
Dynamics of hydrogen shift reactions between peroxy radicals†
Imon
Mandal
 a,
Christopher David
Daub
b,
Rashid
Valiev
b,
Theo
Kurtén
*b and
R.
Benny Gerber
*ac
a,
Christopher David
Daub
b,
Rashid
Valiev
b,
Theo
Kurtén
*b and
R.
Benny Gerber
*ac
aThe Fritz Haber Center for Molecular Dynamics, Institute of Chemistry, The Hebrew University of Jerusalem, Jerusalem 91904, Israel. E-mail: robertbenny.gerber@mail.huji.ac.il
bDepartment of Chemistry, University of Helsinki, Helsinki 00014, Finland. E-mail: theo.kurten@helsinki.fi
cDepartment of Chemistry, University of California, Irvine, California 92697, USA
First published on 30th December 2024
Abstract
Peroxy radicals are key intermediates in many atmospheric processes. Reactions between such radicals are of particular interest as they can lead to accretion products capable of participating in new particle formation (NPF). These reactions proceed through a tetroxide intermediate, which then decomposes to a complex of two alkoxy radicals and O2, with spin conservation dictating that the complex must be formed in the triplet state. The alkoxy complex can follow different pathways e.g. hydrogen(H)-shift reactions, dissociation reactions etc., but the details of the full processes are not yet fully understood. This paper establishes the microscopic mechanisms of the H-shift and other associated pathways in the context of a self-reaction between methoxy radicals, with focus on the roles of the singlet and triplet states involved. Dynamics in time is explored by two methods: the multireference XMS-CASPT2 and very recently developed mixed reference spin–flip TDDFT (MRSF-TDDFT). The metadynamics method is used to compute energetics. The XMS-CASPT2 and the MRSF-TDDFT dynamics simulations yield similar results. This would be very encouraging for future simulations for large radicals, since MRSF-TDDFT simulations enjoy the advantages of linear response theory. Our calculations demonstrate that the reaction between methoxy radicals, though initiated on the triplet state, leads to products predominantly on the singlet surface, following efficient intersystem crossing (ISC). The computed branching ratio between H-shift and dissociation channels agrees well with experiment.
Introduction
The most abundant atmospheric oxidant biradical, molecular oxygen, oxidises organic molecules primarily by creating radical intermediates. Oxidation is first initiated by some reactive oxidants (e.g. OH radical, O3etc.) and then oxygen addition occurs. Organic molecule oxidation is essential to metabolism, food spoilage,1 production of electricity and transportation fuel, and for the formation and degradation of atmospheric pollutants.2 One of the crucial and abundant classes of intermediates during these processes are the organic peroxy radicals (RO2), which form when a carbon-centered radical (R) combines with molecular oxygen. Their reactions have been studied across a broad range of chemical fields, including atmospheric chemistry,3–6 combustion,7 and polymer chemistry.8 Due to their relative stability, RO2 radicals can accumulate at high concentrations in the atmosphere and react via various competing degradation and aggregation pathways which play an essential role in the formation of tropospheric secondary organic aerosols (SOA).9 These tropospheric aerosols and other fine particulate matter pose severe respiratory and cardiovascular health risks in polluted areas as they are the main drivers of air pollution related mortality.10 Despite the health and climate impact of secondary organic aerosol (SOA), the gas-phase processes involved in the formation of SOA remain incompletely understood.In the atmosphere, peroxy radicals can undergo both uni- and bimolecular reactions, with unimolecular H-shift channels of complex RO2 gaining recent attention as possible routes to very highly oxidized, low-volatility products participating in the formation of aerosol particles.11 Nevertheless, bimolecular reactions are the main fate of most RO2 in most atmospheric conditions.3 In polluted conditions, the dominant bimolecular reactant is nitrogen monoxide (NO), while in cleaner conditions, reactions with hydroperoxy radicals (HO2), OH radicals and other peroxy radicals can play a role. Peroxy radical self-and cross-reactions (RO2 + R′O2), while relatively minor overall sinks for RO2, are intriguing especially for aerosol chemistry, as they are one of the very few gas-phase processes potentially leading to accretion products– compounds with more carbon atoms than in the original precursor molecules (e.g. hydrocarbons).
Despite numerous experimental and computational studies, many details about RO2 + R′O2 reactions are still poorly understood. There exists a general consensus that the reaction proceeds through a singlet tetroxide (RO4R′) intermediate,12–18 which has also been observed experimentally in a IR matrix study.19 Computational studies by us and others suggest12,13,16,17 that this tetroxide inevitably decomposes into a complex of two alkoxy radicals and molecular oxygen. For the formation of the tetroxide, or the overall reaction from RO2 + R′O2 to (RO⋯ R′O) + O2, to be thermodynamically possible, the O2 must be formed in its triplet ground state.12 Spin conservation thus requires that the two doublet alkoxy radicals must also be coupled as a triplet. Modelling of the tetroxide decomposition reaction is extremely challenging due to the presence of four unpaired electrons coupled as an overall open-shell singlet.16 An accurate description of open-shell singlet states requires the use of expensive and nontrivial multireference methods.20 As the O2 becomes irrelevant for subsequent reactions after dissociating from the (RO⋯R′O) + O2 complex, most of our work on RO2 + R′O2 reaction channels has focused on the remaining 3(RO⋯R′O) complex.13,21 We note, however, that recent experiments suggest that for some systems, RO⋯O2 reactions may also be important.22
For simple peroxy radicals, the 3(RO⋯R′O) complex has three reaction channels available. First, the alkoxy radicals may simply dissociate, leading to the ‘radical’ or ‘dissociation’ channel generating RO + R′O radicals. Second, if at least one of the two alkoxy radicals is primary or secondary, a H-shift may happen, leading to the ‘molecular’ channel (yielding RC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and R′OH products). Third, an intersystem crossing (ISC) to a singlet state may occur, allowing subsequent recombination to a peroxide (ROOR′) accretion product. For more complex RO2, unimolecular in-complex alkoxy radical reactions open up even more pathways, including recombination to ether or ester type accretion products.23 From an aerosol formation perspective, the most interesting RO2 + R′O2 products are those containing more than about 15 carbon atoms (as well as multiple oxygen-containing functional groups), as this has been shown to be the threshold for efficient participation in new-particle formation.24 However, due to the computational expense of modelling such large systems, much of the modelling work by us and others has focused on smaller model systems, especially the simplest possible case of CH3O2 + CH3O2. For this system, experiments3,25,26 indicate a room-temperature branching ratio of 63–65% for the molecular channel CH3OH + HCHO, and 35–37% for the radical channel CH3O + CH3O, with the CH3OOCH3 peroxide dimer possibly present in trace amounts,25,27 but never confirmed in further studies.3 However, a dimer yield of 10 ± 5% has recently been reported for the self-reaction of ethyl peroxy radicals (CH3CH2OO).15
O and R′OH products). Third, an intersystem crossing (ISC) to a singlet state may occur, allowing subsequent recombination to a peroxide (ROOR′) accretion product. For more complex RO2, unimolecular in-complex alkoxy radical reactions open up even more pathways, including recombination to ether or ester type accretion products.23 From an aerosol formation perspective, the most interesting RO2 + R′O2 products are those containing more than about 15 carbon atoms (as well as multiple oxygen-containing functional groups), as this has been shown to be the threshold for efficient participation in new-particle formation.24 However, due to the computational expense of modelling such large systems, much of the modelling work by us and others has focused on smaller model systems, especially the simplest possible case of CH3O2 + CH3O2. For this system, experiments3,25,26 indicate a room-temperature branching ratio of 63–65% for the molecular channel CH3OH + HCHO, and 35–37% for the radical channel CH3O + CH3O, with the CH3OOCH3 peroxide dimer possibly present in trace amounts,25,27 but never confirmed in further studies.3 However, a dimer yield of 10 ± 5% has recently been reported for the self-reaction of ethyl peroxy radicals (CH3CH2OO).15
On the computational side, the trajectory model by Franzon predicted,28 based on the ωB97xD/jul-cc-pVTZ binding energy, a rate coefficient of 7.56 × 1010 s−1 for the dissociation of 3(CH3O⋯CH3O) into CH3O + CH3O at 298 K. Hasan et al. also reported21 rate coefficients for the dissociation and the H-shift on the triplet surface, with a similar quantum chemical method, as 6.21 × 1013 s−1 and 5.42 × 108 s−1, respectively, when using elementary transition state theory for the rate calculation (test calculations using variational transition state theory but a lower-level quantum chemical method suggest that variational effects likely lower the rate by at most a factor of 3, while tunneling effects are unlikely to be important due to the relatively low barrier). However, the former number is likely unreliable due to the failure of the detailed balance approximation for this system, as discussed by Franzon.28 Regardless of which theory-derived dissociation rate coefficient we pick, these numbers are in seeming contradiction with the experiments, as they suggest that the relative branching ratios of the radical and molecular channels should be at least 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, rather than the experimentally observed ∼35:65. Furthermore, the ISC rate coefficients computed for the 3(CH3O⋯CH3O) system by Valiev et al.13 from the ground triplet state (T1) to the first four excited singlet states (s1–S4) are in the range of 108–1013 s−1, while internal conversion (IC) rate coefficients from the excited singlet states to s1 are 1012–1014 s−1, suggesting that ISC to the singlet surface should out-compete all triplet-surface reactions by orders of magnitude or occur at least at similar rates. If recombination to the peroxide dimer were the sole fate of the 1(CH3O⋯CH3O) system after the ISC, one dominant product should thus actually be CH3OOCH3 – which has not been experimentally reported at all. Overall, this suggests that a more detailed investigation of the dynamics of the (CH3O⋯CH3O) system on both the triplet and singlet surfaces is required to understand the observed product branching ratios, as well as the seeming discrepancy between computational and experimental results.
:1, rather than the experimentally observed ∼35:65. Furthermore, the ISC rate coefficients computed for the 3(CH3O⋯CH3O) system by Valiev et al.13 from the ground triplet state (T1) to the first four excited singlet states (s1–S4) are in the range of 108–1013 s−1, while internal conversion (IC) rate coefficients from the excited singlet states to s1 are 1012–1014 s−1, suggesting that ISC to the singlet surface should out-compete all triplet-surface reactions by orders of magnitude or occur at least at similar rates. If recombination to the peroxide dimer were the sole fate of the 1(CH3O⋯CH3O) system after the ISC, one dominant product should thus actually be CH3OOCH3 – which has not been experimentally reported at all. Overall, this suggests that a more detailed investigation of the dynamics of the (CH3O⋯CH3O) system on both the triplet and singlet surfaces is required to understand the observed product branching ratios, as well as the seeming discrepancy between computational and experimental results.
Until now, computational studies13,21 have investigated the energetics of the possible reaction channels, calculating quantities such as activation energies (barrier heights) based on energy differences between various minima and transition states (saddle points) and rates associated with these pathways. However, to the best of our knowledge, the dynamics of the (CH3O⋯CH3O) intermediates formed in methyl peroxy radical self-reactions have not been directly explored previously. The interaction and involvement of two CH3O radicals in an open-shell system necessitates the use of computationally expensive multi-reference methods, making the simulation of long trajectories in sufficient number to achieve statistical significance prohibitively time-consuming (this becomes even more daunting for larger peroxy systems).
In this study, we use three different methods to investigate the fate of the weakly bound (CH3O⋯CH3O) complexes by simulating molecular dynamics (MD) trajectories on triplet and singlet surfaces. First, we show that multi-reference trajectories using extended multi-state complete active space perturbation theory at the second order (XMS-CASPT2) in the triplet state do not lead to H-shifts. However, similar trajectories initiated in the singlet state corroborate the branching ratio obtained in experiments.3,25 We also employ the MRSF-TDDFT method, which can generate multi-configurational singlet and triplet states in a single calculation. This methodology does not require any prior knowledge of the reaction as it does not require the definition of a fixed active space as in the above-mentioned multireference methods. While the focus of the present paper is on methoxy radical self-reactions, we note that this methodology would be even more beneficial for large polyatomic radicals. Then, to sample the complete free energy surface, metadynamics simulations have been performed, using multi-reference CASSCF to calculate energies and gradients on the singlet surface, and single-reference DFT on the triplet surface.
The outline of the paper is as follows: in the Computational methods section, we discuss in detail and separately the three methods that we employed for MD simulations. The Results and discussion section details the similarities and differences between the different simulations. Finally, in the Conclusions section, we compare our findings resulting from the different methodologies, and present some new insights into the dynamics of the peroxy radical reactions.
Computational methods
XMS-CASPT2 simulations
The adiabatic ab initio molecular dynamic simulations were performed using Newton-X29 and BAGEL software.30 The energy gradients in both triplet and singlet electronic states were calculated using XMS-CASPT231 and the tzvpp basis set in BAGEL. Then these were used in the integration of Newton's equations using the Newton-X software. We considered 6 electrons in 4 molecular orbitals (MOs) for both the electronic states. The selected 4 MOs are lone-pairs, mainly located only on the oxygen atoms with the main contribution from 2p-AO (atomic orbitals) of O atoms. Their energies are almost quasi-degenerate. The active MOs are shown in Fig. S1 (ESI†). The initial geometries for the dynamical simulations were obtained based on the optimized ground triplet electronic state at the XMS-CASPT2(6e,4o) level. Then, starting geometries were obtained from this using the Wigner distribution (WD) at 100 K based on the Hessian calculation of harmonic oscillators, whereas the initial velocities were generated using the Maxwell–Boltzmann distribution (MBD) at 300 K. We used a lower temperature than 300 K for the geometry because the vibrational amplitudes in the WD sampling are much larger than in the MBD sampling, likely due to errors in treating low-amplitude motions as harmonic oscillators.29 The initial velocities correspond to initial kinetic energies of 7–9 kcal mol−1, which roughly agrees with the average kinetic energy of this system at 300 K.10 trajectories were run for 200 fs using a 0.5 fs timestep for integration at constant energy (NVE ensemble) on the triplet surface, but no reactions were observed. Therefore, we performed additional simulations on the singlet surface. Based on the very rapid spin–flip rates computed in our previous study, we used the same geometry (the optimized triplet geometry) as the starting point,13 and generated 100 initial geometries and velocities using the same method we used for the triplet state. Finally, trajectories were simulated from each of the 100 different initial configurations with constant energy (NVE ensemble) using 0.5 fs timestep for integration up to a maximum of 400 fs. However, the active space became unstable after the H-shift occurred, leading to an early termination of these MD simulations in many cases.
MRSF-TDDFT calculations
One of the most difficult challenges in electronic structure theory is to develop a cost-effective method that has a balanced description of dynamical and non-dynamical (static) correlation effects. While single-reference density functional theory (DFT) has been highly successful and widely used, it struggles with open-shell cases. Multi-reference theories (e.g. CASSCF) on the other hand, can accurately account for non-dynamical correlation, and are thus essential for electronically degenerate situations commonly arising in open-shell species. However, the absence of dynamical correlations in these theories requires supplementary calculations, e.g. perturbation methods (such as the above mentioned XMS-CASPT2, MR-MP232etc.) or configuration interaction methods, making these impractical for large systems or time-consuming tasks.In general, single-reference time-dependent density functional theory (TDDFT) with linear response is the most practical and popular methodology for studying excited states. Some of its limitations, including the poor description of multi-reference electronic states, can be addressed by the spin–flip (SF)-TDDFT.33 SF-TDDFT uses an open-shell high-spin triplet reference (Ms = +1,|αα〉) as reference to generate the singlet states and (Ms = 0) triplet states as one of its response states as well as various configurations including important doubly excited ones, but suffers spin contamination due to missing configurations. Introduction of a second reference (Ms = −1,|ββ〉) provides an alternative way of expanding the response space by generating a new mixed reference state that has an equi-ensemble density of the Ms = +1 and Ms = −1 components of a triplet state:34
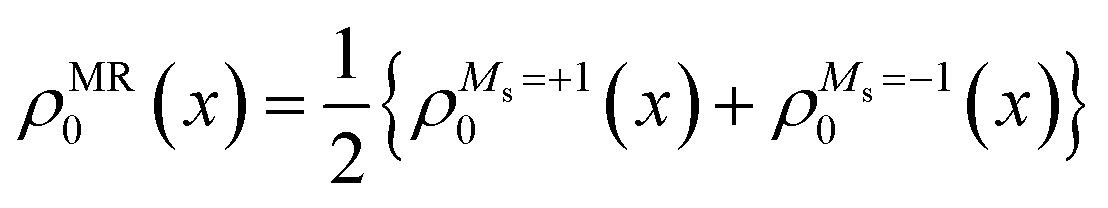 | (1) |
The two references are transformed to a single hypothetical reference by the two spinor-like open-shell orbitals whose one-particle reduced density matrix (RDM) satisfies the idempotent condition. Finally, application of linear response on the mixed hypothetical reference yields a mixed reference spin–flip (MRSF)-TDDFT.35,36 MRSF-TDDFT provides a method of generating the multi-configurational singlet and triplet states attempting to balance the dynamical and non-dynamical electron correlations simultaneously. This multistate method (i.e., yielding results for several states in one computation) does not require an active-space selection, and thus can be used without prior knowledge of the reaction. Moreover, MRSF-TDDFT not only mitigates the problem of spin contamination in SF-TDDFT but also simplifies the identification of the spin states, as here the singlet and triplet response states are generated by different response equations. This method naturally combines the advantage of multi-reference features (strong correlation) without complex and expensive multi-reference orbital optimization while retaining the practicality of linear response theory. Semi-emperical multirefernce methods are also available with lower computational cost but they are less trusted due to their emperical nature of the potential.37,38
The ab initio molecular dynamic simulations using MRSF-TDDFT on the ground triplet state and the first singlet state were performed in GAMESS(US)39,40 using BHHLYP/6-31G(d) functional/basis set. MRSF-TDDFT adopts a collinear (one-component) formalism, therefore the configurations obtained by different SF transitions couple only via the exact exchange. Thus, we use the BHHLYP functional which has 50% exact exchange. The simulation integration timestep was always 0.5 fs initiated with different velocities from MBD with initial kinetic energy 7–9 kcal mol−1 changing the BATHT parameter. In these constant energy simulations, to avoid complete dissociation of the (CH3O⋯CH3O) complex, we applied harmonic restraining potentials (force constant 100 kcal mol−1 Å−2) with a reference distance between carbon atoms in the two methyl fragments of 0.25 nm. Since this density based method with insufficient correlation description does not produce sufficiently attractive potential between the dissociated fragments, to obtain the reacting conformations for hydrogen shift reactions the restraining potential is necessary. In contrast, for XMS-CASPT2 trajectories incorporating better dynamical correlations from wave function based first-principles method, the potential energy surfaces are described more accurately, avoiding dissociation in all instances. These 10 (5 each), maximum 2 ps long constant energy simulations were started from the weakly bound (CH3O⋯CH3O) geometries optimized in singlet and triplet states. Another 5 simulations were initiated from an initial geometry of (CH3O⋯CH3O) where the distance between the oxygen atoms was 2.35 Å. In total, 30 simulations were performed on the singlet and triplet surfaces, 15 simulations on each surface. The limited sampling of the initial geometries and small number of trajectories are sufficient to demonstrate comparison of the potentials with XMS-CASPT2 leading to products in singlet and triplet states. The 2 ps timescales of the simulations are also enough to capture all hydrogen shift events. After the H-shift process, SCF convergence failure occurs in some trajectories, leading to an early termination of these MD simulations. The high spin triplet state with restricted open shell Kohn–Sham determinant was always taken as the reference state.
Metadynamics simulations
The presence of high barriers can pose a challenge for exploring the dynamics of chemical reactions using ab initio methods. To supplement the constant energy trajectories, we have used well-tempered metadynamics.41–43 This method entails the modification of the interatomic potential energy U0(r) by the addition of a bias potential V(s,t) dependent on one or more collective variables s(r) = (s1,s2,…sn). The bias potential gradually introduces repulsive Gaussians along the collective variable(s) coordinates as a function of time. The form of the bias potential is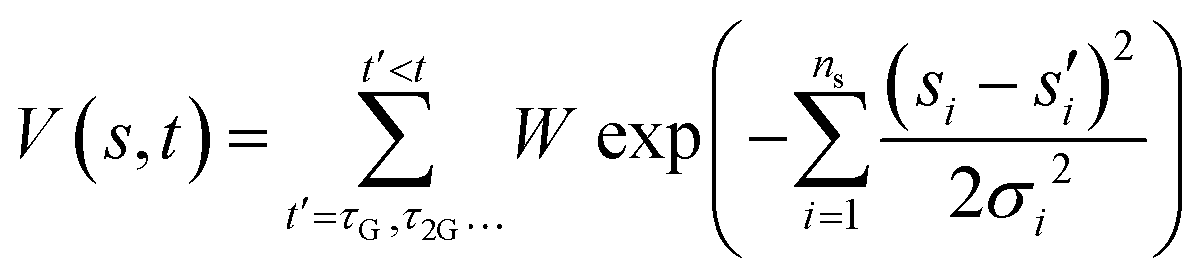 | (2) |
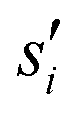 are the current and previous values of si when Gaussians were deposited, σi is the width of the Gaussians in each si, and W is the height of the Gaussians. In contrast to standard metadynamics, in well-tempered metadynamics W is history dependent as well,
are the current and previous values of si when Gaussians were deposited, σi is the width of the Gaussians in each si, and W is the height of the Gaussians. In contrast to standard metadynamics, in well-tempered metadynamics W is history dependent as well,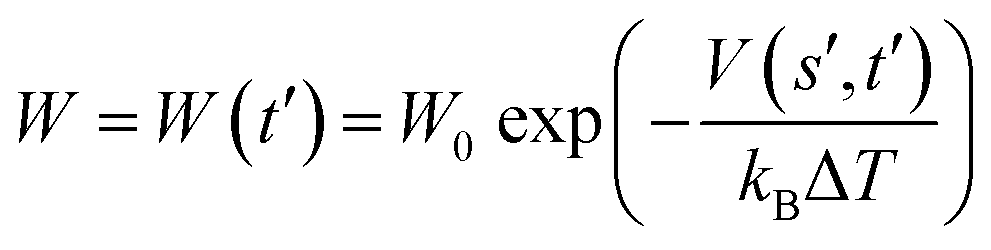 | (3) |
The collective variables s = (s1,s2,…sn) chosen must allow the simulation to be biased effectively in order to explore all of the relevant phase space.45 One limitation is that the variables must be continuous and differentiable. To distinguish bond-making and -breaking events, collective variables of the form
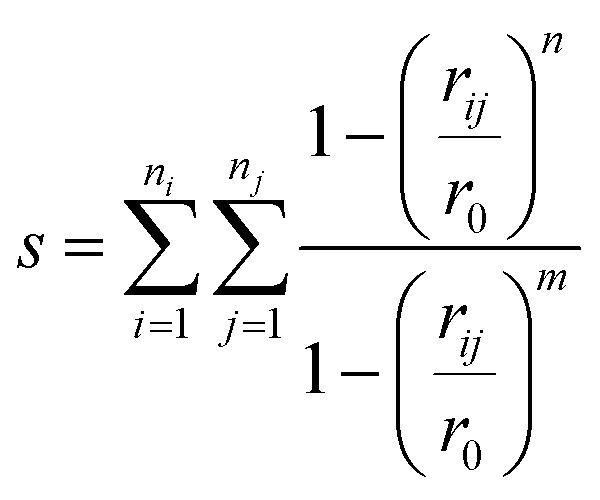 | (4) |
In this study, we use only two collective variables. The first, s1, determines if there is a bond between the two radical oxygen atoms in the two methoxy radicals to form a CH3OOCH3 peroxide dimer
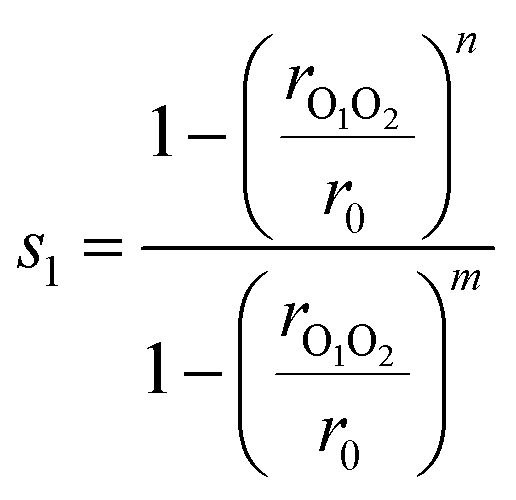 | (5) |
The second variable s2 determines if a H-shift has occurred to form a CH3OH + HCHO closed shell system
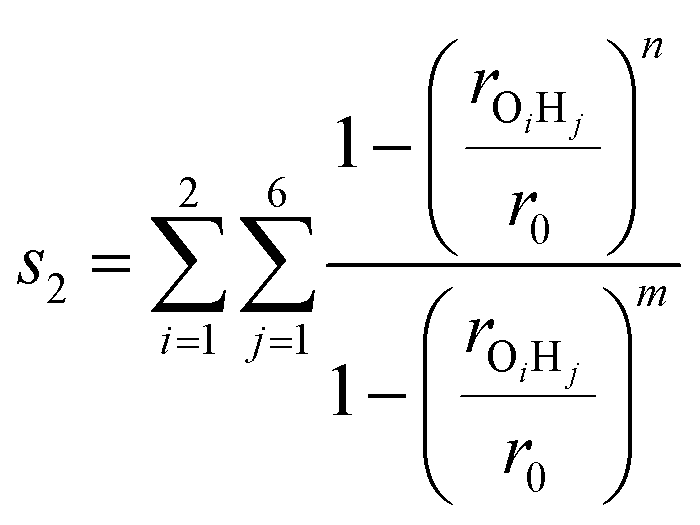 | (6) |
The sums include both of the oxygens, as well as all of the methyl hydrogens, in both of the initial methoxy radicals.
In Table 1 we list all of the parameters used in the metadynamics simulations. The results are described in the following subsections.
| s 1: n, m r0 (nm) | s 2: n, m r0 (nm) | τ G (fs) | W 0 (kcal mol−1) | γ | σ 1 | σ 2 | |
|---|---|---|---|---|---|---|---|
| Singlet | 6, 12, 0.25 | 8,16, 0.15 | 12 | 0.717 | 40 | 0.060 | 0.030 |
| Triplet | 8,16, 0.14 | 24 | 0.239 | 25 | 0.015 |
In this part of the work we used ORCA version 5.0.3 to compute the potential energies and the gradients.46 For propagating the trajectories using metadynamics, we used the ABIN code47 along with PLUMED version 2.7.2.48 The simulation timestep was always 0.48 fs = 20 a.u. and the system temperature T = 300 K.
Although ordinarily simulations of an isolated gas-phase system would suggest constant energy simulations,49 metadynamics requires the use of a thermostat to prevent the temperature from rising when the bias is applied. Here we used a Nosé–Hoover chain thermostat with 4 chains and a relaxation time of 0.25 ps for the singlet surface and 0.1 ps for the triplet surface. One final complication is that in a long simulation, the weakly bound (CH3O⋯CH3O) complex may dissociate, and thus any reactions would not be observed. To prevent this, we applied a harmonic restraining potential with a force constant of 239.0 kcal mol−1 nm−2 when 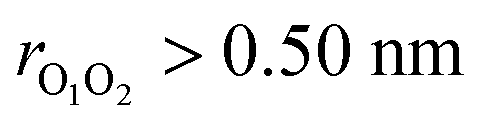 (triplet), and 478.0 kcal mol−1 nm−2 when
(triplet), and 478.0 kcal mol−1 nm−2 when 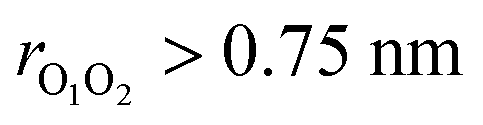 (singlet). We generated a single trajectory on both triplet and singlet surfaces. The simulation on the triplet surface started from the weakly bound (CH3O⋯CH3O) complex and the simulation on the singlet surface started from the dimethyl peroxide (CH3OOCH3) (the reason for this choice is provided in the Results and discussions section).
(singlet). We generated a single trajectory on both triplet and singlet surfaces. The simulation on the triplet surface started from the weakly bound (CH3O⋯CH3O) complex and the simulation on the singlet surface started from the dimethyl peroxide (CH3OOCH3) (the reason for this choice is provided in the Results and discussions section).
Results and discussions
XMS-CASPT2 trajectories show H-shift occurring on the singlet surface
The MD simulations on the triplet surface indicate that the system remain totally unreactive, as the distance between the two carbon atoms of the two methoxy fragments does not increase from the starting value. However, this lack of reaction might be due to the relatively high activation barrier of the H-shift, which was calculated with DFT methods to be 6.75 kcal mol−1 on the triplet state.21 Therefore, we simulated trajectories on the singlet surface. The MD trajectories on the singlet surface reveal that among 100 trajectories, 65 trajectories result in the H-shift reaction, 19 in the dissociation of two CH3O radicals, while 16 remain bound in the complex, but unreactive. The calculated branching ratio of H-shift to dissociation is thus 77% to 23%, which matches the experimental 65% and 35% branching ratio trend reasonably well.3,25 The average time of reaction for the H-shift is 133.56 ± 36.67 fs. The potential energy difference between 1(CH3O⋯CH3O) and 1(CH3OH⋯CH2O) is high (>50 kcal mol−1). Therefore, when the reaction takes place during a trajectory, the kinetic energy increases, and the potential energy drops by the same amount (Fig. S2, ESI†). The error in total energy is a few kcal mol−1 during this simulation, primarily due to the numerical error from the Velocity Verlet algorithm. During dissociation, this huge jump is not observed (Fig. S2, ESI†). However, no CH3OOCH3 peroxide dimer formation is observed, consistent with experiments. Fig. 1(a) shows the bond length between the oxygen atom (O) and the hydrogen atom (H) which shifts from one CH3O radical to the other during one representative trajectory. The continuous increase in the carbon–carbon (C–C) bond length during the dissociation process is shown in Fig. 1(b) for one representative simulation. Thus, multireference XMS-CASPT2 reveals that H-shift of (CH3O⋯CH3O) complexes occurs rapidly on the singlet surface after the (equally rapid) ISC process of 3(CH3O⋯CH3O).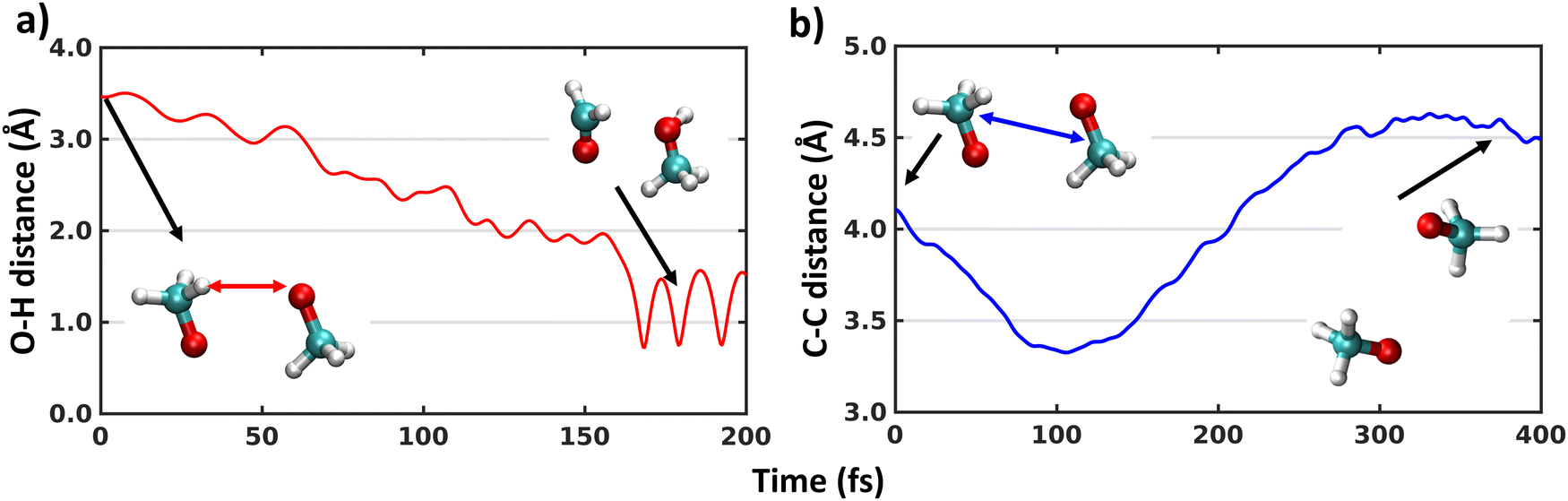 | ||
| Fig. 1 Time evaluation of O–H (a) and C–C (b) bond lengths along two representative trajectories where H-shift and dissociation occurs on the singlet surface. The starting and ending geometries of the (CH3O⋯CH3O) complexes and the colour coded bond lengths are shown in the inset. | ||
MRSF-TDDFT trajectories also reveal H-shift occurrence on the singlet surface
Similar to the above mentioned XMS-CASPT2 simulations, the MRSF-TDDFT trajectories on the triplet surface also remain totally unreactive. Thus, we performed singlet surface trajectories. Since our previous XMS-CASPT2 simulations on singlet surface require prior knowledge about the product to choose the active space, here we have used MRSF-TDDFT dynamics trajectory calculation, which does not require active space selection. Among 15 trajectories initiated from three different structures and five different velocities on singlet surface, all show a H-shift reaction. Due to the harmonic restraining potentials applied on the two carbons of the (CH3O⋯CH3O) complex preventing the fragments from separating too far apart from each other, there is a limitation on simulating the dissociation channel, and we can thus not compute branching ratios between the H-shift and dissociation as we did for the XMS-CASPT2 trajectories. Here, the H-shift takes place with a range of total trajectory time from 100 to 800 fs (except for one trajectory where transient CH3OOCH3 peroxide dimer formation takes place, as discussed in the next paragraph).In the (CH3O⋯CH3O) system, there are 6 possible O–H pairs which can show an intermolecular hydrogen shift between the radicals. Fig. 2 shows the O–H distance for the O–H pair where this H-shift occurs for two representative trajectories initiated from two different sets of structures. Fig. 2(a) depicts the bond lengths initiated from a structure where two O atoms are furthest away, and Fig. 2(b) starts from a structure where two O atoms are very close. The timescales of the H-shift, as indicated by the O–H bond reaching the average value ∼1 Å, do not depend on the starting structure. A concomitant drop in the potential energy and rise of the kinetic energy have been observed during the H-shift, similar to what was obtained with XMS-CASPT2 simulations (Fig. S2 and S3, ESI†). We also analysed O–O distances to investigate any indication of CH3OOCH3 peroxide dimer formation. In Fig. 2(a), transient CH3OOCH3 peroxide dimer formation is observed for a few fs (black circle), but the peroxide dissociates immediately, and the system then undergoes the H-shift after 1.4 ps. These changes are corroborated with the changes in the potential and kinetic energies in Fig. S3a and c (ESI†). Due to the very few degrees of freedom in the (CH3O⋯CH3O) system, the energy released in the peroxide formation is not properly distributed, and the system reverts back to dissociated state. Thus, for larger alkoxy radical systems, more peroxide dimer formation is expected, as also observed in experiments.15,22,23 No other trajectories indicate CH3OOCH3 peroxide dimer formation with MRSF-TDDFT simulations, consistent with experimental detection3,25 and the multi-reference calculations described in the previous section. Thus, MRSF-TDDFT trajectories also reveal that H-shift of (CH3O⋯CH3O) complexes occurs on the singlet surface after the ISC process from 3(CH3O⋯CH3O), consistent with our previous multi-reference XMS-CASPT2 simulations.
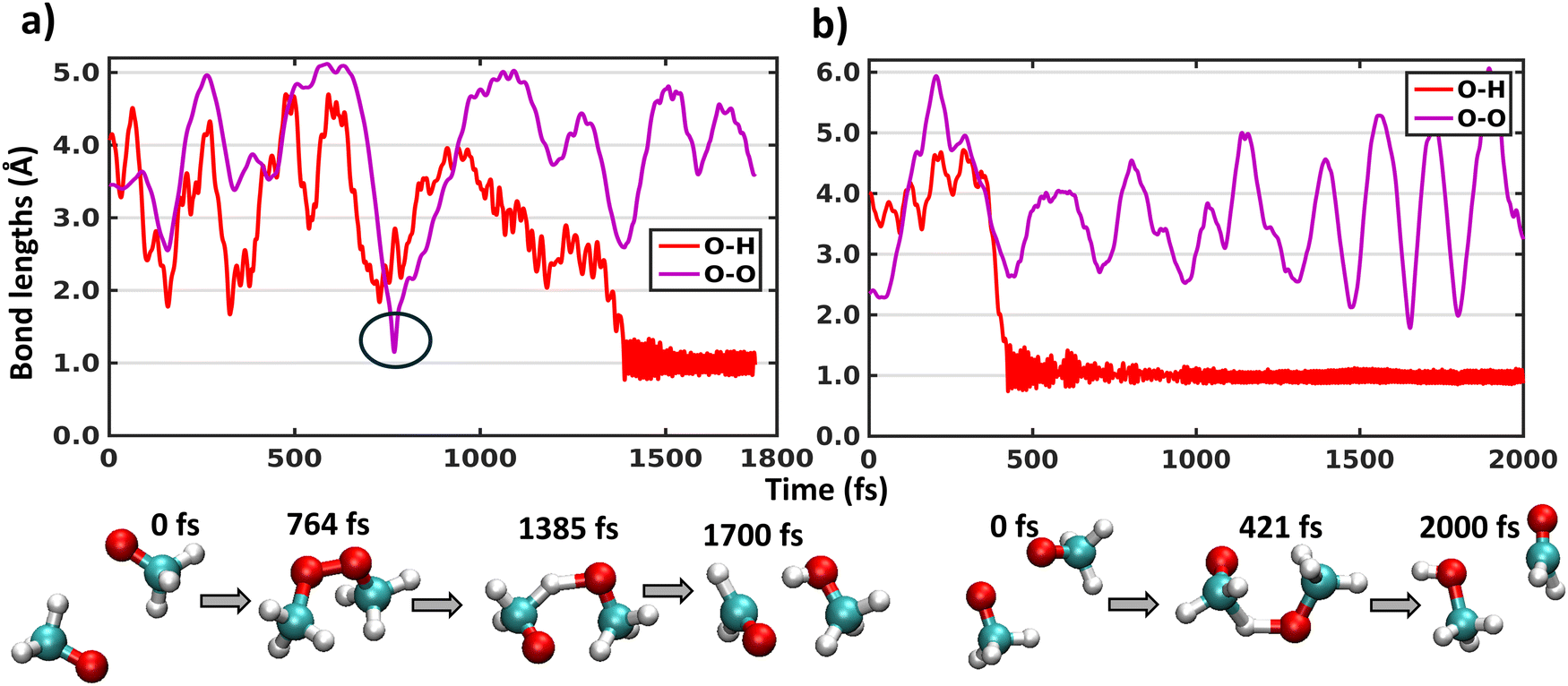 | ||
| Fig. 2 Time evaluation of O–H and O–O bond lengths along two representative trajectories initiated from two different starting geometries where H-shift occurs on the singlet surface. Snapshots from the trajectories with mentioned time are provided in the last row. | ||
Metadynamics on triplet and singlet surfaces
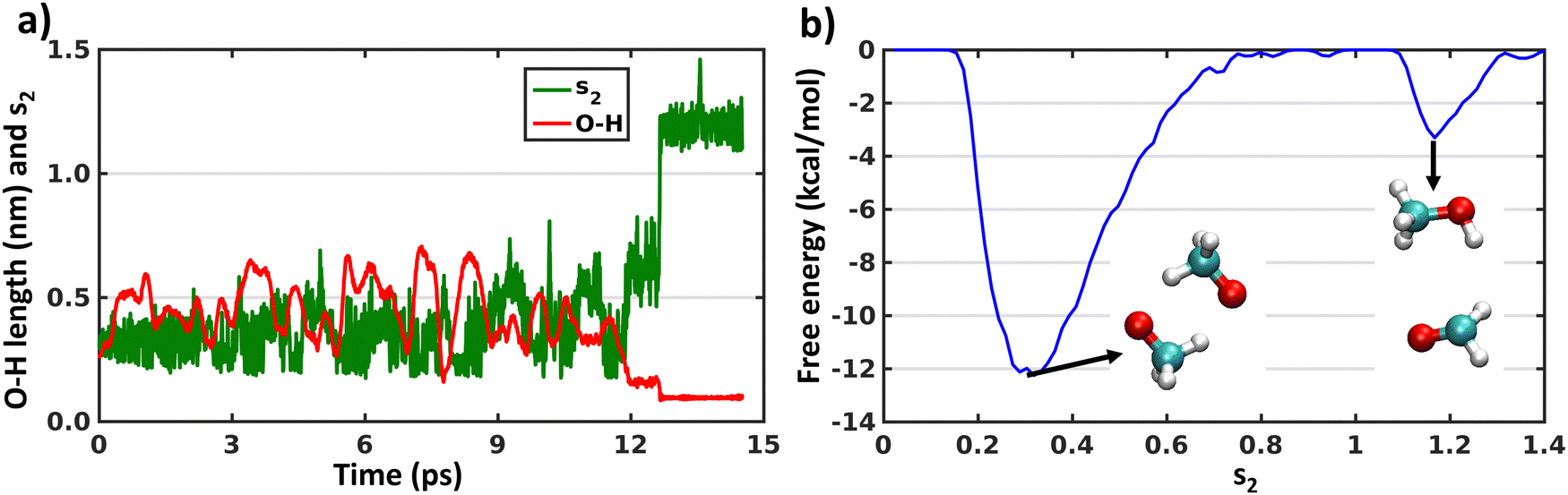 | ||
| Fig. 3 (a) Time evaluation of collective variable (s2) and O–H bond lengths (one among six where H-shift occurs from one CH3O radical to other) along the metadynamics trajectory on the triplet surface. (b) Free energy surface computed by metadynamics simulation on the triplet surface, biasing the s2 collective variable. The total length of the trajectory was 14 ps. The geometries of both minima of the (CH3O⋯CH3O) system are shown in the inset. | ||
Singlet surface
As the XMS-CASPT2 simulations did not reveal peroxy dimer formation (as expected after ISC process), next we perform metadynamics to explore the complete free energy surface on the singlet state. We have already discussed above in the Introduction that describing the reactions of a 1(CH3O⋯CH3O) system on the singlet surface, including formation of the peroxide bond and H-shift reactions, requires the use of multi-reference methods. Therefore, we have used CASSCF with an active space of 6 electrons in 6 orbitals and a cc-pVDZ basis set (Fig. S4, ESI†). A larger active space was chosen than the above XMS-CASPT2 calculations to account for more extensive electronic correlation within the active space with the lower level CASSCF method. The initial configuration was the CH3OOCH3 peroxide dimer, optimized at the same level of theory. This simulation will help us understand the fate of the CH3OOCH3 dimer, whether it remains stable or converts to other products. In Fig. 4 we show the evolution of the collective variables s1 and s2 defined above and the length of the O–H bond that forms during the simulation. It can be seen that the metadynamics bias readily leads the system to decompose into a complex of two methoxy radicals after a few ps. The peroxide reforms and decomposes a few times (s1 evaluation in Fig. 4(a)), before finally overcoming the barrier for the H-shift, indicated by the sudden increase of s2 to a value greater than 1 (Fig. 4(b)). After 40 ps, continued simulation does not lead to any further reactions, owing to the very deep energy minimum of the closed–shell CH3OH + HCHO system. The free energy surface obtained by the metadynamics simulation is shown in Fig. 4(c). It is known that, for CASSCF level of theory, use of an active space including only the electrons and orbitals of the oxygen atoms is sufficient to qualitatively describe the CH3OOCH3 system energetics.16,50 Our metadynamics simulation indicates that the free energy barrier for breaking the peroxide bond is approximately 25 kcal mol−1 (from CH3OOCH3 peroxide dimer minima to dissociated minima through the highest energy of −15 kcal mol−1). This calculated value is somewhat below the bond dissociation energy of ∼40 kcal mol−1 for CH3OOCH3, however some of this difference is due to an entropy boost associated with making two molecules from one.51 However, the energy barrier to form CH3OOCH3 from the dissociated form is 5 kcal mol−1. Fig. 4(c) also shows that the barrier for the hydrogen shift is around 10–15 kcal mol−1 (from any dissociated configuration of (CH3O⋯CH3O) with s1 < 0.6 to H-shifted product CH3OH + HCHO assuming the maximum energy of 0 kcal mol−1), which is mostly an overestimation of the reaction barriers due to the CASSCF level of theory or overestimation of dissociated CH3O radicals minima as discussed below. Overall, the main result of the metadynamics calculations is that on the singlet surface of (CH3O⋯CH3O) a minimum exists at the CH3OOCH3 peroxide dimer, but during the course of time the system moves to the more stable minimum of CH3OH + HCHO. Another minimum also appears with dissociated CH3O radicals on the free energy surface. However, this minimum is likely to be overestimated due to the over-sampling of configurations with dissociated radicals, combined with the insensitivity of s1 (eqn (5)) when the radicals are far apart. On the triplet state only metadynamics is able to overcome the energetic barrier to show the hydrogen shift reaction, corroborating the static DFT calculations.21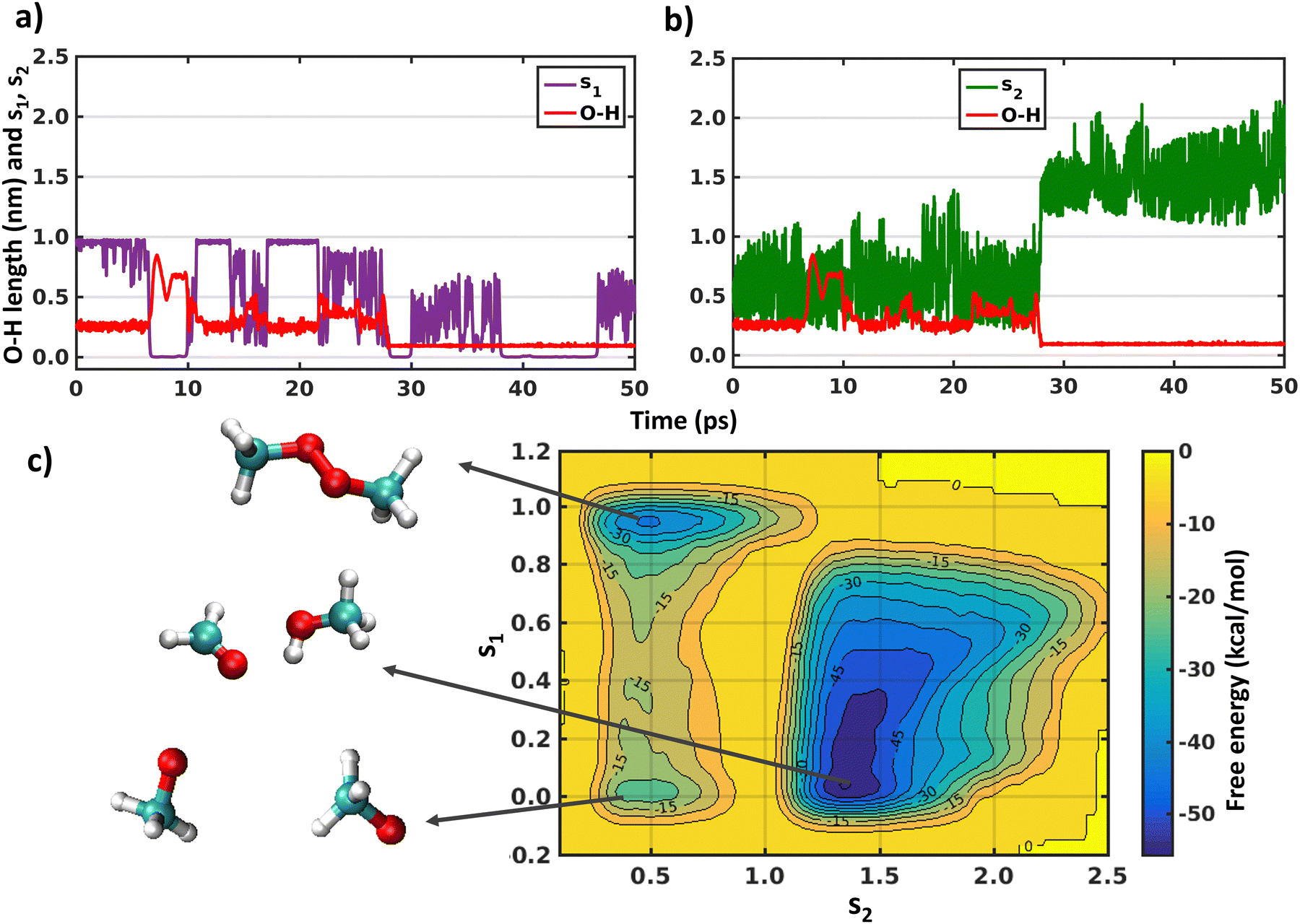 | ||
| Fig. 4 (a) and (b) Time evaluation of collective variables (s1 and s2) and O–H bond lengths (one among six where H-shift occurs from one CH3O radical to other) along the metadynamics trajectory on the singlet surface. (c) Free energy surface as a function of s1 and s2 over the course of metadynamics simulations on the singlet surface. The total length of the trajectory was 148 ps. All the geometries of all three minimas of the (CH3O⋯CH3O) complexes are shown in the inset. | ||
Conclusions
All three methods used in the dynamical simulations of the weakly bound (CH3O⋯CH3O) complexes reveal that H-shift occurs from one CH3O radical to the other radical on the singlet surface. Multi-reference XMS-CASPT2 constant energy trajectories indicate that the branching ratio of the H-shift (forming CH3OH + HCHO) to the dissociation to 2 CH3O radicals is close to the experimental value of 65:35.3,25 The mixed reference SF-TDDFT method, capable of generating multi-configurational singlet and triplet states in one computation, also reveals that the H-shift reaction takes place only on the singlet surface. The triplet surface remains unreactive in both these methods. None of these simulations show any stable CH3OOCH3 peroxide dimer formation on the singlet surface, which also corroborates with experimental results.3
The MRSF-TDDFT methodology, unlike the previously mentioned multi-reference methods, requires no prior knowledge of the reaction, since no selection of an active space is needed. Although this manuscript focuses on methoxy radical self-reactions, this approach would be more advantageous for studying large polyatomic radicals where few prior experimental or theoretical results may exist.
Metadynamics results explore the whole free energy surface of the (CH3O⋯CH3O) system, indicating that it includes not only H-shift and dissociation products but also the CH3OOCH3 peroxide dimer as stable minima. When the dynamical simulation is initiated from the CH3OOCH3 dimer, eventually it converts to the H-shifted product, and never reverts back, as this is by far the most stable minimum. On the triplet surface, only the metadynamics simulation was able to overcome the potential barrier to show the occurrence of the H-shift reaction. Thus, if the H-shift reaction has to occur on the triplet surface, it would be less feasible (or at least slower) than on the singlet surface. However, for larger peroxy systems, H-shifts from (RO⋯RO) (R = larger than CH3) complexes on the triplet surface could have much lower barriers, and thus be competitive. Moreover, formation of peroxy dimers (ROOR) is known to occur experimentally for large enough R. This may be due to interactions between other parts of the large R group,15,22,23 which could enhance the peroxide formation. In some important ways, therefore, the simple methoxy system is unfortunately not a representative model of the larger peroxy systems, and distribution of products between alcohol + aldehyde, peroxide dimer and dissociated radicals (as well as further reaction channels) is likely to be strongly system specific.
In summary, our study including dynamical simulations with different methods demonstrates two main conclusions, a chemical one and another computational one. Chemically, H-shift and other associated reactions from the weakly bound (CH3O⋯CH3O) complex takes place on the singlet surface, following very efficient ISC from the initial triplet. This removes the previously observed discrepancy between the rates calculated based on the triplet states for the H-shift and dissociation and the experimentally observed branching ratio. Computationally, the close agreement in the dynamical simulations between those performed with computationally expensive and time-consuming multi-reference XMS-CASPT2 and more cost-effective MRSF-TDDFT should prove very useful for future studies of reactions between larger peroxy radicals. This conclusion may have important implications also for reactions between atmospheric radicals of other types.
Author contributions
T. K. and R. B. G. conceptualised the idea. I. M., R. V. and C. D. D. performed the MRSF-TDDFT, XMS-CASPT2 dynamical and metadynamical simulations respectively and analysed the data. All authors wrote the manuscript.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. K., C. D. D. and R. V. are supported by the Jane and Aatos Erkko foundation. T. K. and R. V. are also supported by the Academy of Finland (especially the Centre of Excellence VILMA; grant number 346369). R. B. G. and I. M. like to thank the Israel Science Foundation, grant number 593/20 for financial support. Computing resources were supplied by Finland's Center for Scientific Computing (CSC).References
- M. Ahmed, J. Pickova, T. Ahmad, M. Liaquat, A. Farid and M. Jahangir, Oxidation of lipids in foods, Sarhad J. Agric., 2016, 32, 230–238 CrossRef.
- C. L. Heald and J. H. Kroll, The fuel of atmospheric chemistry: Toward a complete description of reactive organic carbon, Sci. Adv., 2020, 6, eaay8967 CrossRef CAS PubMed.
- J. J. Orlando and G. S. Tyndall, Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 2012, 41, 6294–6317 RSC.
- Y. Qian, T. K. Roy, A. W. Jasper, C. A. Sojdak, M. C. Kozlowski, S. J. Klippenstein and M. I. Lester, Isomer-resolved unimolecular dynamics of the hydroperoxyalkyl intermediate (˙QOOH) in cyclohexane oxidation, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2401148121 CrossRef CAS PubMed.
- T. S. Dibble and J. Chai, Advances in Atmospheric Chemistry, World Scientific, 2016, vol. 1, pp. 185–269 Search PubMed.
- L. Vereecken, P. T. M. Carlsson, A. Novelli, F. Bernard, S. S. Brown, C. Cho, J. N. Crowley, H. Fuchs, W. Mellouki, D. Reimer, J. Shenolikar, R. Tillmann, L. Zhou, A. Kiendler-Scharr and A. Wahner, Theoretical and experimental study of peroxy and alkoxy radicals in the NO3-initiated oxidation of isoprene, Phys. Chem. Chem. Phys., 2021, 23, 5496–5515 RSC.
- M. J. Goldman, W. H. Green and J. H. Kroll, Chemistry of Simple Organic Peroxy Radicals under Atmospheric through Combustion Conditions: Role of Temperature, Pressure, and NO Level, J. Phys. Chem. A, 2021, 125, 10303–10314 CrossRef CAS PubMed.
- L. M. Smith, H. M. Aitken and M. L. Coote, The Fate of the Peroxyl Radical in Autoxidation: How Does Polymer Degradation Really Occur?, Acc. Chem. Res., 2018, 51, 2006–2013 CrossRef CAS PubMed.
- J. H. Kroll and J. H. Seinfeld, Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 2008, 42, 3593–3624 CrossRef CAS.
- M. Shrivastava, C. D. Cappa, J. Fan, A. H. Goldstein, A. B. Guenther, J. L. Jimenez, C. Kuang, A. Laskin, S. T. Martin, N. L. Ng, T. Petaja, J. R. Pierce, P. J. Rasch, P. Roldin, J. H. Seinfeld, J. Shilling, J. N. Smith, J. A. Thornton, R. Volkamer, J. Wang, D. R. Worsnop, R. A. Zaveri, A. Zelenyuk and Q. Zhang, Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 2017, 55, 509–559 CrossRef.
- F. Bianchi, T. Kurtén, M. Riva, C. Mohr, M. P. Rissanen, P. Roldin, T. Berndt, J. D. Crounse, P. O. Wennberg, T. F. Mentel, J. Wildt, H. Junninen, T. Jokinen, M. Kulmala, D. R. Worsnop, J. A. Thornton, N. Donahue, H. G. Kjaergaard and M. Ehn, Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 2019, 119, 3472–3509 CrossRef CAS PubMed.
- R. Lee, G. Gryn'ova, K. U. Ingold and M. L. Coote, Why are sec-alkylperoxyl bimolecular self-reactions orders of magnitude faster than the analogous reactions of tert-alkylperoxyls? The unanticipated role of CH hydrogen bond donation, Phys. Chem. Chem. Phys., 2016, 18, 23673–23679 RSC.
- R. R. Valiev, G. Hasan, V. T. Salo, J. Kubecka and T. Kurten, Intersystem Crossings Drive Atmospheric Gas-Phase Dimer Formation, J. Phys. Chem. A, 2019, 123, 6596–6604 CrossRef CAS PubMed.
- L. Chen, Y. Huang, Y. Xue, Z. Jia and W. Wang, OH-initiated atmospheric degradation of hydroxyalkyl hydroperoxides: mechanism, kinetics, and structure–activity relationship, Atmos. Chem. Phys., 2022, 22, 3693–3711 CrossRef CAS.
- H. Yue, C. Zhang, X. Lin, Z. Wen, W. Zhang, S. Mostafa, P.-L. Luo, Z. Zhang, P. Hemberger, C. Fittschen and X. Tang, Dimeric Product of Peroxy Radical Self-Reaction Probed with VUV Photoionization Mass Spectrometry and Theoretical Calculations: The Case of C2H5OOC2H5, Int. J. Mol. Sci., 2023, 24, 3731 CrossRef CAS PubMed.
- V.-T. Salo, J. Chen, N. Runeberg, H. G. Kjaergaard and T. Kurtén, Multireference and Coupled-Cluster Study of Dimethyltetroxide (MeO4Me) Formation and Decomposition, J. Phys. Chem. A, 2024, 128, 1825–1836 CrossRef CAS PubMed.
- V.-T. Salo, R. Valiev, S. Lehtola and T. Kurtén, Gas-Phase Peroxyl Radical Recombination Reactions: A Computational Study of Formation and Decomposition of Tetroxides, J. Phys. Chem. A, 2022, 126, 4046–4056 CrossRef CAS PubMed.
- G. Ghigo, A. Maranzana and G. Tonachini, Combustion and atmospheric oxidation of hydrocarbons: Theoretical study of the methyl peroxyl self-reaction, J. Chem. Phys., 2003, 118, 10575–10583 CrossRef CAS.
- P. Ase, W. Bock and A. Snelson, Alkylperoxy and alkyl radicals. 1. Infrared spectra of CH3O2 and CH3O4CH3 and the ultraviolet photolysis of CH3O2 in argon + oxygen matrixes, J. Phys. Chem., 1986, 90, 2099–2109 CrossRef CAS.
- J. Olsen, D. L. Yeager and P. Jørgensen, Adv. Chem. Phys., 1983, 1–176 CAS.
- G. Hasan, V. T. Salo, R. R. Valiev, J. Kubecka and T. Kurtén, Comparing Reaction Routes for (RO⋯OR′) Intermediates Formed in Peroxy Radical Self- and Cross-Reactions, J. Phys. Chem. A, 2020, 124, 8305–8320 CrossRef CAS PubMed.
- S. E. Murphy, J. D. Crounse, K. H. Møller, S. P. Rezgui, N. J. Hafeman, J. Park, H. G. Kjaergaard, B. M. Stoltz and P. O. Wennberg, Accretion product formation in the self-reaction of ethene-derived hydroxy peroxy radicals, Environ. Sci.: Atmos., 2023, 3, 882–893 CAS.
- O. Peräkylä, T. Berndt, L. Franzon, G. Hasan, M. Meder, R. R. Valiev, C. D. Daub, J. G. Varelas, F. M. Geiger, R. J. Thomson, M. Rissanen, T. Kurtén and M. Ehn, Large Gas-Phase Source of Esters and Other Accretion Products in the Atmosphere, J. Am. Chem. Soc., 2023, 145, 7780–7790 CrossRef PubMed.
- L. Dada, D. Stolzenburg, M. Simon, L. Fischer, M. Heinritzi, M. Wang, M. Xiao, A. L. Vogel, L. Ahonen, A. Amorim, R. Baalbaki, A. Baccarini, U. Baltensperger, F. Bianchi, K. R. Daellenbach, J. DeVivo, A. Dias, J. Dommen, J. Duplissy, H. Finkenzeller, A. Hansel, X.-C. He, V. Hofbauer, C. R. Hoyle, J. Kangasluoma, C. Kim, A. Kürten, A. Kvashnin, R. Mauldin, V. Makhmutov, R. Marten, B. Mentler, W. Nie, T. Petäjä, L. L. J. Quéléver, H. Saathoff, C. Tauber, A. Tome, U. Molteni, R. Volkamer, R. Wagner, A. C. Wagner, D. Wimmer, P. M. Winkler, C. Yan, Q. Zha, M. Rissanen, H. Gordon, J. Curtius, D. R. Worsnop, K. Lehtipalo, N. M. Donahue, J. Kirkby, I. El Haddad and M. Kulmala, Role of sesquiterpenes in biogenic new particle formation, Sci. Adv., 2023, 9, eadi5297 CrossRef CAS PubMed.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi, J. Troe and I. Subcommittee, Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 2006, 6, 3625–4055 CrossRef CAS.
- C. H. Zhang, C. L. Li, W. J. Zhang, X. F. Tang, L. Pillier, C. Schoemaecker and C. Fittschen, Rate constant and branching ratio of the reaction of ethyl peroxy radicals with methyl peroxy radicals, Phys. Chem. Chem. Phys., 2023, 25, 17840–17849 RSC.
- H. Niki, P. Maker, C. Savage and L. Breitenbach, Fourier-transform infrared studies of the self-reaction of CH3O2 radicals, J. Phys. Chem., 1981, 85, 877–881 CrossRef CAS.
- L. Franzon, Simple Physical Model for the Estimation of Irreversible Dissociation Rates for Bimolecular Complexes, J. Phys. Chem. A, 2023, 127, 5956–5966 Search PubMed.
- M. Barbatti, M. Bondanza, R. Crespo-Otero, B. Demoulin, P. O. Dral, G. Granucci, F. Kossoski, H. Lischka, B. Mennucci, S. Mukherjee, M. Pederzoli, M. Persico, M. Pinheiro Jr, J. Pittner, F. Plasser, E. Sangiogo Gil and L. Stojanovic, Newton-X Platform: New Software Developments for Surface Hopping and Nuclear Ensembles, J. Chem. Theory Comput., 2022, 18, 6851–6865 CrossRef CAS PubMed.
- BAGEL, Brilliantly Advanced General Electronic-structure Library. https://www.nubakery.org under the GNU General Public License.
- T. Shiozaki, BAGEL: Brilliantly Advanced General Electronic-structure Library, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1331 Search PubMed.
- J. Kalinowski, M. Räsänen, P. Heinonen, I. Kilpeläinen and R. B. Gerber, Isomerization and Decomposition of a Criegee Intermediate in the Ozonolysis of Alkenes: Dynamics Using a Multireference Potential, Angew. Chem., Int. Ed., 2014, 53, 265–268 CrossRef CAS PubMed.
- Y. Shao, M. Head-Gordon and A. I. Krylov, The spin–flip approach within time-dependent density functional theory: Theory and applications to diradicals, J. Chem. Phys., 2003, 118, 4807–4818 CrossRef CAS.
- S. Lee, M. Filatov, S. Lee and C. H. Choi, Eliminating spin-contamination of spin-flip time dependent density functional theory within linear response formalism by the use of zeroth-order mixed-reference (MR) reduced density matrix, J. Chem. Phys., 2018, 149, 104101 CrossRef PubMed.
- Y. Horbatenko, S. Sadiq, S. Lee, M. Filatov and C. H. Choi, Mixed-Reference Spin-Flip Time-Dependent Density Functional Theory (MRSF-TDDFT) as a Simple yet Accurate Method for Diradicals and Diradicaloids, J. Chem. Theory Comput., 2021, 17, 848–859 CrossRef CAS PubMed.
- W. Park, K. Komarov, S. Lee and C. H. Choi, Mixed-Reference Spin-Flip Time-Dependent Density Functional Theory: Multireference Advantages with the Practicality of Linear Response Theory, J. Phys. Chem. Lett., 2023, 14, 8896–8908 CrossRef CAS PubMed.
- D. Shemesh and R. B. Gerber, Molecular Dynamics of Photoinduced Reactions of Acrylic Acid: Products, Mechanisms, and Comparison with Experiment, J. Phys. Chem. Lett., 2018, 9, 527–533 CrossRef CAS PubMed.
- D. Shemesh, S. L. Blair, S. A. Nizkorodov and R. B. Gerber, Photochemistry of aldehyde clusters: cross-molecular versus unimolecular reaction dynamics, Phys. Chem. Chem. Phys., 2014, 16, 23861–23868 RSC.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery Jr, General atomic and molecular electronic structure system, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- M. S. Gordon and M. W. Schmidt, in Theory and Applications of Computational Chemistry, ed. C. E. Dykstra, G. Frenking, K. S. Kim and G. E. Scuseria, Elsevier, Amsterdam, 2005, pp. 1167–1189 Search PubMed.
- A. Barducci, M. Bonomi and M. Parrinello, Metadynamics, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 826–843 CAS.
- A. Barducci, G. Bussi and M. Parrinello, Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method, Phys. Rev. Lett., 2008, 100, 020603 CrossRef PubMed.
- A. Laio and F. L. Gervasio, Metadynamics: a method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science, Rep. Prog. Phys., 2008, 71, 126601 CrossRef.
- P. Tiwary and M. Parrinello, A Time-Independent Free Energy Estimator for Metadynamics, J. Phys. Chem. B, 2015, 119, 736–742 CrossRef CAS PubMed.
- G. Fiorin, M. L. Klein and J. Hénin, Using collective variables to drive molecular dynamics simulations, Mol. Phys., 2013, 111, 3345–3362 CrossRef CAS.
- F. Neese, Software update: The ORCA program system—Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- D. Hollas, J. Suchan, M. Ončák and P. Slaviček, PHOTOX/ABIN: Pre-release of version 1.1, 2019, https://github.com/PHOTOX/ABIN Search PubMed.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, PLUMED 2: New feathers for an old bird, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
- R. Halonen, I. Neefjes and B. Reischl, Further cautionary tales on thermostatting in molecular dynamics: Energy equipartitioning and non-equilibrium processes in gas-phase simulations, J. Chem. Phys., 2023, 158, 194301 CrossRef CAS PubMed.
- R. R. Valiev, R. T. Nasibullin, S. Juttula and T. Kurten, Fast estimation of intersystem crossing rate constants of radical pairs, New J. Chem., 2024, 48, 18314–18319 Search PubMed.
- R. D. Bach and H. B. Schlegel, Bond Dissociation Energy of Peroxides Revisited, J. Phys. Chem. A, 2020, 124, 4742–4751 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03862b |
| This journal is © the Owner Societies 2025 |