
Amyloid inspired single amino acid (phenylalanine)-based supramolecular functional assemblies: from disease to device applications
Subrat
Vishwakarma
a,
Om Shanker
Tiwari
 b,
Ruchi
Shukla
a,
Ehud
Gazit
*bc and
Pandeeswar
Makam
*a
b,
Ruchi
Shukla
a,
Ehud
Gazit
*bc and
Pandeeswar
Makam
*a
aDepartment of Chemistry, Indian Institute of Technology (BHU), Varanasi, UP 221005, India. E-mail: pandeeswar.chy@itbhu.ac.in
bThe Shmunis School of Biomedicine and Cancer Research, The George S. Wise Faculty of Life Sciences, Tel Aviv University, Tel Aviv 6997801, Israel
cSagol School of Neuroscience, Tel Aviv University, Tel Aviv 6997801, Israel. E-mail: ehudga@tauex.tau.ac.il
First published on 25th November 2024
Abstract
In the evolving landscape of biomolecular supramolecular chemistry, recent studies on phenylalanine (Phe) have revealed important insights into the versatile nature of this essential aromatic amino acid. Phe can spontaneously self-assemble into fibrils with amyloid-like properties linked to the neurological disorder phenylketonuria (PKU). Apart from its pathological implications, Phe also displays complex phase behavior and can undergo structural changes in response to external stimuli. Its ability to co-assemble with other amino acids opens up new possibilities for studying biomolecular interactions. Furthermore, Phe's coordination with metal ions has led to the development of enzyme-mimicking catalytic systems for applications in organic chemistry, environmental monitoring, and healthcare. Research on L and D enantiomers of Phe, particularly on bio-MOFs, has highlighted their potential in advanced technologies, including bioelectronic devices. This review provides a comprehensive overview of the advancements in Phe-based supramolecular assemblies, emphasizing their interdisciplinary relevance. The Phe assemblies show great potential for future therapeutic and functional biomaterial developments, from disease treatments to innovations in bionanozymes and bioelectronics. This review presents a compelling case for the ongoing exploration of Phe's biomolecular supramolecular chemistry as a fundamental framework for developing sustainable and efficient methodologies across various scientific disciplines.
 Subrat Vishwakarma | Mr. Subrat Vishwakarma obtained his MSc degree from the Indian Institute of Technology (BHU), Department of Chemistry, India. After that, he joined as a PhD student under the supervision of Dr. Pandeeswar Makam, Assistant Professor, Department of Chemistry, Indian Institute of Technology (BHU), Varanasi, India. His research interests include extensively bio-inspired supramolecular chemistry, biomimetics, bionanozymes and bionanotechnology. |
 Om Shanker Tiwari | Dr Om Shanker Tiwari obtained his MSc and PhD degrees from the Indian Institute of Science Education and Research (IISER), Pune, India, in 2020 under the guidance of Prof. Krishna N. Ganesh. During his PhD research at the IISER Pune, he worked extensively to understand the assembly of amino acids, peptide nucleic acids, and diPhe-based peptides and their various applications. Subsequently, he joined Prof. Ehud Gazit's research group at Tel Aviv University, Israel as a post-doctoral researcher. His research interests include bio-inspired minimalistic peptides and metabolite supramolecular nanoassemblies for optical, mechanical, conducting, electrochemical, catalytic, and biological applications. |
 Ruchi Shukla | Ms. Ruchi Shukla obtained her MSc degree from the Department of Chemistry at Gorakhpur University, India. In 2022, she joined as a PhD student under the supervision of Dr. Pandeeswar Makam, Assistant Professor, Department of Chemistry, Indian Institute of Technology (BHU), India. Her research interests focus on biomolecular supramolecular chemistry and its applications. |
 Ehud Gazit | Prof. Ehud Gazit is a Professor and Endowed Chair at both the Faculties of Life Sciences and Engineering at Tel Aviv University. He earned his BSc (summa cum laude) from Tel Aviv University's Special University Program for Outstanding Students and his PhD (with distinction) from the Weizmann Institute of Science. After completing his post-doctoral studies at the Massachusetts Institute of Technology, he became a faculty member at Tel Aviv University in 2000. His research focuses on molecular self-assembly, bio-inspired materials, neurodegenerative diseases, and metabolic disorders. Professor Gazit has published over 350 articles and is the inventor of over 100 patents. He was recently named the 2023 International Solvay Chair in Chemistry. |
 Pandeeswar Makam | Dr Pandeeswar Makam is an assistant professor in the Department of Chemistry at the Indian Institute of Technology (BHU). He obtained his MS (2011) and PhD (2016) from the Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), India. He conducted postdoctoral studies (2016–2020) under the Centre for Nanoscience and Nanotechnology Post-Doctoral Fellowship program at Tel Aviv University, Israel. In 2020, he joined the Department of Chemistry at IIT (BHU) as an assistant professor. His research interests lie at the interface of chemistry, biology, and materials science, encompassing areas such as biomaterials chemistry, bionanozymes, bio-inspired supramolecular chemistry, biomimetics, bioelectronics and bionanotechnology. |
1. Introduction
Biomolecular supramolecular chemistry (BSC) is a branch of chemistry that studies the interactions and nanoassemblies of biomolecules, including proteins, nucleic acids, polysaccharides, and lipids.1–4 It utilizes various weak non-covalent interactions, such as hydrogen bonds, hydrophobic forces, van der Waals forces, electrostatic interactions, etc., to attain the desired structures and functions. The most intriguing aspect of these systems is the dynamic nature of non-covalent bonding in supramolecular nanoassemblies, which results in nanostructures with adaptive, self-healing, and stimulus-responsive capabilities.5,6 Protein nanoassemblies are some of the most intriguing classes of supramolecular biomacromolecule organizations.7,8 Proteins spontaneously attain complex quaternary structures composed of polypeptide nanoassemblies through non-covalent interactions, occasionally supplemented by disulphide-covalent connections.9,10 These entities display various morphologies, dimensions, and functionalities, including enzyme catalysis, signal transduction, molecular recognition, self-assembly, and self-organization.11 The BSC aims to gain insight into the molecular mechanisms and functionalities underlying various biological processes. Also, drawing inspiration from biological macromolecular systems, the BSC's fundamental goal is to further the creation of novel biomimetic design methodologies by utilizing small biomolecular systems.12 This innovative minimalistic bioinspired approach has opened new avenues for research and applications in various disciplines, including but not limited to nanotechnology, biotechnology, catalysis, medicine, and materials science.13,14In contemporary times, amyloid fibrils have emerged as highly appreciated protein supramolecular nanoassemblies.15,16 Amyloid fibrils constitute complex, highly ordered biomolecular supramolecular assemblies, primarily composed of β-sheet structures. These entities emerge through the self-assembly process of a variety of proteins and polypeptides, including most notable amyloid-beta (Aβ1–42) and Tau.17,18 Central to numerous amyloid-associated diseases, these fibrils originate from protein/polypeptide misfolding phenomena occurring both within and outside cellular environments. This aberrant folding process culminates in forming insoluble aggregates and plaques, significantly contributing to cellular dysfunction and eventual cell death.5,19,20 Recently, amyloid deposits have been implicated in over 30 distinct pathological conditions, with neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, and prion diseases representing some of the most prominent examples.19,20 Amyloid fibrils are distinguished by their unique morphological properties, including a twisted, elongated fibrous shape with diameters ranging from 5 to 20 nm and a composition predominantly rich in β-sheets.21 This structural configuration facilitates their specific interaction with dyes, such as thioflavin-T (ThT), eliciting a characteristic fluorescence that is indispensable for the real-time monitoring of fibril formation. This monitoring process yields critical insights into the dynamics of amyloid aggregation. The intricate structure of Aβ1–42 has sparked significant interest in nanotechnology, attributed to its remarkably stable and well-defined fibrillar architecture.22–24 In pursuit of elucidating the mechanisms underpinning the formation of amyloid fibrils and harnessing their potential applications, researchers have adopted reductionist methodologies. These methodologies focus on examining shorter peptide sequences that mimic the amyloidogenic properties of Aβ1–42. Research findings indicate that truncated variants of Aβ1–42, such as KLVFFAE, as well as diverse hepta-, hexa-, and pentapeptides, possess the capability to self-assemble into amyloid fibrils (Fig. 1). These fibrils exhibit structural and physical attributes akin to those of the full-length Aβ1–42 peptide, thereby not only advancing our comprehension of the molecular underpinnings of amyloid fibril formation but also laying the groundwork for the development of amyloid-inspired nanomaterials.25–28
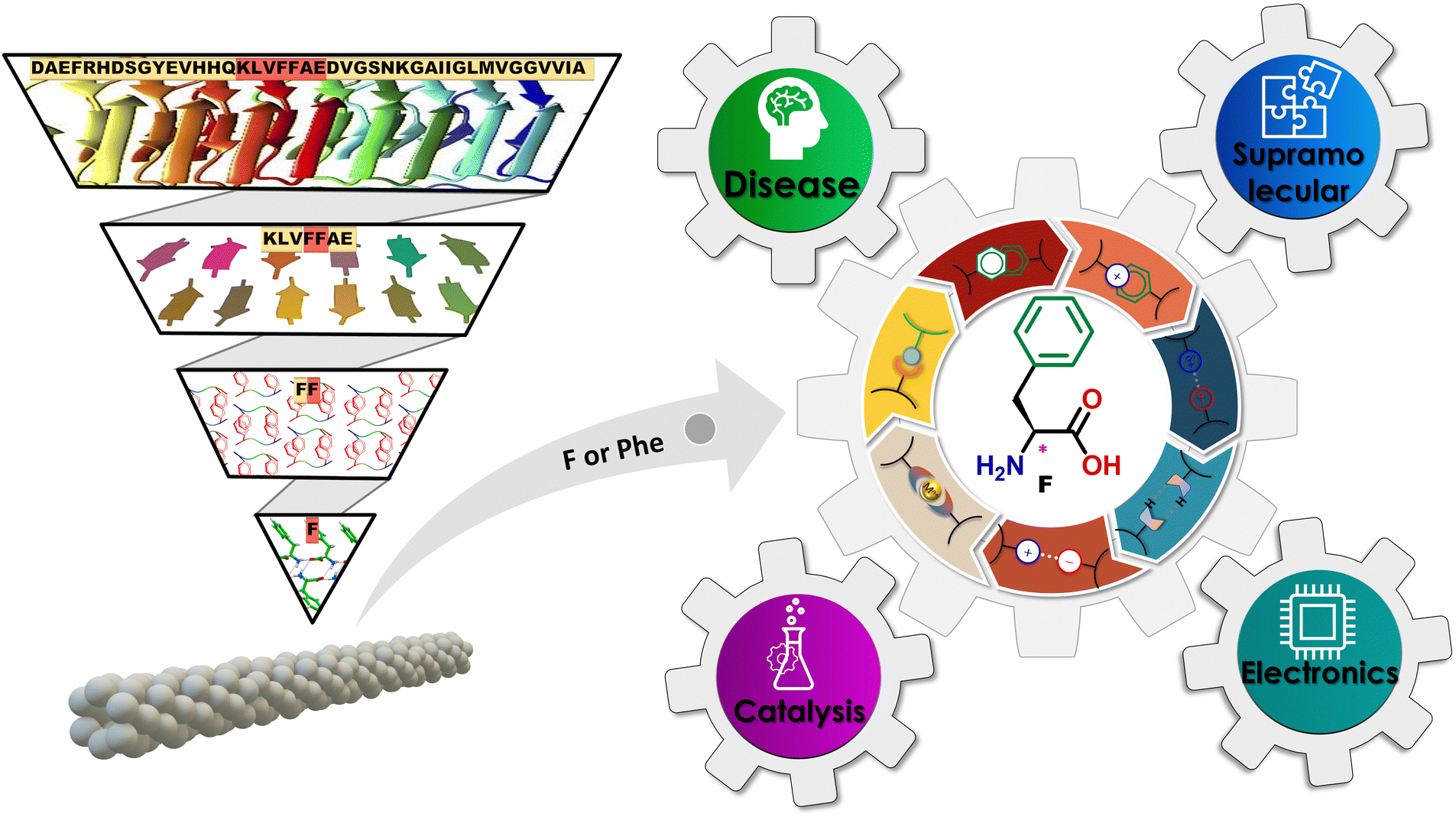 | ||
| Fig. 1 Schematic illustration of the amyloid Aβ (1–42) peptide inspired minimalistic (from peptide fragments to single amino acid F (or Phe)) approach for forming amyloid fibrils. The structure of Phe and its tunable non-covalent supramolecular interactions were explored for a wide spectrum of applications including disease pathology, catalysis, electronic devices, and supramolecular chemistry. | ||
Building on this line of inquiry, Gazit et al. made a pivotal discovery that the Phe dipeptide (Phe–Phe) is a primary recognition element of the β-amyloid polypeptide, thus highlighting its significance when compared to the larger peptides previously mentioned.29,30 Remarkably, this dipeptide demonstrated the ability to self-assemble, forming amyloid-like nanotubular fibril nanoassemblies when dissolved in an aqueous solution. These assemblies replicate not only the luminescence characteristics of amyloids but also their mechanical stiffness, further underlining the potential of such structures. As an extension of their pioneering work, Gazit et al. unveiled that even a single amino acid, Phe, can undergo self-assembly to form fibrils that exhibit amyloid-like biophysical, biochemical, and cytotoxic properties that are mainly associated with neurological disease phenylketonuria (PKU).31 This discovery opens a new vista for exploring metabolic diseases, showcasing the potential of minimalistic approaches in understanding amyloid assembly and interactions. The journey through simplified peptide and amino acid systems has provided valuable insights into the molecular mechanisms underpinning amyloid formation and paved new pathways for exploring therapeutic strategies against amyloid-associated disease.21
Extending beyond pathological aspects, the recent studies on Phe supramolecular assemblies have revealed their ability to form various polymorphic structures under different temperature scenarios, showcasing the complex phase behavior of this amino acid. These structures, particularly supramolecular fibrils, demonstrate significant adaptability to nanostructural transformation in response to various external stimuli, highlighting their tunable self-assembly. Moreover, Phe's interaction with other amino acids leads to unique co-assembly capabilities, providing a broad platform for studying biomolecular interactions. All these findings have paved the way for the development of strategies aimed at preventing the formation of Phe amyloids, showing promise in the treatment of phenylketonuria (PKU). More intriguingly, the capacity of Phe to engage in metal coordination has been explored, revealing new possibilities for its use, especially in creating catalytic systems that mimic enzymes. This property has broadened the applications of Phe-based supramolecular assemblies in fields like organic chemistry, environmental monitoring, and healthcare, placing these assemblies at the intersection of biology, materials science, and nanotechnology. In addition, research into the L and D enantiomers of Phe, especially in forming biological metal–organic frameworks (bio-MOFs) with different metal ions, has further expanded the potential uses of these assemblies. Their application in advanced electronic devices illustrates the fusion of biological and technological progress, marking the beginning of a new era in bioelectronic innovation.32 Thus, Phe supramolecular assemblies are gaining recognition as a crucial area of study, poised to influence the development of future therapeutics and functional materials. This review aims to provide a comprehensive overview of the current state of research on Phe-based advanced supramolecular functional nanoassemblies, highlighting their interdisciplinary significance and technological relevance (Fig. 1). Through a detailed analysis, this review outlines a promising direction for the application of minimalistic biomolecular supramolecular chemistry, spanning from disease treatment to the fabrication of cutting-edge bioelectronic devices, advocating for a future where sustainable and efficient methodologies are paramount.
2. Phe supramolecular amyloid formation and its implications in disease pathology
Phe is an aromatic amino acid that possesses a unique ability to self-assemble into amyloid-like fibrils, leading to the formation of highly organized, supramolecular β-sheet-rich fibrillar structures traditionally associated with amyloid formation.33,34 Elevated levels of Phe, as observed in the genetic disorder phenylketonuria (PKU), facilitate Phe accumulation in tissues and the brain.35 This amplifies the risk of Phe aggregating into amyloid fibrils, which can interfere with normal cellular functions. They achieve this by disrupting cellular membranes, hindering protein folding mechanisms, and triggering oxidative stress, all contributing to neurotoxicity. In the context of PKU, where individuals cannot metabolize Phe efficiently, leading to its toxic accumulation, the structural characteristics of Phe-driven amyloid formation become a significant concern. This process is implicated in the neurological symptoms tied to PKU, such as cognitive deficits, developmental delays, and neurodegeneration. Consequently, investigating the amyloidogenic potential of Phe highlights the critical need to monitor and manage Phe levels in PKU patients.36 Doing so could mitigate these harmful effects and prevent amyloid formation, offering fresh insights into therapeutic strategies for addressing the disease's long-term complications. In this direction, Gazit and coworkers were the first to conduct in vivo biomedical studies on the formation of amyloid-like fibrillar nanoassemblies of Phe at pathologically relevant concentrations.31 These structures are relevant to pathological phenomena and the development of advanced functional nanomaterials. The amyloid formation process of Phe involves the intermolecular stacking of its aromatic benzyl side rings and hydrogen bonding interactions among amino (–NH2) and acid (–COOH) groups, forming stable fibrillar aggregates. Visualized in transmission electron microscopy (TEM) images (Fig. 2a(i)), these fibrils show long, thin, and elongated structures, typically observed in amyloid assemblies. The scale bar of 1 μm emphasizes the nanoscale dimensions of these fibrils. Furthermore, scanning electron microscopy (SEM) (Fig. 2a(ii)) images demonstrate that these similar fibrils are also found under physiological conditions, such as in human blood serum, suggesting their pathological relevance, especially in conditions like phenylketonuria (PKU), a metabolic disorder associated with elevated Phe levels.37 The amyloidogenic nature of these fibrils is corroborated further by Congo red staining, a hallmark diagnostic dye for identifying amyloid structures. When observed under polarized light (Fig. 2a(iii)), Congo red-stained Phe fibrils exhibit characteristic birefringence, a signature feature of amyloid fibrils, attributable to their ordered, cross-β sheet architecture. This observation is further substantiated by confocal microscopy (Fig. 2a(iv)), where fibrils stained with Thioflavin T (ThT),38–40 an amyloid-specific dye, display pronounced fluorescence, emphasizing the densely organized structure of Phe fibrils. Interestingly, Phe fibrils demonstrate intrinsic autofluorescence over an excitation range of 370 to 420 nm (Fig. 2b),41 offering a non-invasive modality for imaging and analyzing their structural organization without reliance on external dyes.42–44 Subsequently, autofluorescence lifetime imaging furnishes a detailed spatial characterization of the Phe fibrils, elucidating their distribution and structural attributes with great detail.45 These analytical techniques are indispensable for a comprehensive understanding of the morphology and interactivity of Phe fibrils within biological environments. The structural organization of Phe fibrils is characterized by a meticulously ordered stacking of β-sheets, as illustrated in the protofilament structure depiction. Precise measurements obtained from X-ray scattering techniques (SAXS/WAXS) shed light on the molecular architecture of the fibrils (Fig. 2c).46 The SAXS/WAXS spectra reveal distinct Bragg peaks (an inter-sheet spacing of 10.3 Å and an inter-strand spacing of 4.6 Å), evidencing the periodic organization of β-sheets and strands within the fibrils (Inset Fig. 2c),47,48 thereby enriching our comprehension of the stable supramolecular assemblies formed by Phe. The investigation into the mechanical properties of these fibrils has unveiled periodic variances in rigidity along their length, a phenomenon of critical importance for discerning the role of Phe fibrils within biological contexts (Fig. 2d). This study offers an understanding of the exceptional stiffness and resilience against degradation of amyloid fibrils; these mechanical attributes are vital for understanding the contribution of such fibrillar aggregates to cellular disruption and disease progression.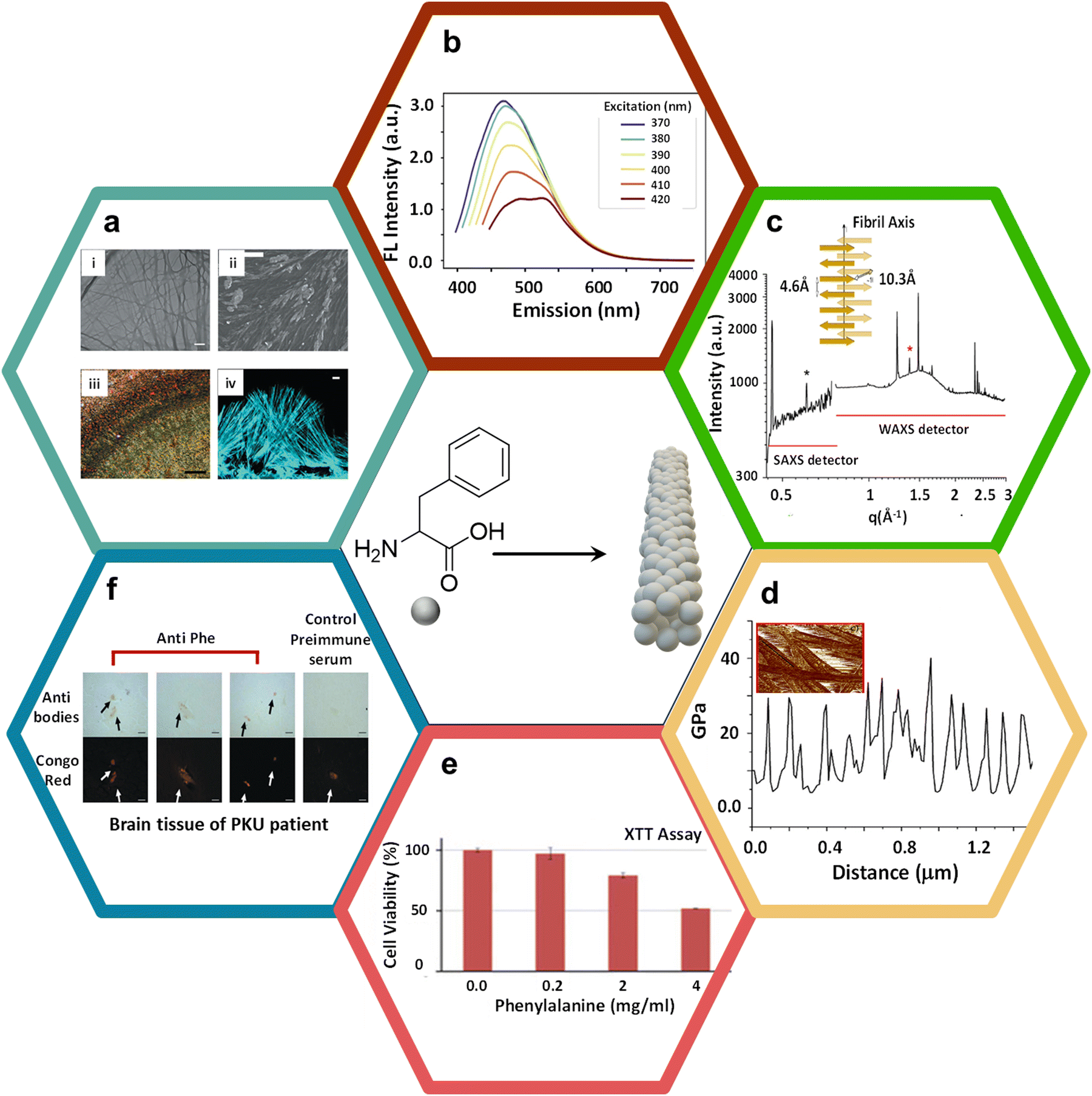 | ||
| Fig. 2 Phe supramolecular amyloid formation, characterization, and its disease pathology. (a) (i) The TEM images display elongated Phe fibrils with a scale bar of 1 μm; (ii) SEM images showing Phe fibrils in human blood serum. The size of the scale bar is 20 μm; (iii) the Phe fibrils were subjected to Congo red staining and subsequently examined under polarized light using a microscope. The size of the scale bar is 500 μm; (iv) confocal microscopy image of fibrils dyed with ThT; scale bar is 10 μm31 (reproduced with permission from ref. 31, Copyright 2020, Nature). (b) Phe nanoassemblies exhibit fluorescence emission over the excitation range of 370 to 420 nm41 (reproduced with permission from ref. 41, Copyright 2021, iScience). (c) The structure of the Phe self-assembly and protofilament demonstrates the spacing between intersheets and interstrands46 (reproduced with permission from ref. 46, Copyright 2020, ACS Appl. Mater. Interfaces 2020). The SAXS/WAXS spectra of Phe nanoassemblies at a concentration of 0.18 M. The spectra presented in their entirety exhibit Bragg reflections resulting from both the intersheet spacing (indicated by the black asterisk, q = 0.61 Å−1, d = 10.3 Å) and the interstrand spacing (indicated by the red asterisk, q = 1.38 Å−1, d = 4.6 Å). (d) Line section through one fibril, showing the periodic variation in stiffness along the fibrils. (e) Phe was dissolved in cell media at a temperature of 90 °C and then gradually cooled. The control (Phe concentration of zero) is a medium without Phe, which underwent similar treatments. The SH-SY5Y cells that had been treated were placed in a medium that included Phe for a duration of 6 hours, after which the XTT reagent was added. The absorbance at a wavelength of 450 nm was measured after a 2.5-hour incubation period49 (reproduced with permission from ref. 49, Copyright 2015, Science Advances). (f) The brain of a patient diagnosed with PKU was subjected to staining using either anti-Phe fibril serum or preimmune serum. The results were then obtained via light microscopy and by subsequently co-staining with Congo red. Depositions positive for Phe were observed within the parietal cortex (reproduced with permission from ref. 31, Copyright 2020, Nature). | ||
The cytotoxic ramifications of Phe fibrils have been scrutinized via a series of cytotoxic assays, notably the XTT assay, as shown in Fig. 2e.49 The assay investigated the response of SH-SY5Y neuroblastoma cells to a gradient of Phe concentrations, elucidating a pronounced, concentration-dependent cytotoxicity at elevated Phe levels. This observation underscores the hypothesis that Phe fibrillar accumulation, a pathological hallmark observed in phenylketonuria (PKU) patients, may induce cellular toxicity, thereby contributing to the neurodegenerative sequelae characteristic of PKU. The toxic aggregation of these fibrils represents a critical etiological factor in PKU pathogenesis, where elevated levels of Phe lead to neurodegenerative consequences. Further corroboration of the pathophysiological significance of Phe fibrils in PKU is provided by histological examination of neural tissues from PKU patients. Immunohistochemical staining techniques, utilizing antibodies specific to Phe fibrils, confirmed their presence within cerebral tissues (Fig. 2f).31 The co-staining with Congo red highlights the deposition of these fibrils in the parietal cortex, a region of the brain associated with cognitive and motor functions.50 This fibrillar accumulation potentially disrupts neuronal functionality, contributing to PKU patients' cognitive detriments. The empirical identification of these fibrils within cerebral tissues underlines the pathological role of Phe self-assembly in the spectrum of neurodegenerative disorders. It highlights the imperative for therapeutic interventions to inhibit or disassemble fibril formation. A deeper analysis of various molecular interactions and packing ordering of Phe self-assemblies is necessary in this regard.
3. Polymorphism in Phe self-assembly
Polymorphism, defined as the capability of a compound to form multiple crystalline structures, is paramount in the solid-state chemistry of Phe self-assembly.51 The Görbitz and co-workers demonstrated the polymorphic diversity in the crystallographic arrangement of Phe, identifying four distinct polymorphs distinguished by their unique space group symmetries, molecular orientations, and the environmental conditions under which they are formed particularly temperature. At ambient temperature (295 K), Polymorph I (Phe-I) crystallizes in the C2 space group with two molecules per unit cell (Z′ = 2), but when cooled to lower temperatures (105/293 K), it crystallizes in the P21 space group with four polymorphs: Phe-I, Phe-II, Phe-III, and Phe-IV.51–53 These molecules per unit cell (Z′ = 4) are shown in (Fig. 3a).54 The molecular arrangement of Phe-I reveals a strong hydrogen-bonded network of carboxyl and amino groups, resulting in characteristic layers of Phe molecules. Polymorph II (Phe-II), depicted in Fig. 3b, crystallizes in the P21 space group with (Z′ = 2) at a temperature of 294 K. The molecular arrangement differs significantly from Phe-I, featuring more intricate hydrogen-bonding interactions and alternate stacking patterns.51,55 This polymorph is particularly noteworthy for its distinctive organization of phenyl rings, contributing to its distinct physicochemical properties compared to Phe-I. These packing variations often result in differences in solubility, stability, and bioavailability, underscoring the importance of polymorphism in the pharmaceutical applications of amino acids. Polymorph III (Phe-III) and Polymorph IV (Phe-IV) have markedly different packing arrangements, with space groups P21 and C2, albeit under the same temperature conditions. As shown in Fig. 3c, Phe-III crystallizes at 100 K with a Z′ value of 4 and exhibits a highly ordered, close-packed structure with significant inter-sheet interactions. This polymorph is notably stable at cryogenic temperatures, positioning it as a valuable model for studying the low temperature behaviours of Phe assemblies. Phe-IV (Fig. 3d) crystallizes at 100 K in the C2 space group, exhibiting a high degree of molecular order. Phe-IV stands out from Phe-III due to its distinct hydrogen-bonding patterns and the arrangement of phenyl rings, resulting in a unique crystalline structure. This polymorph has garnered interest for its potential uses in solid-state materials, attributed to its thermal stability and rigidity. The polymorphism in Phe self-assembly highlights the intricate relationship between molecular arrangement, temperature, and crystallographic symmetry. These differences in crystal structure are not only of pathological significance but also hold practical implications for developing therapeutic strategies, functional materials, and pharmaceutical formulations. Polymorphic transitions can impact solubility, bioavailability, and mechanical properties, making it essential to comprehend and regulate polymorphism in Phe to optimize its utilization in various biomedical and technological applications.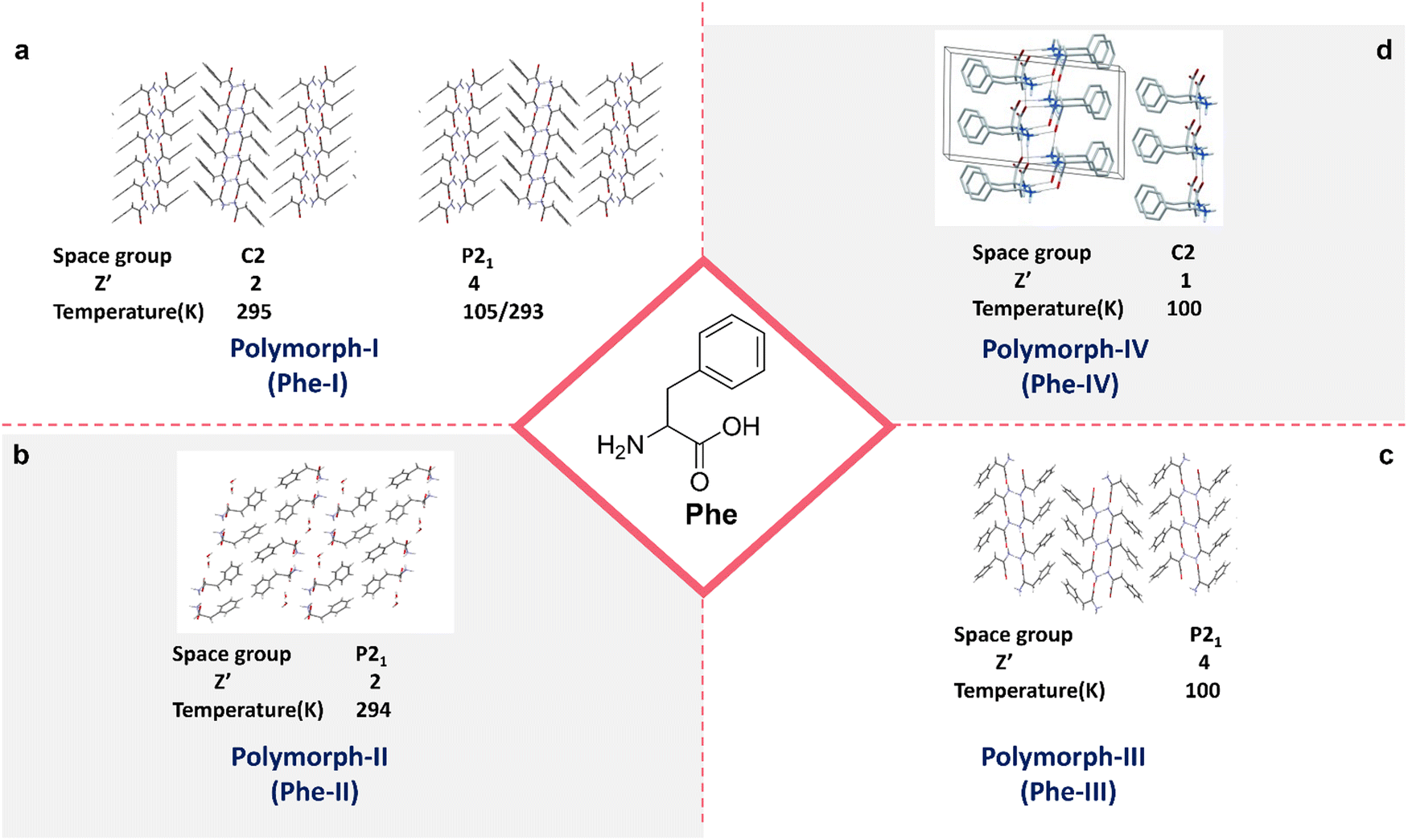 | ||
| Fig. 3 Polymorphism in Phe crystal packing; (a) first polymorphs of Phe represented by Phe-I (reproduced with permission from ref. 52, Copyright 2014, John Wiley & Sons).52 (b) Second polymorphs of Phe represented by Phe-II51 (reproduced with permission from ref. 51. Copyright 2013 J. Phys. Chem. C). (c) Third polymorphs of Phe represented by Phe-III (reproduced with permission from ref. 52, Copyright 2014, John Wiley & Sons).52 (d) Fourth polymorphs of Phe represented by Phe-IV (reproduced with permission from ref. 52, Copyright 2014, John Wiley & Sons).52 | ||
4. Phe amyloid fibril inhibition strategies for therapeutics
Previous sections established that the single amino acid Phe self-assembles into toxic amyloid fibrils, a characteristic shared with other peptides associated with neurodegenerative diseases. This self-assembly is linked to disorders such as phenylketonuria (PKU), where abnormal accumulation of Phe fibrils in the brain can lead to cognitive deficits.56 Consequently, inhibiting Phe fibril formation is a critical therapeutic strategy for addressing PKU.57 An illustrative overview in Fig. 4 presents various strategies to hinder Phe's fibrillization, offering insights into potential therapeutic approaches. One prominent method involves utilizing D-phenylalanine (D-Phe), the enantiomer of Phe, which also forms fibrils in isolation. However, when Phe and D-Phe are mixed in equimolar amounts, fibril formation is disrupted, leading to flake-like structures instead of fibrils (Fig. 4a).58–60 These structures segregate out of the solution and do not contribute to amyloid aggregation, indicating a potential therapeutic avenue where racemic mixtures of Phe and D-Phe could limit fibril-related toxicity. Polyphenolic compounds, such as epigallocatechin-3-gallate (EGCG) and tannic acid (TA), have also demonstrated the ability to inhibit Phe fibrillization.24 SEM images (Fig. 4b and c) validate the inhibition of fibril formation, showing amorphous aggregates rather than the linear fibrils typically seen in amyloid assemblies. These compounds likely interfere with the hydrogen-bonding interactions that drive Phe aggregation, offering a chemical approach to halt fibrillogenesis. Crown ethers like 18-Crown-6 (18C6) and 15-Crown-5 (15C5) serve as another class of inhibitors.61 These cyclic ether molecules can chelate amino groups and disrupt the electrostatic interactions stabilizing Phe fibrils. SEM images (Fig. 4d and e) show that 18C6 is particularly effective, reducing the formation of fibrils more than 15C5. Crown ethers may act by binding to Phe and blocking its aggregation sites, suggesting their utility as fibrillogenesis inhibitors in PKU therapy. The study conducted by Chakraborty et al. delves into the fascinating self-assembly dynamics of phenylalanine (Phe) when introduced into a mixture of divalent (Zn2+, Cd2+, and Hg2+) and trivalent metal ions (Al3+, Ga3+, and In3+) (Fig. 4f).62,63 This process culminates in the formation of a variety of metastable intermediate states and diverse morphological structures. Initially, the assembly process is characterized by the emergence of droplets and microspheres. This early-phase formation is largely attributed to the robust hydrophobic interactions between Phe molecules and the coordination between the Phe and metal ions. As the process unfolds, these microspheres transform intermediate structures, including vesicles, flower-like formations, and toroids. The interaction between Phe and the solvent drives this evolution. Notably, the study highlights that the spherical nanoaggregates that form in the presence of trivalent metal ions exhibit greater stability and longevity. This is suggested to be the result of the stronger hydrophobic interactions characteristic of systems involving trivalent metals as opposed to their divalent counterparts. In contrast, monovalent ions like Na+, Mg2+, and Ca2+ do not have this effect. These findings suggest that specific metal ions can strategically alter Phe assembly pathways, preventing fibril formation. Another interesting study demonstrates that a beer concentration of 1.67% is sufficient to disassemble Phe assemblies effectively. This indicates that, at this specific concentration, beer can disrupt the self-assembled structures of Phe, leading to their breakdown (Fig. 4g).64 This study shows the ability of beer to induce the disassembly of these assemblies. Additionally, the pharmaceutical compound doxycycline has shown promise in inhibiting the crystallization of Phe fibrils (Fig. 4h), confirming that increasing doses of doxycycline reduce the crystallinity of Phe fibrils, suggesting that this antibiotic could be repurposed as an amyloid-modulating agent.65 The interference with well-ordered fibril structures highlights doxycycline's potential to modulate amyloidogenesis. Previous studies have provided strategies to inhibit the toxic amyloid-like assemblies formed by Phe, which are implicated in the pathogenesis of phenylketonuria (PKU). A recent study shows the therapeutic potential of Morin hydrate by using the in vivo phenylketonuria mouse model (Fig. 4i).66 The results demonstrated that treatment with Morin hydrate led to marked improvements in both cognitive and motor functions, key areas affected by phenylalanine toxicity. Moreover, a notable reduction in the number of phenylalanine deposits in the brain was observed, indicating that Morin hydrate effectively disrupted the pathological accumulation of Phe assemblies. Interestingly, while the overall Phe levels in the mice remained elevated, the administration of Morin hydrate resulted in a recovery of critical biochemical markers, including dopaminergic, adrenergic, and neuronal markers, all of which are typically impaired in PKU. This result suggests that the neurotoxic effects of Phe in PKU may be directly linked to its aggregation into toxic assemblies rather than merely its elevated levels. The ability of Morin hydrate to inhibit Phe aggregation without directly lowering systemic Phe concentrations implies that the pathological assemblies formed by Phe play a central role in the disease mechanism of PKU. | ||
Fig. 4 Inhibition strategies for Phe fibril assemblies; (a) D-Phe as an inhibitor58 (reproduced with permission from ref. 58, Copyright 2014 Sci. Rep.). (b) and (c) EGCG and TA as inhibitors24 (reproduced with permission from ref. 24, Copyright 2018 Commun. Chem.). (d) and (e) 18C6 and 15C5 crown ethers as inhibitors61 (reproduced with permission from ref. 61, Copyright 2016 J. Phys. Chem. B). (f) Divalent (Zn2+, Cd2+, and Hg2+) and trivalent (Al3+, Ga3+, and In3+) metal ions as potential inhibitors, showing the formation of a variety of morphologies including droplets, spheres, vesicles, flower-like shapes, and toroids62 (reproduced with permission from ref. 62, Copyright 2022 J. Phys. Chem. Lett.). (g) Common beer as an inhibitor64 (reproduced with permission from ref. 64, Copyright 2022, Elsevier). (h) Effect of the drug deoxy cycline on the in situ formation of Phe fibrils; X-ray scattering 2D patterns of pure 20 mM Phe (left column) and mixed Phe:Doxy, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.005 (central column) and 1:0.05 molar ratios (right column). Images show SAXS (top row) and WAXS (bottom row) patterns65 (reproduced with permission from ref. 65, Copyright 2015 Sci. Rep.). (i) Morrin hydrate as potential theurapeutic agent in the in vivo mouse model66 (reproduced with permission from ref. 66, Copyright 2024 biophyschem). :0.005 (central column) and 1:0.05 molar ratios (right column). Images show SAXS (top row) and WAXS (bottom row) patterns65 (reproduced with permission from ref. 65, Copyright 2015 Sci. Rep.). (i) Morrin hydrate as potential theurapeutic agent in the in vivo mouse model66 (reproduced with permission from ref. 66, Copyright 2024 biophyschem). | ||
These strategies—ranging from enantiomeric mixtures and polyphenols to crown ethers, metal ions, doxycycline, beer, and morin hydrate—represent a comprehensive toolkit for combating Phe fibril formation. These findings deepen our understanding of Phe amyloid inhibition and open the door for therapeutic developments to mitigate PKU and related neurodegenerative conditions.
5. Effect of external stimuli on Phe self-assembly
Phe's self-assembly behavior is intricately influenced by many structural features, including hydrophobic interactions, hydrogen bonding, ionic interactions, and overall conformation. These characteristics do not operate in isolation; they are significantly modulated by external factors such as pH, solvent types, temperature, and ionic strength. For instance, an elevation in temperature can weaken hydrophobic interactions, whereas alterations in pH and the nature of the solvent can have profound effects on hydrogen bonding and ionic interactions. Additionally, changes in the external environment can lead to modifications in the conformation of Phe molecules. Grasping the nuanced relationship between the structural attributes of Phe and external stimuli is fundamental for mastering its self-assembly process. Such insights are invaluable across various domains, including disease therapeutics, materials science, biotechnology, and drug delivery. They are instrumental in designing and engineering materials that possess precisely tailored properties.5.1. Effect of pH
The pH plays a crucial role in the self-assembly of Phe fibrils, as detailed in the research conducted by Kailash Chandra Jena and co-workers.67 The self-assembly of Phe exhibits variations based on its ionic states. At neutral pH, where Phe exists predominantly in a zwitterionic form, the self-assembled nanostructures display fibrillar morphologies, as depicted in Fig. 5a. The central region in the images shows the formation of fibrils under zwitterionic conditions, with the fibrillar structures stabilized by intermolecular interactions, particularly aromatic π–π stacking. In contrast, at low (cationic) and high (anionic) pH levels, the self-assembled structures appear mainly flake-like (Fig. 5a). This distinction implies that electrostatic interactions present under extreme pH conditions hinder the formation of fibrils by disrupting the π–π stacking interactions. At high pH, the –NH2 and –COO− terminals lead to non-zwitterionic interactions, facilitating the formation of extended ladder-like structures. However, introducing extra counterions can destabilize these elongated structures, reorganizing them into condensed, pore-like aggregates. These observations underscore the critical role of electrostatic and aromatic interactions in determining the morphology of Phe self-assemblies. | ||
| Fig. 5 Effect of external stimuli on the Phe self-assembly; (a) effect of pH on the self-assembly of Phe; in acidic and alkaline media, Phe forms flakes; at neutral pH it forms fibrillar morphologies67(reproduced with permission from ref. 67, Copyright 2019, RSC Advances). (b) (i)–(iii) SEM image of Phe with Wo = 1, Wo = 3 and Wo = 20; the images show an enlarged view of the Phe morphologies68 (reproduced with permission from ref. 68, Copyright 2022, IUCrJ) (b) (iv)–(vi) FE-SEM images of phenylalanine self-assembly in water 1:9 water methanol, 1:1 water methanol, and water as solvents43 (reproduced with permission from ref. 43, Copyright 2017, Materials Science and Engineering C). (c) Effect of temperature on Phe at 60 mM concentration. (d) Effect of ionic strength on Phe at 72 mM concentration58 (reproduced with permission from ref. 58, Copyright 2014, Scientific Reports). | ||
5.2. Effect of solvent
The solvent environment, particularly the presence of water, plays a crucial role in determining the morphology of Phe assemblies. Research indicates that bound water environments promote the formation of plate-like Phe crystals. In contrast, the presence of free water leads to the formation of amyloid fibrils (Fig. 5b(i–iii)).68 This underlines the significance of water in the self-assembly process, as free water stabilizes early-stage oligomers and facilitates the vertical stacking required for fibril formation. Furthermore, the dielectric constant of the solvent significantly influences the morphology.43 As Fig. 5b(iv) illustrates, Phe self-assembles into well-defined fibrils in water (a high dielectric constant medium). In a 1:1 mixture of methanol and water, fibrils with a 300–400 nm width are observed (Fig. 5b(v)). With an increased proportion of methanol to 9:1, the fibrils become fused with reduced widths of 100–200 nm (Fig. 5b(vi)). This suggests that controlling the solvent's dielectric constant can offer precise control over the self-assembly patterns of Phe, providing an opportunity to investigate the effects of solvent polarity on resulting morphologies.
5.3. Effect of temperature
Temperature is another critical factor that affects Phe's self-assembly. Higher temperatures lead to a significant increase in particle size by causing the formation of larger nanoassemblies (Fig. 5c).58,69 This is primarily attributed to enhanced hydrophobic interactions, which facilitate the aggregation of Phe molecules into larger structures. A study demonstrated that elevated temperatures promote the creation of a four-fold tube-like structure, as evidenced by simulation experiments.70 Conversely, lower temperatures encourage lateral growth, reducing aromatic ring exposure to water molecules, thereby inhibiting fibril formation.5.4. Effect of ionic strength
The surrounding medium's ionic strength further influences Phe's self-assembly behavior.58 An increase in ionic strength leads to a growth in particle size due to the reduction of surface charges caused by the presence of salts (Fig. 5d). This decrease in charge weakens the electrostatic repulsion between Phe molecules, facilitating their aggregation. The figure depicts a notable trend: the particle size increases significantly as the ionic strength rises from 0 to 1000 mM, highlighting the impact of salt concentration on the self-assembly process.Overall, the self-assembly of Phe is exceptionally responsive to various external factors, including pH, solvent, temperature, and ionic strength. The delicate interplay of electrostatic interactions, hydrophobic effects, and the dielectric properties of the environment ultimately dictates the structure and stability of the resulting assemblies. Gaining a comprehensive understanding of these influences yields valuable insights into the underlying principles of Phe self-assembly. It may guide future studies aiming to manipulate these assemblies for disease theragnostic and specific applications.
6. Co-assembly characteristics of Phe with other amino acids
Phe exhibits several structural attributes that enhance its ability to co-assemble effectively with other amino acids, thus facilitating the creation of stable and varied nanostructures.71 The zwitterionic nature of amino acids, prevalent at neutral pH, allows for the electrostatic interaction between the carboxylate ions (–COO−) situated at the C-terminal end of one Phe molecule and the protonated amino group (–NH3+) at the N-terminal end of another.72 This interaction is further augmented by Phe's nonpolar benzene ring side chain, which engages in hydrophobic interactions with other nonpolar amino acids, markedly amplifying the co-assembled structures’ overall stability. Moreover, the capability of Phe's amino and carboxyl groups to form hydrogen bonds with other amino acids significantly bolsters the co-assembled structures’ robustness. Alongside the weak van der Waals, forces between Phe and the atoms of different amino acids play a crucial role in stabilizing the co-assembled structures.73 Collectively, these structural characteristics not only support the co-assembly of Phe with a broad spectrum of amino acids but also guarantee the generation of tunable nanostructures, highlighting the intricate synergy of forces that contribute to the stabilization and functionality of co-assembled structures.In exploring the co-assembly behaviour of Phe with a range of amino acids, the research conducted by Gazit et al. revealed a fascinating spectrum of supramolecular structures, as detailed in Fig. 6.74,75 Phe demonstrates a versatile ability to co-assemble with various amino acids, paving the way for the creation of a multitude of nanoscale structures, distinguished primarily by their solid-state interlayer separation distances. This phenomenon is evident when Phe is co-assembled with methionine (Met) and valine (Val), where it forms flake-like two-dimensional structures, showcasing a robust interaction among these amino acids.76 In a contrasting scenario, combining Phe with alanine (Ala), aspartic acid (Asp), and glycine (Gly) yields both flakes and fibrils, pointing towards the occurrence of self-sorted structures. This diversity is further enriched when Phe interacts with isoleucine (Ile) and leucine (Leu), leading to the emergence of nano-spherical morphologies, which highlights how amino acids with similar hydrophobic tendencies naturally gravitate toward forming unique spherical constructs. On the other hand, co-assembly with proline (Pro) produces two distinctly different one-dimensional fibrils, an outcome likely linked to Pro's unique ring structure influencing the assembly dynamics. This intricate variation in co-assembly behaviors underscores the significant role of interlayer separation distances among amino acids. Amino acids with similar features synergize, co-assembling into integrated structures, whereas those with differing interlayer distances tend to self-sort, retaining their distinct characteristics. Notably, Phe exhibits a strong affinity for co-assembly with amino acids like Met and Ile, facilitating the formation of distinct supramolecular structures. Conversely, amino acids such as Gly and Ala, which lean towards self-sorting, show minimal co-assembly with Phe. The underlying mechanisms of these co-assembly processes and resultant structures are further elucidated through advanced techniques like electron microscopy and mass spectrometry, offering a window into the atomic-level interactions and structural formation at play. Later, the same research group conducted a detailed exploration into the co-assembly of L-histidine (L-His) with various aromatic amino acids such as phenylalanine (Phe), tyrosine (Tyr), and tryptophan (Trp), encompassing both enantiomeric forms. The collaborative study focused on how L-His co-assembled with Phe, revealing the creation of stable fibrillar supramolecular structures.77 This fascinating outcome was meticulously verified through advanced high-resolution imaging techniques, including optical microscopy, high-resolution scanning electron microscopy (HR-SEM), and atomic force microscopy (AFM). Optical microscopy, in particular, displayed the emergence of plate-like colorless crystals, showcasing a highly organized structure stemming from their interactive processes. Moreover, a time-dependent analysis executed through HR-SEM underscored the enduring stability of these supramolecular assemblies, which remained unchanged over time, attesting to their robustness. A thermodynamic examination employing isothermal titration calorimetry (ITC) demonstrated that the interplay between L-His and Phe is spontaneous, underscored by a significant entropy change (ΔS 71.48 J mol−1 K−1). This points to a strong affinity of L-His toward Phe, laying the groundwork for spontaneous and energetically favorable interaction. Further corroboration of the stability and precise formation of these assemblies was provided by electrospray ionization mass spectrometry (ESI-MS) analysis and high-resolution mass spectrometry (HR-MS), where the observed masses aligned perfectly with the theoretical calculations, thereby substantiating the co-assembly process. Notably, the findings revealed that the co-assembly's morphologies are conserved across different chiral forms of Phe, suggesting that the chirality of Phe does not markedly impact the structural integrity of the resulting assemblies.
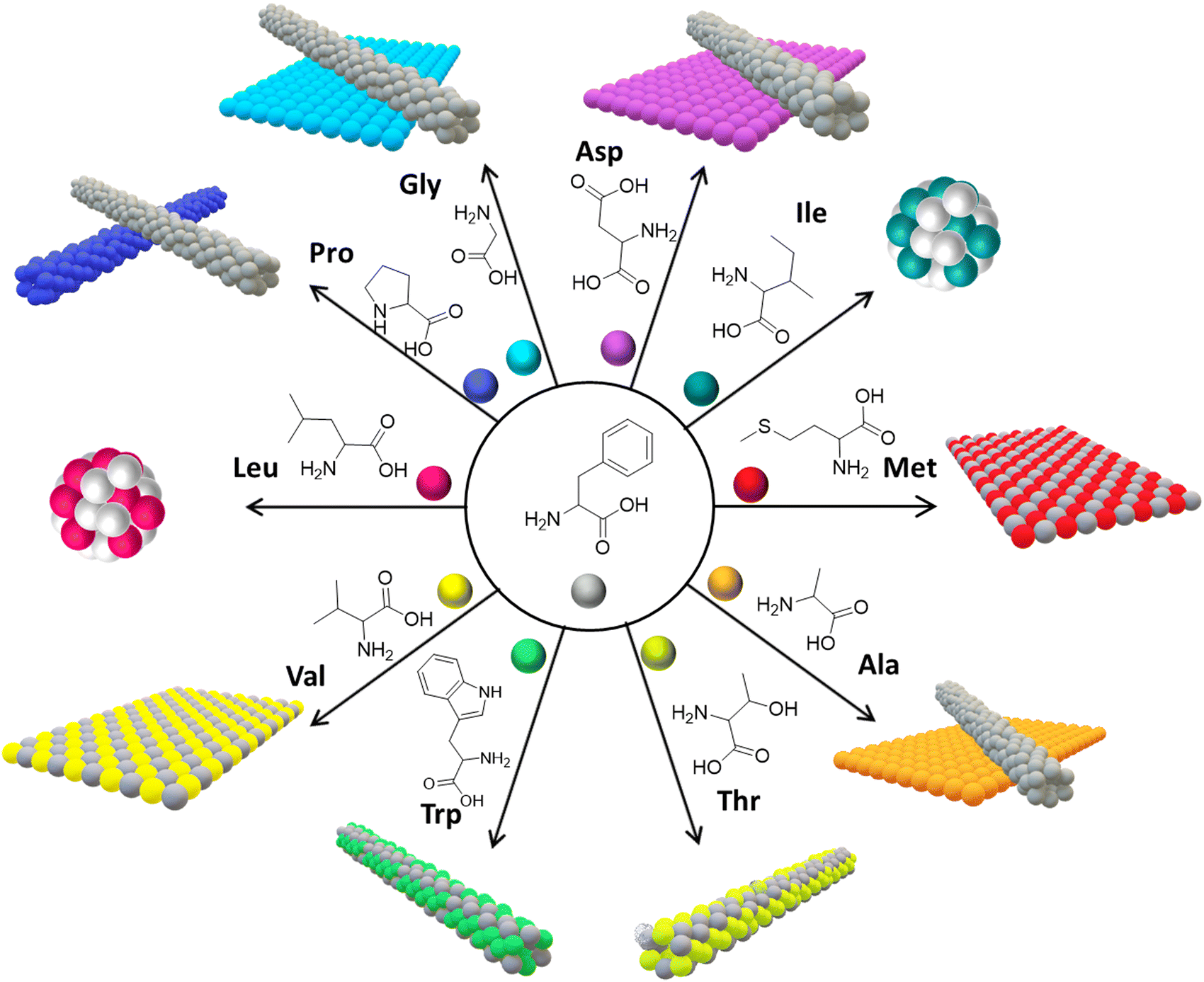 | ||
| Fig. 6 Co-assembly of Phe with other amino acids. 2D nanomorphologies are observed when Phe co-assembled with Met and Val; a mixture of self-segregated 1D and 2D nanostructures was observed when Phe co-assembled with Gly, Ala and Asp; spherical nanomorphologies were observed in the case of Iso and Leu; 1D nanomorphologies were found in the case of Trp, Thr and Pro (reproduced with permission from ref. 76, Copyright 2019, American Chemical Society).76 | ||
In a similar line, Wangoo et al. delved into the cooperative assembly of aliphatic chain amino acids (ACAAs) such as Ala, leu, Ile, and Val with Phe.78 The methodology encompassed mixing these amino acids in a solution, facilitating their interaction and the formation of novel structural arrangements. Given the zwitterionic nature of amino acids, these interactions were efficiently promoted, culminating in hybrid nanostructures distinct from the formation typically associated with Phe alone. Methods including transmission electron microscopy (TEM) and Thioflavin T (ThT) assays were pivotal in characterizing these evolved structures.79 Remarkably, this co-assembly significantly transformed the morphology of Phe nanofibrils, notably diminishing the characteristic β-sheet-like structures linked with amyloid formation. This transition suggests that ACAAs can disrupt the process of Phe aggregate formation, a critical factor behind its toxic effects. The crux of fibril formation inhibition is believed to be the resultant competitive interactions among the amino acids, where ACAAs likely impede the stacking interactions essential for the formation of toxic Phe aggregates. Modulating these interactions hints at a promising route to mitigate the cytotoxic impacts of Phe fibrils, implicated in disorders such as phenylketonuria (PKU). Consequently, these findings highlight the potential of co-assembly strategies as therapeutic avenues to manage amyloid toxicity-associated conditions. Integrating ACAAs into treatment plans could feasibly diminish the detrimental effects of Phe aggregates in PKU patients, possibly ushering in enhanced health outcomes.
Consequently, deepening our understanding of how Phe interacts with other aromatic amino acids advances our knowledge of molecular assembly. It paves the way for the design of tailored and modular nanomaterials. This progress is promising for the innovation in functional nanomaterials and points toward novel therapeutic strategies for treating conditions like phenylketonuria (PKU), thereby connecting molecular insights directly with practical applications.
7. Catalytic metallo-Phe supramolecular assemblies for environmental and healthcare applications
The amino (–NH2) and carboxylic acid (–COOH) functional groups, along with the phenyl side chain of Phe, possess multiple structural features that facilitate effective interactions with metal ions.80 Notably, the carboxylic acid group (–COOH) at the C-terminal end can deprotonate, forming coordinate bonds with metal ions.81–83 This feature is complemented by the amino group (–NH2) at the N-terminal end, which can also establish coordination with metal ions by donating lone-pair electrons. Another critical aspect of Phe's structure is its benzene ring side chain, which can engage in pi-stacking interactions, significantly bolstering the stability of the resultant Phe–metal complex. Furthermore, Phe's specific stereochemistry (L/D) plays a crucial role in dictating the stereoselectivity of its interactions with metal ions, thereby highlighting the sophisticated nature of these biochemical processes. Consequently, Danafer and coworkers reported the synthesis and applications of chrysin-loaded-Phe-coated iron oxide magnetic nanoparticles (IOMNs), represented as chrysin@Phe@IOMNs.84 This study investigated the structural, magnetic, and cytotoxic features of (IOMNs) coated with Phe. Interestingly, it was found that resultant Phe-coated IOMNs are hemocompatible and biocompatible. The findings indicate that the chrysin@Phe@IOMNs exhibit superior anticancer properties compared to free chrysin, mainly attributed to the continuous release of the drug. The results showed that the chrysin@Phe@IOMNs effectively deliver drug chrysin to breast cancer cells. Interestingly, Phe-supported Fe3O4@SiO2 core/shell magnetic nanoparticles (MNPs) can be used as a catalyst for the preparation of new chromenes (4) through the reaction of p-nitrobenzaldehyde (1), 4-hydroxy coumarin (2), and 2-hydroxy naphthalene-1,4-dione (3) in ethanol/H2O (Fig. 7a).85 This approach has several advantages, including using an enhanced catalyst, the capacity to recover the catalyst, low catalyst loading, rapid reaction kinetics, user-friendly operation, a high atom economy, exceptional product yields, and environmental sustainability.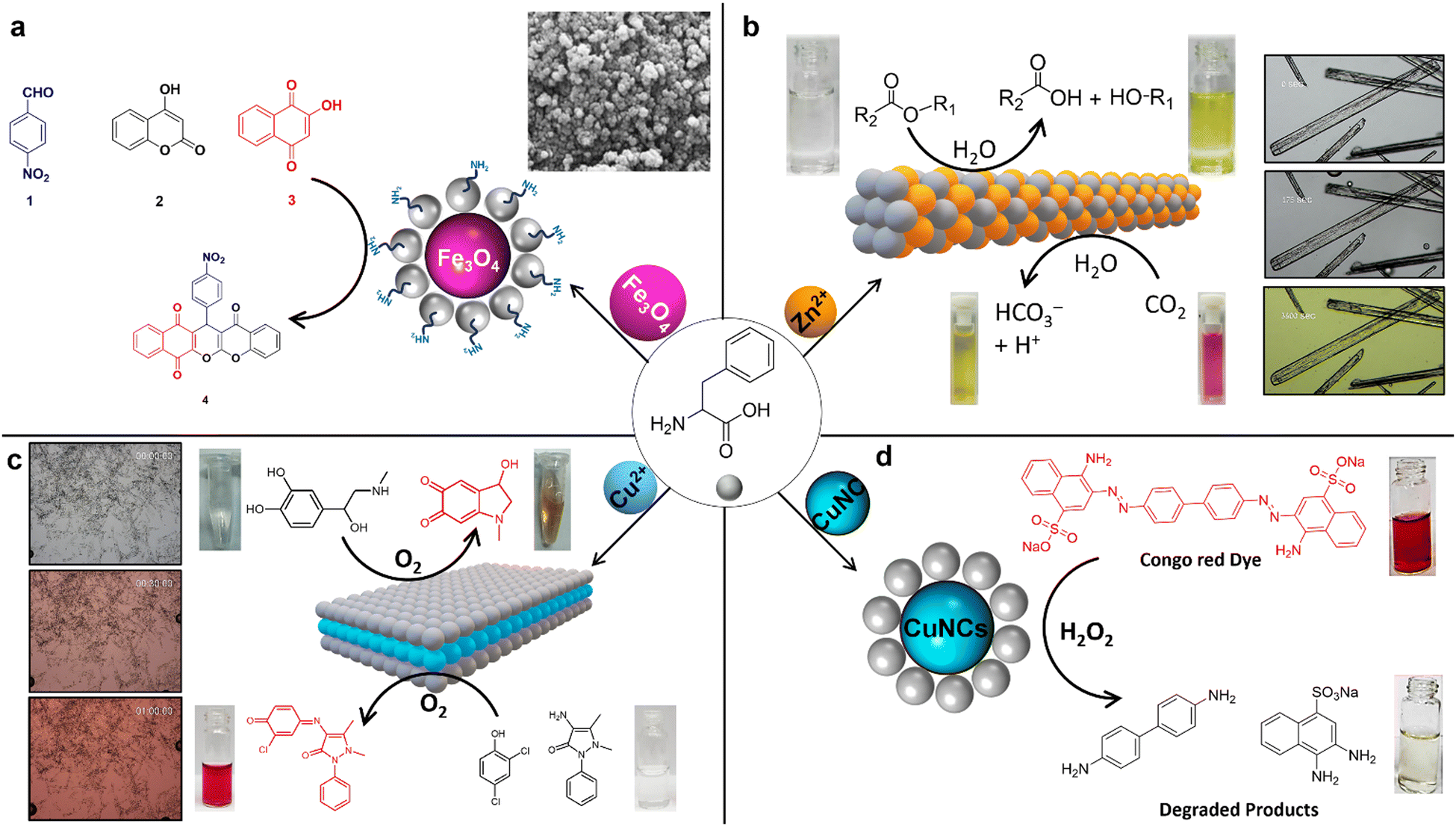 | ||
| Fig. 7 Catalytic metallo-Phe nanoassemblies; (a) Phe-supported Fe3O4@SiO2 acts as a catalyst for the synthesis of chromenes85 (reproduced with permission from ref. 85, Copyright 2018, Springer Nature). (b) Carbonic anhydrase mimicking Phe-Zn bionanozyme and its catalytic esterase and CO2 hydration activity (reproduced with permission from ref. 21 and 86, Copyright 2019, Springer Nature).86 (c) Laccase mimicking Phe-Cu bionanozyme for the catalytic initiation of reaction between 2,4-DP and 4-AP conversion of phenols, and catalytic colorimetric detection of dopamine and L-DOPA87 (reproduced with permission from ref. 87, Copyright 2022, Springer Nature). (d) Phe stabilized copper nanoclusters (CuNCs) for catalytic Congo red dye degradation88 (reproduced with permission from ref. 88, Copyright 2022, Elsevier).88 | ||
In a compelling study, Gazit and colleagues uncovered an enzyme-like catalytic activity within the carbonic anhydrase (CA II) paradigm, achieved using a single amino acid, Phe.86 This discovery underscores a novel metabolite amyloid strategy whereby zinc (Zn2+) ions naturally bind with Phe at a 1:2 stoichiometric ratio. This interaction leads to the forming of a robust, layered, supramolecular, amyloid-like ordered structure, as confirmed through X-ray crystallography. A standout feature of the Phe-Zn single crystals is their exceptional CA II enzyme-like catalytic prowess in promoting ester hydrolysis and CO2 hydration reactions (Fig. 7b). Remarkably, within the scope of artificial short peptide-based hydrolases, Phe-Zn has emerged as the smallest yet most efficient, boasting an esterase catalytic efficiency of 76.54 M−1 s−1. This efficiency is coupled with unparalleled substrate specificity and stereoselectivity. Furthermore, Phe-Zn demonstrates impressive catalytic longevity over numerous cycles and substantial thermal catalytic stability, effectively maintaining function up to 573.15 K. The swift catalytic transformation of carbon dioxide into bicarbonate (HCO3−) via Phe-Zn underscores its potential for a broad range of applications in environmental and biotechnological fields, signaling a groundbreaking approach to designing robust, minimalist artificial enzymes.
The recent work conducted by Makam et al. elegantly demonstrated a straightforward yet innovative method for the spontaneous coordination of Phe with copper ions (Cu2+), leading to the formation of atomic-thick, hierarchical two-dimensional (2D) layered van der Waals (vdW) Phe-Cu crystals.87 This breakthrough leverages the ultrathin 2D framework rich in redox-active Cu2+/Cu+ sites, allowing the Phe-Cu nanosheets to exhibit remarkable capabilities to mimic the enzyme laccase-like biocatalytic oxidation activity, as well as providing ultrasensitive detection of crucial phenolic compounds that are both environmentally toxic and biologically significant (Fig. 7c). The thorough experimental and computational studies have shed light on the intricate mechanistic underpinnings governing the catalytic prowess of the Phe-Cu bionanozyme. Among its attributes, the Phe-Cu bionanozyme stands out for its simplicity and cost-effectiveness—being approximately 5400 times more economical—10 times higher efficiency, and 36 times greater sensitivity when compared with natural laccase enzymes. Furthermore, the resilience of the Phe-Cu bionanozyme is nothing short of remarkable, retaining its stability and reusability under different extreme conditions, ranging from various pH levels and ionic strengths (ensuring its functionality in diverse settings including seawater) to varied temperatures, prolonged storage periods, and the presence of organic solvents. These exceptional features of the Phe-Cu bionanozyme make it highly promising for applications across environmental, industrial, and healthcare sectors, showcasing its versatility and potential impact in these fields. Therefore, Gazit et al.'s advent of this single-amino-acid bionanozyme approach represents a significant stride forward, offering a compelling alternative to the more traditional avenues of protein engineering and nanozyme production.
The study conducted by Hong-Wu Zhang et al. introduces a cutting-edge technique for influencing the chirality of ligands during the creation of supramolecular hydrogels.89 By pairing Phe with copper ions Cu(II), they discovered that this method exhibits superior selectivity over the use of other metal ions. Notably, the research revealed that the gel-forming capability of the Phe-Cu(II) supramolecular metallogelator decreases as the enantiomeric excess of Phe is reduced. This indicates the critical role of the ligand's specific arrangement, either as D- or L-forms, in the gelation process, thus emphasizing chirality's significant impact on the properties of supramolecular materials. Furthermore, these insights pave the way for developing revolutionary chiral sensing platforms. The study opens the possibility of visually detecting chiral molecules through a transition from sol (liquid) to gel triggered by variations in ligand chirality. This offers a groundbreaking approach in sensing applications, facilitating the straightforward identification of chiral substances without resorting to sophisticated instrumentation and thus streamlining the process. In another study, Mukherjee et al. utilized the Phe as a template to synthesize blue-emitting copper nanoclusters (CuNCs) under acidic conditions (pH-4).90 Interestingly, as the pH is increased to 12, the emission color shifts from blue (410 nm) to cyan (490 nm), indicating a significant change in the optical properties of the CuNCs. This pH-induced transformation highlights the tunability of the nanoclusters' photoluminescence, which is a crucial feature for them to investigate further their potential application as a CO2 sensor. The CuNCs exhibit sensitivity to CO2, acting as an efficient probe for detecting dissolved CO2 in the form of bicarbonate ions (HCO3−). The study reports a low limit of detection (LOD) of 60 mM, demonstrating their potential for environmental monitoring. Additionally, the CuNCs show peroxidase-like enzymatic activity, which was tested using o-phenylenediamine (OPD) as a substrate under physiological conditions. This dual functionality enhances the applicability of CuNCs in both sensing and catalysis. Furthermore, Qing et al. discovered that Phe-stabilized copper nanoclusters (Phe-CuNCs) offer a promising approach to the specific elimination of the environmental pollutant Congo red, as depicted in Fig. 7d.88 Under natural lighting conditions, Phe-CuNCs function effectively as facilitators for the capture and transfer of electrons to the surface of Congo Red, aiding in its reduction. Remarkably, this study highlighted the capability of Phe-CuNCs to selectively eradicate bacteria, including E. coli and S. aureus, while posing no harm to normal or tumor cells, even at elevated concentrations. This dual function suggests the significant potential of Phe-CuNCs for treating microbial infections, positioning them as a versatile tool in environmental cleanup and healthcare applications. These conventional methods are often marred by their labor-intensive, chemically intricate, and costly nature, requiring elaborate experimental setups, harsh reaction conditions, and intricate fabrication steps.
8. Metallo-Phe supramolecular assemblies for advanced device technology
Another study of interactions between Phe and metal nanoparticles, focusing on gold (Au), has unveiled promising pathways for advancing optoelectronic technologies (Fig. 8a).91 This exciting area of research was notably advanced by N. A. Kotov and coworkers, who delved into the remarkable sensitivity of self-assembled multilayers of gold nanoparticles (Au NPs), which were modified with enantiopure Phe, to circularly polarized light (CPL). They discovered that when Phe is combined with Au NPs, it forms chiral nanoscale membranes. These membranes can distinguish between right-handed circularly polarized (RCP) and left-handed circularly polarized (LCP) light, producing a photocurrent under RCP that is dramatically up to 2.41 times higher compared to LCP. This dramatic increase in sensitivity stems from the intense plasmonic coupling within the Au nanoparticles. This coupling mechanism efficiently facilitates electron ejection, where the electrons find themselves trapped at the interface between the membrane and the electrolyte due to the substantial Phe layer encasing the nanoparticles. Consequently, a notable polarization-dependent ion accumulation, as demonstrated by the current behaviors in response to RCP and LCP, is illustrated in Fig. 8a(i and ii). Mimicking biological systems, such as the specialized polarization vision of certain marine animals, suggest that these L/D-Phe-Au NP structures could act as a technological parallel in polarization detection, bridging the gap between bio-inspired design and optoelectronic applications.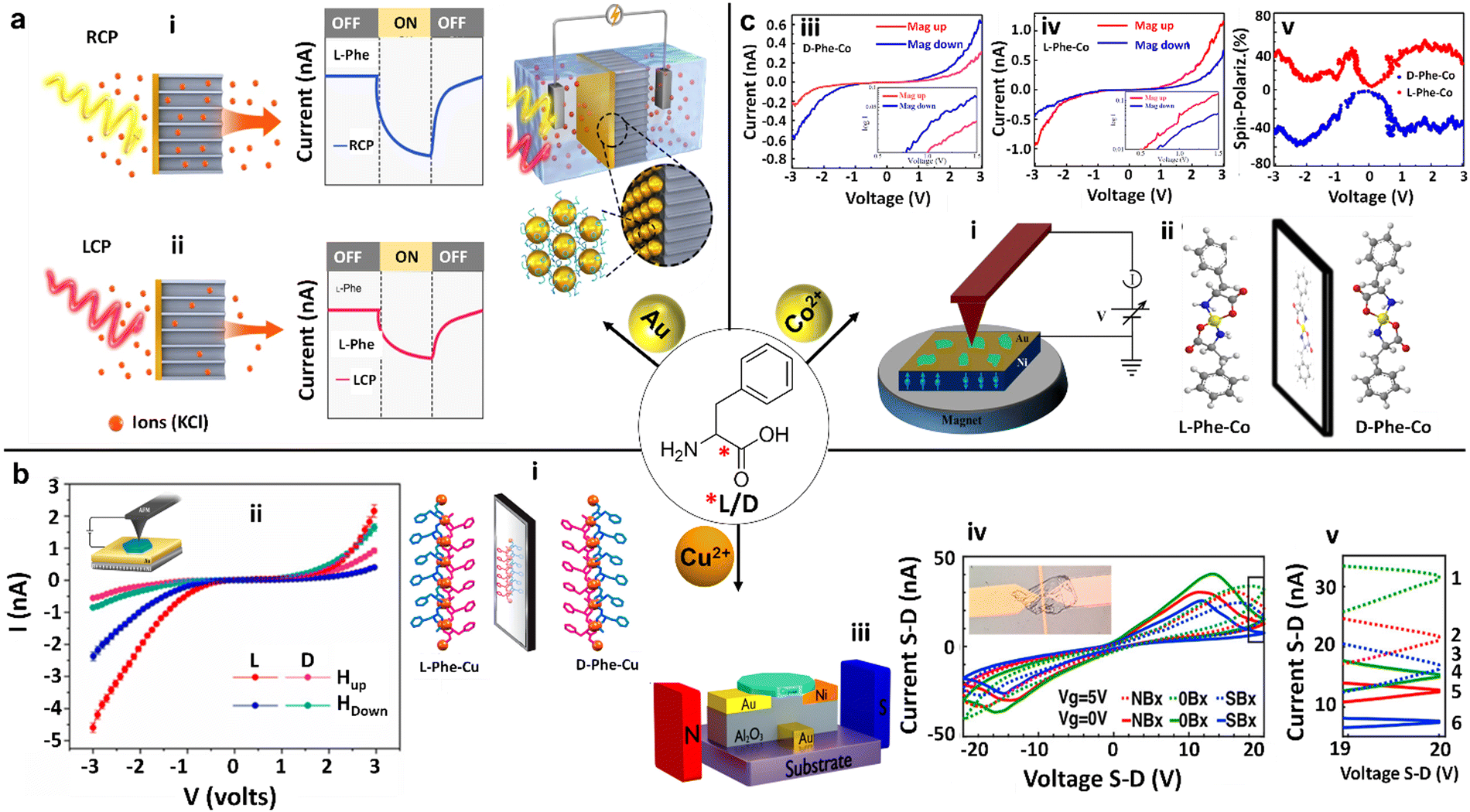 | ||
| Fig. 8 Chiral Phe-metal supramolecular assemblies for advanced electronic device technology. (a) Schematic illustration of an electrochemical cell consisting of a membrane embedded with a chiral L/D-Phe functionalized gold (Au) nanoparticles (NPs) nanofilm, along with its characteristics for photoinduced current generation. The photoinduced current generated across the membranes was greater under Right-Handed Circularly Polarized (RCP) illumination compared to Left-Handed Circularly Polarized (LCP) illumination in nanofilms made from L-Phe-Au NPs. Conversely, the opposite was observed for nanofilms derived from D-Phe-Au NPs (reproduced with permission from ref. 91, Copyright 2022, Springer Nature). (b) (i) Single crystal X-ray diffraction (SCXRD) structure of L/D-Phe-Cu, showing the non-superimposable mirror image enantiomeric coordination organization. (ii) (inset diagram) Schematic diagram of the magnetic conductive-probe atomic force microscopy (mCP-AFM) measurement setup on L/D-Phe-Cu single crystals, along with their corresponding current–voltage (I–V) plots influenced by the substrate magnetization direction (Hup or Hdown). These measurements demonstrate the chirality-induced spin selectivity (CISS) of charge-carrier transmission (reproduced with permission from ref. 92, Copyright 2020, American Chemical Society). (iii) Sketch of the bottom gate device and the measurement setup for D-Phe-Cu crystals. (iv) I–V dependency at two different gate voltages (0 V represented by solid lines and 5 V represented by dashed lines) and three varying magnetic fields for each gate voltage (up, down, and none). Inset: microscopy image of the bottom gate device, featuring a D-Phe-Cu crystal positioned between nickel and gold pads. (v) Close-up view of a narrow voltage band of the I–V curves, as indicated in (iv), highlighting the six different double states (reproduced with permission from ref. 93, Copyright 2021, American Chemical Society). (c) (i) Schematic diagram of the mCP-AFM measurement setup for L/D-Phe-Co single crystals. (ii) The SCXRD structure of D-Phe-Co and L-Phe-Co crystals exhibiting non-superimposable mirror images. (iii) and (iv) I–V plots of L-Phe-Co and D-Phe-Co crystals respectively, under the influence of the substrate magnetization direction (Mag up or Mag down), showcasing the CISS of charge-carrier transmission for both L/D-Phe-Co enantiomers. Insets show the corresponding curves as a log–log plot. (v) The absolute value of the calculated spin polarization for both L/D-Phe-Co enantiomers. The blue curves represent the magnet's north pole pointing downward, while the red curves correspond to the north pole pointing upward (reproduced with permission from ref. 94, Copyright 2023, Royal Society of Chemistry). | ||
The discovery made by Naaman and coworkers concerning the interaction between L/D-Phe and metals, specifically exemplified in bioinspired metal–organic frameworks such as L/D-Phe-Cu crystals, presents noteworthy potential for driving technological advancements (Fig. 8b).92 Featuring prominently in these advancements are chiral crystals that stand out for their ability to conduct electrons in a spin-selective manner at room temperature—a breakthrough essential for the evolution of spintronics. Spintronics is an innovative field aimed at exploiting the spin of electrons to revolutionize electronic devices. Delving deeper into the research, it was discovered that both D- and L-enantiomers of Phe-Cu crystals demonstrate selective conduction of electrons based on spin (Fig. 8b(i and ii). This phenomenon, confirmed through meticulous current–voltage (I–V) measurements, reveals a dependency on the chirality of the crystal, the influence of external magnetic fields, and the bias direction. What adds to the intriguing nature of these findings are the unique magnetic behaviors exhibited by these chiral crystals—they possess weak ferromagnetic properties and ferroelectric properties. A fascinating aspect of their magnetism is their thermal activation, transitioning from an antiferromagnetic state at colder temperatures to a ferromagnetic one upon exceeding 50 K. This transition is attributed to an indirect exchange interaction facilitated by Cu(II) ions within the chiral Phe lattice. A significant technological advancement emerged from this research, marking the development of a spin transistor utilizing D-Phe-Cu chiral crystals, as illustrated in Fig. 8b(iii).93 Remarkably, this device demonstrates memristor-like properties by harnessing both charge and spin trapping to engender multi-level electronic states. Intriguingly, the spin transistor's functionality is influenced by external magnetic fields, which modulate the magnetization of the source, thus affecting the device's drain–source currents (Fig. 8b(iv and v)). The elegance and straightforward design of these bio-organic spin transistors and their multi-level functionality render them highly suitable for seamless integration into organic electronics and bioelectronics. Moreover, their capacity to maintain stable operation under ambient conditions significantly enhances their applicability in practical settings. This research intimates that similar strategic approaches could be applied to a broad spectrum of chiral materials, potentially ushering in an era of innovative spintronic devices adept at manipulating electron spin and magnetic properties on a nanoscopic scale. Building upon their initial insights from L/D-Phe-Cu crystals, the research team expanded their focus to the enantiomeric L/D-Phe-Co metal–organic crystals (MOCs), which have demonstrated remarkable magnetic characteristics (Fig. 8c(i)).94 These include antiferromagnetic interactions between the Co(II) ions within the lattice and the ability for long-range spin transport. A notable breakthrough was discovering the chirality-induced spin selectivity (CISS) effect. This phenomenon leads to spin polarization in the range of 35–45% at room temperature, as observed through magnetic conductive-probe atomic force microscopy techniques (Fig. 8c(iii–v)). Such findings underscore the efficacy of Phe-metal interactions in achieving efficient spin polarization, an essential component in the progression of spintronic devices. The MOCs exhibit electrical and magnetic properties that render them compelling candidates for applications geared toward manipulating electron spins. Specifically, integrating paramagnetic ions with chiral molecular structures in the L/D-Phe-Co crystals (Fig. 8c(ii)) fosters efficient spin conduction and transport across considerable distances, setting the stage for their utilization in future technological advancements. This harmonious blend of chirality, magnetism, and conductivity within L/D-Phe-metal frameworks heralds a new frontier in the evolution of data storage solutions, sensing technologies, and various other domains that depend on the principles of spin-based electronics.
9. Conclusions and outlook
In the quest to elucidate the mechanisms underpinning the formation of Aβ1–42 amyloid fibrils and to harness their potential for biomedical and technological applications, researchers have increasingly turned to reductionist methodologies. These approaches focus on analyzing truncated peptide sequences that emulate the amyloidogenic properties of Aβ1–42. The findings of such research studies have revealed that truncated variants of Aβ1–42, encompassing diverse hepta-, hexa-, penta-, and di-peptides, as well as the single amino acid phenylalanine (Phe), possess the innate capability to self-assemble into amyloid fibrils. Remarkably, these fibrils share structural, physical, and chemical properties with the full-length Aβ1–42 peptide, thereby not only enhancing our understanding of the molecular foundations of amyloid fibril formation but also establishing a foundation for the development of amyloid-inspired minimalistic biomolecular nanomaterials. Of particular interest is the observation that elevated levels of Phe, as seen in the genetic disorder phenylketonuria (PKU), lead to its accumulation in tissues and the brain. This phenomenon has directed a significant portion of research efforts toward studying the supramolecular assembly of Phe and its characteristics. In-depth investigations have underscored the pathological significance of Phe fibrils in PKU and characterized their amyloidogenic nature through various imaging and analytical techniques. These studies have demonstrated that the fibrils exhibit concentration-dependent cytotoxicity on neuroblastoma cells, implicating them in PKU's neurodegenerative dimensions (Table 1). To counteract these adverse effects, extensive research has been dedicated to exploring the molecular organization of Phe and evaluating diverse strategies to inhibit its amyloid self-assembly process. A notable advancement in this area has been the identification of four distinct polymorphs of Phe, denoted as Phe-I, Phe-II, Phe-III, and Phe-IV. Each is characterized by unique space group symmetries, molecular orientations, and formation conditions, particularly temperature. The polymorphism observed in Phe self-assembly carries significant implications for the stability and potential applications of this compound in solid-state materials, as well as offering clues for the inhibition and dissolution of toxic Phe amyloid fibrils and plaques. Building on these insights, various strategies for inhibiting the formation of Phe amyloids have been demonstrated, encompassing the use of small molecules, metal ions, and amino acids in in vitro and in vivo models. Furthermore, Phe supramolecular assemblies have been shown to exhibit diverse stimuli-responsive structural transformations. These structural, chemical, and physical characteristics of Phe have thus garnered attention for their potential in a wide range of biomedical and technological applications. Phe molecules inherent carboxyl and amino functionalities enable spontaneous coordination with metal ions, exhibiting enzyme-like catalytic properties that have been exploited in organic reactions, environmental remediation, and biomarker detection applications. Most notably, the enantiomeric forms of Phe (L and D) have been shown to form mirror-image bio-MOFs in the presence of various metal ions, leading to their application in advanced electronic devices, including chiral light detection devices, chiral-induced spin selective (CISS) conduction effects, and spintronic devices. This comprehensive review provides an in-depth analysis of the recent multifaceted aspects of Phe supramolecular assemblies, covering their morphology, organization, structural studies, and wide-ranging applications in chemistry, biology, and nanotechnology.| S. no. | Supramolecular assembly | Molecules | Structural characteristics | Implications | Ref. |
|---|---|---|---|---|---|
| 1 | Phe self-assembly | Phe | Amyloid fibrils | Phenylketonuria (PKU) disease | 31 |
| 2 | Phe-polymorphism | Phe | Polymorph Phe-I, Phe-II, Phe-III, and Phe-IV with unique space group symmetries and molecular orientations | Thermal phase change behavior for therapeutics and solid-state materials | 51, 52 and 54 |
| 3 | Phe amyloid fibril inhibition | Phe + D-Phe | 2D flakes | PKU therapeutics | 58 |
| Phe + EGCG | Phe fibril disrupted aggregates | 24 | |||
| Phe + TA | Phe fibril disrupted aggregates | ||||
| Phe + 18C6 | Phe fibril disrupted aggregates | 61 | |||
| Phe + 15C5 | Phe fibril disrupted aggregates | ||||
| Phe + Mn+ | Metastable spherical aggregates | 62 | |||
| Beer | Phe fibril disrupted aggregates | 64 | |||
| Phe + Morin hydrate | Potential in vivo PKU therapeutic agent | 66 | |||
| 4 | Phe stimuli-responsive assembly | Phe vs. pH | 2D flakes in acidic and basic media and 1D fibrils in neutral medium | pH tunable Phe self-assembly and nanostructures | 67 |
| Phe vs. (H2O: MeOH) | At various H2O/MeOH solvent ratios, Phe fibrils transformed into fused fibrils with a micrometer diameter | Solvent tunable Phe self-assembly and nanostructures | 43 | ||
| Phe vs. ionic strength | The ionic strength of the medium is proportional to the size of Phe assemblies | Ionic strength tunable Phe self-assembly and nanostructures | 58 | ||
| Phe vs. temperature | The temperature of the medium is proportional to the size of Phe assemblies | Temperature tunable Phe self-assembly and nanostructures | |||
| 5 | Phe co-assembly with amino acids | Phe + Gly/L-Ala/L-Asp) | The self-segregated mixture of 1D fibrils and 2D sheets | Amino acid co-assembly tunable Phe self-assembly and nanostructures | 76 |
| Phe + L-Trp/L-Thr | Co-assembled 1D fibrils | ||||
| Phe + L-Met/L-Val | Co-assembled 2D fibrils | ||||
| Phe + L-Pro | The self-segregated mixture of 1D fibrils | ||||
| Phe + L-Leu/L-Ile | Co-assembled spheres | ||||
| 6 | Phe-Metal assembly | Phe + Fe3O4 | Spheres | Catalytic multicomponent organic synthesis | 85 |
| Phe + Zn2+ | 1D crystals | Carbonic anhydrase mimicking esterase and CO2 hydration activity for biological and environmental remediation | 86 | ||
| Phe + Cu2+ | 2D crystals | Laccase mimicking phenol oxidation activity for environmental remediation and biomarker detection | 87 | ||
| Phe + CuNCs | Spheres | Peroxidase mimicking activity for environmental pollutant dye degradation | 88 | ||
| 7 | Chiral Phe-metal assembly | Phe + Au | Chiral film deposited on the electrode | Chiral light detection device | 91 |
| L/D-Phe + Cu2+ | Chiral 2D crystals | Chirality-induced spin selective conduction device | 92 | ||
| L/D-Phe + Co2+ | Chiral 2D crystals | Chirality-induced spin selective conduction device | 94 | ||
| D-Phe + Cu2+ | Chiral 2D crystals | Spintronic device | 95 |
Hence, the exploration and application of Phe supramolecular assemblies herald an era of significant promise, marking a synergistic intersection across nanotechnology, biomedicine, and materials science. Investigations into the Phe's propensity for amyloid fibril formation, especially in the context of PKU pathology, have expanded our understanding of neurodegenerative diseases and propelled forward the development of cutting-edge nanomaterials and healthcare interventions. In the healthcare sector, the in-depth elucidation of Phe's amyloidogenic characteristics and its role in the pathology of PKU will pave the way for novel therapeutic strategies. These include targeted interventions aimed at mitigating the concentration-dependent cytotoxicity of Phe fibrils and exploring many small biomolecular (or metabolites) co-assembly strategies designed to inhibit the formation of amyloid fibrils. Additionally, the enzyme-like catalytic properties demonstrated by Phe supramolecular assemblies underscore their potential applicability in catalysis, suggesting their utility in organic synthesis, degradation of persistent organic pollutants, development of biosensors, and many more. Looking to the future, incorporating Phe supramolecular assemblies into bioelectronic devices represents a pioneering frontier. The potential of bio-MOFs and the enantiomeric forms of Phe to facilitate the development of chiral electronic devices heralds unprecedented opportunities in electronics. Such advancements promise to revolutionize our interaction with electronic devices by enhancing their sensitivity, selectivity, and versatility, thereby setting a new standard in the field.
In conclusion, exploring the intricate molecular realm of Phe supramolecular assemblies represents merely the initial stages of a promising scientific journey. Phe, distinguished by its unique self-assembly characteristics and broad functional potential, is poised to catalyze significant breakthroughs across various scientific and technological fields. Ongoing research endeavors aimed at deciphering the complex nature and exploiting the capabilities of these assemblies portend a future wherein Phe-based materials and technologies emerge as pivotal contributors to innovation. Such advancements are anticipated to address critical challenges within healthcare, environmental sustainability, and advanced manufacturing sectors, thereby heralding a new scientific discovery and technological development era.
Author contributions
The manuscript was written with contributions from all authors. All authors have approved the final version of the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. M. acknowledges the support of the Science and Engineering Research Board (SERB) of India (SRG/2021/002423) and IIT (BHU) (IIT(BHU)/Budget/2021-22/19-(01)/9221). S. V. thanks the UGC-India and IIT (BHU) for the research fellowship. R. S. thanks the DST-INSPIRE PhD fellowship (IF210343). E. G. thanks the Airforce research laboratories (AFRL) for support. This material is based on work supported by the Air Force Office of Scientific Research under the award number FA8655-21-1-7004. Any opinions, findings, conclusions, or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the United States Air Force.References
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2015, 15, 13–26 CrossRef.
- S. Shaham-Niv, P. Rehak, L. Vuković, L. Adler-Abramovich, P. Král and E. Gazit, Isr. J. Chem., 2017, 57, 729–737 CrossRef CAS.
- J. L. Jiménez, E. J. Nettleton, M. Bouchard, C. V. Robinson, C. M. Dobson and H. R. Saibil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9196–9201 CrossRef.
- A. J. Baldwin, R. Bader, J. Christodoulou, G. E. MacPhee, C. M. Dobson and P. D. Barker, J. Am. Chem. Soc., 2006, 128, 2162–2163 CrossRef CAS PubMed.
- R. Song, X. Wu, B. Xue, Y. Yang, W. Huang, G. Zeng, J. Wang, W. Li, Y. Cao, W. Wang, J. Lu and H. Dong, J. Am. Chem. Soc., 2019, 141, 223–231 CrossRef CAS PubMed.
- K. Tao, P. Makam, R. Aizen and E. Gazit, Science, 2017, 358, eaam9756 CrossRef.
- T. Vijayakanth, S. Dasgupta, P. Ganatra, S. Rencus-Lazar, A. V. Desai, S. Nandi, R. Jain, S. Bera, A. I. Nguyen, E. Gazit and R. Misra, Chem. Soc. Rev., 2024, 53, 3640–3655 RSC.
- T. O. Omosun, M. C. Hsieh, W. S. Childers, D. Das, A. K. Mehta, N. R. Anthony, T. Pan, M. A. Grover, K. M. Berland and D. G. Lynn, Nat. Chem., 2017, 9, 805–809 CrossRef CAS.
- C. S. Sevier and C. A. Kaiser, Nat. Rev. Mol. Cell Biol., 2002, 3, 836–847 CrossRef CAS.
- Y. Liu, L. Zhang and W. Wei, Int. J. Nanomed., 2017, 12, 659–670 CrossRef CAS.
- Q. Liu, A. Kuzuya and Z. G. Wang, iScience, 2023, 26, 105831 CrossRef CAS.
- Y. Wang, Y. Yin, S. Rencus-Lazar, K. Cai, E. Gazit and W. Ji, ChemSystemsChem, 2022, 4, 1–7 CrossRef.
- Y. Wang, S. Rencus-Lazar, H. Zhou, Y. Yin, X. Jiang, K. Cai, E. Gazit and W. Ji, ACS Nano, 2024, 18, 1257–1288 CrossRef CAS PubMed.
- K. Tao, H. Wu, L. Adler-Abramovich, J. Zhang, X. Fan, Y. Wang, Y. Zhang, S. A. M. Tofail, D. Mei, J. Li and E. Gazit, Prog. Mater. Sci., 2024, 142, 101240 CrossRef CAS.
- S. S. Yadav, P. K. Padhy, A. K. Singh, S. Sharma, N. Tanu, S. Fatima, A. Sinha, R. Tariq, N. Varsha, S. K. Sharma and S. Priya, Mater. Adv., 2024, 5, 4078–4090 RSC.
- A. Bansal, M. Schmidt, M. Rennegarbe, C. Haupt, F. Liberta, S. Stecher, I. Puscalau-Girtu, A. Biedermann and M. Fändrich, Nat. Commun., 2021, 12, 1013 CrossRef CAS PubMed.
- S. Peña-Díaz, W. P. Olsen, H. Wang and D. E. Otzen, Adv. Mater., 2024, 2312823, 1–11 Search PubMed.
- S. Abdelrahman, M. Alghrably, J. I. Lachowicz, A.-H. Emwas, C. A. E. Hauser and M. Jaremko, Molecules, 2020, 25, 5245 CrossRef CAS PubMed.
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS.
- C. A. Ross and M. A. Poirier, Nat. Med., 2004, 10, S10 CrossRef PubMed.
- S. A. Levkovich, E. Gazit and D. Laor Bar-Yosef, Trends Microbiol., 2021, 29, 251–265 CrossRef CAS PubMed.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS.
- M. Schmidt, C. Sachse, W. Richter, C. Xu, M. Fändrich and N. Grigorieff, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 19813–19818 CrossRef CAS.
- S. Shaham-Niv, P. Rehak, D. Zaguri, A. Levin, L. Adler-Abramovich, L. Vuković, P. Král and E. Gazit, Commun. Chem., 2018, 1, 25 CrossRef.
- Y. Shen, L. Posavec, S. Bolisetty, F. M. Hilty, G. Nyström, J. Kohlbrecher, M. Hilbe, A. Rossi, J. Baumgartner, M. B. Zimmermann and R. Mezzenga, Nat. Nanotechnol., 2017, 12, 642–647 CrossRef CAS.
- M. Török, Sci. Rep., 2022, 12, 21095 CrossRef PubMed.
- S. Das, R. S. Jacob, K. Patel, N. Singh and S. K. Maji, Biomacromolecules, 2018, 19, 1826–1839 CrossRef CAS PubMed.
- W. Wang, B. He, T. Xiao, M. Xu, B. Liu, Y. Gao, Y. Chen, J. Li, B. Ge, J. Ma and H. Ge, Adv. Funct. Mater., 2023, 33, 1–17 Search PubMed.
- A. S. Cohen and E. Calkins, Nature, 1959, 183, 1202–1203 CrossRef CAS PubMed.
- P. Makam and E. Gazit, Chem. Soc. Rev., 2018, 47, 3406–3420 RSC.
- L. Adler-Abramovich, L. Vaks, O. Carny, D. Trudler, A. Magno, A. Caflisch, D. Frenkel and E. Gazit, Nat. Chem. Biol., 2012, 8, 701–706 CrossRef CAS.
- W. Ji, B. Xue, Z. A. Arnon, H. Yuan, S. Bera, Q. Li, D. Zaguri, N. P. Reynolds, H. Li, Y. Chen, S. Gilead, S. Rencus-Lazar, J. Li, R. Yang, Y. Cao and E. Gazit, ACS Nano, 2019, 13, 14477–14485 CrossRef CAS.
- D. Banik, S. Kundu, P. Banerjee, R. Dutta and N. Sarkar, J. Phys. Chem. B, 2017, 121, 1533–1543 CrossRef CAS.
- P. Chakraborty and E. Gazit, ChemNanoMat, 2018, 4, 730–740 CrossRef CAS PubMed.
- Z. Mohammadzadeh, L. Sharifi, A. Fatholahpour and E. Bazshahi, Int. Breastfeed. J., 2024, 19, 12 CrossRef.
- C. Turgeon, K. Casas, R. Flanagan, A. White, D. Peck, G. B. Pino, A. S. Jones, D. Gavrilov, D. Oglesbee, M. J. Schultz, S. Tortorelli, D. Matern and P. L. Hall, Mol. Genet. Metab. Rep., 2024, 40, 101110 CrossRef CAS.
- M. Cleary, F. Trefz, A. C. Muntau, F. Feillet, F. J. van Spronsen, A. Burlina, A. Bélanger-Quintana, M. Gizewska, C. Gasteyger, E. Bettiol, N. Blau and A. MacDonald, Mol. Genet. Metab., 2013, 110, 418–423 CrossRef CAS PubMed.
- S. Shaham-Niv, Z. A. Arnon, D. Sade, A. Lichtenstein, E. A. Shirshin, S. Kolusheva and E. Gazit, Angew. Chem., Int. Ed., 2018, 57, 12444–12447 CrossRef CAS.
- D. Pinotsi, A. K. Buell, C. M. Dobson, G. S. Kaminski Schierle and C. F. Kaminski, ChemBioChem , 2013, 14, 846–850 CrossRef CAS.
- D. Pinotsi, L. Grisanti, P. Mahou, R. Gebauer, C. F. Kaminski, A. Hassanali and G. S. Kaminski Schierle, J. Am. Chem. Soc., 2016, 138, 3046–3057 CrossRef CAS PubMed.
- Z. A. Arnon, T. Kreiser, B. Yakimov, N. Brown, R. Aizen, S. Shaham-Niv, P. Makam, M. N. Qaisrani, E. Poli, A. Ruggiero, I. Slutsky, A. Hassanali, E. Shirshin, D. Levy and E. Gazit, iScience, 2021, 24, 102695 CrossRef CAS.
- M. Ziaunys and V. Smirnovas, PeerJ, 2019, 7, e6518 CrossRef.
- P. Singh, S. K. Brar, M. Bajaj, N. Narang, V. S. Mithu, O. P. Katare, N. Wangoo and R. K. Sharma, Mater. Sci. Eng., C, 2017, 72, 590–600 CrossRef CAS.
- P. Singh, N. Wangoo and R. K. Sharma, Soft Matter, 2020, 16, 4105–4109 RSC.
- S. K. De, A. Maity and A. Chakraborty, Langmuir, 2021, 37, 5022–5033 CrossRef CAS.
- D. Zaguri, S. Shaham-Niv, P. Chakraborty, Z. Arnon, P. Makam, S. Bera, S. Rencus-Lazar, P. R. Stoddart, E. Gazit and N. P. Reynolds, ACS Appl. Mater. Interfaces, 2020, 12, 21992–22001 CrossRef CAS PubMed.
- T. D. Do, W. M. Kincannon and M. T. Bowers, J. Am. Chem. Soc., 2015, 137, 10080–10083 CrossRef CAS PubMed.
- R. N. Rambaran and L. C. Serpell, Prion, 2008, 2, 112–117 CrossRef PubMed.
- S. Shaham-Niv, L. Adler-Abramovich, L. Schnaider and E. Gazit, Sci. Adv., 2015, 1, e1500137 CrossRef PubMed.
- S. Shaham-Niv, L. Adler-Abramovich, L. Schnaider and E. Gazit, Sci. Adv., 2015, 1, 1–7 Search PubMed.
- P. A. Williams, C. E. Hughes, A. B. M. Buanz, S. Gaisford and K. D. M. Harris, J. Phys. Chem. C, 2013, 117, 12136–12145 CrossRef CAS.
- F. S. Ihlefeldt, F. B. Pettersen, A. von Bonin, M. Zawadzka and C. H. Görbitz, Angew. Chem., Int. Ed., 2014, 53, 13600–13604 CrossRef CAS PubMed.
- E. Mossou, S. C. M. Teixeira, E. P. Mitchell, S. A. Mason, L. Adler-Abramovich, E. Gazit and V. T. Forsyth, Acta Crystallogr., Sect. C: Struct, Chem, 2014, 70, 326–331 CrossRef CAS.
- M. D. King, T. N. Blanton and T. M. Korter, Phys. Chem. Chem. Phys., 2012, 14, 1113–1116 RSC.
- E. Mossou, S. C. M. Teixeira, E. P. Mitchell, S. A. Mason, L. Adler-Abramovich, E. Gazit and V. T. Forsyth, Acta Crystallogr., Sect. C: Struct, Chem, 2014, 70, 326–331 CrossRef CAS.
- B. G. Anand, K. Dubey, D. S. Shekhawat and K. Kar, Sci. Rep., 2017, 7, 1–9 CrossRef CAS PubMed.
- E. Gazit, FEBS J., 2005, 272, 5971–5978 CrossRef CAS PubMed.
- V. Singh, R. K. Rai, A. Arora, N. Sinha and A. K. Thakur, Sci. Rep., 2014, 4, 3875 CrossRef PubMed.
- S. Bera, B. Xue, P. Rehak, G. Jacoby, W. Ji, L. J. W. Shimon, R. Beck, P. Král, Y. Cao and E. Gazit, ACS Nano, 2020, 14, 1694–1706 CrossRef CAS.
- S. Mushnoori, K. Schmidt, V. Nanda and M. Dutt, Org. Biomol. Chem., 2018, 16, 2499–2507 RSC.
- D. Banik, R. Dutta, P. Banerjee, S. Kundu and N. Sarkar, J. Phys. Chem. B, 2016, 120, 7662–7670 CrossRef CAS.
- D. Bagchi, A. Maity, S. K. De and A. Chakraborty, J. Phys. Chem. Lett., 2022, 13, 10409–10417 CrossRef CAS PubMed.
- D. Bagchi, A. Maity, S. K. De and A. Chakraborty, J. Phys. Chem. B, 2021, 125, 12436–12445 CrossRef CAS PubMed.
- D. Banik, P. Banerjee, G. Sabeehuddin and N. Sarkar, Chem. Phys. Lett., 2017, 687, 44–53 CrossRef CAS.
- A. De Luigi, A. Mariani, M. De Paola, A. Re Depaolini, L. Colombo, L. Russo, V. Rondelli, P. Brocca, L. Adler-Abramovich, E. Gazit, E. Del Favero, L. Cantù and M. Salmona, Sci. Rep., 2015, 5, 15902 CrossRef CAS PubMed.
- S. Shaham-Niv, A. Ezra, D. Zaguri, S. R. Shotan, E. Haimov, H. Engel, T. Brider, L. Simhaev, H. M. Barr, L. Adler-Abramovich and E. Gazit, Biophys. Chem., 2024, 308, 107215 CrossRef CAS PubMed.
- D. Tomar, S. Chaudhary and K. C. Jena, RSC Adv., 2019, 9, 12596–12605 RSC.
- Q. Liu, J. Wang, X. Huang, H. Wu, S. Zong, X. Cheng and H. Hao, IUCrJ, 2022, 9, 370–377 CrossRef CAS.
- D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS PubMed.
- S. Uyaver, H. W. Hernandez and M. Gokhan Habiboglu, Phys. Chem. Chem. Phys., 2018, 20, 30525–30536 RSC.
- W. Liyanage and B. L. Nilsson, Langmuir, 2016, 32, 787–799 CrossRef CAS.
- A. S. Rosa, A. C. Cutro, M. A. Frías and E. A. Disalvo, J. Phys. Chem. B, 2015, 119, 15844–15847 CrossRef CAS.
- L. C. Sögütoglu, M. Lutz, H. Meekes, R. de Gelder and E. Vlieg, Cryst. Growth Des., 2017, 17, 6231 CrossRef.
- H. Ren, L. Wu, L. Tan, Y. Bao, Y. Ma, Y. Jin and Q. Zou, Beilstein J. Nanotechnol., 2021, 12, 1140–1150 CrossRef CAS PubMed.
- M. Moazeni, F. Karimzadeh and A. Kermanpur, Soft Matter, 2018, 14, 4996–5007 RSC.
- S. Bera, S. Mondal, Y. Tang, G. Jacoby, E. Arad, T. Guterman, R. Jelinek, R. Beck, G. Wei and E. Gazit, ACS Nano, 2019, 13, 1703–1712 CrossRef CAS PubMed.
- O. S. Tiwari, R. Aizen, M. Meli, G. Colombo, L. J. W. Shimon, N. Tal and E. Gazit, ACS Nano, 2023, 17, 3506–3517 CrossRef CAS PubMed.
- P. Singh, S. K. Pandey, A. Grover, R. K. Sharma and N. Wangoo, Mater. Chem. Front., 2021, 5, 1971–1981 RSC.
- C. Xue, T. Y. Lin, D. Chang and Z. Guo, R. Soc. Open Sci., 2017, 4, 160696 CrossRef.
- D. Van der Helm, M. B. Lawson and E. L. Enwall, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 2411–2418 CrossRef CAS.
- Y. Shimazaki, M. Takani and O. Yamauchi, Dalton Trans., 2009, 7854 RSC.
- V. Subramaniyam, P. V. Ravi and M. Pichumani, J. Mol. Struct., 2022, 1251, 131931 CrossRef CAS.
- K. Tao, A. A. Orr, W. Hu, P. Makam, J. Zhang, Q. Geng, B. Li, J. M. Jakubowski, Y. Wang, P. Tamamis, R. Yang, D. Mei and E. Gazit, J. Mater. Chem. A, 2021, 9, 20385–20394 RSC.
- H. Nosrati, E. Javani, M. Salehiabar, H. Kheiri Manjili, S. Davaran and H. Danafar, J. Mater. Res., 2018, 33, 1602–1611 CrossRef CAS.
- J. Safaei-Ghomi, F. Eshteghal and H. Shahbazi-Alavi, J. Iran. Chem. Soc., 2018, 15, 661–669 CrossRef CAS.
- P. Makam, S. S. R. K. C. Yamijala, K. Tao, L. J. W. Shimon, D. S. Eisenberg, M. R. Sawaya, B. M. Wong and E. Gazit, Nat. Catal., 2019, 2, 977–985 CrossRef CAS.
- P. Makam, S. S. R. K. C. Yamijala, V. S. Bhadram, L. J. W. Shimon, B. M. Wong and E. Gazit, Nat. Commun., 2022, 13, 1505 CrossRef CAS PubMed.
- Y. Wang, Y. Tan, Y. Ding, L. Fu and W. Qing, Colloids Surf., A, 2022, 654, 130072 CrossRef CAS.
- J. S. Shen, G. J. Mao, Y. H. Zhou, Y. B. Jiang and H. W. Zhang, Dalton Trans., 2010, 39, 7054–7058 RSC.
- S. Shekhar, R. Sarker, P. Mahato, S. Agrawal and S. Mukherjee, Nanoscale, 2023, 15, 15368–15381 RSC.
- J. Cai, W. Zhang, L. Xu, C. Hao, W. Ma, M. Sun, X. Wu, X. Qin, F. M. Colombari, A. F. de Moura, J. Xu, M. C. Silva, E. B. Carneiro-Neto, W. R. Gomes, R. A. L. Vallée, E. C. Pereira, X. Liu, C. Xu, R. Klajn, N. A. Kotov and H. Kuang, Nat. Nanotechnol., 2022, 17, 408–416 CrossRef CAS PubMed.
- A. K. Mondal, N. Brown, S. Mishra, P. Makam, D. Wing, S. Gilead, Y. Wiesenfeld, G. Leitus, L. J. W. Shimon, R. Carmieli, D. Ehre, G. Kamieniarz, J. Fransson, O. Hod, L. Kronik, E. Gazit and R. Naaman, ACS Nano, 2020, 14, 16624–16633 CrossRef CAS.
- N. Goren, T. K. Das, N. Brown, S. Gilead, S. Yochelis, E. Gazit, R. Naaman and Y. Paltiel, Nano Lett., 2021, 21, 8657–8663 CrossRef CAS.
- T. Kumar Das, A. K. Mondal, O. S. Tiwari, P. Makam, G. Leitus, E. Gazit, F. Claudio and R. Naaman, Phys. Chem. Chem. Phys., 2023, 25, 22124–22129 RSC.
- N. Goren, T. K. Das, N. Brown, S. Gilead, S. Yochelis, E. Gazit, R. Naaman and Y. Paltiel, Nano Lett., 2021, 21, 8657–8663 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |