
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioorthogonally activated probes for precise fluorescence imaging
Youxin
Fu†
 ab,
Xing
Zhang†
bc,
Luling
Wu
d,
Miaomiao
Wu
b,
Tony D.
James
*de and
Run
Zhang
*b
ab,
Xing
Zhang†
bc,
Luling
Wu
d,
Miaomiao
Wu
b,
Tony D.
James
*de and
Run
Zhang
*b
aCollege of Science, Nanjing Forestry University, Nanjing, 210037, P. R. China
bAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, St Lucia, Queensland 4072, Australia. E-mail: r.zhang@uq.edu.au
cCollege of Ecology and Environment, Nanjing Forestry University, Nanjing, 210037, P. R. China
dDepartment of Chemistry, University of Bath, Bath BA2 7AY, UK. E-mail: t.d.james@bath.ac.uk
eSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, China
First published on 18th November 2024
Abstract
Over the past two decades, bioorthogonal chemistry has undergone a remarkable development, challenging traditional assumptions in biology and medicine. Recent advancements in the design of probes tailored for bioorthogonal applications have met the increasing demand for precise imaging, facilitating the exploration of complex biological systems. These state-of-the-art probes enable highly sensitive, low background, in situ imaging of biological species and events within live organisms, achieving resolutions comparable to the size of the biomolecule under investigation. This review provides a comprehensive examination of various categories of bioorthogonally activated in situ fluorescent labels. It highlights the intricate design and benefits of bioorthogonal chemistry for precise in situ imaging, while also discussing future prospects in this rapidly evolving field.
 Youxin Fu | Youxin Fu is an Assistant Professor at the College of Science, Nanjing Forestry University (NJFU). He received his BSc (2014) and MSc (2017) from the East China University of Science and Technology. He took up a Research Assistant position at the Australian Institute for Bioengineering and Nanotechnology, The University of Queensland (AIBN, UQ), in 2018. He pursued a PhD (2019–2023) at the University of Groningen under the supervision of Prof. Ben L. Feringa and Prof. Wiktor Szymanski, focusing on developing visible light-induced ultrafast photoclick reaction systems. His current research at NJFU focuses on developing new biobased photoclick chemistry systems. |
 Xing Zhang | Xing Zhang is an Assistant Professor at the College of Ecology and Environment, Nanjing Forestry University. She graduated from the School of Environmental and Chemical Engineering of Shanghai University in 2021 with a PhD in Engineering. From 2018 to 2020, she was an exchange doctoral student at the Australian Institute for Bioengineering and Nanotechnology, The University of Queensland (AIBN, UQ), Australia. In 2022, she joined Department of Bioengineering, University of Illinois Urbana-Champaign as a postdoctoral researcher to work on per- and polyfluoroalkyl substances (PFAS). At the Nanjing Forestry University, her research focuses on visual detection of environmental organic pollutants. |
 Luling Wu (left) and Tony D. James (right) | Luling Wu (left) is an EPSRC postdoctoral research fellow at the University of Bath. He obtained his PhD in 2021 supported by the China Scholarship Council (CSC) and University of Bath. His research focuses on the design and synthesis of novel fluorescent probes for disease diagnosis applications. Tony D. James (right) is a Professor at the University of Bath and a Fellow of the Royal Society of Chemistry. He was a Wolfson Research Merit Award holder (2017–2022) and was awarded the Daiwa Adrian Prize (2013), the CASE Prize (2015), the MSMLG Czarnik Award (2018) and Frontiers in Chemistry Diversity Award (2020). His h-index is 91 (Google Scholar) and has been listed by Clarivate as a Highly Cited Researcher since 2022 (selfie taken in August 2024 at the University of Bath Lake). |
 Miaomiao Wu | Miaomiao Wu completed her master's degree in the Frontier Institute of Science and Technology, Xi’an Jiaotong University in 2019. She is now a PhD student in the Australian Institute for Bioengineering and Nanotechnology, The University of Queensland (AIBN, UQ). Her current research interest is in exploring the growth mechanism of two-dimensional nanostructures and building a novel nano-platform for imaging and improving the efficiency of cancer treatment. |
 Run Zhang | Run Zhang is a Senior Research Fellow at the Australian Institute for Bioengineering and Nanotechnology, The University of Queensland (AIBN UQ). He received his PhD in Analytical Chemistry from the Dalian University of Technology in 2012. He worked at Macquarie University (MQ) as a Postdoc (2012) and as an independent MQ Research Fellow (2013–2015). He joined AIBN UQ in 2016 and received ARC DECRA (2017–2019) and NHMRC Emerging Leadership (2020) fellowships. He now leads a Sensing and Imaging Research Group at AIBN UQ, working on the development of responsive molecules/nanomaterials for biosensing and imaging, early disease detection and treatment, and food/agricultural/environmental applications. |
1. Introduction
Molecular bioimaging1,2 using advanced imaging techniques, such as fluorescence imaging,3,4 Raman imaging,5–7 magnetic resonance imaging (MRI),8,9 positron emission tomography (PET),10 photoacoustic imaging,11 and single-photon emission computed tomography (SPECT),12 profoundly impacts biomedical research, disease diagnosis and treatment monitoring, and clinical diagnostics. Among these methods, fluorescence imaging is particularly interesting because it offers us the ability to quantitatively visualize and understand molecular processes in living systems from cellular organelles to entire organisms with high spatiotemporal resolution. The development of fluorescent probes,3 pivotal tools for fluorescence imaging, plays a key role in advancing fluorescence imaging techniques, enabling a deeper understanding of biological processes. Over recent decades, many fluorescent probes have been successfully developed for detecting and tracking cellular dynamics, molecular interactions, and tissue architecture.Traditional fluorescent probes,3,13 involving the direct use of emissive dyes as well as dye conjugated biomolecules, such as antibodies, have been widely adopted for staining the cells and tissues for fluorescence imaging.14 These probes, while effective, often encounter challenges associated with nonspecific binding within cellular and tissue environments, resulting in unexpected signals with compromised accuracy and specificity in fluorescence analysis and imaging. The ability to differentiate genuine signals from nonspecific noise is crucial for acquiring reliable and meaningful information in fluorescence imaging, particularly within the complex context of biological systems.15 Photobleaching of the fluorophores represents another major concern in conventional fluorescence imaging.16 Prolonged exposure to light excitation causes irreversible degradation of fluorophores, restricting the duration of imaging experiments for monitoring the evolution of biomarkers.17 Moreover, autofluorescence18 originating from endogenous fluorescent molecules within cells and tissues poses another challenge in fluorescence analysis and imaging. The presence of intrinsic fluorophores, such as flavins19 and lipofuscins,20 produces intense background signals that overlap with those emitted by the fluorescent probes. As such discriminating specific probe signals from intrinsic background autofluorescence is pivotal for achieving reliable imaging outcomes.
There is a growing emphasis on the development of advanced fluorescent probes with enhanced specificity, minimized nonspecific interactions, and improved photostability.21,22 Innovative strategies explored for the design of new generation probes include the incorporation of responsive elements into probes and the design of alternative fluorophores with superior photophysical properties, such as responsive metal complexes,23 aggregation-induced emission (AIE) probes,24–27 and near-infrared (NIR) II fluorescent dyes.28,29 In brief, while fluorescent probes have contributed significantly to the field of bioimaging, recognizing and mitigating the limitations of traditional probes are crucial for developing and validating new probes that can detect and image biomolecules in living cells and organisms.
Bioorthogonal chemistry30–32 refers to reactions occurring within biological settings without interruption of biomolecule synthesis or biochemical processes. Pioneered by the Carolyn Bertozzi group in 2003,33 the term likely stems from the mathematical concept of orthogonality, indicating independent variation. Over the past two decades, bioorthogonal chemistry has evolved into a crucial tool for biological research. The key criteria of bioorthogonal reactions include operation under physiological conditions, selective and high-yield product formation, resilience to biological agents, rapid product formation at low concentrations, and stable product generation. Moreover, bioorthogonal chemistry involves functional groups that are not naturally present in biological systems, ensuring compatibility within complex environments. This methodology has significantly advanced biological studies by facilitating precise investigation of cellular processes while maintaining biological integrity. Notably, bioorthogonal chemistry, exemplified by copper-catalyzed azide–alkyne cycloaddition (CuAAC),34–37 Staudinger ligation,3 inverse-electron-demand Diels–Alder (IEDDA) reactions,38–40 and 1,2-aminothiol-2-cyanobenzothiazole (CBT) click reactions,41,42 has garnered enormous attention for selective biomolecule labeling,43 targeted imaging,44 and drug delivery45in vivo over recent years.
A signalling unit (chemical reporter) and a bioorthogonal click reaction handle are the two key components of bioorthogonally activated probes.46 The majority of reported bioorthogonal probes are designed by directly incorporating a click reaction handle into fluorescent dyes.31,43,47,48 Following the bioorthogonal reaction the stringent washing to remove any unreacted bioorthogonal probes is essential to reduce background fluorescence. Bioorthogonally activated probes have been subsequently developed to enhance the imaging precision and streamline sample pre-treatment procedures.23,44 These responsive probes exhibit weak fluorescence initially and can be activated by bioorthogonal reactions, resulting in an increase in the fluorescence signal. This kind of selective activation enables the probes to emit fluorescence only in regions where the bioorthogonal reaction occurs. As a result, there is no need for additional washing process in the molecular fluorescence imaging of cells, tissues, and living organisms that are stained with bioorthogonally activated probes. Such bioorthogonally activated probes are particularly useful in scenarios where extensive washing of the samples is not feasible.
In this review, we provide a comprehensive overview of the advancements in bioorthogonally activated fluorescent probe technology over the last decade, with particular focus on applications in precise, wash-free, and in situ biological imaging. We summarize the activated probes based on the types of bioorthogonal reactions, providing a succinct introduction to each reaction in each subsection. Subsequently, we outline recent research progress, identify the limitations of each bioorthogonal reaction, and discuss key future directions. This is followed by a comprehensive summary, emphasizing both the challenges and opportunities faced by bioorthogonally activated probes in achieving precise in situ biological imaging.
2. Bioorthogonally activated probes generated by different activation mechanisms
The advancement of bioorthogonal activated probes strongly relies on the underlying mechanisms of bioorthogonal activation. Since 2003, eight distinct categories of bioorthogonal activation have been identified and successfully adopted for the development of activated probes for precision bioimaging (Scheme 1). These mechanisms of bioorthogonal activation include Staudinger ligation, Cu(I)-catalyzed azide–alkyne cycloadditions (CuAAC), inverse-electron-demand Diels–Alder reactions (IEDDA), 1,2-aminothiol-2-cyanobenzothiazole (CBT) click reactions, strain promoted sydnone alkyne cycloadditions (SPSAC), nitrile imine–alkyne cycloadditions, photoclick reactions, and bioorthogonal nanozyme-catalyzed reactions. In this section, we will provide insights into advances in the development of bioorthogonally activated probes based on the eight bioorthogonal activation mechanisms as well as specific probes for precise fluorescence imaging.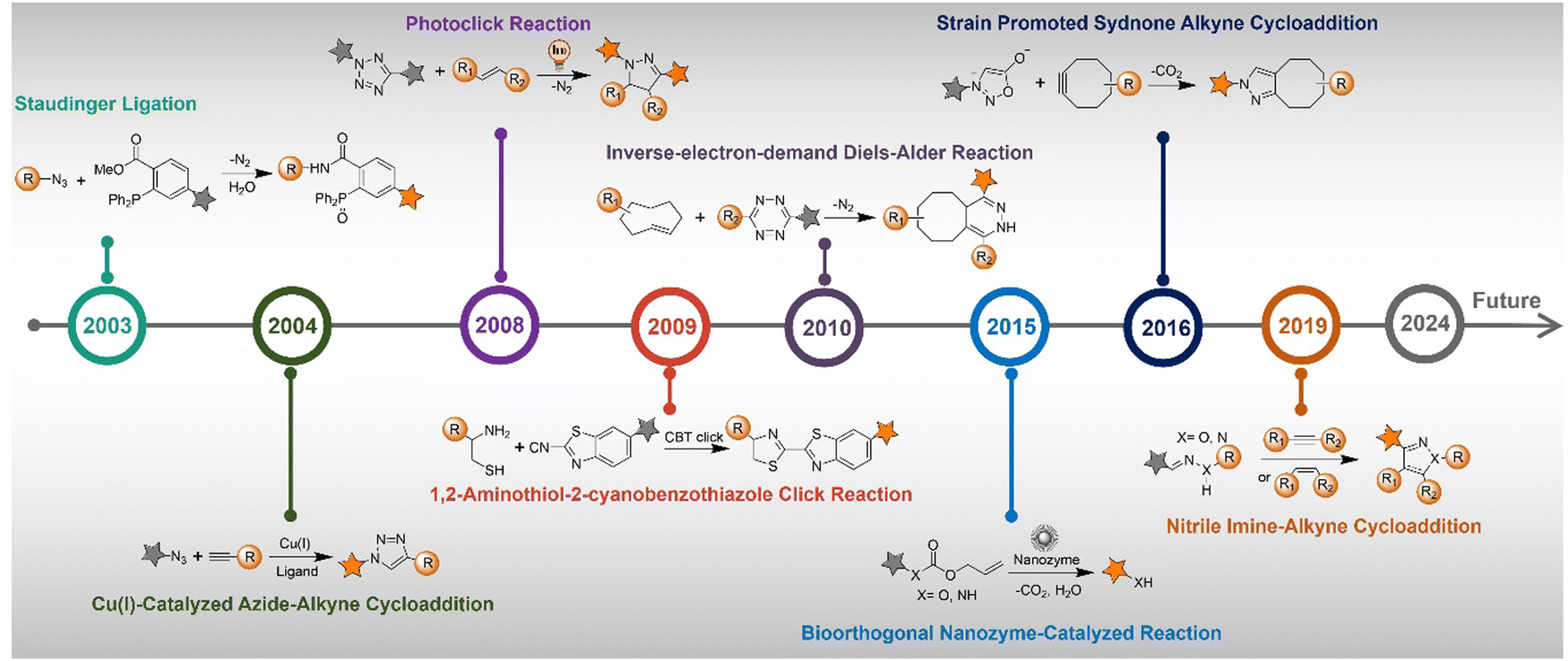 | ||
| Scheme 1 A timeline of development of the bioorthogonally activated probes based on eight activation reactions. | ||
2.1. Staudinger ligation
Staudinger ligation49–51 is an established bioorthogonal reaction derived from the classic Staudinger reduction.52 This bioorthogonal reaction involves the use of organic azides and triphenylphosphine-functional groups as the key components. The underlying mechanism of this bioorthogonal reaction is depicted in Fig. 1A. Diverging from the traditional Staudinger reduction pathway, the intermediate aza-ylide reacts with an adjacent ester, yielding a phosphorane that subsequently collapses to yield a stable amide linkage and a phosphine oxide. The second-order rate constant (k2) for this reaction is approximately ∼10−3 M−1 s−1 in acetonitrile. During the reaction, the phosphine undergoes oxidation, forming a phosphine oxide, while the ester is cleaved, releasing methanol. These concurrent processes have been strategically exploited in developing advanced phosphine probes, activated via Staudinger ligation. | ||
| Fig. 1 Fluorescent probes based on Staudinger ligation. (A) Proposed mechanism of Staudinger ligation, (B) chemical structure and cell imaging of Glu-HT-Me, adapted with permission from ref. 53. Copyright 2022, American Chemical Society; (C) chemical structure and cell imaging of Se-NR, adapted with permission from ref. 54. Copyright 2019, Royal Society of Chemistry; (D) chemical structure and cell imaging of DCX-TPP, adapted with permission from ref. 55. Copyright 2019, Science Direct. | ||
The Staudinger ligation is known for being bioinert and biocompatible since both reactants are entirely nonbiological, which enables selective ligation across all biological compartments, distinguishing it from aldehyde or ketone condensation reactions. Popularized by Bertozzi's work,49,56,57 this reaction has evolved into a standard approach for the development of bioorthogonal probes and the modulation of biological processes. Herein, we examine the latest advancements in using Staudinger ligation-based bioorthogonal probes for precise in situ imaging of living organisms.
Chemical reactions that can occur within specific cellular environments contribute to the investigation of biological processes and the engineering of cell functions. A landmark achievement in this realm was reported in 2000 by the Bertozzi group,49 introducing a chemical transformation that enables the precise formation of covalent adducts among intricately functionalized biopolymers within the cellular matrix. This transformative ligation, inspired by the Staudinger reaction, facilitates the formation of amide bonds through the coupling of an azide moiety with a custom-engineered triarylphosphine. Both reactive units, i.e., azide and triarylphosphine, are synthetic and chemically orthogonal to endogenous cellular constituents. By harnessing metabolic pathways, azides can be incorporated into cell surface glycoconjugates via a synthetic azidosugar. The azide then selectively reacts with a biotinylated triarylphosphine, yielding stable cell-surface adducts. This research provided a new pathway for the simple engineering of cell surfaces using bioorthogonal reactions.
Apart from the glycoconjugate for cell surface modification, cellular glucose uptake plays a pivotal role in energy generation and is implicated in various metabolic disorders. To understand the details of the cellular glucose uptake process, Liang and coworkers53 developed a bioorthogonal fluorescent probe, consisting of two parts Glu-HT-Me and azido-glucose, to light up the glucose uptake in live cells (Fig. 1B). The fluorescence of Glu-HT-Me was quenched due to photo-induced electron transfer (PeT) and the fluorescence was significantly increased (20-fold enhancement) when activated by internalized azido-glucose within 25 min. The fluorescence “OFF” to “ON” response allowed for monitoring glucose uptake dynamics in a wash-free manner. More interestingly, this bioorthogonal light-up fluorescent probe was able to discriminate cancer cells from normal cells due to the different glucose flux in anticancer drug treatment, glycolysis, and transport. The potential application of this probe in tracking glucose uptake fluctuations in a doxycycline-inducible K-rasG12V oncogenic cell system was confirmed.
In addition to the bioorthogonal reaction in a cellular environment, bioorthogonal moieties have been used for the development of activatable photosensitizers and fluorescent probes for reactive biomolecule detection. In 2019, the Liu group54 synthesized a π-extended Se-rhodamine (Se-NR, Fig. 1C) that is capable of efficiently producing singlet oxygen (ΦΔ = 0.35) upon irradiation with red light. Leveraging precise control over the spirocyclization of Se-NR, a bioorthogonally activatable photosensitizer (Se-NR-Az) was developed by masking the amine group as an azide. This Se-NR-Az exhibited low cytotoxicity even under red light irradiation. However, the bioorthogonal reaction of Se-NR-Az with a phosphine resulted in the formation of a photocytotoxic compound, i.e., Se-NR. This bioorthogonally activatable photosensitizer was then used for the controlled inhibition of the growth of HeLa cells by producing reactive oxygen species (ROS).
In another example, the triarylphosphine bioorthogonal moiety has been used by Zhang and coworkers for the development of an activatable probe (DCX-TPP) for the detection of nitroxyl (HNO), a type of ROS derived from nitric oxide (NO) through reduction and protonation (Fig. 1D).55 The probe DCX-TPP was developed by combining a bioorthogonal HNO-recognition moiety, the diphenylphosphinobenzoyl group, with a NIR fluorophore via an ester linker. Initially non-fluorescent, HNO-catalyzed oxidation led to the activation of the probe and the emission of red fluorescence. The probe DCX-TPP could be used to detect 2–80 μM HNO within 10 min with a detection limit (LoD) of 0.05 μM and was able to visualize endogenous HNO in living cells.
In summary, the classic Staudinger ligation uses an azide, a small and biocompatible functional group, seamlessly integrated into biomolecules, for selective reactions with phosphine. However, its sluggish kinetics frequently lead to suboptimal labeling efficiency. While electron-rich phosphines can accelerate the bioorthogonal reaction, the rapid oxidation of phosphines under physiological conditions represents a significant challenge to the ligation process. As such, high concentrations of phosphine reagents are often required to ensure efficient bioorthogonal reactions. However, excess phosphine regents can result in an increase in the background signal during fluorescence imaging, causing false positive results. While the Staudinger ligation remains one of the most important activation mechanisms in the development of bioorthogonal probes, more recent efforts have focused on exploring faster ligation techniques in various scenarios.
2.2. Copper(I)-catalyzed azide–alkyne cycloaddition
The azide and terminal alkyne functional groups, being small and primarily engaged in weak dipolar interactions, exert minimal influence on the functional properties of the molecules to which they are attached. Both azides and alkynes exhibit low reactivity in biological systems, enabling them to traverse intact cells before being triggered by specific conditions or catalysts. The Huisgen 1,3-dipolar cycloaddition58 of organic azides and alkynes, yielding 1,2,3-triazoles, is typically too slow for biological applications (Scheme 2A). However, the introduction of Cu(I) catalysts (Scheme 2B), pioneered by Meldal and Sharpless, significantly accelerates triazole formation up to 100 million-fold.34–36,59 The Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) facilitates the formation of 1,4-disubstituted triazoles exclusively, mimicking native peptide-based connectors in size and geometry and offering superior stability to thermal and chemical cleavage.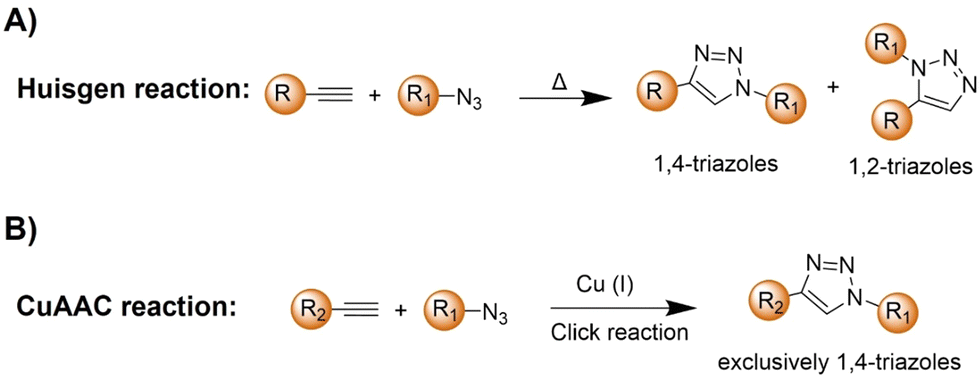 | ||
| Scheme 2 Illustration of the (A) Huisgen reaction and (B) CuAAC reaction. | ||
CuAAC is modular, compatible with aqueous conditions, and largely by-product-free, facilitating successful click reactions. By using various catalysts, including simple Cu(I) salts, small-molecule Cu-binding ligands, and diverse materials, the application of CuAAC has been confirmed in organic synthesis,60 polymer functionalization,61 drug discovery,62 and chemical biology.63 While naturally occurring azides are rare, labelling of proteins, lipids, oligonucleotides, and cell surface moieties with azides or alkynes can be achieved through benign chemical transformations or metabolic incorporation of labeled metabolites, enabling their conjugation to orthogonally labeled partners.
The adoption of CuAAC for bioorthogonal bioconjugation has been significantly advanced by the development of water-soluble Cu(I)-binding ligands, mitigating the limitation of Cu(I)-mediated ROS formation through Fenton reaction with endogenous H2O2. These ligands stabilize the Cu(I) oxidation state, accelerating the CuAAC reaction while minimizing ROS production through the Fenton reaction. Ligands with additional features, such as ROS scavenging, have expanded the use of CuAAC for more demanding biological applications, including in situ labeling of living cellular surfaces, tagging enzymes or biomolecules in complex cellular mixtures, rapid click reactions within cellular compartments, and versatile dye labeling of diverse cellular structures for super-resolution microscopy. Herein, we provide a comprehensive overview of CuAAC reaction applications in the realm of in situ precision imaging over the last decade, categorized by the types of probes developed.
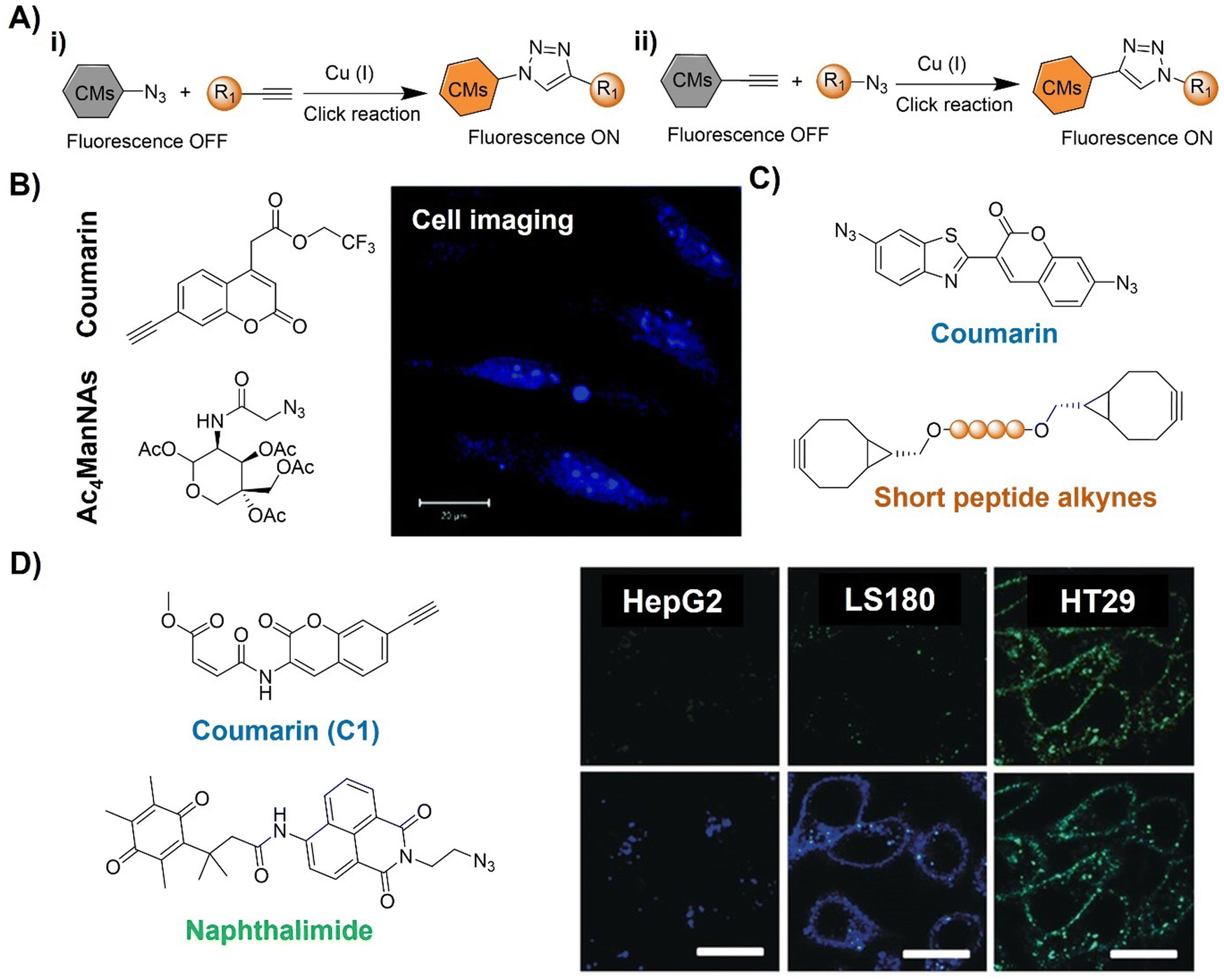 | ||
| Fig. 2 Coumarin fluorophore-based bioorthogonal probes using copper(I)-catalyzed azide–alkyne cycloaddition. (A) Illustration of the CuAAC activated coumarin probes; (B) the structure and cell imaging of the alkynyl modified coumarin, adapted with permission from ref. 64. Copyright 2014, The Royal Society of Chemistry; (C) the structures of bis-azide coumarin and biscyclooctynylated peptides; (D) the structures of the profluorescent coumarin derivative (C1) and azido-naphthalimide and bioimaging in different cell lines, adapted with permission from ref. 65. Copyright 2017, Wiley-VCH GmbH. | ||
A bis-azide fluorogenic probe for two-point bioorthogonal tagging was developed by Kele et al.66 for labeling biscyclooctynylated peptides (Fig. 2C). This probe exhibited robust fluorescence enhancement with a polar hexapeptide containing two cyclooctyne motifs. Theoretical computation revealed that the fluorescence “off–on” response is dependent on dihedral angles between the probe's aromatic core. Amongst the screened biscyclooctynylated peptides, a polar hexapeptide demonstrated near-maximal fluorogenicity. These bis-azides offered a new approach to develop bioorthogonal probes for specific, fluorogenic two-point labeling of proteins containing two genetically encoded cyclooctyne motifs.
Similarly, Liu and colleagues65 developed a doubly caged heterodifunctional linker, a profluorescent coumarin derivative (C1) with orthogonal dual “click” motifs, for functionalization of protein/antibody bioconjugates (Fig. 2D). The core molecule, C1 contains both CuAAC and Michael addition sites on a coumarin skeleton for a “click” style reaction with azide- and thiol-functionalized precursors, respectively. Click reactions of the ethynyl of C1 with naphthalimide QNAM-N3 catalyzed by Cu(I) catalysis and the C1's alkenyl amide/ester unit with a thiol functionalized anti-carcinoembryonic antigen (ACEA) produced QNAM-C1-ACEA and switched on coumarin's emission at 435 nm. The quinone-caged naphthalimide exhibited weak fluorescence, while the emission could be switched on (λem = 530 nm) after a quinone oxidoreductase (NQO1)-triggered cleavage of the quinone cage. Moreover, overlapping of coumarin's emission with the absorption of naphthalimide's allowed FRET upon excitation at 365 nm, facilitating the ratiometric fluorescence response of QNAM-C1-ACEA for analysis and imaging of NQO1. This double-caging strategy and modular/fluorogenic fabrication of functional protein/antibody conjugates offers versatility, potentially integrating with other bioorthogonal reactivity methods.
Cu is an essential element in the body and is linked to neurodegenerative disorders, notably Alzheimer's disease, marked by Aβ aggregation and Cu accumulation. In addition to the exogenous Cu catalyst, the Qu group67 found that Cu accumulation in Aβ plaques can effectively catalyze an azide–alkyne bioorthogonal cycloaddition for fluorescence activation of coumarin and drug synthesis in living cells, a transgenic AD model of Caenorhabditis elegans CL2006, and brain slices of triple transgenic AD mice (Fig. 3A). The as-synthesized drug destroyed Aβ-Cu aggregates, alleviating paralysis and locomotion defects. This in situ approach is a self-triggered drug synthesis method for AD therapy, a pioneering approach utilizing in situ Cu for click reactions in physiological environments.
 | ||
| Fig. 3 Coumarin fluorescent probes based on the copper(I)-catalyzed azide–alkyne cycloaddition. (A) The structure, flow cytometry detection and worm in situ imaging of the coumarin-Aβ-Cu system, adapted from ref. 67, under the license CC BY-NC 3.0 DEED; (B) the structure, flow cytometry detection and in situ cell imaging of the coumarin–MCNs–Cu system, adapted with permission from ref. 68. Copyright 2020, American Chemical Society; (C) the structure and in situ cell imaging of the coumarin–DNAzyme–Cu system, adapted from ref. 69, under the license CC BY-NC 3.0 DEED; and (D) the structure and in situ cell imaging and plumule imaging of the coumarin–MCNs/GSH–Cu(II) system, adapted with permission from ref. 70. Copyright 2021, Elsevier B.V. | ||
Taking advantage of enhanced stability and biosafety, heterogeneous Cu nanoparticles (CuNPs) have emerged as promising contenders for facilitating the CuAAC reaction. Research conducted by Qu et al.68 resulted in a NIR light promoted CuAAC reaction using a biocompatible heterogeneous copper nanocatalyst, which was developed for investigating both photodynamic and photothermal effects in vitro and in vivo (Fig. 3B). Specifically, upon NIR light irradiation, Cu nanoparticle attached mesoporous carbon nanospheres (MCNs-Cu) produce reactive oxygen species (ROS) and heating in a local microenvironment through photodynamic and photothermal (η = 50.6%) effects, respectively, which promote the conversion of inert Cu(0) to Cu(I), expediting the catalytic CuAAC process. This synergistic effect resulted in enhanced activation of a profluorophore (3-azido-7-hydroxycoumarin), as observed in cellular and murine models under NIR irradiation. In addition to the use of CuNPs as a catalyst for CuAAC reactions, further research from the same group also used DNAzyme-augmented bioorthogonal catalysis for targeted synergistic cancer therapy.69 The design of a DNAzyme leveraged DNA structure and substrate interaction enabled precise imaging in situ and efficient prodrug activation in cancer cells for synergistic cancer therapy (Fig. 3C).
CuAAC reaction activated 3-azido-7-hydroxycoumarin has also been explored for the development of fluorescent probes for the detection of glutathione (GSH), antimicrobial resistant bacteria, and foodborne pathogens. In 2021, Song and colleagues70 devised a GSH-triggered MSN (mesoporous silica nanoparticle) nanoreactor featuring a Cu(II)-BTC shell, where Cu(I), generated through in situ reduction by GSH, serves as a catalyst (Fig. 3D). This catalytic activity expedites the bioorthogonal reaction between an alkyne and azide, thereby enhancing the fluorescence of 7-hydroxycoumarin and 4-ethynylanisole loaded within the MSNs. Evaluation of these nanoreactors for GSH detection, spanning cellular to wheat plumule levels, demonstrated their robust biocompatibility and potential for detecting metal ions in plants. As such, the monitoring of intracellular bioorthogonal reactions could advance precision chemistry in the design of intelligent platforms for biomarker detection as well as targeted drug synthesis for precision medicine.
Antibiotic resistance, stemming from the ongoing overuse of antibiotics, poses a global threat to human and animal health, environmental stability, food and nutrition security, economic development, and societal equity and safety. Detection of pathogens, particularly antibiotic-resistant bacteria, has contributed to addressing this threat. CuAAC activated probes based on coumarin derivatives have been applied for foodborne pathogen detection. For example, Gan et al.71 reported a simple, sensitive on-site bioassay for foodborne pathogens employing the Phage@DNAzyme probe (3-azido-7-hydroxycoumarin, Fig. 4A). The probe displayed specificity towards bacterial strains (E. coli as the model) and catalytic efficiency for fluorescent CuAAC click reactions. Synergistic signal amplification using a gold nanoparticle modified slide and Phage@DNAzyme catalysis enabled rapid, sensitive, on-site bacterial measurement with a limit of detection (LOD) of 50 CFU mL−1 in 30 minutes, which could be detected using a smartphone. Using a CuAAC reaction, Yu et al.72 developed a rapid bioorthogonal noncanonical amino acid (BONCAT) labelling method for the detection of antimicrobial-resistant bacterial cells in an aquatic environment (Fig. 4B). Using this system the fluorescence of 3-azido-7-hydroxycoumarin was switched on after a CuAAC reaction with the L-homopropargylglycine incubated E. coli. In the presence of an antibiotic (e.g., chloramphenicol), the antibiotic-resistant E. coli could be easily identified within 20 min of coincubation with 3-azido-7-hydroxycoumarin.
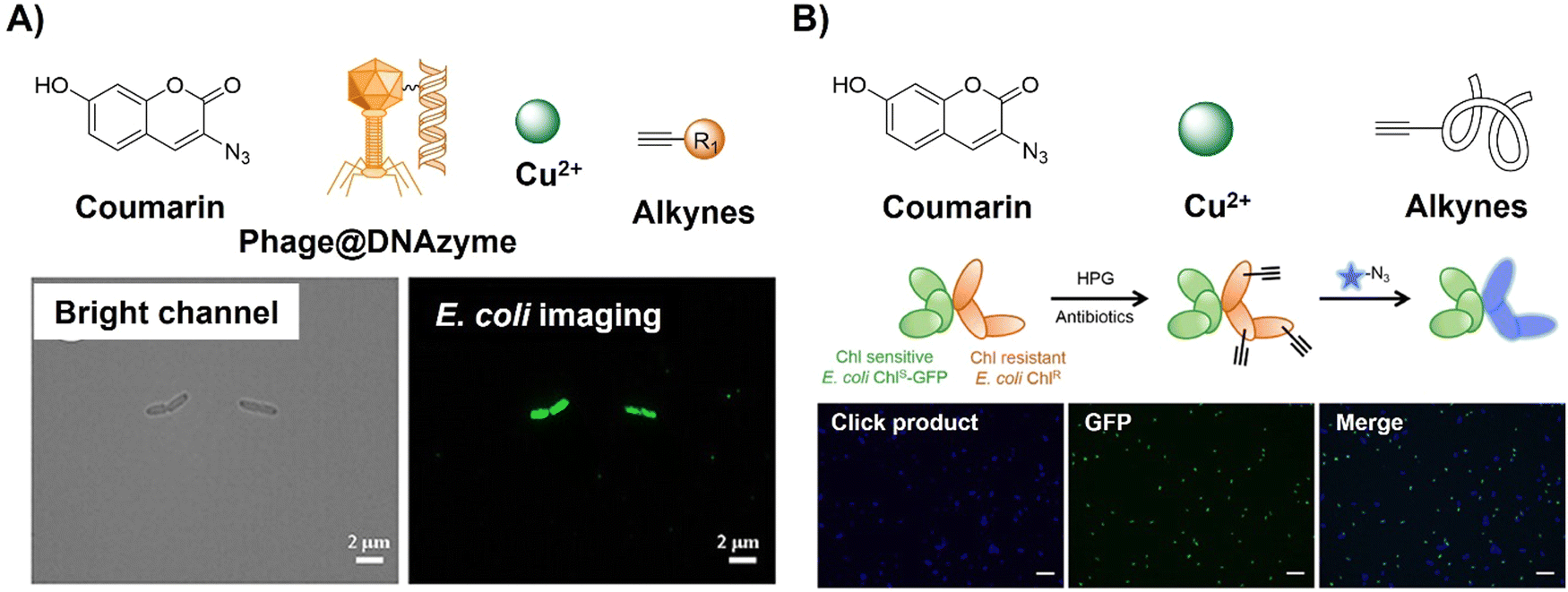 | ||
| Fig. 4 Coumarin fluorophore-based bioorthogonal probes obtained using the copper(I)-catalyzed azide–alkyne cycloaddition. (A) The structure and E. coli imaging of the Phage@DNAzyme probe, adapted with permission from ref. 71. Copyright 2023, American Chemical Society; (B) the structure and selective E. coli labeling of the azido-coumarin system, adapted with permission from ref. 72. Copyright 2022, American Chemical Society. | ||
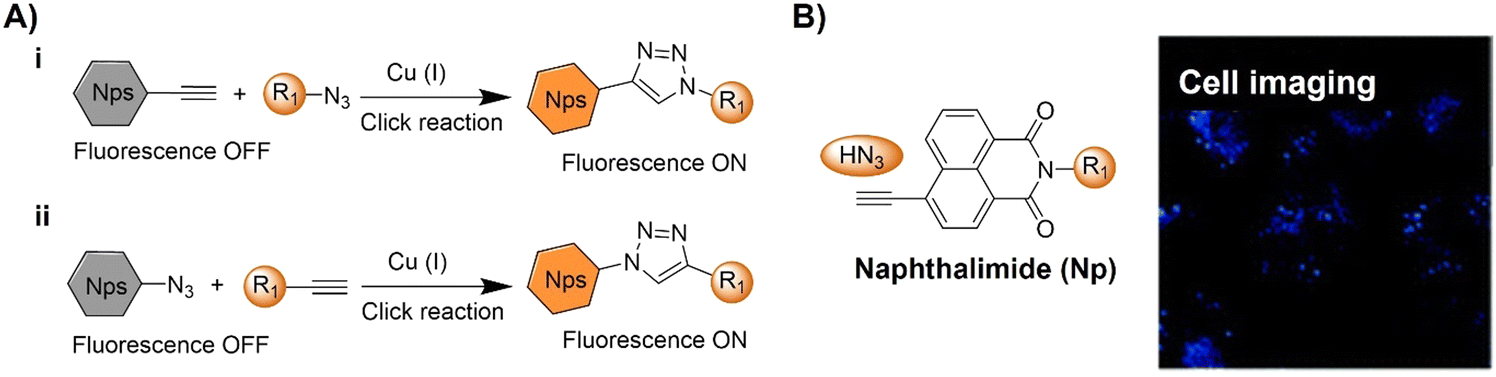 | ||
| Fig. 5 Bioorthogonal fluorescent probes based on the copper(I)-catalyzed azide–alkyne cycloaddition with a naphthalimide fluorophore. (A) Illustration and (B) chemical structure of the CuAAC activated naphthalimide probe for cell imaging. Adapted with permission from ref. 73. Copyright 2013, The Royal Society of Chemistry. | ||
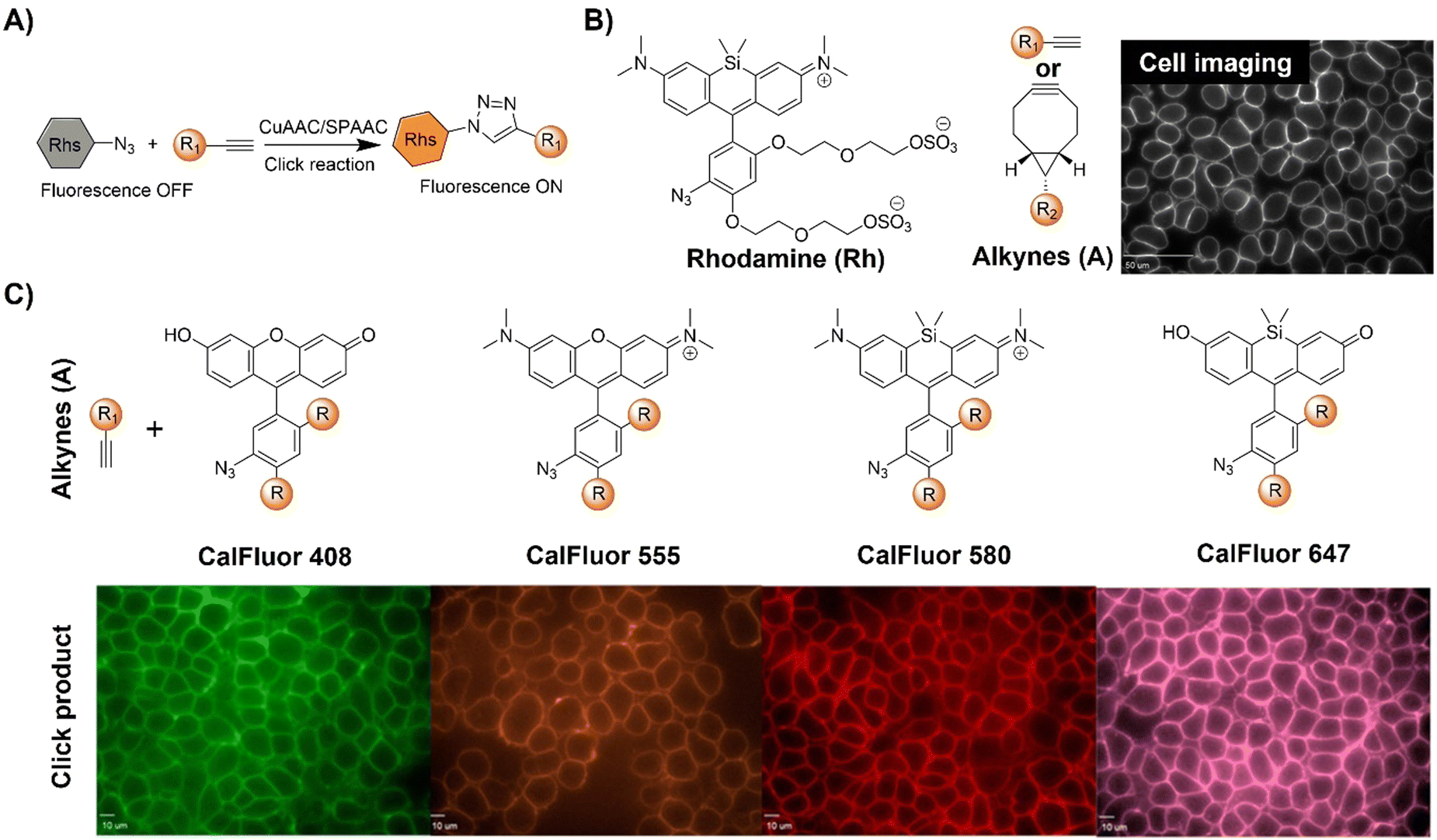 | ||
| Fig. 6 Bioorthogonal fluorescent probes based on the copper(I)-catalyzed azide–alkyne cycloaddition with rhodamine derivatives. (A) Illustration of the CuAAC activated rhodamine probe; (B) the structure and the wash-free mammalian cell surface labeling of the Si-rhodamine, adapted with permission from ref. 74. Copyright 2014, National Academy of Sciences; (C) structures of CalFluors 488, 555, 580, and 647 and their ability to enable the no-wash labeling of cell-surface glycoproteins on HEK 293T cells, adapted with permission from ref. 75. Copyright 2015, American Chemical Society. | ||
In a biological environment, bioorthogonal fluorescent probes spanning the visible spectrum offer precise visualization of metabolically labeled molecules. Toward this end, Bertozzi et al.75 reported the development of highly fluorogenic azide probes based on the PeT fluorescence response mechanism (Fig. 6B). Activated by click chemistry to form triazoles, these probes exhibited remarkable fluorescence enhancement, up to 283-fold, with a ΦF of 0.74. Exploiting a similar PeT mechanism, bioorthogonal probes were developed using diverse fluorophores including bodipy, cyanines, and pyrazolines. Termed CalFluors, these probes with emission from green to far-red wavelengths enabled the sensitive detection of biomolecules without washing steps. Through bioorthogonal reactions, visualization of alkyne-labeled biomolecules (glycans, DNA, RNA, and proteins) in various cellular contexts, including zebrafish and mouse brain tissue slices, was achieved. Further modification of rhodamine derivatives enabled Zhang et al.76 to develop a platform to evaluate protein engineering outcomes via quantification of extracellular alkyne-tagged metabolites using a fluorogenic click reaction. This method facilitated the screening and optimization of alkyne-based biosynthetic tools. For example, the probes for JamB, a membrane-bound bifunctional desaturase/acetylenase, exhibited approximately 20-fold enhanced activity in E. coli.
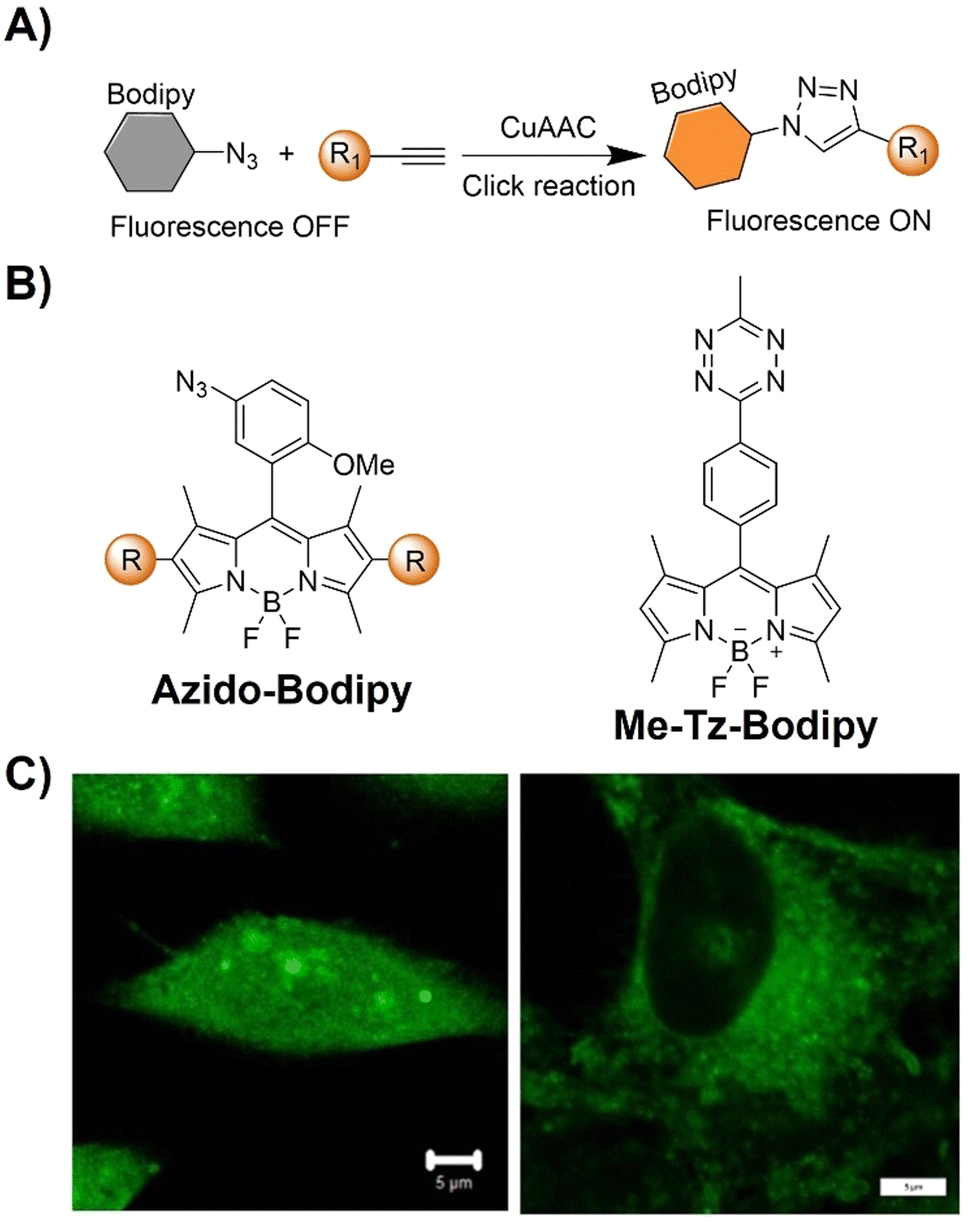 | ||
| Fig. 7 Bioorthogonal fluorescent probes based on the copper(I)-catalyzed azide–alkyne cycloaddition with bodipy derivatives. (A) Illustration of the CuAAC activated Bodipy probe for cell imaging; chemical structure (B) and cell imaging (C) of azido-Bodipy and Me-Tz-Bodipy. Adapted with permission from ref. 78. Copyright 2023, Wiley-VCH GmbH. | ||
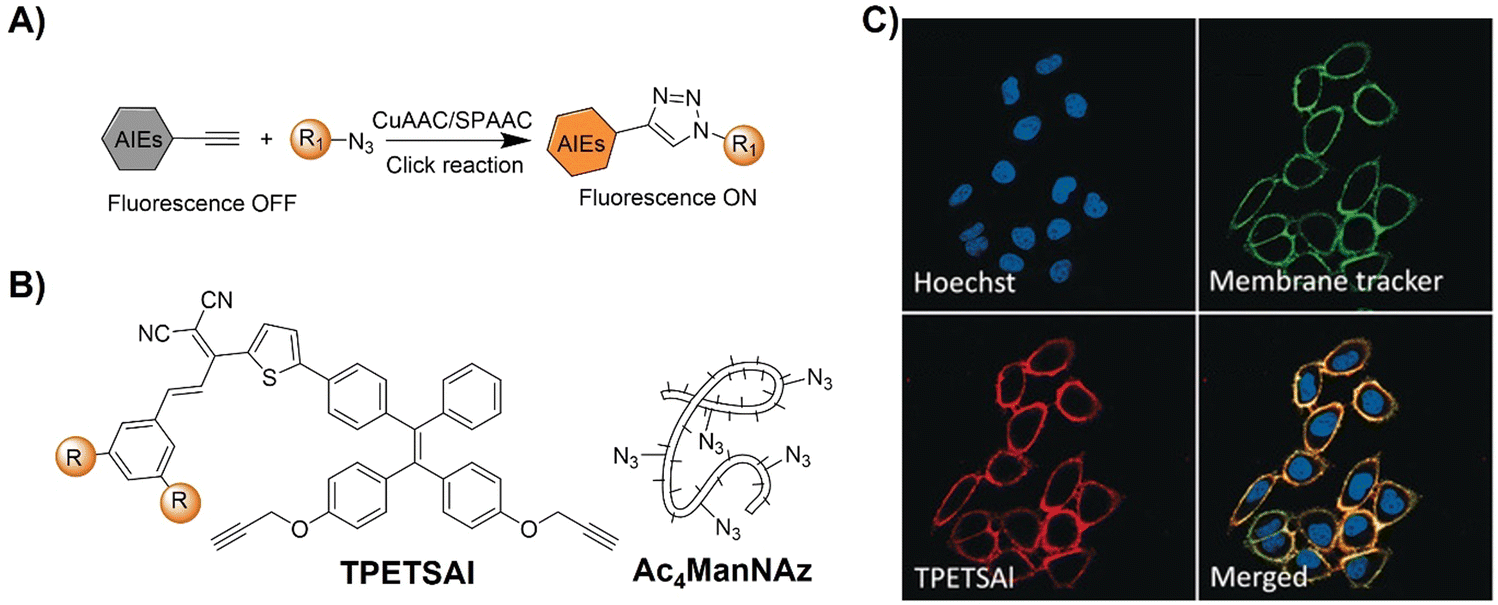 | ||
| Fig. 8 Bioorthogonal probes for fluorescence imaging based on the copper(I)-catalyzed azide–alkyne cycloaddition with AIEgens. (A) Illustration of the CuAAC activated AIEgen probe for cell imaging; chemical structure (B) and (C) co-stain of TPETSAI labeled HeLa cells with membrane tracker CellMask Green. Adapted with permission from ref. 80. Copyright 2016, Wiley-VCH GmbH. | ||
Incorporating bioorthogonal chemical groups into synthetic glycans followed by click reactions with fluorescent probes is pivotal in monitoring the metabolic glycoengineering bioprocesses of unnatural glycans in living systems. Following the success of their prior work, the Liu group81 devised a dual-targeted metabolic precursor, cRGD-S-Ac3ManNAz, selectively targeting αvβ3 integrin-overexpressing MDA-MB-231 cancer cells for internalization through receptor mediated endocytosis and subsequent generation of azide-tagged glycans (Ac3ManNAz) upon the GSH-mediated cleavage of the disulfide bond on cRGD-S-Ac3ManNAz. A bioorthogonal fluorescent probe, TPEBAI, was also developed for specific cancer cell labelling (Fig. 9A). TPEBAI was initially non-fluorescent in aqueous media but exhibited light-up fluorescence upon azide click reaction, enabling selective cancer cell imaging. TPEBAI's robust 1O2 production also allowed for image-guided cancer cell ablation under light irradiation. Normal cells that feature low αvβ3 integrin expression and intracellular GSH levels showed minimal TPEBAI labeling and negligible toxicity under light irradiation, offering precise cancer cell imaging and ablation capabilities.
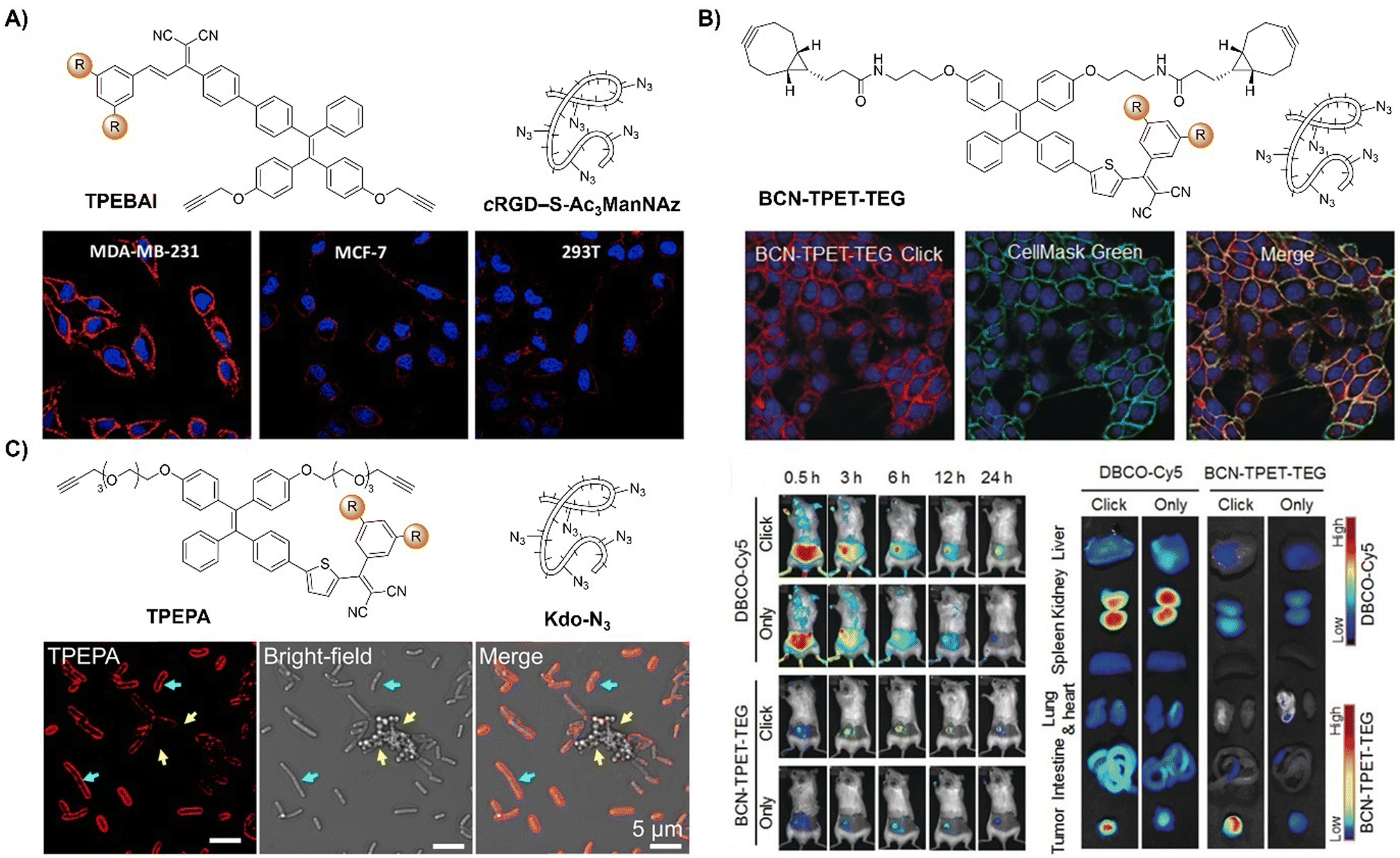 | ||
| Fig. 9 AIE probes for bioorthogonal reaction based on the copper(I)-catalyzed azide–alkyne cycloaddition. (A) The structure and confocal images of TPEBAI labeled MDA-MB-231/MCF-7/293T cells. Adapted with permission from ref. 81. Copyright 2018, American Chemical Society; (B) the structure, overlay images of 4T1 cells and in vivo imaging of BCN-TPET-TEG. Adapted with permission from ref. 82. Copyright 2018, Wiley-VCH GmbH; (C) the structure and TPEPA-selective metabolic labeling of Gram-negative (E. coli) and Gram-positive bacteria (S. aureus) with Kdo-N3 addition. Adapted with permission from ref. 83. Copyright 2020, American Chemical Society. | ||
As a result of the covalent ligation, bioorthogonal labeling using activated probes generally exhibits a better performance in imaging and drug delivery in tumors than active targeting strategies. For instance, Liu et al.82 developed a light-up fluorescent probe (BCN-TPET-TEG, BCN = bicyclo[6.1.0]non-4-yne) utilizing a AIEgen for in vivo bioorthogonal fluorescence tumor labeling (Fig. 9B). BCN-TPET-TEG exhibited low background fluorescence in aqueous media and minimal non-specific interaction with normal tissues. With a broad visible absorption spectrum (400–600 nm) and bright NIR emission (λem = 700 nm), emission of BCN-TPET-TEG was rapidly switched on after reacting with azide groups present on tumor cells, facilitating swift tumor-specific imaging. Moreover, the photosensitizing capacity enables effective image-guided photodynamic tumor therapy.
In addition to the tumor specific labelling and ablation through PDT, AIE based bioorthogonal probes have been developed for specific bacteria discrimination and biofilm eradication by Liu's group.83 TPEPA represents an example of a bioorthogonal fluorescence turn-on probe for this purpose (Fig. 9C). This probe exhibited excellent water solubility and minimal fluorescence in water-based environments. Upon reaction with azide groups engineered onto bacteria through metabolic processes, emission of TPEPA at 680 nm was switched on, enabling fluorescence imaging and discrimination of bacteria, i.e., Gram-negative (G−) E. coli bacteria with Kdo-N3 and Gram-positive (G+) S. aureus bacteria with D-Ala-N3. ROS generation under light irradiation enabled the bioorthogonally activated probe TPEPA to be used as a photosensitizer for the precise ablation of bacteria. Leveraging cell surface components of pathogenic bacteria for specific G+ and G− bacterial discrimination, Liu et al.84 then devised a fluorescence-based Gram-staining method using bioorthogonal fluorescence turn-on probes TPPB and BPTZ (Fig. 10A). Metabolic incorporation of Kdo-N3 and Ala-CP into bacterial surface membranes allowed for selective labeling via strain-promoted alkyne–azide cycloaddition (SPAAC) reaction and Diels–Alder reaction with inverse electronic demand (DARinv). TPPB targets characteristic LPS in G− bacteria, while BPTZ labels both G+ and G− bacteria, enabling clear differentiation with high selectivity and sensitivity (Fig. 10B).
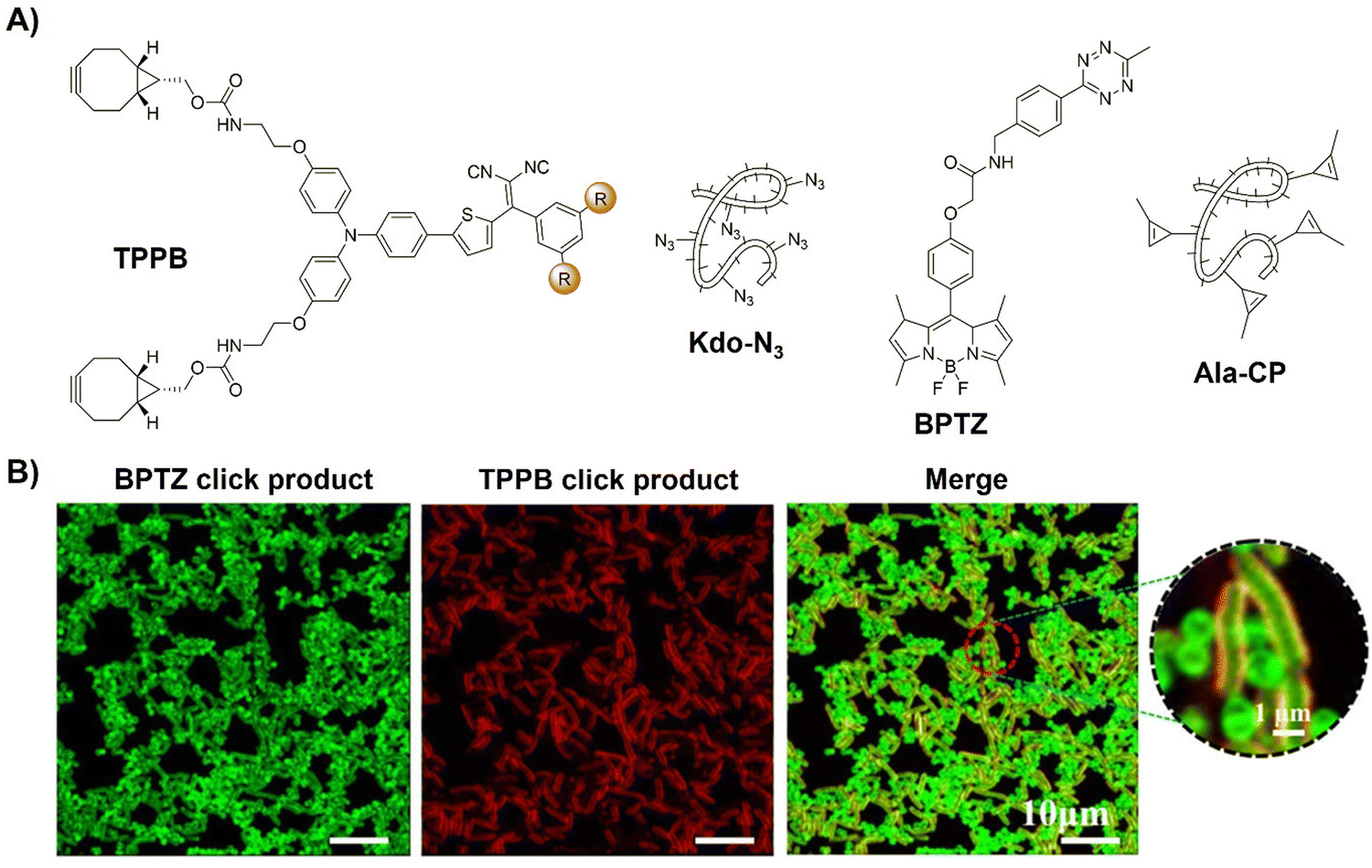 | ||
| Fig. 10 Bioorthogonal probes for bacterial imaging based on the copper(I)-catalyzed azide–alkyne cycloaddition with an AIEgen. (A) The structures of TPPB, Kdo-N3, BPTZ and Ala-CP, (B) metabolic dual labeling of G− bacteria (E. coli) and G+ bacteria (S. aureus) after being co-cultured with solution and then treated with TPPB, Kdo-N3, BPTZ and Ala-CP. Adapted with permission from ref. 84. Copyright 2021, American Chemical Society. | ||
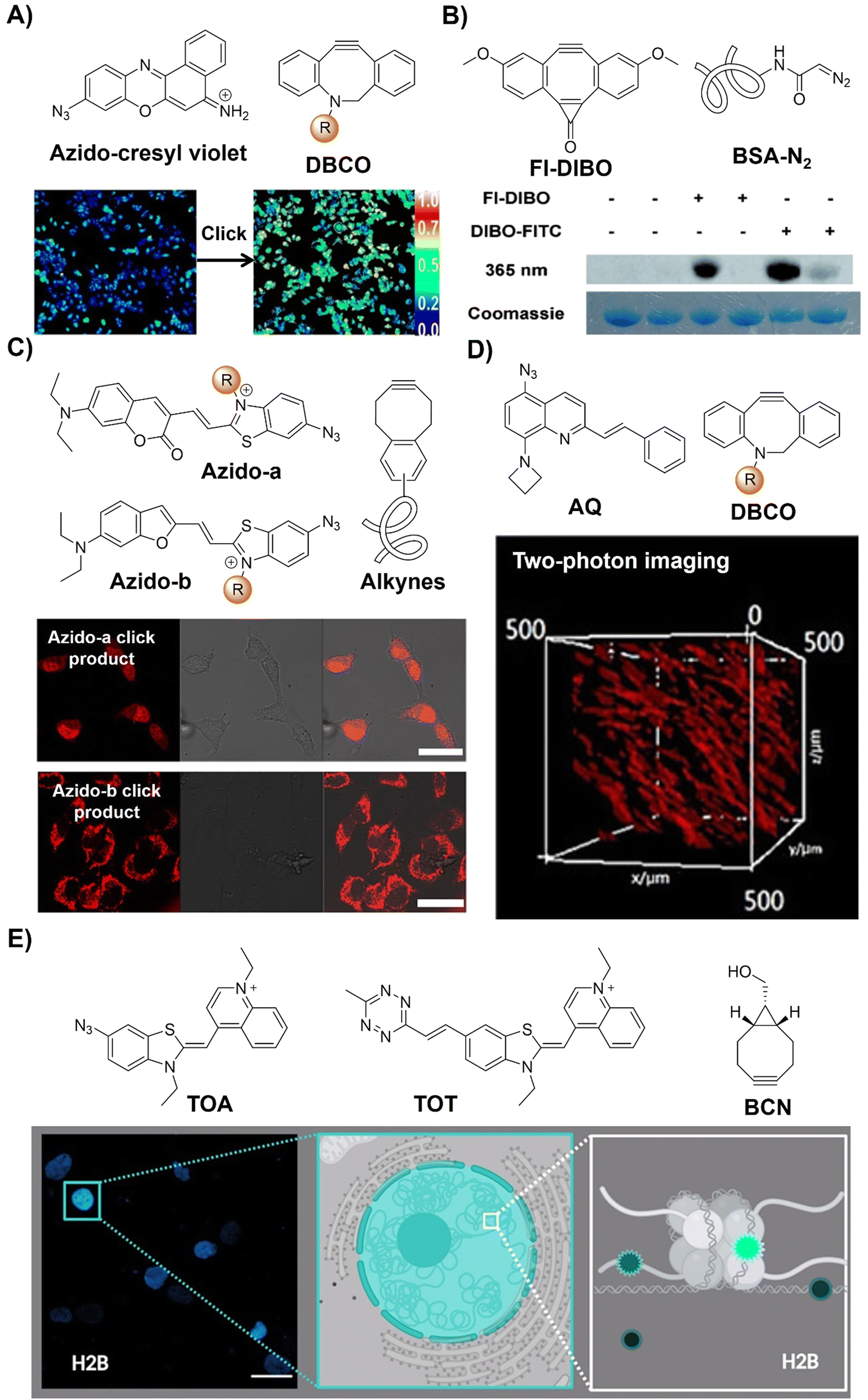 | ||
| Fig. 11 Bioorthogonal probes based on the copper(I)-catalyzed azide–alkyne cycloaddition with other fluorophores. (A) The structure of azido-cresyl violet and ratiometric fluorescence imaging before and after click reaction. Adapted with permission from ref. 85. Copyright 2015, American Chemical Society; (B) the structure of FI-DIBO/BSA-N2 and target protein labeling. Adapted with permission from ref. 86. Copyright 2015, Wiley-VCH GmbH; (C) the structure and cell imaging of azido-a/b. Adapted with permission from ref. 87. Copyright 2016, American Chemical Society; (D) the structure and two-photon cell imaging of AQ. Adapted with permission from ref. 88. Copyright 2020, Wiley-VCH GmbH; and (E) the structures and nucleus imaging of TOA and TOT, adapted from ref. 89, licensed under CC-BY 4.0. | ||
In 2015, Boons and colleagues86 introduced Fl-DIBO, a dibenzocyclooctyne derivative fused with a cyclopropenone moiety, capable of undergoing rapid strain-promoted cycloaddition reactions without catalysts (Fig. 11B). Fl-DIBO reacted rapidly with azides, nitrones, nitrile oxides, and mono- and disubstituted diazo derivatives. While reactions with certain compounds resulted in products of low quantum yield, monosubstituted diazo reagents produced 1H-pyrazole derivatives exhibiting about 160-fold fluorescence enhancement over Fl-DIBO, with over a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold increase in brightness. Fl-DIBO was then used for the precise background-free fluorescence labeling of diazo-tagged proteins, demonstrating the potential of diazo derivatives as biomolecule tags.
000-fold increase in brightness. Fl-DIBO was then used for the precise background-free fluorescence labeling of diazo-tagged proteins, demonstrating the potential of diazo derivatives as biomolecule tags.
Red-emitting fluorescent benzothiazolium dimethine dyes with azide groups have also been employed for bioorthogonal conjugation to alkyne targets by Kele et al.87 Upon the CuAAC reaction of weakly fluorescent probes with alkyne-modified oligonucleotides, the corresponding triazolic products exhibit visible excitation and NIR emission with large Stokes shifts and high quantum yields (a ΦF of up to 0.7). For the alkyne functionalized targets, incorporating DNA with the benzocyclooctyne motif through rigid (ethnynyl) or flexible (ethyl) linkers enabled copper-free click reaction labelling of DNA strands. In vitro evaluation using HeLa cells transfected with cyclooctynylated-DNA confirmed cellular uptake without toxicity, enabling specific tagging of target DNA with negligible background fluorescence, facilitating fluorescent cell imaging (Fig. 11C). Similarly, another two bioorthogonal probes with quinolinium units and azide or Tz motifs on DNA-sensitive frames (TOA and TOT respectively) were developed by the Németh group89 to detect DNA–protein interactions (Fig. 11E).
Bioorthogonal activation and precise imaging in vivo are important for the investigation of bioprocesses but remain challenging due to the limited tissue penetration of excitation light. In this context, Zhu et al.88 developed a series of azide-based light-up bioorthogonal probes, leveraging a weakly fluorescent 8-aminoquinoline (AQ) scaffold for in vivo two-photon fluorescence imaging (Fig. 11D). These probes, upon click reaction with alkynes, exhibited remarkable fluorescence enhancement (up to 1352-fold). By introducing various styryl groups into the AQ scaffold, the probes exhibited excellent two-photon properties (δ = 542 GM at 780 nm). Extensive bioimaging experiments validated the versatility of these probes for live cell/zebrafish imaging, eliminating the need for washing steps. Notably, in vivo two-photon imaging experiments showcased the superior performance of these bioorthogonal probes over conventional fluorophores, boasting a high signal-to-noise ratio and deep tissue penetration.
In summary, the CuAAC reaction has been widely employed for developing bioorthogonally activated probes using fluorophores like coumarin, naphthalimide, rhodamine, bodipy, and AIEgens, facilitating precise in situ imaging in bacteria and live cells. However, its translation into in vivo applications faces challenges. Direct utilization of Cu(I) in living systems can induce the generation of ROS, leading to cellular toxicity, which restricts the application of CuAAC bioorthogonal activation for in vivo use. Efforts to enhance biocompatibility focus on developing water-soluble ligands to accelerate CuAAC while scavenging ROS to mitigate cytotoxicity. Although Cu(I) remains the preferred catalyst, exploration of alternative transition metals such as Ru,90 and Rh91 has been pursued, predominantly in organic solvents, impeding their biological applicability.
Efforts to bypass transition-metal catalysts entirely have gained momentum. Notably, the development of Cu-free SPAAC reactions30,31,48,92–95 forming triazole products has emerged. Incorporating alkynes within strained cyclooctyne systems achieves Cu-free modification, enhancing biocompatibility, albeit with reaction rates comparable to Staudinger ligation and slower than CuAAC. Improving reaction kinetics involves integrating electron-withdrawing fluorine atoms into cyclooctyne reagents, exemplified by difluorinated cyclooctyne (DIFO),96 significantly enhancing reactivity for in vivo bioorthogonal imaging of azide-labeled sugars. Initial in vivo bioimaging applications in organisms such as Caenorhabditis elegans and zebrafish embryos were studied using DIFO and azides.97 Further development of cyclooctyne analogues with improved water solubility and kinetics aims to enhance azide–alkyne cycloaddition efficacy in living organisms.
Despite addressing slow kinetics in Staudinger ligation and the potential toxicity of CuAAC, SPAAC's performance remains suboptimal due to reduced reaction rates, susceptibility to side reactions, and reliance on sterically larger connecting groups that compromise functionality. Consequently, research persists in identifying fully biocompatible accelerating ligands for CuAAC, in order to facilitate rapid intracellular reactions without inducing biological stress or interfering with endogenous copper-binding biomolecules.
2.3. Inverse-electron-demand Diels–Alder reaction
The bioorthogonal Tz ligation38,39,98,99 describes a chemical reaction involving a 1,2,4,5-Tz (Tz) and an alkene or alkyne dienophile, yielding a dihydropyridazine or pyridazine conjugate via a [4+2]/retro [4+2] cycloaddition sequence (Scheme 3). This reaction, known as the IEDDA reaction,100 stands out for its rapid kinetics, occurring without the need for catalysis and yielding nitrogen gas as the sole byproduct. Initially utilized in organic synthesis for their swift reactivity with strained dienophiles, Tzs found application in bioorthogonal reactions in 2008.38 This breakthrough involved reactions with derivatives of trans-cyclooctene (TCO) and norbornene by the groups of Fox38 and Weissleder,101 respectively. Subsequently, there has been a proliferation of chemical diversity for both Tzs and their dienophile reaction partners, allowing for fine-tuning of reaction rates, stability, and the design of ‘minimal’ reporter molecules. The IEDDA reaction of Tz ligation is accelerated by electron-withdrawing groups on the Tz.102,103Tzs are commonly synthesized through the condensation of Pinner salts or nitriles with hydrazine, often facilitated by Lewis acids, thiols, or sulfur catalysts.104 Alternatively, milder protocols have been developed for directly introducing intact Tz groups via palladium-catalyzed coupling reactions of aryl halides or aryl boronic acids.104 Carboxylic esters also serve as handles for the preparation of unsymmetrical and monosubstituted Tzs.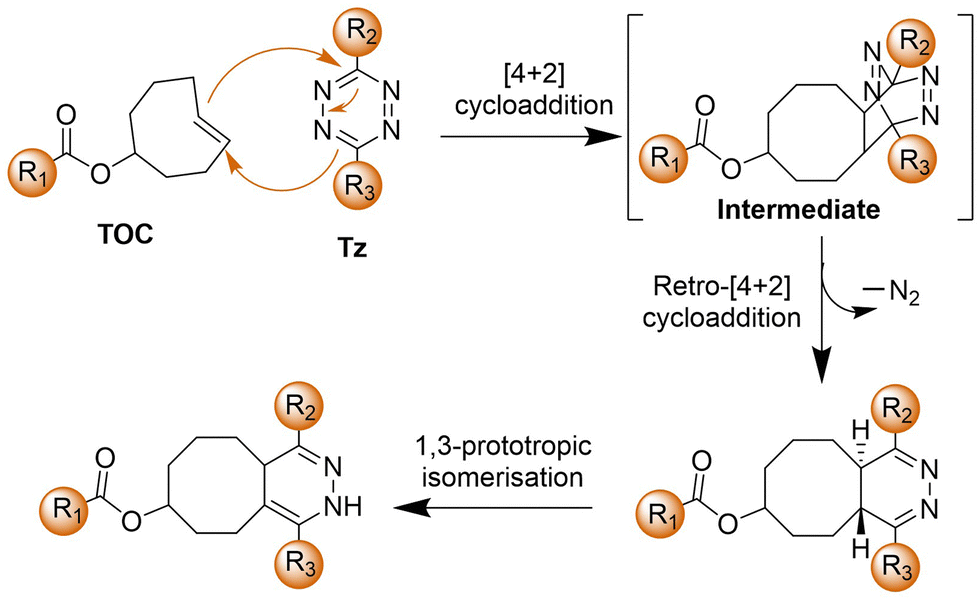 | ||
| Scheme 3 The proposed mechanism of the IEDDA reaction. | ||
Various dienophiles have been developed for Tz ligation. TCO, prepared via flow photochemistry, exhibits unmatched reactivity with Tzs, engaging in the most rapid bioorthogonal reactions known under aqueous conditions.105 Highly strained cyclooctynes such as BCN also react rapidly, yielding aromatic conjugates useful for workflows requiring defined stereochemistry. Cyclopropenes offer a complementary approach, especially beneficial for genetic encoding and metabolic incorporation. Norbornene, known for its good kinetics at a low cost, has proven advantageous for scale-up and materials applications. Smaller dienophiles such as cyclobutenes, azetidines, unstrained olefins, and isonitriles also react efficiently with Tzs, albeit often requiring large excesses of one reagent due to slower kinetics. Triazines can serve as surrogates for Tzs, offering advantages of smaller size and greater stability.104,106
Additionally, Tz also acts as a fluorescence quencher, with its mechanism varying based on the design of the bioorthogonal probes. The common mechanism involves non-radiative dissipation of molecular energy to quench the emission of the fluorophores, where the excited state energy of the fluorophore is released through vibrations, heat, or interactions with the surrounding environment. However, after the IEDDA reaction, these energy release pathways are removed, restoring fluorescence. As a result of the “off–on” fluorescence response, Tz-conjugated bioorthogonal fluorescent probes could be used for wash-free molecular imaging. Varieties of Tz-modified fluorophores use diverse quenching mechanisms, including electronic energy transfer (EET) such as Förster resonance energy transfer (FRET), Dexter, and through-bond energy transfer (TBET), monochromophoric design, and new fluorophore formation post-IEDDA. FRET exploits donor–acceptor excited state interactions, requiring spectral overlap. Dexter-type EET involves electron hopping between molecules, needing donor–acceptor wavefunction overlap. TBET occurs via a rigid π-system linker. Monochromophoric design relies on dark-state Tz quenching. Post-reaction, optically forbidden transitions shift to allowed states, allowing for changes in the fluorescence signal enabling molecular imaging. Research has highlighted that new fluorophore formation post-IEDDA offers high turn-on ratios. Optimizing energy transfer efficiency from a fluorophore to a quencher is crucial in EET strategies. These varied fluorescence response approaches enable flexibility for the development of bioorthogonally activated probes for precise in situ imaging.
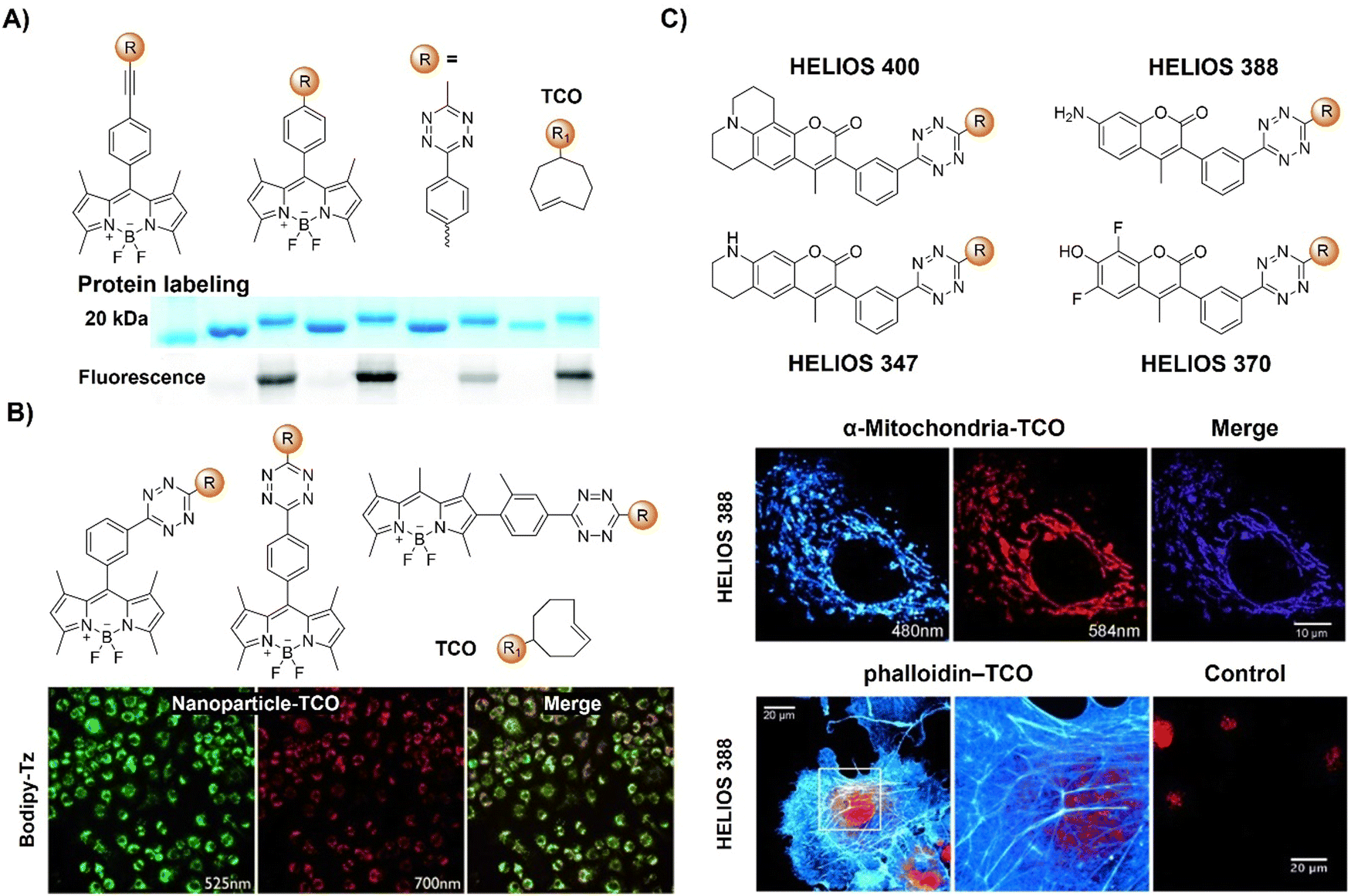 | ||
| Fig. 12 Fluorescent probes based on the inverse-electron-demand Diels–Alder reaction using the TBET fluorescence response mechanism. (A) The structure and protein labeling of the Tz modified bodipy, adapted with permission from ref. 107. Copyright 2014, The Royal Society of Chemistry; (B) the structures and cell imaging of the bodipy-p-Tz derivatives, adapted with permission from ref. 108. Copyright 2013, Wiley-VCH GmbH; (C) the structures of the HELIOS probes and the wash-free fluorogenic imaging of intracellular targets, adapted with permission from ref. 109. Copyright 2014, Wiley-VCH GmbH. | ||
Another bodipy-Tz platform was reported by Weissleder and colleagues for the development of bioorthogonal turn on probes (Fig. 12B).108 After reaction with TCO, the π conjugation of Tz-Bodipy with para- and metaTz exhibited a 900 and 1600-fold increase in fluorescence, respectively. The significant enhancement in fluorescence facilitated the use of the probe for bioorthogonal wash-free labelling and imaging of fixed and live cells. Following this success, the same group developed a series of fluorescent probes with emissions ranging from blue to green, showing fluorescence enhancement surpassing 11000-fold with a ΦF of up to 0.49 (Fig. 12C).109 These “off–on” response probes were engineered using the coumarin fluorochrome with the bioorthogonal TCO-Tz chemistry platform. Leveraging highly efficient TBET, these probes exhibit significant enhancement in brightness (2500–11000-fold enhancement depending on chemical structures and emission of probes). The probes enabled wash-free, fluorogenic imaging of various targets such as cell-surface receptors in cancer cells, mitochondria, and the actin cytoskeleton in a few seconds, featuring minimal background signal and negligible non-specific binding.
To address the limitation of synthetic methods for preparation of Tz-based bioorthogonal probes, Devaraj et al.110 introduced an elimination-Heck cascade reaction for the in situ synthesis of (E)-3-substituted 6-alkenyl-1,2,4,5-Tz derivatives, including 24 compounds with Tz conjugation (Fig. 13A). These derivatives, including fluorescein and bodipy-based fluorescent probes, are suitable for live-cell imaging, exhibiting strong fluorescence turn-on response (up to 400-fold) upon reacting with dienophiles like cyclopropenes and TCO. In another research conducted by the same group,111 a fluorogenic Tz-mediated transfer (TMT) reaction employing the 7-azabenzonorbornadiene derivative was reported for the detection of oligonucleotides with high sensitivity and sequence specificity (Fig. 13B). The key requirement for achieving signal enhancement is the template-driven turnover of antisense probes that was facilitated by spontaneous diazine release post-Tz ligation. Using a highly quenched alkenyl-fluorogenic Tz achieved an over 100-fold fluorescence increase via the TMT reaction. RNA-templated transfer reactions with antisense probes enabled mir-21 detection with high sensitivity at low-picomolar levels, suitable for distinguishing single-base mismatches.
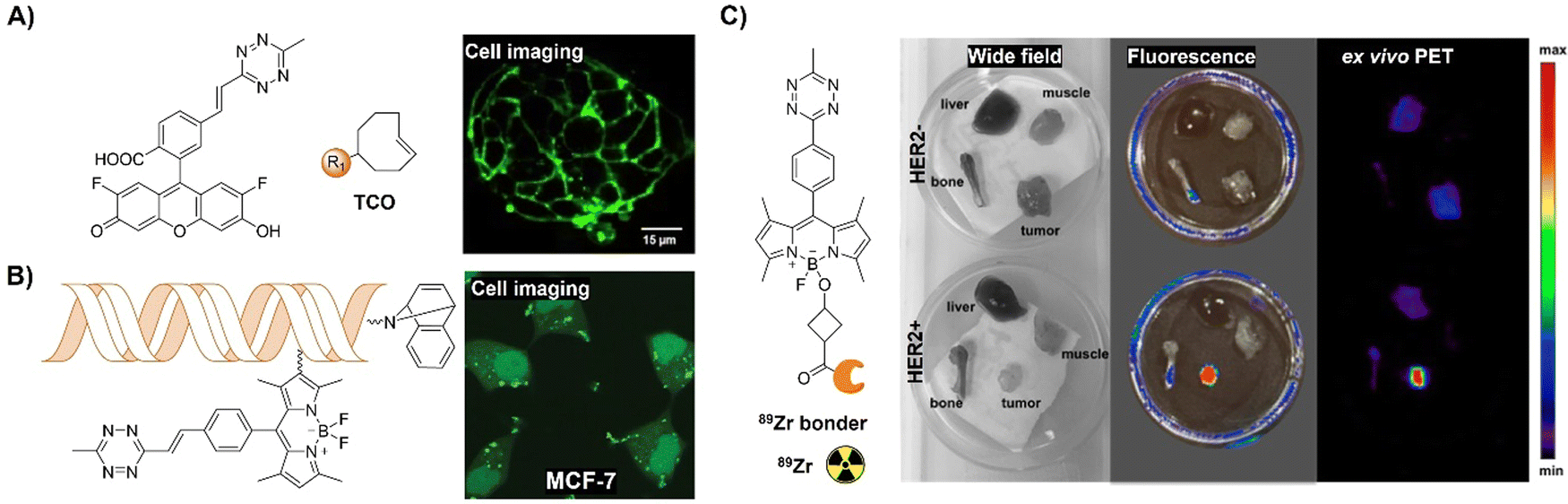 | ||
| Fig. 13 Fluorescent probes based on the inverse-electron-demand Diels–Alder reaction using the TBET fluorescence response mechanism. (A) The structure and LS174T cell imaging of the alkenyl-Tz modified fluorescein-based fluorescent probe, adapted with permission from ref. 110. Copyright 2014, Wiley-VCH GmbH; (B) structures of the Tzs and dienophiles used and live-cell miRNA detection in the human cancer MCF-7 cell line. Adapted with permission from ref. 111. Copyright 2014, American Chemical Society; (C) the structure of the bioorthogonal fluorogenic PET probe and the wide field, fluorescence and ex vivo PET images acquired for the muscle, liver, bone, and tumor, adapted with permission from ref. 112. Copyright 2016, American Chemical Society. | ||
Multi-modality imaging, such as fluorescence-PET, using multiple functionalized probes is an emerging approach for better understanding the biological processes in living organisms. Linking deferoxamine (DFO) with Bodipy-Tz, Weissleder et al.112 reported a bioorthogonal labeling method to create PET/fluorescence mAb imaging probes. The chelate linker, utilizing TCO-Tz chemistry, integrated DFO for 89Zr PET imaging and a bodipy for fluorescence imaging. Fluorescence activation (<3 min) during biomolecule–fluorophore–chelator conjugation allowed for real-time monitoring and precise quantification via absorbance measurements (Fig. 13C). In mice with tumor xenografts, the probe exhibited high target specificity in PET and robust ex vivo fluorescence imaging.
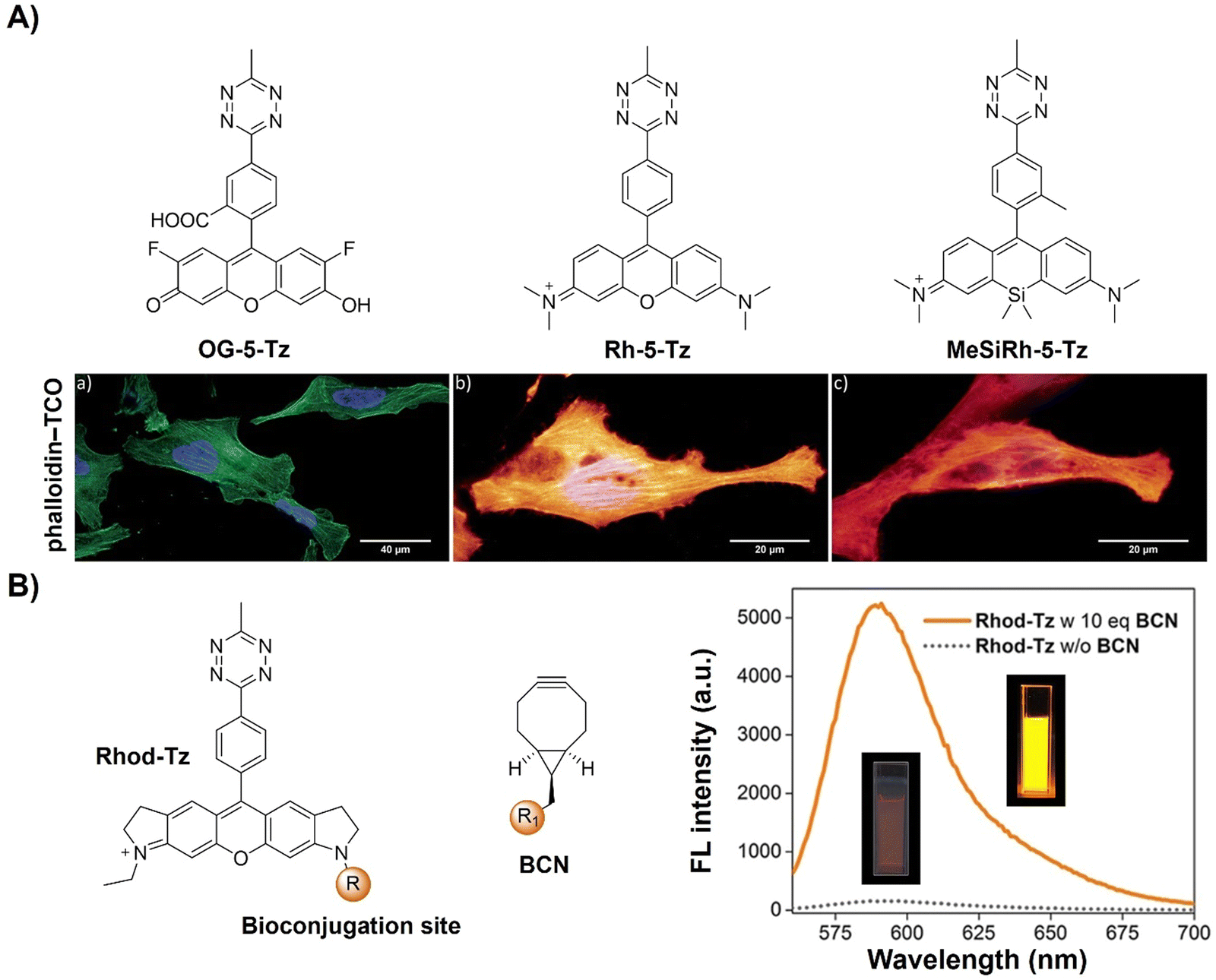
A series of water soluble fluorogenic xanthene Tz derivatives (Fig. 14A) spanning a broad emission spectrum (521–666 nm), encompassing green (fluorescein Oregon Green), yellow (tetramethyl-rhodamine), and far-red (Si-rhodamine), were synthesized by Wombacher et al.113 as bioorthogonal probes for wash-free fluorescence imaging of proteins in live cells. Using these fluorescent probes for selective protein labelling achieved up to 109-fold enhancement in fluorescence upon bioorthogonal IEDDA reaction (DAinv), allowing for wash-free fluorescence microscopic imaging of the actin cytoskeleton in fixed cells and visualization of mitochondrial and nuclear proteins in living cells.
 | ||
| Fig. 14 Fluorescent probes based on the inverse-electron-demand Diels–Alder reaction using the TBET fluorescence response mechanism. (A) The structures of green- to far-red-emitting fluorogenic xanthene Tzs and wash-free fixed cell confocal imaging of actin cytoskeleton, adapted with permission from ref. 113. Copyright 2017, The Royal Society of Chemistry; (B) the structures of Rhod-Tz and BCN and emission spectra of Rhod-Tz with/without BCN addition, adapted with permission from ref. 114. Copyright 2017, Wiley-VCH GmbH. | ||
In addition, the bifunctional rhodamine fluorophore has also been employed for the development of bioorthogonally activated probes. For instance, Wombacher's group reported a bifunctional rhodamine-based fluorescent probe for a DNA-templated assay (Fig. 14B).114 The bifunctional probe (Rhod-Tz-NHS) was prepared and the proximity effect on the reactivity of DAinv-bioorthogonal chemistry was investigated using different dienophiles with different reactivities. Stable dienophiles with poor kinetics gained reactivity through DNA-templated proximity, generating fluorescence signals exclusively from proximity-induced reactions. Up to 34-fold enhancement of the fluorescence signal was obtained within 40 min, with negligible background reactivity at micromolar concentrations. This proximity concept addressed stability challenges in bioorthogonal chemistry, maintaining crucial rapid reaction rates for cellular applications. A subsequent study conducted by the same research group uncovered a dark intermediate in the fluorogenic reaction between Rhod-Tz and TCO, which will be discussed below.115
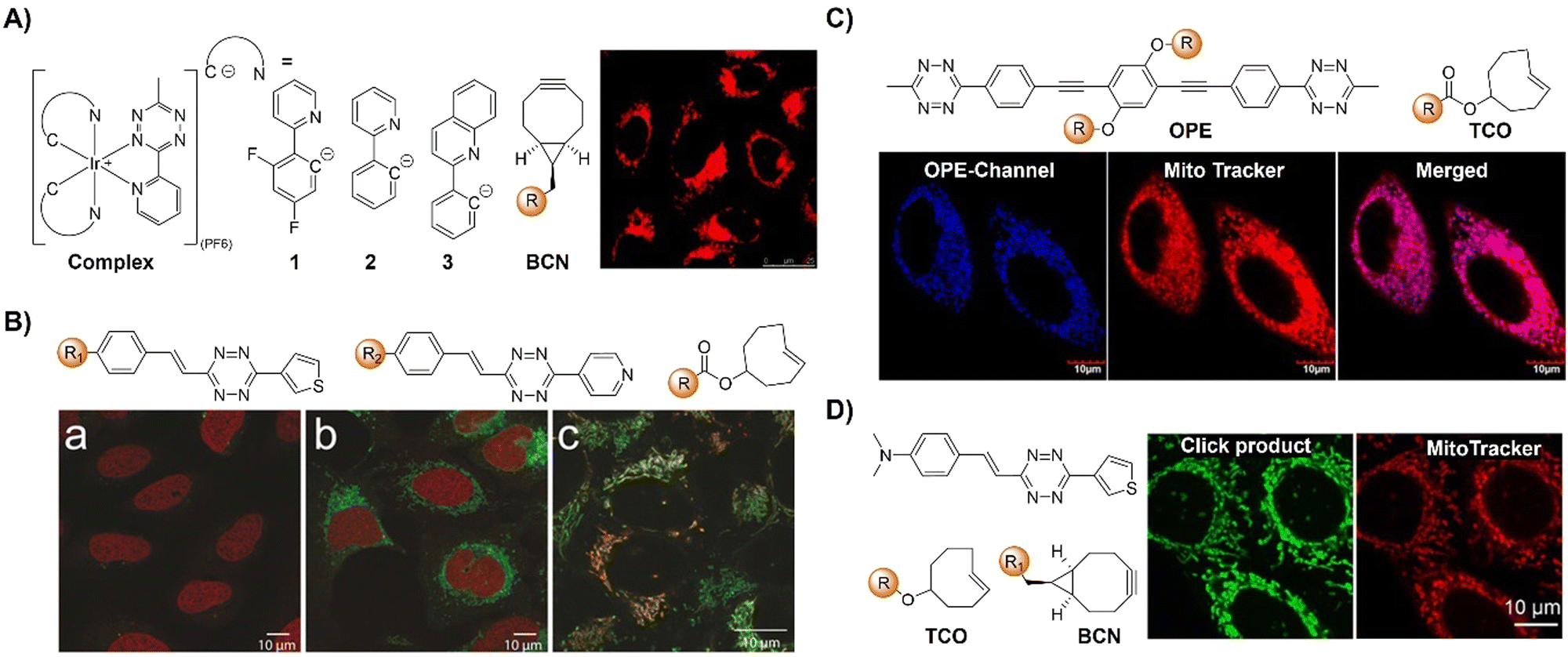
The Tz unit can quench emission from luminophores via FRET, TBET, or PeT, with quenching efficiency influenced by factors like the distance between the donor and acceptor, dipole orientation, spectral overlap, and electron transfer thermodynamics. Unlike the above probes with fluorescence emission from the singlet excited state (S1), metal complexes like iridium(III) and ruthenium(II) complexes emit from the triplet excited state (T1) characterized by large Stokes shift, prolonged emission lifetime, and excellent photostability.116,117 Therefore, a number of responsive probes have been successfully developed for luminescence bioassays and bioimaging. For example, Lo's group118 developed a bioorthogonal luminescence probe based on a luminescent iridium(III) complex bearing a single built-in Tz for protein labelling and cell imaging (Fig. 15A). The probe was prepared by coordinating iridium(III) with a Tz-containing pyridyl ligand, resulting in a weakly luminescent iridium(III) complex with a 60-fold higher reactivity than the ligand itself. These complexes were stable upon co-incubation with biothiols but exhibited significant emission enhancement upon treatment with BCN-OH. HaloTag technology facilitated protein conjugation, with BCN-C6-Cl enabling phosphorogenic coupling. For example, GST-HaloTag treated with BCN-C6-Cl prior to incubation with complex 3 exhibited increased emission intensity, as evidenced by a luminescence band only present in the BCN-C6-Cl-loaded SDS-PAGE gel. The application of complex 3 was demonstrated for detection of BCN-modified endogenous HaloTag proteins in HeLa cells.
 | ||
| Fig. 15 Bioorthgonal probes based on the inverse-electron-demand Diels–Alder reaction using the TBET fluorescence response mechanism. (A) The structures of the Ir complex and BCN and LSCM images of nontransfected HeLa cells, adapted with permission from ref. 118. Copyright 2017, The Royal Society of Chemistry; (B) the structures of a microtubule-selective Taxol-Tet probe and a mitochondria-selective TPP-Tet probe and confocal microscope images of U2OS cells, adapted with permission from ref. 119. Copyright 2017, Wiley-VCH GmbH; (C) the structure of OPE and colocalization analysis of OPE-Mito (top) and OPE (bottom) with Mito Tracker in MCF-7 cells, adapted with permission from ref. 120. Copyright 2018, American Chemical Society; and (D) the structures of electron-donating substituents linked via a π-system fluorophore-Tz and strained dienophiles and fluorogenic labeling of live HeLa cells, adapted with permission from ref. 121. Copyright 2019, Wiley-VCH GmbH. | ||
Reactions with nonfluorescent raw materials to produce highly fluorescent products through bioorthogonal activation are interesting, particularly for fluorescence labelling and in situ imaging of living organisms. Vrabel's research119 revealed how the conformation of the non-fluorescent TCO dienophile directly influences the fluorescent product formation in IEDDA reactions with 1,2,4,5-Tzs (Fig. 15B). Among the evaluated dienophiles, rapid reaction with the axial TCO isomer led to the formation of fluorescent 1,4-dihydropyridazine products with up to 91-fold enhancement. By adjusting the Tz's substitution pattern, the photochemical properties of the corresponding products could be easily tuned to form a new type of fluorophore with intriguing emission properties. This fluorogenic reaction was then applied for rapid labeling of the intracellular compartments of live cells, indicating its potential for advancing IEDDA applications in various fields, including live-cell imaging.
Fluorescence labelling and imaging of cellular organelles, like the mitochondria, are highly desirable for understanding bioprocesses at a subcellular level. To achieve this goal, Wang et al.120 reported a molecular guided approach for delivering functional molecules to mitochondria via intracellular bioorthogonal reactions (Fig. 15C). Oligo(p-phenyleneethynylene) (OPE) that was modified with Tz groups for bioorthogonal click reaction exhibited weak emission. Under laser irradiation and upon bioorthogonal reaction with Mito-TCO bearing triphenylphosphine (TPP) for mitochondrial targeting, a fluorescent Intra-Mito product formed in the cytoplasm could accumulate in the mitochondria, enabling precise mitochondrial labelling and imaging. Unlike most commonly used fluorescent dyes, such as rhodamine 110 and fluorescein for cell labelling, this bioorthogonal approach and the fluorescent product exhibited high stability for the long-term fluorescence imaging of live cells. Another example of TPP guided mitochondrial imaging post-bioorthogonal reaction was reported by Vrabel and colleagues (Fig. 15D).121 In this system, Tzs with electron-donating substituents were linked to the strained bicyclononyne dienophile via a π-system. The produced fluorescent pyridazine dyes resulted in 100–900-fold enhancement of fluorescence intensity upon bioorthogonal reaction. The resulting fluorophores exhibited large Stokes shift and relatively high fluorescence quantum yield for fluorescence labelling and imaging. Reaction of the bioorthogonal probe with TPP linked bicyclononyne enabled the fluorescent product to accumulate in the mitochondria for fluorescence imaging.
The majority of fluorophore-Tz conjugate (FLTz) strategy-based bioorthogonal probes have been developed using a donor–acceptor-type energy transfer mechanism. However, the effective switching on of fluorescence after click reaction and shifting of a probes’ emission wavelength are challenging. To address this, Park and coworkers122 introduced a monochromophoric design strategy to expand the emission color range of FLTzs while maintaining a high fluorescent turn-on/off ratio (Fig. 16A). Indolizine-based emission-tunable Seoul-Fluor (SF) was used as a model fluorophore system for these probes. By enhancing electronic coupling between Tz and π-conjugated systems of the indolizine core, the emission of SF-Tz conjugates (SFTzs) was efficiently quenched. Subsequently an over 1000-fold fluorescence enhancement with a ΦF of up to 0.68 after the IEDDA reaction with TCO was observed. Colorful SFTzs with consistent turn-on/off ratios were then developed across various emission wavelengths. Demonstrating the applicability of the bioorthogonal probes, fluorescence bioimaging of microtubules and mitochondria in live cells was achieved in a wash-free manner, offering a reliable strategy for developing versatile bioorthogonal fluorogenic probes with excellent turn-on ratios, regardless of the emission wavelength.
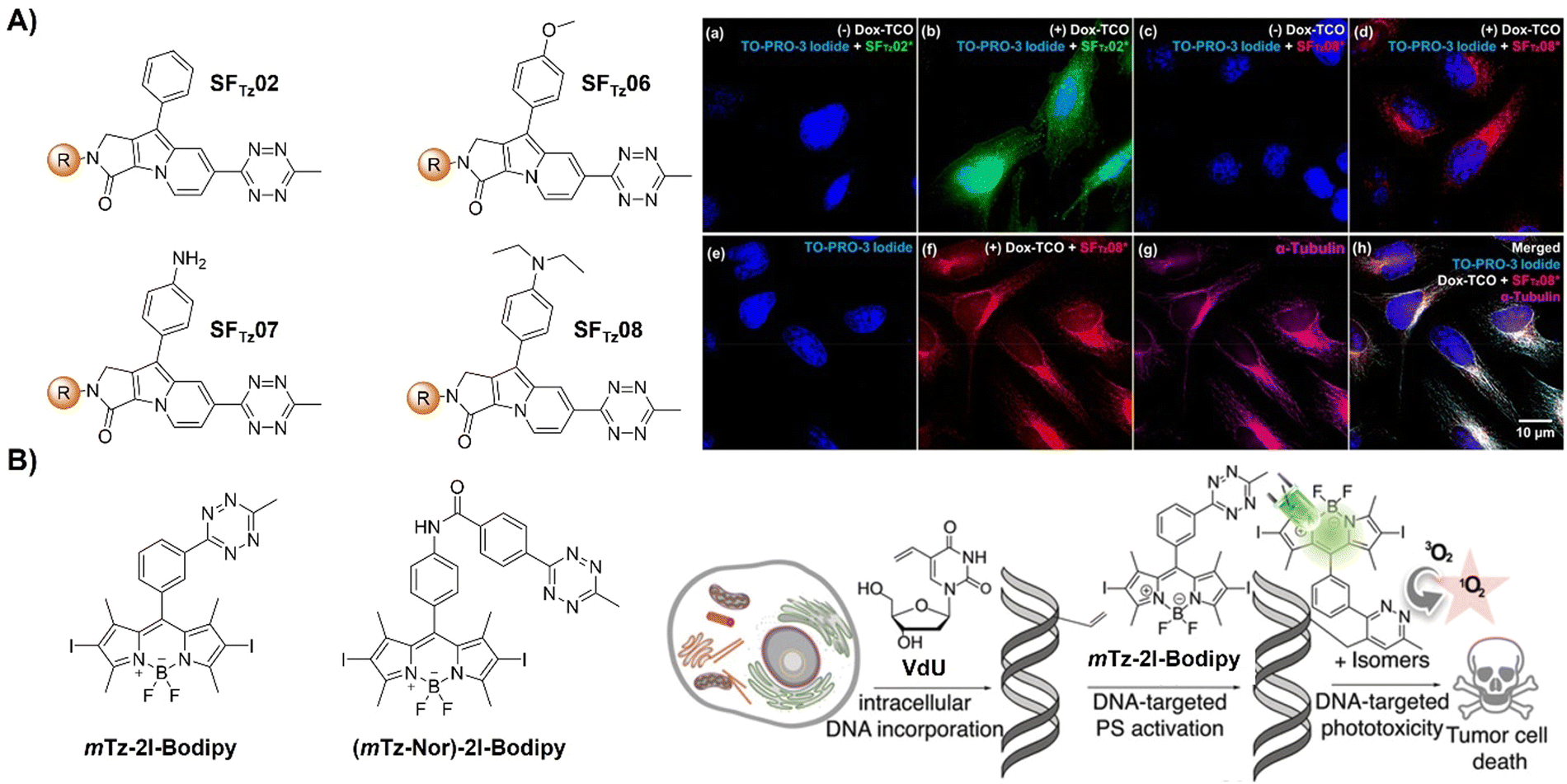 | ||
| Fig. 16 Bioorthogonal probes designed based on the inverse-electron-demand Diels–Alder reaction using the TBET fluorescence response mechanism. (A) Molecular structures of SF-Tz conjugates (SFTzs) and fluorogenic bioorthogonal imaging of innate microtubules with SFTz under fixed-cell conditions, adapted with permission from ref. 122. Copyright 2018, American Chemical Society and (B) the structures of the bioorthogonal, activatable, halogenated bodipy-Tz derivatives and outline of the DNA-targeted bioorthogonal strategy for activating the phototoxicity, adapted with permission from ref. 123. Copyright 2019, Wiley-VCH GmbH. | ||
Activation of AIEgen-based bioorthogonal probes allows for the ROS (e.g., singlet oxygen 1O2) generation for photodynamic therapy (PDT).80–83 Previous research conducted by Weissleder and colleagues has demonstrated the formation of highly luminescent probes after bioorthogonal Bodipy/Tz activation.108 Taking advantage of halogenated Bodipy's photosensitizing performance, Vázquez and colleagues123 developed a series of Bodipy bioorthogonal turn-on probes for precisely generating 1O2 by modifying the 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene core (Fig. 16B). These halogenated Bodipy-Tz probes exhibited non-toxicity to HeLa cells and “off–on” fluorescence response in cell imaging as well as effective photosensitization (k ≈ 3 × 10−3 s−1, ΦΔ ≈ 0.50) for inhibiting cancer cell growth and were obtained through intracellular IEDDA with suitable dienophiles. Another example of site specific bioorthogonal photosensitization for PDT was reported by Lo and colleagues.124 In this system, the bodipy-based dye, distyryl boron dipyrromethene (DSBDP), was equipped with two functional tags, including 1,2,4,5-Tz and alkyne groups (Fig. 17A). These tags could selectively bind to TCO and azide groups, respectively, which were introduced onto the membrane of A431 cells through pre-targeting with TCO-GE11 or metabolic glycoengineering with Ac4ManNAz. Upon bioorthogonal conjugation, the photosensitizer exhibited increased cellular uptake, ROS generation, and photocytotoxicity. The bioorthogonal reaction was confirmed in tumor-bearing nude mice, illustrating the potential of tumor treatment through bioorthogonal activation and light irradiation.
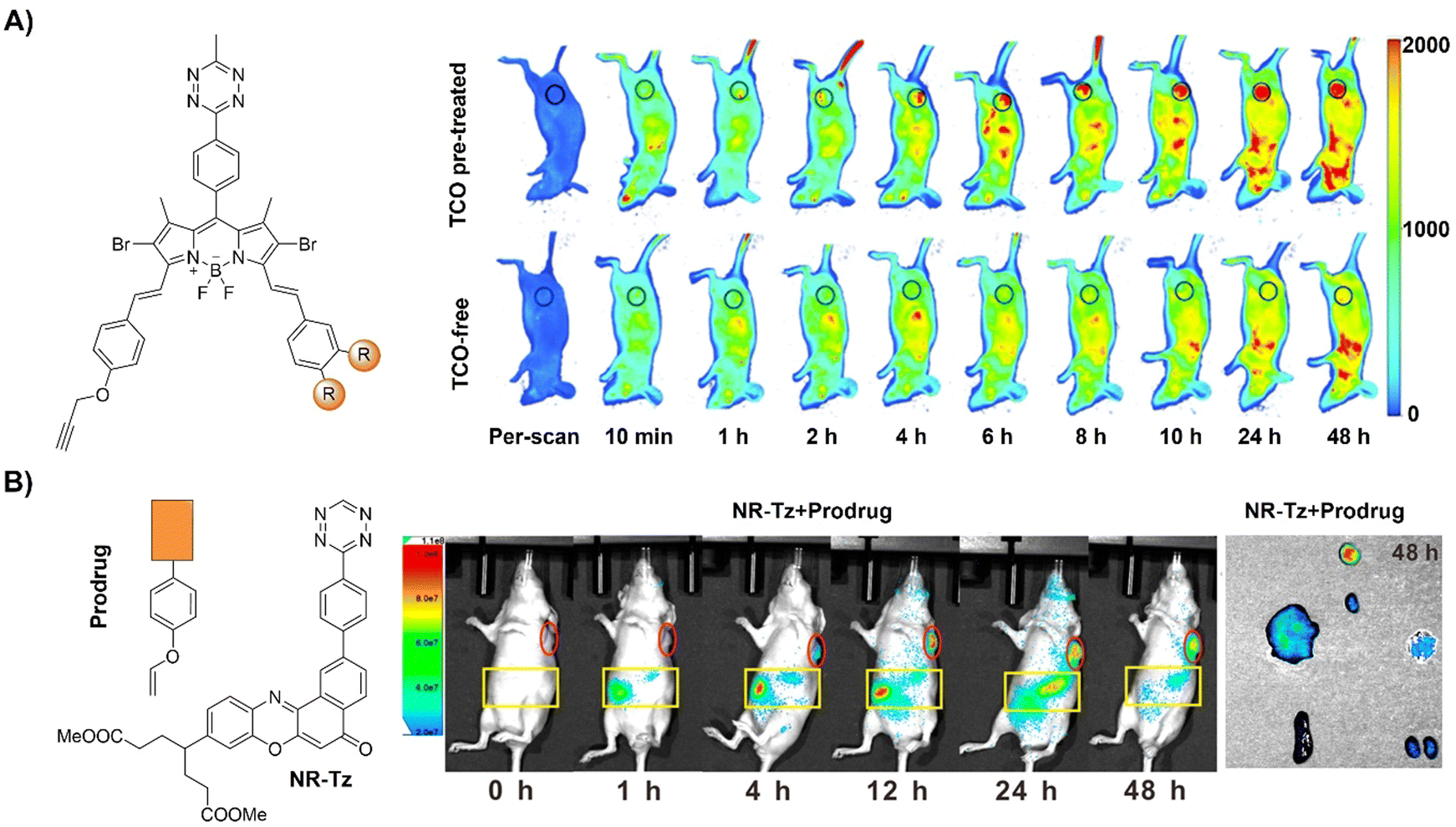 | ||
| Fig. 17 Fluorescent probes developed based on the inverse-electron-demand Diels–Alder reaction using the TBET fluorescence response mechanism. (A) The structure of DSBDP and fluorescence images of A431 tumor-bearing nude mice, adapted with permission from ref. 124. Copyright 2019, The Royal Society of Chemistry and (B) the structures of NR-Tz and the prodrug and in vivo fluorescence images of tumor and abdominal regions of tumor-bearing mice. Ex vivo images of dissected tumors and major organs at 48 h, adapted with permission from ref. 125. Copyright 2019, American Chemical Society. | ||
In addition to bioorthogonal activation for photosensitization, bioorthogonal prodrug activation for tumor imaging and treatment was achieved by Wu and colleagues.125 In this system, a vinyl ether-masked camptothecin (CPT) and a Tz tethered to a NIR fluorophore (NR) were prepared and then loaded into lipid nanoparticles separately (Fig. 17B). Accumulation of these lipid nanoparticles in tumors and the subsequent release of the CPT prodrug and NIR-Tz molecules by the tumor triggered IEDDA reaction facilitated chemotherapy and formation of NIR-Dz with the removal of TBET quenching for tumor fluorescence imaging. This nanosystem is particularly interesting as it allows for monitoring drug release and tumor treatment simply using a bioorthogonal reaction.
Achieving precise signal transduction requires control over the spatial and temporal dynamics of the production of signaling molecules. Phosphatidic acid (PA), a versatile lipid second messenger, exhibits diverse actions influenced by upstream stimuli, biosynthetic origins, and production sites. However, elucidating how cells regulate local PA production to modulate signaling outcomes remains a challenge, as PA biosynthesis sites lack subcellular precision in visualization. In the research lead by Baskin,126 a rapid chemoenzymatic method for imaging physiological PA production by phospholipase D (PLD) enzymes was reported (Fig. 18A). Bulky TCO-containing alcohols served as the nucleophilic site in a transphosphatidylation reaction of PLD's lipid substrate, making TCO-containing lipids suitable for fluorescence labelling through an IEDDA reaction with a fluorogenic Tz reagent. Fluorescent reporter lipids were initially generated in the plasma membrane, and rapid internalization of the fluorescent reporter was observed, suggesting potential applications in probing intracellular phospholipid transport mechanisms. By focusing on the initial 10 s of the IEDDA reaction, precise subcellular localization of endogenous PLD activity triggered by physiological G protein-coupled receptor and receptor tyrosine kinase agonists was possible, implying the capability to track lipid trafficking pathways and the physiological and pathological effects of localized PLD signaling.
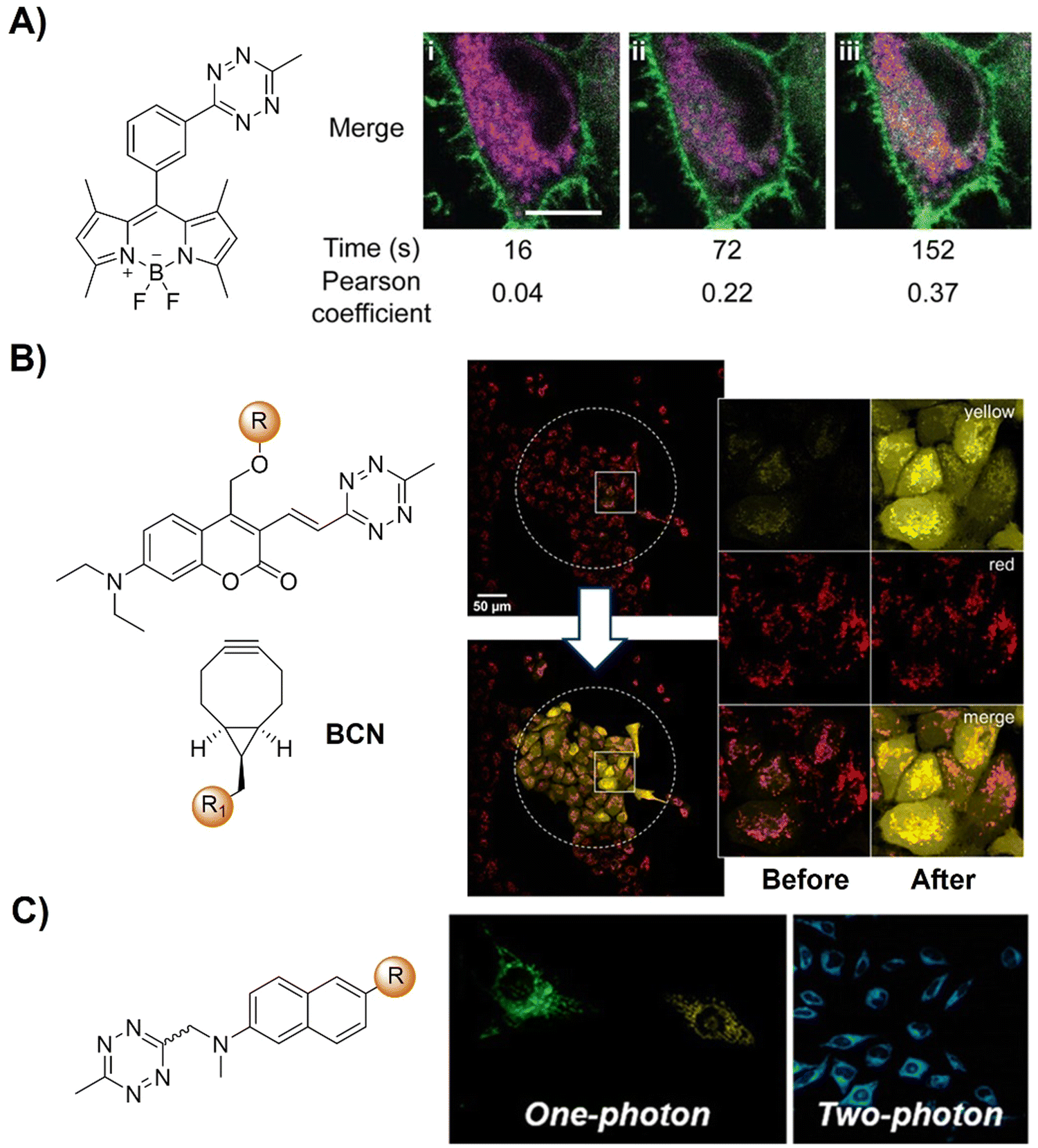 | ||
| Fig. 18 Bioorthogonal probes developed for fluorescence imaging based on the inverse-electron-demand Diels–Alder reaction using the TBET fluorescence response mechanism. (A) The structure of the fluorogenic Tz–Bodipy conjugate and representative images of HeLa cells, adapted with permission from ref. 126. Copyright 2019, National Academy of Sciences; (B) the structures of the model photocages and BCNs and cell imaging after irradiation of the central area (marked by a dotted circle) with the built-in blue lamp (460–500 nm, 5 s), adapted with permission from ref. 127. Licensed under CC-BY; and (C) the structural modification of naphthalene-based two- photon fluorogenic dyes and one-photon/two-photon fluorogenic imaging, adapted with permission from ref. 128. Copyright 2020, American Chemical Society. | ||
The concept of a clickable photocage, involving light to uncage the fluorophore, is an interesting research area for fluorescence bioassays and imaging. A proof-of-concept study for the bioorthogonal click reaction-activatable vinyl Tz-derivatized coumarin photocage system was presented by Kele and coworkers in 2020 (Fig. 18B).127 Both experimental data and theoretical calculations indicated that the presence of the bioorthogonal Tz motif effectively quenched the excited state of coumarin, necessary for photolysis, thereby disabling photoresponsivity (both photocaging and fluorescence). Upon transformation of the Tz moiety in a bioorthogonal click reaction, the sensitivity to light was fully restored. Leveraging bioorthogonal reactions for highly specific cellular targeting, these conditionally activatable photocages offer additional spatial and temporal control over caged compound release. The application of this probe was demonstrated in live cells using a fluorogenic, conditionally activatable construction, which became light-sensitive only when cells were pretargeted with a mitochondria-directed, complementary bioorthogonal function.
Two-photon fluorescent probes129,130 that can be excited by two NIR photons have been widely used in fluorescence biosensing and imaging, including the development of two-photon bioorthogonally activated probes.88 D–A-type naphthalene fluorophores with moderate-to-good multicolor fluorescence in both one- and two-photon excitation processes are the most widely explored fluorophores for the development of two photon fluorescent probes. Building upon these fluorophores, Park's group128 developed a series of Tz conjugated multicolor fluorogenic two photon probes (Fig. 18C). These probes exhibited robust turn-on fluorescence response (up to 70-fold) and rapid kinetics with TCO in both one- and two-photon fluorescence imaging processes, akin to other FRET-based quenched fluorophores with Tz. Modification of either the naphthalene fluorophore with morpholine or TCO with TPP allowed for the development of bioorthogonal probes for the precise wash-free fluorescence imaging of lysosomes and mitochondria, respectively, using one-photon fluorescence microscopy.120,121 TPP-TCO-assisted two photon microscopic (TPM) imaging of HeLa cells exhibited rapid turn-on wash-free fluorescence in mitochondria.
Biomarker detection plays an important role in the diagnosis of various diseases, while traditional methods based on ELISA and western blot techniques are time consuming and labor intensive. The fast affinity induced reaction sensor (FAIRS) technique introduced by Wang's group131 has the potential to address the limitations of traditional methods (Fig. 19A). The probes are comprised of a sandwich ELISA antibody pair, where one antibody is conjugated with a Tz based bodipy fluorophore and another is conjugated with azabenzonorbornadiene (AN) for fluorogenic click reaction. Used for IL-6 detection in blood serum and the cell culture medium, FAIRS achieved high sensitivity within 6.5 ± 1.0 minutes, promising broad applicability in medical and clinical diagnostics for rapid biomarker detection.
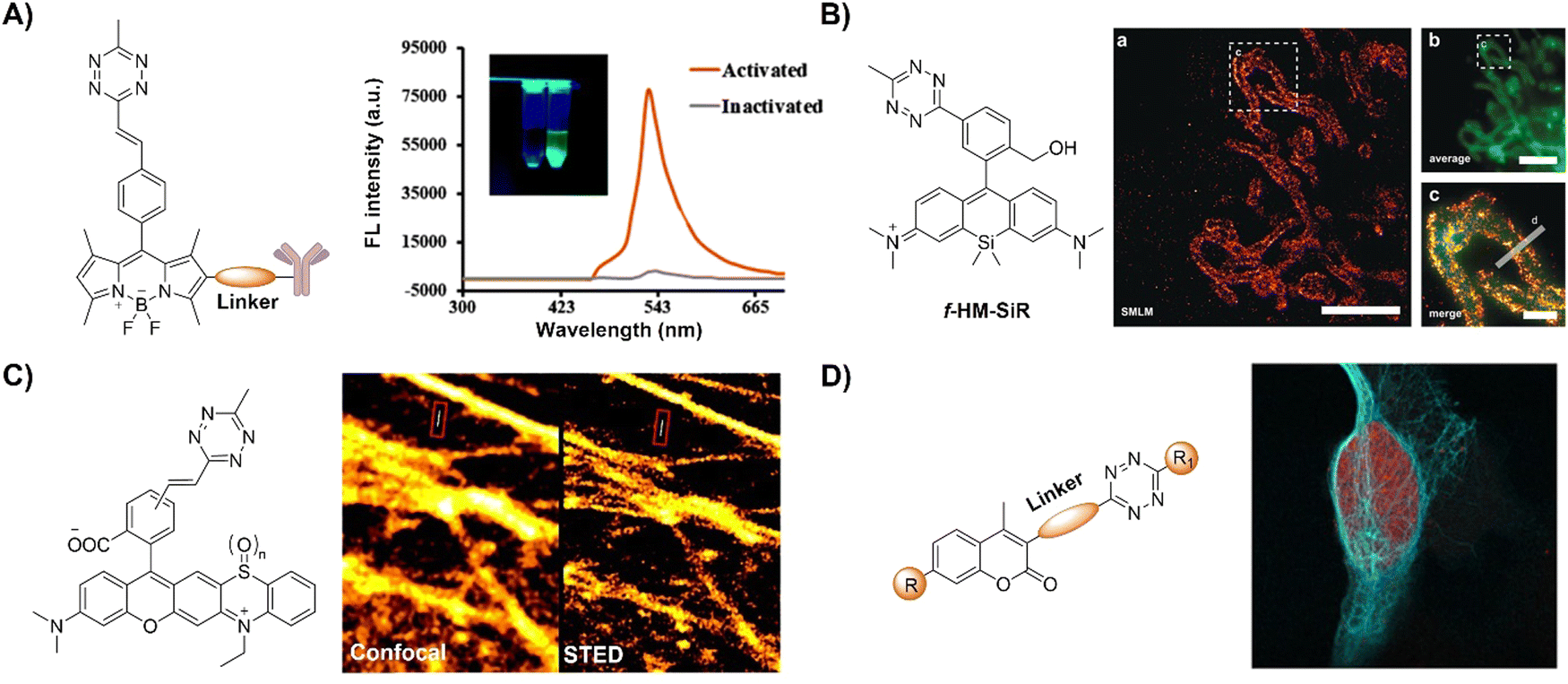 | ||
| Fig. 19 Fluorescent probes designed based on the inverse-electron-demand Diels–Alder reaction using the TBET fluorescence response mechanism. (A) The structure of the Tz conjugated capture antibody and fluorescence emission spectra in the inactivated/activated state, adapted with permission from ref. 131. Copyright 2020, American Chemical Society; (B) the structure of the f-HM-SiR and live-cell SMLM imaging, adapted with permission from ref. 132. Copyright 2020, Wiley-VCH GmbH; (C) the structures of Tz-functionalized rhodaphenothiazines and confocal/stimulated emission depletion microscopy (STED) super-resolution imaging, adapted with permission from ref. 133. Copyright 2020, The Royal Society of Chemistry; and (D) the general formula of coumarin-Tz probes and live cell confocal imaging, adapted with permission from ref. 134. Copyright 2020, Wiley-VCH GmbH. | ||
Super-resolution imaging techniques like single-molecule localization microscopy (SMLM) require active control of fluorescent probes’ emissive and dark states. This could be achieved by high power laser illumination, together with redox reagents or UV exposure. Despite high efficiency, the use of UV light irradiation causes phototoxicity for living cells and organisms, thus not suitable for microscopic imaging. Using an established spontaneously blinking silicon rhodamine (SiR) scaffold, Wombacher and coworkers132 introduced a unique fluorescent probe combining fluorogenic and pH-mediated self-blinking properties, enhancing live-cell localization for SMLM (Fig. 19B). The Tz moiety quenches the fluorescence before reaction, facilitating fluorogenic bioconjugation to dienophile-modified cellular targets (e.g.TPP-BCN) for mitochondria. The probe, f-HM-SiR, boasted superior cell permeability, brightness, and photostability, crucial for live-cell imaging. The self-blinking properties of SiR enabled live-cell SMLM without stabilizing buffers or high excitation power, facilitating long-term, high-resolution intracellular dynamics imaging.
In addition to pH-dependent spontaneous blinking, Kele and colleagues133 devised bioorthogonally applicable Tz-functionalized rhoda phenothiazines, elucidating their light-assisted photooxidation-based fluorogenic properties for stimulated emission depletion microscopy (STED) super resolution imaging (Fig. 19C). The experimental findings confirmed efficient sensitization of 1O2 generation upon green or orange light illumination. This led to self-oxidation of the probes, yielding intensely fluorescent sulfoxide products. Notably, bioconjugated pyridazine exhibited significantly enhanced photooxidizability compared to free Tzs, resulting in a two-orders of magnitude fluorescence increase. This enabled the selective photoactivation of conjugated probes that were used in actin filament labeling with minimal background fluorescence under wash-free conditions. The “ClickOx (clicked, oxidized)” probes used the same green excitation laser for both probe photooxidation and product excitation, offering added value for STED super-resolution microscopy by excitation at 552 nm with a 660 nm depletion laser.
Through-bond energy transfer (TBET)-based Tz probes typically exhibit exceptional fluorescence turn-on responses with various dienophiles, often achieving several thousand-fold enhancement of fluorescence response. For instance, HELIOS (hyperemissive ligation-initiated orthogonal sensing) probes, a series of coumarin-based Tzs, exhibited an outstanding 11000-fold signal enhancement with TCO dienophiles.109 Despite these impressive properties, systematic studies on the design of these coumarin-based probes are essential for developing high quality bioorthogonal probes for precise fluorescence imaging. Towards this aim, Vrabel and coworkers134 synthesized and systematically investigated the photophysical properties of seventeen coumarin-Tz probes that fluoresce upon reaction with TCO- and BCN-containing molecules (Fig. 19D). The obtained data confirmed that attaching an azetidine or N,N-dimethylpiperazine moiety to the coumarin scaffold improves the fluorescence quantum yield. Click reaction with TPP-TCO, TPP-BCN or TCO-docetaxel and BCN-docetaxel allowed for imaging of mitochondria and microtubules in cancer cells.
Abiotic chemical reactions in specific organelles exhibit promise for advancing drug delivery and cell biology and can be achieved by the development of site-specific bioorthogonal probes. In 2021, Vrabel et al.135 reported a structurally redesigned bioorthogonal Tz reagent, enabling the spontaneous accumulation within live mammalian cell mitochondria (Fig. 20A). Optimizing attributes like lipophilicity and positive charge facilitated efficient mitochondrial targeting. Compared to TCO-caged prodrugs, these compounds exhibited a 50-fold potency increase upon mitochondrial release and over 100-fold enhancement versus unmodified compounds. The transformation of common bioorthogonal reagents into organelle-specific probes in this study thus offers potential for subcellular-level prodrug activation and biological process manipulation using purely chemical methods.
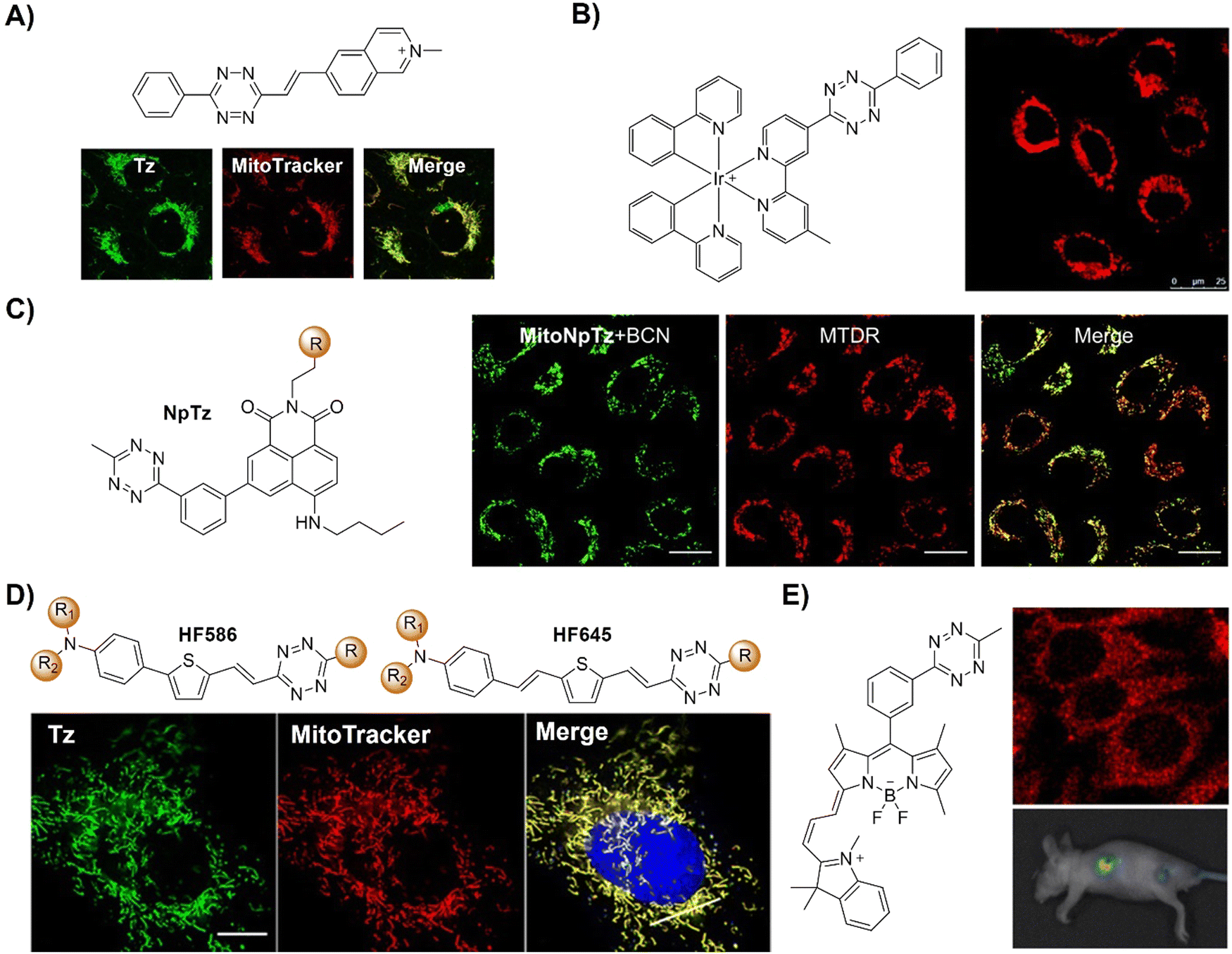 | ||
| Fig. 20 Fluorescent probes based on the inverse-electron-demand Diels–Alder reaction using the TBET fluorescence response mechanism. (A) The structure and Mito imaging of the redesigned Tz probe, adapted with permission from ref. 135, under the license CC-BY-NC-ND; (B) structures and confocal cell imaging of Ir-Tz, adapted with permission from ref. 136. Copyright 2021, The Royal Society of Chemistry; (C) the structures of the naphthalimide tetrazines (Np-Tzs) and representative images of A549 cells, adapted from ref. 137, under the license CC BY-NC 3.0 DEED; (D) the structures of Huaxi-Fluor probes and their application in in situ cell imaging, adapted with permission from ref. 138. Copyright 2021, Wiley-VCH GmbH; and (E) the structure of the bodipy-Tz core and its application in cell and mice imaging, adapted with permission from ref. 139. Copyright 2022, The Royal Society of Chemistry. | ||
The extensive photophysical characteristics of iridium(III) polypyridine complexes have been employed for biomolecular sensing and imaging. Typically, these complexes possess prolonged triplet excited states, making them exceptional photosensitizing agents for 1O2 generation for PDT. Expanding on their prior research, Lo et al.136 introduced iridium(III) Tz complexes with diverse phosphorogenic responses and photosensitization attributes upon bioorthogonal reactions with strained alkynes and alkenes (Fig. 20B). These systems were employed for organelle-specific staining and applications involving controlled photocytotoxicity. In addition to commonly used bodipy, coumarin, and rhodamine frameworks, a series of naphthalimide Tzs,137 such as LysoNpTz and MitoNpTz, were developed by New as bioorthogonal probes for fluorescence imaging of lysosome and mitochondria using traditional morpholine and TPP as the targeting units (Fig. 20C).
Considering the tissue penetration of light, highly fluorogenic Tz bioorthogonal probes emitting at NIR wavelengths are desirable for biomedical imaging. To this aim, Wu et al.138 introduced fluorogenic Huaxi-Fluor probes where the Tz moiety serves as a bioorthogonal partner, quencher, and core of the fluorophore precursor (Fig. 20D). The reaction products, pyridazine derivatives with efficient ICT via a thiophene-containing π-bridge, exhibit a large Stokes shift and high quantum yield in situ. Modulating the conjugation length and electron pull/push capability fine tunes the emission maximum from 556 to 728 nm. Upon bioorthogonal reaction, a hundred-fold fluorescence increase was achieved, enabling wash-free fluorescence cell imaging. Attaching morpholine and TPP with BCN enabled NIR fluorescence imaging of subcellular structures, such as lysosomes and mitochondria, respectively. The application of this bioorthogonal probe was further demonstrated in tumor imaging of a mice model.
Conjugation of Bodipy-Tz with hemi-cyanine led to greater than 221 nm redshift of the emission wavelength for NIR emission. Xu and colleagues139 thus synthesized an efficient NIR turn-on probe, emitting at 731 nm through indolium incorporation into the bodipy-Tz core (Fig. 20E). A norbornene-modified glucosamine derivative was also prepared for metabolic glycoengineering. Combined together, these derivatives enable rapid and sensitive cell imaging in vitro and in vivo. The IEDDA reaction leads to a significant fluorescence increase, enabling tumor-specific imaging in tumor bearing mice. Cyanine dyes generally feature NIR emission with high quantum yields. In 2022, Wang and coworkers140 reported a “torsion-induced disaggregation (TIDA)” phenomenon in a NIR Tz-based cyanine probe (Fig. 21A). Upon Tz-TOC ligation, the cyanine undergoes TIDA, shifting its heptamethine chain from S-trans to S-cis conformation. This shift prevents the aggregation of the products of the probe's bioorthogonal reaction, leading to fluorescence enhancement. In addition to imaging in live cancer cells, the Tz-cyanine probe has been used for sensitive tumor imaging in a mouse model. On the other hand, another work conducted by Wu and colleagues141 introduced a molecular design strategy to create Tz-based far-red/NIR fluorogenic dyes (Fig. 21B). By substituting the amino-Tz group into the meso-positions of various fluorophores, including rhodamine, Si-rhodamines and cyanine, NIR fluorogenic dyes with remarkable turn-on ratios (≥100-fold) were obtained by harnessing the PeT mechanism. These probes exhibit excellent biocompatibility and photostability, and are well-suited for imaging specific intracellular targets and tumors in vivo.
 | ||
| Fig. 21 Bioorthogonal probes based on the inverse-electron-demand Diels–Alder reaction using the TBET fluorescence response mechanism. (A) The structure of the Tz-cyanine core and its application in cell and mice imaging, adapted from ref. 140, under the license CC BY 4.0 DEED; (B) molecular structures and wash-free bioimaging, mice imaging of the representative tetrazine-based probes, adapted with permission from ref. 141. Copyright 2022, Wiley-VCH GmbH; (C) the structure of TMR-Tz-NO and its application in plasma membrane super-resolution imaging in living HeLa cells, adapted from ref. 142, Copyright 2022, American Chemical Society; and (D) the structure of the CFSE-Tz probe and its application in in situ cell imaging, adapted with permission from ref. 143. Copyright 2024, Wiley-VCH GmbH. | ||
Detection and imaging of specific reactive biomolecules like nitric oxide (NO), one of the key RNS and gasotransmitters, in a localized area in biological samples is essential for understanding their biological functions in their native environment. On the basis of the bioorthogonal concept, i.e., the IEDDA reaction between Tz and strained alkyne BCN, Xiao and colleagues142 proposed a versatile toolbox for NO detection and imaging of targeted organelles in live cells and zebrafish (Fig. 21C). In their system, rhodamine-o-phenylenediamine with a Tz unit was used to create the general probe TMR-Tz-NO, in which o-phenylenediamine served as the NO sensing unit and Tz served as the bioorthogonal moiety for anchoring the probe in targeted organelles like mitochondria, lysosomes, and membranes through tethering the BCN moiety to various targeting groups (TPP, morpholine, and Ac4ManN). This toolbox enabled NO imaging in subcellular locations, as demonstrated by zebrafish experiments for tracking inflammation-induced NO production in vivo.
Survival, growth, and differentiation of live cells are heavily influenced by the uptake of exogenous nutrients, and the cellular internalization of these exogenous species can be monitored using bioorthogonal probes. For example, employing IEDDA chemistry, the Kasteren group143 recently reported a bioorthogonal dual fluorogenic probe for monitoring internalization of extracellular nutrition (Fig. 21D). The non-fluorescent carboxyfluorescein-diacetate-succinimidyl ester (CFSE) was conjugated with Tz, a secondary quencher. In this probe system, the diacetate esters can be cleaved by esterase after the probe's internalization into live cells, the succinimidyl ester served as the protein anchoring motif for cytosolic retention, and Tz could stain the internalized substrate through the IEDDA reaction, generating enhanced fluorescence for imaging. This advancement facilitated the monitoring of uptake of diverse dienophile-containing nutrients in live primary immune cell populations through flow cytometry and live-cell microscopy.
Bioorthogonal chemistry also promoted lipid research by enabling the investigations of lipid uptake and processing in living systems while minimizing detectable pendant group-related biological effects. For example, Kasteren and colleageus144 introduced sterculic acid, a 1,2-cyclopropene-containing oleic acid analogue, as a bioorthogonal probe for studying unsaturated free fatty acids in live cells (Fig. 22A). The cyclopropene moiety reacted readily with Tz-fluorophore conjugates, facilitating fatty acid localization via confocal microscopy. This reaction remained orthogonal to subsequent copper catalyzed Huisgen ligation (CCHL) reactions, enabling the simultaneous study of multiple biomolecules. Further research using sterculic acid as a PTM (post-translational modification) for protein modification suggested that this probe is suitable for investigating protein oleoylation via SDS-PAGE and chemical proteomic analysis. This approach, combined with a broadly applicable synthesis of cyclopropene-containing fatty acids, enabled visualization of unsaturated lipids using live-cell microscopy and proteomics.
 | ||
| Fig. 22 Bioorthogonal probes based on the inverse-electron-demand Diels–Alder reaction using the TBET fluorescence response mechanism. (A) The structure of Tz-fluorophore conjugate/sterculic acid and its application in cell imaging, adapted from ref. 144, under the license CC BY-NC 4.0 DEED; (B) the structure of acridine–Tz conjugate (H1-TzB)/vinyl-DNA and wash-free imaging of nucleic acids in HeLa cells, adapted with permission from ref. 145. Copyright 2022, Wiley-VCH GmbH; (C) the structure of the Tz-quenched fluorophore and its application in RNA labelling in live cells, adapted from ref. 146, Copyright 2022, American Chemical Society; (D) the structure of the KIzTz probe and its application in wash-free cell imaging, adapted with permission from ref. 147. Copyright 2021, Elsevier B.V; (E) the structure of the Tz-modified tetraphenylene and its application in wash-free cell imaging, adapted from ref. 148, Copyright 2022, The Royal Society of Chemistry; and (F) the structure of the naphthalimide-Tz probe and its application in bioimaging in live HeLa cells, adapted from ref. 149, Copyright 2022, The Royal Society of Chemistry. | ||
Selective bioorthogonal labelling of nucleic acids contributes significantly to investigating their chemical biology in situ, while steric hindrance from densely packed bioorthogonal functional groups in chromatin could block the chemical modification of nucleic acids in living cells. To overcome this obstacle, Luedtke and coworkers145 introduced a dual enhancement strategy for nucleic acid-templated reactions (Fig. 22B). This approach employed a fluorogenic intercalating agent capable of undergoing IEDDA with DNA containing 5-vinyl-2′-deoxyuridine (VdU) or RNA containing 5-vinyl-uridine (VU). Reversible high-affinity intercalation of the acridine-Tz conjugate “PINK (Probe for Imaging Nucleosidic AlKene)” dramatically accelerated the reaction rate of Tz-alkene IEDDA on duplex DNA by 60000-fold (590 M−1 s−1) in comparison with non-templated reactions. Moreover, loss of Tz-acridine fluorescence quenching enabled highly fluorogenic detection under wash-free conditions, facilitating dynamic live-cell imaging of acridine-modified nucleic acids during cell division. Using this bioorthogonal labelling approach, imaging of nucleic acids in HeLa cells was achieved.
Apart from DNA, the development of bioorthogonal probes for RNA labelling is particularly promising for advancing disease diagnosis and treatment monitoring. To facilitate the RNA labelling in live cells, which was not achieved by conventional methods, Qian and colleagues146 introduced a fluorogenic system for precise mRNA labelling and imaging in live cells by combining hybridization chain reaction (HCR) with proximity-induced bioorthogonal chemistry (Fig. 22C). This strategy offered spatial resolution of low-expression targets, activating Tz-quenched fluorophores through HCR-directed bioorthogonal reactions for fluorescence turn-on and amplification. Additionally, a biocompatible nucleic acid delivery system derived from black phosphorus nanosheets (BPNSs) was employed for efficient imaging of cytosolic mRNAs in live cells. The platform's flexibility in targeting RNA of interest facilitated its use as a versatile tool for RNA tracking in living cells.
Most Tz-based bioorthogonally activated fluorogenic probes encounter aggregation-caused quenching (ACQ) issues. To address these issues, AIEgens have been explored for the development of bioorthogonal probes for fluorescence imaging using IEDDA. For example, Kim and colleagues147 found that Tz could quench the emission of the Kaleidolizine (KIz) AIEgen through TBET, and thus bioorthogonally activated probes KIzTz were designed and synthesized (Fig. 22D). Bioorthogonal click reaction with TPP-TCO, together with AIE, facilitated wash-free fluorescence imaging of mitochondria in live cells using KIzTz. In another example, Tian and coworkers148 devised a series of novel Tz-modified tetraphenylenes (TPEs) as bioorthogonally activated AIE fluorogenic probes (Fig. 22E). Both fluorescence and AIE properties were quenched by Tzvia a TBET mechanism and then reactivated upon Tz-to-pyridazine conversion through the IEDDA reaction. The resulting cycloadducts exhibited significant fluorescence enhancement, large Stokes shift, high fluorescence quantum yield, and AIE activity. Adjusting the length and position of the π-linker finely tunes a probe's photophysical properties, while an excessively planar π-linker induced AIE-to-ACQ transformation. Bi-tetrazyl-substituted probes exhibited higher turn-on ratios due to a “double-quenched” function. Using double-clickable linkers, fluorescent macrocycles were formed, enabling organelle-specific bioorthogonal wash-free imaging in live cells suitable for applications in biomedicine.
Most naphthalimide derivatives, particularly unsubstituted naphthalimides, exhibit ACQ, while some naphthalimide derivatives with appropriate substitution exhibit AIE properties. For example, Tian and coworkers149 introduced a range of TBET-based naphthalimide-Tz probes with broad bioorthogonal applicability and fluorescence activation properties (Fig. 22F). The IEDDA cycloadducts exhibited exceptional photophysical traits, demonstrating AIE characteristics. Modifying the π-bridge linker between the naphthalimide and Tz enabled precise control over emission, spanning from blue to red, while also influencing the AIE attributes. These Tz-based probes facilitated effective wash-free fluorogenic protein labeling and high signal-to-noise ratio mitochondrial imaging (click reaction with BCN-TPP) in live cells.
Two-photon excitation, by shifting absorption to the red, significantly enhances the signal-to-noise ratio and reduces photodamage, enabling imaging approximately 10 times deeper than with confocal microscopy. Despite this, effective two-photon excitable fluorogenic probes are scarce. Mahuteau-Betzer et al.150 introduced fluorogenic probes comprising a two-photon excitable fluorophore and a Tz quenching moiety (Fig. 23A). These probes exhibit impressive kinetics (up to 1.1 × 103 M−1 s−1) and emit in the red with moderate to high turn-on ratios. The best candidate achieved a substantial 13.8-fold turn-on fluorescence response under one-photon excitation, exceeding the 3-fold threshold for high contrast wash-free live-cell imaging. Notably, under two-photon excitation, an 8.0-fold turn-on was observed. The high two-photon brightness (>300 GM) of the resulting adduct allowed for the use of low laser power suitable for in vivo imaging.
 | ||
| Fig. 23 Inverse-electron-demand Diels–Alder reaction-based bioorthogoal probes using the TBET fluorescence response mechanism. (A) The structure of a two-photon excitable Tz-fluorophore conjugate and its application for the imaging of live A549 cells, adapted from ref. 150, under the license CC BY 3.0 DEED; (B) the structure of a C-DIPY-Tz conjugate and wash-free imaging in live cells, adapted with permission from ref. 151. Copyright 2023, Wiley-VCH GmbH; (C) the structure of the Tz-functionalized Cy3 derivative and its application in STED imaging in live cells, adapted from ref. 152, under the license CC BY 3.0 DEED; (D) the structure of the Tz-xanthone (PaX) core fluorophore probe and its application in confocal/super resolution imaging, adapted from ref. 153, under the license CC-BY 4.0; (E) the structure of the Tz-isonitrile probe and its application in wash-free cell imaging, adapted from ref. 154, Copyright 2024, Wiley-VCH GmbH; and (F) the structure of the AIE-Tz probe (MBTPA-Tz as an example) and its application in bioimaging, adapted from ref. 155, Copyright 2024, under the license CC BY 4.0 DEED. | ||
Carbon-dipyrromethenes (C-DIPYs) are a new class of fluorophores exhibiting intense emissions from green to NIR.156 Using C-DIPY as a signaling unit, Ng and colleagues151 synthesized a water-soluble bioorthogonal probe via a two-step procedure from a known carbonyl pyrrole dimer (Fig. 23B). Similar to other probes, the emission of this probe was quenched by a Tz moiety at pH 7.4 PBS by TBET and could be switched on after click reaction with BCN derivatives, resulting in up to 6.6-fold restoration of fluorescence. This bioorthogonal activation was demonstrated in U-87 MG human glioblastoma cells, with fluorescence intensity increasing by 8.7-fold post-incubation with the BCN derivative. Interestingly, in contrast to previous reports on C-DIPYs that are mitochondria targeting due to their cationic nature, this probe preferential localizes in lysosomes, which could be attributed to an energy-dependent endocytic cellular uptake pathway.
Cell surface and intracellular labelling and imaging can be achieved by the development of bioorthogonally activated probes that are cell membrane impermeable and permeable, respectively. Kele and coworkers152 synthesized three series of Tz-functionalized Cy3 derivatives using varied design approaches and explored how the bioorthogonal unit influences the fluorescence of yellow-emitting indocyanines (Fig. 23C). The employed strategies included direct conjugation, phenylene-linked units (TBET design) and vinylene-conjugated systems (π-extended direct conjugation), each comprising membrane-permeable and non-permeable derivatives. Fluorogenic characteristics upon reaction with BCN were assessed via IEDDA and the data indicated that Tz quenching efficiency is dependent on the distance between Tz and the fluorophore, confirming the efficacy of internal conversion-based modulation over through-bond energy transfer. The directly conjugated probes effectively labeled genetically bioorthogonalized extra- and intracellular proteins in live cells for STED super-resolution imaging of target structures.
Harnessing Tz dyads’ inherent excited state quenching to control fluorophore activation, Hell and colleagues153 reported a series of highly compact, click compatible and photoactivatable bioorthogonal probes for super resolution imaging (Fig. 23D). In this probe system the quencher and bioorthogonal click unit Tz was conjugated with the photoactivatable xanthone (PaX) core fluorophore. These probes were able to be used in most nanoscopies, including STED, PALM, and MINFLUX. Leveraging MINFLUX's ultimate resolution, precise visualization of vimentin filament substructure in cellulose was achieved through incorporating clickable unnatural amino acids in the filament monomers’ head domain with molecular-scale precision.
Creating fluorogenic probes that can simultaneously label multiple targets in live cells is of great importance for a better understanding of the complex cellular processes. The emerging [4+1] cycloaddition between Tz and isonitriles offers potential as a bioorthogonal tool for multiplex labelling and fluorescence cell imaging. For instance, the Wu group154 introduced a range of Tz-functionalized bioorthogonal probes for multiplexing and live cell bioimaging (Fig. 23E). By integrating pyrazole adducts into the fluorophore frameworks, the post-reacted probes exhibited impressive fluorescence turn-on ratios, achieving up to 3184-fold enhancement in fluorescence. These modification strategies are adaptable for the development of probes using other fluorophores, enabling the design of probes showing a broad emission spectrum ranging from 473 to 659 nm. In imaging applications, these probes were found to be able to simultaneously label multiple targets in live cells for wash-free fluorescence imaging.
The impact of molecular aggregation on emission properties was included in the design of Tz-functionalized bioorthogonal probes with increased emission enhancement ratio (IAC/IBC, where IAC and IBC stand for fluorescence intensities after and before click reactions, respectively) for labeling and imaging of multiple cellular organelles. As an example of AIEgen-based probes, Tang and coworkers155 demonstrated that an exceptionally high IAC/IBC can be achieved in aggregated systems when Tz is combined with AIEgens (AIE-Tz, Fig. 23F). Tz enhanced its quenching efficiency upon aggregation, remarkably reducing background emissions. Subsequent click reactions induced changes in Tz's chemical structure and prompted AIE, resulting in significantly enhanced IAC/IBC. The potential of the ultra-fluorogenic systems for selective imaging of multiple organelles was confirmed in the mitochondria, plasma membranes, and lipid droplets of live cells.
Previous research conducted by the Lo group has confirmed that the iridium(III) complex with a built-in Tz moiety could serve as a bioorthogonal probe for luminescence biolabeling and imaging.118 Together with the excellent photo- and sono-sensitizing performance of metal complexes, Cai and colleagues created activatable “off–on” sonosensitizers with enhanced sonodynamic activity and immunogenic cell death (ICD) induction capability for precise and potent tumor sono-immunotherapy (Fig. 24A).157 The non-luminescent iridium(III) complexes that are loaded into pH sensitive human serum albumin (HSA) nanoparticles could be released in an acidic microenvironment found in tumors and then activated and anchored to cancer cell membranes after click reaction with BCN receptors. With ultrasound irradiation, cytotoxic ROS was produced for tumor cell ablation through oxidative stresses. Membrane damage induced highly immunogenic PANoptosis, boosting ICD to elicit robust systemic antitumor immunity against primary/distant tumors and lung metastasis. Based on a similar approach, a ruthenium(II) complex with a built-in Tz moiety was developed by the same group for bioorthogonal activation and sonodynamic therapy with boosted antitumor immunity (Fig. 24B).158
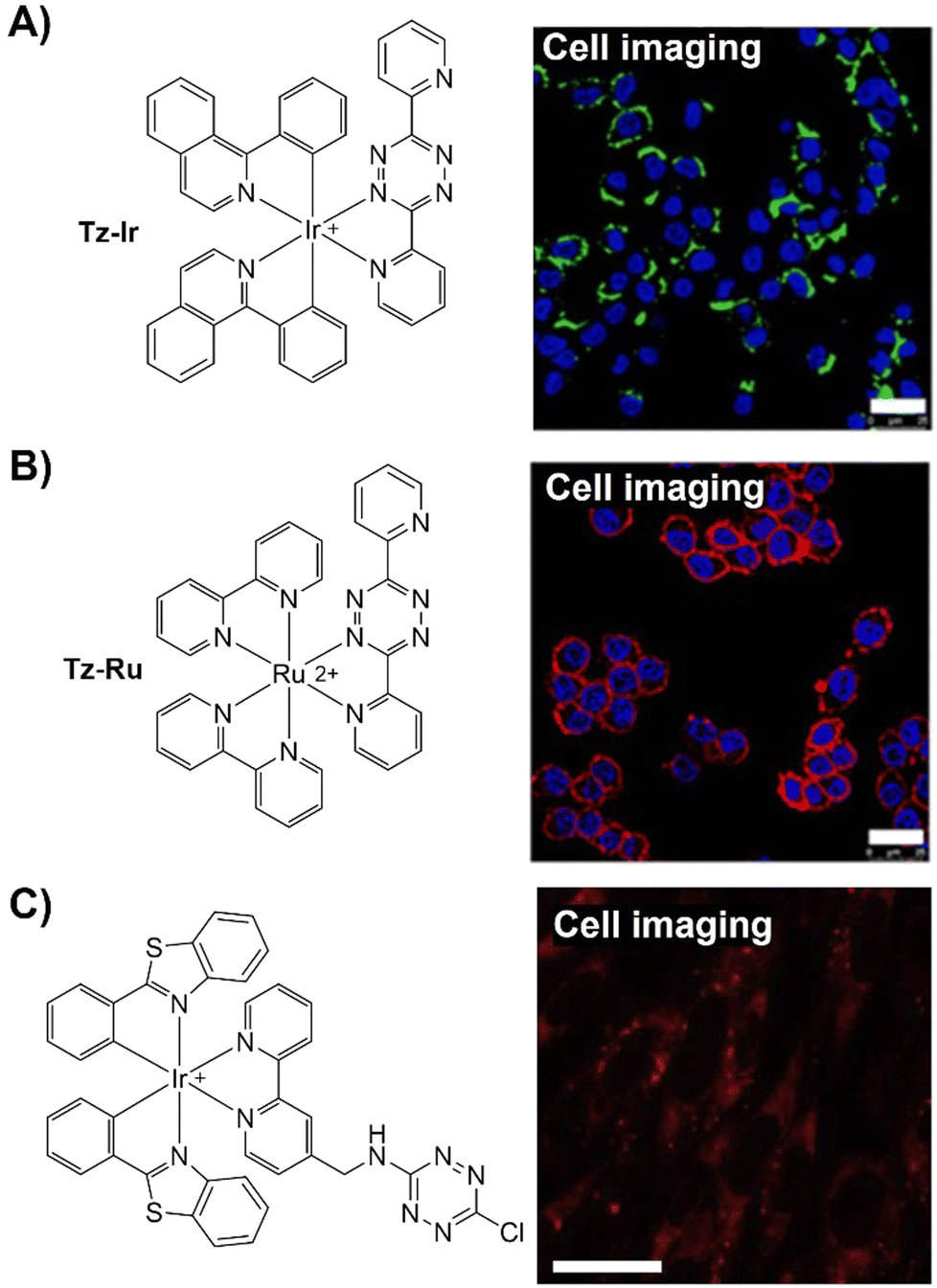 | ||
| Fig. 24 Transition metal complex-based bioorthogonal probes based on the inverse-electron-demand Diels–Alder reaction using the TBET fluorescence response mechanism. (A) The structure of a Tz–Ir complex and its application in in situ cell imaging, adapted with permission from ref. 157. Copyright 2024, Elsevier Ltd; (B) the structure of a Tz–Ru complex and wash-free imaging in live cells, adapted with permission from ref. 158. Copyright 2024, American Chemical Society; and (C) the structure of the Tz-Ir complex and its application in cell imaging, adapted with permission from ref. 159. Copyright 2016, Elsevier Ltd. | ||
On the other hand, the Huang group160 reported a highly sensitive and specific “labeling after recognition” sensing strategy where luminophore labeling and then biological recognition were proposed (Fig. 25A). Based on the FRET from the iridium(III) complex (donor) to rhodamine (acceptor) a bioorthogonal probe was developed for luminescence and lifetime detection and imaging of caspase-3. The donor and acceptor FRET pair was linked using a caspase-3 sensitive linker, the DEVD tetrapeptide. From the luminescence lifetime analysis, it was found that the iridium(III) complex's emission lifetime was quenched by FRET to the rhodamine and then restored after caspase-3 promoted DEVD cleavage. Imaging of caspase-3 catalytic activity in apoptotic cells was achieved via photoluminescence lifetime imaging microscopy, confirming intracellular bioorthogonal bis-labeling and catalytic cleavage dynamics. This study presents a typical example of the “labeling after recognition” sensing strategy and a luminescence lifetime based bioorthogonal probe.
 | ||
| Fig. 25 Inverse-electron-demand Diels–Alder reaction-based bioorthogonal probes using the FRET fluorescence response mechanism. (A) The structure of the Tz-rhodamine derivative and photoluminescence lifetime confocal microscopic images of living HeLa cells, adapted with permission from ref. 160. Copyright 2019, American Chemical Society; (B) the structure of the Tz–terbium complex and luminescence imaging in zebrafish, adapted with permission from ref. 161. Copyright 2020, The Royal Society of Chemistry; (C) the structure of Mag-S-Tz and its application in cell imaging, adapted with permission from ref. 162. Copyright 2016, American Chemical Society; (D) the structure of the Tz-quenched RhoVR derivative and its application in confocal imaging, adapted with permission from ref. 163. Copyright 2022, American Chemical Society; (E) the structure of the Tz tag and its application in wash-free S. aureus bacteria imaging, adapted with permission from ref. 164. Copyright 2020, American Chemical Society; and (F) the structure of the Tz-caged probe and its application in bioimaging in live HSF cells adapted with permission from ref. 165. Copyright 2022, The Royal Society of Chemistry. | ||
In comparison with metal complexes with prolonged emission lifetime at hundreds of nanoseconds, rare earth lanthanide chelates generally have a longer emission lifetime at microsecond levels. The prolonged emission lifetime makes the developed bioorthogonal probes suitable for background-free time-resolved luminescence bioassay and imaging as the lifetime of autofluorescence is generally less than 10 ns. In this context, Wong et al.161 introduced a stable, biocompatible terbium complex, Tb-1, enabling bioorthogonal ligation with engineered cell-surface glycans, yielding responsive luminescence (Fig. 25B). They strategically incorporated a bioorthogonal luminescence resonance energy transfer (LRET) quencher into a tripodal terbium complex with low toxicity. Upon the IEDDA reaction, the electronic structure of the quencher changed, resulting in enhanced terbium emission intensity. The prolonged lifetime (approximately 1 ms) and bright emission (Φ ∼ 10%) allowed for background-free luminescence imaging in A549 cancer cells and zebrafish.
Metal ions, such as sodium (Na+), potassium (K+), magnesium (Mg2+), iron (Fe3+) and calcium (Ca2+), are essential for biological functions, thus monitoring levels of essential metal ions is crucial for disease diagnosis. Bioorthogonally activated probes have been developed for visualizing localization of these metal ions, such as Mg2+. The Buccella group162 devised a two-step method to localize a fluorescent Mg2+ sensor within specific organelles (Fig. 25C). This method involved a rapid reaction between a Tz-functionalized pro-sensor (Mag-S-Tz) and a genetically encoded HaloTag fusion protein. Protein conjugation preserved the sensor's metal-binding properties, with a suitable dissociation constant (Kd = 3.1 mM) for detecting intracellular Mg2+ concentration. This bioorthogonally activated probe could detect Mg2+ in target organelles of HEK 293T cells, offering improved spatial resolution without interference.
Electric potential differences across lipid bilayers are fundamental in cellular physiology. In contrast to plasma membranes, transmembrane potentials of intracellular organelle bilayers, including those of the mitochondria, lysosomes, nuclei, and the endoplasmic reticulum (ER), are difficult to investigate using traditional methods like patch-clamp electrophysiology. Recently, Miller's group163 introduced LUnAR RhoVR (Ligation Unquenched for Activation and Redistribution Rhodamine-based Voltage Reporter) for optically monitoring endoplasmic reticulum (ER) membrane potential changes in living cells (Fig. 25D). By pairing Tz-quenched RhoVR with TCO-conjugated ceramide (Cer-TCO) for ER targeted labelling, specific and bright fluorescence was observed, enabling voltage-sensitive imaging and validation of plasma-ER membrane functional coupling through a K+ diffusion mediated change of ER membrane potential.
Both BCN and TCO are essential linkers for the IEDDA reaction with Tz in bioorthogonal “click” ligation. To enhance the availability of TCO reagents, Zanda and colleagues164 synthesized a series of TCO linkers derived from trans,trans-1,5-cyclooctadiene for biomolecule conjugation (Fig. 25E). These linkers facilitated bioorthogonal ligation with Tz tags, offering linker flexibility crucial for stability and ligation efficiency, as exemplified by BSA. Labelling of biomolecules in cellular lysates and live bacteria like S. aureus was used to demonstrate their application in bioorthogonal ligation, showing promise for live-cell imaging. Moreover, the short PEG linker TCO enabled surface decoration of monoclonal antibodies like rituximab and obinutuzumab with functional handles and minimal mAb aggregation.
Wu and coworkers165 devised a series of fluorescent probes by exploring isonitrile-Tz click-to-release mechanism-based bioorthogonal activation (Fig. 25F). Efficient quenching of canonical fluorophores was observed across the visible spectrum using the tBuTz-cage, with substantial fluorescence activation upon isonitrile-induced cleavage. DNA and RNA templated chemistry accelerated this bioorthogonal cleavage reaction at low concentrations, suggesting potential for miRNA detection. Orthogonality between the tBuTz-caged probe cleavage and iPrTz-caged probe and TCO species cleavage was demonstrated, facilitating their use in live cells. This research using two orthogonal cleavage reactions in live cells has provided options for bioorthogonal fluorogenic probes in multi-target detection.
The distance between Tz and the fluorophore is one of the key points that should be taking into consideration when developing bioorthogonal probes for fluorescence labelling and imaging.144,166–168 On the basis of this consideration, Wombacher's group169 synthesized a series of fluorescence bioorthogonal probes with a close proximity Tz-fluorophore conjugate for wash-free live cell super resolution imaging (Fig. 26A). These probes were designed using a series of red and far-red fluorogenic Tz dyes (HDyes) for bioorthogonal labelling. The short distance between the Tz and fluorophores allowed for effective fluorescence quenching through an energy transfer process. HDyes displayed high fluorescence and was suitable for wash-free live-cell imaging, which was demonstrated in two-color labeling experiments and STED super-resolution imaging of UAA-labeled target proteins. The modular synthetic approach yielded a self-blinking HDye, useful for long-term live-cell STED imaging and super-resolution optical fluctuation imaging (SOFI).
 | ||
| Fig. 26 Fluorescent probes based on the inverse-electron-demand Diels–Alder reaction using the FRET fluorescence response mechanism. (A) The structures of HDyes and live-cell confocal and STED imaging of COS-7 cells, adapted from ref. 169, under the license CC-BY-NC-ND 4.0; (B) the structure of Tz-bodipy and wash-free imaging in live HeLa cells, adapted with permission from ref. 170. Copyright 2020, Wiley-VCH GmbH; (C) the structure of the peptide–Tz conjugate and its application in cell imaging, adapted from ref. 171, under the license CC BY 4.0 DEED; and (D) the structure of Tz-substituted bodipy (Tz-NIR-AZA as an example) and its application in HeLa Kyoto cell imaging, adapted with permission from ref. 172. Copyright 2023, Elsevier Ltd. | ||
Typically, small molecules lack specificity in targeting cellular compartments, often distributing across multiple areas or randomly without achieving desired localization. Vázquez et al.170 introduced a modular peptide-based platform separating the halogenated Bodipy photosensitizer (PS) and the quencher Tz, enhancing photochemical properties and enabling spatial activation within specific organelles upon peptide conjugation (Fig. 26B). Various dual-labeled peptides, differing only in peptidic vectors but sharing the same PS/quencher pair, controlled confined photodynamic effects via bioorthogonal in situ activation in specific subcellular organelles. For example, Tz-bodipy, activated specifically in the cytoplasmic membrane through bioorthogonal IEDDA, exhibited superior PDT performance, enhancing photodynamic effects at lower concentrations without dark toxicity. Compatible with common bioconjugation strategies, this versatile cysteine-based approach facilitated the exploration of diverse subcellular compartments and peptidic functionalities, advancing bioorthogonally activatable PS applicability.
In addition to the PDT,157,158 bioorthogonal chemistry has also enabled the delivery of a drug in the form of a prodrug into specific diseased tissues with minimal drug release in other tissues or side effects.125,171,173 For instance, Wu's group171 introduced 3-vinyl-6-oxymethyl-Tz (voTz) as a reagent for crafting Tz-caged prodrugs and selectively labeling peptides, generating bioorthogonally activatable peptide-prodrug conjugates (Fig. 26C). These stable prodrugs were bound specifically to target cells, aiding cellular uptake. Subsequent bioorthogonal cleavage reactions activated the prodrug, significantly inhibiting tumor cell growth while ensuring remarkable biosafety for normal cells. In vivo trials using a tumor-bearing mice model confirmed the therapeutic efficacy and biosafety of this prodrug design strategy.
Bioorthogonal probes with NIR emission can also be obtained using bodipy derivatives as fluorophores. For example, Wu and coworkers172 explored the use of Tz-substituted BF2-azadipyrromethene probes (bodipy derivatives) and model Tzs for their reactivity with strained alkyne and alkene dienophiles, aiming to identify the optimal probe for cellular imaging applications (Fig. 26D). BCN exhibited superior reactivity, with reactions finishing within 15 min. The Tz-substituted BF2-azadipyrromethene displayed intense fluorescence at 715 nm as the cycloaddition progressed in an aqueous solution, with a remarkable 43-fold enhancement. Such good efficiency prompted its application in bioorthogonal cell imaging, allowing for dynamic imaging of the reaction in subcellular regions of live cells upon pre-incubation with a 2-deoxy-glucose-alkyne dienophile.
 | ||
| Fig. 27 Inverse-electron-demand Diels–Alder reaction-based bioorthogonal probes for fluorescence imaging. (A) The fluorogenic reaction between styrene/Tz and its application for selective labeling of E. coli cells, adapted from ref. 174, under the license CC BY 3.0 DEED; (B) the structure of Tz–bodipy, fluorescence quenching via the TICT process and its application in cell imaging, adapted with permission from ref. 175. Copyright 2022, The Royal Society of Chemistry; (C) fluorogenic reaction between TCO and 1,2,4-triazines and its application in cell imaging in live U2OS cells, adapted from ref. 176, under the license CC BY 3.0 DEED; (D) the structure of ICPr-resorufin/Tz and its application in zebrafish imaging, adapted with permission from ref. 177. Copyright 2018, American Chemical Society; and (E) the structures of the TCO-caged prodrug (TCO-Dox) and Tz-bearing NapK(Tz)YF and in vivo mice imaging, adapted from ref. 178 under the license CC BY 4.0 DEED. | ||
Recently, 1,2,4-triazines have emerged as versatile dienes in IEDDA reactions with strained dienophiles. In comparison with Tz, 1,2,4-triazines exhibit lower reactivity towards both TCO and BCN although they are highly stable under biological conditions. After a systematic investigation to understand the mechanism of reaction of various 1,2,4-triazines with TCOs in the IEDDA reaction, Vrabel's group discovered a strong dependence on the substitution patterns of both substrates (Fig. 27C).176 This exploration led to the discovery of new N-alkyl pyridinium triazines with exceptional properties for bioconjugate chemistry. An efficient, modular synthetic strategy was then used to construct diverse heterobifunctional probes based on the pyridinium 1,2,4-triazine scaffold. The fluorogenic nature (up to 50-fold enhancement with 650 nm emission) of the reactions of these heterodienes with TCOs allowed for fluorogenic cell labeling.
Dissociative bioorthogonal reactions enable precise control over the release of bioactive agents and reporter probes. Franzini et al.177 identified 3-isocyanopropyl (ICPr) substituents as removable masking groups within biological systems (Fig. 27D). The 3-isocyanopropyl derivatives are able to react with Tzs to yield 3-oxopropyl groups for bioorthogonal labelling. The study highlighted rapid reaction kinetics, achieving near-complete liberation of phenols and amines under physiological conditions. Compatibility with living organisms was confirmed by the release of the resorufin fluorophore and the mexiletine drug in zebrafish embryos implanted with Tz-modified beads. Due to their benefits including ease of synthesis, swift kinetics, diverse leaving groups, high release yields, and structural compactness, ICPr derivatives have emerged as appealing chemical caging moieties for applications in chemical biology and drug delivery.
A synergistic prodrug activation system was developed by Gao and coworkers178 using a combination of EISA and Tz/TCO bioorthogonal decaging reactions (Fig. 27E). The system achieved spatiotemporal targeting and selective activation of prodrugs within cancer cells, crucial for enhancing chemotherapeutic selectivity. A Tz moiety was linked to the EISA motif to trigger inverse-electron-demand Diels–Alder (inv-DA) reaction-mediated chemical decaging of a TCO-caged effector molecule intracellularly. Cancer cells with elevated phosphatase levels underwent intracellular EISA viaTz-bearing NapK(Tz)YF, leading to significant Tz accumulation and subsequent liberation of the TCO-caged prodrug (TCO-Dox), inducing cancer cell death while sparing normal cells. The increased emission of the bioorthogonal reaction allowed for imaging of cancer cells as well as the Dox release. This approach enhanced the selectivity of Dox delivery for cancer cells, offering potential for reducing adverse drug effects and demonstrating efficacy in a xenografted cervical cancer mice model.
The IEDDA reaction between Tz and TCO offers precise control over bioactive agents and imaging probe release. For example, Bernardes et al.179 employed a pre-targeted activation strategy using single-walled carbon nanotubes (SWCNTs) bearing Tzs (Tz@SWCNTs) and a TCO-caged molecule to deliver active effector molecules for tumor imaging (Fig. 28A). Optimized in vivo fluorescence imaging was achieved using a new fluorogenic NIR probe, tHCA, that could be activated via bioorthogonal chemistry, enabling tumor imaging in mice. This approach allowed for selective Dox prodrug activation for tumor chemotherapy and real-time fluorescence imaging in living cells. Combining tHCA with a pre-targeted bioorthogonal approach achieved non-invasive, high target-to-background ratio tumor visualization in a xenograft mice model, highlighting the potential of Tz-functionalized SWCNTs for targeted bioorthogonal decaging with minimal off-site activation.
 | ||
| Fig. 28 Bioorthogonal probes for fluorescence imaging based on the inverse-electron-demand Diels–Alder reaction. (A) The structure and cell imaging of tHCA/Tz@SWCNTs, adapted from ref. 179, under the license CC BY 4.0 DEED; (B) the structure of the Tz-prodrug and its application in cell imaging, adapted with permission from ref. 180. Copyright 2022, The Royal Society of Chemistry; (C) the structure of TCO-HCA/Gal-Tz and its application in cell imaging, adapted with permission from ref. 181. Copyright 2024, Wiley-VCH GmbH; and (D) the structures of the CyPVE/Tzs and in vivo fluorescence images of tumor-bearing nude mice, adapted with permission from ref. 182. Copyright 2020, Wiley-VCH GmbH. | ||
In another example, a bioorthogonally activated drug delivery system (DDS) was obtained by Gao's research group,180 and the bioorthogonal prodrug activation allowed for drug release in a precise imaging manner (Fig. 28B). This self-reporting bioorthogonal prodrug activation system was developed using fluorescence emission to interpret activation events. Tz, acting within ROS-responsive supramolecular assemblies, served both as a fluorescence quencher and as a prodrug activator. The subsequent IEDDA reaction with trans-cyclooctene-paclitaxel (TCO-PTX) facilitated the release of fluorescence and active drugs concurrently, enabling a linear relationship for quantifying PTX release. This fluorescence-based visualizable drug delivery system is promising for elucidating drug delivery processes and determining prodrug activation efficacy.
Senescent cell targeted bioorthogonal Tz ligation has contributed to the clearance of these cells in the body, slowing down the aging process as well as promoting anticancer treatment. For example, Wang's group181 presented a senolytic approach using senescent cell-sensitive bioorthogonal Tz ligation. In this system, a galactose (Gal) moiety was engineered into dihydrotetrazine, serving as both a recognition element for senescence-associated β-galactosidase (SA-β-gal) and a caging group for Tz (Fig. 28C). Gal-Tz efficiently released fluorescent hemicyanine (HCA) for the precise imaging and release of Dox from TCO-caged prodrugs for targeting and eliminating senescent HeLa and A549 cells over non-senescent cells with a 16.44 senolytic index. This approach was then used for the selective activation and delivery of proteolysis-targeting chimeras (PROTACs) as senolytics, potentially offering an efficient intervention for cell senescence by degrading targeted proteins catalytically. This research established bioorthogonal Tz ligation as a viable method for selectively removing senescent cells.
Bioorthogonal prodrug activation using exogenous click reactions also offers a solution for precise drug release with fluorescence turn-on response for efficient monitoring, overcoming challenges of current approaches based on endogenous receptors or overexpressed enzymes. For example, prodrug activation was developed by Wang and colleagues,182 in which the drug release was achieved synergistically by using a bioorthogonal reaction and the acidic tumor microenvironment (Fig. 28D). This approach uses a Tz-functionalized pH-sensitive polymer micelle, which disassembles in an acidic environment, triggering a bioorthogonal click reaction to activate PDT. The ROS generation could be specifically activated by Tzs through a bioorthogonal click reaction to trigger NIR fluorescence signals for precise tumor imaging and phototoxicity for cancer cell ablation, allowing for imaging-guided PDT. This design ensures Tz groups remain inert in circulating micelles but become active upon micellular disassembly in tumors, enabling specific prodrug activation. This method, exploiting the dysregulated pH common in cancers, offers a universal approach for bioorthogonal prodrug activation in acidic tumor microenvironments.
In another research, Ng and coworkers183 developed a versatile bioorthogonal strategy enabling targeted delivery and site-specific activation of photosensitizers for precise tumor imaging and antitumoral PDT (Fig. 29A). This approach featured an isonitrile-caged distyryl boron dipyrromethene-based photosensitizer, NC-DSBDP, activated through converting the meso ester substituent to carboxylate via the [4+1] cycloaddition with a Tz derivative. Using galactose or GE11 peptide-conjugated Tzs (gal-Tz and GE11-Tz), cancer cells expressing the asialoglycoprotein receptor or epidermal growth factor receptor were selectively labeled. Internalized NC-DSBDP underwent ester-to-carboxylate transformation triggered by these Tzs, activating fluorescence for imaging and ROS generation within the target cells for cancer cell ablation. In vivo, bioorthogonal activation led to effective tumor eradication in nude mice.
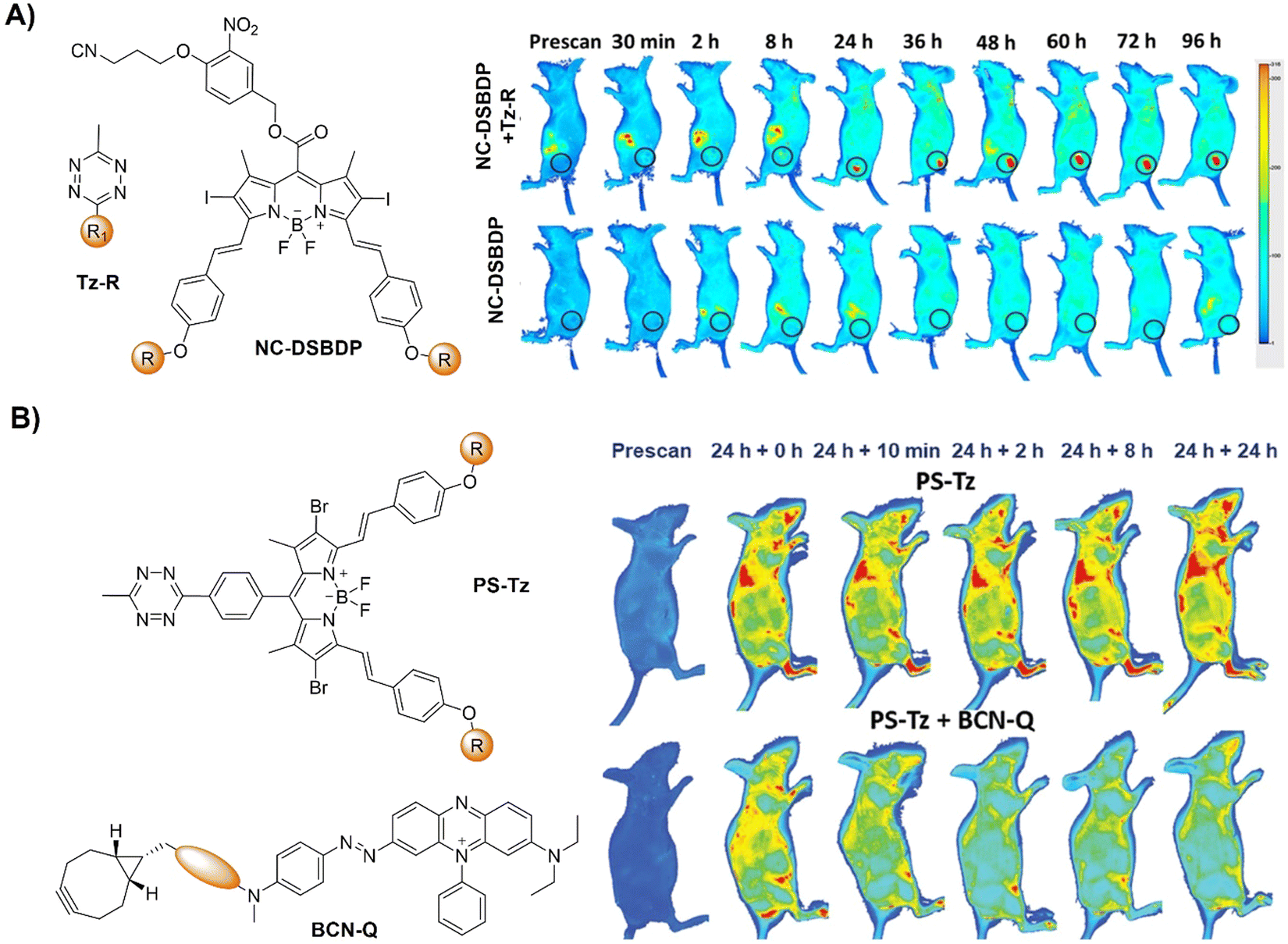 | ||
| Fig. 29 Bioorthogonal probes for in vivo fluorescence imaging based on the inverse-electron-demand Diels–Alder reaction. (A) The structures of Tzs and NC-DSBDP and in vivo fluorescence images of A431 tumor-bearing nude mice, adapted with permission from ref. 183. Copyright 2023, Elsevier B.V. and (B) the structures of BCN-Q and PS-Tz and NIR fluorescence images of the nude mice, adapted with permission from ref. 184. Copyright 2023, Wiley-VCH GmbH. | ||
Despite the successful translation of PDT in cancer therapy, precisely controlling the administration of photosensitizers into a body is extremely hard, thus leading to side effects as the excess photosensitizers in the body could be activated for ROS generation under sunlight irradiation. The patients are thus required to stay in the dark for a few days after treatment. Interesting research led by the Ng group184 reported a bioorthogonal strategy to mitigate photosensitivity post-PDT (Fig. 29B). In this protocol, BCN-Q, consisting of a BCN moiety that swiftly reacts with the Tz substituent of the photosensitizer PS-Tz, and the BHQ-3 quencher deactivate the PS-Tz photosensitizer through a bioorthogonal click reaction. As such the BHQ-3 quencher efficiently reduces the emission of PS-Tz and the generation of ROS via FRET in PBS and diverse cancer cell lines. In nude mice, BCN-Q injection effectively neutralizes the residual photosensitizer without compromising PDT efficacy, evidenced by minimal skin damage post-treatment. This approach thus offered a direct “click-and-quench” mechanism, circumventing the need for tumor-specific photosensitizers and potentially enhancing the clinical viability of PDT by mitigating photosensitivity.
Spatiotemporal multiplexed labelling and imaging of multiple cellular targeted species, enabled by bioorthogonally activated probes, facilitates comprehensive profiling of the functions of these species in live cells. Carlson and coworkers185 reported scission-accelerated fluorophore exchange (SAFE), a technique enabling spatiotemporal multiplexed immunofluorescence imaging of live cells (Fig. 30A). SAFE leveraged rapid IEDDA bioorthogonal click chemistry to swiftly remove immunofluorescence signals from labeled cells’ surfaces. Strategies include multiparameter analysis (n ≥ 14), non-disruptive imaging of murine peripheral blood mononuclear cells and bone marrow cells to study cellular differentiation, and longitudinal multiplexed imaging of bone marrow progenitor cells maturing into neutrophils over 6 days and real-time multiplexed cycling of living mouse hepatic tissues. SAFE is expected to have broad utility for investigating physiological dynamics in living systems.
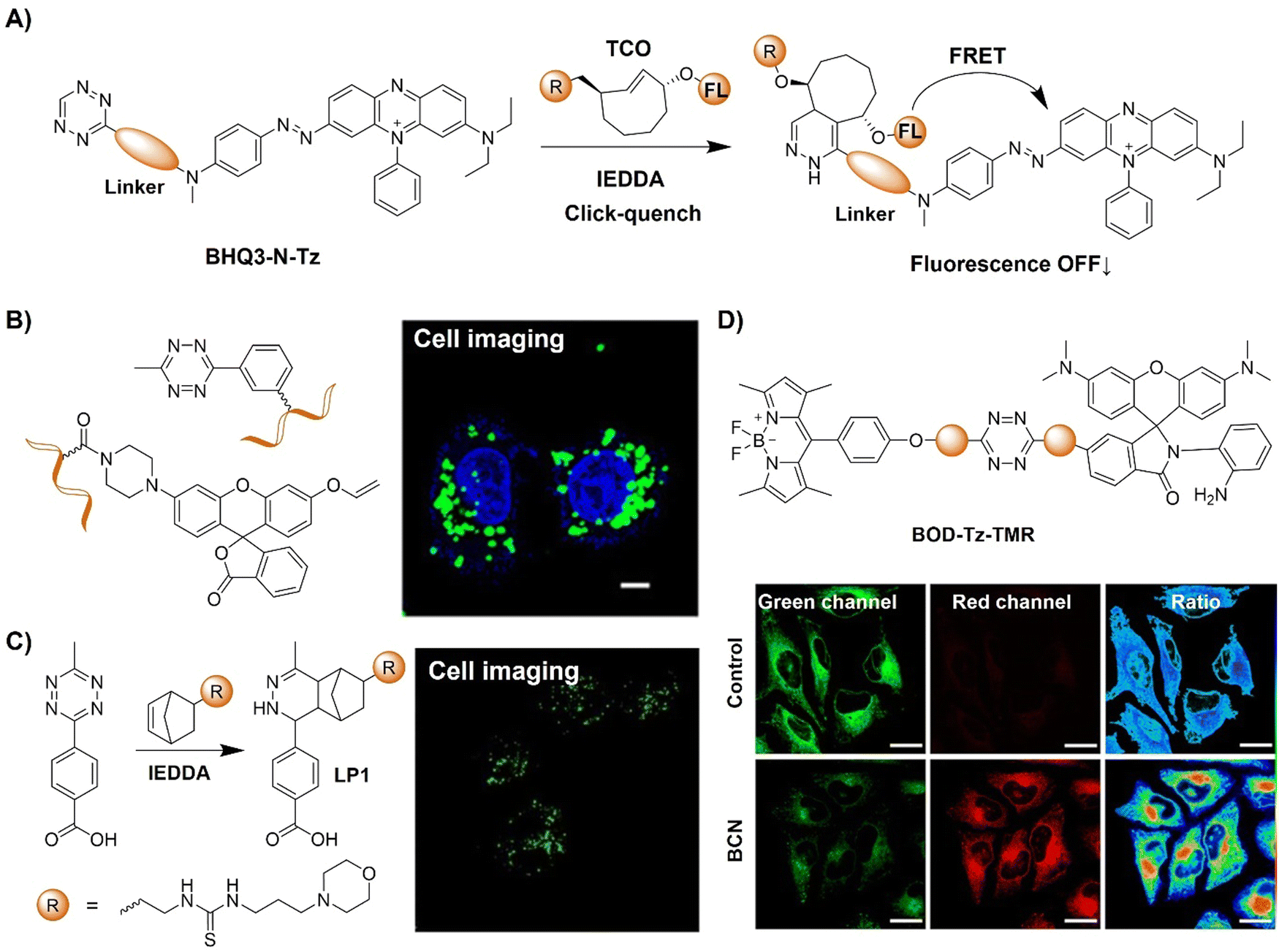 | ||
| Fig. 30 Fluorescent probes designed for cell imaging based on the inverse-electron-demand Diels–Alder reaction. (A) Schematic illustration of the SAFE system; (B) the structures of reaction precursors (Tz and vinyl ether-caged fluorophore) and mRNA imaging in live cells, adapted with permission from ref. 186. Copyright 2022, American Chemical Society; (C) bioorthogonal reaction of TE-norbornenes and bioimaging of the MCF-7 cells, adapted with permission from ref. 187. Copyright 2022, Elsevier B.V; and (D) the structure of BOD-Tz-TMR and ratio imaging of HeLa cells, adapted with permission from ref. 188. Copyright 2023, American Chemical Society. | ||
Bioorthogonal reactions have been particularly useful for nucleic acid detection and imaging due to their specificity and adjustable reaction kinetics. For instance, Jiang and colleagues186 reported a fluorogenic sensor for the precise RNA imaging in live cells by integrating a bioorthogonal reaction with a DNA cascade reaction (Fig. 30B). In this probe system, two DNA hairpin probes were synthesized using Tzs or vinyl ether-caged fluorophores. Upon targeted mRNA-induced catalytic hairpin assembly, spatial proximity facilitated efficient bioorthogonal reactions, unmasking the vinyl ether group to activate fluorescence. This sensor exhibited high signal-to-noise ratios (∼30 fold) and sensitive mRNA detection (4.6 pM detection limit). The “click to release” mechanism allowed for minimal background signals in biological environments for mRNA imaging in live cells and differentiated mRNA expression levels in tumor and normal cells. In another study led by Rentmeister,189 a benzylic linker promoted methyltransferase catalyzed norbornene transfer reaction was employed for rapid bioorthogonal Tz ligation for the specific labelling of DNA and RNA in cell lysate.
A fluorescent probe for in situ lysosome labeling and lysosomal pH sensing was developed by Xu and coworkers187 using fluorescence bioorthogonal reaction between 4-(6-methyl-1,2,4,5-tetrazin-3-yl) benzoic acid (TE) and norbornenes (NC). By pre-treatment of cells with morpholine-modified norbornene (MP-NC), lysosomes rapidly became decorated with functional substrates for bioorthogonal reactions (Fig. 30C). With the introduction of 4-(6-methyl-1,2,4,5-tetrazin-3-yl) benzoic acid (TE), lysosome-targeted probe LP1 was constructed in live cells within 10 min, emitting bioorthogonal fluorescence at 515 nm.
A Tz-based bioorthogonal probe for NO detection was developed by Xiao and coworkers188 and the application of this probe for the detection of NO in atherosclerosis (AS) was demonstrated (Fig. 30D). AS is characterized by plaque formation in blood vessels, leading to severe cardiovascular conditions with varied levels of NO. In this probe system (BOD-Tz-TMR), bodipy and rhodamine with a well-documented NO sensing unit were linked using a Tz moiety, resulting in emission quenching of both fluorophores. Click BCN reaction allowed for switching on of the fluorescence of Bodipy, and sensing of NO led to the restoration of the rhodamine's emission. The bioorthogonal reactions with effective FRET from bodipy to rhodamine enabled the ratiometric fluorescence detection and imaging of NO. This sensor selectively targeted AS plaques, visualizing endogenous NO at two lesion stages in AS mouse models. Ratiometric signals confirmed NO involvement during AS, confirming increased NO generation with lesion progression. This bioorthogonally activated probe thus offered a useful approach for targeted delivery of small molecular probes to AS plaques for reactive biomarker identification and detection.
The rapid kinetics inherent in Tz ligation make it suitable for applications in the precise fluorescence imaging of live cells. However, the stability of Tzs in aqueous solutions or in the presence of redox species varies (e.g. thiols and ROS), with the most reactive Tzs often being the least stable. For example, Li's research has identified that Tz could respond to superoxide (O2˙−), the primary ROS in mammalian cells.99 Using 1,2,4,5-Tz as a superoxide-responsive trigger, a series of fluorescent probes for O2˙− detection using various fluorophores with emission color modulation were developed. These probes exhibit exceptional specificity and fluorescence amplification for O2˙− detection, enabling organelle-resolved multiplexed imaging. Abnormal O2˙− generation in myocardial ischemia/reperfusion injury and high-content screening for superoxide homeostasis mediators has been investigated using these probes.
To tackle challenges associated with the poor stability of Tz, various strategies have been proposed.190 For instance, the Fox group introduced a photochemical variant to this reaction, resulting in a new type of photoclick reaction named photo-induced Tz linkage (photo-IEDDA).191–194 Dihydrotetrazine (DHTz) was used as a precursor for this strategy, along with photosensitizers (such as methylene blue) that generate 1O2 under light irradiation (>600 nm). These species can oxidize DHTzin situ to form Tz, which can then react with TCO to form a cycloadduct. Additionally, disubstituted Tzs, particularly monomethyl Tzs, exhibit improved stability without significantly compromising reactivity. Alternatively, Prescher et al. have used 1,2,4-triazines in IEDDA reactions, which exhibit enhanced stability compared to the corresponding Tzs while maintaining satisfactory kinetics amongst various dienophiles.195 On the other hand, Vrabel et al.196 introduced triazinium ligation, a bioorthogonal conjugation based on N1-alkyl 1,2,4-triazinium salt reaction with strained alkynes. Optimized alkylation of 1,2,4-triazines yielded key triazinium reagents, facilitating tunable reaction rates. Alkylation boosted reactivity by three orders of magnitude, which was validated by DFT calculations. tert-Butylation has emerged as the optimal modification, offering highly reactive and stable salts. Triazinium ligation, orthogonal to strain-promoted azide–alkyne cycloaddition, efficiently modifies biomolecules, exhibiting enhanced cell permeability and intracellular labeling efficiency.
Optimization of dienophile structures strikes a balance between improved stability and reactivity. TCO, which has been extensively studied as a dienophile, can isomerize to less reactive cis-cyclooctenes, posing a challenge for bioorthogonal reactions.197 To address this, the Fox group devised dioxolane-fused trans-cyclooctenes with increased stability and reduced propensity for isomerization. Other dienophiles employed in IEDDA-mediated ligations include norbornenes, cyclooctynes, and methylcyclopropenes. The Bonger research group introduced vinylboronic acids as strained dienophiles, notable for their water solubility and hydrolytic stability as well as high reactivity.198,199 These compounds engaged in rapid in vivoTz ligation with stable precursors by coordination of the boronic acids to the phenol group, followed by swift intramolecular IEDDA reactions. With the continued effort of researchers, we are confident that the IEDDA reaction will find even more applications in a broader range of scenarios.
2.4. 1,2-Aminothiol-CBT click reaction
The biocompatible click reaction between D-cysteine and 2-cyanobenzothiazole (2-CBT), inspired by fireflies, has received considerable interest in the development of bioluminescent probes for bioassays and imaging. In nature, the reaction between D-cysteine and 2-CBT is the final step in the synthesis of firefly luciferin (D-luciferin, D-Luc). In the presence of luciferase, Mg2+, and ATP, D-Luc undergoes oxidation, emitting bioluminescence. In 2009, Rao and colleagues introduced a method to label proteins with terminal cysteines using CBT probes.41 This reaction,41,200–202 exhibiting click-like properties, proved to be simple and modular and required no catalysts or chromatographic purification. Its natural origin ensured excellent biocompatibility, with both reactants displaying good water solubility and selectivity.Furthermore, the second-order rate constant, measured as 9.1 M−1 s−1, surpassed that of the Huisgen click reaction by two orders of magnitude.42 Subsequent studies elucidated the mechanism of this click reaction. As illustrated in Scheme 4, the reaction between D-cysteine and 6-amino-2-cyanobenzothiazole (NCBT) at pH 7.4 shows the sulfhydryl group's higher nucleophilicity than the amino group. This leads to the formation of intermediate 1, followed by the formation of a thiazole ring structure and the release of ammonia gas, resulting in the formation of the final product amino luciferin. During the past few decades, the CBT-Cys click reaction has found diverse applications in protein labeling, molecular imaging (encompassing optical imaging, magnetic resonance imaging, nuclear imaging, and photoacoustic imaging), tumor therapy, and various other fields. This section will focus on the latest advancements in this click reaction for in situ bioimaging, aiming to provide profound and comprehensive insights for its further implementation.
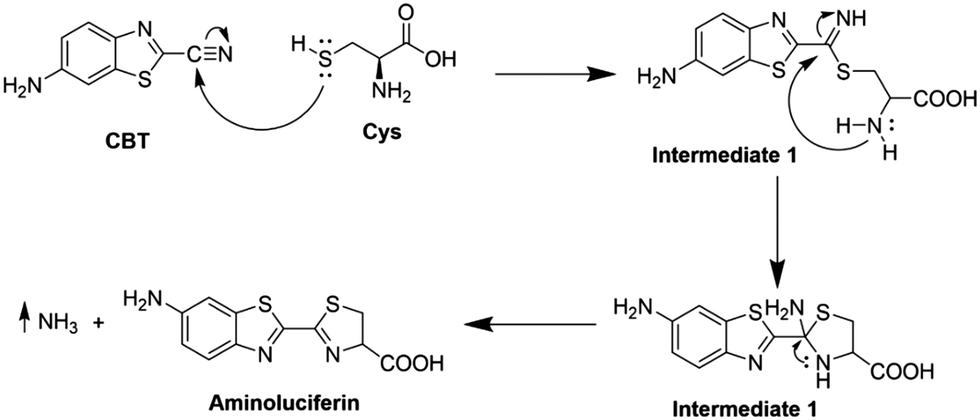 | ||
| Scheme 4 The proposed mechanism of the 1,2-aminothiol-CBT click reaction. | ||
The exploration of in vivo molecular imaging is fundamental to unraveling the complexities of health, injury, aging, and disease by identifying specific biochemical processes like enzymatic activity, small-molecule fluxes, and post-translational modifications. As such probes that can be used for dual analyte detection in situ in living organisms are more desirable for understanding the multiple biochemical processes in a complicated environment. To this end, the Chang group203 developed a strategy for dual-analyte detection in live animals by leveraging the real-time generation of firefly luciferin from two complementary caged precursors sensitive to distinct biochemical targets (Fig. 31A). In this strategy, Peroxy Caged Luciferin-2 (PCL-2), a boronic acid probe is responsive to hydrogen peroxide (H2O2), and a peptide-based probe, z-Ile-Glu-ThrAsp-D-Cys (IETDC) can be activated by active caspase 8. Upon encountering the corresponding stimuli, PCL-2 released 6-hydroxy-2-cyanobenzothiazole (HCBT), and IETDC liberated D-cysteine, resulting in the formation of firefly luciferin in situ and emitting bioluminescence only when both triggers are concurrently active. This system operates akin to AND-type molecular logic gates, providing insights into the simultaneous presence of H2O2 and caspase 8. Selective imaging of either H2O2 or caspase 8 activity was successfully demonstrated in both laboratory settings and live subjects. Furthermore, application of PCL-2 and IETDCin vivo unveiled a simultaneous surge in H2O2 and caspase 8 activity during acute inflammation in live mice. In another study, the Dubikovskaya group204 introduced a biocompatible in vivo ligation reaction based on a selective reaction between D-cysteine and CBTin vivo, producing a luciferin substrate for bioluminescence imaging of firefly luciferase in mice (Fig. 31B). This “split luciferin” ligation reaction was used to investigate the click reactions of 6-amino/hydroxyl-CBT with D-cysteine and L-cysteine and revealed the high reactivity of CBTs towards D-cysteine. Utilizing bioluminescence imaging, visualizing the production of the luciferin substrates in live mice was achieved. Application of this reaction for real-time noninvasive imaging of apoptosis associated with caspase -3/-7 activity was also demonstrated.
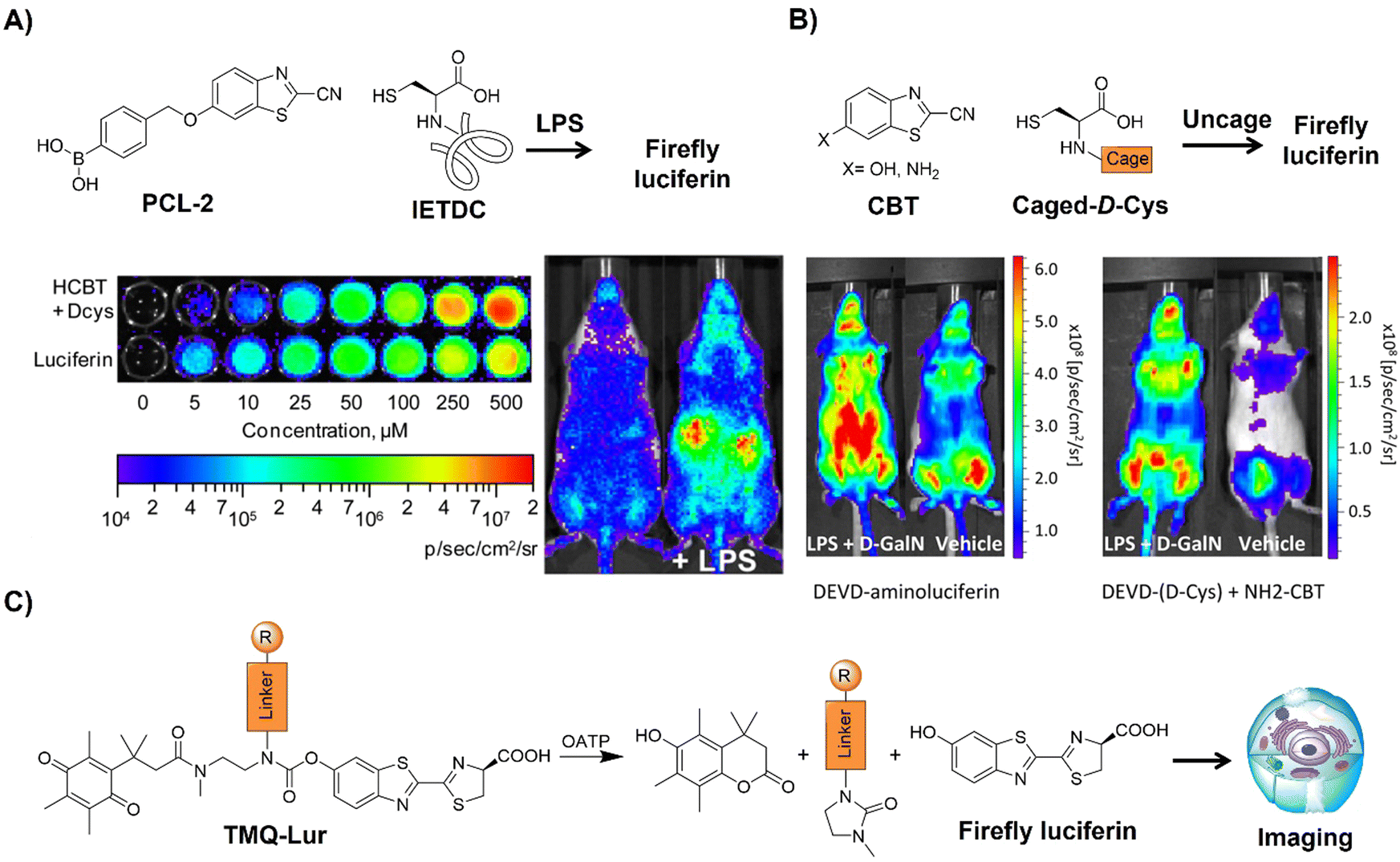 | ||
| Fig. 31 Bioluminescent probes designed for imaging based on the 1,2-aminothiol-CBT click reaction. (A) Illustration of simultaneous detection of H2O2 and caspase 8 activity through release of HCBT and D-cysteine and in situ formation of firefly luciferin and representative image of PC3M-luc cells/mice. Adapted with permission from ref. 203. Copyright 2013, American Chemical Society. (B) Schematic illustration of the “split luciferin” ligation reaction and representative image of mice under different conditions. Adapted with permission from ref. 204. Copyright 2017, American Chemical Society. (C) Schematic illustration of the wash-free luminogenic probes for detecting OATP activity. Adapted with permission from ref. 205. Copyright 2016, American Chemical Society. | ||
Through click reaction of CBT with D-cysteine, Cali and coworkers205 developed wash-free luminogenic probes for detecting the activity of organic anion-transporting polypeptides (OATP), pivotal in drug absorption, distribution, metabolism, and excretion (ADME) processes (Fig. 31C). This approach employed self-cleavage of trimethyl lock quinone-luciferin (TMQ-Lur) tethered to fluorescein as a recognized transporter substrate. Cellular uptake triggered quinone reduction and released luciferin via intramolecular lactonization, yielding detectable bioluminescence signals upon introduction of luciferase. The signal intensity corresponds to the activity of the transporter, enabling wash-free imaging, as demonstrated using HEK293 cells overexpressing OATP1B1*1a and OATP1B3. These results underscore the viability of measuring transporter activity via bioluminescence, with the potential for developing diverse transporter assays by simply altering the substrate.
Controlled self-assembly of small molecules within living systems offers extensive potential for applications in biology and medicine, such as in synthesizing supramolecules and nano/microstructures, both in solution and within living cells. Bioorthogonal reactions based on CBTs and D-cysteine facilitate the controllable self-assembly of fluorescent small molecules to nanoparticles in the body. For example Rao's research200 employed an optimized first-order 1,2-aminothiol-CBT click reaction to control the self-assembly of fluorescent small molecules (Fig. 32). Imaging caspase-3/-7 activity in human tumor xenograft mouse models undergoing chemotherapy was demonstrated as one of the practical applications of this self-assembly system. Validation of the proposed mechanism was achieved using a series of control probes, and the self-assembled nanoaggregates were directly observed both in vitro and in vivo using super-resolution fluorescence microscopy. This bioorthogonal macrocyclization and nano-aggregation, triggered by caspase-3/-7 activity, facilitated effective monitoring of tumor therapy response in vivo and exhibited promise for imaging caspase-3/-7 activity in various diseases, including neurodegenerative diseases and organ-transplant rejection.
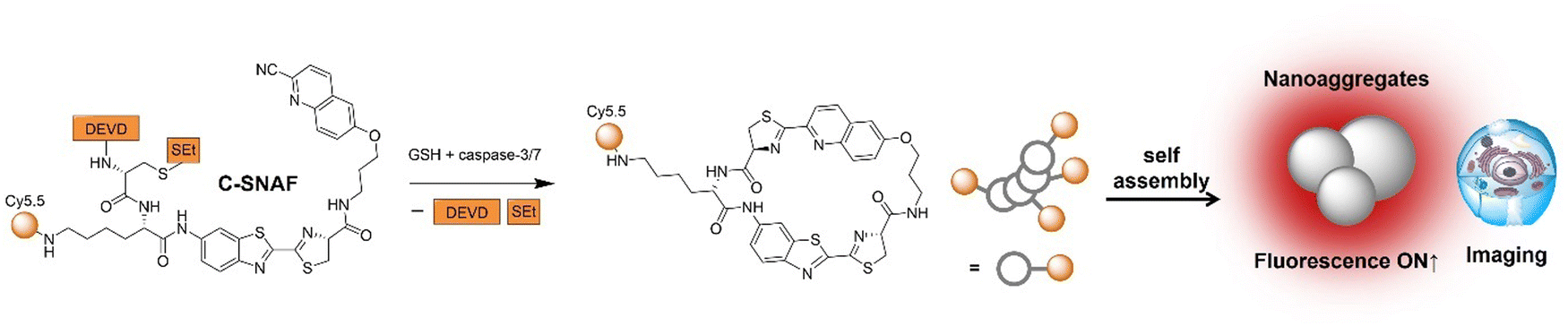 | ||
| Fig. 32 Schematic illustration of the mechanism of probe C-SNAF for caspase-3/7 activity analysis and imaging. | ||
In addition to the probes for the analysis of caspase-3/7 activity, CBT-D-cysteine click condensation reactions have been used for the development of probes for detecting intracellular furin activity by the Liang group (Fig. 33A).206 The probe was developed using a dual quenching approach by combining a click condensation reaction to form the single quenched probe DABCYL-FITC-CBT. This strategy enabled enhanced sensitivity in detecting intracellular furin activity. Under physiological conditions, DABCYL-FITC-CBT underwent reduction-controlled condensation to form DABCYL-FITC-CBT-NPs, with the fluorescence intensity reducing by 1/2.8 of its original intensity. Upon furin cleavage, the dual-quenched DABCYL-FITC-CBT-NPs exhibited an 11-fold fluorescence “turn-on” compared to the single-quenched control probe FITC-DABCYL. Live cell imaging exhibited a 6.3-fold fluorescence “turn-on” for detecting intracellular furin activity. This versatile dual-quenching strategy is interesting and has the potential for enhanced sensitivity in detecting various intracellular enzyme activities. Subsequently, Liang and coworkers207 developed a probe using AIEgens and a furin-controlled CBT click reaction for furin activity assays in live cells (Fig. 33B). Upon reduction and furin hydrolysis, dimer formation occurred, self-assembling into nanoparticles and intensifying the fluorescence through enhanced fluorogen aggregation. Compared to single AIE molecules lacking CBT groups, the fluorescence signals were boosted 1.7 and 3.4-fold in vitro and in living cells, respectively.
 | ||
| Fig. 33 Fluorescent probes designed for bioimaging based on the 1,2-aminothiol-CBT click reaction. (A) Schematic illustration and confocal fluorescence and overlay images of MDA-MB-468 cells of the single/dual quenched nanoparticles. Adapted with permission from ref. 206. Copyright 2018, American Chemical Society. (B) Schematic illustration of a furin-controlled dual AIEgen for enhanced fluorescence sensing of furin activity. Adapted with permission from ref. 207. Copyright 2017, The Royal Society of Chemistry. (C) Schematic illustration of the formation of AIEgen-based nanostructures by enzyme-mediated intracellular reduction and condensation of D2P1 and 3CBT and their applications in in vitro and in vivo imaging. Adapted with permission from ref. 208. Copyright 2021, Wiley-VCH GmbH. | ||
Using a similar TPE-based AIEgen, Liu's group208 recently reported an enzyme-mediated AIEgen intracellular polymerization strategy to enhance drug localization within tumors (Fig. 33C). This approach used an AIEgen-peptide conjugate (D2P1) and cyanobenzothiazole-cysteine (3CBT) to form nanoaggregates in tumor cells via a rapid condensation reaction. Tumor-specific cathepsin protease cleavage triggered polymerization, promoting D2P1 accumulation in tumors for fluorescence activation. This process disrupted cellular motility and inhibited cancer cell proliferation. Moreover, D2P1 is able to generate ROS, boosting tumor PDT efficacy in vivo.
In addition to the analysis based on fluorescence intensity, fluorophore aggregation offers another approach for bioanalysis and imaging, i.e., fluorescence lifetime imaging. For instance, Cui and colleagues209 introduced a reduction-sensitive fluorescent probe, exhibiting altered emission lifetime under reducing conditions as a result of the fluorophore aggregation of the CBT scaffold (Fig. 34A). Remarkable changes in lifetime occur in FITC emission when the compound interacts with glutathione (GSH), indicating the necessity of both a reduction-sensitive disulfide bond and reducing agent for lifetime shift. During FLIM imaging a lifetime decrease was detected after activation by GSH, suggesting the potential of the probe for lifetime imaging of the fluorophore's self-aggregation in cells. In 2020, the Rao group210 introduced in situ polymerization of activatable bioorthogonal small molecules in response to changes in the reducing environment in vivo (Fig. 34B). A carbohydrate linker and CBT-Cys condensation reaction-based small molecule scaffold were developed for rapid condensation upon physicochemical changes, forming pseudopolysaccharides. Evaluation of the fluorescence and photoacoustic properties before and after transformation confirmed in situ polymerization post-local and systemic administration in living mice, highlighting the potential of this probe for dual-modality optical imaging.
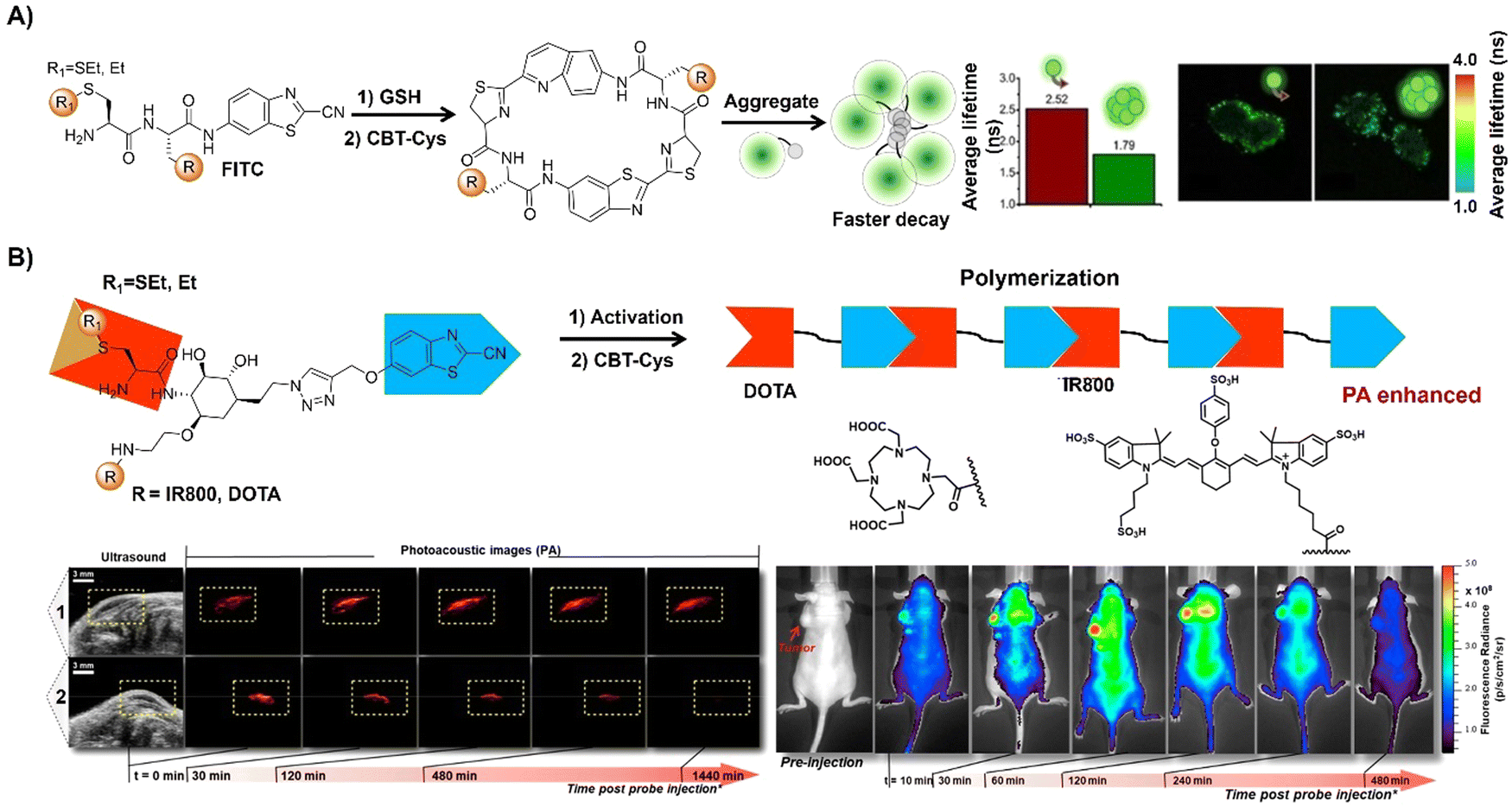 | ||
| Fig. 34 Bioluminescent probes developed by exploring the 1,2-aminothiol-CBT click reaction. (A) Schematic illustration and cell imaging of a reduction-sensitive fluorescent probe obtained viaCBT-Cys reaction. Adapted with permission from ref. 209. Copyright 2020, Wiley-VCH GmbH. (B) Schematic illustration of an in situ polymerization activated probe viaCBT-Cys reaction and ultrasound and photoacoustic images of a subcutaneously injected probe in living mice and longitudinal fluorescence imaging of tumor-bearing mice. Adapted with permission from ref. 210. Copyright 2020, American Chemical Society. | ||
Apart from the probes developed based on CBT-D-cysteine initialized assembly, disassembly of self-quenched nanoparticles has also been adopted for the development of probes by Liang's group. For instance, in 2015, Liang and coworkers211 developed an enzyme-driven disassembly of self-quenched NIR fluorescent nanoparticles, enabling targeted detection of enzyme activity in vitro and in vivo (Fig. 35A). Through strategic design, the NIR probe's fluorescence was initially suppressed via reduction-controlled condensation and nanoparticle assembly, only to be activated upon NP disassembly by furin. Leveraging this enzymatic mechanism, precise NIR detection of furin was achieved in vitro and for live cell imaging. Additionally, the disassembly approach-based probe facilitated discriminative NIR imaging of MDA-MB-468 tumors in nude mice, suggesting the potential for tumor-targeted imaging in clinical applications. Expanding on this research, the same group then developed a NIR probe, Cys(StBu)-Lys(Biotin)-Lys-Lys(Cy5.5)-CBT, to generate self-quenched nanoparticles (1-NPs) for in vitro and in vivo tumor-targeted imaging (Fig. 35B).212 Biotinylated 1-NPs exhibited active uptake by biotin receptor-overexpressing tumor cells via receptor-mediated endocytosis. Intracellular proteolytic cleavage dismantled 1-NPs, yielding the fluorescent “turn-on” molecular probe Lys(Cy5.5)-Luciferin-Lys(Biotin)-Lys-OH (1-D-cleaved). This nanoprobe with NIR fluorescence facilitated tumor-targeted imaging in a nude mouse model.
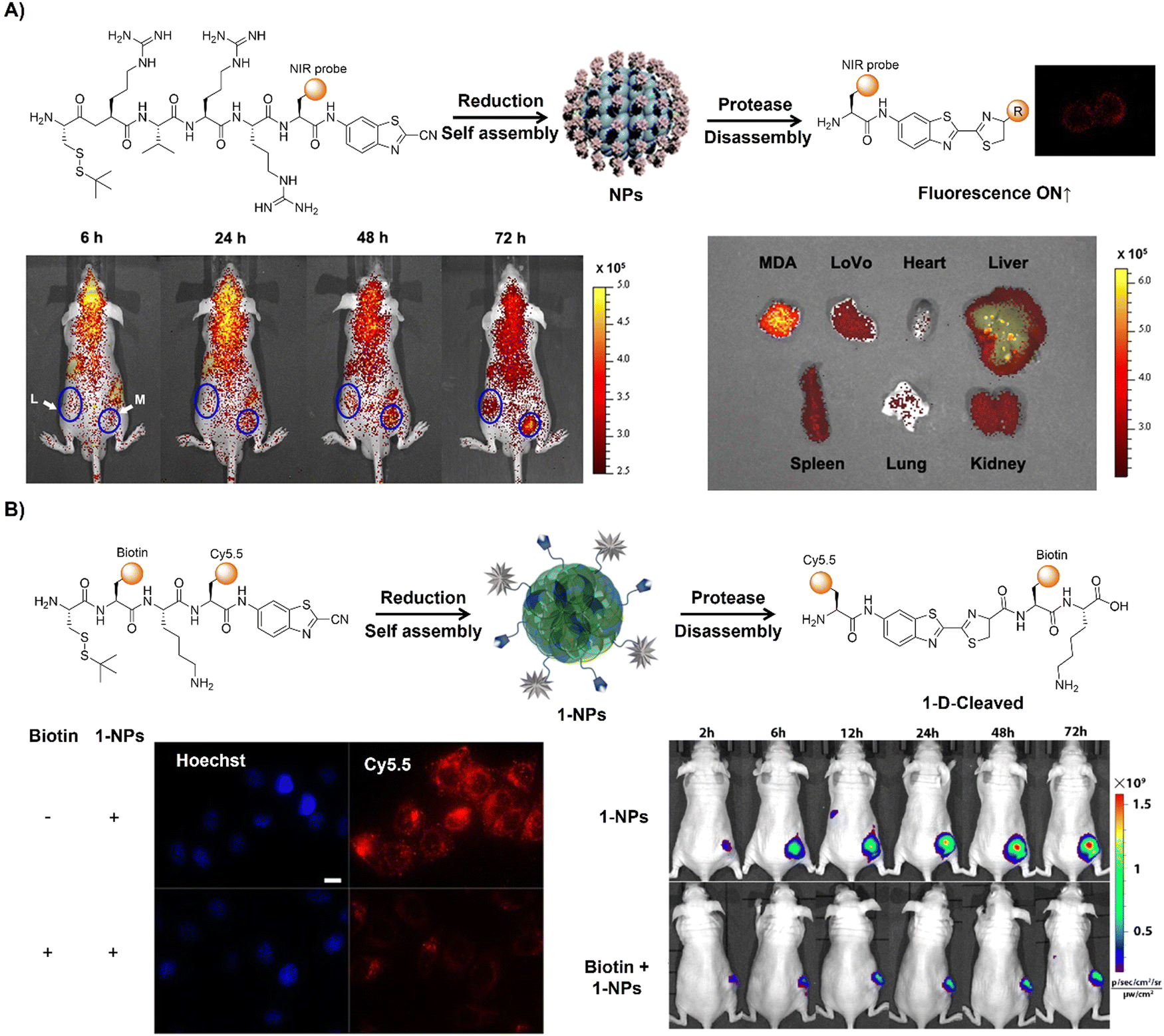 | ||
| Fig. 35 Fluorescent probes developed for in vitro and in vivo imaging based on the 1,2-aminothiol-CBT click reaction. (A) Illustration of the enzyme-driven disassembly of self-quenched NIR fluorescent nanoparticles; in vivo active furin-overexpression-tumor-targeted imaging and ex vivo fluorescence images of different organs from tumor-bearing nude mice. Adapted with permission from ref. 211. Copyright 2015, American Chemical Society. (B) Schematic illustration of reduction-controlled self-assembly of 1-NPs to turn the NIRF signal “Off” and subsequent protease-controlled disassembly of 1-NPs to turn the NIRF signal “On” with fluorescence and overlay images of biotin receptor-positive HeLa cells and time-course fluorescence imaging of nude mice xenografted with HeLa tumor cells. Adapted with permission from ref. 212. Copyright 2017, American Chemical Society. | ||
In brief, we have summarized the progress made in using CBT-Cys click reactions for in situ bioimaging in this section. The CBT-Cys click reaction relies on the condensation between CBT and D-Cys, as observed in fireflies. The mechanism has been proposed and verified experimentally, showcasing numerous advantages over conventional click reactions. This click reaction has exceptional biocompatibility because the reaction has natural origins. This reaction could be easily modulated by changes in pH, reduction, or enzymes under physiological conditions. More importantly, its second-order rate constant is high at 9.19 M−1 s−1, surpassing that of CuAAC by two orders of magnitude. Furthermore, the resulting amphiphilic oligomeric products tend to self-assemble into nanostructures, a unique trait not seen in other click reactions. The versatility of the CBT-Cys click reaction facilitates a wide range of biomedical applications through modification of the chemical structures of the reactants. Under the influence of pH, reduction, or enzymes, the CBT-Cys click condensation reaction has successfully been applied in various fields, including protein labeling, molecular imaging (covering optical, magnetic resonance, nuclear, and photoacoustic imaging) of tumors, preparation of oligomeric nanostructures, and cancer therapy. The development of “smart” small molecular precursors enables precise control over intracellular spatiotemporal assembly. It is expected that this reaction will facilitate the development of novel dendrimer-based probes to enhance imaging contrast (e.g., MRI and fluorescence imaging) by covalently attaching the probes at the periphery. Moreover, as molecular imaging advances towards multimodality, integrating multiple imaging modes into a single platform using the CBT-Cys click reaction holds promise for more accurate disease diagnosis through multimodal molecular imaging.
2.5. Strain promoted sydnone alkyne cycloaddition
Sydnones, a family of mesoionic compounds, have recently emerged as valuable components for click chemistry and bioorthogonal reactions. Reactions of sydnones with cyclooctynes, known as strain promoted sydnone–alkyne cycloaddition (SPSAC, Scheme 5),213,214 enable the synthesis of pyrazoles with intriguing properties. Sydnones offer the advantage of functionalizing mesoionic scaffolds at multiple positions, allowing for fine-tuning of their reactivity. This section will focus on the latest advancements in in situ activated bioimaging utilizing the sydnone–cyclooctyne reaction over the past few years, aiming to provide comprehensive and profound insights for its further implementation.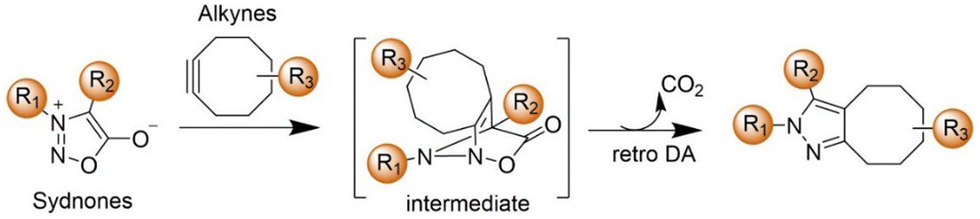 | ||
| Scheme 5 Proposed mechanism of the SPSAC reaction. | ||
In 2014, Taran and colleagues synthesized a series of sydnone derivatives and investigated their reactivities towards BCN through a Cu-free cycloaddition reaction.213 It was found that 4-chloro-sydnones could react with BCN more than 10 times faster than azide-based click reactions. The high reactivity of chlorosydnones was then applied to fluorescence labelling of proteins using rhodamine-linked BCN (BCN-TAMRA). In 2016, the same group214 reported the synthesis and dynamic reactivity of 4-fluorosydnones, an exceptional category of mesoionic dipoles exhibiting remarkable responsiveness towards both copper-catalyzed and strain-promoted cycloaddition reactions with alkynes (Fig. 36A). These mesoionic compounds were synthesized through electrophilic fluorination of σ-sydnone Pd(II) precursors in the presence of Selectfluor. Their interactions with both terminal and cyclic alkynes involve rapid and highly selective transformations, yielding 5-fluoro-1,4-pyrazoles with unprecedented bimolecular rate constants exceeding 104 m−1 s−1. Detailed kinetic investigations were conducted to elucidate the underlying reaction mechanism, further underlining the significance of 4-fluorosydnones, exemplified by their successful radiolabeling with [18F]Selectfluor. On the basis of these studies, the same group then synthesized a range of styryl-pyridinium-based sydnones tailored for wash-free protein labeling and in-cell imaging (Fig. 36B).215 These “turn-on” sydnone probes exhibited high reactivity towards cyclooctynes, facilitating clean reactions with rate constants ranging from 10 to 100 M−1 s−1. Using these probes, highly effective protein labeling and cellular imaging applications were successfully demonstrated.
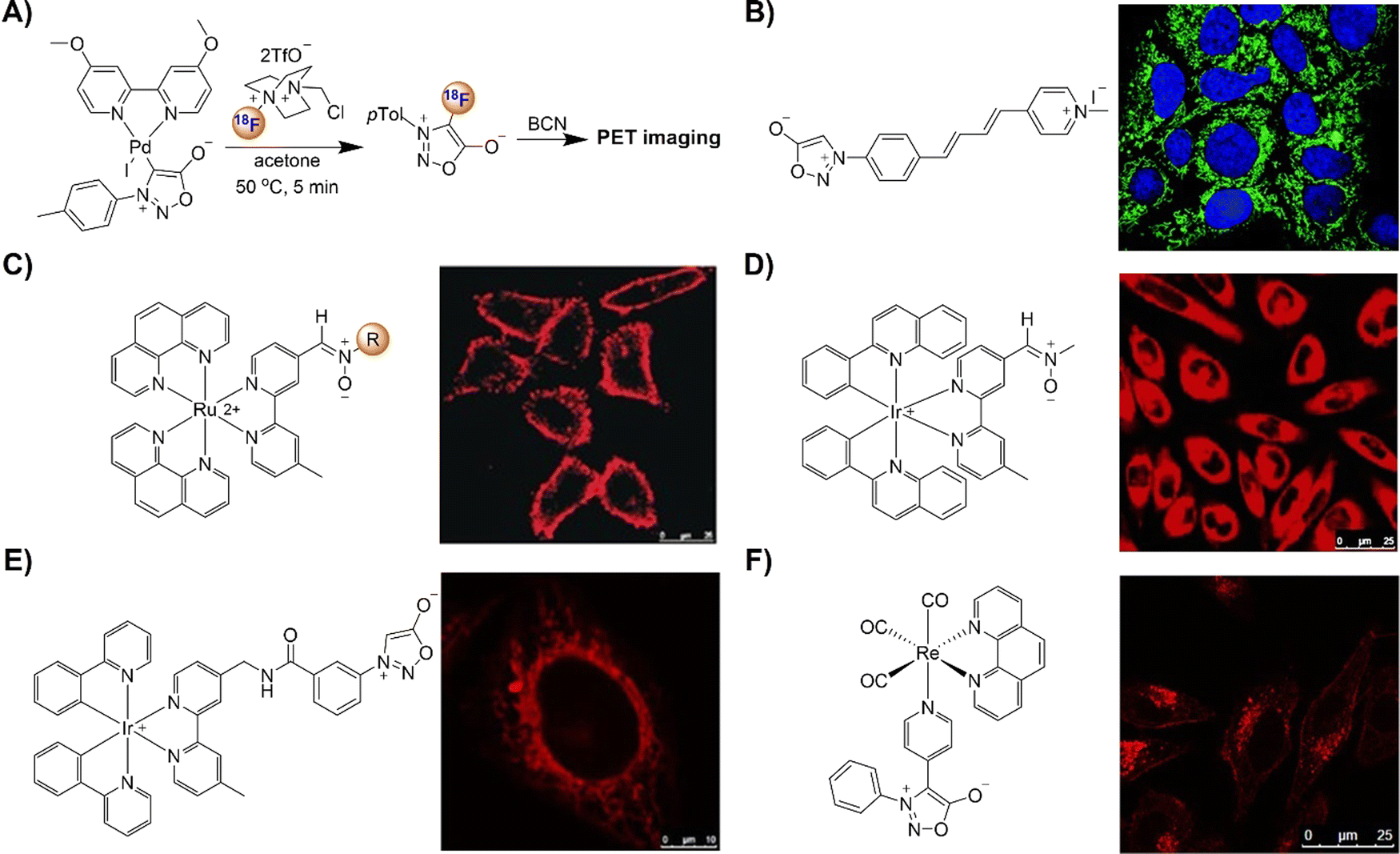 | ||
| Fig. 36 Metal complex-based bioorthogonal probes for cell imaging based on the strain promoted sydnone alkyne cycloaddition. (A) Preparation and use of [18F]sydnone; (B) the structure of 6-sydnone-profluorophores and confocal cell imaging, adapted with permission from ref. 215. Copyright 2019, The Royal Society of Chemistry; (C) the structure of the Ru nitrone complex and LSCM images of the HeLa cells, adapted with permission from ref. 216. Copyright 2016, Wiley-VCH GmbH; (D) the structure of the Ir nitrone complex and LSCM images of the CHO-K1 cells, adapted with permission from ref. 217. Copyright 2016, Wiley-VCH GmbH; (E) the structure of the Ir sydnone complex and LSCM images of the HeLa cells, adapted with permission from ref. 218. Copyright 2018, Wiley-VCH GmbH; and (F) the structure of the Re polypyridine sydnone complex and LSCM images of the HeLa cells, adapted with permission from ref. 219. Copyright 2020, Wiley-VCH GmbH. | ||
The Lo group216 introduced a series of ruthenium(II) polypyridine complexes featuring a nitrone group as phosphorogenic bioorthogonal probes (Fig. 36C). Initially weakly emissive due to rapid C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) –N isomerization of the nitrone moiety, these complexes exhibited significant emission enhancement upon strain-promoted alkyne–nitrone cycloaddition (SPANC) with BCN-modified substrates. By incorporating a dicationic ruthenium(II) polypyridine unit at the α-C-position and a phenyl ring at the N-position of the nitrone, reaction kinetics were remarkably accelerated, up to 278-fold higher than other acyclic nitrone-BCN reactions. Remarkably, these complexes facilitated specific cell membrane/cytosol staining upon labeling with an exogenous substrate, BCN-modified decane, in live cells. Using a similar SPANC approach, the same research group designed three iridium(III) complexes incorporating a nitrone moiety as phosphorogenic bioorthogonal probes (Fig. 36D).217 Similar to the luminescence quenching of the ruthenium(II) polypyridine complex,216 isomerization of the C = N group quenched the emissions of these iridium(III) complex-based probes, while emission enhancement was obtained upon cycloaddition with strained cyclooctynes. Likewise, the cationic iridium(III) center promoted the reaction kinetics of nitrone with BCN. These nitrone complexes also served as phosphorogenic bioorthogonal labels and imaging agents for cyclooctyne-modified proteins, offering promising prospects for advancing phosphorogenic bioorthogonal probes and imaging reagents. Subsequently, the same research group employed the transformation of the highly polar mesoionic sydnone ring into less polar pyrazoles to create luminescent probes for the SPSAC reaction in bioorthogonal labeling and live cell imaging (Fig. 36E).218 The developed iridium(III) sydnone complexes readily reacted with strained alkyne derivatives, resulting in significant emission enhancement (up to 24.9 fold) and extended lifetime. These complex-based probes were successfully utilized to label alkyne-modified proteins and ceramide molecules within live cells.
–N isomerization of the nitrone moiety, these complexes exhibited significant emission enhancement upon strain-promoted alkyne–nitrone cycloaddition (SPANC) with BCN-modified substrates. By incorporating a dicationic ruthenium(II) polypyridine unit at the α-C-position and a phenyl ring at the N-position of the nitrone, reaction kinetics were remarkably accelerated, up to 278-fold higher than other acyclic nitrone-BCN reactions. Remarkably, these complexes facilitated specific cell membrane/cytosol staining upon labeling with an exogenous substrate, BCN-modified decane, in live cells. Using a similar SPANC approach, the same research group designed three iridium(III) complexes incorporating a nitrone moiety as phosphorogenic bioorthogonal probes (Fig. 36D).217 Similar to the luminescence quenching of the ruthenium(II) polypyridine complex,216 isomerization of the C = N group quenched the emissions of these iridium(III) complex-based probes, while emission enhancement was obtained upon cycloaddition with strained cyclooctynes. Likewise, the cationic iridium(III) center promoted the reaction kinetics of nitrone with BCN. These nitrone complexes also served as phosphorogenic bioorthogonal labels and imaging agents for cyclooctyne-modified proteins, offering promising prospects for advancing phosphorogenic bioorthogonal probes and imaging reagents. Subsequently, the same research group employed the transformation of the highly polar mesoionic sydnone ring into less polar pyrazoles to create luminescent probes for the SPSAC reaction in bioorthogonal labeling and live cell imaging (Fig. 36E).218 The developed iridium(III) sydnone complexes readily reacted with strained alkyne derivatives, resulting in significant emission enhancement (up to 24.9 fold) and extended lifetime. These complex-based probes were successfully utilized to label alkyne-modified proteins and ceramide molecules within live cells.
Following the success of ruthenium(II) and iridium(III) complex-based SPSAC bioorthogonal labelling, Lo's group then synthesized phosphorogenic rhenium(I) polypyridine sydnone complexes to expand the bioorthogonal toolkit (Fig. 36F).219 Upon SPSAC with bicyclo[6.1.0]non-4-yn-9-ylmethanol (BCN-OH), these complexes displayed 8.8 to 17.3 fold enhancement in emission. Upon conjugation with BCN-modified bovine serum albumin (BCN-BSA), the emission enhancement of the rhenium(I) complex increased dramatically, reaching up to 38.9 times with lifetimes extended from 1.80 to 4.71 μs. Due to their low cytotoxicity and remarkable emission enhancement capabilities, these complexes were applied for bioimaging of lysosomes in live HeLa cells.
In 2018, the Friscourt group220 reported the development of sydnone-modified coumarins as fluorogenic click reagents (Fig. 37A). The sydnone functionality quenched the fluorescence of the methoxycoumarin dye due to a nonemissive TICT state, with a low quantum yield of 0.004. Upon rapid strain-promoted cycloaddition with cyclooctyne BCN, these reagents produced highly fluorescent pyrazole cycloadducts with emission characteristics similar to 7-methoxycoumarin. Fluorescence quantum yields increased significantly (up to 0.66 and up to 132-fold enhancement), allowing for highly specific wash-free protein labeling.
 | ||
| Fig. 37 Fluorescent probes developed for bioimaging based on the strain promoted sydnone alkyne cycloaddition. (A) The structures and protein labeling of sydnone-modified coumarins, adapted with permission from ref. 220. Copyright 2018, American Chemical Society; (B) the structure of Naph-Syd and fluorescence imaging of ManDIBAC, adapted with permission from ref. 221. Copyright 2019, Elsevier Ltd; and (C) the structure of the nitrone-modified 1,8-naphthalimide and mitochondrial labeling in live cells, adapted with permission from ref. 222. Copyright 2022, Wiley-VCH GmbH. | ||
The Liang group introduced a fluorogenic probe, Naph-Syd, featuring an off–on fluorescence response mechanism for cell imaging. The probe was developed by incorporating a sydnone functionality into a 1,8-naphthlimide fluorophore (Fig. 37B).221 Sydnone played pivotal roles in (i) quenching the fluorescence of 1,8-naphthlimide through a PeT mechanism and (ii) click reaction with BCN derivatives. Upon rapid bioorthogonal cycloadditions with strained alkynes, pyrazole products were formed, leading to a remarkable 300-fold enhancement in 1,8-naphthlimide fluorescence, enabling fluorescence imaging in live cells under metal-free, wash-free conditions. Moreover, the probe exhibited simultaneous turn-on labeling of multiple targets by combining fluorogenic Tz cycloadditions with strained alkenes.
In another study of SPANC reaction-based fluorescence cell labelling, Tian and coworkers222 developed a bioorthogonal turn-on probe utilizing nitrone-modified 1,8-naphthalimide (Fig. 37C). The rearrangement products resulting from nitrone–alkyne cycloadducts exhibited 252-fold fluorescence enhancement, along with a substantial Stokes shift and high ΦF, up to 0.35. The high specificity of this SPANC reaction enabled effective protein labeling in vitro and mitochondrial labeling in living cells with a high signal-to-noise ratio under catalyst-free, wash-free conditions.
In addition to the labelling of proteins and live cells, SPSAC based bioorthogonal tags for labelling and tracking nucleosides and oligonucleotides in live cells have also been investigated by Wagenknecht and colleagues (Fig. 38A).223 Investigation of SPSAC kinetics with model substrates revealed rapid reactions with cyclooctyne probes (up to k = 0.59 M−1 s−1), yielding a 70-fold fluorescence enhancement. Sydnones were integrated into both 2′-deoxyuridines at position 5 and 7-deaza-2′-deoxyadenosines at position 7, and the modified nucleosides were then synthetically incorporated into single-stranded DNAs, allowing for postsynthetic labeling with cyclooctyne probes both in solution and in live HeLa cells.
 | ||
| Fig. 38 Strain promoted sydnone alkyne cycloaddition-based bioorthogonal probes for fluorescence imaging. (A) The structures of the sydnone-modified 2′-deoxyuridines and confocal imaging of the HeLa cells, adapted with permission from ref. 223. Copyright 2021, Wiley-VCH GmbH; (B) the structure of the iminosydnone probe and comparison of the distribution of the two released products in fixed vs. living cells after reaction, adapted with permission from ref. 224. Copyright 2020, The Royal Society of Chemistry; (C) the structure of the COS probe and fluorescence images of reactions, adapted with permission from ref. 225. Copyright 2021, Wiley-VCH GmbH; and (D) the structures of the sydnonimines and fluorescence images of reactions in HeLa cells, adapted with permission from ref. 226. Copyright 2023, Elsevier Ltd. | ||
Probes based on fluorescence turn on click-and-release reactions enable the multicolor labelling of samples under biological conditions. For example, the Taran group224 synthesized a series of iminosydnone probes that can efficiently cleave and unquench two distinct fluorophores upon bioorthogonal reaction with cycloalkynes, resulting in remarkable fluorescence enhancements (Fig. 38B). These versatile iminosydnones served as fluorogenic cleavable linkers, enabling the cellular tracking of the two products generated from the click and release reaction. Examination of cell distributions revealed distinct cytoplasmic localization of the two released products, confirming their separation and movement within cells prior to precipitation. The compatibility of this reaction was validated in fluorescence labelling of fixed or living cells through monitoring the release of the double turn-on fluorescence signals. In another research conducted by the same group, release of isocyanates in live cells was also achieved using bioorthogonal click-and-release reactions, where the reactivity constants reached up to 1000 M−1 s−1.227
Inspired by the strain-promoted cycloaddition reaction involving 1,3-dithiolium-4-olates (DTOs) and cyclooctyne or benzyne derivatives, Liang and colleagues devised a carbonyl sulfide (COS)-based system for controlled release of H2S (Fig. 38C).225 This system was activated by a bioorthogonal click-and-release reaction, enabling precise control over H2S release in both PBS and live cells. Incorporating a diphenylamino substituent onto one phenyl ring of DTO facilitated simultaneous activation of a new thiophene fluorophore, enabling spatiotemporal monitoring of intracellular H2S release using green fluorescence signals. Beyond its bioorthogonal and visualization capabilities, this versatile platform allowed for targeted delivery of H2S to mitochondria. The Liang group226 also explored the mitochondrial targeting capabilities of sydnones and sydnonimines (Fig. 38D). Experimental findings confirmed that both sydnones and sydnonimines exhibited significant mitochondrial distribution, while incorporation of a phenyl group into the C4 position of sydnones markedly diminished their affinity for mitochondria. Leveraging the mitochondrial targeting properties and click-and-release reaction of sydnonimine, the anticancer efficacy of celecoxib delivered into mitochondria was evaluated in HeLa and HepG2 cells.
In summary, bioorthogonal chemistry based on mesoionic sydnones has experienced significant advances since their discovery in 1935. Initially acknowledged for their biological significance, their reactivity was only fully understood following Huisgen's work in 1962. However, harsh reaction conditions and inadequate control over regioselectivity impeded further exploration of their potential for cycloaddition reactions. Nonetheless, recent studies have achieved notable breakthroughs, generating increased interest within the chemical community. Sydnones, especially when combined with strained cycloalkynes, have emerged as potent tools for bioorthogonal chemistry, facilitating in situ bioimaging applications. Despite being larger in size compared to azides, sydnones offer adjustable reactivity through strategic substitution, enabling the development of ultra-fast reactions crucial for in vivo studies. For instance, Taran's research213 conducted using 4-chloro-sydnones resulted in a more than tenfold increase in reactivity towards BCN compared to conventional azides, proving invaluable for fluorescence protein labeling, particularly for proteins present in low abundance. Furthermore, advancements such as iminosydnones227 and highly strained alkynes214 hold promise for further acceleration, with reaction rates exceeding 12000 M−1 s−1, thereby enhancing their suitability for bioimaging. Despite notable advancements, challenges persist, notably, the lack of regiocontrol in the presence of unsymmetrical arynes. Addressing these challenges will undoubtedly propel future progress in this distinctive field of mesoionics.
2.6. Nitrile imine–alkyne cycloaddition
The 1,3-dipolar cycloaddition between nitrile imines and alkenes/alkynes is known to be a rapid bioconjugation reaction for labeling proteins or nucleic acids (Scheme 6).228 Since nitrile imines are inherently unstable, these dipoles are typically generated in situ through different methods like tetrazole photolysis, hydrazone oxidation, or chlorohydrazone hydrolysis. The remarkable reactivity and robustness of nitrile imines have recently sparked interest in their use for designing fluorogenic click probes. In this section, we will evaluate the applications of nitrile imine–alkene/alkyne 1,3-dipolar cycloaddition for in situ activated bioimaging.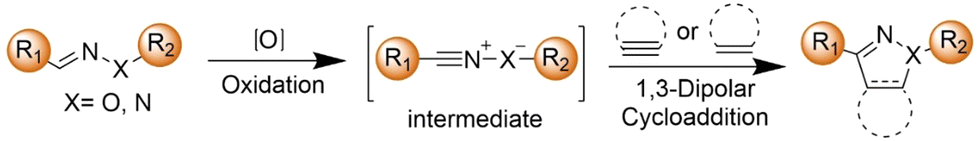 | ||
| Scheme 6 Proposed mechanism of nitrile imine–alkyne cycloaddition. | ||
The Tian group228 introduced 4-oxime-1,8-naphthalimide (NP-Oxm) as a bioorthogonal turn-on probe for the fluorogenic labelling of proteins (Fig. 39A). Using this probe, highly reactive naphthalimide-nitrile oxide was produced in situ, which served as the bioorthogonal handle for 1,3-dipolar cycloaddition with alkenes or alkynes. Upon cycloaddition with alkenes, various 4-isoxazoline-naphthalimides (NP-Dmf, NP-Str, and NP-Nob) exhibited a 5- to 13-fold increase in fluorescence. Notably, the reaction products, 4-isoxazolyl-naphthalimides (NP-BCN and NP-DIBO), resulting from strain-promoted alkyne–nitrile oxide cycloaddition, demonstrated photoactivatable fluorogenic properties via isoxazole–oxazole photoisomerization, boasting over 250-fold fluorescence enhancement with a high fluorescence quantum yield (ΦF > 0.4). Furthermore, NP-Oxm facilitated highly specific protein labeling in a tunable emission manner. This photoactivatable bioorthogonal turn-on strategy provided a useful toolkit for chemical biology investigations, enabling precise visualization of biomolecules in living cells.
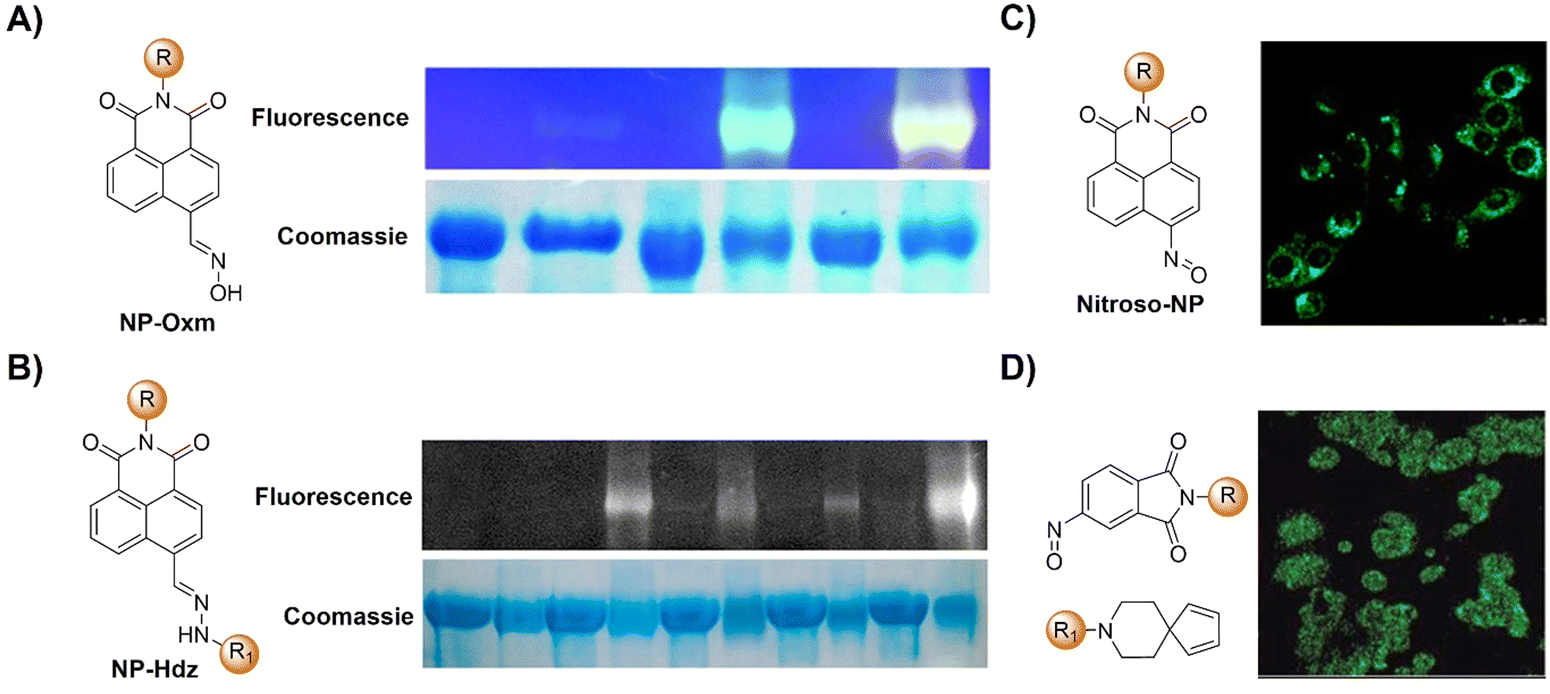 | ||
| Fig. 39 Fluorescent probes based on the nitrile imine–alkyne cycloaddition. (A) The structure and protein labeling of NP-Oxm, adapted with permission from ref. 228. Copyright 2019, The Royal Society of Chemistry; (B) the structure and protein labeling of NP-Hdz, adapted with permission from ref. 229. Copyright 2020, Wiley-VCH GmbH; (C) the structure of Nitroso-NP and fluorescence images of reactions, adapted with permission from ref. 230. Copyright 2021, American Chemical Society; and (D) the structure of the nitroso-modified naphthylene and fluorescence images of reactions, adapted with permission from ref. 231. Copyright 2019, Wiley-VCH GmbH. | ||
In 2020, the same research group devised and synthesized four 4-hydrazone-1,8-naphthalimides as fluorogenic click probes (Fig. 39B).229 These compounds produced nitrile imines in situ that can react with BCNvia a highly fluorescent turn-on strain-promoted 1,3-dipolar cycloaddition, yielding 4-pyrazol-1,8-naphthalimides. These derivatives showed remarkable characteristics including large Stokes shift (up to 152 nm), high ΦF (up to 0.85), and substantial fluorescence enhancement (up to 196.9-fold). These hydrazone probes were effectively employed for the fluorogenic labeling of alkyne modified BSA proteins, demonstrating their potential as valuable tools for protein visualization and detection.
Following this success, the same research group230 then synthesized a series of nitroso-modified naphthylene fluorophores for bioorthogonal nitroso–diene Diels–Alder reactions (Fig. 39C). Upon photoactivation, these probes exhibited remarkable properties, i.e., formation of bridged cycloadducts between 4-nitroso-1,8-naphthlimide or 5-nitroso-naphthalene-1-sulfonamide and cyclic dienes led to aziridine-containing rearrangement products. These products displayed remarkable fluorescence enhancement, with an increase of fluorescence intensity up to 3604-fold, accompanied by large Stokes shifts of up to 188 nm, a high ΦF of up to 0.92, superior photostability, and distinct solvatochromic effects. The cycloadducts formed between nitroso compounds and multiple methyl-substituted cyclopentadiene via [2+2] cycloaddition underwent spontaneous rearrangement to produce fluorescent hydroxylamine products. These nitroso–diene probe pairs were effectively utilized for selective fluorogenic labeling of diene-modified proteins and for wash-free organelle-specific mammalian live-cell imaging in real time, in a spatiotemporally controlled manner.
In another study, Jiang and coworkers231 harnessed the fluorogenic reactivity of nitroso-based bioorthogonal Diels–Alder reactions to develop the fluorogenic nitroso probe (Fig. 39D). This reaction facilitated the generation of a twisted “donor–acceptor–donor” skeleton, crucial for effective separation of HOMO/LUMO electron densities, thereby enabling the development of probes with thermally activated delayed fluorescence (TADF) properties. This unique feature enabled selective activation of Point of Interest (POI) within specific cell compartments while leaving other labeled molecules intact. The precise spatial and temporal control afforded by this fluorogenic labeling system holds promise for monitoring POI dynamics in live cells across various applications. Expanding beyond model protocols, they then devised an efficient “click-like” fluorogenic nitroso–diene pair probe for general, controllable protein labeling in live cells. Proof-of-concept studies underscored the optimized probe's potency for fluorogenic protein labeling with minimal background signals under UV irradiation. The utility of this bioorthogonal reaction in living mice was assessed for noninvasive in vivo fluorescence imaging.
2.7. Photoclick reaction
Photoclick reactions232–234 synergize the benefits of photochemistry235 and classical click chemistry.34–37 Unlike standard click reactions occurring between molecules in their ground state, in photoclick reactions, the partners remain chemically inert until exposure to specific light wavelengths. This illumination activates one of the partners, triggering the formation of new covalent bonds. This property enables the synthesis of diverse organic structures under mild, metal-free conditions, with precise control. Moreover, light irradiation offers non-invasive characteristics, affording superior spatial resolution and temporal control over reactive intermediate generation. These attributes are particularly advantageous for bioorthogonal transformations, making photoclick chemistry an integral part of the toolbox for surface functionalization,236 polymer conjugation,237,238 photocross-linking,239 and bioorthogonal labeling.92,240,241Over the past few decades, researchers have explored an expanding array of photoclick reactions. These encompass diverse reactions such as the photoinduced cycloaddition of tetrazoles and alkenes (NITEC),242–244 UV-light initiated thiol–ene and thiol–yne reactions,127,245,246 light-triggered azide–alkyne cycloadditions,247,248 diarylsydnone–alkene photoclick reaction (photo-DASAC),249 photo-induced azirine–alkene cycloaddition,250 light-triggered oxime ligation reactions,251 and light-mediated coupling of acylsilanes with indoles,252 along with visible-light-induced 9,10-phenanthrenequinone-electron rich alkene (PQ-ERA) photocycloadditions.253–255 Notably, certain photoclick reactions yield products with excellent fluorescence properties, exemplified by NITEC, photo-DASAC, and PQ-ERA. Leveraging the fluorescence responses, these reactions have been extensively used in precise labeling and imaging of biological systems. In this section, we aim to evaluate the evolution of photoclick reactions specifically for in situ activated bioimaging.
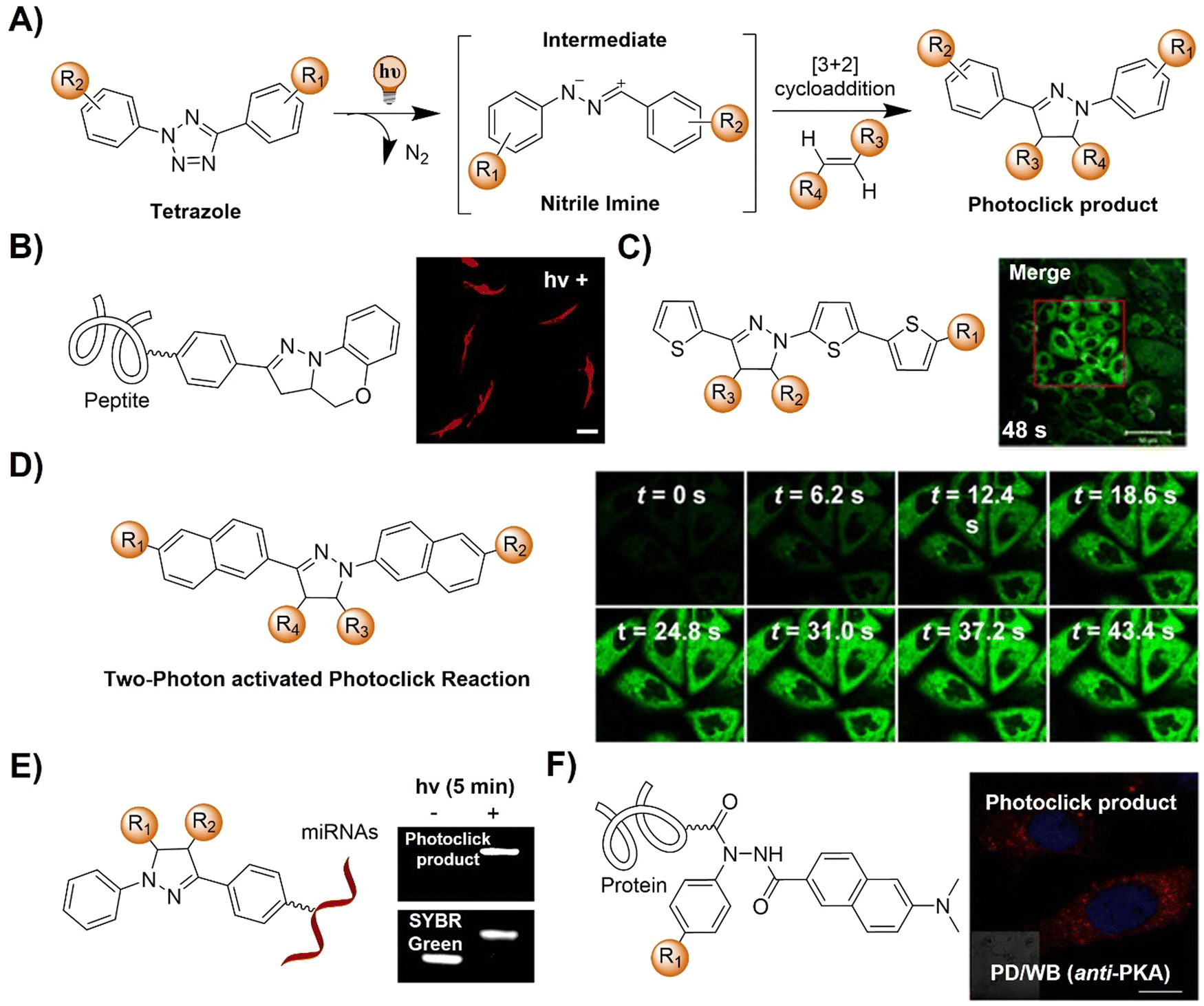 | ||
| Fig. 40 Fluorescent probes based on the light-triggered nitrile imine-mediated tetrazole–ene cycloaddition. (A) Proposed mechanism of the NITEC photoclick reaction; (B) chemical structure of the Tet(II)-GFRGD photoclick product and morphology of hMSCs encapsulated in Tet(II)-GFRGD gels, adapted with permission from ref. 258. Copyright 2013, American Chemical Society; (C) the structure and in situ cell imaging of the oligothiophene-based tetrazole photoclick product, adapted with permission from ref. 259. Copyright 2013, The Royal Society of Chemistry; (D) the structure and in situ CHO cell imaging of the naphthalene-tetrazole photoclick product, adapted with permission from ref. 260. Copyright 2013, American Chemical Society; (E) the structure and HepG2 cell imaging of the miRNA–tetrazole photoclick product, adapted with permission from ref. 261. Copyright 2016, American Chemical Society; (F) the structures and live HepG2 cell imaging of the two-photon tetrazole conjugates, adapted with permission from ref. 240. Copyright 2016, Wiley-VCH GmbH. | ||
In 2013, the Zhang group258 introduced photodegradable hydrogels for three-dimensional cell encapsulation (Fig. 40B). These hydrogels, formed by short peptides with a biaryl-substituted tetrazole phototrigger, offer precise spatial and temporal control over cellular microenvironments. Upon light exposure, the phototrigger underwent rapid intramolecular photoclick ligation, yielding a highly fluorescent pyrazoline, facilitating precise imaging. Tet(I)-GFF and Tet(II)-GFRGD gels, incorporating these triggers, exhibited robust mechanical strength and biocompatibility, supporting extended cell culture. Triggered activation disrupted hydrogel assembly, modulating microenvironments for cells both within and on the gel. This approach is promising for biomedical applications, offering targeted control over cellular environments critical for various research and therapeutic endeavors.
Most photoclick reactions, activated by UV light (300–365 nm), face limitations in biological systems due to phototoxicity. To address this, Lin's group synthesized oligothiophene-based tetrazoles to minimize phototoxicity. For example, a terthiophene-tetrazole exhibited excellent photoreactivity under 405 nm laser (ring rupture quantum yield: 0.16, Fig. 40C).259 On the other hand, a water-soluble terthiophene–tetrazole exhibited faster kinetics with a fumarate dipolarophile (k2 = 1299 ± 110 M−1 s−1), enabling real-time, spatiotemporally controlled precise imaging of microtubules in live mammalian cells. Given the widespread availability of 405 nm lasers in fluorescence microscopes, this class of thiophene-tetrazoles activated by 405 nm lasers have potential for the broader adoption of photoclick chemistry in cell biology research.
To further advance the application of NITEC to living organisms with enhanced spatiotemporal precision, the Lin group devised naphthalene-based tetrazoles activated efficiently via two-photon excitation using a 700 nm femtosecond pulsed laser (Fig. 40D).260 In this two-photon bioorthogonal probe, a water-soluble, cell-permeable naphthalene-based tetrazole that exhibits a significant two-photon cross-section of 3.8 GM was identified for the cycloaddition reaction with acrylamide. This two-photon-triggered, fluorogenic photoclick chemistry was successfully employed for site-selective labeling of alkene-encoded proteins in vitro and for visualizing ligand-bound microtubules in cultured cells. With genetic methods enabling the incorporation of unnatural amino acids containing reactive alkene dipolarophiles in both mammalian cells and Drosophila melanogaster fruit flies, this technique offers a new approach to dissect protein function in living organisms with exceptional spatiotemporal precision.
MicroRNAs (miRNAs) are pivotal in post-transcriptional gene regulation, yet identifying their interactions with target genes in cells remains challenging due to untagged miRNAs. Zhang et al.261 introduced photoclickable miRNAs, pre-tagged with molecular handles, maintaining intracellular functionality while allowing attachment of probes via photoclick reactions (Fig. 40E). Upon transfection, these miRNAs formed functional complexes that can effectively suppress target gene expression. Target genes bound to photoclickable miRNAs were tagged for pull-down and identification. Using miR-106a, miR-27, and miR-122, their intracellular function matched intact miRNAs, surpassing biotinylated versions. Attachment of biotin handles to complexes facilitated pull-down and analysis, unveiling a regulatory pathway in miR-122-mediated cell apoptosis in HepG2 cells.
Harnessing the fluorescence quenching capability of tetrazoles, Yao and coworkers240 designed and synthesized a series of tetrazole-Bodipy conjugates (Fig. 40F). Emission of these compounds was quenched, while upon irradiation and protein labeling, their emission was switched on. In water, an 80-fold fluorescence enhancement was observed post-photoactivation due to a reaction with acetic acid, with similar effects observed in the presence of proteins like HSA and BSA. Staurosporine (STS)-labeled tetrazole-bodipy enabled selective imaging of endogenous protein kinase A (PKA) in HepG2 cells. Intense fluorescence emission in the cell cytoplasm persisted after multiple wash cycles, indicating the formation of a robust covalent bond.
In another study, Zhang and colleagues262 fabricated pyrazoline-based two-photon probes with diverse functionalities for two-photon fluorescence imaging of protease in apoptosis (Fig. 41A). Using this probe system, photoclick reaction of naphthalene-substituted tetrazoles with alkenes was employed to generate naphthalene-substituted pyrazoline, a highly fluorescent two-photon fluorophore, in situ under mild and biocompatible conditions. Various types of two-photon probes, such as those targeting the mitochondria and lysosomes, as well as a complex probe incorporating both a targeting ligand (cRGD) and a short peptide substrate for caspase-3 activation, were developed. Notably, these probes exhibited selective activation by caspase-3 in cells or tumor tissue, enabling 3D imaging of caspase-3 distribution in tumor tissue. The versatility of this method in incorporating multiple functionalities into two-photon probes through a gentle photoclick protocol holds promise for constructing diverse imaging probes for targeted tumor imaging.
 | ||
| Fig. 41 Bioorthogonal probes for fluorescence cell imaging based on the light-triggered nitrile imine-mediated tetrazole–ene cycloaddition. (A) The structure and 3D two-photon imaging of the naphthalene-substituted tetrazole photoclick product, adapted with permission from ref. 262. Copyright 2016, The Royal Society of Chemistry; (B) the structure and HeLa cell imaging of the biaryl-tetrazole-modified peptide photoclick product, adapted with permission from ref. 263. Copyright 2017, The Royal Society of Chemistry; (C) the structure and E. coli cell labeling of the styrene-tetrazole photoclick product, adapted with permission from ref. 264. Copyright 2017, American Chemical Society; and (D) the structure and cell imaging of the bodipy-tetrazole photoclick product, adapted with permission from ref. 265. Copyright 2018, Wiley-VCH GmbH. | ||
Following this success, the same research group263 devised alkaline phosphatase (ALP)-guided self-assembly using a biaryltetrazole-modified peptide precursor, Tet-Gly-DPhe-DTyr(H2PO3). The peptide precursor could form the photo-responsive hydrogelator Tet-GDFDY upon ALP-mediated dephosphorylation (Fig. 41B). This enabled enzyme-instructed self-assembly-photo-induced disassembly (EISA-PIDA) processes on ALP-overexpressing cancer cell membranes. Tet-GDFDY exhibited rapid fluorescence activation under brief light exposure, facilitating real-time monitoring of EISA-PIDA on HeLa cells. Additionally, photo-triggered disassembly of Tet-GDFDY disrupted pericellular nanofibers, potentially offering a mechanism for combating EISA-induced cell death.
To enhance the fluorogenicity of the alkene-tetrazole reaction for protein labelling, the Gou group264 explored the use of styrene as a dipolarophile for alkene-tetrazole reaction. Remarkably, an over 30-fold increase was obtained in fluorescence quantum yield for the reaction product in aqueous environments (Fig. 41C). Mechanistic investigations suggested that this enhancement stems from inadequate protonation of the styrene-tetrazole reaction product. This discovery offered valuable insights for optimizing alkene-tetrazole reactions in biological research. The efficacy of the styrene-tetrazole reaction for fluorogenic protein labeling was demonstrated both in vitro and in vivo through genetic incorporation of a styrene-containing unnatural amino acid. The combination of styrene with various tetrazole derivatives promises the enhanced and diversified application of alkene-tetrazole chemistry in live-cell real-time imaging.
The development of photoclick reaction initialized responsive probes for the detection of biomolecules, such as ROS, was reported by the Lin group (Fig. 41D).265 Three bodipy-tetrazoles featuring para-, meta-, or ortho-linkages between the bithiophene-tetrazole and the bodipy fluorophore were synthesized and their photoclick and sensing reactions were investigated. These compounds exhibited remarkable reactivity towards dimethyl fumarate in vitro in photoclick reactions. Intriguingly, the fluorescence of bodipy experienced a significant decrease post photoclick reaction; yet, this fluorescence loss could be reversed by treatment with chemical oxidants like H2O2. A water-soluble bodipy-tetrazole derivative was also developed, serving as an “off–on” fluorescent probe for the detection of H2O2. This probe facilitated the initial bioorthogonal in situ generation at microtubules and subsequent detection of H2O2 within HeLa cells.
For rapid bioorthogonal labelling of proteins in live cells, the same research group employed a bioinspired approach for designing sterically shielded tetrazoles for bioorthogonal photoinduced cycloaddition with alkene dipolarophiles (Fig. 42A).266 The resulting photogenerated nitrile imine exhibited significant enhancement of selectivity for cycloaddition over nucleophilic addition. Computational and structural analyses indicated that adjacent N-Boc-pyrrole groups provided a steric shield, raising the activation energy to prevent thiolate addition. In situ studies revealed a notably prolonged half-life of 102 s for the shielded nitrile imine compared to its unshielded counterpart. Leveraging the environmentally sensitive pyrazoline fluorophore produced by the cycloaddition, the shielded tetrazole enabled the creation of a fluorescent sensor for monitoring ligand-induced conformational transitions of glutamine binding protein (QBP). Additionally, due to its rapid kinetics and exceptional selectivity, the shielded nitrile imine efficiently labeled an alkene encoded GCGR in live mammalian cells at a concentration as low as 500 nM within 1 min, surpassing the efficiency of optimized Tz reagents. Apart from protein labelling, the “photoclick” method for nucleic acid labeling was explored by Zhang and coworkers in 2019,267 in which fluorescence “switch-on” labeling of synthesized DNA with coumarin-fused tetrazole probes was achieved (Fig. 42B). In this nucleic acid labeling system, DNA and electron-rich alkenes could quench pyrazoline fluorescence while minimally affecting the emission of coumarin fluorophores. CTz-SO3, a water-soluble coumarin-fused tetrazole probe designed in this research, could precisely label and visualize 5-vinyl-2′-deoxyuridine (VdU)-incorporated cellular DNA in real time without fixation or DNA denaturation.
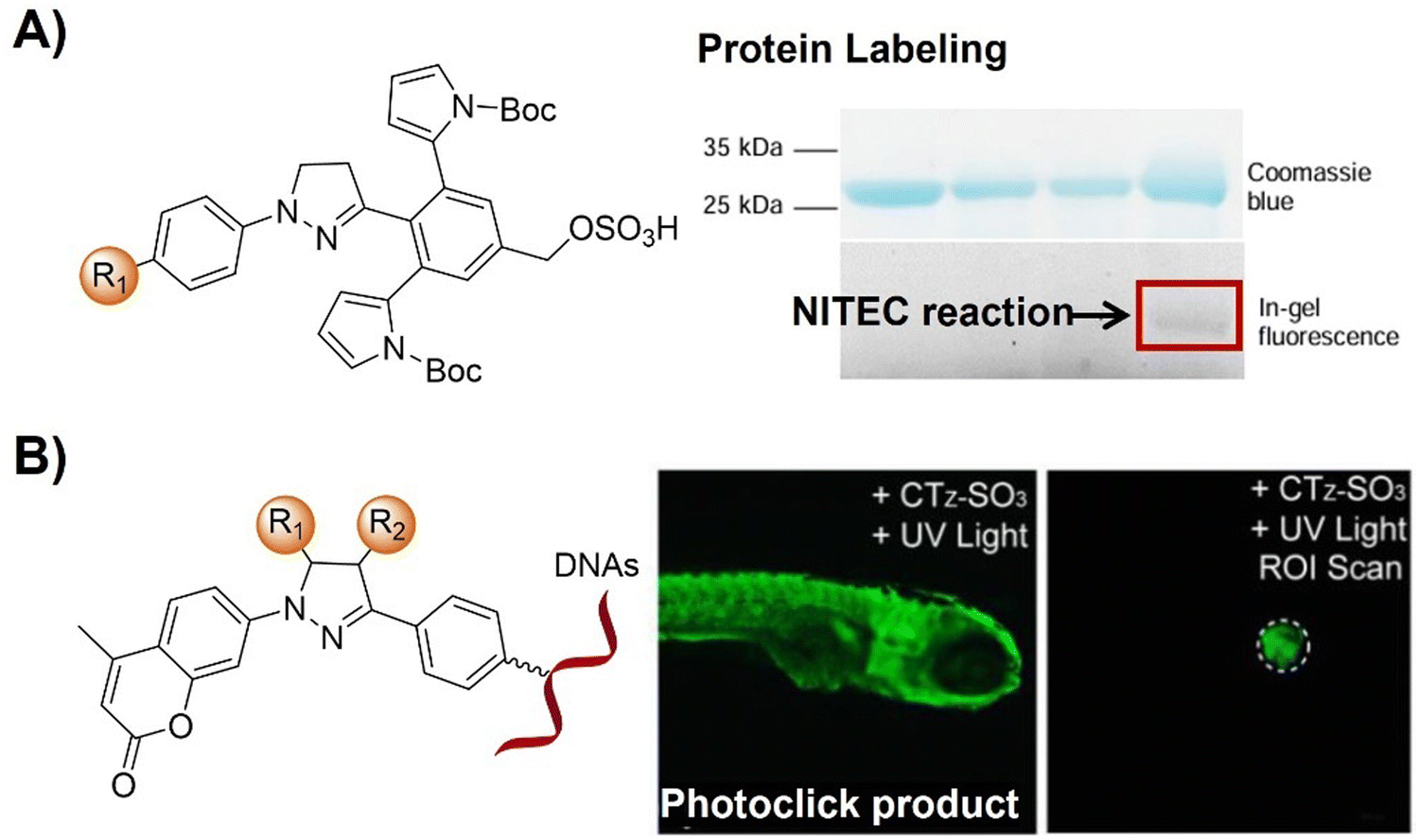 | ||
| Fig. 42 Fluorescent probes for protein labelling and in vivo imaging based on the light-triggered nitrile imine-mediated tetrazole–ene cycloaddition. (A) The structure and protein labeling of the sterically shielded nitrile imine photoclick product, adapted with permission from ref. 266. Copyright 2018, American Chemical Society; (B) the structure and live zebrafish imaging of the coumarin-fused tetrazole photoclick product, adapted with permission from ref. 267. Copyright 2019, American Chemical Society. | ||
In addition to protein and nucleic acid labelling, intramolecular photoclick reactions with fluorogenic probes also enabled the labelling of subcellular structures, like lysosomes and mitochondria. For example, Huang et al.268 devised a straightforward strategy for designing tetrazole-based photoactivatable fluorophores (Mt-Tet and Ly-Tet) that target cellular mitochondria and lysosomes guided by TPP and morpholine, respectively, and exhibit turn on fluorescence via intramolecular photoclick reaction (Fig. 43A). Upon mild photoirradiation, robust fluorescence activation occurred within ∼1 min in vitro and ∼3 min in live cells, allowing for real-time imaging of subcellular organelles. These probes exhibited minimal fluorescence prior to activation, resulting in a reduced background signal compared to standard fluorescence labeling. The spatiotemporally controllable nature of these probes enabled the observation of organelle dynamics by super-resolution Stochastic Optical Reconstruction Microscopy (STORM) imaging.
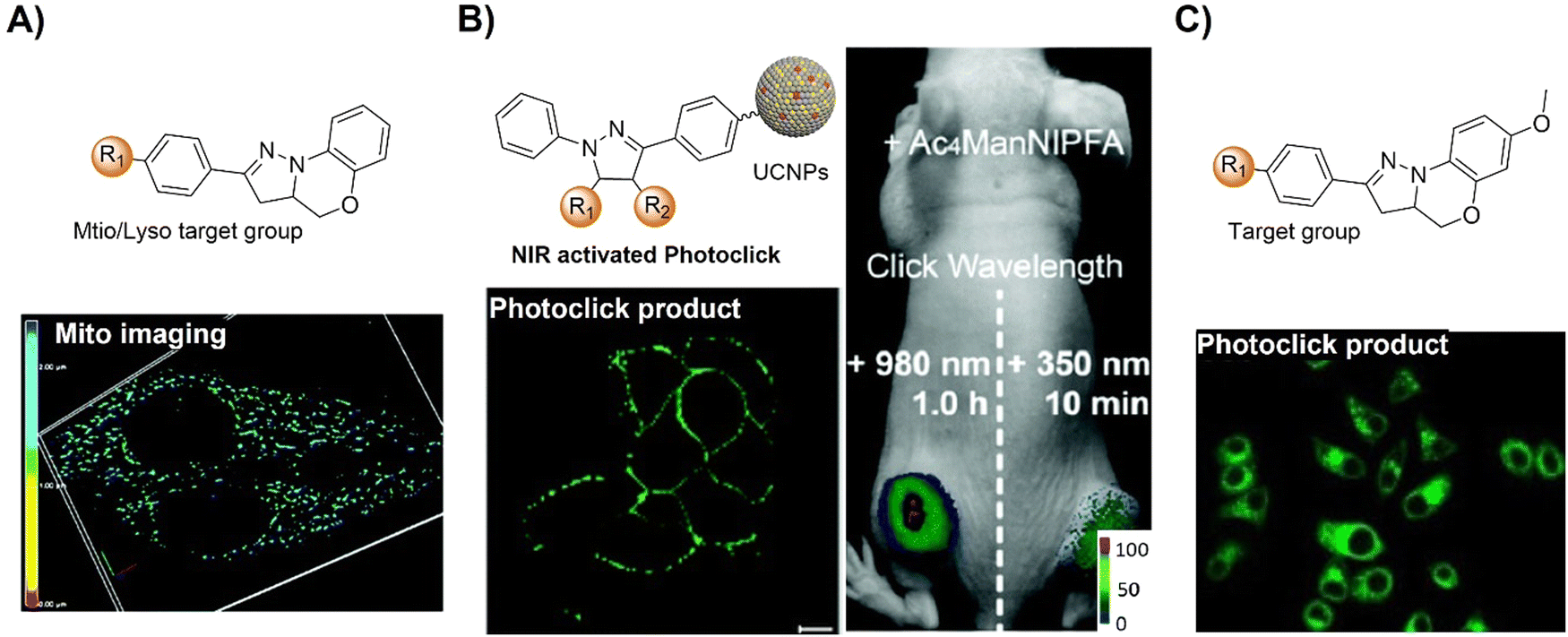 | ||
| Fig. 43 Bioorthogonal probes for fluorescence bioimaging based on the light-triggered nitrile imine-mediated tetrazole–ene cycloaddition. (A) The structure and super-resolution STORM imaging of the intramolecular NITEC photoclick product, adapted with permission from ref. 268. Copyright 2021, The Royal Society of Chemistry; (B) the structure, in vitro and in vivo imaging of the NIR light-activated NITEC photoclick product, adapted with permission from ref. 269. Copyright 2020, The Royal Society of Chemistry; and (C) the structure and endoplasmic reticulum (ER) fluorescence labeling of the fluorescent pyrazoline, adapted with permission from ref. 270. Copyright 2022, Wiley-VCH GmbH. | ||
Sialic acid plays a crucial role in profiling cell activities across various biological and pathological contexts. To understand the biological functions of sialic acids under endogenous conditions, photoclick based bioorthogonal probes have been developed for their precise labelling by Zhang and colleagues.269 With this probe system, upconverting nanoparticles (UCNPs) that can be activated by NIR light while emitting UV-visible light were used to prepare the nanoprobe (tetrazole-UCNP) for spatiotemporal labeling of sialic acids (Fig. 43B). Combined with stable N-alkene-D-mannosamine (Ac4ManNIPFA), this strategy facilitated real-time imaging of metabolically synthesized alkene sialic acids on cell surfaces through fluorogenic cycloaddition. In addition, spatially selective sialic acid visualization was achieved in specific tumor tissues of mice under NIR light activation, enabling in situ controllable labeling in complex biological systems.
Effective labelling of decarboxylases enables direct monitoring of oxidative decarboxylation in situ in biological samples. In 2020, Schwaneberg and coworkers271 devised a robust 96-well photoclick assay for quantifying styrene derivatives and other alkene products resulting from decarboxylation reactions in the micromolar range. This method features user-friendly handling and fluorescence read-out that facilitate high-throughput screening of decarboxylase variant libraries within hours. The assay requires minimal instrumentation, leveraging commonly available UV light irradiation. To demonstrate the application of this method, analysis of the specificity for different substrates of OleT-BM3R fusion enzyme was evaluated, holding promise for enhancing substrate selectivity, H2O2 resistance, and optimizing electron transfer efficiency in OleT and other decarboxylases.
An et al.270 introduced a photoactivatable fluorogenic azide–alkyne click (PFAAC) reaction based on the azide-tetrazole probe platform. Upon light irradiation, selective photolysis of azide and tetrazole rendered these probes nonfluorescent, generating photoactivatable fluorescent reporters post-click reaction (Fig. 43C). This chemistry enabled specific detection of AAC reactions with a remarkable over 1000-fold “turn-on” ratio in a spatiotemporally controlled manner, validated in live cell imaging for organelle labeling. Incorporating a chemical linker into the azide-tetrazole probe expanded PFAAC's utility, allowing for dual-labeling of biomolecules, as confirmed in endoplasmic reticulum (ER) fluorescence labeling with pyrazoline and cy5 dyes. Given the widespread use of click chemistry, PFAAC holds broad potential for biomolecular imaging, tracking, and detection.
In summary, since 2008, the NITEC photoclick reaction has gained increasing attention in various biological applications. Unlike other click reactions, such as CuAAC and IEDDA, NITEC offers unique features like substrate stability in physiological environments, low toxicity, and fluorogenicity enabling efficient sensing and imaging of specific biomolecules. While primarily used for bioorthogonal protein labeling and wash-free in situ imaging, tetrazoles have also modified nucleic acid structures and have been involved in the development of biomaterials like hydrogels. Despite optimizations, drawbacks like UV-restricted activation wavelengths and collateral reactions with biological nucleophiles have hindered their biological utility. Strategies to shift activation wavelengths towards the NIR or explore photocatalysis with UCNPs have shown some promise. Addressing collateral reactions, especially with the formation of undesired pyrazoline, requires modification of the tetrazole scaffold. However, such reactions can also be harnessed for applications like studying protein–protein interactions. With increasing interest in photoclick chemistry and the versatility of light-generated intermediates, photoactivatable tetrazoles hold immense potential across various research fields, from biology to materials science, with scope for further optimization and exploration in novel applications.
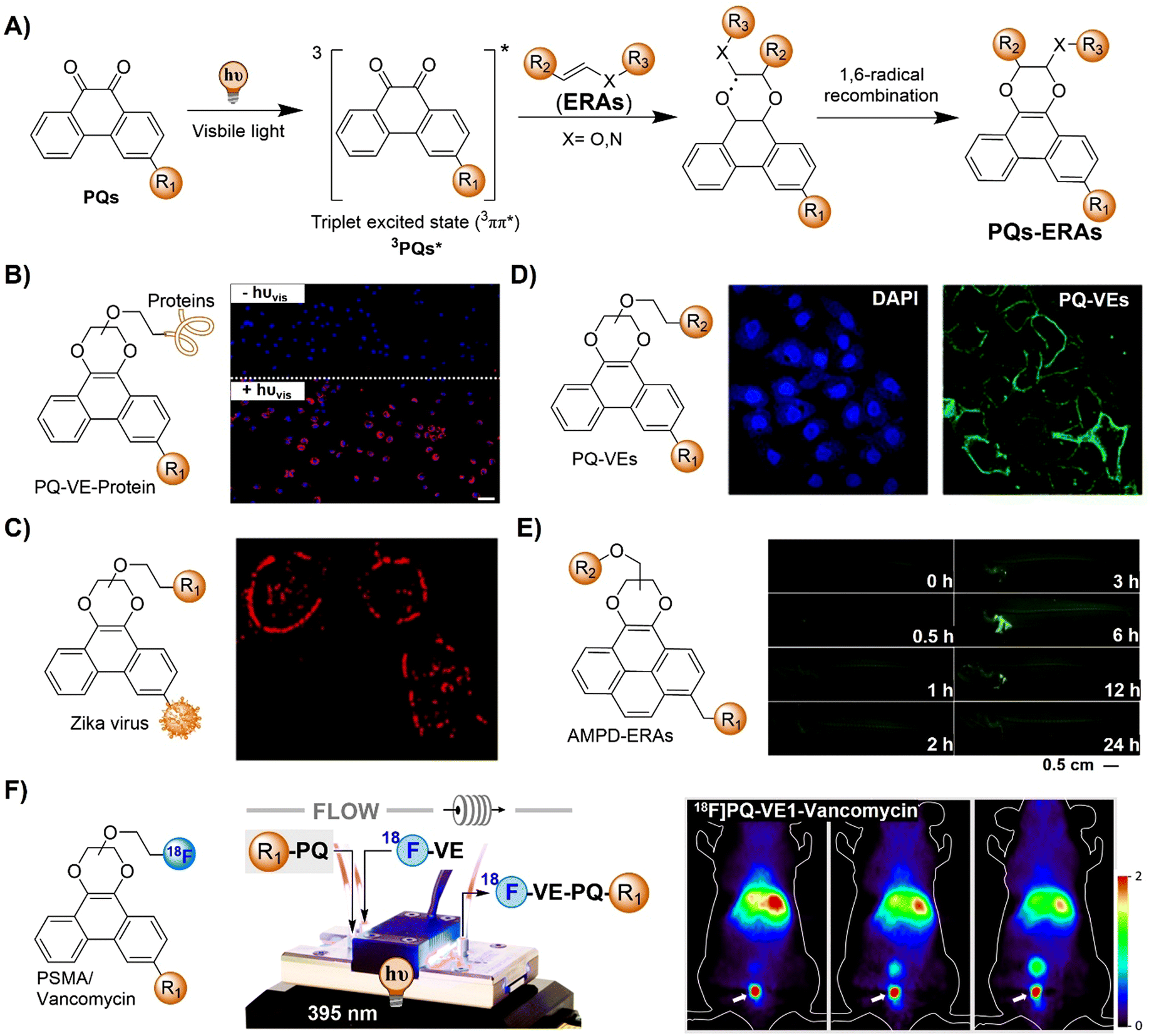 | ||
| Fig. 44 PQ-ERA photocycloaddition reaction based bioorthogonal probes for fluorescence imaging. (A) Proposed mechanism of the PQ-ERA photoclick reaction; (B) spatial labeling of cetuximab on the surface of A549 cells with pretagged VE that was further labeled through the PQ-ERA photocycloaddition reaction, adapted with permission from ref. 254. Copyright 2018, American Chemical Society; (C) PQ-ERA photocycloaddition for the labeling and visualization of the Zika virus, adapted with permission from ref. 275, licensed under CC-BY 4.0; (D) sequential photoirradiation-orthogonal reporter tagging (SPORT) for cell labelling viaPQ-ERA photocycloaddition, adapted with permission from ref. 276, under the license CC BY-NC 3.0 DEED; (E) the structure and in vivo imaging of the AMPD probe on glass catfish; adapted with permission from ref. 277. Copyright 2024, Elsevier B.V; and (F) short-lived 18F-PET tracers prepared viaPQ-ERA photoclick reaction and their application in PET imaging, adapted from ref. 253, licensed under CC-BY-NC-ND 4.0. | ||
In 2018, Zhang and colleagues addressed the limitations of the mercury lamp light source by employing a suitable high-power LED light source.254 Using this hand-held LED lamp, the PQ-ERA photoclick reaction could be executed within a short time frame of 5 min, achieving high yields (up to 93%) even in an aqueous environment (Fig. 44B). The application of the PQ-ERA photoclick reaction system was demonstrated for labeling cell membranes in living cells, underscoring its biocompatibility and potential for biological applications (Fig. 44B). Shortly after, Zheng et al. introduced the PQ-ERA bioorthogonal cycloaddition technique to construct a fluorogenic probe in 2022275 for labeling and visualization of the Zika virus through a straightforward process mediated by visible light (450 nm, Fig. 44C).
Based on the PQ-ERA photoclick reaction, Xie and coworkers276 introduced a method, namely sequential photoirradiation-orthogonal reporter tagging (SPORT), for cell labelling (Fig. 44D). This technique enabled the controlled labeling of surface biomolecules in live cells using visible LED light. Introducing metabolic precursors into live cells triggered a bioorthogonal reaction to convert azide/alkyne functional groups into a photo-active functional group known as PQ. Subsequently, the PQ groups underwent visible light-induced photoclick reactions with vinyl ether-biotin using a white light LED. The effectiveness of this approach was demonstrated by selective labeling of a variety of biomacromolecules, including sialome, mucin-type O-glycome, phospholipids and membrane proteins, highlighting the SPORT strategy for precise investigations of biomolecules within complex biological systems.
Multimodal bioorthogonal small molecule probes also contribute significantly to tracking drugs in living organisms. For instance, Bai and colleagues277 introduced 1-(azidomethyl) pyrene-4,5-dione (AMPD), a versatile probe incorporating adjacent dione structures at the pyrene core for drug target detection in vivo (Fig. 44E). AMPD selectively interacts with ERA-labeled drugs under blue LED light, triggering specific in situ fluorescence for in vivo tracking and flow cytometric sorting. The methyl azide group enhances target protein enrichment via click chemistry. With exceptional biocompatibility, AMPD facilitated the visual tracking in live animals, tissues, cells, and gels, expediting drug discovery by improving comprehension of in vivo drug–target interactions. Beyond its utility in optical imaging, the PQ-ERA photoclick reaction has diversified its applications across a spectrum of molecular imaging techniques, including PET imaging. The Feringa group pioneered the development of an expedited photoclick method for synthesizing short-lived 18F-PET tracers, leveraging the PQ-ERA photoclick reaction (Fig. 44F).253 The corresponding precursors are readily accessible synthetically and can be tailored with various targeting moieties. Using a flow photo-microreactor, the photoclick reaction could be completed within 60 s, facilitating the rapid generation of clinically relevant tracers for prostate cancer, bacterial infection imaging and in vivo PET imaging,278 thus underscoring the practicality of this approach.
The PQ-ERA reaction, as an emerging photoclick system, is particularly favored due to its excellent biocompatibility, use of visible light, and the inherent fluorescence properties of the products. This has led to its widespread adoption for high spatiotemporal resolution light-activated bioimaging.279 Nevertheless, the light to be used for activation of the click reaction at about 400 nm or even visible light exhibits limited tissue penetration depth, impeding its application for in vivo deep tissue photoclick labelling and imaging. Moreover, the reaction rate of the PQ-ERA photoclick reaction is relatively low, approximately 1 M−1 s−1, necessitating stronger light sources and longer illumination periods, potentially leading to phototoxicity concerns.
To overcome these challenges, Feringa's group recently introduced a series of modified PQ-ERA systems. Employing triplet optimization,272,273,280 triplet–triplet energy transfer (TTET),274 and upconversion nanoparticles (UCNPs),281 they successfully engineered ultrafast PQ-ERA reactions with reaction rates (k2) reaching up to 1970 M−1 s−1. These reactions were induced by long-wavelength visible light/NIR light, extending up to 980 nm. While the biocompatibility of these enhanced systems requires further refinement, these probes exhibit promise for the biological application of the PQ-ERA photoclick reaction.
 | ||
| Fig. 45 Bioorthogonal probes for fluorescence imaging based on the diarylsydnone–alkene photoclick reaction. (A) Proposed mechanism of the photo-DASAC photoclick reaction; (B) fluorescence of different photo-DASAC photoclick products, under excitation at 365 nm; (C) selective photoinduced fluorogenic labeling of proteins; adapted with permission from ref. 249. Copyright 2018, American Chemical Society; (D) the structure of the photo-DASAC product and its precise labelling of living A549 cells, adapted with permission from ref. 283. Copyright 2019, The Royal Society of Chemistry; and (E) the structure of the DASyd-BCN and in situ fluorescence imaging of live A549 cells, adapted with permission from ref. 284. Copyright 2019, The Royal Society of Chemistry. | ||
Following this success, the Yu group283 introduced rapid photoclick reactions between DASyd and strained alkynes, specifically BCN, illustrated in Fig. 45D. The reaction between DASyd and BCN exhibited efficient and selective progress under 405 nm light irradiation. Although competing cycloaddition products emerged from the non-light-activated DASyd-alkyne pathway, their occurrence was noticeably slower compared to the photo-DASAC process (Fig. 45D). The slow thermal-mediated cycloaddition reaction was ascribed to steric repulsion induced by the adjacent ortho-diaryl units on the twisted DASyd, hindering the non-light-activated DASAC pathway. To highlight the biocompatibility of this reaction, selective labeling of proteins was achieved on the surfaces of A549 cells (Fig. 45D). Subsequently, the same group engineered a set of photoactivatable β-diarylsydnone-L-alanines (DASAs) known for their outstanding photoreactivity and heightened fluorescence turn-on when interacting with alkenes through a photo-DASAC reaction in a biocompatible setting (Fig. 45E).284 The resulting fluorescent pyrazoline–alanine exhibited impressive environmental sensing properties, enhancing its probe capabilities. Integrating the DASA residue onto the side chain of linear peptides led to the formation of macrocyclic peptides through the in situ photo-cyclization with the alkene residue. These peptides demonstrated fluorogenic translocation across live cell membranes.
 | ||
| Fig. 46 Bioorthogonal probes developed using other photoclick reactions for fluorescent protein labelling and cell imaging. (A) Proposed mechanism and (B) fluorescence changes, protein labeling and cell imaging of the photo-DAFEx photoclick reaction, adapted with permission from ref. 285. Copyright 2023, The Royal Society of Chemistry. | ||
In summary, photoclick reactions offer high spatial-temporal control and rapid reaction kinetics, making them valuable for precise optical imaging. However, several limitations need to be considered when designing photoclick reaction-based bioorthogonal probes. For instance, photoclick reactions are generally triggered by UV or short-wavelength visible light (violet or blue light, ranging from 380 to 465 nm), which are potentially highly phototoxic in biological studies. The limited penetration ability of short-wavelength light hampers the application of photoclick reaction in deep biological tissue and for in vivo imaging. Although two-photon excitation and UCNP NITEC reactions have been developed for cell imaging, the efficiency of photoclick reaction remains low requiring prolonged exposure to high-intensity laser illumination, diminishing the practical value. In terms of upconversion luminescence-based photoclick reactions, the toxicity of UCNPs presents another concern for biological studies in vivo. Future research in this domain will need to concentrate on developing photoclick systems with high biocompatibility and activity triggered by long-wavelength visible light and NIR light, to enhance their applicability for in situ precision imaging, both in vitro and in vivo.
2.8. Bioorthogonal nanozyme-catalyzed reactions
The incorporation of transition metal catalysts into nanoscaffolds enables the development of nanocatalysts that replicate crucial enzymatic behavior. These catalysts, known as bioorthogonal nanozymes,286 engage in bioorthogonal chemistry not accessible to natural living systems (Scheme 7).287,288 This unique capability allows bioorthogonal nanozymes to function as on-site “factories” for producing bioactive molecules as needed. Co-engineering the transition metal catalysts and the nanometric scaffold is essential to create highly effective bioorthogonal nanozymes. In this section, we will provide an overview of recent advances in the field, with a specific focus on the application of bioorthogonal nanozymes for in situ imaging. | ||
| Scheme 7 Illustration of bioorthogonal nanozyme-catalyzed reactions. | ||
Artificial nanoscale catalysts with high turnover are excellent alternatives for initializing bioorthogonal activation for specific labelling and imaging of cellular molecules. In 2015, Meggers and coworkers289 engineered cytochrome P450 monooxygenase enzyme variants that are capable of efficiently removing protecting groups from propargylic and benzylic ethers for bioorthogonal activation (Fig. 47A). These modified cytochrome P450 monooxygenases deprotected compounds in vitro and within living E. coli cells, releasing fluorescent alcohols that are suitable for in situ bioimaging. Results of this research demonstrated the potential of bioorthogonal enzyme/protecting group pairs for targeted release of imaging agents or catalytic activation of prodrugs at specific sites.
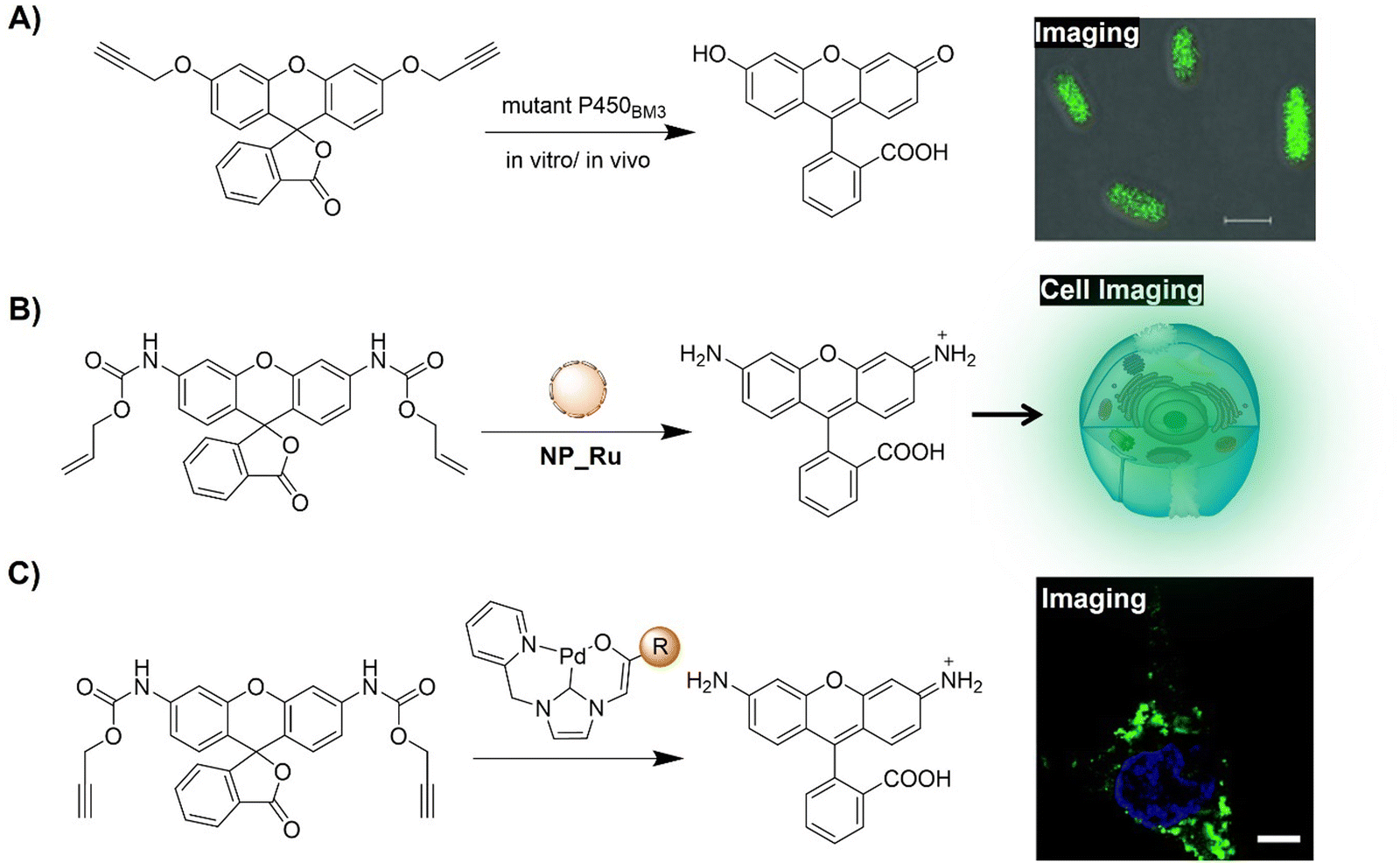 | ||
| Fig. 47 Bioorthogonal nanozyme-catalyzed reaction-based probes for fluorescence cell imaging. (A) Mutant P450BM3 catalyzed decaging of Proc-FITC and bioimaging of E. coli, adapted with permission from ref. 289. Copyright 2015, Wiley-VCH GmbH; (B) schematic illustration of NP_Ru catalyzed decaging of Proc-rhodamine; and (C) Pd catalyzed decaging of Proc-rhodamine and confocal microscopy images of PC-3 cells, adapted with permission from ref. 290. Copyright 2017, The Royal Society of Chemistry. | ||
Bioorthogonal catalysis has been widely adopted in investigating intracellular chemistry, while delivering and controlling synthetic catalytic systems within cells remains challenging. To tackle this issue, Rotello and colleagues291 developed a bioorthogonal nanozyme based on AuNPs, employing transition metal catalysis for in situ imaging and therapy (Fig. 47B). These nanozymes with protein size and surface features offered promise for diverse in vitro and in vivo applications. Controlling their activity via CB[7] host–guest interactions with AuNP ligands’ benzyl headgroups has enabled efficient and reversible catalysis regulation. This strategy could be further used to promote therapeutic applications like prodrug activation to treat non-cancerous chronic diseases. The gated platform ensures controllable catalyst loading, reducing interference and protecting against poisoning, release, distribution, or clearance. The application of this biomimetic and bioorthogonal system was then demonstrated in imaging and prodrug systems through triggered cleavage of allylcarbamates for activating fluorescence and propargyl groups for the release prodrug inside living cells, respectively.
Traditional methods involving Pd catalysts are limited by their poor cellular selectivity, requirement for stoichiometric or excessive Pd complex, and potential cellular toxicity. Therefore, targeted localization of Pd catalysts within cells would advance their applications in biological investigations. Bradley et al.290 expanded Pd-catalyzed chemistry in living systems by introducing a method for delivering and tracking an active organometallic complex within mammalian cells (Fig. 47C). These water-soluble and traceable catalysts, based on Pd(II)–carbene complexes linked to a fluorescently labeled homing peptide, enabled targeted cell delivery. This biocompatible homogeneous Pd catalyst, built on a previously reported carbene Pd ligand coupled to a peptide, exhibited low-cytotoxicity and significant catalytic activity within mammalian cells, allowing for application in bioorthogonal activation of a profluorophore for prostate cancer cell identification and imaging.
Bioorthogonal catalysts are also able to be employed to manipulate the fate of exo-/endo-genous molecules in complicated biological systems. For instance, Broceta and coworkers292 engineered a heterogeneous catalytic system, unlocking previously inaccessible chemical traits of Au-NPs within biological contexts (Fig. 48A). In this catalytic system, bioorthogonal uncaging of various cytotoxic precursors was achieved via unexplored gold reactivity, offering a safe avenue for therapeutic activation through nonbiological routes. Moreover, this solid-supported catalyst could promote the localized release of fluorescein within the brain of a zebrafish, enabling precise in situ imaging. Results of this research enabled chemical modulation of the activity of bioorthogonal reagents in living organisms.
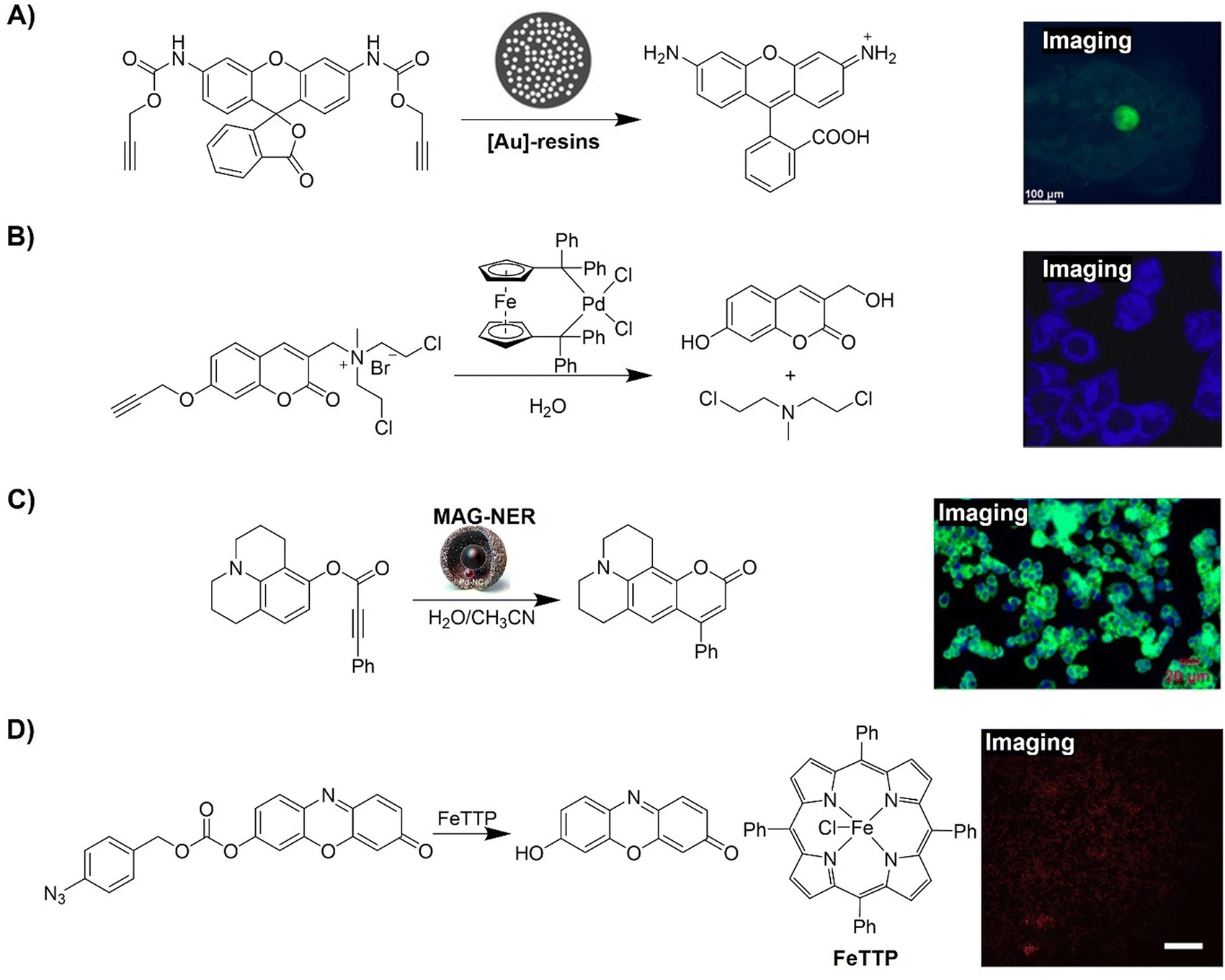 | ||
| Fig. 48 Fluorescent probes for cell and in vivo imaging developed using bioorthogonal catalyzed reactions. (A) [Au]-resin catalyzed decaging of Proc-rhodamine and precise in situ imaging of zebrafish brain, adapted with permission from ref. 292. Copyright 2017, Wiley-VCH GmbH; (B) Pd complex catalyzed activation of the coumarin fluorophore for cell imaging and releasing the anticancer drug for therapy, adapted with permission from ref. 293. Copyright 2017, Elsevier Ltd; (C) MAG-NER catalyzed carbocyclization to activate the fluorescence of nonfluorescent substrates for precise imaging applications, adapted with permission from ref. 294. Copyright 2020, American Chemical Society; and (D) conversion of the profluorophore pro-Res through the FeTTP catalytic reduction of aryl azides, adapted with permission from ref. 295. Copyright 2020, Elsevier Inc. | ||
Manipulating biological processes by bioorthogonal bond-cleavage reactions facilitates in vivo therapeutic applications. For example, Wu and colleagues293 devised a bioorthogonally activated prodrug for precise bioimaging and therapy, comprising a Pd-mediated cleavable propargyl, a coumarin fluorophore, and a potent anticancer agent (Fig. 48B). In vitro studies revealed that the Pd complex triggered propargyl cleavage, initiating a cascade of reactions to activate the coumarin fluorophore for imaging and releasing the anticancer drug for therapy. These components were encapsulated in phospholipid liposomes, forming a two-component bioorthogonal nanosystem. Internalized by HeLa cells, the nanosystem exhibited strong intracellular fluorescence and effectively inhibited tumor growth in a mouse model. The success of this research validated the potential of appropriately formulated bioorthogonal systems for in vivo applications, offering a new approach to develop imaging-guided prodrug delivery systems.
Remote control of the catalytic activity of nanoreactors by magnetic fields is promising for advancing bioorthogonal chemistry in biological studies. In research led by Lee,294 MAG-NERs, hollow-silica-based nanoreactors that are responsive to magnetic induction, were engineered for bioorthogonal organic synthesis in live cells (Fig. 48C). Catalytic metal (Pd) growth within the silica's confines, guided by magnetic fields, accumulated at the Fe3O4–core interface. Pd nanoclusters at this interface catalyzed carbocyclization to activate the fluorescence of nonfluorescent substrates under physiological conditions. MAG-NERs enabled bioorthogonal catalysis at ambient temperature in vitro under remote operation. Customization with diverse catalytic nanoclusters enhanced the activity with a biofriendly, remotely controllable magnetic field, advancing biomedical and biological precision imaging applications.
In addition to the remote magnetic field, integrating bioorthogonal catalysts into thermal responsive nanoparticle platforms yields bioorthogonal “nanozymes” that are controllable via endogenous or exogenous thermal stimuli. In research led by Rotello,295 thermoresponsive nanozymes were prepared by confining porphyrin (FeTTP) supramolecular assemblies within gold nanoparticle monolayers (Fig. 48D). Retaining stimuli-responsive behavior in biological settings, these assemblies enabled localized therapy and in situ imaging. Upon heating, the confined assemblies within the scaffold reversibly dissociated, restoring catalytic activity. This gating mechanism, driven by confined assembly dynamics, resulted in nanozymes with thermoresponsive behavior in complex biological systems. This thermal responsive and reversibly activated system was then used for activating antibiotic based prodrugs for bacterial biofilm elimination.
In another example of bioorthogonal activation of prodrug therapeutics, Rotello and coworkers296 demonstrated selective intracellular activation of molecules using endogenously activated bioorthogonal nanozymes (Fig. 49A). In this research, AuNP ligand structures were able to facilitate protein corona formation, which in turn controlled the catalytic activity of the nanozyme. A hard “irreversible” corona deactivated nanozymes via aggregation and steric hindrance, while a soft “reversible” corona partially reduced the activity of nanozymes. Proteolytic degradation of the corona by endogenous proteases in endosomes and lysosomes restored catalytic activity. Engineering ligand monolayers on nanoparticles yielded a selective intracellular activation system (without tetra(ethylene glycol) (TEG) and an always-on system (with TEG).
 | ||
| Fig. 49 Fluorescent probes for cell imaging developed using bioorthogonal catalyzed reactions. (A) AuNP nanozyme-catalyzed decaging of Proc-rhodamine and precise in situ HeLa cell imaging, adapted with permission from ref. 296. Copyright 2020, American Chemical Society; (B) schematic illustration of the Biot-Ru catalyzed activation of the coumarin fluorophore for precise in situ cell imaging; and (C) Mn(I) catalyzed C–H labeling of complex peptides with bodipy probes for immune cell membrane imaging, adapted from ref. 297, licensed under CC BY 4.0. | ||
Bioorthogonal activation of artificial metalloenzymes has also been adopted for recreating the reaction networks of signal processing between cells and organisms in protocells. For example, Walther and colleagues298 engineered all-DNA protocells hosting an artificial metalloenzyme catalyzing olefin metathesis, triggering downstream morphogenetic responses of varying complexity (Fig. 49B). The enzyme uncaged a pro-fluorescence signal, generating a self-reporting metabolite, weakening DNA duplex interactions and fostering growth, intraparticular adaptation, and interparticle fusion. These processes that mimic cellular adaptation and adhesion offered insight into life-like behavior via abiotic bioorthogonal chemical and mechanical transformations in synthetic protocells.
Late stage bioorthogonal diversification of amino acids and peptides has been employed for drug discovery and molecular bioimaging. In a research led by Ackermann,297 a modular linchpin strategy was engineered for stereo-divergent manganese(I)-catalyzed C–H labeling of complex peptides with bodipy probes (Fig. 49C). Validating manganese(I) catalysis, sensitive molecular rotors for monitoring immune cell membrane changes were obtained. A cholesterol-responsive probe emitting bright fluorescence was also developed, facilitating rapid screening for CD8+ T cell modulators. This divergent C–H activation strategy promises efficient development of activatable fluorophores for real-time, physiological imaging of cell function.
Bioorthogonal transformation of imaging and therapeutic substrates via transition metal catalysts (TMCs) offers versatile biomedical applications. In research involving bioorthogonal transformations in complex biological environments, Sasmal and coworkers299 introduced an organoiridium complex to uncage allyloxycarbonyl-protected amines under biologically relevant conditions and within living cells (Fig. 50A). This uncaging chemistry demonstrated potential applications in diagnosis and treatment by controlled activation of the profluorophore (Rho-alloc) and the prodrug (caged Dox) in HeLa cells. The fluorescence intensity increased up to 56-fold over 8 h, offering a valuable tool for diverse biological and therapeutic applications. In another work, Rotello et al.300 employed a polymeric scaffold to encapsulate transition metal catalysts (TMCs), yielding bioorthogonal “polyzymes” for activating intracellular anticancer therapeutics (Fig. 50B). In this “polyzyme” system, the hydrophobic interior of these polymers protected TMCs from deactivation in biological environments, without the clearance issues seen with inorganic nanoparticle analogues. Polyzymes301 exhibited efficient intracellular activation of pro-dyes and prodrugs in vitro, indicative of their potential for disease diagnosis and treatment through on-site generation of imaging and therapeutic agents (Fig. 50C).
 | ||
| Fig. 50 Fluorescent probes for cell imaging developed using metal-based catalysts. (A) Ir complex catalyzed decaging of a profluorophore (Rho-alloc) and a prodrug (caged Dox) in HeLa cells, adapted with permission from ref. 299. Copyright 2021, American Chemical Society; (B) the Ru-polyzyme-catalyzed activation of Proc-rhodamine for precise in situ cell imaging, adapted with permission from ref. 300. Copyright 2020, Wiley-VCH GmbH; (C) the Pd-polyzyme-catalyzed activation of Proc-rhodamine for precise in situ HeLa cell imaging, adapted with permission from ref. 301. Copyright 2022, American Chemical Society. | ||
Precise localization of bioorthogonal catalysis is crucial to enhance therapeutic efficacy and minimize off-target effects. Towards this end, Rotello et al.302 integrated biomimetic nanozymes with red blood cells (RBCs) as nanozymes for the selective targeting of bacterial infections (Fig. 51A). Utilizing non-immunogenic RBC-hitchhiked nanozymes, this approach selectively eliminated pathogenic bacterial infections. Toxins from bacteria induced hemolysis, leading to nanozyme accumulation and activation at sites of infection. These accumulated nanozymes subsequently released antibiotics, acting as in situ “dye and drug factories” for imaging and eradicating preformed biofilms without harming non-virulent bacteria. This approach exhibited potential for the treatment of recurrent bacterial infections, including chronic wounds and medical device-associated infections, offering targeted therapy without microbe-specific probes. By leveraging cellular interactions, this strategy encouraged diverse biomedical applications, providing a modular platform for imaging and therapeutics across various diseases.
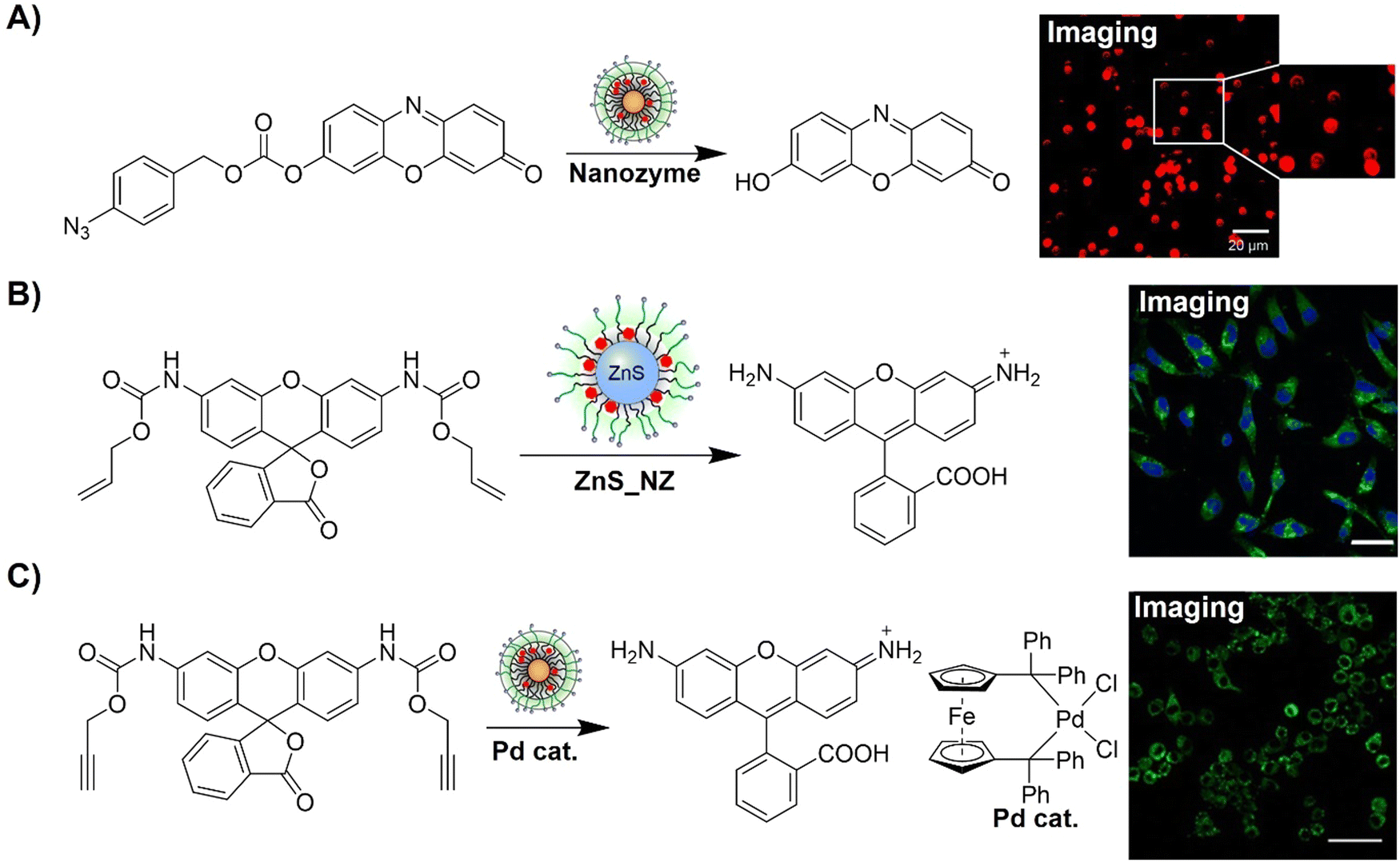 | ||
| Fig. 51 Fluorescent probes for cell imaging developed using nanozyme-catalyzed activation. (A) Conversion of the profluorophore pro-Res through the nanozyme catalytic reduction of aryl azides for cell imaging, adapted with permission from ref. 302. Copyright 2021, The Royal Society of Chemistry; (B) the ZnS_NZ catalyzed activation of Proc-rhodamine for precise in situ cell imaging, adapted with permission from ref. 303. Copyright 2022, American Chemical Society; and (C) the Pd-nanozyme catalyzed activation of Proc-rhodamine for precise in situ cell imaging, adapted with permission from ref. 304, licensed under CC-BY-NC-ND 4.0. | ||
Building on this research, Rotello et al.303 then devised a strategy utilizing surface-functionalized ZnS nanoparticles to achieve both enhanced bioorthogonal catalysis and biodegradability (Fig. 51B). Ligand-mediated acceleration of catalysis facilitated the uncaging of allyl-caged molecules by ruthenium catalysts, with released thiols acting as nucleophiles to accelerate the rate-determining step. Unlike enzyme-mimicking nanozymes such as FeS, ZnS-supported nanozymes underwent chemical transformations inaccessible to natural enzymes. This study highlighted the active involvement of nanoscaffolds as cofactors in bioorthogonal catalysis, surpassing non-degradable gold nanoparticle nanozymes for fluorophore and drug release in vitro. ZnS nanozymes provided a biocompatible platform for in situ production of imaging and therapeutic agents, facilitating clinical translation of bioorthogonal catalysis and enabling localized therapies with minimal side effects and avoidance of particle accumulation.
Macrophage migration to tumor sites via chemoattractant gradients offers an active targeting strategy for cancer therapy. Taking this fact into consideration, Rotello et al.304 introduced bioorthogonal “nanozymes” with TMCs to transform profluorophores and prodrugs into in situ imaging agents and chemotherapeutics within macrophages, promoting macrophage-based drug delivery (Fig. 51C). These nanozymes, embedded in self-assembled monolayer-coated gold nanoparticles, solubilize and stabilize TMCs. Intracellularly localized nanozyme-loaded macrophages retained activity after prolonged incubation, maintaining migratory ability toward tumor chemoattractants and efficiently killing cancer cells. This macrophage-based delivery approach offered tunable dosages and delivery rates, minimizing off-target toxicity while activating prodrugs at tumor sites.
Understanding cellular functions requires detailed analysis of subcellular proteomes in precise spatial and temporal contexts. Chen et al.305 introduced CAT-Prox, a bioorthogonal and photocatalytic decaging-enabled proximity labelling strategy, for spatiotemporally resolved mitochondrial proteome profiling in living cells (Fig. 52A). In this bioorthogonal and photocatalytic system, Ir(ppy)2bpy served as a bioorthogonal and mitochondria-targeting catalyst to control the release of azidobenzyl-caged quinone methide for protein labelling. CAT-Prox exhibited mitochondria specificity in live HeLa and RAW264.7 cells, enriching approximately 300 proteins with 70% specificity. For applications using lipopolysaccharide (LPS)-stimulated RAW264.7 macrophages, CAT-Prox dynamically dissected mitochondrial proteomes. By integrating photocatalytic decaging with proximity-based protein labelling, CAT-Prox exhibited excellent performance as a catalytic and nongenetic alternative for live-cell proteome analysis.
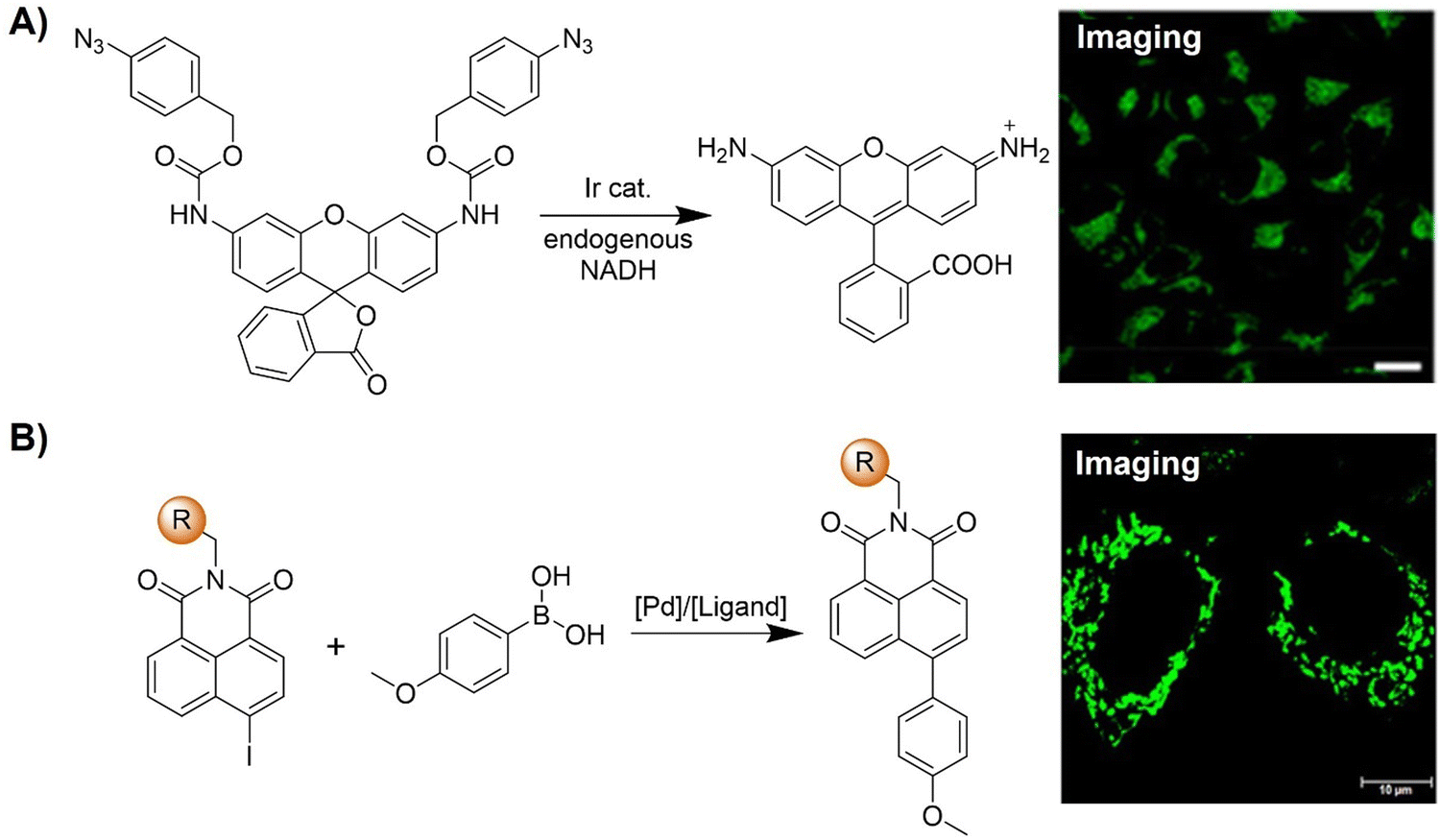 | ||
| Fig. 52 Fluorescent probes developed for cell imaging using metal complex-based bioorthogonal activation. (A) Evaluation of Ir catalytic activity by PAB-rhodamine decaging in mitochondria in living cells through the CAT-Prox strategy, adapted with permission from ref. 305. Copyright 2021, American Chemical Society; (B) the Pd catalyzed fluorophore activation via Suzuki–Miyaura reaction for precise in situ cell imaging, adapted with permission from ref. 306. Copyright 2022, Elsevier B.V. | ||
Tian and colleagues306 developed a versatile method for the in situ assembly of ICT-based light-up fluorophores using bioorthogonal Suzuki–Miyaura cross-coupling (Fig. 52B). This approach was applied to five fluorophore scaffolds, including naphthalimide, coumarin, naphthalene sulfonate, nitrobenzoxadiazole, and acetonaphthone, for the development of bioorthogonally activated probes for fluorescence imaging. The iodine group served as the bioorthogonal handle, quenching fluorescence by blocking ICT when strategically positioned. Upon Suzuki–Miyaura reaction, multicolor fluorophores were generated in situ, exhibiting significant fluorescence enhancement (up to 5277-fold) and high ΦF (up to 0.82). Structure–property relationships of 1,8-naphthalimide probes were systematically explored, enabling fine-tuning of the photophysical properties. This bioorthogonal Suzuki–Miyaura cross-coupling was then applied for peptide modification, fluorogenic protein labeling, and live-cell mitochondria imaging with a high signal-to-noise ratio, representing a valuable expansion of the chemical biology toolbox for diverse biomedical applications.
In summary, bioorthogonal nanozymes represent a groundbreaking approach for biological investigations, enabling the localized synthesis of fluorescent probes and drugs at disease sites. This technique is particularly beneficial for precise in situ imaging and targeted drug delivery under challenging conditions such as tumors, bacterial infections, and during inflammation, where conventional treatments pose risks including off-target effects. Furthermore, nanozymes offer extensive therapeutic potential due to their adaptability and versatile design possibilities. Manipulating various components like nanoparticles, ligands, and TMCs enhances adaptability and enables fine-tuning for future clinical applications.
Ensuring the long-term stability and efficacy of nanozymes is paramount, enabling high drug yields with minimal doses and adaptability to biological changes. Addressing potential cyto- and genotoxicity concerns arising from nanoscaffolds and TMCs is crucial for clinical translation, emphasizing the development of biodegradable scaffolds and non-toxic metal catalysts. Advances in organocatalysis may provide alternatives as well as low toxicity bioorthogonal TMCs based on metals such as silver and manganese. Enhancing the controllability of nanozymes can mitigate safety concerns, although reliable on/off switchable nanozymes have yet to be validated in vivo. Comprehensive studies on the distribution and metabolic pathways of nanozyme components in complex physiological environments are essential for clinical translation. Research efforts should concentrate on comprehensively understanding the complete lifecycle of nanozymes in vivo to effectively address safety concerns.
In brief, bioorthogonal nanozymes facilitate the creation of in situ probes and drug carriers, heralding a new era of enzyme-mimicking therapeutics. Their modular design holds promise for significant advancements in disease detection, deep tissue imaging, and precision therapy for biomedical applications.
3. Conclusion and outlook
Over the past two decades, we have witnessed a remarkable surge in the advancement of bioorthogonal chemistry, signifying a dynamic evolution that challenges longstanding assumptions in both biology and medicine. Particularly notable is the profound impact on fluorescence labeling, which has achieved a significant milestone through the integration of bioorthogonal ligation reactions with sophisticated molecular biological techniques. This symbiotic relationship allows for selective and biocompatible transformations, elevating our ability to scrutinize biological processes with unparalleled precision.This review offers a comprehensive examination of diverse types of bioorthogonally activated in situ fluorescent labels. These probes, endowed with both bioorthogonal and fluorogenic properties, exhibit exceptional efficacy and precision in tagging specific biological targets with complementary chemical reporters. Despite the rapid expansion of the repertoire of bioorthogonal reactions, designing optimal bioorthogonal chemical probes tailored for specific biological systems remains a formidable challenge.
Primarily, there is an urgent need to devise strategies enabling diverse tagging, crucial for facilitating multicolor imaging. While certain orthogonal reactions, such as SPAAC-CuAAC, and specific combinations like the photoclick reaction with SPAAC, permit dual labeling and have achieved intricate multicolor imaging, further advancements in additional orthogonal reactions are still required. Additionally, while current bioorthogonal reactions enable selective labeling of intracellular structures, their predominant application typically involves tagging extracellular targets such as receptors or glycans. To extend the utility of bioorthogonal tagging to intracellular and in vivo scenarios, there is a pressing need for the development of non-toxic, membrane-permeable labels applicable within bioorthogonal contexts. Furthermore, in addition to the continuous optimization of the performance of the existing systems, it is noteworthy that there are ample opportunities for developing novel types of bioorthogonal click reactions based on innovative reactant structures that allow for wash-free in situ precise imaging applications. We are confident that there are more unexplored avenues that need investigation. Through discovering new families of clickable molecules that are capable of meeting the increasingly diverse needs of in vivo applications, it is important to promote current bioorthogonal click chemistry for further advancements in this field.
In conclusion, bioorthogonal chemistry has revolutionized biological labeling and imaging, significantly broadening our horizons within complex physiological environments. The ongoing development of both existing and emerging bioorthogonally activated fluorescent probes holds immense promise for deepening our understanding of living systems. This heightened understanding facilitates improved comprehension of biomolecular processes and aids in the design of more effective drugs, particularly for targeted treatments such as those for tumors. With the relentless pace of innovations and developments, we firmly believe that the future of bioorthogonally activated probes for precise biolabeling and bioimaging is exceptionally promising.
Data availability
Data sharing is not applicable to this article as no new data were created or analyzed in this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the National Health and Medical Research Council (APP1175808). Y. F. is thankful for the generous financial support from the Natural Science Foundation of Jiangsu Province (Grant no. BK20240660). T. D. J. wishes to thank the University of Bath and the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University (2020ZD01) for support. Facilities and assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy and Microanalysis (CMM) and Queensland Node of the Australian National Fabrication Facility (ANFF-Q), the University of Queensland are also acknowledged.References
- M. L. James and S. S. Gambhir, Physiol. Rev., 2012, 92, 897–965 CrossRef CAS.
- R. Weissleder, Science, 2006, 312, 1168–1171 CrossRef CAS.
- D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Chem. Soc. Rev., 2017, 46, 7105–7123 RSC.
- Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC.
- H. Abramczyk and B. Brozek-Pluska, Chem. Rev., 2013, 113, 5766–5781 CrossRef CAS PubMed.
- Q. Yang, Y. Wu, J. Chen, M. Lu, X. Wang, Z. Zhang, H. Xiong, J. Choo and L. Chen, Coord. Chem. Rev., 2024, 507, 215768 CrossRef CAS.
- S. Lee, H. Dang, J.-I. Moon, K. Kim, Y. Joung, S. Park, Q. Yu, J. Chen, M. Lu, L. Chen, S.-W. Joo and J. Choo, Chem. Soc. Rev., 2024, 53, 5394–5427 RSC.
- R. B. Buxton, Introduction to Functional Magnetic Resonance Imaging, Cambridge University Press, 2009 Search PubMed.
- Q. Meng, M. Wu, Z. Shang, Z. Zhang and R. Zhang, Coord. Chem. Rev., 2022, 457, 214398 CrossRef CAS.
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed.
- S. Mallidi, G. P. Luke and S. Emelianov, Trends Biotechnol., 2011, 29, 213–221 CrossRef CAS PubMed.
- Y. Seo, C. Mari and B. H. Hasegawa, Semin. Nucl. Med., 2008, 38, 177–198 CrossRef.
- A. C. Sedgwick, L. Wu, H.-H. Han, S. D. Bull, X.-P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian and J. Yoon, Chem. Soc. Rev., 2018, 47, 8842–8880 RSC.
- T. Nakajima, M. Mitsunaga, N. H. Bander, W. D. Heston, P. L. Choyke and H. Kobayashi, Bioconjugate Chem., 2011, 22, 1700–1705 CrossRef CAS PubMed.
- M. Y. Berezin and S. Achilefu, Chem. Rev., 2010, 110, 2641–2684 CrossRef CAS PubMed.
- A. P. Demchenko, Methods Appl. Fluoresc., 2020, 8, 022001 CrossRef CAS PubMed.
- Q. Zheng, M. F. Juette, S. Jockusch, M. R. Wasserman, Z. Zhou, R. B. Altman and S. C. Blanchard, Chem. Soc. Rev., 2014, 43, 1044–1056 RSC.
- M. Monici, Biotechnol. Annu. Rev., 2005, 11, 227–256 CAS.
- P. F. Heelis, Chem. Soc. Rev., 1982, 11, 15–39 RSC.
- S. A. Schnell, W. A. Staines and M. W. Wessendorf, J. Histochem. Cytochem., 1999, 47, 719–730 CrossRef CAS PubMed.
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed.
- P. Gao, W. Pan, N. Li and B. Tang, Chem. Sci., 2019, 10, 6035–6071 RSC.
- K. K. W. Lo, Acc. Chem. Res., 2020, 53, 32–44 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- M. Giel and Y. Hong, Aggregate, 2023, 4, e336 CrossRef CAS.
- A. L. Antaris, H. Chen, K. Cheng, Y. Sun, G. Hong, C. Qu, S. Diao, Z. Deng, X. Hu, B. Zhang, X. Zhang, O. K. Yaghi, Z. R. Alamparambil, X. Hong, Z. Cheng and H. Dai, Nat. Mater., 2016, 15, 235–242 CrossRef CAS PubMed.
- S. Zhu, R. Tian, A. L. Antaris, X. Chen and H. Dai, Adv. Mater., 2019, 31, 1900321 CrossRef PubMed.
- S. L. Scinto, D. A. Bilodeau, R. Hincapie, W. Lee, S. S. Nguyen, M. Xu, C. W. am Ende, M. G. Finn, K. Lang, Q. Lin, J. P. Pezacki, J. A. Prescher, M. S. Robillard and J. M. Fox, Nat. Rev. Methods Primers, 2021, 1, 30 CrossRef CAS PubMed.
- R. E. Bird, S. A. Lemmel, X. Yu and Q. A. Zhou, Bioconjugate Chem., 2021, 32, 2457–2479 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- H. C. Hang, C. Yu, D. L. Kato and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14846–14851 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- N. K. Devaraj, S. Hilderbrand, R. Upadhyay, R. Mazitschek and R. Weissleder, Angew. Chem., Int. Ed., 2010, 49, 2869–2872 CrossRef CAS PubMed.
- K. S. Yang, G. Budin, T. Reiner, C. Vinegoni and R. Weissleder, Angew. Chem., Int. Ed., 2012, 51, 6598–6603 CrossRef CAS PubMed.
- H. Ren, F. Xiao, K. Zhan, Y. Kim, H. Xie, Z. Xia and J. Rao, Angew. Chem., Int. Ed., 2009, 48, 9658–9662 CrossRef CAS PubMed.
- Y. Zhu, X. Zhang, Q. You and Z. Jiang, Bioorg. Med. Chem., 2022, 68, 116881 CrossRef CAS PubMed.
- V. Rigolot, C. Biot and C. Lion, Angew. Chem., Int. Ed., 2021, 60, 23084–23105 CrossRef CAS PubMed.
- P. Shieh and C. R. Bertozzi, Org. Biomol. Chem., 2014, 12, 9307–9320 RSC.
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS PubMed.
- H. Zhao, Y. He, Y. Lo, H. Song and J. Lu, TrAC, Trends Anal. Chem., 2023, 169, 117388 CrossRef CAS.
- W. Huang and S. T. Laughlin, Cell Chem. Biol., 2024, 31, 409–427 CrossRef CAS PubMed.
- Kenry and B. Liu, Trends Chem., 2019, 1, 763–778 CrossRef CAS.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS.
- M. Köhn and R. Breinbauer, Angew. Chem., Int. Ed., 2004, 43, 3106–3116 CrossRef.
- C. Bednarek, I. Wehl, N. Jung, U. Schepers and S. Bräse, Chem. Rev., 2020, 120, 4301–4354 CrossRef CAS PubMed.
- H. Staudinger and J. Meyer, Helv. Chim. Acta, 1919, 2, 635–646 CrossRef CAS.
- Z. Liang, H. Pang, G. Zeng and T. Chen, Anal. Chem., 2022, 94, 8293–8301 CrossRef CAS.
- W. Lv, S. Chi, W. Feng, T. Liang, D. Song and Z. Liu, Chem. Commun., 2019, 55, 7037–7040 RSC.
- C. X. Zhang, M. H. Xiang, X. J. Liu, F. Wang, R. Q. Yu and J. H. Jiang, Talanta, 2019, 193, 152–160 CrossRef CAS.
- A. S. Cohen, E. A. Dubikovskaya, J. S. Rush and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 8563–8565 CrossRef CAS.
- G. A. Lemieux, C. L. De Graffenried and C. R. Bertozzi, J. Am. Chem. Soc., 2003, 125, 4708–4709 CrossRef CAS PubMed.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 633–645 CrossRef.
- R. S. Silva and D. E. Nicodem, J. Photochem. Photobiol., A, 2004, 162, 231–238 CrossRef CAS.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- M. Meldal, Macromol. Rapid Commun., 2008, 29, 1016–1051 CrossRef CAS.
- X. Jiang, X. Hao, L. Jing, G. Wu, D. Kang, X. Liu and P. Zhan, Expert Opin. Drug Discovery, 2019, 14, 779–789 CrossRef CAS PubMed.
- L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- L. Rong, L. H. Liu, S. Chen, H. Cheng, C. S. Chen, Z. Y. Li, S. Y. Qin and X. Z. Zhang, Chem. Commun., 2014, 50, 667–669 RSC.
- G. Liu, G. Shi, H. Sheng, Y. Jiang, H. Liang and S. Liu, Angew. Chem., Int. Ed., 2017, 56, 8686–8691 CrossRef CAS PubMed.
- O. Demeter, E. A. Fodor, M. Kállay, G. Mez, K. Németh, P. T. Szabó and P. Kele, Chem. – Eur. J., 2016, 22, 6382–6388 CrossRef CAS PubMed.
- Z. Du, D. Yu, X. Du, P. Scott, J. Ren and X. Qu, Chem. Sci., 2019, 10, 10343–10350 RSC.
- Y. You, F. Cao, Y. Zhao, Q. Deng, Y. Sang, Y. Li, K. Dong, J. Ren and X. Qu, ACS Nano, 2020, 14, 4178–4187 CrossRef CAS.
- Y. You, H. Liu, J. Zhu, Y. Wang, F. Pu, J. Ren and X. Qu, Chem. Sci., 2022, 13, 7829–7836 RSC.
- F. Zeng, Y. Pan, X. Luan, Y. Gao, J. Yang, Y. Wang and Y. Song, Sens. Actuators, B, 2021, 345, 130382 CrossRef CAS.
- H. You, M. Wang, S. Wang, J. Xu, S. Hu, T. Li, Z. Yu, D. Tang and N. Gan, Anal. Chem., 2023, 95, 11211–11218 CrossRef CAS PubMed.
- K. L. Wang, J. X. Zhang, D. Min, J. L. Lv, D. F. Liu and H. Q. Yu, Environ. Sci. Technol., 2022, 56, 15685–15694 CrossRef CAS PubMed.
- Y. Zhou, Y. W. Yao, Q. Qi, Y. Fang, J. Y. Li and C. Yao, Chem. Commun., 2013, 49, 5924–5926 RSC.
- P. Shieh, M. S. Siegrist, A. J. Cullen and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 5456–5461 CrossRef CAS PubMed.
- P. Shieh, V. T. Dien, B. J. Beahm, J. M. Castellano, T. Wyss-Coray and C. R. Bertozzi, J. Am. Chem. Soc., 2015, 137, 7145–7151 CrossRef CAS PubMed.
- X. Zhu, P. Shieh, M. Su, C. R. Bertozzi and W. Zhang, Chem. Commun., 2016, 52, 11239–11242 RSC.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- S. K. Leong, Y. Chen, J. Hsiao, C. Tsai and J. Shie, ChemBioChem, 2023, 24, e202300522 CrossRef CAS PubMed.
- C. Yan, J. Dai, Y. Yao, W. Fu, H. Tian, W.-H. Zhu and Z. Guo, Nat. Protoc., 2023, 18, 1316–1336 CrossRef CAS.
- Y. Yuan, S. Xu, X. Cheng, X. Cai and B. Liu, Angew. Chem., Int. Ed., 2016, 55, 6457–6461 CrossRef CAS PubMed.
- F. Hu, Y. Yuan, W. Wu, D. Mao and B. Liu, Anal. Chem., 2018, 90, 6718–6724 CrossRef CAS PubMed.
- F. Hu, D. Mao, Kenry, X. Cai, W. Wu, D. Kong and B. Liu, Angew. Chem., Int. Ed., 2018, 57, 10182–10186 CrossRef CAS.
- M. Wu, G. Qi, X. Liu, Y. Duan, J. Liu and B. Liu, Chem. Mater., 2020, 32, 858–865 CrossRef CAS.
- X. Liu, G. Qi, M. Wu, Y. Pan and B. Liu, Chem. Mater., 2021, 33, 9213–9220 CrossRef CAS.
- H. Fu, Y. Li, L. Sun, P. He and X. Duan, Anal. Chem., 2015, 87, 11332–11336 CrossRef CAS PubMed.
- F. Friscourt, C. J. Fahrni and G. Boons, Chem. – Eur. J., 2015, 21, 13996–14001 CrossRef CAS PubMed.
- Á. Eördögh, J. Steinmeyer, K. Peewasan, U. Schepers, H. A. Wagenknecht and P. Kele, Bioconjugate Chem., 2016, 27, 457–464 CrossRef.
- Y. Dou, Y. Wang, Y. Duan, B. Liu, Q. Hu, W. Shen, H. Sun and Q. Zhu, Chem. – Eur. J., 2020, 26, 4576–4582 CrossRef CAS.
- A. Kormos, A. Egyed, J. M. Olvany, Á. Szatmári, A. Biró, Z. Csorba, P. Kele and K. Németh, Chemosensors, 2022, 10, 37 CrossRef CAS.
- B. C. Boren, S. Narayan, L. K. Rasmussen, L. Zhang, H. Zhao, Z. Lin, G. Jia and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 8923–8930 CrossRef CAS PubMed.
- T. Horneff, S. Chuprakov, N. Chernyak, V. Gevorgyan and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 14972–14974 CrossRef CAS PubMed.
- C. D. McNitt, H. Cheng, S. Ullrich, V. V. Popik and M. Bjerknes, J. Am. Chem. Soc., 2017, 139, 14029–14032 CrossRef CAS PubMed.
- N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952–959 CrossRef CAS PubMed.
- X. Ai, L. Lyu, Y. Zhang, Y. Tang, J. Mu, F. Liu, Y. Zhou, Z. Zuo, G. Liu and B. Xing, Angew. Chem., Int. Ed., 2017, 56, 3031–3035 CrossRef CAS PubMed.
- H. L. Chan, L. Lyu, J. Aw, W. Zhang, J. Li, H. H. Yang, H. Hayashi, S. Chiba and B. Xing, ACS Chem. Biol., 2018, 13, 1890–1896 CrossRef CAS PubMed.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS.
- S. T. Laughlin, J. M. Baskin, S. L. Amacher and C. R. Bertozzi, Science, 2008, 320, 664–667 CrossRef CAS PubMed.
- J. Shum, L. C. Lee, M. W.-L. Chiang, Y. Lam and K. K. Lo, Angew. Chem., Int. Ed., 2023, 62, e202303931 CrossRef CAS PubMed.
- X. Jiang, M. Li, Y. Wang, C. Wang, Y. Wang, T. Shen, L. Shen, X. Liu, Y. Wang and X. Li, Nat. Commun., 2023, 14, 1401 CrossRef CAS.
- A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- N. K. Devaraj, R. Upadhyay, J. B. Haun, S. A. Hilderbrand and R. Weissleder, Angew. Chem., Int. Ed., 2009, 48, 7013–7016 CrossRef CAS.
- E. J. L. Stéen, P. E. Edem, K. Nørregaard, J. T. Jørgensen, V. Shalgunov, A. Kjaer and M. M. Herth, Biomaterials, 2018, 179, 209–245 CrossRef PubMed.
- X. Fan, Y. Ge, F. Lin, Y. Yang, G. Zhang, W. S. C. Ngai, Z. Lin, S. Zheng, J. Wang, J. Zhao, J. Li and P. R. Chen, Angew. Chem., Int. Ed., 2016, 55, 14046–14050 CrossRef CAS PubMed.
- Y. Fang, A. S. Hillman and J. M. Fox, Top. Curr. Chem., 2024, 382, 15 CrossRef CAS PubMed.
- A. Darko, S. Wallace, O. Dmitrenko, M. M. Machovina, R. A. Mehl, J. W. Chin and J. M. Fox, Chem. Sci., 2014, 5, 3770–3776 RSC.
- S. J. Siegl, R. Dzijak, A. Vázquez, R. Pohl and M. Vrabel, Chem. Sci., 2017, 8, 3593–3598 RSC.
- A. Wieczorek, T. Buckup and R. Wombacher, Org. Biomol. Chem., 2014, 12, 4177–4185 RSC.
- J. C. T. Carlson, L. G. Meimetis, S. A. Hilderbrand and R. Weissleder, Angew. Chem., Int. Ed., 2013, 52, 6917–6920 CrossRef CAS.
- L. G. Meimetis, J. C. T. Carlson, R. J. Giedt, R. H. Kohler and R. Weissleder, Angew. Chem., Int. Ed., 2014, 53, 7531–7534 CrossRef CAS.
- H. Wu, J. Yang, J. Šečkutė and N. K. Devaraj, Angew. Chem., Int. Ed., 2014, 53, 5805–5809 CrossRef CAS PubMed.
- H. Wu, B. T. Cisneros, C. M. Cole and N. K. Devaraj, J. Am. Chem. Soc., 2014, 136, 17942–17945 CrossRef CAS PubMed.
- L. G. Meimetis, E. Boros, J. C. Carlson, C. Ran, P. Caravan and R. Weissleder, Bioconjugate Chem., 2016, 27, 257–263 CrossRef CAS PubMed.
- A. Wieczorek, P. Werther, J. Euchner and R. Wombacher, Chem. Sci., 2017, 8, 1506–1510 RSC.
- P. Werther, J. S. Möhler and R. Wombacher, Chem. – Eur. J., 2017, 23, 18216–18224 CrossRef CAS PubMed.
- F. Hild, P. Werther, K. Yserentant, R. Wombacher and D. P. Herten, Biophys. Rep., 2022, 2, 100084 CAS.
- R. Zhang and J. Yuan, Acc. Chem. Res., 2020, 53, 1316–1329 CrossRef CAS PubMed.
- M. Wu, Z. Zhang, J. Yong, P. M. Schenk, D. Tian, Z. P. Xu and R. Zhang, Top. Curr. Chem., 2022, 380, 29 CrossRef CAS PubMed.
- T. S. M. Tang, H. W. Liu and K. K. W. Lo, Chem. Commun., 2017, 53, 3299–3302 RSC.
- A. Vázquez, R. Dzijak, M. Dračínský, R. Rampmaier, S. J. Siegl and M. Vrabel, Angew. Chem., Int. Ed., 2017, 56, 1334–1337 CrossRef PubMed.
- J. Wang, L. Zhou, H. Sun, F. Lv, L. Liu, Y. Ma and S. Wang, Chem. Mater., 2018, 30, 5544–5549 CrossRef CAS.
- S. J. Siegl, J. Galeta, R. Dzijak, M. Dračínský and M. Vrabel, ChemPlusChem, 2019, 84, 493–497 CrossRef CAS PubMed.
- Y. Lee, W. Cho, J. Sung, E. Kim and S. B. Park, J. Am. Chem. Soc., 2018, 140, 974–983 CrossRef CAS PubMed.
- G. Linden, L. Zhang, F. Pieck, U. Linne, D. Kosenkov, R. Tonner and O. Vázquez, Angew. Chem., Int. Ed., 2019, 58, 12868–12873 CrossRef CAS PubMed.
- X. Guo, R. C. H. Wong, Y. Zhou, D. K. P. Ng and P. C. Lo, Chem. Commun., 2019, 55, 13518–13521 RSC.
- X. Xie, B. Li, J. Wang, C. Zhan, Y. Huang, F. Zeng and S. Wu, ACS Mater. Lett., 2019, 1, 549–557 CrossRef CAS.
- D. Liang, K. Wu, R. Tei, T. W. Bumpus, J. Ye and J. M. Baskin, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 15453–15462 CrossRef CAS PubMed.
- M. Bojtár, K. Németh, F. Domahidy, G. Knorr, A. Verkman, M. Kállay and P. Kele, J. Am. Chem. Soc., 2020, 142, 15164–15171 CrossRef PubMed.
- D. Kim, J. H. Lee, J. Y. Koo, H. M. Kim and S. B. Park, Bioconjugate Chem., 2020, 31, 1545–1550 CrossRef CAS PubMed.
- L. Wu, J. Liu, P. Li, B. Tang and T. D. James, Chem. Soc. Rev., 2021, 50, 702–734 RSC.
- X. Wang, P. Li, Q. Ding, C. Wu, W. Zhang and B. Tang, J. Am. Chem. Soc., 2019, 141, 2061–2068 CrossRef CAS PubMed.
- J. Liu, M. A. A. Abdullah, L. Yang and J. Wang, Anal. Chem., 2020, 92, 647–653 CrossRef CAS PubMed.
- P. Werther, K. Yserentant, F. Braun, N. Kaltwasser, C. Popp, M. Baalmann, D. Herten and R. Wombacher, Angew. Chem., Int. Ed., 2020, 59, 804–810 CrossRef CAS PubMed.
- A. Kormos, D. Kern, A. Egyed, B. Söveges, K. Németh and P. Kele, Chem. Commun., 2020, 56, 5425–5428 RSC.
- J. Galeta, R. Dzijak, J. Obořil, M. Dračínský and M. Vrabel, Chem. – Eur. J., 2020, 26, 9945–9953 CrossRef CAS PubMed.
- R. Dzijak, J. Galeta, A. Vázquez, J. Kozák, M. Matoušová, H. Fulka, M. Dračínský and M. Vrabel, JACS Au, 2021, 1, 23–30 CrossRef CAS PubMed.
- P. K. K. Leung, L. C. C. Lee, H. H. Y. Yeung, K. W. Io and K. K. W. Lo, Chem. Commun., 2021, 57, 4914–4917 RSC.
- M. E. Graziotto, L. D. Adair, A. Kaur, P. Vérité, S. R. Ball, M. Sunde, D. Jacquemin and E. J. New, RSC Chem. Biol., 2021, 2, 1491–1498 RSC.
- W. Mao, J. Tang, L. Dai, X. He, J. Li, L. Cai, P. Liao, R. Jiang, J. Zhou and H. Wu, Angew. Chem., Int. Ed., 2021, 60, 2393–2397 CrossRef CAS PubMed.
- Z. He, T. Ishizuka, Y. Hishikawa and Y. Xu, Chem. Commun., 2022, 58, 12479–12482 RSC.
- X. Zhang, J. Gao, Y. Tang, J. Yu, S. S. Liew, C. Qiao, Y. Cao, G. Liu, H. Fan, Y. Xia, J. Tian, K. Pu and Z. Wang, Nat. Commun., 2022, 13, 3513 CrossRef CAS PubMed.
- W. Mao, W. Chi, X. He, C. Wang, X. Wang, H. Yang, X. Liu and H. Wu, Angew. Chem., Int. Ed., 2022, 61, e202117386 CrossRef CAS PubMed.
- L. Zhou, C. Liu, Y. Zheng, Z. Huang, X. Zhang and Y. Xiao, Anal. Chem., 2022, 94, 15678–15685 CrossRef CAS PubMed.
- Y. Wang, D. Torres-Garcia, T. P. Mostert, L. Reinalda and S. I. van Kasteren, Angew. Chem., Int. Ed., 2024, e202401733 CAS.
- K. Bertheussen, M. van de Plassche, T. Bakkum, B. Gagestein, I. Ttofi, A. J. C. Sarris, H. S. Overkleeft, M. van der Stelt and S. I. van Kasteren, Angew. Chem., Int. Ed., 2022, 61, e202207640 CrossRef CAS.
- M. O. Loehr and N. W. Luedtke, Angew. Chem., Int. Ed., 2022, 61, e202112931 CrossRef CAS PubMed.
- Z. Chen, W. T. Wang, W. Wang, J. Huang, J. Y. Liao, S. Zeng and L. Qian, ACS Appl. Mater. Interfaces, 2022, 14, 44054–44064 CrossRef CAS PubMed.
- S. K. Choi, Y. Lee, S. E. Yoon, H. Choi, J. Kim, J. H. Kim, S. Lee, W. Kim and E. Kim, Sens. Actuators, B, 2021, 340, 129966 CrossRef CAS.
- Y. Teng, R. Zhang, B. Yang, H. Yang, X. Li, D. Yin, X. Feng and Y. Tian, J. Mater. Chem. B, 2022, 10, 8642–8649 RSC.
- Y. Wang, Y. Teng, H. Yang, X. Li, D. Yin and Y. Tian, Chem. Commun., 2022, 58, 949–952 RSC.
- M. Auvray, D. Naud-Martin, G. Fontaine, F. Bolze, G. Clavier and F. Mahuteau-Betzer, Chem. Sci., 2023, 14, 8119–8128 RSC.
- L. K. B. Tam, P. Lo, P. C. K. Cheung and D. K. P. Ng, Chem. – Asian J., 2023, 18, e202300562 CrossRef CAS PubMed.
- E. Albitz, K. Németh, G. Knorr and P. Kele, Org. Biomol. Chem., 2023, 21, 7358–7366 RSC.
- A. Aktalay, R. Lincoln, L. Heynck, M. A. do, R. B. F. Lima, A. N. Butkevich, M. L. Bossi and S. W. Hell, ACS Cent. Sci., 2023, 9, 1581–1590 CrossRef CAS.
- Y. Deng, T. Shen, X. Yu, J. Li, P. Zou, Q. Gong, Y. Zheng, H. Sun, X. Liu and H. Wu, Angew. Chem., Int. Ed., 2024, 63, e202319853 CrossRef CAS PubMed.
- S. Segawa, X. Ou, T. Shen, T. Ryu, Y. Ishii, H. H. Y. Sung, I. D. Williams, R. T. K. Kwok, K. Onda, K. Miyata, X. He, X. Liu and B. Z. Tang, Aggregate, 2024, e499 CrossRef CAS.
- H. Zhang, J. Liu, Y.-Q. Sun, M. Liu and W. Guo, J. Am. Chem. Soc., 2020, 142, 17069–17078 CrossRef CAS PubMed.
- X. Xu, S. Shabiti, X. Zhang, J. Zheng, N. Liang, Z. Wang, S. Yu, Y. Wang, S. Jiang, Z. Pan, W. Li and L. Cai, Nano Today, 2024, 56, 102270 CrossRef CAS.
- X. Xu, J. Zheng, N. Liang, X. Zhang, S. Shabiti, Z. Wang, S. Yu, Z.-Y. Pan, W. Li and L. Cai, ACS Nano, 2024, 18, 9413–9430 CrossRef CAS PubMed.
- S. P. Y. Li, A. M. H. Yip, H. W. Liu and K. K. W. Lo, Biomaterials, 2016, 103, 305–313 CrossRef CAS PubMed.
- Q. Wu, K. Y. Zhang, P. Dai, H. Zhu, Y. Wang, L. Song, L. Wang, S. Liu, Q. Zhao and W. Huang, J. Am. Chem. Soc., 2020, 142, 1057–1064 CrossRef CAS PubMed.
- J. Zheng, Q. Zhan, L. Jiang, D. Xing, T. Zhang and K. L. Wong, Inorg. Chem. Front., 2020, 7, 4062–4069 RSC.
- J. J. Gruskos, G. Zhang and D. Buccella, J. Am. Chem. Soc., 2016, 138, 14639–14649 CrossRef CAS PubMed.
- P. E. Z. Klier, A. M. M. Gest, J. G. Martin, R. Roo, M. X. Navarro, L. Lesiak, P. E. Deal, N. Dadina, J. Tyson, A. Schepartz and E. W. Miller, J. Am. Chem. Soc., 2022, 144, 12138–12146 CrossRef CAS PubMed.
- B. Longo, C. Zanato, M. Piras, S. Dall’angelo, A. D. Windhorst, D. J. Vugts, M. Baldassarre and M. Zanda, Bioconjugate Chem., 2020, 31, 2201–2210 CrossRef CAS PubMed.
- X. Zhang, H. Xu, J. Li, D. Su, W. Mao, G. Shen, L. Li and H. Wu, Chem. Commun., 2022, 58, 573–576 RSC.
- H. E. Murrey, J. C. Judkins, C. W. am Ende, T. E. Ballard, Y. Fang, K. Riccardi, L. Di, E. R. Guilmette, J. W. Schwartz, J. M. Fox and D. S. Johnson, J. Am. Chem. Soc., 2015, 137, 11461–11475 CrossRef CAS PubMed.
- M. Işık and M. A. Kısaçam, J. Org. Chem., 2024, 89, 6513–6519 CrossRef PubMed.
- G. Beliu, A. J. Kurz, A. C. Kuhlemann, L. Behringer-Pliess, M. Meub, N. Wolf, J. Seibel, Z.-D. Shi, M. Schnermann, J. B. Grimm, L. D. Lavis, S. Doose and M. Sauer, Commun. Biol., 2019, 2, 261 CrossRef.
- P. Werther, K. Yserentant, F. Braun, K. Grußmayer, V. Navikas, M. Yu, Z. Zhang, M. J. Ziegler, C. Mayer, A. J. Gralak, M. Busch, W. Chi, F. Rominger, A. Radenovic, X. Liu, E. A. Lemke, T. Buckup, D. P. Herten and R. Wombacher, ACS Cent. Sci., 2021, 7, 1561–1571 CrossRef CAS PubMed.
- G. Linden and O. Vázquez, Chem. – Eur. J., 2020, 26, 10014–10023 CrossRef CAS PubMed.
- X. He, J. Li, X. Liang, W. Mao, X. Deng, M. Qin, H. Su and H. Wu, Nat. Commun., 2024, 15, 2831 CrossRef CAS PubMed.
- C. Caulfield, D. F. O’Shea and D. Wu, Tetrahedron, 2023, 138, 133387 CrossRef CAS.
- S. S. Matikonda, D. L. Orsi, V. Staudacher, I. A. Jenkins, F. Fiedler, J. Chen and A. B. Gamble, Chem. Sci., 2015, 6, 1212–1218 RSC.
- X. Shang, X. Song, C. Faller, R. Lai, H. Li, R. Cerny, W. Niu and J. Guo, Chem. Sci., 2017, 8, 1141–1145 RSC.
- L. Chen, F. Li, Y. Li, J. Yang, Y. Li and B. He, Chem. Commun., 2022, 58, 298–301 RSC.
- S. J. Siegl, R. Dzijak, A. Vázquez, R. Pohl and M. Vrabel, Chem. Sci., 2017, 8, 3593–3598 RSC.
- J. Tu, M. Xu, S. Parvez, R. T. Peterson and R. M. Franzini, J. Am. Chem. Soc., 2018, 140, 8410–8414 CrossRef CAS.
- Q. Yao, F. Lin, X. Fan, Y. Wang, Y. Liu, Z. Liu, X. Jiang, P. R. Chen and Y. Gao, Nat. Commun., 2018, 9, 5032 CrossRef PubMed.
- H. Li, J. Conde, A. Guerreiro and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2020, 59, 16023–16032 CrossRef CAS PubMed.
- Y. Zhao, Q. Yao, J. Chen, R. Zhang, J. Song and Y. Gao, Biomater. Sci., 2022, 10, 5662–5668 RSC.
- M. Chang, Y. Dong, H. Xu, A. B. Cruickshank-Taylor, J. S. Kozora, B. Behpour and W. Wang, Angew. Chem., Int. Ed., 2024, 63, e202315425 CrossRef CAS PubMed.
- Y. Dong, Y. Tu, K. Wang, C. Xu, Y. Yuan and J. Wang, Angew. Chem., Int. Ed., 2020, 59, 7168–7172 CrossRef CAS PubMed.
- J. Xiong, E. Y. Xue, Q. Wu, P. C. Lo and D. K. P. Ng, J. Controlled Release, 2023, 353, 663–674 CrossRef CAS PubMed.
- E. Y. Xue, C. Yang, Y. Zhou and D. K. P. Ng, Adv. Sci., 2023, 2306207 Search PubMed.
- J. Ko, M. Wilkovitsch, J. Oh, R. H. Kohler, E. Bolli, M. J. Pittet, C. Vinegoni, D. B. Sykes, H. Mikula, R. Weissleder and J. C. T. Carlson, Nat. Biotechnol., 2022, 40, 1654–1662 CrossRef CAS PubMed.
- S. J. Zhao, P. Zheng, Z. Wu and J. H. Jiang, Anal. Chem., 2022, 94, 2693–2698 CrossRef CAS PubMed.
- G. Feng, Z. Li, P. Zhai, M. Ying, Z. Xu, C. Yang, X. Wang, B. Dong, K. T. Yong and G. Xu, Sens. Actuators, B, 2022, 371, 132577 CrossRef CAS.
- L. Zhou, Z. Wang, L. Wang, X. Zhang and Y. Xiao, J. Am. Chem. Soc., 2023, 145, 28296–28306 CrossRef CAS PubMed.
- F. Muttach, N. Muthmann, D. Reichert, L. Anhäuser and A. Rentmeister, Chem. Sci., 2017, 8, 7947–7953 RSC.
- X. Wang, S. S. Liew, J. Huang, Y. Hu, X. Wei and K. Pu, J. Am. Chem. Soc., 2024, 146(32), 22689–22698 CrossRef CAS PubMed.
- A. Jemas, Y. Xie, J. E. Pigga, J. L. Caplan, C. W. Am Ende and J. M. Fox, J. Am. Chem. Soc., 2022, 144, 1647–1662 CrossRef CAS PubMed.
- J. E. Rosenberger, Y. Xie, Y. Fang, X. Lyu, W. S. Trout, O. Dmitrenko and J. M. Fox, J. Am. Chem. Soc., 2023, 145, 6067–6078 CrossRef CAS PubMed.
- H. Zhang, W. S. Trout, S. Liu, G. A. Andrade, D. A. Hudson, S. L. Scinto, K. T. Dicker, Y. Li, N. Lazouski, J. Rosenthal, C. Thorpe, X. Jia and J. M. Fox, J. Am. Chem. Soc., 2016, 138, 5978–5983 CrossRef CAS PubMed.
- C. Wang, H. Zhang, T. Zhang, X. Zou, H. Wang, J. E. Rosenberger, R. Vannam, W. S. Trout, J. B. Grimm, L. D. Lavis, C. Thorpe, X. Jia, Z. Li and J. M. Fox, J. Am. Chem. Soc., 2021, 143, 10793–10803 CrossRef CAS PubMed.
- D. N. Kamber, Y. Liang, R. J. Blizzard, F. Liu, R. A. Mehl, K. N. Houk and J. A. Prescher, J. Am. Chem. Soc., 2015, 137, 8388–8391 CrossRef CAS PubMed.
- V. Šlachtová, S. Bellová, A. La-Venia, J. Galeta, M. Dračínský, K. Chalupský, A. Dvořáková, H. Mertlíková-Kaiserová, P. Rukovanský, R. Dzijak and M. Vrabel, Angew. Chem., Int. Ed., 2023, 62, e202306828 CrossRef PubMed.
- R. Selvaraj and J. M. Fox, Curr. Opin. Chem. Biol., 2013, 17, 753–760 CrossRef CAS PubMed.
- S. Eising, F. Lelivelt and K. M. Bonger, Angew. Chem., Int. Ed., 2016, 55, 12243–12247 CrossRef CAS PubMed.
- S. Eising, A. H. J. Engwerda, X. Riedijk, F. M. Bickelhaupt and K. M. Bonger, Bioconjugate Chem., 2018, 29, 3054–3059 CrossRef CAS PubMed.
- D. Ye, A. J. Shuhendler, L. Cui, L. Tong, S. S. Tee, G. Tikhomirov, D. W. Felsher and J. Rao, Nat. Chem., 2014, 6, 519–526 CrossRef CAS PubMed.
- X. Ai, C. J. H. Ho, J. Aw, A. B. E. Attia, J. Mu, Y. Wang, X. Wang, Y. Wang, X. Liu, H. Chen, M. Gao, X. Chen, E. K. L. Yeow, G. Liu, M. Olivo and B. Xing, Nat. Commun., 2016, 7, 10432 CrossRef CAS PubMed.
- T. C. Do, J. W. Lau, C. Sun, S. Liu, K. T. Kha, S. T. Lim, Y. Y. Oon, Y. P. Kwan, J. J. Ma, Y. Mu, X. Liu, T. J. Carney, X. Wang and B. Xing, Sci. Adv., 2022, 8, eabq2216 CrossRef CAS PubMed.
- G. C. Van De Bittner, C. R. Bertozzi and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 1783–1795 CrossRef CAS PubMed.
- A. Godinat, H. M. i Park, S. C. Miller, K. Cheng, D. Hanahan, L. E. Sanman, M. Bogyo, A. Yu, G. F. Nikitin, A. Stahl and E. A. Dubikovskaya, ACS Chem. Biol., 2013, 8, 987–999 CrossRef CAS PubMed.
- D. Mustafa, D. Ma, W. Zhou, P. Meisenheimer and J. J. Cali, Bioconjugate Chem., 2016, 27, 87–101 CrossRef CAS PubMed.
- Z. Hai, J. Wu, D. Saimi, Y. Ni, R. Zhou and G. Liang, Anal. Chem., 2018, 90, 1520–1524 CrossRef CAS PubMed.
- X. Liu and G. Liang, Chem. Commun., 2017, 53, 1037–1040 RSC.
- G. Qi, X. Liu, L. Shi, M. Wu, J. Liu and B. Liu, Adv. Mater., 2022, 34, 2106885 CrossRef CAS PubMed.
- K. A. Schleyer, B. D. Datko, B. Burnside, C. Cui, X. Ma, J. K. Grey and L. Cui, ChemBioChem, 2020, 21, 2196–2204 CrossRef CAS PubMed.
- L. Cui, S. Vivona, B. R. Smith, S. R. Kothapalli, J. Liu, X. Ma, Z. Chen, M. Taylor, P. H. Kierstead, J. M. J. Fréchet, S. S. Gambhir and J. Rao, J. Am. Chem. Soc., 2020, 142, 15575–15584 CrossRef CAS PubMed.
- Y. Yuan, J. Zhang, Q. Cao, L. An and G. Liang, Anal. Chem., 2015, 87, 6180–6185 CrossRef CAS PubMed.
- J. Jiang, Z. Zhao, Z. Hai, H. Wang and G. Liang, Anal. Chem., 2017, 89, 9625–9628 CrossRef CAS PubMed.
- L. Plougastel, O. Koniev, S. Specklin, E. Decuypere, C. Créminon, D. A. Buisson, A. Wagner, S. Kolodych and F. Taran, Chem. Commun., 2014, 50, 9376–9378 RSC.
- H. Liu, D. Audisio, L. Plougastel, E. Decuypere, D. A. Buisson, O. Koniev, S. Kolodych, A. Wagner, M. Elhabiri, A. Krzyczmonik, S. Forsback, O. Solin, V. Gouverneur and F. Taran, Angew. Chem., Int. Ed., 2016, 55, 12073–12077 CrossRef CAS PubMed.
- L. Plougastel, M. R. Pattanayak, M. Riomet, S. Bregant, A. Sallustrau, M. Nothisen, A. Wagner, D. Audisio and F. Taran, Chem. Commun., 2019, 55, 4582–4585 RSC.
- T. S. M. Tang, H. W. Liu and K. K. W. Lo, Chem. – Eur. J., 2016, 22, 9649–9659 CrossRef CAS PubMed.
- L. C. Lee, J. C. Lau, H. Liu and K. K. Lo, Angew. Chem., Int. Ed., 2016, 55, 1046–1049 CrossRef CAS PubMed.
- L. C. Lee, H. M. Cheung, H. Liu and K. K. Lo, Chem. – Eur. J., 2018, 24, 14064–14068 CrossRef CAS PubMed.
- J. Shum, P. Zhang, L. C. Lee and K. K. Lo, ChemPlusChem, 2020, 85, 1374–1378 CrossRef CAS PubMed.
- C. Favre and F. Friscourt, Org. Lett., 2018, 20, 4213–4217 CrossRef CAS PubMed.
- Z. Shao, C. Zhang, X. Zhu, Y. Wang, W. Xu, Y. Chen, X. Wang, H. Zhu and Y. Liang, Chin. Chem. Lett., 2019, 30, 2169–2172 CrossRef CAS.
- Y. Teng, H. Yang, X. Li, Y. Wang, D. Yin and Y. Tian, Chin. J. Chem., 2022, 40, 209–214 CrossRef CAS.
- K. Krell, B. Pfeuffer, F. Rönicke, Z. S. Chinoy, C. Favre, F. Friscourt and H. Wagenknecht, Chem. – Eur. J., 2021, 27, 16093–16097 CrossRef CAS PubMed.
- M. Riomet, K. Porte, A. Wijkhuisen, D. Audisio and F. Taran, Chem. Commun., 2020, 56, 7183–7186 RSC.
- Y. Chen, R. Zhao, C. Tang, C. Zhang, W. Xu, L. Wu, Y. Wang, D. Ye and Y. Liang, Angew. Chem., Int. Ed., 2022, 61, e202112734 CrossRef CAS PubMed.
- W. Xu, H. Yu, R. Zhao and Y. Liang, Bioorg. Med. Chem. Lett., 2023, 81, 129129 CrossRef CAS PubMed.
- M. Ribéraud, K. Porte, A. Chevalier, L. Madegard, A. Rachet, A. Delaunay-Moisan, F. Vinchon, P. Thuéry, G. Chiappetta, P. A. Champagne, G. Pieters, D. Audisio and F. Taran, J. Am. Chem. Soc., 2023, 145, 2219–2229 CrossRef PubMed.
- Y. Tian, X. Li and D. Yin, Chem. Commun., 2019, 55, 12865–12868 RSC.
- X. Li, Y. Wang, H. Yang, D. Yin and Y. Tian, Eur. J. Org. Chem., 2020, 4296–4300 CrossRef.
- Y. Tian, H. Yang, X. Li, Y. Wang, Y. Teng and D. Yin, Org. Lett., 2021, 23, 3782–3787 CrossRef CAS PubMed.
- B. Li, X. Zhou, P. Yang, L. Zhu, Y. Zhong, Z. Cai, B. Jiang, X. Cai, J. Liu and X. Jiang, Adv. Sci., 2019, 6, 1802039 CrossRef PubMed.
- G. S. Kumar and Q. Lin, Chem. Rev., 2021, 121, 6991–7031 CrossRef CAS PubMed.
- B. D. Fairbanks, L. J. Macdougall, S. Mavila, J. Sinha, B. E. Kirkpatrick, K. S. Anseth and C. N. Bowman, Chem. Rev., 2021, 121, 6915–6990 CrossRef CAS PubMed.
- M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930–5938 CrossRef CAS PubMed.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, 2006 Search PubMed.
- T. Pauloehrl, G. Delaittre, V. Winkler, A. Welle, M. Bruns, H. G. Börner, A. M. Greiner, M. Bastmeyer and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2012, 51, 1071–1074 CrossRef CAS PubMed.
- S. Arumugam, S. V. Orski, J. Locklin and V. V. Popik, J. Am. Chem. Soc., 2012, 134, 179–182 CrossRef CAS PubMed.
- W. Feng, L. Li, C. Yang, A. Welle, O. Trapp and P. A. Levkin, Angew. Chem., Int. Ed., 2015, 54, 8732–8735 CrossRef CAS PubMed.
- V. X. Truong, F. Li and J. S. Forsythe, Biomacromolecules, 2018, 19, 4277–4285 CrossRef CAS PubMed.
- Z. Li, L. Qian, L. Li, J. C. Bernhammer, H. V. Huynh, J. S. Lee and S. Q. Yao, Angew. Chem., Int. Ed., 2016, 55, 2002–2006 CrossRef CAS PubMed.
- G. S. Kumar, S. Racioppi, E. Zurek and Q. Lin, J. Am. Chem. Soc., 2022, 144, 57–62 CrossRef CAS PubMed.
- W. Song, Y. Wang, J. Qu and Q. Lin, J. Am. Chem. Soc., 2008, 130, 9654–9655 CrossRef CAS PubMed.
- Y. Wang, W. J. Hu, W. Song, R. K. V. Lim and Q. Lin, Org. Lett., 2008, 10, 3725–3728 CrossRef CAS PubMed.
- R. K. V. Lim and Q. Lin, Acc. Chem. Res., 2011, 44, 828–830 CrossRef CAS PubMed.
- Z. Liu, Q. Lin, Y. Sun, T. Liu, C. Bao, F. Li and L. Zhu, Adv. Mater., 2014, 26, 3912–3917 CrossRef CAS PubMed.
- F. Zhang, Z. Zhang, L. Deng, H. Guo, T. Xia, W. Mao and J. Zhang, Org. Lett., 2023, 25, 872–876 CrossRef CAS PubMed.
- S. C. Ritter and B. König, Chem. Commun., 2006, 4694–4696 RSC.
- B. J. Adzima, Y. Tao, C. J. Kloxin, C. A. DeForest, K. S. Anseth and C. N. Bowman, Nat. Chem., 2011, 3, 256–259 CrossRef CAS PubMed.
- L. Zhang, X. Zhang, Z. Yao, S. Jiang, J. Deng, B. Li and Z. Yu, J. Am. Chem. Soc., 2018, 140, 7390–7394 CrossRef CAS PubMed.
- R. K. V. Lim and Q. Lin, Chem. Commun., 2010, 46, 7993–7995 RSC.
- V. X. Truong and C. Barner-Kowollik, ACS Macro Lett., 2021, 10, 78–83 CrossRef CAS PubMed.
- C. Stuckhardt, M. Wissing and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 18605–18611 CrossRef CAS PubMed.
- Y. Fu, H. Helbert, N. A. Simeth, S. Crespi, G. B. Spoelstra, J. M. van Dijl, M. van Oosten, L. R. Nazario, D. van der Born, G. Luurtsema, W. Szymanski, P. H. Elsinga and B. L. Feringa, J. Am. Chem. Soc., 2021, 143, 10041–10047 CrossRef CAS PubMed.
- J. Li, H. Kong, L. Huang, B. Cheng, K. Qin, M. Zheng, Z. Yan and Y. Zhang, J. Am. Chem. Soc., 2018, 140, 14542–14546 CrossRef CAS PubMed.
- A. Schönberg and A. Mustafa, Nature, 1944, 153, 195 CrossRef.
- G. Bertrand and C. Wentrup, Angew. Chem., Int. Ed. Engl., 1994, 33, 527–545 CrossRef.
- A. Padwa, S. Nahm and E. Sato, J. Org. Chem., 1978, 43, 1664–1671 CrossRef CAS.
- M. He, J. Li, S. Tan, R. Wang and Y. Zhang, J. Am. Chem. Soc., 2013, 135, 18718–18721 CrossRef CAS PubMed.
- P. An, Z. Yu and Q. Lin, Chem. Commun., 2013, 49, 9920–9922 RSC.
- Z. Yu, T. Y. Ohulchanskyy, P. An, P. N. Prasad and Q. Lin, J. Am. Chem. Soc., 2013, 135, 16766–16769 CrossRef CAS PubMed.
- J. Li, L. Huang, X. Xiao, Y. Chen, X. Wang, Z. Zhou, C. Zhang and Y. Zhang, J. Am. Chem. Soc., 2016, 138, 15943–15949 CrossRef CAS PubMed.
- M. Zhou, J. Hu, M. Zheng, Q. Song, J. Li and Y. Zhang, Chem. Commun., 2016, 52, 2342–2345 RSC.
- Z. Zhou, X. Xie, Q. Yi, W. Yin, A. A. Kadi, J. Li and Y. Zhang, Org. Biomol. Chem., 2017, 15, 6892–6895 RSC.
- X. Shang, R. Lai, X. Song, H. Li, W. Niu and J. Guo, Bioconjugate Chem., 2017, 28, 2859–2864 CrossRef CAS PubMed.
- P. An, T. M. Lewandowski and Q. Lin, ChemBioChem, 2018, 19, 1326–1333 CrossRef CAS PubMed.
- P. An, T. M. Lewandowski, T. G. Erbay, P. Liu and Q. Lin, J. Am. Chem. Soc., 2018, 140, 4860–4868 CrossRef CAS PubMed.
- Y. Wu, G. Guo, J. Zheng, D. Xing and T. Zhang, ACS Sens., 2019, 4, 44–51 CrossRef CAS PubMed.
- S. Liu, H. Su, L. Bu, J. Yan, G. Li and J. Huang, Analyst, 2021, 146, 1369–1375 RSC.
- Y. Wu, J. Zheng, D. Xing and T. Zhang, Nanoscale, 2020, 12, 10361–10368 RSC.
- M. Xiao, Y. Zhang, R. Li, S. Li, D. Wang and P. An, Chem. – Asian J., 2022, 17, e202200634 CrossRef CAS PubMed.
- U. Markel, P. Lanvers, D. F. Sauer, M. Wittwer, G. V. Dhoke, M. D. Davari, J. Schiffels and U. Schwaneberg, Chem. – Eur. J., 2021, 27, 954–958 CrossRef CAS PubMed.
- Y. Fu, N. A. Simeth, R. Toyoda, R. Brilmayer, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2023, 62, e202218203 CrossRef CAS PubMed.
- Y. Fu, G. Alachouzos, N. A. Simeth, M. Di Donato, M. F. Hilbers, W. J. Buma, W. Szymanski and B. L. Feringa, Chem. Sci., 2023, 14, 7465–7474 RSC.
- Y. Fu, G. Alachouzos, N. A. Simeth, M. Di Donato, M. F. Hilbers, W. J. Buma, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2024, e202319321 CAS.
- J. Zheng, R. Yue, R. Yang, Q. Wu, Y. Wu, M. Huang, X. Chen, W. Lin, J. Huang, X. Chen, Y. Jiang, B. Yang and Y. Liao, Front. Bioeng. Biotechnol., 2022, 10, 940511 CrossRef PubMed.
- F. Wang, H. Kong, X. Meng, X. Tian, C. Wang, L. Xu, X. Zhang, L. Wang and R. Xie, RSC Chem. Biol., 2022, 3, 539–545 RSC.
- F. Shen, Y. Zhang, G. Luan, K. Zhang, Z. Wang, Y. Luo, Y. Hou and G. Bai, Chin. Chem. Lett., 2024, 109646 CrossRef CAS.
- G. B. Spoelstra, S. N. Blok, L. Reali Nazario, L. Noord, Y. Fu, N. A. Simeth, F. F. A. IJpma, M. van Oosten, J. M. van Dijl, B. L. Feringa, W. Szymanski and P. H. Elsinga, Eur. J. Nucl. Med. Mol. Imaging, 2024, 51, 2583–2596 CrossRef CAS PubMed.
- J. Li, H. Kong, C. Zhu and Y. Zhang, Chem. Sci., 2020, 11, 3390–3396 RSC.
- A. M. Doze, Y. Fu, M. Di Donato, M. F. Hilbers, G. Luurtsema, P. H. Elsinga, W. J. Buma, W. Szymanski and B. L. Feringa, Chem. Sci., 2024, 15, 11557–11563 RSC.
- Y. Fu, K. Wu, G. Alachouzos, N. A. Simeth, T. Freese, M. Falkowski, W. Szymanski, H. Zhang and B. L. Feringa, Adv. Funct. Mater., 2023, 33, 2306531 CrossRef CAS.
- I. A. Cherepanov and S. K. Moiseev, Adv. Heterocycl. Chem., 2020, 131, 49–164 CrossRef CAS.
- X. Zhang, X. Wu, S. Jiang, J. Gao, Z. Yao, J. Deng, L. Zhang and Z. Yu, Chem. Commun., 2019, 55, 7187–7190 RSC.
- Z. Yao, X. Wu, X. Zhang, Q. Xiong, S. Jiang and Z. Yu, Org. Biomol. Chem., 2019, 17, 6777–6781 RSC.
- L. Deng, C. Zhang, B. Li, J. Fu, Z. Zhang, S. Li, X. Zhao, Z. Su, C. Hu and Z. Yu, Chem. Sci., 2023, 3630–3641 RSC.
- R. Huang, C. Hirschbiegel, V. Lehot, L. Liu, Y. A. Cicek and V. M. Rotello, Adv. Mater., 2024, 36, 2300943 CrossRef CAS PubMed.
- S. Fedeli, J. Im, S. Gopalakrishnan, J. L. Elia, A. Gupta, D. Kim and V. M. Rotello, Chem. Soc. Rev., 2021, 50, 13467–13480 RSC.
- V. Sebastian, M. Sancho-Albero, M. Arruebo, A. M. Pérez-López, B. Rubio-Ruiz, P. Martin-Duque, A. Unciti-Broceta and J. Santamaría, Nat. Protoc., 2021, 16, 131–163 CrossRef CAS PubMed.
- C. Ritter, N. Nett, C. G. Acevedo-Rocha, R. Lonsdale, K. Kräling, F. Dempwolff, S. Hoebenreich, P. L. Graumann, M. T. Reetz and E. Meggers, Angew. Chem., Int. Ed., 2015, 54, 13440–13443 CrossRef CAS PubMed.
- E. Indrigo, J. Clavadetscher, S. V. Chankeshwara, A. Megia-Fernandez, A. Lilienkampf and M. Bradley, Chem. Commun., 2017, 53, 6712–6715 RSC.
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y. C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597–603 CrossRef CAS PubMed.
- A. M. Pérez-López, B. Rubio-Ruiz, V. Sebastián, L. Hamilton, C. Adam, T. L. Bray, S. Irusta, P. M. Brennan, G. C. Lloyd-Jones, D. Sieger, J. Santamaría and A. Unciti-Broceta, Angew. Chem., Int. Ed., 2017, 56, 12548–12552 CrossRef PubMed.
- B. Li, P. Liu, H. Wu, X. Xie, Z. Chen, F. Zeng and S. Wu, Biomaterials, 2017, 138, 57–68 CrossRef CAS PubMed.
- J. Lee, S. Dubbu, N. Kumari, A. Kumar, J. Lim, S. Kim and I. S. Lee, Nano Lett., 2020, 20, 6981–6988 CrossRef CAS PubMed.
- R. Cao-Milán, S. Gopalakrishnan, L. D. He, R. Huang, L. S. Wang, L. Castellanos, D. C. Luther, R. F. Landis, J. M. V. Makabenta, C. H. Li, X. Zhang, F. Scaletti, R. W. Vachet and V. M. Rotello, Chem, 2020, 6, 1113–1124 Search PubMed.
- X. Zhang, Y. Liu, S. Gopalakrishnan, L. Castellanos-Garcia, G. Li, M. Malassiné, I. Uddin, R. Huang, D. C. Luther, R. W. Vachet and V. M. Rotello, ACS Nano, 2020, 14, 4767–4773 CrossRef CAS PubMed.
- N. Kaplaneris, J. Son, L. Mendive-Tapia, A. Kopp, N. D. Barth, I. Maksso, M. Vendrell and L. Ackermann, Nat. Commun., 2021, 12, 3389 CrossRef CAS PubMed.
- A. Samanta, V. Sabatino, T. R. Ward and A. Walther, Nat. Nanotechnol., 2020, 15, 914–921 CrossRef CAS PubMed.
- N. Singh, A. Gupta, P. Prasad, P. Mahawar, S. Gupta and P. K. Sasmal, Inorg. Chem., 2021, 60, 12644–12650 CrossRef CAS PubMed.
- X. Zhang, R. F. Landis, P. Keshri, R. Cao-Milán, D. C. Luther, S. Gopalakrishnan, Y. Liu, R. Huang, G. Li, M. Malassiné, I. Uddin, B. Rondon and V. M. Rotello, Adv. Healthcare Mater., 2021, 10, 2001627 CrossRef CAS PubMed.
- R. Huang, C. M. Hirschbiegel, X. Zhang, A. Gupta, S. Fedeli, Y. Xu and V. M. Rotello, ACS Appl. Mater. Interfaces, 2022, 14, 31594–31600 CrossRef CAS PubMed.
- A. Gupta, R. Das, J. M. Makabenta, A. Gupta, X. Zhang, T. Jeon, R. Huang, Y. Liu, S. Gopalakrishnan, R. C. Milán and V. M. Rotello, Mater. Horizons, 2021, 8, 3424–3431 RSC.
- X. Zhang, S. Lin, R. Huang, A. Gupta, S. Fedeli, R. Cao-Milán, D. C. Luther, Y. Liu, M. Jiang, G. Li, B. Rondon, H. Wei and V. M. Rotello, J. Am. Chem. Soc., 2022, 144, 12893–12900 CrossRef CAS PubMed.
- R. Das, J. Hardie, B. P. Joshi, X. Zhang, A. Gupta, D. C. Luther, S. Fedeli, M. E. Farkas and V. M. Rotello, JACS Au, 2022, 2, 1679–1685 CrossRef CAS PubMed.
- Z. Huang, Z. Liu, X. Xie, R. Zeng, Z. Chen, L. Kong, X. Fan and P. R. Chen, J. Am. Chem. Soc., 2021, 143, 18714–18720 CrossRef CAS PubMed.
- X. Li, H. Yang, Y. Teng, Y. Wang, D. Yin and Y. Tian, Chin. Chem. Lett., 2022, 33, 4223–4228 CrossRef CAS.
Footnote |
| † Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |