
Don’t forget the trans: double bond isomerism radical-acetylene growth reactions affect the primary stages of PAH and soot formation†
Patricia D.
Kelly
 *a,
Jack A.
Turner
a,
Oisin J.
Shiels
a,
Gabriel
da Silva
b,
Stephen J.
Blanksby
c,
Berwyck L. J.
Poad
c and
Adam J.
Trevitt
*a
*a,
Jack A.
Turner
a,
Oisin J.
Shiels
a,
Gabriel
da Silva
b,
Stephen J.
Blanksby
c,
Berwyck L. J.
Poad
c and
Adam J.
Trevitt
*a
aSchool of Chemistry and Molecular Bioscience, University of Wollongong, Wollongong, NSW, Australia. E-mail: paddy@uow.edu.au; adamt@uow.edu.au
bDepartment of Chemical Engineering, The University of Melbourne, Melbourne, Victoria, Australia
cCentral Analytical Research Facility and the School of Chemistry and Physics, Queensland University of Technology, Brisbane, Queensland, Australia
First published on 28th November 2024
Abstract
In combustion, acetylene is a key species in molecular-weight growth reactions that form polycyclic aromatic hydrocarbons (PAHs) and ultimately soot particles. Radical addition to acetylene generates a vinyl radical intermediate, which has both trans and cis isomers. This isomerism can lead to profound changes in product distributions that are as yet insufficiently investigated. Herein, we explore acetylene addition to substituted trans-vinyl radicals, including potential rearrangement to cis structures, for eight combustion-related hydrocarbon radicals calculated using a composite method (G3X-K). Of these eight systems, the phenyl trans- and cis-Bittner–Howard HACA (hydrogen abstraction, C2H2 Addition) process, where acetylene successively adds to a phenyl radical via a β-styryl intermediate, is simulated using a unified Master Equation model. Including the trans-Bittner–Howard pathway changes the products significantly at all simulated temperatures (550–1800 K) and pressures (5.33 × 102–107 Pa), relative to a cis-only model. Typically, naphthalene remains the dominant product, but its abundance decreases at higher temperatures and pressures. For example, at 1200 K and 105 Pa, its branching ratio decreases from 78.5% to 62.9% when the trans pathway is included. At higher temperatures this decrease corresponds to the formation of alternative C10H8 isomers, including the cis product benzofulvene with 8% maximum abundance at 1200 K and 5.33 × 102 Pa, and E-2-ethynyl-1-phenylethylene, a trans product with 26% maximum abundance at 1800 K, with little pressure dependence. At higher pressures, our model predicts a range of C10H9 radicals, including resonance-stabilised radicals (RSRs). The impact of trans-vinyl radical chemistry in reactive environments means that they are essential to accurately describe combustion reactions and inhibit soot formation.
Introduction
Molecular-weight growth radical chemistry is central to understanding combustion chemistry and it results in the formation of polycyclic aromatic hydrocarbon (PAH) species that eventually seed the formation of soot particles. Both PAHs and soot particles are widely known to contribute strongly to the negative health impact of polluted air1–4 – so accurate models are crucial to predict and mitigate these impacts. Although the process of soot formation by molecular-weight growth has been qualitatively understood for decades,5–8 the precise chemical mechanisms remain a focus of contemporary experimental and computational research.4,7–9As computational methods have increased in power, a greater scope of chemical species have been included in models of hydrocarbon molecular-weight growth.7,8,10–17 Numerous models implicate the reaction between a hydrocarbon radical and an unsaturated closed–shell co-reactant, such as acetylene.18–20 Acetylene is one of the most abundant small hydrocarbons implicated in the formation of larger species.6,21 Radical addition to the acetylene triple bond yields odd-electron adducts with a vinyl (sp2) radical that can interconvert between trans and cis isomeric forms. Scheme 1 shows an example of such an adduct, the β-styryl radical, which has trans (Z-βS)‡ and cis (E-βS) forms that can easily isomerise. However, as shown in Scheme 1, when a second acetylene molecule adds to the radical intermediate, any subsequent trans/cis isomerism between the trans Z-E-SVY and the cis Z-Z-SVY is “locked out” by the high rotational barrier of the double bond. This effect has been noted for vinyl radicals in the solution phase, where differences in reactivity between trans and cis isomers are often observed.22,23 Though many acetylene-radical reactions feature trans and cis vinyl radical adducts, including the Bittner–Howard pathway in the well-known hydrogen abstraction, C2H2 addition (HACA) mechanism, few studies have explicitly examined pathways leading from the trans reactive intermediate.5,24,25
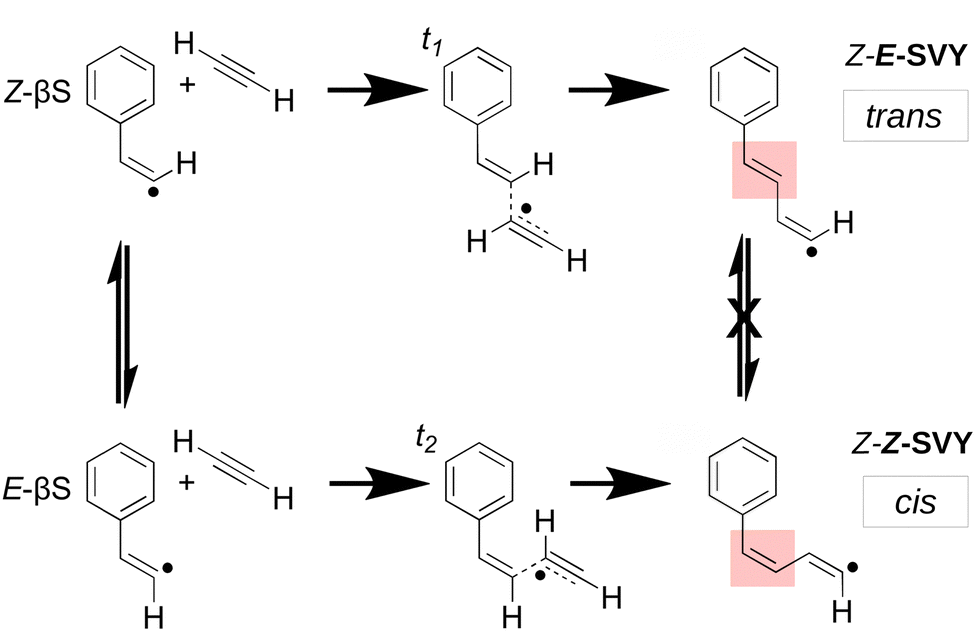 | ||
| Scheme 1 Acetylene addition to both (trans) Z- and (cis) E-β-styryl radicals via transition states t1 and t2, beginning the trans and cis-Bittner–Howard pathways. Z- and E-β-styryl radicals can rapidly interconvert by the movement of one hydrogen atom, whereas the adducts are unable to easily isomerise via rotation of the highlighted double bond. | ||
Scheme 2 shows the general phenyl HACA mechanism, long considered the key process for the growth of benzene to naphthalene and beyond in flames. This mechanism comprises multiple pathways; among these are the Bittner–Howard, Frenklach and modified Frenklach pathways.5,6,25,26 Both the Bittner–Howard and modified Frenklach pathways involve secondary C2H2 addition to a C8H7 radical isomer. Recent work by Mebel et al. argues that these pathways are unrealistic under typical low-pressure flame conditions due to the instability of C8H7 radicals above 1650 K at 1 atm, leading to a low equilibrium concentration of C8H7 relative to C6H5 + C2H2.25 Experiments and further modelling by Green et al.27 focused specifically on these low-pressure, low-temperature conditions, and predicted that ca. 70% of naphthalene produced in the HACA reaction arises via the Bittner–Howard pathway at 700 K, 10 torr at a reaction time of 1 ms. However, modelling by Green et al. of the Bittner–Howard pathway incorporated the radical intermediates calculated by Mebel et al., but these did not appear to include the trans-Bittner–Howard pathway.
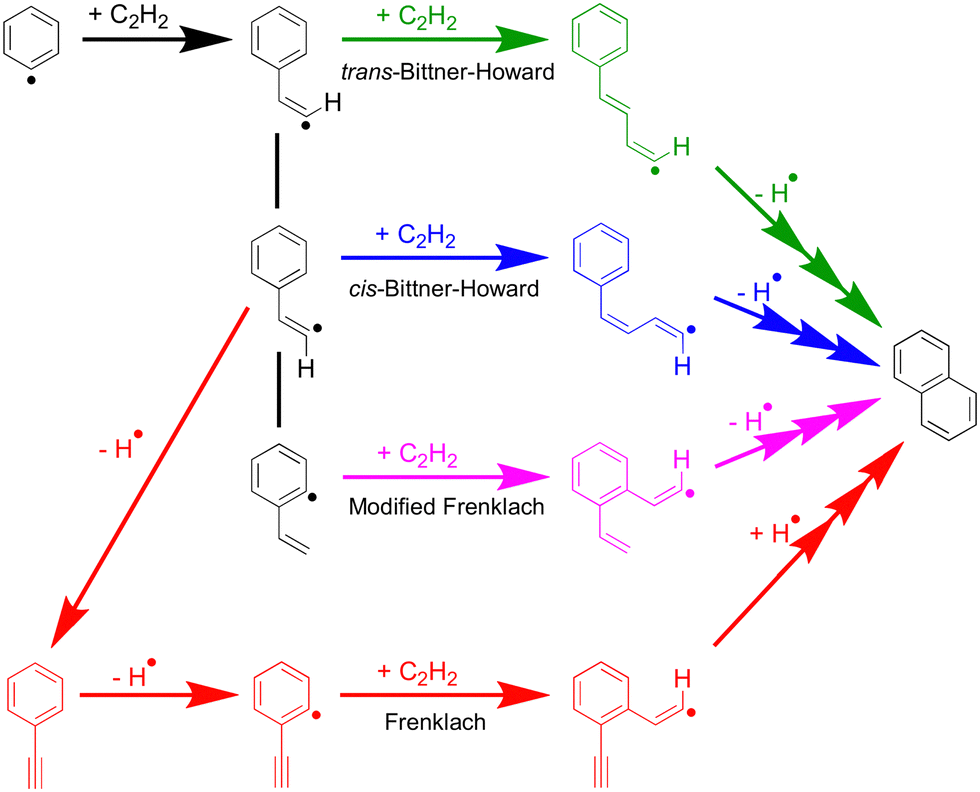 | ||
| Scheme 2 Simplified pathways of the general phenyl HACA mechanism. Green is trans-Bittner–Howard, blue is cis-Bittner–Howard, pink is modified Frenklach and red is Frenklach. Ring closing in the green pathway is inhibited by the high barrier for rotation about the double bond. Multiple-headed arrows indicate a pathway with multiple intermediates. | ||
An important class of molecules in combustion and soot formation are resonance-stabilised radicals (RSRs) and these readily react with other radicals or closed–shell species, while also being relatively long-lived.4,7,8,28–30 The prior work by Mebel25 predicts a variety RSRs arising from HACA reactions, including hydronaphthyl RSRs from both the cis-Bittner–Howard and modified Frenklach pathways. However, within the cis-Bittner–Howard mechanism the only RSR present is an intermediate that leads directly to naphthalene formation, and is only predicted to have significant yield at 100 atm and temperatures below ca. 1000 K. At the lower pressures studied by Green et al., naphthalene is expected to form via so-called “formally direct” pathways, involving well-skipping over RSR intermediates due to the lack of collisional stabilisation.27 Such well-skipping to form naphthalene is not possible in the trans-Bittner–Howard pathway,24,25 however, and this indicates that the inclusion of this pathway may have a large effect on product branching ratios.
In this paper, a detailed theoretical reaction rate model and analysis of both the trans- and cis-Bittner–Howard HACA pathways is reported for an extensive set of C10H9 and C10H8 isomers. Branching ratios for all intermediates and products are reported at temperatures from 550 to 1800 K, and pressures from 5.33 × 102 to 107 Pa. An important conclusion of this paper is that trans/cis isomerism has a significant outcome on predicted branching ratios over large temperature and pressure ranges, and compared with prior modelling the C10H9 RSRs and alternative C10H8 isomers (such as benzofulvene) are much more abundant.
Methods and model description
Simulation of the trans- and cis-Bittner–Howard pathways was achieved using a master equation (ME) model within the MultiWell 2020 program.31–33 The model contains 38 intermediate C10H9 minima (or wells), 78 transition states and 10 dissociation channels including the 2 possible entry channels. This model is a more comprehensive simulation of the secondary reaction than previously published studies. Gaussian 1634 was used to calculate the electronic energy of each structure at the G3X-K level of theory,35 with transition states located by redundant coordinate scans and verified by visual inspection of the imaginary frequency. In cases where visual inspection alone could not unambiguously identify connectivity of minima and transition states, intrinsic reaction coordinate (IRC) calculations were also used to confirm this connectivity. The same model chemistry was also used to calculate a trans pathway for a broad variety of acetylene-radical reactions.RRKM theory is adopted for k(E) calculations, on the basis of sums and densities of states. To calculate the density of states for each stationary point, a RRHO (rigid rotor + harmonic oscillator) model was augmented with hindered rotor potentials to replace the harmonic oscillator descriptions for sufficiently facile bond rotations. Hindered rotor potentials were calculated using relaxed coordinate scans with a step size of 10 degrees. The energies at each point were then fit with a Fourier series comprising nineteen cosine and sine terms. Moments of inertia were estimated at a single point. Collisional energy transfer was described using a single-exponential-down model with parameters based on those of toluene in a He bath gas.36,37 Toluene was chosen as it is a chemically similar species to most of the C10H9 isomers with established parameters, and the effect of altering these parameters is less significant than the uncertainty introduced by errors in the relative energies.25
In total, sixty different sets of initial conditions were simulated, with each simulation including 107 Monte Carlo trials. These conditions described the reaction at six different temperatures between 550 K and 1800 K, and five different pressures between 5.33 × 102 Pa (4 torr) and 107 Pa (7.5 × 104 torr), for two different initiating reactions. The lowest pressure and temperature values were chosen to correspond to concurrent photoionisation mass spectrometry experiments,38 discussed in the ESI† and to be further detailed in a future publication. The initial formation of both trans and cis isomers was incorporated by running simulations starting as chemically activated populations of both initial wells, which can subsequently interconvert, isomerise and dissociate independently. Each of the sixty total simulations were calculated to 1000 collisions, which was sufficient for most of the excess energy to be dissipated or to completely dissociate the C10H9 intermediates. Tertiary addition of another acetylene molecule was deemed to be negligible on this timescale. Species with an sp2 radical and an α-hydrogen were summed together after calculation due to the very low interconversion barrier between these species. Following ref. 39 and 40, we assume that intramolecular vibrational energy redistribution (IVR) is complete in less than a few picoseconds.
To determine the relative contribution of the trans and cis pathways to the overall reaction, the partition functions for entry transition states t1 and t2 were calculated, as labelled in Scheme 1. Transition state theory provides the following equation:41
| k(T) = FwZt(T)e−βEt |
Results and discussion
This first section contains a general overview analysis of trans/cis isomerism chemistry of vinyl radical intermediates involved in acetylene molecular-weight growth chemistry considering a range of fundamental combustion-related hydrocarbon radicals. Of these radicals, the reaction of the phenyl radical with acetylene via the Bittner–Howard pathway was selected for further analysis using a detailed ME model spanning a range of pressure and temperatures, and this is examined in more detail further below. With this detailed ME model at hand, the branching ratio of each species was calculated as a function of pressure and temperature – this allows for comparison to previous studies in the literature. Finally, the contribution of the trans pathway is assessed by calculating the change in branching ratio when it is included and compared with when it is omitted.The trans/cis isomerism in hydrocarbon radical-acetylene reactions
As a starting point, to assess the significance of the trans reaction within the context of acetylene molecular-weight growth reactions, a series of analogous pathways across a range of fundamental hydrocarbon radicals were calculated using the G3X-K method, and these are shown together in Fig. 1. This figure depicts the energy landscape for the catalytic vinylacetylene formation pathway for the RSRs propargyl (CHCCH2) and allyl (CH2CHCH2), together with selected saturated (methyl (CH3), ethyl (C2H5) and propyl (C3H7)) and unsaturated (ethenyl (CH2CH), phenyl (c-C6H5) and ethynyl (C2H)) non-resonance-stabilised radicals. As these pathways do not involve significant R-group interactions, it is a reasonably straightforward comparison. Fig. 1(a) depicts the energy of the primary addition (n = 1) of acetylene to the radical, which forms the adduct complex via a single transition state. For most cases, the transition-state energy is positive with respect to the separated reactants, except for the ethynyl radical case, which is slightly submerged. Fig. 1(b) is a portion of the secondary (n = 2) addition of acetylene to the primary radical adduct. This n = 2 pathway contains a series of H atom shifts, followed by an interconversion between linear and branched isomers that form via a three-membered cyclic intermediate. If viable, this pathway is an alternative pathway to form vinylacetylene, rather than from the reaction of liberated H atoms with acetylene.42 Across both Fig. 1(a) and (b), most intermediates and transition states have relative energies that do not strongly depend on the R group. The differences between each case are most apparent for the primary addition step (Fig. 1(a)) and in the final regeneration of the starting radical (Fig. 1(b)). These points are marked with a black downward arrow, and their spread in energy values is attributed to the varying radical stabilisation energies (RSEs) of each radical, with relatively stable conjugated radicals such as allyl and propargyl having the largest RSE values.43 More localised radicals such as ethynyl, and to a lesser extent ethenyl (vinyl) and phenyl, exhibit the opposite trend and have the highest values. Indeed, Fig. 1(b) shows similar n = 2 energetics across all R groups as the n = 1 adduct always bears an sp2 radical site, and therefore the RSE for each adduct is similar. Furthermore, intermediates along the catalytic vinylacetylene formation pathway have similar energy (relative to their respective n = 1 adducts separated from acetylene), as the interaction with the R group is in most cases quite limited.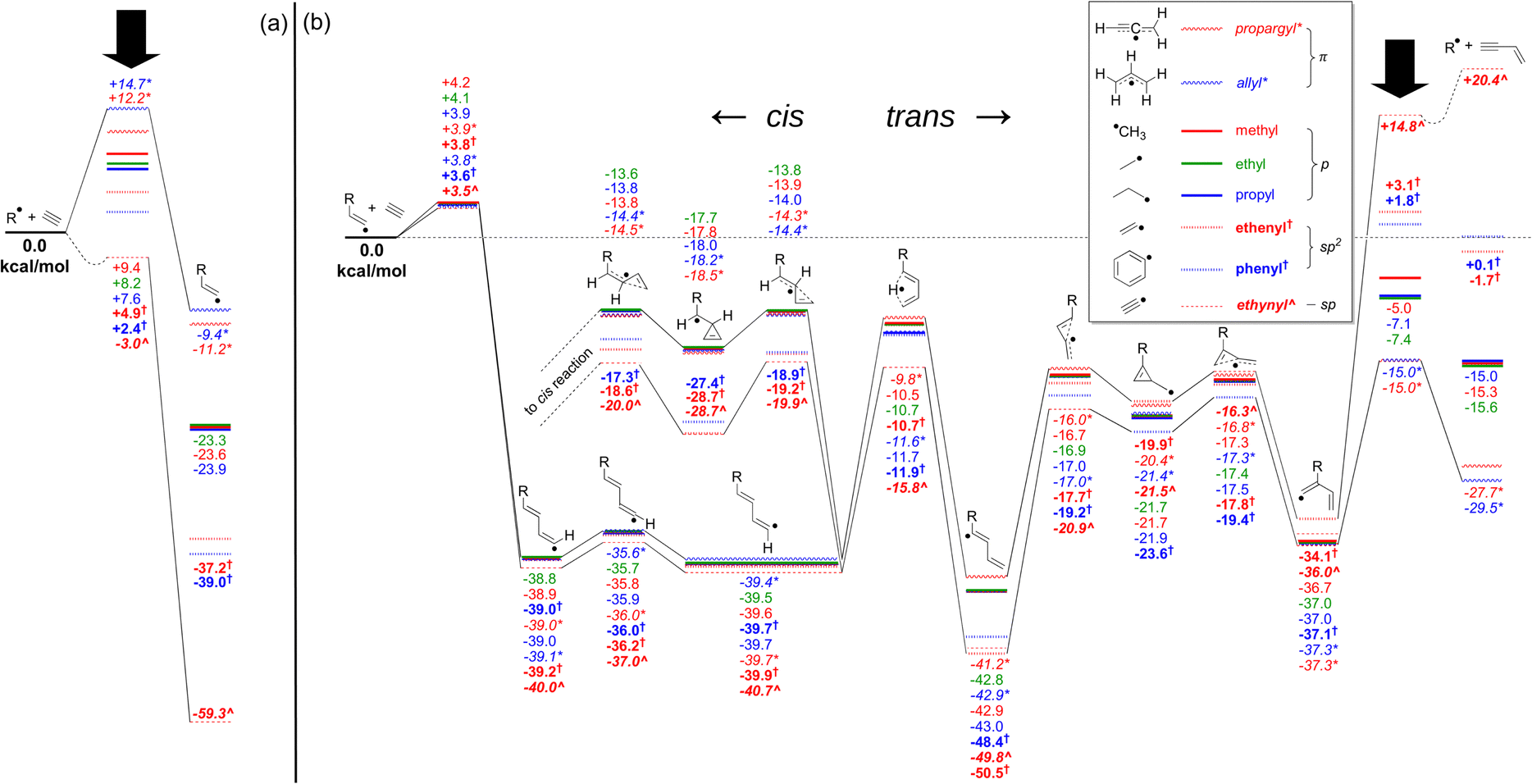 | ||
| Fig. 1 G3X-K energetics, in kcal mol−1, of selected pathways of (a) primary, and (b) trans secondary addition to various radicals. Catalytic vinylacetylene generation progresses to the right, competing with the isomerisation pathway to the cis reaction branching back to the left (the products of which are not shown). Solid arrows indicate the initial formation and final dissociation step. The colours, symbols and text styles are matched to the radical species depicted in the legend. Dotted wells in the adduction or fragmentation step indicate the existence of a pre- or post-reactive complex, the calculation of which was outside the scope of this study. | ||
The primary isomerisation step that connects the trans to the cis pathway also involves a three-member cyclic intermediate and is also only weakly affected by the R-group. Of these, the highest-energy barrier resides between −13.6 and −20.0 kcal mol−1, relative to the separated reactants. The small differences are mostly attributed to resonance stabilisation of the aforementioned three-member cyclic intermediate in cases where the precursor radical site is sp2 or sp (e.g. the phenyl radical), which does not occur for p- or π-type precursor radicals. Therefore, this pathway is more facile in the phenyl case than for most others. Nonetheless, in all cases the three-member cyclic intermediate geometry likely indicates a low density of vibrational states – and so entropic effects may allow higher-energy trans pathways to remain relevant. If these entropic effects allow the trans reaction to contribute significantly even in cases where the interconversion is stabilised, such as for the phenyl radical in the Bittner–Howard pathway, our recommendation is that it must also be included for the non-stabilised cases.
Since the reactions of the phenyl radical (the solid blue pathway in Fig. 1) are implicated in many soot formation models, this pathway is selected for further scrutiny and modelled in detail. To ascertain if the trans pathway of the Bittner–Howard reaction leads to significantly different products, a detailed chemical model is presented below, with branching ratios assessed as a function of pressure, temperature, and trans/cis isomerism.
Product formation in the Bittner–Howard reaction
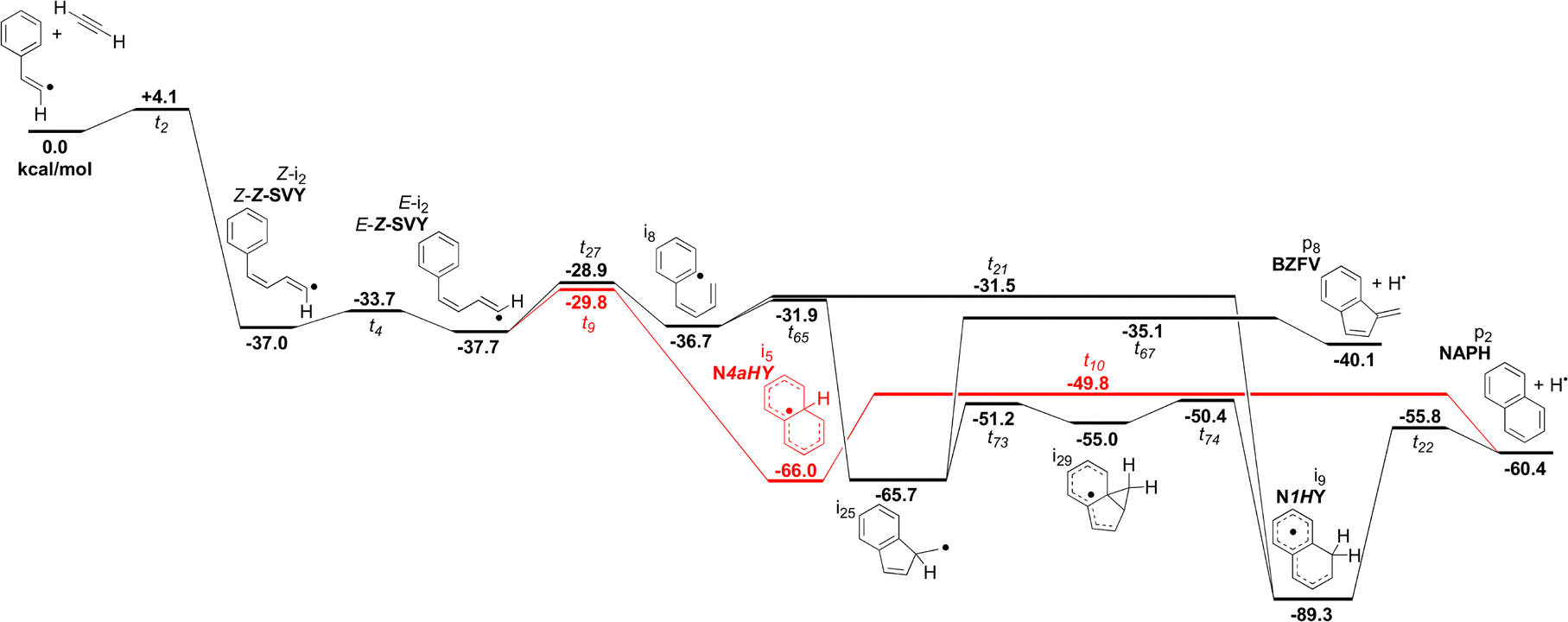
The simplest definition of the Bittner–Howard reaction is a naphthalene-forming reaction between phenyl and acetylene where a second acetylene adds to a β-styryl radical (Scheme 1). In this section, we expand this definition to include the full surface and range of products for the reaction of the β-styryl radical with acetylene. This broader scope includes the trans- and cis-Bittner–Howard variants that depend on the stereoisomerism of the β-styryl radical site. Together, these variants comprise a significant portion of the C10H9 surface. Fig. 2 presents all minima, transition states, reactants and products of this surface modelled in this work. The starting β-styryl radicals are enclosed in red ellipses. For each of these points, the energy relative to separated reactants is provided in the ESI† (Table S2). The left side of Fig. 2 is the trans pathway, with the cis pathway on the right (separated by the dashed curved line). Nine connecting transition states are noted between the trans and cis pathways, most of which are high in energy or are linked to uncommon intermediates. These transition states involve rotation about the α–β bond, either directly or via an intermediate. Once one of the two adducts form, several rearrangements can take place – the most important of which are shown in more detail later. Some rearrangements lead to dissociation, either to re-form the β-styryl radicals, H-loss (C10H8) products (blue rectangles), or the previously described vinylacetylene loss pathway (green diamond). Intermediates and products discussed in the text are labelled with a bold abbreviation.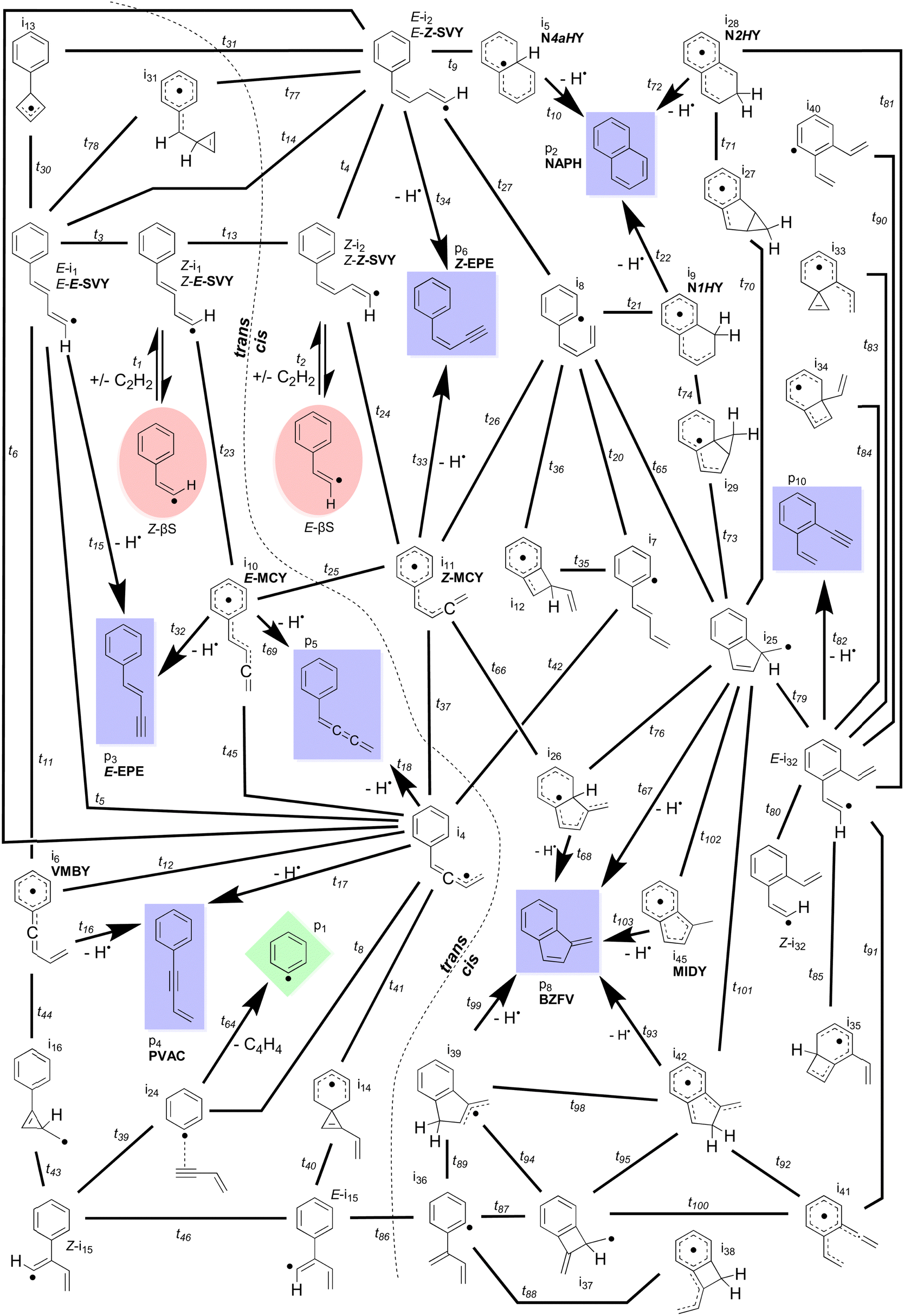 | ||
| Fig. 2 Full network of the modelled intermediates, transition states and exit channels of the C10H9 surface in the Bittner–Howard reaction. Each bold line corresponds to an isomerisation transition state, with arrows depicting attachment and/or detachment. The starting β-styryl C8H7 isomers are enclosed in red ellipses, C10H8 isomers in blue rectangles, and C6H5 in a green diamond. A dotted line approximately divides the trans- (left) and cis-Bittner–Howard (right) pathways. Intermediates and products discussed in the text are labelled with a bold abbreviation, e.g.NAPH. | ||
The points on the surface described in Fig. 2 form the basis of the ME model described in this work, which predicts branching ratios for each species. Of the thirty-four C10H9 intermediates and seven C10H8 products, Scheme 3 lists the fourteen most abundant in the ME model. These species include bicyclic or monocyclic structures, with the bicyclic radicals resulting from the cis pathway, and the monocyclic species mostly from the trans pathway. Aside from the closed–shell C10H8 isomers (top), various open–shell C10H9 species are present, including both of the n = 2 adducts as well as a variety of RSRs. The variation in branching ratios of these species under different conditions is discussed below.
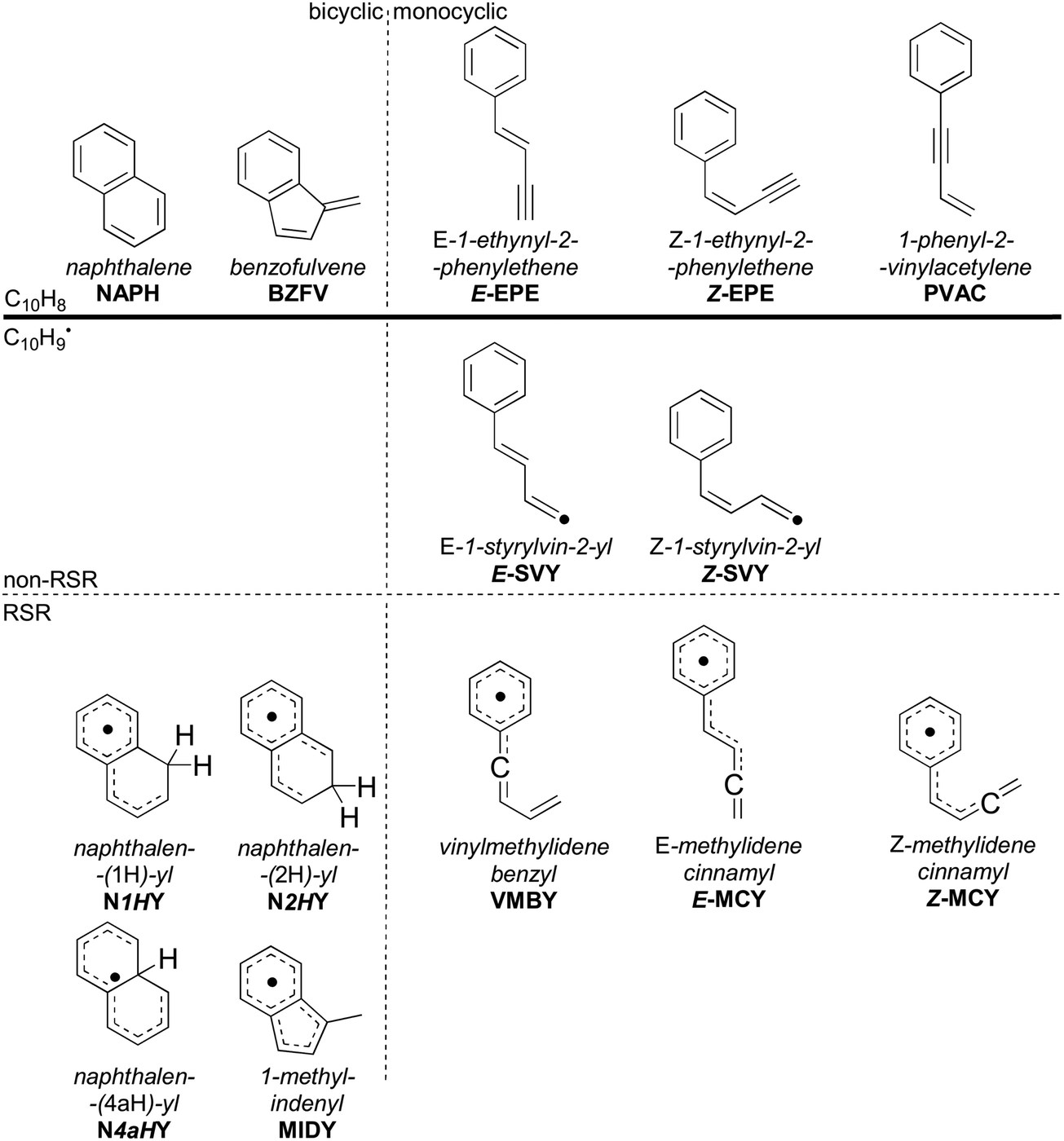 | ||
| Scheme 3 Modelled species with a branching ratio exceeding 1% in any set of simulated pressure and temperature conditions, including the abbreviations used in the text. Species are divided by mass, number of rings, and presence of resonance-stabilised radical sites. | ||
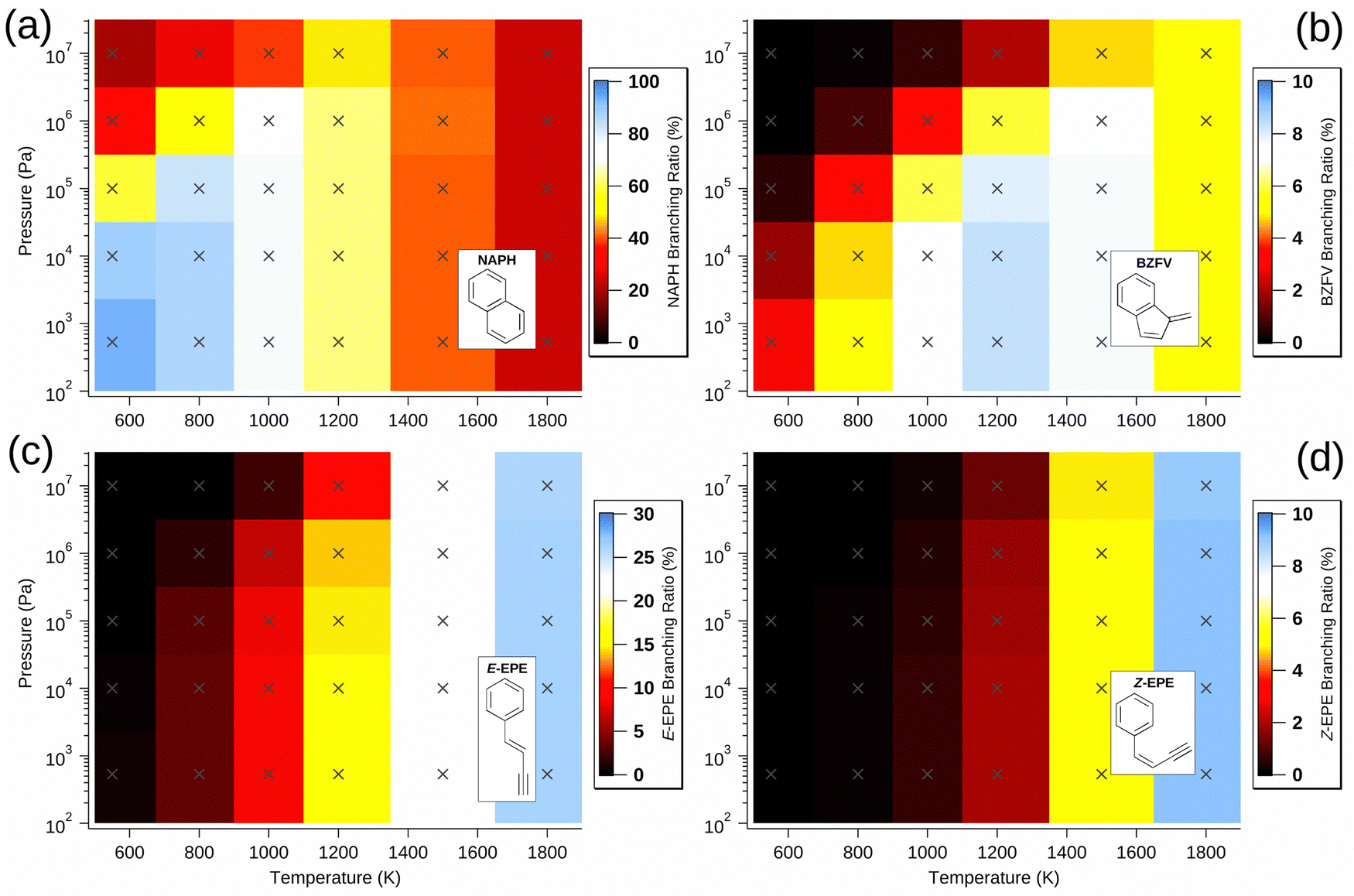 | ||
| Fig. 3 Predicted branching ratio of (a) NAPH, (b) BZFV, (c) E-EPE and (d) Z-EPE, as a function of pressure and temperature. Crosses designate points at which the model was calculated. Colour is used to indicate the value of the branching ratio at these pressure and temperature coordinates – note that the intensity scales are different for each plot. | ||
Though most non-NAPH C10H8 isomers are produced in greater abundance as temperature increases, with only a weak pressure dependence, benzofulvene (BZFV) is most prevalent at low pressure and intermediate temperature and this is due to its production from the cis-Bittner–Howard pathway and its relative stability compared to the monocyclic isomers. Since it is a product of the cis pathway along with NAPH, BZFV is formed under generally similar conditions, but the lower threshold energy for formation of NAPH relative to BZFV results in preference for the former at the lowest temperatures. However, the integrated density of states of BZFV grows more rapidly than that of NAPH as temperature increases, somewhat offsetting this effect at higher temperature. At conditions of 5.33 × 102 Pa and 1200 K, BZFV has a predicted branching ratio exceeding 8% – despite not appearing to be implicated in previous studies.
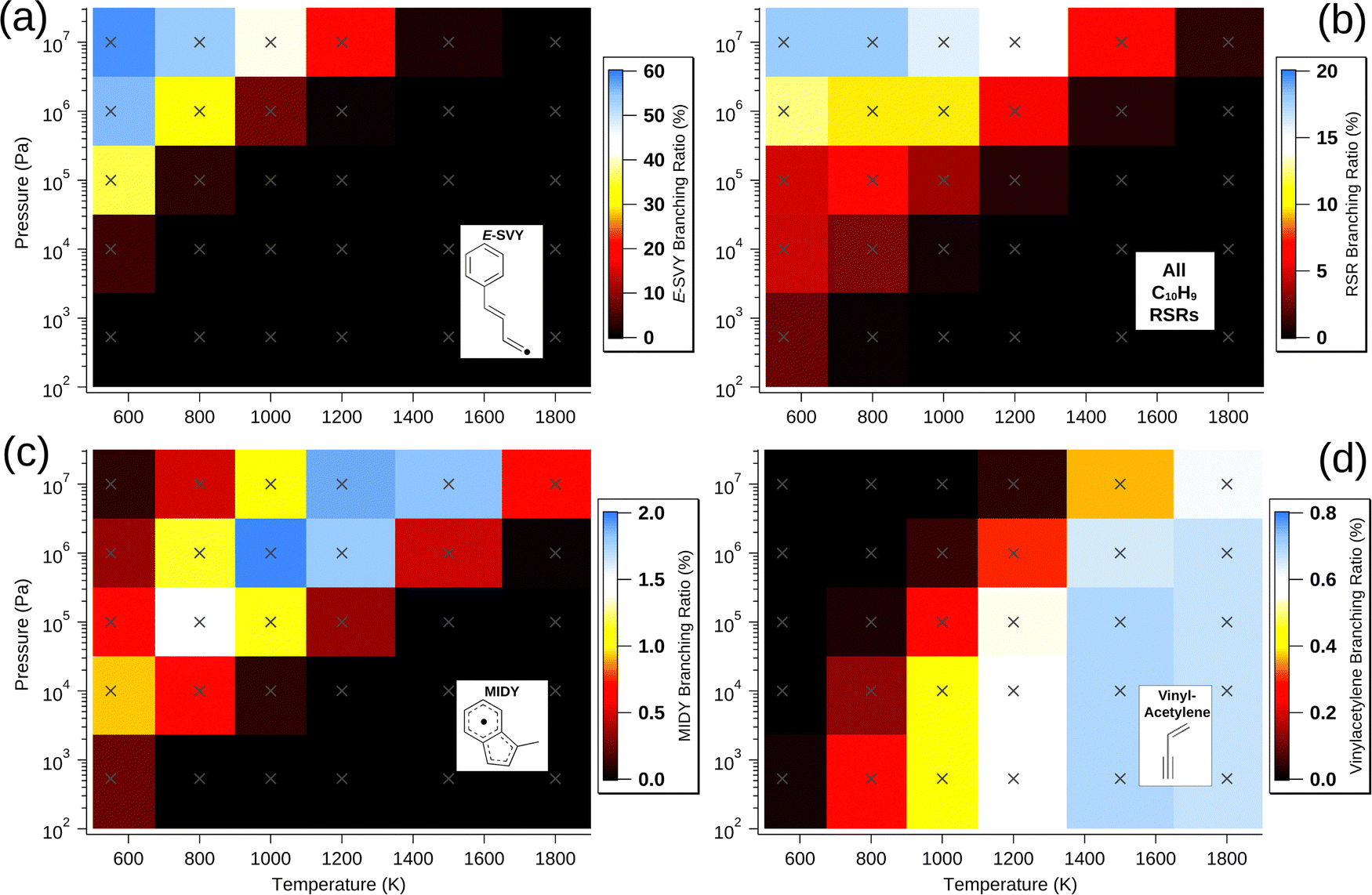 | ||
| Fig. 4 Predicted branching ratio of (a) E-SVY, (b) all RSRs, (c) MIDY and (d) vinylacetylene (with phenyl co-fragment), as a function of pressure and temperature. Crosses designate points at which the model was calculated. Colour is used to indicate the value of the branching ratio at these pressure and temperature coordinates – note that the intensity scales are different for each plot. | ||
Aside from the E- and Z-SVY adduct radicals, all of the radical intermediates that are present in appreciable (>1%) quantities are RSRs. These are vinylmethylidene benzyl (VMBY), MIDY, naphthalen-(1H)-yl (N1HY), naphthalen-(2H)-yl (N2HY), naphthalen-(4aH)-yl (N4aHY), E-methylidene cinnamyl (E-MCY) and Z-methylidene cinnamyl (Z-MCY) and Fig. 4(b) shows the total branching ratio for these and all other RSRs summed together. Together, these RSRs comprise 18% of all products at 107 Pa and 800 K. Generally, as expected, higher pressure and lower temperature are conditions that favour these species, as with all C10H9 radicals in the model. However, the dependence on both pressure and temperature is much weaker than for E- and Z-SVY. RSRs are present at abundances exceeding 1% for sixteen of the thirty simulated pressures and temperatures, indicating that the Bittner–Howard pathway may often contribute to RSR-based molecular-weight growth.
Of note among RSRs is also MIDY, shown in Fig. 4(c). MIDY is a substituted indenyl radical – the indenyl radical is much less prone to decomposition at intermediate temperatures compared with many other fuel-derived radicals.30 Indenyl radicals have been studied as a target for RSR-based molecular-weight growth,4,8,30 and therefore the formation of MIDY in the Bittner–Howard pathway should not be overlooked. Interestingly, MIDY is less present ca. 550 K and 107 Pa, likely due to the many-step process required for its formation and competition with other radicals that are more stable at lower temperature. Though our analysis predicts MIDY has a branching ratio of only a few percent at most, its relative persistence at higher temperatures indicates that the Bittner–Howard pathway in the phenyl HACA reaction can contribute to RSR-based molecular-weight growth even under these conditions.
A minor channel that is neither C10H8 nor C10H9 is the loss of vinylacetylene to regenerate phenyl radicals. As discussed above, this bimolecular product channel is the trans-specific final step of a radical-catalysed acetylene dimerisation that is possible for a wide variety of radicals beyond just the phenyl radical (with β-styryl intermediate). The branching ratio of this vinylacetylene formation in the Bittner–Howard case is shown in Fig. 4(d). The maximum branching ratio of ca. 0.7% is achieved between 1500 and 1800 K and from pressures ranging from 5.33 × 102 to 106 Pa, with a somewhat weak dependence in this region. Though vinylacetylene production via this mechanism appears uncommon in the Bittner–Howard case, equivalent pathways for p-type alkyl radicals may be more likely and should not be ignored in future studies.
Key trans- and cis-Bittner–Howard pathways
Both the trans- and cis-Bittner–Howard pathways are present on the C10H9 landscape, have similar starting internal energies, and pathways for interconversion. Importantly, the inclusion of the trans-Bittner–Howard pathway measurably changes the distribution of products. The key pathways for both the trans and cis reactions must therefore be considered – particularly with regard to the most likely rearrangement between them.The trans-Bittner–Howard pathways with the greatest contribution to our model are shown in Fig. 5. In prior studies, the trans addition to β-styryl radicals is either absent,27 mentioned briefly,24,25 or assumed to isomerise to the cis configuration with no explicit pathway provided.5 However, the formation of E-styrylvinyl radicals (E-SVY), as well as of the monocyclic C10H8 isomers E-EPE and 1-phenyl-2-vinylacetylene (PVAC), is competitive with the most favourable cis isomerisation pathway, which proceeds over t78, i31 and t77 towards NAPH (shown in blue in Fig. 5). This cis isomerisation pathway is analogous to those shown in Fig. 1 for a variety of R-groups. The reaction rate across the transition states t77 and t78 is inhibited by their low density of states. Furthermore, the presence of two “gatekeeper” transition states rather than one significantly slows the rate of isomerisation from E-SVY to Z-2-styrylvinyl (Z-SVY). If the isomerisation continues across t77 to the cis portion of the surface, however, return is unlikely, and the reaction proceeds similarly to the cis case.
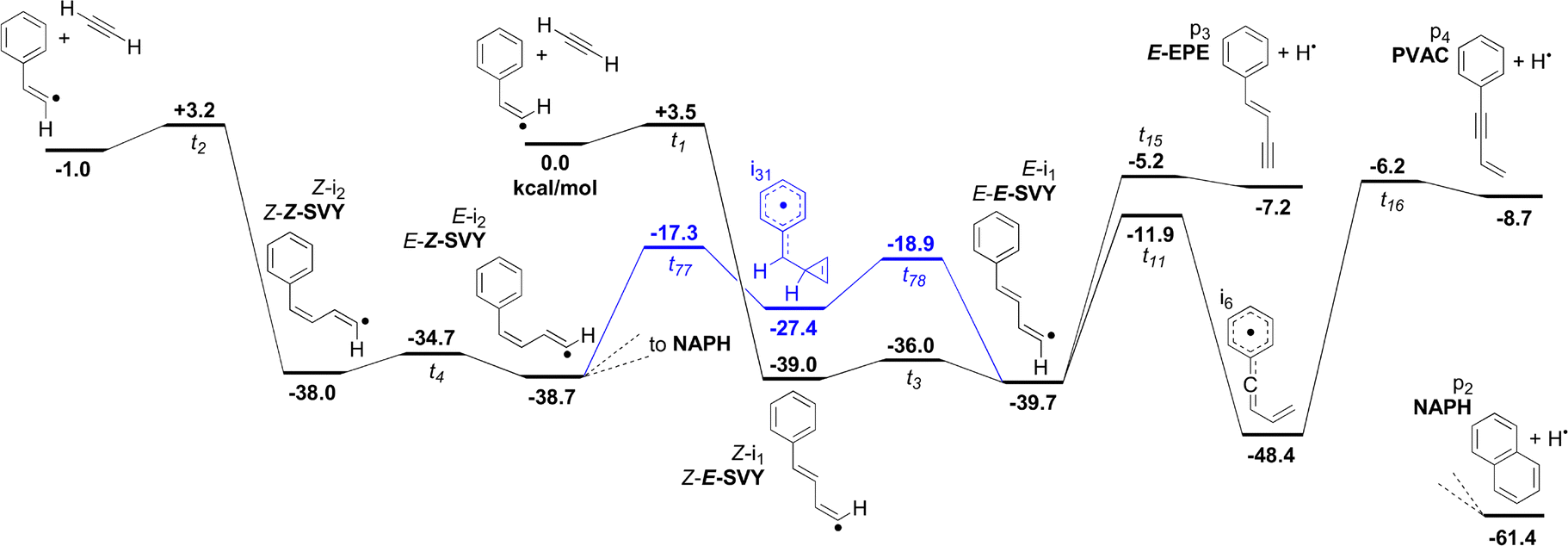 | ||
| Fig. 5 Selected pathways from the trans precursor towards the formation of E-EPE, PVAC, VMBY and E-SVY, as well as the most facile isomerisation to join to the cis surface. The trans/cis isomerisation pathway is shown in blue. NAPH is shown for comparison – see Fig. 6 for further detail. | ||
The cis addition yields three pathways that lead to NAPH formation, presented in Fig. 6. Highlighted in red is a previously reported pathway.25 The black pathways are competitive with the red pathway and are a source of both the BZFV and NAPH products. In particular, BZFV is an alternative product to the ring expansion pathway leading to NAPH. As discussed above, BZFV production maximises at moderate temperatures and low pressures, with a branching ratio of ca. 8% at 5.33 × 102 Pa and 1200 K. In contrast, NAPH production is most favoured at low temperature. This difference is attributed to a more relaxed (more loose) BZFV transition state (t67), which has a strong positive temperature dependence. Though the BZFV branching ratio decreases above 1200 K, NAPH has a faster rate of relative decrease, such that BZFV is ca. 23% of NAPH production at 1800 K and 5.33 × 102 Pa. The overall decrease in NAPH and BZFV within only the cis addition pathway is the result of competition between redissociation and with formation of monocyclic C10H8 isomers, particularly Z-EPE.
 | ||
| Fig. 6 Selected pathways from the cis precursor towards formation of NAPH and BZFV. The pathway modelled by Mebel et al.25 is shown in red. Note that the cis-β-styryl radical is chosen as the reference energy, leading to slightly offset energies relative to Fig. 5. | ||
Quantifying the significance of the trans pathway
To ascribe a value to the significance of the trans-vinyl radical addition chemistry within our model, the branching ratio for each species can be compared to when the cis reaction is alone considered. This leads to the following formula for the sum of absolute differences (SAD):where i is the chemical species, ϕi,mod is the modelled branching ratio averaged over both entry pathways, and ϕi,cis is the branching ratio of the cis reaction only. The prefactor of
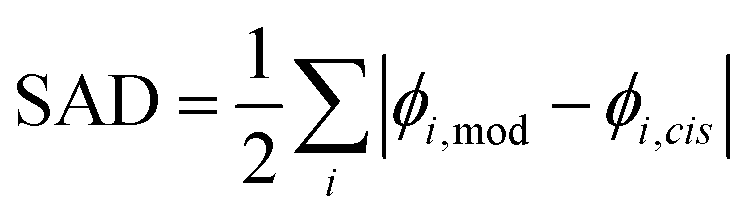
 is included to avoid double-counting, since any decrease in the branching ratio at a particular species must lead to an equivalent increase over some combination of other species. The SAD is thus a value between 0 (no impact) and 1 (wholly dominant) and this quantifies the importance of including the trans pathway. In particular, comparing the combined model with the cis-only model accounts for the effects of the post-adduction surface as well as the relative probabilities of trans and cis addition.
is included to avoid double-counting, since any decrease in the branching ratio at a particular species must lead to an equivalent increase over some combination of other species. The SAD is thus a value between 0 (no impact) and 1 (wholly dominant) and this quantifies the importance of including the trans pathway. In particular, comparing the combined model with the cis-only model accounts for the effects of the post-adduction surface as well as the relative probabilities of trans and cis addition.
Fig. 7 shows the SAD calculated for each temperature and pressure condition. The solid black line represents the maximum potential contribution of the trans reaction, assuming the trans reaction produces totally different products than the cis reaction. This upper bound is derived from the relative population of the trans and cis entry pathways, as calculated from the relative partition functions of the trans and cis transition states. The dotted black line represents a simpler model that ignores the molecular structure, instead only using the energetics of the separated trans and cis reactants. These bounds are discussed in more detail in the Methods & Model Description section. At all temperatures and pressures considered, the inclusion of the trans reaction significantly alters the branching ratios. The amount of change increases with increasing pressure, as earlier intermediates are quenched and stabilise such that this prevents the interconversion between trans and cis pathways. Increasing the temperature has a varying effect, depending on pressure. At the lowest pressures considered, increasing the temperature monotonically increases the effect of the trans reaction. This increase is attributed to the relatively low density of states of key transition states separating the trans and cis reactions, compared with competing pathways. Therefore, entropic effects cause trans/cis interconversion to be disfavoured.
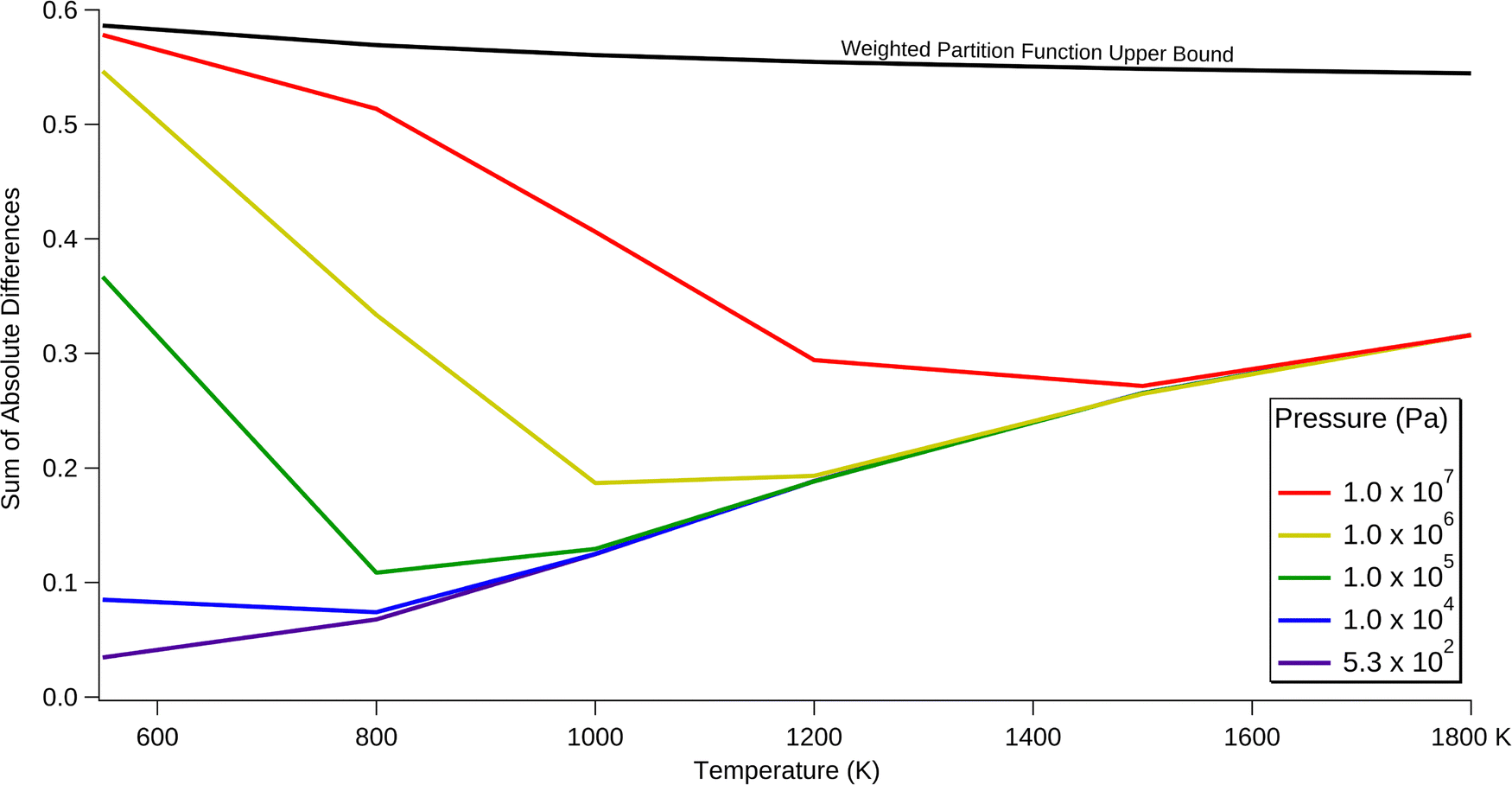 | ||
| Fig. 7 Impact of the trans chemical pathways. The sum of absolute differences (SAD) as a function of pressure and temperature. The lower limit of the blue area is drawn at 5.33 × 102 Pa (4 torr). The weighted partition function upper bound depicts the maximum possible SAD (using the density-of-states weighting scheme between trans and cis starting points as discussed in the methods & model description). | ||
At moderate to high pressures, increasing the temperature first decreases the SAD to the point where it approaches the lowest-pressure case, and then it increases in parallel with this lowest-pressure case. This inversion is possibly due to the decreasing efficiency of collisional deactivation as the temperature of the bath gas increases. Importantly, the trans reaction is most significant at 550 K and 107 Pa, in which it approaches the upper bound of 0.59. Almost all of this is attributed to collision-stabilised E-SVY. Even at atmospheric pressure the trans reaction has a SAD between 0.1 and 0.4 across all considered temperatures, with a maximum at the lowest temperature of 550 K. Thus, it is necessary to include the trans reaction when modelling the Bittner–Howard pathway, regardless of the pressure and temperature.
The SAD value is also useful for testing the robustness of the model to small changes in barrier height. This sensitivity analysis was performed at 1200 K and 105 Pa, corresponding to approximately the midpoint of the temperature and pressure parameters used in this work. By tweaking the energies of the “gatekeeper” transition states between the trans and cis pathways – that is, t77 and t78 – the change in the SAD can be measured. When the barrier heights of these transition states were lowered by 1 kcal mol−1, the SAD decreased from 0.188 to 0.152. A 1 kcal mol−1 increase led to the SAD increasing to 0.229. In either case, the SAD remains the same order of magnitude, leaving the broader argument of this work unchanged. Therefore, while the quantitative values of the SAD may not be perfectly accurate, it can be concluded that the trans reaction must be included in models of aromatic molecular-weight growth chemistry pertinent to the primal stages of soot formation.
Conclusion
The Bittner–Howard pathway, despite its inclusion in HACA models for decades, has concealed complexity that has been underexplored in prior modelling literature. From this expanded modelling study, such pathways are expected to be most prominent at temperatures below 1600 K and pressures equal to or exceeding 105 Pa and these conditions correspond to the greatest quantity and variety of C10H9 radical isomers, including various RSRs. Furthermore, C10H8 comprises a greater diversity than previously described, even at moderate temperatures, including benzofulvene (BZFV) as well as 3 related monocyclic species: E- and Z-2-ethynyl-1-phenylethene (E-EPE and Z-EPE), and 1-phenyl-2-vinylacetylene (PVAC). Though naphthalene (NAPH) is still the C10H8 isomer with the greatest branching ratio below 1800 K, other isomers are competitive from temperatures as low as 800–1000 K, much lower than predicted in earlier studies. Much of the difference can be attributed to the inclusion of trans chemistry in the model, which is most prominent at high pressure and low temperature – contributing the majority of the branching ratio at 550 K and pressures above 106 Pa. Even at the least favourable conditions for the relative contribution of the trans reaction, its inclusion still alters the predicted branching ratios by 4%.Due to the similar ionisation energies of the most abundant predicted C10H8 isomers, experimental verification of the model presented in this work with photoionisation mass spectrometric techniques44 may be challenging. Furthermore, increasing the temperature to encompass more of the model parameter space for the reaction of β-styryl radicals may allow for an enhanced identification of C10H8 isomers.
Finally, many of the trans pathways in the model, including the initial adduction step, do not strongly interact with the phenyl ring such that exchanging the phenyl group with different fundamental substituents results in generally similar energy landscapes, including the isomerisation pathway that connects with the cis reaction. The main differences arise when each radical R group is free, with energies correlated with the RSE of each. Radicals similar to the phenyl radical may therefore have similar acetylene addition reactions that are strongly affected by the inclusion of trans-Bittner–Howard-type pathways. Future Master Equation models of such systems should include these trans pathways. Ultimately, the impact of trans-vinyl radical intermediates on reactive radical chemistry demonstrates that they are necessary for the accurate modelling of combustion reactions and soot formation.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. J. T., G. d. S. and S. J. B. acknowledge financial support for this research from the Australian Research Council (ARC) through the Discovery Project scheme (DP170101596 and DP240100612). The authors also acknowledge the generous allocation of computing resources by the NCI National Facility (Canberra, Australia) under Merit Allocation Scheme. P. D. K., J. A. T. and O. J. S. acknowledge receipt of an Australian Government Research Training Program Scholarship.References
- B. J. Finlayson-Pitts and J. N. PittsJr., Tropospheric air pollution: ozone, airborne toxics, polycyclic aromatic hydrocarbons, and particles, Science, 1997, 276, 1045–1051 CrossRef CAS.
- K.-H. Kim, S. A. Jahan, E. Kabir and R. J. C. Brown, A review of airborne polycyclic aromatic hydrocarbons (PAHs) and their human health effects, Environ. Int., 2013, 60, 71–80 CrossRef CAS.
- I.-Y. L. Hsieh, G. P. Chossière, E. Gençer, H. Chen, S. Barrett and W. H. Green, An Integrated Assessment of Emissions, Air Quality, and Public Health Impacts of China's Transition to Electric Vehicles, Environ. Sci. Technol., 2022, 56, 6836–6846 CrossRef CAS PubMed.
- K. O. Johansson, M. P. Head-Gordon, P. E. Schrader, K. R. Wilson and H. A. Michelsen, Resonance-stabilized hydrocarbon-radical chain reactions may explain soot inception and growth, Science, 2018, 361, 997–1000 CrossRef CAS PubMed.
- J. D. Bittner and J. B. Howard, Composition profiles and reaction mechanisms in a near-sooting premixed benzene/oxygen/argon flame, Proc. Combust. Inst., 1981, 18, 1105–1116 CrossRef.
- M. Frenklach, D. W. Clary, W. C. Gardiner and S. E. Stein, Detailed kinetic modeling of soot formation in shock-tube pyrolysis of acetylene, Proc. Combust. Inst., 1984, 20, 887–901 CrossRef.
- E. Reizer, B. Viskolcz and B. Fiser, Formation and growth mechanisms of polycyclic aromatic hydrocarbons: A mini-review, Chemosphere, 2022, 291, 132793 CrossRef CAS PubMed.
- H. Jin, W. Yuan, W. Li, J. Yang, Z. Zhou, L. Zhao, Y. Li and F. Qi, Combustion chemistry of aromatic hydrocarbons, Prog. Energy Combust. Sci., 2023, 96, 101076 CrossRef.
- V. V. Kislov, A. I. Sadovnikov and A. M. Mebel, Formation Mechanism of Polycyclic Aromatic Hydrocarbons beyond the Second Aromatic Ring, J. Phys. Chem. A, 2013, 117, 4794–4816 CrossRef CAS.
- B. Jursic and Z. Zdravkovski, DFT study of the Diels–Alder reactions between ethylene with buta-1,3-diene and cyclopentadiene, J. Chem. Soc., Perkin Trans. 2, 1995,(6), 1223–1226 RSC.
- R. P. Lindstedt and G. Skevis, Benzene formation chemistry in premixed 1,3-butadiene flames, Proc. Combust. Inst., 1996, 26, 703–709 CrossRef.
- P. M. Woods, T. J. Millar, A. A. Zijlstra and E. Herbst, The Synthesis of Benzene in the Proto-planetary Nebula CRL 618, Astrophys. J., 2002, 574, L167 CrossRef CAS.
- J. A. Miller and S. J. Klippenstein, The Recombination of Propargyl Radicals and Other Reactions on a C6H6 Potential, J. Phys. Chem. A, 2003, 107, 7783–7799 CrossRef CAS.
- F. Zhang, B. Jones, P. Maksyutenko, R. I. Kaiser, C. Chin, V. V. Kislov and A. M. Mebel, Formation of the Phenyl Radical [C6H5(X2A1)] under Single Collision Conditions: A Crossed Molecular Beam and ab Initio Study, J. Am. Chem. Soc., 2010, 132, 2672–2683 CrossRef CAS PubMed.
- E. Wang and J. Ding, Reaction between the i-C4H5 radical and propargyl radical (C3H3): A theoretical study, Chem. Phys. Lett., 2021, 768, 138407 CrossRef CAS.
- E. Georganta, R. K. Rahman, A. Raj and S. Sinha, Growth of polycyclic aromatic hydrocarbons (PAHs) by methyl radicals: Pyrene formation from phenanthrene, Combust. Flame, 2017, 185, 129–141 CrossRef CAS.
- X. Shi, Q. Wang and A. Violi, Reaction pathways for the formation of five-membered rings onto polyaromatic hydrocarbon framework, Fuel, 2021, 283, 119023 CrossRef CAS.
- T. Yu and M. C. Lin, Kinetics of the phenyl radical reaction with ethylene: An RRKM theoretical analysis of low and high temperature data, Combust. Flame, 1995, 100, 169–176 CrossRef CAS.
- A. B. Vakhtin, D. E. Heard, I. W. M. Smith and S. R. Leone, Kinetics of reactions of C2H radical with acetylene, O2, methylacetylene, and allene in a pulsed Laval nozzle apparatus at T = 103 K, Chem. Phys. Lett., 2001, 344, 317–324 CrossRef CAS.
- H. Jin, L. Ye, J. Yang, Y. Jiang, L. Zhao and A. Farooq, Inception of Carbonaceous Nanostructures via Hydrogen-Abstraction Phenylacetylene-Addition Mechanism, J. Am. Chem. Soc., 2021, 143, 20710–20716 CrossRef CAS PubMed.
- J. Zádor, M. D. Fellows and J. A. Miller, Initiation Reactions in Acetylene Pyrolysis, J. Phys. Chem. A, 2017, 121, 4203–4217 CrossRef.
- L. A. Singer and N. P. Kong, Vinyl Radicals. Stereoselectivity in Hydrogen Atom Transfer to Equilibrated Isomeric Vinyl Radicals, J. Am. Chem. Soc., 1966, 88, 5213–5219 CrossRef CAS.
- H. F. Piedra and M. Plaza, Photochemical halogen-bonding assisted generation of vinyl and sulfur-centered radicals: stereoselective catalyst-free C(sp2)–S bond forming reactions, Chem. Sci., 2023, 14, 650–657 RSC.
- V. V. Kislov, N. I. Islamova, A. M. Kolker, S. H. Lin and A. M. Mebel, Hydrogen Abstraction Acetylene Addition and Diels−Alder Mechanisms of PAH Formation:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) A Detailed Study Using First Principles Calculations, J. Chem. Theory Comput., 2005, 1, 908–924 CrossRef CAS PubMed.
A Detailed Study Using First Principles Calculations, J. Chem. Theory Comput., 2005, 1, 908–924 CrossRef CAS PubMed. - A. M. Mebel, Y. Georgievskii, A. W. Jasper and S. J. Klippenstein, Temperature- and pressure-dependent rate coefficients for the HACA pathways from benzene to naphthalene, Proc. Combust. Inst., 2017, 36, 919–926 CrossRef CAS.
- T. Yang, T. P. Troy, B. Xu, O. Kostko, M. Ahmed, A. M. Mebel and R. I. Kaiser, Hydrogen-Abstraction/Acetylene-Addition Exposed, Angew. Chem., Int. Ed., 2016, 55, 14983–14987 CrossRef CAS.
- T.-C. Chu, Z. J. Buras, M. C. Smith, A. B. Uwagwu and W. H. Green, From benzene to naphthalene: direct measurement of reactions and intermediates of phenyl radicals and acetylene, Phys. Chem. Chem. Phys., 2019, 21, 22248–22258 RSC.
- A. E. Long, S. S. Merchant, A. G. Vandeputte, H.-H. Carstensen, A. J. Vervust, G. B. Marin, K. M. Van Geem and W. H. Green, Pressure dependent kinetic analysis of pathways to naphthalene from cyclopentadienyl recombination, Combust. Flame, 2018, 187, 247–256 CrossRef CAS.
- L. Zhao, W. Lu, M. Ahmed, M. V. Zagidullin, V. N. Azyazov, A. N. Morozov, A. M. Mebel and R. I. Kaiser, Gas-phase synthesis of benzene via the propargyl radical self-reaction, Sci. Adv., 2021, 7, eabf0360 CrossRef CAS.
- J. Hanfeng, X. Lili, H. Junyu, Y. Jiuzhong, Z. Yan, C. ChuangChuang, P. Yang and A. Farooq, A chemical kinetic modeling study of indene pyrolysis, Combust. Flame, 2019, 206, 1–20 CrossRef.
- J. R. Barker, T. L. Nguyen, J. F. Stanton, C. Aieta, M. Ceotto, F. Gabas, T. J. D. Kumar, C. G. L. Li, L. L. Lohr, A. Maranzana, N. F. Ortiz, J. M. Preses, J. M. Simmie, J. A. Sonk and P. J. Stimac, MultiWell-2020 Software Suite, K. R. Barker, University of Michigan, Ann Arbor, Michigan, USA, 2020 Search PubMed.
- J. R. Barker, Multiple-Well, multiple-path unimolecular reaction systems. I. MultiWell computer program suite, Int. J. Chem. Kinet., 2001, 33, 232–245 CrossRef CAS.
- J. R. Barker, Energy transfer in master equation simulations: A new approach, Int. J. Chem. Kinet., 2009, 41, 748–763 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision C.01), Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- G. da Silva, G3X-K theory: A composite theoretical method for thermochemical kinetics, Chem. Phys. Lett., 2013, 558, 109–113 CrossRef CAS.
- H. Hippler, J. Troe and H. J. Wendelken, Collisional deactivation of vibrationally highly excited polyatomic molecules. II. Direct observations for excited toluene, J. Chem. Phys., 1983, 78, 6709–6717 CrossRef CAS.
- T. Lenzer, K. Luther, K. Reihs and A. C. Symonds, Collisional energy transfer probabilities of highly excited molecules from kinetically controlled selective ionization (KCSI). II. The collisional relaxation of toluene: P(E′,E) and moments of energy transfer for energies up to 50000 cm−1, J. Chem. Phys., 2000, 112, 4090–4110 CrossRef CAS.
- D. L. Osborn, P. Zou, H. Johnsen, C. C. Hayden, C. A. Taatjes, V. D. Knyazev, S. W. North, D. S. Peterka, M. Ahmed and S. R. Leone, The multiplexed chemical kinetic photoionization mass spectrometer: a new approach to isomer-resolved chemical kinetics, Rev. Sci. Instrum., 2008, 79, 104103 CrossRef.
- A. Kushnarenko, E. Miloglyadov, M. Quack and G. Seyfang, Intramolecular vibrational energy redistribution in HCCCH2X (X = Cl, Br, I) measured by femtosecond pump–probe experiments in a hollow waveguide, Phys. Chem. Chem. Phys., 2018, 20, 10949–10959 RSC.
- S. Karmakar and S. Keshavamurthy, Intramolecular vibrational energy redistribution and the quantum ergodicity transition: a phase space perspective, Phys. Chem. Chem. Phys., 2020, 22, 11139–11173 RSC.
- R. D. Levine, Molecular Reaction Dynamics, Cambridge University Press, Cambridge, 2005 Search PubMed.
- M. C. Smith, G. Liu, Z. J. Buras, T.-C. Chu, J. Yang and W. H. Green, Direct Measurement of Radical-Catalyzed C6H6 Formation from Acetylene and Validation of Theoretical Rate Coefficients for C2H3 + C2H2 and C4H5 + C2H2 Reactions, J. Phys. Chem. A, 2020, 124, 2871–2884 CrossRef CAS.
- J. Hioe and H. Zipse, Radical Stability—Thermochemical Aspects, in Encyclopedia of Radicals in Chemistry, Biology and Materials ed. C. Chatgilialoglu and A. Studer, John Wiley & Sons, Ltd., 2012, Online DOI:10.1002/9781119953678.rad012.
- O. J. Shiels, M. B. Prendergast, J. D. Savee, D. L. Osborn, C. A. Taatjes, S. J. Blanksby, G. da Silva and A. J. Trevitt, Five vs. six membered-ring PAH products from reaction of o-methylphenyl radical and two C3H4 isomers, Phys. Chem. Chem. Phys., 2021, 23, 14913–14924 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03554b |
| ‡ The trans form is Z in this case as the radical site has lower priority than the H atom. |
| This journal is © the Owner Societies 2025 |