
The effect of acceptor and donor doping on the electronic properties of the half-Heusler TiNiSn
Ronit
Eshel
 *a,
David
Fuks
a,
Yaniv
Gelbstein
a and
Daniel
Rabin
ab
*a,
David
Fuks
a,
Yaniv
Gelbstein
a and
Daniel
Rabin
ab
aDepartment of Materials Engineering, Ben-Gurion University of the Negev, Beer Sheva, 84105, Israel. E-mail: ronit.eshel1@gmail.com
bNRCN, P.O. Box 9001 Beer-Sheva 84190, Israel
First published on 28th November 2024
Abstract
Optimizing the electronic transport properties of thermoelectric compounds is commonly achieved by either donor or acceptor atom doping to increase the conduction of the appropriate carrier type, electrons or holes, respectively. Enhancing carrier mobility and carrier concentration will both lead to optimized electronic properties. In this work the effect of various dopants on the electronic properties of TiNiSn was explored, by modeling the doping of an ideal compound on the Ti-sublattice with acceptor or donor elements. Using ab initio DFT calculations and a set of analytical expressions of transport properties, the temperature dependencies of the electronic properties were calculated, to examine possible n-type or p-type dopants to be used in further experimental studies.
1. Introduction
Climate change challenges are drawing further attention to renewable energy solutions. One of the key strategies is waste heat recovery. The demand for alternative energy technologies to reduce the dependence on fossil fuels leads to new research frontiers, including energy harvesting via the direct recovery of waste heat and its conversion into useful electrical energy.1 Nowadays about 50% of industrial energy is wasted as heat.2 Therefore, extensive research is conducted on thermoelectric (TE) materials to obtain an optimized solution for converting heat into electricity, creating an opportunity as a renewable energy. The basis of transforming heat to energy lays in the ability to exploit a temperature gradient to electrical power. TE materials' performance is widely characterized using the TE figure of merit, ZT = α2σT/k, where α is the Seebeck coefficient, σ is the electrical conductivity, k is the thermal conductivity, and T is the temperature.3 The power-factor of a material, PF = α2σ,4 which refers to the product of the electrical conductivity and Seebeck coefficient, measures its ability to extract electrical power from temperature differences.4,5 In the ideal case, the Seebeck coefficient is linked to (i) the relative position of Fermi energy (EF) with respect to the top of the valence band (VB) or to the bottom of the conduction band (CB) in the density of states (DOS) and determines the sign of the Seebeck coefficient, and to (ii) the carrier concentration that will vary with temperature. Lv et al.6 describe the Seebeck coefficient, electrical conductivity and PF variation with the change in carrier concentration. As suggested, the increase in carrier concentration derives an increase in conductivity along with a decrease in the Seebeck coefficient. Also, it is suggested that doping is an effective way to improve the thermoelectric performance when the optimal carrier concentration is achieved.6The PF is useful to determine the optimum electronic properties, since α and σ are parameters most strongly dependent on the carrier concentration, which is effectively demonstrated by the energy levels located at the band edge, known as the effective density of states (DOS).4 The other quantity that is involved in the definition of the figure of merit (ZT) is the thermal conductivity, k, which is less dependent on the concentration of the charge carriers since it is often dominated by the lattice contribution.4 Thus, the carrier concentration that yields the maximum PF for a given material is usually close to that which gives the highest figure of merit.4 The thermopower, or Seebeck coefficient (α), can be thought of as the energy per carrier over temperature. To gain a more effective thermopower, it is necessary to dope the semiconductors with either donor or acceptor states to allow extrinsic conduction of the appropriate carrier type, electrons or holes, respectively.1 Optimal thermoelectric generator (TEG) configuration requires n-type node and p-type node with suitable thermal coefficient, to reduce the risk of thermal expansion differential in the device.7 Recent advancements in TE materials suggested the manipulation of the electronic band structure for enhancing the Seebeck coefficient.5
By the end of the 1980's a new class of intermetallic compounds XNiSn (X = Ti, Zr, Hf) was shown to demonstrate unusual transport properties.8 Half-Heusler (HH) compounds are ternary intermetallic compounds with a typical XYZ formula, and a structure composed of a combination of zinc blende (YZ) and rock-salt (XZ), with the F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m (#216) cubic space group.9 HH alloys are promising TE materials for mid-to-high temperature power generation applications, but a mechanically robust TE module requires both n- and p-type nodes with comparable thermal expansion coefficients. It is highly desirable to have efficient p-type forms of HH alloys to accompany their already very good n-type structures. HH were found to exhibit a semiconducting behavior with absolute Seebeck coefficients as large as 200 to 400 µV K−1 at room temperature,10 and allow easy doping of either impurity or parent elements, improving their electronic properties.11 At present, n-type half-Heusler alloys are based on TiNiSn, and p-type half-Heusler alloys are based on TiCoSb, and so finding adequate p-type TiNiSn compound is valuable. Specifically, TiNiSn is a HH compound, well known for its high thermal stability, semiconductor-like electrical properties, as well as cost-effectiveness,12 making it a viable candidate for engineering of electronic properties, to create n-type and p-type semiconductors with enhanced electrical conductivity. Several methods have been previously used such as isoelectronic substitution of the Ti-atom by heavy atoms (Zr,Hf) to create point defects; or doping with elements rich or deficient in electrons at the Ti/Ni/Sn sub-lattices to increase the concentration of charge carriers (electrons or holes).2,13 Doping of the TiNiSn ternary HH compound may improve the electronic properties by controlling the type of charge carriers, the shape of DOS in the vicinity of the Fermi energy, EF, and engineering of the band gap (Eg). While TiNiSn is well known as an efficient n-type semiconductor, a p-type HH compatible material, is scarce.14 It was previously demonstrated that defects in TiNiSn, such as exchange anti-site defects, Schottky defects and others, may lead to a decrease of the bandgap, simultaneously stimulating a p-type conductivity in this material.15 Another way of achieving a more effective thermopower and increasing the PF is by doping the Ti-sublattice with acceptor or donor elements, thus engineering the band structure and creating a higher DOS near Fermi level.
3m (#216) cubic space group.9 HH alloys are promising TE materials for mid-to-high temperature power generation applications, but a mechanically robust TE module requires both n- and p-type nodes with comparable thermal expansion coefficients. It is highly desirable to have efficient p-type forms of HH alloys to accompany their already very good n-type structures. HH were found to exhibit a semiconducting behavior with absolute Seebeck coefficients as large as 200 to 400 µV K−1 at room temperature,10 and allow easy doping of either impurity or parent elements, improving their electronic properties.11 At present, n-type half-Heusler alloys are based on TiNiSn, and p-type half-Heusler alloys are based on TiCoSb, and so finding adequate p-type TiNiSn compound is valuable. Specifically, TiNiSn is a HH compound, well known for its high thermal stability, semiconductor-like electrical properties, as well as cost-effectiveness,12 making it a viable candidate for engineering of electronic properties, to create n-type and p-type semiconductors with enhanced electrical conductivity. Several methods have been previously used such as isoelectronic substitution of the Ti-atom by heavy atoms (Zr,Hf) to create point defects; or doping with elements rich or deficient in electrons at the Ti/Ni/Sn sub-lattices to increase the concentration of charge carriers (electrons or holes).2,13 Doping of the TiNiSn ternary HH compound may improve the electronic properties by controlling the type of charge carriers, the shape of DOS in the vicinity of the Fermi energy, EF, and engineering of the band gap (Eg). While TiNiSn is well known as an efficient n-type semiconductor, a p-type HH compatible material, is scarce.14 It was previously demonstrated that defects in TiNiSn, such as exchange anti-site defects, Schottky defects and others, may lead to a decrease of the bandgap, simultaneously stimulating a p-type conductivity in this material.15 Another way of achieving a more effective thermopower and increasing the PF is by doping the Ti-sublattice with acceptor or donor elements, thus engineering the band structure and creating a higher DOS near Fermi level.
Impurity doping introduces donor or acceptor elements into the pure system and is expected to change the carriers' concentration (electrons or holes respectively), which may impact the electronic properties of the host material. Donor impurities include elements with a higher valence-electron (VE) than the parent atom,16 creating donor levels below the CB and shift Fermi level higher than in the host compound, and increasing carrier density. Introduction of acceptor impurities will result in a lower VE count, and empty states will be created just above the VB, where electrons from the VB will populate the acceptor states, exciting holes as the charge carriers.16
DOS plot indicates the distribution of states of electrons as a function of energy and can be calculated using density functional theory (DFT).17 The DOS plot may describe the calculated local DOS (LDOS) for each of the elements in the compound or the partial DOS (PDOS) for each of the orbitals. There are several factors that can influence the electronic properties of the HH compound, namely the crystallographic sites (X/Y/Z) and the orbital characters (s/p/d/f), but still only several of them contribute meaningfully to the valence states: most of the variation in an orbital character is accounted for by the X-d, Y-d, and Z-p components alone.18
Such impurity induced scheme in TiNiSn is one of the ways used for engineering the compound to attain a p-type transport character or optimize the n-type material. An important question is which elements can be used to dope the X-sublattice in TiNiSn ternary HH compounds, to optimize the PF of the doped compound, by controlling the type of charge carriers, shape of the DOS in the vicinity of the Fermi energy, and engineering of the band gap. We further explore what is the contribution of the Ti-atom and its corresponding d-states alongside the substitutional atom doping element, to the DOS, and to the electronic transport properties. This research offers a computational methodology to examine various doping elements, based on ab initio DFT calculations and an analytical model.19 Our novelty is in applying first-principles calculations to investigate the contribution of doping elements on the X-atom positions, their respective d-states and their specific role in enhancing the electronic transport properties, by displaying the fine features of the DOS in the vicinity of the Fermi energy. For TiNiSn, this investigation of impurity-induced control by acceptor or donor elements is still not well systematically explored.
2. Methodology
This work uses ab initio DFT calculations integrated with an analytical electronic transport model19 to calculate the electronic properties of a TiNiSn-based material through simulation of a doped Ti-sublattice. We used 4-stages in our work: (i) selection of 6 acceptor/donor elements; (ii) DOS calculation for a 96-atom supercell, using ab initio DFT, replacing one Ti-atom with one atom from the list of doping elements; (iii) calculation of Seebeck coefficient (α) and electrical conductivity (σ), using an analytical electronic transport model, previously reported and demonstrated experimentally,15,19,20 to determine the PF; (iv) investigation of DOS plots from DFT, analyzing the d-states in Ti-atom alongside dopant X-atom.2.1 Acceptor and donor elements selection
An ideal thermoelectric couple should have nearly identical composition for the two legs, allowing no significant mismatch in n- and p-type properties, such as: mechanical, thermal fatigue or thermal expansion.21 In this work we focus on TiNiSn compound as the basis for the substitution of elements on the Ti-sublattice, which may affect the electronic properties, by introducing states and increasing the charge carrier's concentration, thus contributing to the electrical conductivity and Seebeck coefficient.22 To investigate the doping of the Ti-sublattice by substitution of acceptor or donor elements, and their effect on the electronic properties, elements were chosen following the criteria: (i) the number of valence electrons compared with Ti, to create electron or hole surplus in the compound; (ii) the electron configuration, specifically electrons in d-orbital; (iii) and previous experimental results using these elements as acceptors or donors in the X-sublattice – all showed improved electronic properties compared to ideal TiNiSn, as described below. Accordingly, the following elements were chosen: three acceptor elements, with one less valence electron than Ti: Sc,13,23 Al14 and Y;24 and three donor elements: Ta,25 V26,27 with one more valence electron than Ti; and Mn with three more VE,2,28,29 All are listed in Table 1.| Element | Group | Donor/acceptor | Atomic number | VE | Electron configuration |
|---|---|---|---|---|---|
| Ti | 4 | N | 22 | 4 | [Ar] 3d2 4s2 |
| Al | 13 | A | 13 | 3 | [Ne] 3s2 3p1 |
| Sc | 3 | A | 21 | 3 | [Ar] 3d1 4s2 |
| Y | 3 | A | 39 | 3 | [Kr] 4d1 5s2 |
| Ta | 5 | D | 73 | 5 | [Xe] 4f14 5d3 6s2 |
| V | 5 | D | 23 | 5 | [Ar] 3d3 4s2 |
| Mn | 7 | D | 25 | 7 | [Ar] 3d5 4s2 |
Experimental results for acceptor elements, Sc,13,23 Al14 and Y24 were previously reported by Kaller et al.13 for substitution of Ti by Sc, who indicated that Sc acts as an electron acceptor, enabling generation of p-type TiNiSn-rich HH compound, and is dominated by chemical disorder. Horyn et al.23 also conducted experiments with modulated Sc doping in (Ti,Sc)NiSn and (Zr,Sc)NiSn and indicated that higher density of states at the Fermi energy contributed by the Sc substitution leads to high positive Seebeck coefficients in the solid solution and may be attractive materials for thermoelectric applications as p-type nodes. Similar results were reported by Rabin et al.14 in research conducted on Al solubility in TiNiSn, and clearly states that the substitution of Ti by Al results in a p-type conductivity. Substitution of X-atom by Y in the compound (Hf, Zr, Y)Ni(Sn, Sb) was reported by Zhou et al.24 and indicated that The Y-doping substantially increased the Seebeck coefficient because of the introduction of hole carriers, in the temperature range of 300–900 K.
The compounds with the donor elements Ta,25 V26,27 and Mn2,28,29 also were reported with improved TE properties. The substitution of Ta in the Ti-lattice was investigated by Karati et al.25 that demonstrated a Ta-doped compound with conductivity increasing as temperature arise. Stadnyk et al.26 investigated the TE power factor of alloys with V doping the Ti-site and found that the power factor of the solid solution markedly exceeds that of the undoped ternary compound. To compare the effect of higher VE count, we chose Mn with three more VE than Ti, in Berry's research.2,29 Modulation doping of n-type half-metallic MnNiSb was carried out into HH TiNiSn compound. The Seebeck coefficient was found to increase in comparison with pure TiNiSn, and the transport properties confirmed a crossover from metallic to semiconducting behavior caused by the defects, and possibly enhanced by other factors such as the Fermi energy level, density of states, and temperature gradient, affecting the Seebeck coefficient.2,29
2.2 Density functional theory (DFT)
In the context of computational materials science, ab initio DFT calculations allow the prediction and calculation of material behavior based on quantum mechanical considerations, without requiring fundamental material properties.4In this study the density functional theory, DFT, was applied to a HH unit cell (space group #216) with impurities. In the DFT calculations the augmented plane wave plus local orbitals (APW + lo) method was used to solve the Kohn–Sham equations of density functional theory, as implemented in the WIEN2k code (Version 21.1).30,31 In this code, the APW + lo method considers all electrons (core and valence) self-consistently in a full-potential treatment.31 The exchange–correlation potential was calculated within the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA).31 The calculations parameters used in this work are similar to those previously reported for (Ti,Al)NiSn and (Ti,Sc)NiSn.14 Therefore, based on our experience, a similarly high degree of reliability of the obtained results is currently expected. The muffin-tin radii (Rmt) were taken to be equal to 2.0 bohr for all atoms. The cutoff parameter RmtKmax = 7 was taken for the basis-set size at each point, where Kmax presents the magnitude of the largest K vector in the wave function expansion. To represent the valence states, a maximal l, lmax, equal to 10 was taken in the expansion of the radial wave functions inside the atomic sphere. The cut-off energy, separating core and valence states, was chosen to be −7.0 Ryd for all atoms, to minimize the leaking of the electron core states into the interstitial region.
A cubic supercell containing 96 atoms for TiNiSn with a HH crystal structure (space group #216) with impurities was used, with a k-mesh of 3 × 3 × 3 points in the irreducible wedge of the Brillouin zone (BZ). Using ab initio DFT, DOS calculation for a 96-atom supercell, replacing one Ti-atom with one atom from the doping elements was carried out.
Fig. 1 describes the doped compound structure. Energy parameters were calculated for doped compounds of (Ti0.97X0.03)NiSn alloy (X = Al, Sc, Y, Mn, Ta, V).
 | ||
| Fig. 1 96-atom supercell, replacing one Ti-atom (blue) with one atom of doping element (pink). | ||
2.3 Transport properties calculation procedure
According to quantum theory, the probability that an electron state of energy will be occupied is given by the Fermi distribution function. The energy, EF, at which the Fermi distribution function is equal to 1/2 is known as the ‘Fermi level’. At T = 0, the number of electrons can be found by integrating the DOS up to Fermi energy; at T ≠ 0, the number of electrons can be found by integrating the DOS times the Fermi–Dirac distribution, that addresses the chemical potential change with the change in T.32 When the Fermi level is close to the conduction band or valence band edge, some electrons or holes respectively, will be able to contribute to the conduction process, making it an n-type or p-type, semiconductor. If the energy gap (Eg) between the conduction band (EC) and the valence band (EV) is small enough for this to occur, the material is an intrinsic semiconductor. Adding donor or acceptor impurities may induce conduction by either electrons in the conduction band, or holes in the valence band; with this extrinsic conduction, the Fermi level is close to the edge of either the conduction band or valence band.4 The Fermi energy (EF) is only defined at absolute zero, while the Fermi level is defined for any temperature, and describes the energy difference including kinetic energy and potential energy.4,32 Using the ab initio DFT analysis, (EV − EF) values for p-type material and (EF − EC) values for n-type material were directly calculated. Further, a set of analytical expressions for the transport properties was applied to calculate the temperature dependencies of the electronic properties, to evaluate possible n-type or p-type compounds to be used in further experimental studies.A procedure for calculations was used to derive the transport properties of the investigated TiNiSn compounds based on the DFT evaluated EF locations, in relation to EV or EC, where EV is the top of the valence band, and EC is the bottom of the conduction band. The values gathered in Table 2 from DFT at absolute zero, are used in the reduced Fermi energy expression (η) defined in eqn (1), and changes with temperature, whereas (EV − EF) is used for p-type semiconductor (as used in eqn (1)); and (EF − EC) is used for n-type semiconductor. η is then used in Fermi integral in eqn (2) in which ξ is the kinetic energy of a charge carrier, changing from 0 to ∞, and fo is the Fermi distribution function, as in eqn (3). The Fermi integral is solved for (n = 1/2), known as the ‘Fermi level’, as in eqn (4), to determine F1/2 and further calculate the electrical conductivity, σ; and then solved for (n = 3/2 + r) and (n = 1/2 + r), here r describes the carriers scattering parameter (r = −1/2 for the case of a predominant acoustic phonons scattering, such as in TiNiSn4,33), to determine F3/2+r and F1/2+r and further calculate Seebeck coefficient, α, as described below.19
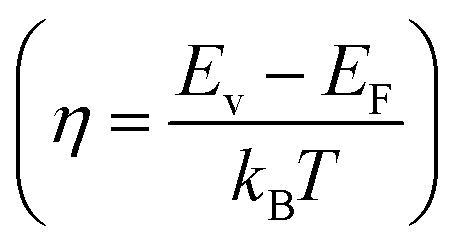 | (1) |
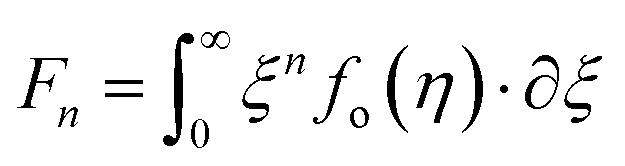 | (2) |
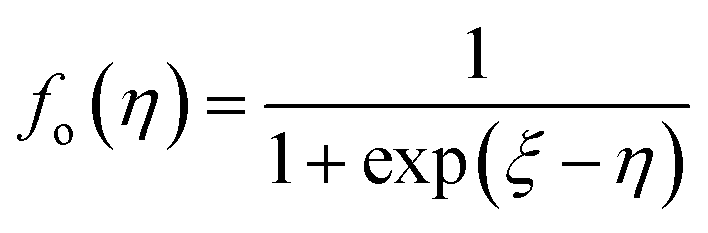 | (3) |
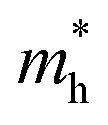 -the hole effective mass, taken as 6 mo for TiNiSn as was previously reported by Zilber et al.;33 and carrier mobility, μ, taken as 11 cm2 V−1 s−1, in agreement with Ren et al.34
-the hole effective mass, taken as 6 mo for TiNiSn as was previously reported by Zilber et al.;33 and carrier mobility, μ, taken as 11 cm2 V−1 s−1, in agreement with Ren et al.34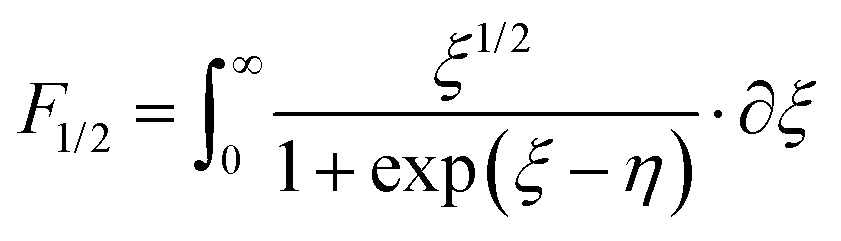 | (4) |
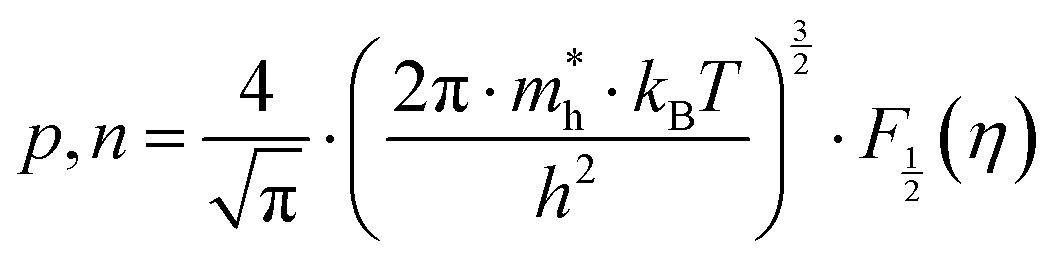 | (5) |
| σ = p·e·µ | (6) |
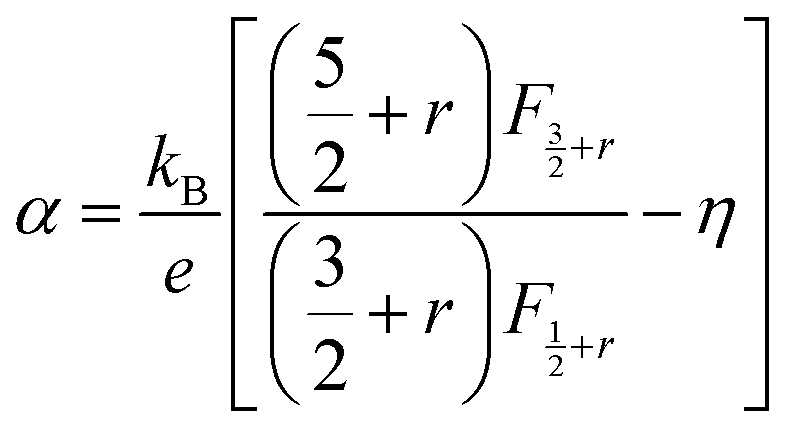 | (7) |
| PF = α2σ. | (8) |
| Compound | Type | DFT calculation of total DOS, for ideal and doped compounds | DFT calculation of total DOS of d-states, for Ti and X | ||
|---|---|---|---|---|---|
| E g (eV) | η, p: Ev − EF (eV), n: EF − EC (eV) | Ti d-states Eg (eV) | X d-states Eg (eV) | ||
| Ideal TiNiSn | p | 0.43 | 0.04 | 0.44 | |
| (Ti0.97Al0.03)NiSn | p | 0.40 | 0.08 | 0.46 | 0.64 |
| (Ti0.97Sc0.03)NiSn | p | 0.35 | 0.14 | 0.46 | 0.49 |
| (Ti0.97Y0.03)NiSn | p | 0.57 | 0.06 | 0.49 | 0.68 |
| (Ti0.97Ta0.03)NiSn | n | 0.43 | 0.14 | 0.44 | 0.49 |
| (Ti0.97V0.03)NiSn | n | 0.41 | 0.13 | 0.44 | 0.38 |
| (Ti0.97Mn0.03)NiSn | n | 0.35 | 0.12 | 0.41 | 0.30 |
3. Results and discussion
Hereby, we will discuss the results of the ab initio DFT calculation, analyze the contribution of acceptor and donor elements as impurities in the form of (Ti0.97X0.03)NiSn, to the parent compound in terms of DOS for HH, and the contribution of specific electron states to the change in DOS, when the substitution occurs on the Ti-sublattice. Table 2 presents the results of calculations for the substitution of one Ti-atom with the doping element in a 96 atom supercell, simulating the composition (Ti0.97X0.03)NiSn.3.1 Seebeck coefficient, electrical conductivity and PF temperature dependence
Using the calculation procedure described above, the calculated temperature dependent electrical conductivity presented in Fig. 2 and Seebeck coefficient in Fig. 3, clearly states an increase in the charge carriers' concentration as compared with the ideal TiNiSn in all doped compounds for all temperatures in range, as shown in both n- and p-type compounds. The positive values of the Seebeck coefficient indicate a p-type conduction, while the negative values indicate an n-type conduction, indicating the acceptor or donor affiliation of the elements.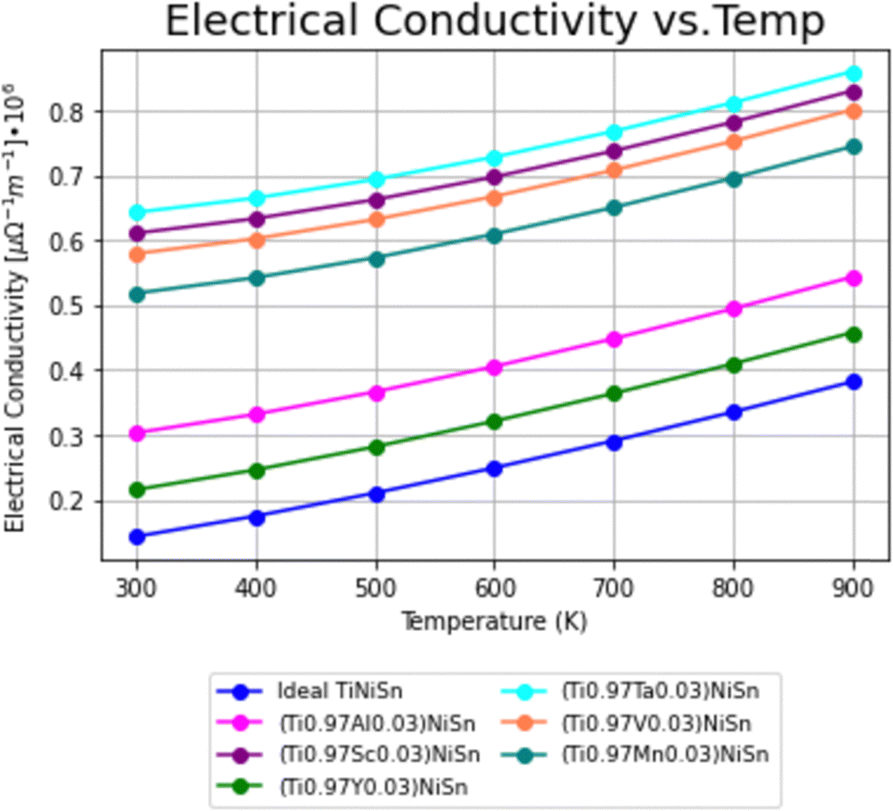 | ||
| Fig. 2 Temperature dependence of electrical conductivity (σ) for ideal TiNiSn and for doped compounds. | ||
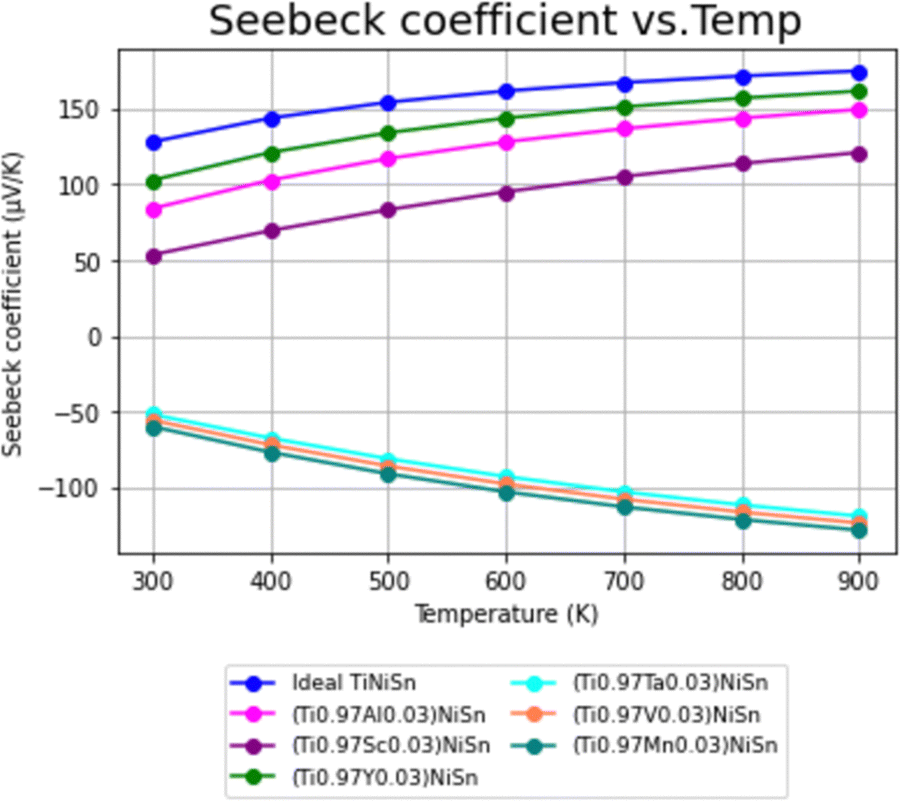 | ||
| Fig. 3 Temperature dependence of Seebeck coefficient (α) for ideal TiNiSn and for doped compounds. | ||
The PF's temperature dependence, calculated from the combination of electrical conductivity and Seebeck coefficient, is described in Fig. 4. PF is slightly favorable for ideal TiNiSn in low temperatures, but the doped compounds are showing slightly higher slope as temperatures increase above 550 K (Fig. 5).
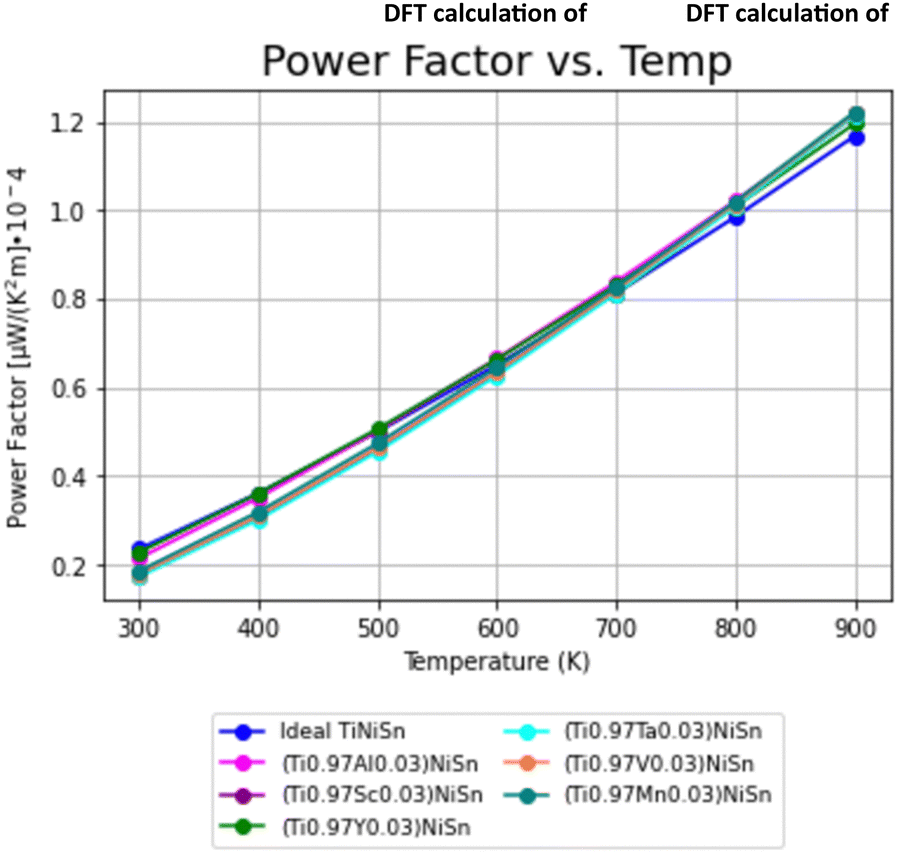 | ||
| Fig. 4 Temperature dependence of power factor (PF) for ideal TiNiSn and for doped compounds. | ||
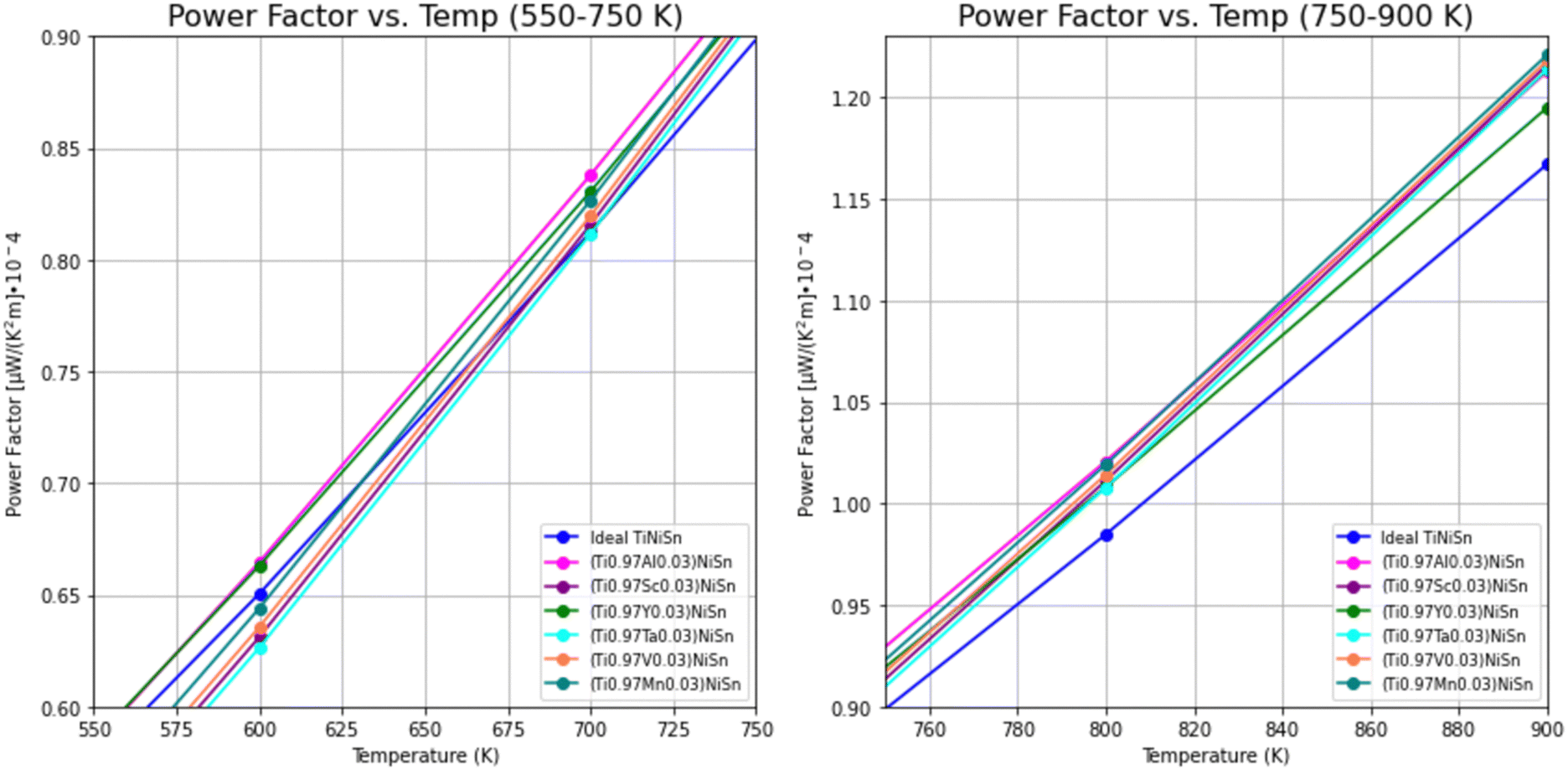 | ||
| Fig. 5 Temperature dependence of power factor for ideal TiNiSn and for doped compounds, in temperatures in the range of 550–900 K. | ||
The trends of PF can be considered as interchanging between the ideal TiNiSn and the doped compounds as the temperature increases, allowing the actual substitution between the Ti-atom and the dopant. At elevated temperatures the atomic fraction, c, of dopants forming the solid solution without its decomposition (which is usually very small) increases. It makes our model of supercell containing c = 0.03 doping atoms more relevant at high temperatures. The fact that the acceptor or donor elements exhibit holes or electrons conduction suggests that TE pairs can be formed.
3.2 Acceptor/donor analysis of orbital contribution
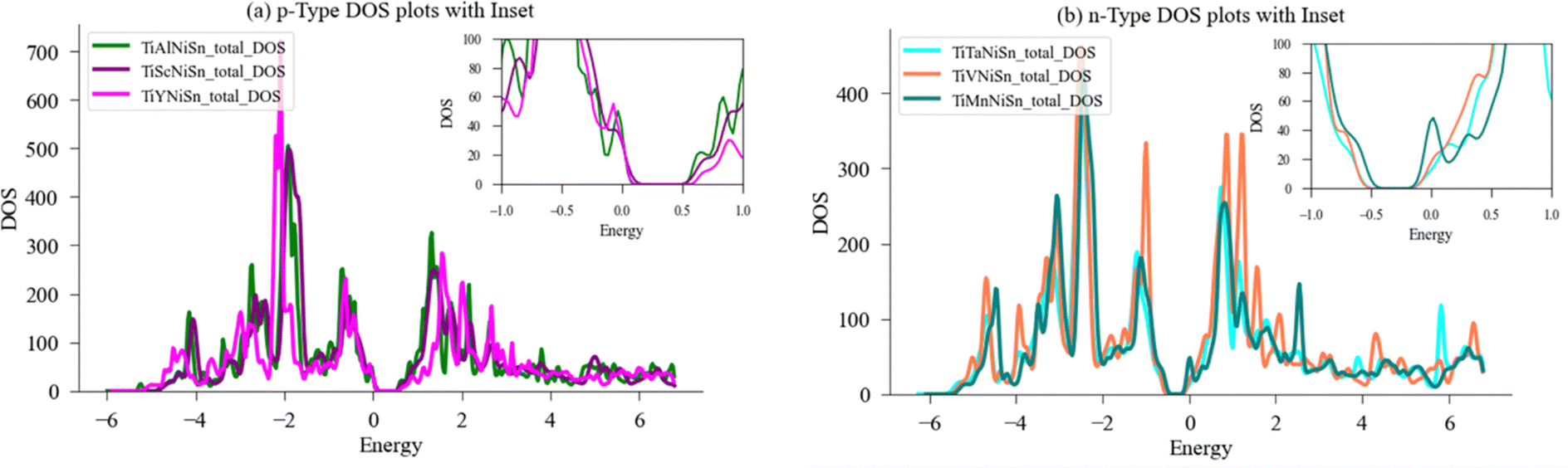 | ||
| Fig. 6 Comparison of DOS for doped TiNiSn in the form of (Ti0.97X0.03)NiSn. (a) Doped with acceptors (X = Al, Sc, Y) forming p-type. (b) Doped with donors (X = Ta, V, Mn) forming n-type. A magnification of the area in the vicinity of Fermi energy, EF, (set to zero) is shown in the inset. | ||
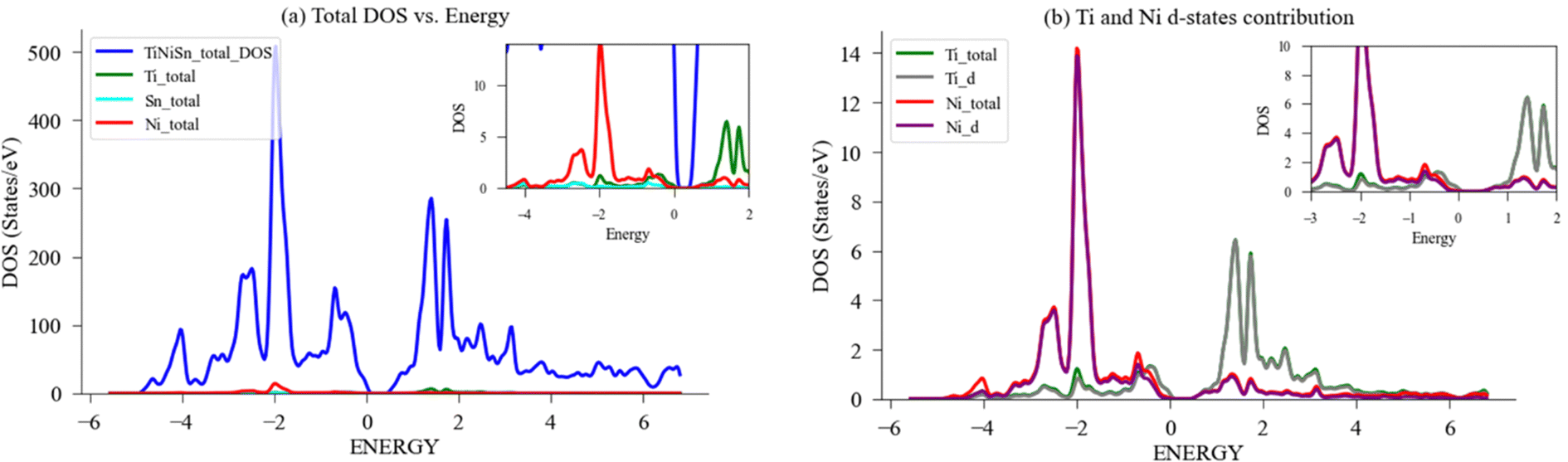 | ||
| Fig. 7 DFT calculation of DOS for ideal TiNiSn. (a) Total and LDOS for 96-atom supercell; (b) Ti, Ni PDOS-d contribution. A magnification of the area in the vicinity of Fermi energy, EF, (set to zero) is shown in the inset. | ||
The local DOS (LDOS) and partial DOS (PDOS) were also calculated for Ti, Ni, Sn as previously demonstrated by Colinet et al.36 and agrees with peaks of Ni States at −2 eV and −0.75 eV and peak of Ti states at 1.3 eV. Fig. 7(a) depicts the total and local DOS of Ti, Ni, Sn, alongside the respective PDOS for Ti-d and Ni-d states in Fig. 7(b). There is full alignment between the contribution of d-states and the respective element, which agrees with previous results that the states below the Fermi level are predominantly of Ni-d character, while the states above the Fermi level are of Ti-d character. This allows to conclude that the d-states contribution to LDOS of Ni and Ti are mainly dominated by their respective d-electrons.36
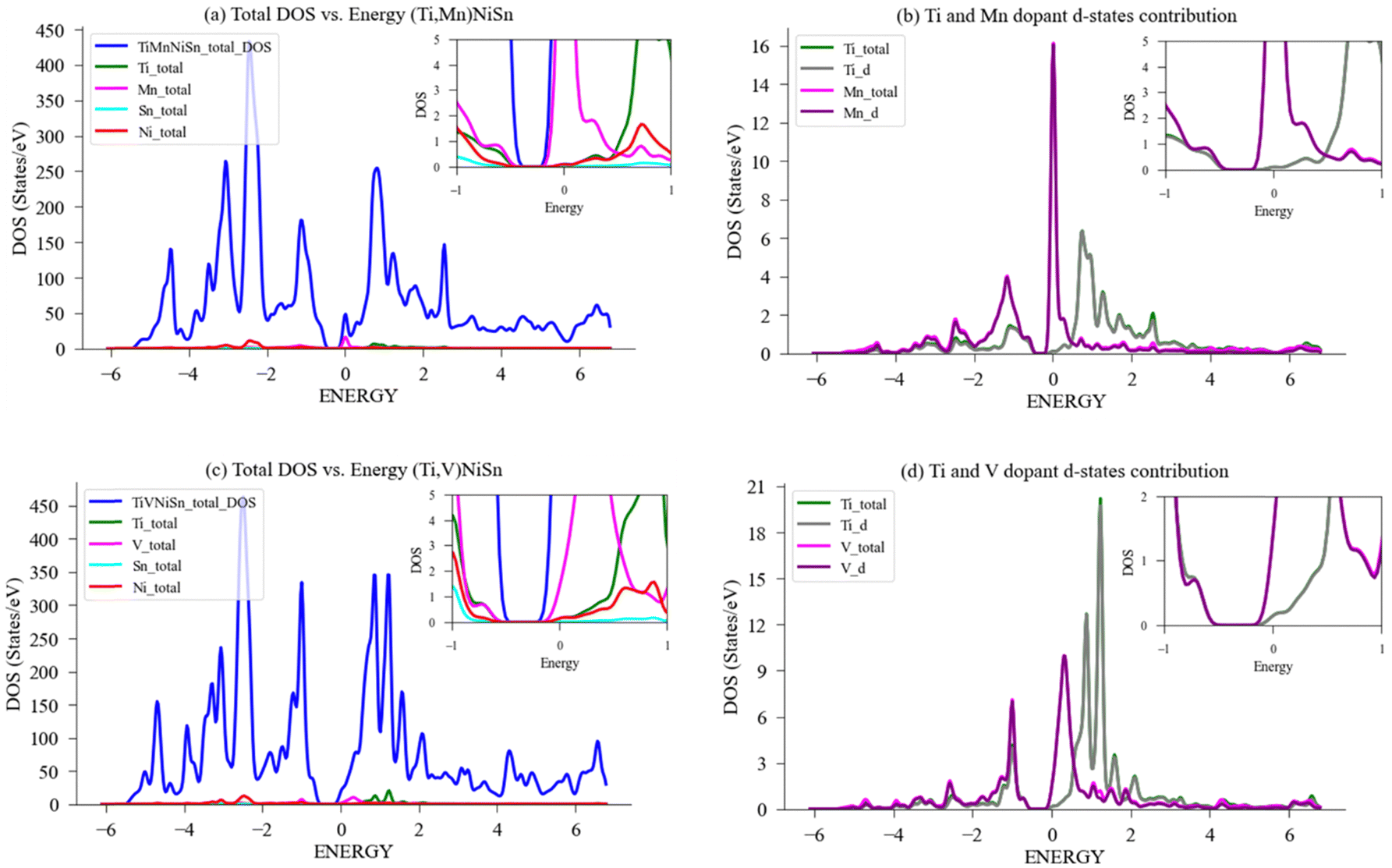 | ||
| Fig. 8 DFT calculation of DOS for (Ti,X)NiSn with donor dopants. (a) Total and LDOS for compound with X = Mn; (b) Ti, Mn PDOS-d contribution; (c) total and LDOS for compound with X = V; (d) Ti, V PDOS-d contribution. A magnification of the area in the vicinity of Fermi energy, EF, (set to zero) is shown in the inset. | ||
Fig. 8(b) exhibits the local and partial DOS calculated for Ti and Mn in a 96-atom supercell structure, and the specific contribution of the d-states to the bandgap of the doped compound. A few key pieces of evidence are shown. First, the bandgap near the Fermi-level narrows from 0.41 eV to 0.30 eV (in Table 2), significantly enabling conductivity through the transition of electrons to the conductivity band; second, the total contribution of the Ti-atom and the d-states of Ti are nearly identical, indicating that the Ti-atom behaviour is dominated by its d-states effect, and the same for Mn; third, the density of states at the Fermi level when comparing the Mn d-states to Ti d-states is significantly higher, therefore allowing a vast of electrons available for conduction. Although it is expected to see the main contribution to the bandgap occurring from electrons residing in the highest orbital (d-orbital), the current investigation found Mn to be the most favourable, as the magnitude of the effect (narrowing of the bandgap and the states near Fermi level), is greater than presented for the other elements. This resides well with this compound showing the highest slope in PF as temperatures arise. Given these attributes, doping the ideal compound with Mn seems favourable compared to the other chosen elements, and we expect to see apparent effect of the electrons in d-orbital to the change in bandgap also in experiments.
Fig. 8(c) exhibit the DOS calculated for Ti-atom and V-atom in a 96-atom supercell structure, and Fig. 8(d) exhibits the specific contribution of the d-states to the bandgap of the doped compound. Again, in the case of V substitution for Ti, the bandgap near the Fermi-level narrows from 0.44 eV to 0.38 eV (in Table 2), the narrowing of the bandgap enables conductivity through easier transition of electrons to the conductivity band. The contribution of Ti-atom and the d-states of Ti are nearly identical, indicating that the Ti-atom behaviour is dominated by its d-states, and the same for V. Additionally, as can be seen in the graph, when comparing the V d-states contribution to the DOS near the Fermi level, it is higher than that of Ti d-states contribution, but less than when comparing the Mn d-states contribution to that of Ti d-states. Again, a higher electrical conductivity is achieved by substituting Ti with V, mainly driven by its d-states contribution vs. the ideal TiNiSn.
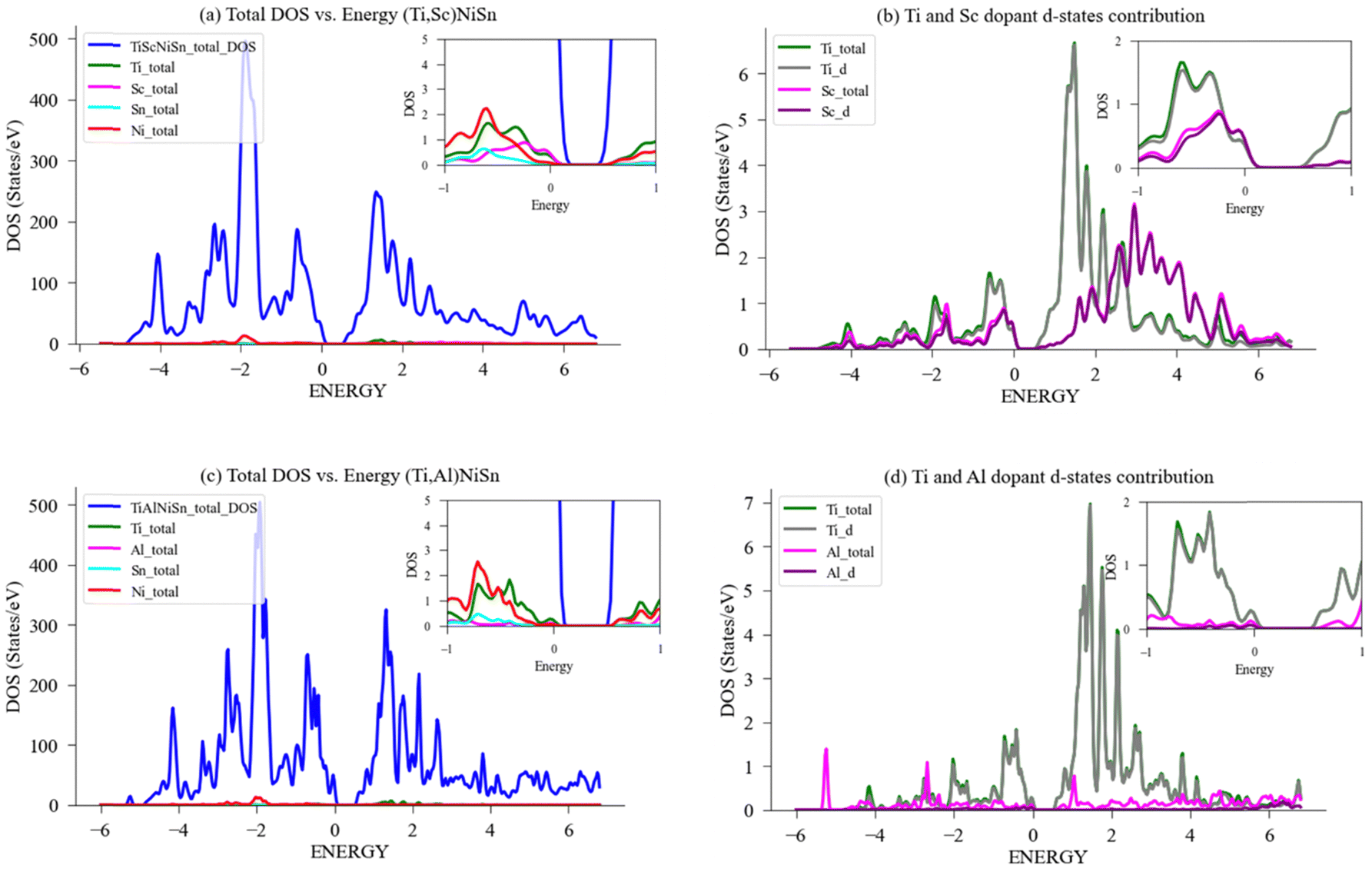 | ||
| Fig. 9 DFT calculation of DOS for (Ti,X)NiSn with acceptor dopants. (a) Total and LDOS for compound with X = Sc; (b) Ti, Sc PDOS-d contribution; (c) total and LDOS for compound with X = Al; (d) Ti, Al PDOS-d contribution. A magnification of the area in the vicinity of Fermi energy, EF, (set to zero) is shown in the inset. | ||
4. Summary and conclusions
In this study we demonstrated that ab initio DFT approach combined with transport properties calculation, may be applied to study the effect of acceptor or donor element substitution of Ti-atom. Our approach was based on the analysis of the contribution of d-states to the DOS in the doped compounds. We considered the valence electron difference between Ti and the acceptor or donor elements and the electron configuration of the elements to simulate the efficiency of doping in delivering a more effective TE conversion, as shown in the calculated power factor of the different compounds.DFT was applied to calculate the DOS of the doped compounds and that of the ideal state, then used in the transport properties calculations to witness trends of PF interchanging between the ideal TiNiSn and the doped compounds as temperature increases. Suggesting effective n-type and p-type TE pairs to be formed from the substitution of Ti with acceptor or donor element, which reside well with experimental results, using similar doped compounds compared to the pure TiNiSn. The analysis of the d-states contribution to DOS, that was suggested in this study, has shown indication of the doping contribution to electronic transport properties in all elements, and specifically the formation of d-states in the (Ti0.97Al0.03NiSn) compound were noticed, which are promoted states caused by an excitation of electrons by the surrounding crystal field of the matrix. Further, a strong indication of n-type compounds using donor elements was found. Specifically, Mn substitution on the Ti sublattice in the composition (Ti0.97Mn0.03NiSn), was found to be most favorable in demonstrating a narrower bandgap than the ideal compound, enabling conductivity through the transition of electrons to the conductivity band. Yet, only moderate indication of p-type compounds was found to generate a favorable bandgap, using the acceptor elements.
It should be mentioned that the analysis suggested in our study, based on the fine features of DOS in the vicinity of the Fermi energy, using bandgap analysis and contribution of d-states to electronic properties, may be used systematically, prior to experimentation, in other acceptor or donor elements, to identify n-type or p-type compounds for thermoelectric devices.
Data availability
The code for WIEN2K can be found at https://www.wien2k.at/index.html. The version of the code employed for this study is version 21.1.31,39Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Israel Science Foundation (ISF), Grant no. 617/24. One of the authors (YG) is the incumbent of the Samuel Ayrton Chair in Metallurgy at BGU.References
- T. M. Tritt and M. A. Subramanian, Thermoelectric materials, phenomena, and applications: A bird's eye view, MRS Bull., 2006, 31, 188–194 CrossRef.
- T. Berry, C. Fu, G. Auffermann, G. H. Fecher, W. Schnelle, F. Serrano-Sanchez, Y. Yue, H. Liang and C. Felser, Enhancing Thermoelectric Performance of TiNiSn half-Heusler Compounds via Modulation Doping, Chem. Mater., 2017, 29, 7042–7048 CrossRef CAS.
- Y. Gelbstein, Morphological effects on the electronic transport properties of three-phase thermoelectric materials, J. Appl. Phys., 2012, 112, 11 CrossRef.
- H. J. Goldsmid, Introduction to Thermoelectricity, Springer Series in Materials Science, Springer-Verlag, Berlin Heidelberg, 2016, vol. 121 Search PubMed.
- J. Zhou, H. Zhu, T. H. Liu, Q. Song, R. He, J. Mao, Z. Liu, W. Ren, B. Liao, D. J. Singh, Z. Ren and G. Chen, Large thermoelectric power factor from crystal symmetry-protected non-bonding orbital in half-Heuslers, Nat. Commun., 2018, 9, 1 CrossRef.
- S. Lv, Z. Qian, D. Hu, X. Li and W. He, A comprehensive review of strategies and approaches for enhancing the performance of thermoelectric module, Energies, 2020, 13, 3142 CrossRef CAS.
- S. Chen and Z. Ren, Recent progress of half-Heusler for moderate temperature thermoelectric applications, Mater. Today, 2013, 16, 387–395 CrossRef CAS.
- G. Zei, S. Matter, F. G. Aliev, N. B. Brandt, V. V. Moshchalkov, V. V. Kozyrkov, R. V. Skolozdra and A. I. Belogorokhov, Gap at the Fermi level in the intermetallic vacancy system RNiSn (R = Ti, Zr, Hf), Z. Phys. B: Condens. Matter, 1989, 75, 167–171 CrossRef.
- M. K. Brod, S. Guo, Y. Zhang and G. J. Snyder, Explaining the electronic band structure of half-Heusler thermoelectric semiconductors for engineering high valley degeneracy, MRS Bull., 2022, 47, 573–583 CrossRef CAS.
- S. J. Poon, T. M. Tritt, Y. Xia, S. Bhattacharya, V. Ponnambalam, A. L. Pope, R. T. Littleton and V. M. Browning, Bandgap features and thermoelectric properties of Ti-based half-Heusler alloys, 18th International Conference on Thermoelectrics, IEEE, 1999, pp. 45–51 Search PubMed.
- J. S. Young and R. G. Reddy, Processing and Thermoelectric Properties of TiNiSn Materials: A Review, J. Mater. Eng. Perform., 2019, 28, 5917–5930 CrossRef CAS.
- K. Kirievsky, Y. Gelbstein and D. Fuks, Phase separation and antisite defects in the thermoelectric TiNiSn half-Heusler alloys, J. Solid State Chem., 2013, 203, 247–254 CrossRef CAS.
- M. Kaller, D. Fuks and Y. Gelbstein, Sc solubility in p-type half-Heusler (Ti1-cScc)NiSn thermoelectric alloys, J. Alloys Compd., 2017, 729, 446–452 CrossRef CAS.
- D. Rabin, D. Fuks and Y. Gelbstein, Al solubility in (Ti1-cAlc)NiSn half-Heusler alloy, Phys. Chem. Chem. Phys., 2019, 21, 7524–7533 RSC.
- K. Kirievsky, D. Fuks and Y. Gelbstein, Composition conserving defects and their influence on the electronic properties of thermoelectric TiNiSn, Phys. Chem. Chem. Phys., 2020, 22, 8035–8047 RSC.
- J. Schmitt, Z. M. Gibbs, G. J. Snyder and C. Felser, Resolving the true band gap of ZrNiSn half-Heusler thermoelectric materials, Mater. Horiz., 2015, 2, 68–75 RSC.
- W. G. Zeier, J. Schmitt, G. Hautier, U. Aydemir, Z. M. Gibbs, C. Felser and G. J. Snyder, Engineering half-Heusler thermoelectric materials using Zintl chemistry, Nat. Rev. Mater., 2016, 1, 6 Search PubMed.
- M. T. Dylla, A. Dunn, S. Anand, A. Jain and G. J. Snyder, Machine Learning Chemical Guidelines for Engineering Electronic Structures in Half-Heusler Thermoelectric Materials, Research, 2020, 2020, 6375171 CrossRef CAS PubMed.
- Y. Gelbstein, Z. Dashevsky and M. P. Dariel, High performance n-type PbTe-based materials for thermoelectric applications, Phys. B, 2005, 363, 196–205 CrossRef CAS.
- A. F. May and G. J. Snyder, Introduction to Modeling Thermoelectric Transport at High Temperatures, Materials, preparation, and characterization in thermoelectrics, 2012, pp. 1–18 Search PubMed.
- M. M. Mallick and S. Vitta, Thermophysical and magnetic properties of p- and n-type Ti-Ni-Sn based half-Heusler alloys, J. Alloys Compd., 2017, 710, 191–198 CrossRef CAS.
- J. Schmitt, Investigation of Acceptor Dopants in ZrNiSn half-Heusler Materials, Dr. rer. nat. thesis, Johannes Gutenberg University of Mainz, 2014 Search PubMed.
- A. Horyn, O. Bodak, L. Romaka, Y. Gorelenko, A. Tkachuk, V. Davydov and Y. Stadnyk, Crystal structure and physical properties of (Ti, Sc)NiSn and (Zr, Sc)NiSn solid solutions, J. Alloys Compd., 2004, 363, 10–14 CrossRef CAS.
- T. J. Zhu, K. Xiao, C. Yu, J. J. Shen, S. H. Yang, A. J. Zhou, X. B. Zhao and J. He, Effects of yttrium doping on the thermoelectric properties of Hf0.6Zr0.4NiSn0.98Sb0.02 half-Heusler alloys, J. Appl. Phys., 2010, 108, 044903 CrossRef.
- A. Karati, S. Mukherjee, R. C. Mallik, R. Shabadi, B. S. Murty and U. V. Varadaraju, Simultaneous increase in thermopower and electrical conductivity through Ta-doping and nanostructuring in half-Heusler TiNiSn alloys, Materialia, 2019, 7, 100410 CrossRef CAS.
- Y. V. Stadnyk, A. M. Goryn, Y. K. Gorelenko, L. P. Romaka and N. A. Mel’nichenko, Thermoelectric power factor of Ti 1-xV xNiSn alloys, Inorg. Mater., 2010, 46, 842–846 CrossRef CAS.
- Y. Stadnyk, A. Horyn, V. V. Romaka, Y. Gorelenko, L. P. Romaka, E. K. Hlil and D. Fruchart, Crystal, electronic structure and electronic transport properties of the Ti1-xVxNiSn (= 00.10) solid solutions, J. Solid State Chem., 2010, 183, 3023–3028 CrossRef CAS.
- T. Graf, C. Felser and S. Parkin, Simple rules for the understanding of Heusler compounds, Prog. Solid State Chem., 2011, 39, 1–50 CrossRef CAS.
- T. Berry, S. Ouardi, G. H. Fecher, B. Balke, G. Kreiner, G. Auffermann, W. Schnelle and C. Felser, Improving thermoelectric performance of TiNiSn by mixing MnNiSb in the half-Heusler structure, Phys. Chem. Chem. Phys., 2017, 19, 1543–1550 RSC.
- K. Schwarz, P. Blaha and G. K. H. Madsen, Electronic structure calculations of solids using the WIEN2k package for material sciences, Comput. Phys. Commun., 2002, 147, 71–76 CrossRef.
- P. Blaha, K. Schwarz, F. Tran, R. Laskowski, G. K. H. Madsen and L. D. Marks, WIEN2k: An APW + lo program for calculating the properties of solids, J. Chem. Phys., 2020, 152, 7 CrossRef.
- D. V. Schroeder, An introduction to thermal physics, Addison Wesley Longman, Weber State University, 1999 Search PubMed.
- T. Zilber, S. Cohen, D. Fuks and Y. Gelbstein, TiNiSn half-Heusler crystals grown from metallic flux for thermoelectric applications, J. Alloys Compd., 2019, 781, 1132–1138 CrossRef CAS.
- W. Ren, H. Zhu, J. Mao, L. You, S. Song, T. Tong, J. Bao, J. Luo, Z. Wang and Z. Ren, Manipulation of Ni Interstitials for Realizing Large Power Factor in TiNiSn-Based Materials, Adv. Electron. Mater., 2019, 5, 7 Search PubMed.
- K. Kirievsky, M. Shlimovich, D. Fuks and Y. Gelbstein, An ab initio study of the thermoelectric enhancement potential in nano-grained TiNiSn, Phys. Chem. Chem. Phys., 2014, 16, 20023–20029 RSC.
- C. Colinet, P. Jund and J. C. Tédenac, NiTiSn a material of technological interest: Ab initio calculations of phase stability and defects, Intermetallics, 2014, 46, 103–110 CrossRef CAS.
- M. Hichour, D. Rached, R. Khenata, M. Rabah, M. Merabet, A. H. Reshak, S. Bin Omran and R. Ahmed, J. Phys. Chem. Solids, 2012, 73, 975–981 CrossRef CAS.
- L. Chaput, J. Tobola, P. Pécheur and H. Scherrer, Electronic structure and thermopower of Ni (Ti0.5Hf0.5) Sn and related half-Heusler phases, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73(4), 045121 CrossRef.
- P. Blaha, K. Schwarz, G. K. H. Madsen, D. Kvasnicka, J. Luitz, R. Laskowski, F. Tran and L. D. Marks, WIEN2k, An Augmented Plane Wave + Local Orbitals Program for Calculating Crystal Properties, Karlheinz Schwarz, Vienna University of Technology, Austria, 2018, ISBN 3-9501031-1-2 Search PubMed.
| This journal is © the Owner Societies 2025 |