
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assignment of IR spectra of ethanol at Brønsted sites of H-ZSM-5 to monomer adsorption using a Fermi resonance model†
Dipanshu
Kumar
 a,
Joachim
Sauer
b,
Alessia
Airi
*cd,
Silvia
Bordiga
d and
Daria Ruth
Galimberti
*a
a,
Joachim
Sauer
b,
Alessia
Airi
*cd,
Silvia
Bordiga
d and
Daria Ruth
Galimberti
*a
aInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: daria.galimberti@ru.nl
bInstitut für Chemie, Humboldt-Universität, Unter den Linden 6, 10117 Berlin, Germany
cINRiM Istituto Nazionale di Ricerca Metrologica, Strada delle cacce 91, I-10135 Turin, Italy. E-mail: a.airi@inrim.it
dChemistry Department, University of Turin, via Gioacchino Quarello 15/A, I-10135 Turin, Italy
First published on 10th December 2024
Abstract
Understanding how alcohol molecules interact with the Brønsted acid sites (BAS) of zeolites is a prerequisite to the design of zeolite catalysts and catalytic processes. Here, we report IR spectra for the adsorption of ethanol on a highly crystalline sample of H-ZSM-5 zeolites exposed to ethanol gas at increasing pressure. We use density functional theory in combination with a FERMI resonance model to assign the measured spectra to a single adsorbed ethanol molecule per BAS. Specifically, we assign the bands at 2450 cm−1 and 1670 cm−1 to a FERMI resonance between the fundamental (Z)O–H stretching band of a single-ethanol-loaded BAS and the first overtone of the (Z)O–H out-of-plane bending. We conclude that adsorbed dimers do not contribute in a noticeable way up to a concentration of almost one ethanol molecule per BAS site. We further show that hybrid functionals (B3LYP) are required to get a close match between the predicted and experimental spectra, whereas commonly used generalized gradient type functionals such as PBE incorrectly describe the potential energy surface. They overestimate the redshift of the OH stretching band on hydrogen bond formation which results in an erroneous assignment of the IR bands.
1. Introduction
Understanding how alcohols interact with the Brønsted Acid Sites (BAS) of zeolites is a prerequisite to designing zeolite catalysts and catalytic processes. While in the past the conversion of methanol1–3 to hydrocarbons, in particular to olefins,4 was in the focus, more recently, there is increasing interest in the interaction of ethanol and higher alcohols with BAS5–10 in connection with biomass conversion.11–13 It is known that methanol6,14 and ethanol5,6 form H bonded complexes with BAS. It is also known that the protonation state of hydrogen-bonded alcohols is loading-dependent.5,15,16 Two alcohol molecules adsorbed on the same BAS are needed to achieve proton transfer to the adsorbed species,17 which is explained by the higher proton affinity of the dimer compared to the monomer.18 However, many aspects of alcohol-BAS interactions are still debated, e.g., the interpretation of the adsorption isotherm and the ratio of adsorbed monomers and dimers at different pressures.5,6Over the years, substantial progress has been made with controlled preparation of zeolite samples and spectroscopic methods.19–22 Vibrational spectroscopies, together with 1H-NMR chemical shifts, have been, and still are, fundamental tools for probing zeolites. In particular, IR spectroscopy has provided crucial insights into zeolite properties and reactivity.23–29 Due to the complexity of the signal, the assignment of spectroscopic signatures such as IR bands or 1H-NMR chemical shifts to relevant surface species requires quantum chemical predictions for atom-scale models of these species.30–34 Computational predictions of IR spectra face two challenges: the correct description of the potential energy surface (PES), i.e., the dependence of the energy on the position of all nuclei, and the description of the vibrational states. For decades, density functional theory (DFT) at the level of the generalized gradient approximation (GGA), for its computational efficiency, remained the standard methodology for describing molecule–surface interactions.3,5,6,9,10,12,13,29,32,35,36 However, this approximation is known to yield too weak and long O–H bonds which are stretched too much on H bond formation.2,6,9,33,37–43 It over stabilizes polar structures and, hence, yields too strong H bonds, too low barriers for proton transfer, and too stable ion-pair structures formed on proton transfer. The overstabilization of H bonded structures is not rectified, but, in contrast, rather amplified when GGA is augmented with a dispersion term.
Regarding the second challenge, the standard protocol follows the double harmonic approximation, which requires only a straightforward calculation of second-order force constants and first derivatives of dipole moments. However, experimental and computational evidence indicates that the IR spectra of adsorbed water and alcohols may possibly feature a Fermi resonance in the characteristic O–H region and, hence, cannot be described in the harmonic approximation.25,26,44,45 For water adsorption, the crucial experiment was the isotope substitution (18O) of water46 which showed no effect on the pair of bands at 2877 cm−1 and 2463 cm−1 and thus clearly supported their assignment to vibrations of the zeolitic BAS. On H bond formation with the adsorbed water molecule, the (Z)O–H stretching is strongly red-shifted and the Fermi resonance with the overtone of the in-plane Si–O–H bending creates an Evans window at the overtone position;37 see ref. 47 for a model calculation. For methanol adsorption, it has been shown that the A–B–C Fermi resonance model48,49 is consistent with the early prediction from MP2 calculations on cluster models, indicating that the (Z)O–H band is red-shifted to 2300–2600 cm−1.14 Also for ethanol adsorption in H-ZSM-5, the presence of the ABC band triad has been proposed.25 The IR studies assigned the ABC triplet of bands at 2980, 1446, and 1391 cm−1 observed for the ethanol loaded zeolite to a Fermi resonance between the (Z)O–H stretching fundamental and the overtones of the in and out of plane Z–O–H bending modes.25,26,45
The Fermi resonance model has been recently questioned based on 2D-IR experiments50 and PBE+D2 calculations at the GGA level.5,50 Hack et al.50 showed that a simple phenomenological model considering the adsorption of a single water molecule and a Fermi resonance is not able to capture all the features of the 2D-IR spectrum measured for a H-ZSM-5 sample at a hydration level of 1.0 H2O/Al, but dimer adsorption should also be considered either as an alternative or together with the Fermi resonance for a single molecule to explain all the bands. However, no spectra have been reported for lower water loading. For ethanol on H-ZSM-5, Alexopoulos et al.5 performed PBE+D2 molecular dynamics simulations and suggested that the 2450 cm−1 band is not a component of the Fermi resonance, but should be rather assigned to the presence of protonated ethanol dimers. As we will show below, this conclusion may be affected by the shortcomings of PBE+D2 for H bonded systems.
Here, we aim to get reliable information on how single ethanol molecules interact with zeolitic BAS. We report FTIR spectra for a well-characterized sample of zeolite H-ZSM-5 exposed to ethanol with increasing pressure from 0.0034 to 0.0135 Torr. This will allow us to examine the possible role of adsorbed ethanol dimers in addition to adsorbed monomers.
With our DFT calculations, we climb the next rung of Jacob's ladder of exchange–correlation functionals. Instead of the GGA-type PBE functional, we use a hybrid functional. Specifically, we use the B3LYP functional augmented with Grimme's D2 dispersion term, which has been shown to be a major improvement compared to PBE+D2.43 For the calculations of the vibrational states we go beyond the double harmonic approximation and apply the model proposed by Iogansen51 to predict the intensities of coupled vibrations resulting from a Fermi resonance between the O–H stretching fundamental and the first overtone of the Si–O–H out-of-plane bending of the BAS engaged in the H bond with ethanol. From the harmonic results for the splitting between the vibrational transitions and their intensities as well as the splitting between the observed bands, we get a simulated spectrum with a compelling assignment of all bands and their intensities in the observed spectrum.
The Fermi resonance model allows us to assign all the bands of the experimental spectrum and to explain the observed changes in the spectra with increasing pressure. We conclude that the FTIR spectrum observed up to almost the concentration of one ethanol per BAS is due to a single adsorbed ethanol molecule per BAS and that adsorbed dimers do not contribute significantly.
2. Experiments
2.1. Synthesis and spectroscopy
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.75 Al2O3:30 SiO2:240 EtOH:2132 H2O, and no organic structure directing agents were used. Crystallization was carried out by hydrothermal treatment in oven at 140 °C for 8 days tumbling at 30 rpm. The obtained powder was filtered and calcined at 550 °C for 7 hours in dry air atmosphere. The acidic form of the zeolite has been obtained by ion exchange of Na+ cations replaced by NH4+ by repeated washing with 1 M solution of NH4NO3 (20 mL solution per g sample) at 80 °C. NH3 is released by calcination at high temperature (500 °C) obtaining the H-ZSM-5 form. The final Si/Al ratio is 15.28 The sample has been characterized by powder X-ray diffraction using a PANalytical PW3050/60 X’Pert PRO MPD diffractometer with Cu anode (Kα = 1.5418 Å) and X’Celerator detector. The pattern confirms the crystal structure of the pure MFI zeolite topology. The morphology of the crystallites has been determined by the analysis of ultra high resolution field emission scanning electron microscopy (UHR FE-SEM) micrographs obtained using an FE-SEM TESCAN S9000G microscope.
:0.75 Al2O3:30 SiO2:240 EtOH:2132 H2O, and no organic structure directing agents were used. Crystallization was carried out by hydrothermal treatment in oven at 140 °C for 8 days tumbling at 30 rpm. The obtained powder was filtered and calcined at 550 °C for 7 hours in dry air atmosphere. The acidic form of the zeolite has been obtained by ion exchange of Na+ cations replaced by NH4+ by repeated washing with 1 M solution of NH4NO3 (20 mL solution per g sample) at 80 °C. NH3 is released by calcination at high temperature (500 °C) obtaining the H-ZSM-5 form. The final Si/Al ratio is 15.28 The sample has been characterized by powder X-ray diffraction using a PANalytical PW3050/60 X’Pert PRO MPD diffractometer with Cu anode (Kα = 1.5418 Å) and X’Celerator detector. The pattern confirms the crystal structure of the pure MFI zeolite topology. The morphology of the crystallites has been determined by the analysis of ultra high resolution field emission scanning electron microscopy (UHR FE-SEM) micrographs obtained using an FE-SEM TESCAN S9000G microscope.
2.2. Experimental spectra
Fig. 1A reports the experimental spectra of H-ZSM-5 loaded with ethanol at increasing pressure (from 0.0034 Torr to 0.0135 Torr) and the resulting adsorption isotherm curve (inset in Fig. 1A) obtained as indicated in the experimental section.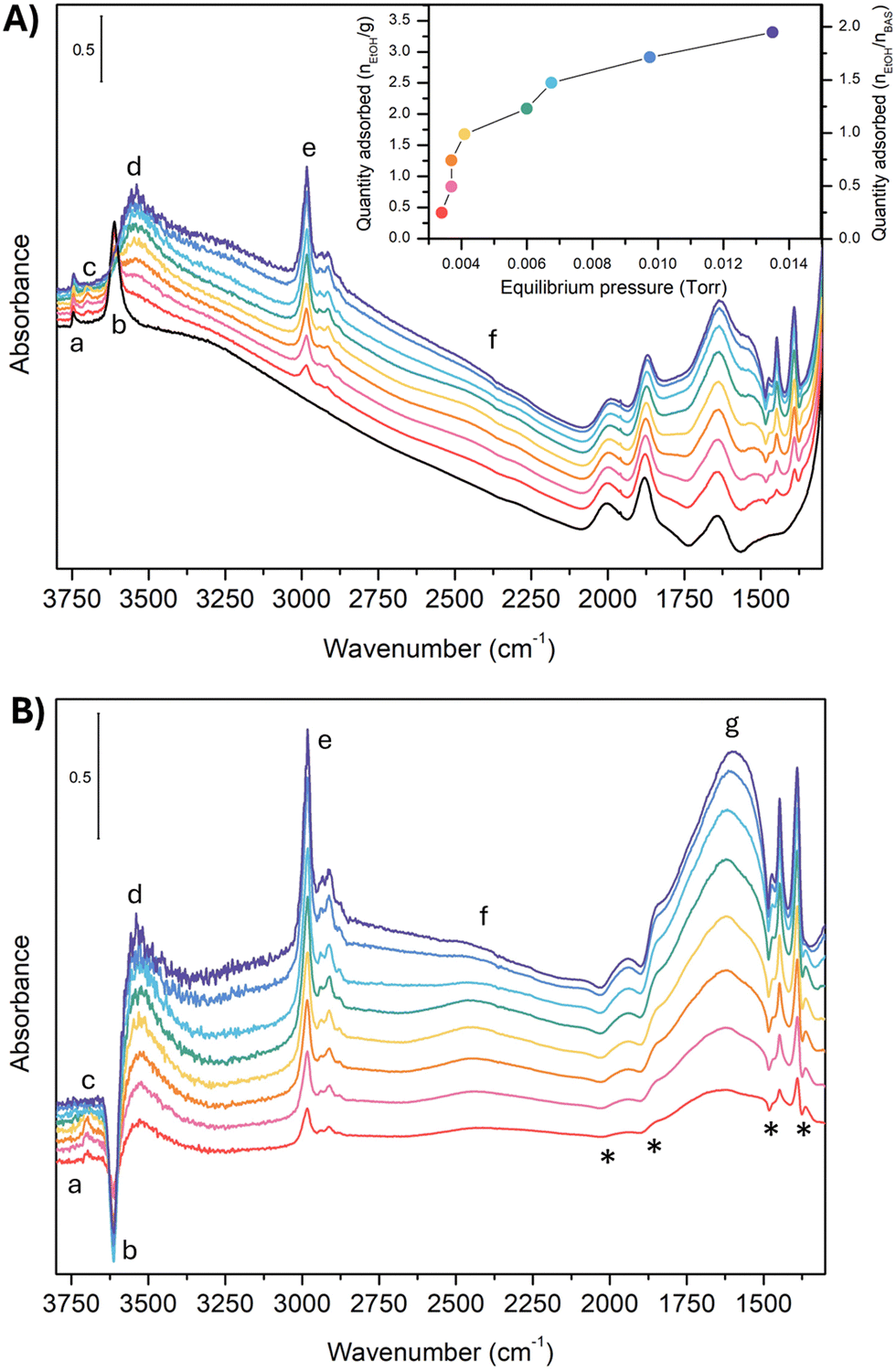 | ||
| Fig. 1 Experimental IR spectra of unloaded H-ZSM-5 (black line) and H-ZSM-5 loaded with ethanol at increasing pressure (from red to blue). Panel (A) measured adsorbance (A). In the inset, the adsorption isotherm. Panel (B) difference spectra at increasing pressure compared to the unloaded zeolite (Adiff). The (*) indicate the artificial, derivative-type bands. The spectra are all reported in arbitrary units. No baseline correction has been introduced. | ||
For each point of the curve, the quantity of adsorbed ethanol per gram of zeolite can also be correlated to the concentration of zeolite BAS, calculated as reported in Section S2 (ESI†). These values will be further discussed later on. The IR spectrum of the unloaded zeolite (black line) shows a first band (a) at 3749 cm−1, attributed to isolated Si–O–H groups at the crystallite external surface and to internal defects.24,44,53–57 The weak intensity of this band for our experimental spectrum guarantees that only a few of these defects are present in our sample. Next to this first peak, we found the strong, sharp band (b) at 3614 cm−1, characteristic of the (Z)O–H stretching mode of the free BAS.44,55 To be noticed is the absence of any features between 3660–3680 cm−1 and above 3750 cm−1, i.e., in the region of the marker bands for the presence of low-acidity OH groups bonded to extra-lattice aluminum and differently coordinated hydroxyl groups.16,24,54–56,58,59
This is an indication that nearly all aluminum should be in the framework (tetracoordinate) positions for our sample. Moving to lower wavenumbers, we found a broad feature between 3500 and 3000 cm−1. This feature is probably an artifact of the asymmetric scattering profile due to the large dimensions of the zeolite crystallites (Fig. S1, ESI†), which are also responsible for the strong sloping of the baseline at high wavenumbers.60 Extra-framework or partially framework-bound aluminum oxo-hydroxo,61–64 H-bonded silanols,65 H-bonded BAS in the framework,34 and Al(OH)n(H2O) sites at the external surface21,66 may contribute to the signal in this spectral region. However, the small intensity in this region demonstrates that, if these defects are present in our sample, they are in negligible quantities. Moreover, we can reasonably exclude the presence of partially in or extra-framework Al species, from the result of the target experiment of ammonia adsorption followed by FT-IR spectroscopy. The absence of any vibrational imprint of the Al-NH3 Lewis pair on the surface (δd mode of ammonium) supports our hypothesis. These results are reported in Section S2 (ESI†).
Between 3000 cm−1 and 2000 cm−1, the unloaded zeolite has no spectral contributions. Between 2000 cm−1 and 1300 cm−1, we find the overtones and combination bands of the skeletal modes of H-ZSM-5,67 whereas the fundamental vibrations fall below 1300 cm−1. In particular, the in-plane and out-of-plane bending modes of the unperturbed BAS67 have been reported at 1042 cm−1 and 305 cm−1, respectively. Because this spectral region has not been measured in our case, we will not further discuss it.
Consider now what happens when we load the zeolite with ethanol. To make the comparison more accessible, we also report the spectral difference compared to the unloaded zeolite (Fig. 1-B). However, looking at the latter, some extra care must be taken. The uptake of molecules into the channel walls causes a slight increase in the lattice parameters, changing the scattering profile and shifting the skeletal modes. Although tiny in absolute terms, the shift and the alteration of the spectral shape is enough to generate in the difference spectra artificial, derivative-type bands, especially for the overtones and combination bands between 2100 cm−1 and 1500 cm−1. In particular, the four negative peaks indicated with an (*) in Fig. 1-B are related to this artifact.29,30
The analysis of the spectra of the loaded zeolite indicates that the ethanol is not interacting with the silanol sites: the silanol marker band (a) is almost unchanged by the increased ethanol loading, i.e., no band can be seen in the difference spectra. In contrast, there are clear markers for adsorption of ethanol on the BAS. The 3614 cm−1 (Z)O–H stretching peak loses intensity (appearance of a negative peak (b) in the difference spectra) and disappears above 0.0060 torr when complete loading is achieved. At the same time, a set of new bands pops up: a small peak at 3695 cm−1 (c) that then suddenly disappears above 0.0060 Torr, the broad feature (d) at 3543 cm−1 characteristic of the stretching of a quasi-free (Et)O–H of an ethanol molecule which accepts an H bond from the zeolitic (Z)O–H group,49 and a set of sharp peaks (e) at 2983 cm−1, 2939 cm−1, 2913 cm−1, and 2876 cm−1 related to the CH2 and CH3 stretching vibrations of ethanol.
Between 2000 cm−1 and 1400 cm−1 the difference spectrum is characterized by a broad and asymmetric feature with many components (g). The dominant component is the fundamental (Z)O–H stretching of the loaded zeolite. It is, however, convoluted with the Z–O–H and Et–O–H bending (the shoulder on the lower frequency side), and the CH2 and CH3 bending modes (the set of sharp peaks). The asymmetric shape of the (Z)O–H stretching, together with the non-linear baseline, and the contributions of the framework overtones make the deconvolution of this feature not straightforward. But we can qualitatively estimate that the (Z)O–H stretching falls around 1670 cm−1 (see Section S3 of the ESI†).
Finally, at around 2450 cm−1 we find a peak (f) which also increases in intensity with increasing ethanol pressure. It is the main focus of this paper, and we will discuss its assignment in detail in Section 4.
To obtain a deeper understanding, Fig. 2 displays the incremental variation of the spectrum dA with the equilibrium ethanol pressure, i.e., each curve is obtained by subtracting the spectrum at a certain pressure the one measured at the previous pressure.
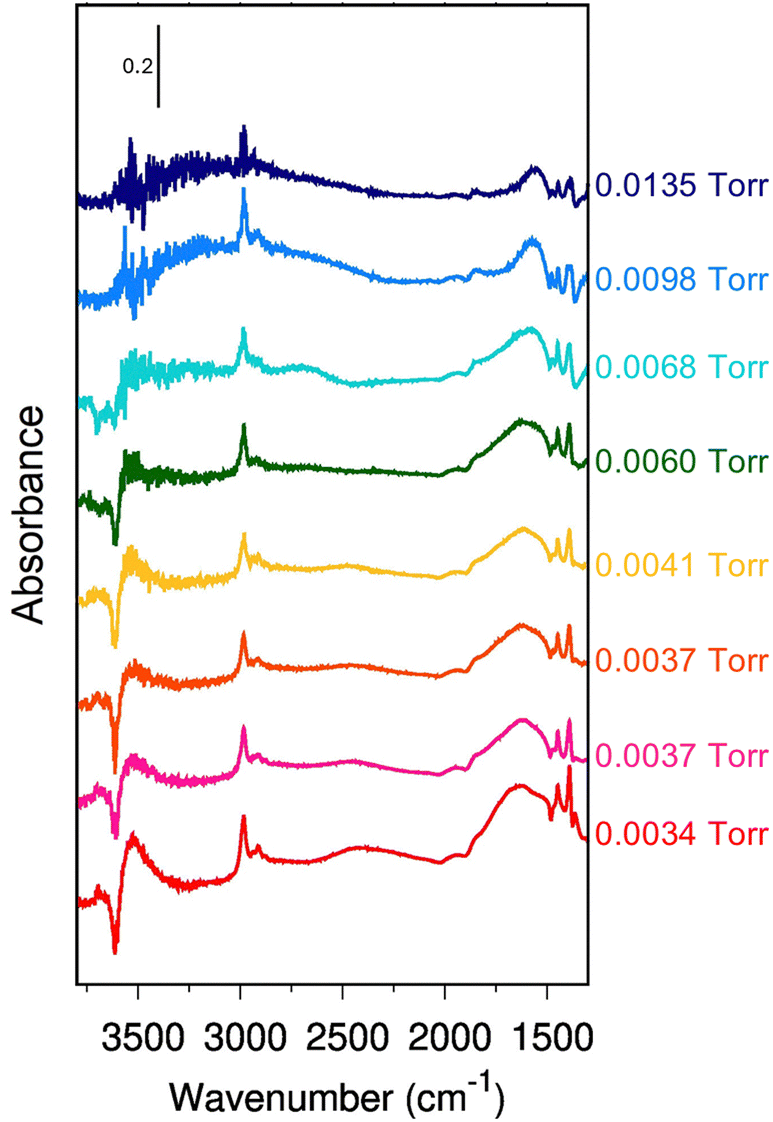 | ||
| Fig. 2 Incremental variation of absorbance with pressure (dA). Increasing pressure from red to blue – see Fig. 1, panel A. On the right side, we report the equilibrium pressure at which each spectrum has been measured. The spectra are all reported in arbitrary units. No baseline correction has been introduced. | ||
A positive band in dA indicates an increase of the total absorbance in the normal spectrum, a flat region that the spectral profile is not changing, and a negative band, a loss of intensity for increasing pressure. Notice that dA (Fig. 2) naturally reduces the artifacts of the scattering profile compared to the difference spectra (Fig. 1-B). Until 0.0041 Torr (red to yellow spectra), all the bands change simultaneously to the same extent, preserving almost exactly the intensity ratio. Between 0.0060 Torr and 0.0067 Torr (green and light blue spectra), the 2450 cm−1 band stops to gain intensity with the increase of the ethanol loading (flat profile in the incremental spectrum). A new contribution pops up instead around 2600 cm−1.
Above 0.0067 Torr (blue spectra), only the C–H stretching bands (e) and the 1560 cm−1 component of the (g) feature still show an increase in intensity. Instead, the incremental profile between 1600 cm−1 and 2000 cm−1 becomes flat, i.e., the 1670 cm−1 band is not growing anymore with the increasing equilibrium pressure. The 3543 cm−1 band seems to follow the same trend. However, the noise of the experimental data does not allow it to be conclusive on this.
3. Theory
3.1. Fermi resonance model
Vibrational configuration interaction (VCI)68–73 would be the method of choice for a proper anharmonic vibrational treatment with the inclusion of all mode couplings, including Fermi-resonance effects, but applications to systems of the size considered here are computationally prohibitive. Another option to include anharmonicity effects are molecular dynamics (MD) simulations from which IR spectra can be obtained by Fourier transformation of the dipole autocorrelation function.5,74–79 Apart from the fact that it is not straightforward to include quantum effects, the computational effort is still orders of magnitude larger than for harmonic force constant calculations. Therefore, up to our knowledge such simulations have only been completed employing GGA-type functionals for adsorption of ethanol in H-ZSM-5.5,79 As a consequence, the IR spectra obtained may suffer from the unrealistically large redshift of O–H stretching vibrations engaged in H bonds.Within the harmonic approximation, Fermi resonances between vibrations cannot be described. However, simple quantum mechanical models51,80,81 can approximate the intensity pattern of coupled vibrations resulting from Fermi resonances. Here, for a single ethanol molecule adsorbed on the Brønsted acid site (BAS) of H-ZSM-5 (1 EtOH/BAS), we adopted the model proposed in ref. 51 to predict the intensities of coupled vibrations resulting from a Fermi resonance between the fundamental (Z)O–H stretching band of the BAS (ν0) and the first overtone of the out-of-plane bending (2γ0). These two vibrations become resonant upon the ethanol molecule adsorption because ν0 is red-shifted by the H bond formation, (Z)O–H(⋯OEt), and γ0 blue-shifted, Z–O–H(⋯OEt).
Differently from previous work on the adsorption of different molecules in H-ZSM-5,49,81 we adopt the simpler AB two-band Fermi resonance model (Fig. 3) instead of the three-band ABC model. Our calculations for ethanol adsorption point to a substantial wavenumber gap (>800 cm−1) between ν0 and the overtone of the Z–O–H⋯OEt in-plane bending (2δ0) which is the “C” component in the ABC model (see Section 3.5 for more details on the position of the fundamental bands).
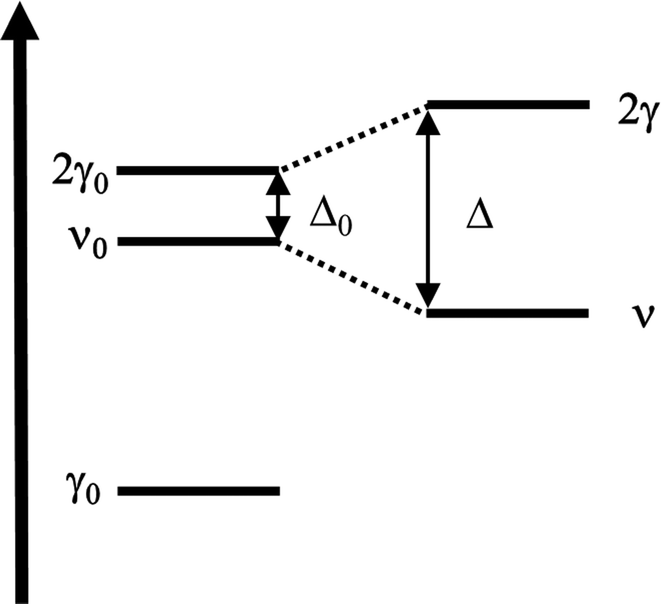 | ||
| Fig. 3 Scheme of the Fermi resonance. | ||
Our model describes the Fermi resonance as a first order perturbation80 of the two quasi degenerate levels ν0 and 2γ0, which yields the eigenvalue problem:
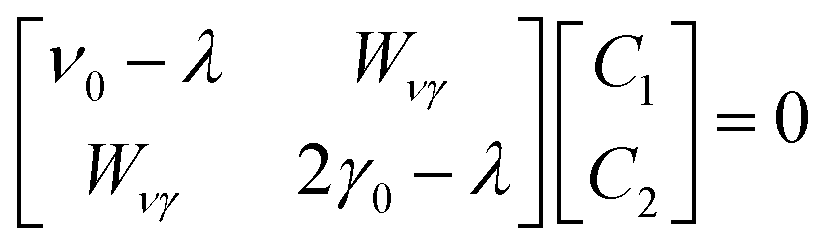 | (1) |
and ψ0ν and ψ02γ are the unperturbed vibrational eigenfunctions of ν0 and 2γ0, respectively.
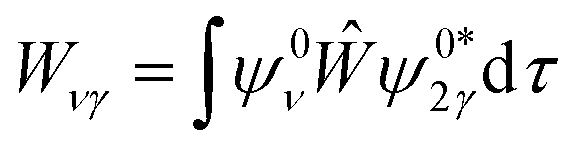
Solving this eigenvalue problem yields:
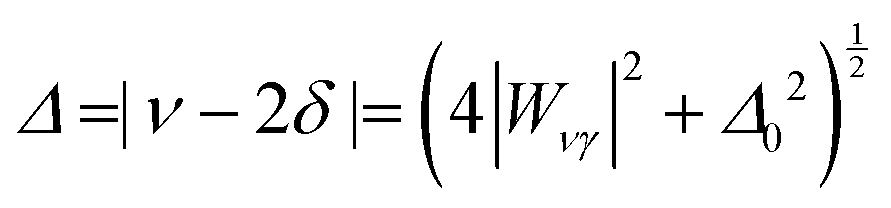
The eigenfunctions of the perturbed states are
| ψν = aψ0ν + bψ02γ | (3) |
| ψν = aψ0ν + bψ02γ | (4) |
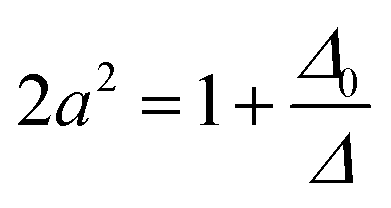 | (5) |
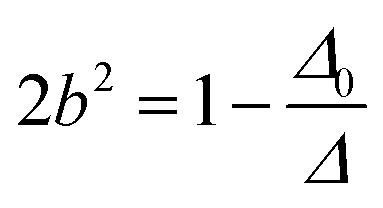 | (6) |
| |Mν| = a|M0ν| + b|M02γ| | (7) |
| |M2γ| = b|M0ν| + a|M02γ| | (8) |
| |Mν| ≈ a|M0ν| | (9) |
| |M2γ| ≈ b|M0ν| | (10) |
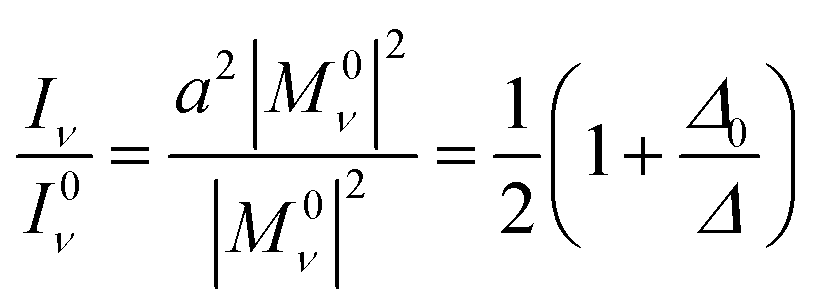 | (11) |
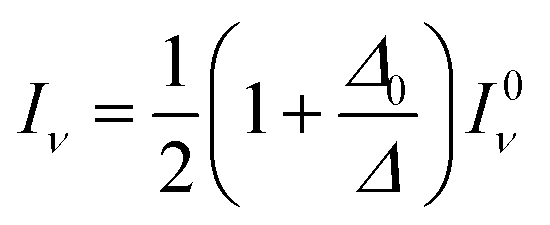 | (12) |
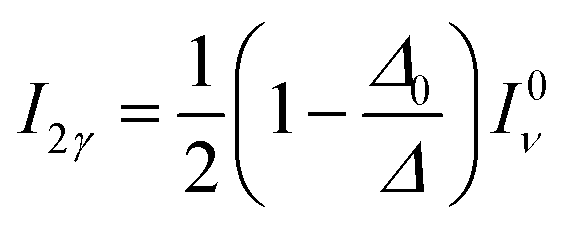 | (13) |
3.2. H-ZSM-5 model
Starting from the structure published in ref. 1, 6 and 82, H-ZSM-5 has been modeled with a single crystallographic unit cell (dimensions a = 20.16 Å, b = 20.03 Å, c = 13.47 Å) that contains 289 atoms, with a Si/Al ratio of 95 (corresponding to one Al per unit cell), and the BAS in the Al12–O20(H)–Si3 “intersection” position (Fig. 4-A). As in ref. 6, in addition to the periodic model, a cluster model consisting of 10TO4 tetrahedra (T = Si, Al) is defined which comprises the most relevant interactions of the ad-molecule with the surface site.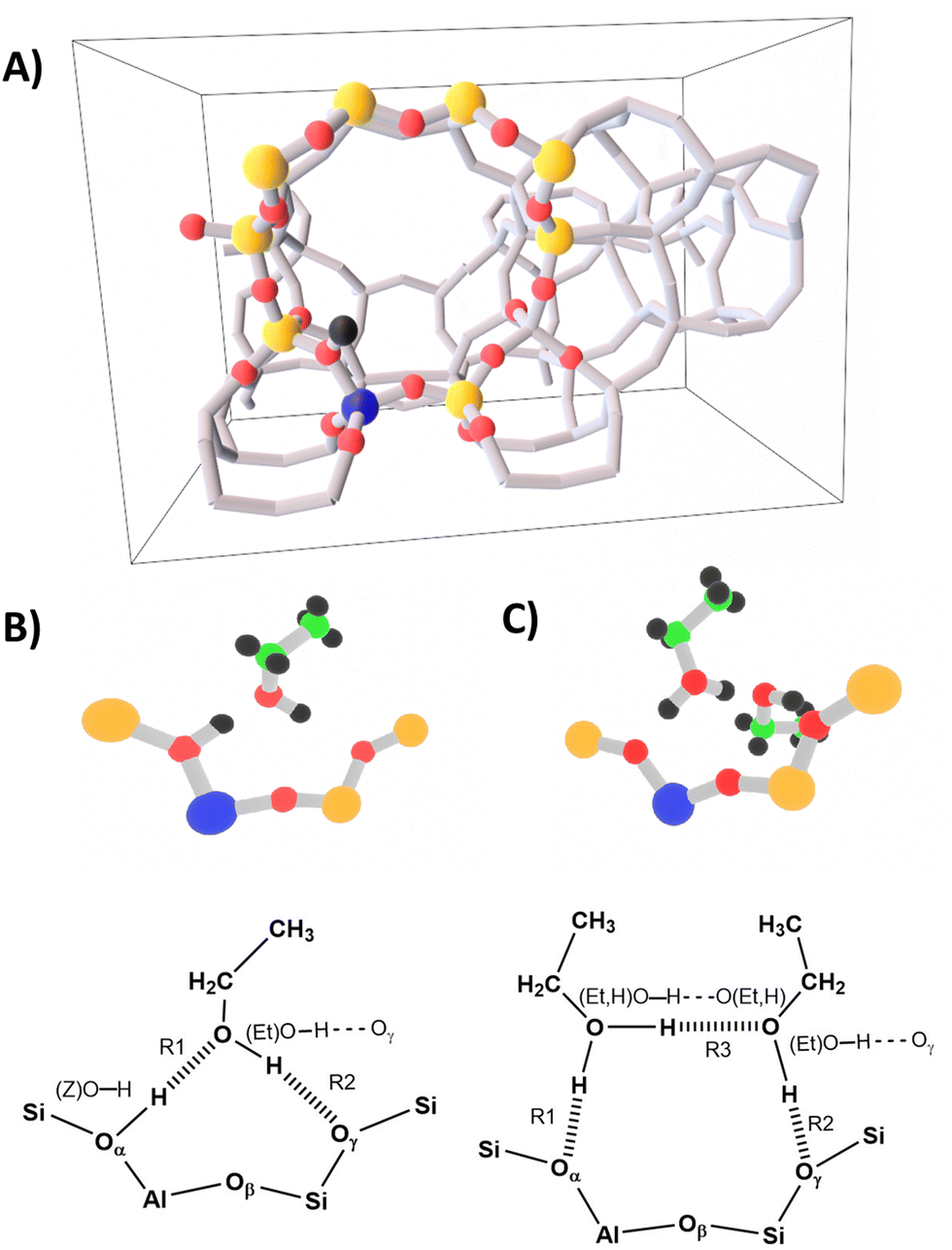 | ||
| Fig. 4 Panel (A) periodic H-ZSM-5 model from ref. 6 with the 10 T cluster shown as ball-and-stick. Panel (B) single ethanol molecule adsorption structure (1 EtOH/BAS), and (panel (C)) ethanol dimer adsorption structure (2 EtOH/BAS). | ||
3.3. Computational methods
To make calculations with hybrid exchange–correlation functionals feasible for periodic models of the size considered here, we performed hybrid high-level QM:low-level QM calculations using the subtractive scheme described in ref. 38, 83 and 84 and implemented in MonaLisa.85,86 Low-level (LL) QM calculations are performed on periodic models, and the energies (E) and forces (F) obtained are corrected with the difference between a high-level (HL) and an LL calculation evaluated for a representative cluster model:| EHL:LL(periodic,cluster) = ELL(periodic) + EHL(cluster) − ELL(cluster) | (14) |
| FHL:LL(periodic,cluster) = FLL(periodic) + FHL(cluster) − FLL(cluster) | (15) |
For the HL calculations the B3LYP93 hybrid functional augmented with D289 has been chosen together with the def2-TZVP basis set.94 The ORCA code95 has been employed. We also tested the ωB97X96 functional as an HL method, but the results are not reported here because they are very similar to the B3LYP results. Moreover, MP2 (in the RI-MP2 version) is used as the HL method to judge the quality of B3LYP+D2 for the systems studied. To eliminate any bias due to basis set differences from the B3LYP vs. PBE comparison, we perform hybrid HL:LL calculations also for PBE+D2 using ORCA with the def2-TZVP basis set as the HL method.
In the following discussion, for simplicity we will refer to the hybrid PBE+D2ORCA:PBE+D2CP2K, B3LYP+D2ORCA:PBE+D2CP2K and MP2ORCA:PBE+D2CP2K, results as PBE+D2, B3LYP+D2 and MP2 results, respectively.
Using as a starting point structures proposed in the past in ref. 6 and 97, we performed hybrid HL:LL structure optimizations. On the energy minimum structures, we computed the IR spectra in double harmonic approximation. The wavenumbers of the fundamental vibrational modes have been obtained using numerical derivatives within the hybrid HL:LL scheme. A partial hessian strategy has been adopted, i.e., only the elements of the hessian related to atoms of the HL cluster have been computed. Finally, to minimize spurious border effects, framework vibrations not involving the BAS OH, have been shifted out of the spectroscopic region of interest to lower wavenumbers by artificially modifying the masses of this part of the system, i.e., only the ethanol molecules and the BAS OH have their real mass. The artificial masses (900.000 a.m.u.) have been chosen in a way that only vibrations belonging to the BAS OH or the ethanol molecules appear between 5 cm−1 and 4000 cm−1. The IR adsorption intensities have been modelled using the atomic polar tensors for the HL cluster only, but computed at the hybrid HL:LL optimized structures. The predicted hybrid PBE+D2 and B3LYP+D2 wavenumbers have been scaled by 0.9804 and by 0.9710, respectively, to effectively account for anharmonic effects and systematic deviations of the harmonic force constants. The scaling factors have been obtained by matching the experimental (Z)O–H stretching mode of the free BAS (3614 cm−1).
For comparison, we reported in the ESI† (Section S8), the wavenumbers obtained using ωB97X and MP2 as HL. The results are not discussed here because they are similar to the B3LYP+D2 ones. The FERMI resonance model requires a certain shift of the fundamental ν0(Z)O–H stretch compared to the 2γ0(Z)O–H and the experimentally measured ν(Z)O–H stretch position. MP2 well predicts these. The B3LYP+D2 shifts are very close to the MP2 ones. Therefore B3LYP+D2 is sufficient to demonstrate that the FERMI-resonance explains the observed spectra, and it can be considered a good compromise between accuracy and computational cost. The ωB97X shows a similar relative position of the bands but with a larger deviation from MP2 shifts. Therefore is less suited than B3LYP+D2 for our purpose.
3.4. DFT results for structures
We investigated the adsorption of a single ethanol molecule at the zeolitic BAS (1 EtOH/BAS, Fig. 4-B) and the formation of a dimer-like structure when a second ethanol molecule is adsorbed (2 EtOH/BAS, Fig. 4-C). Table 1 shows the results obtained for the adsorption energies and bond distances.| PBE+D2 | B3LYP+D2 | MP2 | |
|---|---|---|---|
| 1 EtOH/BAS | |||
| (Z)O–H | 113.5 (6.3) | 107.4 (0.2) | 107.2 |
| R1 | 132.2 (−8.6) | 140.4 (−0.4) | 140.8 |
| O⋯O | 245.6 (−2.4) | 247.6 (−0.4) | 248.0 |
| (Et)O–H | 98.6 (1.0) | 97.2 (−0.4) | 97.6 |
| R2 | 209.7 (−9.1) | 216.8 (−2.0) | 218.8 |
| O⋯O | 308.3 (−7.6) | 313.9 (−2.0) | 315.9 |
| 2 EtOH/BAS | |||
| (Et,H)O–H⋯Oz | 104.9 (1.4) | 103.8 (0.3) | 103.5 |
| R1 | 149.5 (0.1) | 153.2 (3.8) | 149.4 |
| O⋯O | 253.9 (1.3) | 255.7 (3.1) | 252.6 |
| (Et,H)O–H⋯O(Et,H) | 108.5 (3.1) | 105.4 (0.0) | 105.4 |
| R3 | 139.6 (−3.8) | 144.3 (0.9) | 143.4 |
| O⋯O | 247.9 (−0.4) | 249.1 (0.8) | 248.3 |
| (Et)O–Hγ | 98.2 (0.9) | 97.0 (−0.3) | 97.3 |
| R2 | 205.3 (−6.6) | 209.3 (−2.6) | 211.9 |
| O⋯O | 289.1 (−2.0) | 287.9 (−3.2) | 291.1 |
Both our MP2, B3LYP+D2, and PBE+D2 calculations predict a “neutral” adsorption complex for a single ethanol molecule, Fig. 4-B, and an ion-pair structure with a protonated ethanol dimer for adsorption of two ethanol molecules on a BAS, Fig. 4-C. In both cases, the adsorbate forms a ring involving a hydrogen bond between the OH group of ethanol and the Oγ oxygen atom of the framework.
In the monomer structure, the ring comprises a very strong hydrogen bond between the BAS (donor) and oxygen of the ethanol (acceptor) and a weaker interaction between the ethanol OH group and Oγ. In the dimer structure, a protonated ethanol molecule acts as a strong hydrogen bond donor to both the (deprotonated) BAS and to the second (neutral) ethanol molecule. In turn, this second ethanol provides a weak bond with Oγ of the zeolite framework. We also tested the arrangement in which the ring is formed with Oβ oxygen for both 1 EtOH/BAS and 2 EtOH/BAS. However, this arrangement is either not stable or is more than 120 kJ mol−1 higher in energy (see the ESI† for more details). Therefore, we will not discuss it any further.
As we already pointed out, it is known that PBE with D2 (or with other dispersion augmentations such as D3) yields significantly shorter intermolecular OH bond distances and longer intramolecular OH bonds compared to hybrid functional or wavefunction methods like MP2.6,33,37,38,43 This is also what we find for ethanol in this work. Compared to MP2, for 1 EtOH/BAS, the (Z)O–H bond distance is 6.3 pm too long with PBE+D2, whereas B3LYP+D2 shows an almost perfect match. The H bond (O⋯O distance) of ethanol with the zeolitic O–H group for 1 EtOH/BAS is 2.4 pm too short with PBE+D2, but only 0.4 pm with B3LYP+D2. The effect is stronger for the H bond between the ethanol OH group and O of the zeolite wall. With PBE+D2, the bond is 9.1 pm shorter than with MP2, whereas with B3LYP+D2, it is only 2.0 pm shorter. This trend is mirrored by the interatomic distances R1 and R2. With PBE+D2, they are 8.6 pm and 9.1 pm shorter than with MP2, whereas with B3LYP+D2, only 0.4 pm and 2.0 pm shorter.
For 2 EtOH/BAS, the superior performance of B3LYP+D2 compared to PBE+D2 is less systematic but still present. The O–H bonds of the protonated ethanol dimer are 1.4 and 3.1 pm too long with PBE+D2 whereas the B3LYP+D2 results agree within 0.3 pm with the MP2 reference. Compared to MP2, for 2 EtOH/BAS, the H bond between the ethanol OH group and O of the zeolite wall is 0.9 pm too long with PBE+D2, whereas only 0.3 pm too short with B3LYP+D2. The R2 and R3 of the protonated ethanol dimer are 6.6 and 3.8 pm too short with PBE+D2, whereas with B3LYP+D2, R2 is only 2.6 pm shorter than the MP2 reference and R3 0.9 longer. The corresponding H bond (O⋯O distances) are an exception to the general trend, i.e., they are better reproduced by PBE+D2 instead of B3LYP+D2 (respectively, for 2.0 pm and 0.4 pm too short with PBE+D2; 3.2 pm too short and 0.8 too long with B3LYP+D2). However, the better performances of PBE+D2 are due, in this case, to the compensation of the errors on intramolecular distances and H bond angles. In fact the (Et)O–Hγ⋯O and the (Et,H)O–H⋯O(Et,H) angles are predicted to be 142.0° and 174.4° by PBE+D2, 137.0° and 172.9° by B3LYP+D2, and 137.5° and 172.9° by the MP2 reference. Another exception from the general trend is the interatomic distance R1. It is only 0.1 pm shorter than the MP2 reference with PBE+D2 but 3.8 pm too long with B3LYP+D2.
Even considering the discussed exceptions, B3LYP+D2 performs much better than PBE+D2 with respect to the MP2 reference. The small deviations of the B3YLP+D2 structures from the MP2 references make us confident that B3LYP+D2 is a good compromise between accuracy and computational cost and should be suitable for assigning the vibrational spectra. Indeed, this is the case, as we will show in the next sections.
3.5. B3LYP+D2 results for vibrational spectra
Fig. 5 shows the comparison of the IR spectra calculated with B3LYP+D2 for ethanol in the gas phase (Fig. 5-A), for a single adsorbed ethanol molecule (using the Fermi model, Fig. 5-B), and an adsorbed ethanol dimer (Fig. 5-C).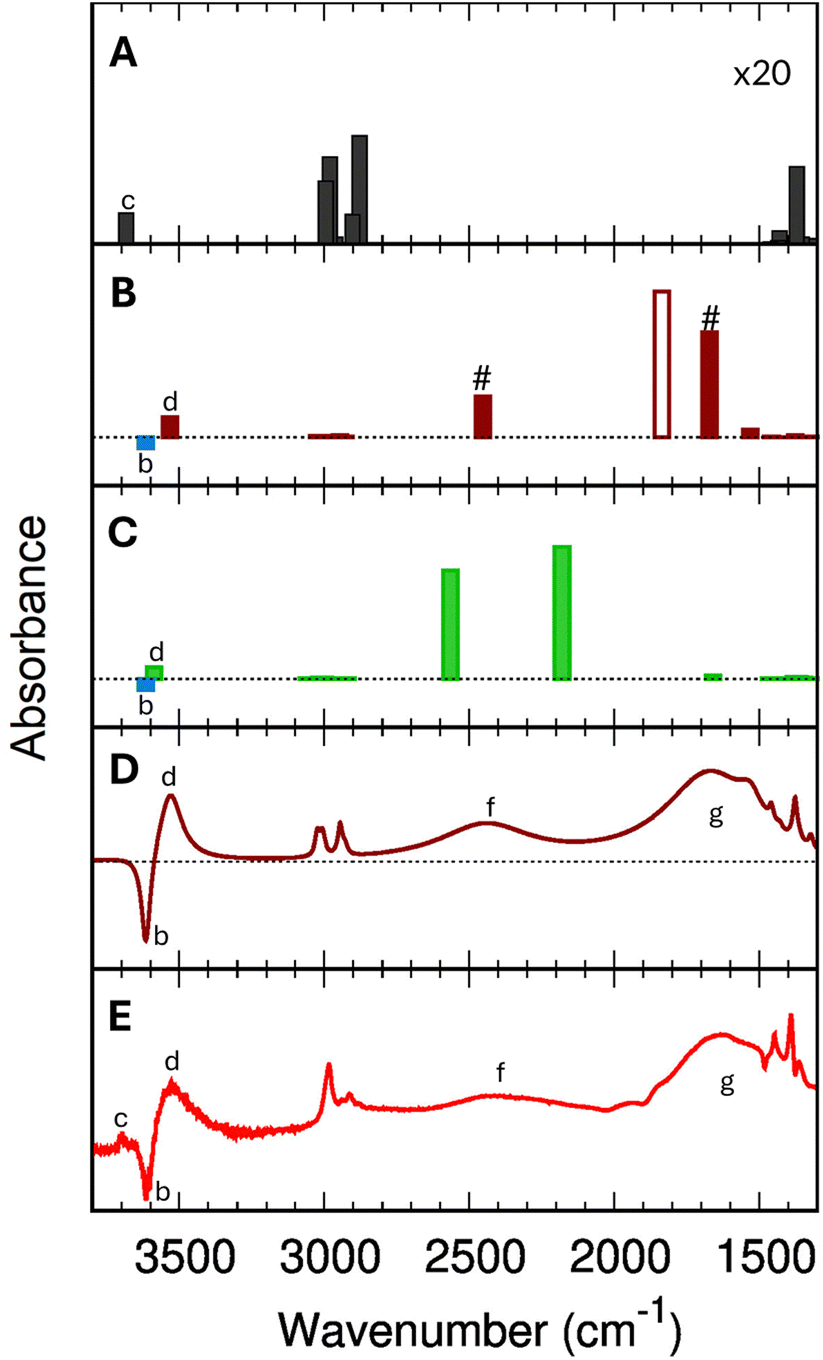 | ||
| Fig. 5 B3LYP+D2 IR spectra (scaled with 0.9710) for isolated gas-phase ethanol molecules (panel (A), intensities multiplied with 20), for single adsorbed ethanol molecules (1 EtOH/BAS, panel (B)), and for the protonated adsorbed dimer (2 EtOH/BAS, panel (C)). For the single adsorbed ethanol (panel (B)), the # indicates the two bands resulting from the Fermi resonance, while the empty bar shows the harmonic (fundamental) (Z)O–H stretching band. The blue bar in panel (B) and (C) marks the position of the (Z)O–H stretching mode of the free BAS (unloaded H-ZSM-5). Panel (D) simulated IR difference spectrum for a single adsorbed ethanol molecule based on B3LYP+D2 results using the Fermi resonance model and assuming a Lorentzian shape with bandwidths given in the ESI,† Section S7. Panel (E) experimental difference spectrum (Adiff) at 0.0034 torr (red trace in Fig. 2). | ||
The calculated wavenumbers (Table 2) have been scaled for 0.9710 to effectively account for anharmonic effects and systematic deviations of the harmonic force constants, see Section S5 of the ESI† for the unscaled numbers. For the two bands resulting from the Fermi resonance (Fig. 5-B, #), we have taken the frequencies from the experiments (2450 and 1670 cm−1, respectively) while we compute the intensities using eqn (12) and (13). For the isolated ethanol (Fig. 5-A), the (Et)O–H stretching band (feature c) is predicted at 3684 cm−1, the five CH2 and CH3 stretching bands are located between 3000 cm−1 and 2880 cm−1 (2997, 2981, 2962, 2905, and 2883 cm−1). All other vibrations, e.g., C–H bending, Et–O–H bending, C–C stretching, fall below 1500 cm−1, out of our range of interest.
| PBE+D2 | B3LYP+D2 | |
|---|---|---|
| sym. = symmetric; asym. = asymmetric; ν = stretching; δ = in plane bending; γ = out of plane bending.a PBE+D2 coupled the (Z)O–H stretching to the Z:O–H in-plane bending and to the Et–O–H bending. We report here the wavenumbers for the modes with, respectively, the strongest stretching and in-plane bending character.b Symmetric and asymmetric stretch for protonated dimer like structure. | ||
| 1 EtOH/BAS | ||
| ν(Et)O–H | 3403 (−217) | 3531 (−157) |
| ν 0(Z)O–H | 1386a (−2228) | 1806 (−1808) |
| 2γ0(Z)O–H | 2224 | 2150 |
| δZ–O–H | 1561a (534) | 1530 (433) |
| 2 EtOH/BAS | ||
| ν(Et)O–H | 3461 | 3564 |
| ν s(O–H)b | 2383 | 2588 |
| ν a(O–H)b | 1910 | 2147 |
| δH–O(Et)–H | 1609 | 1662 |
The harmonic IR spectrum of a single adsorbed molecule (1 EtOH/BAS, Fig. 5-B) is globally much more intense compared to the gas-phase ethanol one due to the charge fluxes involved in the H-bond.98 The (Et)O–H stretching mode red-shifts to 3531 cm−1 (feature d) due to the interaction with the zeolite walls, just below the wavenumber characteristic of the O–H stretching of the unloaded zeolite (3614 cm−1, feature b). As expected, the CH2 and CH3 stretching bands are only slightly shifted compared to the gas phase molecule (3024, 3007, 2999, 2945, and 2928 cm−1). Because of the strong H bond, the (Z)O–H stretching band (fundamental ν0, Fig. 5-B, empty bar) undergoes a significant red-shift of 1810 cm−1 compared to the unloaded zeolite (3614 cm−1, b). The Z–O–H in-plane and out-of-plane bending, instead, are blue-shifted to 1530 cm−1 and 1075 cm−1, respectively.
In the 2 EtOH/BAS IR spectrum (Fig. 5-C), the stretching wavenumber of the terminal (Et)O–H⋯Oγ group (3564 cm−1, feature d) is slightly higher than that of the one of the single adsorbed molecule (3531 cm−1), but below of the free (Et)O–H of the gas phase molecule (3684 cm−1, feature c) and of the O–H stretching of the unloaded zeolite (3614 cm−1, feature b). The characteristic symmetric and antisymmetric OH donor stretching vibrations of the protonated ethanol are located at 2588 cm−1 and 2147 cm−1. The H–O(Et)–H bending of the protonated ethanol is predicted at 1662 cm−1, other features of the spectrum above 1500 cm−1 are the CH2 and CH3 stretching region situated, as for the other systems, between 3060 cm−1 and 2910 cm−1.
3.6. B3LYP+D2 compared to PBE+D2 results
The differences between the PBE+D2 and B3LYP+D2 bond distances significantly impact the spectroscopic predictions (Fig. 6). Let's consider 1 EtOH/BAS. Neither PBE+D2 nor B3LYP+D2 harmonic spectra for 1 EtOH/BAS explain the experimental feature (f). However, B3LYP+D2 has the correct position for band (g) to provide a good fitting if we use the Fermi-resonance model, whereas PBE+D2 does not.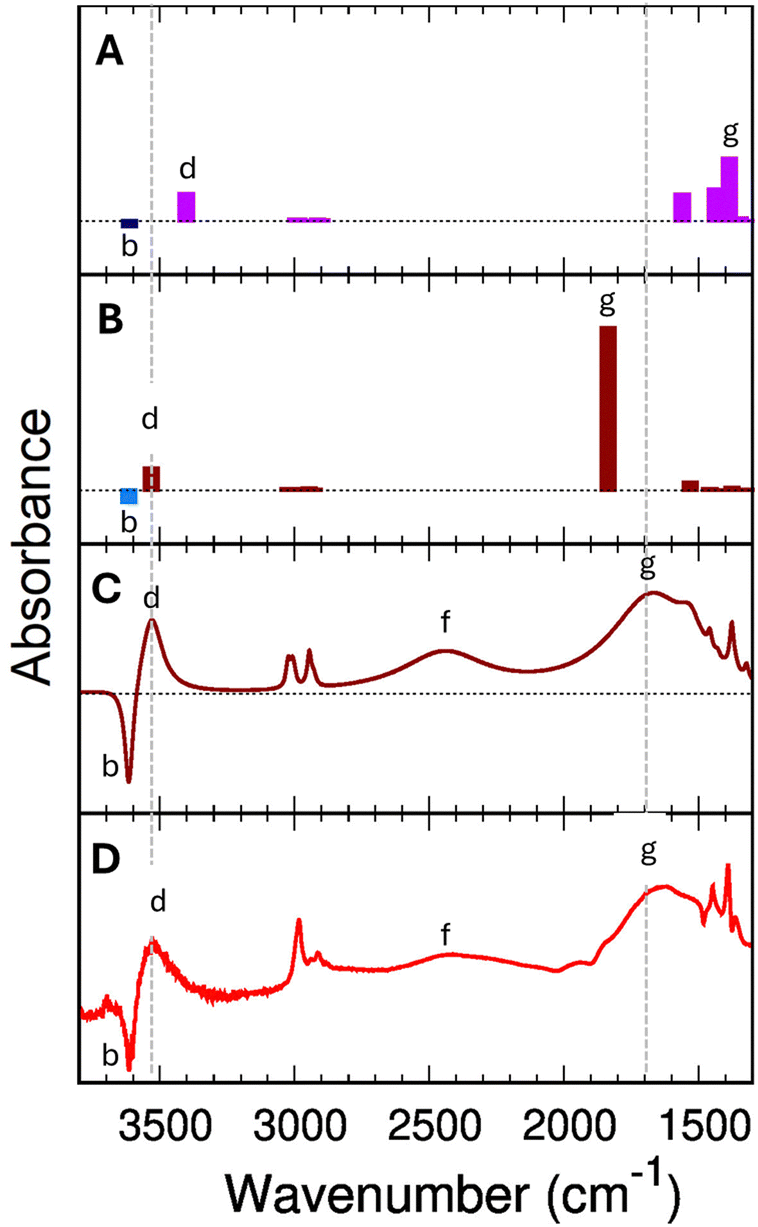 | ||
| Fig. 6 Panel (A): predicted PBE+D2 infrared spectra for the single ethanol (1 EtOH/BAS). Panel (B) predicted B3LYP+D2 infrared spectra for a single ethanol (1 EtOH/BAS, B). The blue bars indicate the position of the (Z)O–H stretching mode of the free BAS (unloaded H-ZSM-5). All the computed wavenumbers have been scaled by 0.9804 for PBE+D2 and 0.9710 for B3LYP+D2. Panel (C) simulated IR difference spectrum for a single adsorbed ethanol molecule based on B3LYP+D2 results and the Fermi resonance model assuming a Lorentzian shape with bandwidths given in the ESI† (Section S7). Panel (D) experimental measured difference spectrum (Adiff) at 0.0034 torr (red line in Fig. 2). | ||
As we have seen in the previous section, the B3LYP+D2 functional predicts the fundamental (Z)O–H stretching (g) as decoupled from other vibrations and positions it at 1806 cm−1.
PBE+D2 shows (Fig. 6-A) an additional 450 cm−1 red-shift compared to B3LYP+D2 (Table 2). The (Z)O–H stretching mode falls in the region of the bending vibrations, and it results heavily coupled with these latter, giving rise to multiple bands between 1350 cm−1 and 1600 cm−1. The projection of the modes on the vibrations of the unloaded zeolite shows that the band with the strongest (Z)O–H stretching component appears at 1386 cm−1 (Fig. 6-A, feature g), followed by a band at 1437 cm−1. The 1386 cm−1 vibration is the mode with the highest IR activity in this spectral region (at least by a factor of two), confirming that the 1386 cm−1 band can be considered the “main” (Z)O–H stretching band. Compared to the experimental value of 1670 cm−1 (band g in Fig. 6-D) the PBE+D2 prediction (1386 cm−1) is significantly lower. However, no other vibrational modes are predicted around 1700 cm−1 that can possibly explain this strong experimental band. The highest wavenumber band below 2000 cm−1 is the Z–O–H in-plane bending, at 1561 cm−1. Therefore, we need to suppose that the 1386 cm−1 PBE+D2 band is indeed the main component of (g) in the experimental spectrum.
The high-frequency region is also not immune to the functional quality. For 1 EtOH/BAS, PBE+D2 predicts the (Et)O–H stretching band at 3403 cm−1 (Fig. 6-A, feature d), 211 cm−1 lower than the (Z)O–H stretching band of the unloaded H-ZSM-5 (feature b, 3614 cm−1). In contrast, the B3LYP+D2 (Fig. 6-B) positions the loaded (Et)O–H stretching for 1 EtOH/BAS at 3531 cm−1 with a b–d gap of 83 cm−1, in good agreement with the experimental b–d gap of about 70 cm−1 (Fig. 6-D).
We conclude that the PBE+D2 predicted wavenumbers are significantly shifted compared to the experimental values. These shifts are not systematic, and even the order of some of the bands is inverted (e.g., the (Z)O–H stretching and the Z–O–H in-plane bending). No scaling factor can reasonably fix this.
One consequence of the artificial shift is that PBE+D2 excludes the Fermi resonance a priori due to the (incorrect) shape of the potential energy surface. The position of the fundamental (Z)O–H stretching band at 1386 cm−1, red-shifted by 290 cm−1 compared to the experimental value, is incompatible with a Fermi resonance. The gap between ν0(Z)O–H and 2γ0(Z)O–H is too large (838 cm−1) to make the Fermi resonance plausible. Indeed ν0(Z)O–H (1386 cm−1) is closer in position to the γ0(Z)O–H fundamental band (1112 cm−1) than its overtone (2224 cm−1). Moreover ν0(Z)O–H would be further red-shift by it, making the comparison with the experimental spectra even worse. Because the problem does not come from the double harmonic approximation but directly from the electronic structure representation, the use of static or MD techniques to include the anharmonicity and coupling effects in the calculation is ineffective.
B3LYP+D2 instead presents a much better match with the experimental values both in the high and low frequencies regions. The B3LYP+D2 prediction for the (Z)O–H stretching (1806 cm−1) is blue-shifted compared to the (g, Fig. 6-B) band in the experimental spectrum (1670 cm1). As we will see in the following, once the Fermi resonance between the (Z)O–H stretching and the Z–O–H out-of-plane bending is taken into account, the comparison between the computed B3LYP+D2 difference spectrum (Fig. 6-C) and the experimental difference spectra at low pressure (Fig. 6-D) is excellent.
4. Assignment of the observed vibrational spectra
Fig. 5-E shows the experimental difference spectrum Adiff at the equilibrium pressure of 0.0034 torr. Compared to the 1 EtOH/BAS (B3LYP+D2) harmonic stick spectrum (Fig. 5-B), the experimental spectrum shows a strong band (g) at lower wavenumbers than the predicted (Z)O–H band (1806 cm−1), whereas band (f) in the experimental spectrum remains unexplained. However, an assignment of all bands of the experimental spectrum is obtained if the Fermi resonance between the shifted (Z)O–H stretching and the overtone of the Z–O–H out-of-plane bending is introduced in our model. The (Z)O–H stretching band (Fig. 5-B, empty bar) is split into two moieties (Fig. 5-B, #) at 2450 cm−1 and 1670 cm−1. The position of the two Fermi bands in our model has been taken from the experiments, while the intensities have been computed by eqn (12) and (13). For a more direct comparison with the experiment, Fig. 5-D shows the computed difference spectrum for 1 EtOH/BAS (using the Fermi model), where we assign a Lorentzian shape to the predicted vibrational transitions. The bandwidths are set around the experimental values, paying attention to preserving the bands' total intensity. The very good agreement leaves no doubt about the assignment to a single adsorbed ethanol molecule. The predicted 1 EtOH/BAS difference spectrum confirms the assignment of the 3614 cm−1 band (b) to the (Z)O–H stretching mode of the free BAS, of the broad feature (d) at 3543 cm−1 to the stretching of (Et)O–Hγ, and a set of sharp peaks (e) around 2900 cm−1 to the CH stretching vibrations of ethanol.There is only one band at 3695 cm−1 (c) which is seen in the experiment but not in the predicted 1 EtOH/BAS spectrum. Our calculations (Fig. 5-A) show that this band can possibly be assigned to the (Et)O–H stretching of a gas phase ethanol, i.e., molecules inside the zeolite, adsorbed on the silicate wall,99,100 but not adsorbed on the BAS or H-bond to other ethanol molecules. In this scenario, the disappearance of this band in the experimental spectra above 0.0041 torr (see Fig. 2 and Section 3.2) would be a marker that all the ethanol molecules are either adsorbed or interacting with other ethanol molecules at these pressures. Further investigations are needed to confirm this assignment that is, however, out of the scope of this discussion.
We can therefore conclude that the theoretical spectrum of a single adsorbed ethanol molecule described using the Fermi resonance model allows us to assign all the vibrational bands in the experimentally measured spectrum.
In the past, differently from the Fermi resonance model for the adsorbed monomer,25,49 the 2450 cm−1 band (f) has been assigned to the symmetric fundamental νs(O–H) stretching mode of protonated ethanol dimer.5 However, both our experimental and computational results do not support this hypothesis. While the match between the experimental spectra and the Fermi corrected 1 EtOH/BAS computed spectrum is straightforward, the dimer hypothesis requires additional assumptions. B3LYP+D2 predicts the symmetric and antisymmetric νs/a(O–H) stretching modes at 2588 cm−1 and 2147 cm−1, respectively. To consider (f) as the signature of the adsorption of a protonated dimer, we need to assume that the νs/a(O–H) stretching modes are by far more anharmonic than the other fundamental vibrational bands, and a stronger scaling is needed to match the experimental spectrum. If we select for these two bands a scaling factor of 0.9273 (instead of 0.9710), the symmetric stretching, νs(O–H), falls at 2450 cm−1, and the antisymmetric one, νa(O–H), at 2083 cm−1. This ad hoc scaling of stretching bands, however, is still not enough to reasonably reproduce the experimental spectrum. From our calculations, νa(O–H) is around 1.22 times stronger in intensity than νs(O–H). However, we cannot recognize in the experimental spectrum any band between 2450 cm−1 and 2000 cm−1 that can be assigned to νa(O–H) (Fig. 6-D). Therefore, to reproduce the experimental results, we need to assume that the anharmonicity further red-shift νa(O–H) band in a way that is convoluted in the (g) feature.
Even after introducing these additional shifts in the model, the dimer hypothesis is not consistent with the system's behavior at low pressure. Consider the evolution of the (f) and (g) intensity ratio at increasing pressure and its physical meaning. The (f) band is present in the experimental spectra already at 0.0034 torr (Fig. 1-B and 2). Due to the nonlinear dependence of the background contribution on the wavenumber, it is not so easy to be quantitative. Still, a fair lower limit for the experimental total absorbance ratio between (f) and (g) is 0.2. In the dimer hypothesis, a ratio of 0.2 implies the presence of at least one 2 EtOH/BAS for every three 1 EtOH/BAS molecules at 0.0034 torr (see the ESI†). At the same time, the disappearance in the incremental variation spectra (Fig. 2) of the 2450 cm−1 feature (f) between 0.006 Torr and 0.00675 Torr (green and light blue spectra) implies, in the dimer hypothesis, that no more dimers are formed above this pressure.
The experimental measurements report that at 0.0060 Torr, when all the BAS are loaded (disappearance of the 3614 cm−1 (Z)O–H stretching band) only around 2.1 mmol g−1 of ethanol have been adsorbed. Considering that this point corresponds to the maximum of the curvature of the experimental isotherm (inset in Fig. 1-A) and the ethanol adsorbed amount is close to our sample BAS concentration (around 1.7 mmol g−1 by ammonia titration reported in Section S2, ESI†), we can identify in this point the formation of the adsorbate monolayer (1.2 EtOH molecules per BAS). This means that at 0.0060 Torr, experimentally we still have essentially only single molecule adsorption. Therefore, we can discharge the hypothesis that 2450 cm−1 is coming only from the dimer.
Instead, a stretching-bending Fermi resonance in the case of single molecule adsorption explains the measured (f)/(g) intensity ratio at 0.0034 torr quite well. Our Fermi model predicts, in fact, an intensity ratio of 0.36, not so far from the experimental value (>0.2) considering the approximations both on the computational and experimental sides. The Fermi resonance model also explains the observed behavior in the incremental difference spectra (Fig. 2). Between 0.0034 Torr and 0.0041 Torr (red to yellow spectra) all the bands are changing in a coordinated manner preserving the intensity ratio because what we are seeing is the progression of the single ethanol molecule adsorption. In the transient region between 0.006 Torr and 0.00675 Torr (green and light blue spectra), we have competition between single molecule and dimer adsorption. At this point the 2450 cm−1 band (a marker of the single molecule adsorption) stops to gain intensity. A new contribution rises above 2600 cm−1 due to the symmetric O–H stretching of the protonated dimer. Above 0.00675 Torr complete loading has been achieved. The formation of a dimer implies the consumption of a single molecule adsorbed. Therefore, while the CH stretching bands and OH bending bends still grow with the uptake of additional ethanol molecules, the dA profile between 1600 cm−1 and 2600 cm−1 becomes flat due to the compensation of the “consumed” single molecule (Z)O–H Fermi resonance with the antisymmetric νa(O–H), stretching and H–O(Et)–H bending of the protonated dimer.
Conclusions
We showed that the measured FTIR spectra for a highly crystalline sample of zeolite H-ZSM-5 exposed to ethanol with increasing pressure (from 0.0034 Torr to 0.0135 Torr) are due to a single adsorbed ethanol molecule per BAS and that adsorbed dimers do not contribute in a noticeable way almost up to a concentration of one ethanol molecule per BAS. The spectrum is dominated by a Fermi resonance between the fundamental (Z)O–H⋯OEt stretching band of the BAS and the first overtone of the Z–O–H⋯OEt out-of-plane bending. We also demonstrated that the commonly used PBE functional yields an erroneous assignment due to an overestimation of the hydrogen bond strength accompanied by an overestimation of the red shift of the zeolitic OH donor bond engaged in hydrogen bonding. In contrast, the spectra predicted with the B3LYP hybrid functional provided an excellent match with the experimental spectra when Fermi resonance couplings are taken into account. However, to describe all the bands of the experimental spectra, the computed harmonic spectra must be corrected for the presence of Fermi resonances. This shows that a proper theoretical description of catalytic processes involving alcohols in zeolites has to go beyond the GGA-type functionals such as PBE and also needs to include mode couplings.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
DK and DRG thank SURF (https://www.surf.nl) for providing computing time and for the support in using the Dutch National Supercomputer Snellius. A. A. and S. B. acknowledge support from the Project CH4.0 under the MUR program “Dipartimenti di Eccellenza 2023-2027” (CUP: D13C22003520001). J. S. has been funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 514934444. Dr Maria Carmen Valsania and Dr Erica Rebba are kindly acknowledged for having performed the FE-SEM measurements.References
- S. Svelle, C. Tuma, X. Rozanska, T. Kerber and J. Sauer, Quantum Chemical Modeling of Zeolite-Catalyzed Methylation Reactions: Toward Chemical Accuracy for Barriers, J. Am. Chem. Soc., 2009, 131, 816–825 CrossRef CAS PubMed.
- P. N. Plessow and F. Studt, Unraveling the Mechanism of the Initiation Reaction of the Methanol to Olefins Process Using ab Initio and DFT Calculations, ACS Catal., 2017, 7, 7987–7994 CrossRef CAS.
- Y. Chen, X. Ma, J. H. Hack, S. Zhang, A. Peng, J. P. Dombrowski, G. A. Voth, A. Tokmakoff, M. C. Kung and H. H. Kung, Molecular Tuning of Reactivity of Zeolite Protons in HZSM-5, J. Am. Chem. Soc., 2024, 146, 10342–10356 CrossRef CAS PubMed.
- B. Vora, J. Q. Chen, A. Bozzano, B. Glover and P. Barger, Various Routes to Methane Utilization—SAPO-34 Catalysis Offers the Best Option, Catal. Today, 2009, 141, 77–83 CrossRef CAS.
- K. Alexopoulos, M.-S. Lee, Y. Liu, Y. Zhi, Y. Liu, M.-F. Reyniers, G. B. Marin, V.-A. Glezakou, R. Rousseau and J. A. Lercher, Anharmonicity and Confinement in Zeolites: Structure, Spectroscopy, and Adsorption Free Energy of Ethanol in H-ZSM-5, J. Phys. Chem. C, 2016, 120, 7172–7182 CrossRef CAS.
- G. Piccini, M. Alessio and J. Sauer, Ab initio study of methanol and ethanol adsorption on Brønsted sites in zeolite H-MFI, Phys. Chem. Chem. Phys., 2018, 20, 19964–19970 RSC.
- F. Chen, M. Shetty, M. Wang, H. Shi, Y. Liu, D. M. Camaioni, O. Y. Gutiérrez and J. A. Lercher, Differences in Mechanism and Rate of Zeolite-Catalyzed Cyclohexanol Dehydration in Apolar and Aqueous Phase, ACS Catal., 2021, 11, 2879–2888 CrossRef CAS.
- M. E. Potter, J. Amsler, L. Spiske, P. N. Plessow, T. Asare, M. Carravetta, R. Raja, P. A. Cox, F. Studt and L.-M. Armstrong, Combining Theoretical and Experimental Methods to Probe Confinement within Microporous Solid Acid Catalysts for Alcohol Dehydration, ACS Catal., 2023, 13, 5955–5968 CrossRef CAS.
- C. Chizallet, C. Bouchy, K. Larmier and G. Pirngruber, Molecular Views on Mechanisms of Brønsted Acid-Catalyzed Reactions in Zeolites, Chem. Rev., 2023, 123, 6107–6196 CrossRef CAS.
- S. Kim, M.-S. Lee, D. M. Camaioni, O. Y. Gutiérrez, V.-A. Glezakou, N. Govind, T. Huthwelker, R. Zhao, R. Rousseau, J. L. Fulton and J. A. Lercher, Self-Organization of 1-Propanol at H-ZSM-5 Brønsted Acid Sites, JACS Au, 2023, 3, 2487–2497 CrossRef CAS PubMed.
- T. Ennaert, J. Van Aelst, J. Dijkmans, R. De Clercq, W. Schutyser, M. Dusselier, D. Verboekend and B. F. Sels, Potential and challenges of zeolite chemistry in the catalytic conversion of biomass, Chem. Soc. Rev., 2016, 45, 584–611 RSC.
- X. Zhou, C. Wang, Y. Chu, J. Xu, Q. Wang, G. Qi, X. Zhao, N. Feng and F. Deng, Observation of an oxonium ion intermediate in ethanol dehydration to ethene on zeolite, Nat. Commun., 2019, 10, 1961 CrossRef PubMed.
- M. Gešvandtnerová, P. Raybaud, C. Chizallet and T. Bučko, Importance of Dynamic Effects in Isobutanol to Linear Butene Conversion Catalyzed by Acid Zeolites Assessed by AIMD, ACS Catal., 2024, 7478–7491, DOI:10.1021/acscatal.4c00736.
- F. Haase and J. Sauer, Interaction of Methanol with Brønsted Sites of Zeolite Catalysts – An Ab initio Study, J. Am. Chem. Soc., 1995, 117, 3780–3789 CrossRef CAS.
- F. Haase, J. Sauer and J. Hutter, Ab initio molecular dynamics simulation of methanol adsorbed in chabazite, Chem. Phys. Lett., 1997, 266, 397–402 CrossRef CAS.
- S. K. Matam, S. A. F. Nastase, A. J. Logsdail and C. Richard, A Catlow, Methanol loading dependent methoxylation in zeolite H-ZSM-5, Chem. Sci., 2020, 11, 6805–6814 RSC.
- J. Sauer, M. Sierka and F. Haase, in Transition State Modeling for Catalysis, ed. D. G. Truhlar and K. Morokuma, American Chemical Society, Washington, 1999, pp. 358–367 Search PubMed.
- J. Sauer, in Handbook of Hydrogen Transfer, ed. R. L. Schowen, J. P. Klinman, J. T. Hynes and H.-H. Limbach, Wiley-VCH, Weinheim, 2006, vol. 2, pp. 685–707 Search PubMed.
- S. Bordiga, I. Roggero, P. Ugliengo, A. Zecchina, V. Bolis, G. Artioli, R. Buzzoni, G. Marra, F. Rivetti, G. Spanò and C. Lamberti, Characterisation of defective silicalites, J. Chem. Soc., Dalton Trans., 2000, 3921–3929, 10.1039/B004794P.
- T. Armaroli, L. J. Simon, M. Digne, T. Montanari, M. Bevilacqua, V. Valtchev, J. Patarin and G. Busca, Effects of crystal size and Si/Al ratio on the surface properties of H-ZSM-5 zeolites, Appl. Catal., 2006, 306, 78–84 CrossRef CAS.
- L. Treps, C. Demaret, D. Wisser, B. Harbuzaru, A. Méthivier, E. Guillon, D. V. Benedis, A. Gomez, T. D. Bruin, M. Rivallan, L. Catita, A. Lesage and C. Chizallet, Spectroscopic Expression of the External Surface Sites of H-ZSM-5, J. Phys. Chem. C, 2021, 125, 2163–2181 CrossRef CAS.
- F. Dubray, E. Dib, I. Medeiros-Costa, C. Aquino, D. Minoux, S. van Daele, N. Nesterenko, J.-P. Gilson and S. Mintova, The challenge of silanol species characterization in zeolites, Inorg. Chem. Front., 2022, 9, 1125–1133 RSC.
- A. Zecchina, G. Spoto and S. Bordiga, Probing the acid sites in confined spaces of microporous materials by vibrational spectroscopy, Phys. Chem. Chem. Phys., 2005, 7, 1627–1642 RSC.
- B.-T. Lønstad Bleken, L. Mino, F. Giordanino, P. Beato, S. Svelle, K. P. Lillerud and S. Bordiga, Probing the surface of nanosheet H-ZSM-5 with FTIR spectroscopy, Phys. Chem. Chem. Phys., 2013, 15, 13363–13370 RSC.
- S. Bordiga, C. Lamberti, F. Bonino, A. Travert and F. Thibault-Starzyk, Probing zeolites by vibrational spectroscopies, Chem. Soc. Rev., 2015, 44, 7262–7341 RSC.
- R. Osuga, T. Yokoi and J. N. Kondo, IR observation of activated ether species on acidic OH groups on H-ZSM-5 zeolites, Mol. Catal., 2019, 477, 110535 CrossRef CAS.
- K. Gołąbek, E. Tabor, V. Pashkova, J. Dedecek, K. Tarach and K. Góra-Marek, The proximity of aluminium atoms influences the reaction pathway of ethanol transformation over zeolite ZSM-5, Commun. Chem., 2020, 3, 25 CrossRef.
- A. Airi, M. Signorile, F. Bonino, P. Quagliotto, S. Bordiga, J. A. Martens and V. Crocellà, Insights on a Hierarchical MFI Zeolite: A Combined Spectroscopic and Catalytic Approach for Exploring the Multilevel Porous System Down to the Active Sites, ACS Appl. Mater. Interfaces, 2021, 13, 49114–49127 CrossRef CAS.
- J. H. Hack, J. P. Dombrowski, X. Ma, Y. Chen, N. H. C. Lewis, W. B. Carpenter, C. Li, G. A. Voth, H. H. Kung and A. Tokmakoff, Structural Characterization of Protonated Water Clusters Confined in HZSM-5 Zeolites, J. Am. Chem. Soc., 2021, 143, 10203–10213 CrossRef CAS PubMed.
- V. Van Speybroeck, K. Hemelsoet, L. Joos, M. Waroquier, R. G. Bell and C. R. A. Catlow, Advances in theory and their application within the field of zeolite chemistry, Chem. Soc. Rev., 2015, 44, 7044–7111 RSC.
- G. Collinge, S. F. Yuk, M.-T. Nguyen, M.-S. Lee, V.-A. Glezakou and R. Rousseau, Effect of Collective Dynamics and Anharmonicity on Entropy in Heterogenous Catalysis: Building the Case for Advanced Molecular Simulations, ACS Catal., 2020, 10, 9236–9260 CrossRef CAS.
- M. Bocus and V. Van Speybroeck, Insights into the Mechanism and Reactivity of Zeolite-Catalyzed Alkylphenol Dealkylation, ACS Catal., 2022, 12, 14227–14242 CrossRef CAS.
- J. Sauer, P. Ugliengo, E. Garrone and V. R. Saunders, Theoretical Study of van der Waals Complexes at Surface Sites in Comparison with the Experiment, Chem. Rev., 1994, 94, 2095–2160 CrossRef CAS.
- H. Windeck, F. Berger and J. Sauer, Spectroscopic Signatures of Internal Hydrogen Bonds of Brønsted-Acid Sites in the Zeolite H-MFI, Angew. Chem., 2023, e202303204 CAS.
- K. Alexopoulos, M. John, K. Van der Borght, V. Galvita, M.-F. Reyniers and G. B. Marin, DFT-based microkinetic modeling of ethanol dehydration in H-ZSM-5, J. Catal., 2016, 339, 173–185 CrossRef CAS.
- J. Sauer, The future of computational catalysis, J. Catal., 2024, 433, 115482 CrossRef CAS.
- M. Krossner and J. Sauer, Interaction of Water with Brønsted Acidic Sites of Zeolite Catalysts. Ab Initio Study of 1:1 and 2:1 Surface Complexes, J. Phys. Chem., 1996, 100, 6199–6211 CrossRef CAS.
- C. Tuma and J. Sauer, A hybrid MP2/planewave-DFT scheme for large chemical systems: proton jumps in zeolites, Chem. Phys. Lett., 2004, 387, 388–394 CrossRef CAS.
- T. J. Goncalves, P. N. Plessow and F. Studt, On the Accuracy of Density Functional Theory in Zeolite Catalysis, ChemCatChem, 2019, 11, 4368–4376 CrossRef CAS.
- F. R. Rehak, G. Piccini, M. Alessio and J. Sauer, Including dispersion in density functional theory for adsorption on flat oxide surfaces, in metal–organic frameworks and in acidic zeolites, Phys. Chem. Chem. Phys., 2020, 22, 7577–7585 RSC.
- F. Berger and J. Sauer, Dimerization of Linear Butenes and Pentenes in an Acidic Zeolite (H-MFI), Angew. Chem., Int. Ed., 2021, 60, 3529–3533 CrossRef CAS PubMed.
- H. Windeck, F. Berger and J. Sauer, Spectroscopic Signatures of Internal Hydrogen Bonds of Brønsted-Acid Sites in the Zeolite H-MFI, Angew. Chem., Int. Ed., 2023, 62, e202303204 CrossRef CAS.
- H. Windeck, F. Berger and J. Sauer, Chemically accurate predictions for water adsorption on Brønsted sites of zeolite H-MFI, Phys. Chem. Chem. Phys., 2024, 26, 23588–23599 RSC.
- C. Pazé, S. Bordiga, C. Lamberti, M. Salvalaggio, A. Zecchina and G. Bellussi, Acidic Properties of H−β Zeolite As Probed by Bases with Proton Affinity in the 118–204 kcal mol−1 Range: A FTIR Investigation, J. Phys. Chem. B, 1997, 101, 4740–4751 CrossRef.
- K. Gołąbek, K. A. Tarach, U. Filek and K. Góra-Marek, Ethylene formation by dehydration of ethanol over medium pore zeolites, Spectrochim. Acta, Part A, 2018, 192, 464–472 CrossRef PubMed.
- F. Wakabayashi, J. N. Kondo, K. Domen and C. Hirose, FT-IR study of H2O Adsorption on H-ZSM-5: Direct evidence for Hydrogen Bonded Adsorption of Water, J. Phys. Chem., 1996, 100, 1442–1444 CrossRef CAS.
- V. V. Mihaleva, R. A. van Santen and A. P. J. Jansen, Quantum chemical calc ulation of infrared spectra of acidic groups in chabasite in the presence of water, J. Chem. Phys., 2004, 120, 9212–9221 CrossRef CAS.
- A. G. Pelmenschikov, J. H. M. C. van Wolput, J. Jänchen and R. A. van Santen, (A,B,C) Triplet of Infrared OH Bonds of Zeolitic H-Complexes, J. Phys. Chem., 1995, 99, 3612–3617 CrossRef CAS.
- A. Zecchina, S. Bordiga, G. Spoto, D. Scarano, G. Spanò and F. Geobaldo, IR spectroscopy of neutral and ionic hydrogen-bonded complexes formed upon interaction of CH3OH, C2H5OH, (CH3)2O, (C2H5)2O and C4H8O with H-Y, H-ZSM-5 and H-mordenite: comparison with analogous adducts formed on the H-Nafion superacidic membrane, J. Chem. Soc., Faraday Trans., 1996, 92, 4863–4875 RSC.
- J. H. Hack, Y. Chen, N. H. C. Lewis, H. H. Kung and A. Tokmakoff, Strong H-bonding from Zeolite Brønsted Acid Site to Water: Origin of the Broad IR Doublet, J. Phys. Chem. B, 2023, 127, 11054–11063 CrossRef CAS PubMed.
- A. Iogansen, Optika i Spektroskopiya, III Izdat, 1967, p. 228 Search PubMed.
- K. Na, C. Jo, J. Kim, K. Cho, J. Jung, Y. Seo, R. J. Messinger, B. F. Chmelka and R. Ryoo, Directing Zeolite Structures into Hierarchically Nanoporous Architectures, Science, 2011, 333, 328–332 CrossRef CAS.
- J. Sauer and A. Bleiber, Internal silanols in zeolites – inferences from quantum chemical calculations, Catal. Today, 1988, 3, 485–492 CrossRef CAS.
- Suwardiyanto, R. F. Howe, E. K. Gibson, C. R. A. Catlow, A. Hameed, J. McGregor, P. Collier, S. F. Parker and D. Lennon, An assessment of hydrocarbon species in the methanol-to-hydrocarbon reaction over a ZSM-5 catalyst, Faraday Discuss., 2017, 197, 447–471 RSC.
- A. A. Gabrienko, I. G. Danilova, S. S. Arzumanov, L. V. Pirutko, D. Freude and A. G. Stepanov, Direct Measurement of Zeolite Brønsted Acidity by FTIR Spectroscopy: Solid-State 1H MAS NMR Approach for Reliable Determination of the Integrated Molar Absorption Coefficients, J. Phys. Chem. C, 2018, 122, 25386–25395 CrossRef CAS.
- S. K. Matam, R. F. Howe, A. Thetford and C. R. A. Catlow, Room temperature methoxylation in zeolite H-ZSM-5: an operando DRIFTS/mass spectrometric study, Chem. Commun., 2018, 54, 12875–12878 RSC.
- I. B. Minova, S. K. Matam, A. Greenaway, C. R. A. Catlow, M. D. Frogley, G. Cinque, P. A. Wright and R. F. Howe, Elementary Steps in the Formation of Hydrocarbons from Surface Methoxy Groups in HZSM-5 Seen by Synchrotron Infrared Microspectroscopy, ACS Catal., 2019, 9, 6564–6570 CrossRef CAS.
- A. Zecchina, S. Bordiga, G. Spoto, D. Scarano, G. Petrini, G. Leofanti, M. Padovan and C. O. Areàn, Low-temperature Fourier-transform infrared investigation of the interaction of CO with nanosized ZSM5 and silicalite, J. Chem. Soc., Faraday Trans., 1992, 88, 2959–2969 RSC.
- I. Kiricsi, C. Flego, G. Pazzuconi, W. O. Parker, Jr., R. Millini, C. Perego and G. Bellussi, Progress toward Understanding Zeolite.beta. Acidity: An IR and 27Al NMR Spectroscopic Study, J. Phys. Chem., 1994, 98, 4627–4634 CrossRef CAS.
- J. M. Chalmers, Handbook of Vibrational Spectroscopy, 2001 DOI:10.1002/0470027320.s3101.
- K. Chen, M. Abdolrhamani, E. Sheets, J. Freeman, G. Ward and J. L. White, Direct Detection of Multiple Acidic Proton Sites in Zeolite HZSM-5, J. Am. Chem. Soc., 2017, 139, 18698–18704 CrossRef CAS PubMed.
- M. Abdolrahmani, K. Chen and J. L. White, Assessment, Control, and Impact of Brønsted Acid Site Heterogeneity in Zeolite HZSM-5, J. Phys. Chem. C, 2018, 122, 15520–15528 CrossRef CAS.
- K. Chen, M. Abdolrahmani, S. Horstmeier, T. N. Pham, V. T. Nguyen, M. Zeets, B. Wang, S. Crossley and J. L. White, Brønsted–Brønsted Synergies between Framework and Noncrystalline Protons in Zeolite H-ZSM-5, ACS Catal., 2019, 9, 6124–6136 CrossRef CAS.
- K. Chen, S. Horstmeier, V. T. Nguyen, B. Wang, S. P. Crossley, T. Pham, Z. Gan, I. Hung and J. L. White, Structure and Catalytic Characterization of a Second Framework Al(IV) Site in Zeolite Catalysts Revealed by NMR at 35.2 T, J. Am. Chem. Soc., 2020, 142, 7514–7523 CrossRef CAS.
- C. Schroeder, V. Siozios, M. Hunger, M. R. Hansen and H. Koller, Disentangling Brønsted Acid Sites and Hydrogen-Bonded Silanol Groups in High-Silica Zeolite H-ZSM-5, J. Phys. Chem. C, 2020, 124, 23380–23386 CrossRef CAS.
- L. Treps, A. Gomez, T. de Bruin and C. Chizallet, Environment, Stability and Acidity of External Surface Sites of Silicalite-1 and ZSM-5 Micro and Nano Slabs, Sheets, and Crystals, ACS Catal., 2020, 10, 3297–3312 CrossRef CAS.
- A. Zecchina, F. Geobaldo, G. Spoto, S. Bordiga, G. Ricchiardi, R. Buzzoni and G. Petrini, FTIR Investigation of the Formation of Neutral and Ionic Hydrogen-Bonded Complexes by Interaction of H-ZSM-5 and H-Mordenite with CH3CN and H2O: Comparison with the H-NAFION Superacidic System, J. Phys. Chem., 1996, 100, 16584–16599 CrossRef CAS.
- M. Neff and G. Rauhut, Toward large scale vibrational configuration interaction calculations, J. Chem. Phys., 2009, 131, 124–129 Search PubMed.
- A. Erba, J. Maul, M. Ferrabone, R. Dovesi, M. Rérat and P. Carbonnière, Anharmonic Vibrational States of Solids from DFT Calculations. Part II: Implementation of the VSCF and VCI Methods, J. Chem. Theory Comput., 2019, 15, 3766–3777 CrossRef CAS.
- R. G. Schireman, J. Maul, A. Erba and M. T. Ruggiero, Anharmonic Coupling of Stretching Vibrations in Ice: A Periodic VSCF and VCI Description, J. Chem. Theory Comput., 2022, 18, 4428–4437 CrossRef CAS.
- D. Mitoli, J. Maul and A. Erba, First-Principles Anharmonic Infrared and Raman Vibrational Spectra of Materials: Fermi Resonance in Dry Ice, J. Phys. Chem. Lett., 2024, 15, 888–894 CrossRef CAS.
- R. Conte, A. Nandi, C. Qu, Q. Yu, P. L. Houston and J. M. Bowman, Semiclassical and VSCF/VCI Calculations of the Vibrational Energies of trans- and gauche-Ethanol Using a CCSD(T) Potential Energy Surface, J. Phys. Chem. A, 2022, 126, 7709–7718 CrossRef CAS PubMed.
- J. M. Bowman, Vibrational Dynamics of Molecules Search PubMed.
- P. L. Silvestrelli, M. Bernasconi and M. Parrinello, Ab initio infrared spectrum of liquid water, Chem. Phys. Lett., 1997, 277, 478–482 CrossRef CAS.
- M.-P. Gaigeot, Theoretical spectroscopy of floppy peptides at room temperature. A DFTMD perspective: gas and aqueous phase, Phys. Chem. Chem. Phys., 2010, 12, 3336–3359 RSC.
- M. Thomas, M. Brehm, R. Fligg, P. Vöhringer and B. Kirchner, Computing vibrational spectra from ab initio molecular dynamics, Phys. Chem. Chem. Phys., 2013, 15, 6608–6622 RSC.
- D. R. Galimberti, A. Milani, M. Tommasini, C. Castiglioni and M.-P. Gaigeot, Combining Static and Dynamical Approaches for Infrared Spectra Calculations of Gas Phase Molecules and Clusters, J. Chem. Theory Comput., 2017, 13, 3802–3813 CrossRef CAS PubMed.
- V. Conti Nibali, S. Pezzotti, F. Sebastiani, D. R. Galimberti, G. Schwaab, M. Heyden, M. P. Gaigeot and M. Havenith, Wrapping Up Hydrophobic Hydration: Locality Matters, J. Phys. Chem. Lett., 2020, 11, 4809–4816 CrossRef CAS.
- A. E. J. Hoffman, W. Temmerman, E. Campbell, A. A. Damin, I. Lezcano-Gonzalez, A. M. Beale, S. Bordiga, J. Hofkens and V. Van Speybroeck, A Critical Assessment on Calculating Vibrational Spectra in Nanostructured Materials, J. Chem. Theory Comput., 2024, 20, 513–531 CrossRef CAS.
- G. Herzberg, Infrared and Raman Spectra of Polyatomic Molecules, Van Nostrand, 1945 Search PubMed.
- S. E. Odinokov and A. V. Iogansen, Torsional γ(OH) vibrations, Fermi resonance [2γ(OH) ⇐ ν(OH)] and isotopic effects in i.r. spectra of H-complexes of carboxylic acids with strong bases, Spectrochim. Acta, Part A, 1972, 28, 2343–2350 CrossRef CAS.
- G. Piccini, M. Alessio and J. Sauer, Ab Initio Calculation of Rate Constants for Molecule–Surface Reactions with Chemical Accuracy, Angew. Chem., Int. Ed., 2016, 55, 5235–5237 CrossRef CAS.
- C. Tuma and J. Sauer, Treating dispersion effects in extended systems by hybrid MP2:DFT calculations—protonation of isobutene in zeolite ferrierite, Phys. Chem. Chem. Phys., 2006, 8, 3955–3965 RSC.
- J. Sauer, Ab Initio Calculations for Molecule–Surface Interactions with Chemical Accuracy, Acc. Chem. Res., 2019, 52, 3502–3510 CrossRef CAS.
- M. Alessio, F. A. Bischoff and J. Sauer, Chemically accurate adsorption energies for methane and ethane monolayers on the MgO(001) surface, Phys. Chem. Chem. Phys., 2018, 20, 9760–9769 RSC.
- F. A. Bischoff, M. Alessio, F. Berger, M. John, M. Rybicki and J. Sauer, Multi-Level Energy Landscapes: The MonaLisa Program, 2019, https://www.chemie.hu-berlin.de/de/forschung/quantenchemie/monalisa/.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- J. VandeVondele and J. Hutter, Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases, J. Chem. Phys., 2007, 127, 114105 CrossRef.
- S. Goedecker, M. Teter and J. Hutter, Separable dual-space Gaussian pseudopotentials, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- F. Neese, Software update: the ORCA program system, version 4.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- F. Neese, Software update: The ORCA program system—Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Systematic optimization of long-range corrected hybrid density functionals, J. Chem. Phys., 2008, 128, 084106 CrossRef.
- S. Abbate, G. Longhi, K. Kwon and A. Moscowitz, The use of cross-correlation functions in the analysis of circular dichroism spectra, J. Chem. Phys., 1998, 108, 50–62 CrossRef CAS.
- D. Galimberti, A. Milani and C. Castiglioni, Infrared intensities and charge mobility in hydrogen bonded complexes, J. Chem. Phys., 2013, 139, 074304 CrossRef.
- M. Rybicki, K. Sillar and J. Sauer, Dual-Site Model for Ab Initio Calculations of Gibbs Free Energies and Enthalpies of Adsorption: Methane in Zeolite Mobile Five (H-MFI), J. Phys. Chem. Lett., 2022, 13, 11595–11600 CrossRef CAS PubMed.
- F. Berger, M. Rybicki and J. Sauer, Molecular Dynamics with Chemical Accuracy—Alkane Adsorption in Acidic Zeolites, ACS Catal., 2023, 13, 2011–2024 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Powder X-Ray diffraction pattern and FE-SEM images. Experimental infrared spectrum of activated H-ZSM-5 in the presence of adsorbed ammonia. Additional tested arrangements for the monomer and the dimer adsorption. Additional discussion on the deconvolution of the infrared spectra. List of the computed theoretical infrared wavenumber and infrared absorption intensities. See DOI: https://doi.org/10.1039/d4cp03861d |
| This journal is © the Owner Societies 2025 |