
Cyclic natural product oligomers: diversity and (bio)synthesis of macrocycles†
Songya
Zhang‡
 ac,
Shuai
Fan‡
d,
Haocheng
He‡
ac,
Jing
Zhu
ac,
Lauren
Murray
efg,
Gong
Liang
ac,
Shi
Ran
d,
Yi Zhun
Zhu
h,
Max J.
Cryle
*efg,
Hai-Yan
He
*d and
Youming
Zhang
*abc
ac,
Shuai
Fan‡
d,
Haocheng
He‡
ac,
Jing
Zhu
ac,
Lauren
Murray
efg,
Gong
Liang
ac,
Shi
Ran
d,
Yi Zhun
Zhu
h,
Max J.
Cryle
*efg,
Hai-Yan
He
*d and
Youming
Zhang
*abc
aCAS Key Laboratory of Quantitative Engineering Biology, Shenzhen Institute of Synthetic Biology, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, China
bHelmholtz International Lab for Anti-infectives, Shandong University-Helmholtz Institute of Biotechnology, State Key Laboratory of Microbial Technology, Shandong University, Qingdao, Shandong 266237, China. E-mail: zhangyouming@sdu.edu.cn
cShenzhen Key Laboratory of Genome Manipulation and Biosynthesis, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, China
dNHC Key Laboratory of Biotechnology for Microbial Drugs, Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100050, China
eDepartment of Biochemistry and Molecular Biology, Monash Biomedicine Discovery Institute, Monash University, Clayton, Victoria 3800, Australia
fEMBL Australia, Monash University, Clayton, Victoria 3800, Australia
gARC Centre of Excellence for Innovations in Peptide and Protein Science, Monash University, Clayton, Victoria 3800, Australia
hSchool of Pharmacy & State Key Lab. for the Quality Research in Chinese Medicine, Macau University of Science and Technology, Macau, China
First published on 25th November 2024
Abstract
Cyclic compounds are generally preferred over linear compounds for functional studies due to their enhanced bioavailability, stability towards metabolic degradation, and selective receptor binding. This has led to a need for effective cyclization strategies for compound synthesis and hence increased interest in macrocyclization mediated by thioesterase (TE) domains, which naturally boost the chemical diversity and bioactivities of cyclic natural products. Many non-ribosomal peptide synthetase (NRPS) and polyketide synthase (PKS) derived natural products are assembled to form cyclodimeric compounds, with these molecules possessing diverse structures and biological activities. There is significant interest in identifying the biosynthetic pathways that produce such molecules given the challenge that cyclodimerization represents from a biosynthetic perspective. In the last decade, many groups have pursued the characterization of TE domains and have provided new insights into this biocatalytic machinery: however, the enzymes involved in formation of cyclodimeric compounds have proven far more elusive. In this review we focus on natural products that involve macrocyclization in their biosynthesis and chemical synthesis, with an emphasis on the function and biosynthetic investigation on the special family of TE domains responsible for forming cyclodimeric natural products. We also introduce additional macrocyclization catalysts, including butelase and the CT-mediated cyclization of peptides, alongside the formation of cyclodipeptides mediated by cyclodipeptide synthases (CDPS) and single-module NRPSs. Due to the interdisciplinary nature of biosynthetic research, we anticipate that this review will prove valuable to synthetic chemists, drug discovery groups, enzymologists, and the biosynthetic community in general, and inspire further efforts to identify and exploit these biocatalysts for the formation of novel bioactive molecules.
 Songya Zhang | Songya Zhang received his Master of Science degree from Shenyang Pharmaceutical University under the supervision of Prof. Huiming Hua. In 2018, he obtained his PhD degree at Albert-Ludwigs-Universität Freiburg in Germany under the supervision of Prof. Andreas Bechthold. Subsequently, he was selected into the Shenzhen High-Level Talent Plan and joined the lab of Prof. Tong Si at the Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences for postdoctoral research. Later, he became an assistant professor in the lab of Prof. Youming Zhang at the Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences. His research is centered around exploring enzymes possessing special catalytic functions and conducting synthetic biology research based on the non-ribosomal peptide derived from natural products. |
 Shuai Fan | Shuai Fan obtained his PhD in 2017 from the Chinese Academy of Medical Sciences & Peking Union Medical College. Subsequently, he joined the Institute of Medical Biotechnology at the Chinese Academy of Medical Sciences, where he has held positions as an assistant researcher and an associate researcher. His primary research focuses on enzymes with novel catalytic mechanisms involved in the biosynthetic pathways of natural products, combining theoretical calculations to elucidate catalytic mechanisms and rational enzyme engineering. |
 Haocheng He | He Haocheng obtained his PhD from Hunan Normal University in 2021, under the supervision of Professor Xia Liqiu. Following his doctoral studies, he joined the laboratory of Professor Zhang Youming at the Shenzhen Institutes of Advanced Technology as a postdoctoral researcher. His research primarily focuses on constructing chassis microorganisms for synthetic biology, aimed at optimizing the production of natural products and enzymes. He Haocheng has accumulated considerable research experience in exploring and regulating the production of natural products, as well as optimizing nitrogenase expression. |
 Max J. Cryle | Max J Cryle obtained his PhD in chemistry from the University of Queensland in 2006. He then moved to the Max Planck Institute for Medical Research in Heidelberg Germany as an HFSP Cross-Disciplinary Fellow and later as an Emmy Noether group leader funded by the DFG. Since 2016 he is an EMBL Australia group leader based within the Biomedicine Discovery Institute at Monash University, where his team focusses on understanding antibiotic biosynthesis (with particular interest in non-ribosomal peptide synthesis) as well as developing new antibiotics. He is also a Chief Investigator in the Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science (CIPPS). |
 Hai-Yan He | Hai-Yan He received his BS in chemistry from Tianjin University. He obtained his PhD in bioorganic chemistry at the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, under the supervision of Prof. Gong-Li Tang. Following a post-doctorate with Katherine Ryan at the University of British Columbia, he began his independent career as a Principal Investigator at the Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences & Peking Union Medical College, China. His research group is devoted to the development of new biocatalysts for production of valuable chemicals and clinical drugs. |
 Youming Zhang | Youming Zhang received his PhD at Ruprecht-Karls-Universität Heidelberg in 1994. After his postdoctoral research in Heidelberg University and European Molecular Biology Laboratory (EMBL), he founded Gene Bridges GmbH (Germany) in 2000 as the co-founder and Chief Scientific Officer. In 2013 he was appointed as the director of the State Key Laboratory of Microbial Technology, Shandong University. He was elected as a member of Academia Europaea (2019) and German National Academy of Science and Engineering (2020). His research is dedicated to development of the Red/ET recombineering and its application in microbial natural product heterologous expression, humanized animal models and tumor targeting bacteria. |
1. Introduction
Natural products (NPs) have historically provided us with a crucial source of unique molecular scaffolds, enabling the development of new therapeutic agents based on their wide range of biological activites.1–3 The cyclization of molecules has been an important strategy in the development for new drugs, and macrocycles are widely present in NPs from various sources. Currently, many NP-derived drugs are focusedon a cyclized scaffold, such as the immunosuppressant cyclosporine, the hormone oxytocin, and numerous antibiotics including vancomycin, daptomycin, erythromycin, and amphotericin. To date, around 70 macrocyclic compounds have been developed as FDA-approved medicines.4Cyclization, a crucial chemical reaction, modifies the activity of natural products by forming a ring structure, of which the size and shape can significantly impact the activity of the molecule, particularly in the design of peptides.5–7 Also, the cyclic nature of the ring is typically essential for the activity, proving highly beneficial in enabling the specific interactions between molecule and its biological target, and improving its pharmacokinetic properties, such as half-life and permeability.7 This makes cyclic peptides highly promising for drug development. Cyclic peptides also have low structural Gibbs free energy, increasing binding affinity and selectivity for target molecules. They can selectively bind to unstable protein surfaces and penetrate cell membranes, thus facilitating target drug development.8,9
The polymerization of molecules as a biosynthetic strategy can bring many advantages,10 for example by generating compounds with high structural complexity and chemical diversity from a much smaller biosynthetic assembly line. Dimerization (as well as higher order multimerization) reactions can also be used to introduce specific functional groups or structural features, whilst at the same time generating enlarged molecules with improved stability or selectivity, which are crucial factors in target binding.11,12 Larger molecules are also able to access multiple binding sites, with examples showing how two separate binding sites on a receptor molecule can be targeted by a dimeric drug, creating much tighter binding than would be possible with two isolated monomeric compounds.13,14 To date, many dimeric natural products have been shown to exhibit enhanced bioactivities when compared to their monomeric form.15–18 Indeed, in some cases, dimeric compounds have been shown to be is active whilst the monomer remains inactive19 (e.g. dimeric lipopeptide fusion inhibitors exhibit significantly greater antiviral potency compared to monomeric derivatives,20,21 and cyclic peptide oligomers display more potent antimicrobial effect than their linear counterparts).22 Due to their broad range of biological activities, some dimeric NPs have been investigated as potential therapeutic agents or have inspired the creation of structural mimics.23–25
This review will focus primarily on cyclodimeric (as well as higher order multimeric) products formed through cyclisation of a dimeric precursor (referred to as macrocyclization), with a subset formed through direct cyclodimerization (the dimerization of two identical monomers to form a cyclic product). Cyclopeptides can be cyclized in various ways, including head-to-tail, head-to-side chain, side chain-to-tail, and side chain-to-side chain. Both de novo design and in vitro evolution are major strategies utilized for current cyclopeptide development.26 A detailed review of cyclic peptide cyclization strategies has been recently published, and will not be discussed in detail here.27–29
In natural product biosynthesis, TE domains play a crucial role in enhancing the chemical diversity and bioactivities of these compounds through cyclization. Typically, TE domains control the final chain length after the assembly of monomers from precursors. Macrocycle formation, often mediated by TE domains, is a common strategy in PKS- and NRPS-mediated natural product assembly lines.30–32 Various types of reactions, including TE-mediated cyclization, cycloaddition, cycloisomerization, esterification, and radical reactions, contribute to the formation of dimeric natural products.11,33 Understanding the enzymes catalysing these reactions within biosynthetic pathways is essential for drug discovery and the development of new biocatalysts for macrocyclization.
Moreover, research has shown that TE domains can mediate diverse atypical cyclization. For example, in teixobactin biosynthesis, the tandem TEs (TE1 and TE2) within the NRPS terminal module (Txo2) collaborate to create an active site for substrate cyclization, with biochemical experiments confirming their essential roles through serine residue acetylation.34 Similarly, the ObiF1 TE domain in obafluorin biosynthesis catalyzes β-lactone ring closure via transthioesterification, influenced by intramolecular interactions with the C-terminal integrated MbtH-like protein (MLP).35 Additionally, the TE domain within curacin A's polyketide synthase mediates a unique termination reaction within module CurM, preceded by sulfonation catalysed by an integrated sulfotransferase (ST) domain.36 Furthermore, the recombinant TE domain StmC/ACP–TE, discovered by the Ge group, showcases tandem reactions, acting as both a thioesterase and cyclase during streptoseomycin biosynthesis.37 Notably, Kobayashi et al. developed a chemoenzymatic approach to synthesize cyclic peptides by exploiting SurE, a protein homologous to a penicillin-binding protein rather than a traditional TE, thereby expanding the scope of chemoenzymatic synthesis of cyclopeptides.38,39 YsfF and YsfFHBT, both with thioesterase activities, are involved in complex biochemical reactions leading to the formation of youssoufene A1, which shows increased inhibition against multidrug-resistant bacteria. It was found that YsfF/YsfFHBT catalyse various reactions, including 6π-electrocyclic ring closure and dimerization.40 These examples collectively highlight the diverse and pivotal roles TE domains play in orchestrating the biosynthesis of natural products.
Currently, many non-ribosomal peptide synthetase (NRPS) and polyketide synthase (PKS) derived cyclodimeric NPs have been identified, with these molecules possessing diverse structures and biological activities. The identification of biosynthetic pathways that produce such molecules is of great interest due to the challenge that cyclization represents: specifically, how do the TE domains in NRPS or PKS assembly lines recognize and accommodate large substrates during catalysis? In the last decade, several groups have pursued identification of such TE domains from NRPS or PKS assembly lines, which has led to new insights to this biocatalytic machinery.41–43 Whilst these have typically not been involved in cyclodimer formation, our team has been one of those that has sought to overcome this by extensively investigating PKS/NRPS synthetases related to such dimerizing TE domains (including those involved in mohangamide and disorazole biosynthesis). Recently, several excellent reviews about TE-related biosynthesis have been published.31,32 However, these works only describe a single example of dimer cyclization. This review focuses on the special class of TE domains involved in the cyclization of dimeric (as well as higher order multimeric) natural product intermediates during NRPS and PKS assembly, including discussion concerning their mechanism.
In this review, we examine key examples of cyclic dimeric (as well as higher order multimeric) natural products. We illustrate the synthetic and biosynthetic routes to these cyclodimeric compounds, including the catalytic mechanism of key biosynthetic enzymes. We also strive to emphasize the gaps in both knowledge and methodology that must be addressed to effectively access these cyclic scaffolds. We limit our discussions to focus primarily on works that generate cyclodimeric natural products including novel analogues, thus largely excluding approaches that lead to the production of high polymer products through cyclo-oligomerization. This review covers the period until July 2023.
2. Cyclodimeric natural products
The cyclization and release reaction are the crucial final step for many natural products biosynthesis pathways. In this regard, thioesterase (TE) domains play a central role, where they exhibit impressive stereospecificity and selectivity that makes them highly valuable biocatalysts for industrial applications involving peptide cyclization. TE domains found in PKS or NRPS pathways commonly facilitate crucial steps of oligomerization and macrocyclization in the biosynthesis of many bioactive natural products.44,45 This section will provide a brief introduction of TE domain structure and the catalytic mechanism, with particular focus on how TE domains involved in cyclodimerization catalyze this transformation during peptide biosynthesis (Fig. 1).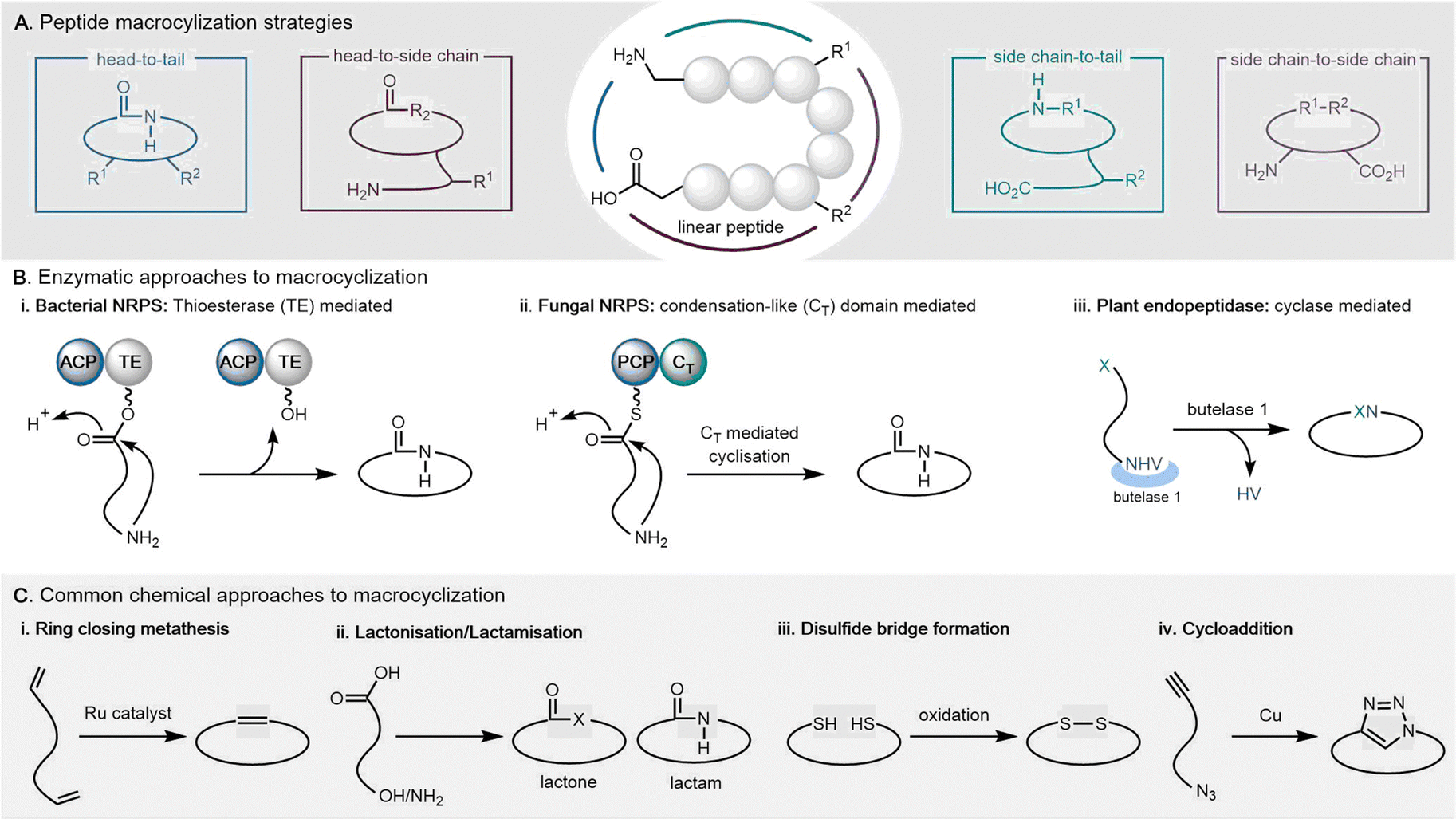 | ||
| Fig. 1 The general cyclisation mechanism of macrocyclic natural products. | ||
2.1 TE domains
Nonribosomal peptide synthetases (NRPS) and polyketide synthases (PKS) are two major types of microbial biosynthesis pathways. These two machineries generate a range of bioactive molecules, including antibacterial, antifungal, immunomodulatory and antitumor agents, all of which have extensive applications in medicine.46–48 PKS and NRPS are both representatives of microbial modular biosynthesis assembly lines. These enzymes utilize simple building blocks (such as two-carbon or three-carbon (acetyl-coenzyme A, malonyl-coenzyme A, propionyl-coenzyme A, and butyryl-coenzyme A) or amino acid units, and through a sequence of modules working in concert, assemble them into long-chain products or macrocyclic precursors with molecular weights of up to several kilodaltons.49 Thioesterase (TE) domains are then typically located in the final module of these systems, where they are responsible for the release of the final product of the assembly line through hydrolysis or through macrocyclization with intramolecular nucleophilic attack by free amino or hydroxyl groups.50 TE domains are responsible for the release of most of the products from both NRPS and PKS pathways. As the last enzyme in the PKS and NRPS assembly lines, the thioesterase controls the recognition and release of the product.31 The recognition of substrates and subsequent catalysis by thioesterase domains are therefore crucial for obtaining the expected product in a manner that does not stall the assembly process, whilst also serving as a control element for peptide modification in trans in certain cases.51To accurately reflect the scope of our investigation, we have clarified that the phylogenetic analysis specifically covers TEs from reported bacterial PKS or NRPSs pathway. We excluded the TEs from fungal PKS/NRPSs which clearly have different evolutionary histories. In some fungal peptide synthetases, the oligomerization is catalysed by the CT domain which will be elaborated in the next section. Interestingly, TE domains with reported iterative catalytic function do not gather in a single cluster (Fig. 2), and the mechanism underpinning their catalysis still requires investigation. It is also apparent that a large number of uncharacterized TE domains remain spread across these different clusters and await characterization.
 | ||
| Fig. 2 Phylogenetic analysis of the TE domain family sequence. Colours represent TE domain function and substrate type (type I or II from NRPS or PKS pathway and others). The characterized TE domain with iterative catalytic function mentioned in this paper was labelled by asterisk. This phylogenetic analysis was based on TE proteins from representative phyla involved in reported biosynthetic pathways. This analysis was conducted using protein data sourced from the reference collections of the Boddy group52 and the Deng group.53 The detailed protein information can be found in the ESI† (Table S1). The sequence alignment and phylogenetic tree was generated by MEGA, the tree was modified by iTOL.54,55 For a more detailed phylogenetic analysis, refer to Caswell et al. (2022).56 | ||
Generally, these thioesterases can be grouped into type I or type II TE domains from NRPS or PKS pathways. Type I TEs are typically integrated into the final module of the biosynthetic assembly line and catalyze product release through hydrolysis or cyclization, whereas type II TEs function independently, often playing a role in proofreading or editing the assembly line intermediates.52,57 (Fig. 2). For example, the TEs from DptD, EndC, Sas19, GlmI and GrsB group together in one cluster, as they are all domains that are found in the final module of an NRPS and are responsible for peptide cyclization (in daptomycin, enduracidin, WS9326, glidomide and gramicidin biosynthesis, respectively). In contrast, PKS-derived intra-module TE domains exist together in different cluster (e.g. TE domains from pikromycin (pikAIV TE), erythromycin (DEBS TE), amphotericin (AmphK TE) and FR-008 (FscF TE) biosynthesis).
Many PKS and NRPS pathways contain stand-alone enzymes known as type II TEs (TEII), which group into one cluster in the phylogenetic tree (Fig. 2).52–56 Type II TEs are not covalently bound to the multienzyme, with recent research suggesting that Type II TEs instead are capable of diverse catalytic functions. Type II TEs belong to the α/β hydrolase family, where their role is typically to hydrolyze thioester-bound residues on the 4′-phosphopantetheine (4′-PPant) arms of acyl carrier proteins (ACPs) or peptidyl carrier proteins (PCPs), such as the TEII BarC from barbamide pathway58,59 or Tcp39 from teicoplanin biosynthesis.51 Some type II TEs have been characterized with the function of chain translocation, such as the type II TEs WS5 (Cal5 or Sas5) and WS20 (Cal20 or Sas20) from the WS9326 pathway and TEII LgnA from legonindolizidin A pathway, where they work as shuttling enzymes for substrates during peptide assembly.52,60 In the case of PnG, this TEII enzyme performs multiple functions in the biosynthesis of phoslactomycin PKS (Pn PKS), including release of an ACP-tethered intermediate as well as proofreading.61
The TE domain of NRPS machineries is responsible for catalyzing the hydrolysis or cyclization reactions of the NRPS polypeptide chain. Studies have shown that NRPS type I thioesterases recognize specific linear peptide-PCP substrates (or even peptide-S-CoA/SNAC substrates) and utilize internal nucleophiles to cyclize the peptide via carbon-to-nitrogen intramolecular lactonization or lactamization.68 As a crucial NRPS catalytic domain, research into the protein structure and enzymatic catalytic function of TE domains has been steadily increasing.
Structural analysis has revealed that TE domains belong to the α/β hydrolase superfamily, which includes enzymes such as lipases, proteases, and esterases.71 The typical α/β hydrolase fold is comprised of a repeating β/α/β motif that forms a seven- or eight-stranded parallel β-sheet wrapped by α-helices on both sides, while several helices on top form the lid region.74,75 In general, the first N-terminal β-strand of the hydrolase fold is absent in TE domain. Instead, the second β-strand serves as the only anti-parallel strand among the remaining six or seven strands in the structure. Sequence alignments showed that lid region significantly differs between TE domains. The differences observed in TE structures mainly lie between the sixth and seventh β-strands, with the α-helices linking these strands displaying diversity in structure, including the insertion of an extra structural domain.76 It has been hypothesized, based on crystal structures and MD simulations, that the lid region opens to accommodate the presentation of thioester substrates. These studies offer detailed insights into the dynamics of the lid region, which is thought to play a role in substrate loading and release.77–79
The structures of SrfTE and FenTE confirm that thioesterases belong to the α/β hydrolase superfamily.71,72 The asymmetric unit of SrfTE reveals the presence of two distinct molecules, each exhibiting closed and open conformations. While SrfTE exists as a dimer in the asymmetric unit, it has been shown to exist as a monomer in a solution environment. In the SrfTE, the lid structure covers three α helices and has two different conformations. When in the “open” conformation, the lid structure folds back, thus exposing the catalytic center. Conversely, in the “closed” conformation, the catalytic center is almost completely covered by the lid. In the Fen TE, the lid structure is the shortest seen for NRPS TE domains, which causes the catalytic center of Fen TE to be exposed, possibly serving to make this TE highly promiscuous. Fully or partly exposed catalytic triads were observed in the related lysosomal palmitoyl protein thioesterases,80 whose substrates are similarly sterically demanding when compared to NRPS peptides.
In terms of the structure of TEs, cyclodimerization is performed by EntF TE and Vlm2, making these of particular interest (Fig. 3A and B). The structure of Vlm2 TE adopts the α/β-hydrolase fold typical of type I TE domains, with a canonical Ser-His-Asp catalytic triad covered by the lid structure.70 The lid of Vlm2 TE is a mobile component that includes an extended loop, three helices (α4–6), a five-residue helix (α7), a long helix (α8), and another short helix (α9), which is thought to have various functions including substrate positioning and solvent exclusion (Fig. 3E). The structure of the EntF PCP–TE di-domain represents the physiological structure before CoA addition to the PCP domain. The PCP and TE domains have a well-defined relative orientation with a primarily hydrophobic interface. The globular core of the TE domain displays two α-helices (α6–α7) protruding from this core, resembling webbed fingers that form a lid covering the TE active site (Fig. 3F). This allows the 4′-PPant arms, which is tethered to Ser 48 of the PCP domain in the holo state and can span up to ∼20 Å, to reach Ser 180 in the EntF TE domain.
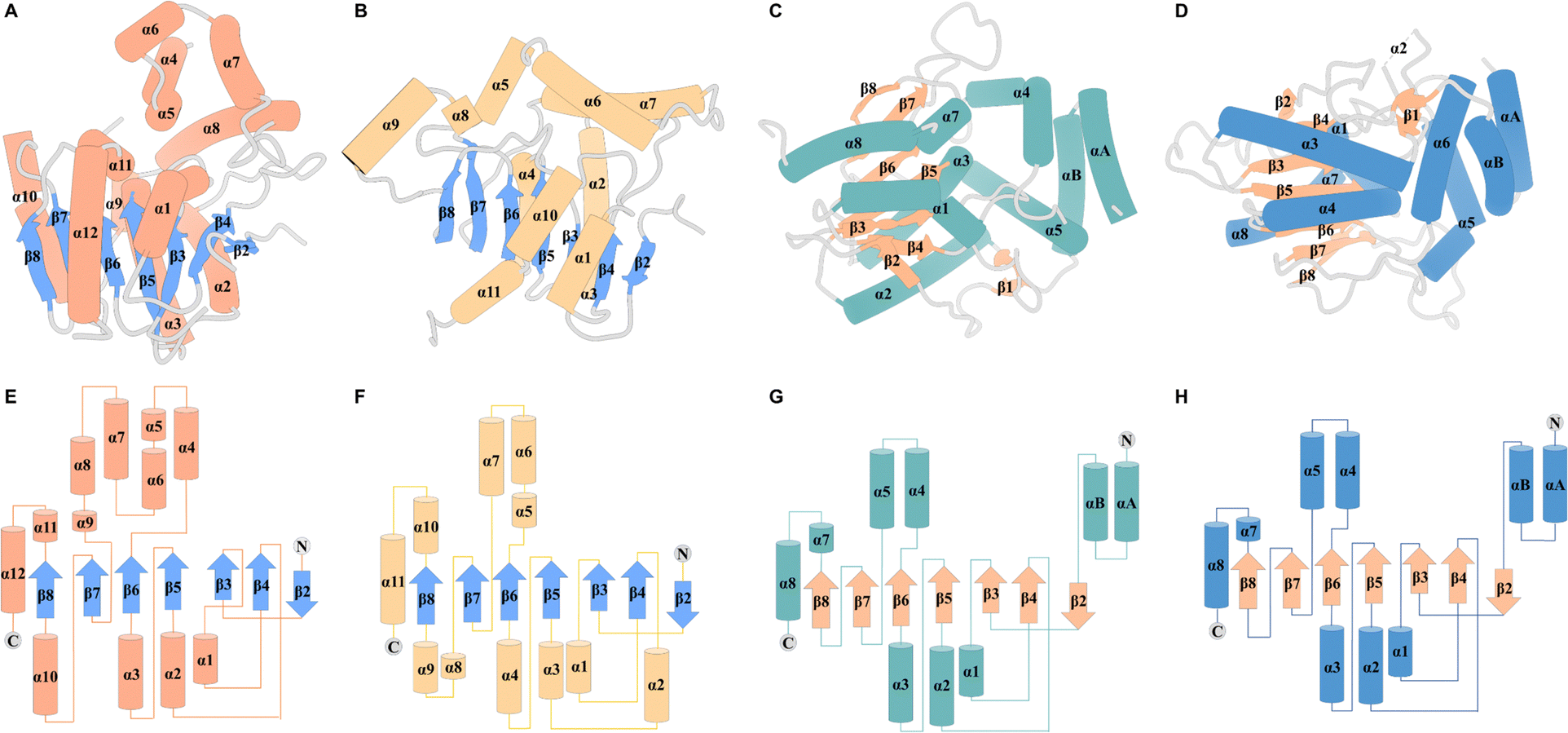 | ||
| Fig. 3 Overall structure of TEs. Structures of Vlm2 TE (A), EntF TE (B), DEBS TE (C) and PICS TE (D). Secondary-structure elements of Vlm2 TE (E), EntF TE (F), DEBS TE (G) and PICS TE (H). | ||
The typical thioesterase mechanism belongs to the Bi–Bi model, where a covalent intermediate is formed between the substrate and the enzyme during the reaction process.56 The first step involves substrate nonribosomal peptide being loaded onto the TE domain by transesterification of the PCP thioester, forming the acetyl–enzyme intermediate.78,81 The second step involves a histidine residue acting as a base to help deprotonate a nucleophile within the substrate (such as the side chain amine, hydroxy and thiol groups of amino acids) that then can attack the acetyl–enzyme complex, forming the tetrahedral intermediate. The tetrahedral intermediate is stabilized by the oxyanion hole by hydrogen bonding from two backbone amide NH groups. Finally, the cyclic product is released from the TE domain, and as a result, the active site of the TE domain is reactivated (Fig. 4).
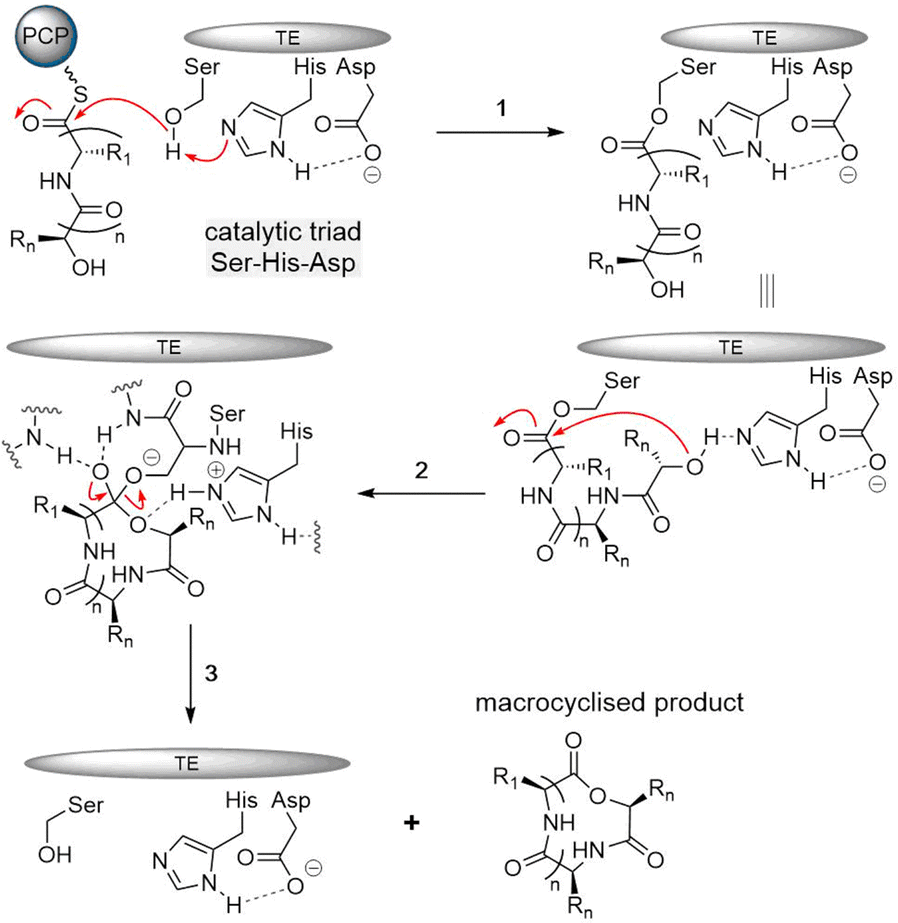 | ||
| Fig. 4 Mechanism of TE-mediated reaction for nonribosomal peptide cyclization. (1) Peptide transfer from PCP to active site serine of TE (2) acyl-TE adduct is attacked by the nucleophilic residue, forming a tetrahedral intermediate that is stabilized by the oxyanion hole. (3) Cyclic product is released and TE domain is reactivated. | ||
The catalytic core of the α/β hydrolase region is highly conserved, usually consisting of a catalytic triad Ser-His-Asp present comprising a nucleophilic serine adjacent to the β5 fold belonging to the GXSXG motif,82 an aspartic acid usually located at the loop following the β7 strand according to its canonical site in α/β hydrolase folds, and finally a histidine positioned on the loop after the last β-fold, which is also highly conserved (Fig. 4).69–71 Interestingly, some TE domains contain a Cys in place of the Ser in catalytic triads.83,84 The backbone amides of the amino acid following the nucleophilic amino acid, as well as another usually located between the β3 fold and the α-helix, form the negatively charged oxyanion hole, which stabilizes the intermediate during the reaction.85
The formation of a linear dimerization intermediate is a prerequisite for cyclodimerization mediated by TE domains. Two possible pathways for the oligomerization of NRPS intermediates by TE domains in valinomycin synthesis have been postulated.70 In the first scenario, ‘forward transfer’, the distal hydroxyl group of the peptidyl-O-TE complex attacks the thioester group in the peptidyl-S-PCP enzyme intermediate, directly forming dipeptidyl-O-TE as a product. In the second scenario, ‘reverse transfer’, the distal hydroxyl group of the peptidyl-S-PCP complex attacks the ester group in the peptidyl-O-TE enzyme intermediate, forming dipeptidyl-S-PCP as a product, which would then be transferred onto the serine of TE domain (Fig. 5). The analysis of the synthetic intermediates identified during valinomycin synthesis has revealed that Vlm TE catalyzes oligomerization through the “reverse transfer” pathway.70
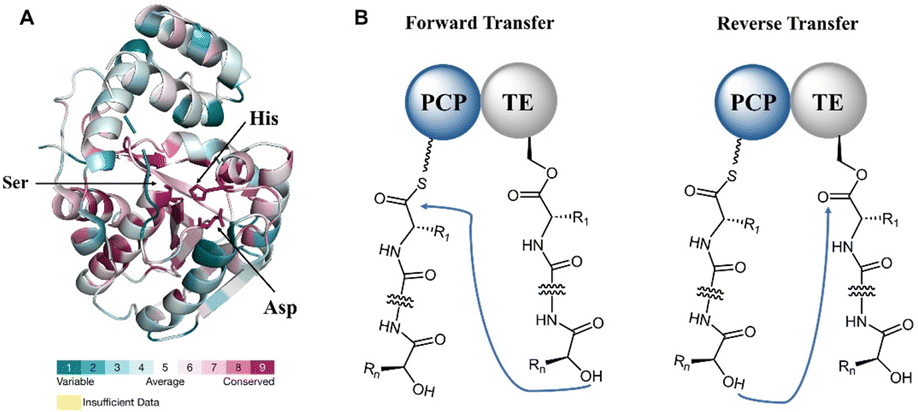 | ||
| Fig. 5 (A) The sequence conservation analysis of TE domain; (B) two scenarios for oligomerization. | ||
Notably, Gln125 of Skyxy-TE is not conserved among other TE enzymes. The catalysis by Skyxy-TE of the triad's His254 might extract an α-proton from the substrate, and Skyxy-TE is capable of utilizing an oxyanion hole to stabilize the enolized intermediate, thereby facilitating the spontaneous epimerization reaction. Nocardicin is a monocyclic β-lactam antibiotic biosynthesised by an NRPS assembly line of five modules, despite only three residues being contained in the final peptide product. In nocardicin biosynthesis, the TE domain NocTE (PDB: 6OJD) has two functions, catalyzing the C-terminal epimerization of an L-Hpg residue to D-Hpg before performing the hydrolytic release of the mature product.86,87 The peptide binding pocket has been resolved by high-resolution crystal structures of the thioesterase domain in ligand-free form and trapped with a fluorophosphonate mimic of the substrate, which has aided in rationalizing the stereoinversion performed by this TE domain.88 The spontaneous epimerization reaction mechanism of NocTE is similar to that of Skyxy-TE. In the substrate of NocTE, the substitution of the hydroxyphenyl moiety stabilizes the carbanion; consequently, the substrate of NocTE can spontaneously epimerize to its stereoisomer (Fig. 6).
 | ||
| Fig. 6 Overall structure of Skyxy-TE (A) and NocTE (B). The catalytic triad is displayed as a green stick model, while the oxyanion hole and key residues are shown as pink stick models. | ||
Both the DEBS TE and PICS TE are classified within the α/β hydrolase family and exhibit a hydrophobic cavity that traverses the protein, along with a hydrophobic dimer interface enriched in leucine residues. The catalytic triad is located in the center of the active site pocket, indicating that substrates must access this pocket during the reaction. The DEBS TE consists of a central β-sheet composed of seven strands, with β2 oriented in antiparallel manner to the remaining strands. Notably, the length of the β7 strand is shorter than β8, and the C-terminus of β7 is twisted 90° to form part of the substrate channel. The β strands are flanked on either side by α helices, two on one side and four on the other. Additionally, at the N-terminal extremity of the TE, two supplementary α-helices constitute the dimer interface.95,98
A two-step mechanism has also been proposed for the TE-mediated formation of macrocyclic polyketides. PKS and NRPS release mechanisms the first step involves the acylation of a linear polyketide thioester at a serine residue that is conserved within the catalytic triad of the PKS TE domains. This step generates an acyl–enzyme intermediate which can be stable for an extended period of time.56 The second step is a nucleophilic attack of an intramolecular hydroxyl group leading to cyclization (Fig. 7), or hydrolysis of the acyl–enzyme intermediate when no efficient intramolecular nucleophile is available.
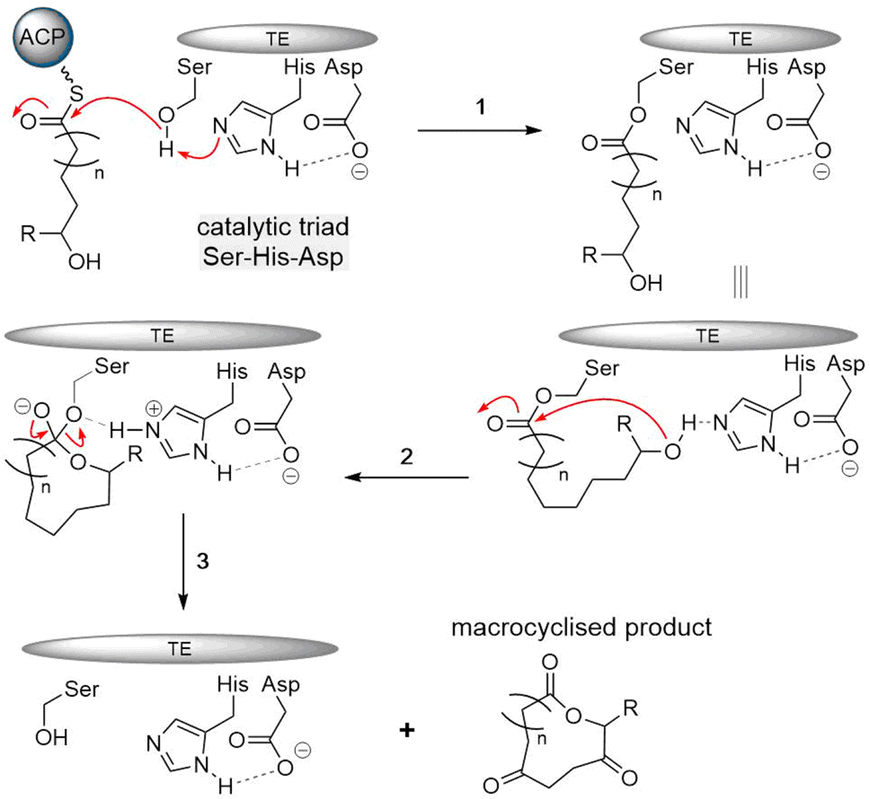 | ||
| Fig. 7 Mechanism of TE-mediated reaction for polyketide cyclization. (1) Peptide transfer from ACP to active site serine of TE (2) acyl-TE adduct is attacked by the nucleophilic residue, forming a tetrahedral intermediate (3) cyclic product is released and TE domain is reactivated. | ||
In the biosynthetic pathway of pikromycin (PIK), PIK-TE-1 demonstrated a higher frequency of “active” conformations, enabling easier transformation into the pre-reaction state necessary for macrocyclization. This transformation was influenced by a hydrogen-bonding interaction with His268 and essential hydrophobic interactions for substrate recognition. Notably, Tyr178 was proposed to widen the exit and facilitate product release in PIK-TE-1. The substrate channel of PIK-TE-1 is crucial for the mechanism and selectivity of macrolactonization, featuring a hydrophobic spacious chamber within the substrate channel, while a hydrophilic barrier at the distal end of the substrate channel induces the long-chain substrate to curl towards the catalytic residue Ser148, thus facilitating macrolactonization. A similar phenomenon exists in Pim TE, where its substrate channel is bifunnel-shaped, with a highly hydrophobic cavity near the catalytic triad, and a hydrophilic region specifically inducing the linear substrate to curl and complete cyclization. Thus, the substrate channel interrupted by a hydrophilic barrier represents a universal mechanism that induces essentially linear hydrophobic substrates to curl into conformations suitable for forming macrolactones.
Computational analysis supported the notion that macrocyclization catalyzed by PIK-TE via re-face nucleophilic attack was thermodynamically and kinetically more favorable than si-face attack.100 Chen et al. combined MD simulations with QM and QM/MM calculations for complexes of DEBS TE and its substrates. The DEBS TE and substrate undergo induced fit aided by hydrogen bonding and hydrophobic interactions. This transition is accompanied by a conformational change in active site pocket of the enzyme, shifting from a “closed” state to an “open” state. Proton abstraction from the substrate is followed by nucleophilic attack on the carbonyl carbon at C1, resulting in the formation of a charged tetrahedral intermediate. The energy barrier for this reaction step is 9.9 kcal mol−1. Finally, the final macrocyclic product is released with a very low barrier of 0.3 kcal mol−1.101
2.2 Cyclodimeric non-ribosomal peptides
Cyclic peptides are currently used with great success in drug therapy, including applications such as the antibiotics vancomycin, daptomycin, dalbavancin and polymyxin B, the hormone analogues octreotide and vasopressin, the immunosuppressant cyclosporine, and the antitumor agents lanreotide and romidepsin.102 As drug molecules, cyclooligomer peptides also have a wide range of biological activities such as anticancer, antiviral, antibacterial and enzyme inhibitors. The cyclization of polypeptide structures is important to maintain the properties of peptide drugs.103–105For NRPS-derived peptides, cyclodimerization mediated by the TE domain further boosts the chemical diversity of the peptide products of these assembly lines. Beyond this, nature employs diverse strategies in constructing macrocyclic molecular scaffolds. For example, P450 oxidases and radical SAM enzymes also can catalyse macrocyclic formation, such as macrocyclic construction in vancomycin and streptide.106,107 Diels–Alder enzymes utilize a cycloaddition reaction to construct the macrocyclic scaffold in pyrroindomycin, whilst the bifunctional oxidase LkcE catalyses a unique combination of amide oxidation and a Mannich reaction to assemble the macrocyclic system in lankacidin.108 Cyclodimerization in NRPS pathway greatly increases the possibilities to access peptides with high structural complexity and bioactivity diversity. Moreover, NRPS modules can be used iteratively to generate larger peptide rings, following the principle of molecular efficiency and atom economy. Understanding how nature generates cyclooligomer peptides from acyclic precursors can provide inspiration for the construction of functional macrocyclic molecules. In this section, we will focus on NRPS-derived NPs that include cyclodimerization mediated by a TE domain in their biosynthesis, and particular focus on the structural characterization and catalytic mechanism of these TE domains.
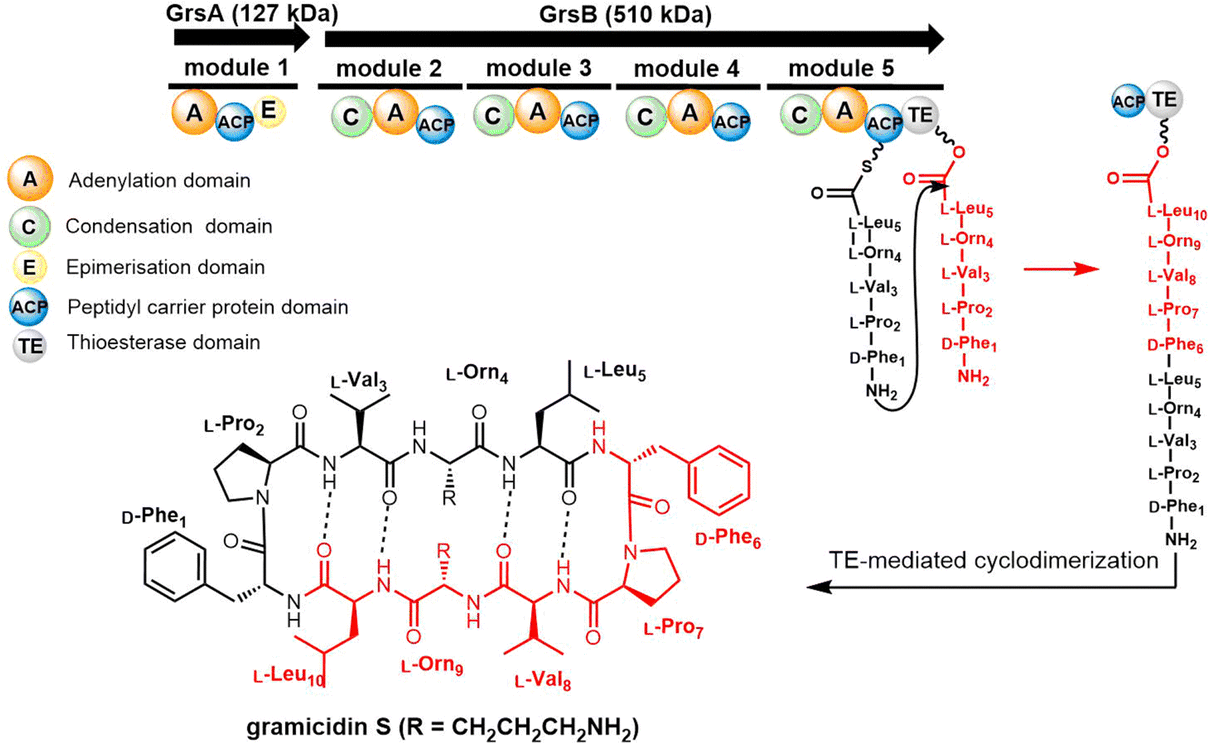 | ||
| Scheme 1 The gramicidin S biosynthesis pathway. The dotted lines are used to indicate the four intramolecular hydrogen bonds found in gramicidin S. | ||
Tyrocidine is a cyclic decapeptide first isolated from the soil bacteria Bacillus brevis in 1940, and is the major constituent of tyrothricin, an antibiotic mixture also containing gramicidin.118 Tyrothricin was an important milestone in antibiotic development, as it was the first antibiotic commercialized for clinical use. Tyrocidine A is active against a broad spectrum of Gram-positive bacteria and acts by disrupting the synthesis of the peptidoglycan layer of bacterial cell walls.119 Tyrocidine has also been shown to be effective at controlling the fungal pathogens.120
Further experiments then demonstrated that the ability of the GrsB TE domain to catalyse the cyclodimerization of peptide substrate decreases with increasing peptide length when these are loaded onto PCP–TE didomain constructs. The GrsB TE was shown to efficiently dimerize or even trimerize 6–15 amino acid containing linear peptides, demonstrating significant flexibility and substrate tolerance in the catalytic pocket of this domain.124 Additional chemoenzymatic studies demonstrated that the GrsB TE can cyclize immobilized linear decapeptide precursors, suggesting its potential use in chemoenzymatic analogue synthesis.125
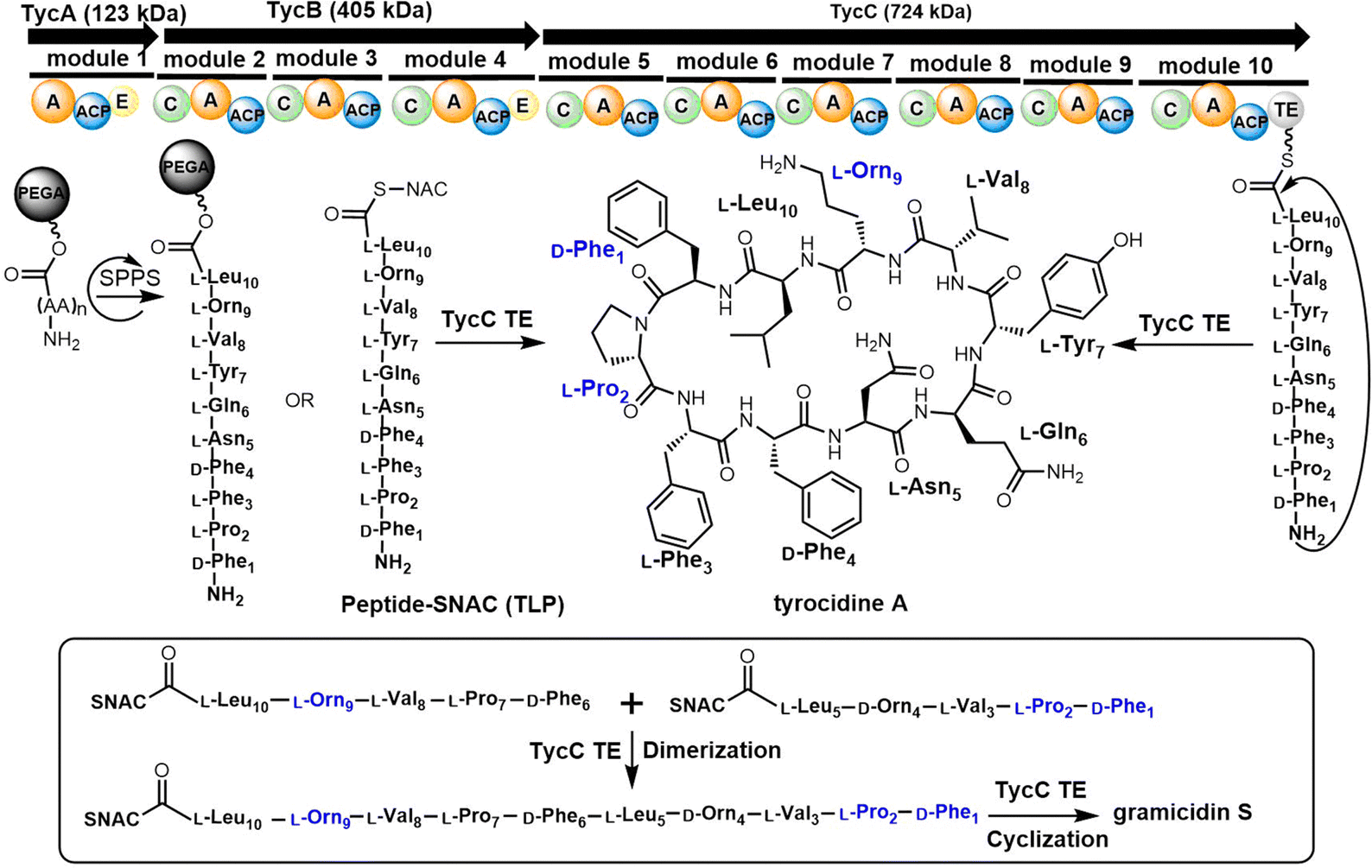 | ||
| Scheme 2 The biosynthesis pathway of tyrocidine A and gramicidin S. | ||
In 1997, the Marahiel lab reported the biosynthetic gene cluster of tyrocidine in Bacillus brevis ATCC 8185. The tyrocidine NRPS synthetase enzymes TycA, TycB and TycC are responsible for peptide assembly of the linear decapeptide.117 The C-terminal TE domain found in TycC (TycC TE) is then responsible for the macrocyclization of the decapeptide to generate tyrocidine A.127 The TycC TE domain was the first excised TE domain (35 kDa) shown to retain cyclization activity. Moreover, it has been demonstrated that the recombinant TycC TE domain is a versatile macrocyclization catalyst, which can generate tyrocidine variants with altered biological activity.127 Whilst the TycC TE domain typically catalyses the cyclization of a decapeptide-thioester to form tyrocidine A, it has been shown that the TycC TE can also catalyse the cyclodimerization of the gramicidin S pentapeptide-SNAC (D-Phe-Pro-Val-Orn-Leu-SNAC) to generate gramicidin S (Scheme 2).126
The TycC TE can further catalyse the macrocyclization of peptide substrates that differ significantly from the linear precursor of tyrocidine. Tyrocidine and gramicidin share structural similarity in both their N-terminal (D-Phe-Pro) and C-terminal residues (Val-Orn-Leu). Biochemical results suggest that TycC TE can cyclize the gramicidin S precursor decapeptide-SNAC (D-Phe-Pro-Val-Orn-Leu-D-Phe-Pro-Val-Orn-Leu-SNAC) and a tyrocidine A precursor (D-Phe-Pro-Phe-D-Phe-Asn-Gln-Tyr-Val-Orn-Leu-SNAC) at a comparable rate to the wildtype peptides. These results suggest that the TycC TE can cyclize different substrates provided that the necessary “recognition residues” near each end of the substrate are present.126 Furthermore, the TycC TE tolerates the replacement of residues 5–8 in peptidyl thioesters, and that it shows comparable catalytic efficiency towards various artificial tyrocidine variants, including glycosylated peptide thioesters.128
To further explore the substrate specificity of the TycC TE, Xie et al. established positional-scanning libraries based on SNAC substrate mimics to determine the catalytic activity of TycC TE towards these libraries (Kcat and Km). In general, TycC TE is less efficient in cyclizing such libraries by a factor of 2- to 82-fold in comparison with the substrate sequence of wild type. Analysis of the catalytic efficiency (Kcat/Km) of these reactions suggested that the N-terminal D-Phe1 and C-terminal Orn9 residues are essential for substrate recognition by the TycC TE domain,129 which is consistent with other reported results.130 It was proposed that the efficient occurrence of TycC TE-mediated cyclization requires the formation of key hydrogen bonding partners between the amine group of the Orn side chain and the carbonyl group of the N-terminal aromatic residue.31 In addition, the results obtained using sub-library O3, where the catalytic efficiency showed the greatest reduction (82-fold), suggest that the side chain of the 4th residue significantly influences TE domain activity (Scheme 2).131
In 2006, Wadhwani et al. synthesized gramicidin S utilizing solid-phase Fmoc chemistry. The sequential synthesis starts from the 2-chlorotrityl (2CT) resin loaded with Fmoc-protected D-phenylalanine, and proceeds using typical SPPS conditions until the linear decapeptide 1 was cleaved from the solid support using TFA/iPr3SiH/H2O. An overall yield of 69% was obtained after peptide cyclisation mediated by PyBOP/HOBT in DCM solution and hydrazine-mediated Dde deprotection (Scheme 3).132
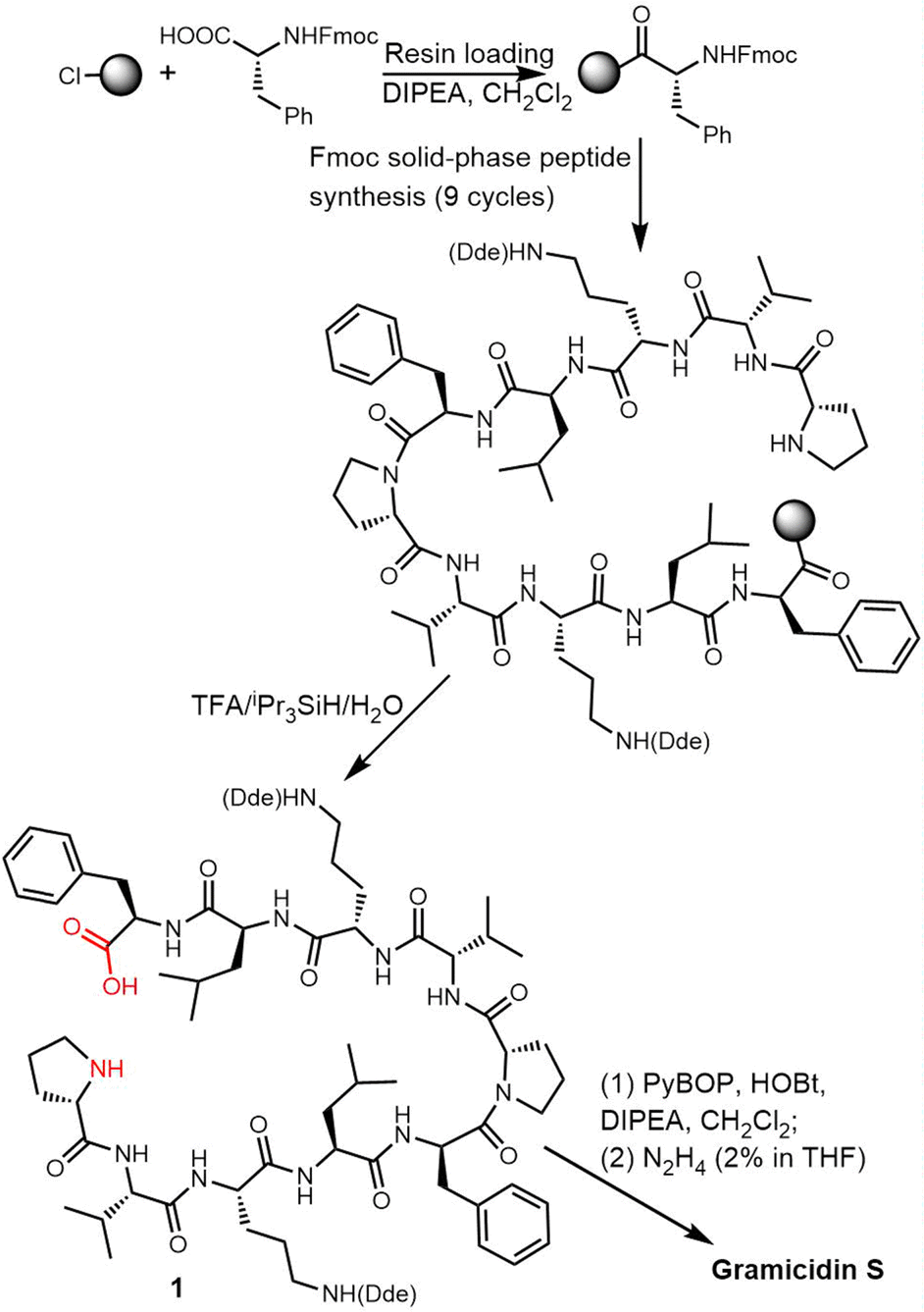 | ||
| Scheme 3 Total synthesis of gramicidin S and its analogues. | ||
Rivera and colleagues developed an on-resin Ugi reaction system for the synthesis of gramicidin S analogues. 2CT resin was used as the solid support for peptide synthesis by solid phase peptide synthesis (SPPS). Next, the intermediate product resin-bound acyclic decapeptide 2 was directly subjected to Ugi macrocyclization to generate a range of gramicidin S analogues (Scheme 4).133,134 Grotenbreg et al. designed an alternate synthesis of gramicidin S via stepwise SPPS using HMPB-resin. After the linear nonapeptide 3 was generated using standard Fmoc-based SPPS, cyclization was performed through the incorporated azide functionality (Scheme 5). After deprotection, the cyclopeptide gramicidin S analogue was obtained in 96% yield.135
 | ||
| Scheme 4 Total synthesis of gramicidin S and its analogues based on Ugi reaction system. | ||
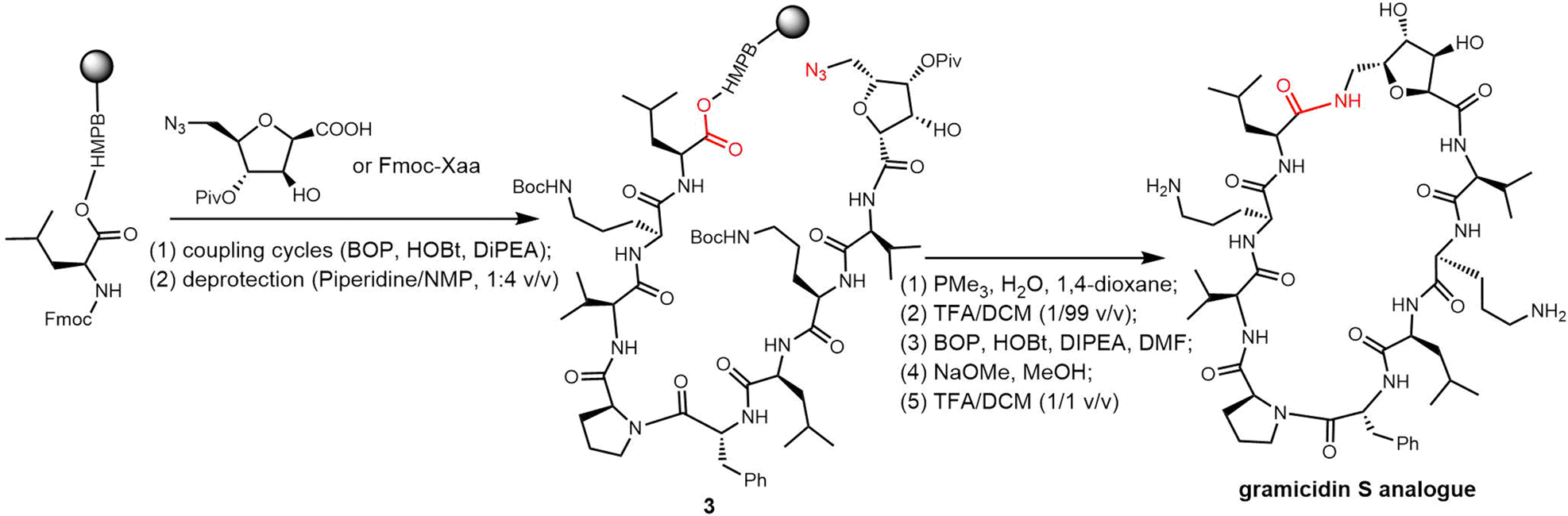 | ||
| Scheme 5 Total synthesis of gramicidin S and its analogues via SPPS using HMPB-resin. | ||
TE domains can be combined with SPPS of linear peptide precursors to cyclize the immobilized peptide, which can greatly enrich the structural diversity of the peptides obtained. As a key feature of the enzymatic synthesis of cyclic peptide molecules via NRPS-mediated biosynthesis is the linkage of an activated linear intermediate peptide to a carrier protein domain via a thioester linkage, this has similarities to SPPS approaches.136 TE domains can act as isolated cyclization catalysts, and further these TE domains can also act on solid-phase linked peptides that contain biomimetic linkers to complete peptide cyclisation, thus enabling the integration of biocatalysis with combinatorial SPPS.137 Kohli and colleagues also synthesized linear peptide SNACs (that mimic the thioester attachment of peptides to the NRPS) to investigate the catalytic function of the TycC TE domain; the results of their study further demonstrate the versatility of this approach for peptide cyclization (Scheme 2).137
Yoon and colleagues recently investigated the macrocyclization of mohangamide A by the TE domain found in the mohangamide NRPS assembly line (Scheme 6). Sequence alignment revealed few differences between the TE domains from Streptomyces sp. SNM55 (WS19T-TE) and S. calvus (Cal19T-TE) (Fig. 2).142 Their biochemical experiments suggested that the mohangamide TE domain displays strict specificity for its substrate, and is only able to catalyse peptide cyclization when both native precursors of mohangamide A are present. This suggests that the structure of the unsymmetrical monomeric peptides is restricted in order to lead to a competent reaction for the WS19T-TE/Cal19T-TE. Further structural investigation is needed to illustrate the detailed catalytic mechanism of this unusual pseudo-dimeric scaffold.143,144
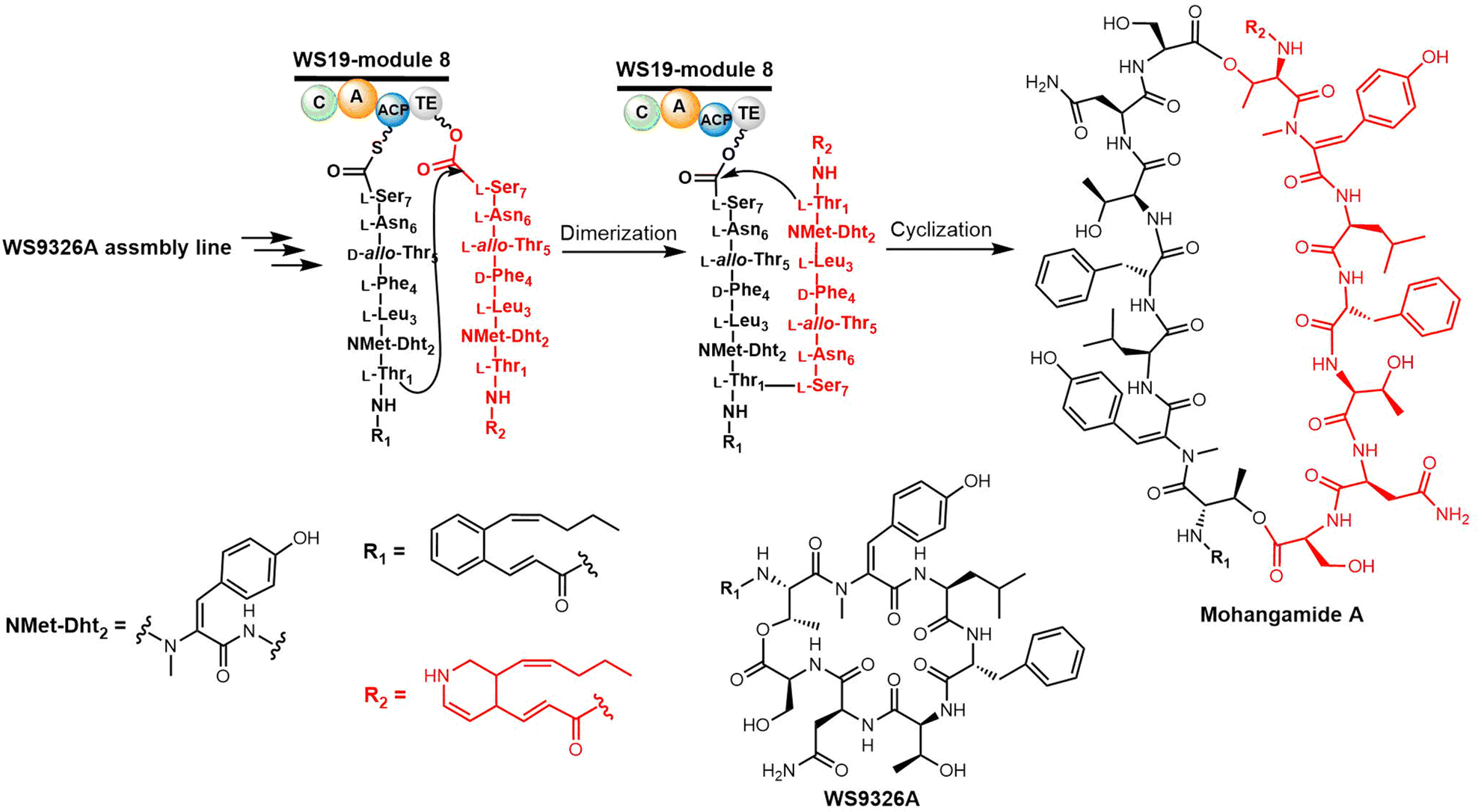 | ||
| Scheme 6 The biosynthetic pathway of mohangamides. | ||
Recently, it was demonstrated that Tyr dehydrogenation occurs during the WS9326A peptide assembly, with the cytochrome P450 Sas16 catalysing the dehydrogenation of the PCP-dipeptide intermediate (Z)-2-pent-1′-enyl-cinnamoyl-Thr-N-Me-Tyr. The P450Sas-mediated incorporation of the double bond follows N-methylation of the Tyr by the N-methyl transferase domain found within the NRPS, and P450Sas appears to be specific for substrates containing the (Z)-2-pent-1′-enyl-cinnamoyl group. The P450Sas structure reveals differences with other P450s involved in the modification of NRPS-associated substrates, including the substitution of the canonical active site alcohol residue with a phenylalanine (F250), which in turn is critical to P450Sas activity and WS9326A biosynthesis. This discovery expanded the repertoire of P450 enzymes that can be used to produce biologically active peptides,145 and raises further questions regarding the specificity of the mohangamide TE domain for the acyl side chains of its substrates.
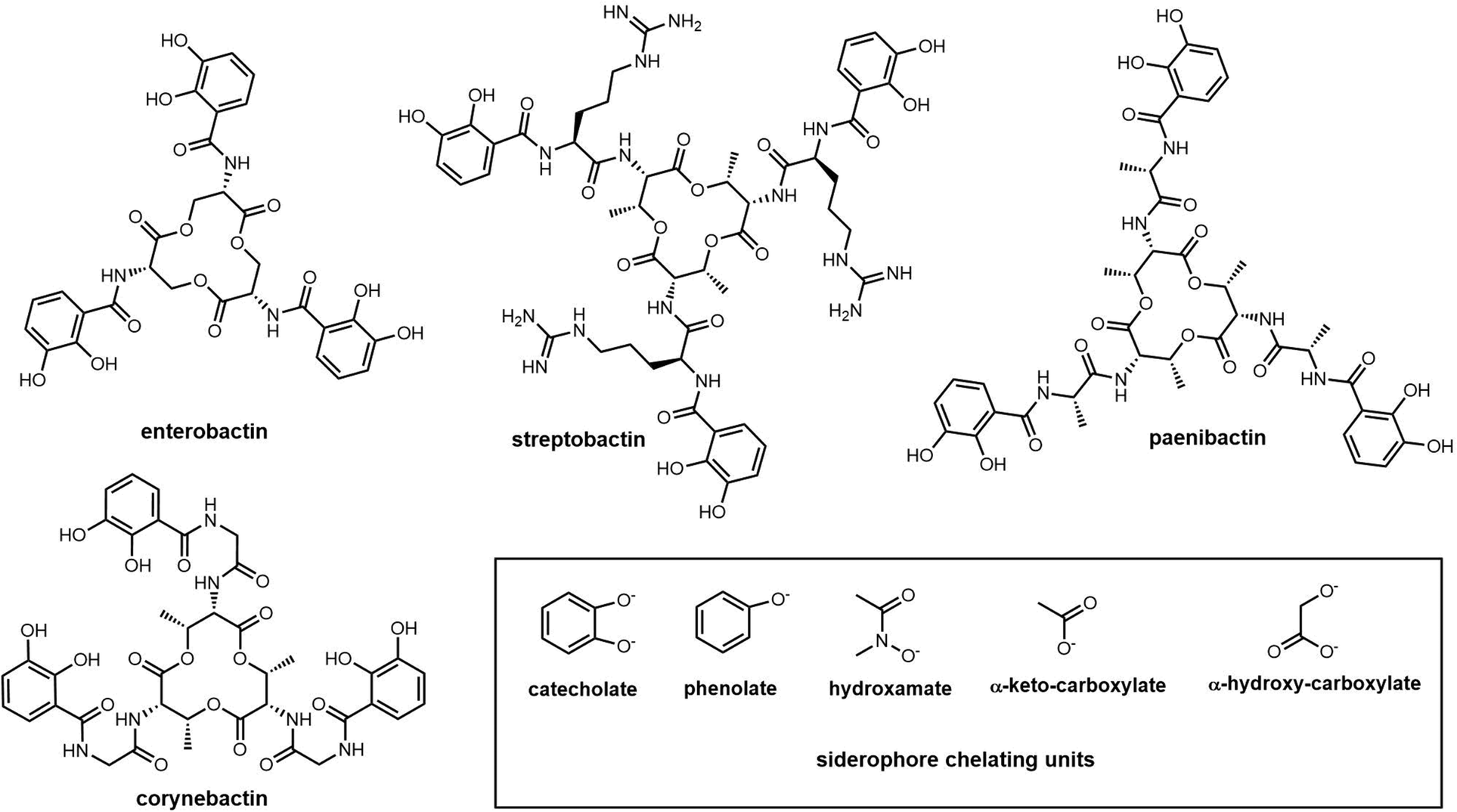 | ||
| Fig. 8 Examples of enterobactin family siderophores and representative siderophore chelating units. | ||
Catechol-type siderophores are widely diverse in their structural composition. In contrast to enterobactin, streptobactin has a cyclic triester-bonded macrocycle scaffold (L-Thr-L-Arg-DHBA)3, which generate a C3 symmetry axis (Fig. 8). Streptobactin was discovered from the marine actinomycete Streptomyces sp. YM5-799, with the corresponding dimer dibenarthin, trimer tribenarthin and monomer benarthin all discovered from the same strain.153 The derivative corynebactin is assembled from the corresponding tri-lactonic scaffold containing the units L-Thr, L-Gly and DHBA (Fig. 8). Corynebactin was discovered from Brevibacterium sp, which are obligate aerobic Gram-positive bacteria found in milk and on human skin.154,155
The enterobactin biosynthetic pathway in E. coli is a well-researched example of a non-linear NRPS assembly line. The enterobactin gene cluster encodes six functional enzymes EntA-F.156,157 The formation of 2,3-dihydroxybenzoic acid (DHBA) requires the activities of EntC, EntB and EntA.156 The three core NRPS enzymes EntE, EntB and EntF comprise the two-module NRPS assembly line, which is responsible for the iterative condensation of three DHBA and three L-serine residues to produce the final iron-chelating product enterobactin.158
EntE is an AMP ligase that activates DHBA through adenylation. EntB, consisting of two domains (C-PCP), accepts the activated dihydroxybenzoate (DHB) and loads this aryl acid on its PCP domain. EntF catalyses the final step in the pathway and comprises four domains (A-C-PCP–TE). In this protein, the A domain initially adenylates a L-Ser residue and transfers it to the adjacent PCP domain. The subsequent C domain catalyses amide bond formation between the 2,3-DHB bound to the upstream EntB Ar-CP domain and the PCP-bound L-Ser residue to generate DHBA-Ser-S-PCP in EntF. The EntF C domain has been demonstrated to possess specificity for L-Ser residue as the downstream (acceptor) substrate.159,160
Cyclotrimerization is required to create the trilactone backbone of enterobactin, which is performed by the final two domains of EntF (PCP–TE) via sequentially transferring oligomer intermediates between the 4′-PPant arm of the PCP domain and the active site serine residue of the TE domain (Scheme 7). The EntF TE domain therefore fulfils roles as both a terminal domain for acyl transfer from the PCP domain and an extended catalyst for enterobactin trimerization. Through three cycles of iterative (2,3-dihydroxybenzoyl)-L-serine (DHB-Ser) synthesis, three DHB-Ser moieties are connected whilst remaining linked to the TE domain by ester bond. Finally, the linear DHB-Ser trimer is cyclized and released from TE domain (Scheme 7).160,161 Structurally, EntF TE is similar to homologous thioesterase domains, such as AB3403-TE, Vlm-TE, FenTE and SrfTE.162 Using solution phase nuclear magnetic resonance (NMR) spectroscopy, Frueh et al. have also successfully elucidated the structure of the apo-PCP–TE didomain (PDB: 2ROQ, 37 kDa) of the enterobactin NRPS synthetase EntF (vide infra).69
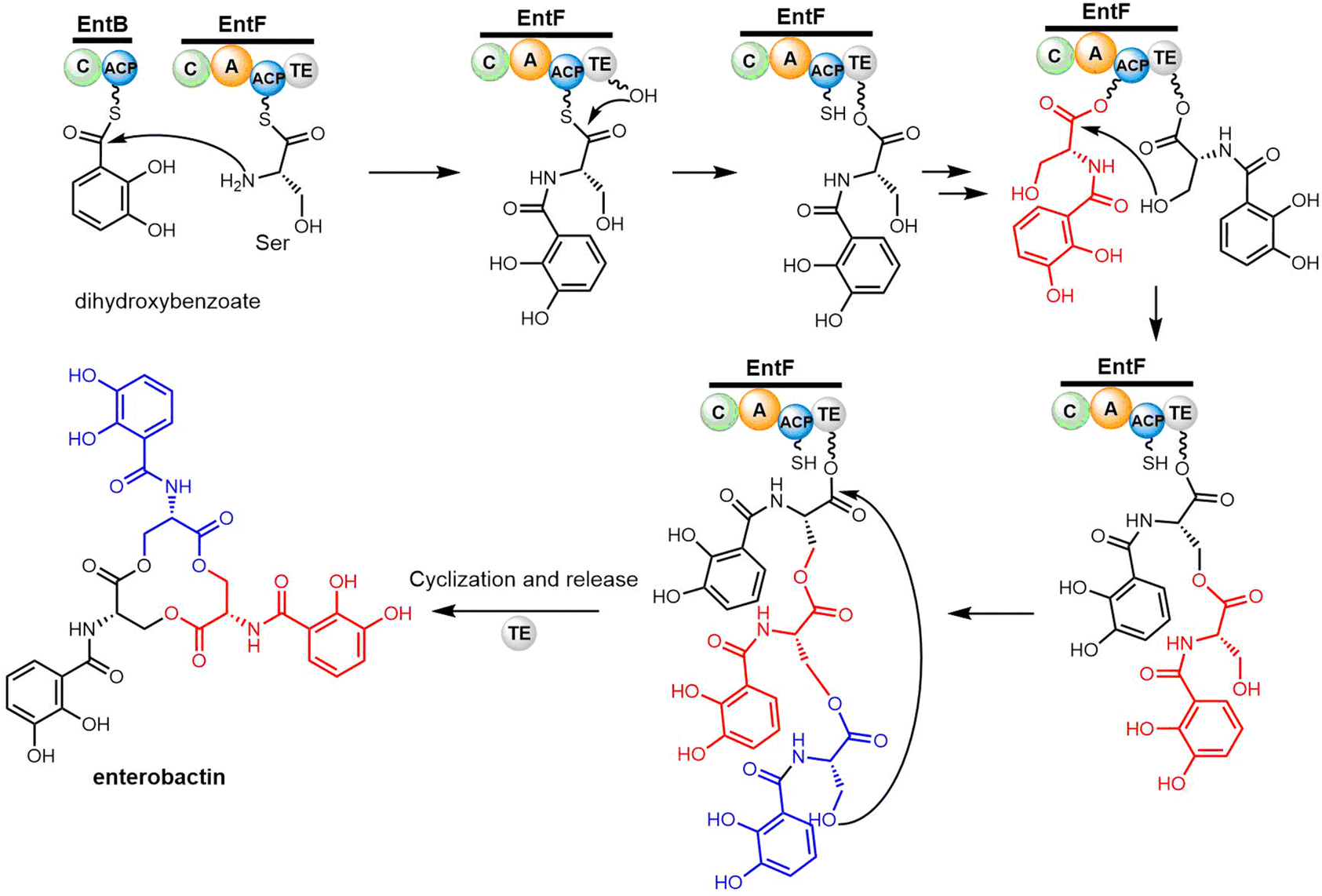 | ||
| Scheme 7 The biosynthetic pathway of enterobactin. | ||
Even though some factors that impact the cyclization and hydrolysis of TE domain mechanism have been investigated for NRPSs, the fundamental structural basis for both iteration and cyclization remain elusive. Whilst multiple sequence alignment of triscatechol siderophore TE domains provides possible clues, the exact mechanism (iteration, hydrolysis, and cyclization) awaits further elucidation.163 Structurally, the conserved Ser-His-Asp catalytic triad (Ser1138, His1271, Asp1165) is vital for EntF-TE thioesterase activity. Ser1138 acts as the nucleophile that binds DHB-Ser precursors. Mutation of Ser1138 to Ala results in complete disruption of substrate oligomerization. Mutation of His1271 to Ala can still generate enterobactin but with an approximately 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold decrease in yield.158,164 Interestingly, the residue Pro1073 is conserved in the EntF and other PCP–TE homologues, and it was recognized to play important role of stabilizing the oxyanion hole in these structures (Fig. 9).165
000-fold decrease in yield.158,164 Interestingly, the residue Pro1073 is conserved in the EntF and other PCP–TE homologues, and it was recognized to play important role of stabilizing the oxyanion hole in these structures (Fig. 9).165
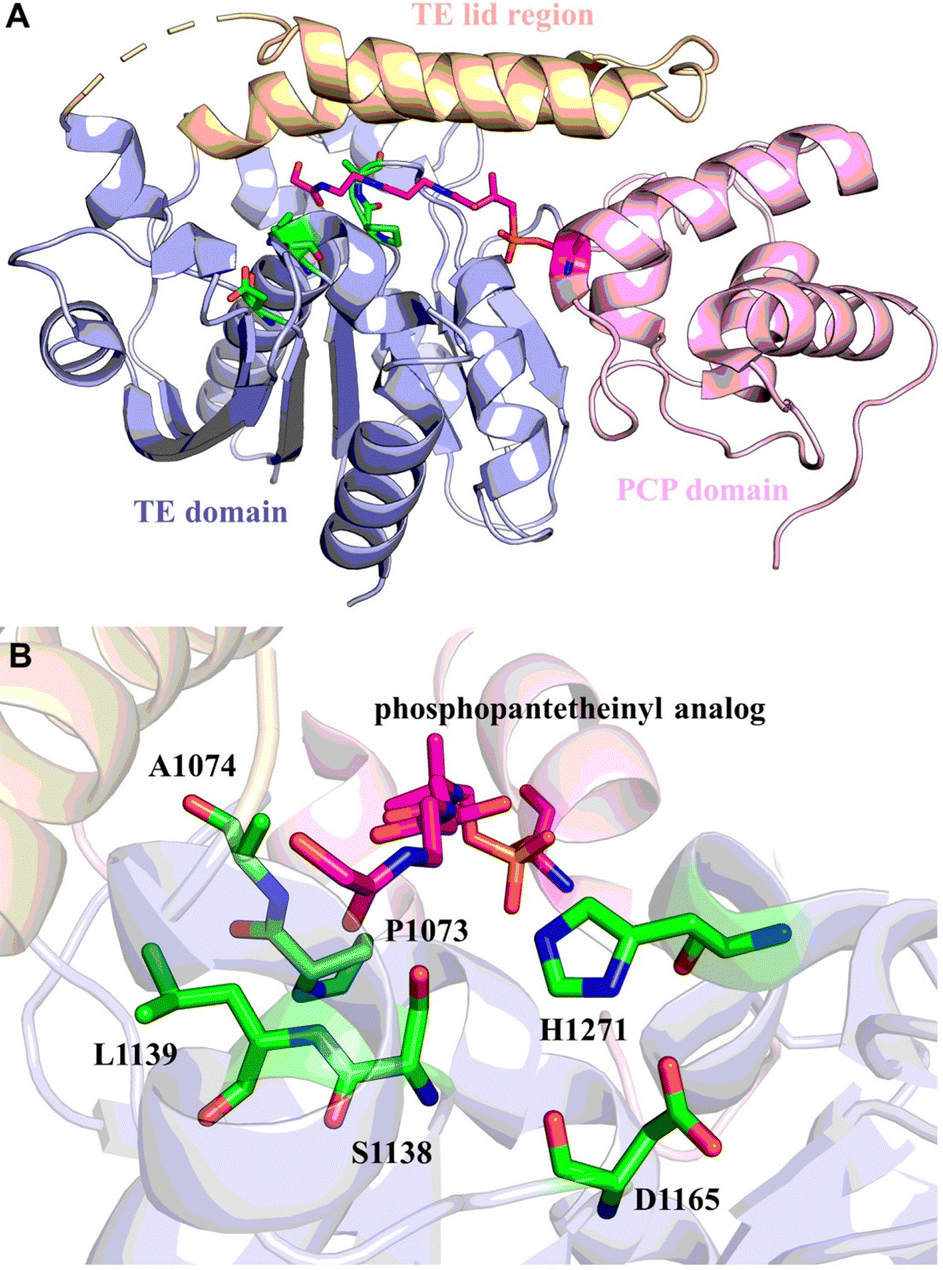 | ||
| Fig. 9 Structure of the PCP–TE di-domain from EntF. (A) The overall structure of the PCP–TE conjugate. The PCP domain (pink) is connected to the TE domain (light blue) through a short linker. The TE lid region is shown in wheat. (B) Interaction of the 4′-PPant arms with the active site of the EntF-TE domain. Residues of the catalytic triad (S1138, H1271, D1165) and oxyanion hole (A1074 and L1139) are shown as green sticks. The phosphopantetheinyl analog is shown as magenta stick. | ||
Iterative biosynthesis facilitates the repeated use of modules and necessitates a “waiting room” for reaction intermediates.164 According to solution NMR and protein interaction analysis, the catalytic pocket formed by PCP–TE di-domains is dynamic. The lid region of the EntF holo-PCP–TE (PDB 3TEJ) domain needs to fluctuate to accommodate various intermediates in the hydrophobic pocket and to remain open to the 4′-PPant arm of the corresponding PCP domain bearing the tripeptide.69 In addition, the process of cyclotrimerization, which is facilitated by the EntF TE, presumably involves an interaction between the two domains, allowing the structure bound to the PCP domain to be transferred into the substrate pocket of the TE domain.165
Protein–protein interactions are also hypothesized to influence the catalytic function of TE domains from specific systems. The Cryle group reported the characterization of the TE domain from the teicoplanin NRPS.51 Based on the results of a series PCP-substrate cleavage activity assays, they found that the activity of the Tcp12-TE domain is dependent on the presence of an unusuallylong N-terminal linker region, which appears to be exclusive to the NRPS machinery seen in GPA biosynthesis. In addition, the longer Tcp12-TE (L-TE) domain of the teicoplanin NRPS has a strong selectivity for the PCP-bound peptide in its optimal cross-linking condition,51 indicating that the TE in GPA biosynthesis acts as a logic gate for the complex X-domain/P450 mediated peptide cyclisation cascade.126,166,167
TE domains are relatively flexible and possess significant conformational diversity. To trap a PCP complex with a TE, Bruner and colleagues utilised the chemical probe α-chloroacetyl-amino-coenzyme A, which is a mimic of the natural thioester substrate targeting the Ser residue in the catalytic triad (Ser1138, His1271, and Asp1165) of TE domains (Fig. 9). After bioconjugation with the PCP domain via the activities of a phosphopantetheinyl transferase, the probe allowed a stable complex of the PCP–TE didomain protein to be trapped, with the resultant structure demonstrating the interaction between the TE and PCP domains.165,168 The Gulick lab revealed that the EntF-TE domain also exhibits significant conformational mobility, as it can adopt multiple positions with respect to other domains based on the analysis of negative-stain electron microscopy.162,169 Based on these structures, it is postulated that the mature PCP domain-bound peptide chain can be transferred to the TE domain via rotation of the linker attached to PCP.162
The total synthesis of enterobactin was first reported in 1977.170 Shanzer et al. developed a synthetic route to the enterobactin cyclic trilactone backbone (4) using an organotin template. This method required fewsynthetic steps, but delivered a relatively low yield of cyclic trilactone (23%).171 In 1997, Ramirez et al. designed a strategy to improve the synthetic yield of enterobactin. Using methyl N-triphenyl-methyl-L-serinate (5) as starting material and a variation on Shanzer's organotin template, they also synthesized the cyclic trilactone (4) intermediate with a much-improved yield (81%) (Scheme 8).172
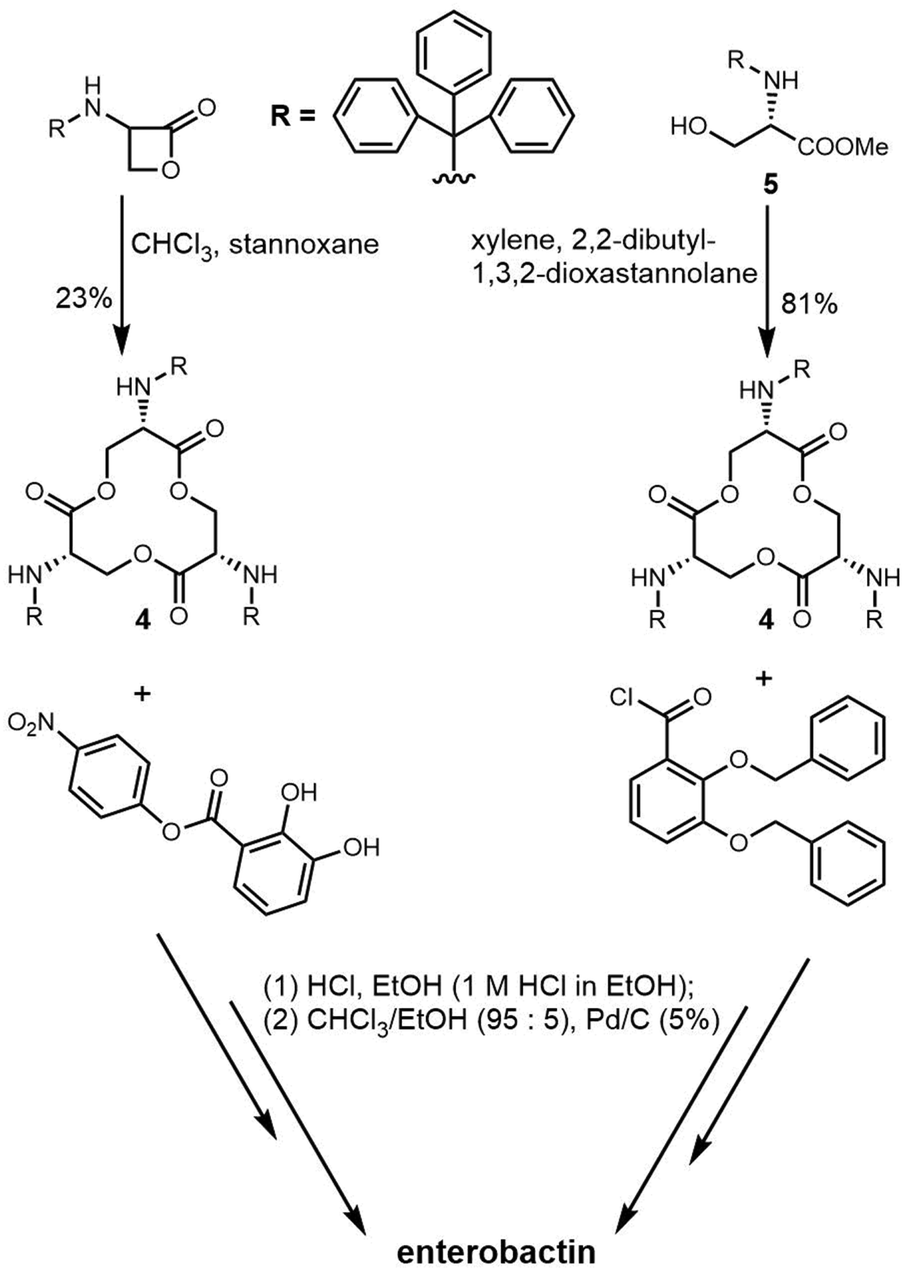 | ||
| Scheme 8 Total synthesis of enterobactin. | ||
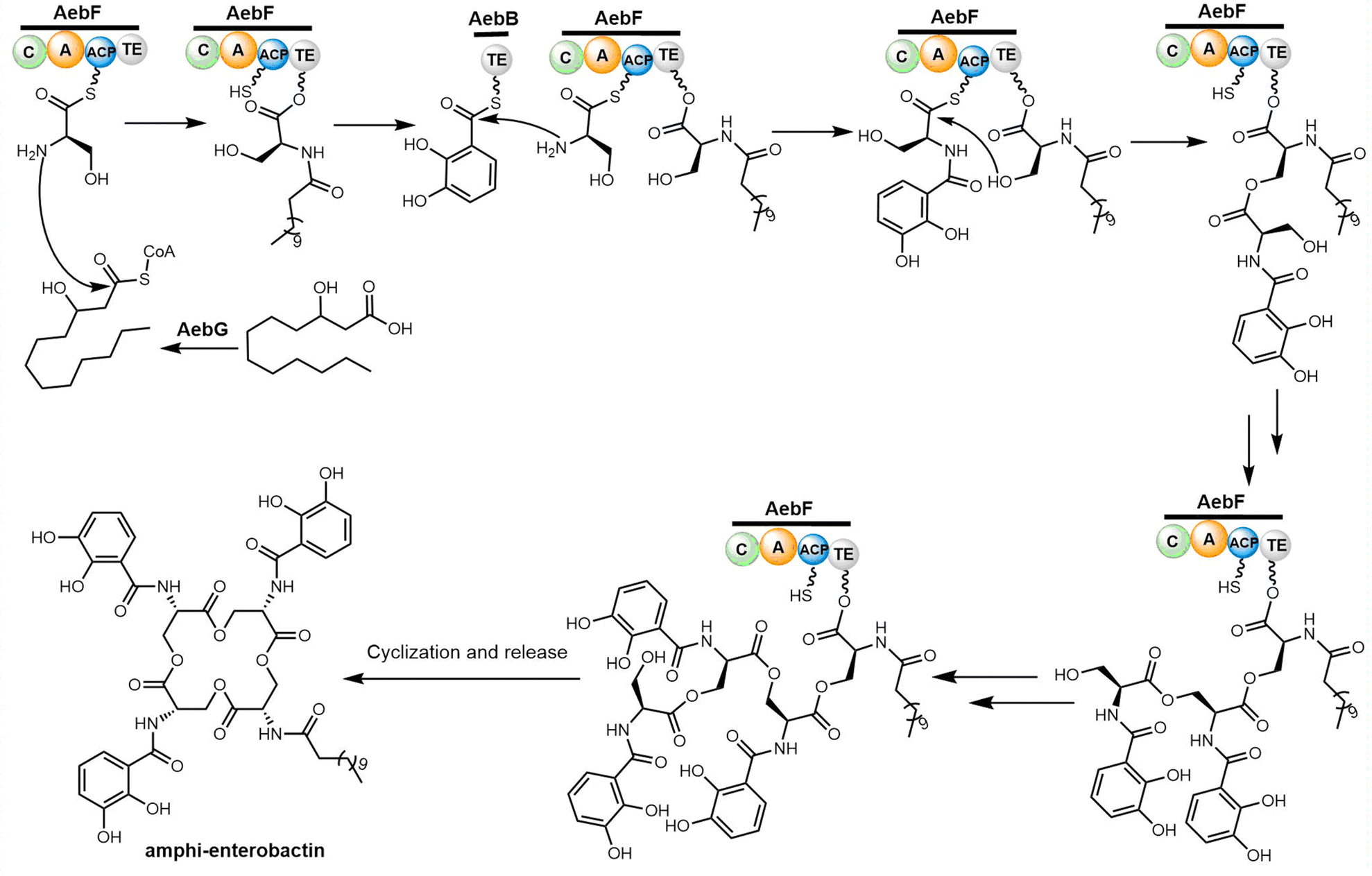 | ||
| Scheme 9 The biosynthetic pathway of amphi-enterobactin. | ||
Through reconstitution and heterologous expression, it was demonstrated that the NRPS enzyme AebF catalyses amide bond formation and cyclization of the tetralactone backbone. The C domain in this single module NRPS performs bifunctional condensation catalytic activity (Scheme 9). The long-chain fatty acid CoA ligase (FACL) AebG is required to activate the fatty acid as a fatty acid CoA thioester. In contrast to the trilactone backbone of enterobactin, the AebF-TE can accommodate the larger 16-member tetralactone backbone of amphi-enterobactin.164,173 Based on the “waiting room” model, the AebF-TE serves as a temporary attachment site for L-Ser-DHBA during amphi-enterobactin assembly.
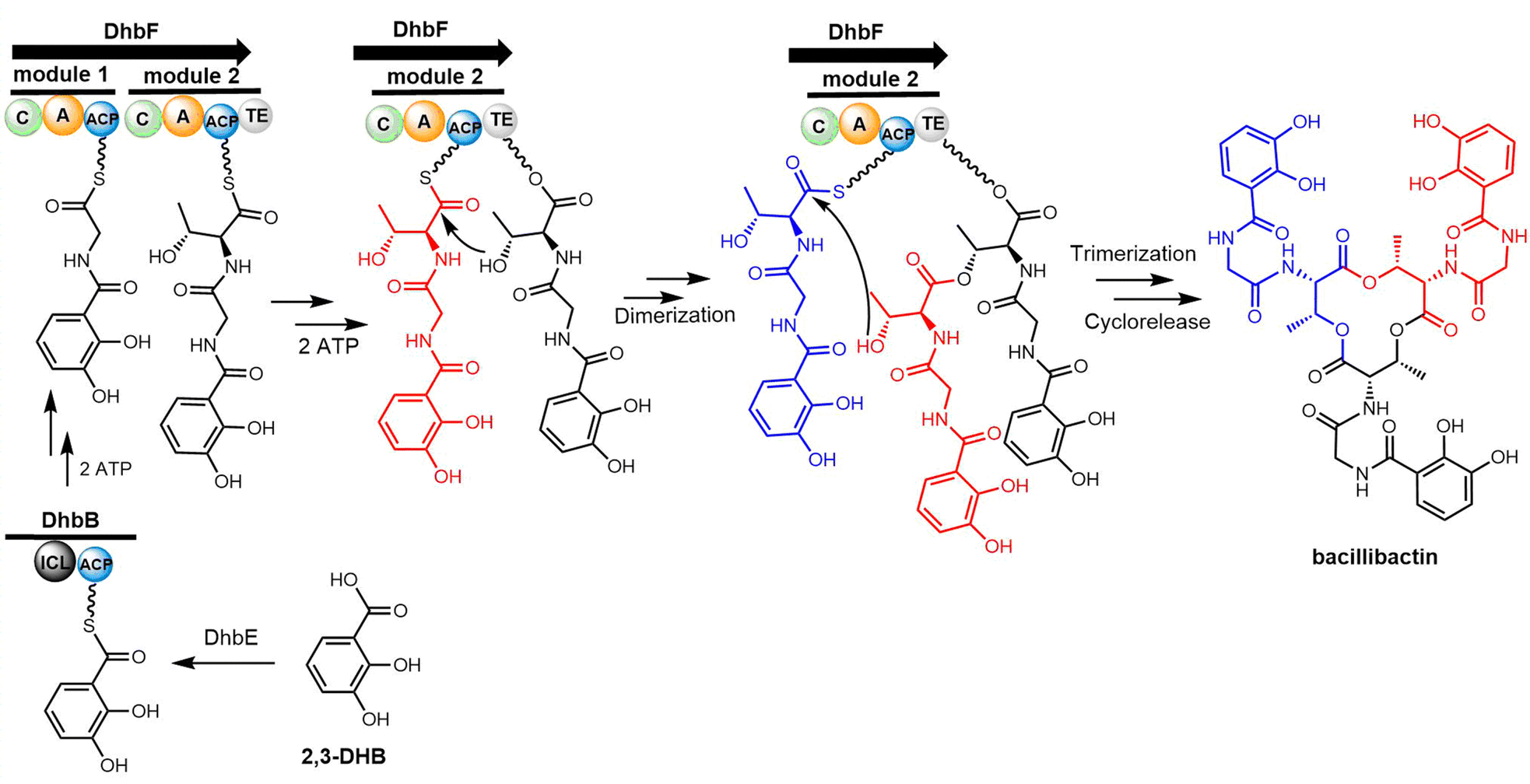 | ||
| Scheme 10 The biosynthetic pathway of bacillibactin. | ||
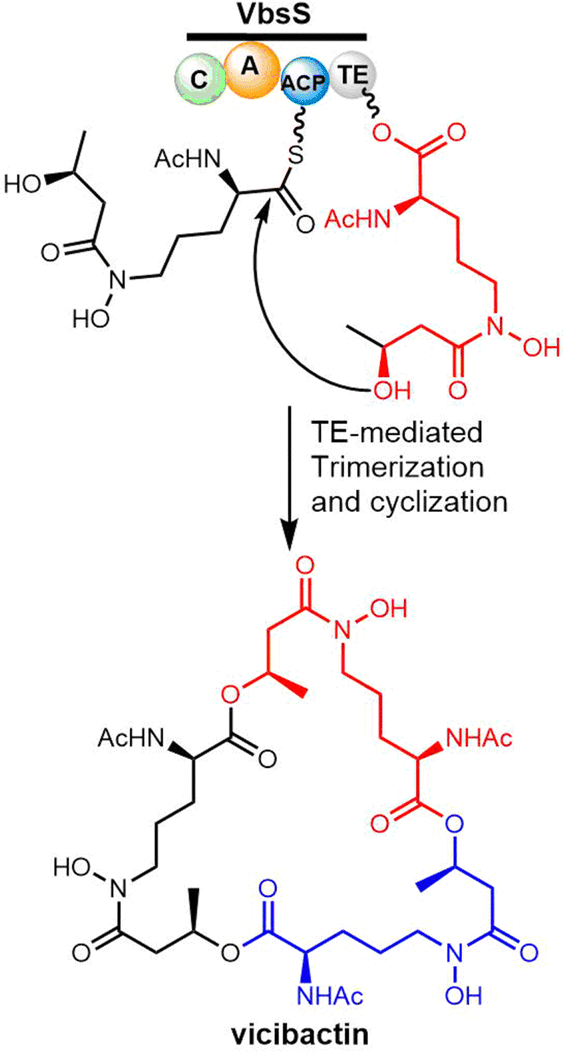 | ||
| Scheme 11 The biosynthetic pathway of vicibactin. | ||
Many studies have reported the genes related to vicibactin biosynthesis and proposed the steps of biosynthetic pathway.180,181 The biosynthesis of vicibactin produced by the symbiotic bacterium Rhizobium leguminosarum involves a cluster of eight genes: vbsG, vbsS, vbsO, vbsA, vbsD, vbsL, vbsC and vbsP.163 Among them, VbsS, a NRPS enzyme (C-A-PCP–TE) plays a crucial role in the assembly of the vicibactin scaffold. VbsS activates L-N5-hydroxy-ornithine and attaches it to the PCP domain via covalent attachment as an acyl thioester. Further, the assembly of vicibactin siderophore involves acylation catalyzed by VbsA, followed by epimerization and acetylation mediated by VbsL. In the last step, trimerization and cyclization are mediated by the TE domain of VbsS (Scheme 11). The cyclotrimerization reaction, catalyzed by VbsS, is similar to that catalyzed by EntF in the biosynthesis of enterobactin.181
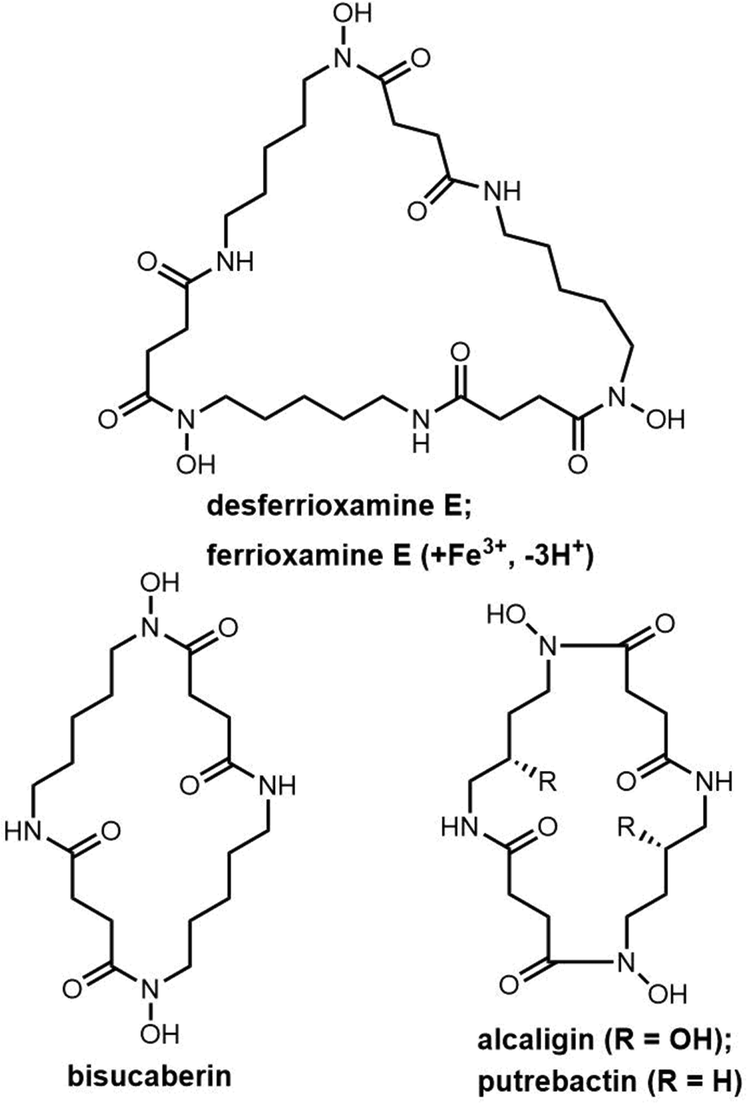 | ||
| Fig. 10 Examples of cyclic siderophores. | ||
The siderophore synthetase DesD, a member of the NIS synthetase superfamily, catalyses the biosynthesis of desferrioxamines in Streptomyces coelicolor M145. DesD iteratively catalyses the ATP-dependent condensation of the repeating units of N-hydroxy-N-succinyl-cadaverine to form the dimer or trimer.182,183 In the biosynthesis of bisucaberin, the BibC multidomain enzyme from Vibrio salmonicida has been identified as catalyzing the macrocyclization of the monomer N-hydroxy-N-succinyl-cadaverine to produce bisucaberin.184 The closely related analogue PubC catalyzes specific macrocyclization and cyclotrimerization in putrebactin biosynthesis (Scheme 12).185
 | ||
| Scheme 12 The biosynthetic route to bisucaberin and putrebactin. | ||
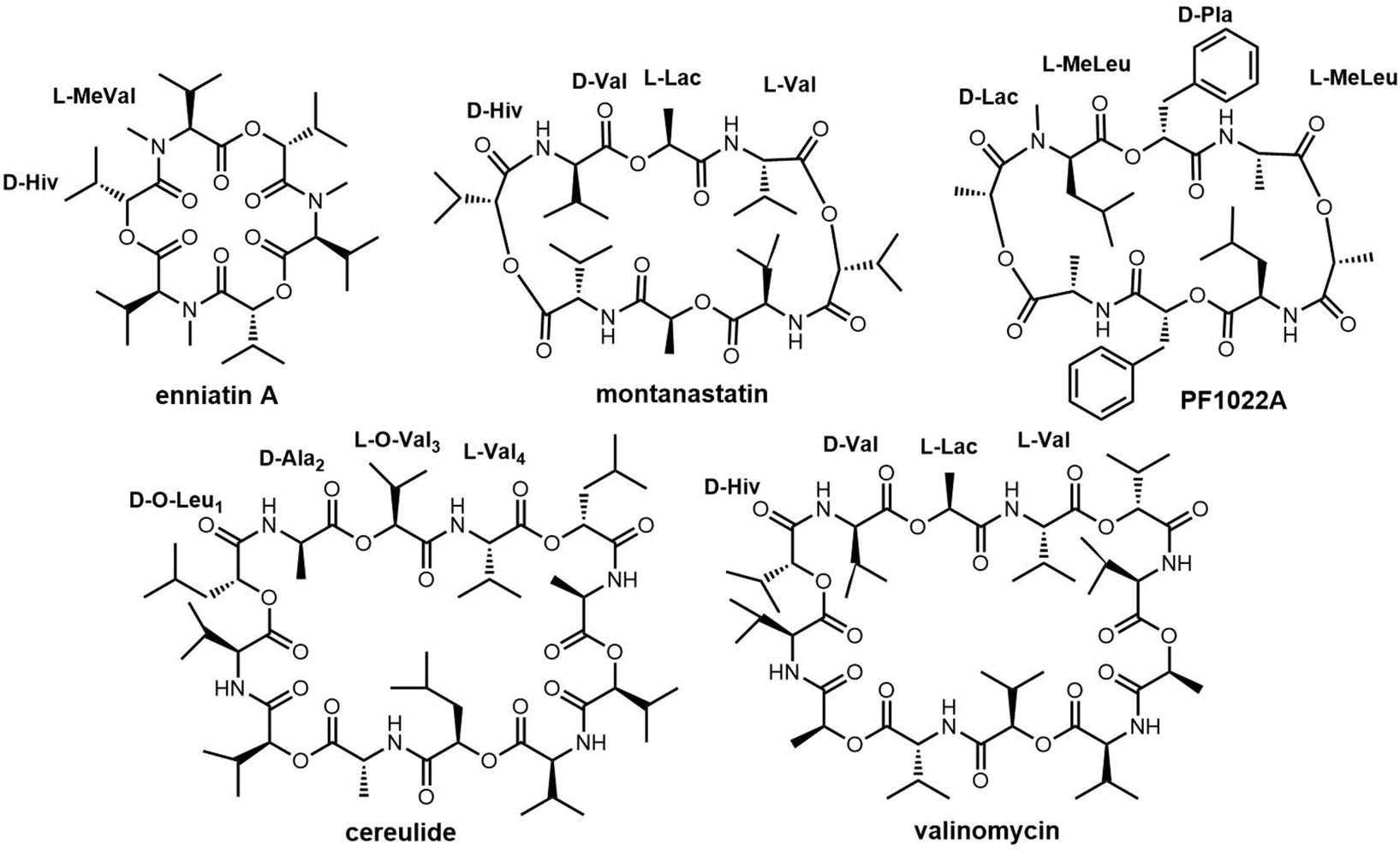 | ||
| Fig. 11 The chemical structures of valinomycin and similar symmetric cyclopeptides. D-Hiv = D-α-hydroxyisovaleric acid; D-Hmp = D-alpha-hydroxy-4-methylpentanoic acid; L-Lac = L-lactic acid; L-MeLeu = L-N-methyl-leucine; D-Pla = D-phenyl-lactic acid. | ||
Cheng et al. first reported the complete biosynthetic gene cluster of valinomycin (termed as vlm) in Streptomyces tsusimaensis ATCC 15141.191 The vlm gene cluster encode two NRPS enzymes: Vlm1 and Vlm2. Vlm1 (370 kDa) includes two modules: A1-KR1-PCP1-C2-A2-PCP2-E2, and Vlm2 (284 kDa) includes another two modules: C3-A3-KR3-PCP3-C4-A4-PCP4-TE, a total of 16 domains constituting 4 modules, each of which is responsible for the incorporation of the building blocks D-α-hydroxyisovaleric acid (D-Hiv), D-valine (D-Val), L-lactate (L-Lac), L-valine (L-Val). The NRPS domain system is homologous to the assembly of related cyclopeptide cereulide (Scheme 13).192
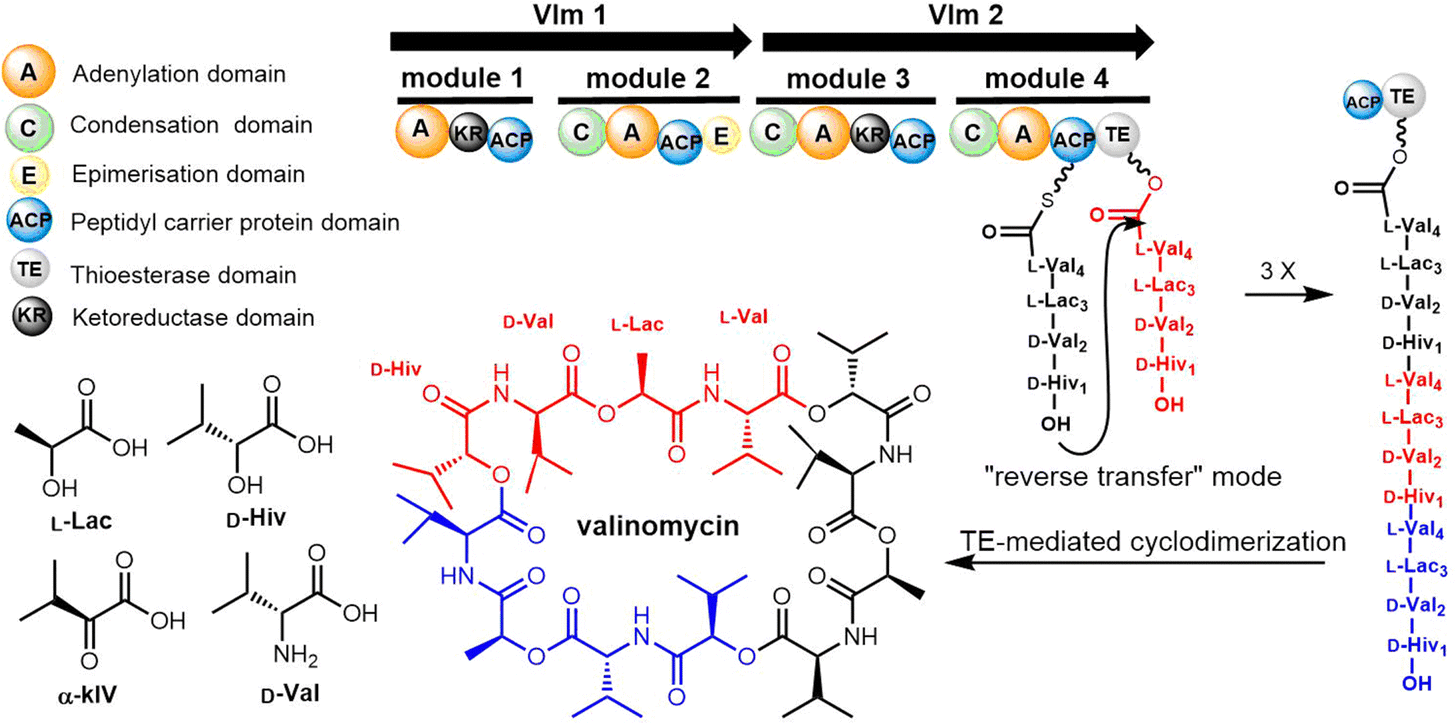 | ||
| Scheme 13 The biosynthetic pathway of valinomycin. | ||
In the valinomycin biosynthesis pathway, the tetrapeptide precursor is synthesised three times, with each monomer synthesis halting at the TE domain. The Vlm2 TE domain catalyses the final oligomerization, cyclization and the release of valinomycin (Scheme 13).193 Montanastatin was discovered as a shunt product of valinomycin. These molecules both comprise the same tetradepsipeptide basic unit, but montanastatin's basic unit only repeats twice, while valinomycin's basic unit repeats three times. Studies have shown that both valinomycin and montanastatin follow the same biosynthetic pathway, which implies that the TE domain loosely controls the basic unit repetition in the valinomycin system.191 It remains unclear how the TE domain can regulate the number of iterations during cyclo-oligomerization.
In 2023, Konno et al. created a substrate peptide-based p-nitrophenyl phosphonate that forms a stable covalent complex with recombinant TycC TE. This method is useful for comprehending substrate identification and substrate-TE interaction during macrocyclization.194 Similarly, recent work by the groups of Schmeing and Chin has provided further structural insights into TE mechanisms, focusing on the Vlm2 TE. By employing genetic code expansion to install the amine-containing analogue 2,3-diaminopropionic acid (Dap) in the active site, they were able to stabilize and trap acyl-TE intermediates. Using this method, the first and last acyl-TE intermediates in the catalytic cycle as Dap conjugates were trapped and resolved, which provides structural insight into the conformational changes in this TE domain that control the oligomerization and cyclization of linear substrates. Moreover, this Dap substitution strategy can be widely used for protein–ligand related characterization or assays. According to this study, the Vlm TE (PDB: 6ECE) lid region could contribute by changing the shape of the depsipeptide chain to encourage intramolecular cyclization. By comparing the protein structure between tetradepsipeptidyl-TEDAP (6ECD) and the dodecadepsipeptidyl-TEDAP (6ECE and 6ECF), significant conformational changes could be observed in the lid region (α4–α8) (Fig. 12). It was proposed that this lid rearrangement is necessary for reorganizing the substrate during the cyclo-oligomerization.70 In addition, by capturing the key intermediate (octadepsipeptidyl-SNAC), their investigation confirmed that Vlm TE mediates peptide oligomerization in the “reverse transfer” mode (Scheme 13), and other NRPS-TEs would also be expected to catalyse the peptide ring dimerization in this manner.
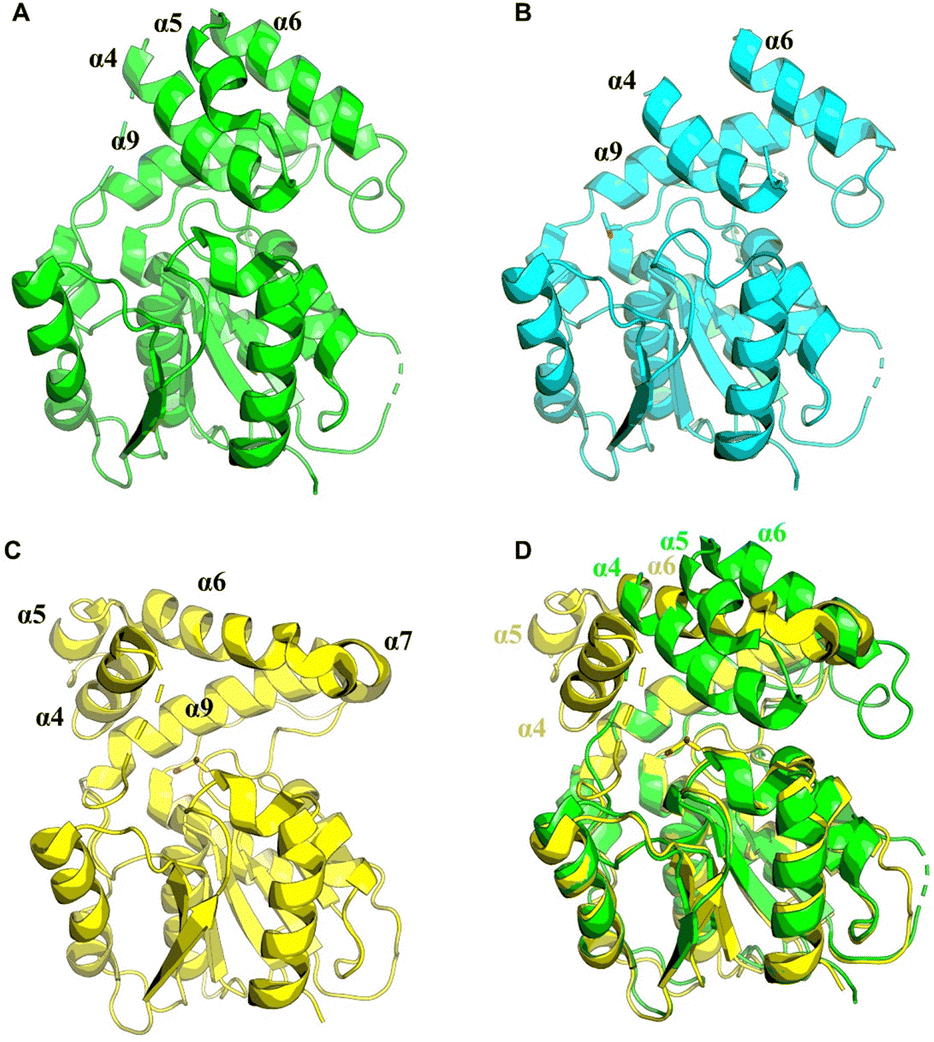 | ||
| Fig. 12 The structural alterations in lid helices α4–α9 among the Vlm TE (A), tetradepsipeptidyl-Vlm TEDAP (B) and dodecadepsipeptidyl-Vlm TEDAP (C). The structures of Vlm2 TE and dodecadepsipeptidyl-Vlm TEDAP were overlaid for comparative analysis (D). | ||
Shemyakin et al. first reported the chemical synthesis of valinomycin in 1963. The open-chain depsipeptide was synthesized using SPPS, which was subsequently cyclized into valinomycin using acid chloride after being cleaved from the resin. Many other reported syntheticroutes follow essentially the same strategy.188,195–197 In 2020, Li and colleagues developed a new biosynthetic method of valinomycin, in which they used a cell-free protein synthesis (CFPS) system to express the entire valinomycin BGC, giving rise to about 37 g L−1 of valinomycin. This demonstrated for the first time that a single-pot CFPS reaction could enable the complete biosynthesis of a complex nonribosomal peptide.198
Cereulide consists of three monomers each comprising the four amino acids D-O-Leu, D-Ala, L-O-Val and L-Val. The BGC of cereulides and isocereulides have been characterized as the ces NRPS from Bacillus cereus.204–206 The cereulide synthetase gene cluster (ces, 24 kb) contains 7 genes: cesH, cesP, cesT, cesA, cesB, cesC and cesD. The biosynthesis of cereulide is closely controlled via post-transcriptional and post-translational regulation.207 The NRPSs CesA and CesB complete the assembly of three repeating tetrapeptide motifs (D-O-Leu-D-Ala-L-O-Val-L-Val) as the cereulide backbone. In addition, CesB2 is a termination module containing the TE domain, which releases the mature nonribosomal peptide by cyclization (Scheme 14).191,208 CesB2 catalyzes the trimerization and macrocyclization of tetrapeptide substrates to finally generate cereulide. Biochemical studies have shown that this TE domain is selective for the nucleophilicity of hydrolyzed deaminopeptidyl-TE intermediates.209
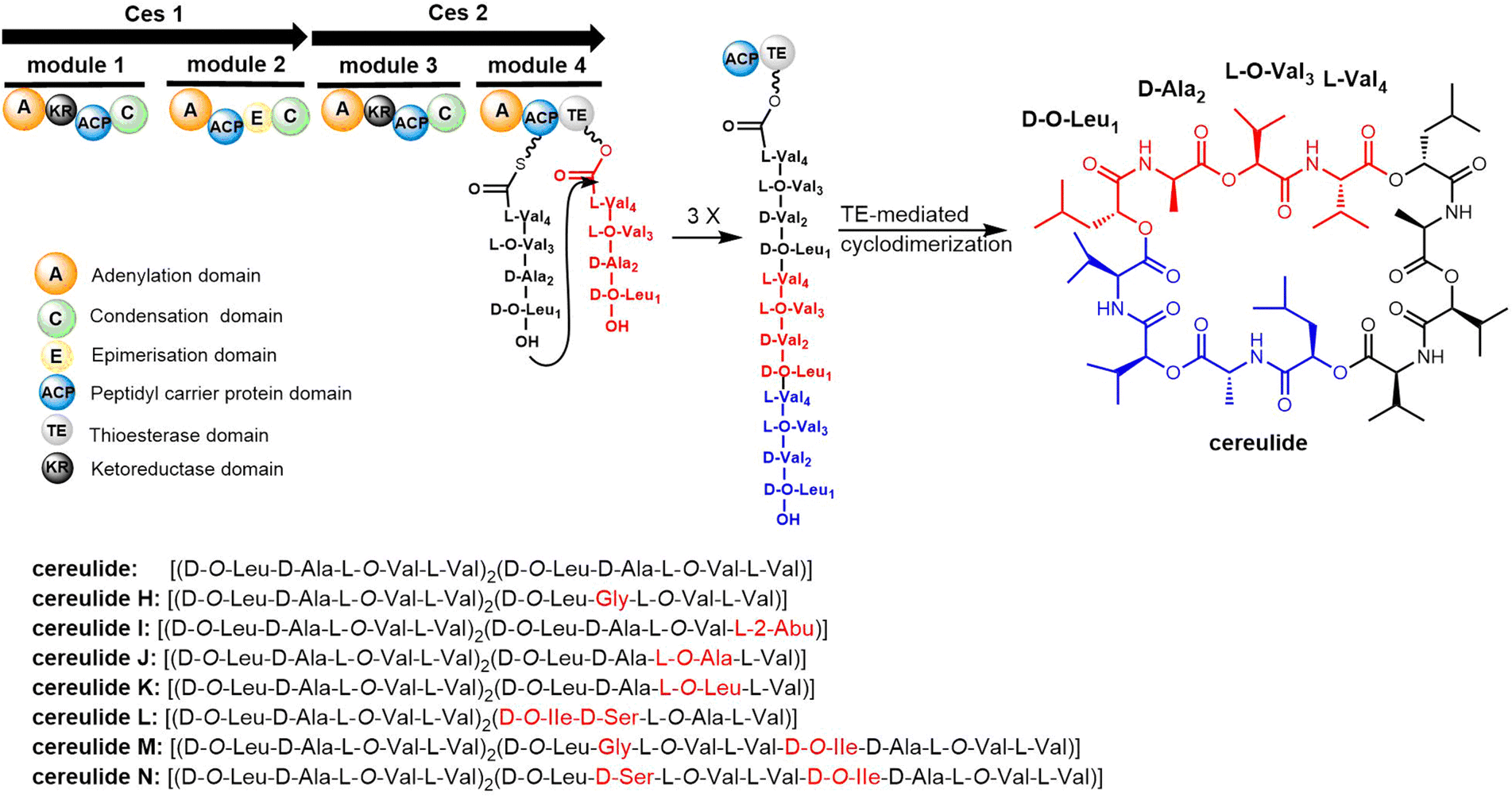 | ||
| Scheme 14 The biosynthetic pathway of cereulides. | ||
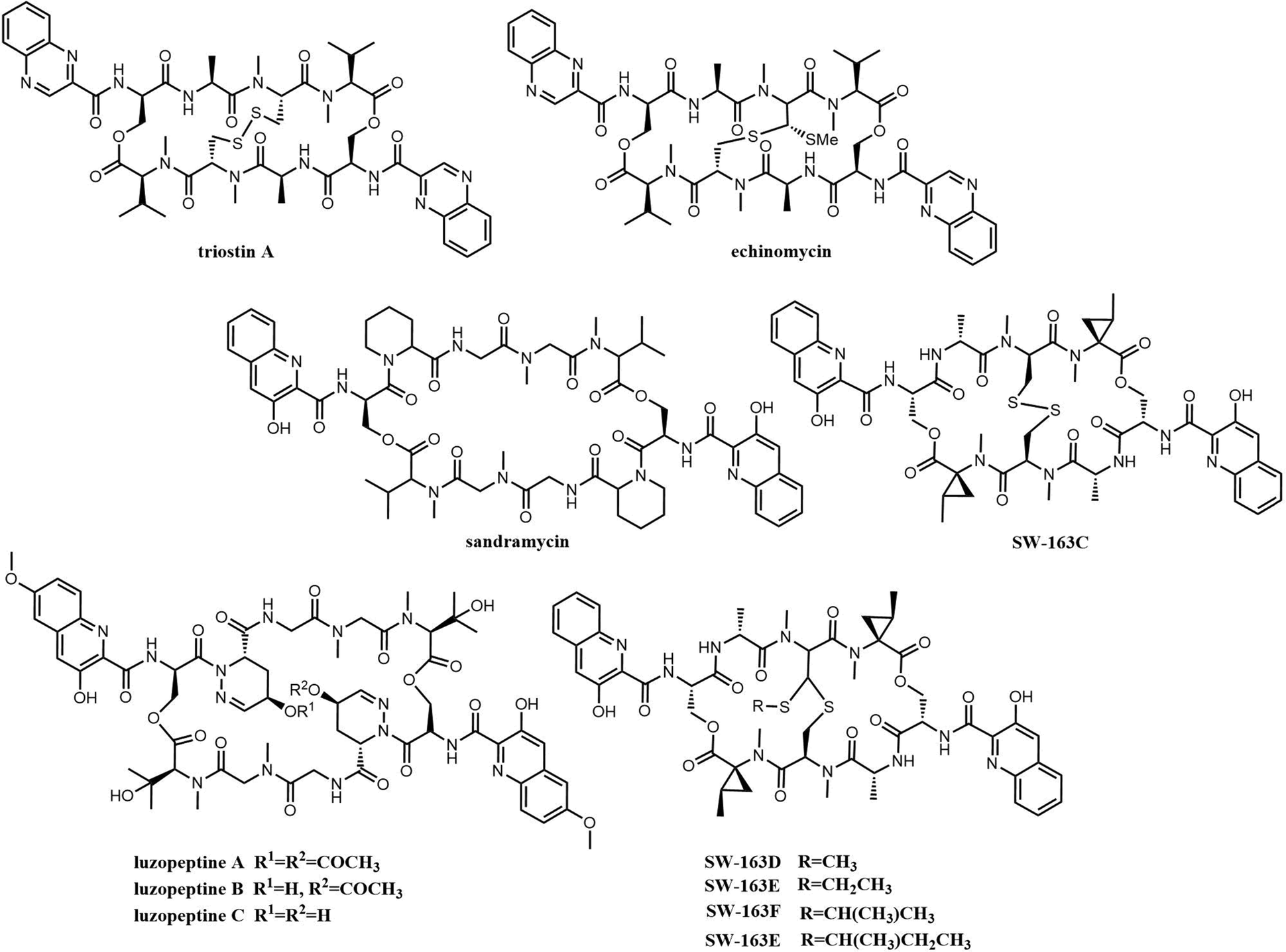 | ||
| Fig. 13 Examples of bisintercalator depsipeptides. | ||
Bisintercalator depsipeptides feature a C2-symmetric macrocyclic scaffold, which is a cyclic structure formed by various amino acids, including two identical planar bicyclic heteroaromatic chromophore moieties that are found at opposite ends of this symmetric peptide scaffold. These chromophore units are the first moieties incorporated during the assembly of these by NRPSs, and is also the reason why these compounds are known as bisintercalators (Fig. 13).211 The chromophore subunits employed in bisintercalators are 3-hydroxy-quinaldic acid (3HQA) and quinoxaline-2-carboxylic acid (QXCA).
This general structure enables bisintercalators to position the two chromophores on the peptide scaffold and in such a manner that they interact deeply with the minor groove of DNA.212 This structure leads to the unwinding and extension of the DNA helix, causing changes in a variety of cellular processes related to replication and transcription and that ultimately result in the death of the affected cells.213
Many of these compounds (thiocoraline, triostin, SW-163 and echinomycin) share high degrees of similarity in terms of chromophores and the number and type of amino acids, which suggests that they originate from similar biosynthetic pathways.214
Echinomycin B, C, D and E have been identified as being produced by the same assembly line, and occur as a consequence of the alternate incorporation of a variety of branched amino acids such as isoleucine indicating a relaxed specificity for some A-domains within the NRPS enzymatic assembly line.218,219 Echinomycin (Quinomycin A) is the most widely studied member of the quinoxaline/quinoline antibiotic group and comprises a C2-symmetric scaffold containing the cross-bridged cyclic octapeptide dilactone core linked by twin quinoxaline/quinoline 2-carbonyl chromophores. Like other bisintercalators, echinomycins show significant inhibitory against cancer stem cells and hypoxia-inducible factor-1 (HIF-1) by inhibition of DNA replication and RNA synthesis.220
The quinomycin biosynthetic gene cluster encodes the enzymes that perform its biosynthesis in two key steps. The first is the synthesis of quinoxalin-2-carboxylic acid (QXCA), which involves not only the secondary metabolic pathway but also the ACP protein from fatty acid metabolism.221–223 Next, the assembly of the echinomycins is performed by an NRPS (Ecm6, Ecm7) that iteratively catalyses the assembly of the linear tetrapeptide peptide precursor. Ecm1 and FabC assist the loading of the chromophore quinoxaline-2-carboxylic acid. The echinomycin TE domain (Ecm TE), located in the final module of Ecm7, is responsible for the dimerization, final cyclization and release of echinomycin (Scheme 15).224
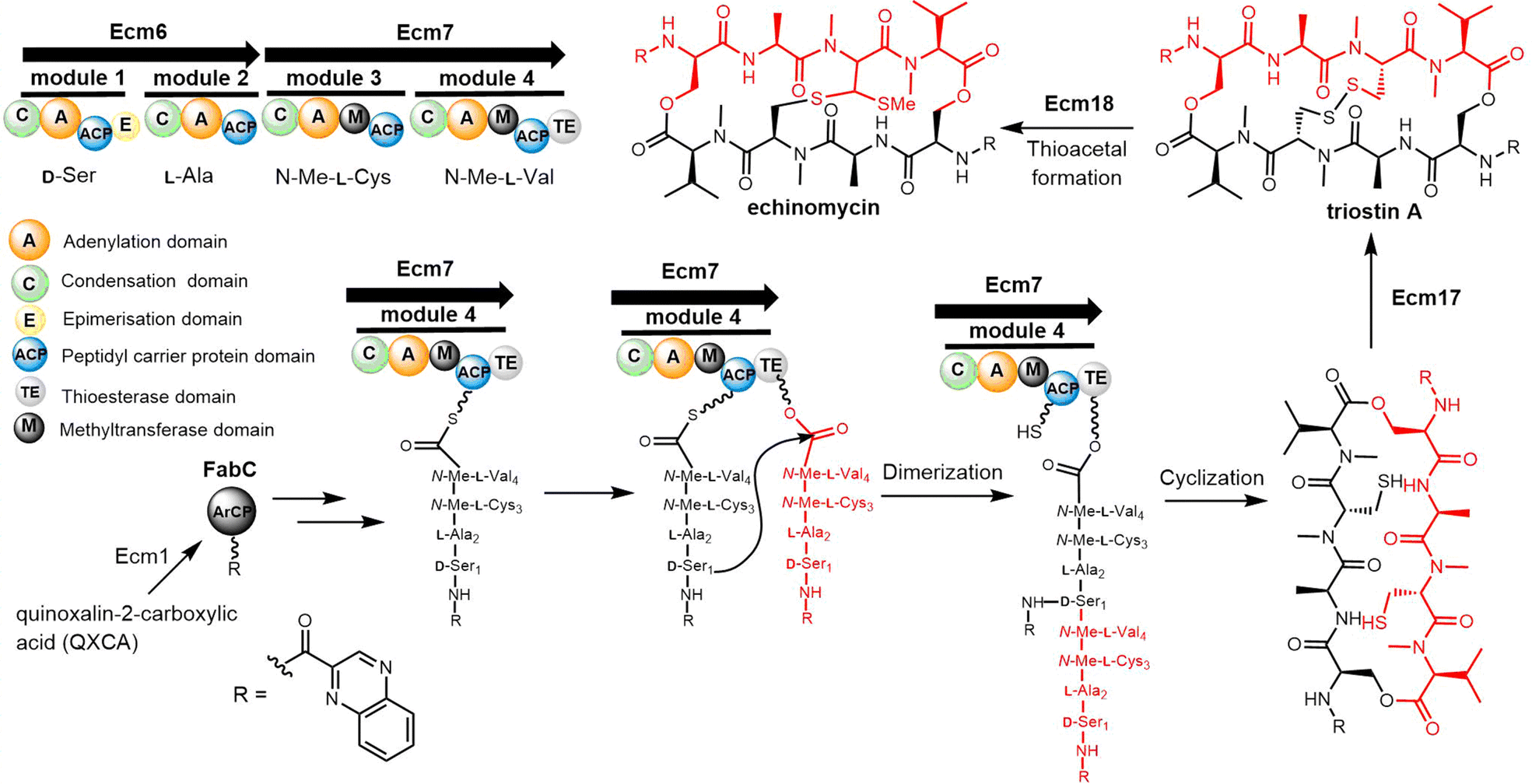 | ||
| Scheme 15 The biosynthesis pathway of echinomycin and triostin A. | ||
Echinomycin and triostin A possess an identical cyclic depsipeptide core that contains C2 symmetry.225 Genome sequencing has demonstrated that triostin A and echinomycin are produced by the same BGC, with the thioacetal bridge-forming enzyme encoded by Ecm18 being responsible for the conversion of triostin A to echinomycin (Scheme 15).222
In the Ecm-TE-mediated cyclization, the reaction has been shown to be limited by the hydrolysis of the product.226,227 Koketsu and colleagues designed a DNA-binding strategy to increase the catalytic efficiency of Ecm TE-catalyzed cyclization.228 This was based on the binding preference of tetrapeptide unit products to a specific DNA sequence, which can inhibit unnecessary product hydrolysis and significantly improve the yield of cyclization products.225
Koketsu et al. synthesized series octapeptide substrates with various substituted amino acid residues and chromophores. Use of these in biochemical assays suggests that the Ecm TE has a relatively broad substrate specificity, and further that the excised Ecm7 TE domain can also catalyse the dimerization of the tetrapeptidyl-SNAC substrates, generating Triostin A.228 In addition, these assays showed that the Ecm TE mediates the cyclization of pyrazine ring-containing analogues at a slower rate (Kcat/Km = 0.046 mM−1 min−1) compared to a higher rate (Kcat/Km = 3.87 mM−1min−1) for the naphthalene ring-containing analogue. This suggests that the ring size of the chromophore has a major influence on substrates recognition by the Ecm TE domain.
Triostin A, by virtue of being a symmetric bicyclic depsipeptide, is an easier synthetic target than echinomycin and a variety of total synthesis methods in solid or liquid phases have been developed to generate triostin A.229–232 In all of these methods, the amino acids are first linked into linear tetradepsipeptides by SPPS, which is followed by a double cyclisation reaction in which the two tetrapeptide chains are first linked to form a linear octapeptide, and then macrocyclization is carried out with a catalyst to obtain the cyclodimeric peptide skeleton.
Sable et al. reported a different method for triostin A synthesis. Here, allyl and alloc-protecting groups were used in the generation of the linear tetradepsipeptides by SPPS, which were then removed using [Pd(PPh3)4] and PhSiH3 in CH2Cl2. Cyclisation was then achieved using N,N′-diisopropylcarbodiimide (DIC)/HOAt in a DMSO/DMF (3:2) solvent mixture to obtain the cyclized intermediate 6, which was cleaved from the resin by treatment with TFA/TIS/CH2Cl2 (10:5:85) in 4.0% overall yield (Scheme 16A). In an alternative route, the 2-quinoxaline carboxylic acid was coupled to the peptide after the cyclization. The desired product was then isolated in 9.1% overall yield (Scheme 16B).230
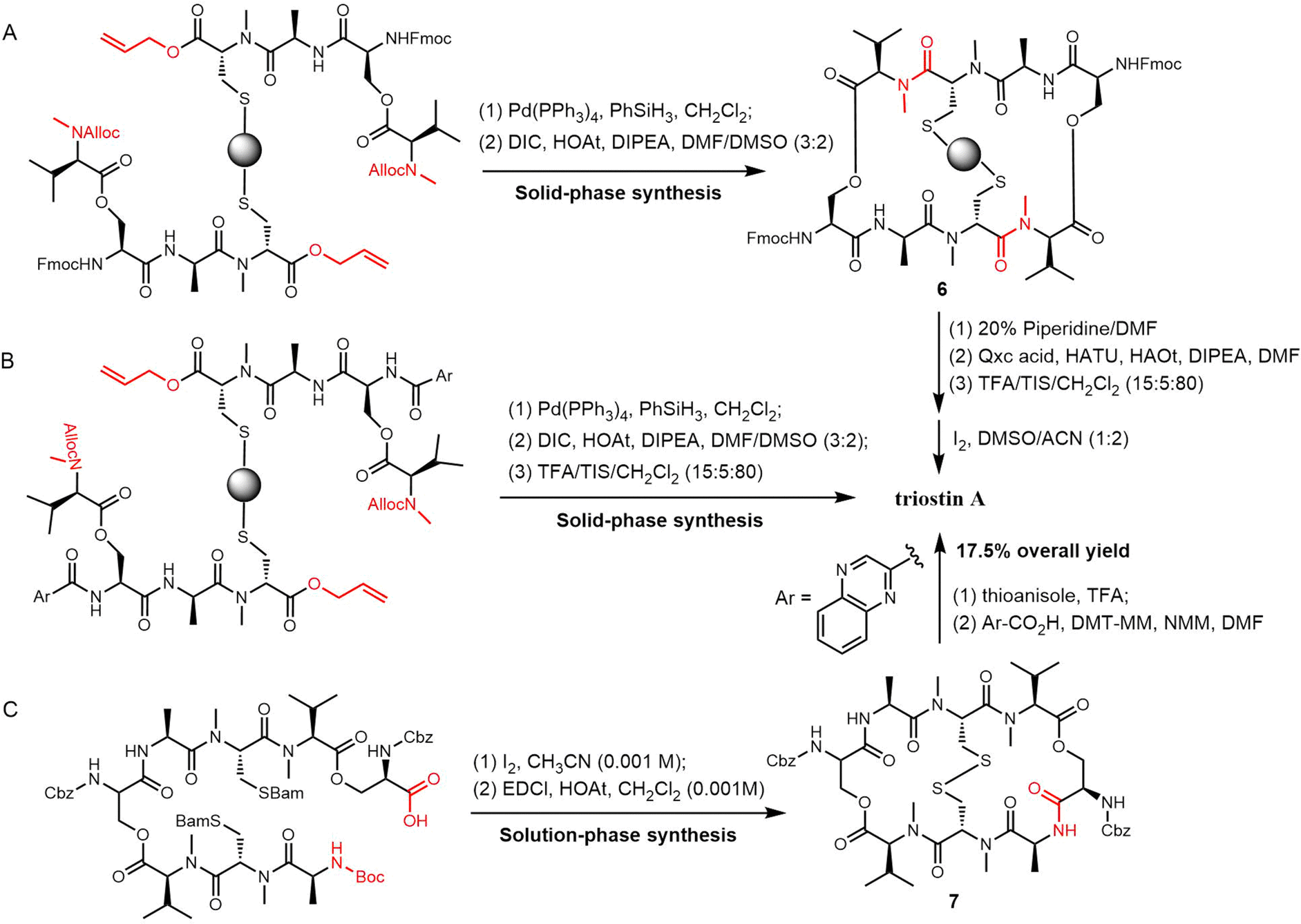 | ||
| Scheme 16 Total synthesis of triostin A with different approaches. | ||
Nagasawa et al. developed a 13-step solution-phase synthesis of triostin A, with cyclization achieved through a macrocyclization reaction. Using iodine for oxidative deprotection of the Bam group on the linear octapeptide forms a disulfide bond, and subsequently, when mixed with EDCI/HOAt at high dilution (0.001 M), the anticipated macrolide 7 was formed with a yield of 48%. Triostin A was then synthesised by removing the Cbz groups on 7 with thioanisole/TFA. This was followed by coupling two 2-quinoxaline carboxylic acid residues, which gave a 17.5% yield of the desired product (Scheme 16C).231
The presence of the thioacetal bridge makes the synthesis of echinomycin and quinomycin more challenging and delayed the first complete synthesis of echinomycin until 2020. In their synthesis, the Ichikawa group prepared the ester 8 and sulfide 9 as key precursors based on their retrosythetic analysis. Simultaneous cyclization and two-directional peptide chain elongation were used to create the C2-symmetrical bicyclic octadecadepsipeptide.233 The sulfur of 10 was oxidized to sulfoxide with m-CPBA, and the sulfoxide was converted into the corresponding chloride using AcCl through the Pummerer rearrangement. Finally, using the nucleophile TMSSMe and ZnCl2 the methylthio group was introduced to generate echinomycin (Scheme 17).233
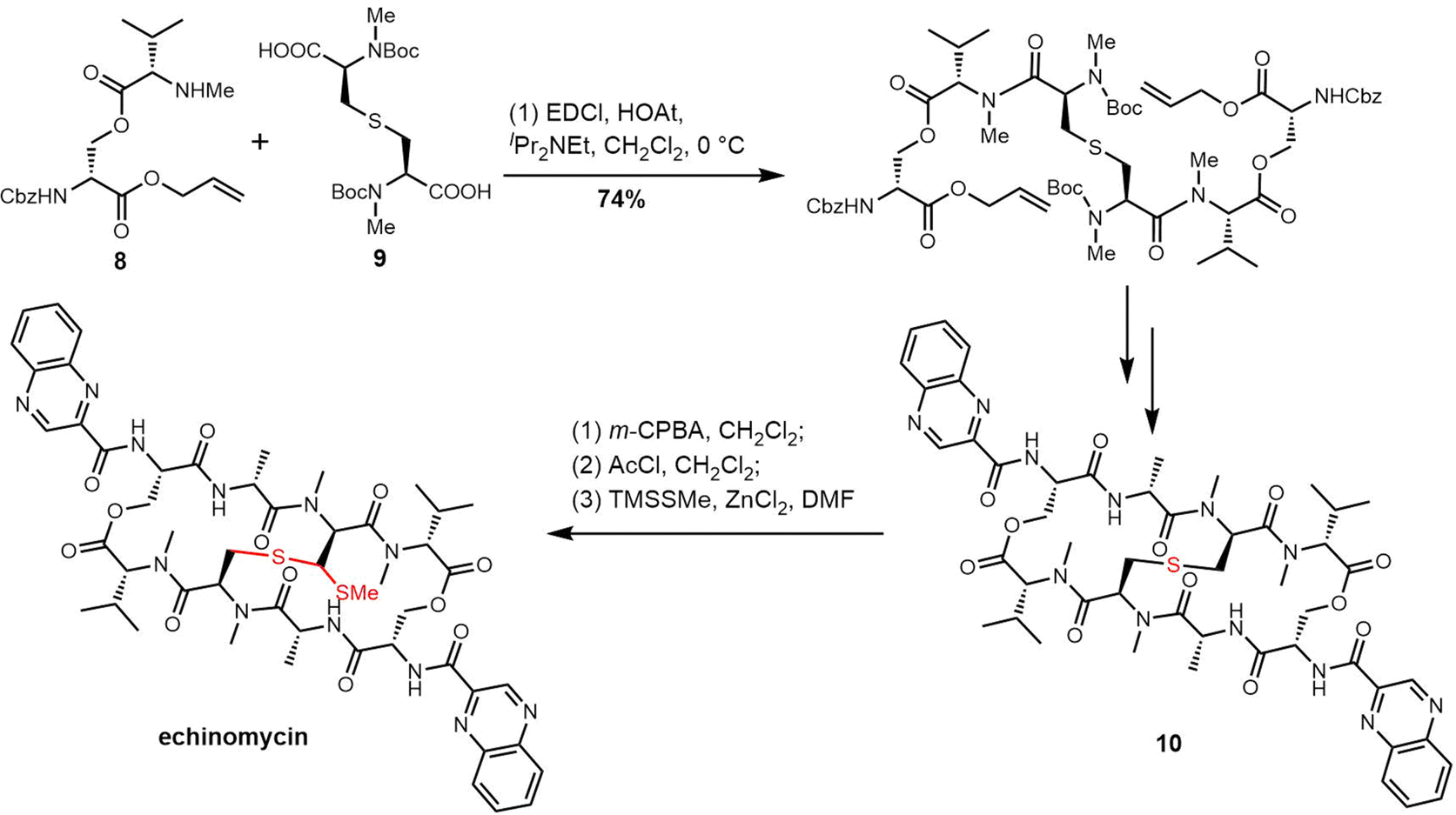 | ||
| Scheme 17 Total synthesis of echinomycin. | ||
SW-163C-E (Fig. 13) is produced by Streptomyces sp., and its biosynthetic pathway is similar to triostin A, thiocoraline and echinomycin. The SW-163 BGC was first annotated in Streptomyces sp. SNA15896, where fifteen genes were shown to be involved in the biosynthesis of SW-163s.223 Amongst these, two NRPSs (Swb16, Swb17) are involved in the assembly of the tetrapeptide chain and the final cyclisation to generate WS-163C. The SAM-dependent methyltransferases Swb8 and radical SAM protein Swb9 are responsible for the conversion of SW-163C to SW-163D and SW-163E-G, respectively.138 In contrast with echinomycin, whose chromophore is quinoxaline, quinoline is found in SW-163-type compounds. In addition to the three common amino acids in the tetrapeptide chain of SW-163-type compounds, they also contain the rare amino acid N-Me-norcoronamic acid.234
 | ||
| Scheme 18 Total synthesis of sandramycin. | ||
The solution-phase total synthesis of sandramycin and its analogues was first completed in 1996 and used a strategy similar to the synthesis of other bisintercalators such as quinomycin. The symmetrical pentadepsipeptides were synthesized first, followed by coupling and macrocyclization of a 32-membered decadepsipeptide at a single secondary amide site to afford the target compound.236
Unlike the sequential peptide coupling approach, Ichikawa et al. recently performed the total synthesis of sandramycin through an Ugi three-component (11, 12, 13) reaction as the key step to obtain a linear pentadepsipeptide 14. Sandramycin was generated by the stepwise coupling of two pentapeptide monomers, with subsequent introduction of the quinaldin chromophores (Scheme 18).237 Chemical synthesis methods mostly use C2 symmetry to synthesize the dimerized backbone of sandramycin, which limits derivatives to those containing C2 symmetry. In 2023, the Ichikawa group further optimized the SPPS approach based on the previous coupling strategy and could synthesize desymmetrized analogues of sandramycin (Scheme 18).238
The Du group identified the biosynthesis gene cluster of luzopeptin A (luz, 48 kb) from Actinomadura luzonensis DSM 43766 and its analogue korkormicins from Micromonospora sp. ATCC 55011 (Scheme 19). Two NRPS enzymes Luz1 and Luz2 are responsible for the assembly of the peptide using the serine, tetrahydropyridazine-3-carboxylic acid (Thp), glycine (2x) and threonine units. The TE domain of Luz2 performs the final release of the product via head-to-tail cyclization (Scheme 19).241 After release, a series post-NRPS modifications are required for the generation of mature luzopeptins. The tailoring enzymes Luz25 (cytochrome P450), Luz26 (cytochrome P450) and Luz27 (membrane acyltransferase) are responsible for the modification of the cyclic peptide to generate the non-proteinogenic amino acid residues β-OH-N-methyl-L-Val and the acyl-substituted tetrahydropyridazine-3-carboxylic acid (Thp), respectively.241
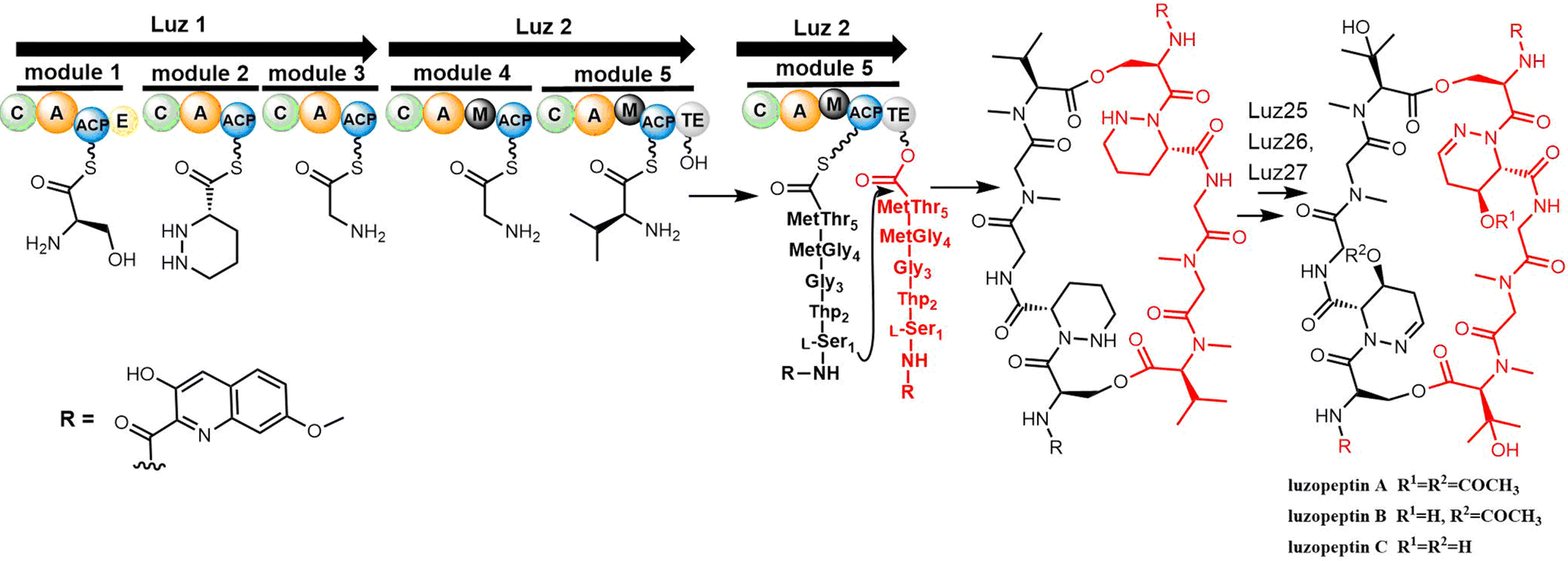 | ||
| Scheme 19 The biosynthetic pathway of luzopeptin A. | ||
As with the synthesis of sandramycin, the synthesis of the luzopeptins commences with the preparation of the linear monomer decadepsipeptide, which is achieved through the coupling of pentadepsipeptides 15 and 16, mediated by EDCI-HOAt (CH2Cl2, 0 °C, 2 h). By using transfer hydrogenolysis (25% aqueous HCO2NH4, 10% Pd/C, EtOH/H2O), the FMOC group and benzyl ester were cleaved, with macrocyclization (EDCI-HOAt, CH2Cl2, 0 °C, 2 h) providing the 32-membered cyclic decadepsipeptide, and finally generating the luzopeptins (Scheme 20).240
 | ||
| Scheme 20 Total synthesis of luzopeptins. | ||
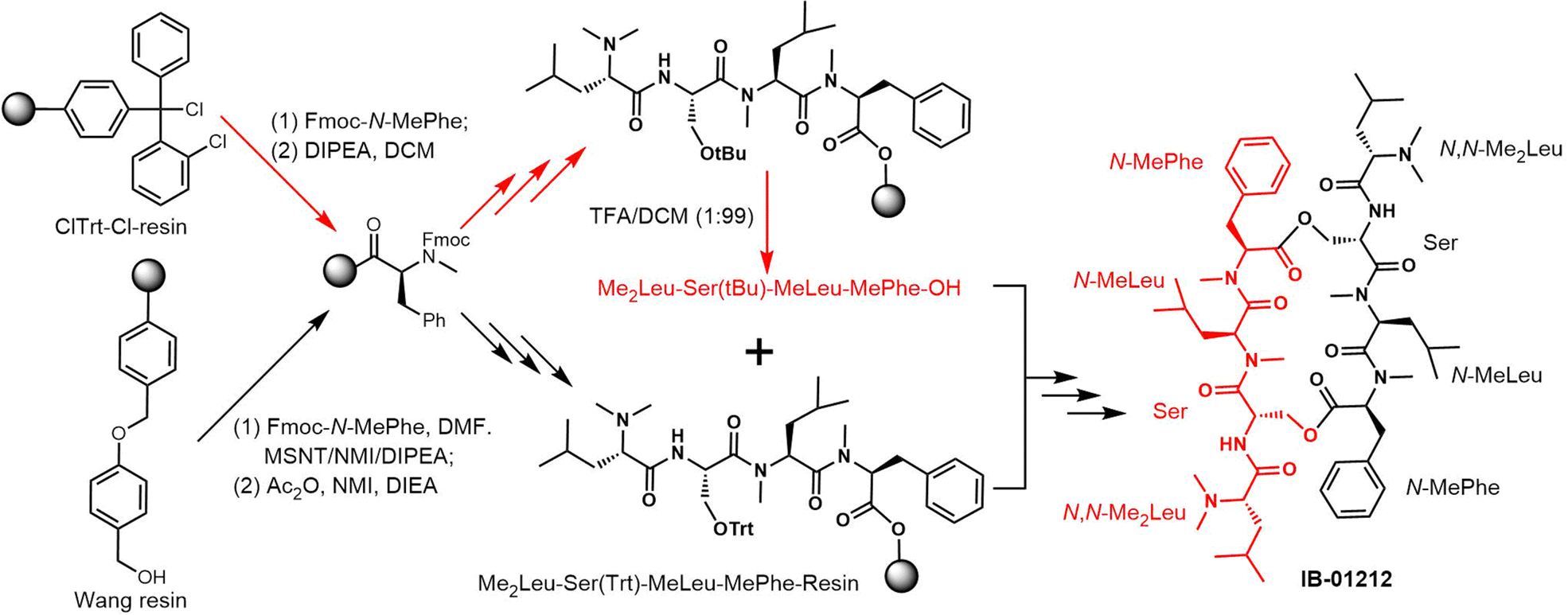 | ||
| Scheme 21 Convergent [4+4] solid-phase synthesis of IB-01212. | ||
 | ||
| Scheme 22 The biosynthetic pathway of thiocoraline. | ||
TioR and TioS are the NRPS enzymes involved in thiocoraline biosynthesis, with TioR comprising two modules (C1-A1-PCP1-E1-C2-A2-PCP2) and TioS two modules (C3-A3-M3-PCP3-C4-A4-M4-PCP4-TE).247 Studies suggest that 3HQA, which is activated by TioJ, is not directly loaded onto the NRPS module but rather onto the fatty acid synthase ACP domain FabC located outside the gene cluster; this phenomenon is similar to that found in the biosynthesis route of echinomycin (Scheme 22).221,248
The TioS TE domain is the first NRPS-derived thioesterase reported to be capable of catalysing the formation of macrothiolactone and macrolactone functionalities in an iterative fashion, which is typical for the assembly of quinoline or quinoxaline carrier compounds. Robbel et al. used TioS PCP–TE to synthesise thiocoraline analogues and showed that the TioS PCP–TE could catalyse the attachment and subsequent cyclisation of tetrapeptide sulphate substrates. By expressing the TE domain of thiocoraline independently and utilizing SNAC-thiophenol mimics, it was also possible to synthesize a range of thiocoraline analogues in vitro using various activated tetrapeptides. These studies highlighted the importance of D-stereochemistry for specific residues to promote cyclization, as opposed to hydrolysis.249 In addition, substrate specificity studies using the TioS PCP–TE showed that the spatial requirement and polarity of the C-terminal amino acid is essential for substrate recognition, ligation and macrocyclization by this TE domain.
Boger and coworkers have reported the total synthesis of thiocoraline using a convergent strategy to prepare the precursor tetradepsipeptide. After deprotection of the amine and carboxylic acid, the key intermediate octadepsipeptide 19 was generated through the coupling of monomer tetradepsipeptides 17 and 18. The ring was closed through a second round of BOC deprotection, Tce ester deprotection and amide formation. 3-Hydroxyquinoline-2-carboxylic acid residues were attached to the scaffold after removing the Cdz-protecting group using TFA-thioanisole at 25 °C for 4 h (Scheme 23).250 Other thiocoraline analogues have been synthesized using a similar strategy.251
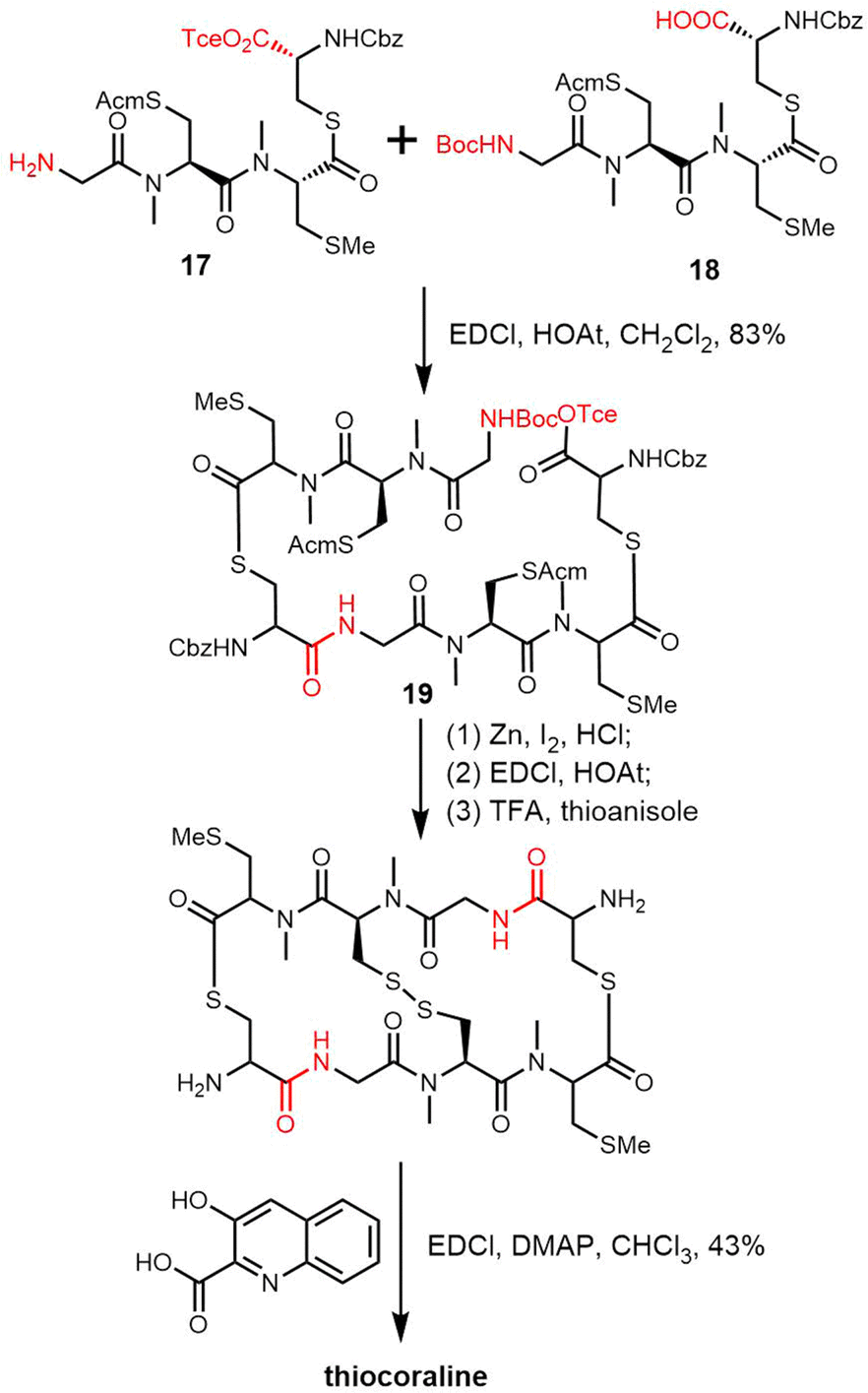 | ||
| Scheme 23 Total synthesis of thiocoraline. | ||
 | ||
| Scheme 24 The biosynthetic pathway of terrequinone A. | ||
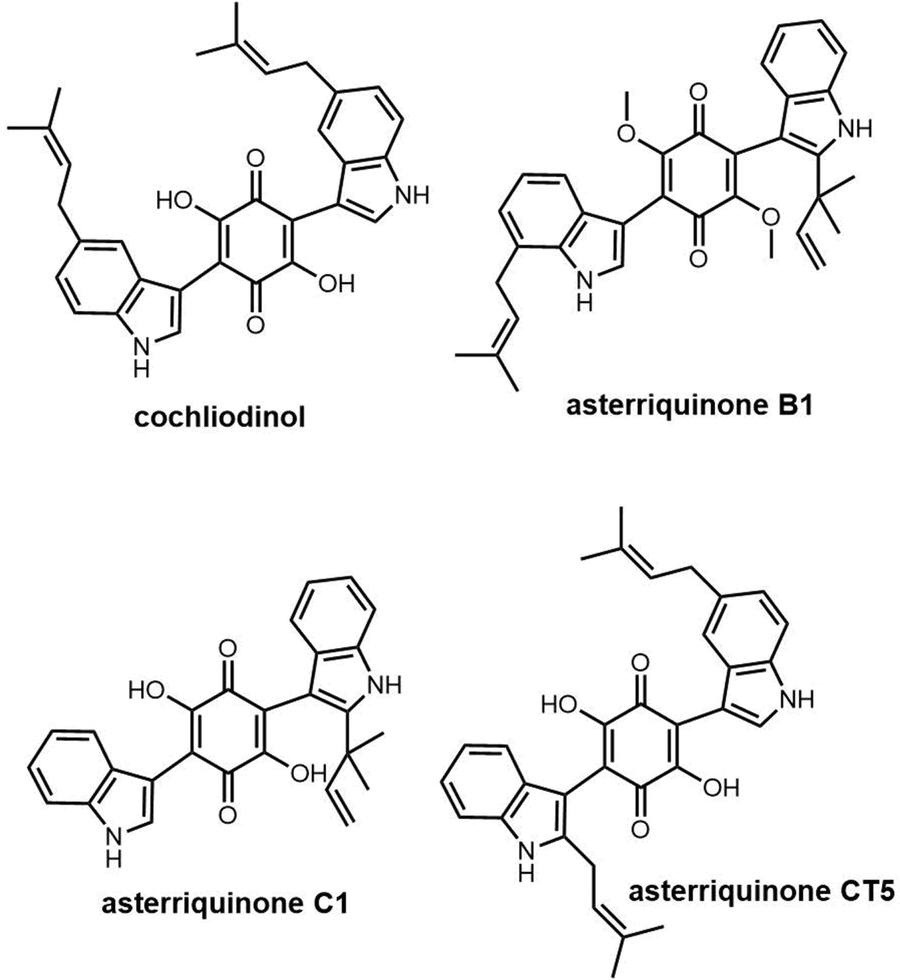 | ||
| Fig. 14 Example of terrequinone A and related compounds. | ||
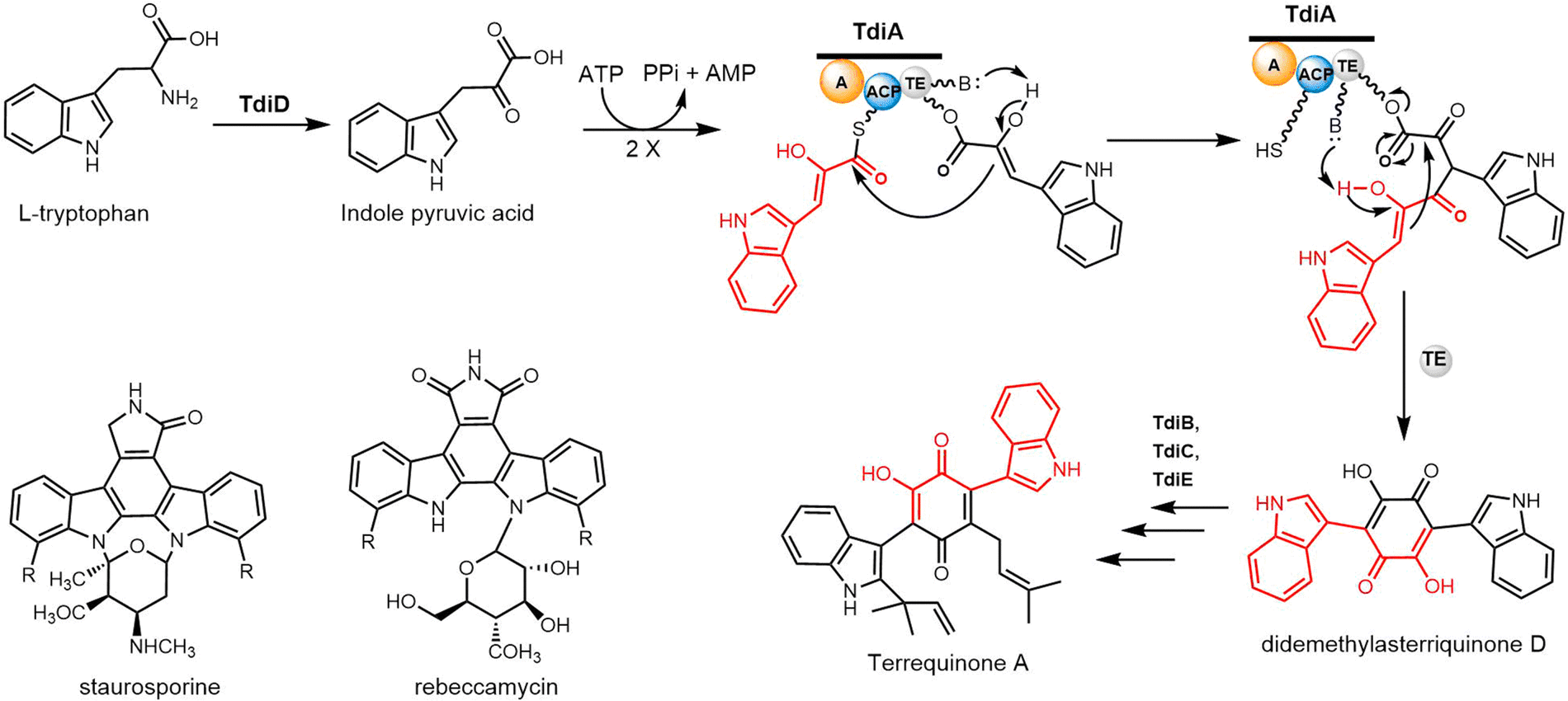
The five genes tdiA–tdiE were identified in Aspergillus nidulans and annotated as the biosynthetic gene cluster of terrequinone A. Walsh and colleagues reconstituted all five proteins (TdiA–TdiE) in E. coli and in doing so achieved the production of terrequinone A.258 The bisindolylbenzoquinone backbone is constructed by indole pyruvic acid (IPA) through dimerization, with the initial substrate IPA derived from tryptophan through the actions of the PLP-dependent transaminase TdiD.
In this pathway, TdiA comprises a single-module NRPSs, which contains three domains responsible for adenylation, substrate tethering and thioester cleavage. The single-module TdiA ends with a thioesterase (TE) domain. Biochemical investigations have suggested that the TdiA TE domain facilitates an intramolecular Claisen condensation reaction, resulting in the symmetric cyclization of two IPA molecules to generate the final product (Scheme 24). The TdiA TE domain can therefore facilitate the formation of carbon–carbon bonds. The mutagenesis of S774A, located within the conserved GXSXGG motif in the TE domain of TdiA, prevented the generation of didemethylasterriquinone D from IPA. This result provides evidence that the TE domain plays a crucial role in facilitating the cyclization of IPA monomers.258
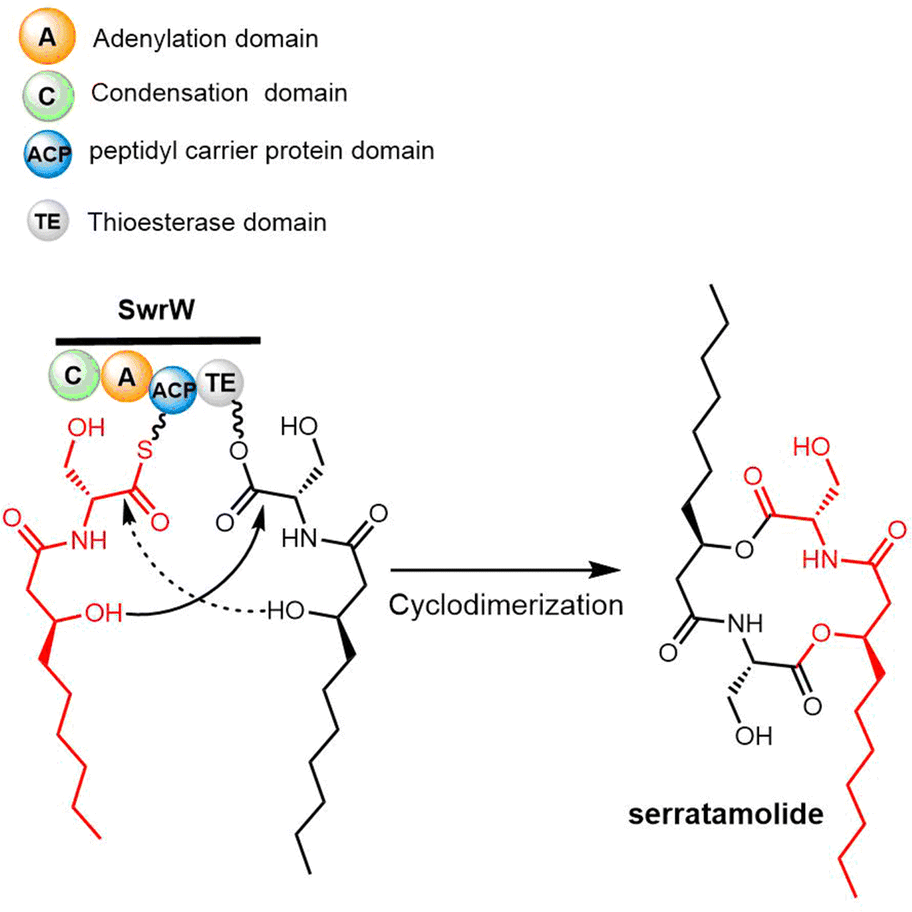 | ||
| Scheme 25 The biosynthetic pathway of serratamolide. | ||
2.3 Cyclodimeric polyketides
Macrodiolides are effective drug candidates with antibacterial capabilities and have drawn great attention due to their distinctive bioactivity. Polyketides make up an important class of macrocyclic natural products. In the class of polyketides, macrolides are the most common form and are primarily biosynthesised by type I PKSs. The most common type of macrolide is a lactone, which is a cyclic ester formed from the condensation of a carboxylic acid and an alcohol. The structural characteristics of macrolides can have a significant impact on their biological activity and other properties.261 Many macrolide-type antibiotics have been used clinically to treat bacterial infections, including erythromycin, clarithromycin, and azithromycin.Dimeric polyketides exhibit a wide range of distinct structural characteristics and have a significant potential to form several new medicinally relevant molecules.11,262,263 Their applications include immunosuppressive drugs, antibiotics, herbicides, fungicides, and active medications against cancer, HIV.264,265 Dimeric polyketides are mainly generated through a dimerization process, including radical reactions, cycloadditions, esterification, and acetal formation.33
Macrodiolides are compounds primarily synthesized via the PKS or hybrid PKS/NRPS pathway and feature a symmetrical or unsymmetrical polyketide scaffold. In PKSs assembly lines, the TE domain is responsible for iteratively synthesising two monomeric precursors before linking these together in a head-to-tail orientation, thus creating a dimer product which serves as the molecular scaffold for the cyclic diolide structure. In this section, we will mostly examine and review polyketides involving macrocyclization in their biosynthesis, with particular interested in considering how the TE domain functions in these processes.
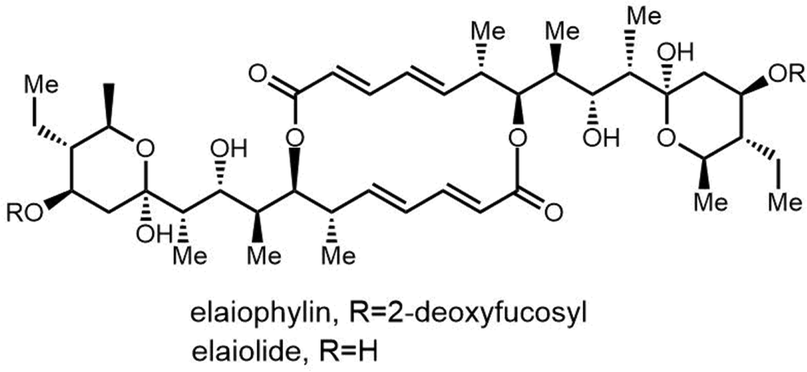 | ||
| Fig. 15 The chemical structure of elaiophylin and its aglycone derivative elaiolide. | ||
Due to its structural complexity and promising biological activity, several groups have completed the synthesis of elaiophylin or aglycon derivatives.273–275 The challenges in such syntheses were the low selectivity of stereospecificity during aldol condensation. Evans et al. developed a cyclic protecting group to restrict the conformational flexibility of the ketone and thus gaining high aldol diastereoselectivity. They first synthesized the hydroxy acid 20 in five steps with carboximide as the starting compound, with cyclodimerization of 20 preformed using a modified Yamaguchi's macrocyclization strategy (Scheme 26). The dimer 21 was converted to 23 in two steps ready for crucial aldol coupling. Dialdehyde 23 was treated with the chlorophenylboryl enolate of protected ethyl ketone 24, providing the bis-aldol adduct 25 as the only detectable product. Finally, deprotection and subsequent cyclization of 25 completed the synthesis of the elaiophylin aglycon elaiolide (Scheme 26).276
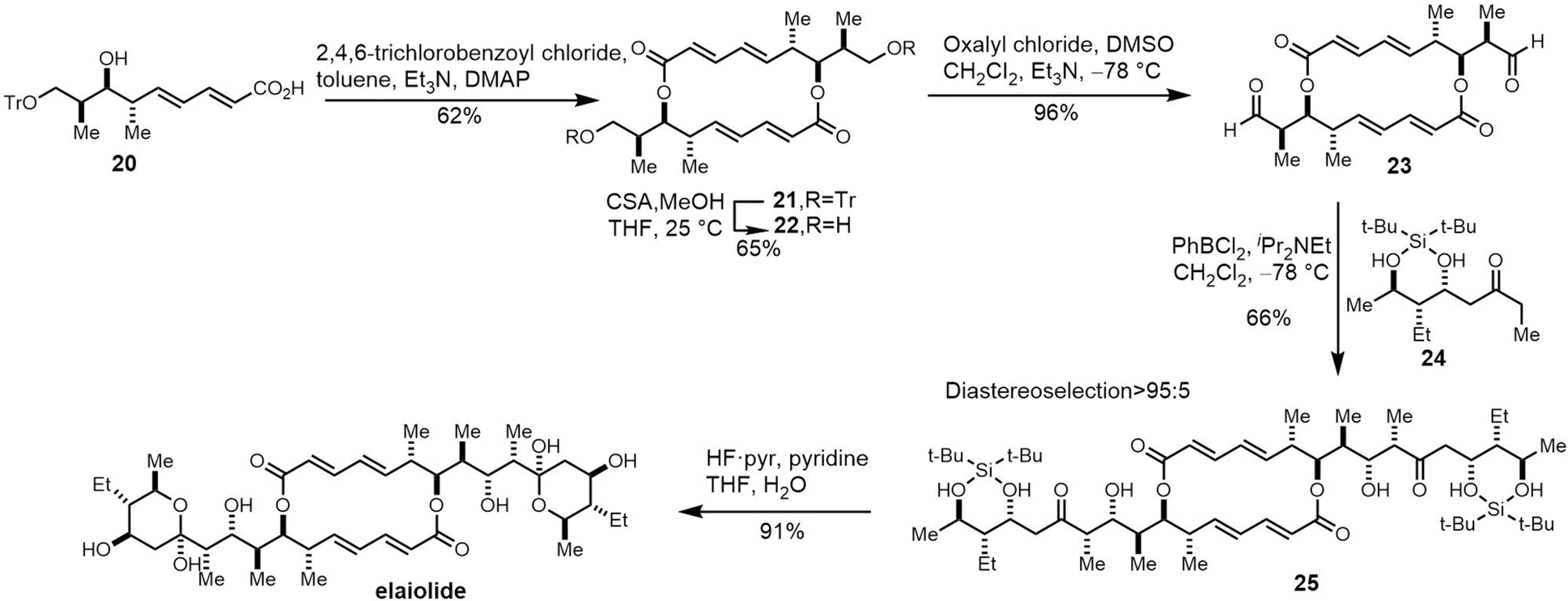 | ||
| Scheme 26 Chemical synthesis of elaiolide. | ||
In 2004, Haydock et al. identified the putative biosynthetic gene cluster of elaiophylin from the elaiophylin-producing strain Streptomyces sp. DSM4137, which was found to contain five PKS genes encoding eight modules, consistent with the assembly of the elaiophylin polyketide chain. The C-terminal TE domain in Ela5 (Ela-TE) was shown to catalyse polyketide chain release and intramolecular lactonization to generate the matured C2 symmetrical macrodiolide (Scheme 27).277
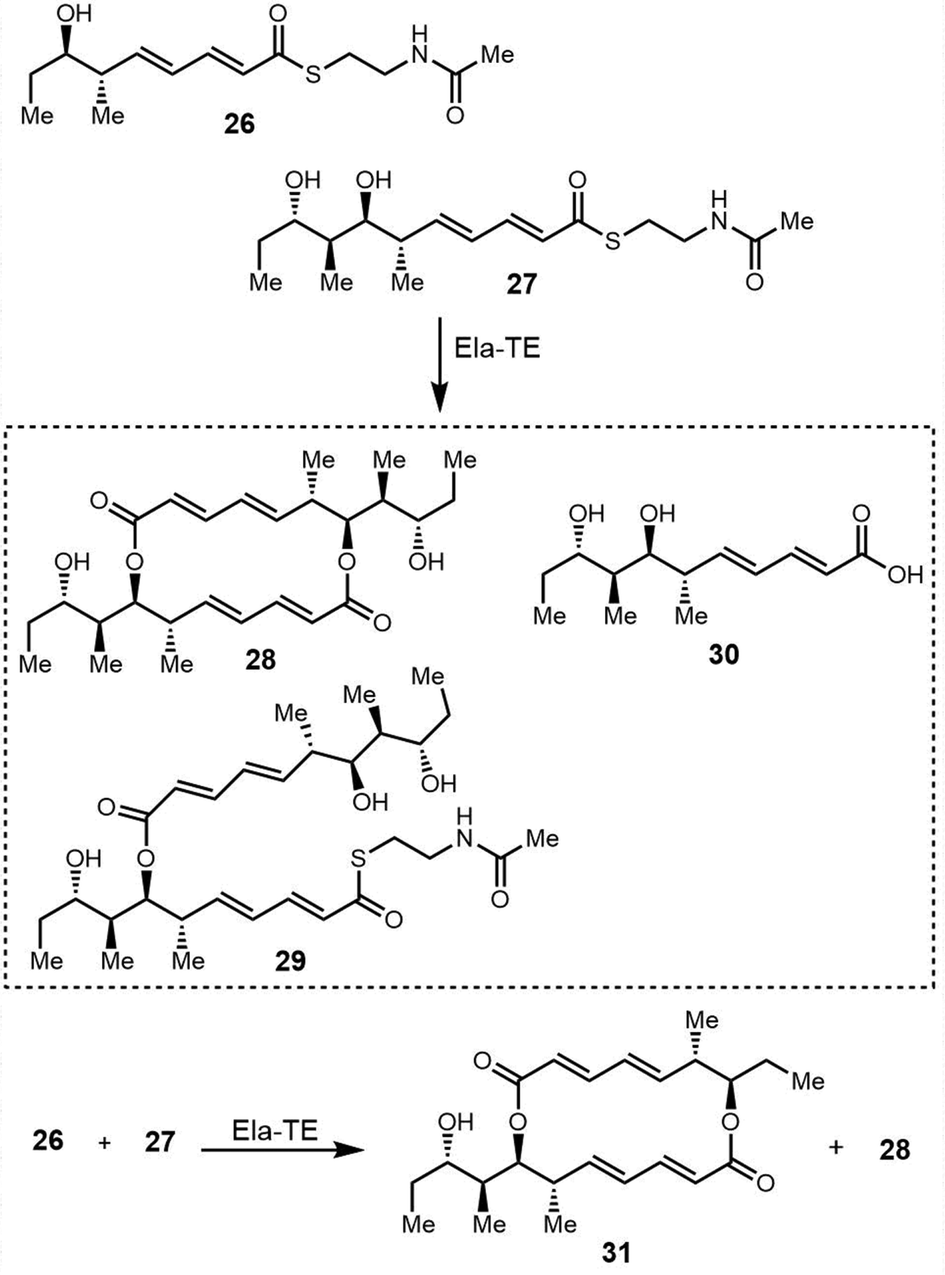 | ||
| Scheme 27 In vitro activities of Ela-TE. | ||
To study the mechanism and selectivity of macrodiolide formation, Leadlay et al. attempted to reconstitute the in vitro activity of the TE domain. Two alternative mechanisms for the formation of symmetrical diolide was proposed, similar to the cyclization steps seen in nonribosomal peptide synthetases. They synthesized SNAC thioesters of tetraketide 26 and pentaketide 27 as substrate mimics, demonstrating that incubation of pentaketide 27 with Ela-TE formed a homodimerized 16-membered decaketide diolide 28. The intermediate 29, a linear dimer linked with SNAC, and hydrolysis product 30 were also detected in these Ela-TE in vitro assays. Ela-TE was also shown to catalyze the conversion of the purified intermediate 29 into the final product 28. When pentaketide 27 and tetraketide 26 were mixed with Ela-TE, the C2-synmmetric product 28, together with a novel “hybrid” macrodiolide 31, could be observed, demonstrating that Ela-TE exhibits substrate flexibility. These results further confirm that the elaiophylin TE iteratively catalyzes two acylation and two deacylation reactions to form the diolide (Scheme 28).278
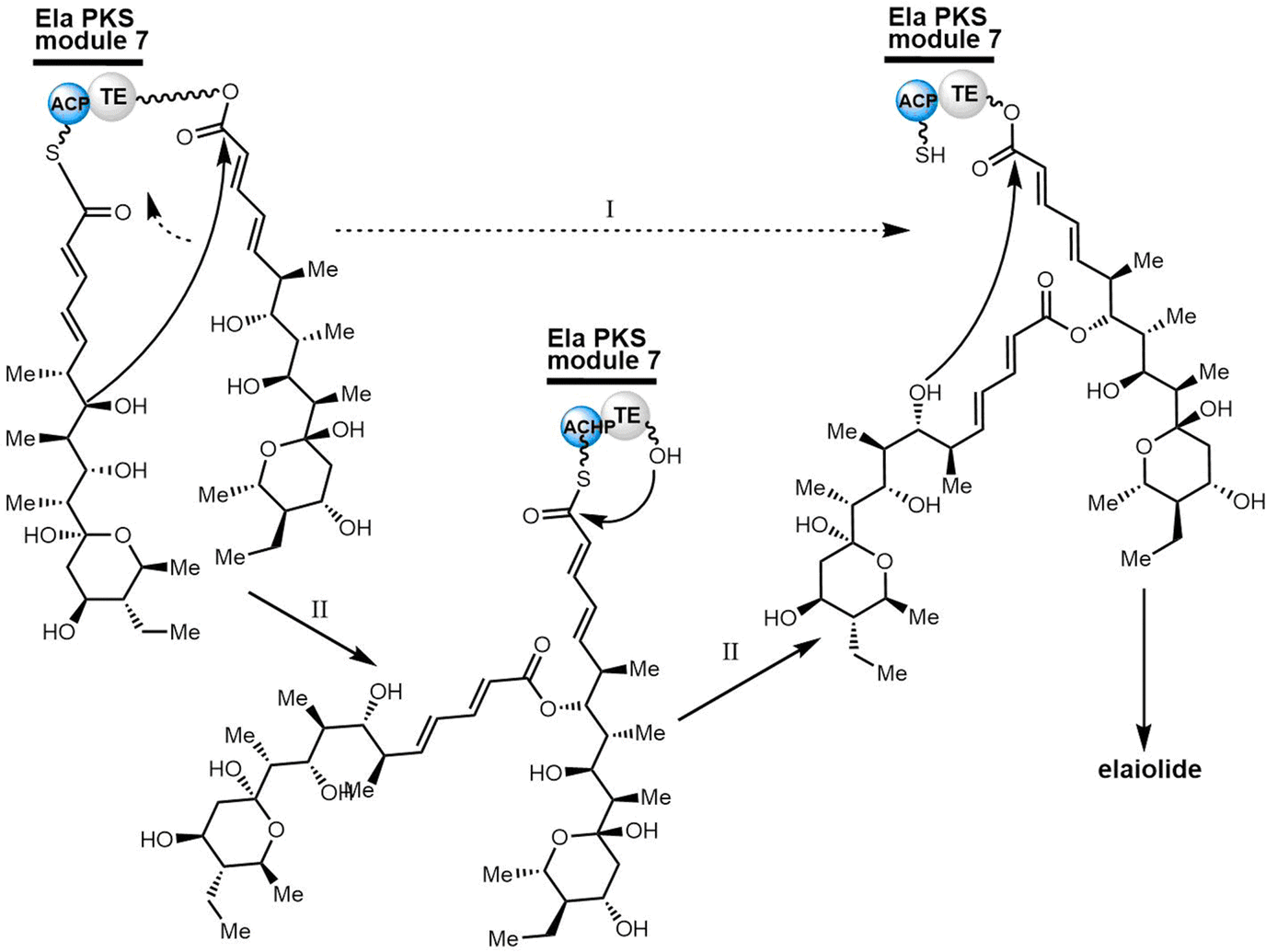 | ||
| Scheme 28 Alternative mechanisms for TE-catalyzed formation of symmetrical diolide. | ||
Interestingly, a recent study has revealed that the biosynthetic pathway of elaiophylin also generates a non-dimerized product known as pteridic acid. This compound exhibits entirely different biological functions from elaiophylin, as it enhances plants' resistance to abiotic stressors. The biosynthesis of pteridic acid also relies on TE enzymes (Scheme 29), possibly due to the broad substrate specificity and catalytic versatility of TE domain.279
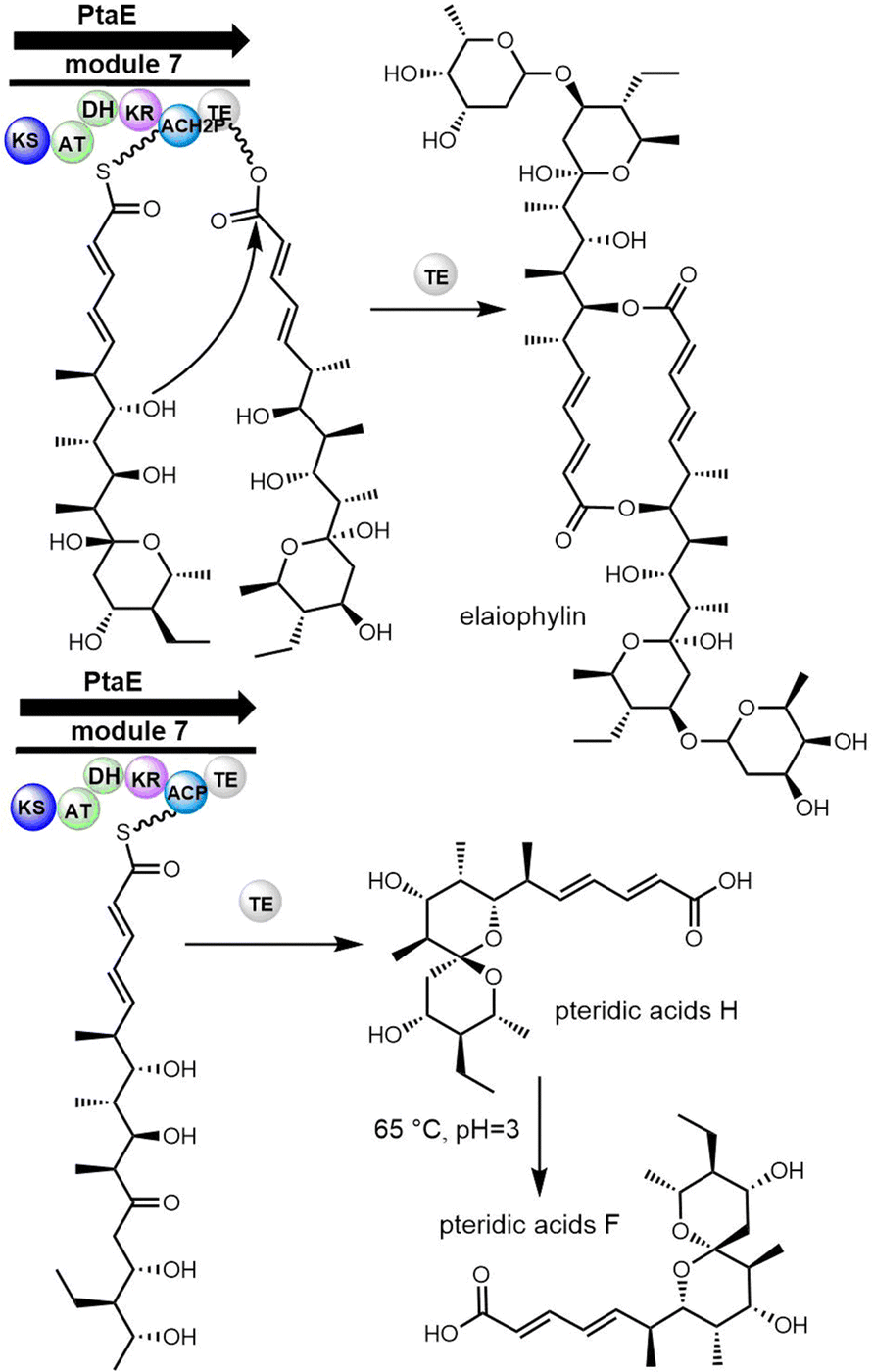 | ||
| Scheme 29 The proposed biosynthetic pathway of pteridic acids and elaiophylin. | ||
6-Deoxyerythronolide B synthase (DEBS) is a modular PKS that catalyses the biosynthesis of 6-deoxyerythronolide B, the macrocyclic core of the antibiotic erythromycin.280 The DEBS assembly line generate more than 50 erythromycin derivatives using combinatorial biosynthesis, which suggests promiscuity of the corresponding TE domain towards different substrates.98,281 Boddy et al. reported that the incubation of the DEBS-TE with the non-natural SNAC thioester mimic 32 generated the 14-membered macrolactone and the hydrolysis product as major products but also minor amounts of C2-symmetrical, head-to-tail dimerized macrodiolide (Scheme 30).53 The observation of linear dimer and the 28-membered macrodiolide is consistent with the reactions catalyzed by Ela-TE.
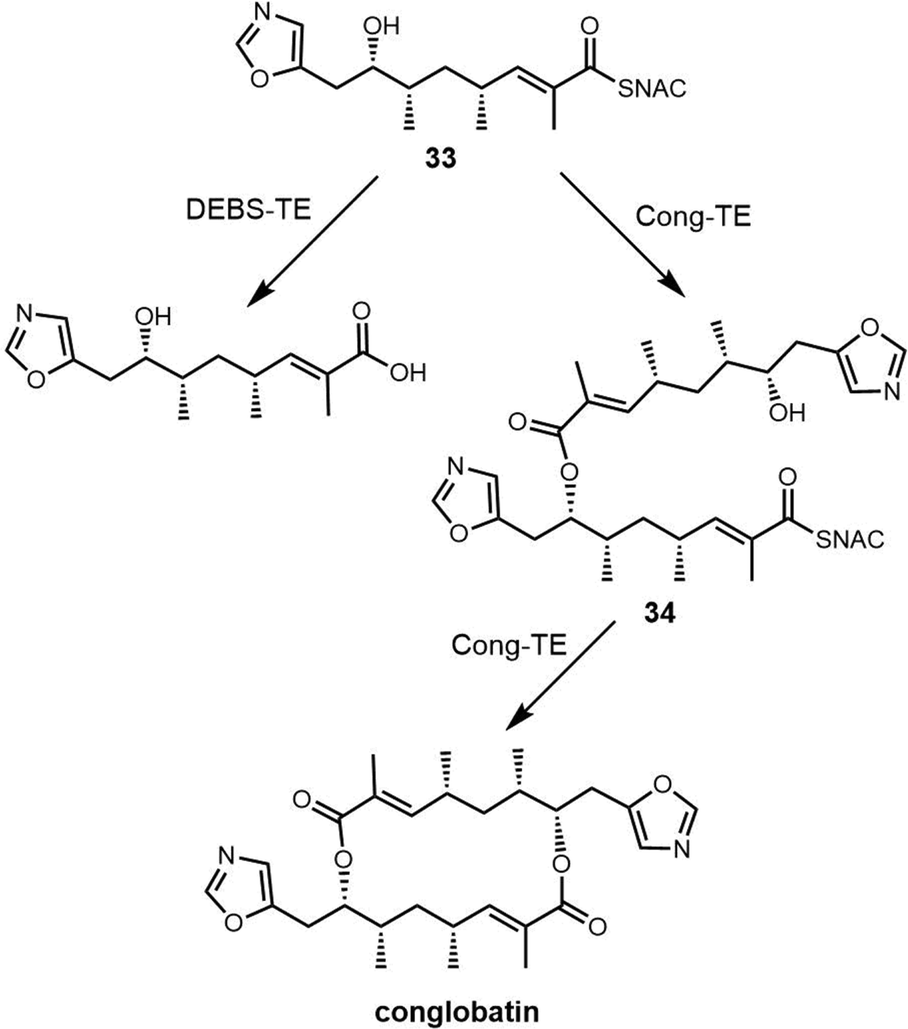 | ||
| Scheme 30 In vitro reconstitution of conglobatin macrodiolide formation. | ||
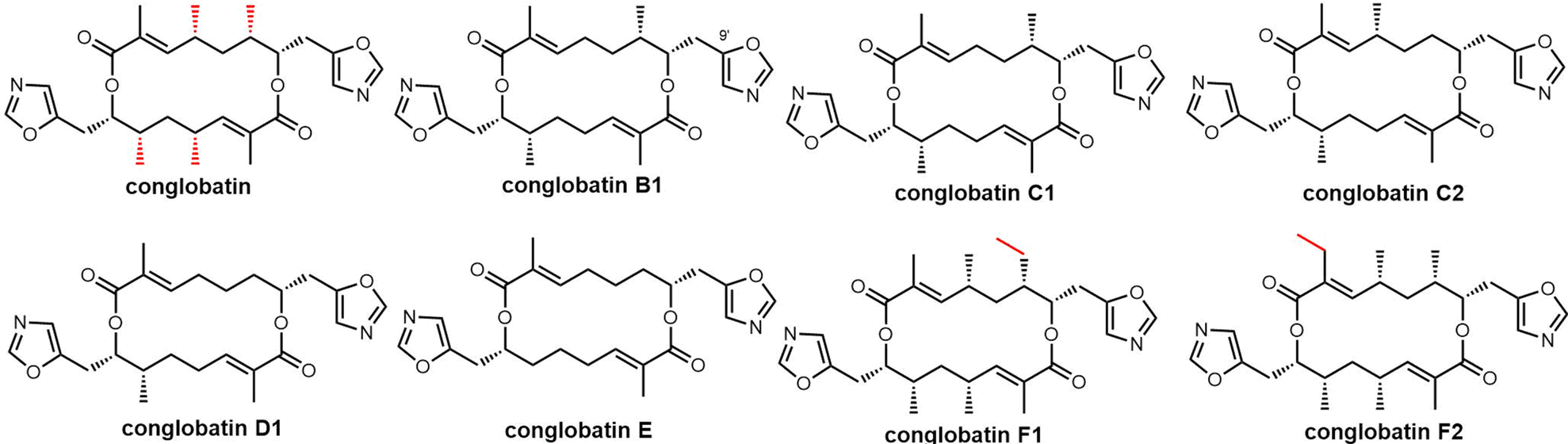 | ||
| Fig. 16 Chemical structure of conglobatin congeners. | ||
In 2015, Leadlay et al. sequenced the conglobatin producer Streptomyces conglobatus and elucidated its biosynthetic gene cluster, suggesting conglobatin was biosynthesized by a PKS-NRPS hybrid. Based on this biosynthetic gene cluster, they reconstituted conglobatin macrodiolide in vitro.264 SNAC thioester 33 was synthesized as a substrate, which when incubated with Cong-TE was converted into the liner dimer 34 and conglobatin. Time course assays showed the production level of conglobatin increased continuously whilst 34 reached a plateau after 2 hours. This suggested that 34 could be an intermediate in diolide formation, which was then demonstrated in the conversion of purified 34 by the Cong-TE into the final product conglobatin (Scheme 31).
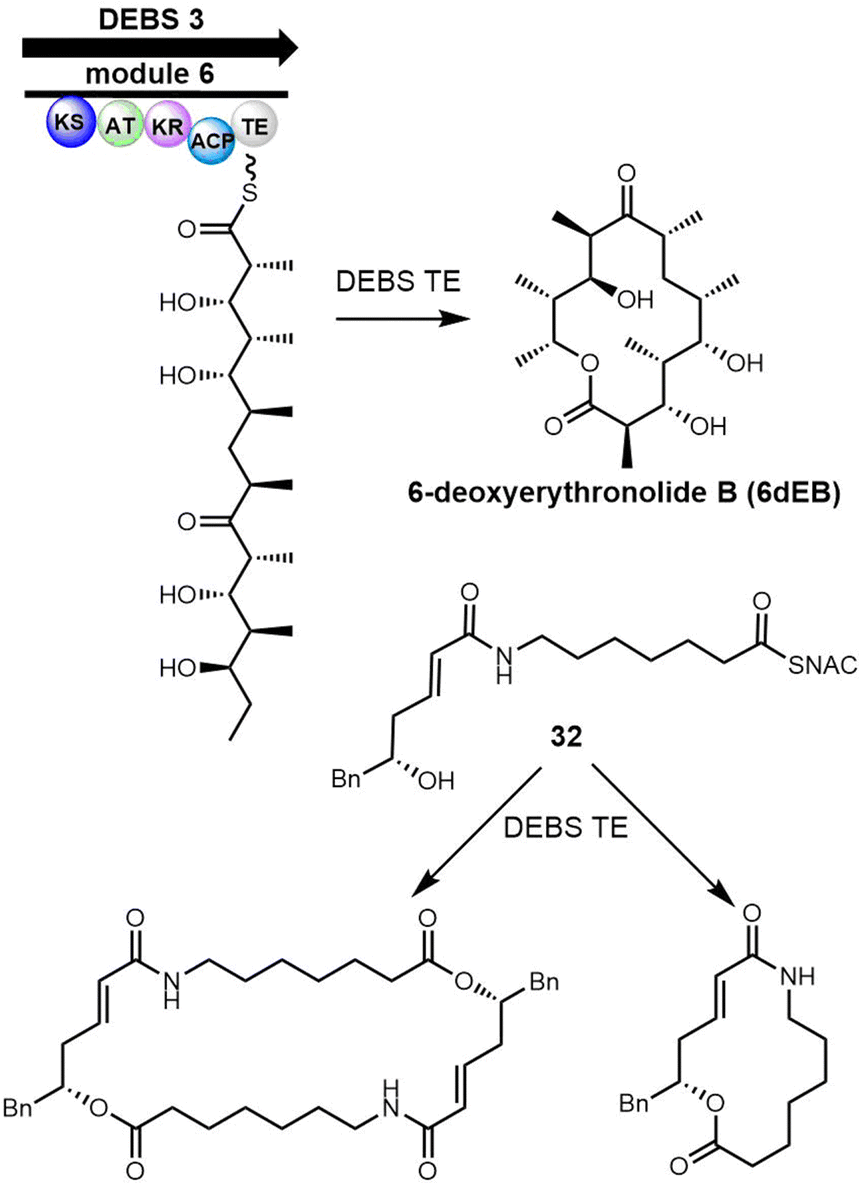 | ||
| Scheme 31 The function of DEBS TE in the biosynthesis of erythromycin. | ||
In an attempt to generate hybrid polyketides with Cong-TE, SNAC thioesters 35 and 36 previously synthesized for use with the elaiophylin TE were also tested as substrates. These experiments demonstrated that when the substrates 34 and 35/36 were co-incubated with Cong-TE, liner dimeric or trimeric products and their corresponding carboxylic acid could be detected. These results reveal significant subtleties exist in the iterative mechanism controlled by macrodiolide TE domains (Scheme 32).
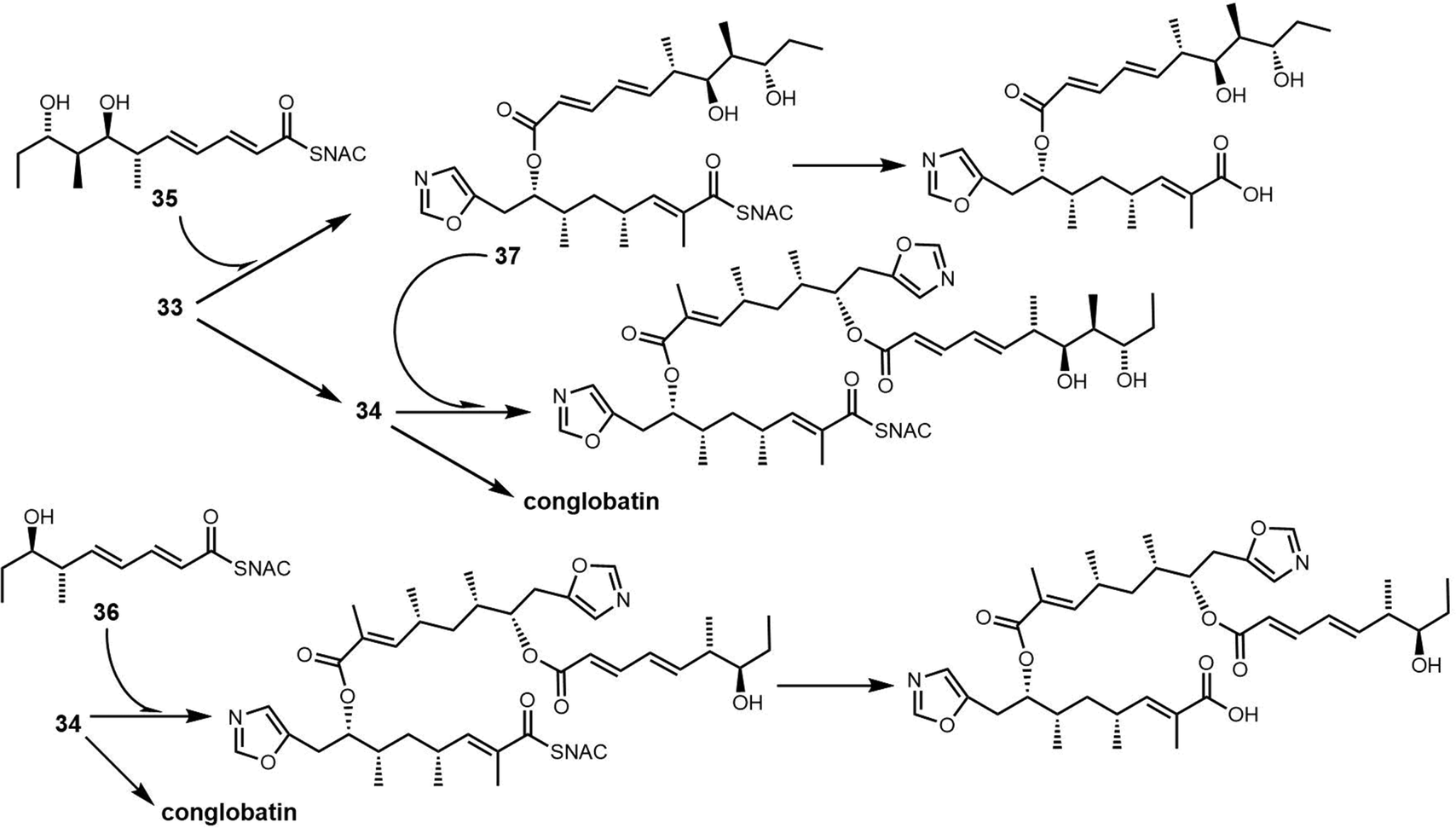 | ||
| Scheme 32 Hybrid polyketide dimers and trimers generated from mixed substrates. | ||
The soil bacterium Streptomyces pactum ATCC 27456 is the producer of many secondary metabolites, including conglobatin, aromatic polyketide NFAT-133 and pactamycin. Several new NFAT-133 derivatives were identified from a mutant of S. pactum, with structural elucidation confirming two of these (TM-127 and TM-128) as hybrids of NFAT-133 and conglobatin (Scheme 33). In vivo gene inactivation confirmed that both biosynthetic gene clusters of NFAT-133 and conglobatin are necessary to produce the hybrids TM-127 and TM-128. The Cong-TE from S. pactum ATCC 27456 was then purified and assayed with NFAT-133 and the SNAC thioester of the conglobatin monomer; compound TM-127 was successfully formed in this assay. In contrast, the assays using inactivated Cong-TE or without Cong-TE were unable to generate TM-127. When Cong-TE was incubated with NFAT-133 and the conglobatin monomer, TM-127 was not produced, demonstrating that both ACP and TE domains are required for such hybrid formation, and further shows that Cong-TE displays broad substrate scope.290
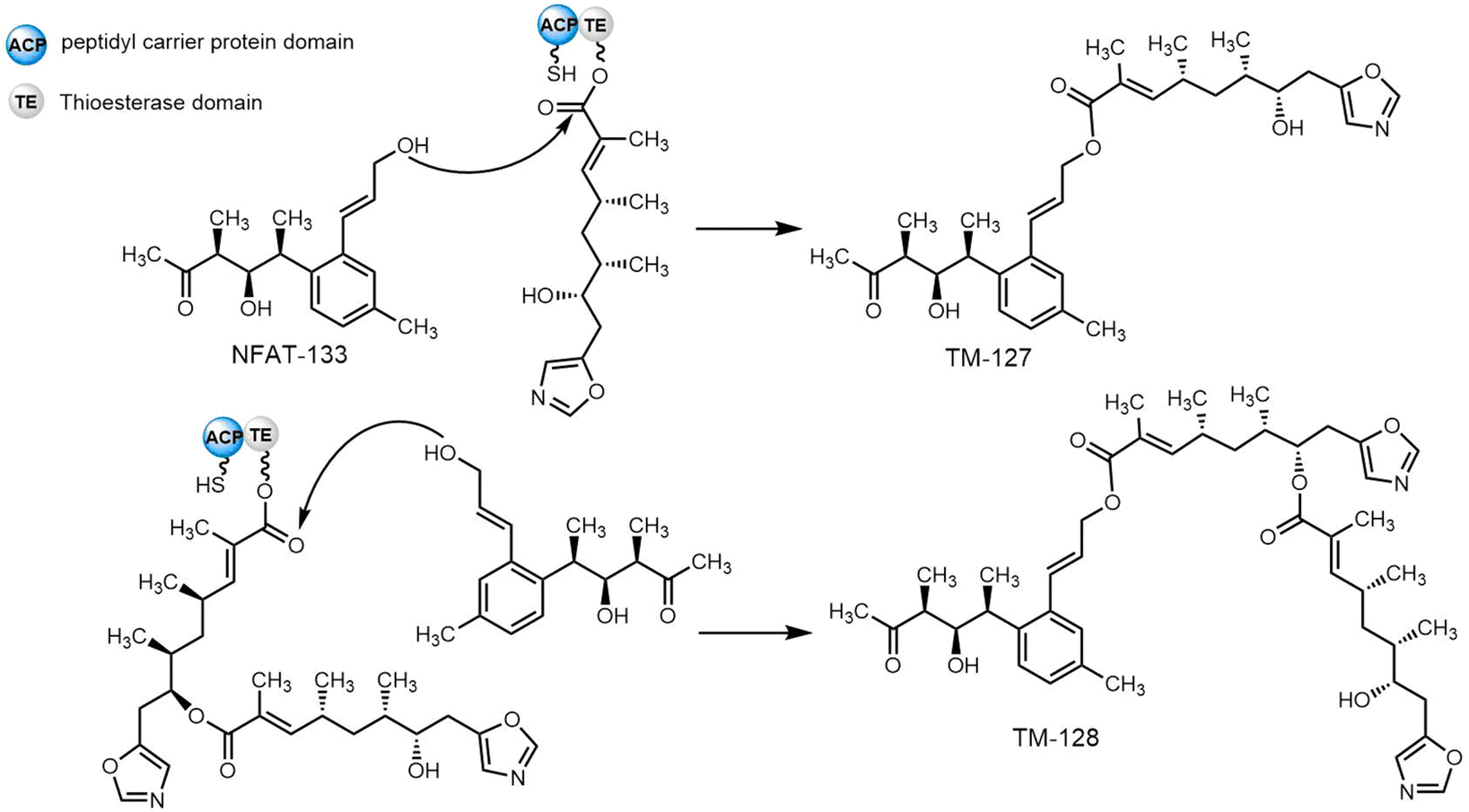 | ||
| Scheme 33 Proposed formation of TM-127 and TM-128 catalyzed by Cong-TE. | ||
Recently, Zhou et al. isolated a series of compounds 38–43 from gene-inactivated mutants of the conglobatin producer S. conglobatus ATCC 31005. Two of these compounds (38/39) are new oxazole-containing esters, whilst 40 is a biosynthetic precursor of 38 and 39 (Fig. 17). Inspired by the formation of 38/39, together with TM-127/TM-128 and considering the substrate scope of Cong-TE, 69 alcohol-containing compounds bearing benzylic, allylic, propargyl, primary, secondary, tertiary, and sulfhydryl hydroxyl groups were tested as substrates for Cong-TE, which was able to form 43 new hybrid esters. When the 43 alcohols that had been accepted by the Cong-TE were individually fed to the strain, 36 of these were successfully incorporated in the Cong NRPS-PKS to yield corresponding oxazole containing esters 44in vivo (Fig. 18).291
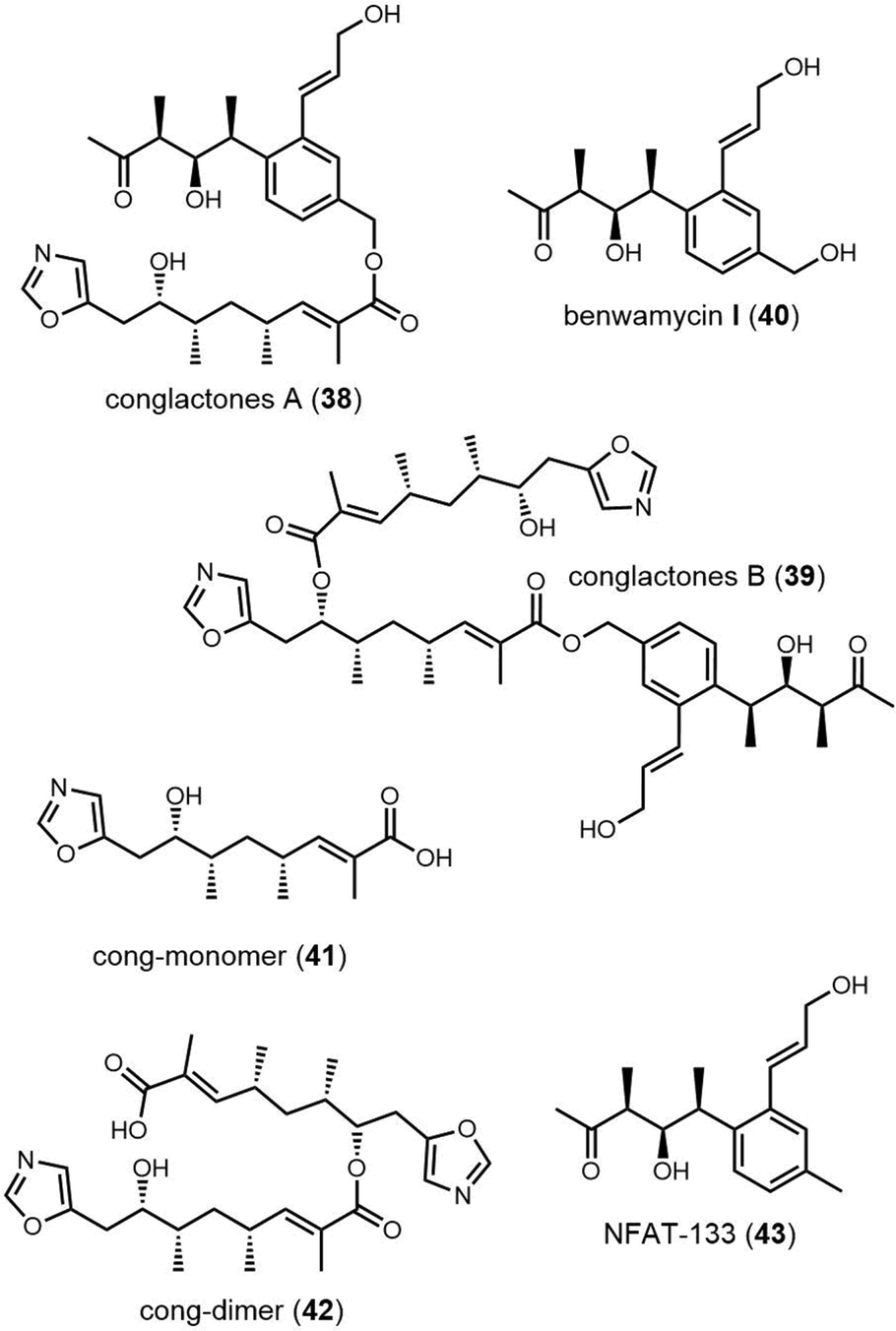 | ||
| Fig. 17 Structures of the compounds 38–43 produced by S. conglobatus. | ||
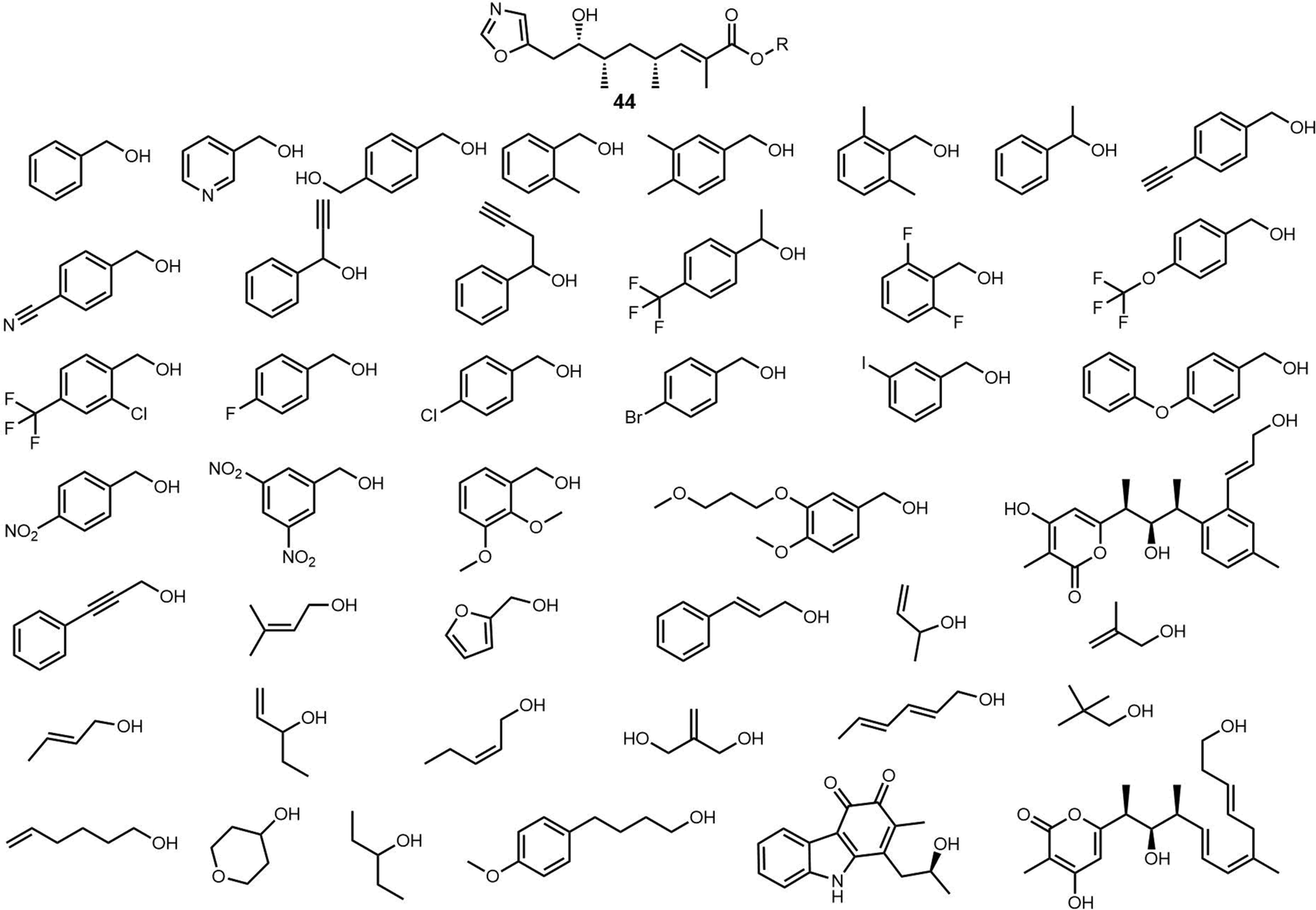 | ||
| Fig. 18 Exotic nucleophilic alcohols accepted by Cong-TE for esterification formation. | ||
Samroiyotmycin A, a C2-symmetric macro diolide displaying activities against the MDR malarial strain P. falciparum K1 and lung carcinoma cell line NCI-H187, was isolated from crude extracts of Streptomyces sp. BCC33756.292 Despite no investigations into the biosynthesis of the samroiyotmycins to date, the dimerization process appears similar to that found in conglobatin biosynthesis. In 2021, Hulme et al. completed the first total synthesis of samroiyotmycin A, not relying on ester-mediated dimerization but rather a one-pot alkyne cross metathesis–ring-closing metathesis (ACM–RCAM) reaction to generate the desired 20-membered macrocyclic framework (Scheme 34).293
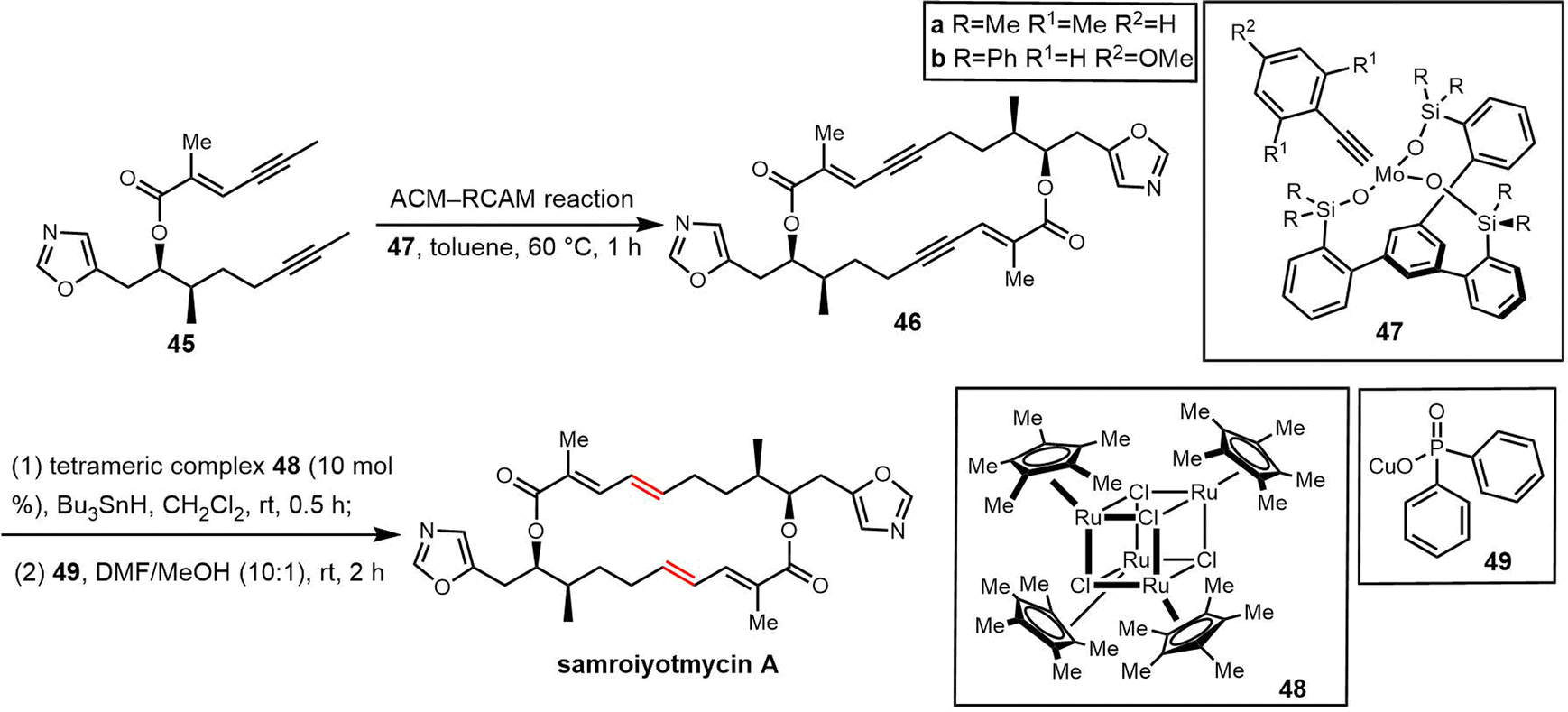 | ||
| Scheme 34 Chemical synthesis of samroiyotmycin A. | ||
This reaction employed two molybdenum alkylidynes (47a/b) endowed with a privileged tripodal silanolate ligand. Treatment of monomer 45 with the complex 47a provided the dimer 46 in good yield by raising the temperature. Under these conditions, higher-order oligomers were also found to be formed via concentration-induced polymerization, and the yield was significantly improved when using a more dilute solution of 45. Treatment of 45 with complex 47b gave full conversion to the desired product 39, but purification of 46 was hindered by fragmentation of the catalyst ligands. Hydrostannation of 46 catalyzed by ruthenium catalyst 48 and subsequent protodestannation in the presence of copper(I) salt 49 led to successful synthesis of the desired (5E,21E) isomer samroiyotmycin A as the major stereoisomer.
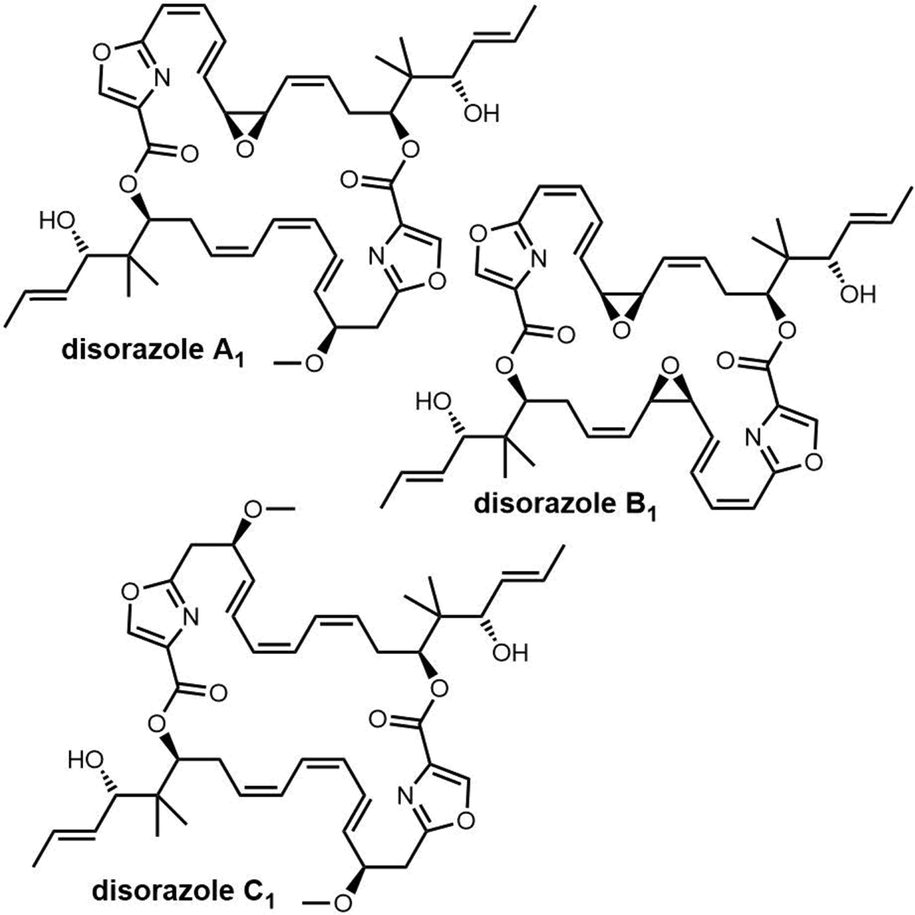 | ||
| Fig. 19 The structure of disorazoles. | ||
Disorazoles are biosynthesized by a trans-AT PKS-NRPS hybrid pathway, with the trans-acyltransferase encoded by a separate gene, dszD(disD). Analysis of the domain organization of this PKS/NRPS reveals that DszC(DisC) contains one NRPS module, utilizing serine to form the oxazole, and two non-extending PKS modules located between NRPS and the TE domain. It has been proposed that a monomer is transferred to the serine residue of the TE domain from the acyl carrier protein before being dimerized with the other monomer loaded on the adjacent acyl carrier protein. Since both non-extending domains contain ACP domains along with the NRPS module, which contains a PCP domain, it is unclear which carrier proteins are involved in the dimerization of monomers (Scheme 35).297
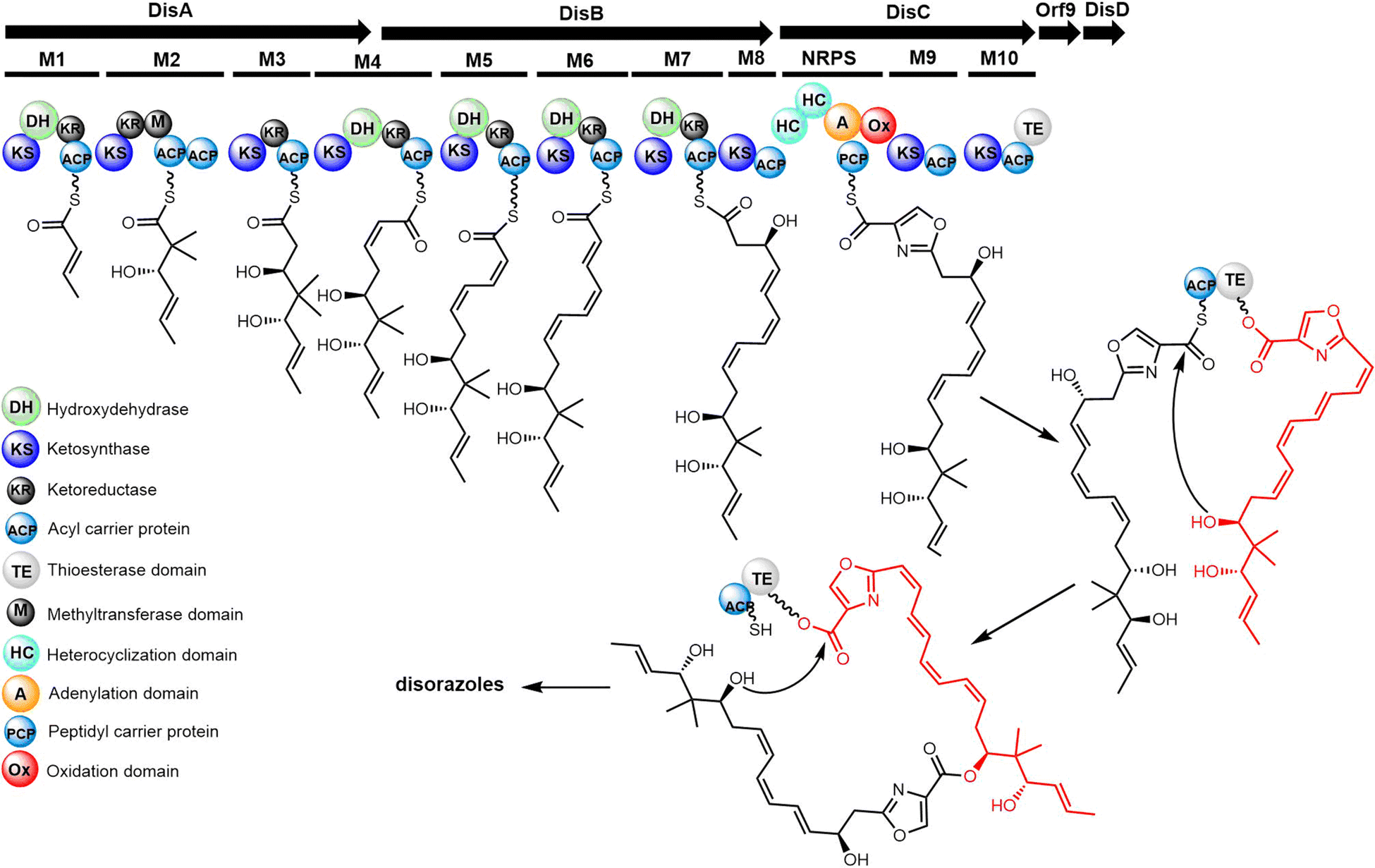 | ||
| Scheme 35 Biosynthesis of disorazoles. | ||
In 2004, Wipf and Graham reported the first asymmetric total synthesis of disorazole C1.298 The dimer was separated into four segments and both segments 50 and 51 were utilized twice for the convergent synthesis. Sonogashira cross-coupling of 50 and 51 formed the protected monomer 52 and subsequent acylation and a second Sonogashira coupling afforded seco-disorazole C1. Selective mono-saponification of 54, followed by a Yamaguchi lactonization provided macrocycle 55. Finally, double alkyne reduction with Lindlar catalyst afforded disorazole C1 (Scheme 36).
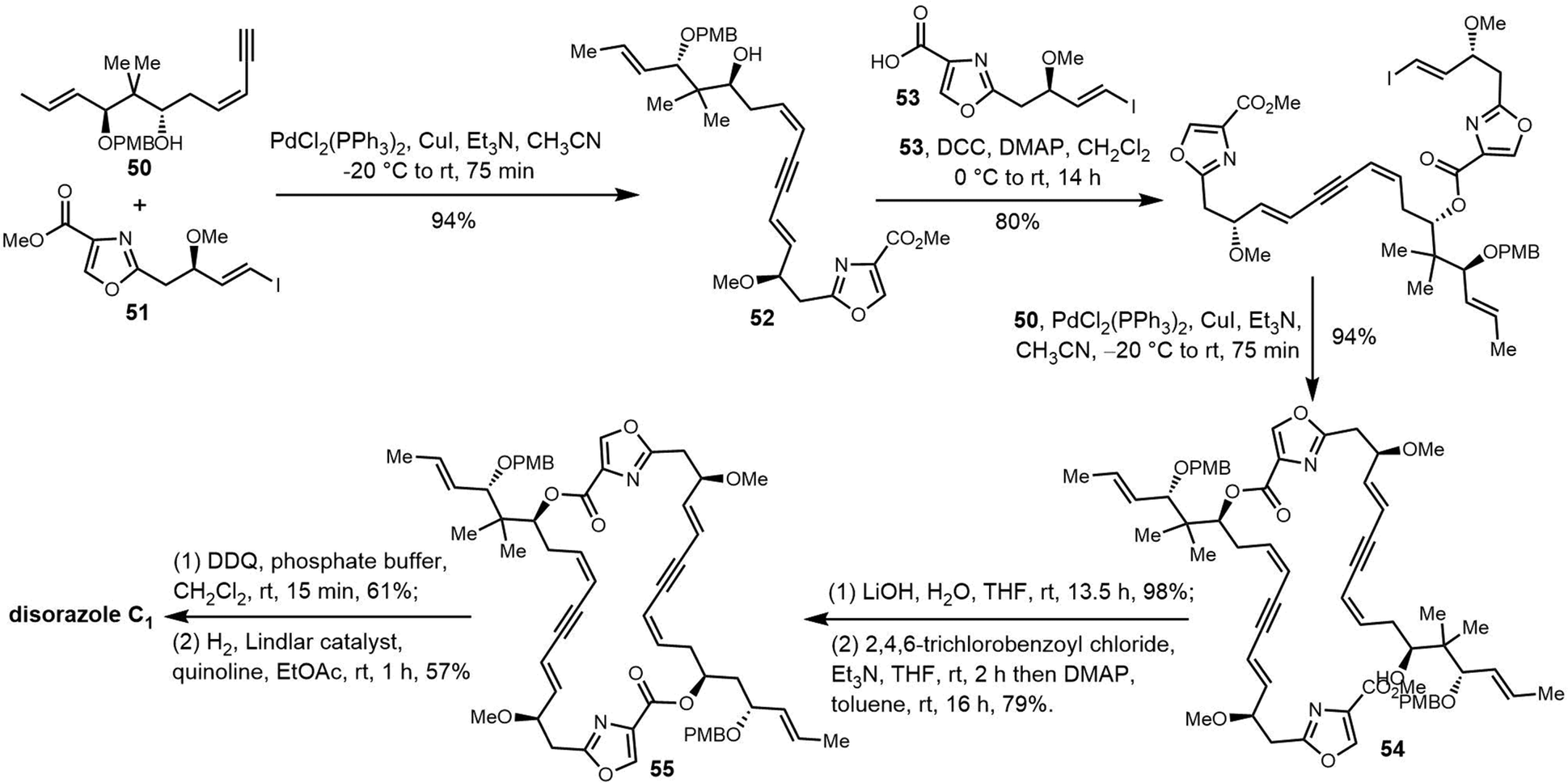 | ||
| Scheme 36 Total synthesis of disorazole C1. | ||
Nicolaou et al. also employed a convergent strategy to synthesize disorazoles A1 and B1.299 Monomers 56, 57 and 58 were constructed by the connection of different building blocks through a Wittig reaction, Suzuki coupling and Stille coupling, with a Sharpless asymmetric epoxidation exploited to synthesize the epoxy vinyl moiety in 57 and 58. Condensation of 56 and 57 by a Yamaguchi esterification provided linear diester 59. Removal of TMS from 59, followed by a Yamaguchi macrolactonization afforded the final product disorazole A1. Using the same strategy, the epoxy monomers 56 and 58 were used to synthesize the C2-symmetrical disorazole B1 (Scheme 37).
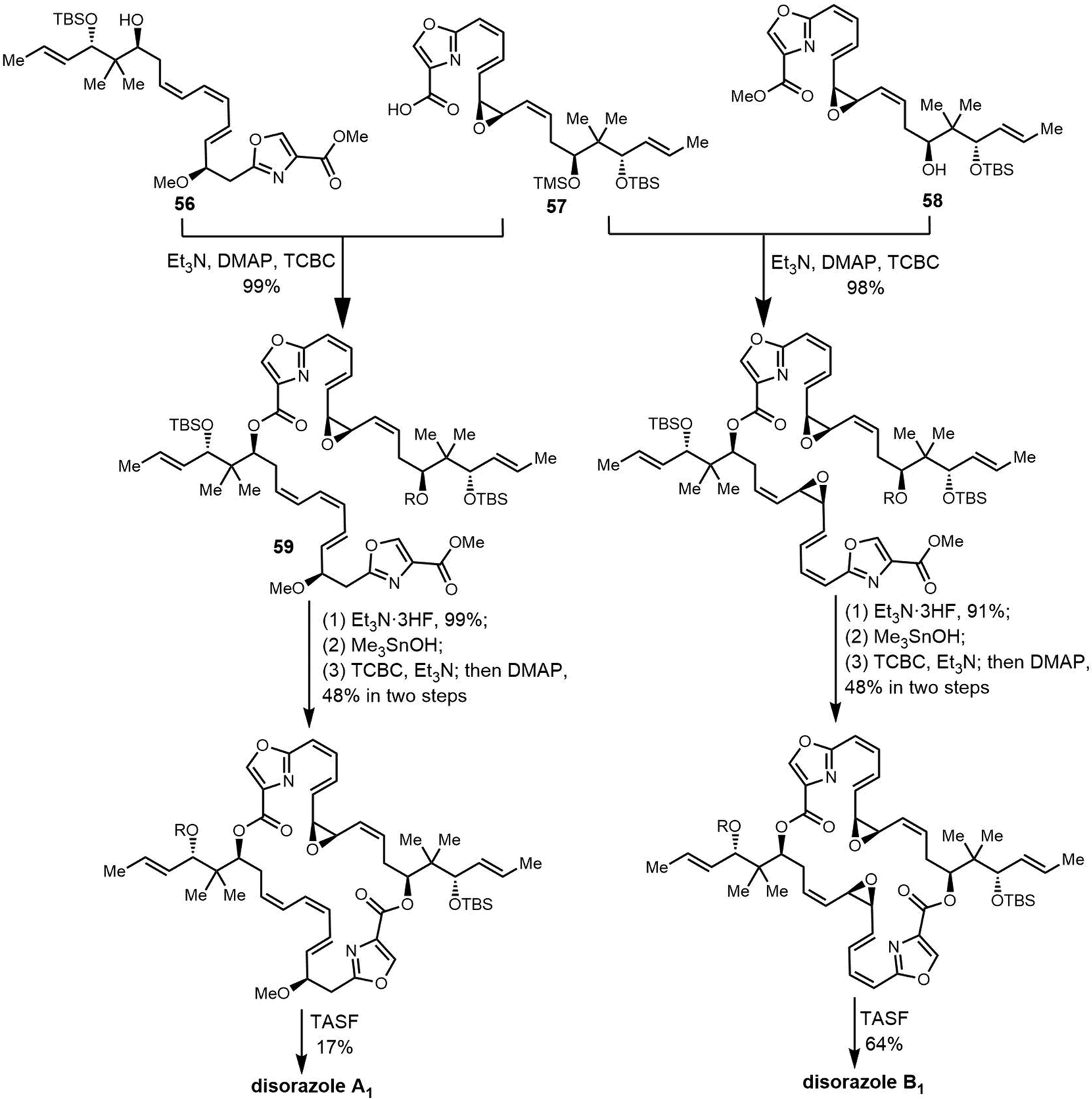 | ||
| Scheme 37 Total synthesis of disorazole A1 and disorazole B1. | ||
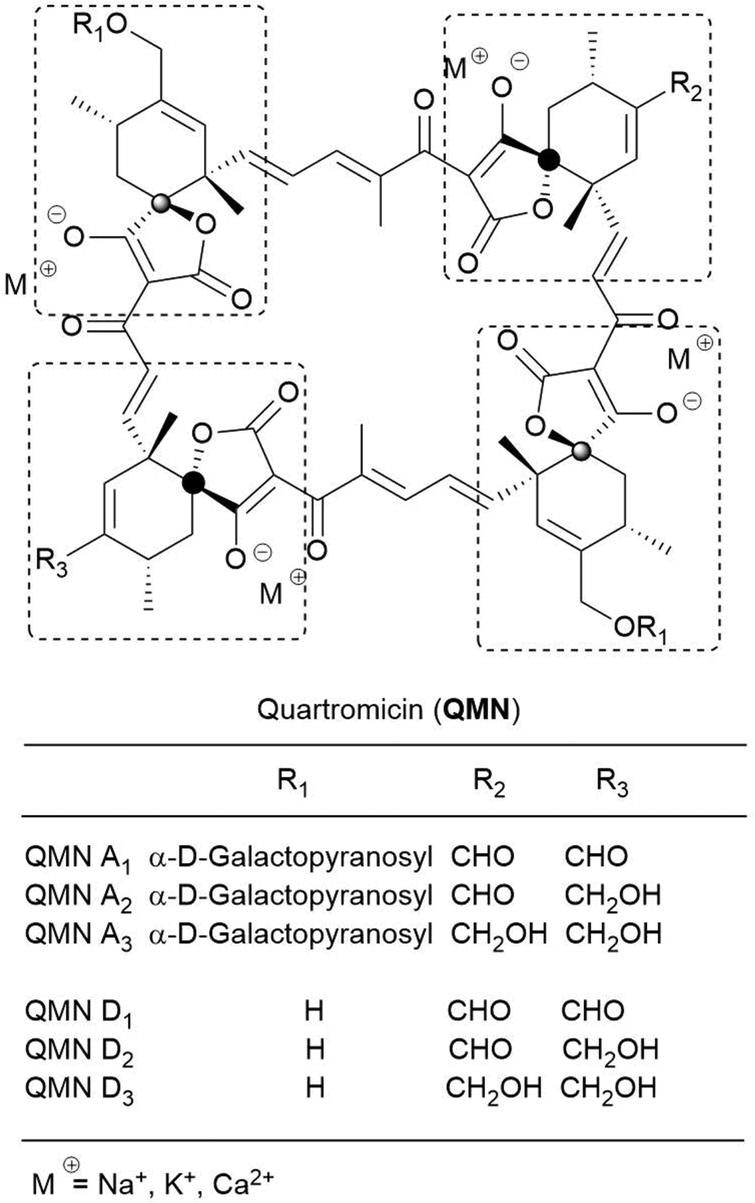 | ||
| Fig. 20 The structure of quartromicin. | ||
In 2012, He et al. reported the biosynthetic gene cluster of quartromicins from the producer A. orientalis No. Q427-8. The QmnA1-QmnA2-QmnA3 PKS assembly is responsible for the synthesis of two polyketide chains through a ‘module skipping’ pathway, after which 3-oxoacyl-ACP synthase III QmnD5 catalyzes the condensation of a glycerate unit and the mature polyketide chains to form tetronates 60 and 61.301 Dehydration of 60–61 relies on QmnD3 and QmnD4 to afford 62 and 63.302 Further biosynthetic studies concerning natural spirotetronate compounds have revealed enzymatic [4+2] reactions are used to form the spirotetronate intermediate.303 A similar intramolecular [4+2] route is anticipated as the source of the four chiral spirotetronate centers found in the quartromicins (Scheme 38).
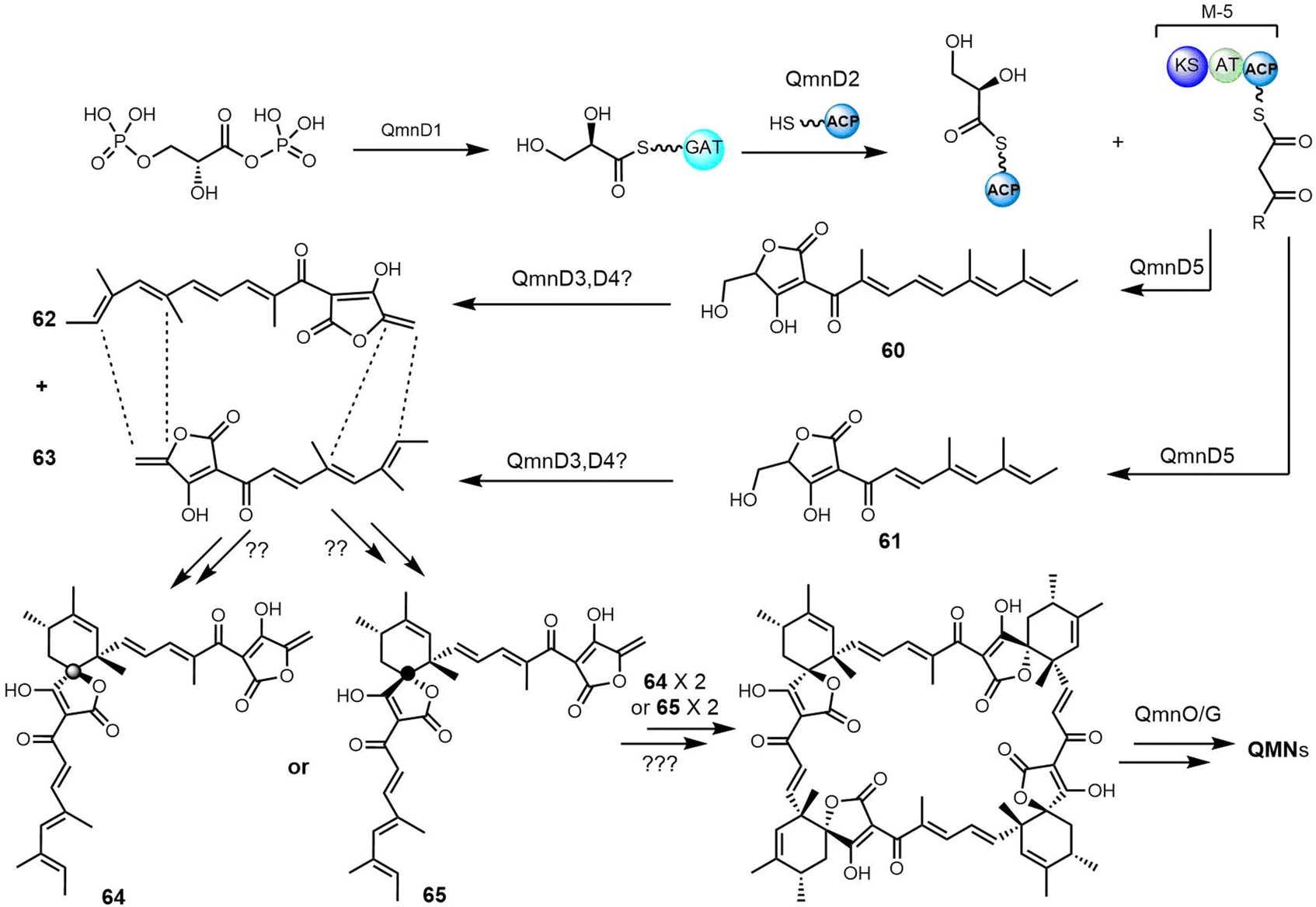 | ||
| Scheme 38 Putative biosynthetic pathway of quartromicins. | ||
Roush et al. completed the diastereoselective synthesis of the endo- and exo-spirotetronate subunits 66 and 67 of the quartromicins through an enantioselective Diels–Alder reaction of an acyclic (Z)-1,3-diene and partially assigned the stereochemical assignment of quartromicins A3 and D3.304,305 They further synthsised the vertical and horizontal bis-spirotetronate (68) units of quartromicins A3 and D3 (Fig. 21).306 However, due to the structural complexity, the total synthesis of quartromicins has not been completed to date.
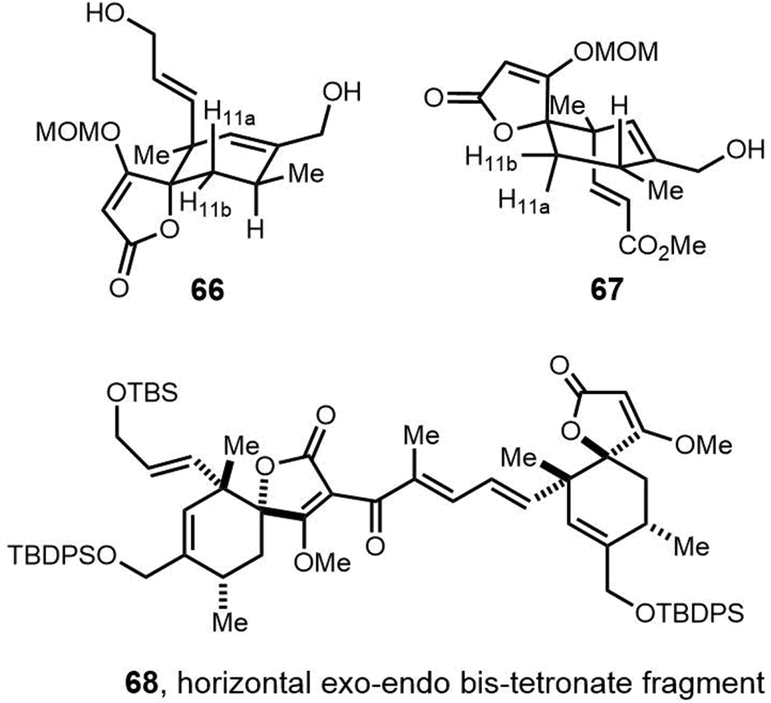 | ||
| Fig. 21 Synthetic spirotetronate subunits for quartromicins. | ||
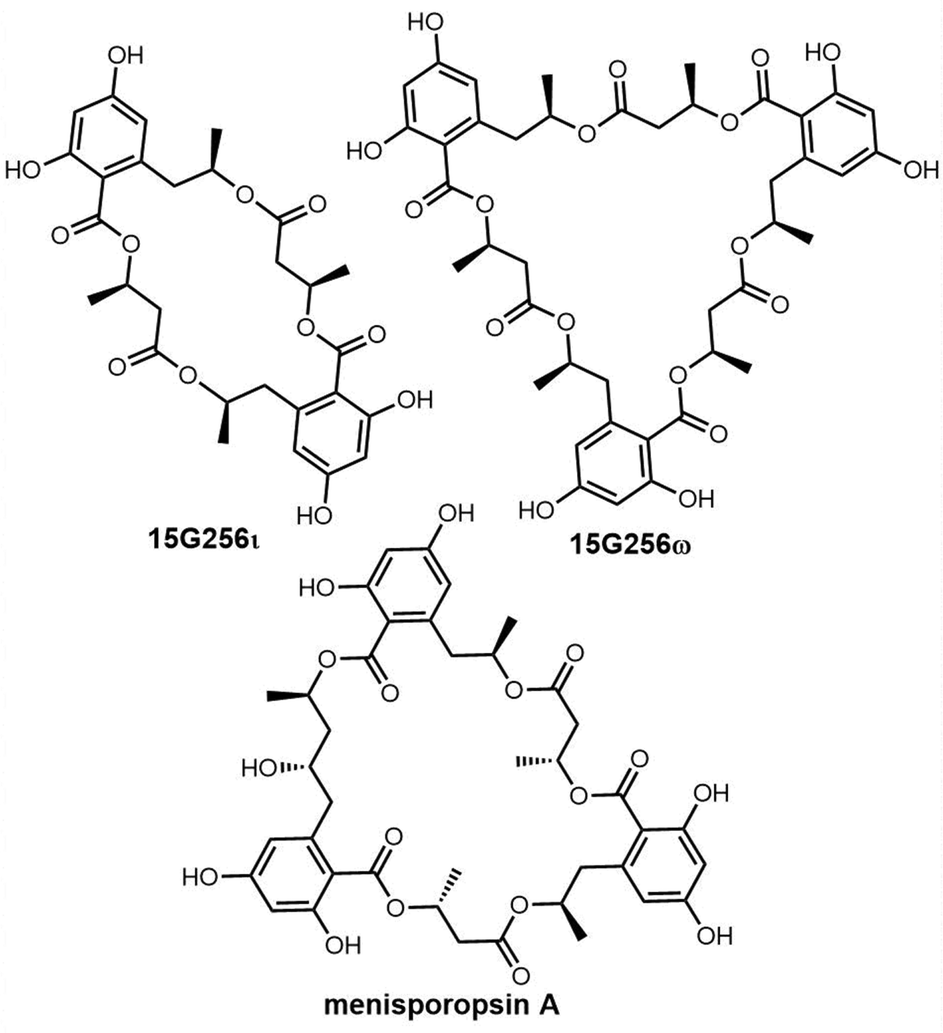 | ||
| Fig. 22 Examples of 15G256 polyesters. | ||
Menisporopsin A, 15G256ι and 15G256ω all contain 3-hydroxybutyric acid and 3,5-dihydroxy-7-(β-hydroxypropyl)-benzoic acid moieties, while menisporopsin A also contains an extra 2,4-dihydroxy-6-(2,4-dihydroxy-n-pentyl)-benzoic acid unit. The menisporopsins exhibit a broad spectrum of cytotoxic, antimycobacterial and antimalarial activity.308,309
Concerning their biosynthesis, a 13C-labeling experiment has demonstrated that the pentalactone scaffold of menisporopsin A is assembled by a polyketide synthase, with all the carbons of menisporopsin A derived from acetate as the sole building block.310
Two polyketide synthases, Men1 (HR-PKS) and Men2 (NR-PKS), were identified in Menisporopsis theobromae BCC 4162 and were shown to be responsible for the biosynthesis of menisporopsin derivatives. These two enzymes are involved in the iterative biosynthesis of the polyketide backbone and the subsequent cyclization of the molecule.
The TE domain in Men2 is responsible for the release of the final product from the PKS assembly line (Scheme 39). The esterification and cyclolactonization reactions required for the synthesis of the menisporopsins could therefore reside in this TE domain of the NR-PKS, which is similar to that of the NRPS catalyzing the elongation and cyclization of trilactone in enterobactin biosynthesis and that of modular PKSs catalyzing macrodiolide formation in elaiophylin (from Streptomyces violaceusniger DSM 413721) and conglobatin biosyntheses (from Streptomyces conglobatus).311
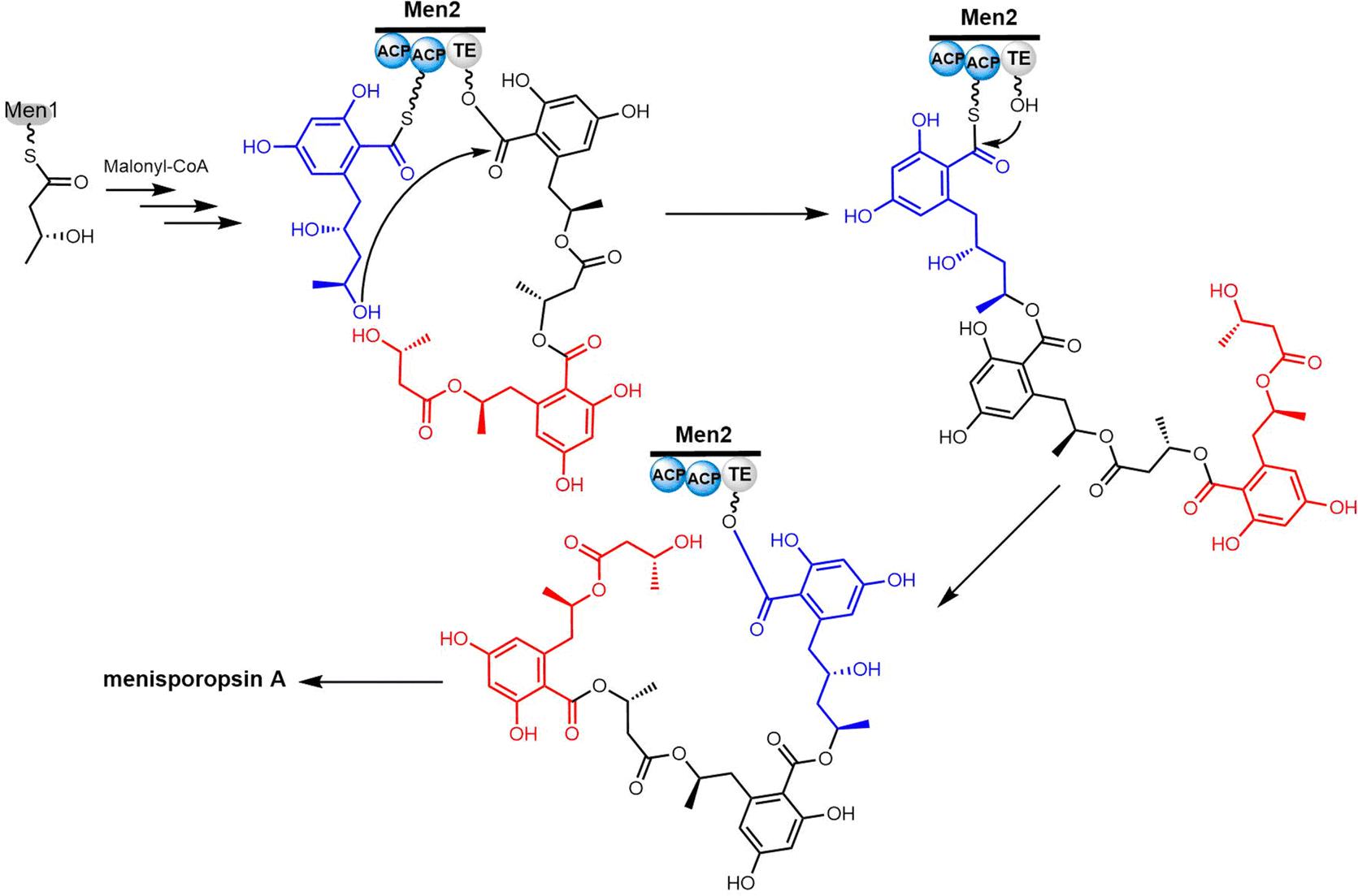 | ||
| Scheme 39 Putative biosynthetic pathway of the menisporopsin A. | ||
 | ||
| Fig. 23 The representative boron-containing natural products. | ||
The boromycins were discovered from the strains Streptomyces antibioticus ETH 28829 and Streptomyces sp. MA4423 and are notable for being the first natural products found that contain the element boron.318,319 Boromycin analogues Tartrolon A and B were isolated from the culture broth of Sorangium cellulosum So ce 678, which contain a similar boron binding region as is found in boromycin and aplasmomycin. The tartrolons exhibit significant antibacterial and cytotoxic activity.314,320
Feeding experiments with 13C-labeled precursors have demonstrated that the biosynthesis of boromycin and aplasmomycin utilise similar assembly pathways.321,322 Recent studies have revealed the biosynthesis of boromycin-related natural products at the genetic level. Elshahawi et al. first identified the tartrolon BGC (trtA–trtJ) from the strain Teredinibacter turnerae T7901.323 Among the trt cluster, trtDEF comprise three large genes that encode trans acyltransferase (AT) type I PKSs. These three multimodular PKS ORFs contain 11 modules in addition to the loading module. No detailed research concerning the mechanism of macrocyclization in this pathway has yet been performed, with the intra-module TE domain in TrtF postulated to be involved with release and cyclization (Scheme 40).324 It has been hypothesized that natural products related to boromycin undergo cyclization catalysed by a TE domain in a process similar to that observed in disorazole biosynthesis; this is due to their similar dimercyclic scaffold.324
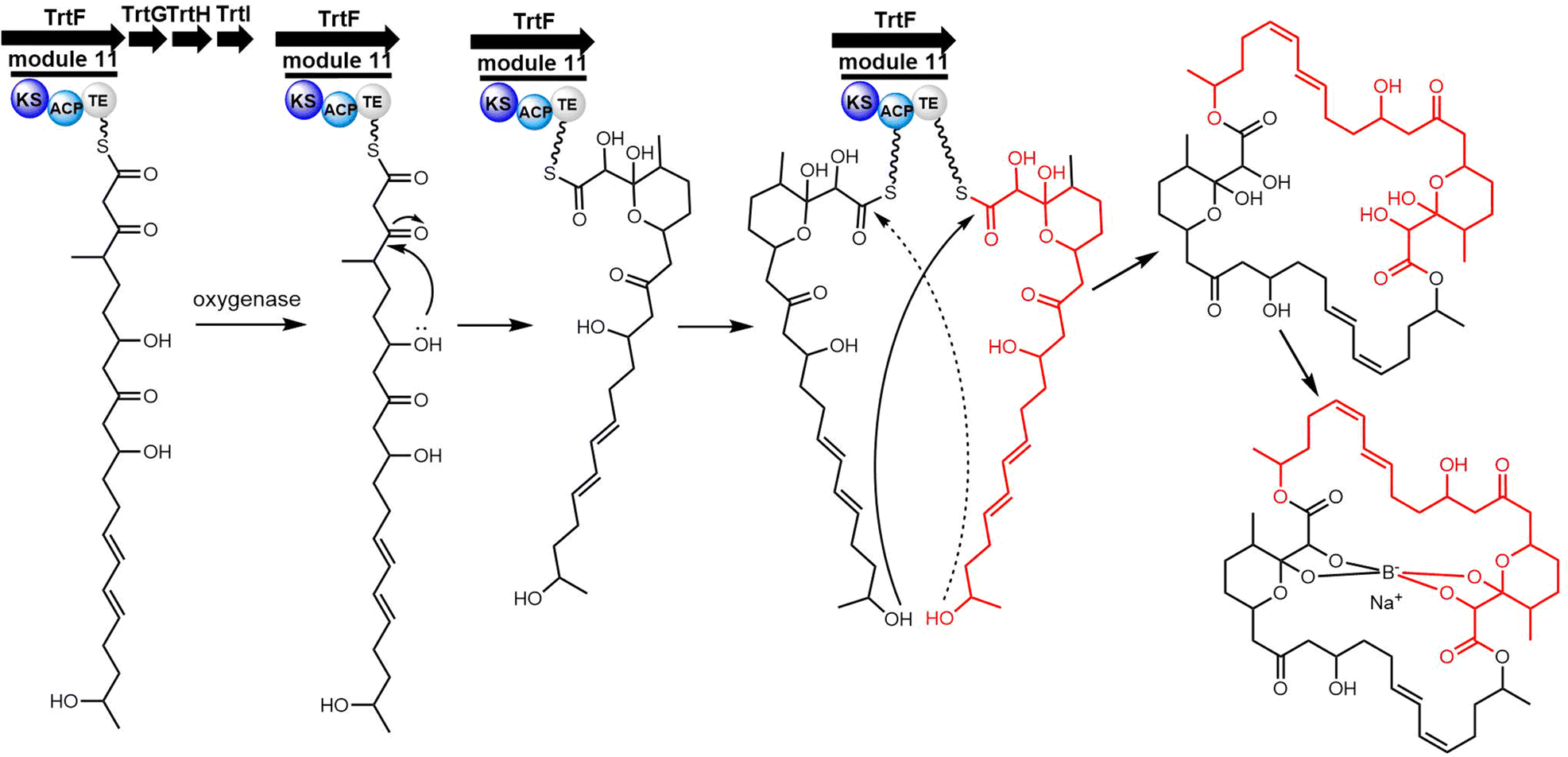 | ||
| Scheme 40 Putative biosynthetic pathway of the tartrolons. | ||
In 2014, Avery et al. completed the chemical synthesis of the symmetrical 36-membered macrodiolide aplasmomycin A via Mukaiyama lactonization strategies. A base-promoted Chan rearrangement method was used to generate the symmetrical 36-membered diolide intermediate during the synthesis. The final product, aplasmomycin A, was generated by biomimetic modification of desboroaplasmomycin A, followed by boration reaction with trimethyl borate. This synthetic strategy is also relevant for the synthesis of boromycin (Scheme 41).325
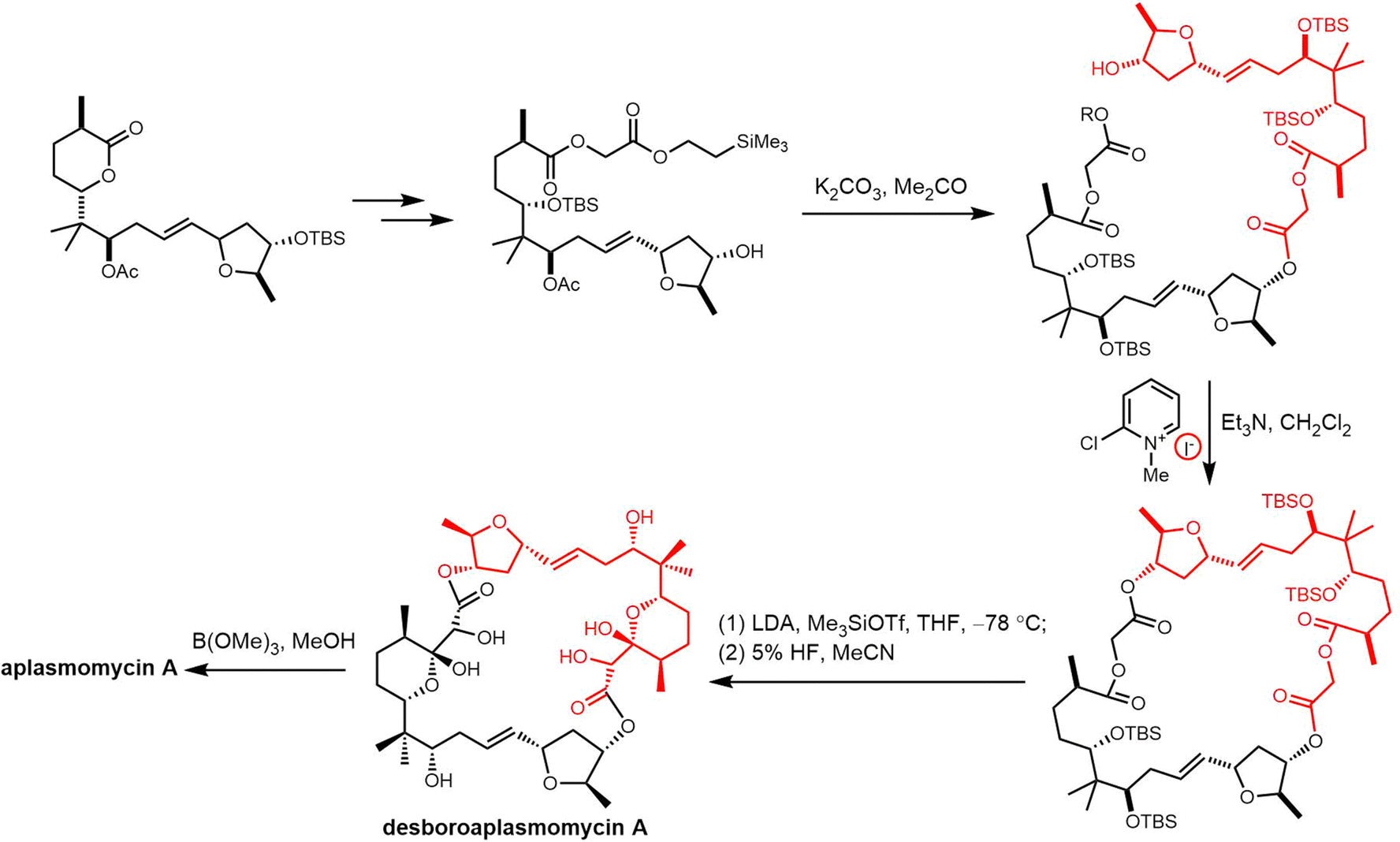 | ||
| Scheme 41 The synthesis of aplasmomycin A. | ||
The pamamycins are group of macrodiolides with antifungal activity isolated from Streptomyces alboniger.329–332 Investigation of S. alboniger IFO 12738 revealed that the derivative pamamycin-607 is the main component in the pamamycin mixture.333,334 The biosynthesis of pamamycin-607 has been investigated by isotopic feeding, which suggested the building blocks of pamamycin-607 were derived from the acetate, propionate, succinate and amino acid (Fig. 24).335
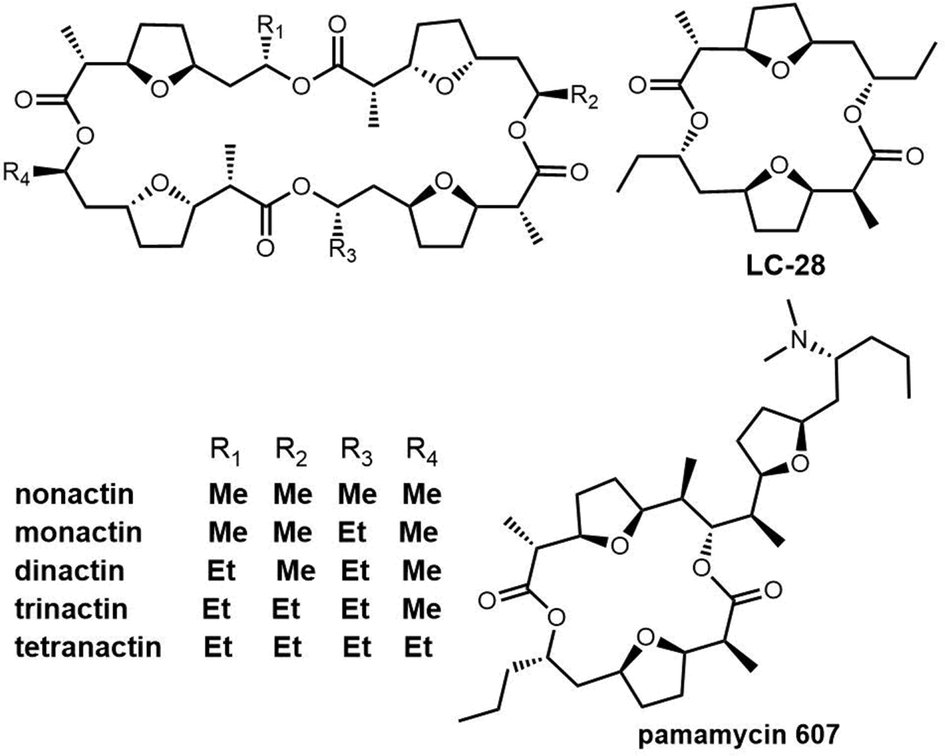 | ||
| Fig. 24 The chemical structures of nonactin and its analogues. | ||
Luzhetskyy et al. recently reported the identification of the nonactin and pamamycin BGCs in S. alboniger DSMZ40043 and S. sp. HKI 118.329 The nonactin BGC contains five KSs-encoding genes, and was characterized from Streptomyces griseus.329 Two KSs, NonJ and NonK, which are highly homologous to the KSs PamJ and PamK in pamamycin biosynthesis, catalyse the C–O condensation reaction of acyl CoA substrates and result in the closure of the macrodiolide ring (Scheme 42).326 Interestingly, L-valine supplementation increased pamamycin production and larger derivatives due to increased availability of CoA thioesters, but negatively affected growth and repressed pamamycin formation in the heterologous host S. albus J1074/R2.334
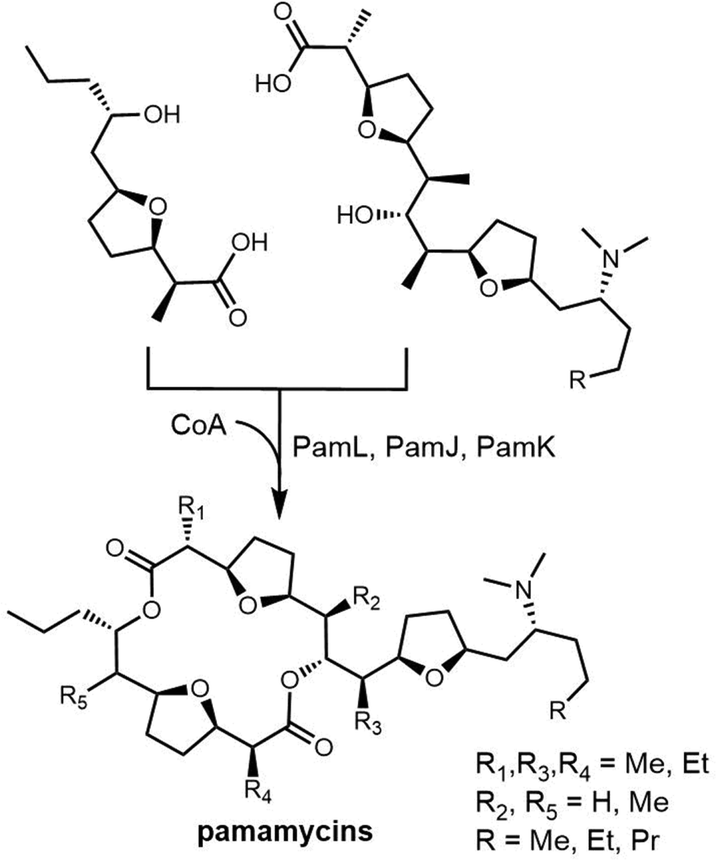 | ||
| Scheme 42 The putative key step in the pamamycin biosynthesis.329 | ||
The total syntheses of the macrotetrolide family antibiotics have been realized through a variety of methods. The key macrocyclic reaction can be performed through macro-lactonization reaction using different catalysts (Scheme 43). Based on retrosynthetic analysis, diverse esterification routes have also been developed to realize the formation of the pamamycin dilactone backbone by coupling two precursor “fragments”.336–342
 | ||
| Scheme 43 The synthetic routes towards pamamycin and derivatives. | ||
3. Alternative approaches to macrocyclization
Natural product biosynthesis contains many strategies to perform the synthesis of carbohydrate-containing macrocycles that extend beyond macrocyclization mediated by TE domains.343 In this section, we will introduce representative examples of macrocyclization, with a focus on the synthesis of macrocyclic molecules that have not been extensively summarized.3.1 Macrocyclization via imine formation
NRPS-mediated peptide synthesis can also be terminated through the reduction of the thioester of the terminal module to liberate an aldehyde, which then undergoes macrocyclization. The nostocyclopeptide class of macrocyclic imines, isolated by the Moore group from the terrestrial cyanobacterium Nostoc sp. ATCC53789, are cyclic heptapeptides, not dimers or oligomers, that differ in the residues present in the terminal position of the peptide.344 Marahiel and co-workers were first to demonstrate that the reductase domain present in the final module of the NRPS machinery was able to liberate the aldehyde heptapeptide product that then spontaneously cyclized to form the nostocyclopeptide products.345 Related classes of cyclic imine heptapeptide natural products isolated from cyanobacteria include koranimine346 and scytonemide A (Scheme 44).347 The facile nature of the macrocyclization has been explored via total synthesis for scytonemide A (Scheme 44),348 and has also shown versatility as a tool in the synthesis of peptide macrocycles more generally.349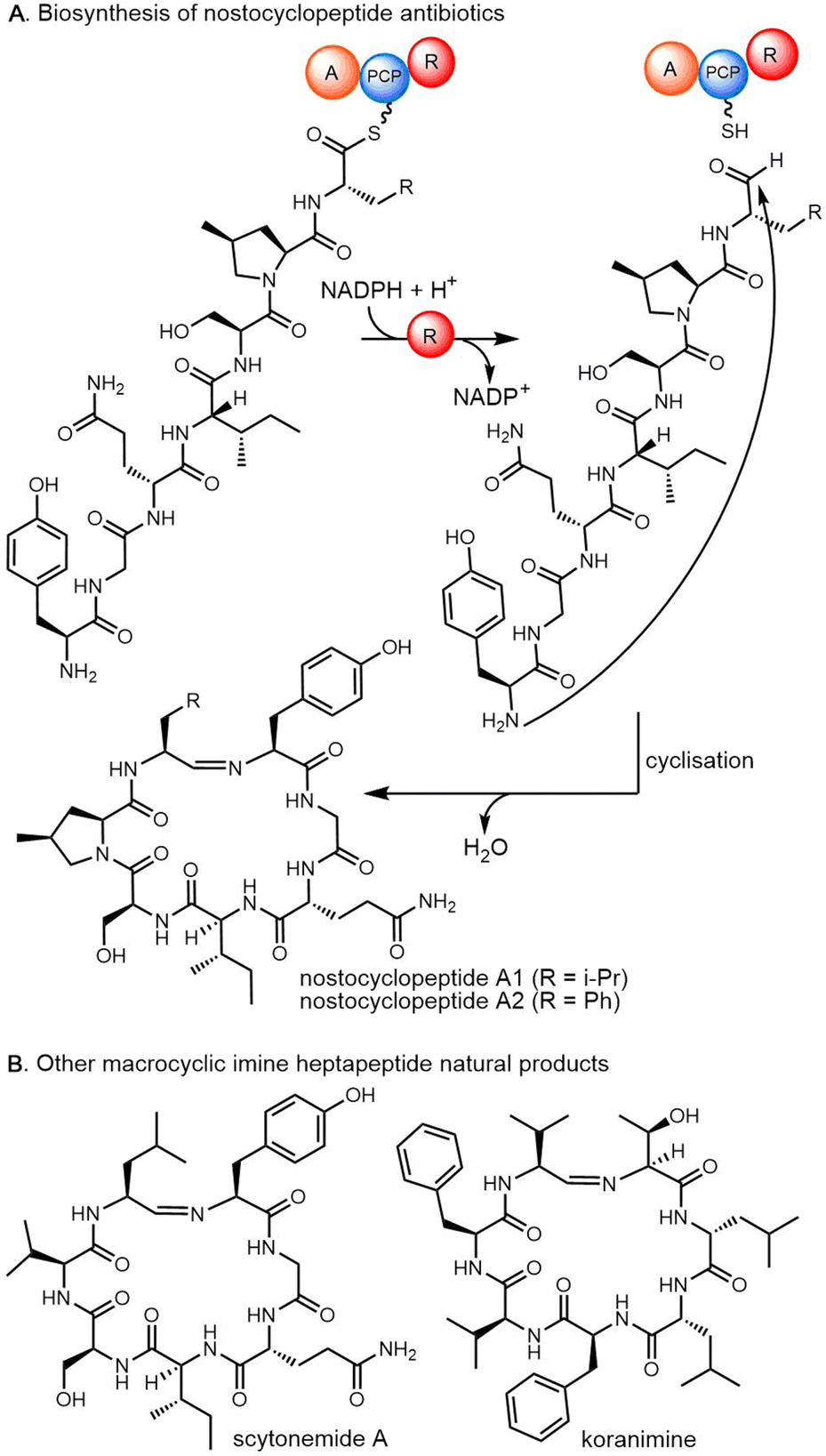 | ||
| Scheme 44 The cyclic imine natural products. (A) total synthesis of scytonemide A; (B) the structure of koranimine and scytonemide A. | ||
3.2 Alchivemycin A
Alchivemycin A (74), which was isolated from a plant-derived strain of Streptomyces, displays a novel polycyclic structure containing a rare 2H-tetrahydro-4,6-dioxo-1,2-oxazine moiety.350 Whilst initial biosynthetic postulates for this PKS-NRPS hybrid relied on NRPS-mediated activation of N-hydroxyglycine,351 recent work from the Tan and Ge groups352 has shown that this moiety is in fact generated by the activity of the FAD-dependent monooxygenase AvmO1 that performs oxygen insertion into the amide bond of a 4-hydroxy-1,5-dihydro-2H-pyrrol-2-one ring in a cyclic precursor molecule 73. The formation of 73 has been demonstrated to commence with intermediate 70 that is itself is proposed to be formed via a [4+2] cycloaddition of the final NRPS-bound intermediate 69 together with tetramic acid formation catalyzed by the Dieckmann cyclase AvmQ.353 Formation of 72 is catalyzed by the enzyme AvmM, which is a novel enzyme class, and involves sequential dehydration to intermediate 71, followed by cyclization through Michael addition to form 72 (Scheme 45).353 The biosynthesis of alchivemycin A (74) therefore represents a fascinating array of novel biosynthetic chemistry.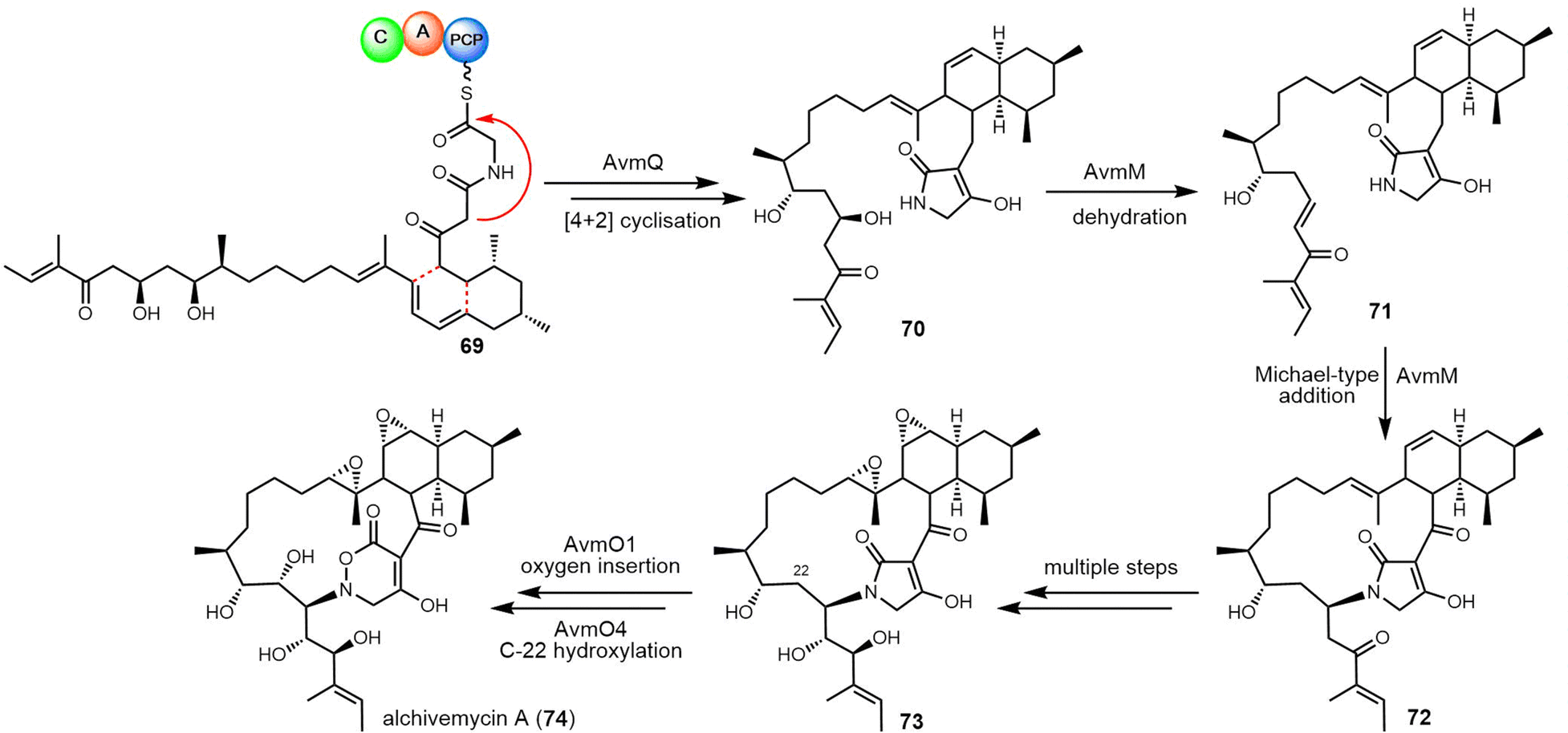 | ||
| Scheme 45 The synthesis of alchivemycin A. | ||
3.3 Butelase-mediated cyclodimerization
Butelase 1 and sortase A are powerful cyclases that can mediate macrocyclization in cyclopeptide synthesis.354,355 The Tam group have reported a biomimetic cyclo-oligomerization synthesis method using butelase 1, which is an Asn/Asp-specific ligase. This method can be used to perform the cyclization of 3–15 amino acids ending with Asn and a His-Val tail (Scheme 46). A one-pot synthesis of dimerized cyclopeptides is also possible through butelase-controlled cyclo-oligomerization. Furthermore, butelase 1 is highly effective at cyclizing macrocycles with more than 200 amino acids and displays a wide substrate recognition range covering L-amino acids and D-amino acids.356,357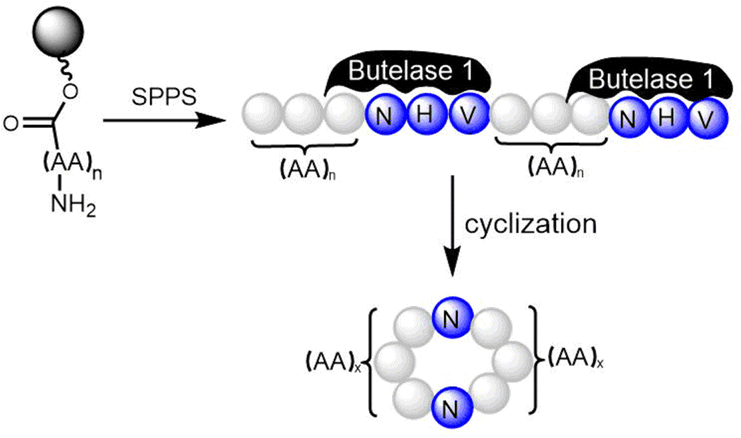 | ||
| Scheme 46 Butelase-mediated cyclization in cyclopeptide synthesis. | ||
3.4 Beauvericin, bassianolide and enniatin
Filamentous fungi are important sources of cyclopeptide natural products. In fungal NRPS systems, the terminal condensation-like (CT) domain is often used to synthesize macrocyclic peptidyl products and is responsible for the release of the cyclopeptide product (Fig. 25). | ||
| Fig. 25 Examples of macrocyclic peptidyl products mediated by the terminal condensation-like (CT) domain. | ||
In comparison with TE domains, CT domains show a different protein fold and different catalytic mechanism. The catalytic triad in the TE domain transfers the peptidyl intermediate to the active site serine, which then attacks the peptidyl-thioester in an intramolecular reaction to complete cyclization. In contrast, CT-catalyzed cyclization does not generate a covalent adduct with the enzyme. Instead, the catalytic histidine in the CT domain (conserved “HHxxxDG” motif) deprotonates the amine nucleophile, which attacks the thioester carbonyl bound to the PCP domain to cyclize and liberate the product.358 In addition, the bacterial TE-catalyzed reactions differ from the fungal CT domain catalysed reactions in that the TE domain can accept peptidyl-S-N-acetylcysteamine (SNAC) mimics, whereas the CT domain needs specific protein–protein interactions with the upstream PCP domain to be competent for catalysis, requiring a specific PCP domain partner. Gao et al. demonstrated that CT domains only recognized peptide substrates tethered to the native PCP domains, while peptidyl-SNAC and peptidyl-CoA substrates were not cyclized.359 The interactions between the PCP and CT domain seems play a “proofreading mechanism” for the correct peptide cyclization and prevent spontaneous hydrolysis reaction.358
Despite sharing the conserved motif with the canonical C domains, CT domain is placed in a separate evolutionary taxonomy due to functional divergence. The structural basis of the unique macrocyclization activity of CT domain has been identified through the analysis of the structural elements of CT domains compared to canonical C domains.359 Specifically, although the protein sequence of CT domains exhibits low similarity to that of canonical C domains (VibH,360 TycC-C6,361 PCP2-C3 didomain362 and SrfA-C363), its overall fold is strikingly similar to these C domains, heterocyclization (Cy) domain and epimerisation (E) domains. CT domains comprise of two largely separate subdomains, with both N-/C-subdomains arranged in a V-shaped manner and adopting the chloramphenicol-acetyltransferase fold (Fig. 26).
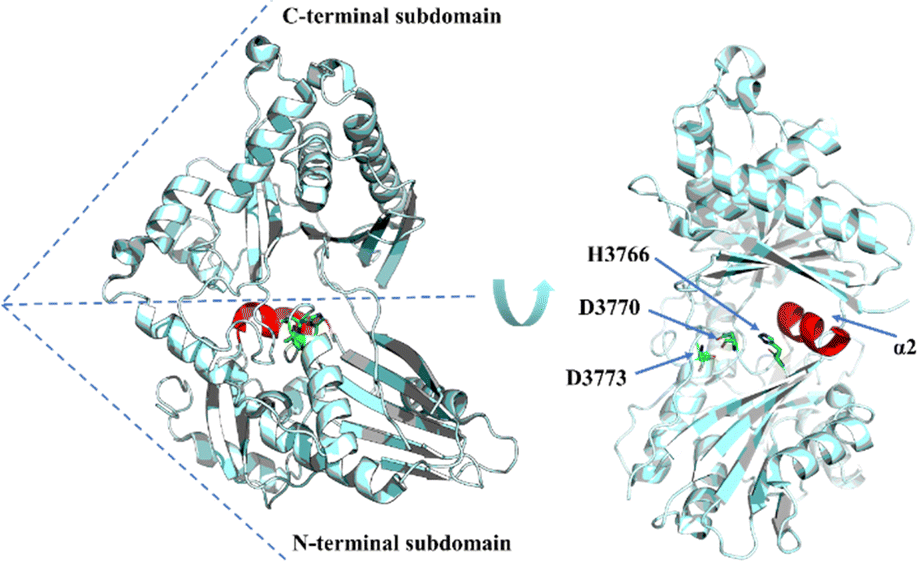 | ||
| Fig. 26 The overall structure of the CT domain. The H3766, D3770, and D3773 are drawn as green sticks, while the α2 helix of CT domain is coloured in red. | ||
However, the main structural feature which distinguishes CT domains from classic C domains is the replacement of the N-terminal loop in C domains with a α1 helix in CT domains. The insertion of this α2 helix in the CT domain results in the compaction of the two subdomains of CT domains close to the acceptor site, ultimately blocking this substrate access channel (Fig. 26). This structural dissimilarity from canonical C domains is noteworthy, as it enables CT domain to have distinctive macrocyclization activities.359
In some fungal depsipeptides synthetases, CT domains are also involved in the catalysis of cyclo-oligomerization. Enniatin (cyclic trimer), beauvericin (cyclic trimer), and bassianolide (cyclic tetramer) are representative cyclo-oligomeric depsipeptides (CODs) that are biosynthesized as dimers, trimers, or tetramers through iterative cyclo-oligomerization by CT domains. There are numerous impressive reviews on these types of peptides from fungi.364–367 In this article, we will briefly summarise some of the previously reviewed material and describe some of the defining characteristics of enniatin, beauvericin, bassianolide, and PF1022A. These cyclopeptides are all assembled via the actions of iterative NRPSs (type B), in which individual modules within the NRPS assembly lines are reused for iterative cycles of peptide assembly.368
The enniatins and beauvericin are mycotoxin CODs found in many fungi, such as Fursarium spp., Verticillium hemipterigenum, Halosarpheia sp., and entomopathogen Beuveria bassiana, amongst others, and exhibit a wide range of biological activities, includes antibacterial, insecticidal, and anticancer activity.369–371 Beauvericin, due to its ionophoric properties, exhibits a similar range of diverse biological activities, including antibiotic, antifungal, insecticidal, and cancer cell antiproliferative and antihaptotactic activity.372–374 As a broad-spectrum anthelmintic, the semisynthetic PF1022A derivative emodepside (bis-para morpholino-PF1022A) has even been brought to market.375
The assembly of enniatin, beauvericin, and bassianolide is performed by an NRPS in each pathway. These NRPSs resemble each other, consisting of a di-module (C1-A1-PCP1, C2-A2-MT-PCP2a-PCP2b-C3) responsible for peptide synthesis (Scheme 47). As an example, the bbBeas encodes a BbBEAS nonribosomal peptide synthetase that was isolated from the strain B. bassiana and confirmed to be responsible for the beauvericin biosynthesis by targeted disruption. BbBEAS utilizes D-2-hydroxyisovalerate (D-Hiv) and L-phenylalanine (Phe) for the iterative synthesis of a predicted N-methyl-dipeptidol intermediate and forms the cyclic trimeric ester beauvericin from this intermediate in an unusual recursive process.376
 | ||
| Scheme 47 (A) The gene cluster organization of bassianolide, beauvericin, PF1022A and enniatin, and (B) their biosynthetic pathway. | ||
Interestingly, these NRPSs uses a specific iterative assembly pathway to form the final cyclic oligomer. It is proposed that A1 and A2 domain adenylate the requisite amino acids (D-α-hydroxycarboxylic acids and various L-amino acids, respectively) and transfer these to PCP1, or PCP2a/PCP2b. PCP2a and PCP2b play alternate roles as acceptors and donor in the condensation process. The Mt-domain catalyses the methylation of the amino acid residue once bound to a PCP domain. The tandem PCP domains in COD synthetases are presumed to store the depsipeptide during the process of elongation (Scheme 47). In this process, the N-terminal C domain (C1) is inactive, while the C2 domain catalyses the condensation of the amino acid acceptor monomers (peptidyl thioester intermediate) attached to PCP2a/PCP2b and the elongated intermediate donors attached to PCP1. The CT domain controls the length of peptide chain, the formation of the ester bond, as well as the macrocyclization and release of the cyclic peptide from the assembly line. As the peptide chain reaches a certain length, the CT domain function shifts from elongation to cyclization, catalysing the attack of the terminal hydroxyl on the thioester, thus releasing the mature cyclopeptide. To date, the exact mechanism of this function switching in CT domains remains enigmatic.192
Recently, Steiniger et al. investigated the combinability of these two types of synthetases (iterative NRPSs and linear NRPSs) and compared the exchange unit variants of four hybrid modules. They demonstrated that the selectivity of the C domain influences the assembly line functionality. These experiments suggested that the PCP-C and C-A domain linker regions significantly influence the iterative peptide assembly process and production titres. Furthermore, they also suggest that swapping parts of the CT domain can be used to uncovered functional aspects of macrocyclization and ring size control in these domains.377 Based on their hybrid synthetase assays, they propose a two-face XU concept for fungal NRPSs engineering, which emphasises the importance of C domain specificity and employs C-A-PCP and A-PCP-C combinatorial engineering methods, which is also potentially transferrable to other NRPS systems from a bioengineering perspective.
PfSYN (350 kDa) is the nonribosomal peptide synthetase that produces PF1022 in an iterative manner.378 PFSYN accepts L-leucine, D-lactate, D-phenyllactate, S-adenosyl-L-methionine (AdoMet) and ATP as substrates to assemble this cyclooctadepsipeptide scaffold.378,379 The Süssmuth group performed a comprehensive investigation into the substrate tolerance of PfSYN, which exhibited broad substrate tolerance including aromatic, heteroaromatic and aliphatic residues. In contrast to the α-hydroxy activating domain of enniatin synthetase (ESYN), this assembly line can activate hydroxy acids derivatives containing para-halogenated phenyl rings and propargyl sidechains.379
Many enniatins have been identified, demonstrating that these cyclodepsipeptide synthetases possess innate flexibility in their substrate specificities and thus can incorporate a wide range of amino and hydroxy acids.370,380 As seen with enniatin biosynthesis, the beauvericin depsipeptide synthetase (BbBEAS, 351 kDa) adopts a similar mechanism,376 although the beauvericin synthetase selectively accepts N-methyl-L-phenylalanine and other aliphatic hydrophobic amino acids as substrates.
In the synthesis of these cyclopeptides the main challenge is the cyclization of the linear tetradepsipeptide precursors. Following solution phase or solid phase peptide synthesis, linear tetradepsipeptide 75 is subjected to Mitsunobu esterification using [DIAD (2 equiv.), PPh3 (3 equiv.), benzene (5 mM)]. These salt-free conditions lead to the isolation of bassianolide, with the yield of tetradepsipeptide 75 macrocyclization increased to 31% through the addition of NaBF4 (Scheme 48).365,381
 | ||
| Scheme 48 Total synthesis of bassianolide. | ||
Verticilide is a 24-membered cyclo-oligomeric depsipeptide consisting of α-hydroxy acid and α-amino acid monomers. It was first isolated from fungus Verticillium sp. FKI-1033 in 2006 by Ōmura et al. Whilst verticilide has no antibiotic activity, it is a candidate for pesticide development.382 In addition, verticilide and its derivatives have been shown to function as an inhibitor of insect RyR, ACAT2, as well as ryanodine receptors.369,383,384 In 2006, Ōmura et al. reported the solution synthesis of verticilide based on Boc/benzyl ester protecting group macrocyclization. After the deprotection of the N- and C-termini and esterification under high dilution, verticilide was generated in a yield of 66% in 13 steps. In this synthesis, PyBrop-mediated N-Me macro-lactamization was employed to construct the acyclic oligomer (Scheme 49).382
 | ||
| Scheme 49 General chemical synthesis step of verticilide and enniatin B. Ghosez's reagent = 1-chloro-N,N,2-trimethyl-1-propenylamine, DMAP = 4-dimethylamino-pyridine, DIPEA = diisopropylethylamine. PyBroP = Bromo-tris-pyrrolidino-phosphonium hexafluorophosphate. | ||
In 2012, Hu et al. reported an optimized flow-synthesis method for the cyclic hexadepsipeptide enniatin B. In this synthesis, the linear monomer hexadepsipeptide 76 was firstly constructed by the coupling of amine salts, with EDCI and Ghosez's reagent (1-chloro-N,N,2-trimethyl-1-propenylamine) used for amide and ester bond formation. In contrast with the reported verticilide synthesis, the macrocyclization step of enniatin B was catalysed using Ghosez's reagent, providing an overall yield of enniatin B of 36% (Scheme 49).385,386
3.5 Cyclodipeptides
Cyclodipeptides (CDPs) contain a characteristic diketopiperazine (DKP) cyclic dimer scaffold, present diverse chemical structures, bioactivities, and are widely present in secondary metabolites from bacteria, fungi, plants and animals.387 Their skeleton comprises a stable six-membered diketopiperazine ring, which is derived from two α-amino acids condensed by peptide bonds. This stable six-membered skeleton helps compounds resist protease hydrolysis and facilitates drug passage through the intestinal and blood-brain barriers.388 DKPs are an important pharmacophore in medicinal chemistry and have attracted increasing interest as drug precursors. Cyclodipeptides are currently being developed as potential anticancer, antibiotic and anti-inflammatory agents, such as plinabulin, phenylahistin and Bicyclomycin.389–391Cyclodipeptide synthases (CDPSs) are the main biosynthetic pathways for DKP biosynthesis. CDPSs use intracellular aminoacyl-tRNA (aa-tRNAs) as substrates to catalyze the cyclization of two amino acids.392 CDPS protein family are usually composed of 200 to 300 amino acid residues. In 2009, the AlbC protein derived from Streptomyces noursei was shown to catalyse the synthesis of cyclo-(L-Phe-L-Leu)-the skeleton to albonoursin, becoming the first CDPS protein whose function had been characterised.393 Many symmetrical cyclodipeptides have been reported to be synthesized by CDPS enzymes, such as mycocyclosin biosynthesized (Rv2775),394 pulcherrimin (YvmC),395 and cyclo(L-Trp-L-Trp) (Amir_4627) (Scheme 50).396
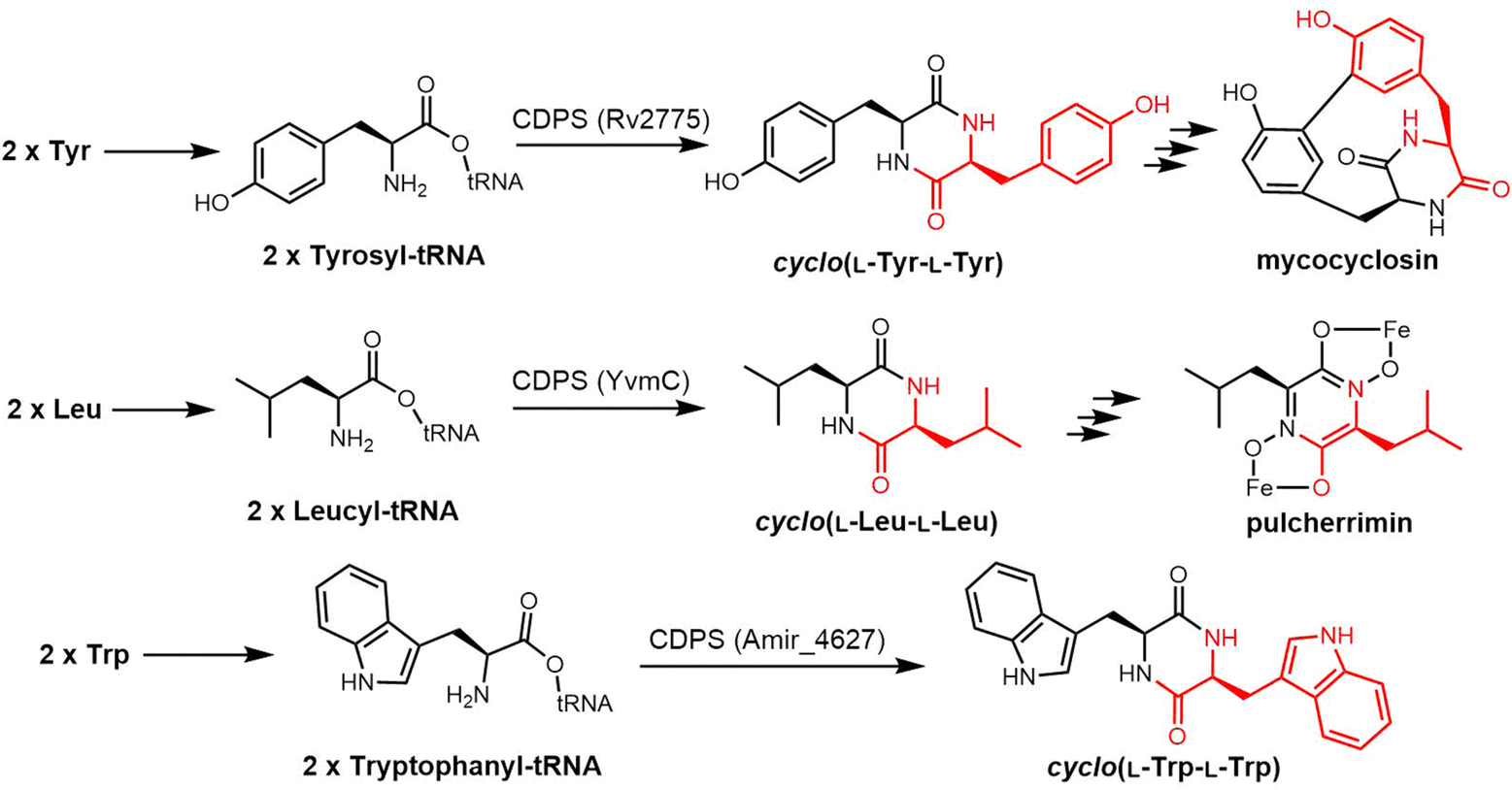 | ||
| Scheme 50 The representative symmetrical cyclodipeptides biosynthesized through CDPS pathway. | ||
Following DKP synthesis, tailoring enzymes encoded in the DKP gene cluster then catalyse modifications of the DKPs to generate diverse structures from the basic cyclic dipeptide skeleton. The post-modified enzymes related to CDPS synthesis that have been reported to include cytochrome P450 monooxygenases (e.g. in mycocyclosin, pulcherrimin and guanitrypmycin),394,395,397 oxidases (in bicyclomycin)398 and methyltransferases (in drimentines),399 amongst other.400 Since there are significant numbers of reviews concerning CDPS biosynthesis, we will not elaborate this pathway further here.392,393,401,402
Beyond the actions of CDPS enzymes, DKPs can also be synthesized via NRPS pathways. Gliovirin, pretrichodermamides, FA2097, aspergillazines, peniciadametizines, acetylaranotin and aspirochlorine are examples of a class of natural products originally derived from the cyclodipeptide cyclo-L-Phe-L-Phe scaffold, which is assembled by an NRPS pathway. The biosynthesis of pretrichodermamide A was recently reported, with this fungal diketopiperazine natural product featuring an α,β-disulfide bridge. Here, the DKP cyclodipeptide backbone of pretrichodermamide A is assembled by the NRPS TdaA (PCP-C-A-PCP-C domain architecture).403,404 The NRPS AtaP from the acetylaranotin pathway, AclP from the aspirochlorine pathway, and Glv21 from the gliovirin pathway all contains a similar NRPS domain architecture (PCP-C-A-PCP-C). These NRPS synthetases are responsible for the assembly of the dipeptide, with the C-terminal CT domain responsible for dimerization and cyclization (Scheme 50). The catalytic core of this domain resembles that of the CT domain in beauvericin biosynthesis.405–407
Indigoidine is a blue pigment found in different types of bacteria and it is a highly effective radical scavenger. Indigoidine exhibits a broad spectrum of medicinal, industrial potential and is biosynthesized through an NRPS pathway,408 where L-Glutamine is the precursor. Indigoidine is assembled by condensing two molecules of L-glutamine by indigoidine synthetase, which is the single-module NPRS BpsA. It comprises of two adenylation (A), one oxidation (Ox), one PCP and one TE domains, with the active holo-BpsA demonstrated to be responsible for the dimerization of two L-glutamine residues to form indigoidine (Scheme 51). Pang et al. reported that the BpsA Ox domains catalyse the key intermediate formation through oxidation.409
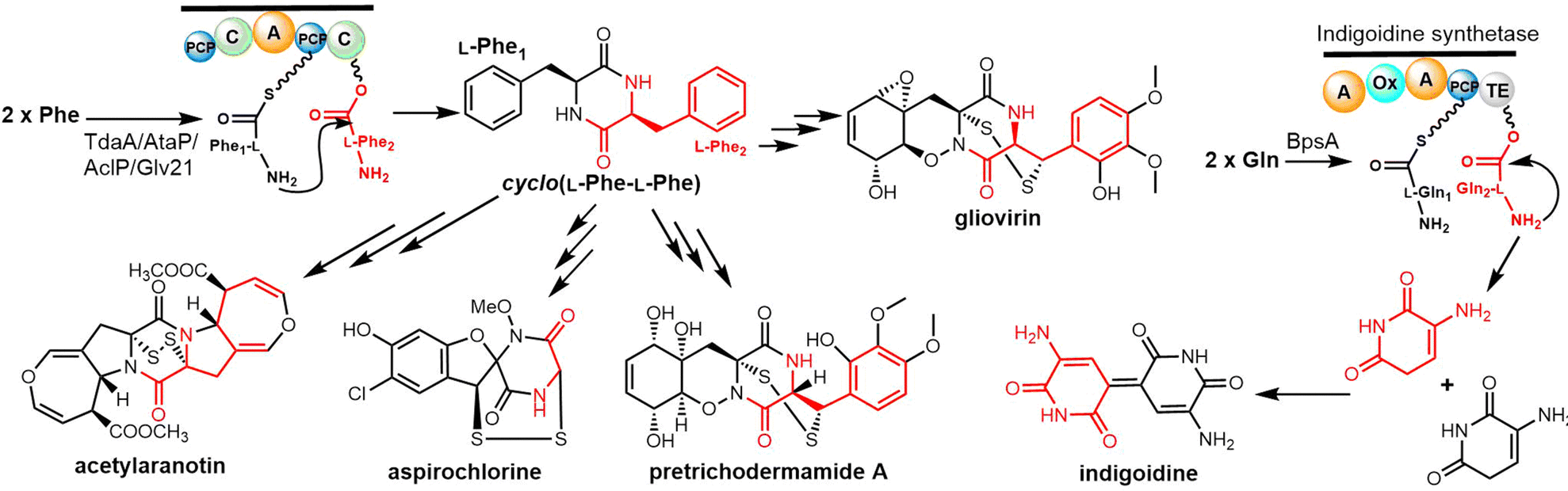 | ||
| Scheme 51 The representative symmetrical cyclodipeptides biosynthesized through NRPS pathway. | ||
3.6 Bipentaromycins
Bipentaromycins are dimeric aromatic polyketides featuring the rare 5/6/6/6/5 pentacyclic carbon skeleton. Cyclodimerization in these molecules is achieved via head-to-tail linkage of the two anthraquinone-based monomers. Recently, the heterologous expression of the bipentaromycin BGC bpa was performed in Streptomyces avermitilis SUKA17. From these experiments, it was proposed that cyclodimerization of an ortho-quinone methide intermediate, either spontaneously or via enzyme catalysis, initiates the 1,4-Michael addition step that leads to the formation of bipentaromycins-like dimerized intermediates (Scheme 52).410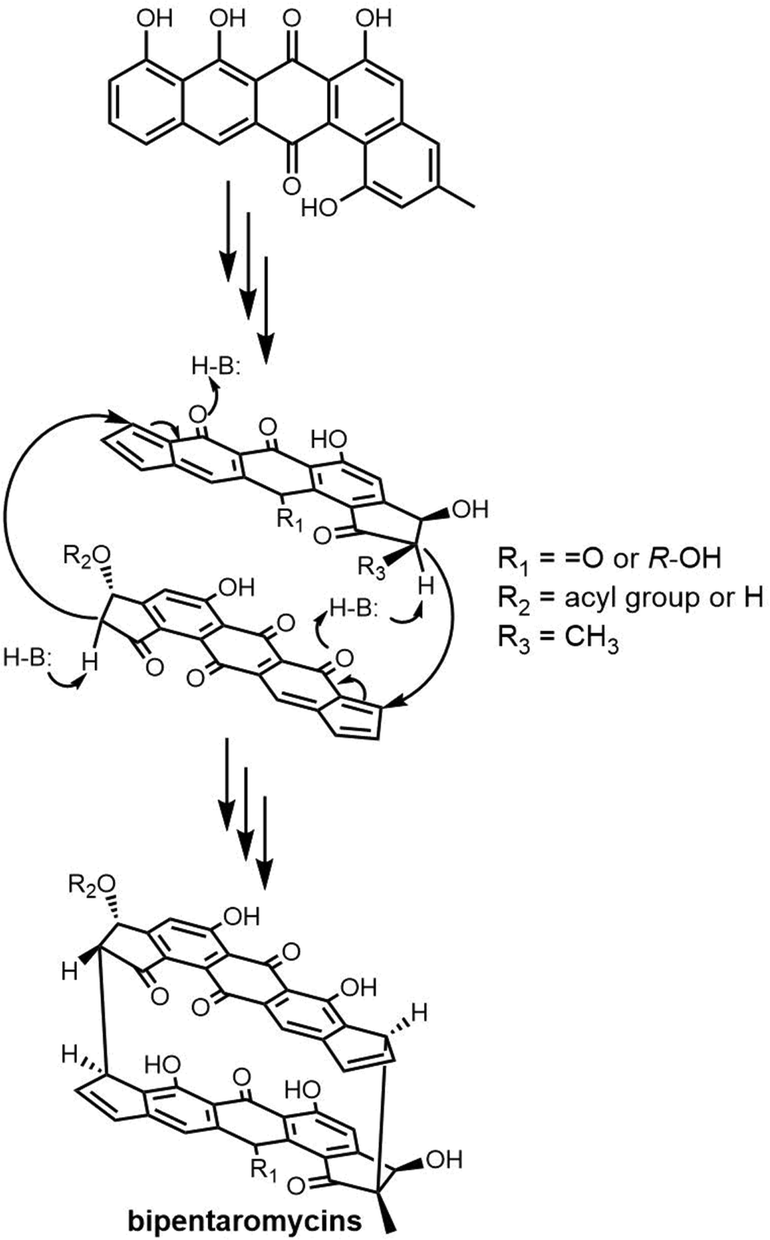 | ||
| Scheme 52 The putative cyclodimerization mechanism of bipentaromycins during biosynthesis. | ||
3.7 Cylindrocyclophanes
The cylindrocyclophanes are a class of cyanobacterial natural products that were first isolated from the terrestrial blue-green alga Cylindrospermum licheniforme. Members of the cylindrocyclophane family display various bioactivities, including antimicrobial activity, antiproliferative activity and cytotoxicity.411–414 The cylindrocyclophanes consist of an unusual [7.7] paracyclophane scaffold, which is structurally derived from the head-to-tail dimerization of alkylresorcinol analogues. Based on isotopic feeding experiments and a retrosynthetic biosynthesis hypothesis,415 the paracyclophane skeleton of the cylindrocyclophanes has been proposed to arise via dimerization of two identical resorcinol fragments, with subsequent electrophilic aromatic alkylation with a nucleophilic ion in the alkyl chain (Scheme 53).416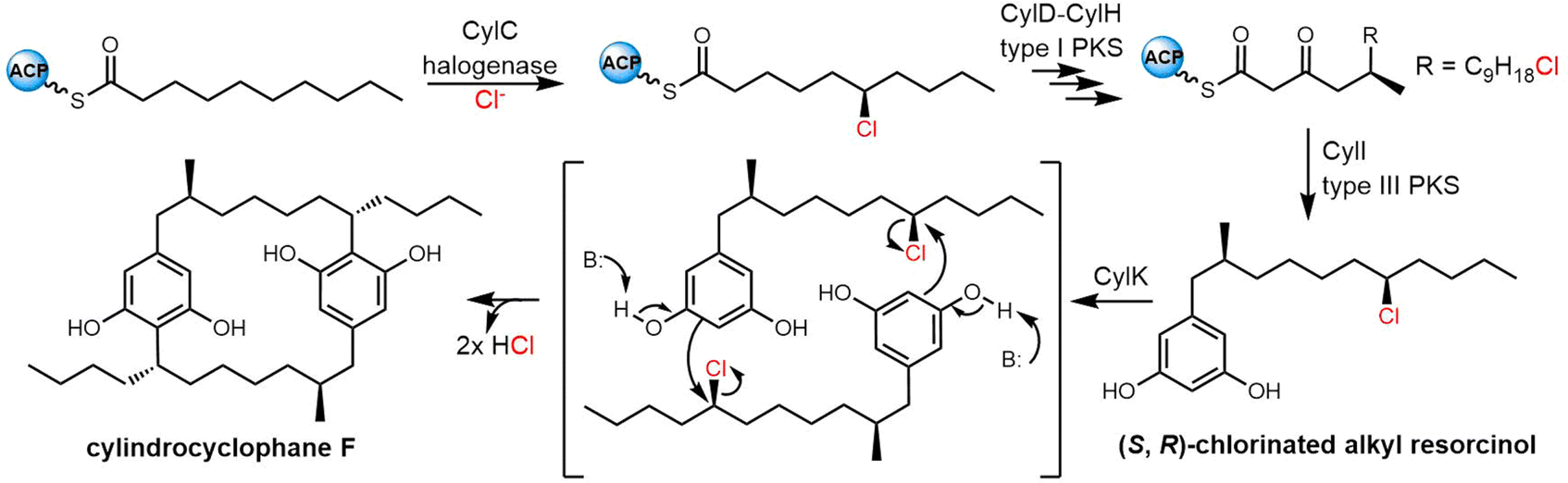 | ||
| Scheme 53 Proposed biosynthetic routes for cylindrocyclophane F. | ||
In the cylindrocyclophane (cyl) cluster from Cylindrospermum licheniforme ATCC 29412, a hemolysin-type calcium-binding protein CylK was biochemically characterized and demonstrated that cyclodimerization reaction uses an alkyl chloride precursor via the SN2 mechanism. The halogenase CylC mediates the chlorination, which was essential for the subsequent nucleophilic displacement of the chloride ion to generate the aryl-alky linkage. A BLAST search has demonstrated that CylC and CylK homologs are widespread in cyanobacterial genomes, offering a possible route to the discovery of new analogues of this compound class (Scheme 53).417
In terms of cylindrocyclophane synthesis, the monomeric precursor thioacetate mesylate 77 was prepared before undergoing cyclodimerization using treatment with NaOMe in MeOH at ambient temperature to afford the corresponding macrocyclic bis (thioether). Oxidation of this bis(thioether) with H2O2 in the presence of (NH4)6Mo7O24·4H2O furnished the macrocyclic bis (sulfone) 78 in 51% overall yield. Finally, the intermediate 78 was then modified to generate the cylindrocyclophanes (Scheme 54).418
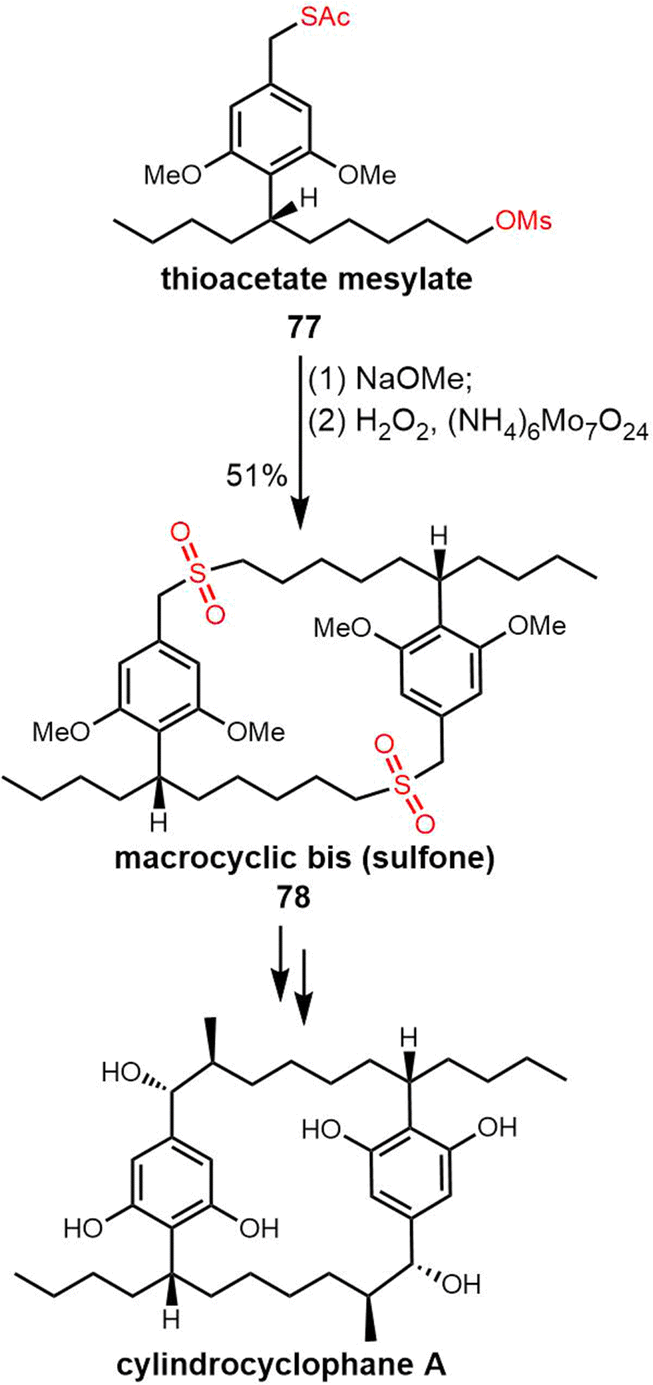 | ||
| Scheme 54 Total synthesis of cylindrocyclophane A. | ||
3.8 Melanthioidine
Tetrahydroisoquinoline alkaloids and dimeric macrocyclic diaryl ethers represent a large family of natural products discovered in plants.419,420 Structurally, these compounds display dimerization using both head-to-tail and head-to-head routes. Tetrahydroisoquinoline alkaloids have been demonstrated to possess a broad range of biological activities including antitumor, anti-inflammatory, anti-hypertensive, anti-plasmodial and antimalarial activity.421Tubocurarine is a naturally dimeric tetrahydroisoquinoline alkaloid isolated from South American plants such as Chondodendron tomentosum.422 Tubocurarine, as a neuromuscular blocking agent, has previously been widely used as a muscle relaxant during surgical procedures. However, this molecule has largely been replaced by safer and more effective drugs such as vecuronium, rocuronium and atracurium in modern procedures.423
Tubocurarine possesses a “head-to-tail”, asymmetrical, quaternary ammonium structure with two ether bridges (8-O-12′ and 11-O-7′) linking the coclaurine monomers. It has been proposed that tubocurarine biosynthesis involves a radical coupling of two enantiomeric monocationic tetrahydrobenzylisoquinolines, the two enantiomers of N-methyl-coclaurine.424 Subsequent methylation of the amine and hydroxyl substituents are then enzymatically performed through the consumption of S-adenosyl methionine (SAM) (Scheme 55).424
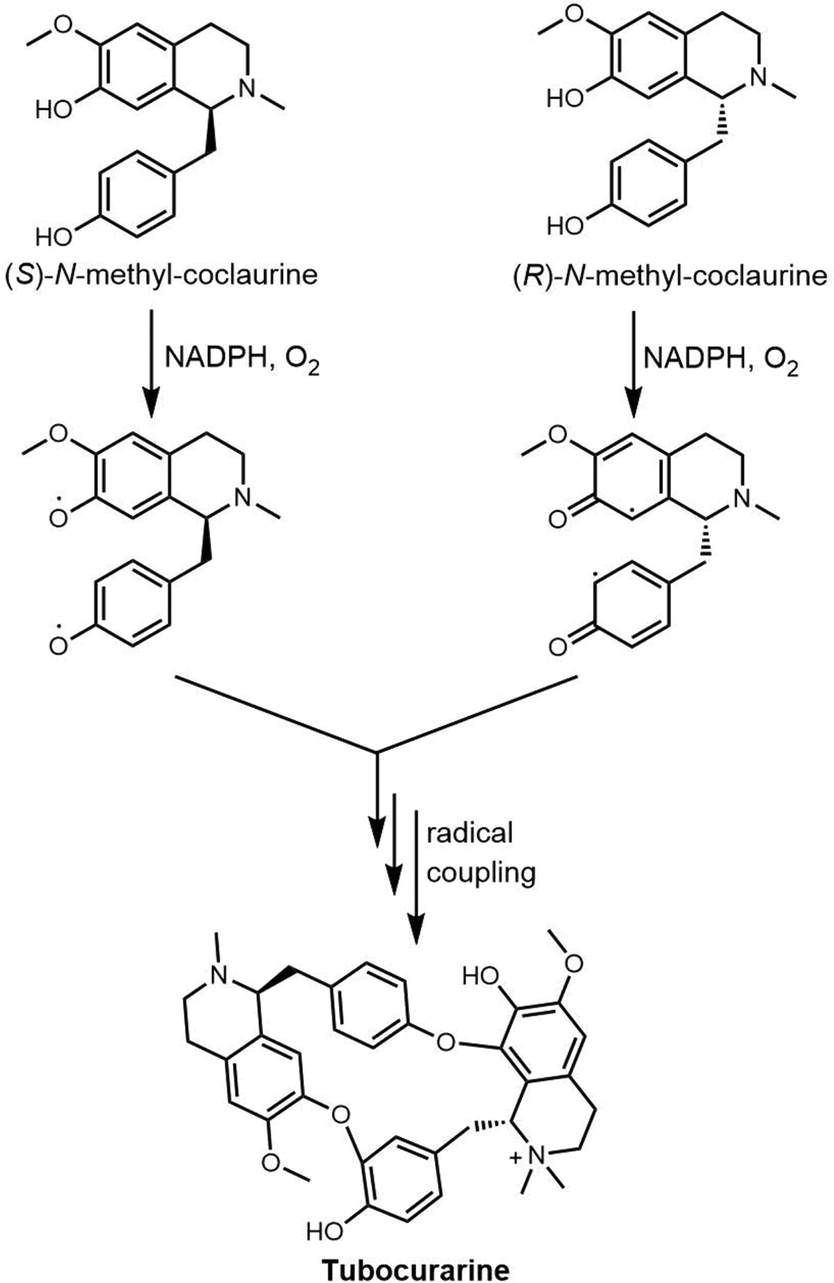 | ||
| Scheme 55 Proposed biosynthetic routes for production of tubocurarine. | ||
In 2016, Wang et al. completed the synthesis of the dimeric macrocyclic alkaloid melanthiodine. The enantioselective phenethyltetrahydroisoquinoline 79 was first synthesised as a monomeric precursor, with subsequent cyclodimerization performed using copper-mediated catalysis. After optimization, the yield of macrocyclic dimer O,O-dibenzylmelanthioidine 80 reached 35% when using the combination of the soluble copper salt CuBr·SMe2 (1 equiv.), Cs2CO3 (3 equiv.), pyridine (0.16 M), 150 °C, 48 h to catalyse this reaction (Scheme 56).421
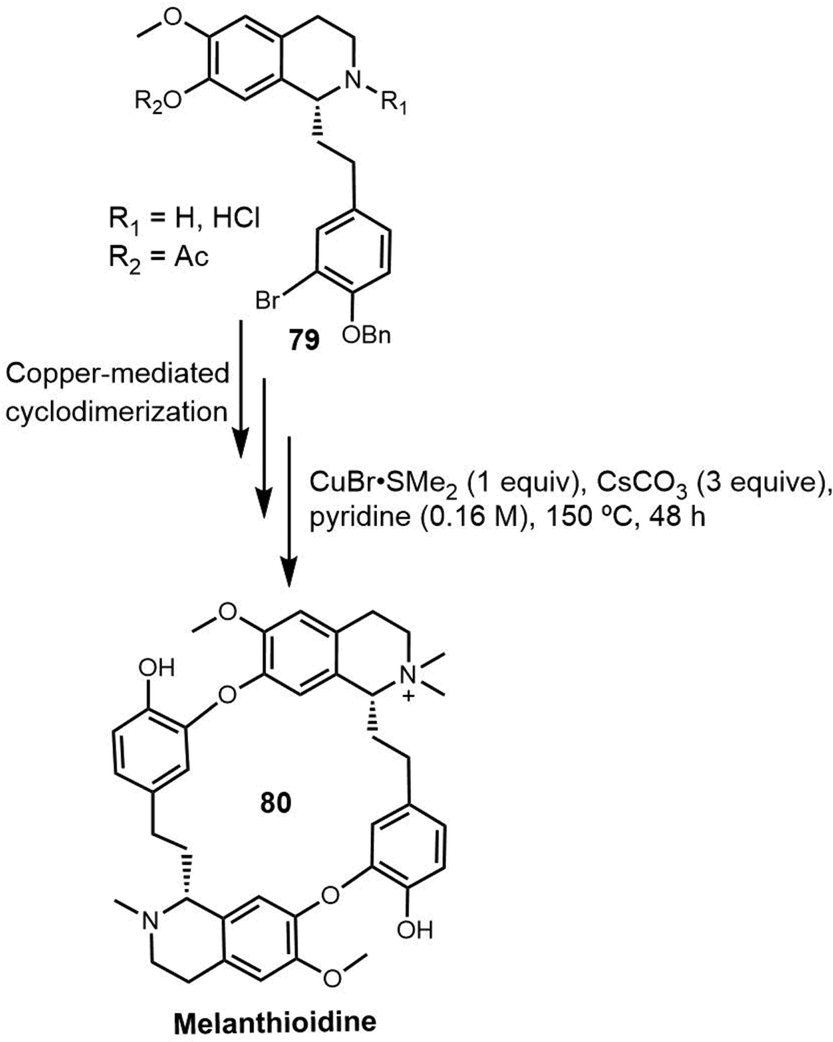 | ||
| Scheme 56 Total synthesis of melanthiodine using copper-mediated cyclodimerization. | ||
3.9 Griffipavixanthone
Griffipavixanthone and garciobioxanthone are group of dimeric xanthones isolated from plants that exhibit a wide range of biological activity, including antitumor and antioxidant properties.425,426 Structurally, unlike other cyclic dimeric PKS products such as gossypol or cyclic dimeric PKS products such as hypericin (both of which are linked by aryl–aryl or C–O bonds), griffipavixanthone and garciobioxanthone possess a unique cyclodimeric backbone consisting of two monomeric tetrahydroxyxanthone units.A Diels–Alder cycloaddition reaction was recognized to be the key step during the biosynthesis of these natural products. Using griffipavixanthone as an example, retrosynthetic analysis suggested that the cyclodimeric structure would be derived from the prenylated xanthone monomer 82, and the core cyclohexene ring would be formed through [4+2] cycloaddition (Scheme 57).427 Oxygenated styrenes are the key intermediates for this cyclization, which is initiated by the formation of a benzyl cation that then undergoes Friedel–Crafts cyclization after adding to the C–C double bond of another styrene molecule.
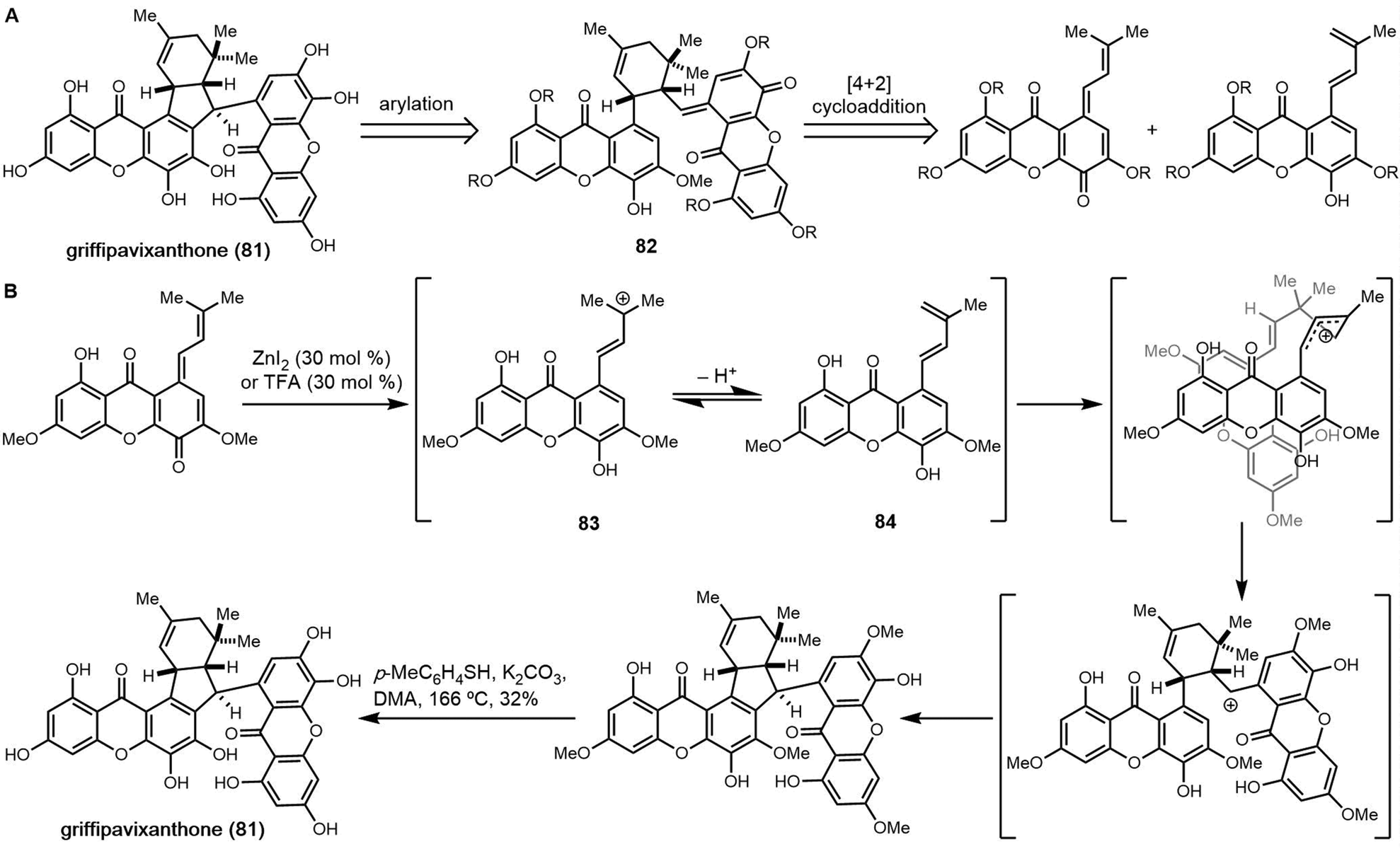 | ||
| Scheme 57 Total synthesis of griffipavixanthone. | ||
In 2017, Porco and co-workers reported the first biomimetic total synthesis of griffipavixanthone (81). The key synthetic step is the formation of a highly strained cyclohexene ring generated through an intermolecular cationic [4+2] cycloaddition reaction. The monomer intermediates vinyl para-quinone methide 83 and an in situ generated isomeric 1,3-butadiene 84 were afforded through isomerization mediated by Lewis or Brønsted acids (Scheme 56).427 In 2018, Schaus, Porco and co-workers achieved the asymmetric synthesis of (+)-griffipavixanthone employing a chiral phosphoric acid-catalyzed cycloaddition.428
3.10 Cyanolide A
Cyanolides are glycosidic macrodiolides possessing C2 symmetry discovered in the cyanobacterium Lyungbya bouillonii that exhibit strong molluscicidal activity against Biomphalaria glabrata (Fig. 27).429 Cyanolide A contains a symmetrical dimeric backbone, with the monomeric unit of cyanolide A derived from a polyketide chain containing five acetate units that is modified by SAM-derived methylation.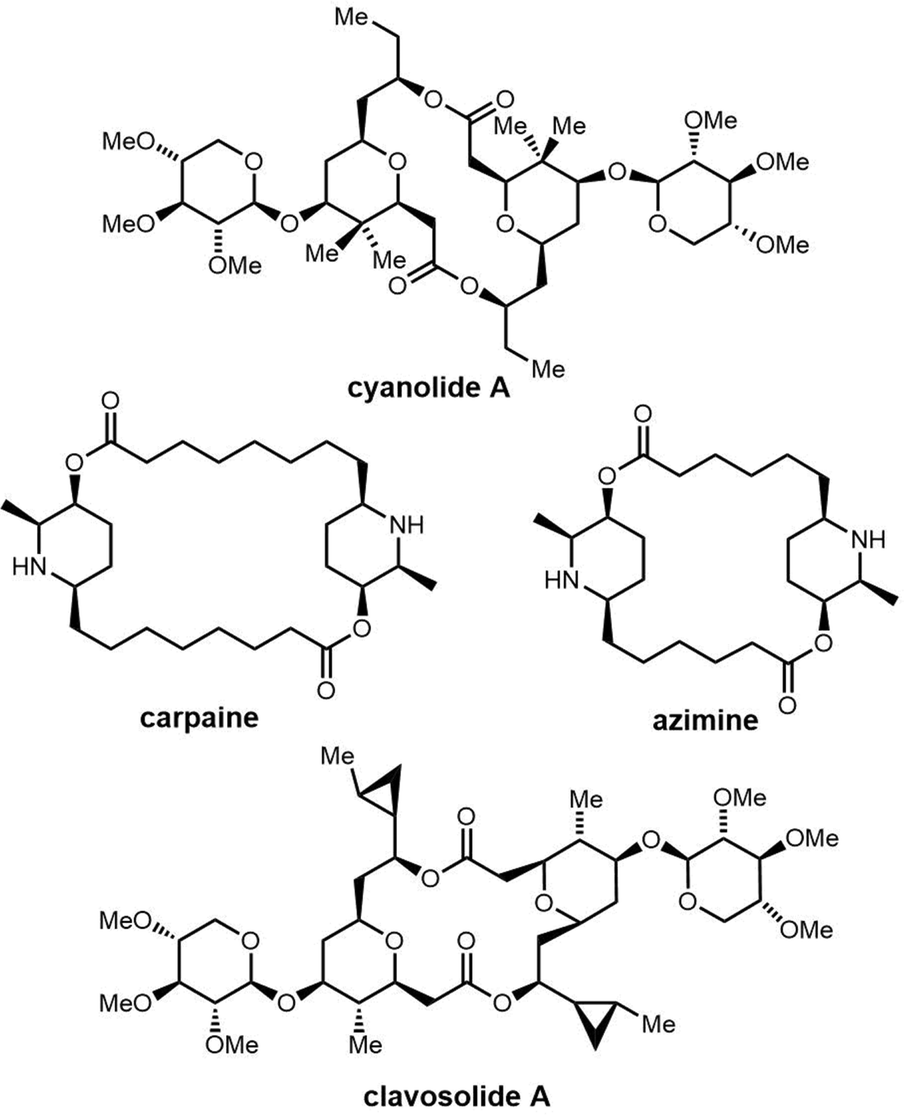 | ||
| Fig. 27 Examples of cyanolides, clavosolides, carpaines and azimines. | ||
Many studies concerning the synthesis of cyanolide A have been reported.430,431 To summarise, two main synthesis routes have been explored, which either perform glycosylation before or after cyclodimerization.
To afford the C2 symmetric diolide, macrocyclization reactions mainly exploit the Shiina lactonization strategy.430–432 The Krische group has reported the most direct six-step total synthesis of cyanolide A starting from neopentyl glycol. The synthesis uses no protecting groups and chiral auxiliaries, with the key intermediate C2-symmetric diol 85 constructed using an asymmetric iridium-catalysed dehydrogenation/allylation strategy (Scheme 58).433
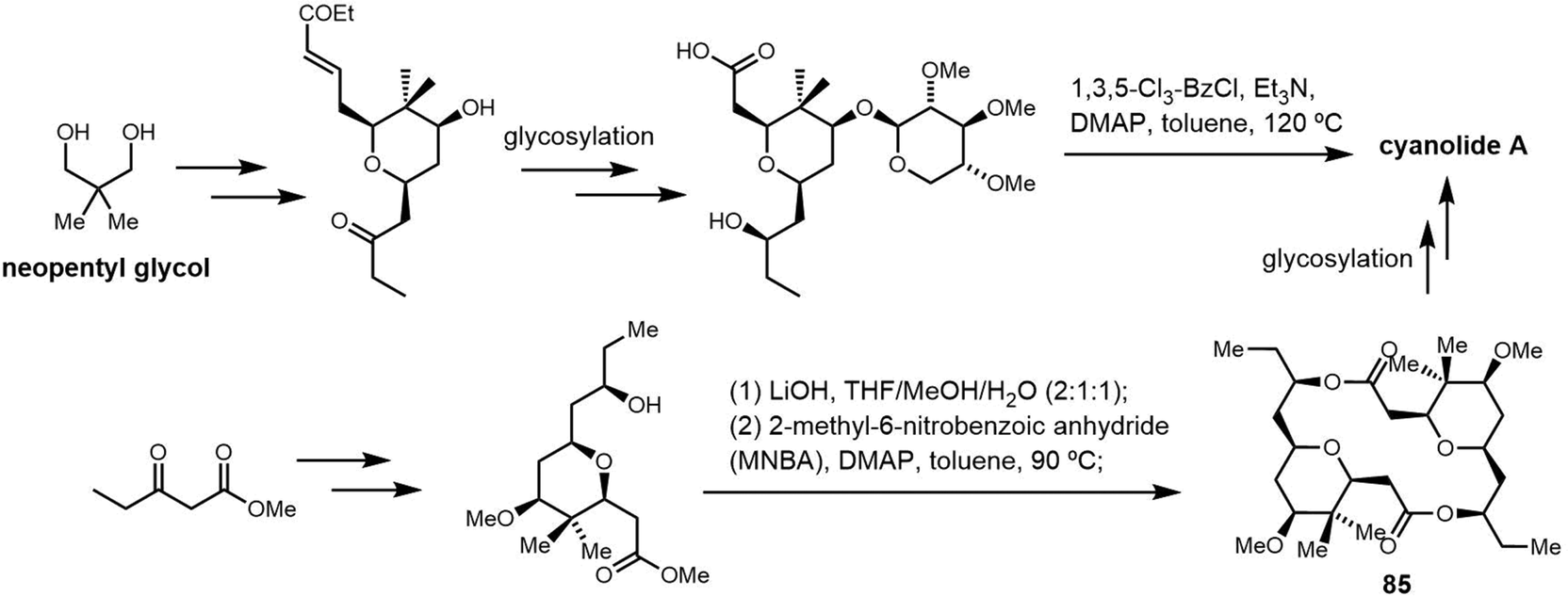 | ||
| Scheme 58 Total synthesis route for cyanolide A. | ||
3.11 Clavosolide
Clavosolide A is a C2-symmetric 20-membered glycosidic macrodiolide discovered from the sponge Myriastra clavosa.434 Clavosolide A and derivatives have been chemically synthesized in a variety of ways.435–438 Recently, Krische and colleagues reported a 7-step total synthesis route for clavosolide A. Following retrosynthetic analysis, the key intermediate hydroxy acid 86 was firstly prepared, followed by macrolactonization to afford clavosolide A in 52% yield (Scheme 59).438 | ||
| Scheme 59 Total synthesis route for clavosolide A. | ||
3.12 Azimine and carpaine
Carpaine and azimines are class of alkaloids found in certain species of plants, such as Carica papaya, Uncaria tomentosa and Uncaria guianensis.439 They are structurally classified as alkaloids, a class of naturally occurring compounds that contain a macrocyclic dilactones containing a 2,3,6-trisubstituted piperidine skeleton.Carpaine has been shown to possess a variety of biological activities, including antimicrobial, antiparasitic, and anti-inflammatory effects. Carpaine has also been investigated for its potential use in the treatment of various diseases, such as cancer, heart disease, and neurodegenerative disorders.440,441 The effects of carpaine may be related to its macrocyclic dilactone structure, a possible cation chelating structure.442 The biosynthesis of carpaine is not well understood, but is hypothesised to begin with the decarboxylation of the amino acid tyrosine to form tyramine, which is then further metabolized to form the alkaloid. The intermediate products of this pathway are believed to be modified and rearranged through additional enzymatic reactions to form the final carpaine molecule.
Taro et al. first reported the retrosynthetic routes of (+)-azimine and (+)-carpaine, where the N-derivatives of azimic acid and carpamic acid were used as starting substrates. Taking the example of (+)-carpaine synthesis, the precursor N-Cbz-carpamic acid was di-lactonized under Yamaguchi macrocyclization condition to generate the cyclodimerized N-Cbz-carpaine in 71% yield. Subsequent deprotection led to the isolation of (+)-carpaine (Scheme 60).443
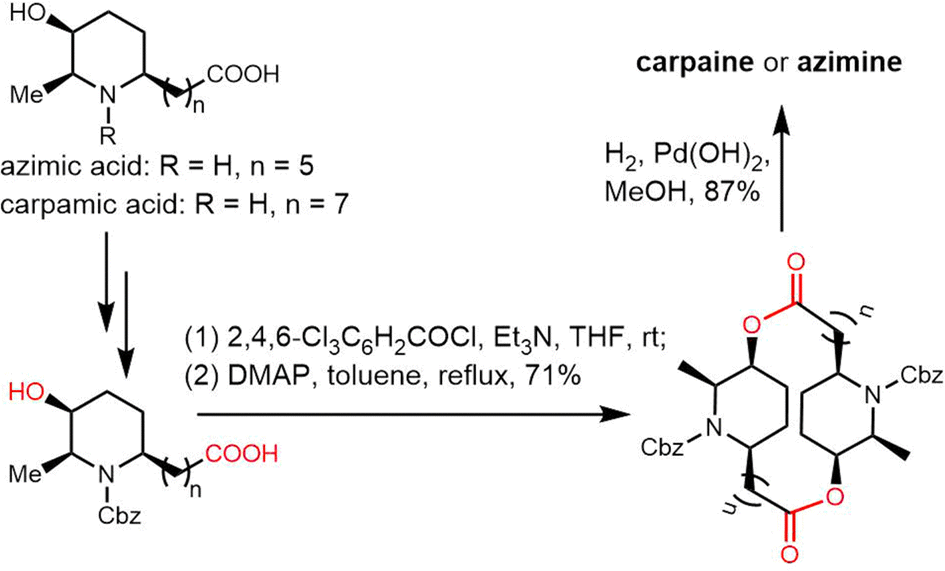 | ||
| Scheme 60 Synthetic routes of (+)-azimine and (+)-carpaine. | ||
3.13 Glycolipids
Macrolactone-containing glycolipids are glycosyl derivatives of lipids widely present in microorganisms. Glycolipids possess C2-symmetrical core structures that are formed through cyclodimerization, with some derivatives containing a C3-symmetrical scaffold.444,445 Representative glycolipids include the glucolipsins, cycloviracins, macroviracin, fattiviracins and arthrobacilin (Fig. 28). To date, few of these compounds have been studied in terms of their biosynthesis.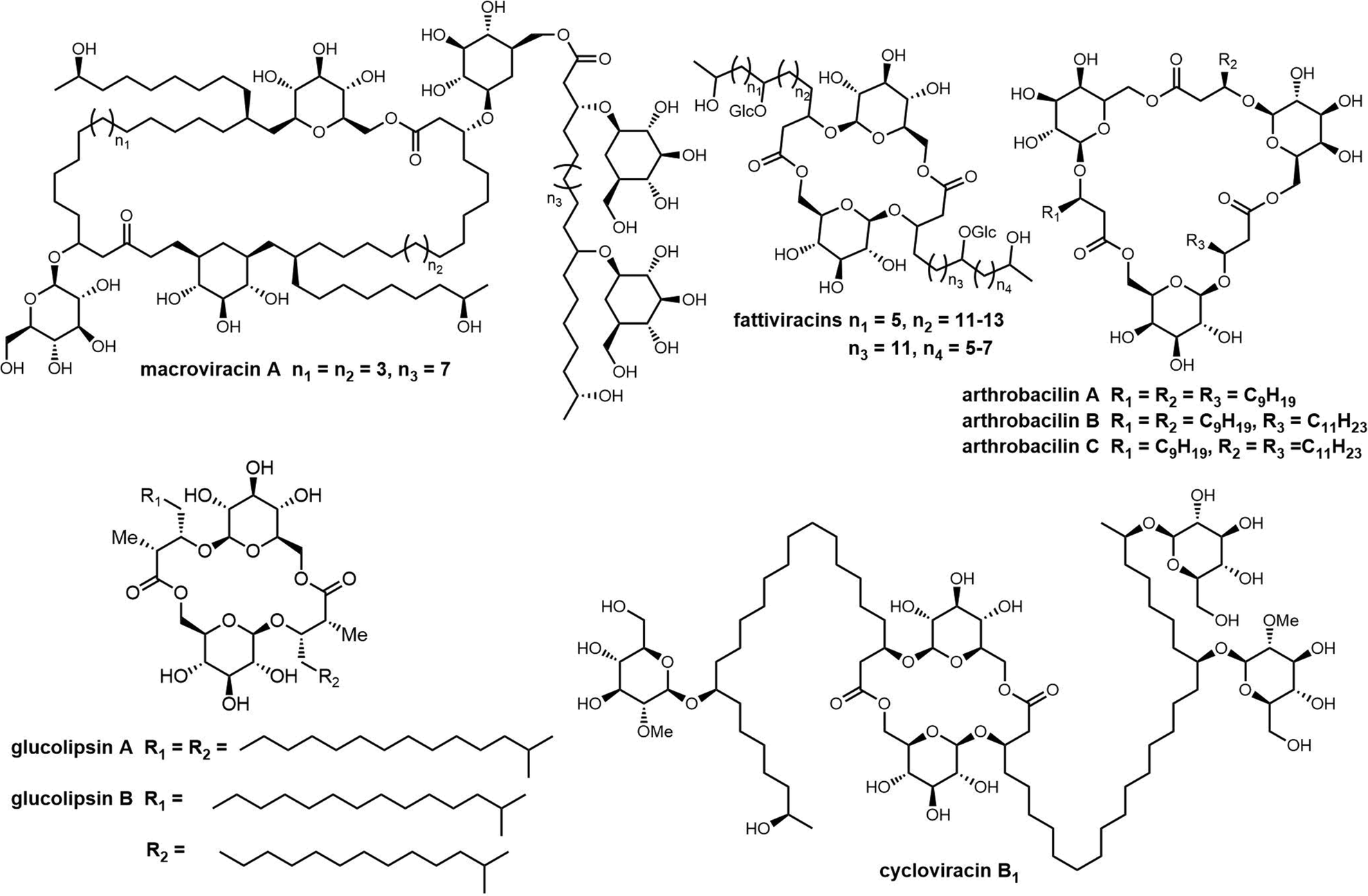 | ||
| Fig. 28 Examples of macrolactone-containing glycolipids. | ||
3.14 Glucolipsins and cycloviracin
Glucolipsin A and B are macrocyclic dilactones produced by Streptomyces purpurogeniscleroticus WC71634 and Nocardia vaccinii WC65712.446 The glucolipsins demonstrate activity in relieving the inhibition of glucokinase by stearoyl-CoA.447 In the glycolipid family, the cycloviracins are glycoside derivatives of the glucolipsins and thus contain a related aglycone. Cycloviracin B1 was discovered to be produced by the actinomycete Kibdelosporangium albatum so. nov. (R761-7) and exhibits significant antiviral activity against influenza A virus, varicella-zoster virus, and human immunodeficiency virus, amongst others.448A synthesis route towards glucolipsin A exploited the cyclodimerization of monomer 87, with the key cyclic dimer intermediate 88 obtained through macrodilactonization, with the reaction using the activating agent 2-chloro-1,3-dimethylimidazolinium chloride (89). The cyclodimerized product 88 was obtained in 54% yield using optimized conditions (Scheme 61).446,449
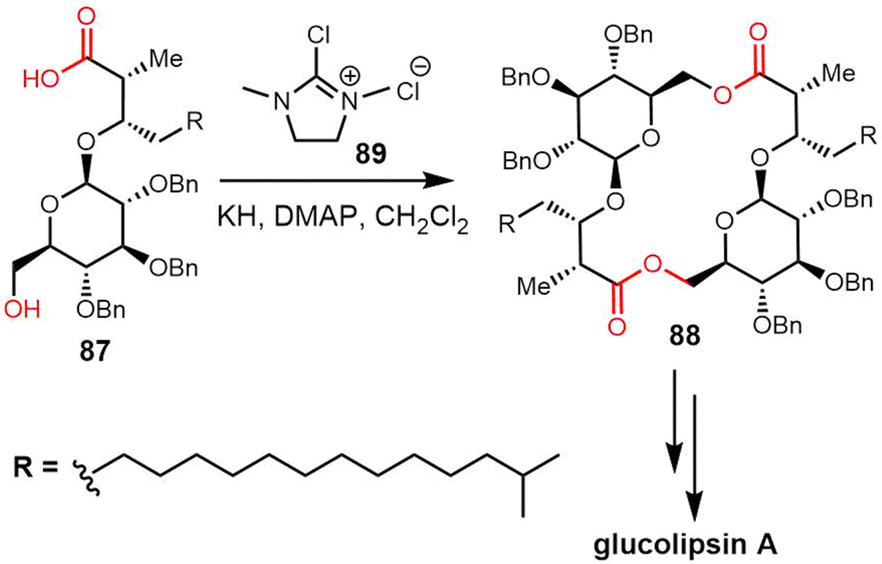 | ||
| Scheme 61 Total synthesis of glucolipsin A. | ||
The synthesis of cycloviracin B was also enabled using 2-chloro-1,3-dimethylimidazolinium chloride (89) in the macrodilactonization step.449–451 Interestingly, it was observed that the addition of KH significantly increases the yield of 90 to 71% (Scheme 62), and from this it was postulated that the presence of potassium cations may structure the precursor molecules in a way that favours cyclodimerization.
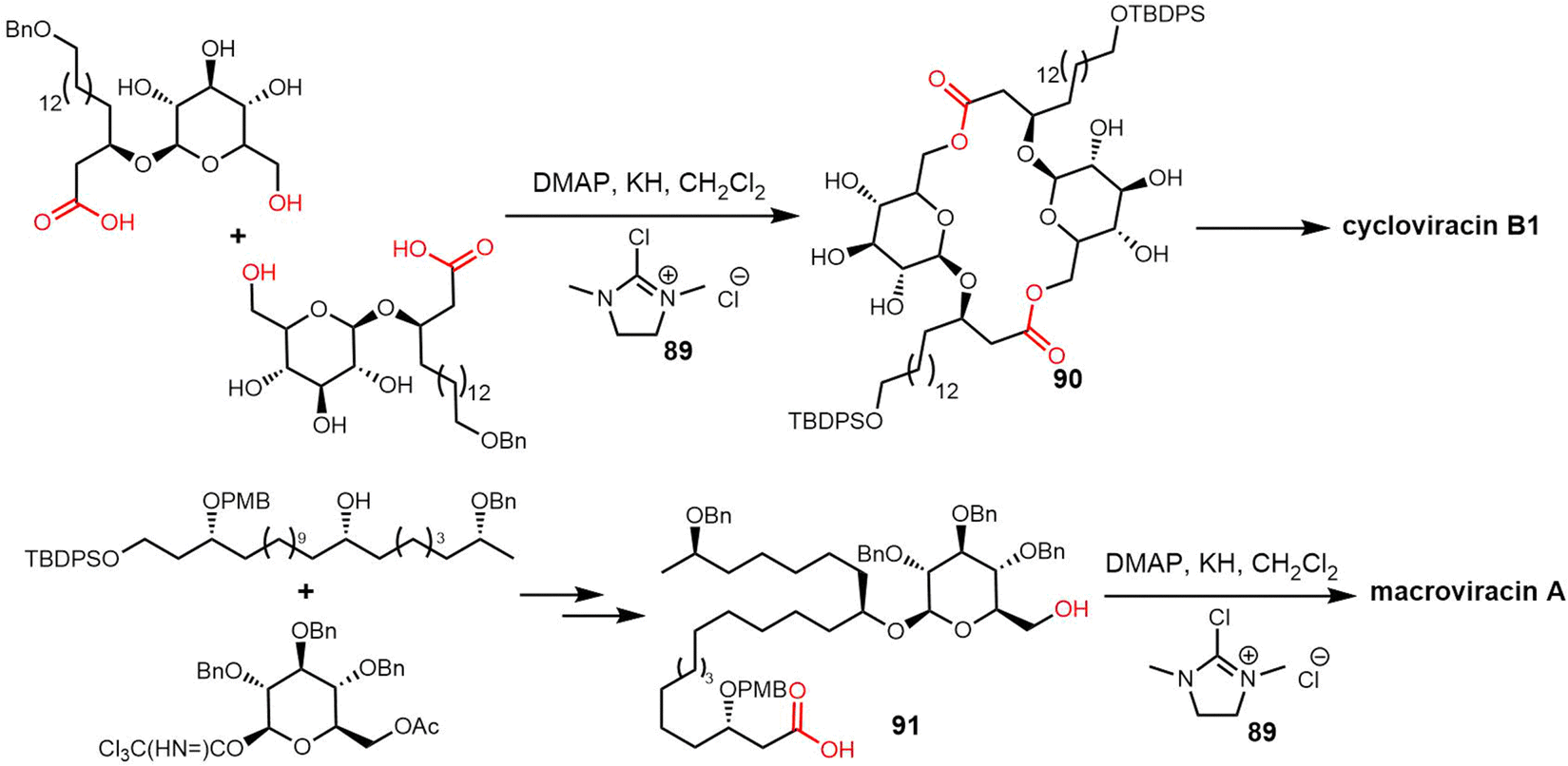 | ||
| Scheme 62 Total synthesis of cycloviracin B and macroviracin A. | ||
3.15 Macroviracin
The macroviracins are a class of C2-symmetric macrocyclic compounds, which were isolated from the mycelium extracts of Streptomyces sp. BA-2836 and classified into eight types of congeners (A–D) by the length (C22 or C24) of the constitutive fatty acids. These cyclic glycolipids exhibit a powerful antiviral activity against HSV-1 and VZV, with their potency is 10 times higher than acyclovir.452 Takahashi and co-workers first reported the synthesis of macroviracin A, which was constructed using the precursor C22-hydroxy carboxylic acid 91 (glucosyl fatty acid) and subsequent intermolecular cyclodimerization. Under optimized conditions (2-chloro-1,3-dimethylimidazolinium chloride, DMC/DMAP, sodium hydride), the dimeric product was obtained in 35% yield, whilst also avoiding the unwanted side products caused by lactonization of the glucosyl fatty acid 91 (Scheme 62).3433.16 Fattiviracins
The fattiviracins are a group of antiviral antibiotics discovered from strain Streptomyces microflavus.453 They exhibit strong antiviral effects against several viruses, such as herpes simplex virus, influenza A virus, and human immunodeficiency virus.454 The fattiviracins consist of macrocyclic diesters, which are created by the linkage of two trihydroxy fatty acids and two D-glucose monomers.455 The water solubility of fattiviracin A1 is facilitated by the presence of four sugar moieties in its structure. This characteristic has the potential to make fattiviracin A1 a promising new antiviral drug with a broad spectrum of activity.4533.17 Arthrobacilin
The arthrobacilins are cyclic glycolipids containing a 27-membered ring scaffold bearing multiple hydroxyl groups. These compounds were discovered from the strain Arthrobactor sp. NR2967,456 and exhibit weak cytotoxic and antibiotic activity.457 In 1994, Garcia et al. reported the synthesis of the arthrobacilins, with analogues synthesized using the monomer ω-hydroxylic acid 92 as the substrate. Treatment of key intermediate 93 with 2-chloro-1,3-dimethylimidazolinium chloride (DCC)/DMAP catalysed the key cyclisation, followed by benzyl deprotection afforded the natural product arthrobacilin A. (Scheme 63).458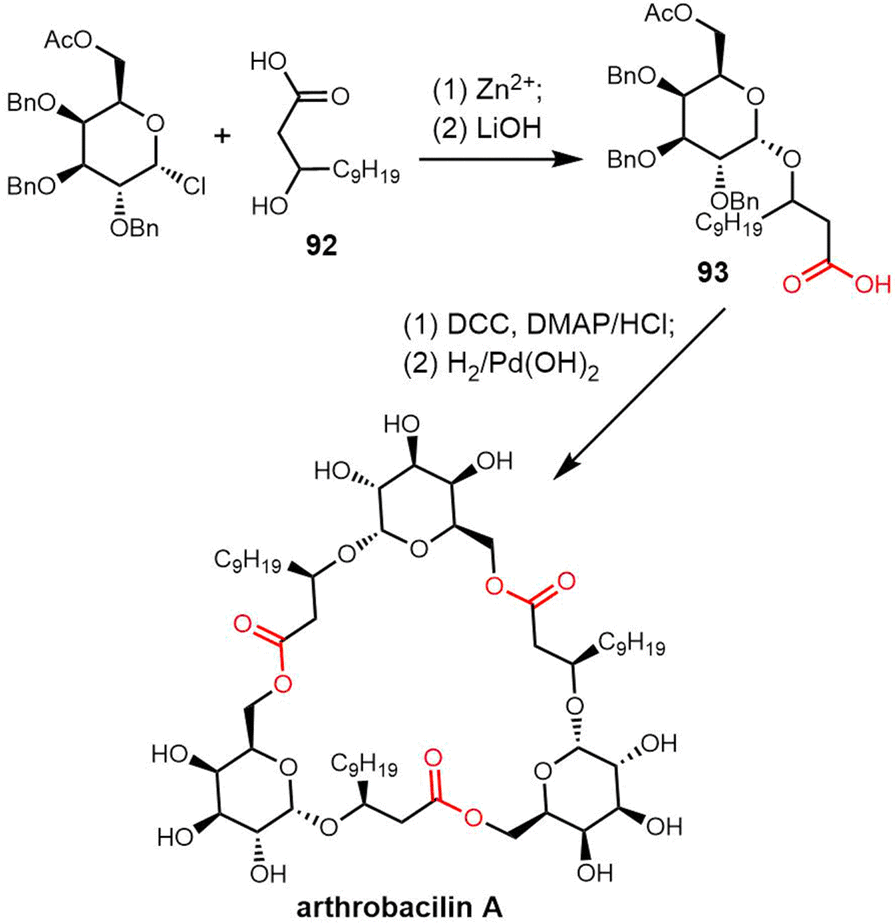 | ||
| Scheme 63 Total synthesis of arthrobacilin A. | ||
4. Chemical routes to macrocyclic scaffolds
Despite recent advances in natural product synthesis, accessing complex cyclodimeric compounds through cyclodimerization and macrocyclization with high synthetic efficiency remains challenging. The specific methods used for cyclodimerization depend on the structure of the target compound and the availability of precursor molecules and reagents. General cyclodimerization approaches were typically developed from classic macrocyclization mechanisms, which mainly include ring-closing metathesis (RCM), multicomponent reactions (MCRs), cycloaddition reactions, macrolactonization, macrolactamization, click reactions and cross-coupling reactions. Thus, diverse macrocyclization methodologies have been developed, with such reactions having been extensively reviewed.459,460Here we instead seek to emphasize the chemical synthesis reactions developed to generate cyclodimeric scaffolds. As most of the synthetic strategies for cyclodimerization and macrocyclization have been described in the above sections on individual cyclodimerized natural products, here we will restrict ourselves to the introduction of representative examples of cyclodimerization or cyclo-oligomerization related to NPs or NP-like small molecules. The chemical synthesis of cyclic supramolecular polymers will not be discussed.
4.1 Lactonization/lactamization
The generation of amide, ester, thioester, disulfide, olefin, or C–C bonds is a common route for achieving peptide cyclization. In cyclopeptide synthesis, the final ring-closing reaction is usually accomplished through lactamization, lactonization, or the generation of a disulfide bridge.27,459Macrocyclic structures with one or more ester connections are known as macrolides. Lactonization often represents an efficient approach for the synthesis of macrolides.461 Porco and co-workers reported distannoxane-catalyzed cyclodimerization, which is mediated by distannoxane transesterification catalysts that facilitate the production of dimeric macrodiolides from various hydroxy esters substrates. They reported that substrate enantioenriched hydroxy esters are readily able to generate the 14- to 22-membered cyclodimerization reaction when using distannoxane transesterification catalysts (Scheme 64).462 Studies have shown that this is also an effective strategy to create stereochemically enriched homodimers from a variety of hydroxy esters monomer pairs. The effectiveness of cyclodimerization reactions has been shown to depend significantly on both microwave power and reaction temperature employed for such reactions.463
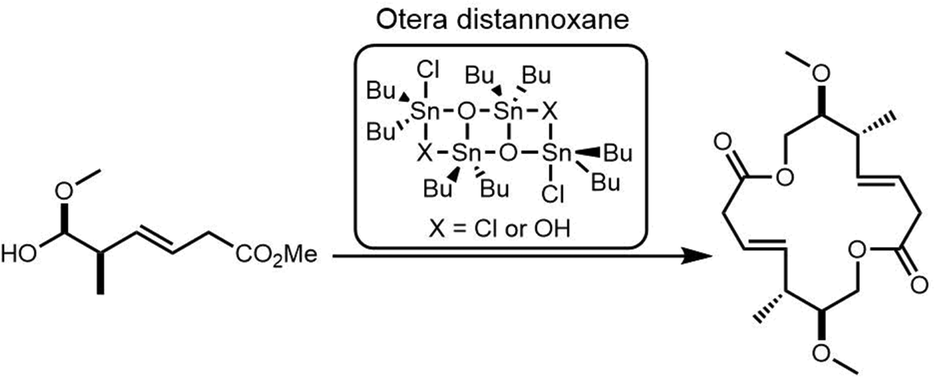 | ||
| Scheme 64 Distannoxane-catalyzed cyclodimerization. | ||
Distannoxane catalysts effectively promote the macrolactonization of seco-ester precursors. Collins and co-workers reported a reaction protocol for macrodiolide synthesis based on seco-acids using Lewis acid Hf (OTf)4 as catalyst, with the only by-product generated in this reaction being water. After optimization, this method was shown to be suitable for the synthesis of the 22-membered macrodiolide 95, and macrocycles 94 and 96 through cyclodimerization (Scheme 65).464
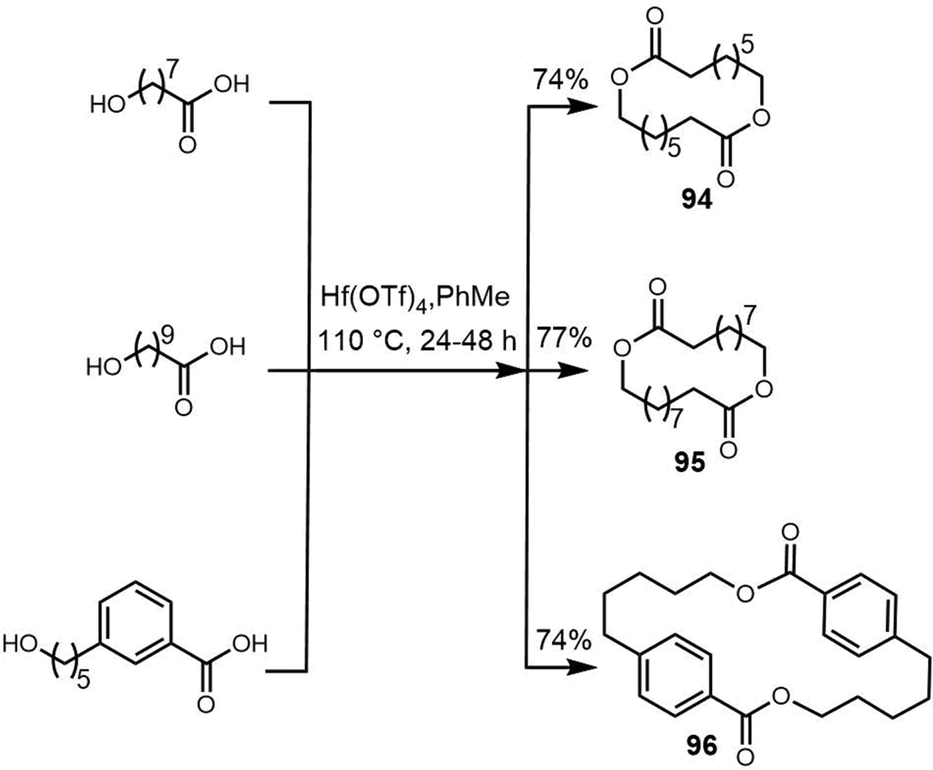 | ||
| Scheme 65 The macrodiolide synthesis based on seco-acids using Lewis acid Hf (OTf)4. | ||
Zhang et al. reported a facile synthesis method for cyclic polyamides through cyclodimerization, with monomer pentafluorophenol esters prepared in advance. After deprotection, the two active groups (NH2 and CO2C6F5) were exposed on both termini, which promoted macro-lactamization by monomers cyclodimerization in an alkaline environment. (Scheme 66).465
 | ||
| Scheme 66 Total synthesis of cyclic polyamides. | ||
Miao et al. investigated the autocatalytic polymerization reaction of tripeptides linked by disulfide bond using (Cys-Xxx-Gly-SEt)2 as the “monomer” starting material. The resulting macrocyclic peptides, containing up to 69 amino acids, displayed autocatalytic kinetics and were formed via native chemical ligation; thiol–thioester exchange “hopping” was likely responsible for this behaviour. This method allows for the straightforward production of thiol-rich macrocyclic oligopeptides, which can be used as a tool for studying functional cyclopeptides (Scheme 67).466
 | ||
| Scheme 67 Autocatalytic polymerization reaction for the synthesis of macrocyclic peptides. | ||
4.2 Multicomponent reactions
Multicomponent reactions (MCRs) have proven to be powerful macrocyclization tools for cyclopeptide synthesis. This method has been developed to synthesize heterocycles, polymers, and natural products, with numerous reviews written about this topic.134,467 Well-known MCRs strategies for cyclodimerization include azide–alkyne cycloadditions such as the “click” reaction, Ugi reaction and Ugi-azide reaction. These methods are especially suitable for the synthesis of cyclic peptide structures, with Ugi multicomponent stapling approaches especially suitable for cyclizing helical peptides (e.g. gramicidin S and sandramycin) as described in the Section 2.2 and Scheme 68. | ||
| Scheme 68 The generation of macrocyclic rings by copper-catalysed azide–alkyne cycloaddition. | ||
Click chemistry has been powerful strategy for chemical backbone synthesis and biomimetic applications.468 Many click reactions have proven to be efficient peptide macrocyclization methods. The azide–alkyne cycloaddition methodology is a valuable technique for generating macrocyclic rings.469 Based on the 1,3-dipolar cycloaddition of azides and alkynes, triazole-containing macrocycles can be readily synthesized. The Liskamp group developed the synthetic route of the ABC ring system of vancomycin through ruthenium-based azide–alkyne cycloaddition (RuAAC). This macrocyclization method exhibited high intramolecular selectivity to mimic the bicyclic formation.470
Jagasia et al. reported a copper-catalysed azide–alkyne cycloaddition reaction (CuAAC) that combined the methods of SPPS and click chemistry. This method can also be applied to perform the head-to-tail cyclodimerization of resin-bound oligopeptides. The advantage of this method is that this reaction was independent of peptide sequence (Scheme 69).471,472
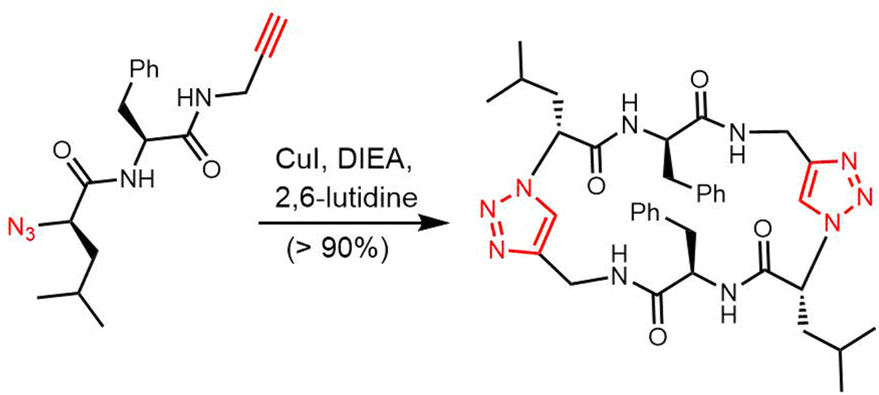 | ||
| Scheme 69 The generation of macrocyclic rings by copper-catalysed azide–alkyne cycloaddition. | ||
The Ghadiri lab has reported a solution-phase synthesis of C2 symmetric cyclic peptide scaffolds through 1,3-dipolar azide–alkyne cycloaddition reactions. This technique utilised a tandem dimerization-macrocyclization approach enabled by facile click reactions of an azido-dipeptide alkyne.473
The azido-alkyne “click reaction” strategy also can be used in the synthesis of cyclodextrin analogues, with the cyclodimerization of an azido-alkyne saccharides performed via Cu-catalyzed dipolar cycloaddition of alkyne and azide functional groups (Scheme 70).474,475 Menand et al. developed an efficient domino Staudinger aza-Wittig reaction to synthesis sugar-aza-crown ethers through the cyclodimerization of C-glycosyl azido aldehydes (Scheme 70).476,477
 | ||
| Scheme 70 The synthesis of cyclodextrin analogues through azido-alkyne “click reaction” strategy. | ||
The exploration of active precursors for efficient cyclodimerization synthesis has been a longstanding objective of organic chemists. Recently, vinylethylene carbonates (VECs) were discovered as promising dipole precursors to iridium-catalysed cascade allylation-macrolactonization reactions.478 Wang and co-workers developed a wide range of C2-symmetric chiral macrodiolides based on VEC substrates and isatoic anhydride analogues (Scheme 71). Using palladium-mediated catalysis, VECs were converted into the key zwitterionic p-allyl palladium intermediate. Subsequently, this intermediate was used to perform diverse reactions including cyclodimerization, which also maintains high diastereo- and enantioselectivity in the final products.479
 | ||
| Scheme 71 The iridium-catalysed cascade allylation-macrolactonization based on vinylethylene carbonates precursors. | ||
4.3 Coupling reactions
Pd-catalysed cross-coupling reactions have been widely applied in many fields related to pharmaceutical development and natural products synthesis.480 Mendive-Tapia et al. reported a cyclodimerization method involving the coupling between tryptophan and L-phenylalanine units to yield the aryl-indole coupled peptide through the Pd-catalysed C–H activation reaction. In addition, the residue number between para-L-Phe and Trp effects the catalytic efficiency of this palladium-catalyzed C–H arylation, with molecules containing short linkers (i, i + 1-meta/para; i + 2-para) tending to afford cyclodimerization. Pd-catalysed C–H activation has also been used to cyclise monomers 97 to generate DKPs 98 with yields of 24% (meta-regiochemistry) and 28% (para-regiochemistry), respectively (Scheme 72).481 | ||
| Scheme 72 Pd-catalysed cross-coupling reactions to cyclopeptide formation. | ||
4.4 Cycloaddition reactions
Cycloaddition reactions are an important way to catalyse the cyclodimerization of unsaturated substrates.482,483 In natural product biosynthesis, the most representative cycloaddition reaction is the Diels–Alder reaction, which catalyses the concerted [4+2] cycloaddition of a precursor diene and a dienophile to form a new 6-membered ring, containing two new C–C bonds. In the case that a heteroatom is involved, this is known as a hetero-Diels–Alder reaction.484–486 Cycloaddition reactions are especially useful for the formation of small-to-medium size cycles and are present in the biosynthesis of many natural products. As a result, many enzymes have been reported as cyclases catalysing [4+2], [2+2] or [6+4] cycloaddition reactions.37,487–489In organic synthesis, cycloaddition reactions are regarded as one of the most effective strategies for constructing cyclic structures. Cycloaddition dimerization obviously differs from the cyclodimerization reaction because cycloaddition involves the interaction between two distinct reactants, usually a conjugated diene and a dienophile, resulting in the creation of a new cyclic molecule. In contrast to cyclodimerization, cycloaddition reaction forms a single cyclic product, not a dimer made of identical or similar reactants. Cycloaddition reaction have also been extensively reviewed.490–493
5. Conclusion and perspectives
With the increasing number of cyclic peptides being developed for use as therapeutic agents, there is great need for efficient and cost-effective synthetic methods for the large-scale synthesis of cyclic molecules. The efficient synthesis of large libraries of cyclic peptides or cyclic peptide-like molecules containing non-natural amino acids (e.g.D- or non-natural amino acids for increased stability and diversity) is also of significant importance.494 However, the efficient cyclisation of peptides using traditional chemical synthesis currently remains a challenge. Many studies have shown that enzymatic domains found in the modular biosynthetic pathways of PKSs and NRPSs can carry out catalytic cyclisation reactions in vitro with a wide range of substrates.4,495 This can enable cyclisation reactions that are challenging to carry out using conventional chemical synthesis.In this review, we summarised the structural traits, distribution, biological functions, and uses of natural products that feature cyclodimeric/cyclomultimeric scaffolds, along with a summary of what is known regarding their biosynthesis and origins. We sought to pay particular attention to the role played by the TE domain of PKS and NRPS machinery in the macrocyclization process. Among these compounds, the most representative examples are gramicidin S and tyrocidine, in which the catalytic mechanism of these TE domain have been relatively well-studied. In addition, we also introduced examples of alternate catalytic cyclization reactions, such as the butelase- and CT-mediated macrocyclization of peptides, as well as the formation of cyclodipeptides mediated by CDPSs and single-module NRPSs. Whilst there are several alternate mechanisms found in nature that perform cyclisation within biosynthetic assembly lines, arguably the most relevant for cyclodimerization is the role of TE domains, given their potential application as biocatalysts. These domains have been widely investigated for their biosynthetic and biocatalytic potential, and whilst we now know significant details concerning the mechanism of cyclisation or dimerization performed by TE domains, there is far less known about the aspects of these domains that control the substrate specificity and positioning that gives rise to their specific products.
Examples of this phenomena can be seen in the activity of the TE domains from gramicidin S (GrsB), tyrocidine (TycC) and WS9326A/mohangamide biosynthesis. The GrsB TE efficiently catalyses the dimerization of assembled pentapeptides to form the cyclic decapeptide gramicidin S, with additional potential for chemoenzymatic analogue synthesis. The TycC TE has been widely studied and cyclises the tyrocidine precursor and a range of modified peptides and is also able to dimerize the gramicidin S pentapeptide, thus emphasizing its versatility. The ability of both TycC TE and GrsB TE to accommodate variations in substrate sequences while maintaining catalytic efficiency suggests a degree of plasticity in their active sites. This plasticity enables these enzymes to recognise and cyclise substrates with similar structural features, even if they are derived from different biosynthetic pathways or synthetically derived.124,125 An excellent example of this substrate promiscuity is the use of the TycC TE domain to generate libraries of tyrocidine derivatives with modified amino acid residues at position 4 of the peptide.131 Kohli et al. noted that the structure of tyrocidine—an amphipathic β-sheet—can also be used to predict the propensity of the TE domain to accept modified residues at P4 in terms of how these might disrupt the structure of the peptide, thus suggesting preorganization of the peptide within the TE must be important for cyclisation. This is in addition to constraints placed on the activity of TE domains (and related PBP-type TE domains) that is seen in the amino acid specificities of such enzymes at the N- and C-termini of the peptide substrates.126,128 The ability of the TycC TE domain to generate the dimeric gramicidin S-structure is also highly significant in this regard, although again there are restraints placed in terms of the amino acids that are accepted at the peptide termini and the structure adopted by the final cyclic peptide in this case. This raises a more general question concerning the importance of preorganization of the substrate for cyclisation by TE domains—something that has largely been investigated in peptide biosynthesis only—and could well call into question the ability to apply TE domains to the cyclisation of substrates that differ significantly from the natural products produced by these enzymes.
Nonetheless, the impressive tolerance of the PBP-type TE SurE for different ring sizes shows that this limitation can be overcome, at least for some systems.39 The TE domain from WS9326A/mohangamide biosynthesis is also a fascinating example of how we are yet to understand the substrate positioning governed by these domains.142,143 In this case, the results of in vitro cyclisation assays clearly show that the differentiating feature in whether the TE domain catalyses a standard cyclisation (to afford WS9326A) or a pseudo-dimerization (to generate mohangamide) is the structure of the acyl unit present on the peptide monomer. Whilst it appears as though mohangamide is likely an unusual minor product of this pathway due to the loading of an unusual dihydropyridine-containing acyl unit, this raises the question of how this seemingly minor alteration to a single building block can completely alter the specificity of the reaction towards dimerization. This requirement for the presence of two different monomers to afford mohangamide is also tantalizing for the generation of controlled dimers via the use of TE domains, as the dihydropyridine-containing acylated peptide appears unable to undergo cyclisation without the presence of the alternate monomer.140 Taken together, these results clearly show that the field requires far more insights into the binding and dynamics of peptides within TE domains in order to understand and exploit such catalytic behaviour.
The interplay of the substrates with the TE domain makes the value of isolated crystal structure without substrate bound of somewhat reduced value, and even the pioneering work of Chin and Schmeing demonstrated the resistance of TE domain complexes to crystallographic investigation, at least from the perspective of explaining the noted cyclisation behaviour of the TE domain studied in this case.70 This strongly suggests that a greater application of computational approaches coupled with in solution biophysical approaches (as have been demonstrated recently for NRPS systems in pioneering work from Mootz and co-workers) is needed to understand these phenomena more completely.496
Despite their challenges, TE domains do offer the major advantage over CT domains in their ability to accept soluble SNACs as substrates, which is a major advantage when exploring the potential for cyclodimerization by such enzymes. Additional examples of TE domains accepting ester substrates provides further support for the value of TE-domains as biocatalysts, which is due to the synthesis of such substrates is significantly simplified because of the improved stability of ester linkages during solid phase peptide synthesis. An important area of future investigation—particularly in regard to pseudo cyclodimerization—is to understand how acyl moieties can be used to control the specific dimerization of two substrates. This would be a very powerful tool for synthetic biology and would have great potential in generating libraries of larger cyclic peptides from a smaller pool of appropriately protected linear intermediates. These findings collectively underscore the versatility and substrate tolerance of these TE domains observed in the NRPS assembly in nature, shedding light on their potential applications in peptide engineering and synthesis.
Data availability
Data availability is not applicable to this article as no new data were created or analyzed in this review article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (no. 82104044 to Songya Zhang, no. T2250710184 to Youming Zhang), Shenzhen Science and Technology Innovation Commission (no. ZDSYS20220303153551001 to Youming Zhang), CAMS Innovation Fund for Medical Sciences (CIFMS, no. 2021-I2M-1-029 to Ran Shi, no. 2022-I2M-2-002 to Hai-Yan He), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (no. 2021-RC350-004 to Hai-Yan He), National Natural Science Foundation of China (no. 22277146 to Hai-Yan He) and National Science Foundation of Beijing Municipality (7244468 to Shuai Fan); This research was also conducted by the Australian Research Council Centre (CE200100012) and funded by the Australian Government.References
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch, The International Natural Product Sciences and C. T. Supuran, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed.
- F. Kopp and M. A. Marahiel, Nat. Prod. Rep., 2007, 24, 735–749 RSC.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- D. G. Jimenez, V. Poongavanam and J. Kihlberg, J. Med. Chem., 2023, 66, 5377–5396 CrossRef PubMed.
- K. Tang, S. Wang, W. S. Gao, Y. H. Song and B. Yu, Acta Pharm. Sin. B, 2022, 12, 4309–4326 CrossRef CAS PubMed.
- H. Zhang and S. Chen, RSC Chem. Biol., 2022, 3, 18–31 RSC.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed.
- Y. U. Kwon and T. Kodadek, Chem. Biol., 2007, 14, 671–677 CrossRef CAS PubMed.
- J. Samanen, F. Ali, T. Romoff, R. Calvo, E. Sorenson, J. Vasko, B. Storer, D. Berry, D. Bennett and M. Strohsacker, et al. , J. Med. Chem., 1991, 34, 3114–3125 CrossRef CAS PubMed.
- J. Sun, H. Yang and W. Tang, Chem. Soc. Rev., 2021, 50, 2320–2336 RSC.
- J. Liu, A. Liu and Y. Hu, Nat. Prod. Rep., 2021, 38, 1469–1505 RSC.
- T. Wezeman, S. Brase and K. S. Masters, Nat. Prod. Rep., 2015, 32, 6–28 RSC.
- A. Paquin, C. Reyes-Moreno and G. Berube, Molecules, 2021, 26, 2340 CrossRef CAS PubMed.
- A. Nudelman, Curr. Med. Chem., 2022, 29, 2751–2845 CrossRef CAS PubMed.
- Y. C. Park, G. Rimbach, C. Saliou, G. Valacchi and L. Packer, FEBS Lett., 2000, 465, 93–97 CrossRef CAS PubMed.
- N. G. M. Gomes, R. B. Pereira, P. B. Andrade and P. Valentao, Mar. Drugs, 2019, 17, 551 CrossRef CAS PubMed.
- K. S. Lam, G. A. Hesler, J. M. Mattei, S. W. Mamber, S. Forenza and K. Tomita, J. Antibiot., 1990, 43, 956–960 CrossRef CAS PubMed.
- G. Berube, Curr. Med. Chem., 2006, 13, 131–154 CrossRef CAS PubMed.
- Z. E. Perlman, J. E. Bock, J. R. Peterson and R. S. Lokey, Bioorg. Med. Chem. Lett., 2005, 15, 5329–5334 CrossRef CAS PubMed.
- R. D. de Vries, K. S. Schmitz, F. T. Bovier, C. Predella, J. Khao, D. Noack, B. L. Haagmans, S. Herfst, K. N. Stearns, J. Drew-Bear, S. Biswas, B. Rockx, G. McGill, N. V. Dorrello, S. H. Gellman, C. A. Alabi, R. L. de Swart, A. Moscona and M. Porotto, Science, 2021, 371, 1379–1382 CrossRef CAS PubMed.
- P. W. Schiller, T. M. Nguyen, C. Lemieux and L. A. Maziak, FEBS Lett., 1985, 191, 231–234 CrossRef CAS PubMed.
- M. L. Huang, S. B. Y. Shin, M. A. Benson, V. J. Torres and K. Kirshenbaum, ChemMedChem, 2012, 7, 114–122 CrossRef CAS PubMed.
- V. Razakantoanina, N. K. Phi Phung and G. Jaureguiberry, Parasitol. Res., 2000, 86, 665–668 CrossRef CAS PubMed.
- I. Takahashi, S. Nakanishi, E. Kobayashi, H. Nakano, K. Suzuki and T. Tamaoki, Biochem. Biophys. Res. Commun., 1989, 165, 1207–1212 CrossRef CAS PubMed.
- J. Skubnik, V. S. Pavlickova, T. Ruml and S. Rimpelova, Biology, 2021, 10, 849 CrossRef CAS PubMed.
- Y. Huang, M. M. Wiedmann and H. Suga, Chem. Rev., 2019, 119, 10360–10391 CrossRef CAS PubMed.
- C. Bechtler and C. Lamers, RSC Med. Chem., 2021, 12, 1325–1351 RSC.
- V. Marti-Centelles, M. D. Pandey, M. I. Burguete and S. V. Luis, Chem. Rev., 2015, 115, 8736–8834 CrossRef CAS PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed.
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496 CrossRef CAS PubMed.
- M. E. Horsman, T. P. Hari and C. N. Boddy, Nat. Prod. Rep., 2016, 33, 183–202 RSC.
- R. F. Little and C. Hertweck, Nat. Prod. Rep., 2022, 39, 163–205 RSC.
- Y. Fan, J. Shen, Z. Liu, K. Xia, W. Zhu and P. Fu, Nat. Prod. Rep., 2022, 39, 1305–1324 RSC.
- D. Mandalapu, X. Ji, J. Chen, C. Guo, W. Q. Liu, W. Ding, J. Zhou and Q. Zhang, J. Org. Chem., 2018, 83, 7271–7275 CrossRef CAS PubMed.
- D. F. Kreitler, E. M. Gemmell, J. E. Schaffer, T. A. Wencewicz and A. M. Gulick, Nat. Commun., 2019, 10, 3432 CrossRef PubMed.
- L. Gu, B. Wang, A. Kulkarni, J. J. Gehret, K. R. Lloyd, L. Gerwick, W. H. Gerwick, P. Wipf, K. Hakansson, J. L. Smith and D. H. Sherman, J. Am. Chem. Soc., 2009, 131, 16033–16035 CrossRef CAS PubMed.
- B. Zhang, K. B. Wang, W. Wang, X. Wang, F. Liu, J. Zhu, J. Shi, L. Y. Li, H. Han, K. Xu, H. Y. Qiao, X. Zhang, R. H. Jiao, K. N. Houk, Y. Liang, R. X. Tan and H. M. Ge, Nature, 2019, 568, 122–126 CrossRef CAS PubMed.
- M. Kobayashi, K. Fujita, K. Matsuda and T. Wakimoto, J. Am. Chem. Soc., 2023, 145, 3270–3275 CrossRef CAS PubMed.
- T. Kuranaga, K. Matsuda, A. Sano, M. Kobayashi, A. Ninomiya, K. Takada, S. Matsunaga and T. Wakimoto, Angew. Chem., Int. Ed., 2018, 57, 9447–9451 CrossRef CAS PubMed.
- Z. Deng, C. Liu, F. Wang, N. Song, J. Liu, H. Li, S. Liu, T. Li, Z. Liu, F. Xiao and W. Li, Angew. Chem., Int. Ed., 2024, e202402010, DOI:10.1002/anie.202402010.
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496 CrossRef CAS PubMed.
- K. A. Bozhuyuk, J. Micklefield and B. Wilkinson, Curr. Opin. Microbiol., 2019, 51, 88–96 CrossRef CAS PubMed.
- A. Koglin and C. T. Walsh, Nat. Prod. Rep., 2009, 26, 987–1000 RSC.
- S. L. Wenski, S. Thiengmag and E. J. N. Helfrich, Synth. Syst. Biotechnol., 2022, 7, 631–647 CrossRef CAS PubMed.
- T. W. Giessen and M. A. Marahiel, FEBS Lett., 2012, 586, 2065–2075 CrossRef CAS PubMed.
- C. T. Walsh, Acc. Chem. Res., 2008, 41, 4–10 CrossRef CAS PubMed.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS PubMed.
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416 RSC.
- K. J. Weissman, Nat. Chem. Biol., 2015, 11, 660–670 CrossRef CAS PubMed.
- S. A. Sieber and M. A. Marahiel, J. Bacteriol., 2003, 185, 7036–7043 CrossRef CAS PubMed.
- M. Peschke, C. Brieke, M. Heimes and M. J. Cryle, ACS Chem. Biol., 2018, 13, 110–120 CrossRef CAS PubMed.
- S. Wang, W. D. G. Brittain, Q. Zhang, Z. Lu, M. H. Tong, K. Wu, K. Kyeremeh, M. Jenner, Y. Yu, S. L. Cobb and H. Deng, Nat. Commun., 2022, 13, 62 CrossRef CAS PubMed.
- T. P. Hari, P. Labana, M. Boileau and C. N. Boddy, ChemBioChem, 2014, 15, 2656–2661 CrossRef CAS PubMed.
- K. Tamura, G. Stecher and S. Kumar, Mol. Biol. Evol., 2021, 38, 3022–3027 CrossRef CAS PubMed.
- I. Letunic and P. Bork, Nucleic Acids Res., 2024, 52, W78–W82 CrossRef PubMed.
- B. T. Caswell, C. C. de Carvalho, H. Nguyen, M. Roy, T. Nguyen and D. C. Cantu, Protein Sci., 2022, 31, 652–676 CrossRef CAS PubMed.
- T. P. Hari, P. Labana, M. Boileau and C. N. Boddy, ChemBioChem, 2014, 15, 2656–2661 CrossRef CAS PubMed.
- Z. Chang, P. Flatt, W. H. Gerwick, V. A. Nguyen, C. L. Willis and D. H. Sherman, Gene, 2002, 296, 235–247 CrossRef CAS PubMed.
- P. M. Flatt, S. J. O'Connell, K. L. McPhail, G. Zeller, C. L. Willis, D. H. Sherman and W. H. Gerwick, J. Nat. Prod., 2006, 69, 938–944 CrossRef CAS PubMed.
- M. S. Kim, M. Bae, Y. E. Jung, J. M. Kim, S. Hwang, M. C. Song, Y. H. Ban, E. S. Bae, S. Hong, S. K. Lee, S. S. Cha, D. C. Oh and Y. J. Yoon, Angew. Chem., Int. Ed., 2021, 60, 19766–19773 CrossRef CAS PubMed.
- K. Geyer, S. Hartmann, R. R. Singh and T. J. Erb, Biochemistry, 2022, 61, 2662–2671 CrossRef CAS PubMed.
- R. D. Sussmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef PubMed.
- A. M. van Wageningen, P. N. Kirkpatrick, D. H. Williams, B. R. Harris, J. K. Kershaw, N. J. Lennard, M. Jones, S. J. Jones and P. J. Solenberg, Chem. Biol., 1998, 5, 155–162 CrossRef CAS PubMed.
- L. Xu, Y. Li, J. B. Biggins, B. R. Bowman, G. L. Verdine, J. B. Gloer, J. A. Alspaugh and G. F. Bills, Appl. Microbiol. Biotechnol., 2018, 102, 2337–2350 CrossRef CAS PubMed.
- B. Shen, L. Du, C. Sanchez, D. J. Edwards, M. Chen and J. M. Murrell, J. Nat. Prod., 2002, 65, 422–431 CrossRef CAS PubMed.
- M. A. Marahiel, Nat. Prod. Rep., 2016, 33, 136–140 RSC.
- T. Izore and M. J. Cryle, Nat. Prod. Rep., 2018, 35, 1120–1139 RSC.
- S. Konno, L. Misson and M. D. Burkart, in Cyclic Peptides: From Bioorganic Synthesis to Applications, ed. J. Koehnke, J. Naismith and W. A. van der Donk, The Royal Society of Chemistry, 2017 10.1039/9781788010153-00033.
- D. P. Frueh, H. Arthanari, A. Koglin, D. A. Vosburg, A. E. Bennett, C. T. Walsh and G. Wagner, Nature, 2008, 454, 903–906 CrossRef CAS PubMed.
- N. Huguenin-Dezot, D. A. Alonzo, G. W. Heberlig, M. Mahesh, D. P. Nguyen, M. H. Dornan, C. N. Boddy, T. M. Schmeing and J. W. Chin, Nature, 2019, 565, 112–117 CrossRef CAS PubMed.
- S. D. Bruner, T. Weber, R. M. Kohli, D. Schwarzer, M. A. Marahiel, C. T. Walsh and M. T. Stubbs, Structure, 2002, 10, 301–310 CrossRef CAS PubMed.
- S. A. Samel, B. Wagner, M. A. Marahiel and L. O. Essen, J. Mol. Biol., 2006, 359, 876–889 CrossRef CAS PubMed.
- J. H. Yu, J. Song, C. B. Chi, T. Liu, T. T. Geng, Z. H. Cai, W. D. Dong, C. Shi, X. Y. Ma, Z. Y. Zhang, X. J. Ma, B. Y. Xing, H. W. Jin, L. R. Zhang, S. W. Dong, D. H. Yang and M. Ma, ACS Catal., 2021, 11, 11733–11741 CrossRef CAS.
- T. L. Bauer, P. C. F. Buchholz and J. Pleiss, FEBS J., 2020, 287, 1035–1053 CrossRef CAS PubMed.
- A. Rauwerdink and R. J. Kazlauskas, ACS Catal., 2015, 5, 6153–6176 CrossRef CAS PubMed.
- D. C. Cantu, Y. Chen and P. J. Reilly, Protein Sci., 2010, 19, 1281–1295 CrossRef CAS PubMed.
- A. Koglin, F. Lohr, F. Bernhard, V. V. Rogov, D. P. Frueh, E. R. Strieter, M. R. Mofid, P. Guntert, G. Wagner, C. T. Walsh, M. A. Marahiel and V. Dotsch, Nature, 2008, 454, 907–911 CrossRef CAS PubMed.
- K. D. Patel, M. R. MacDonald, S. F. Ahmed, J. Singh and A. M. Gulick, Nat. Prod. Rep., 2023, 40, 1550–1582 RSC.
- J. B. Scaglione, D. L. Akey, R. Sullivan, J. D. Kittendorf, C. M. Rath, E. S. Kim, J. L. Smith and D. H. Sherman, Angew. Chem., Int. Ed., 2010, 49, 5726–5730 CrossRef CAS PubMed.
- G. Calero, P. Gupta, M. C. Nonato, S. Tandel, E. R. Biehl, S. L. Hofmann and J. Clardy, J. Biol. Chem., 2003, 278, 37957–37964 CrossRef CAS PubMed.
- B. R. Miller and A. M. Gulick, Methods Mol. Biol., 2016, 1401, 3–29 CrossRef CAS PubMed.
- H. Nakamura, J. X. Wang and E. P. Balskus, Chem. Sci., 2015, 6, 3816–3822 RSC.
- L. Serino, C. Reimmann, P. Visca, M. Beyeler, V. D. Chiesa and D. Haas, J. Bacteriol., 1997, 179, 248–257 CrossRef CAS PubMed.
- S. T. Pullan, G. Chandra, M. J. Bibb and M. Merrick, BMC Genomics, 2011, 12, 175 CrossRef CAS PubMed.
- T. P. Korman, J. M. Crawford, J. W. Labonte, A. G. Newman, J. Wong, C. A. Townsend and S. C. Tsai, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6246–6251 CrossRef CAS PubMed.
- N. M. Gaudelli and C. A. Townsend, Nat. Chem. Biol., 2014, 10, 251–258 CrossRef CAS PubMed.
- N. M. Gaudelli, D. H. Long and C. A. Townsend, Nature, 2015, 520, 383–387 CrossRef CAS PubMed.
- K. D. Patel, F. B. d'Andrea, N. M. Gaudelli, A. R. Buller, C. A. Townsend and A. M. Gulick, Nat. Commun., 2019, 10, 3868 CrossRef PubMed.
- C. Khosla, Y. Tang, A. Y. Chen, N. A. Schnarr and D. E. Cane, Annu. Rev. Biochem., 2007, 76, 195–221 CrossRef CAS PubMed.
- C. D. Campbell and J. C. Vederas, Biopolymers, 2010, 93, 755–763 CrossRef CAS PubMed.
- H. Wang, C. Li, B. Zhang, H. He, P. Jin, J. Wang, J. Zhang, X. Wang and W. Xiang, J. Biotechnol., 2015, 204, 1–2 CrossRef CAS PubMed.
- B. Julien, S. Shah, R. Ziermann, R. Goldman, L. Katz and C. Khosla, Gene, 2000, 249, 153–160 CrossRef CAS PubMed.
- B. Shen, Curr. Opin. Chem. Biol., 2003, 7, 285–295 CrossRef CAS PubMed.
- K. J. Weissman, Methods Enzymol., 2009, 459, 3–16 CAS.
- P. Argyropoulos, F. Bergeret, C. Pardin, J. M. Reimer, A. Pinto, C. N. Boddy and T. M. Schmeing, Biochim. Biophys. Acta, 2016, 1860, 486–497 CrossRef CAS PubMed.
- D. L. Akey, J. D. Kittendorf, J. W. Giraldes, R. A. Fecik, D. H. Sherman and J. L. Smith, Nat. Chem. Biol., 2006, 2, 537–542 CrossRef CAS PubMed.
- J. J. Gehret, L. Gu, W. H. Gerwick, P. Wipf, D. H. Sherman and J. L. Smith, J. Biol. Chem., 2011, 286, 14445–14454 CrossRef CAS PubMed.
- S. C. Tsai, L. J. Miercke, J. Krucinski, R. Gokhale, J. C. Chen, P. G. Foster, D. E. Cane, C. Khosla and R. M. Stroud, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14808–14813 CrossRef CAS PubMed.
- S. C. Tsai, H. X. Lu, D. E. Cane, C. Khosla and R. M. Stroud, Biochemistry, 2002, 41, 12598–12606 CrossRef CAS PubMed.
- T. Shi, L. Liu, W. Tao, S. Luo, S. Fan, X.-L. Wang, L. Bai and Y.-L. Zhao, ACS Catal., 2018, 8, 4323–4332 CrossRef CAS.
- X. P. Chen, T. Shi, X. L. Wang, J. T. Wang, Q. H. Chen, L. Q. Bai and Y. L. Zhao, ACS Catal., 2016, 6, 4369–4378 CrossRef CAS.
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CrossRef CAS PubMed.
- D. Yu, F. Xu, J. Zi, S. Wang, D. Gage, J. Zeng and J. Zhan, Metab. Eng., 2013, 18, 60–68 CrossRef CAS PubMed.
- G. Li Petri, S. Di Martino and M. De Rosa, J. Med. Chem., 2022, 65, 7438–7475 CrossRef CAS PubMed.
- L. Y. Chia, P. V. Kumar, M. A. A. Maki, G. Ravichandran and S. Thilagar, Int. J. Pept. Res. Ther., 2023, 29, 7 CrossRef CAS PubMed.
- K. R. Schramma, L. B. Bushin and M. R. Seyedsayamdost, Nat. Chem., 2015, 7, 431–437 CrossRef CAS PubMed.
- A. Greule, T. Izore, D. Iftime, J. Tailhades, M. Schoppet, Y. Zhao, M. Peschke, I. Ahmed, A. Kulik, M. Adamek, R. J. A. Goode, R. B. Schittenhelm, J. A. Kaczmarski, C. J. Jackson, N. Ziemert, E. H. Krenske, J. J. De Voss, E. Stegmann and M. J. Cryle, Nat. Commun., 2019, 10, 2613 CrossRef PubMed.
- L. C. Cai, Y. M. Yao, S. K. Yeon and I. B. Seiple, J. Am. Chem. Soc., 2020, 142, 15116–15126 CrossRef CAS PubMed.
- G. Gause and M. Brazhnikova, Nature, 1944, 154, 703 CrossRef.
- W. W. Umbreit, The Search for New Antibiotics. G. F. Gause. Yale University Press, New Haven, Conn, 1960. 97 pp. Illus. $4.75, 1960 Search PubMed.
- L. H. Kondejewski, S. W. Farmer, D. S. Wishart, R. E. W. Hancock and R. S. Hodges, Int. J. Pept. Protein Res., 1996, 47, 460–466 CrossRef CAS PubMed.
- M. Wenzel, M. Rautenbach, J. A. Vosloo, T. Siersma, C. H. M. Aisenbrey, E. Zaitseva, W. E. Laubscher, W. van Rensburg, J. C. Behrends, B. Bechinger and L. W. Hamoen, mBio, 2018, 9, e00802–e00818 CrossRef CAS PubMed.
- K. P. Dimick, Proc. Soc. Exp. Biol. Med., 1951, 78, 782–784 CrossRef CAS PubMed.
- R. Sarges and B. Witkop, J. Am. Chem. Soc., 1965, 87, 2011–2020 CrossRef CAS PubMed.
- K. O. Pedersen and R. L. M. Synge, Acta Chem. Scand., 1948, 2, 408–413 CrossRef CAS.
- B. F. Erlnager and L. Goode, Nature, 1954, 174, 840–841 CrossRef CAS PubMed.
- H. D. Mootz and M. A. Marahiel, J. Bacteriol., 1997, 179, 6843–6850 CrossRef CAS PubMed.
- R. J. Dubos and R. D. Hotchkiss, J. Exp. Med., 1941, 73, 629–640 CrossRef CAS PubMed.
- P. J. Loll, E. C. Upton, V. Nahoum, N. J. Economou and S. Cocklin, Biochim. Biophys. Acta, 2014, 1838, 1199–1207 CrossRef CAS PubMed.
- M. Rautenbach, A. M. Troskie, J. A. Vosloo and M. E. Dathe, Biochimie, 2016, 130, 122–131 CrossRef CAS PubMed.
- M. Krause and M. A. Marahiel, J. Bacteriol., 1988, 170, 4669–4674 CrossRef CAS PubMed.
- J. Kratzschmar, M. Krause and M. A. Marahiel, J. Bacteriol., 1989, 171, 5422–5429 CrossRef CAS PubMed.
- F. Pourmasoumi, S. Y. T. De, H. Y. Peng, F. Trottmann, C. Hertweck and H. Kries, ACS Chem. Biol., 2022, 17, 2382–2388 CrossRef CAS PubMed.
- K. M. Hoyer, C. Mahlert and M. A. Marahiel, Chem. Biol., 2007, 14, 13–22 CrossRef CAS PubMed.
- X. M. Wu, X. Z. Bu, K. M. Wong, W. L. Yan and Z. H. Guo, Org. Lett., 2003, 5, 1749–1752 CrossRef CAS PubMed.
- J. W. Trauger, R. M. Kohli, H. D. Mootz, M. A. Marahiel and C. T. Walsh, Nature, 2000, 407, 215–218 CrossRef CAS PubMed.
- R. M. Kohli, J. Takagi and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1247–1252 CrossRef CAS PubMed.
- H. Lin, D. A. Thayer, C. H. Wong and C. T. Walsh, Chem. Biol., 2004, 11, 1635–1642 CrossRef CAS PubMed.
- R. M. Kohli, J. W. Trauger, D. Schwarzer, M. A. Marahiel and C. T. Walsh, Biochemistry, 2001, 40, 7099–7108 CrossRef CAS PubMed.
- J. W. Trauger, R. M. Kohli and C. T. Walsh, Biochemistry, 2001, 40, 7092–7098 CrossRef CAS PubMed.
- G. Xie, M. Uttamchandani, G. Y. Chen, X. Bu, S. S. Lin, K. M. Wong, W. Yan, S. Q. Yao and Z. Guo, Bioorg. Med. Chem. Lett., 2002, 12, 989–992 CrossRef CAS PubMed.
- P. Wadhwani, S. Afonin, M. Ieronimo, J. Buerck and A. S. Ulrich, J. Org. Chem., 2006, 71, 55–61 CrossRef CAS PubMed.
- F. E. Morales, H. E. Garay, D. F. Munoz, Y. E. Augusto, A. J. Otero-Gonzalez, O. Reyes Acosta and D. G. Rivera, Org. Lett., 2015, 17, 2728–2731 CrossRef CAS PubMed.
- L. Reguera and D. G. Rivera, Chem. Rev., 2019, 119, 9836–9860 CrossRef CAS PubMed.
- G. M. Grotenbreg, M. S. Timmer, A. L. Llamas-Saiz, M. Verdoes, G. A. van der Marel, M. J. van Raaij, H. S. Overkleeft and M. Overhand, J. Am. Chem. Soc., 2004, 126, 3444–3446 CrossRef CAS PubMed.
- L. Du and L. Lou, Nat. Prod. Rep., 2010, 27, 255–278 RSC.
- R. M. Kohli, C. T. Walsh and M. D. Burkart, Nature, 2002, 418, 658–661 CrossRef CAS PubMed.
- O. E. Zolova, A. S. Mady and S. Garneau-Tsodikova, Biopolymers, 2010, 93, 777–790 CrossRef CAS PubMed.
- S. Zhang, Y. Chen, J. Zhu, Q. Lu, M. J. Cryle, Y. Zhang and F. Yan, Nat. Prod. Rep., 2023, 40, 557–594 RSC.
- M. Bae, H. Kim, K. Moon, S. J. Nam, J. Shin, K. B. Oh and D. C. Oh, Org. Lett., 2015, 17, 712–715 CrossRef CAS PubMed.
- C. W. Johnston, M. A. Skinnider, M. A. Wyatt, X. Li, M. R. Ranieri, L. Yang, D. L. Zechel, B. Ma and N. A. Magarvey, Nat. Commun., 2015, 6, 8421 CrossRef CAS PubMed.
- S. Zhang, J. Zhu, D. L. Zechel, C. Jessen-Trefzer, R. T. Eastman, T. Paululat and A. Bechthold, ChemBioChem, 2018, 19, 272–279 CrossRef CAS PubMed.
- M. S. Kim, M. Bae, M. C. Song, S. Hwang, D. C. Oh and Y. J. Yoon, Org. Lett., 2022, 24, 4444–4448 CrossRef CAS PubMed.
- Z. Yu, S. Vodanovic-Jankovic, M. Kron and B. Shen, Org. Lett., 2012, 14, 4946–4949 CrossRef CAS PubMed.
- S. Zhang, L. Zhang, A. Greule, J. Tailhades, E. Marschall, P. Prasongpholchai, D. J. Leng, J. Zhang, J. Zhu, J. A. Kaczmarski, R. B. Schittenhelm, O. Einsle, C. J. Jackson, F. Alberti, A. Bechthold, Y. Zhang, M. Tosin, T. Si and M. J. Cryle, Acta Pharm. Sin. B, 2023, 13, 3561–3574 CrossRef CAS PubMed.
- M. L. Guerinot, Annu. Rev. Microbiol., 1994, 48, 743–772 CrossRef CAS PubMed.
- M. Miethke and M. A. Marahiel, Microbiol. Mol. Biol. Rev., 2007, 71, 413–451 CrossRef CAS PubMed.
- L. D. Loomis and Kenneth N. Raymond, Inorg. Chem., 1991, 30, 906–911 CrossRef CAS.
- L. M. Bogomolnaya, R. Tilvawala, J. R. Elfenbein, J. D. Cirillo and H. L. Andrews-Polymenis, mBio, 2020, 11, e00528–e00520 CrossRef PubMed.
- J. R. Pollack and J. B. Neilands, Biochem. Biophys. Res. Commun., 1970, 38, 989–992 CrossRef CAS PubMed.
- I. G. O'Brien and F. Gibson, Biochim. Biophys. Acta, 1970, 215, 393–402 CrossRef PubMed.
- I. G. O'Brien, G. B. Cox and F. Gibson, Biochim. Biophys. Acta, 1971, 237, 537–549 CrossRef PubMed.
- Y. Matsuo, K. Kanoh, J. H. Jang, K. Adachi, S. Matsuda, O. Miki, T. Kato and Y. Shizuri, J. Nat. Prod., 2011, 74, 2371–2376 CrossRef CAS PubMed.
- S. Zajdowicz, J. C. Haller, A. E. Krafft, S. W. Hunsucker, C. T. Mant, M. W. Duncan, R. S. Hodges, D. N. Jones and R. K. Holmes, PLoS One, 2012, 7, e34591 CrossRef CAS PubMed.
- H. Budzikiewicz, A. Bossenkamp, K. Taraz, A. Pandey and J. M. Meyer, Z. Naturforsch., C: J. Biosci., 1997, 52, 551–554 CrossRef CAS.
- I. G. Young, L. Langman, R. K. Luke and F. Gibson, J. Bacteriol., 1971, 106, 51–57 CrossRef CAS PubMed.
- R. K. Luke and F. Gibson, J. Bacteriol., 1971, 107, 557–562 CrossRef CAS PubMed.
- A. M. Gehring, I. Mori and C. T. Walsh, Biochemistry, 1998, 37, 2648–2659 CrossRef CAS PubMed.
- D. E. Ehmann, J. W. Trauger, T. Stachelhaus and C. T. Walsh, Chem. Biol., 2000, 7, 765–772 CrossRef CAS PubMed.
- E. D. Roche and C. T. Walsh, Biochemistry, 2003, 42, 1334–1344 CrossRef CAS PubMed.
- A. M. Gehring, K. A. Bradley and C. T. Walsh, Biochemistry, 1997, 36, 8495–8503 CrossRef CAS PubMed.
- E. J. Drake, B. R. Miller, C. Shi, J. T. Tarrasch, J. A. Sundlov, C. L. Allen, G. Skiniotis, C. C. Aldrich and A. M. Gulick, Nature, 2016, 529, 235–238 CrossRef CAS PubMed.
- Z. L. Reitz, M. Sandy and A. Butler, Metallomics, 2017, 9, 824–839 CrossRef CAS PubMed.
- C. A. Shaw-Reid, N. L. Kelleher, H. C. Losey, A. M. Gehring, C. Berg and C. T. Walsh, Chem. Biol., 1999, 6, 385–400 CrossRef CAS PubMed.
- Y. Liu, T. Zheng and S. D. Bruner, Chem. Biol., 2011, 18, 1482–1488 CrossRef CAS PubMed.
- K. Haslinger, M. Peschke, C. Brieke, E. Maximowitsch and M. J. Cryle, Nature, 2015, 521, 105–109 CrossRef CAS PubMed.
- M. Peschke, M. Gonsior, R. D. Sussmuth and M. J. Cryle, Curr. Opin. Struct. Biol., 2016, 41, 46–53 CrossRef CAS PubMed.
- A. M. Gulick and C. C. Aldrich, Nat. Prod. Rep., 2018, 35, 1156–1184 RSC.
- B. R. Miller, E. J. Drake, C. Shi, C. C. Aldrich and A. M. Gulick, J. Biol. Chem., 2016, 291, 22559–22571 CrossRef CAS PubMed.
- E. J. Corey and S. Bhattacharyya, Tetrahedron Lett., 1977, 3919–3922 CrossRef CAS.
- A. Shanzer and J. Libman, J. Chem. Soc., Chem. Commun., 1983, 846–847, 10.1039/c39830000846.
- R. J. A. Ramirez, L. Karamanukyan, S. Ortiz and C. G. Gutierrez, Tetrahedron Lett., 1997, 38, 749–752 CrossRef CAS.
- H. K. Zane, H. Naka, F. Rosconi, M. Sandy, M. G. Haygood and A. Butler, J. Am. Chem. Soc., 2014, 136, 5615–5618 CrossRef CAS PubMed.
- D. L. McRose, O. Baars, M. R. Seyedsayamdost and F. M. M. Morel, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7581–7586 CrossRef CAS PubMed.
- A. M. Jelowicki and A. Butler, J. Biol. Inorg. Chem., 2022, 27, 565–572 CrossRef CAS PubMed.
- J. J. May, T. M. Wendrich and M. A. Marahiel, J. Biol. Chem., 2001, 276, 7209–7217 CrossRef CAS PubMed.
- Y. P. Wen, X. C. Wu, Y. Teng, C. D. Qian, Z. J. Zhan, Y. H. Zhao and O. Li, Environ. Microbiol., 2011, 13, 2726–2737 CrossRef CAS PubMed.
- Q. H. Wu, K. Throckmorton, M. Maity, M. G. Chevrette, D. R. Braun, S. R. Rajski, C. R. Currie, M. G. Thomas and T. S. Bugni, J. Nat. Prod., 2021, 84, 136–141 CrossRef CAS PubMed.
- H. N. Patel, R. N. Chakraborty and S. B. Desai, FEMS Microbiol. Lett., 1988, 56, 131–134 CrossRef CAS.
- J. R. Heemstra, C. T. Walsh and E. S. Sattely, J. Am. Chem. Soc., 2009, 131, 15317–15329 CrossRef CAS PubMed.
- R. A. Carter, P. S. Worsley, G. Sawers, G. L. Challis, M. J. Dilworth, K. C. Carson, J. A. Lawrence, M. Wexler, A. W. Johnston and K. H. Yeoman, Mol. Microbiol., 2002, 44, 1153–1166 CrossRef CAS PubMed.
- N. Kadi, D. Oves-Costales, F. Barona-Gomez and G. L. Challis, Nat. Chem. Biol., 2007, 3, 652–656 CrossRef CAS PubMed.
- J. Yang, V. S. Banas, K. D. Patel, G. S. M. Rivera, L. S. Mydy, A. M. Gulick and T. A. Wencewicz, J. Biol. Chem., 2022, 298, 102166 CrossRef CAS PubMed.
- N. Kadi, L. Song and G. L. Challis, Chem. Commun., 2008, 5119–5121, 10.1039/b813029a.
- N. Kadi, S. Arbache, L. J. Song, D. Oves-Costales and G. L. Challis, J. Am. Chem. Soc., 2008, 130, 10458–10459 CrossRef CAS PubMed.
- H. Brockmann and G. Schmidt-Kastner, Chem. Ber., 1955, 88, 57–61 CrossRef CAS.
- H. Brockmann, M. Springor, G. Traxler and I. Hofer, Naturwissenschaften, 1963, 50, 689 CrossRef CAS.
- M. M. Shemyakin, N. A. Aldanova, E. I. Vinogradova and M. Y. Feigina, Tetrahedron Lett., 1963, 1921–1925 CrossRef CAS.
- S. H. Huang, Y. S. Liu, W. Q. Liu, P. Neubauer and J. Li, Microorganisms, 2021, 9, 780 CrossRef CAS PubMed.
- Z. Su, X. Ran, J. J. Leitch, A. L. Schwan, R. Faragher and J. Lipkowski, Langmuir, 2019, 35, 16935–16943 CrossRef CAS PubMed.
- Y. Q. Cheng, ChemBioChem, 2006, 7, 471–477 CrossRef CAS PubMed.
- D. A. Alonzo and T. M. Schmeing, Protein Sci., 2020, 29, 2316–2347 CrossRef CAS PubMed.
- J. Jaitzig, J. Li, R. D. Sussmuth and P. Neubauer, ACS Synth. Biol., 2014, 3, 432–438 CrossRef CAS PubMed.
- S. Konno, M. Tanaka, T. Mizuguchi, H. Toyokai, A. Taguchi, A. Taniguchi and Y. Hayashi, J. Pept. Sci., 2023, 30, e3532 CrossRef PubMed.
- B. F. Gisin, R. B. Merrifield and D. C. Tosteson, J. Am. Chem. Soc., 1969, 91, 2691–2695 CrossRef CAS PubMed.
- Y. L. Dory, J. M. Mellor and J. F. Mcaleer, Tetrahedron Lett., 1989, 30, 1695–1698 CrossRef CAS.
- J. R. Dawson, Y. L. Dory, J. M. Mellor and J. F. McAleer, Tetrahedron, 1996, 52, 1361–1378 CrossRef CAS.
- L. Zhuang, S. H. Huang, W. Q. Liu, A. S. Karim, M. C. Jewett and J. Li, Metab. Eng., 2020, 60, 37–44 CrossRef CAS PubMed.
- N. Agata, M. Mori, M. Ohta, S. Suwan, I. Ohtani and M. Isobe, FEMS Microbiol. Lett., 1994, 121, 31–34 CAS.
- V. Walser, M. Kranzler, C. Dawid, M. Ehling-Schulz, T. D. Stark and T. F. Hofmann, Molecules, 2022, 27, 872 CrossRef CAS PubMed.
- M. A. Andersson, P. Hakulinen, U. Honkalampi-Hamalainen, D. Hoornstra, J. C. Lhuguenot, J. Maki-Paakkanen, M. Savolainen, I. Severin, A. L. Stammati, L. Turco, A. Weber, A. von Wright, F. Zucco and M. Salkinoja-Salonen, Toxicon, 2007, 49, 351–367 CrossRef CAS PubMed.
- N. Agata, M. Ohta and M. Mori, Curr. Microbiol., 1996, 33, 67–69 CrossRef CAS PubMed.
- F. Carlin, M. Fricker, A. Pielaat, S. Heisterkamp, R. Shaheen, M. S. Salonen, B. Svensson, C. Nguyen-The and M. Ehling-Schulz, Int. J. Food Microbiol., 2006, 109, 132–138 CrossRef CAS PubMed.
- S. Marxen, T. D. Stark, A. Rutschle, G. Lucking, E. Frenzel, S. Scherer, M. Ehling-Schulz and T. Hofmann, Sci. Rep., 2015, 5, 10637 CrossRef PubMed.
- M. Ehling-Schulz, N. Vukov, A. Schulz, R. Shaheen, M. Andersson, E. Martlbauer and S. Scherer, Appl. Environ. Microbiol., 2005, 71, 105–113 CrossRef CAS PubMed.
- M. Ehling-Schulz, M. Fricker, H. Grallert, P. Rieck, M. Wagner and S. Scherer, BMC Microbiol., 2006, 6, 20 CrossRef PubMed.
- G. Lucking, E. Frenzel, A. Rutschle, S. Marxen, T. D. Stark, T. Hofmann, S. Scherer and M. Ehling-Schulz, Front. Microbiol., 2015, 6, 1101 Search PubMed.
- D. A. Alonzo, N. A. Magarvey and T. M. Schmeing, PLoS One, 2015, 10, e0128569 CrossRef PubMed.
- G. W. Heberlig and C. N. Boddy, J. Nat. Prod., 2020, 83, 1990–1997 CrossRef CAS PubMed.
- G. J. Quigley, G. Ughetto, G. A. van der Marel, J. H. van Boom, A. H. Wang and A. Rich, Science, 1986, 232, 1255–1258 CrossRef CAS PubMed.
- U. Keller and F. Schauwecker, Prog. Nucleic Acid Res. Mol. Biol., 2001, 70, 233–289 CAS.
- H. Chen and D. J. Patel, J. Mol. Biol., 1995, 246, 164–179 CrossRef CAS PubMed.
- J. Y. Park, Y. S. Ryang, K. Y. Shim, J. I. Lee, H. S. Kim, Y. H. Kim and S. K. Kim, Int. J. Biochem. Cell Biol., 2006, 38, 244–254 CrossRef CAS PubMed.
- J. Fernandez, L. Marin, R. Alvarez-Alonso, S. Redondo, J. Carvajal, G. Villamizar, C. J. Villar and F. Lombo, Mar. Drugs, 2014, 12, 2668–2699 CrossRef PubMed.
- R. Corbaz, L. Ettlinger, E. Gaumann, W. Kellerschierlein, F. Kradolfer, L. Neipp, V. Prelog, P. Reusser and H. Zahner, Helv. Chim. Acta, 1957, 40, 199–204 CrossRef CAS.
- T. Yoshida, K. Katagiri and S. Yokozawa, J. Antibiot., 1961, 14, 330–334 CAS.
- C. Zhang, L. X. Kong, Q. Liu, X. Lei, T. Zhu, J. Yin, B. R. Lin, Z. X. Deng and D. L. You, PLoS One, 2013, 8, e56772 CrossRef CAS PubMed.
- J. Shoji, R. Konaka, K. Kawano, N. Higuchi and Y. Kyogoku, J. Antibiot., 1976, 29, 1246–1248 CrossRef CAS PubMed.
- T. Yoshida and K. Katagiri, J. Bacteriol., 1967, 93, 1327–1331 CrossRef CAS PubMed.
- L. G. May, M. A. Madine and M. J. Waring, Nucleic Acids Res., 2004, 32, 65–72 CrossRef CAS PubMed.
- G. Schmoock, F. Pfennig, J. Jewiarz, W. Schlumbohm, W. Laubinger, F. Schauwecker and U. Keller, J. Biol. Chem., 2005, 280, 4339–4349 CrossRef CAS PubMed.
- A. P. Praseuth, C. C. C. Wang, K. Watanabe, K. Hotta, H. Oguri and H. Oikawa, Biotechnol. Prog., 2008, 24, 1226–1231 CrossRef CAS PubMed.
- K. Watanabe, K. Hotta, M. Nakaya, A. P. Praseuth, C. C. C. Wang, D. Inada, K. Takahashi, E. Fukushi, H. Oguri and H. Oikawa, J. Am. Chem. Soc., 2009, 131, 9347–9353 CrossRef CAS PubMed.
- M. Sato, T. Nakazawa, Y. Tsunematsu, K. Hotta and K. Watanabe, Curr. Opin. Chem. Biol., 2013, 17, 537–545 CrossRef CAS PubMed.
- K. Watanabe, K. Hotta, A. P. Praseuth, M. Searcey, C. C. C. Wang, H. Oguri and H. Oikawa, ChemBioChem, 2009, 10, 1965–1968 CrossRef CAS PubMed.
- J. Grunewald, S. A. Sieber and M. A. Marahiel, Biochemistry, 2004, 43, 2915–2925 CrossRef PubMed.
- C. C. Tseng, S. D. Bruner, R. M. Kohli, M. A. Marahiel, C. T. Walsh and S. A. Sieber, Biochemistry, 2002, 41, 13350–13359 CrossRef CAS PubMed.
- K. Koketsu, H. Oguri, K. Watanabe and H. Oikawa, Chem. Biol., 2008, 15, 818–828 CrossRef CAS PubMed.
- J. Tulla-Puche, E. Marcucci, M. Fermin, N. Bayo-Puxan and F. Albericio, Chem. – Eur. J., 2008, 14, 4475–4478 CrossRef CAS PubMed.
- G. A. Sable and D. Lim, J. Org. Chem., 2015, 80, 7486–7494 CrossRef CAS PubMed.
- K. Hattori, K. Koike, K. Okuda, T. Hirayama, M. Ebihara, M. Takenaka and H. Nagasawa, Org. Biomol. Chem., 2016, 14, 2090–2111 RSC.
- P. K. Chakravarty and R. K. Olsen, Tetrahedron Lett., 1978, 19, 1613–1616 CrossRef.
- K. Kojima, F. Yakushiji, A. Katsuyama and S. Ichikawa, Org. Lett., 2020, 22, 4217–4221 CrossRef CAS PubMed.
- C. Maruyama, Y. Chinone, S. Sato, F. Kudo, K. Ohsawa, J. Kubota, J. Hashimoto, I. Kozone, T. Doi, K. Shin-Ya, T. Eguchi and Y. Hamano, Biomolecules, 2020, 10, 210 CrossRef PubMed.
- J. A. Matson, K. L. Colson, G. N. Belofsky and B. B. Bleiberg, J. Antibiot., 1993, 46, 162–166 CrossRef CAS PubMed.
- D. L. Boger, J. H. Chen and K. W. Saionz, J. Am. Chem. Soc., 1996, 118, 1629–1644 CrossRef CAS.
- K. Katayama, K. Nakagawa, H. Takeda, A. Matsuda and S. Ichikawa, Org. Lett., 2014, 16, 428–431 CrossRef CAS PubMed.
- Y. Komatani, K. Momosaki, A. Katsuyama, K. Yamamoto, R. Kaguchi and S. Ichikawa, Org. Lett., 2023, 25, 543–548 CrossRef CAS PubMed.
- M. Konishi, H. Ohkuma, F. Sakai, T. Tsuno, H. Koshiyama, T. Naito and H. Kawaguchi, J. Antibiot., 1981, 34, 148–159 CrossRef CAS PubMed.
- D. L. Boger, M. W. Ledeboer, M. Kume, M. Searcey and Q. Jin, J. Am. Chem. Soc., 1999, 121, 11375–11383 CrossRef CAS.
- X. J. Shi, L. M. Huang, K. H. Song, G. Y. Zhao, Y. Liu, L. X. Lv and Y. L. Du, Angew. Chem., Int. Ed., 2021, 60, 19821–19828 CrossRef CAS PubMed.
- L. J. Cruz, M. M. Insua, J. P. Baz, M. Trujillo, R. A. Rodriguez-Mias, E. Oliveira, E. Giralt, F. Albericio and L. M. Canedo, J. Org. Chem., 2006, 71, 3335–3338 CrossRef CAS PubMed.
- L. J. Cruz, C. Cuevas, L. M. Canedo, E. Giralt and F. Albericio, J. Org. Chem., 2006, 71, 3339–3344 CrossRef CAS PubMed.
- A. V. Mayorov, M. Y. Cai, E. S. Palmer, Z. H. Liu, J. P. Cain, J. Vagner, D. Trivedi and V. J. Hruby, Peptides, 2010, 31, 1894–1905 CrossRef CAS PubMed.
- J. P. Baz, L. M. Canedo, J. L. F. Puentes and M. V. S. Elipe, J. Antibiot., 1997, 50, 738–741 CrossRef PubMed.
- F. Romero, F. Espliego, J. P. Baz, T. G. DeQuesada, D. Gravalos, F. DelaCalle and J. L. FernadezPuertes, J. Antibiot., 1997, 50, 734–737 CrossRef CAS PubMed.
- F. Lombo, A. Velasco, A. Castro, F. de la Calle, A. F. Brana, J. M. Sanchez-Puelles, C. Mendez and J. A. Salas, ChemBioChem, 2006, 7, 366–376 CrossRef CAS PubMed.
- S. Mori, S. K. Shrestha, J. Fernandez, M. A. San Milian, A. Garzan, A. H. Al-Mestarihi, F. Lombo and S. Garneau-Tsodikova, Biochemistry, 2017, 56, 4457–4467 CrossRef CAS PubMed.
- L. Robbel, K. M. Hoyer and M. A. Marahiel, FEBS J., 2009, 276, 1641–1653 CrossRef CAS PubMed.
- D. L. Boger, S. Ichikawa, W. C. Tse, M. P. Hedrick and Q. Jin, J. Am. Chem. Soc., 2001, 123, 561–568 CrossRef CAS PubMed.
- N. Bayo-Puxan, A. Fernandez, J. Tulla-Puche, E. Riego, C. Cuevas, M. Alvarez and F. Albericio, Chem. – Eur. J., 2006, 12, 9001–9009 CrossRef CAS PubMed.
- J. He, E. M. K. Wijeratne, B. P. Bashyal, J. X. Zhan, C. J. Seliga, M. P. X. Liu, E. E. Pierson, L. S. Pierson, H. D. VanEtten and A. A. L. Gunatilaka, J. Nat. Prod., 2004, 67, 1985–1991 CrossRef CAS PubMed.
- C. Sanchez, I. A. Butovich, A. F. Brana, J. Rohr, C. Mendez and J. A. Salas, Chem. Biol., 2002, 9, 519–531 CrossRef CAS PubMed.
- H. Onaka, S. Taniguchi, Y. Igarashi and T. Furumai, Biosci., Biotechnol., Biochem., 2003, 67, 127–138 CrossRef CAS PubMed.
- S. Shimizu, Y. Yamamoto, J. Inagaki and S. Koshimura, Gan, 1982, 73, 642–648 CAS.
- A. Kaji, R. Saito, M. Nomura, K. Miyamoto and N. Kiriyama, Anticancer Res., 1997, 17, 3675–3679 CAS.
- P. Schneider, M. Weber, K. Rosenberger and D. Hoffmeister, Chem. Biol., 2007, 14, 635–644 CrossRef CAS PubMed.
- C. J. Balibar, A. R. Howard and C. T. Walsh, Nat. Chem. Biol., 2007, 3, 584–592 CrossRef CAS PubMed.
- S. Thies, B. Santiago-Schubel, F. Kovacic, F. Rosenau, R. Hausmann and K. E. Jaeger, J. Biotechnol., 2014, 181, 27–30 CrossRef CAS PubMed.
- H. Li, T. Tanikawa, Y. Sato, Y. Nakagawa and T. Matsuyama, Microbiol. Immunol., 2005, 49, 303–310 CrossRef CAS PubMed.
- C. T. Walsh, Science, 2004, 303, 1805–1810 CrossRef CAS PubMed.
- H. Liu, R. N. Ottosen, K. M. Jennet, E. B. Svenningsen, T. F. Kristensen, M. Biltoft, M. R. Jakobsen and T. B. Poulsen, Angew. Chem., Int. Ed., 2021, 60, 18734–18741 CrossRef CAS PubMed.
- P. Steib and B. Breit, Angew. Chem., Int. Ed., 2018, 57, 6572–6576 CrossRef CAS PubMed.
- Y. J. Zhou, A. C. Murphy, M. Samborskyy, P. Prediger, L. C. Dias and P. F. Leadlay, Chem. Biol., 2015, 22, 745–754 CrossRef CAS PubMed.
- J. Ma, Z. Wang, H. Huang, M. Luo, D. Zuo, B. Wang, A. Sun, Y. Q. Cheng, C. Zhang and J. Ju, Angew. Chem., Int. Ed., 2011, 50, 7797–7802 CrossRef CAS PubMed.
- M. Arai, J. Antibiot., 1960, 13, 46–50 CAS.
- M. Arai, J. Antibiot., 1960, 13, 51–56 CAS.
- M. Gui, M. X. Zhang, W. H. Wu and P. Sun, Molecules, 2019, 24, 3840 CrossRef CAS PubMed.
- M. G. Nair, A. Chandra, D. L. Thorogood, E. Ammermann, N. Walker and K. Kiehs, J. Agric. Food Chem., 1994, 42, 2308–2310 CrossRef CAS.
- A. Fang, G. K. Wong and A. L. Demain, J. Antibiot., 2000, 53, 158–162 CrossRef CAS PubMed.
- C. Wu, Y. Tan, M. Gan, Y. Wang, Y. Guan, X. Hu, H. Zhou, X. Shang, X. You, Z. Yang and C. Xiao, J. Nat. Prod., 2013, 76, 2153–2157 CrossRef CAS PubMed.
- J. Ji, K. Wang, X. Meng, H. Zhong, X. Li, H. Zhao, G. Xie, Y. Xie, X. Wang and X. Zhu, Cancers, 2022, 14, 5812 CrossRef CAS PubMed.
- K. Toshima, K. Tatsuta and M. Kinoshita, Tetrahedron Lett., 1986, 27, 4741–4744 CrossRef CAS.
- D. Seebach, H. F. Chow, R. Jackson, K. Lawson, M. Sutter, S. Thaisrivongs and J. Zimmermann, J. Am. Chem. Soc., 1985, 107, 5292–5293 CrossRef CAS.
- H. Nakamura, K. Arata, T. Wakamatsu, Y. Ban and M. Shibasaki, Chem. Pharm. Bull., 1990, 38, 2435–2441 CrossRef CAS.
- D. A. Evans and D. M. Fitch, J. Org. Chem., 1997, 62, 454–455 CrossRef CAS PubMed.
- S. F. Haydock, T. Mironenko, H. I. Ghoorahoo and P. F. Leadlay, J. Biotechnol., 2004, 113, 55–68 CrossRef CAS PubMed.
- Y. Zhou, P. Prediger, L. C. Dias, A. C. Murphy and P. F. Leadlay, Angew. Chem., 2015, 127, 5321–5324 CrossRef PubMed.
- Z. Yang, Y. Qiao, N. C. Konakalla, E. Strobech, P. Harris, G. Peschel, M. Agler-Rosenbaum, T. Weber, E. Andreasson and L. Ding, Nat. Commun., 2023, 14, 7398 CrossRef PubMed.
- H. Zhang, Y. Wang, J. Wu, K. Skalina and B. A. Pfeifer, Chem. Biol., 2010, 17, 1232–1240 CrossRef CAS PubMed.
- Q. Xue, G. Ashley, C. R. Hutchinson and D. V. Santi, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11740–11745 CrossRef CAS PubMed.
- J. W. Westley, C. M. Liu, R. H. Evans and J. F. Blount, J. Antibiot., 1979, 32, 874–877 CrossRef CAS PubMed.
- H. J. Lacey, T. J. Booth, D. Vuong, P. J. Rutledge, E. Lacey, Y. H. Chooi and A. M. Piggott, J. Antibiot., 2020, 73, 756–765 CrossRef CAS PubMed.
- Z. Ju, W. Zhou, H. A. Alharbi, D. C. Howell and T. Mahmud, ACS Chem. Biol., 2021, 16, 2641–2650 CrossRef CAS PubMed.
- C. P. Bold and K.-H. Altmann, Tetrahedron, 2024, 156, 133908 CrossRef CAS.
- L. Lizzadro, O. Spiess, S. Reinecke, M. Stadler and D. Schinzer, Molecules, 2024, 29, 1123 CrossRef CAS PubMed.
- L. Lizzadro, O. Spiess, W. Collisi, M. Stadler and D. Schinzer, ChemBioChem, 2022, 23, e202200458 CrossRef CAS PubMed.
- W. Huang, M. Ye, L. R. Zhang, Q. D. Wu, M. Zhang, J. H. Xu and W. Zheng, Mol. Cancer, 2014, 13, 150 CrossRef PubMed.
- W. Huang, Q. D. Wu, M. Zhang, Y. L. Kong, P. R. Cao, W. Zheng, J. H. Xu and M. Ye, Cancer Lett., 2015, 356, 862–871 CrossRef CAS PubMed.
- W. Zhou, P. Posri and T. Mahmud, ACS Chem. Biol., 2021, 16, 270–276 CrossRef CAS PubMed.
- W. Zhang, M. Yang, W. Li, L. Zhou, Y. Shen, S. P. Wang, J. M. Gao, H. W. Lin, J. Qi and Y. Zhou, J. Agric. Food Chem., 2023, 71, 7459–7467 CrossRef CAS PubMed.
- A. Dramae, S. Nithithanasilp, W. Choowong, P. Rachtawee, S. Prabpai, P. Kongsaeree and P. Pittayakhajonwut, Tetrahedron, 2013, 69, 8205–8208 CrossRef CAS.
- E. Yiannakas, M. I. Grimes, J. T. Whitelegge, A. Furstner and A. N. Hulme, Angew. Chem., Int. Ed., 2021, 60, 18504–18508 CrossRef CAS PubMed.
- R. Jansen, H. Irschik, H. Reichenbach, V. Wray and G. Höfle, Liebigs Ann. Chem., 1994, 1994, 759–773 CrossRef.
- Y. A. Elnakady, F. Sasse, H. Lunsdorf and H. Reichenbach, Biochem. Pharmacol., 2004, 67, 927–935 CrossRef CAS PubMed.
- C. D. Hopkins and P. Wipf, Nat. Prod. Rep., 2009, 26, 585–601 RSC.
- R. Carvalho, R. Reid, N. Viswanathan, H. Gramajo and B. Julien, Gene, 2005, 359, 91–98 CrossRef CAS PubMed.
- P. Wipf and T. H. Graham, J. Am. Chem. Soc., 2004, 126, 15346–15347 CrossRef CAS PubMed.
- K. C. Nicolaou, G. Bellavance, M. Buchman and K. K. Pulukuri, J. Am. Chem. Soc., 2017, 139, 15636–15639 CrossRef CAS PubMed.
- M. Tsunakawa, O. Tenmyo, K. Tomita, N. Naruse, C. Kotake, T. Miyaki, M. Konishi and T. Oki, J. Antibiot., 1992, 45, 180–188 CrossRef CAS PubMed.
- H. Y. He, H. X. Pan, L. F. Wu, B. B. Zhang, H. B. Chai, W. Liu and G. L. Tang, Chem. Biol., 2012, 19, 1313–1323 CrossRef CAS PubMed.
- L. F. Wu, H. Y. He, H. X. Pan, L. Han, R. Wang and G. L. Tang, Org. Lett., 2014, 16, 1578–1581 CrossRef CAS PubMed.
- Z. Tian, P. Sun, Y. Yan, Z. Wu, Q. Zheng, S. Zhou, H. Zhang, F. Yu, X. Jia, D. Chen, A. Mandi, T. Kurtan and W. Liu, Nat. Chem. Biol., 2015, 11, 259–265 CrossRef CAS PubMed.
- W. R. Roush and D. A. Barda, Org. Lett., 2002, 4, 1539–1542 CrossRef CAS PubMed.
- W. R. Roush, C. Limberakis, R. K. Kunz and D. A. Barda, Org. Lett., 2002, 4, 1543–1546 CrossRef CAS PubMed.
- T. K. Trullinger, J. Qi and W. R. Roush, J. Org. Chem., 2006, 71, 6915–6922 CrossRef CAS PubMed.
- G. Schlingmann, L. Milne and G. T. Carter, Tetrahedron, 2002, 58, 6825–6835 CrossRef CAS.
- V. Ruanglek, S. Chokpaiboon, N. Rattanaphan, S. Madla, P. Auncharoen, T. Bunyapaiboonsri and M. Isaka, J. Antibiot., 2007, 60, 748–751 CrossRef CAS PubMed.
- M. Chinworrungsee, P. Kittakoop, M. Isaka, P. Maithip, S. Supothina and Y. Thebtaranonth, J. Nat. Prod., 2004, 67, 689–692 CrossRef CAS PubMed.
- P. Wattana-amorn, P. Juthaphan, M. Sirikamonsil, A. Sriboonlert, T. J. Simpson and N. Kongkathip, J. Nat. Prod., 2013, 76, 1235–1237 CrossRef CAS PubMed.
- W. Bunnak, P. Wonnapinij, A. Sriboonlert, C. M. Lazarus and P. Wattana-Amorn, Org. Biomol. Chem., 2019, 17, 374–379 RSC.
- W. Pache and H. Zähner, Arch. Mikrobiol., 1969, 67, 156–165 CAS.
- J. Kohno, T. Kawahata, T. Otake, M. Morimoto, H. Mori, N. Ueba, M. Nishio, A. Kinumaki, S. Komatsubara and K. Kawashima, Biosci., Biotechnol., Biochem., 1996, 60, 1036–1037 CrossRef CAS PubMed.
- M. Perez, C. Crespo, C. Schleissner, P. Rodriguez, P. Zuniga and F. Reyes, J. Nat. Prod., 2009, 72, 2192–2194 CrossRef CAS PubMed.
- Y. Shimizu, Y. Ogasawara, A. Matsumoto and T. Dairi, J. Antibiot., 2018, 71, 968–970 CrossRef CAS PubMed.
- W. Moreira, D. B. Aziz and T. Dick, Front. Microbiol., 2016, 7, 199 Search PubMed.
- A. Cotto-Rosario, E. Y. D. Miller, F. G. Fumuso, J. A. Clement, M. J. Todd and R. M. O’Connor, Microorganisms, 2022, 10, 2260 CrossRef CAS PubMed.
- S. I. Elshahawi, A. E. Trindade-Silva, A. Hanora, A. W. Han, M. S. Flores, V. Vizzoni, C. G. Schrago, C. A. Soares, G. P. Concepcion, D. L. Distel, E. W. Schmidt and M. G. Haygood, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E295–304 CrossRef CAS PubMed.
- F. Surup, D. Chauhan, J. Niggemann, E. Bartok, J. Herrmann, M. Keck, W. Zander, M. Stadler, V. Hornung and R. Muller, ACS Chem. Biol., 2018, 13, 2981–2988 CrossRef CAS PubMed.
- H. Irschik, D. Schummer, K. Gerth, G. Hofle and H. Reichenbach, J. Antibiot., 1995, 48, 26–30 CrossRef CAS PubMed.
- T. S. S. Chen, C.-J. Chang and H. G. Floss, J. Am. Chem. Soc., 1981, 103, 4565–4568 CrossRef CAS.
- T. S. S. Chen, C. J. Chang and H. G. Floss, J. Org. Chem., 1981, 46, 2661–2665 CrossRef CAS.
- J. C. Yang, R. Madupu, A. S. Durkin, N. A. Ekborg, C. S. Pedamallu, J. B. Hostetler, D. Radune, B. S. Toms, B. Henrissat, P. M. Coutinho, S. Schwarz, L. Field, A. E. Trindade-Silva, C. A. Soares, S. Elshahawi, A. Hanora, E. W. Schmidt, M. G. Haygood, J. Posfai, J. Benner, C. Madinger, J. Nove, B. Anton, K. Chaudhary, J. Foster, A. Holman, S. Kumar, P. A. Lessard, Y. A. Luyten, B. Slatko, N. Wood, B. Wu, M. Teplitski, J. D. Mougous, N. Ward, J. A. Eisen, J. H. Badger and D. L. Distel, PLoS One, 2009, 4, e6085 CrossRef PubMed.
- S. I. Elshahawi, A. E. Trindade-Silva, A. Hanora, A. W. Han, M. S. Flores, V. Vizzoni, C. G. Schrago, C. A. Soares, G. P. Concepcion, D. L. Distel, E. W. Schmidt and M. G. Haygood, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E295–E304 CrossRef CAS PubMed.
- M. A. Avery, S. C. Choudhry, O. P. Dhingra, B. D. Gray, M. C. Kang, S. C. Kuo, T. R. Vedananda, J. D. White and A. J. Whittle, Org. Biomol. Chem., 2014, 12, 9116–9132 RSC.
- H. J. Kwon, W. C. Smith, A. J. Scharon, S. H. Hwang, M. J. Kurth and B. Shen, Science, 2002, 297, 1327–1330 CrossRef CAS PubMed.
- H. J. Kwon, W. C. Smith, L. Xiang and B. Shen, J. Am. Chem. Soc., 2001, 123, 3385–3386 CrossRef CAS PubMed.
- Y. Wu and Y. P. Sun, Org. Lett., 2006, 8, 2831–2834 CrossRef CAS PubMed.
- Y. Rebets, E. Brotz, N. Manderscheid, B. Tokovenko, M. Myronovskyi, P. Metz, L. Petzke and A. Luzhetskyy, Angew. Chem., Int. Ed., 2015, 54, 2280–2284 CrossRef CAS PubMed.
- P. A. McCann and B. M. Pogell, J. Antibiot., 1979, 32, 673–678 CrossRef CAS PubMed.
- M. Natsume, J. Tazawa, K. Yagi, H. Abe, S. Kondo and S. Marumo, J. Antibiot., 1995, 48, 1159–1164 CrossRef CAS PubMed.
- B. M. Pogell, Cell. Mol. Biol., 1998, 44, 461–463 CAS.
- M. Natsume, S. Kondo and S. Marumo, J. Chem. Soc., Chem. Commun., 1989, 1911–1913, 10.1039/c39890001911.
- L. Glaser, M. Kuhl, J. Stegmuller, C. Ruckert, M. Myronovskyi, J. Kalinowski, A. Luzhetskyy and C. Wittmann, Microb. Cell Fact., 2021, 20, 111 CrossRef PubMed.
- M. Hashimoto, H. Komatsu, I. Kozone, H. Kawaide, H. Abe and M. Natsume, Biosci., Biotechnol., Biochem., 2005, 69, 315–320 CrossRef CAS PubMed.
- G. B. Ren and Y. Wu, Org. Lett., 2009, 11, 5638–5641 CrossRef CAS PubMed.
- O. Germay, N. Kumar and E. J. Thomas, Tetrahedron Lett., 2001, 42, 4969–4974 CrossRef CAS.
- E. J. Jeong, E. J. Kang, L. T. Sung, S. K. Hong and E. Lee, J. Am. Chem. Soc., 2002, 124, 14655–14662 CrossRef CAS PubMed.
- P. Fischer, A. B. Garcia Segovia, M. Gruner and P. Metz, Angew. Chem., Int. Ed., 2005, 44, 6231–6234 CrossRef CAS PubMed.
- G. Hanquet, X. Salom-Roig and S. Lanners, Chimia, 2016, 70, 20–28 CrossRef CAS PubMed.
- P. Fischer, M. Gruner, A. Jager, O. Kataeva and P. Metz, Chemistry, 2011, 17, 13334–13340 CrossRef CAS PubMed.
- P. A. Bartlett, J. D. Meadows and E. Ottow, J. Am. Chem. Soc., 1984, 106, 5304–5311 CrossRef CAS.
- J. Xie and N. Bogliotti, Chem. Rev., 2014, 114, 7678–7739 CrossRef CAS PubMed.
- T. Golakoti, W. Y. Yoshida, S. Chaganty and R. E. Moore, J. Nat. Prod., 2001, 64, 54–59 CrossRef CAS PubMed.
- F. Kopp, C. Mahlert, J. Grunewald and M. A. Marahiel, J. Am. Chem. Soc., 2006, 128, 16478–16479 CrossRef CAS PubMed.
- B. S. Evans, I. Ntai, Y. Chen, S. J. Robinson and N. L. Kelleher, J. Am. Chem. Soc., 2011, 133, 7316–7319 CrossRef CAS PubMed.
- A. Krunic, A. Vallat, S. Mo, D. D. Lantvit, S. M. Swanson and J. Orjala, J. Nat. Prod., 2010, 73, 1927–1932 CrossRef CAS PubMed.
- T. A. Wilson, R. J. Tokarski, 2nd, P. Sullivan, R. M. Demoret, J. Orjala, L. H. Rakotondraibe and J. R. Fuchs, J. Nat. Prod., 2018, 81, 534–542 CrossRef CAS PubMed.
- L. R. Malins, J. N. deGruyter, K. J. Robbins, P. M. Scola, M. D. Eastgate, M. R. Ghadiri and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 5233–5241 CrossRef CAS PubMed.
- Y. Igarashi, Y. Kim, Y. In, T. Ishida, Y. Kan, T. Fujita, T. Iwashita, H. Tabata, H. Onaka and T. Furumai, Org. Lett., 2010, 12, 3402–3405 CrossRef CAS PubMed.
- H. Komaki, N. Ichikawa, A. Oguchi, M. Hamada, E. Harunari, S. Kodani, N. Fujita and Y. Igarashi, Stand. Genomic Sci., 2016, 11, 85 CrossRef PubMed.
- H. J. Zhu, B. Zhang, L. Wang, W. Wang, S. H. Liu, Y. Igarashi, G. Bashiri, R. X. Tan and H. M. Ge, J. Am. Chem. Soc., 2021, 143, 4751–4757 CrossRef CAS PubMed.
- H. J. Zhu, B. Zhang, W. Wei, S. H. Liu, L. Xiang, J. Zhu, R. H. Jiao, Y. Igarashi, G. Bashiri, Y. Liang, R. X. Tan and H. M. Ge, Nat. Commun., 2022, 13, 4499 CrossRef CAS PubMed.
- G. K. Nguyen, A. Kam, S. Loo, A. E. Jansson, L. X. Pan and J. P. Tam, J. Am. Chem. Soc., 2015, 137, 15398–15401 CrossRef CAS PubMed.
- C. J. Podracky, C. An, A. DeSousa, B. M. Dorr, D. M. Walsh and D. R. Liu, Nat. Chem. Biol., 2021, 17, 317–325 CrossRef CAS PubMed.
- X. Y. Hemu, X. H. Zhang and J. P. Tam, Org. Lett., 2019, 21, 2029–2032 CrossRef CAS PubMed.
- G. K. T. Nguyen, X. Hemu, J. P. Quek and J. P. Tam, Angew. Chem., Int. Ed., 2016, 55, 12802–12806 CrossRef CAS PubMed.
- X. Gao, S. W. Haynes, B. D. Ames, P. Wang, L. P. Vien, C. T. Walsh and Y. Tang, Nat. Chem. Biol., 2012, 8, 823–830 CrossRef CAS PubMed.
- J. Zhang, N. Liu, R. A. Cacho, Z. Gong, Z. Liu, W. Qin, C. Tang, Y. Tang and J. Zhou, Nat. Chem. Biol., 2016, 12, 1001–1003 CrossRef CAS PubMed.
- T. A. Keating, C. G. Marshall, C. T. Walsh and A. E. Keating, Nat. Struct. Biol., 2002, 9, 522–526 CAS.
- S. A. Samel, G. Schoenafinger, T. A. Knappe, M. A. Marahiel and L. O. Essen, Structure, 2007, 15, 781–792 CrossRef CAS PubMed.
- T. Izore, Y. T. Candace Ho, J. A. Kaczmarski, A. Gavriilidou, K. H. Chow, D. L. Steer, R. J. A. Goode, R. B. Schittenhelm, J. Tailhades, M. Tosin, G. L. Challis, E. H. Krenske, N. Ziemert, C. J. Jackson and M. J. Cryle, Nat. Commun., 2021, 12, 2511 CrossRef CAS PubMed.
- A. Tanovic, S. A. Samel, L. O. Essen and M. A. Marahiel, Science, 2008, 321, 659–663 CrossRef CAS PubMed.
- L. Zhang, C. Wang, K. Chen, W. Zhong, Y. Xu and I. Molnar, Nat. Prod. Rep., 2023, 40, 62–88 RSC.
- S. Sivanathan and J. Scherkenbeck, Molecules, 2014, 19, 12368–12420 CrossRef PubMed.
- F. Kopp and M. A. Marahiel, Nat. Prod. Rep., 2007, 24, 735–749 RSC.
- R. Sussmuth, J. Muller, H. von Dohren and I. Molnar, Nat. Prod. Rep., 2011, 28, 99–124 RSC.
- H. D. Mootz, D. Schwarzer and M. A. Marahiel, ChemBioChem, 2002, 3, 490–504 CrossRef CAS PubMed.
- S. M. Batiste, D. J. Blackwell, K. Kim, D. O. Kryshtal, N. Gomez-Hurtado, R. T. Rebbeck, R. L. Cornea, J. N. Johnston and B. C. Knollmann, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4810–4815 CrossRef CAS PubMed.
- L. Richter, F. Wanka, S. Boecker, D. Storm, T. Kurt, O. Vural, R. Sussmuth and V. Meyer, Fungal Biol. Biotechnol., 2014, 1, 4 CrossRef PubMed.
- M. Urbaniak, A. Waskiewicz and L. Stepien, Toxins, 2020, 12, 765 CrossRef CAS PubMed.
- E. Dorschner and H. Lardy, Antimicrob. Agents Chemother., 1968, 8, 11–14 CAS.
- R. L. Hamill, C. E. Higgens, H. E. Boaz and M. Gorman, Tetrahedron Lett., 1969, 10, 4255–4258 CrossRef.
- A. Logrieco, A. Moretti, G. Castella, M. Kostecki, P. Golinski, A. Ritieni and J. Chelkowski, Appl. Environ. Microbiol., 1998, 64, 3084–3088 CrossRef CAS PubMed.
- A. Harder, L. Holden-Dye, R. Walker and F. Wunderlich, Parasitol. Res., 2005, 97(Suppl 1), S1–S10 CrossRef PubMed.
- Y. Xu, R. Orozco, E. M. Wijeratne, A. A. Gunatilaka, S. P. Stock and I. Molnar, Chem. Biol., 2008, 15, 898–907 CrossRef CAS PubMed.
- C. Steiniger, S. Hoffmann and R. D. Sussmuth, Cell Chem. Biol., 2019, 26, 1526–1534 e1522 CrossRef CAS PubMed.
- W. Weckwerth, K. Miyamoto, K. Iinuma, M. Krause, M. Glinski, T. Storm, G. Bonse, H. Kleinkauf and R. Zocher, J. Biol. Chem., 2000, 275, 17909–17915 CrossRef CAS PubMed.
- J. Muller, S. C. Feifel, T. Schmiederer, R. Zocher and R. D. Sussmuth, ChemBioChem, 2009, 10, 323–328 CrossRef PubMed.
- A. Prosperini, H. Berrada, M. J. Ruiz, F. Caloni, T. Coccini, L. J. Spicer, M. C. Perego and A. Lafranconi, Front. Public Health, 2017, 5, 304 CrossRef PubMed.
- S. M. Batiste and J. N. Johnston, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14893–14897 CrossRef CAS PubMed.
- S. Monma, T. Sunazuka, K. Nagai, T. Arai, K. Shiomi, R. Matsui and S. Omura, Org. Lett., 2006, 8, 5601–5604 CrossRef CAS PubMed.
- T. Ohshiro, D. Matsuda, T. Kazuhiro, R. Uchida, K. Nonaka, R. Masuma and H. Tomoda, J. Antibiot., 2012, 65, 255–262 CrossRef CAS PubMed.
- K. Shiomi, R. Matsui, A. Kakei, Y. Yamaguchi, R. Masuma, H. Hatano, N. Arai, M. Isozaki, H. Tanaka, S. Kobayashi, A. Turberg and S. Omura, J. Antibiot., 2010, 63, 77–82 CrossRef CAS PubMed.
- D. X. Hu, M. Bielitza, P. Koos and S. V. Ley, Tetrahedron Lett., 2012, 53, 4077–4079 CrossRef CAS.
- M. M. Shemyakin, Y. A. Ovchinnikov, A. A. Kiryushkin and V. T. Ivanov, Tetrahedron Lett., 1963, 4, 885–890 CrossRef.
- A. D. Borthwick, Chem. Rev., 2012, 112, 3641–3716 CrossRef CAS PubMed.
- M. Teixidó, E. Zurita, M. Malakoutikhah, T. Tarragó and E. Giralt, J. Am. Chem. Soc., 2007, 129, 11802–11813 CrossRef PubMed.
- M. Millward, P. Mainwaring, A. Mita, K. Federico, G. K. Lloyd, N. Reddinger, S. Nawrocki, M. Mita and M. A. Spear, Invest. New Drugs, 2012, 30, 1065–1073 CrossRef CAS PubMed.
- Y. Yamazaki, K. Tanaka, B. Nicholson, G. Deyanat-Yazdi, B. Potts, T. Yoshida, A. Oda, T. Kitagawa, S. Orikasa, Y. Kiso, H. Yasui, M. Akamatsu, T. Chinen, T. Usui, Y. Shinozaki, F. Yakushiji, B. R. Miller, S. Neuteboom, M. Palladino, K. Kanoh, G. K. Lloyd and Y. Hayashi, J. Med. Chem., 2012, 55, 1056–1071 CrossRef CAS PubMed.
- M. Nishida, Y. Mine, T. Matsubara, S. Goto and S. Kuwahara, J. Antibiot., 1972, 25, 582–593 CrossRef CAS PubMed.
- P. Belin, M. Moutiez, S. Lautru, J. Seguin, J. L. Pernodet and M. Gondry, Nat. Prod. Rep., 2012, 29, 961–979 RSC.
- M. Gondry, L. Sauguet, P. Belin, R. Thai, R. Amouroux, C. Tellier, K. Tuphile, M. Jacquet, S. Braud, M. Courcon, C. Masson, S. Dubois, S. Lautru, A. Lecoq, S. Hashimoto, R. Genet and J. L. Pernodet, Nat. Chem. Biol., 2009, 5, 414–420 CrossRef CAS PubMed.
- M. W. Vetting, S. S. Hegde and J. S. Blanchard, Nat. Chem. Biol., 2010, 6, 797–799 CrossRef CAS PubMed.
- M. J. Cryle, S. G. Bell and I. Schlichting, Biochemistry, 2010, 49, 7282–7296 CrossRef CAS PubMed.
- T. W. Giessen, A. M. von Tesmar and M. A. Marahiel, Biochemistry, 2013, 52, 4274–4283 CrossRef CAS PubMed.
- J. Liu, X. Xie and S. M. Li, Angew. Chem., Int. Ed., 2019, 58, 11534–11540 CrossRef CAS PubMed.
- N. M. Vior, R. Lacret, G. Chandra, S. Dorai-Raj, M. Trick and A. W. Truman, Appl. Environ. Microbiol., 2018, 84, e02828–e02817 CrossRef CAS PubMed.
- T. Yao, J. Liu, E. Jin, Z. Liu, H. Li, Q. Che, T. Zhu, D. Li and W. Li, iScience, 2020, 23, 101323 CrossRef CAS PubMed.
- M. Moutiez, E. Schmitt, J. Seguin, R. Thai, E. Favry, P. Belin, Y. Mechulam and M. Gondry, Nat. Commun., 2014, 5, 5141 CrossRef CAS PubMed.
- Y. Tingting, Z. Jingxing, L. Jing, L. Huayue and L. Wenli, Chin. J. Org. Chem., 2019, 39, 328–338 CrossRef.
- J. F. Gonzalez, I. Ortin, E. de la Cuesta and J. C. Menendez, Chem. Soc. Rev., 2012, 41, 6902–6915 RSC.
- J. Fan, H. Ran, P. L. Wei, Y. Li, H. Liu, S. M. Li, Y. Hu and W. B. Yin, Angew. Chem., Int. Ed., 2023, 62, e202217212 CrossRef CAS PubMed.
- H. Liu, J. Fan, P. Zhang, Y. Hu, X. Liu, S. M. Li and W. B. Yin, Chem. Sci., 2021, 12, 4132–4138 RSC.
- Y. Tsunematsu, N. Maeda, M. Yokoyama, P. Chankhamjon, K. Watanabe, K. Scherlach and C. Hertweck, Angew. Chem., Int. Ed., 2018, 57, 14051–14054 CrossRef CAS PubMed.
- C. J. Guo, H. H. Yeh, Y. M. Chiang, J. F. Sanchez, S. L. Chang, K. S. Bruno and C. C. Wang, J. Am. Chem. Soc., 2013, 135, 7205–7213 CrossRef CAS PubMed.
- P. D. Sherkhane, R. Bansal, K. Banerjee, S. Chatterjee, D. Oulkar, P. Jain, L. Rosenfelder, S. Elgavish, B. A. Horwitz and P. K. Mukherjee, ChemistrySelect, 2017, 2, 3347–3352 CrossRef CAS.
- M. R. Ghiffary, C. P. S. Prabowo, K. Sharma, Y. Yan, S. Y. Lee and H. U. Kim, ACS Sustainable Chem. Eng., 2021, 9, 6613–6622 CrossRef CAS.
- B. Pang, Y. Chen, F. Gan, C. Yan, L. Jin, J. W. Gin, C. J. Petzold and J. D. Keasling, J. Am. Chem. Soc., 2020, 142, 10931–10935 CrossRef CAS PubMed.
- C. Huang, H. Cui, H. Ren and H. Zhao, JACS Au, 2023, 3, 195–203 CrossRef CAS PubMed.
- B. S. Moore, J. L. Chen, G. M. L. Patterson, R. E. Moore, L. S. Brinen, Y. Kato and J. Clardy, J. Am. Chem. Soc., 1990, 112, 4061–4063 CrossRef CAS.
- B. S. Moore, J.-L. Chen, G. M. L. Patterson and R. E. Moore, Tetrahedron, 1992, 48, 3001–3006 CrossRef CAS.
- S. Luo, H. S. Kang, A. Krunic, G. E. Chlipala, G. Cai, W. L. Chen, S. G. Franzblau, S. M. Swanson and J. Orjala, Tetrahedron Lett., 2014, 55, 686–689 CrossRef CAS PubMed.
- H. T. Bui, R. Jansen, H. T. Pham and S. Mundt, J. Nat. Prod., 2007, 70, 499–503 CrossRef CAS PubMed.
- S. C. Bobzin and R. E. Moore, Tetrahedron, 1993, 49, 7615–7626 CrossRef CAS.
- T. P. Martins, C. Rouger, N. R. Glasser, S. Freitas, N. B. de Fraissinette, E. P. Balskus, D. Tasdemir and P. N. Leao, Nat. Prod. Rep., 2019, 36, 1437–1461 RSC.
- H. Nakamura, E. E. Schultz and E. P. Balskus, Nat. Chem. Biol., 2017, 13, 916–921 CrossRef CAS PubMed.
- K. C. Nicolaou, Y. P. Sun, H. Korman and D. Sarlah, Angew. Chem., Int. Ed., 2010, 49, 5875–5878 CrossRef CAS PubMed.
- A. N. Kim, A. Ngamnithiporn, E. Du and B. M. Stoltz, Chem. Rev., 2023, 123, 9447–9496 CrossRef CAS PubMed.
- A. R. Battersby, R. B. Herbert and F. Šantavý, Chem. Commun., 1965, 415–416 RSC.
- J. Wang and G. Evano, Org. Lett., 2016, 18, 3542–3545 CrossRef CAS PubMed.
- O. Wintersteiner and J. D. Dutcher, Science, 1943, 97, 467–470 CrossRef CAS PubMed.
- W. C. Bowman, Br. J. Pharmacol., 2006, 147(Suppl 1), S277–S286 CAS.
- S. Panjikar, J. Stoeckigt, S. O'Connor and H. Warzecha, Nat. Prod. Rep., 2012, 29, 1176–1200 RSC.
- Y.-J. Xu, S.-G. Cao, X.-H. Wu, Y.-H. Lai, B. Tan, J. Pereira, S. Goh, G. Venkatraman, L. J. Harrison and K.-Y. Sim, Tetrahedron Lett., 1998, 39, 9103–9106 CrossRef CAS.
- S. Feng, Y. Jiang, J. Li, S. Qiu and T. Chen, Nat. Prod. Res., 2014, 28, 81–85 CrossRef CAS PubMed.
- K. D. Reichl, M. J. Smith, M. K. Song, R. P. Johnson and J. A. Porco, Jr., J. Am. Chem. Soc., 2017, 139, 14053–14056 CrossRef CAS PubMed.
- M. J. Smith, K. D. Reichl, R. A. Escobar, T. J. Heavey, D. F. Coker, S. E. Schaus and J. A. Porco, Jr., J. Am. Chem. Soc., 2019, 141, 148–153 CrossRef CAS PubMed.
- A. R. Pereira, C. F. McCue and W. H. Gerwick, J. Nat. Prod., 2010, 73, 217–220 CrossRef CAS PubMed.
- G. C. Tay, M. R. Gesinski and S. D. Rychnovsky, Org. Lett., 2013, 15, 4536–4539 CrossRef CAS PubMed.
- H. Kim and J. Hong, Org. Lett., 2010, 12, 2880–2883 CrossRef CAS PubMed.
- W. Che, Y. Z. Li, J. C. Liu, S. F. Zhu, J. H. Xie and Q. L. Zhou, Org. Lett., 2019, 21, 2369–2373 CrossRef CAS PubMed.
- A. R. Waldeck and M. J. Krische, Angew. Chem., Int. Ed., 2013, 52, 4470–4473 CrossRef CAS PubMed.
- M. R. Rao and D. J. Faulkner, J. Nat. Prod., 2002, 65, 386–388 CrossRef CAS PubMed.
- J. B. Baker, H. Kim and J. Hong, Tetrahedron Lett., 2015, 56, 3120–3122 CrossRef CAS PubMed.
- G. Peh and P. E. Floreancig, Org. Lett., 2012, 14, 5614–5617 CrossRef CAS PubMed.
- C. S. Barry, J. D. Elsworth, P. T. Seden, N. Bushby, J. R. Harding, R. W. Alder and C. L. Willis, Org. Lett., 2006, 8, 3319–3322 CrossRef CAS PubMed.
- J. M. Cabrera and M. J. Krische, Angew. Chem., Int. Ed., 2019, 58, 10718–10722 CrossRef CAS PubMed.
- K. Jadhav, K. Ahir, S. Desai, S. Desai and S. Acharya, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2022, 1213, 123533 CrossRef CAS PubMed.
- G. Rall, T. Smalberger, H. De Waal and R. Arndt, Tetrahedron Lett., 1967, 8, 3465–3469 CrossRef.
- H. Rapoport and H. D. Baldridge, Jr., J. Am. Chem. Soc., 1951, 73, 343–346 CrossRef CAS.
- V. Zunjar, R. P. Dash, M. Jivrajani, B. Trivedi and M. Nivsarkar, J. Ethnopharmacol., 2016, 181, 20–25 CrossRef CAS PubMed.
- T. Sato, S. Aoyagi and C. Kibayashi, Org. Lett., 2003, 5, 3839–3842 CrossRef CAS PubMed.
- R. C. R. Jala, S. Vudhgiri and C. G. Kumar, Carbohydr. Res., 2022, 516, 108556 CrossRef CAS PubMed.
- A. M. Abdel-Mawgoud and G. Stephanopoulos, Synth. Syst. Biotechnol., 2018, 3, 3–19 CrossRef PubMed.
- A. Furstner, J. Ruiz-Caro, H. Prinz and H. Waldmann, J. Org. Chem., 2004, 69, 459–467 CrossRef PubMed.
- J. Qian-Cutrone, T. Ueki, S. Huang, K. A. Mookhtiar, R. Ezekiel, S. S. Kalinowski, K. S. Brown, J. Golik, S. Lowe, D. M. Pirnik, R. Hugill, J. A. Veitch, S. E. Klohr, J. L. Whitney and S. P. Manly, J. Antibiot., 1999, 52, 245–255 CrossRef CAS PubMed.
- M. Tsunakawa, N. Komiyama, O. Tenmyo, K. Tomita, K. Kawano, C. Kotake, M. Konishi and T. Oki, J. Antibiot., 1992, 45, 1467–1471 CrossRef CAS PubMed.
- A. Furstner, J. Mlynarski and M. Albert, J. Am. Chem. Soc., 2002, 124, 10274–10275 CrossRef PubMed.
- A. Furstner, M. Albert, J. Mlynarski, M. Matheu and E. DeClercq, J. Am. Chem. Soc., 2003, 125, 13132–13142 CrossRef PubMed.
- A. Furstner, M. Albert, J. Mlynarski and M. Matheu, J. Am. Chem. Soc., 2002, 124, 1168–1169 CrossRef PubMed.
- S. Takahashi, M. Hosoya, H. Koshino and T. Nakata, Org. Lett., 2003, 5, 1555–1558 CrossRef CAS PubMed.
- K. Yokomizo, Y. Miyamoto, K. Nagao, E. Kumagae, E. S. Habib, K. Suzuki, S. Harada and M. Uyeda, J. Antibiot., 1998, 51, 1035–1039 CrossRef CAS PubMed.
- M. Uyeda, Actinomycetologica, 2003, 17, 57–66 CrossRef CAS.
- E. S. Habib, K. Yokomizo, K. Murata and M. Uyeda, J. Antibiot., 2000, 53, 1420–1423 CrossRef CAS PubMed.
- T. Ohtsuka, Y. Itezono, N. Nakayama, M. Kurano, N. Nakada, H. Tanaka, S. Sawairi, K. Yokose and H. Seto, Tetrahedron Lett., 1992, 33, 2705–2708 CrossRef CAS.
- M. Wietz, M. Mansson, J. S. Bowman, N. Blom, Y. Ng and L. Gram, Appl. Environ. Microbiol., 2012, 78, 2039–2042 CrossRef CAS PubMed.
- D. M. Garcia, H. Yamada, S. Hatakeyama and M. Nishizawa, Tetrahedron Lett., 1994, 35, 3325–3328 CrossRef CAS.
- V. Marti-Centelles, M. D. Pandey, M. I. Burguete and S. V. Luis, Chem. Rev., 2015, 115, 8736–8834 CrossRef CAS PubMed.
- A. Gradillas and J. Perez-Castells, Angew. Chem., Int. Ed., 2006, 45, 6086–6101 CrossRef CAS PubMed.
- G. P. Dinos, Br. J. Pharmacol., 2017, 174, 2967–2983 CrossRef CAS PubMed.
- Q. Su, A. B. Beeler, E. Lobkovsky, J. A. Porco, Jr. and J. S. Panek, Org. Lett., 2003, 5, 2149–2152 CrossRef CAS PubMed.
- A. B. Beeler, D. E. Acquilano, Q. Su, F. Yan, B. L. Roth, J. S. Panek and J. A. Porco, Jr., J. Comb. Chem., 2005, 7, 673–681 CrossRef CAS PubMed.
- M. de Leseleuc and S. K. Collins, Chem. Commun., 2015, 51, 10471–10474 RSC.
- Q. Zhang, X. Cui, S. Lin, J. Zhou and G. Yuan, Org. Lett., 2012, 14, 6126–6129 CrossRef CAS PubMed.
- X. Miao, A. Paikar, B. Lerner, Y. Diskin-Posner, G. Shmul and S. N. Semenov, Angew. Chem., Int. Ed., 2021, 60, 20366–20375 CrossRef CAS PubMed.
- L. A. Wessjohann, D. G. Rivera and O. E. Vercillo, Chem. Rev., 2009, 109, 796–814 CrossRef CAS PubMed.
- C. D. Hein, X. M. Liu and D. Wang, Pharm. Res., 2008, 25, 2216–2230 CrossRef CAS PubMed.
- G. Chouhan and K. James, Org. Lett., 2011, 13, 2754–2757 CrossRef CAS PubMed.
- X. Yang, L. P. Beroske, J. Kemmink, D. T. Rijkers and R. M. Liskamp, Tetrahedron Lett., 2017, 58, 4542–4546 CrossRef CAS.
- R. Jagasia, J. M. Holub, M. Bollinger, K. Kirshenbaum and M. G. Finn, J. Org. Chem., 2009, 74, 2964–2974 CrossRef CAS PubMed.
- S. Punna, J. Kuzelka, Q. Wang and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2215–2220 CrossRef CAS PubMed.
- J. H. van Maarseveen, W. S. Horne and M. R. Ghadiri, Org. Lett., 2005, 7, 4503–4506 CrossRef CAS PubMed.
- K. D. Bodine, D. Y. Gin and M. S. Gin, J. Am. Chem. Soc., 2004, 126, 1638–1639 CrossRef CAS PubMed.
- N. Pietrzik, D. Schmollinger and T. Ziegler, Beilstein J. Org. Chem., 2008, 4, 30 Search PubMed.
- M. Menand, J. C. Blais, J. M. Valery and J. Xie, J. Org. Chem., 2006, 71, 3295–3298 CrossRef CAS PubMed.
- A. Bordes, A. Poveda, N. Fontelle, A. Arda, J. Guillard, Y. B. Ruan, J. Marrot, S. Imaeda, A. Kato, J. Desire, J. Xie, J. Jimenez-Barbero and Y. Bleriot, Carbohydr. Res., 2021, 501, 108258 CrossRef CAS PubMed.
- W. Fu, L. Wang, Z. Yang, J. S. Shen, F. Tang, J. Zhang and X. Cui, Chem. Commun., 2020, 56, 960–963 RSC.
- Q. Xiong, L. Xiao, X. Q. Dong and C. J. Wang, Org. Lett., 2022, 24, 2579–2584 CrossRef CAS PubMed.
- R. Emadi, A. Bahrami Nekoo, F. Molaverdi, Z. Khorsandi, R. Sheibani and H. Sadeghi-Aliabadi, RSC Adv., 2023, 13, 18715–18733 RSC.
- L. Mendive-Tapia, A. Bertran, J. García, G. Acosta, F. Albericio and R. Lavilla, Chem. – Eur. J., 2016, 22, 13114–13119 CrossRef CAS PubMed.
- J. Garcia-Lacuna, G. Dominguez and J. Perez-Castells, ChemSusChem, 2020, 13, 5138–5163 CrossRef CAS PubMed.
- K. E. Ylijoki and J. M. Stryker, Chem. Rev., 2013, 113, 2244–2266 CrossRef CAS PubMed.
- S. B. Needleman and M. Chang Kuo, Chem. Rev., 1962, 62, 405–431 CrossRef CAS.
- H. Oikawa and T. Tokiwano, Nat. Prod. Rep., 2004, 21, 321–352 RSC.
- K. Klas, S. Tsukamoto, D. H. Sherman and R. M. Williams, J. Org. Chem., 2015, 80, 11672–11685 CrossRef CAS PubMed.
- H. Wang, Y. Zou, M. Li, Z. Tang, J. Wang, Z. Tian, N. Strassner, Q. Yang, Q. Zheng, Y. Guo, W. Liu, L. Pan and K. N. Houk, Nat. Chem., 2023, 15, 177–184 CrossRef CAS PubMed.
- Q. Zheng, Y. Gong, Y. Guo, Z. Zhao, Z. Wu, Z. Zhou, D. Chen, L. Pan and W. Liu, Cell Chem. Biol., 2018, 25, 718–727 e713 CrossRef CAS PubMed.
- Z. Zhang, C. S. Jamieson, Y. L. Zhao, D. Li, M. Ohashi, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2019, 141, 5659–5663 CrossRef CAS PubMed.
- B. Briou, B. Ameduri and B. Boutevin, Chem. Soc. Rev., 2021, 50, 11055–11097 RSC.
- M. H. Cao, N. J. Green and S. Z. Xu, Org. Biomol. Chem., 2017, 15, 3105–3129 RSC.
- K. Takao, R. Munakata and K. Tadano, Chem. Rev., 2005, 105, 4779–4807 CrossRef CAS PubMed.
- G. Dominguez and J. Perez-Castells, Chem. Soc. Rev., 2011, 40, 3430–3444 RSC.
- P. J. Salveson, A. P. Moyer, M. Y. Said, G. Gökce, X. Li, A. Kang, H. Nguyen, A. K. Bera, P. M. Levine, G. Bhardwaj and D. Baker, Science, 2024, 384, 420–428 CrossRef CAS PubMed.
- V. Sarojini, A. J. Cameron, K. G. Varnava, W. A. Denny and G. Sanjayan, Chem. Rev., 2019, 119, 10318–10359 CrossRef CAS PubMed.
- A. L. Feldberg, F. Mayerthaler, J. Ruschenbaum, J. Kroger and H. D. Mootz, Angew. Chem., Int. Ed., 2024, e202317753, DOI:10.1002/anie.202317753.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cs00909a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |