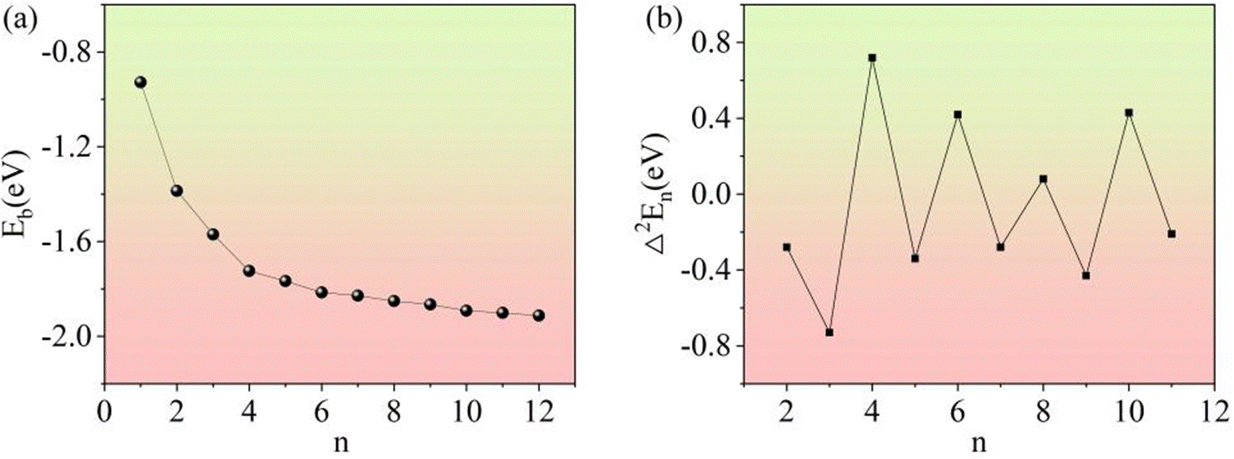
3.3.1. Relative stability of (Li2S)n clusters.
In general, the stability of a cluster can be judged by the average binding energy (Eb) and the second-order energy difference (Δ2En).39,40 The variation in the Eb with cluster size is shown in Fig. 13a. As the cluster size increases, Eb monotonically decreases, which is consistent with previous results.22 The second-order energy difference can be defined as:| | | Δ2En = En+1 + En−1 − 2En | (2) |
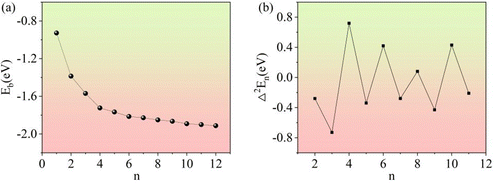 |
| | Fig. 13 The average binding energies (a) and second-order energy differences (b) of the clusters. | |
As shown in Fig. 13b, the second-order energy difference clearly shows the relative stability of (Li2S)n (n = 1–12) clusters. In the interval of n = 2–11, Δ2En shows an alternating parity pattern. Clusters containing an even number of S atoms have a relatively higher Δ2En than those containing an odd number of S atoms. This phenomenon is known as the parity oscillation effect.41 It can be observed that the highest peak of second-order energy difference corresponds to the (Li2S)4 cluster.
3.3.2. Electronic properties of (Li2S)n clusters.
The electronic properties of (Li2S)n (n = 1–12) cluster structures were calculated. These included the DOS, the energy gap (Egap) between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), ionization potential (IP), electron affinity (EA), electronegativity (χM), and chemical hardness (η).
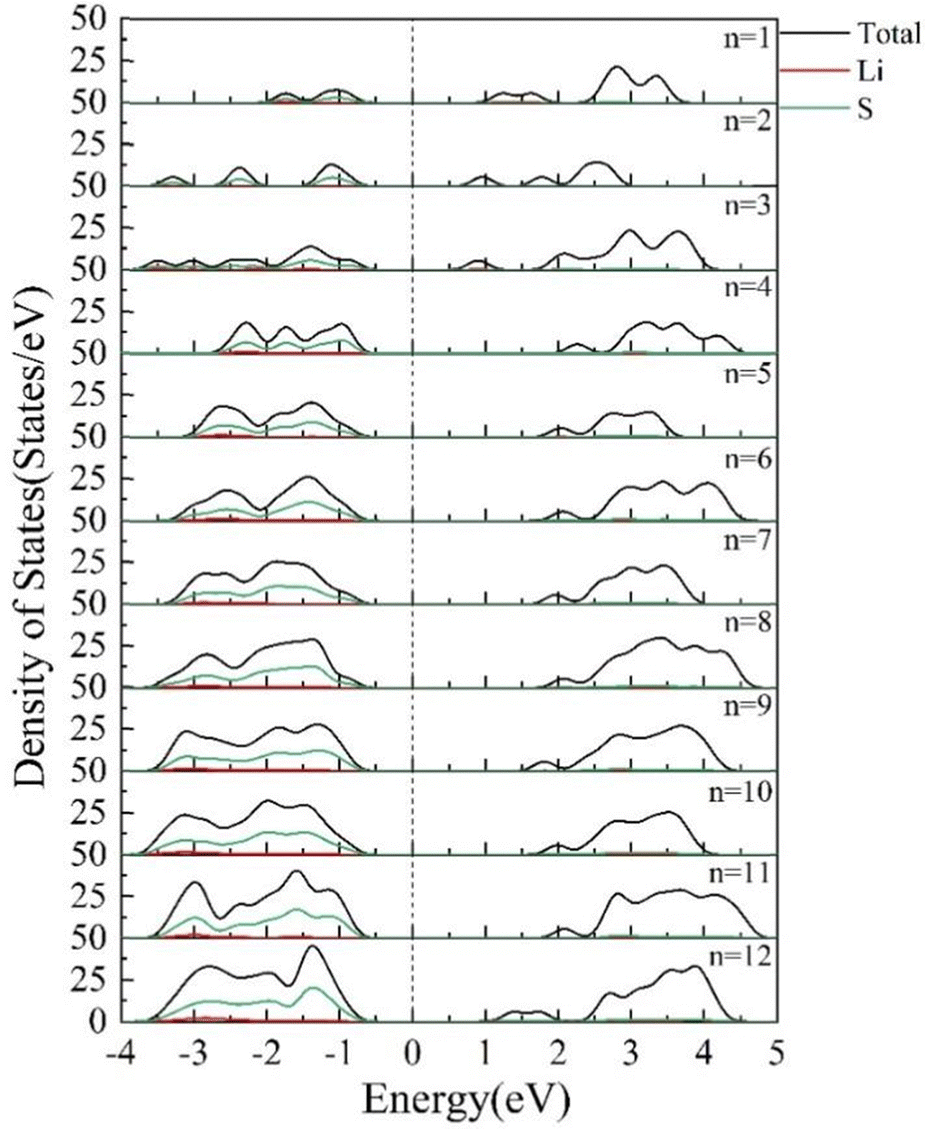
The DOS of the clusters reflects the distribution of the number of electrons in a certain energy level range.42 If the number of energy levels in the given interval is higher, the DOS is higher. The DOS of (Li2S)n (n = 1–12) clusters is shown in Fig. 14. As the cluster size increases, the peaks on either side of the Fermi energy level become larger. The bandwidth gradually broadens from 1 eV for Li2S to 3 eV for (Li2S)12. The peaks between −4 and −1 eV are mainly due to the 3p state of the S atoms. The peak due to the 3p state of the S atom moves away from the Fermi energy level when n < 11.
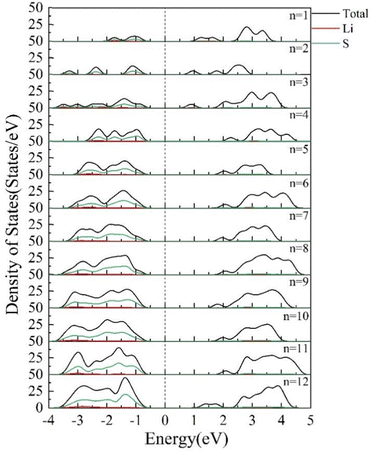 |
| | Fig. 14 Densities of states of (Li2S)n (n = 1–12) clusters. | |
The energies (EHOMO and ELUMO) of the HOMO and LUMO levels reflect the ability of a cluster to gain or lose electrons.43 In Table 5, it can be seen that the EHOMO of the clusters was the smallest (absolute maximum) when n = 4. The Egap between EHOMO and ELUMO is an important parameter in characterizing the electronic structure and stability of a cluster. Egap corresponds to the ability of an electron to jump from an occupied orbital to a vacant orbital. To some extent, it also represents the ability of the molecule to participate in chemical reactions.44 Higher energy gaps mean that higher energies are required to change the electronic structure of the clusters. The corresponding clusters are more chemically stable and less chemically reactive. In Table 5, it can be seen that the Egap of the clusters gradually decreased from n = 1 to 3, and then stabilized for n ≥ 4.
Table 5 Orbital energy levels of (Li2S)n (n = 1–12) clusters
| Cluster |
E
LUMO (eV) |
E
HOMO (eV) |
E
gap (eV) |
| Li2S |
−1.385 |
−3.549 |
2.164 |
| (Li2S)2 |
−1.614 |
−3.496 |
1.882 |
| (Li2S)3 |
−1.831 |
−3.593 |
1.762 |
| (Li2S)4 |
−1.213 |
−4.391 |
3.178 |
| (Li2S)5 |
−1.179 |
−4.143 |
2.964 |
| (Li2S)6 |
−1.051 |
−4.037 |
2.986 |
| (Li2S)7 |
−1.020 |
−3.895 |
2.875 |
| (Li2S)8 |
−0.787 |
−3.768 |
2.982 |
| (Li2S)9 |
−1.022 |
−3.757 |
2.735 |
| (Li2S)10 |
−0.844 |
−3.744 |
2.900 |
| (Li2S)11 |
−0.758 |
−3.751 |
2.993 |
| (Li2S)12 |
−1.326 |
−3.627 |
2.300 |
Ionization potential (IP) is an important parameter that reflects the interdependence between the cluster size and electronic structure.45 The IP of a system can be defined as:
where
N is the number of electrons,
E(
N) is the energy of the (Li
2S)
n cluster, and
E(
N − 1) is the energy of the cluster after losing an electron. Here, it is assumed that the spatial structure of the ionization system remains unchanged.
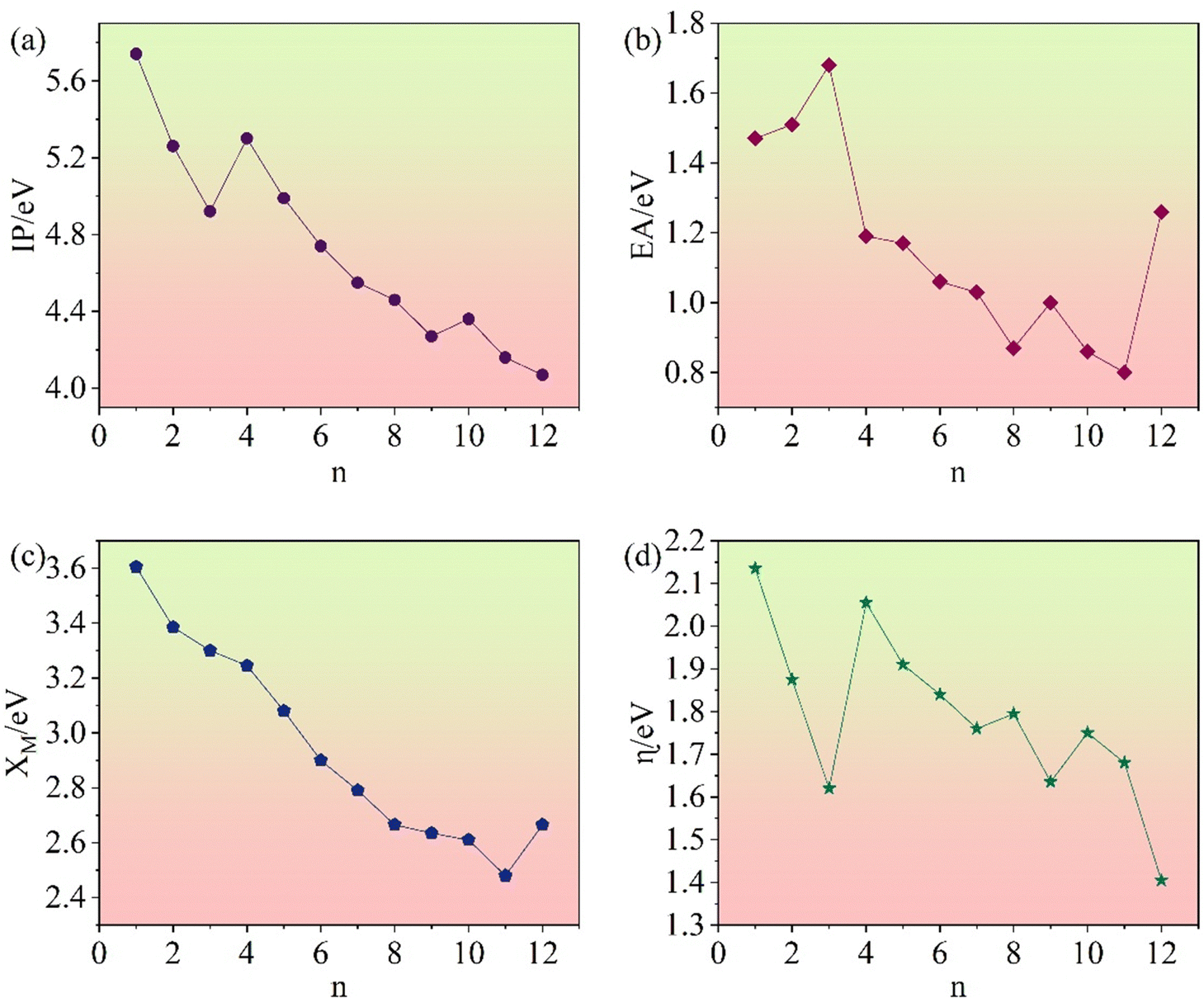
Fig. 15a illustrates the IPs for
n = 1–12 (Li
2S)
n clusters. They show an overall decreasing trend with increasing size.
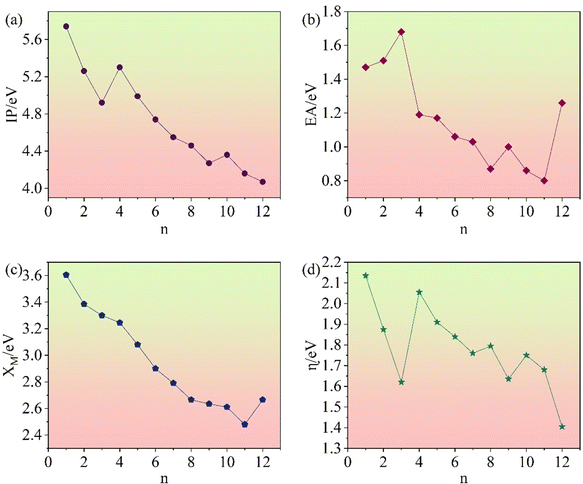 |
| | Fig. 15 Ionization potentials (a), electron affinities (b), electronegativities (c), and chemical hardnesses (d) of (Li2S)n (n = 1–12) clusters. | |
Electron affinity (EA) refers to the energy that needs to be surmounted for an atom or molecule to gain an electron.46 EA can be defined as:
The lower the EA, the easier it is to gain an electron. Fig. 15b illustrates the variation in the EAs of (Li2S)n clusters with size. They show a general decreasing trend. Among the homologues, (Li2S)3 has the highest EA value.
Electronegativity (χM) combines the IP and the EA.47 It indicates the relative strength of the ability to attract electrons when forming a chemical bond between two different atoms.48 The electronegativity of atoms and molecules can be estimated according to eqn (6):
| | 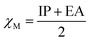 | (5) |
Fig. 15c shows the variation of the electronegativity of (Li2S)n with cluster size. In general, it decreases with increasing cluster size.
Chemical hardness (η) can be used to characterize the stability of clusters.49 Theoretically, η can be calculated from the IP and the EA:
| | 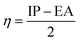 | (6) |
The calculated chemical hardnesses of (Li2S)n clusters are shown in Fig. 15d. The η of (Li2S)n decreases with increasing cluster size.
By investigating IP, EA, χM, and η, it became apparent that these properties of (Li2S)n (n = 1–12) generally decrease with increasing cluster size. This trend may be influenced by various factors, such as size effects, surface effects, and internal interactions. These findings further our understanding of the structures and properties of (Li2S)n clusters, and provide theoretical guidance for application in various fields.
4. Conclusions
In summary, the structures and electronic properties of 2D, bulk, and cluster Li2S have been predicted by IM2ODE and DFT methods. Novel monolayer and double-layer hexagonal structures of 2D Li2S have been predicted. The double-layer structure is more stable than the single-layer structure and has a wider band gap. These structures are predicted to be stable by phonon spectral calculations. The predicted structures of 3D Li2S encompass three structures in the MP database and various novel structures. They also include a sandwich structure similar to that of 1T-MoS2 and double-layer hexagonal structures of 2D Li2S. These 3D Li2S structures have a wide range of band gaps. A stable new 3D structure with the space group P63mc was found. With increasing number of atoms, the (Li2S)n clusters converge into a cage-like structure, and the average binding energy decreases. The second-order energy difference shows an odd–even oscillatory pattern. The ionization potentials, electron affinities, electronegativities, and chemical hardnesses decrease with increasing number of atoms. These findings should provide greater theoretical understanding of the properties and behavior of new 2D, 3D, and cluster functional materials.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21703158, 21873073 and 12404265) and the Basic Research Project of Wenzhou City (L2023005, L20240001 and L20240002). We gratefully acknowledge HZWTECH for providing computation facilities.
References
- M. Y. Wang, J. J. Mao, Y. D. Pang, X. L. Zhang, Z. X. Yang, Z. S. Lu and S. T. Yang, Theoretical investigation of synergistically boosting the anchoring and electrochemical performance of lithiophilic/sulfiphilic transition metal carbides for lithium–sulfur batteries, Nanoscale, 2024, 16, 462–473 RSC
 .
.
- T. Hakari, A. Hayashi and M. Tatsumisago, Highly utilized lithium sulfide active material by enhancing conductivity in all-solid-state batteries, Chem. Lett., 2015, 44, 1664–1666 CrossRef CAS .
- S. Q. Li, D. Leng, W. Y. Li, L. Qie, Z. H. Dong, Z. Q. Cheng and Z. Y. Fan, Recent progress in developing Li2S cathodes for Li–S batteries, Energy Storage Mater., 2020, 27, 279–296 CrossRef .
- A. Hayashi, R. Ohtsubo, T. Ohtomo, F. Mizuno and M. Tatsumisago, All-solid-state rechargeable lithium batteries with Li2S as a positive electrode material, J. Power Sources, 2008, 183, 422–426 CrossRef CAS .
- M. Tatsumisago, Glassy materials based on Li2S for all-solid-state lithium secondary batteries, Solid State Ionics, 2004, 175, 13–18 CrossRef CAS .
- H. L. Ye, M. Li, T. C. Liu, Y. G. Li and J. Lu, Activating Li2S as the lithium-containing cathode in lithium–sulfur batteries, ACS Energy Lett., 2020, 5, 2234–2245 CrossRef CAS .
- X. Li, M. X. Gao, W. B. Du, B. Ni, Y. H. Wu, Y. F. Liu, C. X. Shang, Z. X. Guo and H. G. Pan, A mechanochemical synthesis of submicron-sized Li2S and a mesoporous Li2S/C hybrid for high performance lithium/sulfur battery cathodes, J. Mater. Chem. A, 2017, 5, 6471–6482 RSC .
- C. C. Wei, C. Yu, L. F. Peng, Z. Q. Zhang, R. N. Xu, Z. K. Wu, C. Liao, W. Zhang, L. Zhang, S. J. Cheng and J. Xie, Tuning ionic conductivity to enable all-climate solid-state Li–S batteries with superior performances, Mater. Adv., 2022, 3, 1047 RSC .
- Z. Pourali, M. R. Yaftian and M. R. Sovizi, Li2S/transition metal carbide composite as cathode material for high performance lithium-sulfur batteries, Mater. Chem. Phys., 2018, 217, 117–124 CrossRef CAS .
- L. W. Yang, Q. Li, Y. Wang, Y. X. Chen, X. D. Guo, Z. G. Wu, G. Chen, B. H. Zhong, W. Xiang and Y. J. Zhong, A review of cathode materials in lithium-sulfur batteries, Ionics, 2020, 26, 5299–5318 CrossRef CAS .
- H. Yu, P. Zeng, H. Liu, X. Zhou, C. M. Guo, Y. F. Li, S. S. Liu, M. F. Chen, X. W. Guo, B. B. Chang, T. J. Wu and X. Y. Wang, Li2S In-situ grown on three-dimensional porous carbon architecture with electron/ion channels and dual active sites as cathodes of Li–S batteries, ACS Appl. Mater. Interfaces, 2021, 13, 32968–32977 CrossRef CAS PubMed .
- L. Chen, N. L. D. Rago, I. D. Bloom and L. L. Shaw, New insights into the electrode mechanism of lithium sulfur batteries via air-free post-test analysis, Chem. Commun., 2016, 52, 9913 RSC .
- Y. Fujita, K. Münch, T. Asakura, K. Motohashi, A. Sakuda, J. Janek and A. Hayashi, Dynamic volume change of Li2S-based active material and the influence of stacking pressure on capacity in all-solid-state batteries, Chem. Mater., 2024, 36, 7533–7540 CrossRef CAS .
- X. B. Meng, D. J. Comstock, T. T. Fister and J. W. Elam, Vapor-phase atomic-controllable growth of amorphous Li2S for high-performance lithium–sulfur batteries, ACS Nano, 2014, 8, 10963–71092 CrossRef CAS PubMed .
- X. Gao, X. L. Zheng, J. Y. Wang, Z. W. Zhang, X. Xiao, J. Y. Wan, Y. S. Ye, L.-Y. Chou, H. K. Lee, J. Y. Wang, R. A. Vilá, Y. F. Yang, P. Zhang, L.-W. Wang and Y. Cui, Incorporating the nanoscale encapsulation concept from liquid electrolytes into solid-state lithium–sulfur batteries, Nano Lett., 2020, 20, 5496–5503 CrossRef CAS .
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Electric field effect in atomically thin carbon films, Science, 2004, 306, 666–669 CrossRef CAS .
- X. C. Yu, S. L. Zhang, H. B. Zeng and Q. J. Wang, Lateral black phosphorene P–N junctions formed via chemical doping for high performance near-infrared photodetector, Nano Energy, 2016, 25, 34–41 CrossRef CAS .
- M. Q. Xie, S. L. Zhang, B. Cai, Y. Huang, Y. S. Zou, B. Guo, Y. Gu and H. B. Zeng, A promising two-dimensional solar cell donor: black arsenic–phosphorus monolayer with 1.54 eV direct bandgap and mobility exceeding 14000 cm2 V−1 s−1, Nano Energy, 2016, 28, 433 CrossRef CAS .
- J. Yang, Y.-L. Jiang, L.-J. Li, E. Muhire and M.-Z. Gao, High-performance photodetectors and enhanced photocatalysts of two-dimensional TiO2 nanosheets under UV light excitation, Nanoscale, 2016, 8, 8170 RSC .
- Y. J. Yang, S. Umrao, S. Lai and S. J. Lee, Large-area highly conductive transparent two-dimensional Ti2CTx film, J. Phys. Chem. Lett., 2017, 8, 859 CrossRef CAS .
- D. Yin, J. Feng, N. R. Jiang, R. Ma, Y. F. Liu and H. B. Sun, Two-dimensional stretchable organic light-emitting devices with high efficiency, ACS Appl. Mater. Interface, 2016, 8, 31166 CrossRef CAS .
- T. Yu, F. Li, C. Y. Liu, S. T. Zhang, H. Y. Xu and G. C. Yang, Understanding the role of lithium sulfide clusters in lithium–sulfur batteries, J. Mater. Chem. A, 2017, 5, 9293–9298 RSC .
- Z. X. Liu, H. Q. Deng, W. Y. Hu, F. Gao, S. G. Zhang, P. B. Balbuena and P. P. Mukherjee, Revealing reaction mechanisms of nanoconfined Li2S: implications for lithium–sulfur batteries, Phys. Chem. Chem. Phys., 2018, 20, 11713–11721 RSC .
- L. M. Suo, Y. J. Zhu, F. D. Han, T. Gao, C. Luo, X. L. Fan, Y.-S. Hu and C. S. Wang, Carbon cage encapsulating nano-cluster Li2S by ionic liquid polymerization and pyrolysis for high performance Li–S batteries, Nano Energy, 2015, 13, 467–473 CrossRef CAS .
- K. Zhang, L. J. Wang, Z. Hu, F. Y. Cheng and J. Chen, Ultrasmall Li2S nanoparticles anchored in graphene nanosheets for high-energy lithium-ion batteries, Sci. Rep., 2014, 4, 6467 CrossRef CAS PubMed .
- Y.-Y. Zhang, W. G. Gao, S. Y. Chen, H. J. Xiang and X.-G. Gong, Inverse design of materials by multi-objective differential evolution, Comput. Mater. Sci., 2015, 98, 51–55 CrossRef CAS .
- J. Lu, S.-H. Wei, Y.-Y. Zhang, D.-Y. Hua and X.-M. Duan, Geometric, electronic and magnetic properties of Aun, Aun−1Pt and Aun−2Pt2 (n = 2–9) clusters: A first-principles study, Comput. Theor. Chem., 2016, 1090, 157–164 CrossRef CAS .
- H.-Z. Chen, Y.-Y. Zhang, X.-G. Gong and H.-J. Xiang, Predicting new TiO2 phases with low band gaps by a multi-objective global optimization approach, J. Phys. Chem. C, 2014, 118, 2333–2337 CrossRef CAS .
- G. Kresse and J. Furthmiiller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS .
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed .
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS .
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed .
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS .
- M. Naseri and S. R. Lin, 2D Li2S monolayer: A global minimum lithium sulfide sandwich, Chem. Phys. Lett., 2019, 722, 58–63 CrossRef CAS .
- Y. Q. Fang, J. Pan, J. Q. He, R. C. Luo, D. Wang, X. L. Che, K. J. Bu, P. Liu, G. Mu, H. Zhang, T. Q. Lin and F. Q. Huang, Structure re-determination and superconductivity observation of bulk 1T MoS2, Angew. Chem., Int. Ed., 2018, 57, 1232–1235 CrossRef CAS PubMed .
- C.-R. Gerhorst, A. Neukirchen, D. A. Klüppelberg, G. Bihlmayer, M. Betzinger, G. Michalicek, D. Wortmann and S. Blügel, Phonons from density-functional perturbation theory using the all-electron full-potential linearized augmented plane-wave method, Electron. Struct., 2024, 6, 017001 CrossRef CAS .
- A. Togo, L. Chaput, I. Tanaka and G. Hug, First-principles phonon calculations of thermal expansion inTi3SiC2, Ti3AlC2, and Ti3GeC2, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 174301 CrossRef .
- O. I. Barkalov, P. G. Naumov, C. Felser and S. A. Medvedev, Pressure-induced transition to Ni2In-type phase in lithium sulfide (Li2S), Solid State Sci., 2016, 61, 220–224 CrossRef CAS .
- K.-H. Lin, H.-T. Chen, H.-L. Chen, J.-S. Lin, S.-P. Ju, C.-F. Tseng, C.-H. Hsu, H.-W. Yang, K.-F. Lin and M.-H. Weng, A density functional theory study on the structure stability of silica nanoclusters, J. Nanosci. Nanotechnol., 2013, 13, 1414–1417 CrossRef CAS .
- P. Chetri, B. Bora and T. K. Baishya, Molecular graph theory based study on Lin cluster: a correlation between physical property and topological descriptors, Bull. Mater. Sci., 2021, 44, 212 CrossRef CAS .
- C. H. Yao and Y. D. Li, Evolution of the structural and electronic properties of AlnP13−n (n = 0–13) clusters, Theor. Chem. Acc., 2022, 141, 53 Search PubMed .
- J. Xiang, X. H. Yan, Y. L. Mao, Y. Xiao and S. H. Wei, Structural, electronic and magnetic properties of small TinAl (n = 1–8) clusters, Int. J. Mod. Phys. C, 2004, 15, 775–782 CrossRef CAS .
- Y. Gao, S. Bulusu and X. C. Zeng, Gold-caged metal clusters with large HOMO–LUMO gap and high electron affinity, J. Am. Chem. Soc., 2005, 127, 15680–15681 CrossRef CAS PubMed .
- K. Krzymiski, P. Malecha, B. Zadykowicz, A. Wróblewska and J. Błaejowski,
1H and 13C NMR spectra, structure and physicochemical features of phenyl acridine-9-carboxylates and 10-methyl-9-(phenoxycarbonyl)acridinium trifluoromethanesulphonates-alkyl substituted in the phenyl fragment, Spectrochim. Acta A, 2011, 78, 401–409 CrossRef .
- M. Svanqvist and K. Hansen, Non-jellium scaling of metal cluster ionization energies and electron affinities, Eur. Phys. J. D, 2009, 56, 199–203 CrossRef .
- Y. Erdogdu and S. Erkoç, Evolution of the electronic structure and properties of charged titanium doped aluminum nanoclusters, Comput. Mater. Sci., 2013, 79, 599–610 CrossRef CAS .
- V. K. Kochnev, Finite element method for atoms, Chem. Phys., 2021, 548, 111197 CrossRef CAS .
- S. Kaya and C. Kaya, A new equation based on ionization energies and electron affinities of atoms for calculating of group electronegativity, Comput. Theor. Chem., 2015, 1052, 42–46 CrossRef CAS .
- R. A. Alvarez B, N. S. Flores-Lopez, G. Calderón-Ayala, R. B. Hurtado, M. Cortez-Valadez and M. Flores-Acosta, First-principles calculations of gold and silver clusters doped with lithium atoms, Phys. E, 2019, 109, 78–83 CrossRef CAS .
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Hongping
Xiao
a,
Yueyu
Zhang
*a,
Hongping
Xiao
a,
Yueyu
Zhang