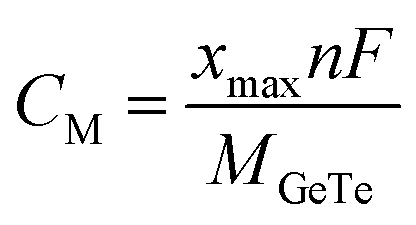First principles study on monolayer GeTe as an anode material for multivalent ion batteries†
Received
14th September 2024
, Accepted 26th November 2024
First published on 27th November 2024
Abstract
Finding suitable anode materials for multivalent ion batteries (MuIBs) is the key to improving theoretical capacity, reducing development costs and enhancing the safety of energy storage batteries. In recent years, monolayer GeTe has been reported as an anode material in monovalent ion batteries, but it has not received much attention in MuIBs. This article uses first principles methods based on density functional theory (DFT) to explore the application prospects of monolayer GeTe with a unique serrated wrinkled layer structure as an anode material for multivalent metal ion (Al3+/Mg2+/Ca2+) batteries. The research results show that Al3+, Mg2+ and Ca2+ have low diffusion barriers (0.47, 0.35 and 0.61 eV) on monolayer GeTe, indicating its excellent diffusion ability and fast charge discharge rate during the charging and discharging process. Reasonable open circuit voltages (0.62, 0.85 and 0.64 V) and theoretical specific capacities higher than those of commercial graphite anode materials (624.6, 446.1 and 446.1 mA h g−1) indicate that monolayer GeTe has the ability to store Al3+/Mg2+/Ca2+. Finally, molecular dynamics simulations (MD) are used to calculate the adsorption energy and density field of ions during their movement on the surface of monolayer GeTe, demonstrating the stable adsorption ability of monolayer GeTe and the strong interaction between the two. This article reveals that monolayer GeTe can be used as a promising candidate anode material for MuIBs.
1. Introduction
Lithium ion batteries (LIBs) have been perceived as the main power source for consumer electronics in the past few years, and they have also been broadly used in hybrid electric vehicles (HEVs) and electric vehicles (EVs) with the growing demand of society.1 For LIBs anodes, commercial graphite with a limited theoretical capacity of 372 mA h g−1 has prompted the exploration of alternative negative materials,1,2 and multivalent ion batteries (MuIBs) have opened up new opportunities. The reason is that inserting the same number of multivalent ions (such as Mg2+, Zn2+, Ca2+ and Al3+) can provide two or three times the capacity compared to monovalent ions (Li+, Na+ and K+),3–7 and as a result, a higher theoretical capacity is obtained (Mg2+: 2205 mA h g−1, Ca2+: 1337 mA h g−1, and Al3+: 2978 mA h g−1).8–10 In addition, high stability, low toxicity, low cost, high security, and high abundance make multivalent cations very promising for metal ion batteries.11,12
In LIBs, silicon (Si) is the most popular lithium alloy material, but germanium (Ge) is theoretically more attractive because it has a higher ion diffusion coefficient (400 times higher than Si) and superior electronic conductivity (Ge: 2.1 S m−1, Si: 1.6 × 10−3 S m−1).13,14 Therefore, Ge can form Li3.75Ge and NaxGe, proving that Ge can also be used as a negative material for LIBs and SIBs.15,16 Similarly, the chalcogenides of sulfur (S), selenium (Se), and tellurium (Te) can also form M2X (M = Li, Na; X = S, Se, Te) phases with Li/Na alloys, indicating their potential as anode materials for LIBs and SIBs.17–19 Te in particular has many advantages, for example, it has the strongest metallic properties among all chalcogenides, resulting in the highest conductivity (S: 5 × 10−22 MS m−1, Se: 1 × 10−10 MS m−1, Te: 2 × 10−4 MS m−1),20 which is beneficial for the rapid diffusion of ions, thereby improving the charging and discharging rate of the battery.
Germanium telluride (GeTe) is a two-dimensional material that combines the excellent characteristics of both Ge and Te elements, which has been widely studied in ferroelectric semiconductors, low-temperature superconductors, and thermoelectrics due to its semiconductor properties with different band gaps and phase transitions in the crystal structure.21–23 In addition, GeTe has three main polymorphic transition phases: α (rhombus), γ (square), and β (cubic). In these phases, α-GeTe exhibits a unique “zigzag” wrinkled layer structure, which is different from the GeS and GeSe analogues (“armchair” structure). The unique “zigzag” atomic arrangement is conducive to the storage and rapid diffusion of Li+/Na+,24 therefore the electrochemical performance is superior to that of GeS and GeSe in the latest research.25 Meanwhile, the density of α-GeTe (6.14 g cm−3) is the highest among reported Ge based lithium ion battery materials (GeS: 4.24 g cm−3, GeS2: 2.94 g cm−3, and GeP3: 3.85 g cm−3), thus providing a higher energy density.26
Inspired by the lithium storage performance of graphene which is twice that of graphite, researchers have also attempted to obtain monolayer GeTe nanosheets through mechanical stripping and laser thinning with physical methods and exfoliation for α-GeTe with chemical preparation.27,28 But the exploration of monolayer GeTe in the field of energy storage is limited to experimental aspects,25,29 and comprehensive predictions in the theoretical field are still relatively scarce, especially in the field of MuIBs where systematic research has not been seen. Therefore, in this article, a first principles approach based on density functional theory (DFT) is adopted to discuss the applicability of monolayer GeTe in aluminum ion batteries (AIBs), magnesium ion batteries (MIBs), and calcium ion batteries (CIBs). We find that Al3+, Mg2+ and Ca2+ have a lower diffusion barrier on monolayer GeTe, indicating that it has a faster ion diffusion rate as a negative material for MuIBs. In terms of theoretical capacity, monolayer GeTe has a reasonable open circuit voltage and high theoretical specific capacity, indicating good practical application prospects. This work not only predicts the potential of monolayer GeTe as an anode material for MuIBs from a theoretical perspective, but also helps researchers provide valuable research in promoting the application of other similar structured materials in various energy storage applications, such as cation and anion batteries.
2. Computational details
Using a first principles method based on density functional theory (DFT) implemented in the DMol3 tools of the Material Studio,30 we used the Perdew–Burke–Ernzerhof (PBE) functional of the generalized gradient approximation (GGA) for exchanging correlation functions.31 The basis set was DNP which contained double-numerical quality with the inclusion of polarization functions,32 the basis file was set to 4.4, and the Brillouin zone used a 6 × 6 × 1 Monkhorst Pack grid for K-point setting. A vacuum thickness of 20 Å was set in the z-axis direction to avoid interlayer interactions, indicating that the structure was reasonable and in line with practical significance. The transition state search used the LST/QST method to calculate the diffusion barrier of Al3+/Mg2+/Ca2+ on the surface of monolayer GeTe along the stable adsorption sites.33 Molecular dynamics simulation (MD) was conducted using the Forcite tools,29 to construct the structure of the MD simulation, two Al3+/Mg2+/Ca2+ were absorbed on the surface of monolayer GeTe with the optimal adsorption site. Using the NVT canonical ensemble, the initial velocity was Random, the temperature range was 300 K, the time step was 1 fs, and the simulation time was 5000 ps, ensuring that Al3+/Mg2+/Ca2+ exhibited diffusion behavior on monolayer GeTe crystals.
3. Results and discussion
3.1 Crystal structure and electronic properties of monolayer GeTe
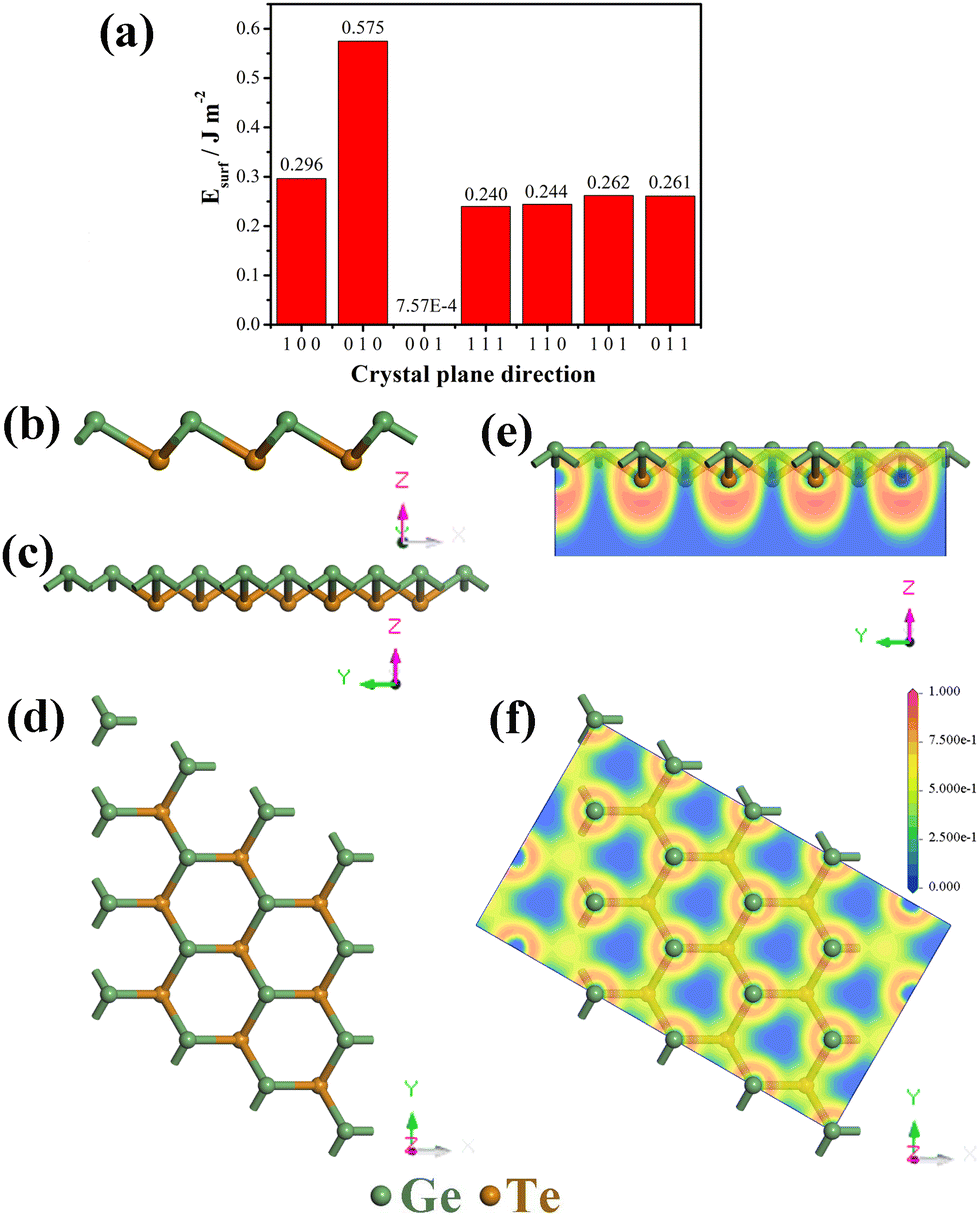
Monolayer GeTe has a unique serrated wrinkled layer structure, and we established a monolayer primitive GeTe structure based on the experimentally obtained α-GeTe crystal parameters. The lattice constant of primitive GeTe is a = b = 4.18 Å, and the space group is R3m(160), which is consistent with previously reported results.34,35 To obtain a stable unit cell structure, the surface energy of different cross-sectional directions of the unit cell is studied. In morphology, surface energy (Esurf) refers to the energy with a certain thickness of the crystal plane, which is the energy difference between the surface and the interior of the crystal,36 and it is used to characterize the stability of the crystal surface. The smaller the surface energy, the more stable the surface, and its formula is defined as:37| | 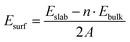 | (1) |
In the formula, Eslab refers to the energy of the crystal surface, Ebulk refers to the energy of the crystal cell, n refers to the ratio of the surface model to the number of atoms in the crystal, A refers to the cross-sectional area of the surface model, and 2 represents the existence of two surfaces in the surface model, which are calculated at once. In order to obtain the most stable crystal cell structure, we calculated the surface energy of seven different crystal plane directions, and the obtained values are shown in Fig. 1a. It can be seen that the Esurf of monolayer primitive GeTe is the lowest in the (001) direction (E(001)surf = 7.57 × 10−4 J m−2). Based on the conclusions obtained, a surface model is constructed for monolayer GeTe in the (001) crystal plane direction, and a vacuum thickness of 20 Å is set to avoid interlayer interactions. At the same time, a 3 × 3 × 1 supercell (referred to as monolayer GeTe, as shown in Fig. 1b–d) is used. We can see that monolayer GeTe has a serrated wrinkled layer structure, where Ge and Te atoms alternate with each other. The unique wrinkled layer structure can provide more ion adsorption sites, thereby increasing its theoretical capacity. In Fig. 1e and f, the local electron function (ELF) is used to analyze the bonding properties of monolayer GeTe structures. According to the valence bond theory, regions with ELF values less than 0.5 represent ionic bonds, while regions with ELF values greater than 0.5 represent covalent bonds.38 Therefore, it can be clearly observed that there are a large number of electrons near Ge and Te atoms, indicating that Ge–Te bonds have strong covalent properties.
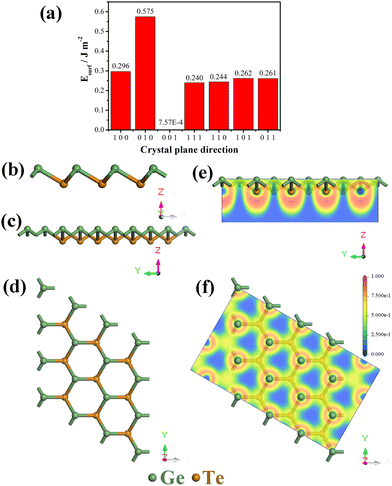 |
| | Fig. 1 (a) Surface energy of monolayer primary GeTe in different crystal plane directions; structure diagram of monolayer GeTe: (b) front view, (c) side view, (d) top view; electron localization function (ELF) of monolayer GeTe: (e) side view, (f) top view. | |
Fig. S1 (ESI†) shows the band structure and corresponding density of states (DOS) plot of a monolayer GeTe crystal obtained through DFT calculations. In band theory, the difference between the valence band maximum (VBM) and the conduction band minimum (CBM) is the band gap. It should be noted that, as stated in previous literature, the traditional GGA-PBE function is not conducive to accurately describing the electronic properties of two-dimensional materials and has an undeniable underestimation of the band gap.39 Correspondingly, a mixing function similar to HSE06 can more accurately predict the band gap of materials.40 Therefore, we added a comparison chart of the band gap of monolayer GeTe under traditional GGA-PBE and HSE06 functions (Fig. S1, ESI†). It can be seen that the band gaps of monolayer GeTe under different functions of GGA-PBE and HSE06 are 1.315 and 1.837 eV, respectively, and they perfectly match the density of states. This result is consistent with previous literature reports that the GGA-PBE function does underestimate the value of band gap, but due to sufficient consideration of the huge computational cost,41 it is not used for monolayer GeTe calculations here. In addition, our goal is not to obtain the precise band gap of monolayer GeTe, but to explore whether monolayer GeTe is a semiconductor with an indirect band gap and good conductivity, thus possessing innate conditions as an anode material in the field of metal ion batteries. Therefore, we perform monolayer GeTe computing tasks based on GGA-PBE functionality.
3.2 Adsorption of Al3+/Mg2+/Ca2+ by monolayer GeTe
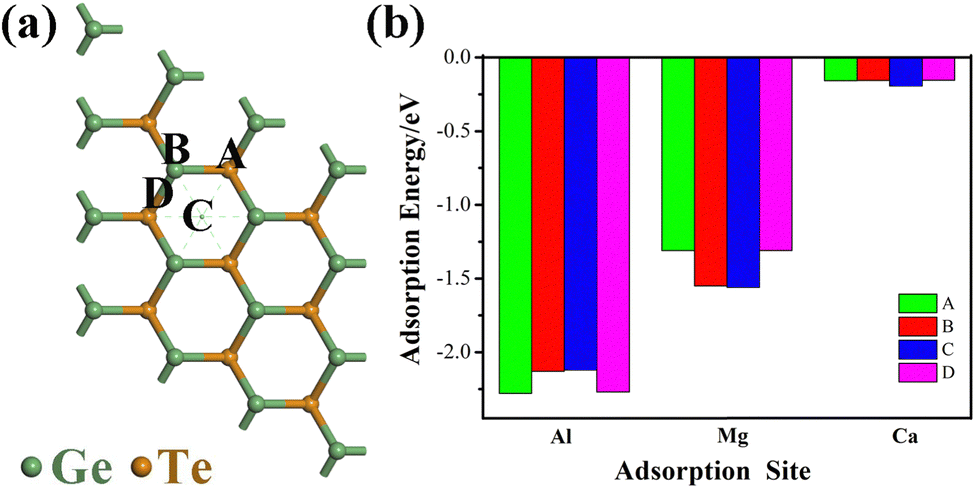
The spontaneous adsorption of ions is an important criterion for determining whether a material can become an anode material for batteries. In order to investigate the optimal adsorption position of metal ions (Al3+/Mg2+/Ca2+) and prevent the influence of ion interaction, we use a 3 × 3 × 1 supercell structure for calculation and consider all possible binding configurations and the most stable binding configuration. Finally, we select three different adsorption sites, as shown in Fig. 2a, point A is located directly above the Te atom, point B is located directly above the Ge atom, point C is located directly above the center point of the hexagonal structure, and point D is located directly above the the Ge–Te bridge. After fully optimizing the three sites, the adsorption energies at four different positions are obtained (Fig. 2b). The adsorption energy is defined as:42| | | Ead = EM+GeTe − EGeTe − EM (M = Al3+/Mg2+/Ca2+) | (2) |
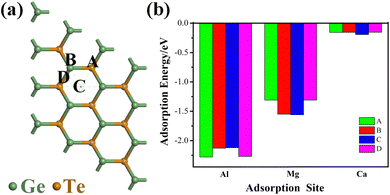 |
| | Fig. 2 (a) Four adsorption sites for metal ions: site A is located directly above the Te atom, site B is located directly above the Ge atom, site C is located directly above the center point of the hexagonal structure, and D is located directly above the Ge–Te bridge; (b) the adsorption energies of Al3+/Mg2+/Ca2+ at four adsorption sites. | |
In the equation, Ead represents the adsorption energy, and when the value is negative, it indicates that the metal ions can effectively adsorb on the monolayer GeTe surface, and the larger the absolute value, the more stable the ion adsorption. EGeTe and EM+GeTe represent the total energy of the system before and after monolayer GeTe adsorption of Al3+/Mg2+/Ca2+ respectively, while EM represents the energy of a single metal ion. In Fig. 2b, it can be seen that the adsorption energies calculated for the four adsorption points are all negative, indicating that the interaction between the adsorbent and the adsorption surface is favorable, and metal ions can be stably adsorbed on the monolayer GeTe surface. By calculating the adsorption energies of Al3+/Mg2+/Ca2+ on sites A (−2.28, −1.31 and −0.16 eV), B (−2.13, −1.55 and −0.16 eV), C (−2.12, −1.56 and −0.20 eV), and D (−2.27, −1.31 and −0.15 eV), the optimal adsorption positions for Al3+/Mg2+/Ca2+ on monolayer GeTe can be obtained as sites A, C and C. The three lower adsorption energy values (−2.28, −1.56 and −0.20 eV) indicate that monolayer GeTe can serve as an anode material for metal ion batteries and exhibit strong interactions with Al3+/Mg2+/Ca2+ on the surface of GeTe, which is beneficial for the stability of the entire structure and charge transfer between the two.
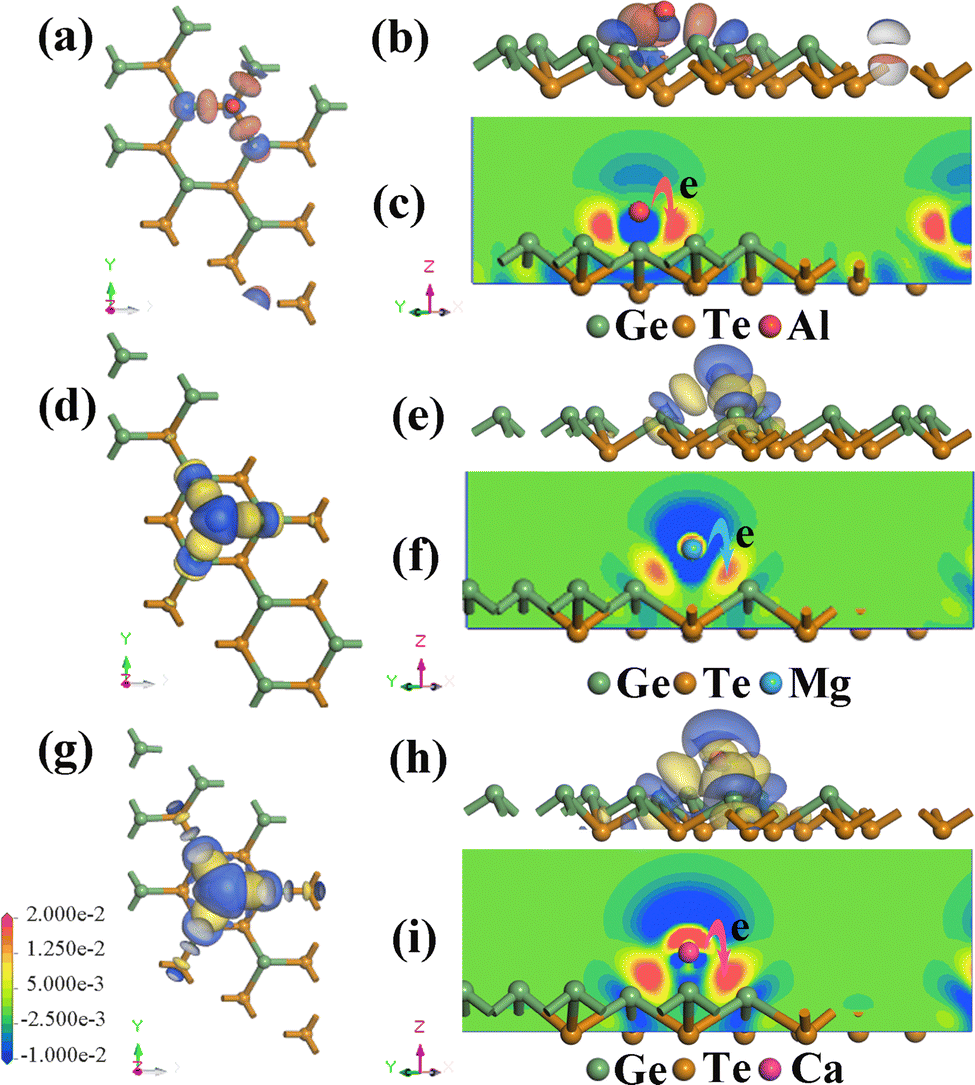
To gain a deeper understanding of the correlation and charge transfer between Al3+/Mg2+/Ca2+ and monolayer GeTe, we calculate the electron density difference, which is calculated through the CASTEP tools, and the formula is defined as follows:
| | | Δρad = ρM+GeTe − ρGeTe − ρM (M = Al3+/Mg2+/Ca2+) | (3) |
In the formula,
ρGeTe and
ρM+GeTe represent the charge density of monolayer GeTe before and after adsorption of Al
3+/Mg
2+/Ca
2+ respectively, and
ρM represents the charge density of individual metal ions. As shown in
Fig. 3, there are different color distributions between metal ions and GeTe, where red represents electron accumulation and blue represents electron dissipation. It can be seen that charge transfer mainly occurs around the Al
3+/Mg
2+/Ca
2+, and electron accumulation occurs at the interface between the Al
3+/Mg
2+/Ca
2+ and the monolayer GeTe (
Fig. 3a–b, d, e and g, h). We can also better demonstrate the transfer of electrons from Al
3+/Mg
2+/Ca
2+ to monolayer GeTe along the
z-axis section (
Fig. 3c, f, and i). To verify the above conclusion, we quantitatively estimate the charge transfer amount using Mulliken charge analysis and display it in
Table 1. It can be seen that Al/Mg/Ca undergoes charge transfer at different adsorption sites, indicating that Al/Mg/Ca is almost completely ionized and exists in the form of cations.
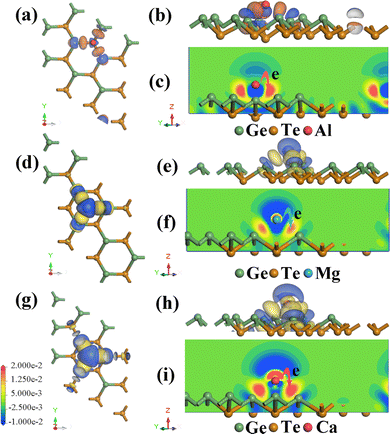 |
| | Fig. 3 Electron density difference of (a)–(c) Al, (d)–(f) Mg and (g)–(i) Ca adsorbed on the surface of monolayer GeTe. (c), (f) and (i) The electron density difference obtained along the z-axis cross-section. | |
Table 1 The adsorption energy (Ead/eV), charge transfer (Q/e), vertical distance (H/Å) and work function (Φ/eV) of Al3+, Mg2+ and Ca2+ at different adsorption sites on monolayer GeTe
| Structural properties |
Adsorption sites |
Al |
Mg |
Ca |
|
E
ad/eV |
A |
−2.28 |
−1.31 |
−0.16 |
| B |
−2.13 |
−1.55 |
−0.16 |
| C |
−2.12 |
−1.56 |
−0.20 |
|
|
|
Q/e |
A |
0.264 |
0.299 |
0.934 |
| B |
0.395 |
0.292 |
0.939 |
| C |
0.391 |
0.357 |
0.935 |
|
|
|
H/Å |
A |
3.089 |
3.448 |
3.429 |
| B |
2.729 |
2.944 |
2.997 |
| C |
1.853 |
2.442 |
2.548 |
|
|
|
Φ/eV |
A |
3.73 |
3.89 |
3.35 |
| B |
4.64 |
3.89 |
3.67 |
| C |
3.56 |
4.05 |
3.43 |
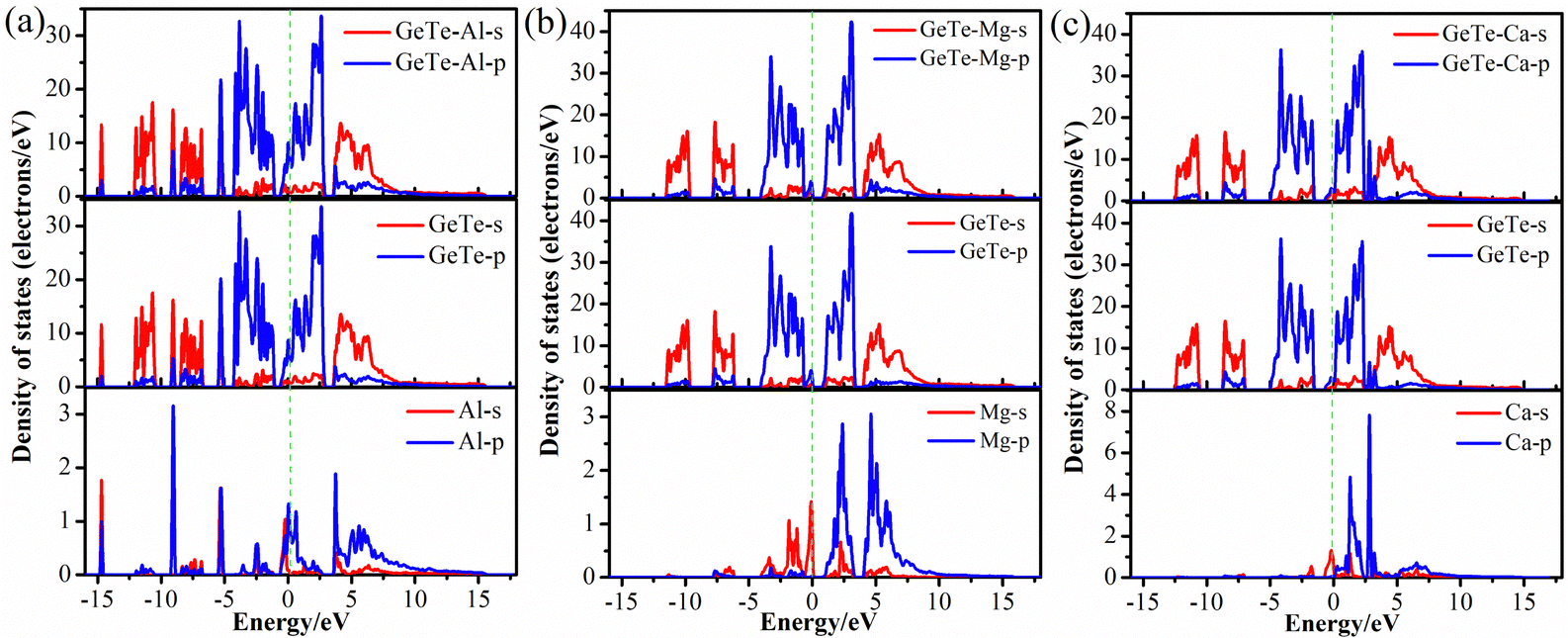
Fig. 4 calculates the PDOS diagram of monolayer GeTe after adsorption of Al3+/Mg2+/Ca2+ to understand the adsorption differences of different metal ions on monolayer GeTe. It can be seen that near the Fermi level, there is a significant overlap between the p orbitals of GeTe and the p orbitals of Al. Similarly, we can see that there is also a significant overlap between the p orbitals of GeTe and the s orbitals of Mg (Ca), indicating a significant hybridization effect. This hybridization phenomenon suggests the existence of interaction between GeTe and metal ion. This overlap usually leads to the transfer of electrons between atomic orbitals, resulting in the formation of new electronic states in the material, and the electronic structure of the material also changes accordingly, thereby affecting the electrochemical properties such as conductivity and stability of the material. It is evident that all three adsorption systems exhibit metallic properties, indicating that the conductivity of monolayer GeTe is significantly improved through the adsorption of metal ions, which is beneficial for the ion diffusion rate of monolayer GeTe materials in batteries.
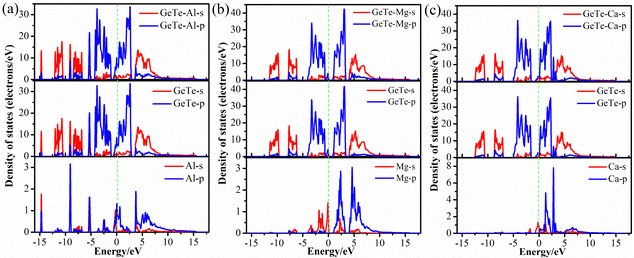 |
| | Fig. 4 Partial density of states of (a) Al3+, (b) Mg2+ and (c) Ca2+ adsorbed on the surface of monolayer GeTe. | |
3.3 Diffusion of Al3+/Mg2+/Ca2+ on the monolayer GeTe surface
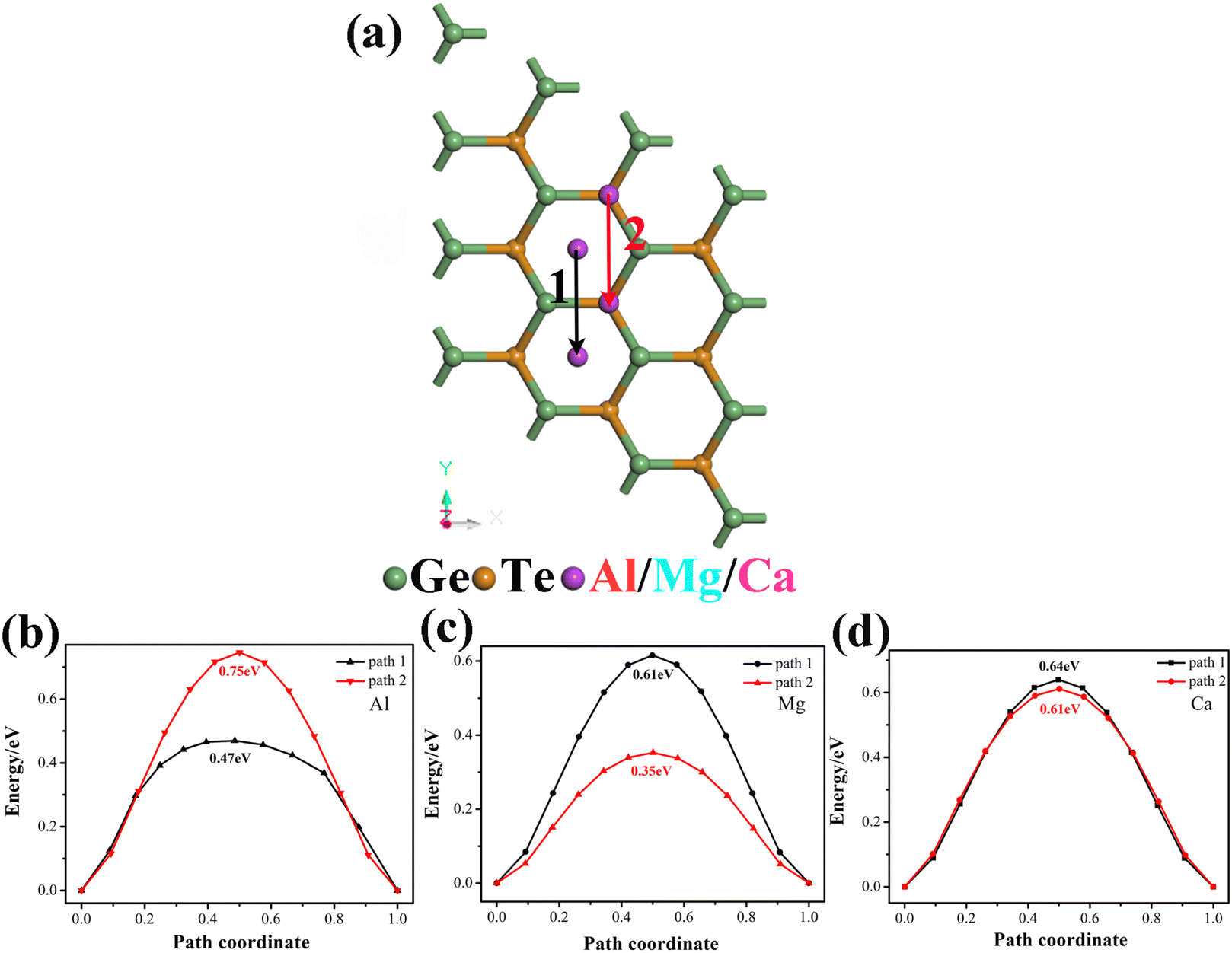
The diffusion rate of ions is another important key factor in the performance of electrode materials, which is closely related to the rate performance of batteries. The diffusion barrier refers to the obstacle to ion diffusion in materials, when ions attempt to diffuse through the lattice, they need to overcome a certain barrier energy to move to a new position, in solid materials, the presence of diffusion barriers can affect the diffusion rate of the material. Fig. 5 shows the diffusion paths and diffusion barriers of Al3+/Mg2+/Ca2+ tested with the LST/QST method. After considering the symmetry and stable adsorption sites of the metal adsorption system, we choose two paths to detect the diffusion barrier of metal ions on monolayer GeTe (Fig. 5a). Path 1 is the migration of metal ions from site C to nearby site C′, while Path 2 is the migration of metal ions from site A to another site A′. For Al3+ (Fig. 5b), the calculated diffusion barriers are 0.75 and 0.47 eV, respectively, obviously, Al3+ preferentially migrates towards path 1. For Mg2+ (Fig. 5c), the diffusion barriers are 0.61 and 0.35 eV, and the same applies to Ca2+ (Fig. 5d), the diffusion barriers are 0.64 and 0.61 eV, and we can see that both Mg2+ and Ca2+ tend to migrate along path 2. Overall, the minimum diffusion barriers of Al3+/Mg2+/Ca2+ on the surface of monolayer GeTe are 0.47, 0.35 and 0.61 eV, respectively. At the same time, we also see that the three diffusion barriers are lower than most previously reported multivalent metal battery materials (Table 2), reflecting the fast ion diffusion ability of monolayer GeTe in AIBs, MIBs and CIBs.
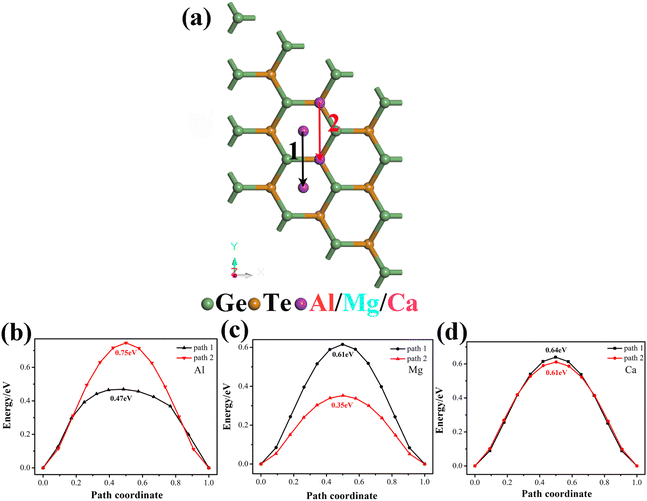 |
| | Fig. 5 (a) Two diffusion paths and the corresponding diffusion barriers of (b) Al3+, (c) Mg2+, and (d) Ca2+ on the monolayer GeTe surface. | |
Table 2 Comparison of diffusion barriers and theoretical capacities of reported two-dimensional materials and monolayer GeTe in aluminum ion batteries (AIBs), magnesium ion batteries (MIBs), and calcium ion batteries (CIBs)
| Material type |
Materials |
AIBs |
MIBs |
CIBs |
| Diffusion barrier (eV) |
Theoretical capacity (mA h g−1) |
Diffusion barrier (eV) |
Theoretical capacity (mA h g−1) |
Diffusion barrier (eV) |
Theoretical capacity (mA h g−1) |
| This work |
GeTe |
0.47 |
624.6 |
0.35 |
446.1 |
0.61 |
446.1 |
| TMDs |
ScS243 |
|
324.29 |
|
324.29 |
|
|
| WS244 |
0.268 |
531.58 |
0.007 |
360.78 |
0.2 |
326.09 |
| VS245 |
0.342 |
78 |
0.416 |
1863 |
|
|
| MoSSe46 |
0.527 |
|
0.37 |
|
|
|
| VS247 |
|
|
|
233 |
|
|
| VSe248 |
|
|
|
|
|
257 |
| MoTe249 |
|
|
|
|
0.56 |
|
| WSe250 |
|
|
|
|
0.2 |
311.63 |
| TiS251 |
|
|
1.2 |
|
|
|
| Heterostructures |
G/MoS252 |
|
|
0.49 |
147.26 |
|
|
| VS2/Ti2CO253 |
|
|
0.34 |
1013 |
|
|
| VS2/Ti2CS253 |
|
|
0.34 |
934 |
|
|
| MoS2/Ti2CS254 |
|
|
0.38 |
121 |
|
|
| MoS2/V2CS254 |
|
|
0.19 |
80 |
|
|
| G/hBN55 |
0.01 |
183 |
|
|
|
|
| h-BP/V2CS256 |
|
|
0.26 |
1219 |
0.11 |
732 |
| MXenes |
V2CS256 |
|
|
0.31 |
753 |
0.12 |
452 |
| V2CSe257 |
|
|
|
|
0.24 |
394.12 |
| Ti2CS258 |
0.47 |
308.74 |
0.46 |
1871.13 |
0.33 |
623.72 |
| Ti2CO259 |
|
552 |
|
570 |
|
487 |
| V2CO259 |
|
540 |
|
550 |
|
480 |
| Nb2CO259 |
|
375 |
|
385 |
|
350 |
| Ti2CO260 |
0.393 |
255 |
0.781 |
1534 |
|
|
| Mo2CS261 |
|
|
0.22 |
800 |
0.26 |
400 |
| Monolayers |
BGe62 |
|
|
0.8 |
1284.6 |
0.52 |
1284.6 |
| GaS63 |
0.034 |
98.76 |
|
|
|
|
| AsP64 |
0.17 |
758.5 |
0.1 |
505.7 |
0.43 |
505.7 |
| BSi65 |
|
|
0.86 |
2749 |
|
|
| Other |
Phosphorene66 |
|
|
0.05 |
|
|
|
| Phosphorene67 |
0.13–0.77 |
1047 |
|
|
|
|
| g-Mg3N268 |
0.937 |
797 |
0.828 |
531 |
1.008 |
1594 |
| g-C3N469 |
|
|
2.25 |
319.2 |
|
|
| TiO270 |
1.53 |
660.62 |
|
|
|
|
Based on the diffusion barrier, the diffusion coefficient of ions can be further estimated:71
| | 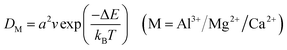 | (4) |
In the formula,
a is the diffusion distance of Al
3+/Mg
2+/Ca
2+ along the corresponding path,
v is the heat induced frequency, usually taken as 10
13 Hz, Δ
E is the diffusion barrier of Al
3+/Mg
2+/Ca
2+ (0.47, 0.35 and 0.61 eV), and
kB and
T represent the Boltzmann constant and system temperature (300 K), respectively. The calculated Al
3+/Mg
2+/Ca
2+ diffusion coefficients of monolayer GeTe at AIBs, MIBs and CIBs are 2.23 × 10
−10, 2.31 × 10
−8 and 1.00 × 10
−12 cm
2 s
−1 respectively, indicating that monolayer GeTe has a fast ion diffusion ability.
3.4 Theoretical capacity and open circuit voltage of monolayer GeTe
Open circuit voltage (OCV) is another important property to evaluate the performance of MuIBs. We consider the reaction formula of monolayer GeTe in a half cell:| | | GeTe + xM + xe− ↔ MxGeTe | (5) |
Before discussing OCV, we should calculate the average adsorption energy (Eave) that can effectively reflect the stability of the adsorption system, Eave is also used to represent the ability of monolayer GeTe to bind Al3+/Mg2+/Ca2+.72 In order to obtain OCV more accurately, it is crucial to evolve Eave by increasing the coverage of adsorbed ions, the calculation formula is as follows:72
| | 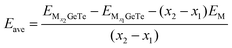 | (6) |
In the formula, M represents Al
3+/Mg
2+/Ca
2+,
x1 and
x2 represent the number of adsorbed Al
3+/Mg
2+/Ca
2+,
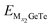
and
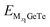
represent the energy of the monolayer GeTe system when the adsorbed ion content is
x2 and
x1 respectively, and
EM represents the energy of a single Al
3+/Mg
2+/Ca
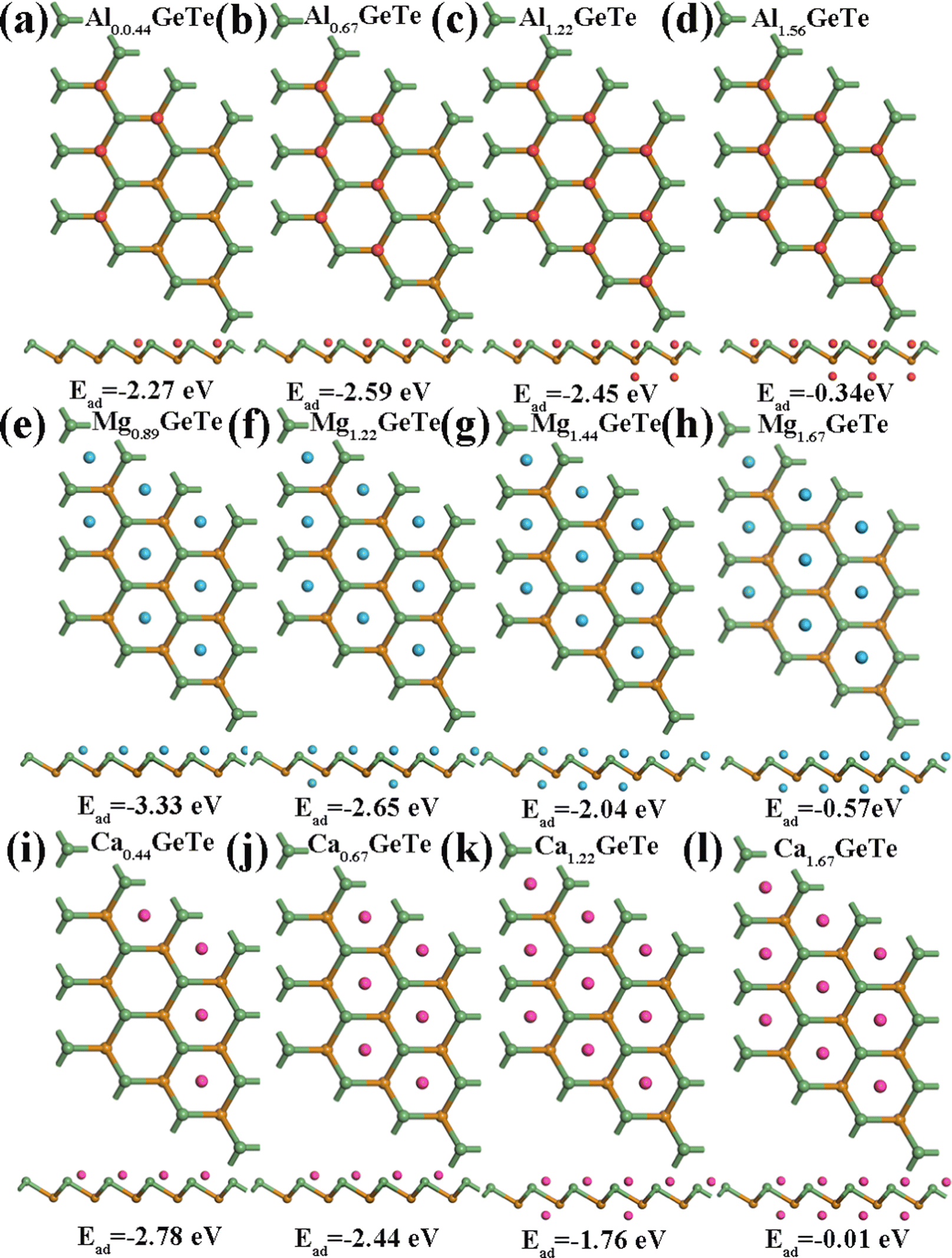
2+ bulk metal. Due to the high symmetry of the monolayer GeTe structure, we consider all double-sided adsorption to achieve higher capacity. According to the conclusion drawn from
Fig. 2, the preferred positions occupied by Al
3+/Mg
2+/Ca
2+ on monolayer GeTe are sites A, C and C, therefore, we sequentially adsorb Al
3+ at site A on the surface of monolayer GeTe, Mg
2+ at site C on the surface of monolayer GeTe, and Ca
2+ at site C on the surface of monolayer GeTe. The typical M
xGeTe (M = Al
3+/Mg
2+/Ca
2+) diagrams are shown in
Fig. 6, corresponding to the average adsorption energy at different metal ion content (
x) in the M
xGeTe system (
Fig. 7a, c and e). As can be seen, with the increase of
x, the average adsorption energy gradually increases due to the shortening of the distance between adjacent metal atoms and the strengthening of their repulsive interactions. Once the adsorption energy increases to 0, it means that no metal ions are adsorbed, thus obtaining the maximum content
x of metal ions in the M
xGeTe system. All average adsorption energies throughout the entire process are negative, thereby preventing the formation of metal dendrites. As the ion content of the adsorption system increases, the trend of OCV variation is plotted in
Fig. 7b, d and f. The OCV formula is defined as follows:
73| | 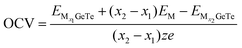 | (7) |
where, M represents Al
3+/Mg
2+/Ca
2+,
x1 and
x2 represent the number of adsorbed Al
3+/Mg
2+/Ca
2+,
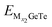
and
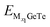
represent the energy of the monolayer GeTe system when the adsorbed ion content is
x2 and
x1 respectively,
z represents the number of metal ions’ valence electrons, and
EM represents the energy of a single Al
3+/Mg
2+/Ca
2+ bulk metal. For the Al
xGeTe system (
Fig. 7b, 0 ≤
x ≤ 1.56), it can be seen that starting from OCV = 0.76 V, as the ion content increases, the OCV of the Al
xGeTe system gradually decreases within the range of 0.76 to 0.11 V, but all are positive, with an average OCV of 0.62 V. For Mg
xGeTe (
Fig. 7d, 0 ≤
x ≤ 1.67) and Ca
xGeTe (
Fig. 7f, 0 ≤
x ≤ 1.67), the OCV ranges are 1.66–0.29 V and 1.39–0.007 V respectively, and decrease significantly with increasing ion content, and the average voltage during the entire discharge process is 0.85 and 0.64 V respectively. The average OCV of the three are within the working voltage range of the commonly used anode material (0.1–1.0 V). Finally, the monolayer GeTe can accommodate 1.56 Al
3+ (Al
1.56GeTe), 1.67 Mg
2+ (Mg
1.67GeTe), and 1.67 Ca
2+ (Ca
1.67GeTe), respectively (
Fig. 7b, d and f). The reasonable OCV and maximum adsorption concentration further demonstrate that monolayer GeTe can serve as an anode material for AIBs, MIBs, and CIBs.
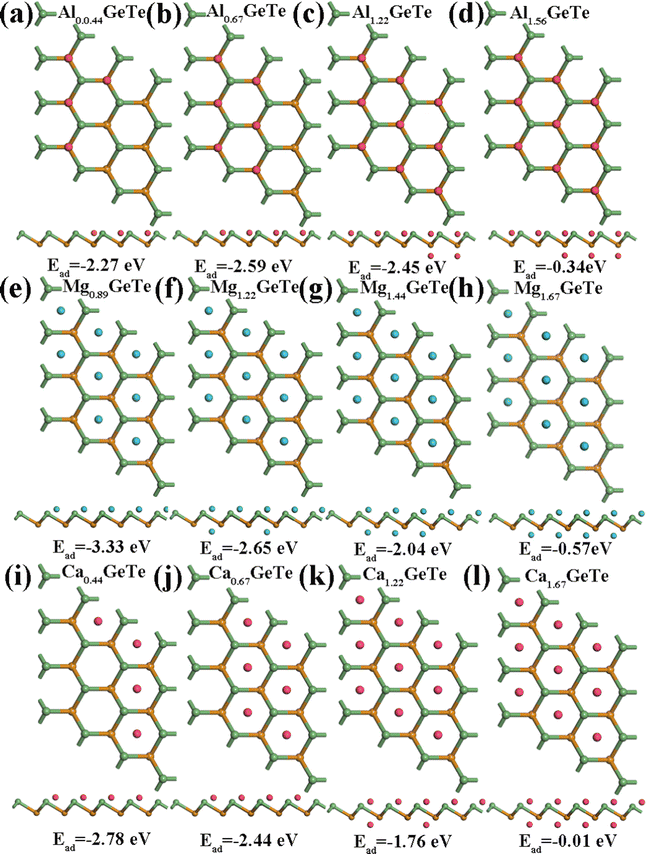 |
| | Fig. 6 Typical MxGeTe (M = Al3+/Mg2+/Ca2+) structural diagram and corresponding average adsorption energy: (a)–(d) are the AlxGeTe diagram, (e)–(h) are the MgxGeTe diagram, and (i)–(l) are the CaxGeTe diagram. | |
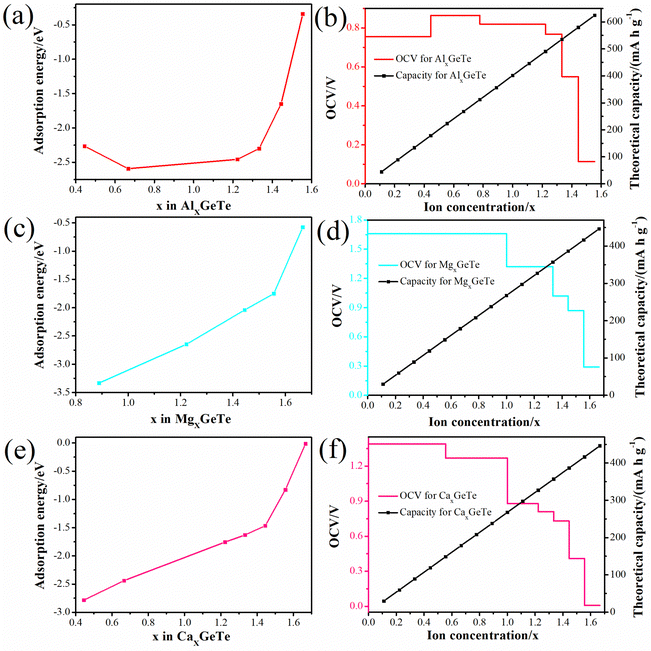 |
| | Fig. 7 The average adsorption energy of monolayer GeTe as a function of (a) Al3+, (c) Mg2+ and (e) Ca2+ content. The OCV and theoretical capacity as a function of (b) Al3+, (d) Mg2+ and (f) Ca2+ content. | |
We determine the maximum theoretical capacity (CM) using the following formula:
| | 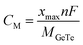 | (8) |
where,
xmax represents the maximum adsorbed ion concentration,
n represents the number of ion valence states (Al
3+/Mg
2+/Ca
2+ are 3, 2 and 2, respectively),
F represents the Faraday constant (26
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
801 mA h mol
−1), and
MGeTe is the molar mass of monolayer GeTe (g mol
−1). Obviously, as the ion concentration increases, the storage capacity of monolayer GeTe on metal ion batteries also increases accordingly. The theoretical capacities calculated according to
eqn (8) are Al
1.56GeTe (624.6 mA h g
−1), Mg
1.67GeTe (446.1 mA h g
−1), and Ca
1.67GeTe (446.1 mA h g
−1) (
Fig. 7b, d and f); it should be pointed out that the capacity of monolayer GeTe is indeed not high, but it exceeds some popular materials reported, including in the field of aluminum ion batteries (ScS
2:
43 324.29, WS
2:
44 531.58, VS
2:
45 78, Ti
2CS
2:
58 308.74, Ti
2CO
2:
59 552, V
2CO
2:
59 540, Nb
2CO
2:
59 375, Ti
2CO
2:
60 255, GaS:
63 98.76 mA h g
−1), magnesium ion batteries (ScS
2:
43 324.29, WS
2:
44 360.78, MoS
2/Ti
2CS
2:
54 121, MoS
2/V
2CS
2:
54 80, Nb
2CO
2:
59 385, g-C
3N
4:
69 319.2 mA h g
−1), and calcium ion batteries (WS
2:
44 326.09, VSe
2:
48 257, WSe
2:
50 311.63, V
2CSe
2:
57 394.12, Nb
2CO
2:
59 350, Mo
2CS
2:
61 400 mA h g
−1), with a higher capacity than commercial graphite (372 mA h g
−1) at the same time.
2 Regardless, our goal is to search for a variety of materials and predict their potential as anode materials for metal ion batteries, providing a new approach for the development of new energy and experimental exploration, and exploring the theoretical capacity of monolayer GeTe in MuIBs can effectively stimulate in-depth research on this graphene-like material family.
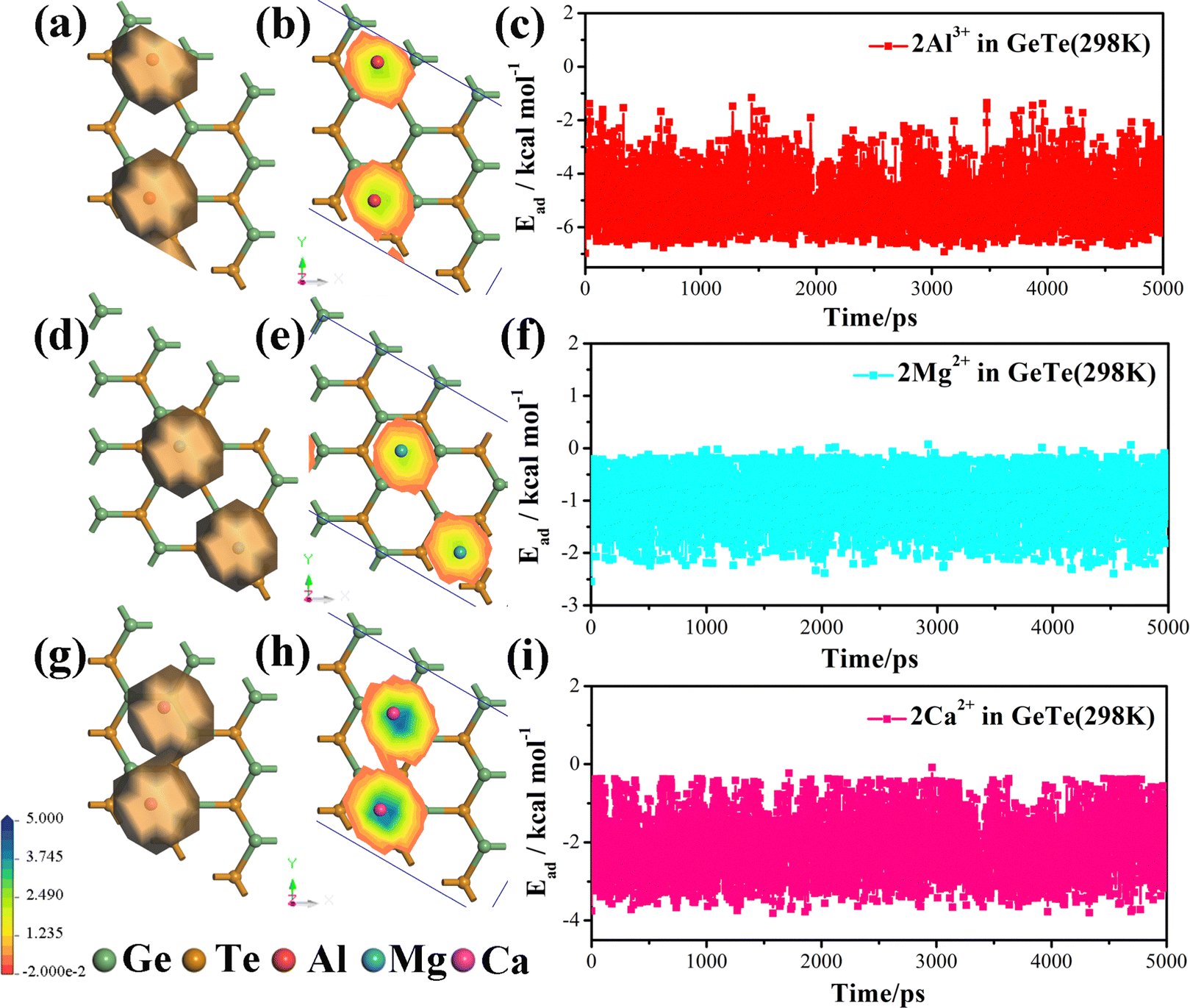
In order to gain a more intuitive understanding of the movement of metal ions, we use molecular dynamics analysis to study the density distribution map of Al3+/Mg2+/Ca2+ on the surface of monolayer GeTe. We construct a model by adsorbing two Al3+/Mg2+/Ca2+ on the surface of monolayer GeTe with the adsorption sites A, C and C, respectively. As shown in Fig. 8, it is evident that Al3+/Mg2+/Ca2+ migrate on the surface of monolayer GeTe and form a local distribution of motion density. The trajectory of ions mainly revolves around the starting position, and the more they deviate from the starting position, the lower the probability of Al3+/Mg2+/Ca2+ being involved. It can be seen that except for slight movement of metal ions, Ge, and Te, there is no significant deformation or bond breaking in the Al3+–GeTe, Mg2+–GeTe and Ca2+–GeTe systems, indicating that the system has good thermal stability at 300 K. To explore whether Al3+, Mg2+ and Ca2+ are stably adsorbed on the surface of monolayer GeTe during the entire movement process, we also calculate the adsorption energy of monolayer GeTe for metal ions in 5000 ps (Fig. 8c, f, and i). It can be seen that the adsorption energies of all three systems are negative, indicating that Al3+/Mg2+/Ca2+ can stably adsorb on the surface of monolayer GeTe during intense movement. In addition, we have also listed the volume expansion values of some common two-dimensional materials, as shown in Table S1 (ESI†). Through comparison, we can see that the volume expansion values of monolayer GeTe in AIBs, MIBs and CIBs are 14.3%, 15.7% and 25.8% respectively. Although they are larger than some other two-dimensional materials such as commercial graphite74 (12%), they are still smaller than Ge75 (≥207%), MoS276 (27%), BSi77 (33%), Si78 (300%), Sn79 (260%) and GeSe280 (200%). Therefore, monolayer GeTe has good thermal stability and lower volume expansion, which provides another supporting condition for monolayer GeTe as an anode material for AIBs, MIBs and CIBs.
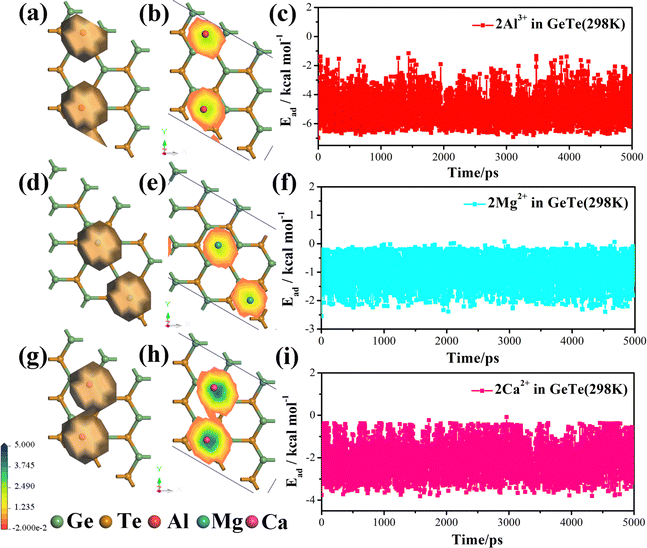 |
| | Fig. 8 Density distribution and adsorption energy at different motion times of (a)–(c) Al3+, (d)–(f) Mg2+ and (g)–(i) Ca2+ on the surface of monolayer GeTe. | |
4. Conclusions
In summary, we investigate the properties of two-dimensional monolayer GeTe as a potential anode for MuIBs. The research results show that monolayer GeTe is beneficial for the stable adsorption of Al3+/Mg2+/Ca2+ (−2.28, −1.56 and −0.20 eV), and the strong interaction between the two is demonstrated through electron density difference and charge transfer. In addition, Al3+, Mg2+ and Ca2+ have low diffusion barriers (0.47, 0.35 and 0.61 eV) on monolayer GeTe, indicating its excellent diffusion ability and fast charge discharge rate during the charging and discharging process. Reasonable open circuit voltages (0.62, 0.85 and 0.64 V) and theoretical specific capacities higher than commercial graphite anode material (624.6, 446.1 and 446.1 mA h g−1) indicate that monolayer GeTe has considerable energy density. Finally, we use molecular dynamics to calculate the adsorption energy and density field of Al3+/Mg2+/Ca2+ during their movement on the surface of monolayer GeTe, and it can be seen that the adsorption energy of the entire process is negative, which means that the adsorption of Al3+/Mg2+/Ca2+ by monolayer GeTe is extremely stable even during long-term ion movement. This proves the strong interaction between the two and the stable adsorption ability of monolayer GeTe once again. It is interesting that although the theoretical capacity (446.1 mA h g−1) in magnesium ion batteries is slightly lower than that in aluminum ion batteries (624.6 mA h g−1), monolayer GeTe has the lowest diffusion barrier (0.35 eV) in magnesium ion batteries and is superior to most reported two-dimensional materials, resulting in a higher diffusion coefficient (2.31 × 10−8 cm2 s−1). The excellent ion diffusion ability is beneficial for providing new ideas for researchers to test the rate performance of monolayer GeTe in experiments in the future. In short, this article reveals that monolayer GeTe can be used as a promising candidate anode material for MuIBs, and provides a new approach for the development and design of other similar structured materials in the field of energy storage in the future.
Author contributions
Junjie Chen: conceptualization, investigation, writing – original draft, formal analysis. Zhiyu Zhou: methodology, formal analysis. Ruidan Zhang: software, formal analysis – review and editing, supervision.
Data availability
The data that support the findings of this study are included within the article and ESI.†
Conflicts of interest
There are no conflicts to declare. No primary research results, software or code have been included and no new data are generated or analysed as part of this review.
Acknowledgements
This work was jointly supported by the National Natural Science Foundation of China (No. 22103013), the Education and Scientific Research Project for Middle-aged and Young Teachers in Fujian Province (No. JAT231194), and the Natural Science Foundation of Fujian Province (No. 2021J01184).
References
- N. Liu, Z. Lu, J. Zhao, M. T. McDowell, H. W. Lee, W. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed.
- L. Wen, F. Li and H. M. Cheng, Adv. Mater., 2016, 28, 4306–4337 CrossRef CAS.
- B. Liu, T. Luo, G. Y. Mu, X. F. Wang, D. Chen and G. Z. Shen, ACS Nano, 2013, 7, 8051–8058 CrossRef CAS.
- K. Sam, J. Mater. Chem. A, 2019, 7, 2922 RSC.
- W. Q. Tang, J. Xuan, H. Z. Wang, S. L. Zhao and H. L. Liu, J. Energy Storage, 2019, 24, 100800 CrossRef.
- M. C. Lin, M. Gong, B. G. Lu, Y. P. Wu, D. Y. Wang, M. Y. Guan, M. Angell, C. X. Chen, J. Yang and B. J. Hwang, Nature, 2015, 520, 324–328 CrossRef CAS PubMed.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS.
- Y. Liang, R. J. Feng, S. Yang, H. Ma, L. Jing and J. Chen, Adv. Mater., 2011, 23, 640–643 CrossRef CAS PubMed.
- H. Chen, F. Guo, Y. J. Liu, T. Q. Huang, B. N. Zheng, N. Ananth, Z. Xu, W. W. Gao and C. Gao, Adv. Mater., 2017, 29, 1605958 CrossRef.
- R. J. Gummow, G. Vamvounis, M. B. Kannan and Y. H. He, Adv. Mater., 2018, 30, 1801702 CrossRef.
- I. Shterenberg, M. Salama, Y. Gofer, E. Levi and D. Aurbach, MRS Bull., 2014, 39, 453–460 CrossRef CAS.
- C. Chowdhury, P. Gain and A. Datta, Phys. Chem. Chem. Phys., 2021, 23, 9466–9475 RSC.
- C. S. Fuller and J. C. Severiens, Phys. Rev., 1954, 96, 21–24 CrossRef CAS.
- C. Yue, Y. J. Yu, Z. G. Wu, X. He, J. Y. Wang, J. T. Li, C. Li, S. T. Wu, J. Li and J. Y. Kang, Nanoscale, 2014, 6, 1817–1822 RSC.
- L. Y. Lim, N. Liu, Y. Cui and M. F. Toney, Chem. Mater., 2014, 26, 3739–3746 CrossRef CAS.
- X. Lu, E. R. Adkins, Y. He, L. Zhong, L. Luo, S. X. Mao, C. M. Wang and B. A. Korgel, Chem. Mater., 2016, 28, 1236–1242 CrossRef CAS.
- Y. Zhang, Q. Zhou, J. Zhu, Q. Yan, S. X. Dou and W. Sun, Adv. Funct. Mater., 2017, 27, 1702317 CrossRef.
- L. Zeng, W. Zeng, Y. Jiang, X. Wei, W. Li, C. Yang, Y. Zhu and Y. Yu, Adv. Energy Mater., 2014, 5, 1401377 CrossRef.
- J. T. Xu, J. M. Ma, Q. H. Fan, S. J. Guo and S. X. Dou, Adv. Mater., 2017, 29, 1606454 CrossRef.
- J. U. Seo, G. K. Seong and C. M. Park, Sci. Rep., 2015, 5, 7969 CrossRef CAS PubMed.
- S. L. Chen, Y. Z. Zhong, J. F. Cai, Z. W. Zhang, F. Gao, S. H. Huo, J. H. Wu, C. Cui, X. J. Tan, G. Q. Liu, D. Fang and J. Jiang, Mater. Today Phys., 2024, 43, 101393 CrossRef CAS.
- Y. W. Tung and M. L. Cohen, Phys. Rev., 1969, 180, 823–826 CrossRef CAS.
- A. I. Lebedev, I. A. Sluchinskaya, V. N. Demin and I. H. Munro, Phase Transitions, 1997, 60, 67–77 CrossRef CAS.
- G. K. Sung and C. M. Park, J. Mater. Chem. A, 2017, 5, 5685–5689 RSC.
- X. H. Liu, Q. T. Ye, R. Z. Yao, B. Chen, W. Liang, Y. S. Liu, Y. P. Liu, D. M. Chen, Y. Q. Wei, D. Li and Y. Chen, Energy Storage Mater., 2023, 63, 103039 CrossRef.
- K. H. Nam, G. K. Sung, J. H. Choi, J. S. Youn, K. J. Jeon and C. M. Park, J. Mater. Chem. A, 2019, 7, 3278–3288 RSC.
- H. Q. Zhao, Y. L. Mao, X. Mao, X. Shi, C. S. Xu, C. X. Wang, S. M. Zhang and D. H. Zhou, Adv. Funct. Mater., 2017, 28, 1704855 CrossRef.
- P. P. Zhang, F. L. Zhao, P. Long, Y. Wang, Y. C. Yue, X. Y. Liu, Y. Y. Feng, R. J. Li, W. P. Hu, Y. Li and W. Feng, Nanoscale, 2018, 10, 15989–15997 RSC.
- T. B. Zeng, D. Feng, Q. M. Peng, Q. Liu, G. C. Xi and G. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 15178–15189 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- T. A. Halgren and W. N. Lipscomb, Chem. Phys. Lett., 1977, 49, 225–232 CrossRef CAS.
- Y. Q. Wei, L. Huang, J. J. Chen, Y. P. Guo, S. Q. Wang, H. Q. Li and T. Y. Zhai, ACS Appl. Mater. Interfaces, 2019, 11, 41374–41382 CrossRef CAS.
- G. X. Ge, Y. W. Zhang, H. X. Yan, J. M. Yang, L. Zhou and X. J. Sui, Appl. Surf. Sci., 2021, 538, 148009 CrossRef CAS.
- L. Guo, C. W. Qi, X. W. Zheng, R. H. Zhang, X. Shen and S. Kaya, RSC Adv., 2017, 7, 29042–29050 RSC.
- M. Jäckle and A. Groß, J. Chem. Phys., 2014, 141, 174710 CrossRef.
- T. Yu, Z. Y. Zhao, L. L. Liu, S. T. Zhang, H. Y. Xu and G. C. Yang, J. Am. Chem. Soc., 2018, 140, 5962–5968 CrossRef CAS PubMed.
- S. Y. Bai, C. Y. Niu, W. Y. Yu, Z. L. Zhu, X. L. Cai and Y. Jia, Nanoscale Res. Lett., 2018, 13, 404 CrossRef PubMed.
- S. K. Wang, C. D. Ren, H. Y. Tian, J. Yu and M. L. Sun, Phys. Chem. Chem. Phys., 2018, 20, 13394–13399 RSC.
- H. Y. Ye, F. F. Hu, H. Y. Tang, L. W. Yang, X. P. Chen, L. G. Wang and G. Q. Zhang, Phys. Chem. Chem. Phys., 2018, 20, 16067–16076 RSC.
- Y. W. Wang, W. Tian, H. J. Zhang and Y. Wang, Phys. Chem. Chem. Phys., 2021, 23, 12288–12295 RSC.
- D. Chakraborty, M. Pandey and P. Johari, Adv. Theory Simul., 2024, 7, 2300486 CrossRef CAS.
- G. R. Vakili-Nezhaad, A. M. Gujarathi, N. Al Rawahi and M. Mohammadi, Mater. Chem. Phys., 2019, 230, 114–121 CrossRef CAS.
- D. S. Wang, Y. H. Liu, X. Meng, Y. J. Wei, Y. Y. Zhao, Q. Pang and G. Chen, J. Mater. Chem. A, 2017, 5, 21370–21377 RSC.
- J. B. Zhang, H. Li, J. Q. Zhou, S. M. Jin and B. B. Chen, Mater. Today Commun., 2022, 32, 103974 CrossRef CAS.
- J. D. Yang, J. X. Wang, X. Y. Dong, L. Zhu, D. W. Hou, W. Zeng and J. F. Wang, Appl. Surf. Sci., 2021, 544, 148775 CrossRef CAS.
- M. Salavati and T. Rabczuluk, Comput. Mater. Sci., 2019, 160, 360–367 CrossRef CAS.
- N. Liu, Y. Y. Feng, X. Li and W. T. Yu, J. Mol. Model., 2024, 30, 119 CrossRef CAS PubMed.
- X. W. Niu and Y. Y. Feng, J. Mol. Model., 2024, 30, 211 CrossRef CAS.
- A. Emly and A. Van der Ven, Inorg. Chem., 2015, 54, 4394–4402 CrossRef CAS.
- J. Q. Qi, Q. Li, M. Y. Huang, J. J. Ni, Y. W. Sui, Q. K. Meng, F. X. Wei, L. Zhu and W. Q. Wei, Colloids Surf., A, 2024, 683, 132998 CrossRef CAS.
- N. Li, Y. R. Li, X. H. Zhu, C. X. Huang and J. Fan, Appl. Surf. Sci., 2021, 543, 148772 CrossRef CAS.
- X. Yuan, Z. H. Chen, B. Huang, Y. P. He and N. G. Zhou, J. Phys. Chem. C, 2021, 125, 10226–10234 CrossRef CAS.
- P. Bhauriyal, G. Bhattacharyya, K. S. Rawat and B. Pathak, J. Phys. Chem. C, 2019, 123, 3959–3967 CrossRef CAS.
- X. Yuan, Z. Y. Zhang, Y. P. He and N. G. Zhou, Phys. Chem. Chem. Phys., 2023, 25, 10011–10021 RSC.
- Y. Z. Wang, Y. H. Ma, Q. F. Zhang, R. Huang, B. L. Gao, Z. W. Li, G. N. Li and F. Liang, Curr. Appl. Phys., 2022, 41, 7–13 CrossRef.
- Y. T. Wang, M. Zhou, L. C. Xu, W. T. Zhao, R. Li, Z. Yang, R. P. Liu and X. Y. Li, J. Power Sources, 2020, 451, 227791 CrossRef CAS.
- Y. Xie, Y. Dall’Agnese, M. Naguib, Y. Gogotsi, M. W. Barsoum, H. L. L. Zhuang and P. R. C. Kent, ACS Nano, 2014, 8, 9606–9615 CrossRef CAS PubMed.
- C. L. Wei, T. M. Fang, X. Tang, K. Jiang and X. M. Liu, Langmuir, 2022, 38, 11732–11742 CrossRef CAS PubMed.
- H. X. Luo, P. Long, J. R. Xiao, X. Q. Dai and Z. Y. Wang, Mater. Today Commun., 2024, 38, 108285 CrossRef CAS.
- M. S. Zyane, H. Rghioui, M. A. Tamerd, A. Achahbar, M. Zanouni and A. Marjaoui, Mater. Today Commun., 2024, 38, 108469 CrossRef CAS.
- X. Y. Zhang, C. Yang, Y. Y. Pan, M. Y. Weng, L. Q. Xu, S. Q. Liu, J. Yang, J. H. Yan, J. Z. Li, B. W. Shi, J. B. Yang, J. X. Zheng, F. Pan and J. Lu, J. Mater. Chem. A, 2019, 7, 14042–14050 RSC.
- J. H. Hao, D. L. Zhang, Z. J. Wang, S. X. Chen, J. H. Xu and Y. F. Wang, Mater. Today Commun., 2024, 38, 108423 CrossRef CAS.
- C. Xiao, X. Q. Tang, J. F. Peng and Y. H. Ding, Appl. Surf. Sci., 2021, 563, 150278 CrossRef CAS.
- A. Sibari, A. El Marjaoui, M. Lakhal, Z. Kerrami, A. Kara, M. Benaissa, A. Ennaoui, M. Hamedoun, A. Benyoussef and O. Mounkachi, Sol. Energy Mater. Sol. Cells, 2018, 180, 253–257 CrossRef CAS.
- J. H. Hao, Z. J. Wang, W. X. Kong, J. Y. Lan, W. B. Li, Y. F. Wang and T. B. Yuan, Surf. Sci., 2023, 728, 122195 CrossRef CAS.
- L. X. Xiong, H. W. Wang, W. Xiong, S. C. Yu and C. Y. Ouyang, RSC Adv., 2019, 9, 27378–27385 RSC.
- J. H. Zhang, G. Liu, H. C. Hu, L. Y. Wu, Q. Wang, X. J. Xin, S. J. Li and P. F. Lu, Appl. Surf. Sci., 2019, 487, 1026–1032 CrossRef CAS.
- W. Q. Tang, J. Xuan, H. Z. Wang, S. L. Zhao and H. L. Liu, J. Energy Storage, 2019, 24, 100800 CrossRef.
- D. H. Seo, Y. U. Park, S. W. Kim, I. Park, R. A. Shakoor and K. Kang, Phys. Rev. B.:Condens. Matter Mater. Phys., 2011, 83, 205127 CrossRef.
- R. Q. Lian, D. S. Wang, Q. F. Yang, D. X. Kan, G. Chen, C. G. Gao and Y. J. Wei, Electrochim. Acta, 2019, 326, 134955 CrossRef CAS.
- X. J. Ye, G. L. Zhu, J. Liu, C. S. Liu and X. H. Yan, J. Phys. Chem. C, 2019, 123, 15777–15786 CrossRef CAS.
- W. J. Zhang, J. Power Sources, 2011, 196, 13–24 CrossRef CAS.
- C. Kim, U. Hwang, S. Lee and Y. K. Han, Nanomaterials, 2023, 13, 2868 CrossRef CAS PubMed.
- M. Mortazavi, C. Wang, J. K. Deng, V. B. Shenoy and N. V. Medhekar, J. Power Sources, 2014, 268, 279–286 CrossRef CAS.
- A. Samad, A. Shafique, U. Schwingenschlögl, Z. W. Ji and G. F. Luo, Chem. Phys. Chem., 2022, 23, e202200041 CrossRef CAS PubMed.
- F. Z. Zhang, Y. Y. Ma, M. M. Jiang, W. Luo and J. P. Yang, Rare Met., 2022, 41, 1276–1283 CrossRef CAS.
- J. B. Cook, E. Detsi, Y. J. Liu, Y. L. Liang, H. Kim, X. Petrissans, B. S. Dunn and S. H. Tolbert, ACS Appl. Mater. Interfaces, 2017, 9, 293–303 CrossRef CAS.
- Q. D. Chen, S. Tang, D. Feng, Y. H. Xie, F. Wu, D. L. Xie, Y. Mei and T. B. A. Zeng, J. Alloys Compd., 2023, 968, 172106 CrossRef CAS.
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Zhiyu
Zhou
a and
Ruidan
Zhang
*b
*a,
Zhiyu
Zhou
a and
Ruidan
Zhang
*b
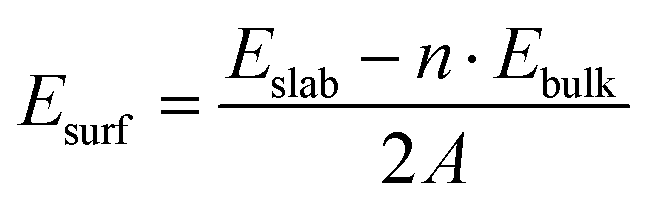
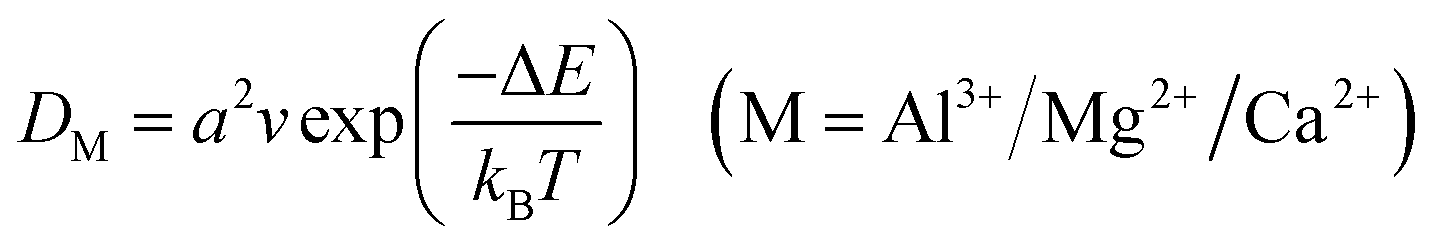
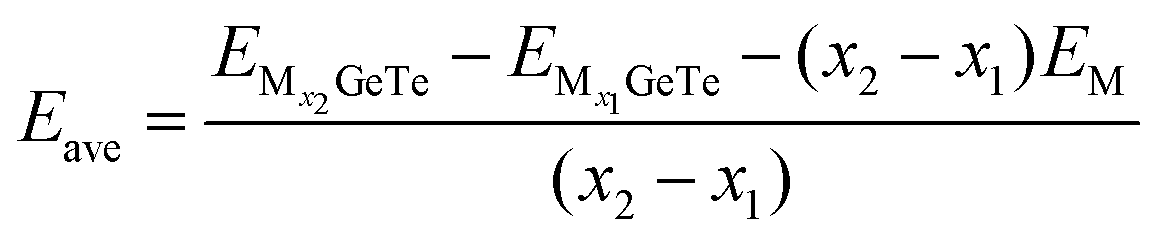
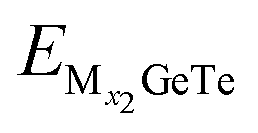 and
and 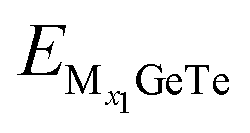 represent the energy of the monolayer GeTe system when the adsorbed ion content is x2 and x1 respectively, and EM represents the energy of a single Al3+/Mg2+/Ca2+ bulk metal. Due to the high symmetry of the monolayer GeTe structure, we consider all double-sided adsorption to achieve higher capacity. According to the conclusion drawn from Fig. 2, the preferred positions occupied by Al3+/Mg2+/Ca2+ on monolayer GeTe are sites A, C and C, therefore, we sequentially adsorb Al3+ at site A on the surface of monolayer GeTe, Mg2+ at site C on the surface of monolayer GeTe, and Ca2+ at site C on the surface of monolayer GeTe. The typical MxGeTe (M = Al3+/Mg2+/Ca2+) diagrams are shown in Fig. 6, corresponding to the average adsorption energy at different metal ion content (x) in the MxGeTe system (Fig. 7a, c and e). As can be seen, with the increase of x, the average adsorption energy gradually increases due to the shortening of the distance between adjacent metal atoms and the strengthening of their repulsive interactions. Once the adsorption energy increases to 0, it means that no metal ions are adsorbed, thus obtaining the maximum content x of metal ions in the MxGeTe system. All average adsorption energies throughout the entire process are negative, thereby preventing the formation of metal dendrites. As the ion content of the adsorption system increases, the trend of OCV variation is plotted in Fig. 7b, d and f. The OCV formula is defined as follows:73
represent the energy of the monolayer GeTe system when the adsorbed ion content is x2 and x1 respectively, and EM represents the energy of a single Al3+/Mg2+/Ca2+ bulk metal. Due to the high symmetry of the monolayer GeTe structure, we consider all double-sided adsorption to achieve higher capacity. According to the conclusion drawn from Fig. 2, the preferred positions occupied by Al3+/Mg2+/Ca2+ on monolayer GeTe are sites A, C and C, therefore, we sequentially adsorb Al3+ at site A on the surface of monolayer GeTe, Mg2+ at site C on the surface of monolayer GeTe, and Ca2+ at site C on the surface of monolayer GeTe. The typical MxGeTe (M = Al3+/Mg2+/Ca2+) diagrams are shown in Fig. 6, corresponding to the average adsorption energy at different metal ion content (x) in the MxGeTe system (Fig. 7a, c and e). As can be seen, with the increase of x, the average adsorption energy gradually increases due to the shortening of the distance between adjacent metal atoms and the strengthening of their repulsive interactions. Once the adsorption energy increases to 0, it means that no metal ions are adsorbed, thus obtaining the maximum content x of metal ions in the MxGeTe system. All average adsorption energies throughout the entire process are negative, thereby preventing the formation of metal dendrites. As the ion content of the adsorption system increases, the trend of OCV variation is plotted in Fig. 7b, d and f. The OCV formula is defined as follows:73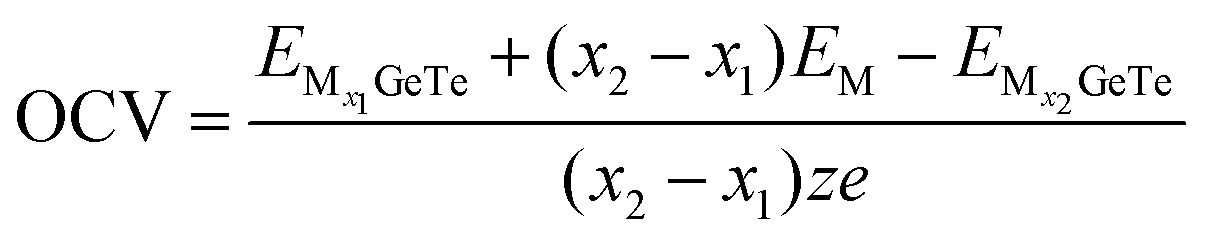
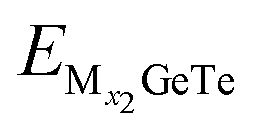 and
and 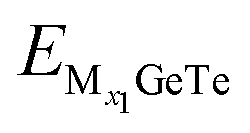 represent the energy of the monolayer GeTe system when the adsorbed ion content is x2 and x1 respectively, z represents the number of metal ions’ valence electrons, and EM represents the energy of a single Al3+/Mg2+/Ca2+ bulk metal. For the AlxGeTe system (Fig. 7b, 0 ≤ x ≤ 1.56), it can be seen that starting from OCV = 0.76 V, as the ion content increases, the OCV of the AlxGeTe system gradually decreases within the range of 0.76 to 0.11 V, but all are positive, with an average OCV of 0.62 V. For MgxGeTe (Fig. 7d, 0 ≤ x ≤ 1.67) and CaxGeTe (Fig. 7f, 0 ≤ x ≤ 1.67), the OCV ranges are 1.66–0.29 V and 1.39–0.007 V respectively, and decrease significantly with increasing ion content, and the average voltage during the entire discharge process is 0.85 and 0.64 V respectively. The average OCV of the three are within the working voltage range of the commonly used anode material (0.1–1.0 V). Finally, the monolayer GeTe can accommodate 1.56 Al3+ (Al1.56GeTe), 1.67 Mg2+ (Mg1.67GeTe), and 1.67 Ca2+ (Ca1.67GeTe), respectively (Fig. 7b, d and f). The reasonable OCV and maximum adsorption concentration further demonstrate that monolayer GeTe can serve as an anode material for AIBs, MIBs, and CIBs.
represent the energy of the monolayer GeTe system when the adsorbed ion content is x2 and x1 respectively, z represents the number of metal ions’ valence electrons, and EM represents the energy of a single Al3+/Mg2+/Ca2+ bulk metal. For the AlxGeTe system (Fig. 7b, 0 ≤ x ≤ 1.56), it can be seen that starting from OCV = 0.76 V, as the ion content increases, the OCV of the AlxGeTe system gradually decreases within the range of 0.76 to 0.11 V, but all are positive, with an average OCV of 0.62 V. For MgxGeTe (Fig. 7d, 0 ≤ x ≤ 1.67) and CaxGeTe (Fig. 7f, 0 ≤ x ≤ 1.67), the OCV ranges are 1.66–0.29 V and 1.39–0.007 V respectively, and decrease significantly with increasing ion content, and the average voltage during the entire discharge process is 0.85 and 0.64 V respectively. The average OCV of the three are within the working voltage range of the commonly used anode material (0.1–1.0 V). Finally, the monolayer GeTe can accommodate 1.56 Al3+ (Al1.56GeTe), 1.67 Mg2+ (Mg1.67GeTe), and 1.67 Ca2+ (Ca1.67GeTe), respectively (Fig. 7b, d and f). The reasonable OCV and maximum adsorption concentration further demonstrate that monolayer GeTe can serve as an anode material for AIBs, MIBs, and CIBs.