
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal complex-based TADF: design, characterization, and lighting devices
Afsaneh
Farokhi†
a,
Sophia
Lipinski†
 b,
Luca M.
Cavinato†
b,
Hashem
Shahroosvand
*a,
Babak
Pashaei
c,
Soheila
Karimi
a,
Sebastiano
Bellani
de,
Francesco
Bonaccorso
de and
Rubén D.
Costa
*b
b,
Luca M.
Cavinato†
b,
Hashem
Shahroosvand
*a,
Babak
Pashaei
c,
Soheila
Karimi
a,
Sebastiano
Bellani
de,
Francesco
Bonaccorso
de and
Rubén D.
Costa
*b
aGroup for Molecular Engineering of Advanced Functional Materials (GMA), Chemistry Department, University of Zanjan, Zanjan, Iran
bTechnical University of Munich, Campus Straubing for Biotechnology and Sustainability, Chair of Biogenic Functional Materials, Schulgasse 22, Straubing 94315, Germany. E-mail: ruben.costa@tum.de
cDepartment of Inorganic Chemistry, Faculty of Chemistry, University of Mazandaran, Babolsar, Iran
dGraphene Labs, Istituto Italiano di Tecnologia, 16163 Genova, Italy
eBeDimensional Spa., 16163 Genova, Italy
First published on 20th November 2024
Abstract
The development of novel, efficient and cost-effective emitters for solid-state lighting devices (SSLDs) is ubiquitous to meet the increasingly demanding needs of advanced lighting technologies. In this context, the emergence of thermally activated delayed fluorescence (TADF) materials has stunned the photonics community. In particular, inorganic TADF material-based compounds can be ad hoc engineered by chemical modification of the coordinated ligands and the type of metal centre, allowing control of their ultimate photo-/electroluminescence properties, while providing a viable emitter platform for enhancing the efficiency of state-of-the-art organic light-emitting diodes (OLEDs) and light-emitting electrochemical cells (LECs). By presenting an overview of the state of the art of all metal complex-based TADF compounds, this review aims to provide a comprehensive, authoritative and critical reference for their design, characterization and device application, highlighting the advantages and drawbacks for the chemical, photonic and optoelectronic communities involved in this interdisciplinary research field.
 Afsaneh Farokhi | Dr Afsaneh Farokhi completed both her BSc (2009) and MSc (2011) studies in Chemistry at the University of Zanjan (ZNU). She received her PhD degree in Inorganic Chemistry from the ZNU in 2017 to investigate the “synthesis, characterization and catalytic activity of asymmetric manganese porphyrin nanocatalysts”. From 2020 to now, Afsaneh have been a postdoctoral researcher under Prof. Hashem Shahroosvand at the ZNU. Her current research interests are mainly focused on the design and synthesis of novel hole transport materials in perovskite solar cells (PSCs). |
 Sophia Lipinski | Sophia Lipinski received her bachelor's degree in chemistry from the Technical University of Munich (TUM) and her master's degree in chemistry from the University of Konstanz. Currently, she is a PhD student in Prof. Costa's group at the Chair of Biogenic Functional Materials at TUM. Her main research focuses on the synthesis and characterization of sustainable d10 transition metal complexes and their implementation in light-emitting electrochemical cells. |
 Luca M. Cavinato | Luca Maria Cavinato got his BSc in Chemistry and Chemical Technologies and his MSc in Industrial Chemistry at the University of Turin followed by one year as Research Associate at IMDEA Materials Institute. Currently, he is a final year PhD Student at the Chair of Biogenic functional Materials at the Technical University of Munich under the supervision of Prof. Dr Costa. His research activity focuses on the study and development of innovative organic and organo-metallic materials for photovoltaic and lightning purposes. |
 Hashem Shahroosvand | Hashem Shahroosvand is a full professor in Inorganic Chemistry at University of Zanjan from 2009, as head of Group for Molecular Engineering of Advanced Functional Materials (GMA). Shahroos appeared in the list of 50 excellent scientists in IRAN by Iran Science Elites Federation from 2017 by now. His research encompasses molecularly engineered hole transport materials (HTMs) for perovskite solar cells (PSCs) and new emitters for light-emitting electrochemical cells (LEC) applications. New directions of cost effective optoelectronic devices based on carbon derivative electrodes are also in progress in his lab. Shahroos also extensively dealing with the philosophical objects in Chemistry. |
 Babak Pashaei | Babak Pashaei (BP) received his PhD in Inorganic Chemistry in 2019 from Zanjan University, Zanjan, Iran, under the guidance of Prof. Shahroosvand. He worked as a postdoctoral researcher in the same lab from 2019 to 2020, focusing on the synthesis of hole transporting materials for application in perovskite solar cells (PSCs). BP is currently Assistant Professor at Mazandaran University, Babolsar, Iran. His research focuses on artificial photosynthesis, light-emitting electrochemical cells, dye-sensitized solar cells, and PSCs. |
 Rubén D. Costa | Rubén D. Costa received his BSc/MSc (2006) and PhD (2010) in Chemistry from the U. Valencia. In 2011–2013, he was Humboldt Postdoc at the U. Erlangen-Nürnberg, working on nanocarbon-based PVs. In 2014, he became Liebig group leader focused on fluorescent proteins and d10-materials for lighting. In 2017, he moved to IMDEA Materials and was appointed as Associate Professor at the U. Waseda in 2018. Since 2020 he is Full Professor at the Technical University of Munich, heading the Chair of Biogenic Functional Materials. His research encompasses fluorescent protein engineering and protein-/d10-complex chemistry to advance sustainable concepts for lighting/PV technologies. |
1. Introduction
Over the past decade, the global energy demand has been growing exponentially because of population growth and industrial development.1 While the transition to renewable energy sources, such as wind, solar and nuclear energy,2,3 ubiquitous cornerstone for long-term sustainable socioeconomic,4,5 the introduction of efficient optoelectronics is necessary to reduce electric power consumption and heat dissipation towards mitigating the energy crisis. As example, Fig. 1a shows the widespread use of electricity at night, as mapped by the NASA Earth Observatory.6 In this context, SSLDs exhibiting 90% higher luminous efficacy (defined as the ratio of luminous flux to power) and 25 times longer lasting lighting performance in comparison with those of traditional incandescent bulbs and fluorescent compact lamps are playing a major role – Fig. 1b and c. SSL technologies can be classified into three categories: (i) inorganic light-emitting diodes (LEDs), (ii) organic light-emitting diodes (OLED), and (iii) light-emitting electrochemical cells (LECs).7 Their basic device structures are depicted in Fig. 2.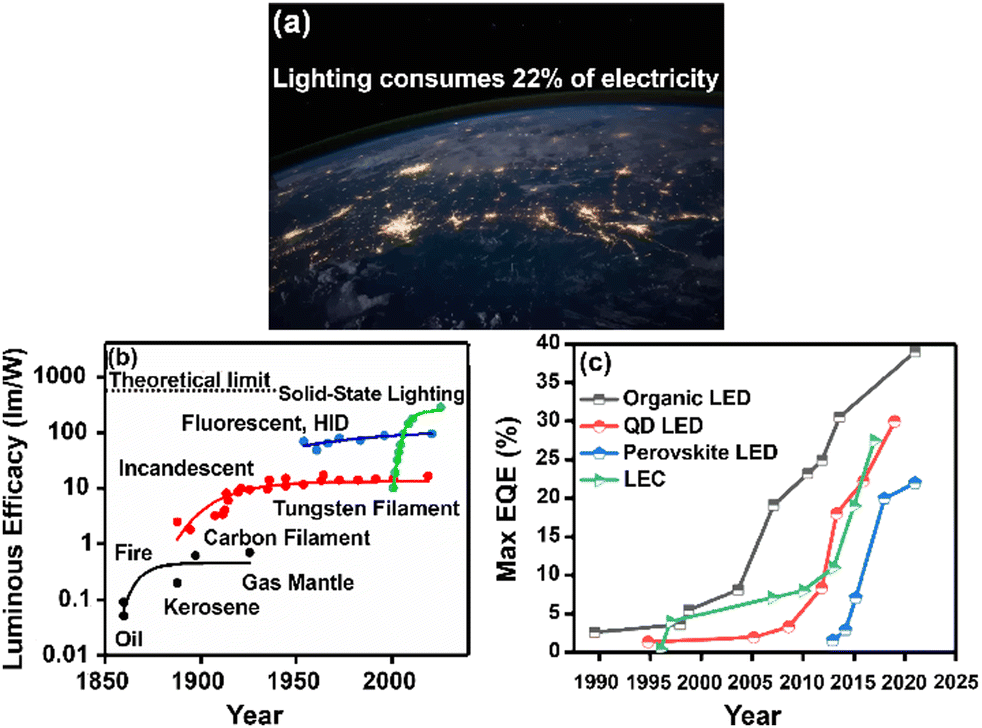 | ||
| Fig. 1 (a) The image of Earth's city lights (credit: image courtesy NASA). (b) Historical evolution of light sources’ performance as a function of time. (c) Comparison of EQE in display technologies based on organic LEDs, quantum dot LEDs, and perovskite LEDs. Data are taken from references as follows: OLEDs, ref. 8–15; QD LEDs, ref. 16–22; and PeLEDs, ref. 23–27, LEC, ref. 28,29. | ||
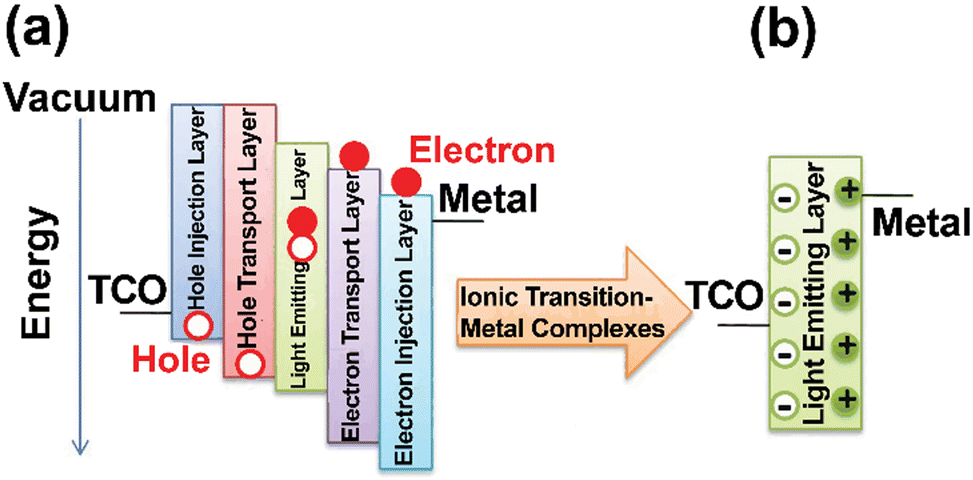 | ||
| Fig. 2 SSLD structures along with the mechanisms. (a) OLED and (b) LEC. Reproduced from ref. 7. Copyright 2019, Royal Society of Chemistry. | ||
Briefly, SSLs are based on semiconducting materials as high-efficiency illumination sources through direct conversion of electric energy into visible light, that is, the electroluminescence concept.30,31 Inorganic LEDs are based on direct band-gap inorganic semiconductors as the light-emitting materials,32–34 such as group III-nitrides35–38 and inorganic phosphors as color down-converting filters.39,40 The basic architecture is based on a three-layered structure of a p-type semiconductor, a n-type semiconductor, and a depletion layer,41–43 leading to a p–n junction diode structure.41–43 Once the LED is forward biased, the electrons at the conduction of band recombine with the holes at the valence band, releasing radiative energy. By doing so, inorganic LEDs can theoretically reach an internal quantum efficiency (IQE) of 100%. The optical emission wavelength can be adjusted by changing active material composition, which determines the optical and electronic properties of the material itself.41,42 Typical examples of active regions are made of Ge,44,45 GaAs,46–48 group-III nitrides (e.g., GaN, InGaN)36,49,50 and phosphides (e.g., InP,51 GaAsP,52 and AlGaInP53).
Even though inorganic LEDs could reach nearly 100% of IQE,54 there are a few problems, such as low color-rendering index (CRI) for white light sources55 and high cost of materials56 and related processing,57,58 in particular over large-areas substrates.59 Nevertheless, the inorganic LED market rules many application fields, such as medical,60–62 optical communication63,64 and display technologies.32,65 For the interested reader, there are several recent reviews about the limitations and challenges of inorganic LEDs.66,67 However, the TADF mechanism has not been applied to this technology and, in turn, it will not be further discussed in this review.
In parallel, the interest on organic molecular materials as emitters has significantly grown since Tang and Van Slyke revealed their electroluminescence behavior in a simple two-layer device based on 8-hydroxyquinoline aluminum (Alq3) as the light-emitting materials and an aromatic diamine as the hole transporting layer.68 Afterwards, intensive research efforts on organic semiconductors led to the strong development of OLEDs – Fig. 2, which use organic molecules and/or compounds as the electroluminescent (EL) materials forming the so-called light-emitting layer (LEL),69 but also for hole/electron injection layers (HIL and EIL), hole/electron transport layers (HTL and ETL) and LEL – Fig. 2a. Thus, their architecture is best described as multilayered active layer placed between a transparent anode, such as indium tin oxide (ITO), and an air-sensitive cathode, such as Ca or Mg.70,71 The charge injection/transporting layers transfer the carriers until they reach each other in the LEL and prevent the hole/electron diffusion toward the electrodes, leading to an efficient exciton confinement at the LEL. Given that exciton generation in fluorescent organic molecules under electrical bias yields 25% singlet excitons and 75% triplet excitons, a significant portion of the electrical energy, namely 75%, is dissipated as heat due to triplet exciton non-radiative deactivation. As a consequence, the theoretical maximum external quantum efficiency (EQE) can reach approximately 5%, considering a typical light out-coupling factor of around 20%.72
Though OLEDs offer the possibility to fabricate lightweight, large-area, flexible and through low-cost solution-based manufacturing processes,73–76e.g., spin coating,77,78 casting,79 and printing techniques.80,81 The manufacturing of commercially available OLEDs currently takes place through high-cost and time-consuming vacuum thermal evaporation,82 significantly increasing the final OLED cost. Moreover, the oxygen sensitivity of organic LELs, as well as the use of chemically reactive electrodes requires rigorous encapsulation. Despite these drawbacks, the performance of OLEDs in terms of stability and efficiency has rapidly increased, making them extremely attractive.73,83 Indeed, todays global market is predicted to rise over 13% during the forecast period 2020–2025, with applications in residential,84,85 commercial86,87 and industrial sectors.88,89 This has been strongly related to (i) the development of emitters going from fluorescent organic molecules/conjugated polymers, phosphorescent transition metal complexes, quantum dots, perovskites, and now TADF molecules, (ii) host:guest active layers,72,90–93 and (iii) device architectures, such as tandem and micro-OLEDs for advanced sensing systems and displays (e.g., portability94,95 and wearability96,97). In the context of the different emitter generations, the reader is invited to check the following reviews along with a short description in Section 2.98 Herein, we will concentrate on metal complex-based TADF emitters applied to OLEDs, highlighting their photo-/electro-luminescence features with respect to the current challenges.
As a final SSL family, LECs shine as the simplest, cheapest, and moderate performing devices.99–102 They consist of a single ion-based LEL sandwiched by two air-stable electrodes – Fig. 2b. The application of electric bias causes a redistribution of ions at the electrode surface allowing carrier injection as well as the growth of the p- and n-doped regions close to each electrode. This generates the so-called p–i–n junction, in which excitons are formed at the neutral (i) region flanked by the p-/n-doped regions, that is, the so-called p–i–n junction. A major advantage of LECs is their easy fabrication using spray coating103,104 on any arbitrary 3D-shape electrode with a good thickness tolerance105 and encapsulation requirements.106 Likewise OLEDs, the LEL of LECs consists of emitters, such as fluorescent organic molecules/conjugated polymers blended with ion polyelectrolytes, phosphorescent ionic transition metal complexes (iTMCs) with ionic liquids, quantum dots with ion polyelectrolytes, perovskites, and more recently neutral and ionic TADF emitters with electrolytes.107–112 Among them, iTMC-based LECs exhibit several advantages over polymeric-LECs,106,113 as they are easily dissolved in polar solvents, which can be consequently processed in form of dispersion/solution through printing methods avoiding phase separation. Indeed, Cu(I), Ru(II), Pt(III), Os(II) and Ir(III) iTMCs used in LECs are mostly ionic and they have achieved high stabilities (>5000 h) and EQEs >10% without using electrolytes.73,106,114–116 Afterwards, small organic molecules have been also revealed to be suitable LEL materials for LECs because of their high photoluminescence quantum yields (PLQYs), simple molecular modifications, and presence of TADF mechanism. Here, design rules for the device composition (emitter, electrolyte, host:guest type and composition, etc.) have led to good brightness levels (2000 cd m−2) at high EQE (up to 27.5%) as achieved by researchers at Umeå and Linköping universities.29 Based on these advances, medical, signing and human services are realistic niches for LECs.117 Excellent reviews about each type of emitters, electrolytes, unconventional architectures, and device modus operando are provided for the interested reader.112,118 Likewise the OLEDs, this review will also concentrate on the design and implementation metal complex-based TADF emitters in the LECs field.
Finally, it is important to stress that organic TADF field has been widely advanced in the last decades. In this context, the interest on metal complex-based TADF is two-fold. On one hand, the presence of a metal ion core can lead to short excited state lifetimes due to the high spin–orbit coupling (SOC) reducing the probability of parasite non-radiative decay pathways, such as exciton-polaron quenching and chemical degradation. On the other hand, purely organic TADF emitters require complex multi-step synthesis that are in antithesis with sustainability requirements.119,120 It is still poorly understood why the device performance are not comparable (OLEDs and LECs) between both families of TADF emitters. Thus, a comprehensive overview is necessary to foster new advances in the near future of metal complex-based TADF emitters.
In view of all of the aforementioned, the scope of this review is a comprehensive overview of the progress of metal complex-based TADF as potential emitters for SSLs. Other reviews have been focused on describing photo-/electro-luminescent behavior of either TADF organic molecules120,121 or all-inorganic TADF emitters,119 while much less attention has been placed on TMCs with a few reviews focused on either device performance or photophysical behavior of particular families.90,122 Herein, we wish to provide to the reader a comprehensive overview, including design rules of metal complex-based TADF emitters and how to characterize them in Section 2 as well as the lessons learnt from their device performance in OLEDs and LECs in Section 3. This is finalized with a conclusion and outlook, highlighting the limitations/challenges in this ever-growing field.
2. TADF emitters
2.1. Physics and designs
Although TADF materials were discovered in the 1960s,91–93 their use in optoelectronics was first proposed by Adachi and co-workers in 2009,123 paving the way toward a new generation of highly efficient OLEDs14,124–128 and LECs.129Fig. 3 shows the number of TADF material-related publications per year from 2011, reflecting how TADF materials influenced both the photonic and optics research communities during the last years.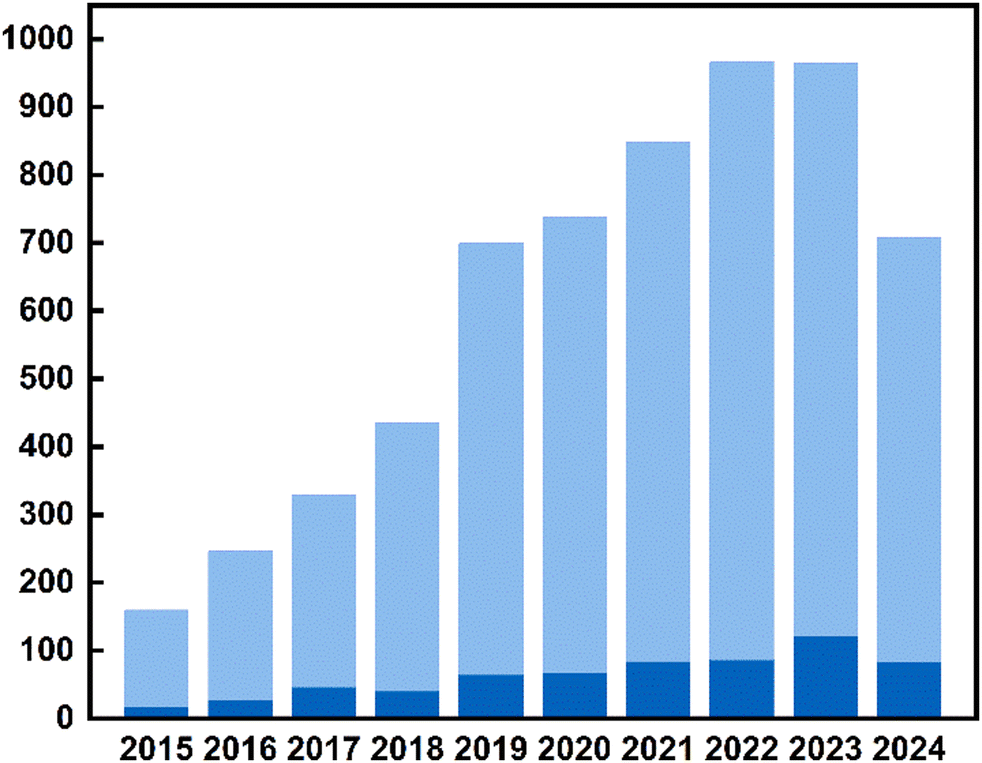 | ||
| Fig. 3 The number of publications related to TADF emitters (light blue) highlighting the metal complex-based TADF emitters (deep blue). | ||
Thanks to their photophysical properties, TADF materials provide several advantages in SSLs. This is best reflected in the OLED evolution – Fig. 4. In the 1st generation OLEDs (until the 90s), green and red fluorescent materials have been used, with a low theoretical IQE of 25% and a short operational lifespan.130 The issues were partially solved with the 2nd generation of OLEDs (up to 00 s), based on phosphorescent Pt(II) and Ir(III) organometallic complexes – Fig. 4. The theoretical IQE improved up to 100% thanks to the high SOC of such materials, and the operational lifespan reached incredibly high values (>50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h). However, some challenges remained unsolved (lack of stable blue emitters), and new ones rose (dependence on rare and expensive elements). Especially the former is particularly relevant because it hinders the development of white OLEDs (WOLEDs) with a high color rendering index (CRI).130 TADF materials constitute the so-called 3rd-generation OLEDs.131 They can upconvert triplet states (T1) into singlet states (S1) through reversible intersystem crossing (rISC) mechanism – Fig. 4, unlocking their theoretical IQE beyond 25% and up to 100% through the (delayed) fluorescence given by the radiative decay of their excited singlet states (i.e., S1 → S0).7,14 Notably, the electroluminescence mechanism leading to theoretical IQE up to 100% is substantial different from those of phosphorescent emitters belonging to the 2nd-generation OLEDs, whose luminescence results from the T1 decay toward S0 (i.e., T1 → S0).9 The theoretical 100% IQE of TADF material-based OLEDs14,132,133 relies on the reduced energy difference between the lowest singlet (i.e., S1) and triplet (i.e., T1) excited states (ΔEST, also referred to singlet–triplet energy splitting) in TADF materials compared to fluorescent molecules. Such optical characteristic promotes the rISC (T1 → S1) effects,134 thus the TADF mechanism. Though TADF is not typically encountered in fluorescent emitters, since their large ΔEST prohibits efficient rISC,135,136 the design of TADF organic emitters has been successful (e.g., Tetrakis-N-carbazoyl-isophthalonitrile).137,138 Here, the energetic features of TADF materials partially resembles the one of phosphorescent emitters,139 in which small ΔEST (but higher compared to TADF materials), the presence of heavy transition metals (such as Ir in Ir(ppy)3, ppy is 2-phenylpyridine) and the involvement of singlet and triplet states belonging to different electronic configurations promote the intersystem crossing (ISC), in accordance with El-Sayed rules136,140,141 The ISC is the isoenergetic radiationless process, involving a transition between the two electronic states with different states spin multiplicity. In phosphorescent emitters, it enables the filling of excited T1 states that provide phosphorescence, acting as light-emitting sources.142
000 h). However, some challenges remained unsolved (lack of stable blue emitters), and new ones rose (dependence on rare and expensive elements). Especially the former is particularly relevant because it hinders the development of white OLEDs (WOLEDs) with a high color rendering index (CRI).130 TADF materials constitute the so-called 3rd-generation OLEDs.131 They can upconvert triplet states (T1) into singlet states (S1) through reversible intersystem crossing (rISC) mechanism – Fig. 4, unlocking their theoretical IQE beyond 25% and up to 100% through the (delayed) fluorescence given by the radiative decay of their excited singlet states (i.e., S1 → S0).7,14 Notably, the electroluminescence mechanism leading to theoretical IQE up to 100% is substantial different from those of phosphorescent emitters belonging to the 2nd-generation OLEDs, whose luminescence results from the T1 decay toward S0 (i.e., T1 → S0).9 The theoretical 100% IQE of TADF material-based OLEDs14,132,133 relies on the reduced energy difference between the lowest singlet (i.e., S1) and triplet (i.e., T1) excited states (ΔEST, also referred to singlet–triplet energy splitting) in TADF materials compared to fluorescent molecules. Such optical characteristic promotes the rISC (T1 → S1) effects,134 thus the TADF mechanism. Though TADF is not typically encountered in fluorescent emitters, since their large ΔEST prohibits efficient rISC,135,136 the design of TADF organic emitters has been successful (e.g., Tetrakis-N-carbazoyl-isophthalonitrile).137,138 Here, the energetic features of TADF materials partially resembles the one of phosphorescent emitters,139 in which small ΔEST (but higher compared to TADF materials), the presence of heavy transition metals (such as Ir in Ir(ppy)3, ppy is 2-phenylpyridine) and the involvement of singlet and triplet states belonging to different electronic configurations promote the intersystem crossing (ISC), in accordance with El-Sayed rules136,140,141 The ISC is the isoenergetic radiationless process, involving a transition between the two electronic states with different states spin multiplicity. In phosphorescent emitters, it enables the filling of excited T1 states that provide phosphorescence, acting as light-emitting sources.142
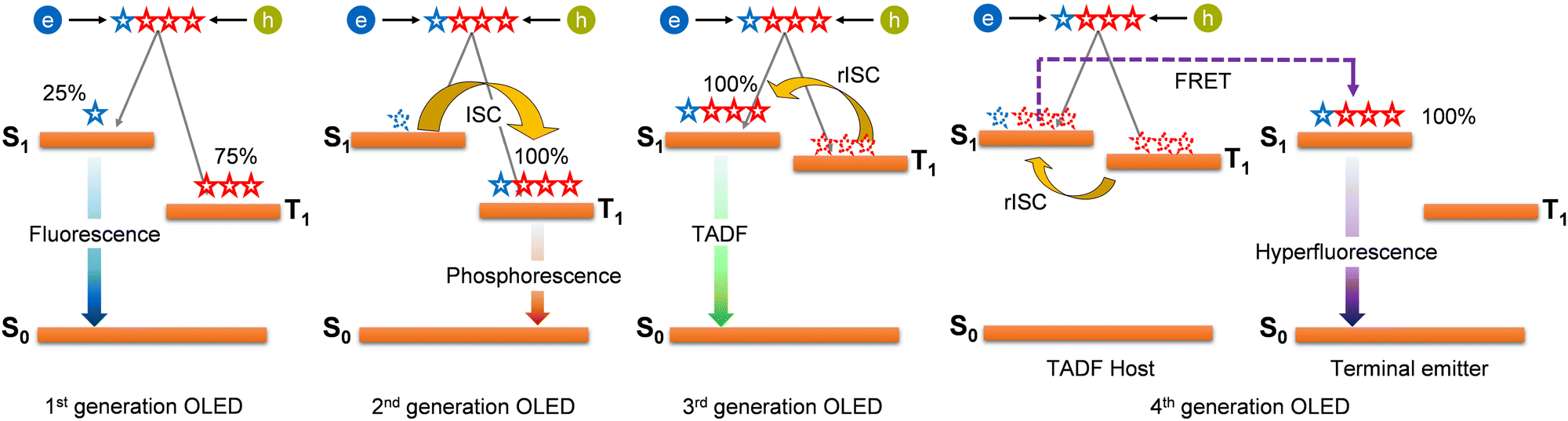 | ||
| Fig. 4 Sketch of the working principles of 1st, 2nd, 3rd, and 4th generation OLEDs. The transition processes of photogenerated excitons for fluorescence, phosphorescence, TADF, and hyperfluorescence are highlighted. | ||
Clearly, the rational design of materials with proper ΔEST (typically <1613 cm−1) enables the competition between phosphorescence and TADF processes to be controlled via ISC and rISC, determining the final nature (TADF or phosphorescence) of the emitters in OLEDs. All TADF molecules contain donor and acceptor moieties, which have electron-rich functional units with deep highest occupied molecular orbital (HOMO) and electron-deficient functional units with a deep lowest unoccupied molecular orbital (LUMO),143,144 respectively. As a result, the donor and acceptor moieties participate in the hole transport and electron transport, respectively.
Based on this rationale, numerous families of small organic molecules have been recently produced and investigated containing spiro derivatives,145 triazines,146 spiro-acridines,147 phenazines and carbazolyl dicyanobenzene (CDCB).14,148 Likewise, dx-TMCs, such as (i) d6 Ir(III), (ii) d8 Pd(0), Pd(II), Pt(II), and Au(III), and (iii) d10 Cu(I), Ag(I), Au(I), and Zn(II), have also shown a similar charge transfer (CT) character that allows measuring TADF emitters.
However, TADF materials development is still relatively new (<25 years; Fig. 3), with stability and color purity needing of further optimization. Moreover, the synthesis of advanced organic materials became more intricate and complex, dramatically influencing cost and scalability. Finally, achieving high-power TADF-based devices remains challenging due to their high degradation rate at high applied current densities.
To overcome these limitations, hyperfluorescence and hyperphosphorescence systems are currently being developed, constituting the 4th generation OLEDs – Fig. 4.149–152 The primary exciton formation is confined to the terminal fluorescence/phosphorescence emitter by controlling the doping concentration ratio of the TADF assist dopant. Thus, the triplet excitons are first upconverted into the S1 state via rISC in the TADF material. Then, they are transferred by Förster resonance energy transfer (FRET) to the final emitter molecule. The idea is based on two key points: (i) fluorescent emitters' high stability and narrow emission spectra, which lead to high color purity, and (ii) TADF molecules’ effective rISC process. Nevertheless, this is in its early stage, facing material design and a limited understanding of the hyperfluorescence mechanism. Demonstrators and performance optimization are yet to be realized. Long-term stability and commercial viability are unknown.153,154
2.2. How to characterize metal complex-based TADF emitters? Models, characterization methods, and assumptions
In general, efficient TADF emitters must meet two key criteria simultaneously: (i) maintaining a small energy gap between the singlet and triplet excited states (ΔEST) and (ii) minimizing non-radiative decay. This ensures that the triplet excited state features a prolonged existence, maximizing the opportunity for triplet harvesting through the thermally activated rISC and, consequently, enhancing the emission yield. The critical factor in maximizing the rISC rate constant is achieving a small ΔEST, as defined by eqn (1), so understanding how to minimize it in molecular structures becomes crucial.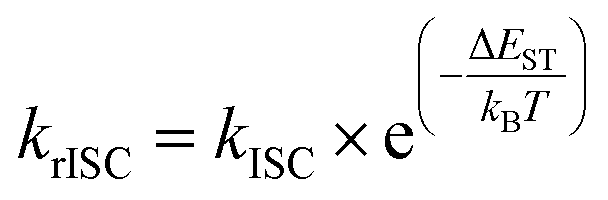 | (1) |
Calculating the energy of the lowest singlet (ES1) and triplet (ET1) excited states involves considering three aspects: (i) the orbital energy (Eorb), (ii) the electron repulsion energy (K), and (iii) the exchange energy (J). These factors affect the HOMO and LUMO energies and topological distribution.155 Thus, they are equally relevant on the singlet and triplet excited states that featured identical electronic configurations. Indeed, the exchange term increases the singlet energy and decreases the triplet energy by an equal amount, as per eqn (2) and (3).124 Consequently, the singlet–triplet energy gap ΔEST is determined by eqn (4), and it is evident that minimizing this gap requires reducing the exchange energy J, calculated using eqn (5).
| ES1 = Eorb + K + J | (2) |
| ET1 = Eorb + K − J | (3) |
| ΔEST = ES1 − ET1 = 2J | (4) |
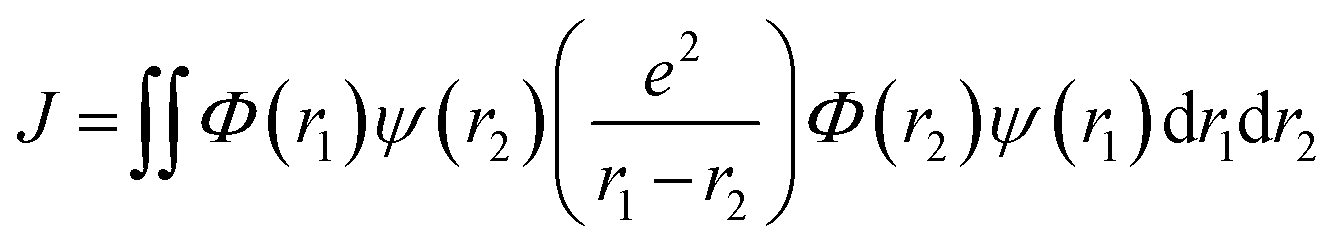 | (5) |
Here, Φ and ψ represent the HOMO and LUMO wavefunctions, respectively, and e is the electron charge. In the context of HOMO–LUMO transitions, the Φ and ψ parameters are replaced with the many-body electronic wavefunction for the singlet and triplet states. This shows that minimizing J reduces the HOMO and LUMO overlapping, which can be achieved initially by spatially separating these frontier orbitals.156
Organic molecules exhibiting TADF typically contain electron acceptor (A) and donor (D) moieties that promote D–A charge transfer (CT) characteristics in the excited state. These molecules are usually composed of D and A units connected via an aromatic bridge, resulting in excited states with strong CT characteristics. The energy difference between the singlet and triplet excited states can be further minimized by twisting the D and A units around the D–A axis, creating near-orthogonal D–A relative orientations, or by increasing the D–A distance through the use of a longer bridge – Section 2.3.157 Similarly, for TMCs, this is related to the occurrence of metal-to-ligand charge-transfer (MLCT) (and ligand-to-ligand charge-transfer (LLCT)) states having frontier orbitals, which are spatially largely separated – Section 2.3. Finally, on the context of solid-state light-emitting devices, in which TADF materials are employed, their properties are intricately tied to the permissibility of the S1 → S0 transition. This is because: (i) the radiative rate kr(S1 → S0) should be maximized for high PLQYs and (ii) short decay times play a crucial role in minimizing roll-off effects and enhancing stability.158,159 In the last decades, numerous researchers have endeavored to comprehend the photophysics of TADF materials by employing a conventional rate equation approach to curve-fit time-resolved photoluminescence decays, wherein each decay process is considered. While this strategy is easy to apply, prior analyses often relied on several a priori assumptions, resulting in significant deviations from experimental observations or, at times, erroneous conclusions. Based on excellent works from several groups (Penfold, Thompson, Adachi, Monkman, Yersin, Tsuchiya, Zysman-Colman, Samuel, etc.),155,160–164 we will consider three cases: (i) kISC ≫ kr(S1 → S0), (ii) kISC ≪ kr(S1 → S0), and (iii) general case with kISC ≅ kr(S1 → S0). Thus, TADF molecules can be categorized into three main groups based on their photophysical features of coordination complexes.
In this situation, the decay time τ(T) of the luminescent molecule is described by eqn (6):
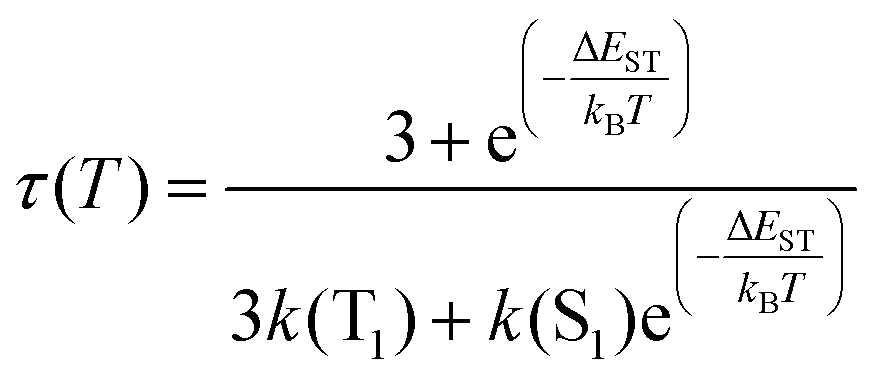 | (6) |
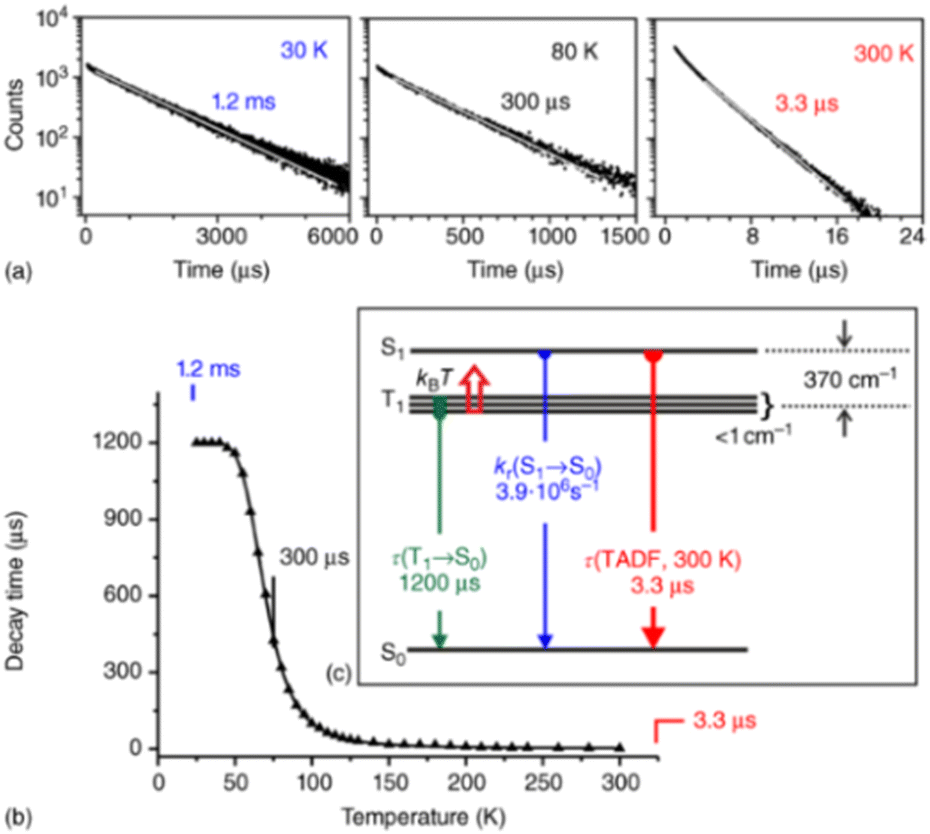 | ||
| Fig. 5 (a) Emission decay profiles of Cu(dppb)(pz2Bph2) (Cu10) (powder) at 30, 80, and 300K recorded after pulsed excitation at λexc = 378 nm and detected at λdet = 540nm. (b) Emission decay time τ versus temperature. The solid line represents a fit of eqn (6) to the experimental τ(T) values fixing τ(T1) = 1.2ms as measured at T = 30 K. The fit parameters are τ(S1) = 180 ns and ΔEST = 370 cm−1, respectively. Reproduced with permission from ref. 128. | ||
In such systems, the rate constants are easily accessible through the system of eqn (7):170
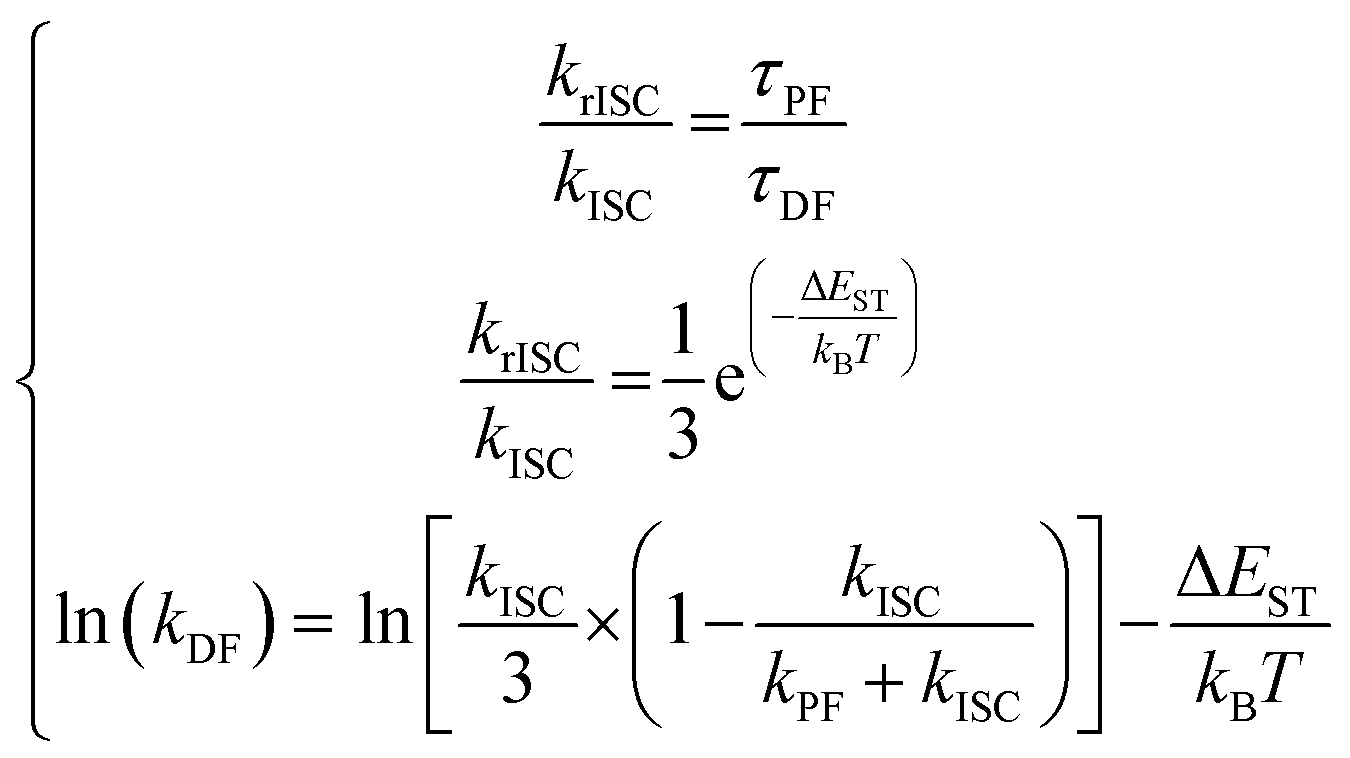 | (7) |
In this context, assuming that kISC is temperature-independent, it is possible to obtain directly the krISC and kDF from the corresponding Arrhenius plots. However, this set of equations holds only within the assumption of unity PLQY, neglecting the non-radiative pathways and, often, they are applied outside their boundary conditions.169–172
In low-emissive systems formally only the general equation can be applied in a wide range of temperature (i.e., 77–300 K):170
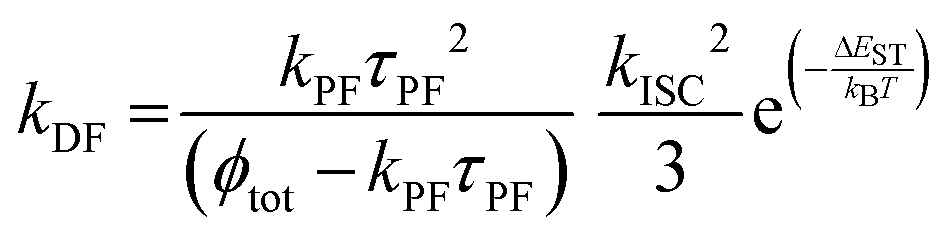 | (8) |
As discussed deeper in the next sections, the photophysical properties are connected to the molecular geometry. For instance, in a family of luminescent Cu(I) complexes, [Cu(DPEPhos)-(N^N)][BF4], (DPEPhos = bis[2 (diphenylphosphino)-phenyl]ether and N^N = sulfur-bridged dipyridyl ligands) the exploration of the influence of the bridging sulfur atom oxidation state, sulfide(II) or sulfone(VI), and also substituents in the 6,6′-sites of the pyridyl rings (R = H, Me, Ph) was carried out.173 It was shown that through these structural modification, various complex geometries can be obtained so that the proton (Cu-DPS, Cu-DPSO2) and phenyl (Cu-Ph-DPS, Cu-Ph-DPSO2) substituted derivatives form monometallic complexes, whereas methyl-substituted derivatives (Cu-Me-DPS, Cu-Me-DPSO2) have a “Goldilocks” degree of steric bulk, which results in bimetallic species. All complexes except Cu-Ph-DPS, were demonstrated to emit through a TADF mechanism, while the photoluminescence characteristics of Cu-Ph-DPS were found to be in line with triplet ligand-centered (3LC) emission. Moreover, the reduction in the long lifetime component on cooling for Cu-Ph-DPS – Fig. 6, indicates that this is not TADF emitter. In addition, in Cu-Me-DPS the relation τ(T) ≈ τph is not reached at 77 K, but lower temperatures are required.
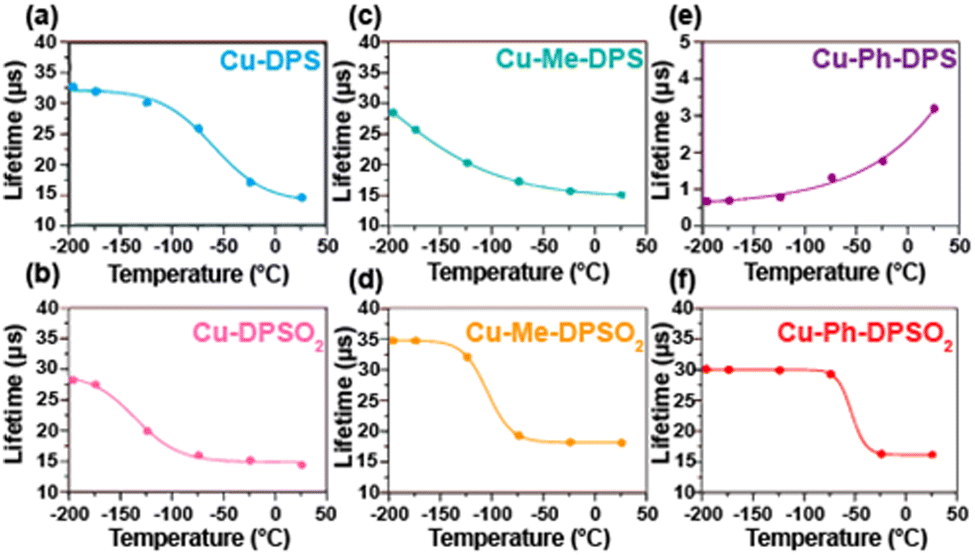 | ||
| Fig. 6 Solid-state photoluminescence lifetimes of (a) Cu-DPS; (b) Cu-DPSO2; (c) Cu-Me-DPS; (d) Cu-Me-DPSO2; (e) Cu-Ph-DPS; and (f) Cu-Ph-DPSO2 from −196 to 25 °C. Samples drop-cast as neat thin-films from MeOH on quartz slides (λexc = 370 nm). Reproduced from ref. 173. Copyright 2019, American Chemical Society. | ||
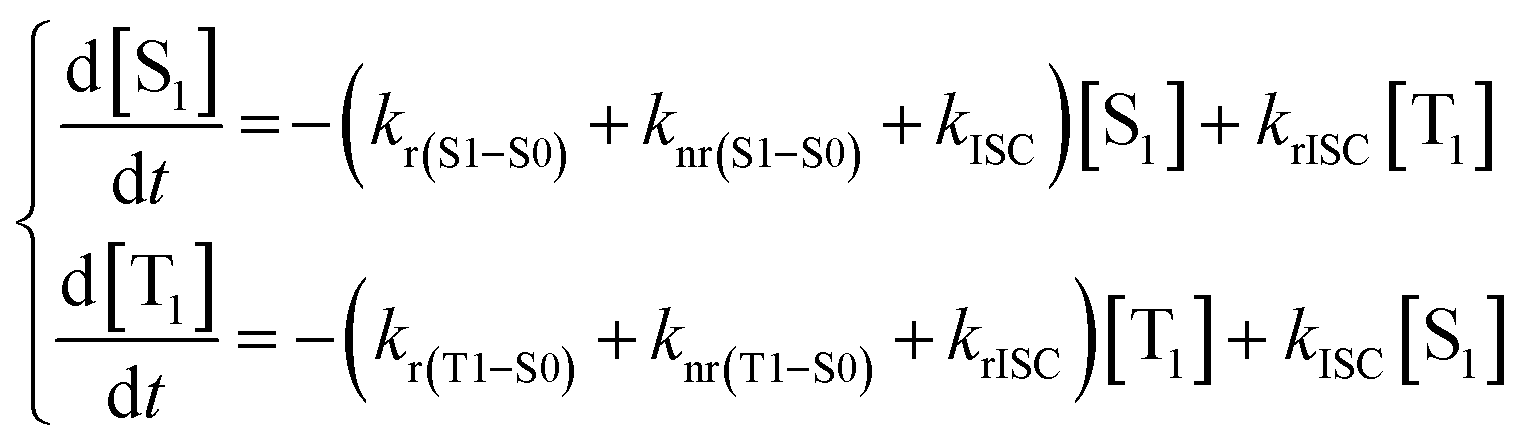 | (9) |
With [S1] and [T1] being the population of the respective excited states. The solution of this system of differential equations provided two different eigenvalues:174
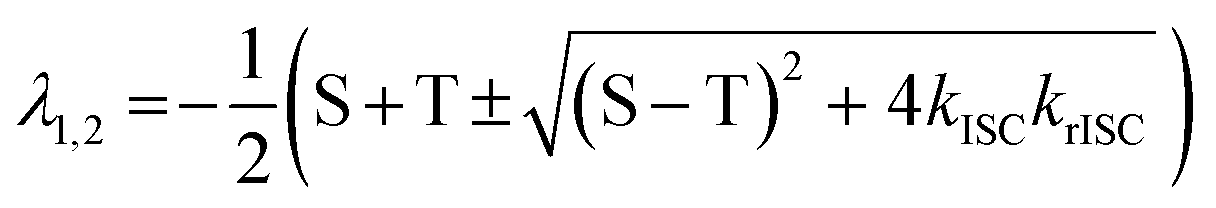 | (10) |
S and T are S = kr(S1–S0) + knr(S1–S0) + kISC and = kr(T1−S0) + knr(T1−S0) + krISC, respectively, which have been condensed for the sake of clarity. In this context, it is possible to extract the relation:
| S + T = kPF + kDF | (11) |
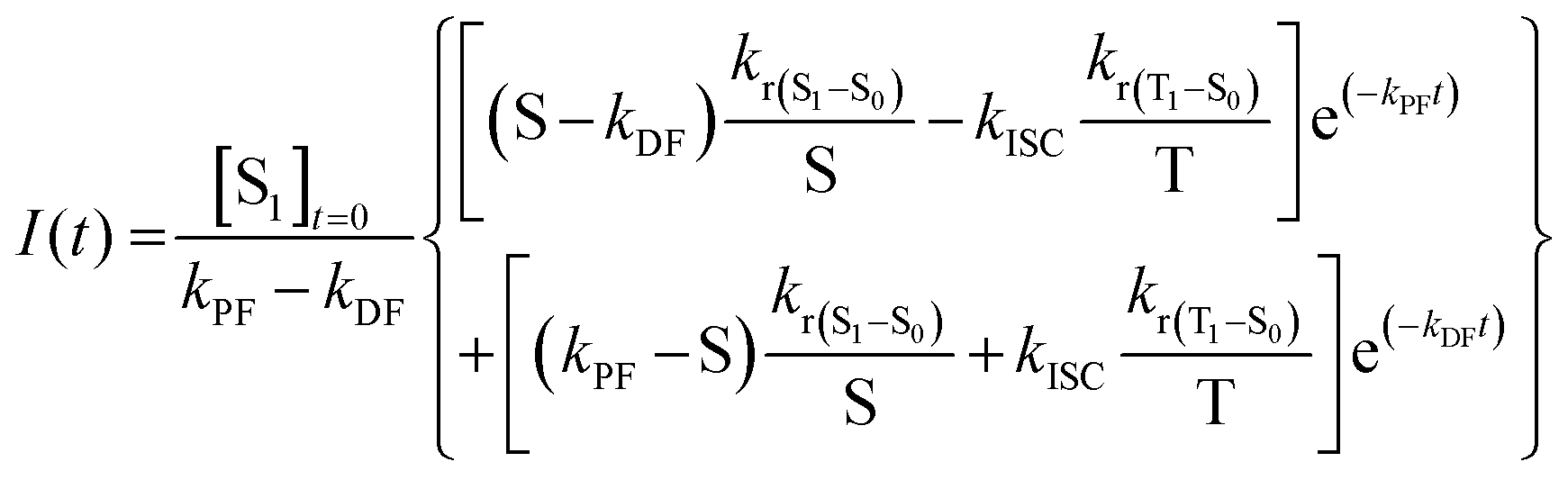 | (12) |
However, despite being the most complete and accurate model equation obtained so far, it is difficult to use it as it is, without any assumptions. Likely, since the T1 level is lower in energy than S1 in conventional TADF emitters, it is possible to assume that S ≫ krISC, which means that at least one of the three parameters (kr(S1–S0) + knr(S1–S0) + kISC) is greater than krISC. Thus, we can simplify eqn (11):
| I(t) = (APF + ADF)e(−kPFt) + Ad[−e(−kPFt) + e(−kDFt)] | (13) |
APF and ADF are the pre-exponential factors of the biexponential curve.174,179Eqn (13) is the one that must be used to fit experimental excited state decay profiles. From APF, ADF, kPF, kDF, and the PLQY at room temperature (Φtot), it is possible to derive the specific quantum efficiency of the prompt fluorescence (ΦPF) and delayed emission taking into account both delayed fluorescence and phosphorescence (ΦDF + Φph):
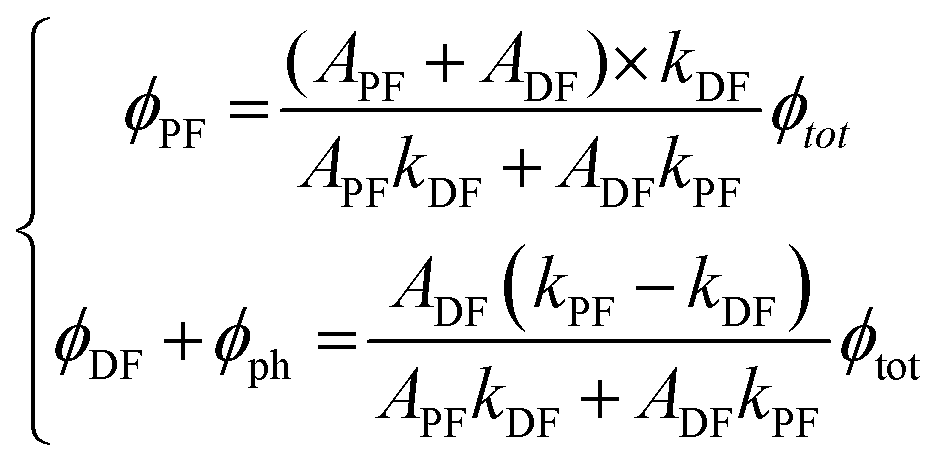 | (14) |
From this point on, it is possible to derive without any further assumptions the exact solutions from S1 and T1 decays:174
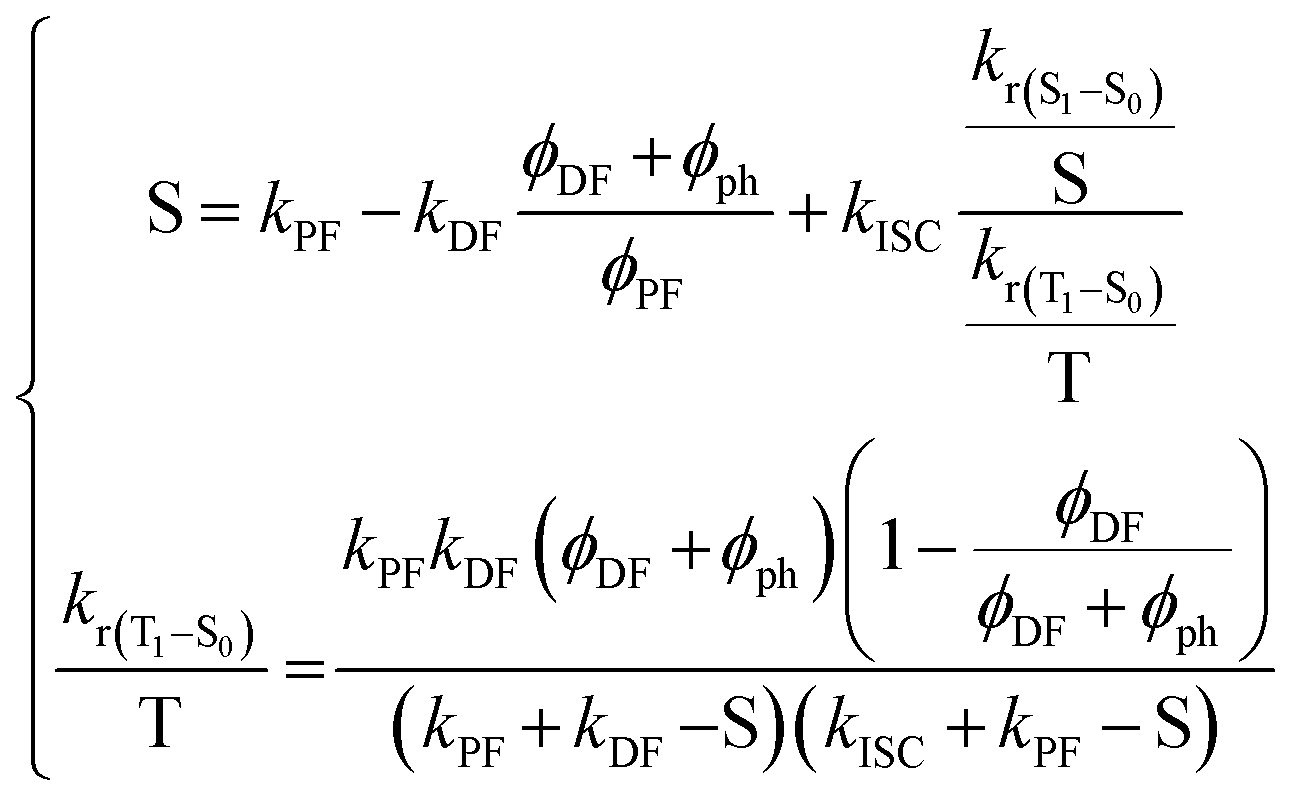 | (15) |
However, due to the presence of terms S and kISC in the equations as both independent and dependent variables, precise rates determination requires a numerical analysis. Nevertheless, in most TADF organic molecules the phosphorescence efficiency is close to zero (Φph ≅ 0), thus, the system of equations simplifies to eqn (16)–(20), which are the most commonly used:174
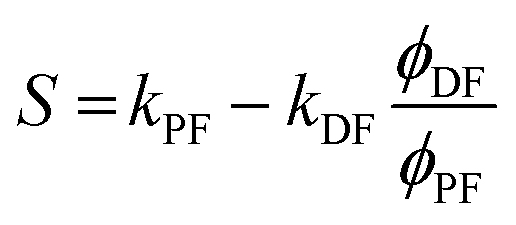 | (16) |
| kr(S1–S0) = kPFϕPF | (17) |
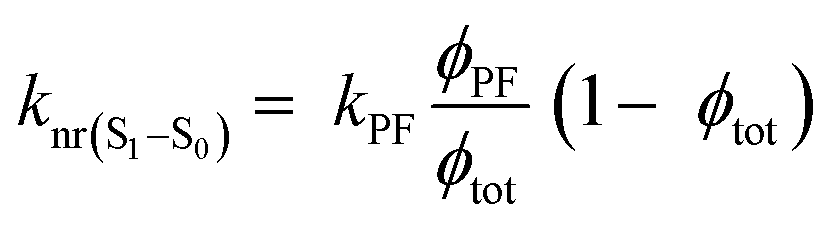 | (18) |
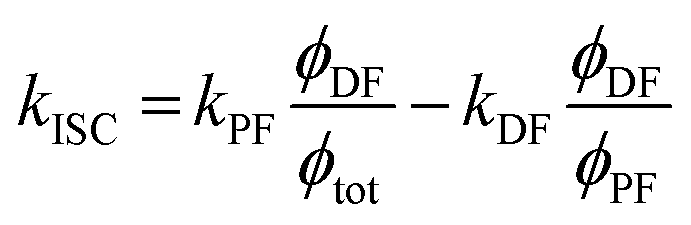 | (19) |
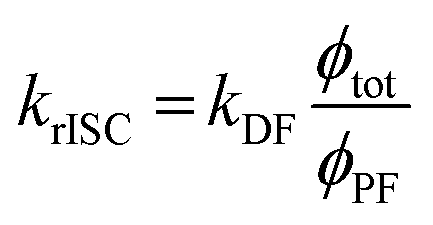 | (20) |
In summary, an exact solution of the kinetics in TADF materials is possible only numerically from steady-state and transient emission spectroscopy experimental data.
Within this case are lying also the metal complex-based TADF emitters with light atoms, such as Al, as demonstrated by the work of Sasabe – see next section.180 In such complexes a prompt fluorescent component is present – Fig. 7.
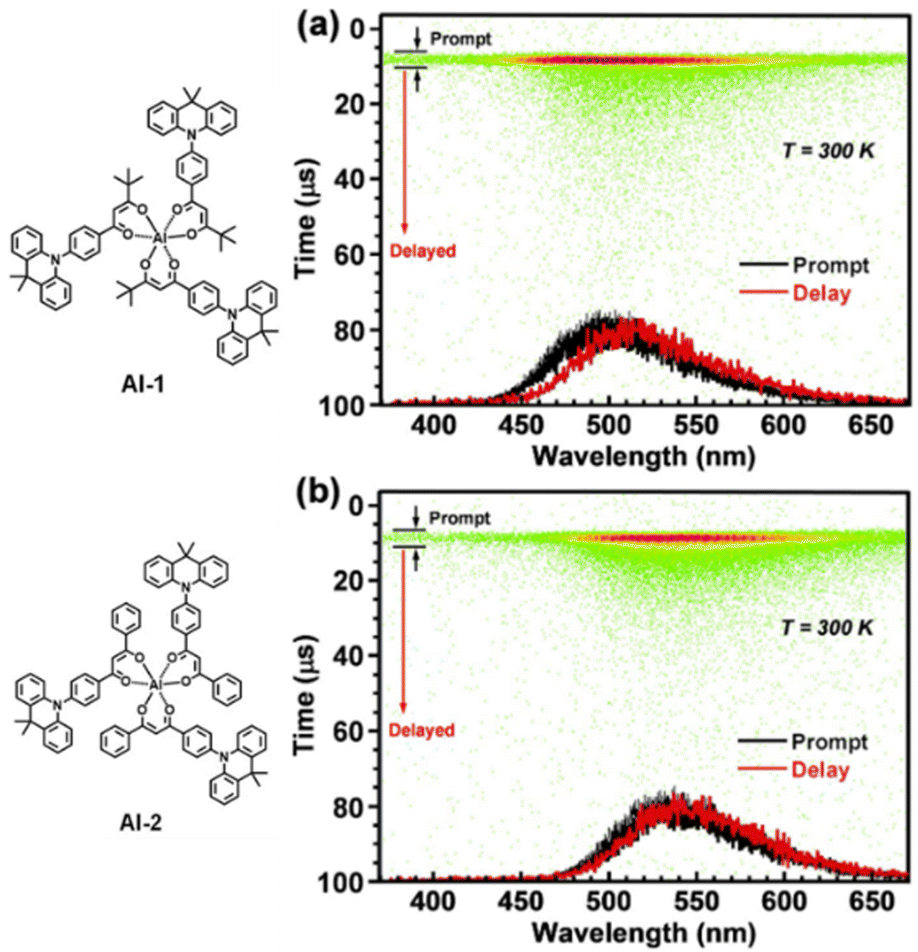 | ||
| Fig. 7 Streak images and prompt and delayed PL spectra at 300 K; green dots represent PL photon counts for (a) 30 wt% Al1-and (b) Al2-doped 4,4′-bis(n-carbazolyl)-1,1′-biphenyl (CBP) film. Reproduced with permission ref. 180. | ||
2.3. Which design rules must be followed for metal complex-based TADF emitters?
In general, metal complex-based TADF emitters are anticipated to possess several distinctive attributes: (i) heightened thermal stability and molecular rigidity through complexation and rigidification, (ii) enhanced PLQY via reinforcing acceptor aptitude and structural rigidity, (iii) anticipated reduction ΔEST by altering the acceptor moiety of conjugated ligands, and (iv) shorten of excited state decay lifetime due to the heavy atom effect (higher SOC). Similar to organic molecules, the golden design rule to follow is the spatial separation of the HOMO and LUMO. However, the dx-metal complexes design is complex due to the energy proximity of various transitions (i.e., MLCT, LLCT, etc.).In addition, it is important to consider that some families of complexes are prone to geometrical changes upon excitation (e.g. flattening distortion, change of coordination geometry/sphere). This results in new non-radiative deactivation pathways. This is reflected in high non-radiative rates and small PLQYs in low-energy emitting metal complexes, hampering their TADF study – Section 2.2.128 Rational design strategies to minimize or even circumvent these undesired geometrical changes will be, therefore, discussed below. Finally, the energy splitting between the singlet and triplet excited states, ΔEST, must be <1613 cm−1/0.2 eV to ensure efficient rISC at room temperature.181 This is mainly achieved by a small spatial overlap of the HOMO and LUMO orbitals and other orbitals involved in the lowest excited state transitions. These requirements are best fulfilled when the lowest excited states are of CT character (e.g., MLCT or LLCT). However, the reduction of ΔEST values below 200–300 cm−1 is not beneficial anymore; instead, such small ΔEST values lead to significantly decreased radiative rates, kr(S1 → S0) representing the third parameter. To obtain highly efficient TADF emitters, the allowedness of the S1 → S0 transition should be as high as possible.
This allows the achievement of short TADF decay times that are highly desired for device applications to avoid any adverse impact on their performances, e.g. roll-off effects, and stability problems – Section 3.128 However, quantum mechanical calculations reveal a close relation between kr(S1 → S0) and ΔEST. In detail, a small overlap of the HOMO and the LUMO benefits a small ΔEST, but results in a small oscillator strength of the S1 → S0 transition and large τTADF – vice versa; Fig. 8.128
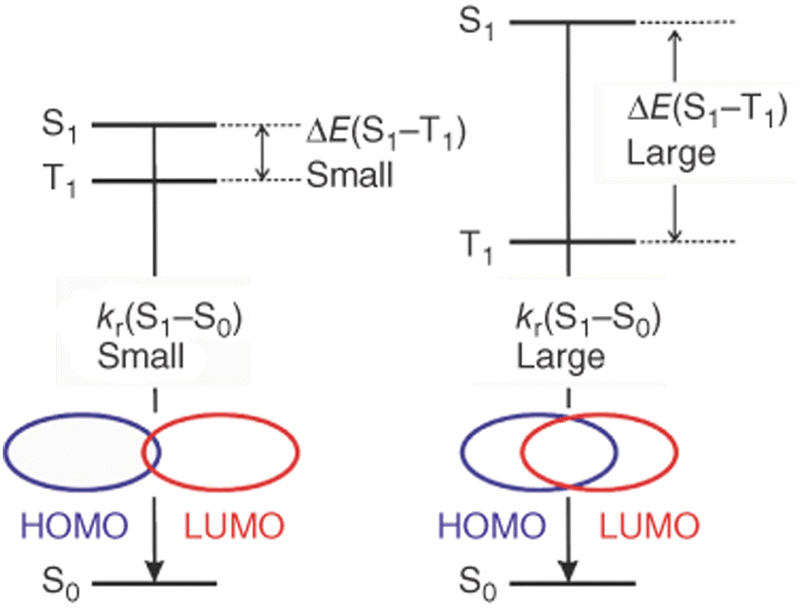 | ||
| Fig. 8 Schematic representation of the relationship between ΔEST, kr(S1 → S0), and the spatial overlap of the HOMO and LUMO – represented with permission ref. 181. | ||
Based on the above concepts and according to the population of the d-orbitals of the metallic core, several design rules have been identified. The following will summarize the molecular strategies to achieve TADF metal complexes up to date.
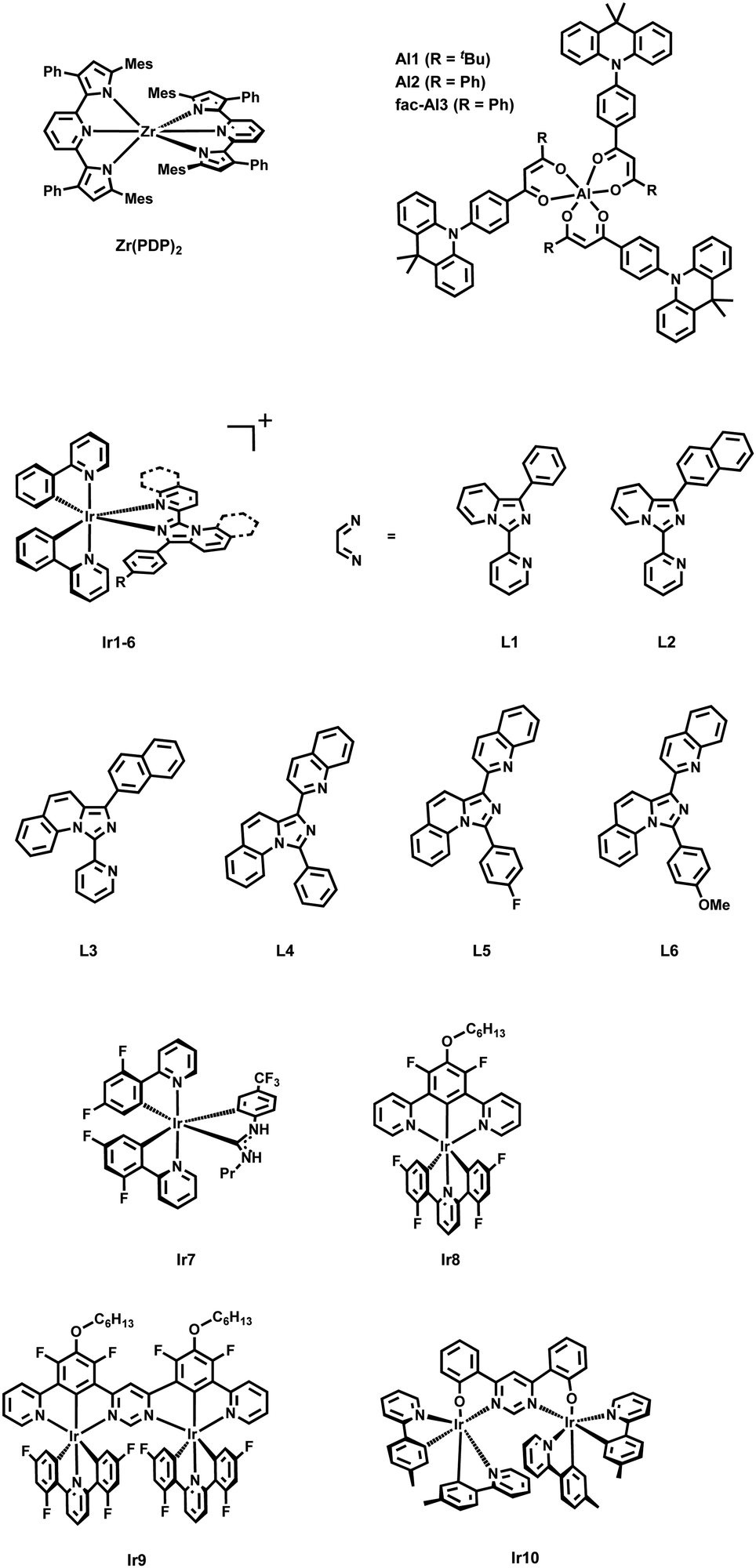 | ||
| Scheme 1 Structural representations of Zr(PDP)2, Al1–3 and Ir1–10. | ||
By contrast with the seminal field of TADF Zr(IV)-based materials, recent advancements have led to the development of mononuclear Al(III) complexes, showing promising properties in lighting devices. In 2020, Kido and Sasabe180 introduced a groundbreaking series of highly luminescent Al(III) complexes, incorporating phenylacridine-modified asymmetric acetylacetonate (acac)-type ligands (Al1 and Al2, Scheme 1). These complexes exhibited PLQYs of up to 79% associated to TADF, outperforming conventional ligand components. Notably, the shorter τDF of the complexes compared to that of the ligand suggests an enhancement in luminescence efficiency upon coordination with Al(III). The discovery of mononuclear Al(III) complexes exhibiting TADF behavior opens up new avenues for the design and development of advanced optoelectronic materials. The exceptional PLQYs and efficient TADF properties of these complexes hold promise for their application in OLEDs (Section 3). Two years later, the same group achieved a TADF Al(III) complex with near-unity PLQY (Al3, Scheme 1).183 They re-designed the chemical configuration of the β-diketone ligand by introducing an additional donor unit. The new complex emitted yellow light with a rapid radiative decay rate of 107 s−1, a brief delay time below 2.0 μs in solid form, and its OLEDs showed EQE surpassing 18%. So far, the design by Sasabe is the only one that has led to highly luminescent and TADF mononuclear complexes for OLED applications. In these examples, the TADF is metal-mediated as the spatial distribution of the ligands is locked so that LLCT and ILCT transitions become dominant and highly emissive. Thus, the D–A ligand design turns out to be a key aspect as in other metal complexes based on Pd(II) and Ag(I) – vide infra.
Despite the novel complexes have not been employed in lighting devices yet, their work is a milestone in the development of TADF Ir(III) complexes. To date, only other two families of mononuclear Ir(III) complexes have been proved to show TADF. First, a cyclometalated Ir(III) complex with two C^N ligands (C^N = 2-(2,4-difluorophenyl)pyridine, F2ppy) and one acyclic diamino carbene (ADC) ancillary ligand (Ir7) reported in 2018 by Teets’ group.190 In this context, it is worth to mention also the theoretical work by Zhang's group that nicely deepens the discussion about the excitate-state dynamic of Ir7 (Scheme 1).191 Indeed, it is very interestingly not only because it is the first example of metal-mediated nucleophilic addition to a coordinated isocyanide followed by orthometalation of the formed ADC in mild conditions, but also due to its highly emissive nature (PLQY of 79% in PMMA) with relatively short lifetime (0.9 μs). Again, it only exists a small overlap between HOMO and LUMO, which leads to small ΔEST. Combined with the proper strength of the SOC, the rate of rISC calculated from the triplet excited state to the singlet excited state is higher than the phosphorescence emission from the former to the ground state. This has been related to efficient TADF, reaching krISC of 1.86 × 109 s−1 that is among the highest rate also compared with tradition organic molecules – Fig. 9. Lastly, Shafikov and Kozhevnikov's groups reported a non-stereogenic dinuclear complexes that show TADF in both, the monomeric and dimeric forms in 2021 (Ir8–9, Scheme 1).192 Importantly, utilizing symmetrical dianionic C^N^C ligands simplified the synthesis process, avoiding the formation of detrimental diastereomers. Moreover, this approach eliminated the need for monodentate ligands, such as chlorides, which may contribute to direct SOC in the T1 state to ground state. In detail, they used 2,6-di(2,4,-difluorophenyl)pyridine as C^N^C ligand and 4,6-difluoro-5-n-hexyloxo-1,3-di(pyridine-2-yl)benzene as N^C^N for the mononuclear complex Ir8 and its pyrimidine derivatives for the dinuclear complex Ir9. At room temperature, they show kr of 1.50 × 105 s−1 and 6.1 × 105 s−1 for Ir8 and Ir9, respectively. These values are remarkably high with respect to other red-emitting Ir(III) complexes. In addition, the unstructured shapes of their emission spectra and their significant overlap with the corresponding S0 → S1 absorption bands indicate a charge-transfer character of the emitting state T1 and its close energetic proximity to the excited singlet states. Gao and Cui's groups demonstrated through detailed theoretical work that the energy of the triplet excited state is only slightly lower than the one of singlet excited (only 2.2 kcal mol−1).193 The d2 orbital is mostly located on the central Ir atom and the phenyl group of the N^C^N ligand, whereas the π3* orbital is spread across the entire length of the N^C^N ligand. This leads to a poor HOMO–LUMO overlap. They also estimated the krISC of Ir8 to be as high as 2.29 × 109 s−1, sufficiently high to guarantee an efficient TADF mechanism at room temperature.
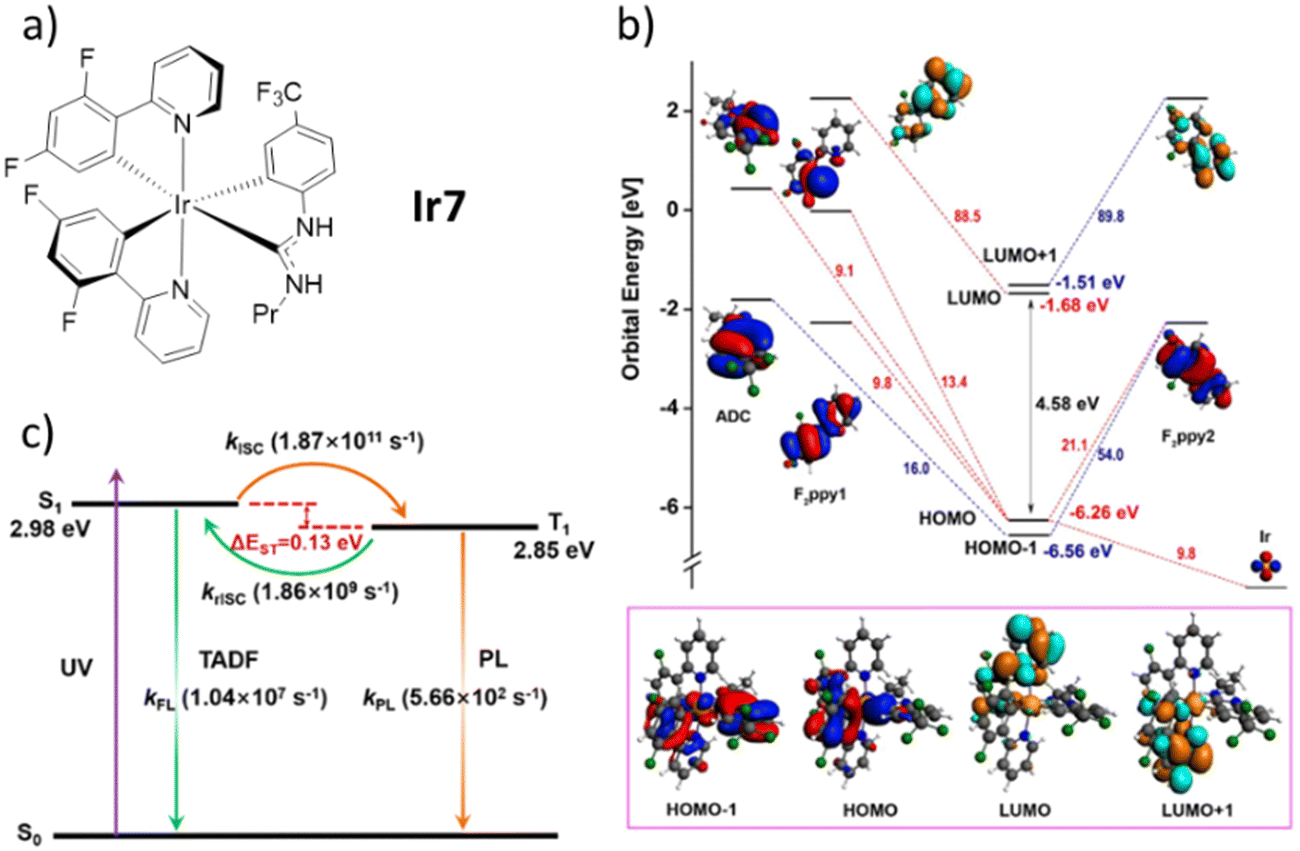 | ||
| Fig. 9 (a) Structure of Ir7. (b) Fragment analysis of frontier molecular orbitals at the S0 minima for compound Ir7. Also shown are the molecular orbital energies (in eV). (c) TADF working mechanism for the Ir7. Reproduced with permission from ref. 190 and 191. | ||
Although the experimental insights for a TADF mechanism of Ir9 were only limited, Kozhevnikov's group, in a recent follow-up paper, confirmed the behavior more in detail in a close-related dinuclear complex (Ir10, Scheme 1) using 4,6-bis(2-hydroxyphenyl)pyrimidine as bis-N^O bridging coordinating ligand.194 In this case, the short linker has been useful also to perform a diasterospecifc reaction, leading to only the rac-ΛΛ or rac-ΔΔ. The temperature-dependent steady-state spectra indicate the presence of two luminescent parts: a broad, featureless higher-energy part prevalent at elevated temperatures, and a lower-energy part with a noticeable vibronic shoulder at lower temperatures. Furthermore, the variation of the observed excited state lifetime with temperature follows a Boltzmann's distribution, confirming the fast equilibrium between the two states. Finally, OLEDs were fabricated (Section 3). These seminal works expand the comprehension of potential emissive routes in mononuclear and dinuclear Ir-complexes, moving beyond the model reliant on SOC. TADF emerges as a probable yet overlooked pathway in such complexes led by the poor HOMO–LUMO overlapping.
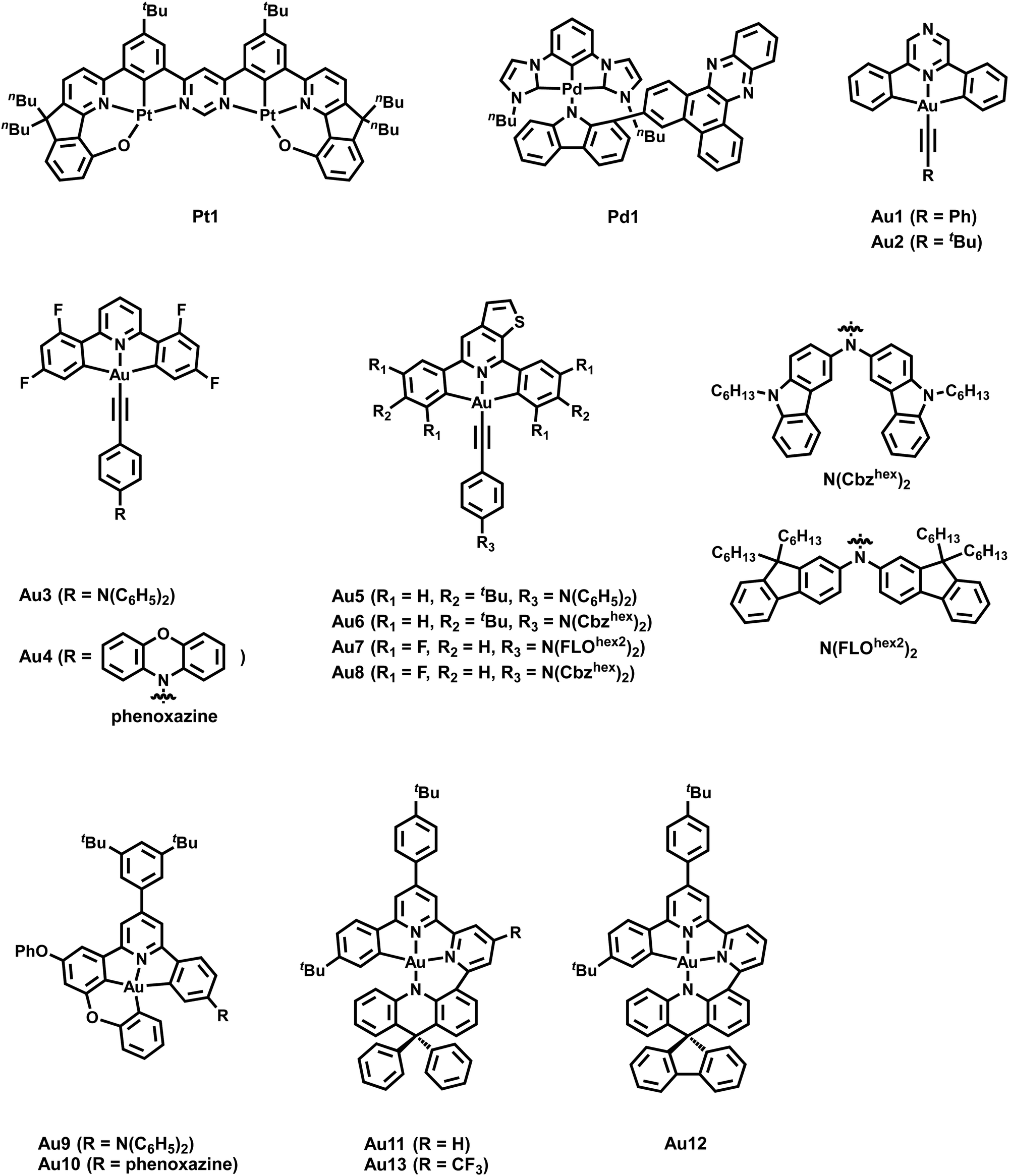 | ||
| Scheme 2 Molecular structures of Pd1, Pt1 and Au1–13. | ||
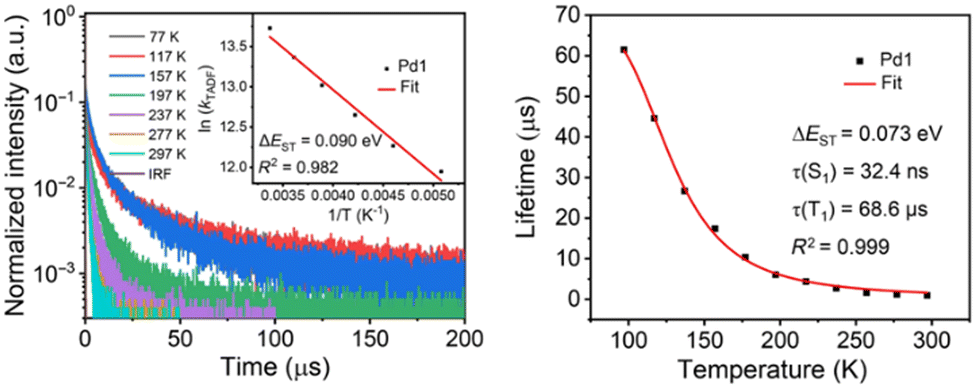 | ||
| Fig. 10 Left: Temperature-dependent transient PL characteristics of Pd1 in PMMA films. Inset: Arrhenius fit of the kDF value vs. temperature. Right: Boltzmann-type fitting of the emission lifetimes of Pd1 in PMMA film at various temperatures. Printed with permission from ref. 200. | ||
| Complex | λ max (298 K)/λmax (77 K) [nm] | PLQY [%] | τ DF [μs] | ΔEST [cm−1] | k r × 105 [s−1] | k nr × 105 [s−1] | Ref. |
|---|---|---|---|---|---|---|---|
| a 5 wt% of Pd1 doped polymethyl methacrylate (PMMA). b 0.1 wt% of Pt1 in Zeonex® films. | |||||||
| Zr(PDP)2 | 595 | 45 | 350 | 1652 | 281 | — | 182 |
| Al1 | 497 | 65 | 4.4 | 1694 | — | — | 180 |
| Al2 | 534 | 79 | 3.9 | 645 | — | — | 180 |
| Al3 | 554 | 92 | <2 | 516 | 210 | 183 | |
| Ir1 | 465, 560 | 6 | 0.54 | <800 | — | — | 189 |
| Ir2 | 475, 560 | 2 | 0.30 | <800 | — | — | 189 |
| Ir3 | 470, 615 | 6 | 0.61 | <800 | — | — | 189 |
| Ir4 | 465, 575 | 12 | 0.68 | <800 | — | — | 189 |
| Ir5 | 465, 575 | 2 | 0.58 | <800 | — | — | 189 |
| Ir6 | 470, 575 | 1 | 0.41 | <800 | — | — | 189 |
| Ir7 | 498/456 | 22 | 0.90 | 1048 | 104 | — | 191 |
| Ir8 | 537/540 | 15 | 1.00 | 800 | 1.5 | 8.5 | 192 |
| Ir9 | 642/645 | 80 | 1.31 | — | 6.1 | 1.5 | 192 |
| Ir10 | 643 | 30 | 0.85 | 556 | 49 | — | 194 |
| Pd1 | 637/— | 89 | 0.97 | 726/589 | 9.2 | 1.1 | 200 |
| Pt1 | 640/— | — | 2.3 | 484 | — | — | 201 |
Likewise, Pt(II) complexes are widely used to exploit their phosphorescent-dominated emissions, but only little is known about their TADF. As revealed by previous studies, the introduction of a second metal center was found to strongly increase the kr constant and concomitantly reduce ΔEST.202,203 Dias and coworkers successfully applied this strategy, whilst obtaining a new dinuclear Pt(II) TADF complex, Pt1 – Scheme 2, with remarkably small energy splitting of 556 cm−1 and short excited state lifetimes in the range of 1–2 μs – Table 1.201 Both, the Pt(II) d-orbital admixtures in the ILCT character of the lowest-energy transition and the small HOMO–LUMO overlap strongly favor the TADF emission in the reported complex. In a subsequent study of the same group, a similar dinuclear Pt-complex was found to display a dual emission involving TADF from S1 state and phosphorescence from the T1 state.204
Finally, Au(III) exhibits low-lying d orbitals as further reflected in its relatively high reduction potential. Consequently, the lowest excited states are mostly LC with small metal contributions and thus, efficient phosphorescence displays the main emissive pathway further favored by its high SOC constant. The concomitant long triplet emission lifetimes limit their performance in OLEDs as roll-off effects are evident.205–212 In 2015, Bochmann and coworkers reported the first Au(III) complexes with TADF at room temperature.213 Alkynyl Au(III) complexes containing pincer-like pyrazine moieties (C^N^C) for the first time, Au1–2 – Scheme 2, showed a blue-shifted emission behavior upon a temperature increase from 77 to 298 K in the solid state. The transitions are characterized by metal-perturbed LLCT nature (π(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh) → π*(C^Npz^C)). Distinct modulations in solution via protonation or the addition of AgOTf or CuOTf lead to enhanced TADF emission arising from the coexistence of high energy TADF and 3IL (C^Npz^C)/3LLCT (X → C^Npz^C) transitions.
CPh) → π*(C^Npz^C)). Distinct modulations in solution via protonation or the addition of AgOTf or CuOTf lead to enhanced TADF emission arising from the coexistence of high energy TADF and 3IL (C^Npz^C)/3LLCT (X → C^Npz^C) transitions.
In the following years, research started to focus on changing the nature of the lowest excited state of cyclometalated (C^N^C/C^C^N) Au(III) complexes to induce efficient TADF. In detail, emissions from 3IL states with rather long-lived excited state lifetimes and small kr rates mainly dominate the emission pathway of cyclometalated Au(III) complexes. To achieve CT nature of the lowest excited state, several strategies to increase (i) the energy of the 3IL states, (ii) the energy difference between the 3IL and 3LLCT states and/or (iii) to lower the energy of the 1/3LLCT states were assessed. In this context, Che et al. reported several cyclometalated Au(III) complexes with donor-substituted amino-alkyl auxiliary ligands.214,215 Due to their limited thermal stability, Che's group focused on alkynyl ligands owing to the stronger Au(III)–Csp(acetylide) bond character, Au3–4 (Scheme 2).216 They hypothesized that the combination of 2,6-bis(2,4-difluorophenyl)pyridine or 2,6-bis(2,4-diterbutylphenyl)pyrazine accepting-pincer ligands (C^N^C) with alkynyl ligands bearing strong donor groups, e.g. p-NPh2, m-NPh2, phenoxazine, provoke higher 3IL states and larger energy separations between 3IL/3LLCT states that can facilitate TADF. The spatially well-separated donor (alkynyl) and acceptor (C^N^C) units led to efficient TADF emitters, Au3–4, which was substantially supported by DFT/TD-DFT calculations. The latter also gives evidence for the high rotational flexibility of the auxiliary ligand, owing to the presence of the CC bond and thus, an increased distance between the two ligands. This results in decreased steric hindrances between the protons of the phenyl moiety (alkynyl ligand) and those of phenyl rings of C^N^C. In the case of Au3, the energy splitting ΔE(S1–T1) was found to strongly relate with the dihedral angle between the C^N^C plane and the phenyl group connected to the CC bond, δ. For instance, ΔEST drastically decreases from 2660 cm−1 (δ = 5.4°) to 182 cm−1 for δ of 101°. In a subsequent work, Yam and coworkers presented several in-depth studies involving DFT/TD-DFT calculations and nanosecond TA measurements to rationalize the design of cyclometalated (C^N^C/C^C^N) Au(III) alkynyl/carbazolyl TADF emitters.217–219 Among other ligands systems, we exemplarily discuss how the nature of the excited states of three alkynyl Au(III) complexes, Au5–8 (Scheme 2), were manipulated to enable TADF. As indicated by the vibronic-structured emission band of Au5 and its relatively long excited state lifetime of 96.1 μs with a small kr of 5.93 × 103 s−1, TD-DFT calculations further confirmed that the emission originates from 3IL states [π → π* (C^NTHPY^C)]. Next, the donor strength of the alkynyl ligands was increased by replacing the triphenylamine unit with a Cz-triphenylamine moiety, Au6–8, to destabilize the HOMO energy. This finally results in achieving energetically lower lying 1/3LLCT [π(fused heterocyclic akynyl) → π* (C^NTHPY^C)] states and thus TADF is induced (Fig. 11) as indicated by short excited state lifetimes of 0.4–5.7 μs and high kr up to 1.00 × 106 s−1. This was further corroborated by temperature-dependent studies of Au6 and Au7, as the same structureless and broad emission band evolved at 77 K, but with a small shoulder around 525 nm that was assigned to 3IL transitions.
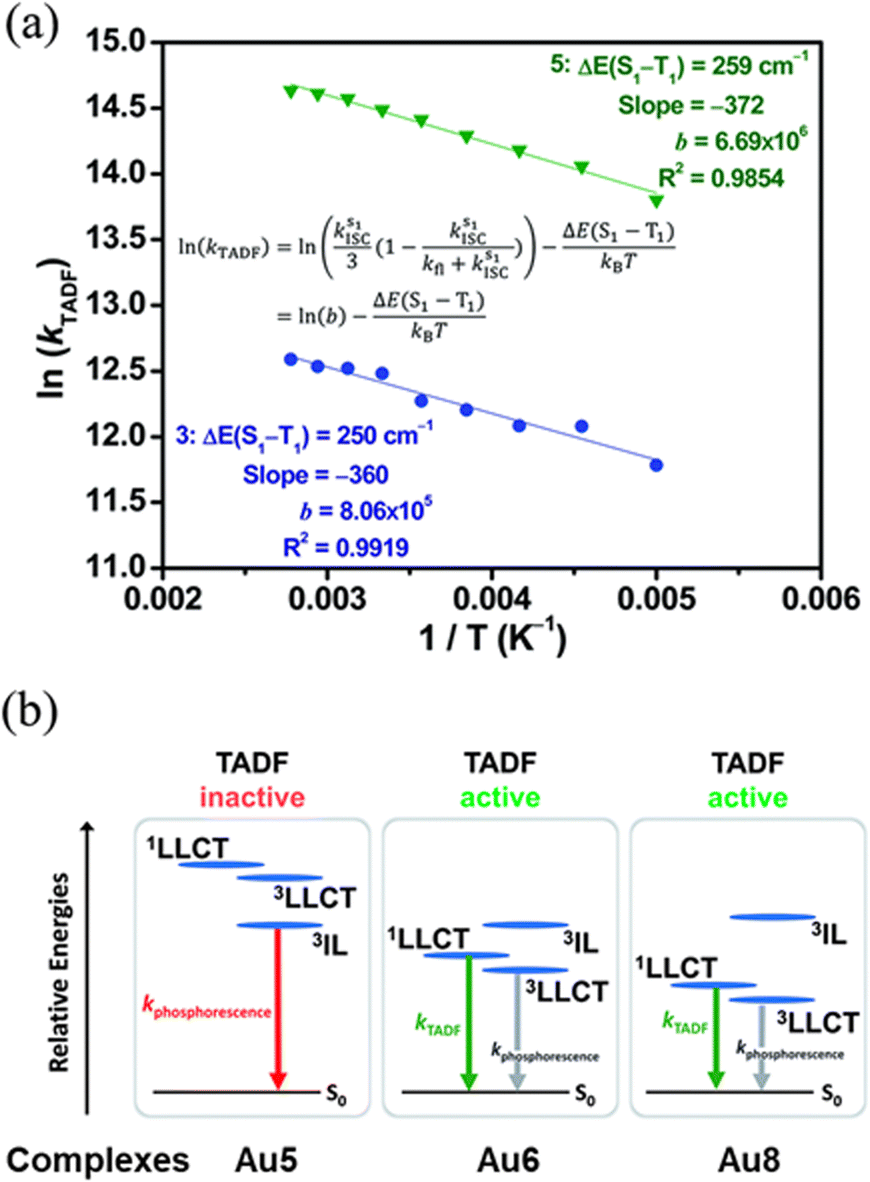 | ||
| Fig. 11 (a) Plot of ln(kDF) vs. 1/T of Au6 () and Au7 () in 5 wt% doped m-N,N′-dicarbazolyl-3,5-benzene (mCP) thin-films monitored at 560 and 570 nm, respectively, and the fits of the temperature-dependent lifetime data from 200 to 360 K. (b) Qualitative state diagram depicting the relative energies of the 1,3LLCT states and the 3IL state of Au5, Au6 and Au8 in the solid state at 298 K. The trend of the relative energies of the states is based on the results from the emission studies. Printed with permission from ref. 217. | ||
The ΔEST was then estimated with an Arrhenius plot derived from a full kinetic scheme for two-level systems – Fig. 11, but only in the range from 200 to 360 K to reduce the contribution of the 3IL excited state in this fitting procedure. The experimentally obtained ΔEST value of 250 cm−1 (Au6, Scheme 2) and 259 cm−1 (Au7, Scheme 2) is in agreement with the one obtained from TD-DFT (ΔEST,calc = 435 cm−1 for Au6). A further increase of the energy splitting between 3IL and 1/3LLCT was achieved by replacing the tert-butyl-substituted cyclometalating ligand in Au6 by the flour-substituted one that gives Au7 and Au8 – Scheme 2, Fig. 11 and Table 2.
| Complex | λ max (298 K)/λmax (77 K) [nm] | PLQY [%] | τ T/τDF [μs] | ΔEST [cm−1] | k r × 105 [s−1] | k nr × 105 [s−1] | Ref. |
|---|---|---|---|---|---|---|---|
| a PMMA thin-film samples, 4 wt% of the complex. b Tris(4-carbazolyl-9-ylphenyl)amine (TCTA): 1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene (TPBi) thin-film samples, 4 wt% of the complex. c mCP thin-film with 5 wt% of the complex. d TCTA thin-film with 8 and 4 wt% for Au9 and Au10, respectively. e Obtained from TD-DFT calculations. | |||||||
| Au1 | 523/530 | 4.5 | — | — | — | — | 213 |
| Au2 | 523/532 | 8.3 | — | — | — | — | 213 |
| Au3 | 577/— | 88 | —/0.85 | — | 10.35 | — | 216 |
| Au4 | 567/— | 65 | —/1.46 | — | 4.45 | — | 216 |
| Au5 | 528, 568, 616/— | 57 | 69.1/— | 0.059 | 0.045 | 217 | |
| Au6 | 562/— | 87 | —/5.7 | 250 | 1.56 | 42.1 | 217 |
| Au7 | 585/ | 70 | —/0.6 | 259 | 5.38 | 2.31 | 217 |
| Au8 | 636/— | 40 | —/0.4 | — | 1.00 | 15.0 | 217 |
| Au9 | 520/— | 82 | —/2.08 | 686e | 3.94 | — | 220 |
| Au10 | 568/— | 71 | —/2.54 | — | 2.80 | — | 220 |
| Au11 | 595/— | 73 | —/1.2 | 1129 | 6.1 | 221 | |
| Au12 | 598/— | 63 | —/2.0 | 1129 | 3.2 | 221 | |
| Au13 | 650/— | 12 | —/1.4 | — | 0.86 | 221 | |
In fact, the stability of metal complexes can be improved by increasing the chelating behavior ligands. The use of rigid tetradentate ligands involving C-donor atom(s) is known to strengthen the metal–ligand bond character, resulting in higher thermal stabilities and restricted excited state structural distortions. Onda and coworkers recently suggested that such rigid structures can also reduce the activation energy barrier for rISC and favor efficient TADF.222 Consequently, Che and others started to work on tetradentate ligand frameworks for Au(III) complexes.223 First, Che's group implemented a microwave-assisted C–H activation step leading to a new synthetic pathway toward tetradentate (C^C^N^C) Au(III) complexes – Au9–10; Scheme 2 and Table 2.
The introduction of the alkoxy group in the 3-position of the phenyl ring of the C^N^C moiety of the C^C^N^C ligand scaffold was considered as key to obtain a superior rigidity as 5-5-6 membered chelating rings are formed. In terms of design rules toward TADF, the incorporation of a donor group, e.g. DMA, Au9, or phenoxazine, Au10, in the phenyl ring trans to the pyridine unit was proven to be paramount for spatially well-separated HOMO and LUMOs and thus, small ΔEST values. Indeed, ΔEST = 686 cm−1 and excited states of 1/3LLCT [π(donor unit) → π* (C^N^C)] character confirmed the presence of TADF. These findings agree well with the lifetimes of ca. 2 μs associated with PLQYs up to 82%, Au9, and radiative decay rates up to 3.94 × 105 s−1 in thin-film states. In 2023, Yam and co-workers demonstrated how to obtain low-energy emitting Au(III) complexes without sacrificing PLQYs and TADF behavior, Au11–13 – Scheme 2.221 The use of the more rigid tetradentate ligand scaffold (e.g., C^C^N^N) instead of the pincer-like ones in combination with strong donating acridinyl moieties resulted in (i) orange to deep-red emitting Au(III) complexes with PLQYs up to 76% and radiative decay rates in the range of 105 s−1, and (ii) decreased ΔEST owing to the stabilization of the S1 states. The latter can be mainly rationalized by the increased energy difference between the IL and ILCT excited states, as already discussed above. In addition, the color tuning was shown to be more effective when introducing electron-withdrawing groups in the pyridine unit instead of incorporating electron-donating groups in the acridinyl moiety.
Over the last two decades, a large number and variety of Cu(I) complexes, including different geometries and nuclearities, have shown TADF. This can be rationalized by the spatially well-separated HOMO and LUMOs. In addition, owing to the energetically high-lying 3d orbitals of Cu(I), they strongly contribute to the nature of the frontier orbitals, and hence, the low-energy transitions are often of CT character, e.g. MLCT, (M+X)LCT or LLCT. In this context, the relatively small SOC of copper plays a key role therein as it is large enough to ensure efficient rISC to populate S1 owing to the involvement of Cu(I) in the frontier orbitals but is small enough to hamper efficient transitions from T1 to S0. However, the choice of ligands and coordination motifs can lead to scenarios, in which the SOC becomes large, and thus, the emission is not only ruled by TADF but also phosphorescence.181 Among mononuclear complexes, linear/two-coordinated and four-coordinated Cu(I) complexes display the most prominent classes, while less attention has been devoted to trigonal/three-coordinated complexes.
Among four-coordinated complexes, heteroleptic cationic as well as their neutral analogs display the most studied representatives. Heteroleptic cationic Cu(I) complexes, [Cu(N^N)(P^P)]+, typically feature chelating diphosphine ligands (P^P) – e.g. dppbz, POP (= bis[2-(dipenylphosphino)phenyl]ether), xantphos = 4,5-bis-(diphenylphosphino)-9,9-dimethylxanthene, and modifications thereof – and bidentate diimine ligands (N^N), e.g. diimines, phenanthroline – Fig. 12.
 | ||
| Fig. 12 Diimine ligands used in Cu-complexes for color tuning. | ||
The HOMO is predominantly of 3d-nature of the Cu(I) orbitals with minor contributions of the P atoms of P^P, while the LUMO is solely located on the π* system of the diimine ligand (N^N).172,225,226 As confirmed by various in-depth TD-DFT/DFT calculations, the lowest excited states are mainly of MLCT character, and in combination with the distinct spatial separation of the HOMO and LUMO, a small energy splitting is obtained, resulting in efficient TADF. In terms of color tunability, the emission colors are adjusted by de-/stabilizing the LUMO energy, since chemical modifications of the diimine ligands are more straightforward compared to diphosphines. The resulting large variety of diimine ligands, e.g. bipyridines, phenanthroline, pyridine/pyrazine-pyrazole, biquinolines, dipyridylamines, and derivatives thereof, gave access to emission colors of the corresponding Cu(I) complexes covering the whole visible and NIR regions – Fig. 12.227
In this context, Costa and coworkers developed a multivariate analysis tool to predict photoluminescent and electroluminescent key parameters, such as PLQYs and λem, for a given molecular structure. This was exploited to refine a highly blue emissive Cu(I) complex for LECs – Fig. 13 and Section 3.228
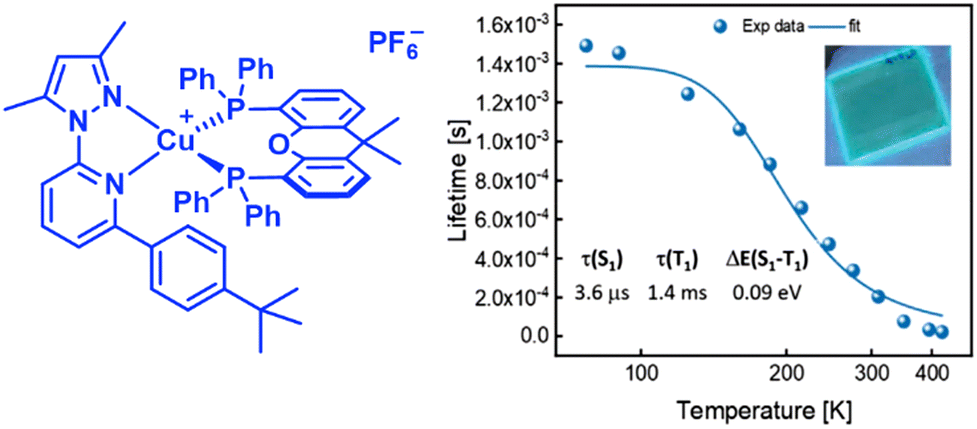 | ||
| Fig. 13 Left: Structural representation of a highly emissive blue-emitting Cu(I) complex. Right: 〈τ〉 vs. temperature plot the corresponding thin-film fitted with eqn (6), thin-film under UV irradiation (λex = 305 nm) is depicted as inset. Reproduced with permission from ref. 228. | ||
As a tremendous number of four-coordinated Cu(I) complexes have been explored over the last two decades, this section mainly focus on the design strategies to unlock TADF emission in these complexes combined with the most prominent examples. Interested readers are highly recommended to refer to the excellent previous reviews targeting emissive Cu(I) complexes as a complement to this review.90,128,172,226,229,230
As noted above, lifetimes in the sub-microsecond range and high PLQYs are paramount for efficient TADF emitters and display a crucial requirement for the validity of the fitting procedures – Section 2.1. In this regard, major attention has been devoted to strategies to minimize the undesired flattening distortion of Cu(I) complexes upon excitation.231 In detail, upon excitation a weakening of the Cu–P σ-bond and a geometrical change from (pseudo-)tetrahedral to square-planar, e.g. Jahn–Teller distortion, due to the partial oxidation of Cu(I), is typically noted. As a consequence, fast non-radiative decay rates are reflected in drastically decreased PLQYs. First, in-depth studies on heteroleptic [Cu(P^P)(N^N)]+ complexes revealed the importance of using chelating and large bite-angle diphosphines like POP and xantphos ligands due to (i) the prevention of ligand redistribution processes and the formation of undesired homoleptic complexes and (ii) improved photophysical properties, especially increased PLQYs – Cu1–2, Scheme 3 and Table 3.232–234 In this regard, Zysman-Colman and coworkers recently studied the influence of large bite angles diphosphines, e.g. POP (102°), xantphos (112°), homoxantphos (102°), as well as their flexibility range on the photophysical and EL properties. The heteroleptic [Cu(homoxantphos)(dmp)]+ complex, Cu3 – Scheme 3, displays the highest PLQYs and is the most interesting candidate in terms of electroluminescence – Section 3.235 Further improvements can be obtained by using matrix materials like cages of rigid microenvironments236–238 or performing changes at the molecular level. For instance, the introduction of bulky groups, e.g. alky, methoxy, or phenyl groups, at the ortho position of the diimine ligands leads to a 5-fold increase of the PLQYs, e.g.Cu2–8 – Scheme 3, since the increased bulkiness of the N^N ligands drastically suppresses structural distortions upon excitation.239 Among others, Constable and Housecraft further expanded the library of substituted diimine ligands – e.g. including alkenyls, akinyls, and thioethers.240–242
 | ||
| Scheme 3 Molecular structures of Cu1–27. | ||
| Complex | λ max (298 K)/λmax (77 K) [nm] | PLQY [%] | τ Ph/τDF [μs] | ΔEST [cm−1] | k r × 104 [s−1] | k nr × 104 [s−1] | Ref. |
|---|---|---|---|---|---|---|---|
| a Ground powder sample. | |||||||
| Cu1 | 541/— | 13 | —/8.2 | — | 1.58 | 10.6 | 243 |
| Cu2 | 556/— | 15 | —/4.5 | — | 3.33 | 18.9 | 243 |
| Cu3 | 576/— | 35 | — | — | — | — | 235 |
| Cu4 | 527/— | 49 | —/13.2 | 244 | |||
| Cu5 | 519/— | 69 | —/20.3 | 244 | |||
| Cu6 | 580/— | 3.0 | —/1.5 | 245 | |||
| Cu7 | 535/— | 43 | —/10.5 | 239 | |||
| Cu8 | 555/575 | 55 | 87/11 | 720 | 5.0 | 4.1 | 239 |
| Cu9 | 530/562 | 80 | 240/14 | 1000 | 5.7 | — | 246 |
| Cu10 | 535/535 | 70 | 1200/3.3 | 370 | 21 | — | 247 |
| Cu11 | 540/540 | 90 | 680/5 | 600 | 18 | 2.0 | 231 |
| Cu12a | 475/490 | 76 | 34/11 | 710 | 6.9 | 2.2 | 248 |
| Cu13 | 575/585 | 73 | 18/21 | — | 4.1 | 1.5 | 248 |
| Cu14 | 463/— | 22 | 45/13 | 2823 | 1.7 | 6.0 | 249 |
| Cu15 | 458/— | 86 | 81/44 | — | 1.9 | 0.3 | 249 |
| Cu16 | 473/483 | 15 | 32/6 | 565 | 2.5 | 14.2- | 250 |
| Cu17 | 474/482 | 73 | 38/14 | — | 5.2 | 1.9 | 250 |
| Cu18 | 503/519 | 86 | 87/13 | 484 | 6.6 | 1.1 | 250 |
| Cu19S/R | 579/580 | 55 | 900/2.5 | 680 | 22 | 18 | 251 |
| Cu20S/R | 606/603 | 53 | 1600/1.7 | 490 | 31 | 27 | 251 |
| Cu21 | 502/505 | 80 | 211/4 | 252 | |||
| Cu22 | 520/524 | 60 | 2300/4.3 | 252 | |||
| Cu23 | 533/537 | 60 | 2400/4.2 | 252 | |||
| Cu24 | 506/513 | 45 | 220/6.6 | 460 | 6.8 | — | 253 |
| Cu25 | 490/498 | 65 | 930/4.1 | 510 | 16 | — | 253 |
| Cu26 | 464/472 | 65 | 270/4.6 | 570 | 14 | — | 253 |
| Cu27 | 465/465 | 65 | 250/5.6 | 630 | 12 | — | 253 |
| Cu28mono | 548/— | 58 | 2450/6.0 | 490 | 180 | 7.0 | 254 |
| Cu28dimer | 577/— | 80 | 2300/1.2 | 390 | 1130 | 67 | 254 |
Yersin and coworkers presented a design strategy to increase the overall rigidity of the coordination sphere. In short, the bulky diphosphine, phanephos, was employed to build a rigid “semicage” around the Cu(I) center in combination with dmp, Cu9 – Scheme 3. Table 3 and Fig. 14. The mutual steric interactions between the P^P and dmp drastically minimize radiationless relaxations that result in high PLQYs up to 80% and record small non-radiative decay rates of 1.4 × 104 s−1 without exceeding the required ΔEST of 1000 cm−1. In addition, the authors noted that the emission scenario at ambient temperature is characterized by a mixture of TADF and phosphorescence. The ratio of S1/T1 was then estimated to be 20 by applying the following equation:246
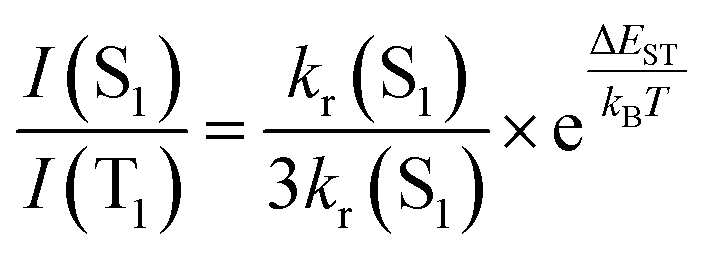 | (21) |
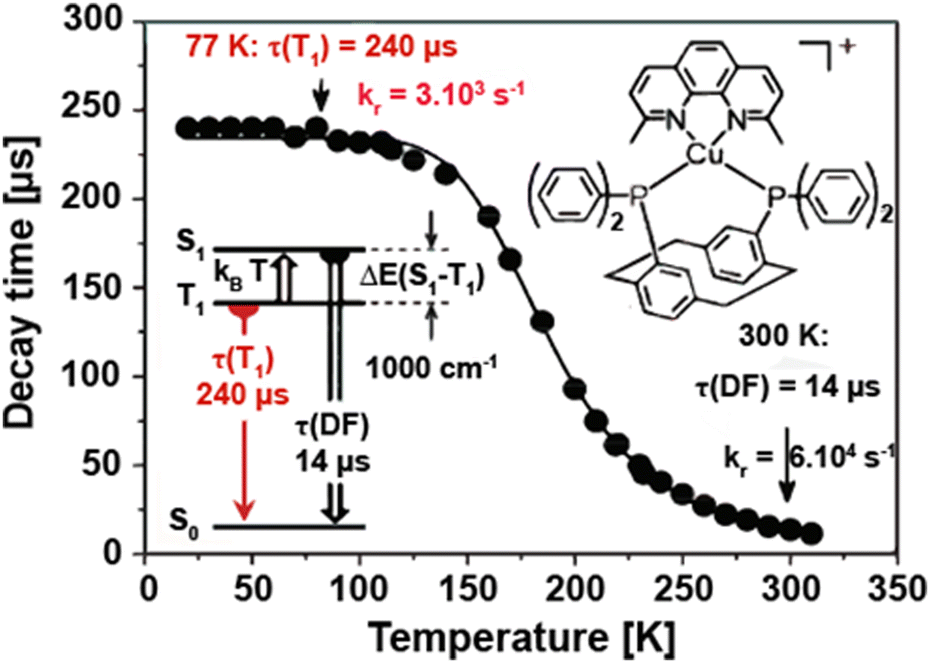 | ||
| Fig. 14 Emission decay time of Cu9 in powder state vs. temperature. The sample was excited with a pulsed UV laser at λex = 355 nm (pulse width 7 ns). The emission was detected at λdet = 550 nm. The solid line represents a fit of eqn (6) to the experimental data with the phosphorescence decay time τ(T1) = 240 μs measured at 77 K. The resulting fit parameters are ΔEST = 1000 cm−1 and τ(S1) = 40 ns, representing the magnitude of the singlet–triplet splitting and the decay time of the spontaneous S1 → S0 fluorescence, respectively. The spontaneous fluorescence is not observed directly due to much faster intersystem crossing. τ(DF) = 14 μs is the decay time of the delayed fluorescence at ambient temperature. Reproduced with permission from ref. 246. Copyright 2013, Royal Society of Chemistry. | ||
However, Cu9 already demonstrates the challenge of achieving high radiative rates, without sacrificing small ΔEST values to obtain efficient TADF. In this context, a neutral heteroleptic, four-coordinated TADF Cu(I) emitters, Cu10 – Scheme 3, with a relatively small ΔEST of 370 cm−1 and short excited state lifetimes of τ = 3.3 μs was presented.247 The small ΔEST value originates from a small overlap of the HOMO and the LUMO. The former features a 3d metal character with significant contribution from the nitrogen atoms of the anionic pyrazole-substituted phenyl borate ligand and the phosphorous atoms of dppbz. The LUMO is solely localized on the phenyl backbone ring of the dppbz ligand. As a consequence of the small HOMO–LUMO overlap and the related small exchange interactions, the S1 → S0 transition is less favorable. Indeed, this is reflected in the calculated oscillator strength of f = 0.0016 and the relatively small fluorescence decay rate of 3.9 × 106 s−1 obtained from the three-state Boltzmann-type fitting – eqn (6).128,247,255
An alternative strategy to obtain high PLQYs, short τTADF while maintain a small ΔEST was also presented by Yersin and coworkers. They designed a new rigid tridentate N,P,P-ligand, 3,5-dimethyl-1-(2-((2-(di-otolyl)phosphanyl)(o-tolyl)-phosphanyl)phenyl)-1H-pyrazole, to achieve a TADF Cu(I) complex with small freedom for distortion in the excited states.231 Further rigidity was achieved by employing the anionic thiophenolate ligand. The resulting TADF Cu(I) complex Cu11 (Scheme 3) exhibits PLQYs up to 90% in the solid state with high TADF decay rates of 1.8 × 105 s−1 without the expected increase of ΔEST. The calculated small ΔEST of 600 cm−1 is favored by (i) the spatially well-separated HOMO (Cu(I) center + thiophenolate) and LUMO (phenyl backbone of the N,P,P ligand) and (ii) the MLCT character of the lowest excited states.
Concerning tri-coordinating Cu(I) complexes, Yersin, Gaillard, and Costa introduced a new family of neutral and cationic three-coordinated Cu(I) complexes bearing bidentate nitrogen-based ligands, e.g. bis-pyridyl, dipyridylamine, phenanthroline, bipyridine, and modifications thereof, and N-heterocyclic carbene (NHC) ligands.248,256,257 The (i) good σ-donating and π-accepting properties of carbene ligands and (ii) the possibility to introduce bulky substituents in the carbene backbone strongly contribute to the high stability of the resulting NHC–Cu(I) complexes.
While the first neutral NHC–Cu(I) complexes that bear bidentate nitrogen-based ligands solely displayed phosphorescence,256,257 Yersin and coworkers shed light on the crucial parameters that rule the TADF behavior in these complexes.248 In short, it was found that ΔEST is marginally affected by electronic substitution on the NHC ligands, but strongly impacted by steric modifications (e.g.iPr vs. Me) – of the phenyl groups bound to the nitrogen atoms of the NHC ligand. For instance, Cu12a shows TADF, while Cu13 only emits from T1. Based on these findings, in-depth TD-DFT/DFT calculations of a model complex, Cu12b, without any substituents in the phenyl group revealed a significant relation between the torsion angle, N-C-Cu-N, and ΔEST – Scheme 3, Fig. 15 and Table 3. In detail, it was found that a planar configuration (torsion angle of 0°) leads to a delocalization of the HOMO – without affecting the LUMO – which then resulted in a better spatial separation of the HOMO and LUMO and a small ΔEST of 540 cm−1. This is in good agreement with the experimentally found value of 740 cm−1 for Cu12a – Table 3. The authors also noted SOC is effective in Cu12a as the phosphorescent lifetime of 34 μs is only three times longer than the one obtained at ambient conditions that correspond to TADF.
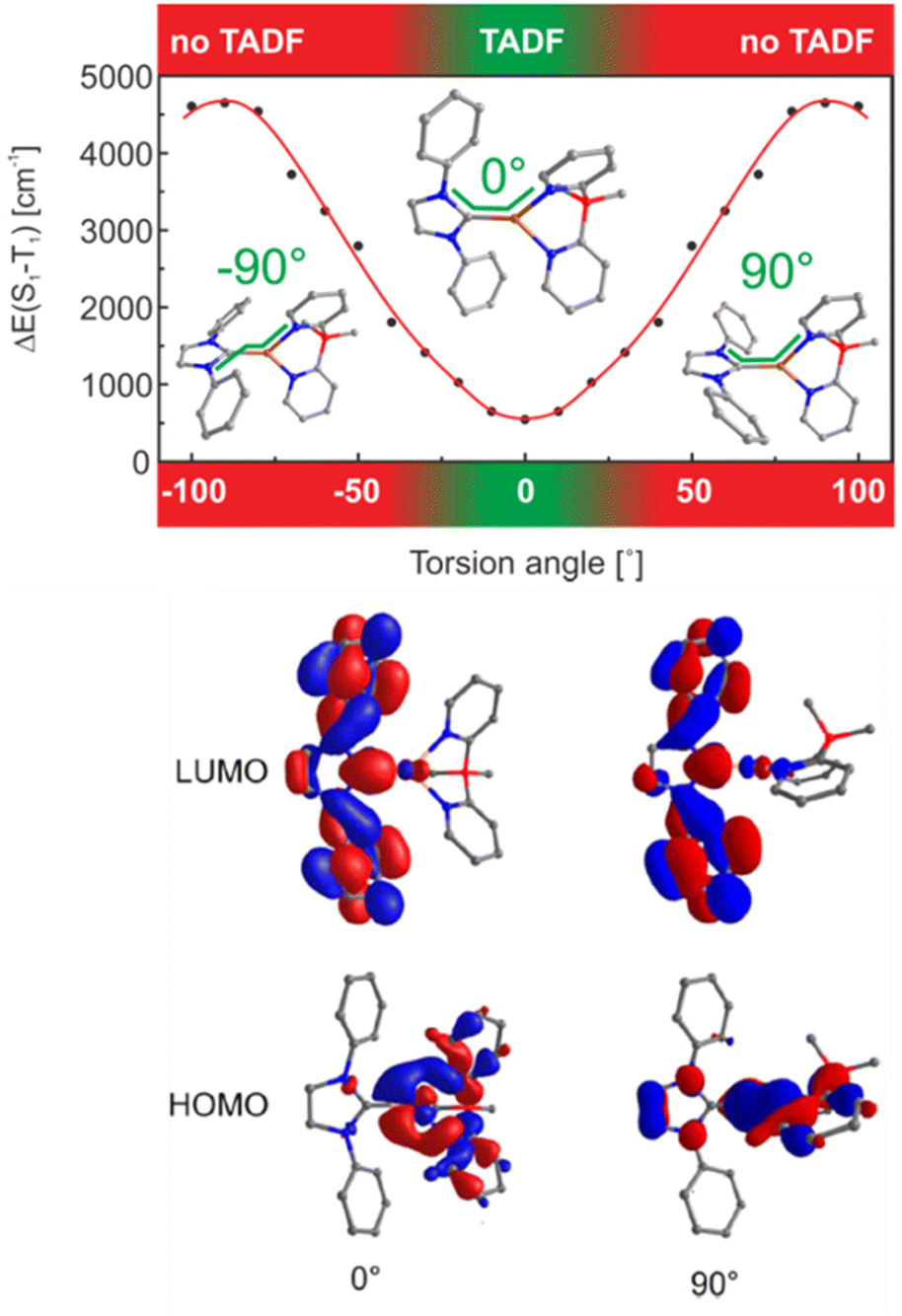 | ||
| Fig. 15 Top: Singlet−triplet splitting ΔE(S1–T1) in dependence of the torsion angle N−C−Cu−N (marked by the green line) as obtained from DFT and TD-DFT calculations on the B3LYP/def2-SVP level of theory. Bottom: HOMOs and LUMOs of model compound Cu12b displayed for a torsion angles of 0° and 90°, respectively. Reproduced with permission from ref. 248. | ||
Gaillard, Costa and coworkers studied the cationic NHC–Cu(I) analogs that feature – instead of the anionic dipyridyl borate ligand – neutral bis-pyridyl ligands with different bridging groups – Table 3. As the change of the ligand as well as the presence of the anion and its interactions with the cationic counterpart can change the electronic situation in the NHC–Cu(I) complex significantly, the findings from Yersin et al. cannot be extrapolated to these systems. However, certain trends have been established. At first, electronic modifications of the NHC ligands were found to marginally impact the photoluminescent properties as well as the nature of lowest excited states. In contrast, they are much more sensitive to electronic and steric changes in the bis-pyridyl ligands.249,257 For instance, the emission color is adjusted by modifying the dpa ligands, since the LUMO is located on the pyridine rings of the dpa, while the HOMO is mainly of metal and N-pz character of the dpa ligand. Further, it was found that (i) shorter Cu–Ccarbene bond distances, (ii) certain substitution patterns in the bis-pyridyl ligand, and (iii) high symmetry breaks, typically lead to higher PLQYs. The latter is related to (i) CH–π interactions between the phenyl group of the NHC ligand and the hydrogen atoms of the bis-pyridyl ligand, (ii) changes of the bridging atom in the bis-pyridyl ligand, e.g. from NH to CMe2 or PPh, and (iii) H–F interaction between the PF6− anion and the hydrogen atoms of the bis-pyridyl ligand. As indicated above, the HOMO and LUMO are spatially well separated and according to DFT/TD-DFT calculations, Cu14 and Cu15 (Scheme 3) display TADF, but with a relatively large energy splitting of 2823 cm−1 (0.35 eV). This value was, indeed, defined as the upper limit for NHC–Cu(I) complexes by Yersin. In a subsequent work, the influence of the bridging group, e.g. CH2, CMe2 or PPh, on the TADF properties was studied, showing a much higher ΔEST values of 4839 cm−1 (0.6 eV) – Cu16–18; Scheme 3.250
The relatively long emission decay times in TADF Cu(I) complexes displays still a challenge in designing efficient TADF emitters suitable for LECs and OLEDs, since roll-off effects on the efficiency are expected to occur already at low current densities.167,252,258–261 In this context, Steffen, Pflaum, and coworkers designed the neutral Cz-based TADF Cu(I) complexes bearing the chiral diphosphine, S- and R-BINAP, Cu19S/R and Cu20S/R, that show record short τTADF in the range of 1.7–2.5 μs and kr constants up to 3.1 × 105 s−1 in the ground state – Scheme 3 and Fig. 16.251 The TADF originates from LLCT states with the carbazolate unit as donor and the (S/R)-BINAP as accepting moiety with admixtures of MLCT. The authors noted a strong dependency of the PLQYs, emission maxima, and TADF properties on environmental changes since strong C–H⋯π interactions in the crystalline state – confirmed by single crystal X-ray and powder X-ray diffraction (PXRD) analysis – were found. Upon grinding of the crystalline samples,the intermolecular C–H⋯π interactions were disrupted with a concomitant reduction of the number of surrounding dipoles. This results in a decreased energy splitting of the 1/3LLCT states with a concomitant increase of the energetic separation between 1/3LLCT and 3LC (BINAP) states (ΔEST of 600 cm−1 (Cu20S/R) in crystalline to 490 cm−1 (Cu20S/R) in the ground state). Consequently, TADF becomes more efficient. In addition, Cu19S/R and Cu20S/R show circularly polarized luminescence (CPL) behavior with dissymmetry factors, glum, of up to ±6.0 × 10−3 in THF and ±2.1 × 10−2 in the solid state – Scheme 3.
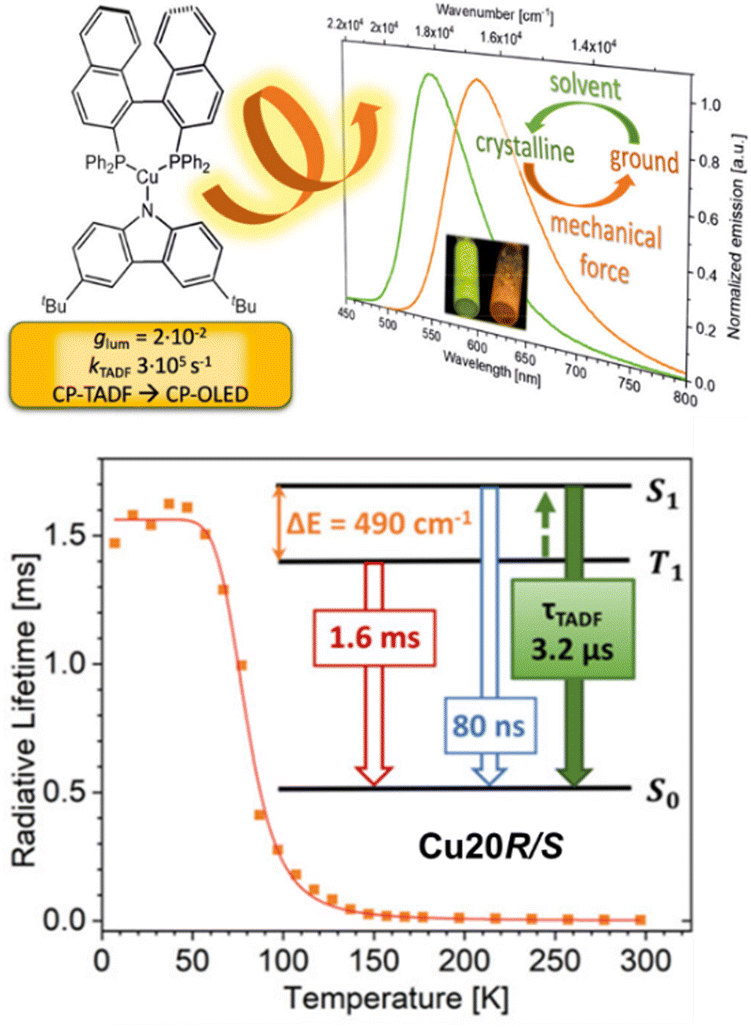 | ||
| Fig. 16 Top: Structural presentation of Cu20R/S and the corresponding emission spectrum of the crystalline and ground powder sample, as well as their UV irradiation, is shown as inset. Bottom: Temperature dependence of the radiative lifetimes of Cu20R/S in the ground solid state and the state diagram visualizing the TADF processes. Reproduced with permission from ref. 251. | ||
TADF can also be achieved in dinuclear Cu(I) complexes as well as in Cu-based clusters.253,262–265 One prominent class displays halogen-bridges Cu(I) complexes, namely [Cu(μ-X)(L^L)]2 with L = P^P or P^N and X = Cl, Br, I.266 Among others, Ueno, Yersin, and coworkers reported a series of blue to green-emitting dinuclear TADF Cu(I) complexes bearing the chelating dppbz or aminophospine ligands with X = I−, Br−, Cl−, – Cu21–27; Scheme 3 and Fig. 17 and Table 3.252,253 While TADF originates from spatially well-separated HOMO and LUMO with the lowest excited states of (M+X)LCT character, the choice of the halogen ion marginally influence TADF properties but the emission color. The ligand field strength increases in the order of I− < Br− < Cl− and leads to a stabilization of the HOMO energy – without drastically affecting the LUMO, resulting in red-shifted emission maxima – Table 3. Interestingly, the TADF decay times are relatively short for Cu(I) complexes, ranging between 4.1 and 6.6 μs at ambient conditions. Still, no clear dependence of the choice of the bridging halogen atom was noted.252,253
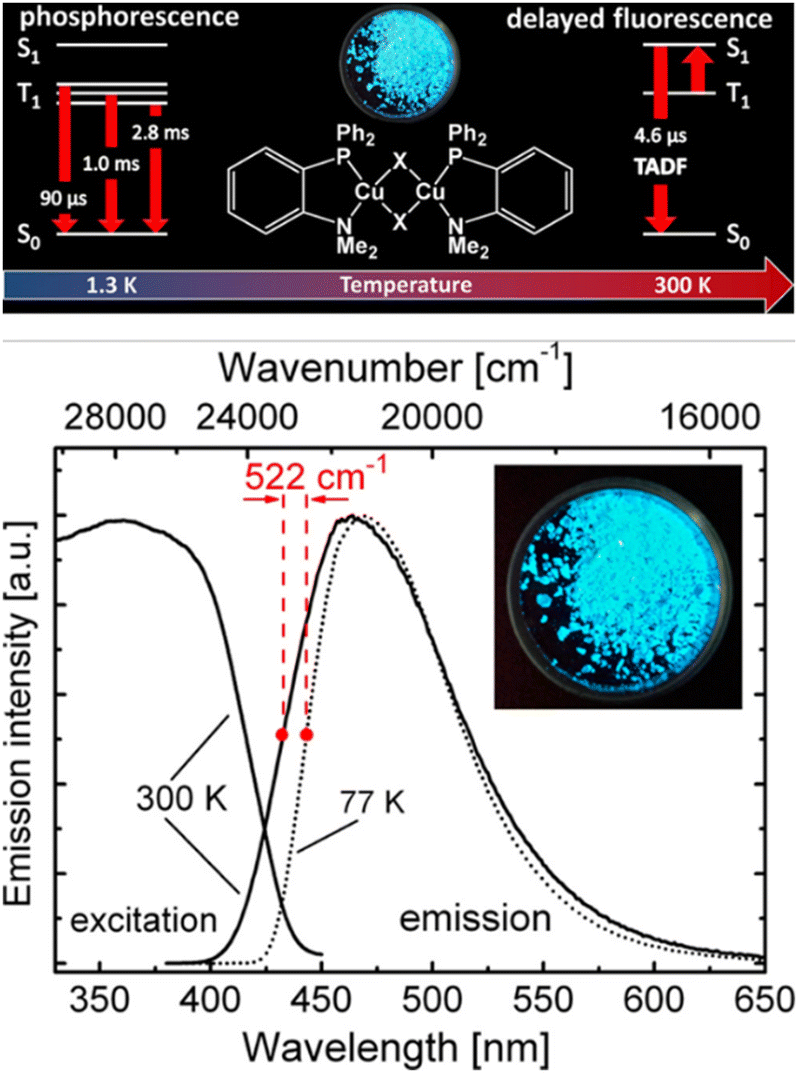 | ||
| Fig. 17 Top: Structural representation of Cu24–26 (X = Cl, Br, I) and corresponding state diagram visualizing the emission mechanisms at 1.3 K and 300 K. Bottom: Emission and excitation spectra of compound Cu26 (X = I) (powder) at ambient temperature (solid line) and 77 K (dotted line). The emission spectra were recorded under excitation at λexc = 360 nm, and the excitation spectra were detected at λdet = 470 nm. The inset shows a photo of the powder sample of compound Cu26 excited at 365 nm. Printed with permission from ref. 253. | ||
In a following study, Yersin et al. demonstrated how to overcome the close relation between a small ΔEST and the related less favorable S1 → S0 – vide infra.254 In detail, the Davidov model was applied to TADF molecules for the first time. It is based on coupling two symmetry-related transition dipole moments of the same molecule. The resulting combinations of the transition dipoles, that is, parallel or antiparallel alignment that could cancel out or double and thus, give a twice as large transition dipole moment as the monomeric unit – Fig. 18. This concept was realized by combining twice the quasi-Cu(I) TADF monomer, Cu28mono, resulting in the Cu(I) dimer Cu28dimer (Scheme 3 and Fig. 18 and Table 3) that displays landmark high radiative rates of 1.13 × 107 s−1 (fcal = 0.0499) and record short τTADF of 1.2 μs without the expected concomitant increase of ΔEST. Indeed, ΔEST was reduced to an unusually small value of 390 cm−1 since the energy of the lowest singlet excited state was lowered for the dimer as well.
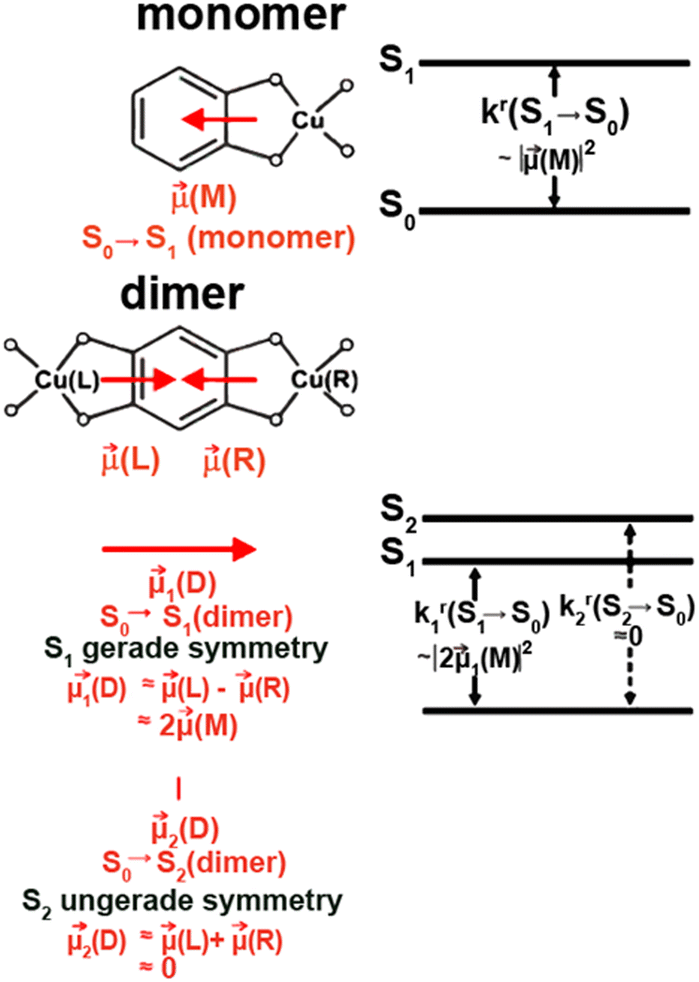 | ||
| Fig. 18 Schematic guiding model for Cu28dimer with a left (L) – right (R) inversion symmetry relation. For this dimer, one finds a distinctly faster rate of the S1 → S0 transition than for the related monomer, Cu28monomer, however, maintaining a small ΔEST energy gap. The model does not provide the sequence of the two singlet states S1 and S2 of the dimer. However, TD-DFT calculations show that the S1(1MLCT) dimer state will be the lower in energy. The triplet states are not discussed in this guiding model. TD-DFT calculations place them energetically below but near to the singlets. kr is the radiative rate that is proportional to the squared transition dipole moment. Reproduced with permissions from ref. 254. Copyright 2019, American Chemical Society. | ||
While Cu(I) complexes have been extensively investigated over the last two decades, significant attention has recently started to be devoted to Ag(I) complexes. Many factors, like the higher oxidation potential of Ag(I) beneficial for possible LEC or OLED applications, and their higher PLQYs trigger the research in this direction.267 However, compared to Cu(I) complexes, the design of efficient TADF Ag(I) complexes is more challenging due to the higher oxidation potential of Ag(I). The related energetically low-lying 4d-orbitals of Ag(I) barely contribute to the frontier orbitals, and thus, the lowest energy electronic transitions are scarcely of 1/3MLCT character. Instead, they are often characterized by LC excited states of3 ππ* (3LC) nature causing long-lived phosphorescence with even slow ISC rates. Therefore, only a few examples of Ag(I) complexes as efficient TADF emitters are known so far.
In 2017, Yersin et al. reported a breakthrough TADF Ag(I) series, [Ag(phen*)(P2-nCB)] Ag1–Ag4 (λem = 575–526 nm), displaying PLQYs up to 100% and record short radiative TADF decay times, τTADF, of 1.4 μs (Ag4) – Scheme 4 and Table 4.268,269 This was achieved by a rational design strategy based on several parameters. A highly electron-donating ligand, the anionic bis(diphenylphosphine)-nido-carborane (P2-nCB), was chosen to destabilize the energetically low-lying metal 4d-orbitals. DFT/TD-DFT calculations confirmed the success of this strategy as the silver contribution to the HOMO was increased to up to 13%. This resulted in lowest excited state transitions of MLL′CT, while the spatial separation of the HOMO and LUMO with L and L′ being different ligands was maintained. Indeed, a relatively small energy separation of 650 cm−1 was found for Ag4 – Fig. 19.
 | ||
| Scheme 4 Molecular structures of Ag1–15 and Zn1–9. | ||
| Complex | λ max (298 K)/λmax (77 K) [nm] | PLQY [%] | τ Ph/τDF [μs] | ΔEST [cm−1] | k r × 104 [s−1] | k nr × 104 [s−1] | Ref. |
|---|---|---|---|---|---|---|---|
| a In doped PMMA films (15 wt%). b In DPEPO films. c In a poly(9-vinylcarbazone) (PVK) matrix. d In doped polystyrene (PS) films (1 wt%). | |||||||
| Ag1 | 575/— | 36 | 270/2.0 | — | 18 | 32 | 269 |
| Ag2 | 562/— | 45 | 310/1.7 | — | 26 | 32 | 269 |
| Ag3 | 537/— | 78 | 804/2.8 | 650 | 28 | 7.9 | 269 |
| Ag4 | 526/— | 100 | 1300/1.4 | 650 | 71 | <2.1 | 269 |
| Ag5 | 479/484 | 70 | —/270, 470 | — | 5.4 | — | 231 |
| Ag6 | 555/— | 70 | 1845/1.9 | 480 | 37 | — | 270 |
| Ag7 | 527/529 | 76 | —/0.65 | 403 | — | — | 271 |
| Ag8 | 472/469 | 87.6 | 24400/6.3 |
1371 | — | — | 272 |
| Ag9 | 471/469 | 89.4 | 26100/6.5 |
1210 | — | — | 272 |
| Ag10 | 573/557 | 62b | 4650/3.5 | 645 | — | — | 273 |
| Ag11 | 487/500 | 56 | —/7.38 | 850 | — | — | 274 |
| Ag12 | 463/482 | 70 | —/8.22 | 890 | — | — | 274 |
| Ag13 | 463/479 | 98 | —/8.03 | 800 | — | — | 274 |
| Ag14 | 514/—c | 45 | —/0.42 | — | 110 | 130 | 275 |
| Ag15 | 472/472d | 100 | —/0.5 | 150 | 200 | <2 | 169 |
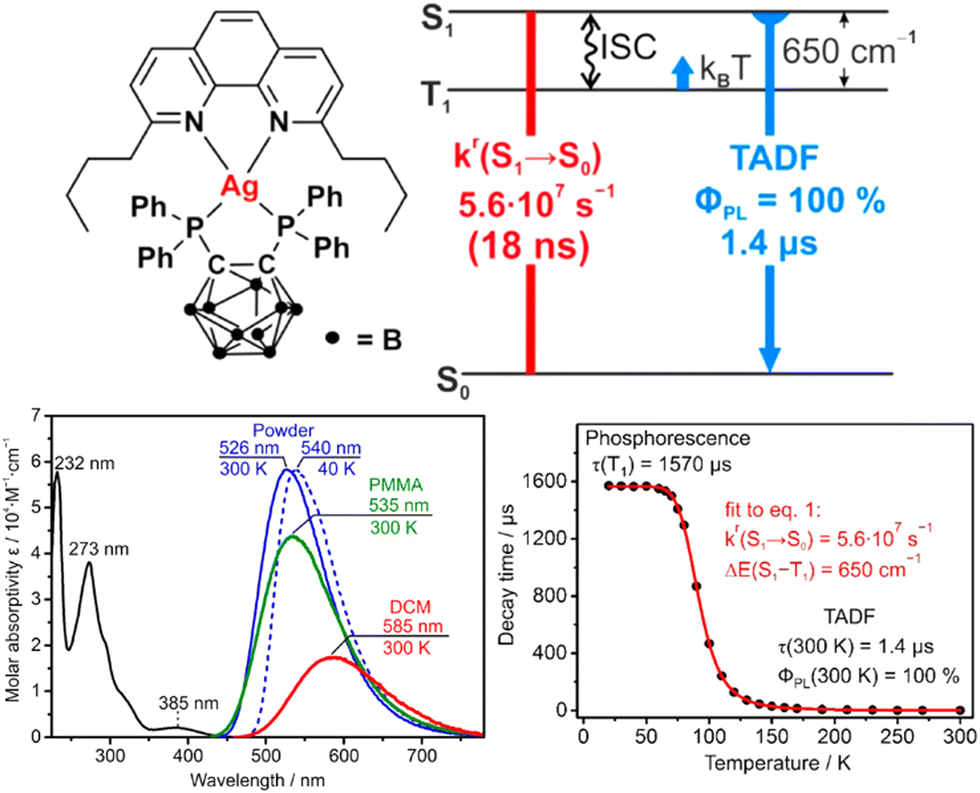 | ||
| Fig. 19 Top: Structural representation of Ag4 and the state energy diagram visualizing the TADF process. Bottom left: Absorption spectrum of the Ag4 measured in dichloromethane (DCM) at 300 K (black) and emission spectra of the Ag(dbp)(P2-nCB) complex shown as colored lines (λexc = 410 nm) measured under different conditions. The DCM solutions had a concentration of ≈10−5 M. The PMMA film was doped with ≈1 wt % complex. Bottom right: Luminescence decay times (τ) of Ag4 powder measured at different temperatures (left) and plotted vs temperature (right). The kr(S1 → S0) of 5.6 × 107 s−1 (18 ns) and the ΔEST of 650 cm−1 result from a fit of eqn (6) to the experimental τ(T) values, with τ(T1) fixed to 1570 μs as determined directly for temperatures below 60 K (plateau). Printed with permission from ref. 268. | ||
Second, the rigidity of this molecular structure turned out to be crucial for high PLQYs and a high radiative decay rate, kr(S1 → S0). This was proven by four phenanthroline derivatives modified either in the 2,9- or 4,7-position, Ag1–4 – Table 4. First, the increasing bulkiness of the phenanthroline ligands from Ag1 to Ag4 resulted in increased oscillator strength of S1 → S0 transitions (f(S1 → S0) = 0.0258 for Ag1 to f(S1 → S0) = 0.0536 for Ag4), while maintaining a relatively small energy separation of ΔEST of 650 cm−1. The obtained radiative decay rates, e.g. kr(S1 → S0) = 7.1 × 105 s−1 for Ag4, is one order of magnitude higher than usually found for Cu(I) complexes with similar ΔEST values.262 Third, the high rigidity also results in strongly increased PLQYs from 36% for Ag1 to 100% for Ag4. Theoretical calculations and single X-ray analysis further confirms this design strategy, since steric interactions between the n-butyl groups in the 2,9-positions of the phenanthroline and the phenyl groups of the P2-nCB ligand are present, minimizing geometrical distortion in the excited state – Fig. 20. Overall, this study presented the first example of an efficient TADF Ag(I) complex, displaying high PLQY with a moderately small ΔEST of 650 cm−1 and high radiative rate, reflected in record short TADF decay times of 1.4 μs.
 | ||
| Fig. 20 Coordination core geometries of complexes Ag1, Ag2, Ag3 and Ag4 as found by single-crystal X-ray diffraction analysis. The given interplanar angles characterize the deviations from the tetrahedral geometry (φ = 90°) (not to be confused with the valence angle of 109° in a tetrahedral molecule such as methane). Represented with permission from ref. 269. | ||
The same strategy was also applied to a neutral, mononuclear Ag(I) complex Ag5 – Scheme 4. Similar to Cu(I) analogous, Yersin and coworkers designed a rigid, tridentate N,P,P-ligand, 3,5-dimethyl-1-(2-((2-(di-o-tolyl)-phosphanyl)-(o-tolyl)-phosphanyl)phenyl)-1H-pyrazole, to minimize structural distortions upon excitation which resulted in PLYQs up to 70% – Table 4. TADF was achieved by a distinct spatial separation of the HOMO (metal halide moiety) and the LUMO (N,P,P-ligand). The silver contribution to the HOMO enabled MLCT transitions.231 In 2018, Yersin and coworkers reported a neutral, dinuclear complex bearing the tetradentate ligand, 1,2,4,5-tetrakis(diphenylphosphino)benzene (tpbz), as a bridging ligand and two terminal P2-nCB ligands, [Ag2(tpbz)(P2-nCB)2] Ag6 – Scheme 4 and Table 4. This complex design is similar to the one employed for Ag1–Ag4, namely, destabilizing the d-orbitals by using the same strongly donating ligand, P2-nCB. Thus, Ag6 shows also efficient TADF with a small ΔEST of 480 cm−1 and PLQY up to 70%. Moreover, Ag6 displays another example of an Ag(I) TADF emitter with an outstanding short TADF decay time of 1.9 μs.270
Lu et al. demonstrated that this strategy can also be applied to a neutral, tetranuclear Ag(I) complexes. In this case, four Ag(I) centers coordinate to two 4,6-dimethyl-2-(5-phenyl-4H-1,2,4-triazol-3-yl)pyrimidine (DMPTP) and three POP moieties, Ag7 – Scheme 4 and Fig. 21.271 The combination of the large bite-angle bis(diphosphine), the two methyl groups as well as the phenyl group at the pyrimidine-triazole ligand introduced a highly rigid environment giving rise to PLQYs of 76%. Moreover, this design leads to a substantial silver metal anion contribution to the HOMO, and thus, the lowest-excited states are of MLCT and LLCT character. The spatial separation of the HOMO and the LUMO, which is mainly located on the DMPTP unit, benefits the small ligand field splitting energy of 403 cm−1, while maintaining a fast decay time of 0.65 μs.
 | ||
| Fig. 21 Synthesis and structural presentation of Ag7 (anions, hydrogen atoms and included solvent molecules have been omitted for clarity). Printed with permission from ref. 271. | ||
A second strategy towards TADF Ag(I) complexes was presented by Lu et al. in 2020 as a coordination-induced TADF mechanism.272 In short, the usually “undesired” LC character of the excited states of Ag(I) complexes was exploited by the coordinating the non-TADF D–A molecule with a large ΔEST of 3791 cm−1 to a silver ion. The corresponding heteroleptic complexes, bearing POP (Ag8) or xantphos (Ag9) as the second chelating ligand – Scheme 4, Fig. 22 and Table 4, show a deep blue emission at 462 nm with PLQYs of 65.9%. The coordination of DMAC-MPyPz to Ag(I) leads to the planarization of the DMAC unit. Thus, the increased molecular rigidity forces a larger twisting in the D–A emitter, resulting in smaller energy splitting between the triplet and the singlet excited states (ΔEST = 1371 cm−1 and 1210 cm−1 for Ag8 and Ag9) and minimizing flattening distortions upon excitation. According to TD-DFT calculations, the S1 → S0 transition is mainly of ILCT character, which explains the negligible Jahn–Teller distortion upon excitation and thus the small non-radiative decay rates and the high PLQY. Again, such high radiative decay rates of 1.9 × 107 s−1 (Ag8) display record values for TADF Ag(I) complexes so far. The potential of this strategy was further confirmed by using different D–A molecule, e.g. a spiro-type diimine ligand.276
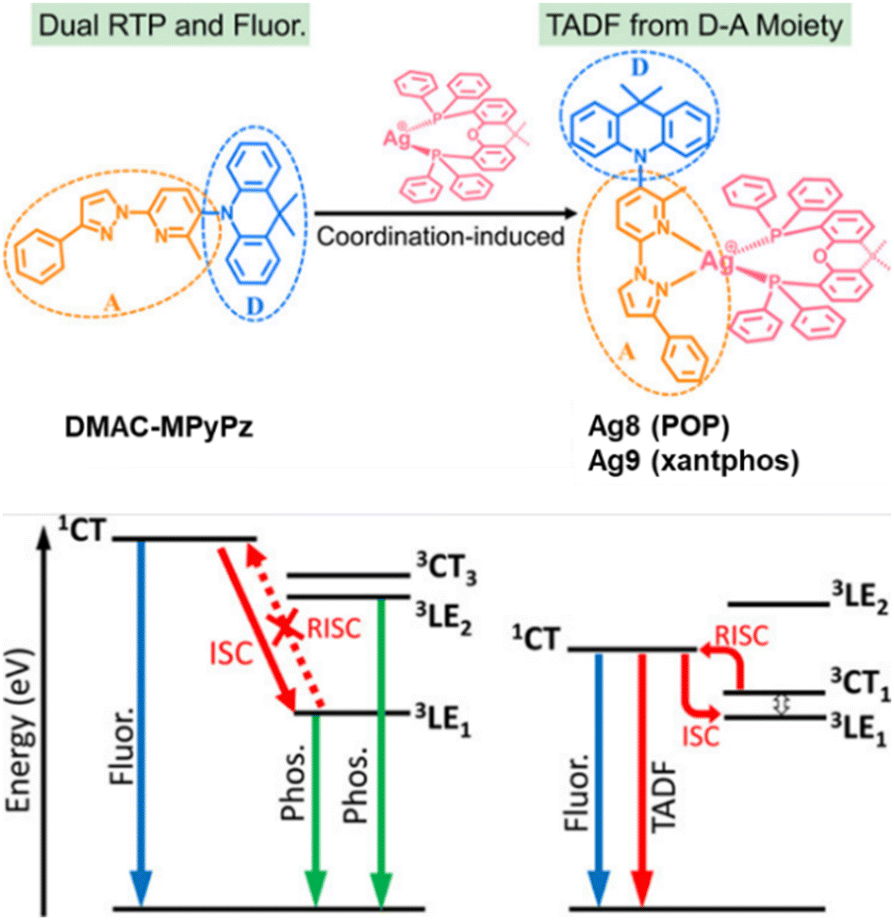 | ||
| Fig. 22 Top: Molecular design strategies for the realization of TADF including the molecular structure of Ag8 and Ag9. Bottom: PL processes for the free DMAC-MPyPz ligand (left) and the corresponding Ag(I) complexes (right). Printed with permission from ref. 272. | ||
Inspired by these findings, Yang's group applied this strategy to cationic Ag(I) complexes.273 Again, a D–A moiety as diimine, in which the pyridine-substituted 1-phenyl-1H-phenanthro[9,10-d]-imidazole (PI) accepting moiety was functionalized with the 10H-phenoxazine (PXZ) donor unit, was combined with POP to yield the orange-emitting Ag(I) complex Ag10 (Scheme 4) with PLQYs up to 62%. Based on temperature-dependent excited state lifetime measurements, the typical TADF behavior for Ag10 in a bis[2-(di(phenyl)phosphino)-phenyl]ether oxide (DPEPO) film with a relatively small ΔEST of 645 cm−1 and τDF of 3.5 μs was found. DFT/TD-DFT aided in assigning the nature of the excited states S1 and T1 to 1/3ILCT. The HOMO and LUMO are located on the PXZ and PIPy units. While Lu and coworkers report the coordination of the D–A unit to the Ag(I) center and its concomitant twisting as a key feature to obtain efficient TADF, Yang et al. claim that the coordination of the silver ion stabilizes the 1ILCT and 3ILCT states which leads to an increased ILCT character of the T1 state and thus, ΔEST decreases.
Hoshino and coworkers prepared a series of blue-emitting three-coordinated Ag(I) bromide complexes, Ag11–Ag13, bearing modified 1,2-bis(bis(2-alkylphenyl)phosphino)benzene ligands – Scheme 4 and Table 4.274 The increased steric hindrance of the alkyl groups gives the binuclear-bridge complex Ag11, while the bulky ethyl- and isopropyl-substituted bis(diphosphines) form the mononuclear species, Ag12 and Ag13. The bulkiness of the alkyl groups directly affects the PLQYs, increasing from 56% for Ag11 to 98% for Ag13 – Table 4. This trend was ascribed to the restricted geometrical distortions owing to increased steric hindrance in the peripheral phenyl groups. All three complexes show TADF behavior with energy splittings around 800–890 cm−1Table 4. Based on DFT/TD-DFT calculations, the TADF emission mainly stems from (σ + X) → π* transitions.
As a final note in both families of Cu(I) and Ag(I) complexes, linear complexes have evolved as substantial alternatives without sacrificing the TADF properties when rational ligand combinations are employed. In detail, Romanov, Bochmann, Credgington, and Thompson et al. found that replacing the halogen ligand with a strong donating ligand, e.g. anionic secondary amines, in carbene metal complexes changes the dominant emissive pathway from phosphorescence to TADF. In this work, we are focusing briefly on the design rules for linear coinage carbene-metal-amide (CMA) complexes showing TADF as they are of particular interest due to their (i) high PLQYs (up to 90–100%), (ii) good color tunability, (iii) fast kISC rates (iv) and short excited state lifetimes in the sub-/microsecond regime.277 For deeper insights, the interested reader is recommended to refer to Thompson s’ recent review.122
In short, the metal ion serves as a linker between the π-donating amide, e.g. diaryl- or dialkylamines or Cz and derivatives thereof, and the π-accepting carbene unit, e.g. NHCs, MAC, CAAC = cyclic (alkyl)(amino)-carbene, CCArC, BZI derivatives. The HOMO is mainly located on the amide unit with small to minor metal contributions (less than 15%) – decreasing in the following order: Cu(I) > Au(I) > Ag(I), while the LUMO is mainly of p-orbital character of the carbon atom of the carbene ligand.169 Despite the minor contribution of the metal ion to the frontier orbitals, it still holds a key position as (i) its d-orbitals serve as a bridge for the CT from the amide to the carbene, and (ii) its high SOC constant increases the efficiency of the ISC/rISC processes (kISC = 1010–1011 s−1) and thus favors TADF. Moreover, the choice of the metal becomes more evident for the extinction coefficients, oscillator strengths, excited state lifetimes, and ΔEST values. While the oscillator strength and extinction coefficients decline in the following order: Au(I) > Cu(I) > Ag(I), τ and ΔEST increase in the order of Ag(I) < Au(I) < Cu(I). The latter tendency correlates well with the Ccarbene–metal–Namide distance: Ag: 4.15 Å, Cu: 3.7 Å, Ag: 4.0 Å, which is a consequence of the decreasing atomic radii: Ag > Au > Cu. In this context, Bochmann and Thompson reported two examples of Ag(I)-based CMA complexes, Ag14 and Ag15 – Scheme 4, with record short lifetimes of 420 and 500 ns and singlet–triplet energy splitting down to 645 cm−1 (Ag14).169,275 As mentioned before, fast radiative TADF decay times are crucial for optoelectronic applications, which are ideally obtained with large oscillator strength f(S1 → S0) (and thus fast kr(S1 → S0)) and small ΔEST. However, a small ΔEST results from a small overlap of the frontier orbitals but lowers kr(S1 → S0) and thus the TADF decay time. In this context, Thompson et al. established a relationship between the degree of spatial overlap between the hole and electron wave functions that describe the excited states and the TADF key parameters.278 In this study, twelve CMA-TADF emitters were prepared– including Cu(I), Ag(I), and Au(I) ions – with high PLQYs (<50%) and short lifetimes (>2 μs). Natural transition orbitals were used to evaluate this overlap, ΛNTO. Both krS1 and De were found to increase exponentially with increasing NTO overlap. The optimal range of the NTO overlap to obtain the best compromise of kr(S1 → S0) and ΔEST was found to range between 0.25–0.3.
As the last representative in this family, Zn(II) displays the most common oxidation state for Zn with excellent redox stability and a substantial variety of different coordination modes. While high-coordination numbers (>4) are well established, low-coordination complexes are less explored due to their high reactivity, but they are of high interest due to the increased Lewis acidity, the atypical reactivity, and the planar geometric configuration. Recently, different design strategies toward such low-coordinated Zn(II) complexes were introduced. In short, strong σ-donating groups combined with sterically demanding ligands can compromise its highly electron-deficient character and protect the metal center from further attachment/coordination of ligands/solvent molecules. Due to the d10 electron configuration of Zn(II) and consequently, the lack of metal-centered excited states, Zn(II) emitters are of high interest for photoluminescent applications. However, the high oxidation potential of Zn(II) leads to the absence of d-orbital contributions in the excited states and hampers the access of MLCT lowest excited states. Thus, the heavy-atom effect of Zn(II) is almost negligible and disfavors efficient the spin–orbit coupling. As a result, most of the emission mechanisms reported for Zn(II) so far, are fluorescent decay pathways, and only a few, but an increasing number of phosphorescent and TADF scenarios are known.
The first Zn(II)-based TADF emitter was introduced by Adachi et al. in 2015.279 A monomeric, homoleptic four-coordinated Zn(II) complex, Zn1, bearing the D-A ligand, PX-BOX-X = bis(2-(benzo[d]oxazol-2-yl)-5-(10H-phenoxazin-10-yl)phenolate), with excited states of ILCT nature shows characteristic TADF features – Table 5. Through the coordination of the D–A ligands, the angle between the donor and acceptor unit was changed with a concomitant decrease of ΔEST from 2500 cm−1 of the free ligand to 484 cm−1 – Zn1; Scheme 4.
| Complex | λ max (298 K)/λmax (77 K) [nm] | PLQY [%] | τ Ph/τDF [μs] | ΔEST [cm−1] | k r × 105 [s−1] | k nr × 105 [s−1] | Ref. |
|---|---|---|---|---|---|---|---|
| a 6 wt% emitter in 3,3′-di(9H-carbazol-9-yl)-1,1′-biphenyl (mCBP) doped films. b Photophysical data of Zn5 (10 wt%) in a PMMA matrix. | |||||||
| Zn1 | 542/— | 78.4 | —/37.8 | 485 | — | — | 279 |
| Zn2 | 480/— | 3 | 4500/24.5 | 1000 | — | — | 280 |
| Zn3 | 452, 540 | 50 | 828/— | 242 | — | — | 281 |
| Zn4 | 577/558 | 10 | 5900/0.74 | 637 | 1.4 × 105 | 1.2 × 106 | 282 |
| Zn5 | 560/556 | 28 | 1940/0.63 | — | 4.5 × 105 | — | 283 |
| Zn6 | 583/568 | 13 | 46/0.17 | — | 7.5 × 105 | — | 283 |
| Zn7 | 580/553 | 49 | 346/0.49 | 452 | 1.2 × 106 | — | 283 |
| Zn8 | 468/— | 12 | — | 2662 | — | — | 284 |
| Zn9 | 508/— | 33 | — | 484 | — | — | 284 |
In 2021, Roesky's group reported a tetrahedral, blue-emitting Zn(II) iminophosphonamide (NPN) complex, Zn2 – Scheme 4, showing TADF in the solid state – Table 5.280 In detail, the twisting of the NPN moiety in the lowest triplet state T1 enables an efficient thermally driven spin–flip from the triplet to the singlet state with an energy splitting of 1000 cm−1 which was obtained from fitting the temperature-dependent excite state lifetimes from 5 to 298 K.
Back in 1985, Crosby et al. reported a series of four-coordinated [Zn(SC6H4-4-R)2(phen)] complexes, e.g.Zn3 (R = Cl, Scheme 4), showing a dual emission character.285–287 While the vibrationally structured high-energy band was assigned to phosphorescence from locally excited 3π–π* states centered on the phenanthroline ligand, the broad and unstructured emission band was reported to stem from interligand thiolate → phen LLCT transitions. Steffen, Marian and coworkers revisited the proposed kinetic scheme by temperature-dependent and time-resolved luminescent measurements. The obtained high kr constant kr = 3.5 × 105 s−1 in combination with high-level DFT/multireference configuration interaction (MRCI) calculations reveals efficient reverse intersystem crossing by up-conversion from 3LLCT to the 1LLCT state – Fig. 23. This leads to efficient population of the 1LLCT state and to TADF as major emission channel.281
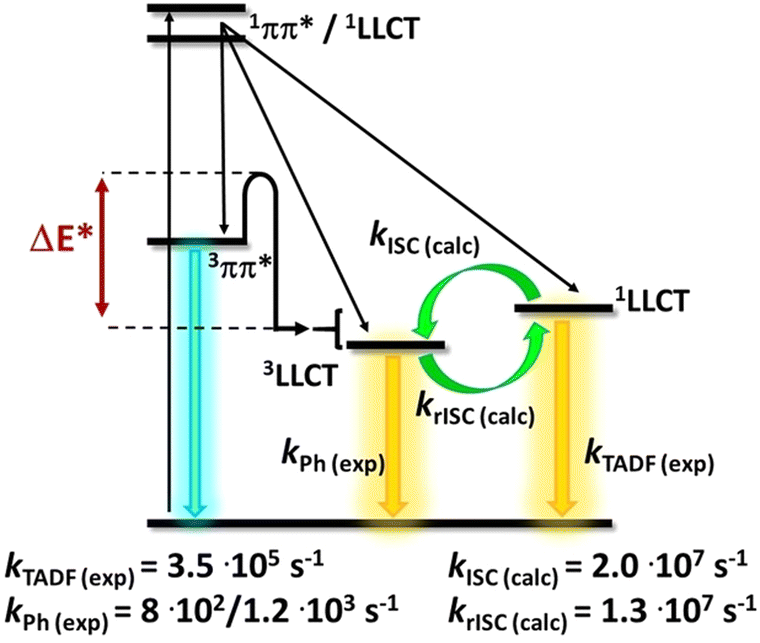 | ||
| Fig. 23 Proposed excited-state decay mechanism upon photoexcitation of Zn3. Printed with permission from ref. 281. | ||
In 2023, Steffen and Marian et al. followed the strategy of a donor-Zn(II)-acceptor molecular system to enter 1/3LLCT states and thus to obtain high radiative rates.282 The presented dimeric Zn(II) carbene complex bearing two electron-rich, chelating benzene-1,2-dithiolate ligands combined with electrophilic carbene ligands – Zn4; Scheme 4, shows moderate brightness (up to 10% in a PMMA matrix with 10 wt%) in the green-yellow range of the electromagnetic spectrum – Table 5. Excited state lifetimes in the range of 472–741 ns in the solid state reveal unprecedented high radiative rates for Zn(II) complexes up to 1.4 × 105 s−1. Variable temperature studies in conjunction with DFT/MRCI calculations disclose TADF as main emission pathway at room temperature origination from the LLCT state. The energy splitting, ΔEST, was estimated to 637 cm−1. The high stability towards moisture, air and irradiation stems from the μ2,k2-bonding mode of the dithiolate ligand that is a key feature for the steric stabilization and protection of the Zn–carbene bond and combined with long-lived triplet states makes Zn4 an ideal candidate for Dexter energy transfer photocatalysis.
Based on a theoretical study on monomeric carbene–zinc–dianionic complexes that focused on the influence of the π-accepting and σ-donor character of different carbenes (NHC, CAAC, CAArC) on the photoluminescent properties – including TADF behavior,288 Steffen and coworkers introduced a new generation of monomeric carbene-zinc-dithiolate complexes displaying characteristic TADF behavior.283 In detail, menthyl-substituted cAAC (MenthcAAC) ligand and derivatives of dithiolate ligands gave three monomeric Zn(II) complexes, Zn5 (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H), Zn6 (RMe) and Zn7 (RtBu) in Scheme 4, while the latter one shows polymorphism. The bright yellow to orange photoluminescence with PLYQs up to 49% in the crystal state and lifetimes in the range of 170–630 ns result in unprecedented high kr constants up to 7.5 × 105 s−1 – Table 5. Time-resolved variable temperature studies reveal a change of the emitting state below 50 K, accompanied by two orders of magnitude decreased radiative rates (kr = 2.5 × 103 s−1). The TADF behavior was finally confirmed by fitting the temperature-dependent excited state lifetime measurements with the Boltzman-type equation (Section 2) yielding an ΔEST of 452 cm−1 for Zn7. According to high-level DFT/MRCI calculations the relative orientation of the two ligands involved in the 1/3LLCT states displays a key parameter to control the TADF properties. For instance, a dihedral angle of 33–36° in Zn7 gives rise to a very efficient rISC rate in the range of 109 s−1 owing to efficient SOC mediated by the sulfur atom.
H), Zn6 (RMe) and Zn7 (RtBu) in Scheme 4, while the latter one shows polymorphism. The bright yellow to orange photoluminescence with PLYQs up to 49% in the crystal state and lifetimes in the range of 170–630 ns result in unprecedented high kr constants up to 7.5 × 105 s−1 – Table 5. Time-resolved variable temperature studies reveal a change of the emitting state below 50 K, accompanied by two orders of magnitude decreased radiative rates (kr = 2.5 × 103 s−1). The TADF behavior was finally confirmed by fitting the temperature-dependent excited state lifetime measurements with the Boltzman-type equation (Section 2) yielding an ΔEST of 452 cm−1 for Zn7. According to high-level DFT/MRCI calculations the relative orientation of the two ligands involved in the 1/3LLCT states displays a key parameter to control the TADF properties. For instance, a dihedral angle of 33–36° in Zn7 gives rise to a very efficient rISC rate in the range of 109 s−1 owing to efficient SOC mediated by the sulfur atom.
He et al. presented an alternative approach toward Zn(II)-TADF emitter involving an intracomplex D–A ion pair that forms an exciplex with TADF behavior. The cationic [Zn(tpy)2]2+ complex displays the accepting group and was combined with a Cz-type counter anion, (4-(9H-carbazol-9-yl)phenyl)-trifluoroborate (CAZ-p-BF3−), or (2-(9H-carbazol-9-yl)-phenyl)trifluoroborate (CAZ-o-BF3−), that serves as donor, Zn8 and Zn9 – Scheme 4 and Table 5.284 Single X-ray analysis show stronger π–π interactions between the donor and acceptor ions for Zn9. They rule the photophysical properties since only Zn9 emits via TADF at room temperature. ΔEST was estimated to 484 cm−1 in the crystalline state for Zn9, while Zn8 exhibits a six-fold larger ΔEST (i.e., 2662 cm−1).
3. TADF dx-metal complexes in solid-state lighting
3.1. OLEDs
TADF technology rapidly developed in recent years, and several companies invested high-cost budget to develop red, green and yellow TADF emitters that can offer a cost-effective substitute to phosphorescent organic light-emitting diodes (PHOLEDs) mainly supplied by Universal Display Corporation (UDC) For instance, Cynora company is developing new emitter materials for SSLDs, as shown for its initial marketable product based on TADF fluorescent blue emitter exhibiting a 15% efficiency improvement over existing blue emitters on the market – Fig. 24a. Kyulux Inc., a world leader in the development of OLED Hyper fluorescence™/TADF technology, announced that WiseChip launches world's first commercial Hyperfluorescence™ OLED demonstrate based on TADF materials designed by Kyulux Inc. The 2.7-inch, 128 × 64-pixel array, yellow type Hyper fluorescence™ OLED can reach up to 220 nits–2.5 times brighter than using general fluorescence materials. Surprisingly, the lifetime of the product is more than 50000 h – Fig. 24b.
 | ||
| Fig. 24 Examples of TADF OLEDs for (a) blue and (b) yellow. Reproduced with permissions from ref. 289. | ||
Recent reviews summarized the progresses of OLEDs based on TADF organic compounds.125,290–294 To complement these reviews, our work aims to show the latest progress achieved in the field OLED based on metal complex based TADF emitters. Accordingly, the following sections will summarize the most relevant complexes applied to OLEDs, highlighting their device performance as well as their photoluminescence features as a complement of Section 2.
To provide a comprehensive view, a final approach to merge TADF and Ir(III) complexes is the integration of TADF interfacial exciplex and phosphorescent ultrathin emitting layers (Ph-UEMLs) made of Ir(III) complexes. In short, monochrome/WOLEDs were structured by directly incorporating single or complementary Ph-UEMLs at the interface of the TADF interfacial exciplex ((mCP)/2,4,6-tris[3-(diphenylphosphinyl)phenyl]-1,3,5-triazine (PO-T2T)) using a simple doping-free technique. The hole-transporting donor layer (mCP) and electron-transporting acceptor layer (PO-T2T) effectively confine carrier recombination to the interface of the mCP/PO-T2T exciplex. Simultaneously, optimizing the thickness of the hole-transporting layer promotes a well-balanced distribution of electrons and holes at the mCP/PO-T2T interface. Additionally, the Ph-UEMLs are sensitized by the TADF interfacial exciplex of mCP/PO-T2T which enables complete exciton utilization through multi-channel energy transfer from the TADF interfacial exciplex to the Ph-UEMLs. These factors significantly contribute to the exceptional performance of the suggested monochrome/WOLEDs. As a result, all monochrome OLEDs in blue, green, orange, and red colors demonstrated a low turn-on voltage of 2.4–2.5 V and achieved outstanding device performance with maximum EQE (EQEmax) exceeding 20%. Particularly, the blue OLED based on bis[2-(4,6-difluorophenyl)pyridinato-C2,N] (picolinato)iridium(III) (FIrpic) achieved a remarkable forward-viewing EQE of 30.21%, which stands among the highest values reported in literature. Furthermore, the fabricated two-, three-, and four-color WOLEDs also delivered exceptional performance with a low turn-on voltage of 2.5 V and high EQEs of 23.47, 22.70, and 23.88% (equivalent to 78.80, 76.70, and 70.89 lm W−1). All white devices exhibited remarkable color stability across a practical luminance range from approximately 5000 to 12000 cd m−2, indicating significant potential for real-world applications. Overall, this study shows that complete exciton utilization can be achieved through multi-channel energy transfer. In this case, Ph-UEMLs sensitized by TADF interfacial exciplex were employed.
The proposed scheme is believed to open a new pathway for developing ultra-simple and high-performance monochrome/WOLEDs.300
Besides the above scenario, Pander et al. have recently presented compelling evidence of TADF as discussed in Section 2. dinuclear, [Ir(N^C)2]2(m-L), displays deep-red luminescence (λem = 643 nm) and a TADF with kr of 3.5 × 105 s−1 associated to a modest ΔEST of around 403 cm−1. An OLED fabricated by incorporating 1 as a dopant in the emitting layer (EML) at 5 wt% exhibits red electroluminescence centered at 625 nm with an EQE value of 5.5% and a maximum brightness of 6300 cd m−2.194 This represents the first work on using TADF based Ir(III) complexes and, in turn, much more needs to be studied to determine the limitations and challenges.
280 cd m−2 – Table 6.
 | ||
| Scheme 5 Molecular structures of Pd2–8, Pt2–14 and Au14–91. | ||
 | ||
| Fig. 25 Luminescence spectra of (a) Pd(tzp-1) (Pd2), (b) Pd(tzp-2) (Pd3), (c) Pd(tzp-3) (Pd4), (d) Pd(tzp-4) (Pd5), (e) Pd(tzp-5) (Pd6), and (f) Pt(tzp-2) (Pt2) at 77 K in 2-MeTHF (dash-dotted lines), at room temperature in CH2Cl2 solution (solid lines) and at room temperature in PMMA film (solid-ball lines). The chemical structure of each emitter is shown in the inset. The peaks in the black circles are MADF emissions. Reproduced with permissions from ref. 198. Copyright 2019, American Chemical Society. | ||
| Complex | OLED Structure | Device | Concentration [%wt] | ELmax [nm] | CIE [x,y] | V on [V] | CEmax [cd A−1] | η p [lm W−1] | L max [cd m−2] at Vapp | η ext [%] | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A: by using a higher band-gap material PYD2 (2,6-bis(9H-carbazol-9-yl)pyridine) to replace PVK: OXD-7 as the host, the emission peak maxima of the device with 60 further blue-shifts to 486 nm with CIE coordinates of (0.21, 0.42). The EQEmax of this device is 15.7% which slightly decreased to 14.4% at 1000 cd m−2. HAT-CN: 1,4,5,8,9,11-hexaazatriphenylenehexacarbonitrile. DMIC-TRZ: {5-(3-(4,6-diphenyl-1,3,5-triazin-2-yl)phenyl)-7,7-dimethyl-5,7-dihydroindeno[2,1-b]carbazole}. ANT-BIZ: {1-(4-(10-([1,10-biphenyl]-4-yl)anthracen-9-yl)phenyl)-2-ethyl-1H-benzo[d]-imidazole}. Al: aluminum. Liq: 8-hydroxyquinolinolato-lithium. TBBD: N4,N4,N4′,N4′-tetra(4-biphenylyl)-biphenyl-4,4′-diamine. o-SFAF: (N-([1,1′-biphenyl]-2-yl)-N-(9,9-dimethyl-9H-fluoren-2-yl)-9,9′-spirobi[fluoren]-2-amine). NPB: 4,4′-bis[N-(1-naphthyl)-N-phenylamino]-biphenyl. PVKH: poly(9-vinylcarbazole). TPD: N,N′-bis(3-methylphenyl)-N,N′-bis(phenyl)-benzidine. PBD: {2-(4-biphenyl)-5-(4-tert-butylphenyl)-1,3,4-oxadiazole}. PFI: perfluorinated ionomer. mCPBC: 9-(3-(9H-carbazol-9-yl)phenyl)-9H-3,9′-bicarbazole. PIM-TRZ: di-[4-(N,N-ditolyamino)-phenyl]cyclohexane. DMFBD-TRZ: 2-(3′-(9,9-dimethyl-9H-fluoren-2-yl)-[1,1′-biphenyl]-3-yl)-4,6-diphenyl-1,3,5-triazine. PCZAC: 9,9-dimethyl-10-(9-phenyl-9H-carbazol-3-yl)−9,10-dihydroacridine. PBICT: 2-phenyl-4,6-bis(12-phenylindolo[2,3-a]carbazole-11-yl)−1,3,5-triazine. DBTTP1: 4-(3-(triphenylene-2-yl)phenyl)dibenzo[b,d]thiophene. TSPO1: diphenyl-4-triphenylsilylphenylphosphineoxide. BCFA: N-([1,1′-biphenyl]-4-yl)-9,9-dimethyl-N-(4-(9-phenyl-9H-carbazol-3-yl)phenyl)-9H-fluoren-2-amine. HT host: 9-[1,1′-diphenyl]-3-yl-9H-3,9′-bicarbazole. ET host: 9-(3′-(9H-carbazol-9-yl)-6-cyano-[1,1′-biphenyl]-3-yl)-9H-carbazole-3-carbonitrile. mCP-2CN: 9-(3-(9H-carbazol-9-yl)phenyl)-9H-carbazole-3,6-dicarbonitrile. DBFPO: dibenzo[b,d]furan-2,8-diylbis(diphenylphosphineoxide). LiQ: 8-hydroxyquinoline lithium. FSFA: N-([1,1′-biphenyl]-2-yl)-N-(9,9-dimethyl-9H-fluoren-2-yl)-9,9′-spirobi[fluoren]-2-amine. FSF4A: N-([1,1′-biphenyl]-2-yl)-N-(9,9-dimethyl-9H-fluoren-2-yl)-9,9′-spirobi[fluoren]-4-amine. DMIC-Cz: 7,7-dimethyl-5-phenyl-2-(9-phenyl-9H-carbazol-3-yl)-5,7-dihydroindeno[2,1-b]carbazole. 3TPyMB: tris(2,4,6-trimethyl-3-(pyridin-3-yl)phenyl)borane. TmPyPB: 1,3,5-tri[(3-pyridyl)phen-3-yl]benzene. PPF: (2,8-bis(diphenyl-phosphoryl)-dibenzo[b,d]furan). Tm3PyP26PyB: 1,3,5-tris(6-(3-(pyridin-3-yl)phenyl)pyridine-2-yl)benzene. | |||||||||||
| Pt2 | HAT-CN/NPD/TCTA/mCBP/Pt2:mCBP/DPPS/Bepp2:Li2CO3/Li2CO3/Al | — | — | 508, 545 | 0.31, 0.61 | — | — | — | 28280 |
8.7 | 198 |
| Pd8 | ITO/HAT-CN (5 nm)/TAPC (30 nm)/TCTA (15 nm)/mCBP (10 nm)/DMIC-TRZ: Pd8 (40 nm)/PO-T2T (20 nm)/ANT-BIZ (30 nm)/Liq (2 nm)/Al (100 nm). | — | 1 | 576 | (0.49, 0.50) | — | 79.1 | 76.4 | 150300 |
28.8 | 200 |
| 3 | 583 | (0.51, 0.48) | 62.7 | 56.3 | 103100 |
25.1 | |||||
| Pd8 | ITO/HAT-CN (5 nm)/TBBD (30 nm)/o-SFAF (15 nm)/DMIC-TRZ: Pd8 (45 nm)/ANT-BIZ (40 nm)/Liq (2 nm)/Al (100 nm) | — | 1 | 575 | (0.49, 0.50) | — | 75.2 | 98.5 | 124800 |
27.5 | 200 |
| Pd7 | ITO/HAT-CN (5 nm)/TBBD (30 nm)/o-SFAF (15 nm)/DMIC-TRZ: Pd7 (45 nm)/ANT-BIZ (40 nm)/Liq (2 nm)/Al (100 nm) | — | 1 | 636 | (0.62, 0.38) | — | 32.1 | 42.0 | 70230 |
31.4 | 200 |
| Pd7 | ITO/HAT-CN (5 nm)/TAPC (30 nm)/TCTA (15 nm)/mCBP (10 nm)/DMIC-TRZ: Pd7 (40 nm)/PO-T2T (20 nm)/ANT-BIZ (30 nm)/Liq (2 nm)/Al (100 nm) | — | 1 | 640 | (0.63, 0.37) | — | 29.6 | 27.5 | 92000 |
30.1 | 200 |
| Pt3 | ITO/NPB/ultrathin layer of Pt3/mCP/4P-NPD/TPBi/LiF/Al | Ulth1 | — | 602 | (0.54, 0.44) | 3.0 | — | 10.6 | 1651 | 13.6 | 301 |
| Ulth2 | 654 | (0.55, 0.44) | 3.5 | 11.3 | 2093 | 15.5 | |||||
| Ulth3 | 664 | (0.57, 0.42) | 6.5 | 7.8 | 826 | 21.9 | |||||
| Pt4 | ITO/NPB/a dopant of Pt4 in CBP/mCP/4P-NPD/TPBi/LiF/Al | Dop1 | — | 526 | (0.33, 0.61) | 4.0 | — | 33.5 | 5448 | 18.7 | 301 |
| Dop2 | 528 | (0.36, 0.59) | 5.0 | 25.7 | 4186 | 20.2 | |||||
| Dop3 | 676 | (0.47, 0.48) | 4.5 | 6.1 | 3093 | 8.6 | |||||
| Dop4 | 654 | (0.35, 0.28) | 7.0 | 4.8 | 1459 | 13.7 | |||||
| Dop5 | 658 | (0.64, 0.36) | 9.0 | 3.1 | 707 | 15.1 | |||||
| Dop6 | 674 | (0.64, 0.36) | 6.0 | 1.6 | 1162 | 9.1 | |||||
| Pt3 | — | Hyb W1 | — | 654 | (0.41, 0.31) | 9.0 | — | 4.0 | 1778 | 12.9 | 301 |
| Pt4 | Hyb W2 | 532 | (0.30, 0.31) | 7.0 | 6.3 | 1541 | 10.5 | ||||
| Pt4 | Hyb W3 | 654 | (0.35, 0.32) | 9.5 | 5.0 | 1412 | 13.7 | ||||
| Pt1 | ITO|PEDOT:PSS Al4083 (30 nm)|mCP: PO-T2T (n: m) co x% 2 (y nm)|PO-T2T (50 nm)|LiF (0.8 nm)|Al (100 nm) | 1 | 5 | 637, 730 | — | — | — | — | — | 2.64 | 204 |
| Pt5 | 2 | 20 | 651, 787 | 0.99 | |||||||
| Pt6 | 3 | 33 | 805 | 0.51 | |||||||
| Pt1 | ITO/HIL 1.3N (45 nm)/PVKH (10 nm)/TPD: PBD (60:40 w/w) co 5% 5 (30 nm)/TPBi (50 nm)/LiF (0.8 nm)/Al (100 nm) |
— | — | 607 | (0.62, 0.37) | 7 | — | — | 11000 |
7.4 | 201 |
| Pt12 | ITO/PEDOT:PSS (50 nm)/Poly-TPD (15 nm)/47% mCP:47% OXD-7:6% PtAu2 complex Pt12 (50 nm)/ETL (40 nm)/LiF (1 nm)/Al (100 nm) |
— | 6 | 525 | (0.27, 0.69) | 4.0 | 80.6 | 42.8 | 10865 |
20.8 | 302 |
| Pt9 | ITO/PEDOT:PSS (50 nm)/Poly-TPD (15 nm)/host materials:PtAu2 complex (50 nm)/BmPyPb (40 nm)/LiF (1 nm)/Al (100 nm) | — | — | 557 | (0.46, 0.54) | 3.2 | 76.2 | 45.2 | 8368 | 21.6 | 302 |
| Pt12 | 525 | (0.29, 0.66) | 4.0 | 80.6 | 42.8 | 10865 |
20.8 | ||||
| Pt13 | ITO/(PEDOT:PSS):PFI (54 nm)/mCPBC:PIM-TRZ: Pt13 2 wt% (50 nm)/DMFBD-TRZ (10 nm)/(Na-An-BI):Liq (30 nm)/Liq (2 nm)/Al (80 nm) | D1 | 2 | 507 | (0.22, 0.63) | 3.3 | 41.0 | 32.5 | 7765 | 13.5 | 303 |
| D2 | 503 | (0.13, 0.63) | 3.0 | 46.9 | 41.9 | 4892 | 16.6 | ||||
| D3 | 528 | (0.38, 0.57) | 3.3 | 32.7 | 25.8 | 3759 | 9.7 | ||||
| Au15 | — | — | — | — | — | 2.6 | 76.3 | 62.7 | 44700 |
26.3 | 304 |
| Au16 | 3.0 | 45.2 | 33.6 | 39540 |
17.9 | ||||||
| Au17 | 2.6 | 87.1 | 75.1 | 73100 |
27.5 | ||||||
| Au18 | ITO/HAT-CN (5 nm)/TAPC (40 nm)/mCBP (10 nm)/Au-emitter:mCBP (20 nm)/PPF (10 nm)/TmPyPb (40 nm)/LiF(1.2 nm)/Al (100 nm) | — | 4 | 566 | (0.47, 0.52) | — | 60.3 | 72.1 | 198000 |
21.3 | 305 |
| Au19 | 10 | 502 | (0.21, 0.51) | 76.7 | 79.3 | 236000 |
26.9 | ||||
| Au20 | 2 | 483 | (0.15, 0.26) | 34.7 | 29.2 | 12500 |
23.6 | ||||
| Au21 | 6 | 503 | (0.20, 0.50) | 72.0 | 63.7 | 147000 |
25.9 | ||||
| Au22 | 10 | 517 | (0.25, 0.58) | 78.2 | 62.3 | 230000 |
24.5 | ||||
| Au23 | 6 | 487 | (0.18, 0.33) | 32.6 | 33.2 | 93000 |
15.1 | ||||
| Au24 | 2 | 632 | (0.61, 0.39) | 18.8 | 22.3 | 68800 |
17.4 | ||||
| Au25 | 10 | 705 | (0.69, 0.30) | 0.58 | 0.61 | 3200 | 9.96 | ||||
| Au21 (6 wt%):BN2 (1 wt%) | 6 | 535 | (0.29, 0.65) | 92.2 | 90.0 | 265000 |
25.3 | ||||
| Au26 | ITO/60 nm PEDOT:PSS/20 nm TAPC/10 nm PCZAC/25 nm PBICT: DBTTP1:Au(I) complex/5 nm TSPO1/40 nm TPBi/1.5 nm LiF/200 nm Al | — | 3 | 539 | (0.36, 0.59) | 4.5 | 85.7 | 76.7 | — | 24.4 | 306 |
| Au27 | 551 | (0.41, 0.56) | 4.8 | 77.5 | 67.7 | 23.3 | |||||
| Au28 | 650 | (0.63, 0.36) | 7.3 | 7.9 | 5.3 | 10.7 | |||||
| Au29 | 655 | (0.63, 0.36) | 7.0 | 7.8 | 5.9 | 11.6 | |||||
| Au30 | 680 | (0.62, 0.36) | 8.2 | 2.8 | 2.1 | 7.0 | |||||
| Au32 | (ITO)/10 nm p-doped (3 wt% NDP series, Novaled AG) (BCFA)/40 nm BCFA/10 nm (HT host): (ET host) (6:4, v/v):20 wt% Au(I) complex:emitter/(mCP-2CN)/30 nm (DBFPO): (LiQ)/LiQ/Al |
— | 20 wt%: 1.5 wt% | 475 | (0.108, 0.160) | 4.5 | — | 36.1 | — | 30.2 | 307 |
| Au33 | 494 | (0.156, 0.496) | 4.6 | 58.4 | 22.8 | ||||||
| Au34 | ITO/HAT-CN (10 nm)/FSFA (120 nm)/FSF4A (5 nm)/(NHC)AuBN: DMIC-Cz: DMIC-TRZ (30 nm)/ANT-BIZ (5 nm)/ANT-BIZ: Liq (1:1, 25 nm)/Liq (2 nm)/Al (100 nm). |
2 | 0.5 | 511 | (0.20, 0.69) | — | 86.1 | — | 2.53 × 105 | 24.8 | 308 |
| Au35 | — | 0.5 | 509 | (0.16, 0.66) | 74.0 | 1.17 × 105 | 24.0 | ||||
| Au36 | 1 | 2 | 540 | (0.16, 0.68) | 97.1 | 2.16 × 105 | 30.3 | ||||
| Au37 | — | 4 | 515 | (0.22, 0.67) | 82.8 | 1.92 × 105 | 24.0 | ||||
| Au38 | — | 1 | 512 | (0.18, 0.69) | 93.5 | 1.51 × 105 | 27.6 | ||||
| Au40 | ITO/HAT-CN (5 nm)/TAPC (30 nm)/TCTA (15 nm)/mCBP (10 nm)/DMIC-TRZ: emitter (45 nm)/PO-T2T (20 nm)/ANT-BIZ (30 nm)/Liq (2 nm)/AL (100 nm) | — | 1 | 531 | (0.37, 0.57) | 2.8 | 75.7 | 63.2 | 3000 | 23.9 | 309 |
| Au5 | ITO/PEDOT:PSS; 40 nm/emissive layer (30 nm)/3TPyMB; 5 nm/TmPyPB; 30 nm/LiF (0.8 nm)/Al (100 nm) | — | 10 | 528 | (0.37, 0.60) | — | 37.0 | 18.2 | — | 10.0 | 217 |
| Au43 | 20 | 532 | (0.42, 0.56) | 30.1 | 18.5 | 8.9 | |||||
| Au6 | 20 | 572 | (0.49, 0.50) | 22.2 | 12.4 | 8.0 | |||||
| Au44 | 20 | 588 | (0.53, 0.48) | 12.3 | 5.2 | 5.2 | |||||
| Au7 | 20 | 616 | (0.60, 0.40) | 7.9 | 4.4 | 5.3 | |||||
| Au46 | ITO/PEDOT:PSS/PVK:OXD-7: Au(III) complex/TPBi/LiF/Al | — | 4 | 509 | (0.29, 0.52) | — | 24.0 | 12.7 | 27400 |
8.3 | 310 |
| 8 | — | (0.30, 0.53) | 36.9 | 20.9 | 34200 |
12.5 | |||||
| 12 | — | (0.31, 0.54) | 43.9 | 22.5 | 53840 |
14.6 | |||||
| 16 | — | (0.32, 0.55) | 44.9 | 23.6 | 57340 |
14.8 | |||||
| Au49 | ITO/PEDOT:PSS/PVK:OXD-7: Au(III) complex/TPBi/LiF/Al | A | 4 | 500 | (0.25, 0.47) | — | 34.9 | 22.5 | 11450 |
12.7 | 310 |
| 8 | — | (0.26, 0.49) | 60.3 | 43.0 | 22750 |
20.6 | |||||
| 12 | — | (0.26, 0.50) | 60.2 | 41.1 | 27900 |
20.9 | |||||
| 16 | — | (0.27, 0.51) | 70.4 | 47.3 | 33740 |
23.8 | |||||
| Au10 | ITO/HAT-CN(5 nm)/TaPc (50 nm)/TCTA: AuIII emitter (10 nm)/TmPyPb (50 nm)/LiF (1.2 nm)/Al (100 nm) | — | 4 | — | 0.38, 0.56 | — | 65.59 | 91.98 | 7300 | 20.84 | 223 |
| 8 | 0.40, 0.55 | 70.82 | 81.73 | 16500 |
22.72 | ||||||
| 16 | 0.43, 0.54 | 77.78 | 94.00 | 22700 |
25.03 | ||||||
| Au60 | ITO/HAT-CN (5 nm)/TAPC (50 nm)/TCTA (10 nm)/AuIII emitter: DPEPO: TCTA (20 nm)/DPEPO (10 nm)/TmPyPB (40 nm)/LiF (1 nm)/Al (100 nm) | — | 4 | 540 | (0.38, 0.57) | — | 82.9 | 91.1 | 31000 |
24.9 | 311 |
| Au61 | 8 | 529 | (0.31, 0.60) | 89.0 | 87.7 | 35100 |
26.2 | ||||
| Au63 | 8 | 537 | (0.34, 0.60) | 92.5 | 104 | 43200 |
27.3 | ||||
| Au60 | ITO/HAT-CN (5 nm)/TAPC (40 nm)/mCBP (10 nm)/BN2:complex Au60:mCBP (20 nm)/PPF (10 nm)/TmPyPB (40 nm)/LiF (1.2 nm)/Al (100 nm) | A: Au60 (6 wt%): BN2 (1 wt%) | Au60 (6 wt%): BN2 (1 wt%) | 538 | (0.33, 0.65) | — | 120.7 | 122.5 | 170000 |
29.4 | 311 |
| Au3 | ITO/HAT-CN(5 nm)/TAPC (40 nm)/TCTA(10 nm)/TCTA:TPBi:Au(III) emitter(10)/TPBi(10 nm)/TmPyPb (40 nm)/LiF (1.2 nm)/Al (100 nm) | — | 2 | — | (0.32, 0.56) | — | 67.0 | 81.0 | 32000 |
21.6 | 216 |
| Au3 | 4 | (0.34, 0.56) | 71.9 | 87.0 | 37000 |
23.1 | |||||
| Au3 | 8 | (0.35, 0.56) | 76.0 | 99.4 | 37500 |
23.1 | |||||
| Au4 | 2 | (0.38, 0.56) | 59.9 | 72.3 | 21300 |
19.5 | |||||
| Au4 | 4 | (0.41, 0.55) | 57.5 | 86.9 | 26300 |
19.7 | |||||
| Au4 | 8 | (0.43, 0.53) | 53.8 | 65.0 | 17200 |
19.5 | |||||
| Au67 | 2 | (0.33, 0.56) | 55.5 | 69.5 | 30900 |
18.2 | |||||
| Au67 | 4 | (0.40, 0.55) | 70.6 | 82.8 | 70300 |
23.4 | |||||
| Au67 | 8 | (0.41, 0.54) | 66.3 | 79.6 | 59100 |
22.2 | |||||
| Au70 | Solution-processed devices, ITO/PEDOT:PSS; 40 nm/x% Au(III):mCP (30 nm)/3TPyMB; 5 nm/TmPyPB; 40 nm/LiF (1 nm)/Al (150 nm) | — | 20 | 612 | (0.61, 0.39) | — | 8.3 | 4.1 | — | 5.5 | 221 |
| Au71 | 604 | (0.59, 0.41) | 22.9 | 16.9 | 12.2 | ||||||
| Au72 | 608 | (0.60, 0.40) | 12.6 | 7.8 | 7.8 | ||||||
| Au75 | 628 | (0.64, 0.36) | 5.0 | 3.2 | 4.5 | ||||||
| Au70 | Vacuum-deposited devices, ITO/α-NPD; 40 nm/TCTA; 5 nm/x% Au70-Au75: m-CBP; 20 nm/Tm3PyP26PyB; 50 nm/LiF (1 nm)/Al (150 nm) | — | 5 | 608 | (0.58, 0.41) | — | 14.1 | 12.6 | 1483 | 9.3 | 221 |
| Au71 | 8 | 600 | (0.57, 0.43) | 23.4 | 21.0 | 2522 | 12.7 | ||||
| Au72 | 5 | 600 | (0.56, 0.44) | 17.8 | 16.0 | 1507 | 9.9 | ||||
| Au73 | 8 | 612 | (0.60, 0.40) | 15.8 | 16.6 | 1867 | 10.8 | ||||
| Au74 | 11 | 612 | (0.60, 0.40) | 13.7 | 12.9 | 1604 | 9.2 | ||||
| Au75 | 2 | 616 | (0.59, 0.40) | 10.5 | 8.3 | 706 | 7.8 | ||||
| Au77 | ITO/α-NPD; 40 nm/TCTA; 5 nm/emissive layer/Tm3PyP26PyB; 50 nm/LiF (1 nm)/Al (150 nm) | — | 14 | 528 | (0.32, 0.61) | — | 43.9 | 38.8 | — | 12.6 | 312 |
| Au78 | 5 | 552 | (0.42, 0.56) | 34.8 | 31.2 | 10.3 | |||||
| Au79 | 14 | 532 | (0.33, 0.61) | 55.5 | 58.1 | 15.7 | |||||
| Au80 | 8 | 544 | (0.39, 0.58) | 53.0 | 41.6 | 15.4 | |||||
| Au81 | 2 | 612 | (0.59, 0.40) | 10.3 | 9.2 | 7.0 | |||||
| Au83 | ITO/PEDOT:PSS (40 nm)/Au83-85:mCP (30 nm)/3TPyMB (5 nm)/TmPyPB (30 nm) LiF (1 nm)/Al (120 nm) | — | 20 | 548 | (0.40, 0.57) | — | 40.0 | 15.7 | 689 | 11.9 | 313 |
| Au84 | 560 | (0.43, 0.54) | 27.1 | 10.4 | — | 8.7 | |||||
| Au85 | 540 | (0.38, 0.57) | 52.6 | 41.3 | 850 | 15.8 | |||||
| Au86 | Solution-processed device, ITO/PEDOT:PSS (40 nm)/Au86-91: mCP (30 nm)/3TPyMB (5 nm)/TmPyPB (40 nm) LiF Al | — | 20 | 500, 528 | (0.29, 0.56) | — | 26.2 | 13.7 | — | 8.4 | 314 |
| Au87 | 520 | (0.29, 0.58) | 38.0 | 17.1 | 11.7 | ||||||
| Au88 | 584 | (0.51, 0.48) | 15.4 | 6.3 | 6.1 | ||||||
| Au89 | 576 | (0.49, 0.50) | 27.7 | 11.4 | 10.0 | ||||||
| Au90 | 560 | (0.44, 0.54) | 36.6 | 18.7 | 11.4 | ||||||
| Au91 | 556 | (0.42, 0.55) | 34.8 | 15.6 | 10.9 | ||||||
| Au87 | Vacuum-deposited device, ITO/PEDOT:PSS (40 nm)/Au87, Au89, Au90 and Au91: mCP (30 nm)/3TPyMB (5 nm)/TmPyPB (40 nm) LiF Al | — | 14 | 528 | (0.33, 0.56) | — | 36.6 | 38.3 | 3314 | 12.0 | 314 |
| Au89 | 5 | 564 | (0.45, 0.52) | 33.7 | 30.3 | 4321 | 12.2 | ||||
| Au90 | 11 | 564 | (0.46, 0.53) | 42.6 | 41.5 | 4442 | 14.9 | ||||
| Au91 | 8 | 544 | (0.40, 0.55) | 46.4 | 41.6 | 2110 | 15.0 | ||||
Li et al. present a novel development of TADF Pd(II) complexes that show ILCT excited states perturbed by small metal contribution. They have successfully developed two complexes (Pd7 and Pd8; Scheme 5) emitting orange and red light with efficiencies of 82% and 89% and lifetimes of 2.19 and 0.97 μs, respectively – Table 7. Through mixed transient spectroscopic and theoretical analyses on one complex, they have identified a metal-perturbed rapid ISC process. OLEDs utilizing these Pd(II) complexes exhibit remarkable EQEmax, ranging from 27.5% to 31.4% – Table 6, with minimal roll-offs down to 1% at 1000 cd m−2. Additionally, the operational stability of the Pd(II) complexes is exceptional, with LT95 values exceeding 220 hours at 1000 cd m−2. This stability stems from the strong σ-donating ligands, the presence of multiple intramolecular noncovalent interactions and in addition from their short emission lifetimes. This research highlights a favorable strategy for creating effective and durable luminescent complexes without using third-row transition metals.200
| Complex | Medium | λ em [nm] | PLQY [%] | τ exp [μs] | k r [s−1] | k nr [s−1] | k ISC [s−1] | k rISC [s−1] | ΔEST [cm−1] | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Pd2 | At RT in DCM | 498, 537, 582 | 0.21 | 30.5 | — | — | — | — | 1202 | 198 |
| Pd3 | 498, 536, 580 | 0.52 | 26.7 | 1153 | ||||||
| Pd4 | 513, 551 | 1.34 | 152.9 | 1839 | ||||||
| Pd5 | 513, 550 | 1.72 | 118.5 | 1887 | ||||||
| Pd6 | 499, 538, 582 | 2.88 | 44.4 | |||||||
| Pt2 | 501, 539, 576 | 50.1 | 15.8 | |||||||
| Pd2 | At RT in PMMA | 461, 498, 536, 580 | — | 89.4 | — | — | — | — | — | 198 |
| Pd3 | 462, 498, 536, 579 | 79.9 | ||||||||
| Pd4 | 457, 513, 548 | 109.9 | ||||||||
| Pd5 | 456, 515, 551 | 57.0 | ||||||||
| Pd6 | 498, 536, 597 | 109.5 | ||||||||
| Pt2 | 500, 537, 579 | 12.2 | ||||||||
| Pd7 | In 5 wt% doped PMMA at 298 K and (77 K) | 637 (634) | 89 | 0.97 (70.6) | 9.2 × 105 | 1.1 × 105 | — | — | — | 200 |
| Pd8 | 598 (594) | 82 | 2.19 (336) | 3.7 × 105 | 8.2 × 104 | |||||
| Pt5 | Toluene Chlorobenzene CH2Cl2 | 579, 617 | 0.16 | 1.2 | 1.3 × 105 | 7.1 × 105 | — | — | — | 204 |
| 578, 635 | 0.51 | 5.0 | 1.0 × 105 | 1.0 × 105 | ||||||
| 568, 641 | 0.34 | 3.9 | 0.9 × 105 | 1.7 × 105 | ||||||
| Pt6 | Toluene Chlorobenzene CH2Cl2 | 495, 514 | 0.82 | 5.3 | 1.6 × 105 | 0.3 × 105 | — | — | — | 204 |
| 514 | 0.85 | 5.7 | 1.5 × 105 | 0.3 × 105 | ||||||
| 512 | 0.73 | 6.4 | 1.1 × 105 | 0.4 × 105 | ||||||
| Pt1 | MCH | 602, 654 | 0.83 ± 0.090 | 2.1 ± 0.1 | 4.0 ± 0.6 × 105 | 0.8 ± 0.5 × 105 | — | — | 201 | |
| 0.68 ± 0.08 | 1.2 ± 0.1 | 5 ± 1 × 105 | 2.6 ± 0.9 × 105 | |||||||
| Toluene | 612 | 0.04 ± 0.01 | 0.11 ± 0.01 | 4 ± 2 × 105 4 ± 2 × 105 | 92 ± 9 × 105 180 ± 9 × 105 | |||||
| 2Me-THF DCM | 618, 664 | 0.02 ± 0.01 | 0.055 ± 0.002 | 4.0 ± 0.6 × 105 | 0.8 ± 0.5 × 105 | |||||
| Pt7 | CH2Cl2 (2 × 10−5 M) | 556 | <0.5 | 7.7 | — | — | — | — | — | 302 |
| Pt8 | 557 | 4 | 4.3 | |||||||
| Pt9 | 555 | 52 | 14.8 | |||||||
| Pt10 | 524 | <0.5 | 7.9 | |||||||
| Pt11 | 525 | 46 | 4.9 | |||||||
| Pt12 | 525 | 81 | 4.0 | |||||||
| Pt7 | Solid | 556 | <0.5 | 8.3 | — | — | — | — | — | 302 |
| Pt8 | 553 | 11 | 9.9 | |||||||
| Pt9 | 553 | 14 | 11.2 | |||||||
| Pt10 | 521 | <0.5 | 10.6 | |||||||
| Pt11 | 530 | 3 | 11.4 | |||||||
| Pt12 | 520 | 8 | 10.9 | |||||||
| Pt7 | PMMA film | 552 | 20 | 26.2 | — | — | — | — | — | 302 |
| Pt8 | 553 | 46 | 15.6 | |||||||
| Pt9 | 551 | 84 | 23.8 | |||||||
| Pt10 | 516 | 33 | 13.9 | |||||||
| Pt11 | 522 | 86 | 12.3 | |||||||
| Pt12 | 520 | 90 | 19.4 | |||||||
| Pt7 | mCP film | 553 | 52 | 28.4 | — | — | — | — | — | 302 |
| Pt8 | 554 | 93 | 14.5 | |||||||
| Pt9 | 552 | 94 | 20.4 | |||||||
| Pt10 | 519 | 59 | 19.1 | |||||||
| Pt11 | 524 | 96 | 8.8 | |||||||
| Pt12 | 521 | 97 | 17.2 | |||||||
| Pt13 | Toluene solution (1 × 10−5 M, 298 K) | 497, 536, 578 | 75 | — | — | — | — | — | — | 303 |
| Pt14 | 527, 564, 612 | 35 | ||||||||
| Au18 | Toluene (2 × 105 M) | 617 | 0.17 | 0.17 | 10.0 | 8.3 | 3.1 × 1010 | 0.8 × 109 | 565 | 305 |
| Au19 | 550 | 0.73 | 0.33 | 22.1 | 8.2 | — | — | — | ||
| Au20 | 485 | 0.72 | 0.36 | 20.0 | 7.8 | — | — | — | ||
| Au21 | 550 | 0.53 | 0.45 | 11.7 | 10.4 | 4.3 × 1010 | 3.0 × 109 | 323 | ||
| Au22 | 570 | 0.60 | 0.36 | 16.6 | 11.1 | 3.9 × 1010 | 1.2 × 109 | 484 | ||
| Au23 | 500 | 0.76 | 0.45 | 16.9 | 5.3 | 9.9 × 1010 | 1.7 × 109 | 645 | ||
| Au24 | 728 | 0.10 | <0.1 | — | — | 9.2 × 1010 | 3.5 × 109 | 484 | ||
| Au25 | 768 | 0.02 | <0.1 | — | — | — | — | — | ||
| Au18 | 6 wt% mCBP | 554 | 0.99 | 0.39 | 25.4 | 0.3 | 3.1 × 1010 | 0.8 × 109 | 565 | 305 |
| Au19 | 504 | 0.99 | 0.31 | 31.9 | 0.3 | — | — | — | ||
| Au20 | 468 | 0.75 | 1.1 | 6.8 | 2.2 | — | — | — | ||
| Au21 | 509 | 0.91 | 0.35 | 26.0 | 2.6 | 4.3 × 1010 | 3.0 × 109 | 323 | ||
| Au22 | 525 | 0.99 | 0.32 | 30.9 | 0.3 | 3.9 × 1010 | 1.2 × 109 | 484 | ||
| Au23 | 466 | 0.82 | 0.66 | 12.4 | 2.7 | 9.9 × 1010 | 1.7 × 109 | 645 | ||
| Au24 | 644 | 0.83 | 0.28 | 29.6 | 6.1 | 9.2 × 1010 | 3.5 × 109 | 484 | ||
| Au25 | 666 | 0.63 | 0.21 | 30.0 | 17.6 | — | — | — | ||
| Au26 | Thin-films | 533 | 0.84 | 0.84 | 26 × 105 | 5.0 × 105 | 21 × 108 | 6.6 × 108 | 323 | 306 |
| Au27 | 553 | 0.87 | 0.87 | 27 × 105 | 2.4 × 105 | 22 × 108 | 4.5 × 108 | 444 | ||
| Au28 | 663 | 0.26 | 0.26 | 8.7 × 105 | 25 × 105 | 7.9 × 108 | 1.5 × 108 | 395 | ||
| Au29 | 666 | 0.22 | 0.22 | 8.5 × 105 | 30 × 105 | 25 × 108 | 2.6 × 108 | 516 | ||
| Au30 | 705 | 0.10 | 0.10 | 6.3 × 105 | 56 × 105 | 3.8 × 108 | 4.7 × 108 | 419 | ||
| Au31 | 750 | — | — | — | — | — | — | — | ||
| Au32 | Zeonex films | 444 | 0.92 | 1.6 | >3.9 × 107 | >0.034 × 107 | >370 × 107 | >65 × 107 | 565/661 | 307 |
| Au33 | 466 | 0.95 | 0.38 | >13 × 107 | >0.066 × 107 | >180 × 107 | >45 × 107 | 645/476 | ||
| Au34 | THF solution (2 × 10−5 M) (298 K) | 511 | 0.88 | 7.9 | — | — | — | — | — | 308 |
| Au35 | 511 | 0.83 | 7.2 | |||||||
| Au36 | 511 | 0.86 | 6.9 | |||||||
| Au37 | 510 | 0.78 | 6.5 | |||||||
| Au38 | 511 | 0.93 | 7.3 | |||||||
| Au39 | 471 | 0.89 | 27.0 | |||||||
| Au34 | 2% PMMA (298 K) | 515 | 0.92 | 5.5 | — | — | — | — | — | 308 |
| Au35 | 514 | 0.90 | 5.8 | |||||||
| Au36 | 513 | 0.91 | 5.5 | |||||||
| Au37 | 514 | 0.87 | 5.6 | |||||||
| Au38 | 514 | 0.88 | 5.9 | |||||||
| Au39 | 473 | 0.84 | 22.8 | |||||||
| Au40 | 10 wt% DPEPO film at 300 K (77 K) | 525 (477) | 66 | 2.9 (48.5) | 2.3 × 105 | 1.2 × 105 | — | — | 561 | 309 |
| Au41 | 478 (478) | 51 | 704 (966) | 7.2 × 102 | 7 × 102 | — | ||||
| Au42 | 529 (529) | 66 | 1.2 (23.4) | 5.5 × 105 | 2.8 × 105 | 506 | ||||
| Au5 | Toluene (298 K) | 524, 564, 606 | 0.029 | 5.7 | 5.14 × 103 | 1.70 × 105 | — | — | — | 217 |
| Au43 | 525, 563, 609 | 0.083 | 3.9 | 2.12 × 104 | 2.35 × 105 | |||||
| Au6 | 606 | 0.066 | 0.3 | 2.19 × 105 | 3.11 × 106 | |||||
| Au44 | 660 | 0.022 | <0.1 | 2.20 × 105 | 9.78 × 106 | |||||
| Au7 | 656 | 0.028 | 2.8 | 1.01 × 104 | 3.47 × 105 | |||||
| Au8 | 740 | 0.012 | 1.6 | 7.25 × 103 | 6.18 × 105 | |||||
| Au5 | Thin-film (298 K) 5 wt% in mCP | 528, 568, 616 | 0.57 | 96.1 | 5.93 × 103 | 4.47 × 103 | — | — | — | 217 |
| Au43 | 528, 568, 616 | 0.52 | 79.3 | 6.56 × 103 | 6.05 × 103 | |||||
| Au6 | 562 | 0.87 | 5.7 | 1.53 × 105 | 2.28 × 104 | |||||
| Au44 | 559 | 0.53 | 13.5 | 3.93 × 104 | 3.48 × 104 | |||||
| Au7 | 585 | 0.70 | 1.3 | 5.38 × 105 | 2.31 × 105 | |||||
| Au8 | 636 | 0.40 | 0.4 | 1.00 × 106 | 1.50 × 106 | |||||
| Au45 | In Toluene, room temperature | (468, 503, 534) | 0.14 | 61.5 | 2 × 103 | — | — | — | — | 310 |
| Au46 | 534 | 0.74 | 1.03 | 7.2 × 105 | ||||||
| 0.32 | 5.6 × 105 | |||||||||
| Au47 | 596 | 0.18 | 43.5 | 3 × 103 | ||||||
| Au48 | (469, 503, 532) | 0.13 | 0.71 | 11.1 × 105 | ||||||
| Au49 | 524 | 0.79 | 0.75 | 9.1 × 105 | ||||||
| Au50 | 511 | 0.68 | 29.4 | 2 × 103 | ||||||
| Au51 | (591, 641, 702) | 0.065 | 1.10 | 6.8 × 105 | ||||||
| Au52 | 527 | 0.75 | 61.5 | 2 × 103 | ||||||
| Au45 | 4 wt% in PMMA thin-film, room temperature | (471, 504, 534) | 0.14 | 51 | 3 × 103 | — | — | — | — | 310 |
| Au46 | 523 | 0.66 | 1.35 | 4.9 × 105 | 209 | |||||
| Au47 | 564 | 0.42 | 0.74 | 5.7 × 105 | 393 | |||||
| Au48 | (471, 504, 534) | 0.13 | 34 | 4 × 103 | — | |||||
| Au49 | 517 | 0.84 | 0.72 | 11.7 × 105 | 318 | |||||
| Au50 | 513 | 0.78 | 0.85 | 9.2 × 105 | — | |||||
| Au51 | (593, 642, 701) | 0.031 | 16.5 | 2 × 103 | — | |||||
| Au52 | 521 | 0.67 | 1.10 | 6.1 × 105 | ||||||
| Au53 | In toluene | 495, 526, 565 498, 526, 567 524, 550 612 493, 521 485, 518, 552 533 580 480, 513, 557 | 0.54 | 93.1 | 5.8 × 103 | — | — | — | — | 223 |
| Au54 | 0.40 | 77.1 | 5.2 × 103 | |||||||
| Au9 | 0.77 | 94.3 | 8.2 × 103 | |||||||
| Au10 | 0.47 | 0.62 | 758 × 103 | |||||||
| Au55 | 0.28 | 225 | 1.2 × 103 | |||||||
| Au56 | 0.26 | 152 | 1.7 × 103 | |||||||
| Au57 | 0.94 | 1.61 | 584 × 103 | |||||||
| Au58 | 0.74 | 0.79 | 937 × 103 | |||||||
| Au59 | 0.003 | 1.04 | 2.88 × 103 | |||||||
| Au53 | In thin-films | 492, 523, 560 | 0.20 | 43.8 | 4.57 × 103 | — | — | — | — | 223 |
| Au54 | 490, 522, 562 | 0.04 | 90.1 | 0.44 × 103 | ||||||
| Au9 | 550 | 0.47 | 56.8 | 8.27 × 103 | ||||||
| Au10 | 568 | 0.89 | 1.69 | 527 × 103 | ||||||
| Au55 | 489, 519, 555 | 0.06 | 147 | 0.41 × 103 | ||||||
| Au56 | 486, 518, 555 | 0.06 | 90.4 | 0.66 × 103 | ||||||
| Au57 | 520 | 0.82 | 2.08 | 394 × 103 | ||||||
| Au58 | 568 | 0.71 | 2.54 | 280 × 103 | ||||||
| Au59 | 482, 515, 552 | 0.06 | 36.01 | 1.67 × 103 | ||||||
| Au53 | 492, 523, 560 | 0.20 | 43.8 | 4.57 × 103 | ||||||
| Au54 | 490, 522, 562 | 0.04 | 90.1 | 0.44 × 103 | ||||||
| Au9 | 550 | 0.47 | 56.8 | 8.27 × 103 | ||||||
| Au10 | 568 | 0.89 | 1.69 | 527 × 103 | ||||||
| Au55 | 489, 519, 555 | 0.06 | 147 | 0.41 × 103 | ||||||
| Au56 | 486, 518, 555 | 0.06 | 90.4 | 0.66 × 103 | ||||||
| Au57 | 520 | 0.82 | 2.08 | 394 × 103 | ||||||
| Au58 | 568 | 0.71 | 2.54 | 280 × 103 | ||||||
| Au60 | Toluene | 547 | 80 | 0.56 | 1.43 × 106 | 0.36 × 106 | 1.52 × 1011 | — | — | 311 |
| Au61 | 537 | 77 | 0.67 | 1.15 × 106 | 0.34 × 106 | 7.14 × 1010 | ||||
| Au62 | 528 | 73 | 19 | 0.038 × 106 | 0.014 × 106 | 4.88 × 1010 | ||||
| Au63 | 532 | 88 | 0.69 | 1.28 × 106 | 0.17 × 106 | 1.00 × 1011 | ||||
| Au60 | 4 wt% in DPEPO/TCTA | 530 | 98 | 0.56 | 1.75 × 106 | 0.036 × 106 | — | — | — | 311 |
| Au61 | 521 | 93 | 0.51 | 1.82 × 106 | 0.14 × 106 | |||||
| Au63 | 528 | 99 | 0.54 | 1.83 × 106 | 0.019 × 106 | |||||
| Au64 | In toluene | 466, 495, 530 | 0.002 | 0.34 | 0.06 × 105 | — | — | — | — | 216 |
| Au3 | 574 | 0.60 | 0.78 | 7.69 × 105 | ||||||
| Au65 | 545 | 0.21 | 1.25 | 1.68 × 105 | ||||||
| Au66 | 562 | 0.49 | 0.80 | 6.13 × 105 | ||||||
| Au4 | 603 | 0.57 | 0.84 | 6.79 × 105 | ||||||
| Au67 | 594 | 0.25 | 0.33 | 7.58 × 105 | ||||||
| Au68 | 632 | 0.02 | 0.20 | 1.00 × 105 | ||||||
| Au69 | 625 | 0.08 | 0.25 | 3.20 × 105 | ||||||
| Au64 | In thin-films | 468, 496, 530 | 0.028 | 22.4 | 0.01 × 105 | — | — | — | — | 216 |
| Au3 | 577 | 0.88 | 0.85 | 10.35 × 105 | ||||||
| Au65 | 546 | 0.29 | 3.78 | 0.77 × 105 | ||||||
| Au66 | 560 | 0.67 | 1.43 | 4.69 × 105 | ||||||
| Au4 | 567 | 0.65 | 1.46 | 4.45 × 105 | ||||||
| Au67 | 568 | 0.80 | 1.19 | 6.72 × 105 | ||||||
| Au68 | 595 | 0.56 | 0.77 | 7.27 × 105 | ||||||
| Au69 | 527, 570, 640 | 0.09 | 0.33 | 2.73 × 105 | ||||||
| Au70 | Toluene (298 K) | 638 | 0.06 | 1.5 | 4.0 × 104 | 6.3 × 105 | — | — | — | 221 |
| Au71 | 615 | 0.08 | 1.0 | 8.0 × 104 | 9.2 × 105 | 1048 | ||||
| Au72 | 625 | 0.11 | 1.3 | 8.5 × 104 | 6.8 × 105 | 968 | ||||
| Au73 | 621 | 0.08 | 0.8 | 1.0 × 105 | 1.2 × 106 | — | ||||
| Au74 | 628 | 0.05 | 0.5 | 1.0 × 105 | 2.0 × 106 | — | ||||
| Au75 | 650 | 0.07 | 1.3 | 5.4 × 104 | 7.2 × 105 | — | ||||
| Au76 | 714 | 4.4 × 10−3 | 0.2 | 2.2 × 104 | 5.0 × 106 | — | ||||
| Au77 | Toluene (298 K) | 532 | 0.23 | 8.1 | 2.8 × 104 | 9.5 × 104 | — | — | 1290 | 312 |
| Au78 | 550 | 0.19 | 7.2 | 2.6 × 104 | 1.3 × 105 | — | ||||
| Au79 | 534 | 0.20 | 10.5 | 1.9 × 104 | 7.6 × 104 | 1290 | ||||
| Au80 | 549 | 0.21 | 4.8 | 4.4 × 104 | 1.7 × 105 | — | ||||
| Au81 | 635 | 0.09 | 5.3 | 1.7 × 104 | 1.7 × 105 | 1452 | ||||
| Au82 | 630 | 0.11 | 5.6 | 2.0 × 104 | 1.6 × 105 | — | ||||
| Au77 | Thin-film (298), in mCP | 531 | 0.77 | 1.9, 22.1 | — | — | — | — | 1290 | 312 |
| Au78 | 546 | 0.75 | 2.9, 18.8 | — | ||||||
| Au79 | 534 | 0.74 | 2.3, 20.6 | 1290 | ||||||
| Au80 | 546 | 0.67 | 2.4, 20.9 | — | ||||||
| Au81 | 604 | 0.30 | 1.6, 8.2 | 1452 | ||||||
| Au82 | 608 | 0.24 | 2.4, 9.7 | — | ||||||
| Au83 | Toluene (298 K) | 607 | 0.040 | 0.5 | 8.0 × 104 | 1.9 × 106 | — | — | — | 313 |
| Au84 | 624 | 0.036 | 0.2 | 1.8 × 105 | 4.8 × 106 | |||||
| Au85 | 584 | 0.091 | 0.5 | 1.8 × 105 | 1.8 × 106 | |||||
| Au83 | Thin flm (298 K), 20 wt% in mCP | 547 | 0.82 | 3.5 | 2.3 × 105 | 5.1 × 104 | — | — | — | 313 |
| Au84 | 543 | 0.68 | 1.1 | 6.2 × 105 | 2.9 × 105 | |||||
| Au85 | 525 | 0.58 | 1.6 | 3.6 × 105 | 2.6 × 105 | |||||
| Au86 | Toluene (298 K) | 540 | 0.15 | 2.0 | 7.50 × 104 | 4.25 × 105 | — | — | — | 314 |
| Au87 | 570 | 0.14 | 0.6 | 2.33 × 105 | 1.43 × 106 | |||||
| Au88 | 659 | 0.003 | 0.04 | 6.98 × 104 | 2.32 × 107 | |||||
| Au89 | 637 | 0.01 | 0.1 | 1.00 × 105 | 9.90 × 106 | |||||
| Au90 | 600 | 0.05 | 0.5 | 1.00 × 105 | 1.90 × 106 | |||||
| Au91 | 599 | 0.07 | 0.5 | 1.20 × 105 | 1.88 × 106 | |||||
| Au86 | Thin flm (298 K), 20 wt% in mCP | 498, 526 | 0.34 | 62.9 | 5.41 × 103 | 1.05 × 104 | — | — | — | 314 |
| Au87 | 501, 524 | 0.59 | 54.7 | 1.08 × 104 | 7.50 × 103 | |||||
| Au88 | 580 | 0.27 | 0.8 | 3.38 × 105 | 9.13 × 105 | |||||
| Au89 | 564 | 0.52 | 1.3 | 4.00 × 105 | 3.69 × 105 | |||||
| Au90 | 548 | 0.66 | 4.8 | 1.38 × 105 | 7.08 × 104 | |||||
| Au91 | 543 | 0.68 | 3.2 | 2.13 × 105 | 1.00 × 105 |
Two solid-state near-infrared (NIR) emitting Pt(II) complexes that appear yellow and green in solution, namely PBSNND (Pt3) and AtFNND (Pt4) – Scheme 5, were successfully utilized alongside the classical blue fluorescent N,N′-di-1-napthalenyl-N,N′-diphenyl-[1,1′:4′,1′′:4′′,1′′′′-quaterthiophene]-4,4′′′-diamine (4P-NPD) to create hybrid WOLEDs. By employing an ultra-thin EML technique, dual emission in yellow and deep red/NIR EL was effectively achieved in Pt3-based hybrid WOLEDs. The most optimal EL performance was observed in a hybrid WOLED configuration with a 1.0 nm ultra-thin phosphorescent emitting layer, and a 4P-NPD layer (2.5 nm) positioned in front of TPBi, the ETL of the device (Hyb W1). Hyb W1 displayed warm white EL with a color coordinate of x,y CIE color coordinates of (0.41, 0.31), a high CRI of 86, and an EQE reaching 12.9% – Table 6. In contrast, by using a singly doped phosphorescent emitting layer, dual emission in green and deep red/NIR emission was achieved in Pt4-based hybrid WOLEDs. With an Pt4 dopant concentration of 5% or 7% by weight, a purplish blue white electroluminescence with x,y CIE color coordinates of 0.30, 0.31, a CRI of 84, and an EQE reaching 10.5% (Table 6). For devices Hyb W3, an authentic white EL was achieved with x,y CIE color coordinates (0.35, 0.32), an exceptional CRI of 88, and a high EQE of 13.7% – Table 6.301
Pander and co-workers conducted the preparation and comprehensive photophysical analysis of a di–Pt(II) complex featuring a ditopic bisN^C^N ligand (Pt1, Pt5 and Pt6) – Scheme 5. This complex demonstrates dual luminescent behavior, emitting both delayed fluorescence and phosphorescence simultaneously. Through a comparison with the mono-Pt(II) counterpart, they establish that the di–Pt(II) complex activates TADF owing to three primary differences when compared to the mono-Pt(II) form: a higher singlet radiative rate constant (kSr), a reduced ΔEST, and an extended phosphorescence decay lifetime (τPH). Their observations align with similar trends observed in other di–Pt(II) complexes, indicating that bimetallic structures create favorable conditions for TADF. Additionally, the diPt(II) complex (Pt5) exhibits a long-wavelength-emissive excimer, leading to NIR electroluminescence at 805 nm in a solution-processed OLED device with a remarkable EQEmax of 0.51% – Table 6.204
A new dinuclear Pt(II) complex (Pt1; Scheme 5) has been synthesized, featuring a ditopic, bis-tetradentate ligand. This ligand provides a planar O^N^C^N coordination environment for each metal ion, with both metals bound to the nitrogen atoms of a bridging pyrimidine group. The complex exhibits strong red luminescence with a PLQY of 83% in deoxygenated methylcyclohexane solution at room temperature, along with a notably short, excited state lifetime of 2.1 ms – Table 7. These exceptional properties stem from an impressively high kr constant of approximately 4 × 105 s−1 – Table 7, and small FWHM of 75 nm in OLED host and outstanding 22 nm in methylcyclohexane. This unique behavior results from efficient thermally activated rISC, facilitated by a small ΔEST of just 556 ± 24 cm−1. In OLED applications, the complex was successfully integrated into solution-processed devices, achieving EQEmax of 7.4% – Table 6. This development marks the well-documented instance of a Pt(II) complex exhibiting TADF at room temperature, as well as being the incorporation of a Pt(II)-based delayed fluorescence emitter into an OLED system.201
Achieving narrow-band emission is essential for enhancing color purity in panel displays. However, this goal presents a significant challenge due to the broad bandwidths typically associated with TADF or phosphorescence in organic and metal–organic materials, arising from multiple CT transitions. In this study, a novel strategy is suggested to realize effective narrow-band emission through the sensitization of LC phosphorescence by significant intermetallic interactions. Compared to the weak phosphorescence observed in mononuclear Pt(II) precursors, the introduction of Pt(II)–Au(I) heteronuclear structures (Pt7–12, Scheme 5) results in a remarkable activation of highly efficient LC phosphorescence through significant d8–d10 interaction, achieving PLQYs of up to 81% in solution and 97% in doped films – Table 7. Through this approach, high-efficiency solution-processed OLEDs producing narrow-band emission have been developed. These OLED devices exhibit impressive EQE of 21.6% and FWHM of 36 nm for yellow electroluminescence, as well as EQE = 20.8% and FWHM = 32 nm for green electroluminescence – Table 6. The incorporation of Pt-Au heteronuclear structures not only enhances ISC from singlet to triplet states but also mitigates non-radiative thermal relaxation processes, leading to a substantial sensitization of LC phosphorescence. Notably, PtAu2 complexes supported by double triphosphine ligands exhibit superior protection against non-radiative deactivation of triplet states compared to PtAu complexes linked by double diphosphine ligands, thereby yielding significantly stronger phosphorescence. The comparison of the aborpstion and emission features of the Pt(II) precursor and the related Pt–Au heteronuclear complexes suggest that the Pt–Au clusters function effectively as sensitizers to caputure light. Finally, this can then be effectively transferred to the naphthalene-acetylide ligands.302
In following, Xu et al. propose a viable molecular develop approach to create an organic electroluminescent emitter with narrowband emission. This approach involves incorporating an innovative multiple resonance TADF fragment into the traditional Pt(II) complex. The solution-processed OLED, utilizing the designed Pt(II) complex BNCz-PPy-Pt-acac (BNCPPt) (Pt13, Scheme 5) as the dopant emitter, demonstrates light green emission with a peak wavelength of 507 nm and a narrow FWHM of 35 nm. This system achieves a EQEmax of 13.5% with minimal efficiency roll-off of just 4.4% at an illumination intensity of 1000 cd m−2 – Table 6. This complex not only incorporates the advantageous features of the multiresonant design, but also preserves the remarkable traits of short lifetime and minimal efficiency reduction of heavy metal complexes. The impressive electroluminescent characteristics exhibited by Pt13 were the foundation for a practical and insightful molecular model in materials research for cutting-edge display technologies. Significantly, this novel design approach bridges the multiresonant strategy with heavy metal atoms, establishes a definitive molecular structure design principle, and introduces a fresh strategy for developing heavy metal complexes with narrowband emission tailored for ultra-high definition displays.303
Despite the fact that the first Au(I) TADF emitter Au(PP)(PS) (Au14, Scheme 5) (PP = 1,2-bis(diphenylphosphino)benzene and PS = 2-diphenylphosphinobenzenethiolate was developed by Osawa et al. in 2013, the presence of rapid ligand exchange in solution precluded its implementation in OLEDs).315 However, very recently, examples of TADF Au(I) complexes have been employed in OLEDs. Here, Credgington, Bochmann, Linnolahtl et al. demonstrated that the new linear donor-bridge-acceptor design that produced very effective rISC to singlets in Cu(I) complexes was also applicable for the design of the first examples of TADF in Au(I) chemistry.316 In this respect, Table 6 list the most important Au(I) complexes reported in literature, together with the OLED structure in which they have been used, as well as the corresponding device metrics. Photophysical and TADF properties of OLED utilizing Au(I) complexes are summarized in Table 7.
Specifically, the complexes (CAAC)AuCz (Au15, Scheme 5), (CAAC)AuNPh2 (Au16, Scheme 5) and (CAAC)AuDTBCz (Au17, Scheme 5) (Cz anion, DTBCz = 3,6-di-tert-butylcarbazole anion) were implemented in all-organic multi-layer OLEDs. The devices consist on a ∼180 nm polymeric hole-transport/electron-blocking layer, poly(9,9-dioctylfluoreneco-N-(4-butylphenyl)diphenylamine) (TFB), deposited on poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS) from toluene solution, a ∼20 nm LEL comprising the corresponding complex, is deposited on PVK host from a dimethylformamide solution. Simultaneously, a layer of about 70 nm thickness comprising bathophenanthroline (BPhen), which acts as an electron-transport/hole-blocking layer, is deposited.
They were also able to implement complex (CAAC)AuCz (Au15, Scheme 5) in a thermal evaporation fabrication routine and demonstrated the viability of these complexes for industrial application. A device with lesser device-to-device variation compared with the solution-processed devices and a champion ηEQE,EL = 26.9% is reported.304 The authors explore mCP (Fig. 26c) and TCP (Fig. 26d) as host materials and found a critical effect on varying the host and the dopant concentration on the properties of the device, so a careful device optimization is mandatory.
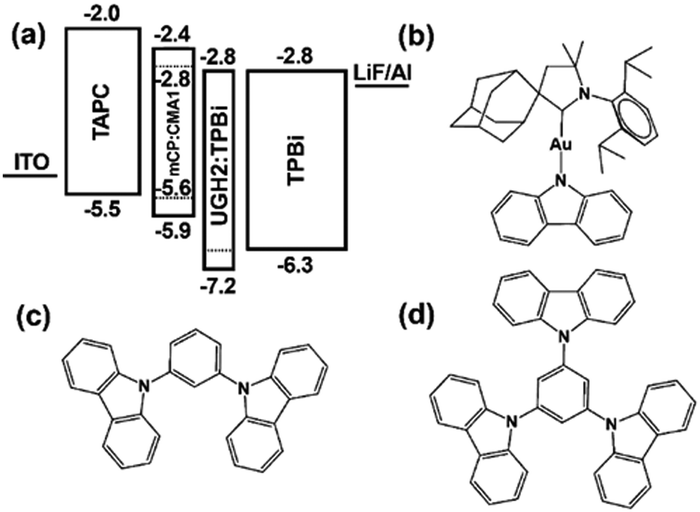 | ||
| Fig. 26 (a) Device architecture of CMA1 OLEDs ITO/1,1′-bis[(di-4-tolylamino)phenyl]cyclohexane (TAPC) (40 nm)/mCP:CMA1(20 nm, varying wt%)/UGH2:TPBi(10 nm, 10 wt% TPBi)/TPBi (40 nm)/LiF(1 nm)/Al (100 nm) HOMO and LUMO energy levels are indicated in eV. (b)–(d) Complex (CAAC)AuCz Au15 (b), mCP (c), and TCP (d). Reproduced with permissions from ref. 304. Copyright 2018, Wiley-VCH. | ||
Lam et al. present a group of structurally straightforward and operationally robust two-coordinate CMA(Au) TADF materials, Au18–25 (Scheme 5), leveraging a CMA molecular framework incorporating sterically bulky pyrazine-/pyridine-/quinoxaline-fused NHC ligands.
These CMA(Au) emitters exhibit thermal stability, establish coplanar or orthogonal alignment between the carbene and amide ligands. They exhibit blue to deep red TADF emissions (466–666 nm) that are characterized by singlet ligand-to-ligand-charge-transfer excited states with emission quantum yields ranging from 0.63 to 0.99 – Table 8, short emission decay lifetimes (below 1.1 μs), and radiative decay rate constants of 0.68 to 3.2 × 106 s−1 in thin-film configurations at ambient temperature.
| Complex | OLED structure | Device | Concentration [wt%] | ELmax [nm] | CIE [x,y] | V on [V] | CEmax [cd A−1] | η p [lm W−1] | L max [cd m−2] at Vapp | EQE [%] | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a To simplify the process, we directly spin-coated a mixture of czpzpy and the starting material [Cu(CH3CN)2(POP)]BF4 as emitting layer, expecting they form the emitter 14 during the solution process. For comparison, we planned to prepare the same EML as Device F by controlling the ratio of two materials (see Experimental section for details), and the resulting device is named as Device G. Other layers of Device G are identical to those of Device F. b Turn-on voltage recorded at a luminance of 1 cd m−2. c Measured at 1000 cd m−2. d PYD2 host. e CBP host. f Defined at brightness of 0.1 cd m−2. A,B: hole-transporting 2,6-mCPy and electron-transporting DPEPO with high triplet energy levels (2.93 eV and 3.0 eV, respectively) are used as hosts, which are named as Device A and Device B, respectively. C: CDBP. D: 30 wt% of TAPC:CDBP. E: 30 wt% of TAPC: mCP. A-1 SC, neat EML chlorobenzene g/g. A-2 SC, neat EML chlorobenzene a/a. A-3 SC, neat EML o-xylene g/g. A-4 SC, neat EML o-xylene a/a. B-1 IJP, neat EML indane a/a. B-2 IJP, neat EML indane/mesitylene a/a. C-1 SC, doped EML toluene g/g. (* SC: spincoated, IJP: inkjet printed; ** first letter indicates conditions for processing, second for drying; g: glovebox, a: air). 2,6-mCPy = 2,6-bis(N-carbazolyl)pyridine. 2,6DCzppy: (2,6-bis(3-(9H-carbazol-9-yl)phenyl)pyridine). B3PYMPM: (4,6-Bis(3,5-di(pyridin-3-yl)phenyl)-2-methylpyrimidine). OTPD: N,N′-bis(4-(6-((3-ethyloxetan-3-yl)methoxy))-hexylphenyl)-N,N′-diphenyl-4,4′-diamine. Cz35PyDCb: 5-(5-(3-(9H-carbazol-9-yl)phenyl)pyridin-3-yl)-5H-pyrido[3,2-b]indole. | |||||||||||
| Cu29 | ITO/PEDOT:PSS (40 nm)/20 wt% of Cu29, Cu30 or Cu31:host (30 nm)/DPEPO (50 nm)/LiF (0.7 nm)/Al (100 nm) | A | — | 519 | (0.24,0.36) | 6.9 | 8.82 | — | 1116 | 3.18 | 317 |
| Cu30 | A | 504 | (0.21,0.30) | 6.9 | 4.07 | 502 | 1.59 | ||||
| Cu31 | A | 508 | (0.21,0.33) | 6.1 | 23.68 | 2033 | 8.47 | ||||
| Cu29 | B | 500 | (0.20,0.29) | 8.7 | 5.15 | 309 | 1.98 | ||||
| Cu30 | B | 484 | (0.17,0.21) | 9.3 | 8.37 | 194 | 3.72 | ||||
| Cu31 | B | 496 | (0.18,0.25) | 11.5 | 3.53 | 139 | 1.42 | ||||
| Cu32 | ITO/PEDOT:PSS/PVK/10 wt% of Cu32: host| 3TPyMB |LiF:Al | C | — | 550 | (0.40,0.52) | 6.4 | 9.7 | — | — | 3.6 | 315 |
| D | 560 | (0.40,0.52) | 5.0 | 17.5 | 6.3 | ||||||
| E | 560 | (0.40,0.53) | 5.0 | 21.3 | 7.8 | ||||||
| Cu33 | ITO/PEDOT:PSS/PLEXCORE UT-314/Cu33:PYD2/3TPyMB/LiF:Al | — | — | — | — | 2.6 | 73 | — | 10.000 (10) | 23 | 318 |
| Cu34 | (ITO)/TAPC/mCP+10% Cu34–Cu38/3TPyMB/LiF/Al | — | — | 527 | (0.30,0.55) | 3.3 | 67.7 | — | — | 21.1 | 319 |
| Cu35 | 517 | (0.29,0.4) | 3.0 | 65.3 | 21.3 | ||||||
| Cu36 | 513 | (0.26,0.51) | 3.1 | 62.4 | 21.2 | ||||||
| Cu37 | 529 | (0.32,0.54) | 3.3 | 69.4 | 22.5 | ||||||
| Cu38 | 515 | (0.26,0.51) | 3.3 | 55.6 | 18.6 | ||||||
| Cu40 | ITO/HAT-CN(5 nm)/TAPC (40 nm)/TCTA (10 nm)/TCTA:DPEPO: Cu40 or Cu11(10 nm)/DPEPO (40 nm)/LiF (1.2 nm)/Al (100 nm) | — | 2 | 521 | (0.33,0.52) | — | 41.9 | 43.8 | 9260 | 16.4 | 231 |
| Cu40 | 4 | 521 | (0.35,0.52) | 39.5 | 41.4 | 8080 | 13.9 | ||||
| Cu40 | 8 | 521 | (0.35,0.52) | 38.7 | 40.5 | 7600 | 13.7 | ||||
| Cu11 | 2 | 535 | (0.35,0.51) | 30.5 | 28.2 | 760 | 10.8 | ||||
| Cu11 | 4 | 535 | (0.36,0.52) | 36.5 | 31.8 | 433 | 12.8 | ||||
| Cu11 | 8 | 535 | (0.37,0.52) | 33.5 | 29.2 | 304 | 11.6 | ||||
| Cu42 | ITO/PEDOT:PSS/Cu42 (20 wt%):czpzpy/DPEPO/TPBi/LiF/Al | Fa | — | 514 | (0.259,0.49) | 5.6 | 17.34 | — | 2939 (14.1) | 6.34 | 320 |
| G | 516 | (0.252,0.49) | 5.6 | 17.53 | 3251 (14.3) | 6.36 | |||||
| Cu43 | ITO/PEDOT:PSS/Cu43 or Cu44:mCP/3TPyMB/LiF:Al | — | — | 490 | (0.17,0.37) | 5.3 | 14.01 | — | 6563 | 5.83 | 321 |
| Cu44 | 501 | (0.21,0.43) | 7.4 | 20.24 | 5579 | 7.42 | |||||
| Cu45 | ITO/PEDOT:PSS/TCTA/Cu45, Cu46, or Cu47 (10 wt%):mCP/TmPyPb/LiF/Al | D1 | — | 544 | (0.37,0.55) | 5.2b | 47.03 | 21.62 | 11010 (11.0) | 14.81 | 322 |
| Cu46 | D2 | 544 | (0.38,0.55) | 5.6b | 35.61 | 14.48 | 5152 (11.0) | 11.17 | |||
| Cu47 | D3 | 544 | (0.38,0.54) | 5.3b | 21.33 | 12.07 | 5242 (9.0) | 6.67 | |||
| Cu48 | ITO/MoO3/TAPC/TCTA/mCP:10% Cu48–50/TmPyP/LiF/Al | — | — | 550 | (0.31,0.54)c | 5.1 | 32.9 | 19.9 | 2339 (17.1) | 10.1 | 323 |
| Cu49 | 520 | (0.31,0.50)c | 5.4 | 20.4 | 11.2 | 2399 (15.9) | 7.3 | ||||
| Cu50 | 550,610 | (0.38,0.51)c | 3.9 | 22.9 | 16.0 | 3256 (15) | 8.3 | ||||
| Cu53 | ITO/MoO3, 10 nm/TAPC, 40 nm/2,6DCzppy: 5 wt% Cu53 (20 nm)/TmPyPB, 50 nm/LiF (1 nm)/Al (100 nm) | R/S-D | 542 | (0.34,0.53) | 4.1 | 78 | 57 | 1990 | 21.7 | 324 | |
| Cu54 | ITO/PEDOT:PSS/EML |TPBi/|LiF/Al | A-1 | — | 553 | — | 3.7 | 24.4 | 15.3 | 42000 |
8.0 | 325 |
| A-2 | 553 | 3.5 | 24.9 | 15.9 | 40000 |
7.8 | |||||
| A-3 | 553 | 3.8 | 24.5 | 15.6 | 40000 |
7.8 | |||||
| A-4 | 552 | 3.4 | 24.6 | 16.6 | 35000 |
7.9 | |||||
| B-1 | 550 | 4.0 | 7.3 | 4.5 | 1600 | 2.1 | |||||
| B-2 | 550 | 3.7 | 15.2 | 8.7 | 3000 | 4.7 | |||||
| C-1 | 552 | 3.7 | 36.4 | 21.8 | 34000 |
11.4 | |||||
| Cu55 | ITO/MoO3/TAPC/mCP:Cu55–57 (10 wt%)/TPBi/LiF/Al | — | — | ≈584 | (0.473,0.495) | — | 24.7 | 21.4 | — | 9.6 | 326 |
| Cu56 | ≈584 | (0.466,0.495) | 32.7 | 28.8 | 12.4 | ||||||
| Cu57 | ≈584 | (0.475, 0.493) | 40.8 | 35.9 | 16.3 | ||||||
| Cu58 | ITO (135 nm)/PEDOT:PSS (40 nm)/20 wt% Cu58, Cu59 or Cu60:80 wt% PYD2 (30 nm)/3TPyMB (50 nm)/LiF (0.8 nm)/Al (100 nm) |
1 | — | 523 | (0.30,0.54) | 8.5 | 24.8 | — | 2342 (22.9) | 7.6 | 327 |
| Cu59 | 2 | 526 | (0.30,0.54) | 7.7 | 20.4 | 2012 (17.7) | 6.2 | ||||
| Cu60 | 3 | 522 | (0.29,0.53) | 7.3 | 27.1 | 2012 (17.7) | 8.3 | ||||
| Cu61 | ITO/PEDOT:PSS/PYD2:Cu(I) complex/DPEPO (10 nm)/TPBi (40 nm)/LiF/Al | — | 4 | 526 | (0.36,0.55) | — | 48.12 | 18.68 | 7350 | 15.17 | 328 |
| Cu61 | 8 | 531 | (0.36,0.56) | 51.52 | 21.29 | 8640 | 16.25 | ||||
| Cu61 | 12 | 535 | (0.37,0.56) | 58.50 | 22.97 | 8650 | 18.46 | ||||
| Cu61 | 20 | 540 | (0.37,0.56) | 45.27 | 17.43 | 9310 | 14.31 | ||||
| Cu62 | 4 | 570 | (0.48,0.51) | 24.51 | 8.21 | 10210 |
8.99 | ||||
| Cu62 | 8 | 578 | (0.50,0.49) | 32.00 | 9.57 | 17238 |
12.41 | ||||
| Cu62 | 12 | 582 | (0.51,0.48) | 35.27 | 10.00 | 17600 |
14.30 | ||||
| Cu62 | 20 | 589 | (0.53,0.47) | 23.67 | 5.99 | 12110 |
8.69 | ||||
| Cu63 | 4d | 614 | (0.54,0.38) | 8.44 | 2.76 | 4880 | 5.95 | ||||
| Cu63 | 8d | 622 | (0.57,0.38) | 12.74 | 8.17 | 5280 | 8.96 | ||||
| Cu63 | 12d | 627 | (0.59,0.38) | 3.53 | 1.12 | 4040 | 2.97 | ||||
| Cu63 | 20d | 634 | (0.61,0.37) | 2.36 | 0.69 | 1690 | 2.27 | ||||
| Cu63 | 4 e | 631 | (0.61,0.38) | 11.26 | 4.10 | 4630 | 10.17 | ||||
| Cu63 | 8 e | 638 | (0.62,0.37) | 6.67 | 2.47 | 4390 | 6.82 | ||||
| Cu63 | 12e | 641 | (0.63,0.37) | 3.72 | 1.12 | 5260 | 4.14 | ||||
| Cu63 | 20e | 647 | (0.64,0.36) | 1.48 | 0.42 | 2660 | 1.85 | ||||
| Cu66 | ITO/PEDOT:PSS/85% BCPO:15% Cu complex/|TPBi/LiF/Al | — | — | ∼530 | (0.32,0.53) | 4 | — | — | 12800 (14.0 V) |
10.5 | 238 |
| Cu4 | ∼530 | (0.33,0.54) | 4 | 7740 (14.5 V) | 9.5 | ||||||
| Cu69 | 70 nm ITO/5 nm HA-TCN/40 nm (TAPC/10 nm mCP/25 nm Cu69/65 nm TPBi/1.5 nm LiQ/100 nm Al) | — | 10 | 537 | — | 3.0f | — | — | 41000 |
17.0 | 329 |
| 40 | 543 | 2.5f | 54000 |
19.4 | |||||||
| 100 | 555 | 2.5f | 41000 |
16.3 | |||||||
| Cu77 | ITO/HAT-CN (5 nm)/NPB (30 nm)/Tris-pcz (15 nm)/CCP (6 nm)/Cu77/PPF (10 nm)/BPPB(25 nm)/Liq (2 nm)/Al (120 nm) | A | 5 | 453 | (0.19,0.22) | 3.9 | — | — | 910 | 11.2 | 330 |
| B | 20 | 459 | (0.20,0.25) | 3.4 | 1730 | 20.6 | |||||
| C | 40 | 468 | (0.20,0.26) | 3.4 | 1570 | 16.6 | |||||
| D | 20 | 457 | (0.18,0.19) | 4.6 | 1540 | 18.1 | |||||
| E | — | 477 | (0.22,0.31) | 3.0 | 940 | 10.3 | |||||
| Cu78 | ITO/MoO3 (2 nm)/TAPC (80 nm)/mCP (10 nm)/emissive layer (20 nm)/TPBi (10 nm)/3TPyMB (60 nm)/LiF (1 nm)/Al (140 nm) | A | 1.0 | 622 | (0.56,0.44) | 2.9 | 30.2 | 30.1 | 16894 |
20.7 | 331 |
| B | 1.5 | 628 | (0.58,0.42) | 3.1 | 31.2 | 28.9 | 17706 |
21.1 | |||
| C | 3.0 | 629 | (0.57,0.43) | 3.1 | 19.9 | 18.4 | 9233 | 17.6 | |||
| Cu79 | ITO/HAT-CN (5 nm)/TAPC (50 nm)/TCTA (10 nm)/CuI complex:host (20 nm)/hole-blocking layer (HBL) (10 nm)/TPBi (40 nm)/LiF (1 nm)/Al (100 nm) | — | 6 | 582 | (0.51,0.48) | — | 44.7 | 46.2 | 222200 |
18.7 | 332 |
| Cu80 | 4 | 521 | (0.27,0.57) | 67.6 | 60.7 | 208000 |
20.6 | ||||
| Cu81 | 2 | 619 | (0.58,0.42) | 22.1 | 22.0 | 155000 |
14.4 | ||||
| Cu83 | 4 | 474 | (0.14,0.22) | 36.4 | 32.6 | 15600 |
23.6 | ||||
| Cu84 | ITO/PEDOT:PSS/TAPC/TAPC:[Cu84][BArF4]/B3PYMPM/LiF/Al | — | — | — | (0.305,0.637) | 2.4 | — | — | 8900 (15 V) | 11.2 | 333 |
| ITO/PEDOT:PSS/TAPC/CBP:[Cu84][BArF4]/B3PYMPM/LiF/Al | — | 4.2 | 19000 |
— | |||||||
| Cu86 | ITO/PEDOT:PSS/complex Cu86/TPBi/LiF/Al | — | — | 564 | (0.43,0.51) | — | — | — | 234 | 7.74 | 334 |
| Cu92 | ITO/PEDOT:PSS (50 nm)/OTPD (4 nm)/PYD2:emitter Cu92 and Cu93(60 nm)/DPEPO (10 nm)/TPBi (40 nm)/LiF (1.2 nm)/Al (100 nm) | — | 24 | ∼560 | (0.43,0.54) | 6.4 | 18.23 | 9.98 | 6800 | 5.91 | 335 |
| Cu93 | 16 | ∼560 | (0.51,0.49) | — | 19.74 | 8.86 | 2520 | 7.96 | |||
| Cu101 | ITO/TAPC (35 nm)/TCTA (5 nm)/mCBP doped with a certain weight percent of compound Cu101 or Cu102 (25 nm)/TmPyPB (50 nm)/LiF (1.5 nm)/Al (150 nm) | A1 | 1 | — | (0.30,0.41) | 3.8 | 22.7 | 19.8 | 13542 |
10.3 | 336 |
| Cu101 | A2 | 5 | — | (0.36,0.42) | 3.6 | 25.9 | 22.3 | 17839 |
13.7 | ||
| Cu101 | A3 | 10 | 494,590 | (0.38,0.42) | 3.5 | 34.1 | 27.1 | 44215 |
16.5 | ||
| Cu101 | A4 | 20 | — | (0.43,0.46) | 3.7 | 26.0 | 20.2 | 41695 |
15.7 | ||
| Cu102 | B1 | 1 | 494 | (0.25,0.42) | 3.7 | 31.5 | 25.4 | 35947 |
12.9 | ||
| Cu102 | B2 | 5 | — | (0.26,0.45) | 3.6 | 31.9 | 26.0 | 42773 |
12.2 | ||
| Cu102 | B3 | 10 | — | (0.30,0.45) | 3.8 | 21.7 | 15.5 | 36478 |
9.2 | ||
| Cu102 | B4 | 20 | — | (0.36,0.48) | 3.8 | 14.8 | 9.4 | 37183 |
5.9 | ||
| Cu103 | ITO/HAT-CN (10 nm)/NPB (10 nm)/TCTA (20 nm)/Host (aza-SBFs):8% CuI (20 nm)/Cz35PyDCb (5 nm)/TmPyPB (50 nm)/Liq (1 nm)/Al (100 nm) | A | — | 552 | — | 4.1 | 16.8 | 13.2 | 4342 | 6.6 | 337 |
| Cu104 | B | 588 | 4.5 | 16.5 | 11.3 | 6740 | 7.6 | ||||
| Cu105 | C | 552 | 4.2 | 21.7 | 17.9 | 7600 | 8.8 | ||||
| Cu106 | D | 540 | 4.2 | 41.9 | 32.9 | 6180 | 16.8 | ||||
| Cu107 | ITO/PEDOT:PSS/PYD2: Cu107/DPEPO (10 nm)/TPBi (40 nm)/LiF (1.2 nm)/Al (100 nm) | — | 2 | — | (0.38,0.45) | — | 6.48 | 3.39 | 1160 | 2.61 | 338 |
| 4 | (0.38,0.48) | 8.39 | 4.01 | 1880 | 3.14 | ||||||
| 8 | (0.38,0.49) | 10.5 | 4.25 | 2500 | 3.80 | ||||||
| Ag10 | ITO/PEDOT:PSS (50 nm)/OTPD (4 nm)/PYD2:emitter Ag10 (60 nm)/DPEPO (10 nm)/TmPyPb (40 nm)/LiF (1.2 nm)/Al (100 nm) | — | 4 | — | (0.44,0.52) | 6.1 | 23.85 | 9.37 | 3300 | 8.46 | 273 |
| 8 | (0.45,0.52) | 6.4 | 23.11 | 9.22 | 3015 | 8.76 | |||||
| 12 | (0.46,0.52) | 6.8 | 19.41 | 7.28 | 1211 | 6.78 | |||||
Investigations using ultrafast spectroscopy on compounds Au18, Au21, Au22, Au23, and Au24 (Scheme 5) unveil a highly effective ISC mechanism exhibiting kISC ranging from 3.1 to 9.9 × 1010 s−1, alongside a rapid rISC process characterized by krISC between 8 and 35 × 108 s−1 – Table 9. The impact of extending the π-conjugation and adopting an orthogonal molecular geometry is evident in diminishing both the ΔE(S1–T1) and S1 transition dipole moment. The OLEDs in blue to NIR spectral range showed exceptional performance metrics, achieving EQE/CE/PE of up to 26.9%, 76.7 cd A−1, and 79.3 lm W−1, respectively – Table 6. Noteworthy electroluminescent properties are observed in vacuum-deposited Au(I) complex based OLEDs, showing exceptionally high brightness levels at 300000 cd m−2, EQEs reaching up to 26.2% with minimal roll-offs down to 2.6% at 1000 cd m−2. Detailed investigations conducted in their laboratory revealed the operational lifetimes of these devices, reaching impressive LT95 values (at L0, of 1000 cd m−2) of up to 2082, 239, 126, and 30.5 h for yellow, green, red, and blue OLEDs, respectively. Furthermore, the NIR OLED demonstrated a LT95 of 768 hours under a consistent driving current density of 30 mA cm−2. Utilizing complex Au21 as the sensitizer and BN2 as the emitter, they successfully developed narrowband TADF-sensitized fluorescence OLEDs. These OLEDs showed remarkable performance, achieving a high EQE of 19% at a luminance of 1000 cd m−2 with minimal efficiency roll-offs ranging from 12% to 25%. Furthermore, ultrapure-green TADF-enhanced fluorescent OLEDs using the CMA(Au) emitter as a sensitizer and a multiresonance terminal emitter achieve impressive EQEs of up to 25.3% – Table 6.305
| Complex | Medium | λem [nm] | PLQY [%] | τexp [μs] | k r [s−1] | k nr [s−1] | k ISC [s−1] | k rISC [s−1] | ΔEST [cm−1] | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a λ exc = 310 nm. b λ exc = 355 nm. c Absolute PL quantum yield in the solid state (error ± 8%). d Energy gap between S1 and T1 levels obtained by temperature dependence of decay time. e T = 300, 77 K, respectively. f T = 300, 77 K, respectively. g Calculated from S1 and T1 of complexes as powders. h Absolute emission quantum in the solid state. Experimental errors are ±5%. i Emission lifetime. Experimental errors are ±5%. j T = 298, 77 K. k T = 298, 77 K. l Energy gap between S1 and T1 determined from the emission spectra at 77 K and 298 K. m Data are given in parentheses is ΦF. n Energy gap between S1 and T1 determined from the emission spectra at 77 K and 298 K. o Double entries for each complex refer to oxygen free (o) and airequilibrated samples (p). p Double entries for each complex refer to oxygen free (o) and airequilibrated samples (p). q This compound exhibits a biexponential decay with a short component of 7.29 μs. r The S1 and T1 energy levels were estimated according to the emission peak onsets at 297 K and 77 K. | ||||||||||
| Cu29 | In degassed CH2Cl2, T = 298 K | 590 | 2.1 | 1.6 | - | — | — | — | — | 317 |
| Cu30 | 536 | 45 | 11.9 | |||||||
| Cu31 | 540 | 30 | 13.3 | |||||||
| Cu29 | Crystalline powder, T = 298 K | 490 | 56 | 20.4 | — | — | — | — | 1452 | 317 |
| Cu30 | 465 | 87 | 12.2 | 1371 | ||||||
| Cu31 | 492 | 75 | 22.8 | 1452 | ||||||
| Cu32 | In the solid state T = 293 K | 521a | 52a | 1.73, 0.33 | — | — | — | — | 309cal | 315 |
| Ag16 | 505a | 32a | 2.22, 0.56 | 199cal | ||||||
| Au14 | 610a | 12a | 1.66, 0.47 | 405cal | ||||||
| Cu32 | In the solid state T = 77 K | 534a | 73a | 847 | — | — | — | — | 309cal | 315 |
| Ag16 | 530a | 72a | 574 | 199cal | ||||||
| Au14 | 630a | 18a | 52 | 405cal | ||||||
| Cu33 | — | — | 92 | 3.6 | — | — | — | — | 726 | 318 |
| Cu34 | In the solid state T = 293 K | 517b | 38c | 4.6b | — | — | — | — | 680d | 319 |
| Cu35 | 512b | 55c | 8.0b | 810d | ||||||
| Cu36 | 473b | 59c | 7.1b | 830d | ||||||
| Cu37 | 487b | 80c | 6.5b | 600d | ||||||
| Cu38 | 486b | 95c | 8.9b | 710d | ||||||
| Cu34 | In the solid state T = 77 K | 507b | 56c | 2500b | 2.2 × 102 | 1.8 × 102 | — | — | — | 319 |
| Cu35 | 500b | 85c | 360b | 2.4 × 103 | 4.2 × 102 | |||||
| Cu36 | 456b | 85c | 100b | 8.5 × 103 | 1.5 × 103 | |||||
| Cu37 | 477b | 85c | 520b | 1.6 × 103 | 2.9 × 102 | |||||
| Cu38 | 487b | 95c | 910b | 1.0 × 103 | 5.5 × 10 | |||||
| Cu19 | Ground Solid State, 295 K | 579 | 55 | 2.5 | 22 × 104 | 18 × 104 | — | — | — | 251 |
| Cu19 | mCP (1 wt%), 295 K | 534 | 64 | 14.3 | 4.5 × 104 | 2.5 × 104 | — | — | — | 251 |
| Cu19 | CzSi (10 wt%), 295 K | 575 | 47 | 2.2 | 21 × 104 | 24 × 104 | — | — | — | 251 |
| Cu19 | UGH-3 (10 wt%), 295 K | 575 | 30 | 2.4 | 13 × 104 | 30 × 104 | — | — | — | 251 |
| Cu19 | PMMA (1 wt%), 295 K | 553 | 47 | 12.0 | 3.9 × 104 | 4.4 × 104 | — | — | — | 251 |
| Cu20 | Ground Solid State, 295 K | 606 | 53 | 1.7 | 31 × 104 | 27 × 104 | — | — | — | 251 |
| Cu20 | mCP (1 wt%), 295 K | 538 | 51 | 149 | 0.34 × 104 | 0.33 × 104 | — | — | — | 251 |
| Cu20 | CzSi (10 wt%), 295 K | 595 | 14 | 1.3 | 11 × 104 | 68 × 104 | — | — | — | 251 |
| Cu20 | UGH-3 (10 wt%), 295 K | 590 | 18 | 1.2 | 15 × 104 | 68 × 104 | — | — | — | 251 |
| Cu20 | PMMA (1 wt%), 295 K | 540 | 46 | 116 | 0.40 × 104 | 0.47 × 104 | — | — | — | 251 |
| Cu39 | Powder at 300 K/77 K | 541/551 | 83/85 | 9/3300 | 9.2 × 104/2.6 × 102 | — | — | — | 231 | |
| Cu40 | Powder at 300 K/77 K | 530/530 | 82/85 | 7/≈420 | 12 × 104/20 × 102 | — | — | — | 231 | |
| Cu11 | Powder at 300 K/77 K | 540/540 | 90/70 | 5/680 | 18 × 104/10 × 102 | — | — | — | 231 | |
| Cu11 | Powder at 300 K/50 K | 540/540 | 90/70 (77 K) | 5/700 | 18 × 104/10 × 102 | 2.0 × 104/4.3 × 102 | — | — | 600 | 231 |
| Cu11 | Doped in PMMA (≈1 wt%) | 544 | 56 | 7 | 8.0 × 104 | 6.3 × 104 | — | — | 231 | |
| Cu11 | Fluid DCM Solution (c ≈ 10−5 M) | 544 | 1 | 4 | 0.25 × 104 | 25 × 104 | — | — | 231 | |
| Cu41 | In the solid state T = 293 K | 495 | 45 | 134 | 3.4 × 103 | 4.1 × 103 | — | — | 1452 | 320 |
| Cu42 | 518 | 98 | 23 | 4.3 × 104 | 8.7 × 102 | 1048 | ||||
| Cu41 | In the solid state T = 77 K | 507 | 71 | 671 | 1.1 × 103 | 4.3 × 102 | — | — | — | 320 |
| Cu42 | 521 | 39 | 521 | 9.5 × 102 | 1.5 × 103 | |||||
| Cu43 | In CH2Cl2, T = 77 K | — | — | — | — | — | — | — | — | 321 |
| Cu43 | In powder, T = 77 K | 501 | — | 593 | — | — | — | — | — | 321 |
| Cu43 | In CH2Cl2, T = 293 K | 518 | 16 | 4.6 | — | — | — | — | — | 321 |
| Cu43 | In powder, T = 293 K | 493 | 60.9 | 145 | — | — | — | — | 968 | 321 |
| Cu44 | In CH2Cl2, T = 77 K | — | — | — | — | — | — | — | — | 321 |
| Cu44 | In powder, T = 77 K | 516 | — | 496 | — | — | — | — | — | 321 |
| Cu44 | In CH2Cl2, T = 293 K | 518 | 19 | 7.2 | — | — | — | — | — | 321 |
| Cu44 | In powder, T = 293 K | 475 | 40.7 | 51 | — | — | — | — | 968 | 321 |
| Cu45 | Solution (in degassed CH2Cl2) | (616e, 543f) | — | — | — | — | — | — | — | 322 |
| Cu46 | (616e, 538f) | — | — | — | — | — | — | — | ||
| Cu47 | (616e, 546f) | — | — | — | — | — | — | — | ||
| Cu45 | Powder, 300 K | 550 | 22.4 | 5.7 | — | — | — | — | 645g | 322 |
| Cu46 | 549 | 18.5 | 5.7 | — | — | — | — | 726g | ||
| Cu47 | 556 | 20.0 | 5.7 | — | — | — | — | 323g | ||
| Cu45 | Powder, 77 K | 568 | 19.3 | 343 | — | — | — | — | — | 322 |
| Cu46 | 560 | 16.2 | 317 | — | — | — | — | — | ||
| Cu47 | 564 | 19.4 | 363 | — | — | — | — | — | ||
| Cu45 | PMMA film | 528 | 33.1 | — | — | — | — | — | — | 322 |
| Cu46 | 525 | 31.7 | — | — | — | — | — | — | ||
| Cu47 | 528 | 31.5 | — | — | — | — | — | — | ||
| Cu48 | In the solid state T = 298 K | 498 | 32h | 2.5i | 1.28 × 105 | — | — | — | 968cal | 323 |
| Cu49 | 511 | 28h | 12.5i | 0.22 × 105 | — | — | — | 968cal | ||
| Cu50 | 527 | 29h | 4.8i | 0.60 × 105 | — | — | — | 1129cal | ||
| Cu48 | In the solid state T = 77 K | 500 | — | 103i | — | — | — | — | — | 323 |
| Cu49 | 517 | — | 714i | — | — | — | — | |||
| Cu50 | 532 | — | 818i | — | — | — | — | |||
| Cu51 | CH2Cl2 (1 × 10−5 mol L−1) at room temperature | 505 | — | — | — | — | — | — | 121 | 324 |
| Cu52 | CH2Cl2 (1 × 10−5 mol L−1) at room temperature | 502 | — | — | — | — | — | — | 153 | 324 |
| Cu53 | CH2Cl2 (1 × 10−5 mol L−1) at room temperature | 500 | — | — | — | — | — | — | 32 | 324 |
| Cu54 | In the solid state as neat powder at room temperature | 550 | 75 ± 5 | 6.9j/21.4k | 0.11 × 10−6 | 0.03 × 10−6 | — | — | — | 325 |
| Cu54 | Neat film at room temperature | 550 | 57 ± 5 | 6.2 | 0.09 × 10−6 | 0.07 × 10−6 | — | — | — | 325 |
| Cu54 | 50% in PMMA at room temperature | 550 | 56 ± 5 | 7.7 | 0.07 × 10−6 | 0.06 × 10−6 | — | — | — | 325 |
| Cu54 | 40% in PYD2 at room temperature | 550 | 62 ± 5 | 6.8 | 0.10 × 10−6 | 0.04 × 10−6 | — | — | — | 325 |
| Cu54 | 20% in PYD2 at room temperature | 550 | 60 ± 5 | 6.7 | 0.09 × 10−6 | 0.06 × 10−6 | — | — | — | 325 |
| Cu55 | In film T = 300 K | 530 | 76 | 19 | 4.0 × 104 | 1.3 × 104 | — | — | (798l, 40.3m) | 326 |
| Cu56 | 523 | 79 | 16 | 4.9 × 104 | 1.4 × 104 | (758l, 290m) | ||||
| Cu57 | 521 | 83 | 11 | 7.5 × 104 | 1.6 × 104 | (-l, 363m) | ||||
| Cu55 | In film T = 77 K | 531 | 82 | 1778 | 4.6 × 102 | 1.0 × 102 | — | — | — | 326 |
| Cu56 | 531 | 83 | 1611 | 5.2 × 102 | 1.0 × 102 | |||||
| Cu57 | 531 | 85 | 294 | 2.9 × 103 | 5.0 × 102 | |||||
| Cu58 | T = 77 K | 509 | 43 ± 5 | 5.5 ± 1 | 8.0 × 104 | 1.0 × 105 | — | — | — | 327 |
| Cu59 | 519 | 29 ± 5 | 16 ± 1 | 1.8 × 104 | 4.4 × 105 | |||||
| Cu60 | 503 | 79 ± 5 | 5.5 ± 1 | 1.4 × 105 | 3.8 × 104 | |||||
| Cu58 | T = 298 | 523 | 67 ± 5 | 158 ± 1 | 4.2 × 103 | 2.1 × 103 | — | — | 718n | 327 |
| Cu59 | 546 | 54 ± 5 | 356 ± 1 | 1.5 × 103 | 1.3 × 103 | 1065n | ||||
| Cu60 | 516 | 82 ± 5 | 209 ± 1 | 3.9 × 103 | 8.4 × 102 | 758n | ||||
| Cu61 | — | — | — | — | — | — | — | — | 1121 | 328 |
| Cu62 | 1621 | |||||||||
| Cu63 | 2073 | |||||||||
| Cu64 | CH2Cl2 at 298 K | 560 | 28.4o/0.84p | 10.9o/0.26p | 2.61 × 104o/3.23 × 104p | 6.56 × 104o/381 × 104p | — | — | 976cal | 238 |
| Cu65 | 568 | 19.7o/0.68p | 9.0o/ | 2.26 × 104o/2.62 × 104p | 8.86 × 104o/382 × 104p | 879cal | ||||
| Cu66 | 568 | 22.9o/0.73p | 0.26p | 2.30 × 104o/3.32 × 104p | 7.70 × 104o/451 × 104p | |||||
| Cu4 | 565 | 22.6 | 10.0o/0.22p | 2.09 × 104 | 6.65 × 104 | 895cal | ||||
| 1081cal | ||||||||||
| Cu64 | CH2Cl2 at 77 K | 546 | — | 321 | — | — | — | — | — | 238 |
| Cu65 | 525 | 173 | ||||||||
| Cu66 | 525 | 178 | ||||||||
| Cu4 | 518 | 211 | ||||||||
| Cu64 | PMMA (1% wt) at 298 K | 532 | 57 | 18.0, 36.5 | — | — | — | — | — | 238 |
| Cu65 | 520 | 51 | 22.8, 57.9 | |||||||
| Cu66 | 520 | 53 | 24.0, 63.0 | |||||||
| Cu4 | 520 | 65 | 25.3, 75.1 | |||||||
| Cu64 | Powders at 298 K | 553 | 41 | 16.3 | — | — | — | — | — | 238 |
| Cu65 | 548 | 15 | 14.9q | |||||||
| Cu66 | 538 | 32 | 17.1 | |||||||
| Cu4 | 527 | 37 | 27.5 | |||||||
| Cu67 | Solution in 2-MeTHF, RT | 448 | 24 | 2.3 | 1.0 × 105 | 3.3 × 105 | — | — | — | 329 |
| Cu68 | 492 | 100 | 1.2 | 8.3 × 105 | <0.083 × 105 | — | ||||
| Cu69 | 542 | 55 | 1.1 | 5.0 × 105 | 4.1 × 105 | 500 | ||||
| Cu70 | 602 | 5 | 0.080 | 6.2 × 105 | 119 × 105 | — | ||||
| Cu71 | 666 | 2 | 0.052 | 3.8 × 105 | 180 × 105 | — | ||||
| Cu72 | — | — | — | — | — | — | ||||
| Cu67 | Solution in 2-MeTHF, 77 K | 424 | — | 9300 | — | — | — | — | — | 361 |
| Cu68 | 428 | 2200 | ||||||||
| Cu69 | 432 | 2100 (62%) and 345 (38%) @430 nm, 181 @520 nm | ||||||||
| Cu70 | 492 | 18 | ||||||||
| Cu71 | 536 | 210 | ||||||||
| Cu72 | — | — | ||||||||
| Cu67 | 1 wt% in PS film, RT | 432 | 80 | 2.6 (47%), 14 (53%) | 0.9 × 105 | 0.23 × 105 | — | — | — | 329 |
| Cu68 | 468 | 100 | 1.3 | 7.7 × 105 | <0.077 × 105 | |||||
| Cu69 | 506 | 90 | 1.4 | 6.4 × 105 | 0.71 × 105 | |||||
| Cu70 | 548 | 78 | 1.2 | 6.5 × 105 | 1.8 × 105 | |||||
| Cu71 | 616 | 30 | 0.75 | 4.0 × 105 | 9.3 × 105 | |||||
| Cu72 | 704 | 3 | 0.19 | 1.6 × 105 | 51 × 105 | |||||
| Cu67 | 1 wt% in PS film, 77 K | 424 | — | 3200 | — | — | — | — | — | 361 |
| Cu68 | 464 | 100 | ||||||||
| Cu69 | 502 | 140 | ||||||||
| Cu70 | 544 | 150 | ||||||||
| Cu71 | 612 | 410 | ||||||||
| Cu72 | 682 | 150 | ||||||||
| Cu67 | Neat solid, RT | 438 | 5 | 0.37 (33%), 1.8 (67%) | 0.38 × 105 | 7.2 × 105 | — | — | — | 329 |
| Cu68 | 474 | 76 | 0.75 | 10.0 × 105 | 3.2 × 105 | |||||
| Cu69 | 492 | 53 | 0.84 | 6.3 × 105 | 5.5 × 105 | |||||
| Cu70 | 550 | 68 | 1.0 | 6.8 × 105 | 3.2 × 105 | |||||
| Cu71 | 616 | 15 | 0.33 | 4.5 × 105 | 26 × 105 | |||||
| Cu72 | 658 | 12 | 0.39 | 3.1 × 105 | 22 × 105 | |||||
| Cu67 | Neat solid, 77 K | 438 | — | 7100 | — | — | — | — | — | 329 |
| Cu68 | 468 | 90 | ||||||||
| Cu69 | 482 | 160 | ||||||||
| Cu70 | 558 | 280 | ||||||||
| Cu71 | 598 | 180 | ||||||||
| Cu72 | 634 | 310 | ||||||||
| Cu73 | 1 wt% doped PS film at RT | 505 | 19 | 0.55 | 3.5 × 105 | 15 × 105 | — | — | — | 330 |
| Cu74 | 560 | 5 | 0.19 | 2.6 × 105 | 50 × 105 | |||||
| Cu75 | 560 | 8 | 0.18 | 4.4 × 105 | 51 × 105 | |||||
| Cu76 | 555 | 12 | 0.82 | 1.5 × 105 | 11 × 105 | |||||
| Cu77 | 458 | 74 | 1.9 | 3.9 × 105 | 1.4 × 105 | |||||
| Cu78 | Toluene (1 × 10−4 M, 300 K) | 638 | 12 | 0.11 | 6.9 × 105 | 3.9 × 105 | — | — | 444 | 331 |
| Cu78 | PS-doped film (with a 5 wt% doping concentration) | 609 | 24 | 0.42 | 6.9 × 105 | 3.9 × 105 | — | — | 444 | 331 |
| Cu78 | CBP:TPBi-doped film (with a 1.5 wt% doping concentration) | 599 | 65 | 0.93 | 6.9 × 105 | 3.9 × 105 | — | — | 444 | 331 |
| Cu79 | Toluene ([Cu] = 1 × 10−4 M) | 624 | 29 | 0.18 | 16.1 × 105 | 39.4 × 105 | — | — | — | 332 |
| Cu80 | Toluene ([Cu] = 5 × 10−5 M) | 555 | 58 | 0.36 | 16.1 × 105 | 11.6 × 105 | — | |||
| Cu81 | Toluene ([Cu] = 1 × 10−4 M) | 660 | 14 | 0.11 | 12.7 × 105 | 78.2 × 105 | 81 | |||
| Cu82 | Toluene ([Cu] = 5 × 10−5 M) | 635 | 15 | 0.12 | 12.5 × 105 | 70.8 × 105 | — | |||
| Cu83 | Toluene ([Cu] = 5 × 10−5 M) | 502 | 74 | 0.55 | 13.5 × 105 | 4.7 × 105 | 81 | |||
| [Cu84][BArf4] | Powdered solid, 298 K | 518 | 50 | — | 5.1 × 104 | 5.1 × 104 | — | — | 581 ± 32 | 333 |
| [Cu84][BF4] | 518 | — | 9.8 | — | — | |||||
| [Cu84][BArf4] | Powdered solid, 77 K | — | 71 | — | 7.1 × 103 | 2.9 × 103 | — | — | — | 333 |
| [Cu84][BF4] | — | 100 | — | — | ||||||
| [Cu84][BArf4] | DCM solution, 298 K | — | 78 | — | — | — | — | — | — | 333 |
| [Cu84][BF4] | — | |||||||||
| [Cu84][BArf4] | DCM solution, 77 K | — | 93 | — | — | — | — | — | — | 333 |
| [Cu84][BF4] | — | |||||||||
| [Cu84][BArf4] | THF solution, 298 K | — | 79 | — | 7.2 × 104 | 1.9 × 104 | — | — | — | 333 |
| [Cu84][BF4] | 525 | — | — | — | ||||||
| [Cu84][BArf4] | THF solution, 77 K | — | 82 | — | — | — | — | — | — | 333 |
| [Cu84][BF4] | — | |||||||||
| Cu85 | In powder state, 297 K | 485 | 0.41 | 36.4 | 1.13 × 104 | — | — | — | 850 | 334 |
| Cu86 | 506 | 0.52 | 48.9 | 1.06 × 104 | (476r) | |||||
| Cu87 | 535 | 0.29 | 20.8 | 1.39 × 104 | 807 (1031r) | |||||
| Cu88 | 515 | 0.18 | 9.0 | 2.04 × 104 | 940 (1035r) 528 (464r) | |||||
| Cu89 | 535 | 0.07 | 10.0 | 0.65 × 104 | 644 (841r) | |||||
| Cu90 | 516 | 0.03 | 4.2 | 0.62 × 104 | 492 (848r) | |||||
| Cu85 | In powder state, 77 K | 473 | — | 73.9 | — | — | — | — | — | 334 |
| Cu86 | 498 | 0.87 | 595 | |||||||
| Cu87 | 536 | — | 772 | |||||||
| Cu88 | 499 | — | 108 | |||||||
| Cu89 | 502 | — | 469 | |||||||
| Cu90 | 508 | — | 462 | |||||||
| Cu91 | 10 wt% doped DPEPO film 298 K/77 K | 534/544 | 62 | 21.4 | — | — | — | — | 887 | 335 |
| Cu92 | 533/545 | 71 | 24.1 | 645 | ||||||
| Cu93 | 565/582 | 48 | 5.8 | 565 | ||||||
| Cu94 | 564/575 | 42 | 4.3 | 403 | ||||||
| Cu95 | Toluene solution | 511 | 37 | 0.58 | 6.4 × 105 | 10.9 × 105 | — | — | — | 336 |
| Cu96 | 517 | 42 | 0.75 | 5.6 × 105 | 7.7 × 105 | |||||
| Cu97 | 507 | 41 | 0.36 | 11.4 × 105 | 16.4 × 105 | |||||
| Cu98 | 513 | 79 | 0.85 | 9.3 × 105 | 2.5 × 105 | |||||
| Cu99 | 521 | 39 | 0.46 | 8.5 × 105 | 13.3 × 105 | |||||
| Cu100 | 527 | 23 | 0.41 | 5.6 × 105 | 18.8 × 105 | |||||
| Cu101 | 549 | 73 | 0.72 | 10.1 × 105 | 3.6 × 105 | |||||
| Cu102 | 554 | 76 | 0.76 | 10.0 × 105 | 3.2 × 105 | |||||
| Cu95 | PMMA films (5 wt%) | 471 | 74 | 2.6 | 2.8 × 105 | 1.0 × 105 | — | — | — | 336 |
| Cu96 | 478 | 82 | 1.9 | 4.3 × 105 | 0.9 × 105 | |||||
| Cu97 | 467 | 68 | 1.9 | 3.6 × 105 | 1.6 × 105 | |||||
| Cu98 | 474 | 84 | 2.1 | 4.0 × 105 | 0.8 × 105 | |||||
| Cu99 | 479 | 71 | 2.1 | 3.4 × 105 | 1.4 × 105 | |||||
| Cu100 | 484 | 78 | 1.6 | 4.9 × 105 | 1.4 × 105 | |||||
| Cu101 | 499 | 73 | 1.6 | 4.6 × 105 | 1.7 × 105 | |||||
| Cu102 | 509 | 89 | 1.2 | 7.2 × 105 | 1.2 × 105 | |||||
| Cu103 | aza-SBFs:CuI of 1.00:0.08 in DMF was measured at 77 K |
517/556/615 | 4.6 | 0.6/5.8/55.8 | — | — | 3.18 × 108 | 7.95 × 104 | 242–1936 | 337 |
| Cu104 | 625 | 52.6 | 18.1 | 4.65 × 108 | 1.78 × 105 | 1290 | ||||
| Cu105 | 580 | 60.4 | 21.1 | 5.05 × 108 | 3.20 × 105 | 1290 | ||||
| Cu106 | 550 | 92.2 | 40.6 | 6.18 × 108 | 2.84 × 105 | 807 | ||||
| Cu107 | Thin-film doped in PYD2 (8 wt%) | 544 | 27 | 3.1 | — | — | — | — | — | 338 |
| Cu107 | Thin-film doped in mCP (8 wt%) | 535 | 20 | 5.5 | — | — | — | — | — | 338 |
| Cu107 | Thin-film doped in PVK (8 wt%) | 545 | 11 | 3.2 | — | — | — | — | — | 338 |
| Cu107 | Thin-film doped in TCTA (8 wt%) | 542 | 10 | 4.3 | — | — | — | — | — | 338 |
| Cu107 | Thin-film doped in CBP (8 wt%) | 537 | 13 | 2.8 | — | — | — | — | — | 338 |
| Ag17 | DPEPO film at 298 and (77 K) | 520/536 | 0.48 | 4.7 | — | — | — | — | 1694 | 273 |
| Ag18 | 524/535 | 0.34 | 7.4 | 1694 | ||||||
| Ag10 | 573/585 | 0.62 | 3.5 | 1290 | ||||||
| Ag19 | 535/557 | 0.50 | 3.7 | 1774 | ||||||
To tackle the issue of efficient red-emitting TADF OLEDs, researchers are exploring molecular design strategies focused on enhancing the PLQY of two-coordinate Au(I) complex through the manipulation of amido ligands, namely [Au(DippPZI)(ACD)] (Au26), [Au(DippPZI)(Cz)] (Au27), [Au(DippPZI)(DPA)] (Au28), [Au(DippPZI)(DPAC)] (Au29), [Au(DippPZI)(DMAC)] (Au30), and [Au(DippPZI)(PXZ)] (Au31) [1,3-bis(2,6-diisopropylphenyl)pyrazinoimidazolium (DippPZI), acridin-9-onide (ACD), cabazolide (Cz), N,N-diphenylamide (DPA), 9,9-diphenylacridinide (DPAC), 9,9-dimethylacridinide (DMAC), and phenoxazinide (PXZ)] – Scheme 5. They demonstrated photoluminescence peak wavelengths spanning 533–750 nm and PLQY values of up to 10% even in the NIR emission region, surpassing energy gap limitations. The λem was observed to correlate with the Eox, a relationship affected by the amido ligand, aligning with the nature of the LLCT transition. As λem increased, the PLQY exhibited a sharp decline, suggesting nonradiative processes dictated by the energy gap law. Quantum chemical calculations and photophysical analyses elucidate the role of radiative control, which enhances significantly as the overlap between hole and electron distributions (Sr(r)) in the excited state increases. It is revealed that Sr(r) increases with the distance between the hole-distribution centroid and the nitrogen atom (dh−N) in an amido ligand. Notably, multilayer OLEDs incorporating the Au(I) complex emitters demonstrate superior performance, pushing the boundaries of the electroluminescence wavelength−EQE space established by previous coinage metal complex devices. These Au(I) complex-based OLEDs achieved a EQEmax of 7.0% at λEL of 680 nm – Table 6, which could be extended up to 706 nm at high concentrations. This study is expected to inform future molecular design approaches aimed at enhancing the efficiency of OLEDs emitting long-wavelength light.306
Likewise, the development of organic blue-emitting devices has encountered challenges in achieving high EQEs, narrow bandwidths, and extended operational lifetimes under intense luminance. A novel material approach has been demonstrated to address these crucial requirements. This innovative strategy leverages linear heteroleptic Au(I) complex exciton harvesters [Au(DippBZI)(Cz)] (Au32) and [Au(DippBZI)(TMCz)] (Au33) in conjunction with multiresonant emitters – Scheme 5. The resultant organic electroluminescence devices deliver blue emissions x/y CIE color coordinates of 0.108, 0.160 and exhibit a notably narrow FWHM value of 20 nm. These devices achieve an impressive EQEmax of up to 30.2% – Table 6. Noteworthy is the sustained EQE performance of 22.2% even at high luminance levels of 2000 cd m−2, surpassing conventional devices using organic exciton harvesters plagued by significant quantum efficiency roll-offs. The mitigation of EQE roll-off is credited to the expeditious transformation of dark triplet excitons into luminescent singlet excitons facilitated by the Au(I) complexes. The investigations, rooted in photophysical assessments and quantum chemical computations, elucidate that the swift capture of triplet excitons is a result of the synergistic interaction between the highly effective triplet-to-singlet conversion aided by the robust SOC within the Au 5d orbitals and the minimal ΔE(ICT–3LE) due to the well-separated natural transition orbital distributions. Furthermore, the advanced device exhibits a substantial tenfold enhancement in operational lifetime compared to control devices. The research attributes these improvements to the distinctive capability of Au(I) complexes in rapidly capturing triplet excitons. Additionally, these complexes facilitate Förster energy transfer to the emitter, effectively curtailing hazardous triplet–triplet Dexter energy transfer. The findings of this study hold promise for driving the commercialization of high-efficiency and stable blue electroluminescence devices, marking a significant leap in the advancement of organic optoelectronics technologies.307
Acceleration of singlet–triplet ISC plays a crucial role in enhancing triplet exciton utilization in emitters, such as (SIPr)AuBN (Au34), (IPr)AuBN (Au35), (BzIPr)AuBN (Au36), (PzIPr)AuBN (Au37), (PyIPr)AuBN (Au38) and (BzIPr)AuBNO (Au39) – Scheme 5. In contrast to their organic parent counterparts, Au(I) emitters achieved approximately a two-fold increase in ISC and rISC rates due to improved SOC. This study outlines a straightforward Au(I) coordination approach to amplify the SOC of green and blue BN(O)-based TADF emitters, leading to a significant increase in the rate constants of the spectroscopically observed ISC process to 3 × 109 s−1 with nearly complete ISC quantum yields. Consequently, the resulting thermally-stable AuI emitters achieved substantial values of delayed fluorescence kr constant, up to 1.3 × 105/1.7 × 105 s−1 in THF/PMMA films, while maintaining narrowband emissions (FWHM = 30–37 nm) and high PLQYs (0.9). Vapor-deposited ultrapure-green OLEDs doped with (BzIPr)AuBN (Au36) demonstrated high luminance of up to 2.53 × 105 cd m−2, along with EQEmax of up to 30.3% – Table 6, narrowband electroluminescence with FWHM of 34 nm and minimal roll-offs at 0.8%, accompanied by extended device lifetimes (LT60) of 1210 h at 1000 cd m−2. This work propose that this uncomplicated and readily applicable metal coordination technique aimed at enhancing triplet exciton utilization could be expanded to various TADF compounds beyond TADF emitters. This expansion opens up a new avenue for practical TADF OLEDs.308
The practical utility of luminescent mononuclear Au(I) complexes as optoelectronic materials has been constrained by their poor stability. In this study, Li et al. present a novel approach to enhance the stability of several novel Au(I) complexes (IPzIDCz (Au40), ImIDCz (Au41) and IPzTPA (Au42) exhibiting TADF – Scheme 5). By employing a highly rigid and groove-like σ-donating aryl ligand, they have fostered the formation of dual Au⋯H C hydrogen bonds, thereby fortifying the stability of the complex. The existence of secondary metal–ligand interactions has been confirmed through single-crystal structure analysis, nuclear magnetic resonance (NMR) spectroscopy, and theoretical simulations. In conjunction with the steric influences of the ligands, the metal ion is enveloped within a highly congested coordination environment. They exhibit notably augmented thermal stability and PLQY values of around 76%. Thus, OLEDs fabricated via vacuum deposition demonstrate promising electroluminescence, achieving EQEmax exceeding 23% and minimal 3.3% efficiency roll-off even at luminance levels of 10000 cd m−2 – Table 6. The estimated LT50 surpassing 77000 h at an initial luminance of 100 cd m−2 underscores the impressive operational stability of the system. This research offers a pathway for designing stable luminescent Au(I) complexes with remarkable potential for practical applications in optoelectronic devices.309
The first TADF Au(III) emitter was developed by Fernandez-Cestau et al. in 2015.213 Remarkably, the utilization of Bis-cyclometallated aryl Au(III) complexes integrated with TADF has enabled the development of solution-processed OLEDs. These OLEDs exhibit impressive figures of merit, including EQEs, CEs, and luminance reaching as far as 23.8%, 70.4 cd A−1, and 57340 cd m−2, respectively. This indicates that Au(III)-TADF emitters demonstrate competitiveness with the top-performing Pt(II) and Ir(III) phosphorescent emitters in terms of EQE and luminance for OLED applications.310
Chan and co-workers present the design and synthesis of a novel group of Au(III) complexes, that is, Au5–8 and Au43–44 featuring fused heterocyclic alkynyl ligands – Scheme 5, exhibiting adjustable emission colors from yellow to red in solid form and demonstrating TADF characteristics. These complexes show notable PLQY of up to 0.87 – Table 10. The increased energy gap between the higher-lying 3IL excited state and the lower-lying LLCT excited state in the complexes results in reduced involvement of the long-lived 3IL state in the emission process of Au6–8 and Au44 – Scheme 5. Consequently, these complexes exhibit heightened emission intensity as the temperature rises, with a rate constant (kr) reaching up to 106 s−1, attributed to TADF. Upon elevating the temperature from 200 to 360 K, a substantial rise in emission intensity is observed without significant alterations in excited-state lifetime, suggesting an escalation in the radiative decay rate. Au(III) complex-based OLEDs fabricated through solution processing have achieved EQEmax of up to 10.0% – Table 6. These devices utilize TADF Au(III) complexes to significantly decrease efficiency roll-offs from around 60% to approximately 17% at a luminance level of 1000 cd m−2. Notably, the ΔEST is estimated to be as small as 242 cm−1, aligning with computational predictions at 403 cm−1. The meticulous selection of the cyclometalating ligand and fused heterocyclic ligand plays a pivotal role in inducing TADF by regulating the energy levels of ILCT and LLCT This study marks a milestone in fabricating TADF Au(III) complex based OLEDs with fused heterocyclic alkynyl ligands capable of emitting yellow to red light.217
| Complex | LEC structure | ELmax [nm] | CIE [x,y] | CEmax [cd A−1] | η p [lm W−1] | L max [cd m−2] at Vapp | EQE [%] | Ref. |
|---|---|---|---|---|---|---|---|---|
| Cu109 | ITO/PEDOT:PSS/[Cu(P^P)^N^N][PF6]:[Emim][PF6] | 595 | — | 0.7 | 0.2 | 65 | 0.4 | 339 |
| Cu110 | 589 | (0.449, 0.532) | 1.1 | 0.4 | 109 | 0.5 | ||
| Cu117 | 593 | 1.3 | 0.4 | 131 | 0.6 | |||
| Cu118 | ITO/EDOT:PSS (80 nm)/EML (100 nm):[Emim][PF6]/Al (100 nm) | — | — | 0.3 | 17 | 340 | ||
| Cu119 | 1.3 | 63 | ||||||
| Cu120 | 0.7 | 37 | ||||||
| Cu121 | 0.6 | 32 | ||||||
| Cu122 | 0.2 | 14 | ||||||
| Cu123 | 0.4 | 22 | ||||||
| Cu124 | 0.9 | 45 | ||||||
| Cu125 | 1.6 | 79 | ||||||
| Cu126 | 1.6 | 80 | ||||||
| Cu127 | 0.9 | 44 | ||||||
| Cu128 | 0.8 | 39 | ||||||
| Cu129 | 1.1 | 56 | ||||||
| Cu130 | — | — | 1.1 | 0.2 | 53 | 0.2 | 341 | |
| Cu131 | 1.5 | 0.5 | 77 | 0.7 | ||||
| Cu132 | ITO/PEDOT:PSS(90 nm)/Cu132: [EMIM][PF6] 4:1 (100 nm)/Al (70 nm) |
557 | — | 5.2 | — | 53 | — | 245 |
| Cu133 | ITO/PEDOT:PSS(80 nm)/Cu133(90 nm)/Al (90 nm) |
580 | — | 0.4 | — | 54 | — | 342 |
| Cu117 | ITO/PEDOT:PSS(80 nm)/Cu117:[EMIM][PF6] 4:1 (120 nm)/Al (90 nm) |
593 | — | 1.3 | — | 131 | — | 339 |
| Cu140 | ITO/PEDOT:PSS/[Cu(P^P)(N^N)][PF6]:[Emim][PF6] 4:1 molar ratio/Al L |
582 | — | 4.5 | 2.3 | 451 | 1.8 | 343 |
| Cu134 | 588 | 0.7 | 0.4 | 33 | 0.30 | |||
| Cu139 | 570 | 4.6 | 2.0 | 462 | 1.7 | |||
| Cu135 | 589 | 1.3 | 0.9 | 130 | 1.20 | |||
| Cu138 | 571 | 1.8 | 0.6 | 92 | 0.6 | |||
| Cu136 | 585 | 0.1 | 0.1 | 13 | 0.05 | |||
| Cu141 | 580 | 1.7 | 0.7 | 87 | 1.1 | |||
| Cu137 | 586 | 0.3 | 0.2 | 34 | 0.15 | |||
| Cu142 | ITO/PEDOT:PSS (40 nm)/EML:LiBF4 (80 nm)/Al | — | — | 1.7 | — | 169 | 0.65 | 235 |
| Cu143 | 2.2 | 223 | 0.84 | |||||
| Cu3 | 4.5 | 452 | 1.85 | |||||
| Cu145 | N/A | 2 | N/A | |||||
| Cu146 | 1.1 | 108 | 0.45 | |||||
| Cu148 | ITO/PEDOT:PSS (70 nm)/active layer (80–100 nm)/Al (90 nm) | 523 | (0.26, 0.38) | 0.02 | 0.03 | 6 | 0.01 | 344 |
| Cu149 | 562 | (0.38, 0.53) | 0.35 | 0.11 | 117 | 0.13 | ||
| Cu151 | 577 | (0.49, 0.49) | 0.04 | 0.03 | 10 | 0.02 | ||
| Cu152 | ITO/PEDOT:PSS(80 nm)/CBP (25 nm)/Cu152(90 nm)/Al (90 nm) | 500 | — | 1.2 | — | 160 | — | 345 |
In 2017, several highly luminescent cyclometalated Au(III) complexes (Au45–52; Scheme 5), containing aryl ligand were reported by Che et al., so that among Au(III) complexes, these complexes exhibit the highest PLQY both in solution and thin-films (0.79 and 0.84 respectively).310 Furthermore, the origination of TADF in some of them that attributed to small ΔEST of 200–500 cm−1 which was confirmed by variable temperature-emission lifetime measurements and DFT calculations, were attributed to the existence of amino group on the ancillary aryl ligand. A plausible explanation for the detection of 3IL emission rather than TADF in Au50 (Scheme 5) could be attributed to the significantly lower energy level of the 3IL excited state compared to the 1LLCT and 3LLCT excited states. This disparity in energy levels is attributed to the substantial exchange energy for the localized IL excited state, thereby leading to emission originating from the 3IL state. When Au(III) aryl emitters (Au46 and Au49; Scheme 5) were tested in solution-processed OLEDs, the sky-blue to green electroluminescence with EQEs, luminance and current efficiencies up to 23.8%, 57340 cd m−2 and 70.4 cd A−1 respectively, were obtained – Fig. 27.
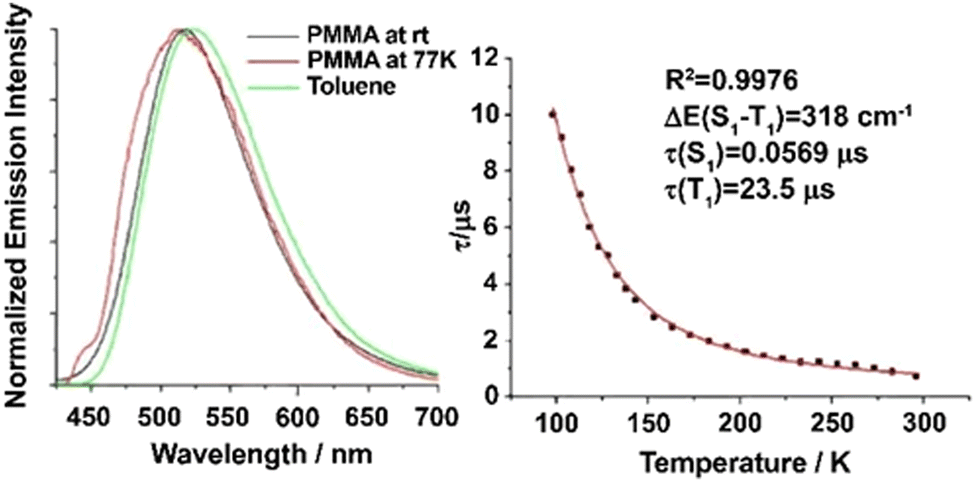 | ||
| Fig. 27 Emission spectra and lifetime data of Au49. (left) Emission spectra of the complex (4 wt%) in PMMA thin-film at rt and 77 K, and in toluene at rt. (right) Plot of emission lifetime monitored at 517 nm against temperature and fitting of the data (red line) to the equation for TADF. Reproduced with permissions from ref. 310. Copyright 2017, Wiley-VCH. | ||
It has been shown that increasing the chelating features of the ligands can improve the stability of metal complexes and metal–ligand bond strength. Therefore, tetradentate ligands are thought to have the ability to induce greater thermal stability and to inhibit structural distortion of complex in the excited state.
The increasing the rigidity of emitters can repress the structural distortion of complex during S1–T1 transformation. Thus, it decreases the activation energy barrier for reverse rISC, resulting in effective TADF. To this end, Zhou et al. reported tetradentate [C^C^N^C] Au(III) complexes containing 5-5-6 membered chelate rings via microwave-induced C–H bond activation – Au53–59; Scheme 5.223 These Au(III) complexes showed robust PL with Φ of 0.94, which is attributed to three emission origins 3IL, 3ILCT and TADF, adjusted by modifying the nature and positions of substituents on the C^C^N^C ligand. The emission mechanism of Au(III) complexes is TADF due to large kr and short τ. The OLED devices constructed with these TADF emitters indicated EQEs of up to 25% and LT95 of up to 5280 h at 100 cd m−2, which is 10 times more effective than tridentate emitters – Table 6.
Key parameters for the commercialization of OLEDs include high EQEmax, minimal efficiency roll-offs, and extended operational lifetime at practical luminances. To meet these criteria simultaneously, maximizing the radiative decay rate constant (kr) is essential. For a TADF emitter, achieving a large S1 → S0 radiative decay rate constant (kSr) alongside a reduced ΔE(S1–T1) is crucial. Several new tetradentate TADF Au(III) complexes (Au60–63; Scheme 5) has been designed to narrow the ΔE(S1–T1) while maintaining a high kSr. They exhibit green emission with nearly unity PLQY and a kr approaching 2 × 106 s−1 in thin-films. Green OLEDs fabricated via vacuum deposition using complexes Au60 and Au63 achieved impressive EQEmax of up to 24% and 27% with efficiency roll-offs of 5.5% and 2.2% at 1000 cd m−2, respectively – Table 6. The EQE remain high even at 10000 cd m−2, reaching 19% (Au60) and 24% (Au63). Complex Au60 demonstrates a remarkable long operational lifetime (LT90) of 1820 hours at 1000 cd m−2, representing one of the most stable TADF-OLEDs to date. The incorporation of a multiresonant-TADF emitter into the OLEDs has led to the realization of high-color-quality displays in the Au60-based device. This integration preserves the inherent narrowband emission of the multiresonant-TADF emitter while significantly enhancing the electroluminescent performance. Notably, these devices exhibit an impressive EQEmax of up to 29.4%, reduced efficiency roll-offs, a maximum luminance of 170000 cd m−2, and a PEmax of 122.5 lm W−1 – Table 6. The exceptional performance of these tetradentate Au(III)-TADF emitters highlights their potential as leading contenders for the development of high-efficiency and operationally stable OLEDs, driving sustainable advancements in display technologies.311
A rare example of Au-complexes, which can be used in next-generation displays and lighting technologies, are donor–acceptor Au(III) acetylide emitters with strong thermal stability and luminescence – Au64–69; Scheme 5.216 Owing to the stabilized LUMO of the 2,6-bis(4-(tert-butyl)phenyl)pyrazine in complex Au68, their absorption bands are red-shifted compared with those including di-deprotonated 2,6-bis(2,4-difluorophenyl)pyridine. Moreover, amino-substituted Au3 and Au65–69 complexes showed wide emission band with color from green to red emission and high PLQYs of up to 60% relative to complex Au64 – Table 11. The investigation in thin-film and DFT/TD-DFT proved that decay occurs through TADF mechanism with PLQY up to 88% and emission τ ≈ 1–2 μs at ambient temperature. OLED based on device fabricated with Au67 indicated an EQEs reaching 23.4% and luminance of 70300 cd m−2 – Table 6.
| Complex | Medium | λ em [nm] | Φ PLQY [%] | τ exp [μs] | k r [s−1] | k nr [s−1] | ΔEST [cm−1] | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Solution concentration = 5.0 × 10−5 mol dm−3. b λ exc = 365 nm. c λ exc = 405 nm. Deaeration was by flow of argon. d Solution concentration = 2.5 × 10−5 mol dm−3. e λ exc = 420 nm. f λ exc = 400 nm. g λ exc = 410 nm. h λ exc = 390 nm. i Solution concentration = 2.5 × 105 mol dm−3. j Solution concentration = 5.0 × 105 mol dm−3. k λ exc = 365 nm. l At 298 K (77 K). | ||||||||
| Cu6 | Powdera | 581b | 3.0b | 1.5b | — | — | — | 339 |
| Cu108 | 587b | 1.7b | 1.3b | |||||
| Cu109 | 575b | 6.2b | 2.9b | |||||
| Cu110 | 581b | 11.1b | 2.9b | |||||
| Cu111 | 648 | 0.5c | 0.185c | |||||
| Cu112 | 647 | 0.5c | 0.251c | |||||
| Cu113 | 664c | 0.5c | 0.096c | |||||
| Cu114 | 632c | 0.9c | 0.579c | |||||
| Cu117 | 517b | 50.3b | 12b | |||||
| Cu115 | 547b | 33.8 | 9.6b | |||||
| Cu116 | 539b | 37.3b | 11.4b | |||||
| Cu6 | Me-THF at 77 K | 610 | — | 16 | — | — | — | 339 |
| Cu108 | 613 | 11 | ||||||
| Cu109 | 610 | 45 | ||||||
| Cu110 | 595 | 31 | ||||||
| Cu111 | 656 | — | ||||||
| Cu112 | 646 | — | ||||||
| Cu113 | 650 | 3 | ||||||
| Cu114 | 652 | 5 | ||||||
| Cu117 | 604 | 42 | ||||||
| Cu115 | 567 | 46 | ||||||
| Cu116 | 551 | 88 | ||||||
| Cu6 | CH2Cl2d solution | 618, 649d | 0.4/0.5 | 43/46 | — | — | 1428 | 339 |
| Cu108 | 620, 650d | 0.5/0.5 | 75/104 | 1492 | ||||
| Cu109 | 618, 646d | 0.7/0.7 | 95/119 | 968–1613 | ||||
| Cu110 | 622, 647d | 0.6/0.6 | 84/99 | 968–1613 | ||||
| Cu111 | — | —/— | —/— | 968–1613 | ||||
| Cu112 | — | —/— | —/— | 968–1613 | ||||
| Cu113 | 667, 697a | —/— | —/— | 1524 | ||||
| Cu114 | 667, 705a | —/— | —/— | 1589 | ||||
| Cu117 | 612, 637d | 0.5/0.5 | 39/39 | 887 | ||||
| Cu115 | 605, 635d | 1.0/1.8 | 27/78 | 968–1613 | ||||
| Cu116 | 606, 635d | 1.6/10.0 | 451/3406 | 968–1613 | ||||
| Cu118 | Solution | 613, 640e | <1 | 0.204 | — | — | — | 340 |
| Cu119 | 613, 641f | <1 | 0.253 | |||||
| Cu120 | 610, 641g | 1 | 0.277 | |||||
| Cu121 | 613, 639e | <1 | 0.334 | |||||
| Cu122 | 605, 635e | <1 | 0.259 | |||||
| Cu123 | 606, 630h | <1 | 0.258 | |||||
| Cu124 | 609, 637h | <1 | 0.231 | |||||
| Cu125 | 610, 633g | 1 | 0.279 | |||||
| Cu126 | 608, 635e | <1 | 0.313 | |||||
| Cu127 | 608, 632e | 1 | 0.315 | |||||
| Cu128 | 605, 630e | <1 | 0.280 | |||||
| Cu129 | 603, 629e | 1 | 0.30 | |||||
| Cu118 | Solution at 77 K | 599 | 5 | 21 | — | — | — | 340 |
| Cu119 | 596 | 6 | 28 | |||||
| Cu120 | 613 | 3 | 16 | |||||
| Cu121 | 563 | 10 | 48 | |||||
| Cu122 | 598 | 9 | 31 | |||||
| Cu123 | 600 | 7 | 33 | |||||
| Cu124 | 593 | 11 | 23 | |||||
| Cu125 | 594 | 15 | 23 | |||||
| Cu126 | 610 | 11 | 13 | |||||
| Cu127 | 588 | 20 | 38 | |||||
| Cu128 | 575 | 20 | 38 | |||||
| Cu129 | 576b | 23 | 44 | |||||
| Cu118 | Powder | 565 | 17 | 3.3 | — | — | — | 340 |
| Cu119 | 570 | 9 | 2.7 | |||||
| Cu120 | 585 | 5 | 1.5 | |||||
| Cu121 | 549 | 30 | 10.2 | |||||
| Cu122 | 564 | 22 | 6.5 | |||||
| Cu123 | 566 | 20 | 6.2 | |||||
| Cu124 | 566 | 19 | 4.7 | |||||
| Cu125 | 566 | 22 | 4.0 | |||||
| Cu126 | 572 | 12 | 2.7 | |||||
| Cu127 | 557 | 21 | 6.0 | |||||
| Cu128 | 552 | 32 | 6.5 | |||||
| Cu129 | 552b | 38 | 9.1 | |||||
| Cu118 | Film | — | 4 | — | — | — | — | 340 |
| Cu119 | 5 | |||||||
| Cu120 | 5 | |||||||
| Cu121 | 5 | |||||||
| Cu122 | 5 | |||||||
| Cu123 | 7 | |||||||
| Cu124 | 6 | |||||||
| Cu125 | 7 | |||||||
| Cu126 | 6 | |||||||
| Cu127 | 5 | |||||||
| Cu128 | 6 | |||||||
| Cu129 | 7 | |||||||
| Cu134 | CH2Cl2 solutioni (non-deaerated/deaerated) | 622, 643 | 0.5/0.7k | 0.057/0.108k | 8.8 × 104/6.5 × 104 | 1.7 × 107/9.2 × 106 | — | 343 |
| Cu135 | 616, 642 | 0.4/0.9k | 0.153/0.338k | 2.6 × 104/2.7 × 104 | 6.5 × 106/2.9 × 106 | |||
| Cu136 | 614, 648j | 0.5/0.5k | 0.039/0.045k | 1.3 × 105/1.1 × 105 | 2.6 × 107/2.2 × 107 | |||
| Cu137 | 615, 632j | 0.4/0.5k | 0.076/0.093k | 5.3 × 104/5.4 × 104 | 1.3 × 107/1.1 × 107 | |||
| Cu138 | 598, 630 | 1.0/1.5k | 0.202/0.730k | 5.0 × 104/2.1 × 104 | 4.9 × 106/1.3 × 106 | |||
| Cu139 | 582, 627 | 0.9/3.3k | 0.228/1.595k | 3.9 × 104/2.1 × 104 | 4.3 × 106/6.1 × 105 | |||
| Cu140 | 597, 629 | 0.8/6.0k | 0.240/2.401k | 3.3 × 104/2.5 × 104 | 4.1 × 106/3.9 × 105 | |||
| Cu141 | 583, 626 | 0.9/9.6k | 0.262/4.987k | 3.4 × 104/1.9 × 104 | 3.8 × 106/1.8 × 105 | |||
| Cu134 | Powderk | 585 | 2.7 | 2.3 | 11.7 × 109 | 423.0 × 109 | — | 343 |
| Cu135 | 571 | 6.3 | 5.1 | 12.4 × 109 | 183.7 × 109 | |||
| Cu136 | 602 | 1.1 | 0.4 | 27.5 × 109 | 2472.5 × 109 | |||
| Cu137 | 556 | 9.6 | 3.3 | 29.1 × 109 | 273.9 × 109 | |||
| Cu138 | 518 | 42.7 | 9.3 | 45.9 × 109 | 61.6 × 109 | |||
| Cu139 | 529 | 58.8 | 9.8 | 60.0 × 109 | 42.0 × 109 | |||
| Cu140 | 558 | 27.5 | 8.7 | 31.6 × 109 | 83.3 × 109 | |||
| Cu141 | 550 | 9.8 | 10.2 | 9.6 × 109 | 88.4 × 109 | |||
| Cu142 | Dilute degassed solutions (ca. 10−5 M) | 550 | 71 | — | — | — | 1532 | 235 |
| Cu143 | 549 | 40 | 1452 | |||||
| Cu3 | 567 | 60 | 1613 | |||||
| Cu144 | 547 | <1 | 1532 | |||||
| Cu145 | 543 | 1 | 1532 | |||||
| Cu146 | 541 | 98 | 1613 | |||||
| Cu147 | 431 | <1 | 1452 | |||||
| Cu148 | Powder 298 K (77 K) | 480 (490)l | 17 | 24.7 (198.4)l | 0.7 × 104 | 3.4 × 104 | 951 | 386 |
| Cu149 | 551 (549)l | 14 | 7.31 (982.4)l | 1.9 × 104 | 11.8 × 104 | 773 | ||
| Cu150 | 546 (537)l | <1 | 11.2 (155.6)l | 0.1 × 104 | 8.8 × 104 | 588 | ||
| Cu151 | 574 (584)l | 38 | 3.0 (244.1)l | 12.7 × 104 | 20.7 × 104 | 500 | ||
| Cu148 | Thin-film 298 K (77 K) | 497 (493)l | 8 | 15.2 (177.0)l | 0.5 × 104 | 6.0 × 104 | — | 344 |
| Cu149 | 540 (537)l | 10 | 61.9 (781.0)l | 0.2 × 104 | 1.5 × 104 | — | ||
| Cu150 | 546 (537)l | <1 | 7.5 (160.3)l | 0.1 × 104 | 1.3 × 104 | — | ||
| Cu151 | 580 (571)l | 21 | 47.6 (944.0)l | 0.4 × 104 | 1.7 × 104 | — | ||
Several novel Au(III) complexes (Au70–76; Scheme 5) have been successfully developed and prepared, featuring tetradentate C∧C∧N∧N ligands with acridinyl groups that exhibit TADF. These complexes emit in orange-red to deep-red hues through incorporation of different substituents onto the acridinyl and pyridine units, exihibiting high PLQYs of up to 0.76 in thin-films. Notably, they demonstrate short τ of ≤2.0 μs and significant kr on the order of 105 s−1 – Table 7. High-performance OLEDs have been successfully fabricated using these complexes, both through solution processing and vacuum deposition. The resulting devices show exceptional EQEmax of 12.2% and 12.7%, respectively – Table 6, ranking among the highest reported values for red-emitting Au(III)-based OLEDs. Moreover, these red-emitting devices exhibit satisfactory operational half-lifetime (LT50) values reaching up to 34058 hours. The operational lifetimes of these devices is strongly influenced by the functional groups attached to the acridinyl units. Incorporating –O– and –S– linkers has been shown to significantly extend the LT50 value by an order of magnitude. The TADF characteristics of the complexes are further supported by the blue shifted emission band and the notable increased emission intensity as the temperature rises. Estimates suggest that there are small ΔEST, with values of approximately 1000 cm−1 observed in toluene and the solid state, respectively. Temperature-dependent ultrafast TA investigations have confirmed the TADF of these complexes, revealing rISC and providing the activation parameters for the first time, shedding light on their excited-state dynamics.221
Several novel Au(III) complexes with tetradentate C^C^N^N ligands containing pyridine and pyrazine have been formulated and prepared (Au77–82; Scheme 5) to exhibit TADF. These complexes featured high PLQYs of up to 0.77 in thin-films – Table 7. In toluene and doped mCP thin-films, their enhanced thermal emissions revealed hypsochromic spectral shifts of approximately 403–1371 cm−1 along with significantly reduced τ values. Utilizing TA spectroscopy, the direct observation of rISC processes enabled the determination of activation parameters. In the solution state, the kr was analyzed using the Boltzmann two-level model, yielding estimated ΔEST of approximately 1290–1452 cm−1 for Au77, Au79, and Au81. By altering the electron-donating – tBu substituent in the cyclometalating ligand to influence the host–guest interactions and molecular anisotropy, the permanent dipole moments of the complexes in Au77–80 (Scheme 5) were adjusted to increase the TDMVs’ θh enhancing the out-coupling efficiency (ηout) in the OLEDs. Consequently, more horizontal-orientated complexes Au79 and Au80 were applied in vacuum-deposited OLEDs reaching promising electroluminescent performance of EQEmax of up to 15.7% and strongly improved ηout by ∼30%, compared to their regioisomers, Au77 and Au78 – Table 6 the based on the more horizontal-orientated complexes Au79 and Au80 showed improvements in ηout by ∼30%, compared to their regioisomers, Au77 and Au78 and exhibited promising electroluminescent performance, achieving EQEmax of up to 15.7% – Table 6. Introducing a -tBu substituent to the carbazolyl groups not only enhanced the operational stability of the OLEDs, but also facilitated horizontal molecular orientation. This study shows the potential of the – tBu substituent in optimizing Au(III) C^C^N^N systems for the advancement of efficient Au(III)-based OLEDs, offering valuable insights for further development efforts.312
Several novel carbazolyl Au(III) dendrimers containing C^C^N ligands (Au83–85; Scheme 5) has been developed and synthesized. Exceptionally high PLQY reaching up to 82% in thin-films, along with large radiative decay rate constants on the order of 105 s−1, have been observed – Table 7. These Au(III) dendrimers demonstrate TADF, supported by time-resolved photoluminescence decay and computational investigations, with estimated ΔEST values of ca. 32 cm−1. OLEDs based on these Au(III) dendrimers were successfully fabricated via solution processing. These OLEDs achieved current efficacies of 52.6 cd A−1, EQEmax of 15.8%, and substantial power efficiency of 41.3 lm W−1 – Table 6. Furthermore, the operational stability of these OLEDs was evaluated, revealing remarkable results. The devices utilizing zero- and second-generation dendrimers showed maximum half-lifetimes of 1305 and 322 h at 100 cd m−2, respectively. This pioneering work demonstrates the application of Au(III) dendrimers in operationally stable solution-processed OLEDs.313
Several new carbazolyl ligands has been synthesized by incorporating various electron-donating and electron-accepting group at the 2- and 3-positions of the ancillary ligand, such as 4-(diphenylamino)aryl, cyano, and diphenylphosphine oxide, to create a novel class of Au(III) complexes (Au86–91; Scheme 5). They are designed to tune the energies of their triplet intraligand and LLCT excited states to enable TADF. This can effectively reduce the τ in the Au(III) complexes by more than two orders of magnitude from ∼80 ms to 0.8 ms. Upon excitation, these complexes exhibit high PLQYs of up to 80% in thin-films, with short τ as low as 1 ms. OLEDs based on these complexes, fabricated using both vacuum deposition and solution processing methods, display promising electroluminescent performance – Table 6. These devices achieve EQEmax of 15.0% and 11.7%, respectively. Importantly, the suppression of the roll-off in solution-processed devices can be notably reduced when TADF is utilized. Specifically, the roll-off decreases from 65% in phosphorescence-based devices (Au86) to just 1% in TADF-based devices (Au89), achieving this improvement at a practical brightness level of 1000 cd m−2. The superior electroluminescent characteristics are attributed to the presence of multichannel radiative decay pathways through both phosphorescence and TADF mechanisms, which effectively reduce emission lifetimes and mitigate the adverse effects of triplet–triplet annihilation in the Au(III) complexes.314
Due to the importance of developing blue-emitting OLEDs, a number of cationic Cu(I) complexes with excellent greenish-blue to blue emission based on POP and N-linked 2-pyridyl pyrazolate diimine ligands, e.g., [Cu(pypz)(POP)]BF4 (Cu29), [Cu(pympz)(POP)]BF4 (Cu30), and [Cu(pytfmpz)(POP)]BF4 (Cu31) (pypz = 1-(2-pyridyl)pyrazole, pympz = 3-methyl-1-(2-pyridyl)pyrazole, and pytfmpz = 3-trifluoromethyl-1-(2-15 pyridyl)pyrazole) were synthesized and characterized – Scheme 6.317 The HOMO–LUMO energy gap is adjustable to obtain blue light emission.346 The electron donating capability of the pypz ligand led to the increase of the HOMO–LUMO energy gap and the shift of the emissions to the blue region through the instability of the LUMO level of the luminescent dopant substances. In addition, different substituents such as CH3 and CF3 on the pyrazole ring of 2-pyridyl pyrazolate ligands were used to adjust the emission efficiencies and colors of the related Cu(I) complexes. The dependence of emission decay time on spectroscopic properties under different temperature was an interesting phenomenon in these complexes.317 The application of these complexes as dopants with various hosts in solution-processed OLEDs showed the values of 23.68 cd A−1 for efficiency and 2033 cd cm−2 for peak brightness for Cu31 as dopant and 2,6-mCPy as host – Table 8.317 Effective confinement of triplet excitons and charge carriers led to these performances. Conversely, the nearly equal LUMO level of the 2,6-mCPy host and dopants of Cu29 or Cu30 results in relatively low efficiencies of OLEDs made of Cu29 and Cu30.317
 | ||
| Scheme 6 Molecular structures of Cu29–107 and Ag16–19. | ||
Several neutral d10 metal complexes containing tetrahedral structures, [Cu(PP)(PS)] (Cu32), [Ag(PP)(PS)] (Ag16), and [Au(PP)(PS)] (Au14), containing two bidentate ligands [PS− = 2-diphenylphosphinobenzenethiolate] and [PP = 1,2 bis(diphenylphosphino)benzene] have been also investigated – Scheme 6.315 In accordance with molecular orbital calculations, the electron-donating property of the PS ligand decreases the participation of metal orbitals in the HOMOs, impeding an efficient MLCT of their excited states and the TADF of metal(I) complexes associated with LLCT transition. Despite the high thermal stability of Cu32, Ag16 and Au14, (decomposition temperatures above 300 °C), the negligible vapor pressure up to thermal decomposition of mentioned complexes hampers their vacuum deposition for the realization of emitting films. Thus, the latter have been realized through wet processes. Fig. 28 illustrates the emission spectra of complexes Cu32, Ag16 and Au14 at 77 and 293 K as well as the electroluminescence spectra and dependence of the EQE on the current density for OLEDs containing Cu32 as a dopant. According to these data, Cu32 exhibits an efficient green TADF that was successfully exploited in OLEDs. Contrary to Cu32, the rapid ligand exchange reaction in case of Au14 causes its instability in solution, while the low solubility of Ag16 in organic solvents restricts its use as luminescent guest molecules.
 | ||
| Fig. 28 (a) Corrected emission spectra of Cu32 (green), Ag16 (blue), and Au14 (red) in the solid state at 293 (solid lines) and 77 (dashed lines) K; λext = 310 nm. The upper inset shows luminescence images of Cu32, Ag16 and Au14 at 293 K; λext = 354 nm. The lower inset shows a two-state model for Cu32. (b) Properties of OLEDs [device A (red), B (green), and C (blue)] containing Cu32 as a dopant. (A) electroluminescence spectra, (B) dependence of EQE on current density. Reproduced with permissions from ref. 315. Copyright 2013, Royal Society of Chemistry. | ||
Binuclear Cu(I) complexes based on N-heterocyclic phosphine were also applied to OLEDs. They consist of a Cu2I2 moiety with a butterfly-like structure as shown in Cu33 – Scheme 6, featuring two ancillary phosphine ligands and an N,P-ligand bridge318 Air stability and lower ΔEST (200 meV) than similar green emitting Cu-compounds are the most relevant features.347 Four-fold bridged Cu(I) centers in this dinuclear Cu(I) complex led to its high thermal stability (Td = 290 °C), which was another advantage of this dinuclear complex. The use of this multi-bridge dinuclear Cu(I) complex as a TADF emitters in OLEDs with an EQE of 23% (Table 8), due to its distinct structure, high processability, high PLQY in PYD2- doped films and low ΔEST. This value is close to the high-performance EQE of devices under vacuum made of Ir(III) complexes. For the sake of completeness, Cu33-based OLEDs are demonstrated that the device indicates a pronounced roll-off of the current efficiency as luminance increases. Imbalance of charge leads to this behavior because hole transport occurs through the emitter itself rather than through the host material. In general, the progression of a novel generation of TADF-based OLEDs using the concept of multi-bridged binuclear Cu(I) complex is being explored as a new paradigm shift.
By considering the dependence of knr and PLQYs on the structural distortions of tetrahedral Cu(I) complexes in the excited state – Section 2, several highly luminescent Cu(I) complexes with a three-coordinate structure without any structural modification in the excited state, (LMe)CuX [X = Cl (Cu34), Br (Cu35), I (Cu36)], (LEt)CuBr (Cu37), and (LiPr) CuBr (Cu38) with bidentate bisphosphine ligands [LMe = 1,2 bis[bis(2-methylphenyl)phosphino]benzene, LEt = 1,2-bis[bis(2-ethylphenyl)-phosphino]benzene, and LiPr = 1,2-bis[bis(2-isopropylphenyl)phosphino]benzene] were introduced – Scheme 6.319 In these complexes, the molecular motions were limited by the presence of varied ortho-substituents on the peripheral phenyl moieties. Furthermore, in complexes Cu34–38 – Scheme 6, the emission arises from (σ + X) → π* (diphosphine ligand) transition. The presence of alkyl substituents at the ortho positions of the peripheral phenyl moieties was determined to have a marginal impact on the electronic excited states. Since the emission of complexes Cu34–38 is associated to a (σ + X) → π* (diphosphine ligand) transition, the photoluminescent characteristics of these complexes are predominantly determined by the diphosphine ligands. Fig. 29 shows their electroluminescence characteristics. The TADF-type OLEDs based on complexes Cu34–38 exhibited a green luminescence with high EQE values ranging from 18.6 to 22.5% – Table 8.
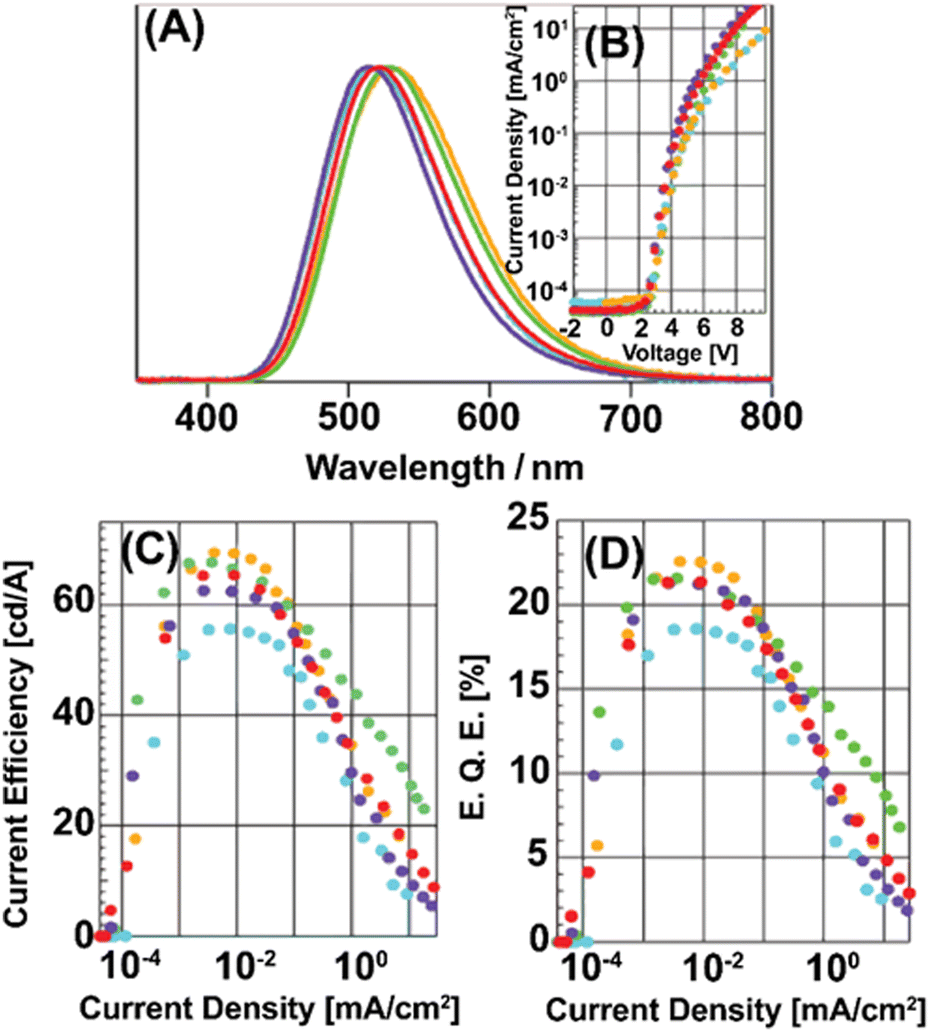 | ||
| Fig. 29 Properties of OLEDs containing complex Cu34 (green line), complex Cu35 (red line), complex Cu36 (purple line), complex Cu37 (orange line) and complex Cu38 (blue line). (A) Electroluminescence spectra; (B) the dependence of the EQE on the current density; (C) I–V characteristics; (D) the dependence of the current efficiency on the current density. Reproduced with permissions from ref. 319. Copyright 2015, Royal Society of Chemistry. | ||
In this line, Pflaum and colleagues recently presented the synthesis of enantiomerically pure Cu(CbzR)[(S/R)-BINAP] [R = H (Cu19), 3,6-tBu (Cu20)] as highly effective TADF emitters (Scheme 3) with impressive radiative rate constants (kDF) close to 3.1 × 105 s−1 originating from 1/3LLCT states, as confirmed by temperature-dependent time-resolved luminescence experiment as well as CPL signal. They exhibit strong C–H⋯π interactions between the ligands and neighboring molecules. For instance, in single crystalline solid state, a dominant phosphorescent component was observed from 3LLCT/Cbz states with maximum peak wavelengths of 564 nm for (Cu19) and 549 nm for (Cu20). However, highly efficient TADF was achieved by grinding complexes. As a result, kr = to 3.1 × 105 s−1 and a shift in the emitting 1/3LLCT states to λmax = 579 (Cu19) and 606 (Cu20) nm was gained – Table 9. The choice of matrix material for fabrication of device is strongly influenced by intermolecular hydrogen bonding on excited state energies. The emission shift to the green region of the electromagnetic spectrum by polar and sterically unprotected mCP or PMMA results in significant decline in kr to <5 × 104 for Cu19 and <0.5 × 104 s−1 for Cu20 – Table 9. Retention of outstanding TADF characteristics of Cu(I) complexes at lower emission energies is achieved by using nonpolar UGH-3 or bulk CzSi. The asymmetry values (glum) for CPL of ±6 × 10−3 (Cu19) and ±5 × 10−3 (Cu20) in solution, and ±1.7 × 10−2 (Cu19) and ±2.1 × 10−2 (Cu20) in the solid state are due to the notable rotational strength of the electronic transitions arising from the chirality of the emitter molecules. These findings highlight the importance of careful device design in achieving efficient generation of Circularly Polarized Electroluminescence (CP EL).251
Neutral Cu(I) and Ag(I) complexes have also been synthesized using a novel rigid tridentate N,P,P ligand called dmpzpp, 3,5-dimethyl-1-(2-((2-(di-o-tolyl)-phosphanyl) (o-tolyl)-phosphanyl)phenyl)-1H-pyrazole. The complexes include Cu(dmpzpp)Br (Cu39), Cu(dmpzpp)I (Cu40), Cu(dmpzpp)SPh (Cu11) (SPh refers to thiophenylato) – Scheme 6. Cu11, which has bulky ligands and shows PLQY = 90% – Table 9, was specifically studied for emission properties in the range of 1.7 to 300 K. At temperatures up to approximately 70 K, Cu11 exhibits only long-lived phosphorescence with a τ = 1 ms, attributed to weak SOC. The zero-field splitting of T1 in the three substrates are less than 1 cm−1. The phosphorescence is primarily caused by spin-vibronic mechanisms, as indicated by estimated individual decay times of 2400, 2250, and 292 μs. As the temperature increases up to 300 K, the significantly reduction in radiative decay time by more than two orders of magnitude to τDF = 5.6 μs was observed. This was attributed to TADF. This rapid decay time is influenced by the small ΔEST of 600 cm−1 or 74 meV and a fast radiative S1 to S0 transition rate = 1.1 × 107 s−1 (equivalent to 91 ns). To fabricate OLEDs, Cu40 and Cu11 were used in cohost device structures. For instance, a green emitting OLED with 2 wt% concentration of Cu40 was constructed which showed values of (0.33, 0.52) for CIE coordinates, 16.4% for EQE and nearly 10000 cd m−2 – Table 8.231
Two highly efficient complexes, [Cu(czpzpy)(PPh3)]BF4 (Cu41) and [Cu(czpzpy)(POP)]BF4 (Cu42) – Scheme 6, were prepared through the use of a novel diamine ligand based on Cz (czpzpy) and two different types of phosphine-based ligand (PPh3 and POP), in which czpzpy = 2-(9H-carbazolyl)-6-(1H-pyrazolyl)pyridine, PPh3 = triphenylphosphine.320 The Oak Ridge Thermal Ellipsoid Plot (ORTEP) diagrams of Cu41 and Cu42 are displayed in Fig. 30. The grafting the functional Cz group into the diamine ligand improves the light-emitting and hole-transporting properties of cuprous complexes. The TADF along with high PLQY close to 100% was achieved for Cu42 at ambient temperature in thin-film – Table 9. In this approach, czpzpy ligand has a dual role, i.e., acting as a ligand to create the luminescent complex as well as a host for the formed emitter in OLEDs due to its effective hole-transporting capability and elevated triplet energy level. The associated electroluminescence properties and quantitative results of Cu42 are presented in Fig. 30.
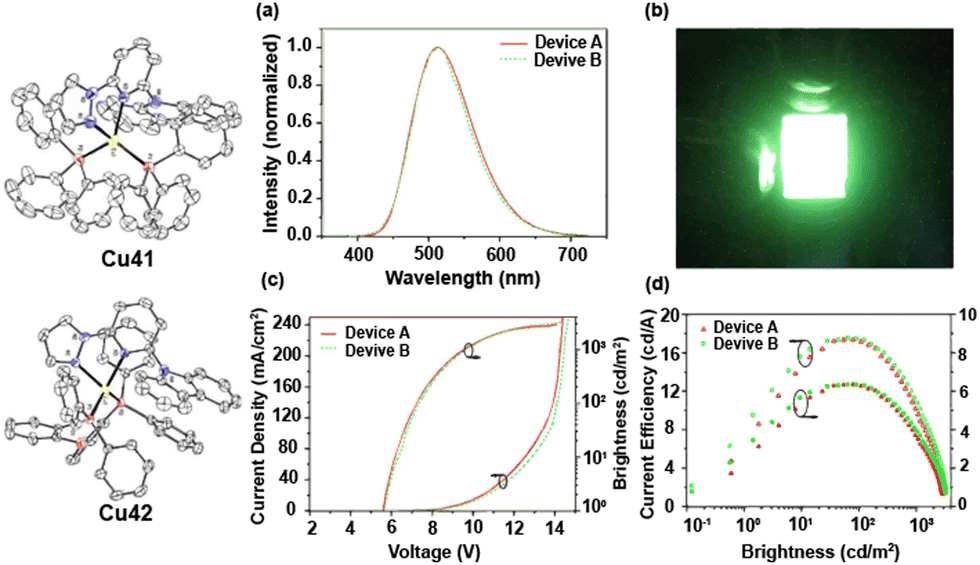 | ||
| Fig. 30 ORTEP diagrams of Cu41 and Cu42 with thermal ellipsoids at 30% probability level. Solvent molecules, the anion, and H atoms are omitted for clarity. (a) Electroluminescence spectra (device a = Cu41, device b = Cu42); (b) photo of Device B; (c) current density–voltage–brightness (I–V–B) curves; (d) plots of EQE and current efficiency vs. brightness. Reproduced with permissions from ref. 320. Copyright 2015, Royal Society of Chemistry. | ||
Preparation of two bluish-green Cu(I) complexes [Cu(PNNA)(POP)]BF4 (Cu43) and [Cu(PNNA)(xantphos)]BF4 (Cu44), (PNNA = 9,9-dimethyl-10-(6-(3-phenyl-1H-pyrazol-1-yl)pyridin-3-yl)-9,10-dihydroacridine) with a functionalized diimine and phosphine ligands were reported as TADF emitters – Scheme 6.321 Here, the use of the donor segment, 9,9-dimethylacridan, on the diimine ligand was due to its excellent hole-transporting ability. Devices with Cu44 as dopant showed the highest efficiency of 20.24 (cd A−1), brightness (5579 cd m−2) and the bluest electroluminescence due to the properties of its rigid and bulky ligands – Table 8.
In classical tetrahedral heteroleptic Cu(I) complexes, three yellowish-green emitting cationic cuprous complexes [Cu(POP)(ECAF)]PF6 (Cu45), [Cu(POP)(EHCAF)]PF6 (Cu46), and [Cu(POP)(PCAF)]PF6 (Cu47), based on POP along with the very bulky and bipolar ancillary ligands 9,9-bis(9-ethylcarbazol-3-yl)-4,5-diazafluorene (ECAF), 9,9-bis(9-ethylhexylcarbazol-3-yl)-4,5-diazafluorene (EHCAF), and 9,9-bis(9-phenylcarbazaol-3-yl)-4,5-diazafluorene (PCAF), were investigated by the group of Zhang – Scheme 6.322 Typicall, ionic Cu(I) complexes must be processed using solution-based techniques. However the ancillary ligands decreases the lattice energies and weaken the electrostatic interaction, allowing the preparation of vacuum-evaporated OLEDs. In addition, the introduction of the 4,5-diazafluorene as electron-transporting and Cz moieties as hole-transporting causes bipolar charge-transporting abilities, balancing the charge carrier transport in the LEL, increasing the OLED performance. Due to the singlet harvesting, triplet harvesting and good thermal stabilities, the OLEDs exhibited a high electroluminescence brighness – 5152–11010 cd m−2; Table 8.255,326,348,349
Three novel effective blue-green neutral halogen-bridged dinuclear Cu(I) complexes constructed from a new bidentate 1,2-bis(diphenylphosphino)-4,5-dimethylbenzene (dpmb) ligand, [Cu(μ-X)dpmb]2 (X = I (Cu48), Br (Cu49), Cl (Cu50)) – Scheme 6, and possessing small ΔEST energy gaps (954–1120 cm−1), were recently reported to investigate the impact of two ortho-methyl electron-donating groups on the diphosphine.323Fig. 31 shows the ORTEP diagrams of complexes Cu48–50. The origin of the emission in Cu48–50 are attributed to the (σ + X) → π* transition, (σ is the Sigma bond between P and Cu). The methyl groups of these complexes significantly create a blue shift of the above transition energy, leading to a blue emission. When these complexes were used as the luminescent dopants with optimum concentration of 10% in TADF-type OLED devices fabricated by vacuum processes, bright green luminescence with EQEmax of 10.1% and current efficiencies of 32.9 cd A−1 were obtained for the case of complexes Cu48 – Table 8. Fig. 31 shows the device characteristics of based on three complexes. The λmax follow the sequence Cu48 < Cu49 < Cu50, aligning with the expected ligand field strengths of the halogen ions in the complexes (I− < Br− < Cl−).350 Indeed, a slight blue shift for the electroluminescent peak wavelength of device Cu48 compared to devices Cu49 and Cu50 was observed. In addition, the order of Cu48 < Cu49 < Cu50 for the FWHM of the devices with each dopant is in line with the ligand field strength order of the halogen ions in the complexes. Probably, the improved electron injection to the EML causing a significant diminish in the turn-on and driving voltage of the device with Cu50. Furthermore, the relatively low efficiency of devices with Cu49 and Cu50 indicates complete confinement of holes and electrons in the EML as well as effective suppression of current-induced exciton quenching because of the rather strong ligand field strength of the halogen ions in the complexes.
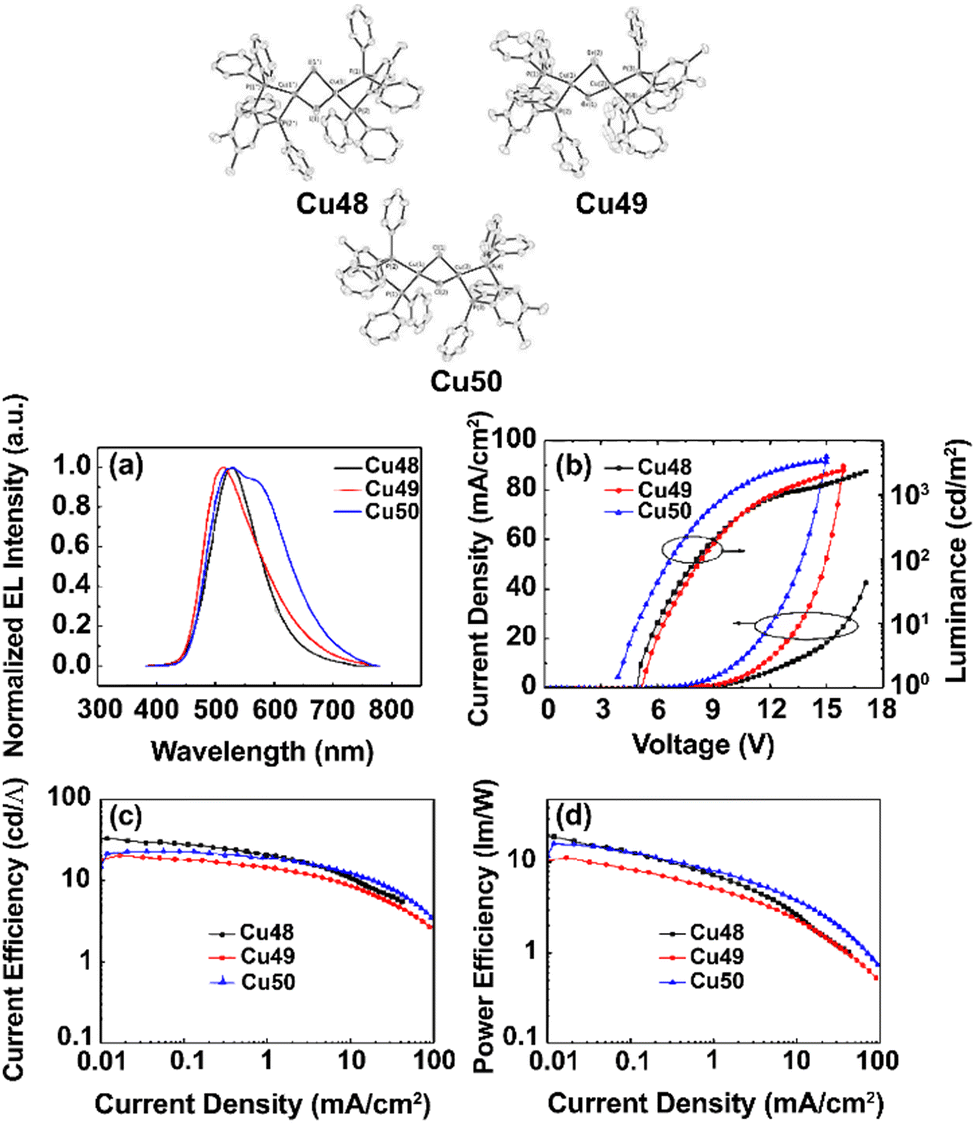 | ||
| Fig. 31 ORTEP diagrams of complexes Cu48–50. (a) Electroluminescence spectra of devices Cu48–50; (b) current density–voltage–luminance characteristics (J–V–L) of devices 17-A3; (e) EQE versus current density curves for devices Cu48–50; (d) power efficiency versus current density for devices Cu48–50. Reproduced with permissions from ref. 323. Copyright 2016, Elsevier. | ||
Similar to Pflaum et al. Zhou and co-workers successfully prepared three chiral Cu(I) chloro-bridged dimers, namely R/S-(BINAP)2Cu(μ-X2) (X = Cl (Cu51), Br (Cu52), and I (Cu53)) – Scheme 6, at room temperature within a short time of 10 min, with yields exceeding 95%. These dimers were based on R/S-2,2′-bis(diphenylphosphino)-1,1′-binaphthalene (R/S-BINAP) ligands showing high dissymmetry factors (g) surpassing 1 × 10−2. Additionally, the small ΔEST and the overlap of theoretical frontier molecular orbitals led to TADF emission mechanism. Notably, when circularly polarized organic light-emitting diodes (CP-OLEDs) were fabricated using the evaporation method with R/S-(BINAP)2Cu(μ-I2) (Cu53), a remarkable EQEmax of 21.7% was achieved – Table 8. Intriguingly, these CP-OLEDs also exhibited mirror-imaged CP EL with |gEL| close to 3.0 × 10−3.324
As a final asset of this family of dinuclear Cu(I) complexes is the possibility to fabricate efficient inkjet-printed OLEDs. As leading example, a highly soluble Cu(I)-NHetPHOS TADF emitter was proposed.325 As for the case of dinuclear NHetPHOS-complexes, Cu54 (Scheme 6) comprises a butterfly-shaped Cu2I2 moiety, a bridging N,P-ligand containing a nitrogen-with heterocycle (NHet), and two extra monodentate P-donors with tris-m-tolylphosphine, P(mTol)3. The OLED with this complex yielded up to 11% EQE in large areas – Table 8.
In a related family of heteroleptic halogen-based Cu(I) complexes, the use of different halogens clearly allowed to balance dual emissions mechanisms (phosphorescence and TADF). In short, a set of mononuclear Cu(I) complexes TTPPCuX (X = Cl, Br, I); TTPP = tridentate phosphine ligand 2,2′ (phenylphosphine-diyl)bis(2,1-phenylene) bis(diphenylphosphine) (Cu55–57; Scheme 6) with stable and rigid tetrahedral geometry were studied.326 The emission spectra of TTPPCuX at ambient temperature gradually blue-shifted by decreasing the ligand-field strength for X, from Cl, Br to I.253 Significantly, by considering the ΔEST and heavy atom effect for SOC improvement at ambient temperature, the activation of the T1 → S0 transition, the balanced dual emissions with a significant phosphorescence fraction of 39%, PLQY of 85%, and ten-fold smaller triplet lifetime and about 50% larger kr were obtained by TTPPCuI – Table 9. This lead to a EQEmax of 16.3% – Table 8, which is among the top values developed for OLEDs with yellow-emitting Cu(I) complexes248,318,348,349,351–355 was achieved with TTPPCuI. Fig. 32 reports the electroluminescence performance of TTPPCuX-based OLEDs.
 | ||
| Fig. 32 Electroluminescence performance of TTPPCuX-based OLEDs. Current density (J) (hollow)–luminance (solid)–voltage curves, electroluminescence spectra at 1000 cd m−2 and device photos at 5 V (inset). Reproduced with permissions from ref. 326. Copyright 2016, Europe PMC. | ||
In 2017, three cationic green emissive binuclear copper complexes containing various groups were systematically studied by Lin et al.327 The cationic copper complexes were [Cu2(pytzph)(POP)2](BF4)2 (Cu58), [Cu2(pytzphcf)(POP)2](BF4)2 (Cu59) and [Cu2(pytzphcz)(POP)2](BF4)2 (Cu60) with tetraimine derivatives and bisphosphine ligands, (pytzph = 6,6′-(1-phenyl-1,2,4-triazole-3,5-diyl)bis(2-methylpyridine), pytzphcf = 6,60′-(1-(4 (trifluoromethyl)phenyl)-1,2,4-triazole-3,5-diyl)bis-(2-methylpyridine), pytzphcz = 9-(4-(3,5-bis(6-methylpyridin-2-yl)-1,2,4-triazol-1-yl)phenyl)-carbazole and POP) – Scheme 6. The small ΔEST 718–1064 cm−1 led to highly efficient green TADF. However, among all these complexes, the highest efficiency in OLEDs was noted for those with Cu60, reaching an EQE of 8.3% – Table 8. Its high PLQY and superior Cz group's hole-conducting properties, and high rigidity led to this excellent device. In the same line, Gary et al. demonstrated excellent properties for rigid ligands, such as: (i) thermal stability,356 (ii) intermolecular interactions, (iii) capability to modulate HOMO and LUMO, (iv) high PLQY,357–360 (v) hydrophilicity,361–365 and (vi) concentration quenching behavior.366–368 As example, the synthesis of three cationic TADF Cu(I) complexes (Cu61–63; Scheme 6)328 using dppnc (7,8-bis(diphenylphosphino)-7,8-dicarba-nido-undecaborate) and neocuproine ligands were reported, highlighting how the neocuproine ligand structure rules the emission color change from green to dark red and, as such, the position of the T1 state.328 Thus the difference between the T1 of the complex and the energy levels of the host material was used to rationalized the decrease of EQE from 18.46% (Cu61) to 10.17% (Cu63) – Table 8. In addition, there was a significant effect of dopant concentrations at different levels between 4 and 20 wt% on the performance of these devices – Table 8. The best device made of 4 wt% Cu(dppnc)-R emitter and CBP showed pure red emission with a value of 10.17% for EQE.
Ligand rigidity has exerted a strong impact on heteroleptic Cu(I) complexes comprising bisphosphines and macrocyclic phenanthroline ligands.369,370 In detail, synthesis and investigations of a series of heteroleptic Cu(I) pseudorotaxanes developed by macrocyclic phenanthroline ligands with various ring sizes and POP, [Cu(mXX)(POP)]+ (mXX = m30 (Cu64), m37 (Cu65) and m42 (Cu66)), was published by Mohankumar and co-workers238 and compared with the standard material [Cu(dmp)(POP)]+, (Cu4), (dmp = 2,9-dimethyl-1,10-phenanthroline) – Scheme 6.371–373 Due to gradual suppression of steric congestion caused by a rise in the macrocycle size, the Cu(I) pseudorotaxane with the largest macrocycle (Cu66) exhibited a higher electrochemical stability compared to small counterparts. However, all of them showed similar TADF parameters – Table 8. Eventually, to get a high-performance OLEDs, Cu66 was chosen as emitter due to its straightforward preparation, high PLQY in PMMA (53%, Table 9) and excellent electrochemical stability, while providing intense bright green electroluminescence that is comparable or even superior to those already reported for green OLEDs containing [Cu(dnbp)(DPEPhos)]+ (dnbp = 2,9-di-n-butylphenanthroline) by Adachi and coworkers.349Fig. 33 shows the electroluminescence spectra of the OLEDs based on Cu66.
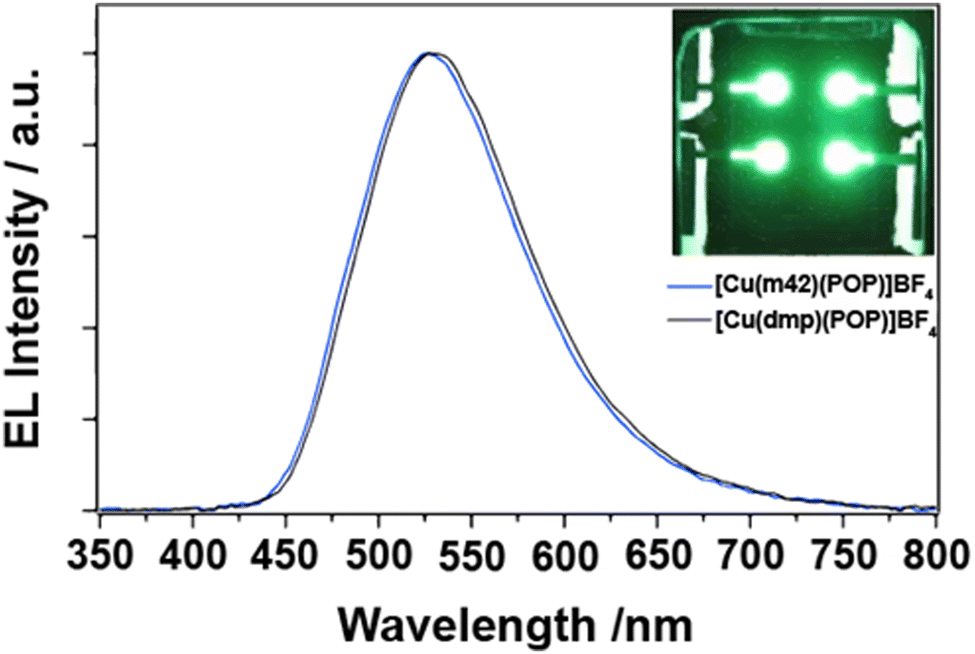 | ||
| Fig. 33 Electroluminescence spectra at 10 V of OLEDs based on Cu66 and Cu4. In the inset is reported a photograph of OLEDs based on Cu66. Reproduced with permissions from ref. 238. Copyright 2018, American Chemical Society. | ||
The design and development of a series of two-coordinate Cu complexes with exceptional luminescence efficiency, comprising nonconventional cyclic amidocarbenes374–377 containing: (MAC*)Cu(CzCN2) (Cu67); (MAC*)Cu(CzCN) (Cu68); (MAC*)Cu(Cz) (Cu69); (DAC*)Cu-(CzCN2) (Cu70); (DAC*)Cu(CzCN) (Cu71, Scheme 6) and (DAC*)Cu(Cz) (Cu72), in which MAC = cyclic monoamido-aminocarbene, DAC = cyclic diamidocarbene and “*” exhibits that 2,6-diisopropylphenyl is an aryl group bonded to N, have been also reported – Scheme 6.329 By selecting thre carbine (acceptor) and carbazolyl (donor), the emission color was systematically tuned from violet (λmax = 432 nm) to deep red (λmax = 704 nm), as shown in Fig. 34 and Table 9. Highly effective TADF, high PLQY (until 100%) and short decay lifetimes (τ ≈ 1 μs) were obtained (Table 9). As illustrated in Fig. 34, small ΔEST of 500 cm−1 and 105–106 s−1 for kr of these complexes are analogous to those of highly effective emitters containing noble metal, such as Pt and Ir.127 Vapor-deposited OLEDs based on (MAC*)Cu(Cz), Cu69 as the green emissive dopants provided a high EQE (19.4%) and brightness (54000 cd m−2) with modest roll-off at high currents – Table 8 and Fig. 35. In addition, the host-free devices containing Cu69 presented highly efficient OLEDs with EQE of 16.3% – Table 8. This result highlights the capability of these complexes as neat emitters for OLED devices.
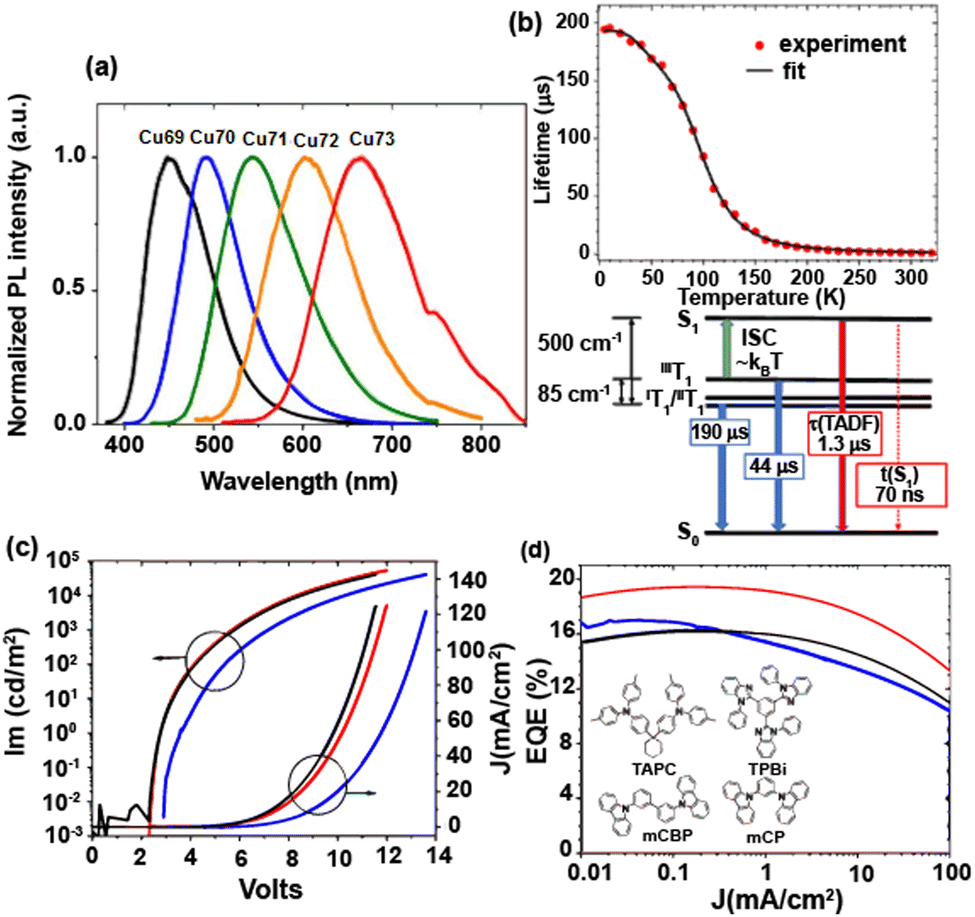 | ||
| Fig. 34 (a) Emission spectra of complexes Cu67–71 in 2-MeTHF at room temperature. Reproduced from ref. 329. (b) Emission lifetime versus temperature of complex Cu69 in the PS film (top). Energy level diagram for complex Cu69 derived from fit to eqn (1) (bottom). Reproduced from ref. 329. (c) EL device characteristics containing complex Cu69 at doping concentrations of 10% (blue), 40% (red), and 100% (black). (B) Current density–voltage–luminance (J–V–L). (d) EQE. Inset: Molecular structures of materials used in the devices. Reproduced from ref. 329. Copyright 2019, American Chemical Society. | ||
 | ||
| Fig. 35 (a) Schematic architecture of the OLEDs structure. (b) Emission from an OLED prototype with [2]+ as a luminophore. (c) Green emission of crystalline [Cu84][BAr4F] under UV (254 nm) light exposure. (d) EQE and current efficiency vs. current density. (e) Current density–luminance–voltage characteristics of the electroluminescence device. (f) Electroluminescence spectra for various driving voltages. Reproduced from ref. 333. Copyright 2019, American Chemical Society. | ||
In particular, TADF in linear two-coordinate coinage metal complexes is greatly influenced by the geometric ligand arrangement. Liu et al. developed a series of CMA CuI TADF emitters (Cu73–77; Scheme 6) and achieved the gradual tuning of configuration from coplanar to orthogonal by introducing various substituents on the indolyl ligands. The inclusion of a 2-CF3-indolyl substituent resulted in a confined twist configuration, characterized by a dihedral angle of approximately 45°, which can be attributed to the combined effects of intramolecular steric hindrance and electronic repulsion. Its blue emission, with a peak at 458 nm, demonstrates a high PLQY value of 0.74 and a short τ value of 1.9 μs. These characteristics indicate a sufficiently fast kr = 3.9 × 105 s−1 and a suppressed knr = 1.4 × 105 s−1 – Table 9. These exceptional luminescent characteristics can be assigned to the optimal overlap of the HOMO and LUMO on CuI d orbitals. This overlap ensures not only a small ΔEST, but also a significant transition oscillator strength, facilitating rapid kr. Both doped and host-free emissive layer designs achieve EQEs above 20% and 10%, respectively – Table 8. These results highlight the potential of the configurationally confined approach in the development of efficient linear CuI TADF emitters in OLEDs.330
In addition, the development of effective red-emitting Cu(I) complexes is a major problem in OLEDs due to weak SOC and a significant excited-state reorganization effect in Cu(I) complexes. Wu et al. successfully developed a red Cu(I) complex known as MAC*-Cu-DPAC (Cu78; Scheme 6), by employing the nonconjugated MAC* ligand and a rigid donor ligand namely, 9,9-diphenyl-9,10-dihydroacridine. The Cu(I) complex demonstrated excellent red emission characteristics with a PLQY of 70% and a sub-μs lifetime. This was made possible by the linear geometry and coplanar conformation of the acceptor and donor ligands, resulting in a high value of 77% for horizontal dipole ratio in the host matrix. The resulting OLEDs incorporating MAC*-Cu-DPAC (Cu78) displayed impressive EQEs, reaching 21.1% at maximum (Tables 8) and 20.1% at 1000 nits. Furthermore, the OLEDs show emission bands peaking around 630 nm and remarkable performances representing the state-of-the-art for red-emitting OLEDs based on coinage metal complexes.331
As a final work, Lam and coworkers focused on addressing the challenges associated with achieving high efficiency and long-term stability in OLEDs with stable two-coordinate CMA-type CuI-TADF emitters (Cu79–83, Scheme 6) with bulky pyrazine-(PzIPr) or pyridine-fused N-heterocyclic carbine (PyIPr*) and Cz ligands. These emitters feature improved bonding interactions of amide-Cu-carbene and exhibit TADF arising from the 1LL′CT(Cz→PzIPr/PyIPr*) excited states, which extends across the blue to red regions of the electromagnetic spectrum, with exceptionally high kr ranging from 1.1 to 2.2 × 106 s−1. They exhibited impressive PLQYs, reaching up to 0.89. Notably, these complexes achieved high value of 2.2 × 106 s−1 for kr at ambient temperature, which was attributed to their narrow ΔEST gap (ranging from 407 to 511 cm−1) and fast kr(S1) (2.6 to 3.1 × 107 s−1). Moreover, the OLEDs were fabricated using these CuI emitters through vapor deposition techniques and demonstrated outstanding EQE, reaching up to 23.6%, and high luminance levels of up to 222200 cd m−2 – Table 8. Notably, these devices exhibited practical operational stability, with LT90 values of 1300 h at 1000 cd m−2 under laboratory conditions. These findings highlight the potential of CuI-TADF emitters as replacements for precious metal phosphors in the OLED industry.332
Olaru and co-workers333 presented highly photo- and electro luminescent clustered structure [Cu4(PCP)3] [A] (Cu84; Scheme 6), (A = BF4− or BArF4− and PCP = 2,6-(PPh2)2C6H3 and ArF = 3,5-(CF3)2C6H3), comprising a Cu4-core along with bulky tridentate carbanionic diphosphine, ligands. This rare cationic organocopper cluster geometry suppressed the non-radiative decays, resulting in a high PLQY in both solution and solid state with narrow emission bands (FWHM of 60 nm and 58 nm, respectively). According to PL decay kinetics and DFT calculations, the complex emission mechanism involves contributions of two phenomena, phosphorescence and TADF over a wide temperature range. The analysis suggests that at 300 K 94% of the total photoluminescence can be attributed to single harvesting, indicating a significant contribution from TADF, whereas the partial TADF involvement decreases to 0.5% at low temperature (77 K), at which phosphorescence prevails. The use of cationic tetranuclear organo-copper cluster [Cu4(PCP)3]+ in OLEDs resulted in bright green emission (8900 cd m−2 at 15 V) with 65 nm FWHM, a relatively high EQEmax (11.2% at 42 cd m−2) and low turn-on voltage (2.4 V at 1 cd m−2) – Table 8. Fig. 35 shows the optoelectronic properties of OLEDs made of the aforementioned complexes. By using a bipolar host material named as CBP, the OLED featured high brightness (19000 cdm−2) with a further increase of the turn-on voltages (4.2 V) – Table 8. This was ascribed to the deeper CBP HOMO level in comparison to TAPC, di-[4-(N,N-di-p-tolyl-amino)-phenyl]cyclohexane, (6.0 eV vs. 5.5 eV) where the hole injection is instead deteriorated.
Recently, Guo et al.334 developed the first efficient TADF-OLEDs based on ultra-soluble Cu(I) halide complexes comprising non-symmetrically substituted bidentate phosphine and PPh3 ligands, [CuX(PPh3)(dpts)], in which dpts = 2-trimethylsilyl-3,4-bis(diphenylphosphine)thiophene, X = I for (Cu85), Br for (Cu86), Cl for (Cu87) and [CuX(PPh3)(dppt)], in which dppt = 3,4-bis(diphenylphosphino)thiophene, X = I for (Cu88), Br for (Cu89), Cl for (Cu90) – Scheme 6. Since the insolubility of neutral TADF materials often impedes the fabrication of solution processed devices via spin coating techniques, the insertion of flexible and bulky trimethylsilyl group into diphosphine ligand served to dramatically increases the solubility, as well as the PLQY (owing to the limitation of nonradiative processes induced by Jahn–Teller distortion of emissive excited states) of Cu(I) halide complexes. Consequently, ultra-soluble TADF materials were proposed for the manufacture of solution-processed devices as well as emitting material inks that are customizable for different print heads through the use of solvent blends with specifically engineered viscosity and surface tension characteristics. All the new four-coordinate mononuclear Cu(I) halide complexes demonstrated deep blue-green to yellowish green emissions in the range of 485–535 nm in powder state at ambient temperature – Table 9; Cu85–87, λex = 360 and for Cu88–90λex = 373 nm. Fig. 36b shows the emission spectra and x/y CIE coordinates for Cu85–90 in powder state at 297 K. They provided high PLQYs ranging from 29 to 55% compared to Cu88–90 (PLQY = 3–18%; Table 9). The emission of the complexes predominantly arises from MLCT, XLCT and intra-ligand transitions. The estimated ΔEST = 464–1035 cm−1 were supported by calculations (492–940 cm−1), revealing that they can exhibit efficient TADF.378 Solution-processed undoped and doped OLEDs containing Cu86 displayed yellow green emission (564 nm) (Fig. 36). Devices doped with CBP and using mCP as host materials exhibited inferior performance compared to the non-doped device, probably because of the inadequate energy level compatibility between CBP, mCP and Cu86. Meanwhile, the undoped device indicated maximum brightness and EQE of 234 cd m−2 and 7.74% respectively (Table 8).
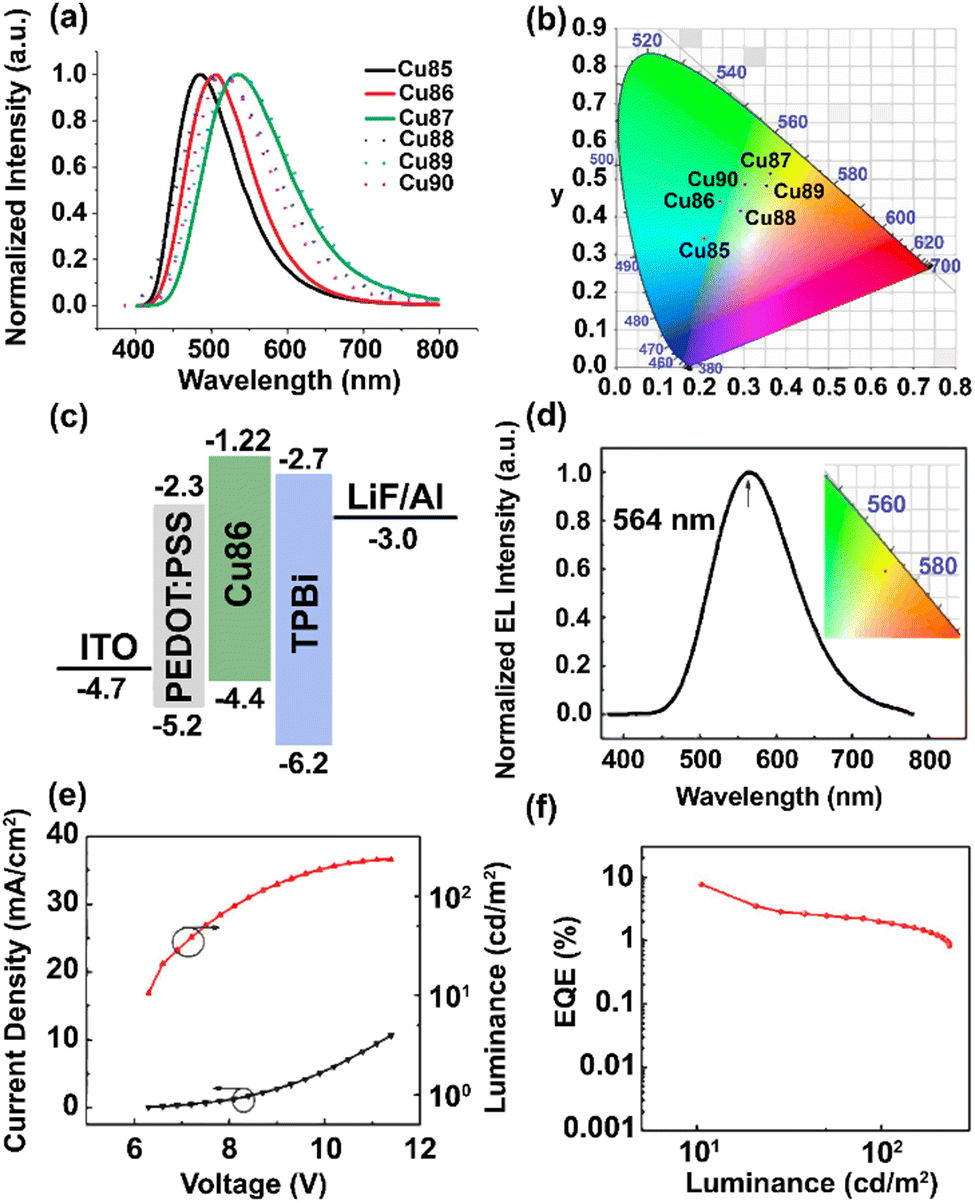 | ||
| Fig. 36 (a) Normalized emission spectra and (b) CIE graph of complexes Cu85–90 in powder state at 297 K. Reproduced from ref. 334. (c) Energy-level diagram of the device based on the complex Cu86; (d) electroluminescence spectra, and the inset is the CIE of electroluminescence spectra; (e) current density–voltage–luminance (J–V–L) characteristics; (f) EQE–luminance characteristics. Reproduced from ref. 334. Copyright 2020, Elsevier. | ||
As explained in Section 2, TADF Cu(I) complexes have been prepared utilizing basic D–A type N∧N ligands and auxiliary phosphine ligands. The coordination of the metal ion induces a reduction in the energy gap for the LC excited states, leading to a switch from fluorescence in the free N∧N ligands to TADF in the complexes (Cu91–94; Scheme 6). Remarkably high PLQYs reaching up to 0.71 at room temperature in doped films (Table 9) have been achieved. Through DFT calculations, the complexes have been found to possess intraligand charge-transfer excited states that primarily localize on the N∧N ligands. The energy level of the excited state of intra-ligand CT is significantly influenced by the N∧N ligand donating strength, thereby dictating the energetic ordering of the 3CT and 3LE states. Consequently, the emission lifetimes of the Cu(I) complexes exhibit substantial variation, exceeding four-fold differences. Furthermore, solution-processed OLEDs fabricated from one of these novel emitters exhibit EQEmax as high as 7.96% (Table 8). It has been observed that a shorter emission lifetime is advantageous for attaining higher EQEs. The promising outcomes of this study, integrated with logical design approaches may serve as a valuable guidance for the progress of Cu-complexes and OLEDs.335
Lately, luminescent complexes of CMA containing group 11 metals (Cu, Ag, Au) have gained significant interest for their remarkable emission efficiency and high radiative decay rates (kr). Chang et al. investigate a set of eight Cu(I) complexes (Cu95–102; Scheme 6) with previously unexplored 1,3-thiazoline carbenes, analyzing their light emission properties for OLEDs. They demonstrated the high-performance EL excimers, whose prevalence of EL can be adjusted from either the monomer (bluish green) or the excimer (orange-red) by altering the emitter's steric hindrance or concentration within vacuum-deposited emissive layers. The radiative rates of the materials exhibit high values, ranging from (2.8–7.2) × 105 s−1 – Table 9. By optimizing the emitter structure and mass fraction, devices based on emitter Cu101 achieved both monomer and excimer electroluminescence, enabling the development of a single-emitter WOLED with high EQE of 16.5% and Lmax exceeding 40000 cd m−2 (Table 8). The broad emission bands of the monomer and excimer contributed to a CRI > 80. These results emphasize the potential of cost-effective copper complexes for lighting devices, offering reduced expenses and simplified device architecture.336
The direct co-deposition of copper halide and organic materials is considered a promising and straightforward approach for manufacturing OLEDs, compared to using pre-synthesized complexes Zhang et al. co-deposited four host aza-9,9-Spirobifluorenes (aza-SBFs) ligands with varying positions of the nitrogen atom in the azafluorene moiety along with Cu(I) iodide (CuI) to make emissive complexes, called α-aza-SBF:CuI (Cu103), β-aza-SBF: CuI (Cu104), γ-aza-SBF:CuI (Cu105) and δ-aza-SBF:CuI (Cu106) – Scheme 6. Interestingly, these co-deposited Cu(I) complexes exhibited both phosphorescent and TADF. Notably, by modifying the situation of the nitrogen atom, the TADF was successfully improved, leading to a high PLQY of 92.2% (Cu106; Table 9). As a result, green emission at 540 nm along with values of 41.9 cd A−1 for current efficiency, value of 32.9 lm W−1 for power efficiency and value of 16.8% for EQE of OLED device fabricated with Cu106 compound as emitter were achieved – Table 8. This study highlight the future potential of utilizing the co-deposition strategy for manufacturing cost-effective and highly efficient OLED devices.337
Cheng and co-workers investigated the luminescent Cu(I) dimer complex Cu2Cl2(P∩N)2 (Cu107; Scheme 6) (P∩N = diphenylphosphanyl-6-methyl-pyridine) as a potential emitter material for OLEDs. This complex exhibits TADF and phosphorescence even at room temperature. By using a solution-processable approach, we fabricated an OLED with the configuration of ITO/PEDOT:PSS/PYD2:Cu107/DPEPO (10 nm)/TPBi (40 nm)/LiF (1.2 nm)/Al (100 nm) that emits warm white light (CIE coordinates (0.38, 0.49)) with a moderate EQE = 3.80 – Table 8. The EQE value obtained for a material doped with Cu107 is lower than predicted, despite the considerable PLQY of 92% for the powder state. The lower EQE value is attributed to the less rigid host materia PYD2l, which enhances unique geometric reorganization upon excitation compared to the powder medium. Consequently, the host material with a doping concentration of 8 wt% shows a PLQY of 27%. When the EQE is normalized to PLQY = 100%, the normalized EQE is found to be 14%. This suggests that designing the molecular structure of the emitter and the host environment more distinctly rigidly can lead to high-performance Cu(I) emitter devices.338
As a final remark for designing TADF OLEDs with Cu-complexes, Wang and co-workers proposed to use Cu(II) acetate as a spin sensitizer to simplify rISC and enhance electroluminescence in TADF-exciplex OLEDs. Notable alterations in the intensity and lifetime of photoluminescence were observed due to the coordination interaction of Cu(II) acetate with exciplex molecules involving intermolecular CT properties. Additionally, the addition of copper acetate as a spin sensitizer was evidenced by magneto-photoluminescence data to promote spin transition in the rISC process, resulting in an approximately 80% enhancement of electroluminescence in spin-sensitized TADF-based OLEDs. These findings demonstrate that utilizing a spin sensitizer such as copper acetate can dominate the spin-forbidden limitation and minimizing the ΔEST in the engineering of TADF materials to promising improve performance of OLED device. This discovery opens up possibilities for developing efficient OLEDs and paves the way to advance the next-generation organic electronics.379
In stark contrast to Cu(I) complexes, Ag(I) complexes have been relatively unexplored yet. Here, Li et al. introduce several new [Ag(N^N)(P^P)]-PF6 complexes (Ag10 and Ag17–19; Scheme 6) that indicate highly effective TADF utilizing usable neutral diamine ligands and readily accessible auxiliary diphosphine chelates. The coordination of the metal ion leads to a reduction in ΔEST, facilitating the effective TADF from the excited state centered on the ligand. PLQY values of up to 62% in doped films were achieved – Table 9. Their high PLQYs in combination with significant delayed fluorescence ratio, allowed the development of solution-processed OLEDs with a remarkable EQEmax of 8.76% – Table 8.273
3.2. LECs
LECs are the simplest and cheapest SSL concept as they combine air-stable electrodes with solution-based fabrications techniques. Over the last years, ionic Cu(I) complexes gained much attention as electroluminescent emitters due to their well-adjustable photophysical properties, such as TADF, emission color, rich molecular design. So far, the best Cu(I) complex-based LECs achieve brightness of up to 452 d m−2, efficiencies up to 4.5 cd A−1 and EQEs of 1.85% for the mid-energy emitting region,235 while blue-emitting Cu(I) complex-based LECs have been close performing with efficiencies of 3.6 cd A and EQE of 1.2%.228 Finally, red Cu(I) complex-based LECs have also led to interesting performances with irradiances of 130 μW cm−2 and day stabilities.380 Recently, the LEC community started to consider Ag(I) complex-based LECs. Although clear design rules for TADF in Ag-complexes are missing and TADF is rarely observed in Ag-complexes, considerably device performance were achieved – especially in the low-energy region (λexc = 645 nm, Irrmax = 36 μW cm−2, t1/2 = 5 h).381,382 Finally, very recently, He et al. introduced the first TADF-LEC with Ir-complexes as dopant as reported in OLEDs. Accordingly, the following will summarize the most relevant complexes applied to LECs, highlighting their device performance (Table 10) as well as their photoluminescence features as a complement of Section 2 (Table 11).Along the evolution of LECs based on [Cu(N^N)(P^P)]+ family, Constable and Housecroft systematically studied the effect of the substituent in N^N ligands on both the photo- and electro-luminescence behaviors. In detail, the family [Cu(P^P)(N^N)][PF6] with P^P = POP or xantphos, N^N = 2,2′-bipyridinebpy (bpy) ligands with methyl and CF3 moieties as substitute in various positions of the bpy ligand, (6,6′-(CF3)2bpy, 6-CF3bpy, 5,5′-(CF3)2bpy, 4,4′-(CF3)2bpy, 6,6′-Me2-4,4′-(CF3)2bpy) (Cu6, Cu108–116; Scheme 7).339 While the TADF in powder was not affected with a ΔEST of 968–1613 cm−1 (TD-DFT calculations), the highest PLQY (50.3%) in powder sample (Table 11) were achieved by [Cu(xantphos)(4,4′-(CF3)2bpy)][PF6]. Among them, only [Cu(POP)(6-CF3bpy)][PF6], [Cu(xantphos)(6-CF3bpy)][PF6] and [Cu(xantphos)(6,6′-Me2-4,4′ (CF3)2bpy)][PF6] were used in LECs devices due to their high PLQY values because of dependency of the efficacy as well as the luminance of the LECs with the PLQY value in thin-film and exhibit yellow electroluminescence in wavelength rang 589–595 nm with maximum EQEs of 0.4%–0.6% were obtained – Table 10. However, CF3-free bpy-based [Cu(P^P)(N^N)][PF6] complexes have better performance in comparison with their incorporating CF3 counterparts due to their lower PLQY in films. By adding CF3 units, the E1/2ox value for the Cu+/Cu2+ process in [Cu(POP)(bpy)]+ and [Cu(xantphos)(bpy)]+ complexes increases to higher potentials (+0.85 to +0.96 V). These identified trends in E1/2ox align with the findings obtained from DFT calculations. The properties of the N^N ligand remarkably modifies the HOMO–LUMO separation, and the largest redshift in the MLCT band is observed in [Cu(P^P)(5,5′-(CF3)2bpy)]+ complex, which is also supported by DFT calculations. The emission energies of [Cu(P^P)(bpy)]+ complexes with groups at the 6,6′-position of bpy do not align with the predicted values based on electronic requirements, such as MO analysis or electrochemical and optical absorption gaps. This is due to the introduction substituents inhibiting tetrahedron flattening distortions related to T1 relaxation and prevents its stabilization. As a result, the T1 state remains at higher energies in complexes with substituents at 6,6′-positions.
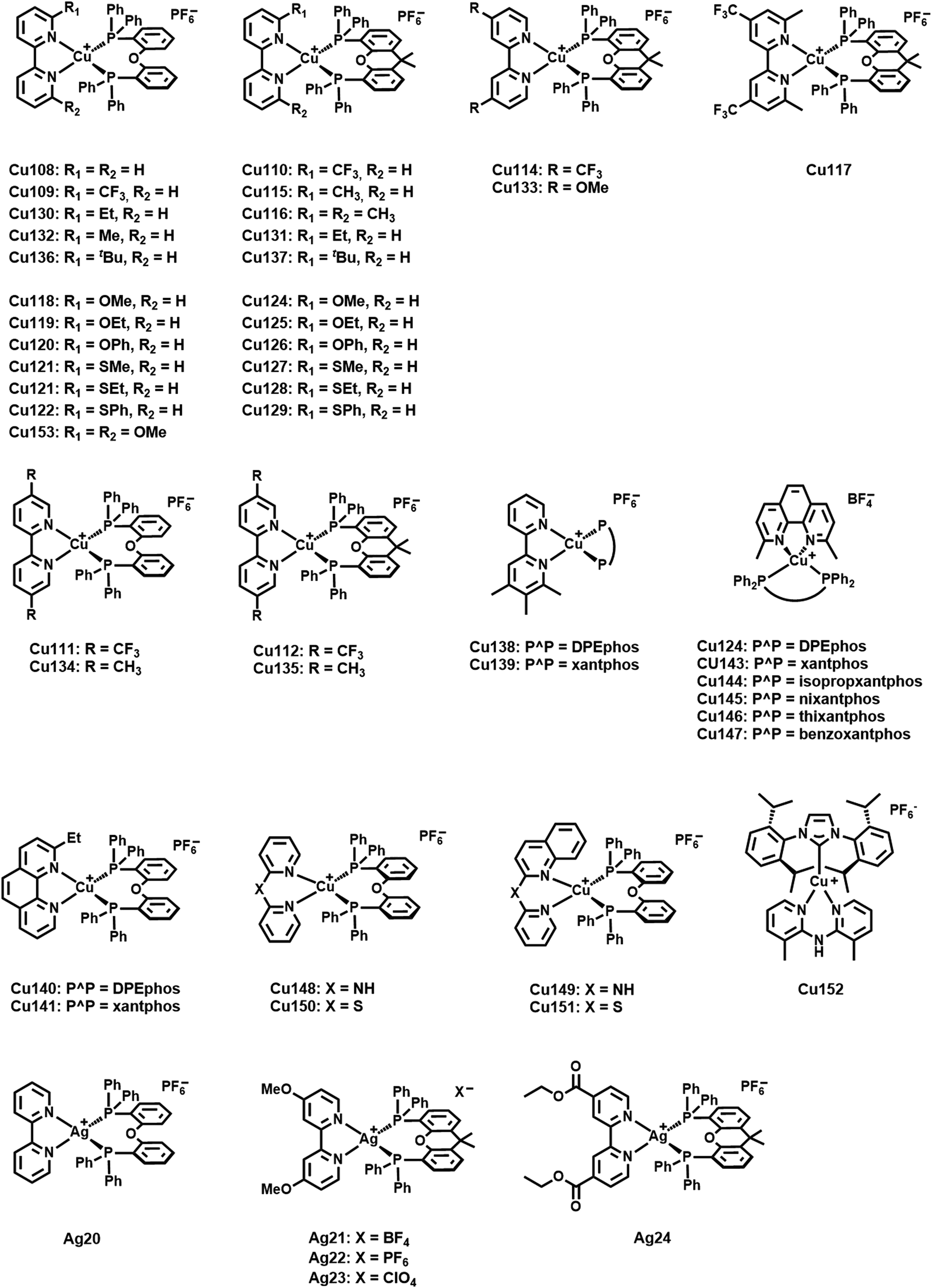 | ||
| Scheme 7 Molecular structures of Cu108–152 and Ag20–24. | ||
In this line, the same authors reported another large series of twelve heteroleptic [Cu(P^P)(N^N)][PF6] complexes – Scheme 7, (Cu118) [Cu(POP)(MeObpy)][PF6], (Cu119) [Cu(POP)(EtObpy)][PF6], (Cu120) [Cu(POP)(PhObpy)][PF6], (Cu121) [Cu(POP)(MeSbpy)][PF6], (Cu122) [Cu(POP)(EtSbpy)][PF6], (Cu123) [Cu(POP)(Phsbpy)][PF6] as well as (Cu124) [Cu(xantphos)(MeObpy)][PF6], (Cu125) [Cu(xantphos)(EtObpy)][PF6], (Cu126) [Cu(xantphos)(PhObpy)][PF6], (Cu127) [Cu(xantphos)(MeSbpy)][PF6], (Cu128) [Cu(xantphos)(EtSbpy)][PF6], (Cu129) [Cu(xantphos)(PhSbpy)][PF6], in which P^P = POP or xantphos, and N^N =(MeObpy)= 6-methoxy-2,2′-bipyridine, (EtObpy) =6-ethoxy-2,2′-bipyridine, (PhObpy) = 6-phenyloxy-2,2′-bipyridine, (MeSbpy) = 6-methylthio-2,2′- bipyridine, (EtSbpy) = 6-ethylthio-2,2′-bipyridine and (PhSbpy) = 6-phenylthio-2,2′-bipyridine, through the addition of different moieties into the 6-substituted site of bpy ligand was performed to investigate the potential of these compounds as emissive dopants for applications in LECs.340 The distorted tetrahedral geometry for Cu(I) and the chelating states for each N^N and P^P ligand in all compounds are established by single crystal structures. In the complexes containing xantphos, the asymmetric bpy ligand with a substituent group at position 6 is located on the “bowl” of xanthene. In addition, these materials are yellow emitters with a PLQY of 38% and an τ value of 10.2 ms in powder state – Table 11. Changing from powder to frozen Me-THF increases the τ value that could suggest the presence of TADF, but further studies were not provided. All of the twelve compounds have also been used in LECs and complexes with alkoxy- or phenyloxy- functionalized ligands showed stable and bright LEC devices that being extremely important for the later progression of electroluminescent devices containing Cu(I) metal ion center. The time evolution of efficacy and luminance for devices using Cu(I) complexes with RObpy and either (a) xantphos or (b) POP ligands are presented in Fig. 37. Furthermore, LECs based on [Cu(P^P)(EtObpy)][PF6] demonstrate boosted values of Lummax, t1/2, ton and efficacy when compared to the state-of-the-art LECs with Cu(I) which include [Cu(P^P)(Etbpy)][PF6] (Cu130 and Cu131; Scheme 7).341
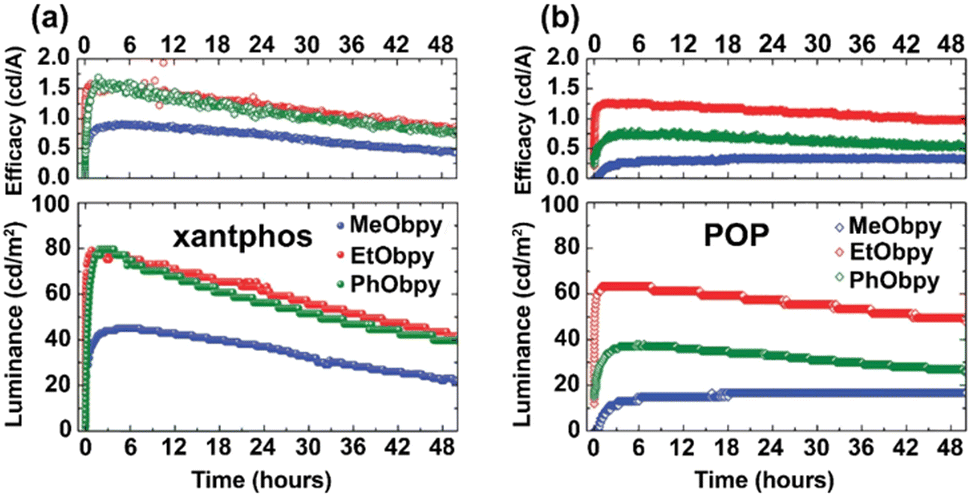 | ||
| Fig. 37 Efficacy (top) and luminance (bottom) versus time for LECs employing Cu(I) complexes with RObpy and either (a) xantphos or (b) POP ligands. LECs were driven at an average current density of 50 A m2. Reproduced from ref. 340. Copyright 2018, Royal Society of Chemistry. | ||
The above examples mainly emphasized the preparation of Cu(I) complexes by attaching one moiety at the ortho and para positions of the bipyridine ligand. However, a few reports have shown the benefits of (i) the substitution at the para position and (ii) a fully substitution at the ortho position. On one hand, Costa et al. conducted a thorough investigation on the impact of electron donor and electron acceptor moieties at the 4-positions of bpy in [Cu(bpy)(xantphos)] complexes.342 They found that the LEC efficiency improved with increasing negativity of the σ-Hammett parameter (σp), which characterizes the σ-donation capability of the substituents The best performing complex, namely the TADF [Cu(4,4′-dimethoxy-2,2′-bipyridine)(POP)]BF4 (Cu133; Scheme 7), achieved 54 cd m−2 at 10 mA of pulsed driving current – Table 10. Very recently, Keller et al. proved that also the para substitution with electron-withdrawing fluorinated groups can lead to improved figures-of-merit.339 As an example, [Cu(6,6′-Me2-4,4′-(CF3)2bpy)(xantphos)]PF6 (Cu117; Scheme 7) exhibited a maximum luminance of 131 cd m−2 as well as a lifetime stability of 2 h – Table 10. The τ value of 12 μs at room temperature and of 42 μs at 77 K along with a S1–T1 energy separation of 1613 cm−1 suggest a TADF emission mechanism – Table 11. On the other hand, the substitution with the methyl moieties at the ortho position relative to the N bipyridine atoms brought to a substantial improvement of the performances when compared to the non-substituted bipyridine. LECs based on [Cu(6,6′-dimethyl-2,2′-bpy)(POP)]PF6 (Cu132; Scheme 7) and the ionic liquid 1-ethyl-3-methylimidazolium hexafluorophosphate [EMIM][PF6] in molar ratio 4:1 achieved 53 cd m−2 at a current density of 10 A m−2, thus achieving 5.2 cd A−1 for copper-based LECs – Table 10.245
The impact of the P^P ligand was also disclosed by several groups. Constable and Housecroft group synthesized and characterized eight heteroleptic [Cu(POP)(N^N)][PF6] and [Cu(xantphos)(N^N)][PF6] complexes (Cu134–141; Scheme 7), [N^N is 5,5′-dimethyl-2,2′-bipyridine (5,5′-Me2bpy),4,5,6-trimethyl-2,2′-bipyridine (4,5,6-Me3bpy), 6-(tert-butyl)-2,2′-bipyridine (6-tBubpy) and 2-ethyl-1,10-phenanthroline (2-Etphen) and P^P is either bis(2-(diphenylphosphino)phenyl)ether (POP, PIN [oxydi(2,1-phenylene)]bis(diphenylphosphane)) or 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (xantphos, PIN (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane))]. This study shows that electronic and steric impacts can result in a remarkable enhancement of PLQY values and the Lmax of the device. Methyl or ethyl units in the 6-position of the bpy ligand improved the PLQY and device performance, while a tert-butyl substitution has a negative impact, leading to lower PLQY and shorter τ values of the complexes. The study found that incorporating 5,5′-Me2bpy leads to Inefficient emitters than 6-Mebpy, indicating that stabilizing the tetrahedral complex geometry with a group in the 6-site of the bpy is more beneficial than the electron-donating impact of two methyl moieties. Replacing the ligand with the analogous phenanthroline, 2-Etphen resulted in high values of luminance but only the complex with xantphos showed longer lifetimes for device. The study also showed that the device with the similar POP complex was significantly less stable with 2-Etphen compared to 6-Etbpy. These yellow to green emitters exhibit tunable photophysical properties, such as emission maxima in the range of 518–602 nm and PLQYs about 1.1–58.8% – Table 11. They also displayed TADF and have shown promising performance in LECs with value of 462 cd m−2 for Lmax and a value of 98 h for half-lifetimes of device – Table 10. The research provides understanding about the effects of alkyl substitution on the photophysical and device characteristics of the complexes, highlighting the importance of a systematic approach to improve the design of the complexes for enhanced device brightness and lifetime.343 Zysman-Colman et al. investigated on the photophysics, electrochemistry, and electroluminescence of seven cationic heteroleptic Cu(I) complexes, namely [Cu(P^P)(dmphen)]BF4 (Cu142–147; Scheme 7) (dmphen = 2,9-dimethyl-1,10-phenanthroline and P^P represents a diphosphine chelate in homoxantphos, isopropxantphos, nixantphos, thixantphos, and benzoxantphos). The main goal was to investigate the influence of the diphosphine ligand's bite angle on the photophysical characteristics of the complexes. Among these complexes, several displayed relatively high PLQYs both in solid-state (35%) and in solution (up to 98%, Table 11). There is a correlation between the powder PLQYs and the % Vbur of the P^P ligand. Among them, Cu3 exhibited a longer device lifetime (L50 of ca. 17 h) compared to the reference Cu(I)-based LEC [Cu(dnbp)(DPEPhos)]+ (1.2 h). A high value for % Vbur results in PLQY values of 35% – Table 10, reaching EQEmax of 4.4% for the devices. Finally, Cu3 exhibited superior ECL performance in comparison to the other complexes. This improvement can be assigned to the high stability of both the oxidized and reduced species, as evidenced by its voltammetric profile. Particularly, complexes with more reversible electrochemistry displayed higher annihilation ECL and exhibited improved LECs.235
To increase the efficiency and reach other colors in TADF-based Cu-complexes based LECs, several authors explored several N^N ligands. As far as we known, there are three mainly families of copper (I) complexes that have been tested in LECs devices: (i) homoleptic and heteroleptic Cu-iTMCs bearing diimine (N^N), diphosphine (P^P) and phosphine-amide (P^N) ligands, (ii) dinuclear Cu-iTMCs and (iii) NHC Cu-iTMCs in which NHC is N-heterocyclic carbene. For instance, the former have shown great performances in green- and yellow devices,172,232 while prior to 2022 blue LECs were only achieved with NHC-based Cu-iTMCs showing stabilities of <5 min at 0.17 cd A−1 and 20 cd m−2.345
This gap of knowledge was particular limiting the development of white LECs. In fact, more efforts were done toward the development of red-emitting Cu(I) complexes that they were mixed with high-energy emitting SMs (e.g., using CBP). In the first report of Cu(I) white LECs, a combination of CBP and [Cu(dcbq)(xantphos)]+ in which dcbq is 4,4′-diethylester-2,2′-biquinoline, only moderate performance were achieved (i.e., 4 cd m−2 at 25 mA).380 In 2022, Costa and Galliard et al. improved the red-emitting component trough the used of pyrazine and pyrimidyl ancillary N^N ligands.171 The best devices reached luminances of 12 cd m−2 associated to an excellent white color quality (i.e., x/y CIE coordinates of 0.31/0.32 and CRI of 90) that is stable over their lifetime.
Despite these encouraging reports, the lack of stable and efficient blue Cu(I) complexes was a huge bottleneck for the field of Cu(I) complex-based white LECs. However, in 2022, Costa et al.228 introduced a novel approach using a large dataset and multivariate analysis to predict the performance of copper complexes ([Cu(N^N)(P^P)]+) in thin-film lighting devices. This method breaks away from traditional trial-and-error approaches that focus on single variables at a time. The key to this approach lies in the correlation between simulated and experimental data based on a multivariate model using data from over 90 Cu-iTMCs applied in LECs, including their X-ray structures, electronic properties, and performance in thin-film devices (emission wavelength, efficiency, etc.). This research achieved two breakthroughs: (i) highly efficient blue LECs using were fabricated successfully, and (ii) the first single-layered white LEC using only Cu-iTMCs was achieved. In particular, [Cu(N^N)(P^P)]+ in which N^N is 2-(4-(tert-butyl)phenyl)-6-(3,5-dimethyl-1H-pyrazol-1-yl)pyridine and P^P is 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (Cu8; Scheme 3) based LECs achieved a maximum efficiency of 3.6 cd A−1, which is considered top-notch for copper-based LECs regardless of color and it is better than state-of-art blue Ir-iTMCs LECs.187
Despite the first fully Cu(I) complex-based white LECs was achieved, the poor performance of the low-energy emitting Cu-iTMCs was still of concern. Thus, Galliard et al.344 focused on improving the performance of red-emitting complexes exploring conventional strategies: (i) extending the π-systems and (ii) introducing S-bridges between heteroaromatic rings. This resulted in the synthesis of two new heteroleptic Cu(I) complexes: 2-(pyridin-2-yl-l2-azanyl)quinoline (CuN2) (Cu149) and 2-(naphthalen-2-ylthio)-quinoline (CuS2) (Cu151) as N∧N, with bis[(2-diphenylphosphino)phenyl] ether as the P∧P ancillary ligand – Scheme 7. These complexes exhibited enhanced PLQY and TADF processes in comparison with their reference complexes: di(pyridin-2-yl)-l2-azane (CuN1) (Cu148) and di(pyridin-2-yl)sulfane (CuS1) (Cu150) shown in Scheme 7. Notably, Cu151 showed the highest PLQY (38% compared to 17%, 14%, and 1% for Cu148, Cu149, and Cu150, respectively, Table 11). However, only Cu149-LECs demonstrated superior performance, with a current density of 0.35 cd A−1 at a luminance of 117 cd m−2 – Table 10, whereas Cu148-LECs performed poorly (0.02 cd A−1 at 6 cd m−2; Table 10), and Cu151-LECs had low performance (0.04 cd A−1 at 10 cd m−2; Table 10). This indicated that traditional chemical engineering strategies did not effectively enhance device efficiency. Capitalizing on the acquired knowledge trough the previously reported multivariate analysis, it has been proposed a non-obvious design consideration that takes into account the ligand polarization features (specifically, clogP and tPSA) governing ε and σ in thin-films and, consequently, the operation mechanism of LECs. This novel design insight was uncovered through a multivariate statistical analysis model that connects electronic parameters and X-ray structural of Cu(I) complexes with their photo-/electro-luminescent behaviors in thin-films, in the context of their applications in lighting and electrochemical impedance spectroscopy (EIS) studies. These analyses revealed a new design rule: the polarizability of the ancillary ligand plays a key role in improving the efficiency of LECs. Thus, it is imperative to consider the polarizability of the ligands in addition to the well-known structural parameters.344 Overall, this methodology can also be applied to understand the limitations in the complex designs beyond the discovery of new examples for a desired color.
In 2016, Costa, Gaillard et al. published a study on a series of mononuclear Cu(I) complexes containing NHC and nitrogen bidentate ligands. These complexes displayed highly favorable luminescence characteristics, demonstrating blue or blue-green emission.249,250 All the complexes showed τ values ranging from 6 to 14 μs at ambient temperature and from 32 to 87 μs at 77 K. According to Thompson et al., a 2- to 3-fold increase in the τ when changing ambient temperature to 77 K is a good indication of a TADF emission process rather than a phosphorescence one.248 The best performing LEC was achieved with [Cu(IPr)(3-Medpa)]PF6, (Cu152; Scheme 7) with a luminance of 20 cd m−2, efficacy of 0.17 cd A−1, and a total emitted energy (Etot) of 4 mJ. This parameter refers to the radiant flux of the device over time, starting from t = 0 when bias was applied until the time it reaches a 1/5 of the maximum irradiance value. Thus, this stability parameters enables a reliable comparison of devices, regardless of their varying luminance levels, as recommended by Bard and colleagues.387 This value was recently improved in a following contribution by decoupling hole and electron transport in a multi-layered architecture, reaching 160 cd m−2, an efficacy of 1.2 cd A−1 and a total emitted energy of 32.7 mJ – Table 10.345
In 2018, Costa and coworkers performed an in-depth study on (i) the molecular design of Cu(I) complexes and their photophysical and electroluminescent properties and (ii) introduced a novel device architecture.388 First, the introduction of two methoxy groups at the 6,6′-position of the bpy ligand led to a two-fold increase of the PLQYs from 6% ([Cu(bpy)(POP)]PF6) (Cu108) to 14% ([Cu(6,6′-(MeO)2-bpy)(POP)]PF6) (Cu153). This molecular modification was also noted in the performance of the pristine devices as the stabilities and efficiencies improved drastically from 0.6 to 1.1 h and 0.1 cd A−1 to 0.3 cd A−1, respectively. Second, the authors addressed the frequently observed issue of Cu(I) complex-based LECs namely the formation of irreversible oxidized species owing to the irreversible oxidation of the employed Cu(I) complexes. In detail, they introduced a multilayered device architecture incorporating 4,4′-bis(9-carbazolyl)-1,1′-biphenyl, 4,4-N,N′-dicarbazole-1,1′-biphenyl as hole injection layer to decouple the hole injection, transport and exciton formation – Fig. 38. Finally, this resulted in 10-fold increased stabilities for both complexes without affecting brightness and efficiency. The success of this multilayered device architecture was further confirmed by other works.171,380
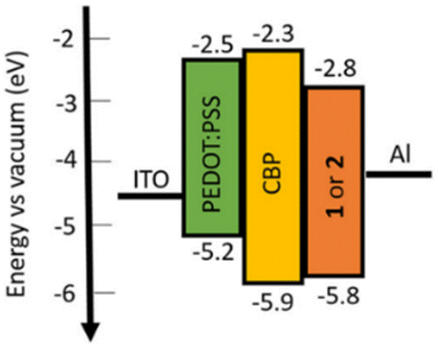 | ||
| Fig. 38 Energy diagram of the multilayer device architecture (1 = Cu108, 2 = Cu153, HOMO and LUMO levels were derived from cyclic voltammetry). Reproduced with permissions from ref. 388. | ||
As an alternative strategy to the ones mentioned above (e.g. molecular modifications, device architecture) in order to overcome the drawbacks of Cu(I) complexes and Cu(I)-LECs, e.g. (i) Jahn–Teller distortion upon excitation, (ii) unforeseen changes from powder to thin-film and (iii) irreversible oxidation behavior of Cu(I) complexes and the related formation of oxidized species in the devices, (iv) lack of efficient blue-emitting Cu(I) complex-based LECs, the research community started to work on Ag(I) as the most natural alternative. This progression was driven by the fact that Ag(I) complexes are known to exhibit (i) high PLQYs, (ii) a higher ligand field splitting energy of Ag compared to Cu(I) facilitating the design of efficient blue-emitting LECs and (iii) high oxidation potentials.267
In 2015, Moudam et al. reported the first ever Ag(I)-based LEC.389 In short, the heteroleptic, tetrahedral Ag(I) complex, [Ag(bpy)(POP)]BF4 (Ag20), was applied in a LEC device (ITO/PEDOT:PSS/Ag20:[BMIM]PF6/LiF:Ag) showing a very broad emission band peaking around 550 nm. A maximum efficacy of nearly 0.45 cd A−1 (5.5 V) with a luminance efficiency of 54 cd m−2 (9.4 V) was obtained without any notes about the device and complex stability itself. Inspired by this, Costa and coworkers performed an in-depth study of a similar Ag(I) complex, namely [Ag(4,4′-dimethoxy-2,2′-bipyridine)(xantphos)]X (X = BF4 (Ag21), PF6 (Ag22), and ClO4 (Ag23)), to elucidate their electrochemical and electroluminescent behavior.382 The pristine device of all three complexes (ITO/PEDOT:PSS/Ag(I)/Al) was found to show an unusual LEC profile with a continuously decreasing voltage profile associated with a luminance of 40 cd m−2, efficacy of 0.2 cd A−1, and very poor stability of 30 s – Fig. 39. A joint study of cyclic voltammetry in solution and EIS and X-ray diffraction (XRD) measurements of the fresh and the post-mortem devices revealed the presence of Ag(0) in solution and in post-mortem devices – Fig. 39. In detail, the authors claimed that upon reduction of the studied Ag(I) complexes, an irreversible decomposition to Ag nanoclusters/nanoparticles occurs in solution, but also in thin-films. Finally, the device stability was improved by using a double-layered device architecture to decouple electron injection and exciton formation giving rise to a 4 order of magnitude improved stability (>80 h) and a brightness of 35 cd m−2, but the broad whitish electroluminescent spectrum (x/y CIE color coordinates of 0.40/0.44) was assigned to an exciplex-like emission. However, both Moudam and Costa did not provide further experimental studies to elucidate the emission mechanism of the employed Ag(I) complexes.
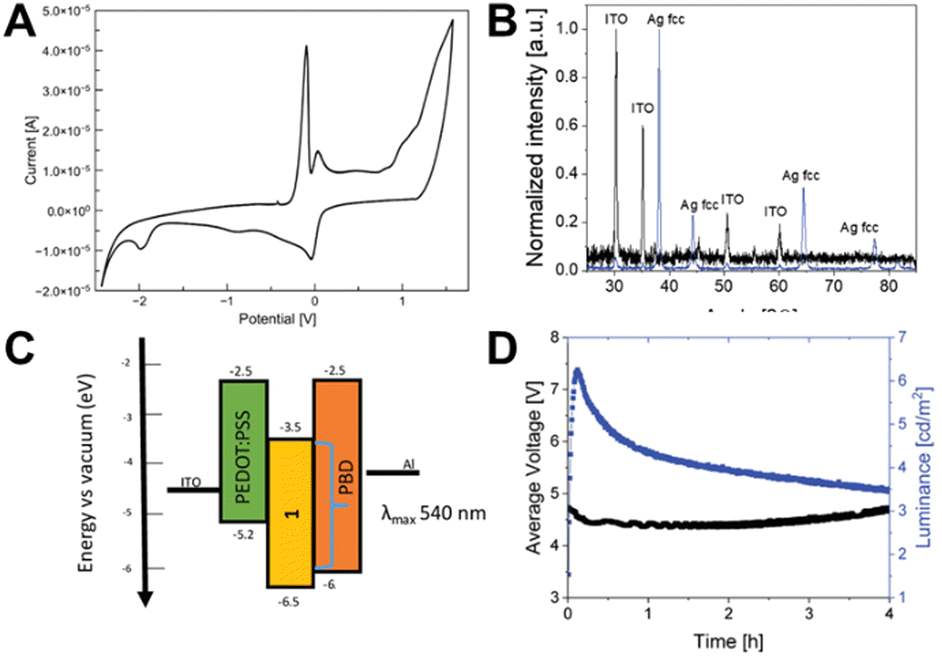 | ||
| Fig. 39 (A) Second scan of the cyclic voltammetry measurement of Ag21 in acetonitrile versus Fc/Fc+. (B) XRD measurements of fresh (blue) and used (black) Ag21 pristine devices. The peaks related to ITO and to metallic silver (fcc crystalline cell) are marked above. (C) Schemes of a multilayered device architecture using PBD as electron injection layer. (D) Average voltage and luminance over time (bottom) of Ag21-LEC devices driven at pulsed current of 15 mA. Reproduced with permissions from ref. 382. | ||
In 2023, a thoughtful complex design strategy to provide the first red-emitting Ag(I) complex, [Ag(debq)(xantphos)]PF6 (Ag24), with PLQYs up to 42%.381 More intriguing, outstanding electrochemical stabilities under reductive conditions were obtained. The authors claimed that the high rigidity of the employed debq ligand combined with Xantphos as large bite-angle diphosphine are paramount for the greatly improved electrochemical stability – Fig. 40. Thus, the resulting first ever reported low-energy emitting Ag(I) complex-based LEC outperformed the state-of-the-art pristine reference by three orders of magnitude (2.5 h). EIS and cyclic voltammetry disclosed the oxidation/hole injection as detrimental process. Applying a multilayered device architecture, namely using CBP as hole transport material, to decouple hole injection/transport and exciton formation lead to a device stability of up to 5 h and irradiances of 35 μW cm−2.
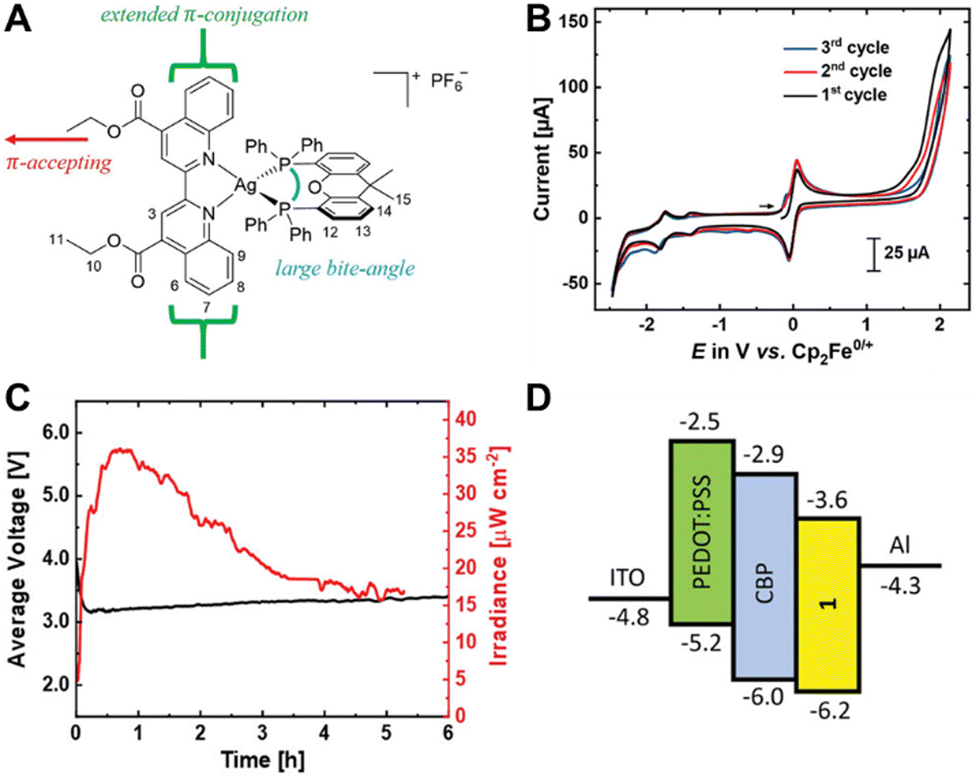 | ||
| Fig. 40 (A) Chemical structure representation of Ag24 including the design strategy. (B) Cyclic voltammogram of Ag24 (10−4 M; 100 mV s−1) inCH3CN/NBu4PF6 (0.1 M), at room temperature using (i) working electrode: platinum, (ii) counter electrode: platinum, and (iii) reference electrode: Ag/AgNO3. (C) Average voltage (black) and irradiance (red) versus time of CBP-Ag24-LEC, driven at pulsed 20 mA. (D) Schematic representation of the energy levels for the multi-layered device (1 = Ag24). Reproduced with permissions from ref. 381. | ||
4. Conclusion and outlook
This review provides a comprehensive summary of a hot topic in the design of TADF emitters. Although major efforts are devoted to organic-TADF materials, metal complexes with TADF are attracting an increasing interest in both OLEDs and LECs as shown in the summary provided in this review (Section 1). More importantly, Section 2 has provided a good overview about the design of metal complex based TADF compounds. They can be ad hoc engineered via chemical modification of coordinated ligand frameworks and/or the type of metal ion center, enabling the control of their photo- and electro-luminescence features in crystalline powder and, ultimately, in thin-films.228 In general, a spatial separation between HOMO and LUMO is required to minimize the energy difference between singlet and triplet states. In particular, several design rules have been disclosed for each family of TADF emitters based on the metal ions. In Al-complexes a strong donor ligand coupled to β-diketone is required. The complexation with Al(III) ions is acting as an acceptor moiety. In contrast, in Ir-based complexes, a strong donor (carbene) is directly coordinated to the metal ions and thus, rISC competes with ISC, despite the high SOC due to the heavy atom effect. In the case of Pd-/Pt-based complexes showing TADF, there is a clear lack of design rules as only few examples are known so far. For instance, the coordination of a D–A ligand was found to unlock TADF in a Pd-complex. The introduction of a second metal center and thus, the formation of a dinuclear Pd-complexes led to the first Pd-complex showing TADF. The main strategy for Au-complexes is based on changing the nature of the lowest excited states from intra-ligand to LL’CT character. In this context, three strategies have been proposed: (i) increasing the energy of the 3IL states, (ii) increasing the energy difference between the 3IL and 3LLCT states and/or (iii) decreasing the energy of the 1/3LLCT states. For instance, the first two aspects were realized by combining 2,6-bis(2,4-difluorophenyl)pyridine or 2,6-bis(2,4-diterbutylphenyl)pyrazine accepting-pincer ligands (C^N^C) with alkynyl ligands bearing strong donor groups, e.g. p-NPh2, m-NPh2, phenoxazine.For d10 transition metal complexes, the geometry must be taken into account when designing the TADF emitter. For example, in linear Cu-, Ag- and Au-based complexes TADF is mainly achieved through a donor–metal–acceptor system with lowest excited states of LL′CT character. Thus, negatively charged donor ligands (e.g., carbazolates) are often combined with strong acceptor ligand like carbenes (e.g. NHCs, MCAs). The TADF scenario in trigonal or tetrahedral Cu(I) complexes has been extensively explored and thus, clear design rules have been estabished. In short, the lowest excited states are mainly of MLCT character since the HOMO and LUMO are spatially well-separated resulting in small ΔEST. However, the opposing relationship between the HOMO–LUMO overlap, ΔEST and oscillator strength of the S1 → S0 transition/τTADF still displays a challenge in this context.
In contrast, the TADF scenario in Ag- and Zn-based complexes becomes much more challenging owing to energetically low-lying d-orbitals. In the case of Ag-complexes, fluorescencence and multi-phosphorescence processes are more frequently noted and only a few examples of unambiguous TADF have been reported. In these cases, TADF was achieved by using a strongly donating ligands, e.g., anionic diphosphines, to destabilize the low-lying d-orbitals resulting in the lowest excited state with a strong MLCT character. There are only a few examples of Zn-complex TADF emitters, but so far, no clear design rules have been established with the exception of the coordination of a D–A ligand that resulted in a TADF Zn-complex, in which the emission originates from LL-′CT excited states. This was realized by designing a donor–Zn–acceptor system including carbenes a acceptor and di-/thiolates as donor ligands.
Although clear design rules are lacking for several families, it has been successfully demonstrated that TADF can be achieved even in those with high SOC constants that are traditionally known as efficient phosphorescent emitters, e.g., Ir-/Pd-/Pt-complexes. This encourages future work to establish clear structure–property relationships and eventually apply them in OLEDs/LECs.
It is also critical to pay attention to the protocols to measure TADF in these families. As dicussed in Section 2, it is important to highlight that the assumptions that can be made depend on the systems studied. For instance, in the case of kISC ≫ kr(S1 → S0), which is the most common for TMCs, it is advisable to use Boltzmann's equation (eqn (6) and (7)) only if the complexes are highly emissive (PLQY > 50%). In the other cases (e.g., TADF Al-based complexes) the assumption kISC ≅ kr(S1 → S0) holds true. Thus, it is better to describe the system using eqn (19).
In this regard, it is also imperative to determine the TADF behavior in powders and thin-films applied to devices to confirm the device trends with respect to EQE values. This is related to the changes in the coordination sphere around the metal ion as the films are mostly processed via solution-based techniques in technologies, such as LECs. Changes on the ligand coordination affect the energy and nature of the excited states, SOC, and the HOMO–LUMO overlap, altering ΔEST and kr. This has been largely neglected in the literature, but it has been shown to be critical for Cu-complexes. For example, efficient and stable blue LECs have recently been achieved by exploiting the information obtained through multivariate analysis of a dataset of >90 contributions.228 Here, not only first-order interactions but also their interplay (second order interactions) must be taken into account to chemically design Cu-complexes with the desired emission features (color and PLQY) in thin-films. Thus, it is highly necessary to get more information on these points to reach a more mature understanding, creating a larger data collection that will allow the application of predictive models based on artificial intelligence algorithms. Likewise, ion electrolytes are commonly used as additives in LECs, which could also affect the TADF behavior of the films due to strong internal electric fields and temperature generation,390–394 with detrimental effects on the TADF performance and device efficiencies. Thus, the design of these complexes and the active layer must be confirmed by spectroscopic studies of the TADF of the active layers applied to devices.
In terms of device performance, most of the effort over the past 20 years has been devoted to Cu(I) complexes, which basically cover two families of TADF emitters. Today, the best performing blue,228 yellow,395 and red380 Cu(I) complex-based LECs have brightness of 180/140/30 cd m−2 associated with stabilities of 25/0.2/20 h, respectively. While they are not competitive for the mid/low energy visible region, they have recently outperformed the Ir(III)-based counterpart in the blue region. OLEDs based on Cu(I) complexes with very high EQE of 23.6% and luminance of 222200 cd m−2 have already been achieved,332 while color tunability has been successfully addressed. Finally, device stability is typically less addressed in OLEDs, while it is a major concern in LECs, in which the electrochemical reversibility of the TADF complexes is critical. This has strongly influenced the hole injection/transport and electron injection/transport processes, respectively, in Cu/Ag complex-based LECs. Here, the device architecture to decouple charge injection/transport and exciton recombination in LECs has successfully prevented device degradation. However, further efforts are needed to provide electrochemically reversible TADF complexes. In this context, Ir-based TADF complexes in LECs as well as the use of Cu-complexes as dopants for hyperfluorescent LECs need to be explored soon. Finally, the use of ionic additives, such as ionic liquids, is a controversial topic regarding their impact on the TADF mechanism in thin-films due to the lack of available data. Therefore, systematic studies are desirable.
Besides the above criticism on the metal complex based TADF emitters for SSL, we need to contextualize the above best performances for OLEDs and LECs with their respective devices with the best organic TADF emitters. In fact, we can conclude that metal complex based TADF emitters are indeed a competitive field. For example, the best EQE and luminance for OLEDs with inorganic TADF emitters have been achieved for Pd-complexes with 31.4% (Pd7) and 150300 cd m−2 (Pd8).200 However, the record of luminance of 265000 cd m−2 has been reported for the Au family (Au25) associated with an EQE of 25.3%.305 These values are very close to the average best OLEDs based on organic TADF emitters.396 Besides the fact that LECs are less performing than OLEDs, the interest in metal complex-based TADF emitters is even more attractive in this field, since LECs based on pristine ionic organic TADF emitters have only exhibited EQEs < 0.1%.397 As comparison, the best LECs with metal complex TADF emitters have shown EQEs of 1.85%.235 However, it is worth noting that recently ionic organic TADF emitters have been successfully stabilized in an ionic host matrix, reaching the unprecedented record value of 10% EQE.398 Thus, a fair comparison with ionic metal TADF compounds is currently lacking, as metal complexes have not been implemented in host:guest schemes. We believe that these outstanding results will soon have a ripple effect throughout the research community.
In light of the above, we hope that this review will help and motivate the community to further advance this exciting field, which is still full of challenges, toward a mature understanding.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.References
- F. Perera, Int. J. Environ. Res. Public Health, 2018, 15, 16 CrossRef PubMed.
- P. A. Owusu and S. Asumadu-Sarkodie, Cogent Eng., 2016, 3, 1167990 CrossRef.
- D. Gielen, F. Boshell, D. Saygin, M. D. Bazilian, N. Wagner and R. Gorini, Energy Strategy Rev., 2019, 24, 38–50 CrossRef.
- T. Güney, Int. J. Sustainalbe Dev., 2019, 26, 389–397 Search PubMed.
- A. Qazi, F. Hussain, N. A. Rahim, G. Hardaker, D. Alghazzawi, K. Shaban and K. Haruna, IEEE Access, 2019, 7, 63837–63851 Search PubMed.
- Y.-H. Kim, H. Cho and T.-W. Lee, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11694–11702 CrossRef CAS.
- B. Pashaei, S. Karimi, H. Shahroosvand, P. Abbasi, M. Pilkington, A. Bartolotta, E. Fresta, J. Fernandez-Cestau, R. D. Costa and F. Bonaccorso, Chem. Soc. Rev., 2019, 48, 5033–5139 RSC.
- C. W. Tang, S. A. VanSlyke and C. H. Chen, J. Appl. Phys., 1989, 65, 3610–3616 CrossRef CAS.
- C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048–5051 CrossRef CAS.
- Y.-L. Chang, Z. Wang, M. Helander, J. Qiu, D. Puzzo and Z. Lu, Org. Electron., 2012, 13, 925–931 CrossRef CAS.
- D. H. Kim, N. S. Cho, H. Y. Oh, J. H. Yang, W. S. Jeon, J. S. Park, M. C. Suh and J. H. Kwon, Adv. Mater., 2011, 23, 2721–2726 CrossRef CAS PubMed.
- Y. H. Kim, C. Wolf, H. Cho, S. H. Jeong and T. W. Lee, Adv. Mater., 2016, 28, 734–741 CrossRef CAS PubMed.
- D. Kabra, L. P. Lu, M. H. Song, H. J. Snaith and R. H. Friend, Adv. Mater., 2010, 22, 3194–3198 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Y. K. Chen, J. Jayakumar, C. M. Hsieh, T. L. Wu, C. C. Liao, J. Pandidurai, C. L. Ko, W. Y. Hung and C. H. Cheng, Adv. Mater., 2021, 33, 2008032 CrossRef CAS.
- V. L. Colvin, M. C. Schlamp and A. P. Alivisatos, Nature, 1994, 370, 354–357 CrossRef CAS.
- H. Mattoussi, L. H. Radzilowski, B. O. Dabbousi, E. L. Thomas, M. G. Bawendi and M. F. Rubner, J. Appl. Phys., 1998, 83, 7965–7974 CrossRef CAS.
- M. Schlamp, X. Peng and A.-L. Alivisatos, J. Appl. Phys., 1997, 82, 5837–5842 CrossRef CAS.
- S. Coe, W.-K. Woo, M. Bawendi and V. Bulović, Nature, 2002, 420, 800–803 CrossRef CAS PubMed.
- S. Coe-Sullivan, J. S. Steckel, W. K. Woo, M. G. Bawendi and V. Bulović, Adv. Funct. Mater., 2005, 15, 1117–1124 CrossRef CAS.
- J. Kwak, W. K. Bae, D. Lee, I. Park, J. Lim, M. Park, H. Cho, H. Woo, D. Y. Yoon and K. Char, Nano Lett., 2012, 12, 2362–2366 CrossRef CAS PubMed.
- X. Dai, Z. Zhang, Y. Jin, Y. Niu, H. Cao, X. Liang, L. Chen, J. Wang and X. Peng, Nature, 2014, 515, 96–99 CrossRef CAS PubMed.
- Z.-K. Tan, R. S. Moghaddam, M. L. Lai, P. Docampo, R. Higler, F. Deschler, M. Price, A. Sadhanala, L. M. Pazos and D. Credgington, Nat. Nanotechnol., 2014, 9, 687–692 CrossRef CAS PubMed.
- Y. H. Kim, H. Cho, J. H. Heo, T. S. Kim, N. Myoung, C. L. Lee, S. H. Im and T. W. Lee, Adv. Mater., 2015, 27, 1248–1254 CrossRef CAS.
- H. Cho, S.-H. Jeong, M.-H. Park, Y.-H. Kim, C. Wolf, C.-L. Lee, J. H. Heo, A. Sadhanala, N. Myoung and S. Yoo, Science, 2015, 350, 1222–1225 CrossRef CAS PubMed.
- K. Lin, J. Xing, L. N. Quan, F. P. G. de Arquer, X. Gong, J. Lu, L. Xie, W. Zhao, D. Zhang and C. Yan, Nature, 2018, 562, 245–248 CrossRef CAS PubMed.
- Z. Chu, Q. Ye, Y. Zhao, F. Ma, Z. Yin, X. Zhang and J. You, Adv. Mater., 2021, 33, 2007169 CrossRef CAS PubMed.
- Y. Yang and Q. Pei, J. Appl. Phys., 1997, 81, 3294–3298 CrossRef CAS.
- S. Tang, A. Sandström, P. Lundberg, T. Lanz, C. Larsen, S. van Reenen, M. Kemerink and L. Edman, Nat. Commun., 2017, 8, 1–9 CrossRef CAS PubMed.
- M. S. Shur and R. Zukauskas, Proc. IEEE, 2005, 93, 1691–1703 CAS.
- I. L. Azevedo, M. G. Morgan and F. Morgan, Proc. IEEE, 2009, 97, 481–510 CAS.
- R. S. Cok, M. Meitl, R. Rotzoll, G. Melnik, A. Fecioru, A. J. Trindade, B. Raymond, S. Bonafede, D. Gomez and T. Moore, J. Soc. Inf. Disp., 2017, 25, 589–609 CrossRef CAS.
- H. Zhang and J. A. Rogers, Adv. Opt. Mater., 2019, 7, 1800936 CrossRef.
- Q. Huang, Y. Zou, S. A. Bourelle, T. Zhai, T. Wu, Y. Tan, Y. Li, J. Li, S. Duhm and T. Song, Nanoscale Horiz., 2019, 4, 924–932 RSC.
- M. Kneissl, T. Kolbe, C. Chua, V. Kueller, N. Lobo, J. Stellmach, A. Knauer, H. Rodriguez, S. Einfeldt and Z. Yang, Semicond. Sci. Technol., 2010, 26, 014036 CrossRef.
- T.-Y. Seong, J. Han, H. Amano and H. Morkoç, III-Nitride based light emitting diodes and applications, Springer, 2013 Search PubMed.
- C. Weisbuch, M. Piccardo, L. Martinelli, J. Iveland, J. Peretti and J. S. Speck, Phys. Status Solidi, 2015, 212, 899–913 CrossRef CAS.
- H. Masui, S. Nakamura, S. P. DenBaars and U. K. Mishra, IEEE Trans. Electron Devices, 2009, 57, 88–100 Search PubMed.
- L. Chen, C.-C. Lin, C.-W. Yeh and R.-S. Liu, Materials, 2010, 3, 2172–2195 CrossRef CAS.
- H. Lin, T. Hu, Y. Cheng, M. Chen and Y. Wang, Laser Photonics Rev., 2018, 12, 1700344 CrossRef.
- G. B. Nair and S. J. Dhoble, The Fundamentals and Applications of Light-Emitting Diodes: The Revolution in the Lighting Industry, Woodhead Publishing, 2020 Search PubMed.
- J. Li, J. Wang, X. Yi, Z. Liu, T. Wei, J. Yan and B. Xue, III-Nitrides Light Emitting Diodes: Technology and Applications, Springer, 2020, pp. 7–18 Search PubMed.
- G. Held, Introduction to light emitting diode technology and applications, Auerbach Publications, 2016 Search PubMed.
- X. Sun, J. Liu, L. C. Kimerling and J. Michel, Opt. Lett., 2009, 34, 1198–1200 CrossRef CAS PubMed.
- J. Jiang, M. Xue, C.-Y. Lu, C. S. Fenrich, M. Morea, K. Zang, J. Gao, M. Cheng, Y. Zhang and T. I. Kamins, ACS Photonics, 2019, 6, 915–923 CrossRef CAS.
- M. Premkumar, M. Arun, S. Sathiya Priya, D. Kalaiarasi and R. Prathipa, Mater. Today: Proc., 2023, 80, 1932–1935 CAS.
- H.-J. Yeh and J. S. Smith, IEEE Photonics Technol. Lett., 1994, 6, 706–708 Search PubMed.
- S. Tsintzos, N. Pelekanos, G. Konstantinidis, Z. Hatzopoulos and P. Savvidis, Nature, 2008, 453, 372–375 CrossRef CAS PubMed.
- H. Morkoc and S. N. Mohammad, Science, 1995, 267, 51–55 CrossRef CAS.
- D.-S. Shin, D.-P. Han, J.-Y. Oh and J.-I. Shim, Appl. Phys. Lett., 2012, 100, 153506 CrossRef.
- W.-C. Chao, T.-H. Chiang, Y.-C. Liu, Z.-X. Huang, C.-C. Liao, C.-H. Chu, C.-H. Wang, H.-W. Tseng, W.-Y. Hung and P.-T. Chou, Commun. Mater., 2021, 2, 96 CrossRef CAS.
- Y. Ozen, T. Sertel, S. S. Cetin and S. Ozcelik, J. Electron. Mater., 2018, 47, 7129–7133 CrossRef CAS.
- W. C. Peng and Y. S. Wu, Appl. Phys. Lett., 2004, 84, 1841–1843 CrossRef CAS.
- E. Dupont, H. Liu, M. Buchanan, S. Chiu and M. Gao, Appl. Phys. Lett., 2000, 76, 4–6 CrossRef CAS.
- T. Khan, P. Bodrogi, Q. T. Vinh and H. Winkler, LED lighting: Technology and perception, John Wiley & Sons, 2015 Search PubMed.
- D. Zhu, D. Wallis and C. Humphreys, Rep. Prog. Phys., 2013, 76, 106501 CrossRef CAS.
- S. N. Mohammad and H. Morkoç, Prog. Quantum Electron., 1996, 20, 361–525 CrossRef CAS.
- M. S. Wong, S. Nakamura and S. P. DenBaars, ECS J. Solid State Sci. Technol., 2019, 9, 015012 CrossRef.
- T. Ayari, S. Sundaram, C. Bishop, A. Mballo, P. Vuong, Y. Halfaya, S. Karrakchou, S. Gautier, P. L. Voss and J. P. Salvestrini, Adv. Mater. Technol., 2019, 4, 1900164 CrossRef CAS.
- M. A. Naeser, R. Zafonte, M. H. Krengel, P. I. Martin, J. Frazier, M. R. Hamblin, J. A. Knight, W. P. Meehan III and E. H. Baker, J. Neurotrauma, 2014, 31, 1008–1017 CrossRef.
- D. R. Opel, E. Hagstrom, A. K. Pace, K. Sisto, S. A. Hirano-ALi, S. Desai and J. Swan, J. Clin. Aesthet. Dermatol., 2015, 8, 36 Search PubMed.
- E. C. P. Leal-Junior, A. A. Vanin, E. F. Miranda, P. D. T. C. De Carvalho, S. Dal Corso and J. M. Bjordal, Lasers Med. Sci., 2015, 30, 925–939 CrossRef PubMed.
- T. Müller, J. Skiba-Szymanska, A. Krysa, J. Huwer, M. Felle, M. Anderson, R. Stevenson, J. Heffernan, D. A. Ritchie and A. Shields, Nat. Commun., 2018, 9, 1–6 CrossRef PubMed.
- C. Chow, C. Yeh, Y. Liu and Y. Liu, IEEE Photon. Soc. Newslett., 2012, 26, 9–13 Search PubMed.
- Y. H. Song, S. H. Choi, W. K. Park, J. S. Yoo, S. B. Kwon, B. K. Kang, S. R. Park, Y. S. Seo, W. S. Yang and D. H. Yoon, Sci. Rep., 2018, 8, 1–6 Search PubMed.
- H. A. Höppe, Angew. Chem., Int. Ed., 2009, 48, 3572–3582 CrossRef.
- N. Guan, X. Dai, A. V. Babichev, F. H. Julien and M. Tchernycheva, Chem. Sci., 2017, 8, 7904–7911 RSC.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- A. Salehi, X. Fu, D. H. Shin and F. So, Adv. Funct. Mater., 2019, 29, 1808803 CrossRef.
- N. T. Kalyani and S. Dhoble, Renewable Sustainable Energy Rev., 2012, 16, 2696–2723 CrossRef.
- T. Oyamada, C. Maeda, H. Sasabe and C. Adachi, Jpn. J. Appl. Phys., 2003, 42, L1535 CrossRef CAS.
- Y. Xu, P. Xu, D. Hu and Y. Ma, Chem. Soc. Rev., 2021, 50, 1030–1069 RSC.
- G. M. Farinola and R. Ragni, Chem. Soc. Rev., 2011, 40, 3467–3482 RSC.
- S. Ho, S. Liu, Y. Chen and F. So, J. Photonics Energy, 2015, 5, 057611 CrossRef.
- K. S. Yook and J. Y. Lee, Adv. Mater., 2014, 26, 4218–4233 CrossRef CAS PubMed.
- C. A. Zuniga, S. Barlow and S. R. Marder, Chem. Mater., 2011, 23, 658–681 CrossRef CAS.
- W. J. Hyun, S. H. Im, O. O. Park and B. D. Chin, Org. Electron., 2012, 13, 579–585 CrossRef CAS.
- K. M. Kuznetsov, M. I. Kozlov, A. N. Aslandukov, A. A. Vashchenko, A. Medved’ko, E. Latipov, A. S. Goloveshkin, D. Tsymbarenko and V. V. Utochnikova, Dalton Trans., 2021, 50, 9685–9689 RSC.
- J. Jeong, D. Mascaro and S. Blair, Org. Electron., 2011, 12, 2095–2102 CrossRef CAS.
- C. S. Buga and J. C. Viana, Adv. Mater. Technol., 2021, 2001016 CrossRef.
- J. Li, L. Xu, C. W. Tang and A. A. Shestopalov, ACS Appl. Mater. Interfaces, 2016, 8, 16809–16815 CrossRef CAS PubMed.
- J.-C. G. Bünzli, S. Comby, A.-S. Chauvin and C. D. Vandevyver, J. Rare Earths, 2007, 25, 257–274 CrossRef.
- M. C. Gather, A. Köhnen and K. Meerholz, Adv. Mater., 2011, 23, 233–248 CrossRef CAS PubMed.
- A. L. Hicks, T. L. Theis and M. L. Zellner, J. Ind. Ecol., 2015, 19, 285–295 CrossRef.
- D. F. de Souza, P. P. F. da Silva, L. F. A. Fontenele, G. D. Barbosa and M. de Oliveira Jesus, Energy Rep., 2019, 5, 409–424 CrossRef.
- T. D. Little, P. Dib, K. Shah, N. Barraford and B. Gallagher, IEEE, 2008, 373–378 Search PubMed.
- A. Sabbar, S. Madhusoodhanan, S. Al-Kabi, B. Dong, J. Wang, S. Atcitty, R. Kaplar, D. Ding, A. Mantooth and S.-Q. Yu, Sci. Rep., 2019, 9, 1–8 CrossRef CAS PubMed.
- S. M. Pawson and M.-F. Bader, Ecol. Appl., 2014, 24, 1561–1568 CrossRef CAS PubMed.
- B. K. Hawes, T. T. Brunyé, C. R. Mahoney, J. M. Sullivan and C. D. Aall, Int. J. Ind. Ergon., 2012, 42, 122–128 CrossRef.
- H. Yersin, R. Czerwieniec, U. Monkowius, R. Ramazanov, R. Valiev, M. Z. Shafikov, W.-M. Kwok and C. Ma, Coord. Chem. Rev., 2023, 478, 214975 CrossRef CAS.
- R. Delorme and F. Perrin, J. Phys. Radium, 1929, 10, 177–186 CrossRef CAS.
- G. N. Lewis, D. Lipkin and T. T. Magel, J. Am. Chem. Soc., 1941, 63, 3005–3018 CrossRef CAS.
- C. Parker and C. Hatchard, Trans. Faraday Soc., 1961, 57, 1894–1904 RSC.
- S. Kim, H. J. Kwon, S. Lee, H. Shim, Y. Chun, W. Choi, J. Kwack, D. Han, M. Song and S. Kim, Adv. Mater., 2011, 23, 3511–3516 CrossRef CAS PubMed.
- B. A. Prabowo, L.-C. Su, Y.-F. Chang, H.-C. Lai, N.-F. Chiu and K.-C. Liu, Sens. Actuators, B, 2016, 222, 1058–1065 CrossRef CAS.
- Y. Jeon, H. R. Choi, M. Lim, S. Choi, H. Kim, J. H. Kwon, K. C. Park and K. C. Choi, Adv. Mater. Technol., 2018, 3, 1700391 CrossRef.
- Y. J. Song, J.-W. Kim, H.-E. Cho, Y. H. Son, M. H. Lee, J. Lee, K. C. Choi and S.-M. Lee, ACS Nano, 2020, 14, 1133–1140 CrossRef CAS PubMed.
- N. Thejo Kalyani and S. J. Dhoble, Renewable Sustainable Energy Rev., 2015, 44, 319–347 CrossRef.
- C. Zhang, R. Liu, D. Zhang and L. Duan, Adv. Funct. Mater., 2020, 30, 1907156 CrossRef CAS.
- S. B. Meier, D. Tordera, A. Pertegas, C. Roldan-Carmona, E. Orti and H. J. Bolink, Mater. Today, 2014, 17, 217–223 CrossRef CAS.
- D. Tordera, S. Meier, M. Lenes, R. D. Costa, E. Ortí, W. Sarfert and H. J. Bolink, Adv. Mater., 2012, 24, 897–900 CrossRef CAS PubMed.
- S. Tang and L. Edman, Photoluminescent Materials and Electroluminescent Devices, Springer, 2017, pp. 375–395 Search PubMed.
- A. Sandström and L. Edman, Energy Technol., 2015, 3, 329–339 CrossRef.
- S. Karimi, H. Shahroosvand, S. Bellani and F. Bonaccorso, J. Phys. Chem. C, 2021, 125, 819–829 CrossRef CAS.
- Q. Zhang, Q. Zhou, Y. Cheng, L. Wang, D. Ma, X. Jing and F. Wang, Adv. Funct. Mater., 2006, 16, 1203–1208 CrossRef CAS.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178–8211 CrossRef CAS PubMed.
- Q. Sun, Y. Li and Q. Pei, J. Disp. Technol., 2007, 3, 211–224 CAS.
- Q. Pei, G. Yu, C. Zhang, Y. Yang and A. J. Heeger, Science, 1995, 269, 1086–1088 CrossRef CAS.
- J.-K. Lee, D. Yoo and M. F. Rubner, Chem. Mater., 1997, 9, 1710–1712 CrossRef CAS.
- M. S. Lowry and S. Bernhard, Chem. – Eur. J., 2006, 12, 7970–7977 CrossRef CAS PubMed.
- J. D. Slinker, J. Rivnay, J. S. Moskowitz, J. B. Parker, S. Bernhard, H. D. Abruña and G. G. Malliaras, J. Mater. Chem., 2007, 17, 2976–2988 RSC.
- E. Fresta and R. D. Costa, J. Mater. Chem. C, 2017, 5, 5643–5675 RSC.
- J. D. Slinker, J. Rivnay, J. A. DeFranco, D. A. Bernards, A. A. Gorodetsky, S. T. Parker, M. P. Cox, R. Rohl, G. G. Malliaras and S. Flores-Torres, J. Appl. Phys., 2006, 99, 074502 CrossRef.
- F. Dumur, D. Bertin and D. Gigmes, Internet J. Nanotechnol., 2012, 9, 377–395 CrossRef CAS.
- J. E. Namanga, H. Pei, G. Bousrez, V. Smetana, N. Gerlitzki and A.-V. Mudring, ACS Appl. Energy Mater., 2020, 3, 9271–9277 CrossRef CAS.
- J. Zhang, L. Zhou, H. A. Al-Attar, K. Shao, L. Wang, D. Zhu, Z. Su, M. R. Bryce and A. P. Monkman, Adv. Funct. Mater., 2013, 23, 4667–4677 CrossRef CAS.
- A. Malewar, Light-emitting electrochemical cell (LECs) – now an efficient and bright device, https://www.techexplorist.com/light-emitting-electrochemical-cell-lecs-now-efficient-bright-device/10428/.
- K. Youssef, Y. Li, S. O'Keeffe, L. Li and Q. Pei, Adv. Funct. Mater., 2020, 30, 1909102 CrossRef CAS.
- Y. Tang, M. Deng, Z. Zhou, C. Kang, J. Wang and Q. Liu, Coord. Chem. Rev., 2024, 499, 215490 CrossRef CAS.
- X.-K. Chen, D. Kim and J.-L. Brédas, Acc. Chem. Res., 2018, 51, 2215–2224 CrossRef CAS PubMed.
- X. Liang, Z.-L. Tu and Y.-X. Zheng, Chem. – Eur. J., 2019, 25, 5623–5642 CrossRef CAS PubMed.
- T.-Y. Li, S.-J. Zheng, P. I. Djurovich and M. E. Thompson, Chem. Rev., 2024, 124, 4332–4392 CrossRef CAS.
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- D. Volz and J. Photonics, Energy, 2016, 6, 020901 Search PubMed.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- Highly efficient OLEDs: Materials based on thermally activated delayed fluorescence, ed. H. Yersin, John Wiley & Sons, 2019 Search PubMed.
- M. Y. Wong and E. Zysman-Colman, in Light-Emitting Electrochemical Cells: Concepts, Advances and Challenges, ed. R. D. Costa, Springer International Publishing, Cham, 2017, pp. 237–266 Search PubMed.
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed.
- C. Adachi, Jpn. J. Appl. Phys., 2014, 53, 060101 CrossRef.
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 42 CrossRef.
- K. Sato, K. Shizu, K. Yoshimura, A. Kawada, H. Miyazaki and C. Adachi, Phys. Rev. Lett., 2013, 110, 247401 CrossRef PubMed.
- H. Yersin, A. F. Rausch and R. Czerwieniec, Phys. Org. Semicond., 2012, 371–424 CAS.
- P. W. Atkins and R. S. Friedman, Molecular quantum mechanics, Oxford University Press, New York, 2011, 4th edn Search PubMed.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of photochemistry, CRC Press, Boca Raton, 2006 Search PubMed.
- R. Ishimatsu, S. Matsunami, K. Shizu, C. Adachi, K. Nakano and T. Imato, J. Phys. Chem. A, 2013, 117, 5607–5612 CrossRef CAS PubMed.
- Y. Im and J. Y. Lee, Chem. Mater., 2014, 26, 1413–1419 CrossRef CAS.
- L.-S. Cui, S.-B. Ruan, F. Bencheikh, R. Nagata, L. Zhang, K. Inada, H. Nakanotani, L.-S. Liao and C. Adachi, Nat. Commun., 2017, 8, 1–8 CrossRef CAS PubMed.
- E. Y.-T. Li, T.-Y. Jiang, Y. Chi and P.-T. Chou, Phys. Chem. Chem. Phys., 2014, 16, 26184–26192 RSC.
- M. Baba, J. Phys. Chem. A, 2011, 115, 9514–9519 CrossRef CAS PubMed.
- P. Ceroni, Chem, 2016, 1, 524–526 CAS.
- T. Tsuzuki and S. Tokito, Adv. Mater., 2007, 19, 276–280 CrossRef CAS.
- T.-Y. Li, J. Wu, Z.-G. Wu, Y.-X. Zheng, J.-L. Zuo and Y. Pan, Coord. Chem. Rev., 2018, 374, 55–92 CrossRef CAS.
- T. Nakagawa, S.-Y. Ku, K.-T. Wong and C. Adachi, Chem. Commun., 2012, 48, 9580–9582 RSC.
- S. Y. Lee, T. Yasuda, H. Nomura and C. Adachi, Appl. Phys. Lett., 2012, 101, 093306 CrossRef.
- G. Méhes, H. Nomura, Q. Zhang, T. Nakagawa and C. Adachi, Angew. Chem., Int. Ed., 2012, 51, 11311–11315 CrossRef PubMed.
- S. Liu, B. Yang, J. Chen, D. Wei, D. Zheng, Q. Kong, W. Deng and K. Han, Angew. Chem., Int. Ed., 2020, 59, 21925–21929 CrossRef CAS PubMed.
- J. Adachi, H. Kakizoe, P. K. D. Tsang and A. Endo, SID Symp. Dig. Tech. Pap., 2019, 50, 95–98 CrossRef CAS.
- C. Adachi, SID Symp. Dig. Tech. Pap., 2013, 44, 513–514 CrossRef CAS.
- A. M. Polgar, C. M. Tonge, C. J. Christopherson, N. R. Paisley, A. C. Reyes and Z. M. Hudson, ACS Appl. Mater. Interfaces, 2020, 12, 38602–38613 CrossRef CAS PubMed.
- H. Nakanotani, T. Higuchi, T. Furukawa, K. Masui, K. Morimoto, M. Numata, H. Tanaka, Y. Sagara, T. Yasuda and C. Adachi, Nat. Commun., 2014, 5, 4016 CrossRef CAS PubMed.
- K. Stavrou, L. G. Franca, A. Danos and A. P. Monkman, Nat. Photonics, 2024, 18, 554–561 CrossRef CAS.
- S. K. Behera and R. D. Costa, J. Mater. Chem. C, 2023, 11, 13647–13656 RSC.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- P. W. Atkins, Quanta: A Handbook of Concepts, Oxford University Press, 1991 Search PubMed.
- B. Milián-Medina and J. Gierschner, Org. Electron., 2012, 13, 985–991 CrossRef.
- S. Sudheendran Swayamprabha, D. K. Dubey, Shahnawaz, R. A. K. Yadav, M. R. Nagar, A. Sharma, F. C. Tung and J. H. Jou, Adv. Sci., 2021, 8, 2002254 CrossRef CAS PubMed.
- Z. D. Popovic and H. Aziz, IEEE J. Sel. Top. Quantum Electron., 2002, 8, 362–371 CrossRef CAS.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- M. N. Berberan-Santos and J. M. M. Garcia, J. Am. Chem. Soc., 1996, 118, 9391–9394 CrossRef CAS.
- R. J. Vázquez, J. H. Yun, A. K. Muthike, M. Howell, H. Kim, I. K. Madu, T. Kim, P. Zimmerman, J. Y. Lee and T. G. Iii, J. Am. Chem. Soc., 2020, 142, 8074–8079 CrossRef PubMed.
- Y. Wada, H. Nakagawa, S. Matsumoto, Y. Wakisaka and H. Kaji, Nat. Photonics, 2020, 14, 643–649 CrossRef CAS.
- N. Haase, A. Danos, C. Pflumm, A. Morherr, P. Stachelek, A. Mekic, W. Brütting and A. P. Monkman, J. Phys. Chem. C, 2018, 122, 29173–29179 CrossRef CAS.
- J. R. Kirchhoff, R. E. Gamache, Jr., M. W. Blaskie, A. A. Del Paggio, R. K. Lengel and D. R. McMillin, Inorg. Chem., 1983, 22, 2380–2384 CrossRef CAS.
- M. Iwamura, H. Watanabe, K. Ishii, S. Takeuchi and T. Tahara, J. Am. Chem. Soc., 2011, 133, 7728–7736 CrossRef CAS PubMed.
- N. Armaroli, G. Accorsi, F. Cardinali and A. Listorti, in Photochemistry and Photophysics of Coordination Compounds I, ed. V. Balzani and S. Campagna, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, pp. 69–115 Search PubMed.
- M. Iwamura, S. Takeuchi and T. Tahara, Acc. Chem. Res., 2015, 48, 782–791 CrossRef CAS PubMed.
- R. Hamze, S. Shi, S. C. Kapper, D. S. Muthiah Ravinson, L. Estergreen, M.-C. Jung, A. C. Tadle, R. Haiges, P. I. Djurovich, J. L. Peltier, R. Jazzar, G. Bertrand, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2019, 141, 8616–8626 CrossRef CAS PubMed.
- D. S. M. Ravinson and M. E. Thompson, Mater. Horiz., 2020, 7, 1210–1217 RSC.
- E. Fresta, G. U. Mahoro, L. M. Cavinato, J.-F. Lohier, J.-L. Renaud, S. Gaillard and R. D. Costa, Adv. Opt. Mater., 2022, 10, 2101999 CrossRef CAS.
- G. U. Mahoro, J. Fernandez-Cestau, J.-L. Renaud, P. B. Coto, R. D. Costa and S. Gaillard, Adv. Opt. Mater., 2020, 8, 2000260 CrossRef CAS.
- C. M. Brown, C. Li, V. Carta, W. Li, Z. Xu, P. H. F. Stroppa, I. D. Samuel, E. Zysman-Colman and M. O. Wolf, Inorg. Chem., 2019, 58, 7156–7168 CrossRef CAS PubMed.
- Y. Tsuchiya, S. Diesing, F. Bencheikh, Y. Wada, P. L. dos Santos, H. Kaji, E. Zysman-Colman, I. D. W. Samuel and C. Adachi, J. Phys. Chem. A, 2021, 125, 8074–8089 CrossRef CAS PubMed.
- D. Y. Kondakov, Philos. Trans. R. Soc. A, 2015, 373, 20140321 CrossRef PubMed.
- Y. Sun, N. C. Giebink, H. Kanno, B. Ma, M. E. Thompson and S. R. Forrest, Nature, 2006, 440, 908–912 CrossRef CAS PubMed.
- G. Schwartz, M. Pfeiffer, S. Reineke, K. Walzer and K. Leo, Adv. Mater., 2007, 19, 3672–3676 CrossRef CAS.
- C.-J. Zheng, J. Wang, J. Ye, M.-F. Lo, X.-K. Liu, M.-K. Fung, X.-H. Zhang and C.-S. Lee, Adv. Mater., 2013, 25, 2205–2211 CrossRef CAS PubMed.
- L. M. Cavinato, K. Yamaoka, S. Lipinski, V. Calvi, D. Wehenkel, R. van Rijn, K. Albrecht and R. D. Costa, Adv. Funct. Mater., 2023, 33, 2302483 CrossRef CAS.
- K. Nakao, H. Sasabe, Y. Shibuya, A. Matsunaga, H. Katagiri and J. J. A. C. Kido, Angew. Chem., 2021, 133, 6101–6106 CrossRef.
- H. Yersin, R. Czerwieniec, M. Z. Shafikov and A. F. Suleymanova, ChemPhysChem, 2017, 18, 3508–3535 CrossRef CAS PubMed.
- Y. Zhang, T. S. Lee, J. M. Favale, D. C. Leary, J. L. Petersen, G. D. Scholes, F. N. Castellano and C. Milsmann, Nat. Chem., 2020, 12, 345–352 CrossRef CAS PubMed.
- M. Matsuya, H. Sasabe, S. Sumikoshi, K. Hoshi, K. Nakao, K. Kumada, R. Sugiyama, R. Sato and J. Kido, Bull. Chem. Soc. Jpn., 2023, 96, 183–189 CrossRef CAS.
- R. D. Costa, F. J. Céspedes-Guirao, E. Ortí, H. J. Bolink, J. Gierschner, F. Fernández-Lázaro and A. Sastre-Santos, Chem. Commun., 2009, 3886–3888 RSC.
- A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971–12979 CrossRef CAS PubMed.
- L. He, L. Duan, J. Qiao, D. Zhang, L. Wang and Y. Qiu, Chem. Commun., 2011, 47, 6467–6469 RSC.
- R. Bai, X. Meng, X. Wang and L. J. A. F. M. He, Adv. Funct. Mater., 2020, 30, 1907169 CrossRef CAS.
- G. Albrecht, C. Geis, J. Herr, J. Ruhl, R. Göttlich and D. Schlettwein, Org. Electron., 2019, 65, 321–326 CrossRef CAS.
- J. M. Herr, C. Rössiger, H. Locke, M. Wilhelm, J. Becker, W. Heimbrodt, D. Schlettwein and R. Göttlich, Dyes Pigm., 2020, 180, 108512 CrossRef CAS.
- H. Na and T. S. Teets, J. Am. Chem. Soc., 2018, 140, 6353–6360 CrossRef CAS PubMed.
- Y.-J. Gao, T.-T. Zhang and W.-K. Chen, J. Phys. Chem. C, 2021, 125, 5670–5677 CrossRef CAS.
- M. Z. Shafikov, R. Martinscroft, C. Hodgson, A. Hayer, A. Auch and V. N. Kozhevnikov, Inorg. Chem., 2021, 60, 1780–1789 CrossRef CAS PubMed.
- L.-Y. Peng, Z.-W. Li, G.-N. Pan, W.-K. Chen, Y.-J. Gao and G. Cui, Phys. Chem. Chem. Phys., 2023, 25, 6454–6460 RSC.
- P. Pander, A. V. Zaytsev, A. Sil, G. V. Baryshnikov, F. Siddique, J. A. G. Williams, F. B. Dias and V. N. Kozhevnikov, Chem. Sci., 2023, 14, 13934–13943 RSC.
- Z. Q. Zhu, T. Fleetham, E. Turner and J. Li, Adv. Mater., 2015, 27, 2533–2537 CrossRef CAS PubMed.
- Z.-Q. Zhu, C.-D. Park, K. Klimes and J. Li, Adv. Opt. Mater., 2019, 7, 1801518 CrossRef.
- P. W. Zach, S. A. Freunberger, I. Klimant and S. M. Borisov, ACS Appl. Mater. Interfaces, 2017, 9, 38008–38023 CrossRef CAS PubMed.
- G. Li, Q. Chen, J. Zheng, Q. Wang, F. Zhan, W. Lou, Y. F. Yang and Y. She, Inorg. Chem., 2019, 58, 14349–14360 CrossRef CAS PubMed.
- Y. She, K. Xu, X. Fang, Y. F. Yang, W. Lou, Y. Hu, Q. Zhang and G. Li, Inorg. Chem., 2021, 60, 12972–12983 CrossRef CAS PubMed.
- J.-G. Yang, X. Feng, N. Li, J. Li, X.-F. Song, M.-D. Li, G. Cui, J. Zhang, C. Jiang and C. J. S. A. Yang, Sci. Adv., 2023, 9, eadh0198 CrossRef CAS PubMed.
- P. Pander, R. Daniels, A. V. Zaytsev, A. Horn, A. Sil, T. J. Penfold, J. G. Williams, V. N. Kozhevnikov and F. B. Dias, Chem. Sci., 2021, 12, 6172–6180 RSC.
- M. Z. Shafikov, R. Daniels, P. Pander, F. B. Dias, J. A. G. Williams and V. N. Kozhevnikov, ACS Appl. Mater. Interfaces, 2019, 11, 8182–8193 CrossRef CAS PubMed.
- V. N. Kozhevnikov, M. C. Durrant and J. A. G. Williams, Inorg. Chem., 2011, 50, 6304–6313 CrossRef CAS PubMed.
- P. Pander, A. V. Zaytsev, A. Sil, J. A. G. Williams, P.-H. Lanoe, V. N. Kozhevnikov and F. B. Dias, J. Mater. Chem. C, 2021, 9, 10276–10287 RSC.
- W.-P. To, G. S.-M. Tong, W. Lu, C. Ma, J. Liu, A. L.-F. Chow and C.-M. Che, Angew. Chem., 2012, 51, 2654–2657 CrossRef CAS PubMed.
- W. P. To, K. T. Chan, G. S. M. Tong, C. Ma, W. M. Kwok, X. Guan, K. H. Low and C. M. Che, Angew. Chem., Int. Ed., 2013, 52, 6648 CrossRef CAS PubMed.
- F. F. Hung, W. P. To, J. J. Zhang, C. Ma, W. Y. Wong and C. M. Che, Chem. – Eur. J., 2014, 20, 8604–8614 CrossRef CAS PubMed.
- R. Kumar, A. Linden and C. J. A. C. Nevado, Angew. Chem., 2015, 127, 14495–14498 CrossRef.
- W.-P. To, G. S. M. Tong, C.-W. Cheung, C. Yang, D. Zhou and C.-M. Che, Inorg. Chem., 2017, 56, 5046–5059 CrossRef CAS PubMed.
- M. Bachmann, O. Blacque and K. Venkatesan, Chem. – Eur. J., 2017, 23, 9451–9456 CrossRef CAS PubMed.
- L. Currie, J. Fernandez-Cestau, L. Rocchigiani, B. Bertrand, S. J. Lancaster, D. L. Hughes, H. Duckworth, S. T. Jones, D. Credgington and T. Penfold, Chem. – Eur. J., 2017, 23, 105–113 CrossRef CAS PubMed.
- B. Y.-W. Wong, H.-L. Wong, Y.-C. Wong, V. K.-M. Au, M.-Y. Chan and V. W. Yam, Chem. Sci., 2017, 8, 6936–6946 RSC.
- J. Fernandez-Cestau, B. Bertrand, M. Blaya, G. A. Jones, T. J. Penfold and M. Bochmann, Chem. Commun., 2015, 51, 16629–16632 RSC.
- W. P. To, D. Zhou, G. S. M. Tong, G. Cheng, C. Yang and C. Che, Angew. Chem., Int. Ed., 2017, 56, 14036–14041 CrossRef CAS PubMed.
- D. Zhou, G. Cheng, G. S. M. Tong and C. M. Che, Chem. – Eur. J., 2020, 26, 15718–15726 CrossRef CAS PubMed.
- D. Zhou, W. P. To, Y. Kwak, Y. Cho, G. Cheng, G. S. M. Tong and C. M. Che, Adv. Sci., 2019, 6, 1802297 CrossRef CAS PubMed.
- C. C. Au-Yeung, L.-K. Li, M.-C. Tang, S.-L. Lai, W.-L. Cheung, M. Ng, M.-Y. Chan and V. W.-W. Yam, Chem. Sci., 2021, 12, 9516–9527 RSC.
- C. E. Johnson, J. Schwarz, M. Deegbey, O. Prakash, K. Sharma, P. Huang, T. Ericsson, L. Häggström, J. Bendix, A. K. Gupta, E. Jakubikova, K. Wärnmark and R. Lomoth, Chem. Sci., 2023, 14, 10129–10139 RSC.
- L.-K. Li, C. C. Au-Yeung, M.-C. Tang, S.-L. Lai, W.-L. Cheung, M. Ng, M.-Y. Chan and V. W. Yam, Mater. Horiz., 2022, 9, 281–293 RSC.
- D. Zhou, W. P. To, G. S. M. Tong, G. Cheng, L. Du, D. L. Phillips and C. M. Che, Angew. Chem., 2020, 132, 6437–6444 CrossRef.
- W.-K. Kwok, L.-K. Li, S.-L. Lai, M.-Y. Leung, W. K. Tang, S.-C. Cheng, M.-C. Tang, W.-L. Cheung, C.-C. Ko, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2023, 145, 9584–9595 CrossRef CAS PubMed.
- M. Saigo, K. Miyata, S. I. Tanaka, H. Nakanotani, C. Adachi and K. Onda, J. Phys. Chem. Lett., 2019, 10, 2475–2480 CrossRef CAS PubMed.
- D. Zhou, W. To, G. Tong, G. Cheng, L. Du, D. Phillips and C. Che, Angew. Chem., Int. Ed., 2020, 59, 6375–6382 CrossRef CAS PubMed.
- K. Nozaki and M. Iwamura, Highly efficient OLEDs: Materials based on thermally activated delayed fluorescence, 2018, pp. 61–91 Search PubMed.
- J. Beaudelot, S. Oger, S. Peruško, T.-A. Phan, T. Teunens, C. Moucheron and G. Evano, Chem. Rev., 2022, 122, 16365–16609 CrossRef CAS PubMed.
- C. E. Housecroft and E. C. J. J. o M. C. C. Constable, J. Mater. Chem. C, 2022, 10, 4456–4482 RSC.
- G. U. Mahoro, E. Fresta, M. Elie, D. di Nasso, Q. Zhang, J.-F. Lohier, J.-L. Renaud, M. Linares, R. Wannemacher, J. Cabanillas-Gonzalez, R. D. Costa and S. Gaillard, Dalton Trans., 2021, 50, 11049–11060 RSC.
- L. M. Cavinato, S. Wölfl, A. Pöthig, E. Fresta, C. Garino, J. Fernandez-Cestau, C. Barolo and R. D. Costa, Adv. Mater., 2022, 34, 2109228 CrossRef CAS PubMed.
- C. Sandoval-Pauker, M. Santander-Nelli and P. Dreyse, RSC Adv., 2022, 12, 10653–10674 RSC.
- H. Takeda, A. Kobayashi and K. Tsuge, Coord. Chem. Rev., 2022, 470, 214700 CrossRef CAS.
- M. Klein, N. Rau, M. Wende, J. Sundermeyer, G. Cheng, C.-M. Che, A. Schinabeck and H. Yersin, Chem. Mater., 2020, 32, 10365–10382 CrossRef CAS.
- A. Kaeser, O. Moudam, G. Accorsi, I. Séguy, J. Navarro, A. Belbakra, C. Duhayon, N. Armaroli, B. Delavaux-Nicot and J.-F. Nierengarten, Eur. J. Inorg. Chem., 2014, 1345–1355 CrossRef CAS.
- R. D. Costa, D. Tordera, E. Ortí, H. J. Bolink, J. Schönle, S. Graber, C. E. Housecroft, E. C. Constable and J. A. Zampese, J. Mater. Chem., 2011, 21, 16108–16118 RSC.
- A. Kaeser, M. Mohankumar, J. Mohanraj, F. Monti, M. Holler, J. J. Cid, O. Moudam, I. Nierengarten, L. Karmazin-Brelot, C. Duhayon, B. Delavaux-Nicot, N. Armaroli and J. F. Nierengarten, Inorg. Chem., 2013, 52, 12140–12151 CrossRef CAS PubMed.
- C. Li, C. F. R. Mackenzie, S. A. Said, A. K. Pal, M. A. Haghighatbin, A. Babaei, M. Sessolo, D. B. Cordes, A. M. Z. Slawin, P. C. J. Kamer, H. J. Bolink, C. F. Hogan and E. Zysman-Colman, Inorg. Chem., 2021, 60, 10323–10339 CrossRef CAS PubMed.
- T. A. Phan, N. Armaroli, A. Saavedra Moncada, E. Bandini, B. Delavaux-Nicot, J. F. Nierengarten and D. J. A. C. Armspach, Angew. Chem., 2023, 135, e202214638 CrossRef.
- M. Matejdes, M. Stöter, R. Czerwieniec, M. Leitl, S. Rosenfeldt, T. Schumacher, J. Albert, M. Lippitz, H. Yersin and J. Breu, Adv. Opt. Mater., 2021, 9, 2100516 CrossRef CAS.
- M. Mohankumar, M. Holler, E. Meichsner, J.-F. Nierengarten, F. Niess, J.-P. Sauvage, B. Delavaux-Nicot, E. Leoni, F. Monti, J. M. Malicka, M. Cocchi, E. Bandini and N. Armaroli, J. Am. Chem. Soc., 2018, 140, 2336–2347 CrossRef CAS PubMed.
- C. L. Linfoot, M. J. Leitl, P. Richardson, A. F. Rausch, O. Chepelin, F. J. White, H. Yersin and N. Robertson, Inorg. Chem., 2014, 53, 10854–10861 CrossRef CAS PubMed.
- M. Meyer, A. Prescimone, E. C. Constable and C. E. Housecroft, Dalton Trans., 2024, 53, 5453–5465 RSC.
- D. Gejsnæs-Schaad, M. Meyer, A. Prescimone, C. E. Housecroft and E. C. Constable, CrystEngComm, 2023, 25, 3000–3012 RSC.
- I. Nohara, C. Wegeberg, M. Devereux, A. Prescimone, C. E. Housecroft and E. C. Constable, J. Mater. Chem. C, 2022, 10, 3089–3102 RSC.
- K. Zhang and D. Zhang, Spectrochim. Acta, Part A, 2014, 124, 341–348 CrossRef CAS PubMed.
- M. Osawa and M. Hoshino, in Highly Efficient OLEDs, ed. H. Yersin, 2018, pp. 119–176 Search PubMed.
- S. Keller, E. C. Constable, C. E. Housecroft, M. Neuburger, A. Prescimone, G. Longo, A. Pertegás, M. Sessolo and H. J. Bolink, Dalton Trans., 2014, 43, 16593–16596 RSC.
- R. Czerwieniec, K. Kowalski and H. Yersin, Dalton Trans., 2013, 42, 9826–9830 RSC.
- R. Czerwieniec and H. Yersin, Inorg. Chem., 2015, 54, 4322–4327 CrossRef CAS PubMed.
- M. J. Leitl, V. A. Krylova, P. I. Djurovich, M. E. Thompson and H. Yersin, J. Am. Chem. Soc., 2014, 136, 16032–16038 CrossRef CAS.
- M. Elie, F. Sguerra, F. Di Meo, M. D. Weber, R. Marion, A. Grimault, J.-F. Lohier, A. Stallivieri, A. Brosseau, R. B. Pansu, J.-L. Renaud, M. Linares, M. Hamel, R. D. Costa and S. Gaillard, ACS Appl. Mater. Interfaces, 2016, 8, 14678–14691 CrossRef CAS PubMed.
- M. Elie, M. D. Weber, F. Di Meo, F. Sguerra, J. F. Lohier, R. B. Pansu, J. L. Renaud, M. Hamel, M. Linares and R. D. Costa, Chem. – Eur. J., 2017, 23, 16328–16337 CrossRef CAS PubMed.
- A. M. T. Muthig, O. Mrózek, T. Ferschke, M. Rödel, B. Ewald, J. Kuhnt, C. Lenczyk, J. Pflaum and A. Steffen, J. Am. Chem. Soc., 2023, 145, 4438–4449 CrossRef CAS PubMed.
- A. Tsuboyama, K. Kuge, M. Furugori, S. Okada, M. Hoshino and K. Ueno, Inorg. Chem., 2007, 46, 1992–2001 CrossRef CAS PubMed.
- M. J. Leitl, F.-R. Küchle, H. A. Mayer, L. Wesemann and H. Yersin, J. Phys. Chem. A, 2013, 117, 11823–11836 CrossRef CAS PubMed.
- A. Schinabeck, J. Chen, L. Kang, T. Teng, H. H. H. Homeier, A. F. Suleymanova, M. Z. Shafikov, R. Yu, C.-Z. Lu and H. Yersin, Chem. Mater., 2019, 31, 4392–4404 CrossRef CAS.
- S. Igawa, M. Hashimoto, I. Kawata, M. Yashima, M. Hoshino and M. Osawa, J. Mater. Chem. C, 2013, 1, 542–551 RSC.
- V. A. Krylova, P. I. Djurovich, M. T. Whited and M. E. Thompson, Chem. Commun., 2010, 46, 6696–6698 RSC.
- V. A. Krylova, P. I. Djurovich, B. L. Conley, R. Haiges, M. T. Whited, T. J. Williams and M. E. Thompson, Chem. Commun., 2014, 50, 7176–7179 RSC.
- P. A. Breddels, P. A. Berdowski, G. Blasse and D. McMillin, J. Chem. Soc., Faraday Trans. 2, 1982, 78, 595–601 RSC.
- D. Felder, J.-F. Nierengarten, F. Barigelletti, B. Ventura and N. Armaroli, J. Am. Chem. Soc., 2001, 123, 6291–6299 CrossRef CAS PubMed.
- Z. A. Siddique, Y. Yamamoto, T. Ohno and K. Nozaki, Inorg. Chem., 2003, 42, 6366–6378 CrossRef CAS PubMed.
- N. C. Giebink and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 235215 CrossRef.
- R. Czerwieniec, M. J. Leitl, H. H. H. Homeier and H. Yersin, Coord. Chem. Rev., 2016, 325, 2–28 CrossRef CAS.
- Y. Okano, H. Ohara, A. Kobayashi, M. Yoshida and M. Kato, Inorg. Chem., 2016, 55, 5227–5236 CrossRef CAS PubMed.
- A. V. Artem’ev, M. P. Davydova, A. S. Berezin, M. R. Ryzhikov and D. G. Samsonenko, Inorg. Chem., 2020, 59, 10699–10706 CrossRef PubMed.
- X.-W. Zhang, C.-H. Huang, M. Yang, X.-L. Chen and C.-Z. Lu, Dalton Trans., 2023, 52, 9893–9898 RSC.
- D. Volz, D. M. Zink, T. Bocksrocker, J. Friedrichs, M. Nieger, T. Baumann, U. Lemmer and S. Bräse, Chem. Mater., 2013, 25, 3414–3426 CrossRef CAS.
- C.-W. Hsu, C.-C. Lin, M.-W. Chung, Y. Chi, G.-H. Lee, P.-T. Chou, C.-H. Chang and P.-Y. Chen, J. Am. Chem. Soc., 2011, 133, 12085–12099 CrossRef CAS PubMed.
- M. Z. Shafikov, A. F. Suleymanova, R. Czerwieniec and H. Yersin, Chem. Mater., 2017, 29, 1708–1715 CrossRef CAS.
- M. Z. Shafikov, A. F. Suleymanova, R. Czerwieniec and H. Yersin, Inorg. Chem., 2017, 56, 13274–13285 CrossRef CAS PubMed.
- M. Z. Shafikov, A. F. Suleymanova, A. Schinabeck and H. Yersin, J. Phys. Chem. Lett., 2018, 9, 702–709 CrossRef CAS PubMed.
- X.-M. Gan, R. Yu, X.-L. Chen, M. Yang, L. Lin, X.-Y. Wu and C.-Z. Lu, Dalton Trans., 2018, 47, 5956–5960 RSC.
- J.-H. Jia, D. Liang, R. Yu, X.-L. Chen, L. Meng, J.-F. Chang, J.-Z. Liao, M. Yang, X.-N. Li and C.-Z. Lu, Chem. Mater., 2020, 32, 620–629 CrossRef CAS.
- T. Teng, K. Li, G. Cheng, Y. Wang, J. Wang, J. Li, C. Zhou, H. Liu, T. Zou, J. Xiong, C. Wu, H.-X. Zhang, C.-M. Che and C. Yang, Inorg. Chem., 2020, 59, 12122–12131 CrossRef CAS PubMed.
- M. Osawa, M. Hashimoto, I. Kawata and M. Hoshino, Dalton Trans., 2017, 46, 12446–12455 RSC.
- A. S. Romanov, S. T. Jones, L. Yang, P. J. Conaghan, D. Di, M. Linnolahti, D. Credgington and M. Bochmann, Adv. Opt. Mater., 2018, 6, 1801347 CrossRef.
- D. Liang, J.-H. Jia, X.-B. Cai, Y.-Q. Zhao, Z.-Q. Wang and C.-Z. Lu, Inorg. Chem. Front., 2022, 9, 6561–6566 RSC.
- A. Ying and S. Gong, Chem. – Eur. J., 2023, 29, e202301885 CrossRef CAS PubMed.
- T.-y Li, J. Schaab, P. I. Djurovich and M. E. Thompson, J. Mater. Chem. C, 2022, 10, 4674–4683 RSC.
- Y. Sakai, Y. Sagara, H. Nomura, N. Nakamura, Y. Suzuki, H. Miyazaki and C. Adachi, Chem. Commun., 2015, 51, 3181–3184 RSC.
- B. Goswami, T. J. Feuerstein, R. Yadav, S. Lebedkin, P. J. Boden, S. T. Steiger, G. Niedner-Schatteburg, M. Gerhards, M. M. Kappes and P. W. Roesky, Chem. – Eur. J., 2021, 27, 15110–15119 CrossRef PubMed.
- N. Lüdtke, J. Kuhnt, T. Heil, A. Steffen and C. M. Marian, ChemPhotoChem, 2023, 7, e202200142 CrossRef.
- O. Mrózek, M. Mitra, B. Hupp, A. Belyaev, N. Lüdtke, D. Wagner, C. Wang, O. S. Wenger, C. M. Marian and A. Steffen, Chem. – Eur. J., 2023, 29, e202203980 CrossRef PubMed.
- M. Mitra, O. Mrózek, M. Putscher, J. Guhl, B. Hupp, A. Belyaev, C. M. Marian and A. Steffen, Angew. Chem., Int. Ed., 2024, 63, e202316300 CrossRef CAS PubMed.
- K. Zhang, X. Meng and L. He, Inorg. Chem., 2023, 62, 2135–2145 CrossRef CAS PubMed.
- G. Crosby, R. Highland and K. Truesdell, Coord. Chem. Rev., 1985, 64, 41–52 CrossRef CAS.
- R. G. Highland and G. A. Crosby, Chem. Phys. Lett., 1985, 119, 454–458 CrossRef CAS.
- R. G. Highland, J. G. Brummer and G. A. Crosby, J. Phys. Chem., 1986, 90, 1593–1598 CrossRef CAS.
- N. Lüdtke, A. Steffen and C. M. Marian, Inorg. Chem., 2022, 61, 20896–20905 CrossRef PubMed.
- Innovation Center for Organic Electronics (INOEL), https://inoel.yz.yamagata-u.ac.jp/research-group/, https://inoel.yz.yamagata-u.ac.jp/yu-flec/tech-result/.
- L. Bergmann, D. M. Zink, S. Bräse, T. Baumann and D. Volz, Top. Curr. Chem., 2016, 374, 22 CrossRef PubMed.
- M. Godumala, S. Choi, M. J. Cho and D. H. Choi, J. Mater. Chem. C, 2016, 4, 11355–11381 RSC.
- B. Wex and B. R. Kaafarani, J. Mater. Chem. C, 2017, 5, 8622–8653 RSC.
- X. Cao, D. Zhang, S. Zhang, Y. Tao and W. Huang, J. Mater. Chem. C, 2017, 5, 7699–7714 RSC.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- X. Yang, X. Zhou, Y. X. Zhang, D. Li, C. Li, C. You, T. C. Chou, S. J. Su, P. T. Chou and Y. Chi, Adv. Sci., 2022, 9, e2201150 CrossRef PubMed.
- A. Thamarappalli, C. S. K. Ranasinghe, J. Jang, M. Gao, P. L. Burn, E. V. Puttock and P. E. Shaw, Adv. Funct. Mater., 2022, 32, 2205077 CrossRef CAS.
- Y. Liu, Y. Xie, Y. Cheng, X. Tian, L. Hua, S. Ying, S. Yan and Z. Ren, Chem. Eng. J., 2023, 455, 140747 CrossRef CAS.
- J. Yan, S. F. Wang, C. H. Hsu, E. H. Shi, C. C. Wu, P. T. Chou, S. M. Yiu, Y. Chi, C. You, I. C. Peng and W. Y. Hung, ACS Appl. Mater. Interfaces, 2023, 15, 21333–21343 CrossRef CAS PubMed.
- J. Yan, D. Y. Zhou, L. S. Liao, M. Kuhn, X. Zhou, S. M. Yiu and Y. Chi, Nat. Commun., 2023, 14, 6419 CrossRef CAS PubMed.
- Y. Miao, G. Wang, M. Yin, Y. Guo, B. Zhao and H. Wang, Chem. Eng. J., 2023, 461, 141921 CrossRef CAS.
- H. T. Kidanu and C.-T. Chen, J. Mater. Chem. C, 2021, 9, 15470–15476 RSC.
- Q. Wang, J.-Y. Wang, H. Zeng, L.-Y. Zhang and Z.-N. Chen, Sci. China: Chem., 2022, 65, 1559–1568 CrossRef CAS.
- Y. Feng, X. Zhuang, Y. Xu, J. Xue, C. Qu, Q. Wang, Y. Liu and Y. Wang, Chem. Eng. J., 2023, 478, 147123 CrossRef CAS.
- P. J. Conaghan, S. M. Menke, A. S. Romanov, S. T. Jones, A. J. Pearson, E. W. Evans, M. Bochmann, N. C. Greenham and D. Credgington, Adv. Mater., 2018, 30, 1870265 CrossRef.
- R. Tang, S. Xu, L. Du, F. F. Hung, T. L. Lam, G. Cheng, K. H. Low, Q. Wan, S. Wu, Y. Chen and C. M. Che, Adv. Opt. Mater., 2023, 11, 2300950 CrossRef CAS.
- S. Avula, B. H. Jhun, U. Jo, S. Heo, J. Y. Lee and Y. You, Adv. Sci., 2024, 11, 2305745 CrossRef CAS PubMed.
- S. Heo, Y. Jung, J. Kim, I. Kim, H. J. Bae, W. J. Son, H. Choi and Y. You, Adv. Opt. Mater., 2022, 10, 2201610 CrossRef CAS.
- S. Cai, G. S. M. Tong, L. Du, G. K. So, F. F. Hung, T. L. Lam, G. Cheng, H. Xiao, X. Chang, Z. X. Xu and C. M. Che, Angew. Chem., 2022, 61, e202213392 CrossRef CAS PubMed.
- X. Feng, J. G. Yang, J. Miao, C. Zhong, X. Yin, N. Li, C. Wu, Q. Zhang, Y. Chen, K. Li and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202209451 CrossRef CAS PubMed.
- W. P. To, D. Zhou, G. S. M. Tong, G. Cheng, C. Yang and C. M. Che, Angew. Chem., 2017, 129, 14224–14229 CrossRef.
- D. Zhou, G. S. M. Tong, G. Cheng, Y. K. Tang, W. Liu, D. Ma, L. Du, J. R. Chen and C. M. Che, Adv. Mater., 2022, 34, e2206598 CrossRef PubMed.
- C. C. Au-Yeung, M. Y. Leung, S. L. Lai, S. C. Cheng, L. K. Li, M. C. Tang, W. K. Kwok, C. C. Ko, M. Y. Chan and V. W. Yam, Mater. Horiz., 2024, 11, 151–162 RSC.
- L. K. Li, W. K. Kwok, M. C. Tang, W. L. Cheung, S. L. Lai, M. Ng, M. Y. Chan and V. W. Yam, Chem. Sci., 2021, 12, 14833–14844 RSC.
- C. Y. Wong, M. C. Tang, L. K. Li, M. Y. Leung, W. K. Tang, S. L. Lai, W. L. Cheung, M. Ng, M. Y. Chan and V. W. Yam, Chem. Sci., 2022, 13, 10129–10140 RSC.
- M. Osawa, I. Kawata, R. Ishii, S. Igawa, M. Hashimoto and M. Hoshino, J. Mater. Chem. C, 2013, 1, 4375–4383 RSC.
- D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. H. Rivett, S. Jones, T. H. Thomas, M. Abdi Jalebi, R. H. Friend, M. Linnolahti, M. Bochmann and D. Credgington, Science, 2017, 356, 159–163 CrossRef CAS PubMed.
- X.-L. Chen, R. Yu, Q.-K. Zhang, L.-J. Zhou, X.-Y. Wu, Q. Zhang and C.-Z. Lu, Chem. Mater., 2013, 25, 3910–3920 CrossRef CAS.
- D. Volz, Y. Chen, M. Wallesch, R. Liu, C. Fléchon, D. M. Zink, J. Friedrichs, H. Flügge, R. Steininger and J. Göttlicher, Adv. Mater., 2015, 27, 2538–2543 CrossRef CAS PubMed.
- M. Osawa, M. Hoshino, M. Hashimoto, I. Kawata, S. Igawa and M. Yashima, Dalton Trans., 2015, 44, 8369–8378 RSC.
- X.-L. Chen, C.-S. Lin, X.-Y. Wu, R. Yu, T. Teng, Q.-K. Zhang, Q. Zhang, W.-B. Yang and C.-Z. Lu, J. Mater. Chem. C, 2015, 3, 1187–1195 RSC.
- D. Liang, X.-L. Chen, J.-Z. Liao, J.-Y. Hu, J.-H. Jia and C.-Z. Lu, Inorg. Chem., 2016, 55, 7467–7475 CrossRef CAS PubMed.
- F. Zhang, Y. Guan, X. Chen, S. Wang, D. Liang, Y. Feng, S. Chen, S. Li, Z. Li and F. Zhang, Inorg. Chem., 2017, 56, 3742–3753 CrossRef CAS PubMed.
- X. Hong, B. Wang, L. Liu, X.-X. Zhong, F.-B. Li, L. Wang, W.-Y. Wong, H.-M. Qin and Y. H. Lo, J. Lumin., 2016, 180, 64–72 CrossRef CAS.
- Y.-H. Zhou, A.-W. Zhang, R.-J. Huang, Y.-H. Sun, Z.-J. Chen, B.-S. Zhu and Y.-X. Zheng, J. Mater. Chem. C, 2023, 11, 1329–1335 RSC.
- M. Wallesch, A. Verma, C. Fléchon, H. Flügge, D. M. Zink, S. M. Seifermann, J. M. Navarro, T. Vitova, J. Göttlicher and R. Steininger, Chem. – Eur. J., 2016, 22, 16400–16405 CrossRef CAS PubMed.
- J. Zhang, C. Duan, C. Han, H. Yang, Y. Wei and H. Xu, Adv. Mater., 2016, 28, 5975–5979 CrossRef CAS PubMed.
- L. Lin, D.-H. Chen, R. Yu, X.-L. Chen, W.-J. Zhu, D. Liang, J.-F. Chang, Q. Zhang and C.-Z. Lu, J. Mater. Chem. C, 2017, 5, 4495–4504 RSC.
- G. K. M. So, G. Cheng, J. Wang, X. Chang, C. C. Kwok, H. Zhang and C. M. Che, Chem. – Asian J., 2017, 12, 1490–1498 CrossRef CAS PubMed.
- S. Shi, M. C. Jung, C. Coburn, A. Tadle, D. Sylvinson MR, P. I. Djurovich, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2019, 141, 3576–3588 CrossRef CAS PubMed.
- H. J. Wang, Y. Liu, B. Yu, S. Q. Song, Y. X. Zheng, K. Liu, P. Chen, H. Wang, J. Jiang and T. Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202217195 CrossRef CAS PubMed.
- A. Ying, Y. H. Huang, C. H. Lu, Z. Chen, W. K. Lee, X. Zeng, T. Chen, X. Cao, C. C. Wu, S. Gong and C. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 13478–13486 CrossRef CAS PubMed.
- R. Tang, S. Xu, T. L. Lam, G. Cheng, L. Du, Q. Wan, J. Yang, F. F. Hung, K. H. Low, D. L. Phillips and C. M. Che, Angew. Chem., Int. Ed., 2022, 61, e202203982 CrossRef CAS PubMed.
- M. Olaru, E. Rychagova, S. Ketkov, Y. Shynkarenko, S. Yakunin, M. V. Kovalenko, A. Yablonskiy, B. Andreev, F. Kleemiss and J. Beckmann, J. Am. Chem. Soc., 2019, 142, 373–381 CrossRef PubMed.
- B.-K. Guo, F. Yang, Y.-Q. Wang, Q. Wei, L. Liu, X.-X. Zhong, L. Wang, J.-K. Gong, F.-B. Li and W.-Y. Wong, J. Lumin., 2020, 220, 116963 CrossRef CAS.
- T. Teng, J. Xiong, G. Cheng, C. Zhou, X. Lv and K. Li, Molecules, 2021, 26, 1125 CrossRef CAS PubMed.
- A. Ruduss, B. Turovska, S. Belyakov, K. A. Stucere, A. Vembris, G. Baryshnikov, H. Agren, J. C. Lu, W. H. Lin, C. H. Chang and K. Traskovskis, ACS Appl. Mater. Interfaces, 2022, 14, 15478–15493 CrossRef CAS PubMed.
- J. Guo, Z. Zhang, P. Wu, J. Zhu, D. Dou, Z. Liao, R. Xia, K. Wang and Z. Wang, J. Lumin., 2021, 239, 118354 CrossRef CAS.
- G. Cheng, D. Zhou, U. Monkowius and H. Yersin, Micromachines, 2021, 12, 1500 CrossRef PubMed.
- S. Keller, F. Brunner, J. M. Junquera-Hernandez, A. Pertegas, M. G. La-Placa, A. Prescimone, E. C. Constable, H. J. Bolink, E. Orti and C. E. Housecroft, ChemPlusChem, 2018, 83, 217–229 CrossRef CAS PubMed.
- M. Alkan-Zambada, S. Keller, L. Martínez-Sarti, A. Prescimone, J. M. Junquera-Hernández, E. C. Constable, H. J. Bolink, M. Sessolo, E. Ortí and C. E. Housecroft, J. Mater. Chem. C, 2018, 6, 8460–8471 RSC.
- S. Keller, A. Pertegás, G. Longo, L. Martínez, J. Cerdá, J. M. Junquera-Hernández, A. Prescimone, E. C. Constable, C. E. Housecroft and E. Ortí, J. Mater. Chem. C, 2016, 4, 3857–3871 RSC.
- M. D. Weber, M. Viciano-Chumillas, D. Armentano, J. Cano and R. D. Costa, Dalton Trans., 2017, 46, 6312–6323 RSC.
- S. Keller, A. Prescimone, M. G. La Placa, J. M. Junquera-Hernandez, H. J. Bolink, E. C. Constable, M. Sessolo, E. Orti and C. E. Housecroft, RSC Adv., 2020, 10, 22631–22644 RSC.
- G. Giobbio, L. M. Cavinato, E. Fresta, A. Montrieul, G. L. Umuhire Mahoro, J. François, J.-L. Renaud, M. Linares, S. Gaillard and R. D. Costa, Adv. Funct. Mater., 2023, 33, 2304668–2304678 CrossRef CAS.
- M. D. Weber, E. Fresta, M. Elie, M. E. Miehlich, J. L. Renaud, K. Meyer, S. Gaillard and R. D. Costa, Adv. Funct. Mater., 2018, 28, 1707423 CrossRef.
- L. Zhang, B. Li and Z. Su, J. Phys. Chem. C, 2009, 113, 13968–13973 CrossRef CAS.
- M. Wallesch, D. Volz, D. M. Zink, U. Schepers, M. Nieger, T. Baumann and S. Bräse, Chem. – Eur. J., 2014, 20, 6578–6590 CrossRef CAS PubMed.
- M. Hashimoto, S. Igawa, M. Yashima, I. Kawata, M. Hoshino and M. Osawa, J. Am. Chem. Soc., 2011, 133, 10348–10351 CrossRef CAS PubMed.
- Q. Zhang, T. Komino, S. Huang, S. Matsunami, K. Goushi and C. Adachi, Adv. Funct. Mater., 2012, 22, 2327–2336 CrossRef CAS.
- D. R. Evans and C. A. Reed, J. Am. Chem. Soc., 2000, 122, 4660–4667 CrossRef CAS.
- J. C. Deaton, S. C. Switalski, D. Y. Kondakov, R. H. Young, T. D. Pawlik, D. J. Giesen, S. B. Harkins, A. J. Miller, S. F. Mickenberg and J. C. Peters, J. Am. Chem. Soc., 2010, 132, 9499–9508 CrossRef CAS PubMed.
- Z. Liu, M. F. Qayyum, C. Wu, M. T. Whited, P. I. Djurovich, K. O. Hodgson, B. Hedman, E. I. Solomon and M. E. Thompson, J. Am. Chem. Soc., 2011, 133, 3700–3703 CrossRef CAS PubMed.
- X. Liu, T. Zhang, T. Ni, N. Jiang, Z. Liu, Z. Bian, Z. Lu and C. Huang, Adv. Funct. Mater., 2014, 24, 5385–5392 CrossRef CAS.
- G. Cheng, G. K.-M. So, W.-P. To, Y. Chen, C.-C. Kwok, C. Ma, X. Guan, X. Chang, W.-M. Kwok and C.-M. Che, Chem. Sci., 2015, 6, 4623–4635 RSC.
- Q. Zhang, X.-L. Chen, J. Chen, X.-Y. Wu, R. Yu and C.-Z. Lu, Dalton Trans., 2015, 44, 10022–10029 RSC.
- X. Li, H. Yan and Q. Zhao, Chem. – Eur. J., 2016, 22, 1888–1898 CrossRef CAS PubMed.
- B. P. Dash, R. Satapathy, E. R. Gaillard, J. A. Maguire and N. S. Hosmane, J. Am. Chem. Soc., 2010, 132, 6578–6587 CrossRef CAS PubMed.
- A. Ferrer-Ugalde, A. González-Campo, C. Viñas, J. Rodríguez-Romero, R. Santillan, N. Farfán, R. Sillanpää, A. Sousa-Pedrares, R. Núñez and F. Teixidor, Chem. – Eur. J., 2014, 20, 9940–9951 CrossRef CAS PubMed.
- R. Núñez, P. Farràs, F. Teixidor, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., 2006, 118, 1292–1294 CrossRef.
- L. Zhu, W. Lv, S. Liu, H. Yan, Q. Zhao and W. Huang, Chem. Commun., 2013, 49, 10638–10640 RSC.
- C. Shi, H. Sun, Q. Jiang, Q. Zhao, J. Wang, W. Huang and H. Yan, Chem. Commun., 2013, 49, 4746–4748 RSC.
- N. S. Hosmane, Boron science: new technologies and applications, CRC Press, 2016 Search PubMed.
- H. Naito, Y. Morisaki and Y. Chujo, Angew. Chem., Int. Ed., 2015, 54, 5084–5087 CrossRef CAS PubMed.
- A. Ferrer-Ugalde, E. J. Juárez-Pérez, F. Teixidor, C. Viñas, R. Sillanpää, E. Pérez-Inestrosa and R. Núñez, Chem. – Eur. J., 2012, 18, 544–553 CrossRef CAS PubMed.
- Y. Ma, S. Liu, H. Yang, Y. Wu, C. Yang, X. Liu, Q. Zhao, H. Wu, J. Liang and F. Li, J. Mater. Chem., 2011, 21, 18974–18982 RSC.
- A. M. Prokhorov, T. Hofbeck, R. Czerwieniec, A. F. Suleymanova, D. N. Kozhevnikov and H. Yersin, J. Am. Chem. Soc., 2014, 136, 9637–9642 CrossRef CAS PubMed.
- J. Guo, D. Liu, J. Zhang, J. Zhang, Q. Miao and Z. Xie, Chem. Commun., 2015, 51, 12004–12007 RSC.
- C. Shi, H. Sun, X. Tang, W. Lv, H. Yan, Q. Zhao, J. Wang and W. Huang, Angew. Chem., 2013, 125, 13676–13680 CrossRef.
- M. Mohankumar, F. Monti, M. Holler, F. Niess, B. Delavaux-Nicot, N. Armaroli, J. P. Sauvage and J. F. Nierengarten, Chem. – Eur. J., 2014, 20, 12083–12090 CrossRef CAS PubMed.
- M. Mohankumar, M. Holler, J. F. Nierengarten and J. P. Sauvage, Chem. – Eur. J., 2012, 18, 12192–12195 CrossRef CAS PubMed.
- N. Armaroli, G. Accorsi, M. Holler, O. Moudam, J. F. Nierengarten, Z. Zhou, R. T. Wegh and R. Welter, Adv. Mater., 2006, 18, 1313–1316 CrossRef CAS.
- D. G. Cuttell, S.-M. Kuang, P. E. Fanwick, D. R. McMillin and R. A. Walton, J. Am. Chem. Soc., 2002, 124, 6–7 CrossRef CAS PubMed.
- S.-M. Kuang, D. G. Cuttell, D. R. McMillin, P. E. Fanwick and R. A. Walton, Inorg. Chem., 2002, 41, 3313–3322 CrossRef CAS PubMed.
- G. A. Blake, J. P. Moerdyk and C. W. Bielawski, Organometallics, 2012, 31, 3373–3378 CrossRef CAS.
- M. Braun, W. Frank, G. J. Reiss and C. Ganter, Organometallics, 2010, 29, 4418–4420 CrossRef CAS.
- T. W. Hudnall and C. W. Bielawski, J. Am. Chem. Soc., 2009, 131, 16039–16041 CrossRef CAS PubMed.
- V. César, N. Lugan and G. Lavigne, Eur. J. Inorg. Chem., 2010, 361–365 CrossRef.
- L. Kang, J. Chen, T. Teng, X.-L. Chen, R. Yu and C.-Z. Lu, Dalton Trans., 2015, 44, 11649–11659 RSC.
- P. Jin, Z. Zhou, H. Wang, J. Hao, R. Chen, J. Wang and C. Zhang, J. Phys. Chem. Lett., 2022, 13, 2516–2522 CrossRef CAS PubMed.
- E. Fresta, M. D. Weber, J. Fernandez-Cestau and R. D. J. A. O. M. Costa, Adv. Opt. Mater., 2019, 7, 1900830 CrossRef CAS.
- S. Lipinski, L. M. Cavinato, T. Pickl, G. Biffi, A. Pöthig, P. B. Coto, J. Fernández-Cestau and R. Costa, Adv. Opt. Mater., 2023, 11, 2203145 CrossRef CAS.
- E. Fresta, J. M. Carbonell-Vilar, J. Yu, D. Armentano, J. Cano, M. Viciano-Chumillas and R. D. J. A. F. M. Costa, Adv. Funct. Mater., 2019, 29, 1901797 CrossRef.
- E. Fresta and R. D. Costa, Adv. Funct. Mater., 2020, 30, 1908176 CrossRef CAS.
- R. Bai, X. Meng, X. Wang and L. He, Adv. Funct. Mater., 2021, 31, 2007167 CrossRef CAS.
- Y.-M. Wang, F. Teng, Y.-B. Hou, Z. Xu, Y.-S. Wang and W.-F. Fu, Appl. Phys. Lett., 2005, 87, 233512 CrossRef.
- M. D. Weber, C. Garino, G. Volpi, E. Casamassa, M. Milanesio, C. Barolo and R. D. Costa, Dalton Trans., 2016, 45, 8984–8993 RSC.
- G. Kalyuzhny, M. Buda, J. McNeill, P. Barbara and A. J. Bard, J. Am. Chem. Soc., 2003, 125, 6272–6283 CrossRef CAS PubMed.
- E. Fresta, G. Volpi, M. Milanesio, C. Garino, C. Barolo and R. D. Costa, Inorg. Chem., 2018, 57, 10469–10479 CrossRef CAS PubMed.
- O. Moudam, A. C. Tsipis, S. Kommanaboyina, P. N. Horton and S. J. Coles, RSC Adv., 2015, 5, 95047–95053 RSC.
- J. Ràfols-Ribé, E. Gracia-Espino, S. Jenatsch, P. Lundberg, A. Sandström, S. Tang, C. Larsen and L. J. A. O. M. Edman, Adv. Opt. Mater., 2021, 9, 2001405 CrossRef.
- M. D. Weber, J. E. Wittmann, A. Burger, O. B. Malcıoğlu, J. Segarra-Martí, A. Hirsch, P. B. Coto, M. Bockstedte and R. D. J. A. F. M. Costa, Adv. Funct. Mater., 2016, 26, 6737–6750 CrossRef CAS.
- E. Fresta, K. Baumgärtner, J. Cabanillas-Gonzalez, M. Mastalerz and R. D. Costa, Nanoscale Horiz., 2020, 5, 473–480 RSC.
- E. Fresta, J. Dosso, J. Cabanillas-Gonzalez, D. Bonifazi and R. D. Costa, ACS Appl. Mater. Interfaces, 2020, 12, 28426–28434 CrossRef CAS PubMed.
- E. Fresta, J. Dosso, J. Cabanillas-González, D. Bonifazi and R. Costa, Adv. Funct. Mater., 2020, 30, 1906830 CrossRef CAS.
- S. Keller, A. Prescimone, H. Bolink, M. Sessolo, G. Longo, L. Martínez-Sarti, J. M. Junquera-Hernández, E. C. Constable, E. Ortí and C. E. Housecroft, Dalton Trans., 2018, 47, 14263–14276 RSC.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- M. Karaman, A. Kumar Gupta, S. Madayanad Suresh, T. Matulaitis, L. Mardegan, D. Tordera, H. J. Bolink, S. Wu, S. Warriner, I. D. Samuel and E. Zysman-Colman, Beilstein J. Org. Chem., 2022, 18, 1311–1321 CrossRef CAS PubMed.
- K. Zhang, X. Pang, Y. Song, Y. Xiu, R. Yu and L. He, Adv. Opt. Mater., 2024, 12, 2400467 CrossRef CAS.
Footnote |
| † Equally contributed. |
| This journal is © The Royal Society of Chemistry 2025 |