
Valley splitting of monolayer Hf3C2O2 by the spin–orbit coupling effect: first principles calculations using the HSE06 methods†
Shiqian
Qiao‡
,
Yang
Zhang‡
,
Shasha
Li
 ,
Lujun
Wei
,
Hong
Wu
and
Feng
Li
*
,
Lujun
Wei
,
Hong
Wu
and
Feng
Li
*
School of Science & New Energy Technology Engineering Laboratory of Jiangsu Provence, Nanjing University of Posts and Telecommunications (NJUPT), Nanjing 210046, China. E-mail: lifeng@njupt.edu.cn
First published on 29th November 2024
Abstract
Electrons have not only charge and spin degrees of freedom, but also additional valley degrees of freedom. The search for valleytronic materials with large valley splits is important for the development of valleytronics. In this work, we applied first principles computations to calculate 1 L Hf3C2O2 at the level of HSE06. When the spin–orbit coupling (SOC) effect is considered, 1 L Hf3C2O2 is an indirect bandgap semiconductor with a bandgap of 0.952 eV. Meanwhile, valley splitting occurs between the conduction bands Γ and K, with a valley splitting value as high as 98.228 meV. Bader charge analysis was used to determine that Hf–O and Hf–C are ionic bonds. The computed elastic constants and phonon spectra proved that 1 L Hf3C2O2 is mechanically and dynamically stable. In addition, the Berry curvature of 1 L Hf3C2O2 is non-zero. In the work the effect of the electronic properties of 1 L Hf3C2O2 was also calculated with respect to the biaxial strain. The results show that the biaxial strain can well regulate the band gap and valley splitting. Finally, we calculated the effects of hole and electron doping on the band gap and valley splitting of 1 L Hf3C2O2. The results show that the band gap and valley splitting are linearly related to the doping concentration. Our study shows that 1 L Hf3C2O2 is a promising two-dimensional valleytronics material.
1. Introduction
The concept of energy valleys has already been mentioned in 3D system materials in 1977.1 An energy valley refers to the extreme point in an energy band, whether it is the valence band or the conduction band, the extreme point is the energy valley, and these positions are generally near the Fermi level. In k-spaces with special symmetric materials, there are multiple energy degenerate valleys. Under certain external conditions, the simplicity of these energy valleys will be broken, so that the electrons in the energy valleys show dynamic polarization effects. From a theoretical point of view, these energy valleys can be viewed as discrete degrees of freedom, which are used to label information. Similar to electron and spin degrees of freedom, there is potential for coding and information storage functions, providing a theoretical basis for the development of next-generation electronic devices.Since graphene has been successfully prepared experimentally, the study of 2D valleytronics has attracted a lot of attention.2 In addition to charge and spin, the electrons have a new degree of freedom called the valley degree of freedom. It is usually the robust to smooth deformation and scattering of long-wave phonons,3 which are important for practical applications. Therefore, the valley index is very popular as a new information carrier. In order to fully realize the valley index, the ultimate goal is to break the simplicity of the energy bands.4
In recent years, there have been many candidate materials for two-dimensional valleytronics. For example, germanene,5 transition metal disulfides,6 the 2D MA2Z4 family7–10 and 2D transition metal carbides and/or nitrides (MXenes).11,12 At present, the most popular and the largest number of 2D valleytronics materials are 2D transition metal sulfides, with a chemical formula of MX2 (M = Cr, Mo, W, etc., and X is the coordination element).2,12–14 Transition metal carbides and/or nitrides, as the largest known family of 2D materials, have received extensive attention due to their layered structures.
A new family of 2D materials (MXenes) consisting of transition metal carbides and/or nitrides are the most promising materials due to their superb properties.15 Ti3C2Tx was the first MXenes to be discovered and has been in the spotlight as a representative member of the MXenes family.12 MXenes are typically prepared by selectively etching out element A from the bulk phase MAX, where A is a group IIIA to VIA element. The general formula is Mn+1XnTx (M is a transition metal atom, X is a carbon/nitrogen atom, and Tx is a halogen element).16 Valleytronics in MXene materials have been reported in recent years. For example, Li and He et al. found that a 2D Janus Cr2COF (∼334 meV) can generate valley polarization.17 Lu and Liu et al. found that Y3N2O2 (∼21.3 meV) and La3N2O2 (∼100.4 meV) also exhibit valley polarization.18 Lan et al. found that W2NSCl (∼83 meV and ∼491 meV) has valley splitting.19 However, reports related to the valleytronics of MXene materials are still rare.
In this work, the HSE06 method is employed to calculate the first principles of 1 L Hf3C2O2. When SOC is considered, the band gap of 1 L Hf3C2O2 is 0.952 eV. At the same time, the valley splitting occurs between the conduction bands Γ and K, with a valley splitting value of 98.228 meV. In addition, the stability of 1 L Hf3C2O2 is calculated. The results show that 1 L Hf3C2O2 is mechanically and dynamically stable. In this work the Berry curvature of 1 L Hf3C2O2 was calculated and is found to be non-zero. The biaxial strain is an effective method to regulate the electronic properties. Therefore, the influence of the electronic properties of 1 L Hf3C2O2 on the biaxial strain is calculated. The results show that the biaxial strain can well regulate the band gap and valley splitting. Our study shows that 1 L Hf3C2O2 is a promising 2D valleytronics material.
2. Computational methods
The first-principles computations are used based on the density-functional theory (DFT)20 in the Vienna Ab initio Simulation Package (VASP).21,22 The projected affixed wave (PAW) method is used to describe the interaction between electrons and ions.23,24 The Perdew–Burke–Ernzerhof (PBE) functional is used to deal with exchange correlation potential in generalized gradient approximation (GGA).25 The GGA+U26 method is used to correct the strongly correlated d-orbital electrons, where the U value is calculated by the linear response method.27 In this paper, parameters U and J are set in an efficient manner. After calculation, the linear response of Hf3C2O2 is shown in Fig. 2(c), where Ueff = (U − J) = 5.295 eV. Meanwhile, the Heyd–Scuseria–Ernzerhof hybrid functional (HSE06) method is used to calculate the electronic structure.28 The cutoff energy is set to 500 eV. The convergence criterion for the force is 0.01 eV Å−1. The convergence standard of energy is 10−6 eV. A vacuum layer of 15 Å is provided in the z-direction. A 15 × 15 × 1 Monkhorst–Pack k-point grid was used to sample the 2D Brillouin zone, where Γ is located at the center of the Brillouin zone.29 The DFT-D3 method was used to correct the interlayer van der Waals forces.30 The phonon spectrum is calculated using the PHONOPY code,31 taking a grid of k points of 3 × 3 × 1 and using a supercell of 4 × 4 × 1. The SOC effect is also considered to calculate the electronic structure of the material.32 The PYPROCAR code was used to draw spin polarized energy bands.33 Finally, the Berry curvature of the whole 2D Brillouin zone is calculated using the Vaspberry code.3. Results and discussion
3.1. Structure and stability
The top and side views of the 1 L Hf3C2O2 crystal structure are shown in Fig. 1(a) and (c). The space group of 1 L Hf3C2O2 is P6/mmm (No. 191). It has a hexagonal honeycomb structure. After optimization, the lattice constant of 1 L Hf3C2O2 is a = b = 3.252 Å. The thickness of the 1 L Hf3C2O2 is 7.645 Å, the bond length of Hf and O atoms is 2.122 Å, the bond length of the uppermost Hf and C atoms is 2.340 Å, and the bond length of the middle layer Hf and C atoms is 2.365 Å. The spatial inversion symmetry of the 1 L Hf3C2O2 crystal structure is broken. The high symmetry points of the Brillouin zone are also shown in Fig. 1(b).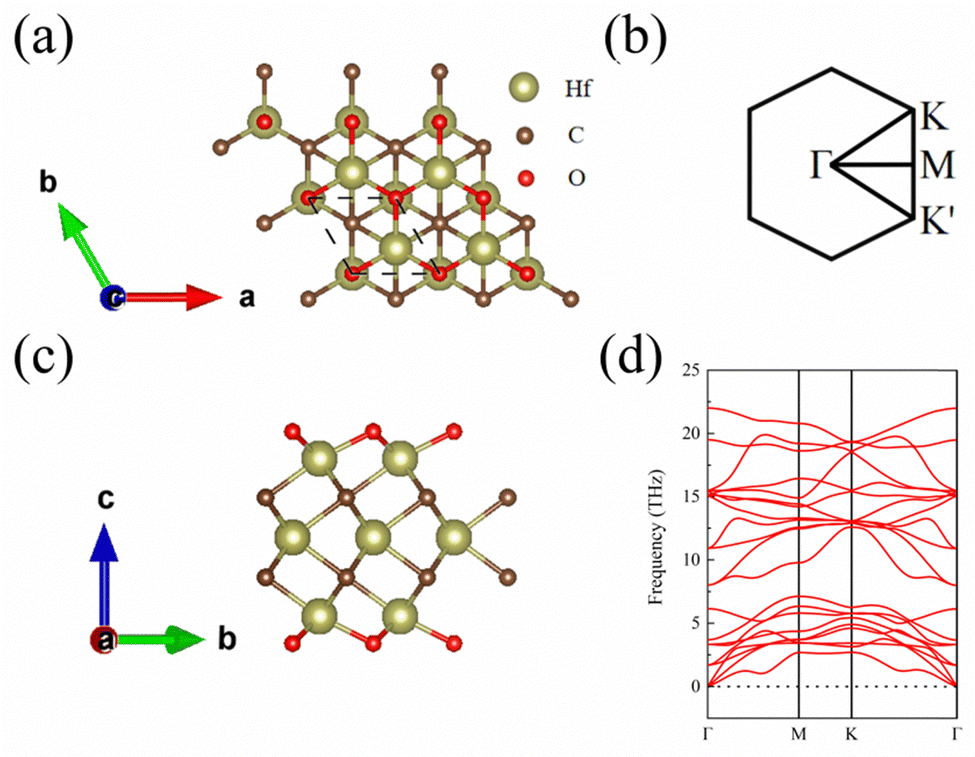 | ||
| Fig. 1 (a) Top view of the 1 L Hf3C2O2 crystal structure. (b) High symmetry points in the Brillouin zone of 1 L Hf3C2O2. (c) Side view of the 1 L Hf3C2O2 crystal structure. (d) Phonon spectrum of 1 L Hf3C2O2. | ||
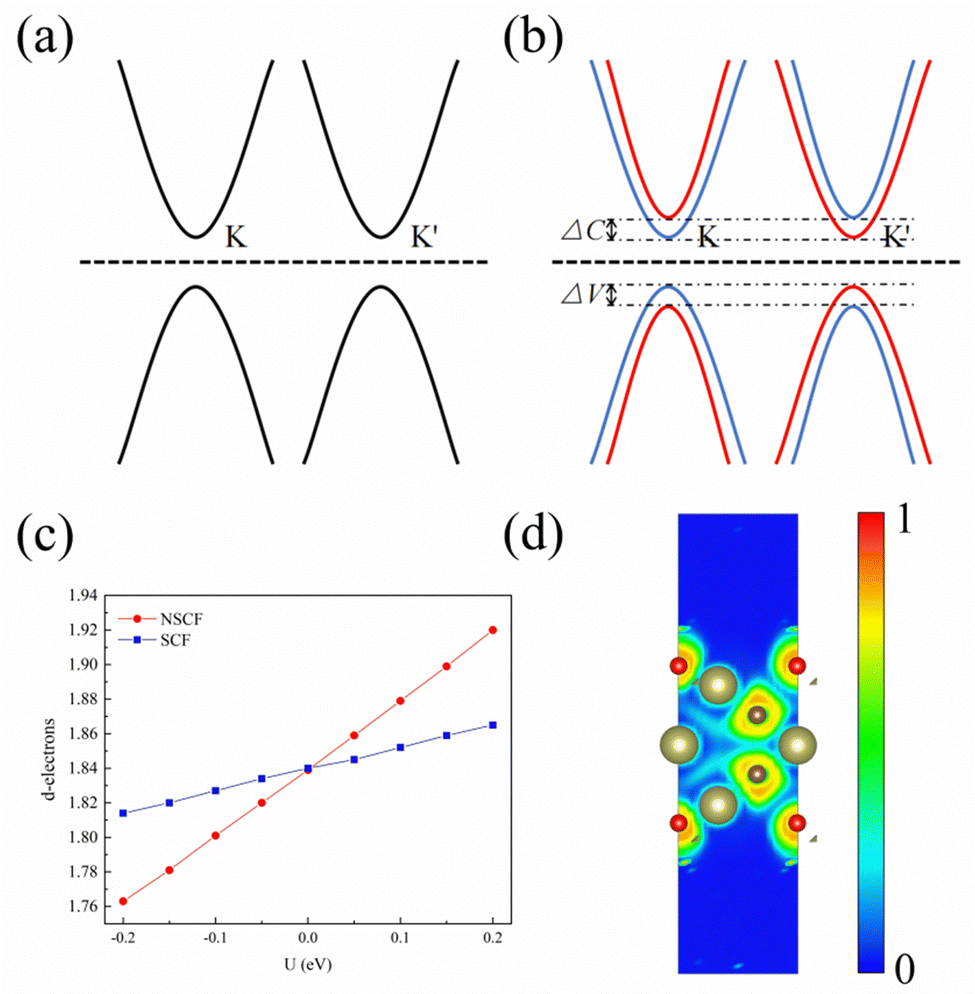 | ||
| Fig. 2 (a) Schematic diagram of energy bands when SOC is not considered. (b) Schematic of energy bands when SOC is considered. (c) Linear response diagram of 1 L Hf3C2O2, with the horizontal coordinates representing the applied effective Coulomb field and the exchange interaction term, and the vertical coordinates representing the number of d-orbital charge occupations of the plus U atom. (d) Plot of the electronic localization function of 1 L Hf3C2O2. | ||
Since the structural stability is important for the applications, in this work, to determine the stability of 1 L Hf3C2O2, we calculated the phonon spectrum and elastic constants. First, in this work the phonon spectrum of the whole Brillouin region along the high symmetric point Γ–M–K–Γ was obtained, as shown in Fig. 1(d). From the phonon spectrum, there is no imaginary frequency in the phonon spectrum of 1 L Hf3C2O2, which indicates that 1 L Hf3C2O2 is dynamically stable. Then, in this work the elastic constants of 1 L Hf3C2O2 are calculated, which are C11 = 331.31 N m−1, C12 = 95.17 N m−1, and C66 = 118.07 N m−1, satisfying the Born criterion:34,35
| C11 > 00, C11 > |C12| |
This indicates that 1 L Hf3C2O2 is mechanically stable. Based on the elastic constant, the Young's modulus and Poisson's ratio of Hf3C2O2 are also calculated. The Young's modulus characterizes the softness or hardness of a material;36 commonly graphene has a Young's modulus of 340 N m−1 and MoS2 has a Young's modulus of 129 N m−1.37 The Young's modulus of 1 L Hf3C2O2 was calculated to be 303.97 N m−1, which indicates that the softness and hardness of 1 L Hf3C2O2 is basically comparable to that of graphene and is tougher than that of MoS2. Poisson's ratio characterizes the ratio of the transverse dimensional change to the longitudinal dimensional change when the material is strained. Most 2D materials have Poisson's ratios within 0–0.5.38 The Poisson's ratio for 1 L Hf3C2O2 is 0.287, whereas it is 0.25 for MoS2 and 0.3 for CrSe2. The results indicate that 1 L Hf3C2O2 has a moderate mechanical response to external loads. Overall, 1 L Hf3C2O2 is mechanically and kinetically stable, which also suggests that 1 L Hf3C2O2 can be synthesized in the experiment.
3.2. Electronic properties and valley splitting
To study the bonding properties of 1 L Hf3C2O2, we used the Bader charge analysis method for charge transfer. The results show that there is a significant charge transfer in 1 L Hf3C2O2. For example, the Hf atom in the middle layer lost 2.04 electrons. The top Hf atom lost 1.52 electrons. The bottom Hf atom lost 1.52 electrons. The C atom in the upper layer gained 1.55 electrons. The lower C atom gained 1.55 electrons. The O atom in the upper layer gained 0.99 electrons. The lower O atom gained 0.99 electrons. In terms of the number of electrons gained and lost, the atoms in 1 L Hf3C2O2 have a large number of electrons gained and lost, and the gain and loss of electrons is more pronounced. This suggests that there is an ionic bond between the Hf atom and the C atom, as well as an ionic bond between the Hf atom and the O atom. In order to determine the bonding properties of 1 L Hf3C2O2, we used the electronic states for analysis, as shown in Fig. 2(d). The electrons are mainly distributed around O and C atoms, with some localized around Hf atoms, suggesting that the Hf–O and Hf–C bonds have ionic properties. This result is consistent with the Bader charge analysis.To study the electronic properties of 1 L Hf3C2O2, we calculated the energy band structure. Fig. 3(a) and (b) are schematic illustrations of valley splitting. Fig. 2(a) is a schematic diagram of the energy bands that are simple when SOC is not considered. When SOC is considered, a large change in the energy bands occurs and the simplicity is broken, producing valley splitting, as shown in Fig. 2(b). ΔC and ΔV denote the size of the conduction and valence band splits, respectively. Therefore, when SOC is not considered, 1 L Hf3C2O2 is an indirect bandgap semiconductor with a bandgap of 0.979 eV, as shown in Fig. 3(a). The top of the valence band is located at point Γ in the Brillouin zone, while the bottom of the conduction band is located at point M in the Brillouin zone. Since transition metals have very strong SOC, the energy band structure of 1 L Hf3C2O2 changes when SOC is considered, as shown in Fig. 3(b). 1 L Hf3C2O2 remains an indirect bandgap semiconductor. The positions of the valence band tops and conduction band bottoms are the same as when SOC is not considered. When SOC is considered, the biggest change is that the simplicity of the energy band is broken, resulting in valley splitting. The valley splitting value between Γ and K is 98.228 meV. It is higher than the valley splitting values of 50 meV for ZrNCl39 and 63 meV for CrSiGeN4,40 and 70 meV for TiBrI.41
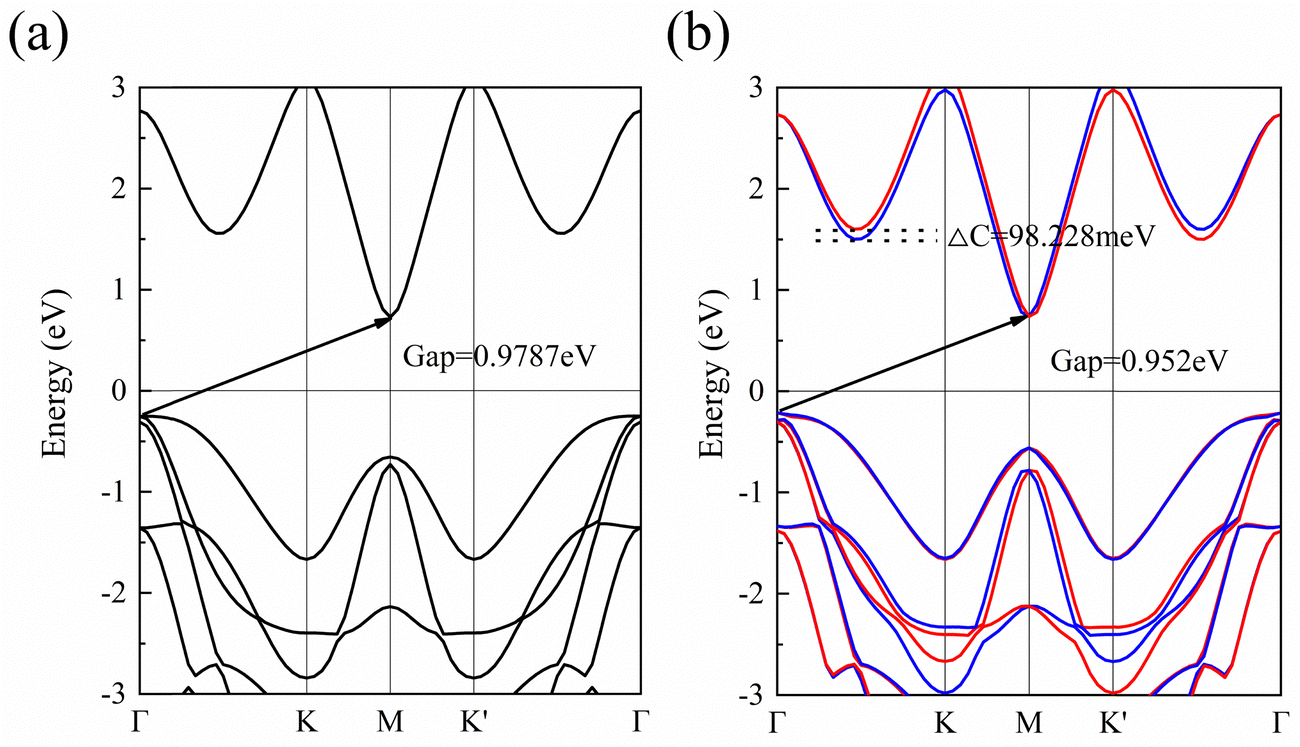 | ||
| Fig. 3 (a) Energy band diagram calculated by the method of HSE06 when SOC is not considered. (b) Energy band diagram calculated by the method of HSE06 when the SOC is considered, where red represents spin up and blue represents spin down. | ||
In this work, we also used the PBE and GGA+U methods for computing the energy bands of 1 L Hf3C2O2, as shown in Fig. 4. Fig. 4(a) and (b) show the energy bands calculated by the PBE method. When SOC is not considered, 1 L Hf3C2O2 is also an indirect bandgap semiconductor with a bandgap of 0.428 eV. The valence band top is similarly located at the Γ point in the Brillouin zone, while the conduction band bottom is also located at the M point in the Brillouin zone.
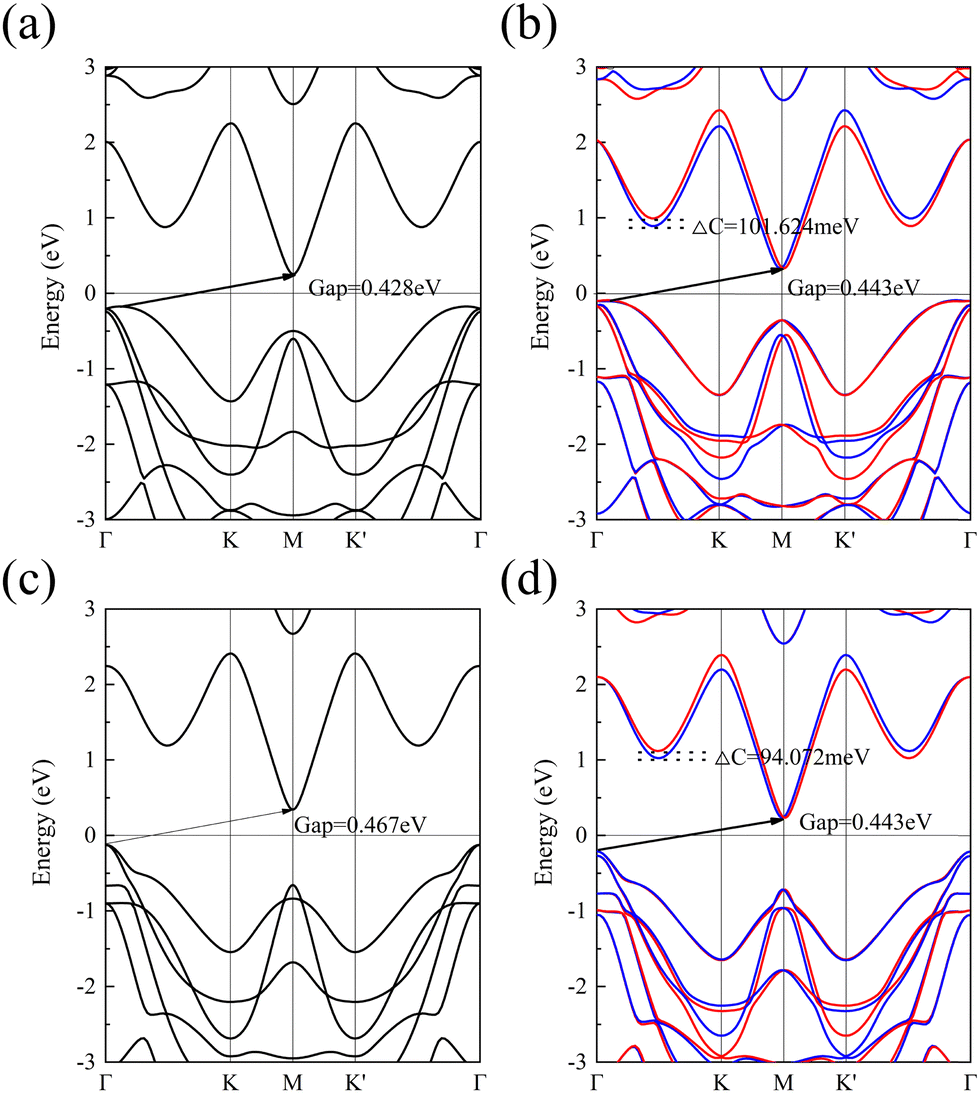 | ||
| Fig. 4 (a) Energy band diagram calculated using the PBE method when SOC is not considered. (b) Energy band diagram calculated using the PBE method when SOC is considered. (c) Energy band diagram calculated using the GGA+U method when SOC is not considered. (b) Energy band diagram calculated using the GGA+U method when SOC is considered. Red represents spin up and blue represents spin down. | ||
When SOC is considered, 1 L Hf3C2O2 remains an indirect bandgap semiconductor with a bandgap of 0.443 eV. The valley splitting was produced between Γ and K, with a valley splitting value of 101.624 meV. Fig. 4(c) and (d) show the energy bands calculated by the GGA+U method. Similarly, when SOC is not considered, 1 L Hf3C2O2 is an indirect bandgap semiconductor with a bandgap of 0.467 eV. The top of the valence band is located at the Γ point in the Brillouin zone, while the bottom of the conduction band is located at the M point in the Brillouin zone. When SOC is considered, 1 L Hf3C2O2 remains an indirect bandgap semiconductor with a bandgap of 0.443 eV. The positions of the top of the valence band and the bottom of the conduction band remain constant. Valley splitting was produced between Γ and K with a valley splitting value of 94.072 meV. Regarding the accuracy of the calculations, the HSE06 method is more accurate than the PBE and GGA+U methods in determining the electronic properties.
In order to study the source of valley splitting in 1 L Hf3C2O2, the local state density and partial state density of 1 L Hf3C2O2 were calculated respectively. As shown in Fig. 5(a), in the localized density of states diagram for 1 L Hf3C2O2 red represents the electron occupation number of the transition metal atom Hf, and green and blue represent the electron occupation numbers of the C and O atoms, respectively. Based on the energy band diagram we know that the valley splitting of 1 L Hf3C2O2 is above the Fermi energy level, so we mainly look at the part of the localized state density above the Fermi energy level. It can be concluded that above the Fermi surface it is mainly occupied by electrons of transition metal Hf atoms. More specifically, we are able to determine that the energy bands above the Fermi surface are mainly occupied by the d orbitals of the transition metal Hf atoms based on the partial state density map of 1 L Hf3C2O2, where yellow, violet, and red represent the number of electrons occupying the d, p, and s orbitals of the transition metal Hf atoms, respectively. In order to determine more clearly the specific form of the contribution of the d-orbitals of Hf atoms to valley splitting, we plotted the projected energy band diagram of the d-orbitals of Hf atoms as shown in Fig. 5(b). Between Γ and K, the valley splitting of the conduction band is mainly contributed by the dx2–y2 orbitals of the Hf atoms. The valley splitting between K′ and Γ is mainly contributed by the dz2 and dx2–y2 and dxy orbitals of the Hf atom.
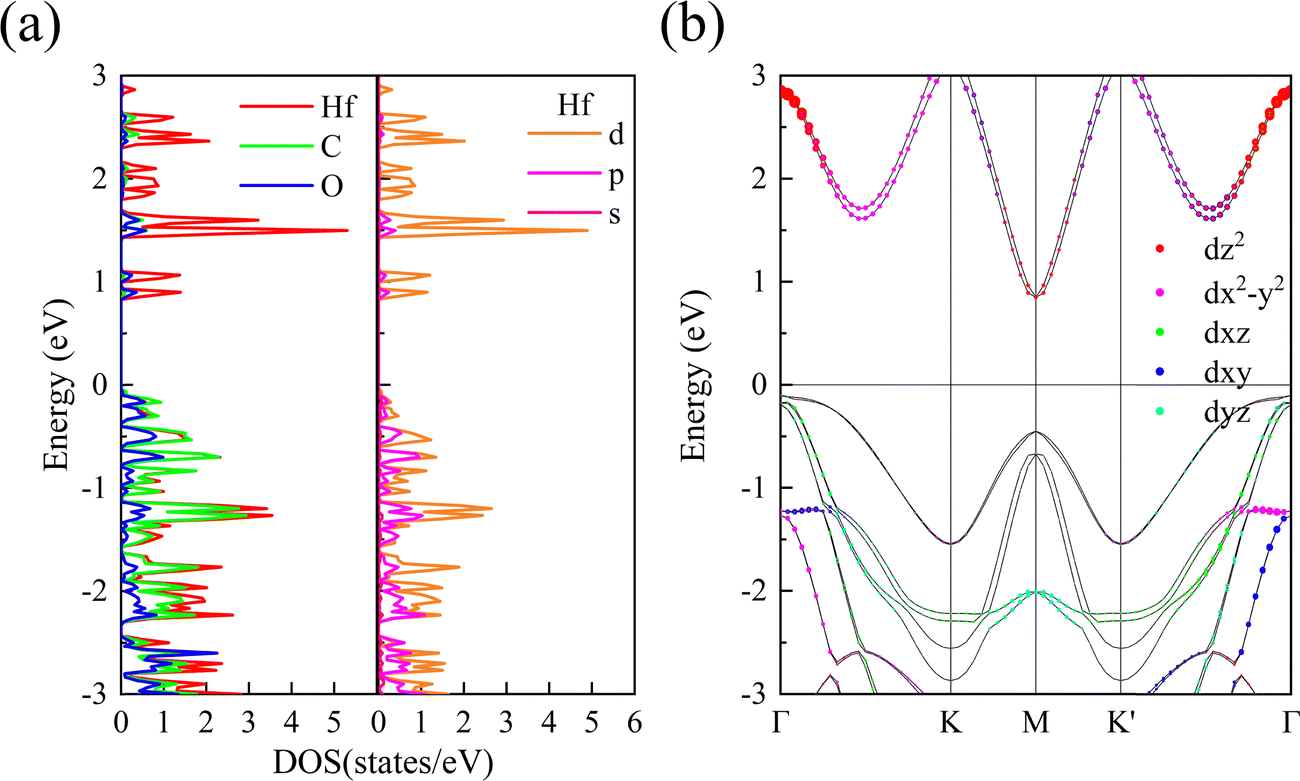 | ||
| Fig. 5 (a) Localized density of states for 1 L Hf3C2O2 and partial wave density of states for the transition metal element Hf. (b) Energy band diagram of the d-orbital projection of 1 L Hf3C2O2. The Fermi energy level is set to 0 eV. | ||
3.3. Berry curvature
Since 1 L Hf3C2O2 breaks spatial inversion symmetry but is protected by temporal inversion symmetry, 1 L Hf3C2O2 should have Berry curvature with opposite directions but equal values. To confirm this, we calculated the out-of-plane Berry curvature of the conduction band where the 1 L Hf3C2O2 valley splitting is located. The Berry curvature can be written as:42where fn denotes the Fermi–Dirac distribution function, νx/νy denotes the velocity operator of the Bloch function along the direction, and ψnk/ψmk denotes the wave function with eigenvalues. Due to the breaking of the inversion symmetry of 1 L Hf3C2O2, the carriers in the K/K′ valley of 1 L Hf3C2O2 have a non-zero Berry curvature along the out-of-plane direction. However, under the protection of time reversal symmetry, the Berry curvature satisfies −Ωn(k) = Ωn(−k). The Berry curvature of K/K′ is equal in magnitude and opposite in direction, as shown in Fig. 6(a). As we can see from the figure, there are alternating areas of dark blue and dark red at K and K′, and the darker the color, the larger the value.
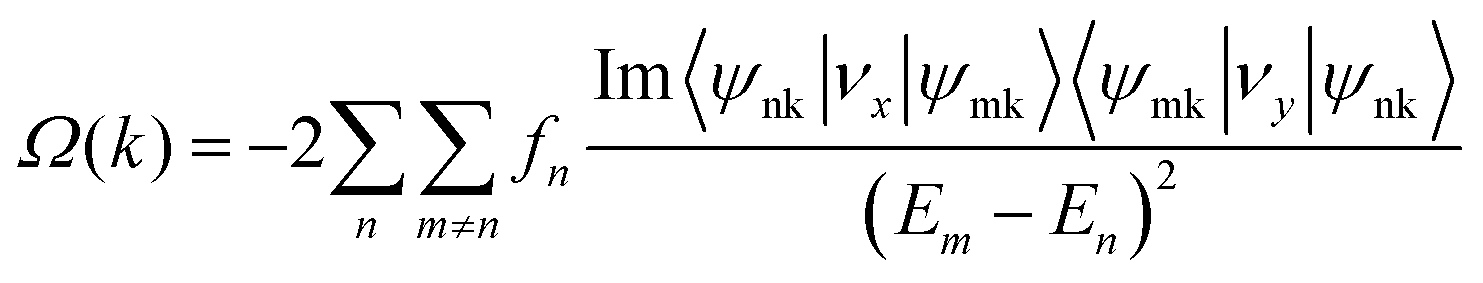
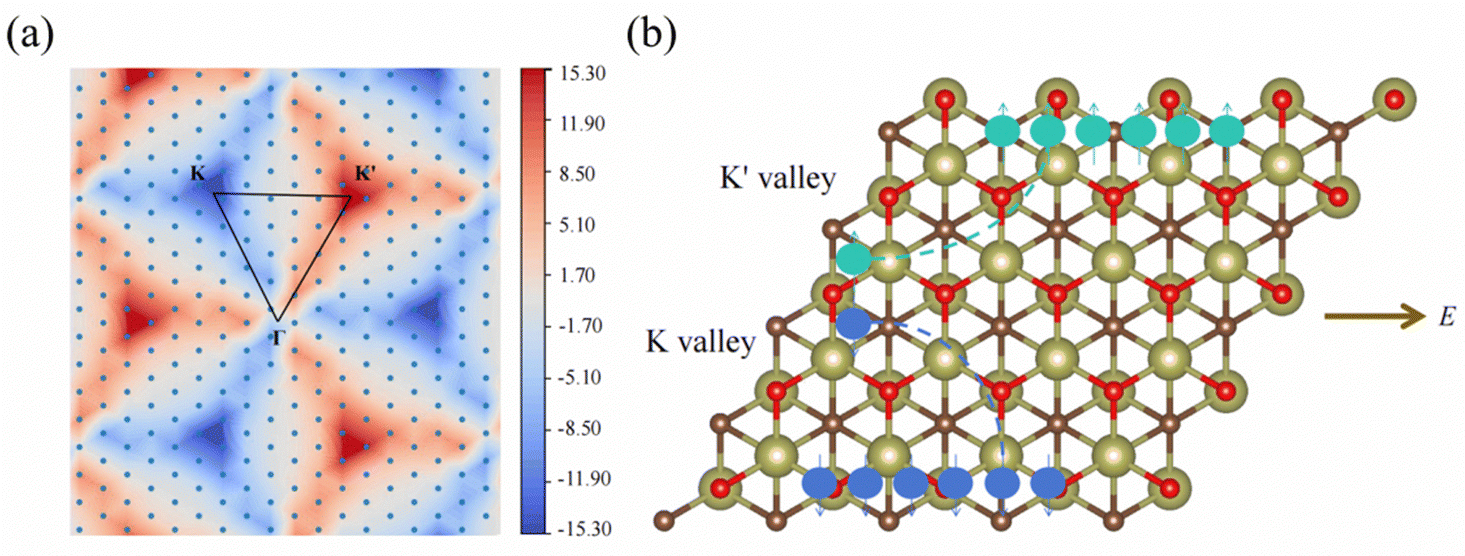 | ||
| Fig. 6 (a) Contour plot of the Berry curvature of 1 L Hf3C2O2 in the whole Brillouin zone. (b) Schematic of the anomalous valley Hall effect in the K/K′ valley, with arrows indicating spin-up and spin-down states and circles indicating holes. | ||
The valley splitting in Hf3C2O2 occurs between Γ–K, not on K/K′. From the band structure, the local maximum between Γ–K is 98.228 meV for valley splitting, while on K/K′, there is a maximum of 208.815 meV. From the graph of Berry curvature, this conclusion can be verified. We can see from the graph of the Berry curvature that on K/K′, the color of the Berry curvature is the darkest, representing the maximum of the Berry curvature, and there is a local value between Γ–K. The Berry curvature corresponds to the band structure.
Due to the non-zero Berry curvature of 1 L Hf3C2O2, when an in-plane electric field E is applied, the Bloch electrons exhibit anomalous velocities perpendicular to the electric field:14
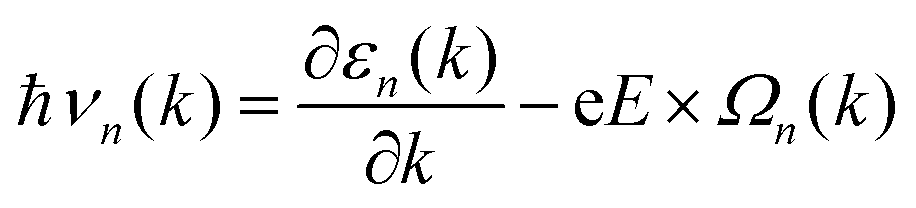
3.4. Biaxial strain
2D materials can often withstand large lattice strains and their properties can be effectively tuned by strain engineering.44 Biaxial strain can be expressed as:where a0 refers to the lattice constant of the material when no biaxial strain is applied and a represents the lattice constant after biaxial strain is applied. A negative value of ε indicates a compressive strain on the material and a positive value of ε indicates a tensile strain on the material. Therefore, the present work also calculates the variation patterns of band gap and valley splitting values under biaxial strain. As shown in Fig. 7(a), the band gap first increases and then decreases with strain. The band gap is maximized at 1% applied strain with a maximum value of 0.962 eV. As shown in Fig. 7(b), the valley splitting of the conduction band increases with the increase of strain. When a strain of 4% is applied, the valley splitting value reaches its maximum, which is 104.358 meV. Therefore, the band gap and valley splitting of 1 L Hf3C2O2 are robust. To reflect the evolution trend of its electronic properties, we plotted the band structure of 1 L Hf3C2O2 under strains of −4% to 4%, as shown in Fig. S1 (ESI†).
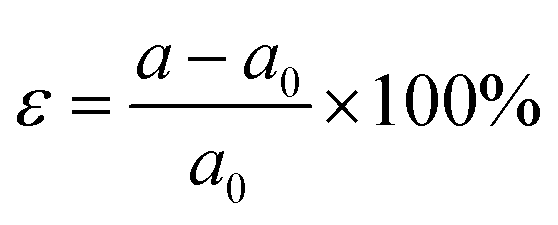
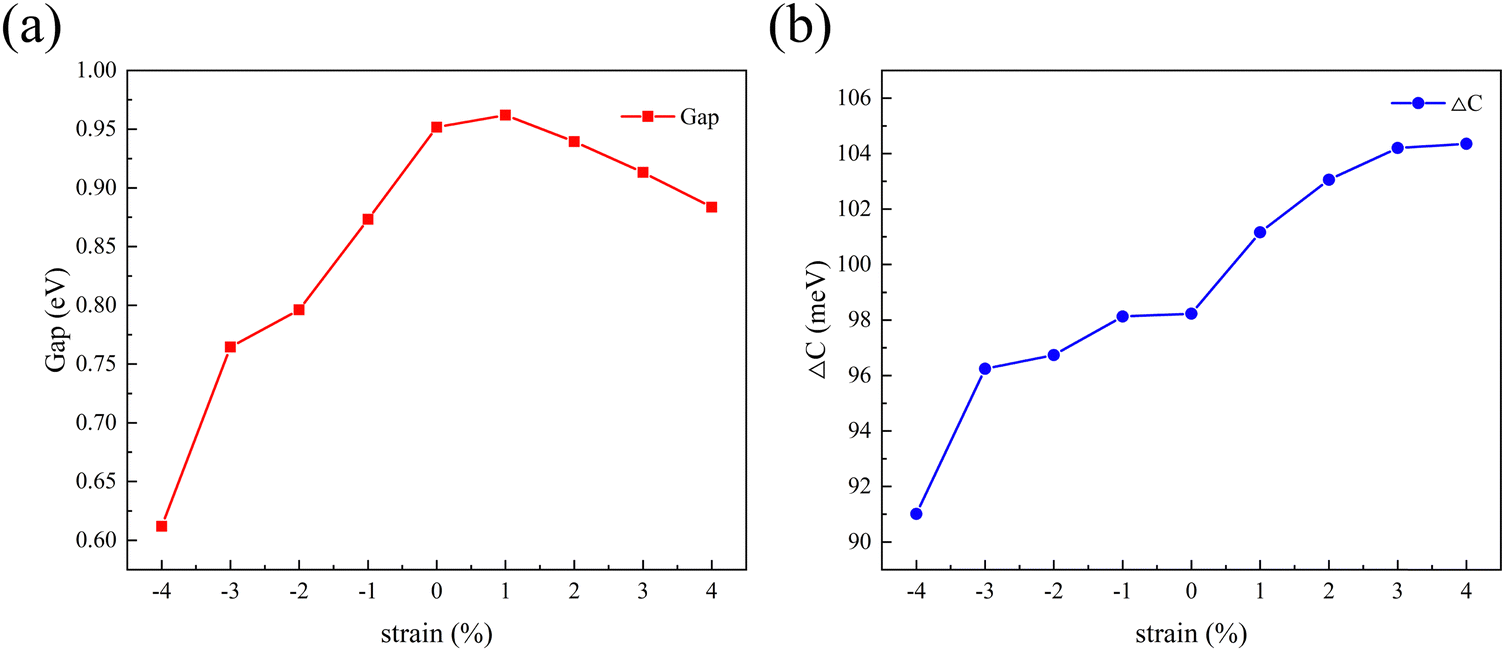 | ||
| Fig. 7 Plots of the variation in (a) band gap and (b) valley splitting values of Hf3C2O2 when biaxial strains from −4% to 4% are applied. | ||
3.5. Doping
To demonstrate the doping effect, the band gaps and valley splitting as a function of the electron and hole doping concentration are investigated. As shown in Fig. 8(a), the band gap increases with increasing hole doping concentration. Instead, the band gap becomes smaller with increasing electron doping concentration. Similarly, the relationship between valley splitting and doping concentration is the same, as shown in Fig. 8(b). The valley splitting increases with more hole doping, while the valley splitting decreases with more electron doping. Therefore, for 1 L Hf3C2O2, hole doping is a feasible way to increase the valley splitting. To reflect the evolution trend of its electronic properties, we plotted the band structure of 1 L Hf3C2O2 with doping concentrations ranging from −0.1 electron per unit cell to 0.1 electron per unit cell in Fig. S2 (ESI†).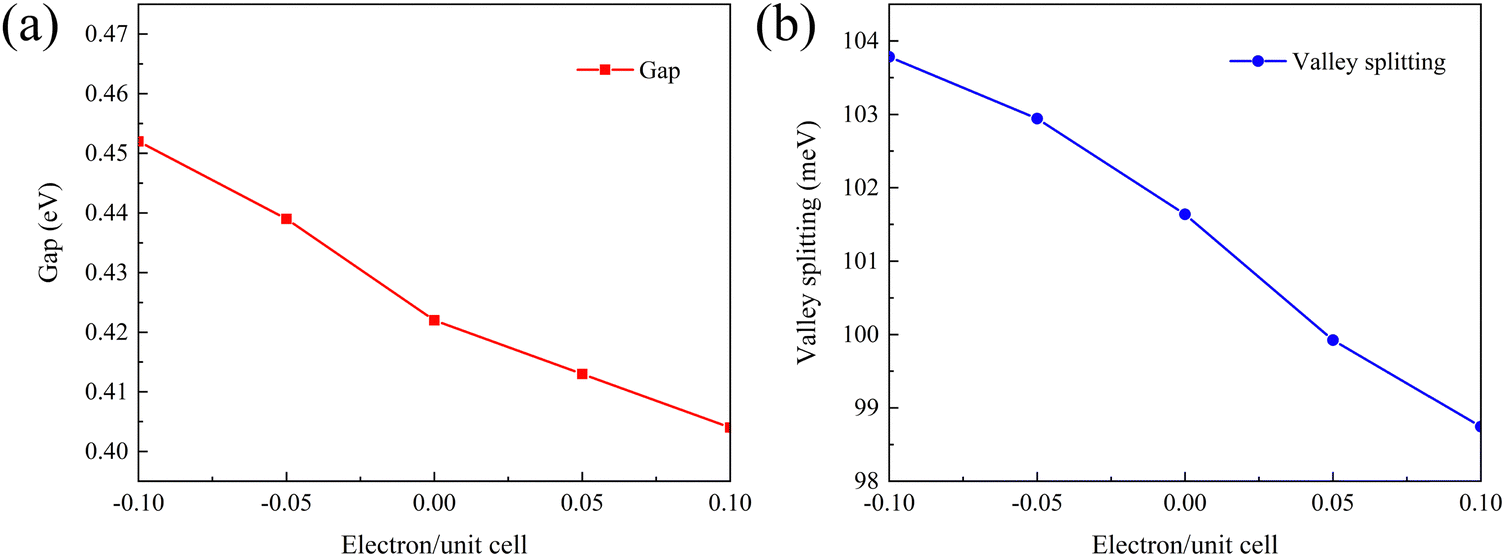 | ||
| Fig. 8 Curves of (a) band gap and (b) valley splitting of 1 L Hf3C2O2 with doping concentration. 0.00 means neither gain nor loss of electrons, a negative number means loss of electrons (hole doping) and a positive number means gain of electrons (electron doping). | ||
4. Conclusions
In this work, we employed the HSE06 method to determine the properties of 1 L Hf3C2O2. Without considering SOC, 1 L Hf3C2O2 is an indirect bandgap semiconductor. When SOC is taken into account, the bandgap of 1 L Hf3C2O2 decreases. The valley splitting value of 98.228 meV occurs between the Γ and K points in the conduction band. Calculations have verified that 1 L Hf3C2O2 exhibits good mechanical and dynamical stability. Additionally, the Berry curvature of 1 L Hf3C2O2 is non-zero, with equal magnitudes but opposite signs at K and K′. As an effective means of tuning electronic properties, biaxial strain shows good stability for the bandgap and valley splitting of 1 L Hf3C2O2 under such conditions. Both the bandgap and valley splitting values decrease with increasing doping concentration. Our research indicates that 1 L Hf3C2O2 is a promising two-dimensional valleytronics material.Data availability
All data that support the findings of this study are included within the article (and any ESI† files).Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Supported by the National Natural Science Foundation of China (Grant No. 61704083 and 12104234), the Natural science fund for colleges and universities in Jiangsu Province (Grant No. 21KJD140005), and the Natural Science Foundation of Jiangsu Province (Grant No. BK20210578). The authors gratefully acknowledge the computing time granted by the Shanghai Supercomputer Centre.References
- F. J. Ohkawa and Y. Uemura, J. Phys. Soc. Jpn., 1977, 43, 907–916 CrossRef CAS.
- J. R. Schaibley, H. Yu, G. Clark, P. Rivera, J. S. Ross, K. L. Seyler, W. Yao and X. Xu, Nat. Rev. Mater., 2016, 1, 1–15 Search PubMed.
- Y. Ma, L. Kou, A. Du, B. Huang, Y. Dai and T. Heine, Phys. Rev. B, 2018, 97, 035444 CrossRef CAS.
- Z. Wang, X. Han and Y. Liang, Phys. Chem. Chem. Phys., 2024, 26, 17148–17154 RSC.
- G. Liu, S. Liu, B. Xu, C. Ouyang and H. Song, J. Appl. Phys., 2015, 118, 124303 CrossRef.
- C. Tan and H. Zhang, Chem. Soc. Rev., 2015, 44, 2713–2731 RSC.
- A. Bafekry, C. Stampfl, M. Naseri, M. M. Fadlallah, M. Faraji, M. Ghergherehchi, D. Gogova and S. Feghhi, J. Appl. Phys., 2021, 129, 155103 CrossRef CAS.
- J.-S. Yang, L. Zhao, L. Shi-Qi, H. Liu, L. Wang, M. Chen, J. Gao and J. Zhao, Nanoscale, 2021, 13, 5479–5488 RSC.
- J. Yuan, Q. Wei, M. Sun, X. Yan, Y. Cai, L. Shen and U. Schwingenschlögl, Phys. Rev. B, 2022, 105, 195151 CrossRef CAS.
- P. Li, X. Yang, Q.-S. Jiang, Y.-Z. Wu and W. Xun, Phys. Rev. Mater., 2023, 7, 064002 CrossRef CAS.
- C. E. Shuck and Y. Gogotsi, Chem. Eng. J., 2020, 401, 125786 CrossRef CAS.
- S. Venkateshalu, M. Shariq, B. Kim, M. Patel, K. S. Mahabari, S.-I. Choi, N. K. Chaudhari, A. N. Grace and K. Lee, J. Mater. Chem. A, 2023, 11, 13107–13132 RSC.
- G.-B. Liu, D. Xiao, Y. Yao, X. Xu and W. Yao, Chem. Soc. Rev., 2015, 44, 2643–2663 RSC.
- X. Xu, W. Yao, D. Xiao and T. F. Heinz, Nat. Phys., 2014, 10, 343–350 Search PubMed.
- Y. Gogotsi and B. Anasori, Journal, 2019, 13, 8491–8494 Search PubMed.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- S. Li, J. He, L. Grajciar and P. Nachtigall, J. Mater. Chem. C, 2021, 9, 11132–11141 RSC.
- J. Lu, R. Liu, F. Yue, X. Zhao, G. Hu, X. Yuan and J. Ren, J. Phys. Chem. Lett., 2022, 14, 132–138 CrossRef PubMed.
- M. Lan, S. Wang, X. Liu, S. Ma, S. Qiao, Y. Li, H. Wu, F. Li and Y. Pu, Phys. Chem. Chem. Phys., 2024, 26, 8945–8951 RSC.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- V. Milman, B. Winkler, J. White, C. Pickard, M. Payne, E. Akhmatskaya and R. Nobes, Int. J. Quantum Chem., 2000, 77, 895–910 CrossRef CAS.
- Y. Zhang and W. Yang, Phys. Rev. Lett., 1998, 80, 890 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505 CrossRef CAS.
- M. Cococcioni and S. De Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035105 CrossRef.
- J. Heyd and G. E. Scuseria, J. Chem. Phys., 2004, 121, 1187–1192 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 15 CrossRef.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- M. Blume and R. Watson, Proceedings of the Royal Society of London. Series A. Mathematical Physical Sciences, 1963, 271, 565–578.
- U. Herath, P. Tavadze, X. He, E. Bousquet, S. Singh, F. Muñoz and A. H. Romero, Comput. Phys. Commun., 2020, 251, 107080 CrossRef CAS.
- Y. Wang, M. Qiao, Y. Li and Z. Chen, Nanoscale Horiz., 2018, 3, 327–334 RSC.
- R. Peng, Y. Ma, B. Huang and Y. Dai, J. Mater. Chem. A, 2019, 7, 603–610 RSC.
- Y. Li, X. Xu, M. Lan, S. Wang, T. Huang, H. Wu, F. Li and Y. Pu, Phys. Chem. Chem. Phys., 2022, 24, 25962–25968 RSC.
- S. Singh, C. Espejo and A. H. Romero, Phys. Rev. B, 2018, 98, 155309 CrossRef CAS.
- D. Çakır, F. M. Peeters and C. Sevik, Appl. Phys. Lett., 2014, 104, 203110 CrossRef.
- P. Zhao, Y. Liang, Y. Ma and T. Frauenheim, Phys. Rev. B, 2023, 107, 035416 CrossRef CAS.
- Y. Li, M. Lan, S. Wang, T. Huang, Y. Chen, H. Wu, F. Li and Y. Pu, Phys. Chem. Chem. Phys., 2023, 25, 15676–15682 RSC.
- Y. Wang, W. Wei, H. Wang, N. Mao, F. Li, B. Huang and Y. Dai, J. Phys. Chem. Lett., 2019, 10, 7426–7432 CrossRef CAS.
- W. Yao, D. Xiao and Q. Niu, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 235406 CrossRef.
- W. Tong, S. Gong, X. Wan and C. Duan, Nat. Commun., 2016, 7, 13612 CrossRef CAS.
- S. Li, W. Wu, X. Feng, S. Guan, W. Feng, Y. Yao and S. A. Yang, Phys. Rev. B, 2020, 102, 235435 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04003a |
| ‡ Shiqian Qiao and Yang Zhang contributed equally to this manuscript. |
| This journal is © the Owner Societies 2025 |