
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDissecting errors in machine learning for retrosynthesis: a granular metric framework and a transformer-based model for more informative predictions
Arihanth Srikar
Tadanki
 a,
H.
Surya Prakash Rao
b and
U. Deva
Priyakumar
*a
a,
H.
Surya Prakash Rao
b and
U. Deva
Priyakumar
*a
aCenter for Computational Natural Sciences and Bioinformatics, International Institute of Information Technology, Hyderabad 500 032, India. E-mail: deva@iiit.ac.in
bTeadus Pharma Pvt. Ltd, Hyderabad, India
First published on 18th February 2025
Abstract
Chemical reaction prediction, encompassing forward synthesis and retrosynthesis, stands as a fundamental challenge in organic synthesis. A widely adopted computational approach frames synthesis prediction as a sequence-to-sequence translation task, using the commonly used SMILES representation for molecules. The current evaluation of machine learning methods for retrosynthesis assumes perfect training data, overlooking imperfections in reaction equations in popular datasets, such as missing reactants, products, other physical and practical constraints such as temperature and cost, primarily due to a focus on the target molecule. This limitation leads to an incomplete representation of viable synthetic routes, especially when multiple sets of reactants can yield a given desired product. In response to these shortcomings, this study examines the prevailing evaluation methods and introduces comprehensive metrics designed to address imperfections in the dataset. Our novel metrics not only assess absolute accuracy by comparing predicted outputs with ground truth but also introduce a nuanced evaluation approach. We provide scores for partial correctness and compute adjusted accuracy through graph matching, acknowledging the inherent complexities of retrosynthetic pathways. Additionally, we explore the impact of small molecular augmentations while preserving chemical properties and employ similarity matching to enhance the assessment of prediction quality. We introduce SynFormer, a sequence-to-sequence model tailored for SMILES representation. It incorporates architectural enhancements to the original transformer, effectively tackling the challenges of chemical reaction prediction. SynFormer achieves a Top-1 accuracy of 53.2% on the USPTO-50k dataset, matching the performance of widely accepted models like Chemformer, but with greater efficiency by eliminating the need for pre-training.
1 Introduction
Retrosynthetic analysis,1 a fundamental problem in organic synthesis, involves predicting the possible reaction precursors given a desired product. Synthesis involves predicting the reaction outcome based on a given precursor, primarily focusing on small to medium sized molecules. In contrast, retrosynthesis can be conceptualized as the inverse of synthesis,2 posing a notably more challenging task. In retrosynthesis, the information provided is generally limited to the molecule of interest, which can be synthesized through multiple feasible pathways or replaced by synthetic equivalents. Conventional retrosynthesis involves the meticulous deconstruction of target molecules into simpler precursors, relying on chemists' expertise in organic chemistry principles and synthetic methodologies. However, this manual process demands extensive knowledge and experience. Computational retrosynthesis addresses these limitations by leveraging extensive reaction databases and algorithms to propose diverse synthetic routes efficiently.The problem of retrosynthesis predictions was initially addressed by reaction-template-based methods such as LHASA3 and SYNTHIA.4 While effective for a limited set of reactions, these methods heavily rely on atom mapping, impacting model performance. Moreover, maintaining template databases posed challenges, leading to their limited adoption. Recent efforts have shifted towards neural network approaches, including template classification5–8 and template re-ranking based on molecular similarity.9 Despite their usefulness, template-based methods suffer from a trade off between generality and specificity, and they struggle to generalize to unseen templates.
To overcome these limitations, template-free approaches have gained prominence and are primarily categorized into graph edit-based and translation-based methods. Graph edit-based methods10–14 model reaction prediction and retrosynthesis as graph transformations, with some incorporating template information in semi-template-based methods.15–18 On the other hand, early neural machine translation methods were based on sequence-to-sequence models such as Recurrent Neural Networks (RNNs),19–21 followed by transformer approaches22–27 which are widely adopted due to their simpler end-to-end training and well-optimized neural architectures from Natural Language Processing (NLP).
Neural machine translation methods rely on SMILES28 representations of molecules for retrosynthesis; however, SMILES lack a bijective mapping to molecular structures, posing a challenge in mapping unique SMILES representations to the same point in the latent space. To overcome this, experimental evidence suggests that data augmentation with chemically equivalent SMILES29 enhances empirical performance, as demonstrated by a study30 showing an 8.1% improvement in uniquely generated molecules through SMILES randomization. Additionally, pre-training models on large datasets with data augmentation is a common practice to improve generalizability and convergence on downstream tasks,31 leading to a notable increase in Top-1 accuracy from 50.7% to 53.3% in single-step retrosynthetic predictions, as evidenced by Chemformer.22 However, pre-training masked language models may reach a point of diminishing returns as the size of the supervised dataset increases significantly,32 prompting questions about the necessity of pre-training. Nevertheless, there has been limited exploration of adapting or modifying this architecture specifically for efficiently improving retrosynthetic analysis.
Current methods for retrosynthetic analysis rely on the USPTO-50k33 dataset, the gold standard for benchmarking model performance. Each reaction within this dataset comprises a single molecule of interest and can contain multiple reactant molecules. The USPTO-50k dataset lacks crucial information necessary for accurately determining product outcomes, such as solvents, catalysts, reagents, and reaction conditions. Additionally, the dataset typically presents only one set of possible reactants for each product of interest, overlooking the possibility of multiple chemically viable reactant molecules capable of producing the same product, thus neglecting alternate pathways.
In this study, we analyze misclassified reactions across various methods, revealing that some predictions are less incorrect than others as assessed by an organic chemistry expert. We identify distinct categories of incorrect predictions related to the set of reactant molecules: (i) complete misidentification, (ii) incorrect stereochemistry,8 (iii) partial incorrectness, and (iv) inaccurate substructure recognition,27 particularly concerning leaving groups. To address this challenge, we propose the Retro-Synth Score (R-SS), a metric focused on recognizing “better mistakes” and ranking methods based on the degree of correctness of their predictions.
Despite the slight enhancement in performance afforded by pre-training, the process is computationally expensive and demands extensive hyper-parameter tuning, as well as the identification of optimal pre-training strategies, which are typically determined empirically. In response to these challenges, we introduce SynFormer, a transformer-based model that matches previous state-of-the-art models under a more comprehensive evaluation methodology, all while achieving a five-fold reduction in training time compared to Chemformer,22 accomplished by eliminating the need for pre-training.
Our main contributions can be summarised as follows:
(1) We propose the Retro-Synth Score (R-SS), a more nuanced and realistic evaluation method that measures the accuracy of models and evaluates the quality of errors.
(2) We propose SynFormer, a sequence-to-sequence model for SMILES with architectural modifications to the original transformer, enabling better generalization and improved performance on single-step retrosynthetic predictions without pre-training.
2 Methods
2.1 Dataset
We evaluate single-step retrosynthesis performance using the Retro-Synth Score (R-SS) on the USPTO-50k dataset, a compilation of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 037 reactions sourced from US patents spanning 1976 to 2016 and originally curated by Lowe.34 While this dataset is fundamental for retrosynthetic analysis, it lacks crucial information such as physical conditions (e.g., temperature) and chemical inputs (e.g., solvents, reagents, and by-products). This absence compromises atom conservation principles and the completeness of reaction data, hindering thorough algorithmic evaluation and potentially mislabeling valid alternatives as incorrect pathways. Additionally, it lacks practical information such as cost, availability, ease of preparation, and regulatory considerations, which are vital factors in selecting molecules.
037 reactions sourced from US patents spanning 1976 to 2016 and originally curated by Lowe.34 While this dataset is fundamental for retrosynthetic analysis, it lacks crucial information such as physical conditions (e.g., temperature) and chemical inputs (e.g., solvents, reagents, and by-products). This absence compromises atom conservation principles and the completeness of reaction data, hindering thorough algorithmic evaluation and potentially mislabeling valid alternatives as incorrect pathways. Additionally, it lacks practical information such as cost, availability, ease of preparation, and regulatory considerations, which are vital factors in selecting molecules.
In this work, we represent molecules as SMILES. The inherent ordering introduced by the SMILES molecular representation poses a challenge, resulting in a many-to-one mapping issue. This characteristic impedes the training of models aiming to precisely match predicted SMILES with reactant SMILES in retrosynthetic analysis, thereby affecting the models' generalization capability. While pre-training and data augmentation techniques offer a potential solution, they increase training time. Recognizing and addressing these dataset limitations are crucial for accurate algorithmic evaluation and effective molecular representation in chemical synthesis studies.
2.2 Retro-Synth Score (R-SS)
Algorithms for retrosynthesis typically assume perfect training data and primarily rely on accuracy to evaluate model correctness.35 However, efforts to address the limitations of accuracy include metrics like MaxFrag accuracy,27 which focuses on the largest fragment match to overcome prediction limitations, and metrics that ignore stereochemistry8,36 for a more relaxed evaluation. Additionally, metrics such as Top-N accuracy, round trip accuracy, coverage, and diversity aim to capture the effectiveness and quality of predictions.36 Despite their usefulness, these metrics individually do not provide a comprehensive assessment of model performance. For instance, Top-N accuracy has been criticized for prioritizing frequently observed answers over chemically meaningful predictions, while increasing suggestions (through beam search or similar methods) may lead to a decrease in valid suggestions and significantly increase the inference time. Similarly, while round-trip accuracy37 is informative, it requires an additional pre-trained forward-synthesis prediction model, introducing complexity and computational overhead.In response, we combine accuracy, stereo-agnostic accuracy, partial correctness, and Tanimoto similarity38 to create a new set of metrics, the computation of which is shown in Fig. 1. These metrics are calculated based on two distinct settings: halogen-sensitive and halogen-agnostic. We define each metric below. Detailed information regarding each metric can be found at Section A.
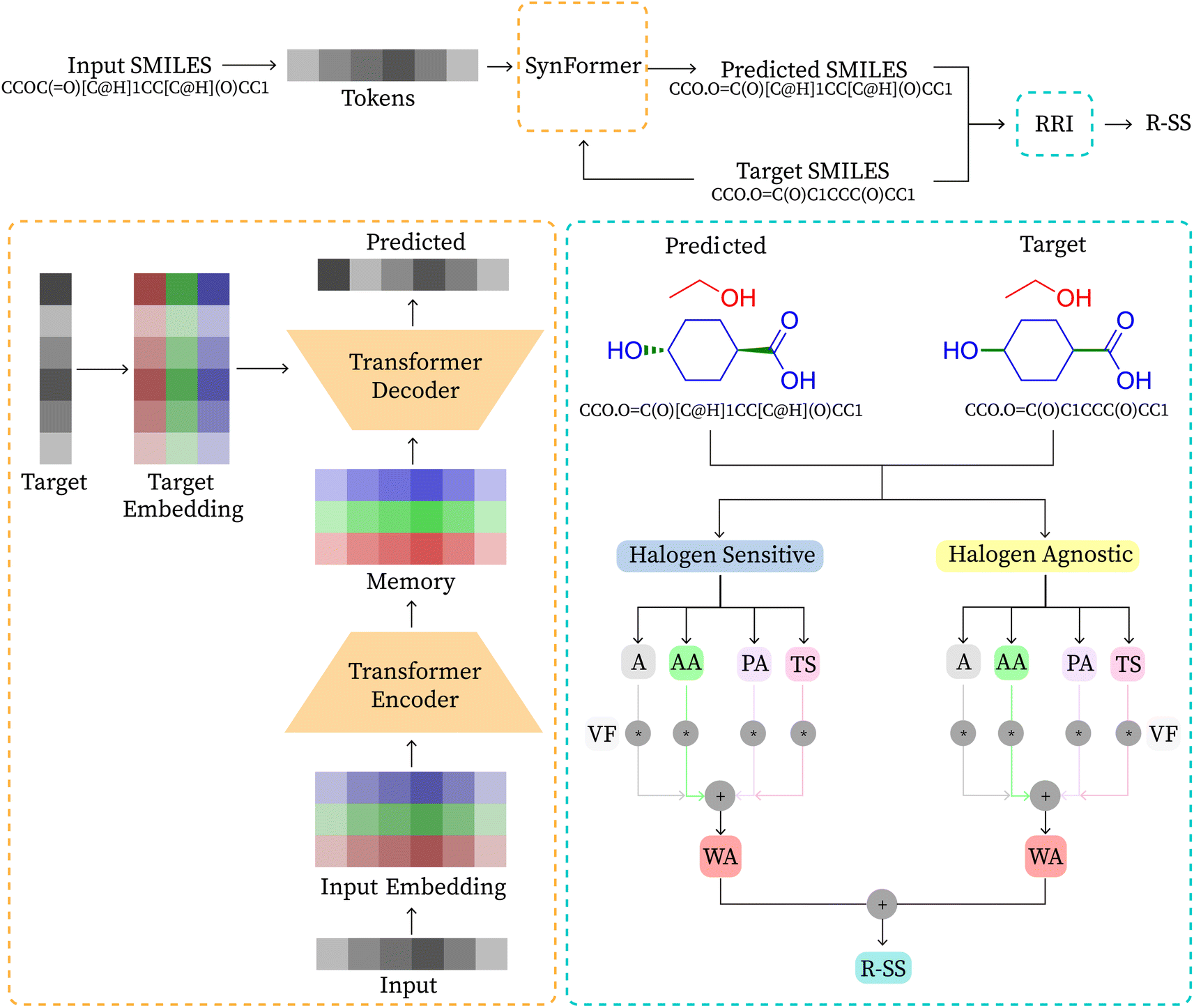 | ||
| Fig. 1 Model architecture for SynFormer and computation of R-SS. Top: the overall flowchart. Bottom left: encoder–decoder transformer architecture of SynFormer. Bottom right: calculation of the Retro-Synth Score (R-SS). | ||
• Similar reactivity: the halogens being replaced must exhibit similar reactivity. Fluorine, chlorine, bromine, and iodine are often interchangeable in reactions involving halogen substitution.
• Reaction conditions: consistency in reaction conditions, such as temperature, pressure, solvent, and catalysts, is essential. Changes in these parameters can influence reaction pathways and product formation. However, datasets like USPTO-50k typically lack this information.
• Substrate compatibility: the substitution should be compatible with the substrate and other functional groups present in the molecule. Some substrates may exhibit selectivity toward specific halogens due to steric or electronic effects. The detailed case-study will show that the examples in the dataset allow for relaxing this constraint.
• By-product consideration: differences in by-products resulting from substitution should not affect the desired product or its downstream synthetic steps. In retrosynthetic analysis, the focus is typically on the main reaction pathway, making by-products irrelevant.
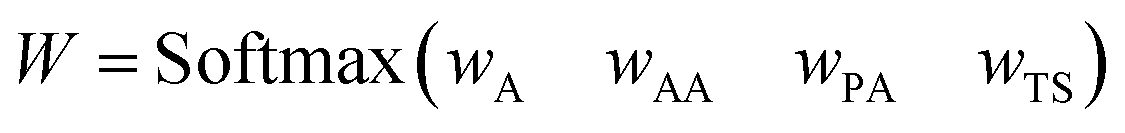 | (1) |
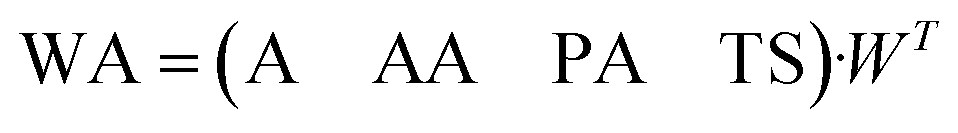 | (2) |
 | (3) |
 | (4) |
| Category | Abbreviation | Expansion | Evaluator |
|---|---|---|---|
| Condition | PT | Pre-trained | |
| HA | Halogen agnostic | ||
| Computed metrics | A | Top-1 accuracy | Perfect match |
| AA | Stereo-agnostic accuracy | Structural match | |
| PA | Partial accuracy | Fraction of perfect match | |
| TS | Tanimoto similarity | Structural similarity | |
| VF | Validity factor | Chemical correctness | |
| Derived metrics | WA | Weighted average | |
| R-SS | Retro-synth score |
The importance of each metric is chosen empirically, where wA = 1, wAA = 2, wPA = 4, and wTS = 1. Tanimoto similarity holds the same weight as accuracy, stereo-agnostic accuracy is twice as important as accuracy, and partial accuracy carries twice the weight of stereo-agnostic accuracy. This weighting reflects how each metric relaxes the evaluation criteria and encompasses different aspects of the chemical context, as explained in their descriptions. Metrics with higher importance indicate a greater likelihood of correctness under a valid chemically alternate route, as demonstrated by a case study. Additionally, we show that varying the weights does not change the trend of the reported results. We demonstrate the application of our metric through an example reaction presented in Table 2.
| P dataset | R dataset | R predicted | HA | A | AA | PA | TS | WA | R-SS |
|---|---|---|---|---|---|---|---|---|---|
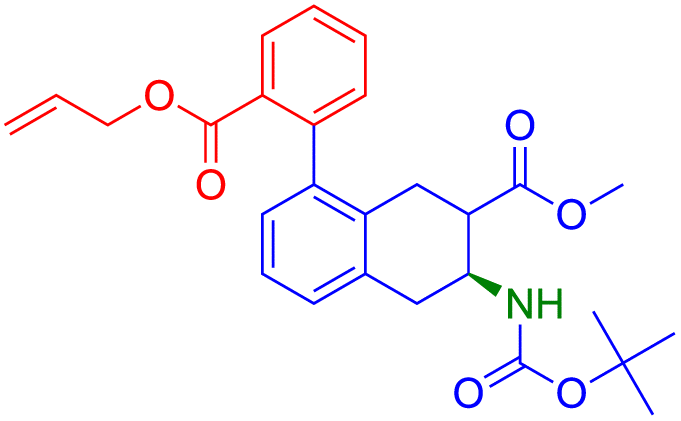
|
|

|
✗ | 0.00 | 0.00 | 0.50 | 0.77 | 0.44 | 0.69 |
| ✓ | 0.00 | 1.00 | 1.00 | 0.77 | 0.95 |
R-SS prioritizes Top-1 accuracy over Top-N due to beam search limitations, where larger beam sizes often produce invalid SMILES and degrade output quality across beams. The Top-1 result is typically the most reliable, and validating multiple outputs is impractical for chemists. R-SS also achieves efficiency by avoiding the need for evaluation across multiple beams for the same molecule and minimizing reliance on transformer pre-training.
2.3 SynFormer
A transformer model41 is a type of deep learning architecture and is primarily used for sequence-to-sequence tasks, such as machine translation and text generation. It consists of multiple layers of attention mechanisms and feed-forward neural networks. In transformer models, each input token is processed independently, allowing for parallel computation and capturing long-range dependencies effectively. Encoder-only models exclusively employ the encoder component of the transformer, extracting relevant features from the input sequence. Conversely, decoder-only models focus on generating the output sequence based on the encoded input. Encoder–decoder models combine both encoder and decoder components, commonly employed in tasks like machine translation and text summarization, where the model first processes the input sequence and then generates the output sequence accordingly. In this study, we adopt the encoder–decoder architecture, as illustrated in Fig. 1, where the decoder produces the reactant SMILES conditioned on the features extracted by the encoder from the product SMILES.Large language models (LLMs) such as BERT,42 BART,43 GPT,44,45 and LLaMA46 have revolutionized self-supervised learning in the field of NLP. Several advancements have been made to improve the performance of these algorithms by making modifications to the original transformer architecture. Positional encoding techniques such as No Position Encoding (NoPE),47 Absolute Position Encoding (APE),41 Rotary Position Encoding (RoPE),48 T5 relative bias,49 and ALiBi50 have been developed to encode positional information effectively. Additionally, studies have been conducted51–55 to compare activation functions such as ReLU,56 ReLU2,51 and various GLU variants like GELU, ReGLU, and Swish to determine the best option for the task.
In this work, we design SynFormer based on the original transformer encoder–decoder architecture, replace positional embeddings with rotary embeddings, and use RELU2 as the activation function. Detailed information regarding the architecture can be found in Section B.
2.4 Model training
We train the model for 1000 epochs with an effective batch size of 96, using the AdamW optimizer. The learning rate is set to 0.001, with betas of 0.9 and 0.999, and a weight decay of 0.1.58 We employ a one-cycle learning rate scheduler with a dividing factor of 10000. Data augmentation is applied at each epoch by shuffling the atom order within molecules, achieved by randomizing SMILES representations with a 50% probability. Additional details on data augmentations are mentioned in Appendix C. The dataset is split into training, testing, and validation sets with an 8:1:1 ratio. All methods in this work are trained, tested, and evaluated using the same data split. For SynFormer, output SMILES for R-SS computation are generated using Top-k sampling with k set to 1 and a temperature of 1.0. For Top-3, Top-5, and Top-10 results, we use beam search with 3, 5, and 10 beams respectively to generate the reactant SMILES. Beam search is used for generating SMILES in Chemformer, Chemformer PT, and Graph2SMILES. SynFormer training is conducted on 4 NVIDIA GeForce RTX 2080 Ti GPUs, taking approximately 13.5 hours.
2.5 Implementation details
SynFormer is implemented using the PyTorch Lightning59 and X-transformers60 frameworks, following an encoder–decoder-based transformer model similar to Chemformer. The model architecture includes 6 transformer layers, each equipped with 8 heads in the multi-attention block, and no bias in the feed-forward network. A bottleneck dimension of 512 and a feed-forward dimension of 2048 is specified, with a dropout rate of 0.1 applied to the layers, attention, and feed-forward components of both the encoder and decoder. The token embedding layers of the encoder and decoder are tied, and residual attention is omitted. Operating on a vocabulary size of 576, the model can accommodate a maximum context length of 512. Tokenization and augmentation of SMILES are adapted from Chemformer, leveraging the PySMILESUtils framework.612.6 Experiments
Graph2SMILES. A graph-based approach consists of a graph encoder, a transformer encoder and SMILES decoder as shown in Fig. 2. They eliminate input-side augmentation by leveraging the permutation invariant graph representation which is then fed to a global attention transformer based encoder with graph-aware positional embeddings to generalise across multiple molecules and finally decoded autoregressively by a transformer decoder. The scale of their architecture is comparable to that of SynFormer, using 6 layers and 8 attention heads with a bottle-neck dimension of 256 and feed forward dimension of 2048 in both encoders and the decoder.
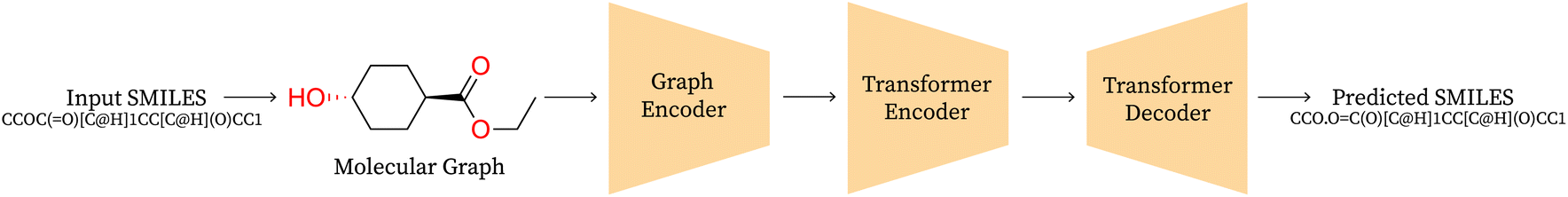 | ||
| Fig. 2 Graph2SMILES architecture. | ||
Chemformer. A sequence-to-sequence based translation approach on SMILES using the BART language model. We utilise both pre-trained and randomly initialised models which are then finetuned on USPTO-50k. We pick the small model as the parameters are comparable to SynFormer, using 6 layers and 8 attention heads with a bottle-neck dimension of 512 and a feed forward dimension of 2048 in the encoder and the decoder.
3 Results and discussions
3.1 Graph vs. language models: evaluating approaches on USPTO-50k
The rise of transformer approaches in retrosynthesis reflects advancements in natural language processing. Unlike graph-based methods, these approaches operate on SMILES, which is not bijectively mapped to the molecular structure and lacks permutation invariance, a key advantage of graphs. Despite these limitations, accuracy remains the most widely used evaluation metric. It measures the correctness of a prediction using stringent string matching of the canonical representation of the set of molecules, making it highly sensitive to even the smallest molecular augmentations, structural changes, and stereochemical alterations.The comparison between Chemformer PT and Graph2SMILES, as shown in Table 3, offers insights beyond simple accuracy metrics. Chemformer PT outperforms Graph2SMILES across all metrics, with a 5.45% higher Retro-Synth Score. However, Graph2SMILES demonstrates better Top-3, Top-5, and Top-10 accuracy from Table 4, indicating its strength in predicting alternate chemical routes.
| Algorithm | HA | A | AA | PA | TS | WA | R-SS |
|---|---|---|---|---|---|---|---|
| Graph2SMILES57 | ✗ | 0.483 | 0.510 | 0.565 | 0.765 | 0.564 | 0.572 |
| ✓ | 0.509 | 0.537 | 0.581 | 0.765 | 0.580 | ||
| Chemformer22 | ✗ | 0.507 | 0.527 | 0.577 | 0.776 | 0.577 | 0.585 |
| ✓ | 0.533 | 0.553 | 0.592 | 0.776 | 0.593 | ||
| Chemformer PT22 | ✗ | 0.533 | 0.545 | 0.599 | 0.786 | 0.598 | 0.605 |
| ✓ | 0.560 | 0.571 | 0.613 | 0.786 | 0.613 | ||
| SynFormer (ours) | ✗ | 0.532 | 0.548 | 0.598 | 0.784 | 0.598 | 0.606 |
| ✓ | 0.558 | 0.575 | 0.613 | 0.784 | 0.613 |
| Algorithm | Top-3 accuracy | Top-5 accuracy | Top-10 accuracy |
|---|---|---|---|
| Chemformer PT | — | 0.611 | 0.617 |
| SynFormer | 0.596 | 0.612 | 0.623 |
| Graph2SMILES | 0.636 | 0.679 | 0.709 |
The trend of leading in Top-1 accuracy is expected from Chemformer PT, given its extensive training on a vast molecular structure database containing 100 million molecules,62 followed by fine-tuning on 50000 reactions in USPTO-50k. Language models excel in all metrics of the R-SS, showcasing better generalization when predicting multiple molecules. This advantage likely stems from input side augmentations, which provide multiple SMILES representations of the same molecule by shuffling the order of atoms and molecules. Studies show that these augmentations, achieved by randomizing SMILES, significantly improve performance by 8.1% in generating molecules.30 We found an improvement of almost 10% in SynFormer's performance (Table 5) through input-side SMILES randomization.
| Algorithm | R-SS |
|---|---|
| SynFormer | |
| With rotary embeddings and ReLU2, and without input side augmentation | 0.551 |
| With GELU activation function | 0.593 |
| With learnt positional embeddings | 0.594 |
| With residuals and cross-attention residuals | 0.596 |
| With rotary embeddings and ReLU2 | 0.606 |
While both graph-based and language-based methods effectively predict alternate chemical routes, they differ in how accessible these routes are. Language models, like Chemformer PT, excel in the Retro-Synth Score, showcasing superior pattern recognition in molecule building for retrosynthesis. Higher Top-1 accuracy, stereo-agnostic accuracy, partial accuracy, and Tanimoto similarity (both halogen-sensitive and halogen-agnostic) reflect Chemformer PT's ability to propose valid alternate pathways. These predictions often require minimal intervention from chemists, as the differences between the predicted and actual reactants from USPTO-50k are usually minor molecular augmentations. In contrast, Graph2SMILES leads in Top-N predictions, offering alternate pathways that often need to be verified by chemists due to significant variations in the predictions. This is because Top-k accuracy utilizes beam search to generate multiple sets of reactant molecules, where each set may differ considerably. Additionally, beam search suffers from declining quality as the number of beams increases, as reflected in the plateauing of Top-N accuracy at around 73% with a beam size of 20.
Despite these differences, the performance gap between graph-based methods and language models is smaller than expected. Graph2SMILES shows robust performance, challenging assumptions about its inferiority to pre-trained language models. Moreover, it eliminates the need for pre-training, raising questions about the necessity of extensive pre-training for models like Chemformer PT. These findings highlight the strengths of graph-based representations while pointing to the efficiency and generalization capabilities of language models.
3.2 Improvement over language model with pre-training
While SynFormer and Chemformer PT share similarities in model size and the use of SMILES with input-side augmentations, they differ significantly in training data volume. Chemformer PT, pre-trained on 2000 times more data than USPTO-50k, has a slight edge in accuracy, outperforming SynFormer by 0.36%. However, this comes at the cost of six times longer training time and greater computational resources. Despite having less exposure to chemical structures, SynFormer achieves a 0.17% higher Retro-Synth Score and leads Chemformer PT in stereo-agnostic accuracy by 0.70% under halogen-agnostic conditions, demonstrating its ability to capture underlying data patterns effectively. SynFormer matches the performance of the pre-trained Chemformer PT with fewer training data across R-SS and metrics like Top-5 and Top-10 accuracy, as shown in Table 4.3.3 Improvement over language model without pre-training
Table 7 presents a summary of the results from comparing SynFormer to Chemformer on the USPTO-50k dataset. SynFormer demonstrates a notable performance improvement of 3.47% over Chemformer, achieved under identical training conditions and with an equal amount of data, all without pre-training. Across all metrics, SynFormer consistently outperforms Chemformer by a significant margin. Additionally, SynFormer exhibits better performance compared to other methods that rely solely on fine-tuning, utilizing only data augmentations without the need for pre-training on extensive reaction databases, thus highlighting its superior generalizability and efficiency.This comparison is particularly significant as it highlights the barrier to training language models. By omitting the pre-training step, it becomes feasible to train the network on USPTO-50k using a much smaller machine with a single GPU and limited RAM. While we acknowledge that pre-training is a one-time process, it is expensive to select the right training strategy and perform hyperparameter tuning.
3.4 Ablation study
Table 5 provides a summary of the Retro-Synth Scores obtained from different variants of SynFormer. These variants include using the ReLU2 activation function, employing rotary embeddings, and omitting residual connections. Interestingly, the results show that ReLU2 outperforms other activation functions, while rotary embeddings surpass learned positional embeddings. Surprisingly, the variant without residual connections demonstrates improved performance in this context. All the above experiments were run with 50% input side augmentation of SMILES unless specified otherwise. The run without any data augmentation did not generalise well on the test set.3.5 R-SS weights
In this paper, we empirically determine the importance of each metric, setting wA = 1, wAA = 2, wPA = 4, and wTS = 1. We demonstrate that the overall trend remains consistent even when the weights are altered in Table 6. Additionally, we compare our chosen weights to scenarios where all metrics are weighted equally and where all metrics except Tanimoto are weighted equally due to differences in the numerical range.3.6 Qualitative results
We show the merit of a relaxed evaluation metric, taking an example for each index that we introduce. Fig. 3 demonstrates examples where accuracy fails to capture valid predictions due to strict string matching of canonical smiles, resulting in incorrect classifications.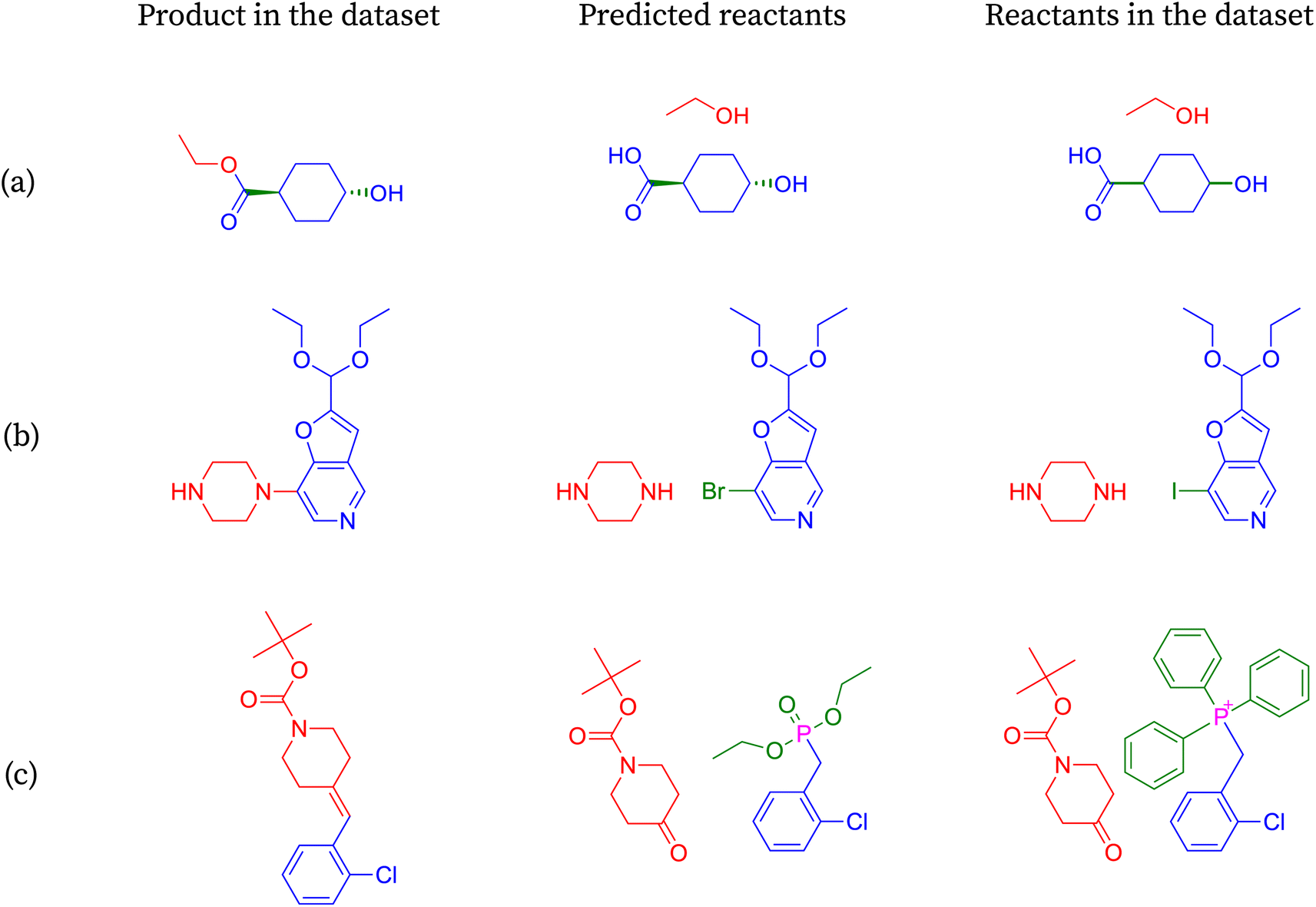 | ||
| Fig. 3 SynFormer prediction output evaluated with the Retro-Synth Score. These reactions are classified as incorrect by accuracy and correct under the introduced metrics, suggesting a valid alternate reaction pathway. The reactions are ordered by metrics (a) stereo-agnostic accuracy, (b) halogen agnostic environment, and (c) partial accuracy. | ||
In reaction shown in Fig. 3a, the predicted molecules differ from the target molecules only in their stereochemistry. The stereochemistry of the predicted molecules matches the input molecule and appears reasonable to infer its correctness. However, due to the stringent string matching of SMILES, this reaction prediction is deemed incorrect according to accuracy, with the reason being ambiguous.
Consider reaction from Fig. 3b, where one of the molecules remains the same while the other differs only in the halogen present between the target and the predicted molecules. In this case, we lack information such as the by-product of the reaction to precisely determine the leaving group. However, the reaction remains chemically viable when iodine is replaced by bromine at the same position. Moreover, the reaction will remain chemically viable when iodine is replaced by any halogen. Once again, accuracy fails to capture chemically equivalent species.
The transformation shown in Fig. 3c involves the olefination of a ketone using either the Wittig or Wittig–Horner reagent. While SynFormer accurately predicts the reactant ketone, it differs in its choice of reagent. SynFormer predicts the Wittig–Horner reagent, whereas the literature specifies the classical Wittig reagent. Both reagents react with the ketone in the presence of a suitable base and under appropriate conditions to produce the target olefinic product. This highlights the nuances that our metric identifies, which are overlooked by standard accuracy measures.
3.7 Case study
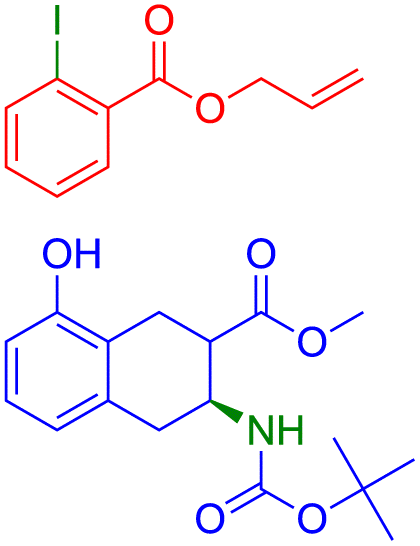
We present a selection of reactions categorized according to our evaluation metrics for a comprehensive analysis of outcomes. The reactions chosen are incorrectly predicted according to accuracy and correctly predicted according to our metrics and evaluated by an expert. We lack information regarding yield or reaction conditions such as solvent, temperature, concentration, and time which are crucial parameters for the success of a reaction and should be considered during the design phase.We analyze SynFormer's predicted reactants alongside the reactants in the dataset, given the product from the dataset. In this section, we showcase the advantages of our introduced metrics and offer insights into the validity of these alternative routes by referencing patent literature and reaction mechanisms.63–71 We analyse the following metrics and conditions: stereo-agnostic accuracy, partial accuracy, and halogen agnostic. Within each of these, inputs are categorized based on the type of reaction needed in retrosynthesis and the complexity of the products.
 | ||
| Fig. 4 Reactions labeled as incorrect by accuracy are identified as correct by stereo-agnostic accuracy. | ||
The reaction in Fig. 4a involved in the above retrosynthesis is esterification.72 In this transformation, alcohol (specifically, ethanol) condenses with a carboxylic acid (4-hydroxycyclohexane-1-carboxylic acid) with concomitant loss of a water molecule. In the reactant, the hydroxy and the carboxylic acid groups are in a stable equatorial conformation and trans to each other. The esterification is generally an acid catalyzed reaction. It is unlikely that the stereochemistry of the acid and hydroxy groups gets disturbed under the reaction conditions. SynFormer accurately predicted the reactant carboxylic acid and the reagent alcohol (ethanol) and the prediction agrees with the reactants in the dataset. Furthermore, SynFormer anticipates that the stereochemistry remains unaltered, a finding consistent with the dataset's reactants, which are agnostic to the stereochemistry of the substituents.
The retrosynthesis depicted in Fig. 4b involves a two-step transformation known as reductive amination, wherein the amine and the aldehyde react.73 Initially, the aldehyde condenses with a primary amine under mildly acidic conditions, forming an imine while releasing water. Subsequent reduction of the imine with hydride reducing agents such as sodium cyanoborohydride or sodium acetoxyborohydride yields the alkylated amine, as observed in the product. The product molecule in this case contains several functional groups, including a secondary amine, ester, and a carbobenzyloxy (Cbz) protected secondary amine, along with methyl and phenyl substitutions on the pyrrolidine ring. SynFormer accurately conducts retrosynthesis at the secondary amine of the product molecule, correctly identifying the primary amine and the substituted benzaldehyde. Its predictions align with the reactants in the dataset, encompassing all aspects, including the stereochemistry of consecutive substitutions at the four stereogenic carbon atoms of the product.
The transformation depicted in Fig. 4c involves the hydrolysis of an ester, typically carried out under basic conditions, yielding the salt of the corresponding carboxylic acid.74 The retrosynthesis of the acid (product) leads to the ester (reactants), aligning well with the reactants in the dataset, except for the stereochemical orientation of two substitutions on the fused cyclohexane ring. While both the product and the reactant structures in the dataset exhibit cis stereochemistry, the predicted reactants show trans stereochemistry. This discrepancy may arise because trans stereochemistry is thermodynamically more stable and could form via isomerization under basic conditions, suggesting that SynFormer accurately predicted the reactant structure.
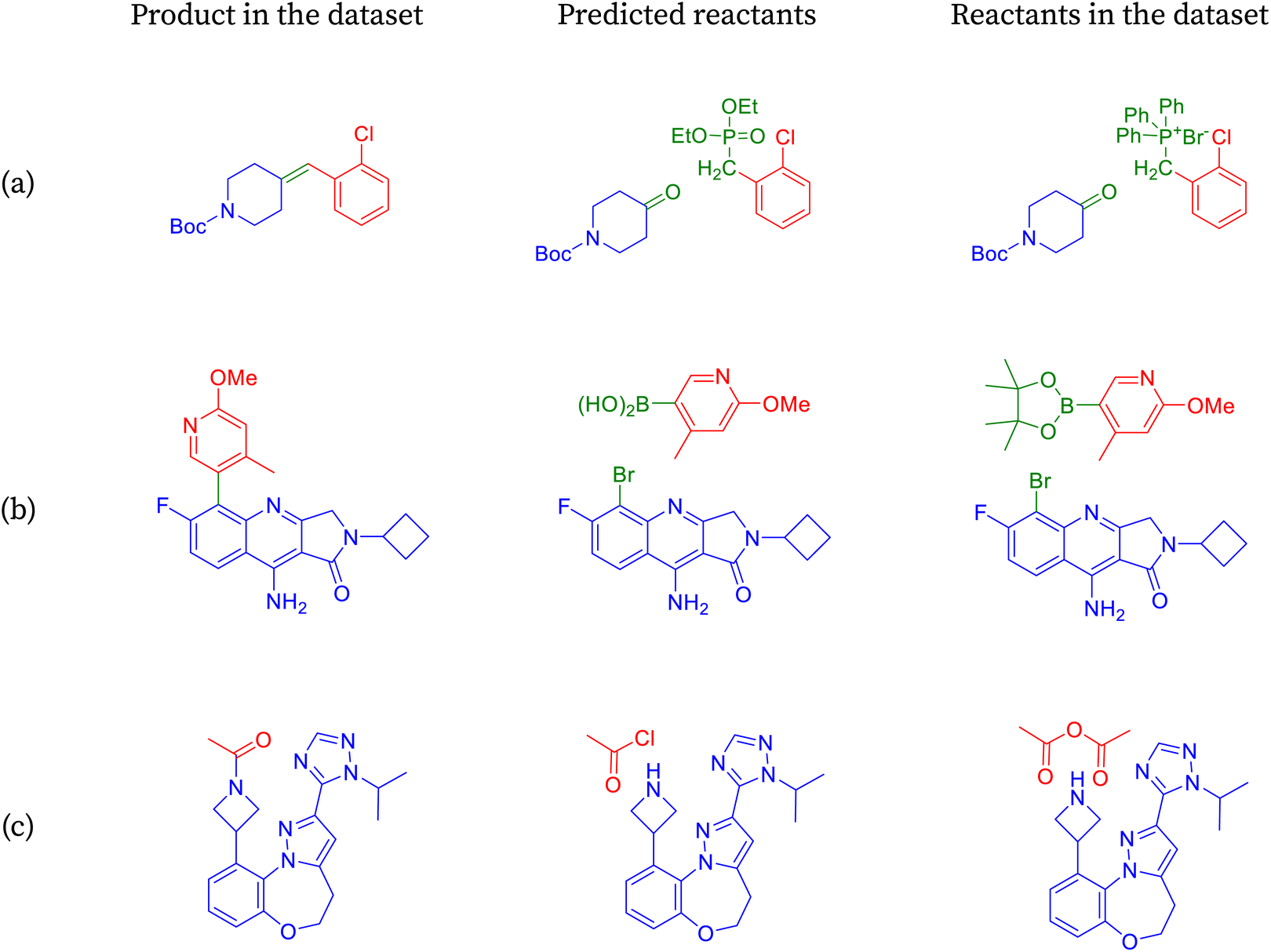 | ||
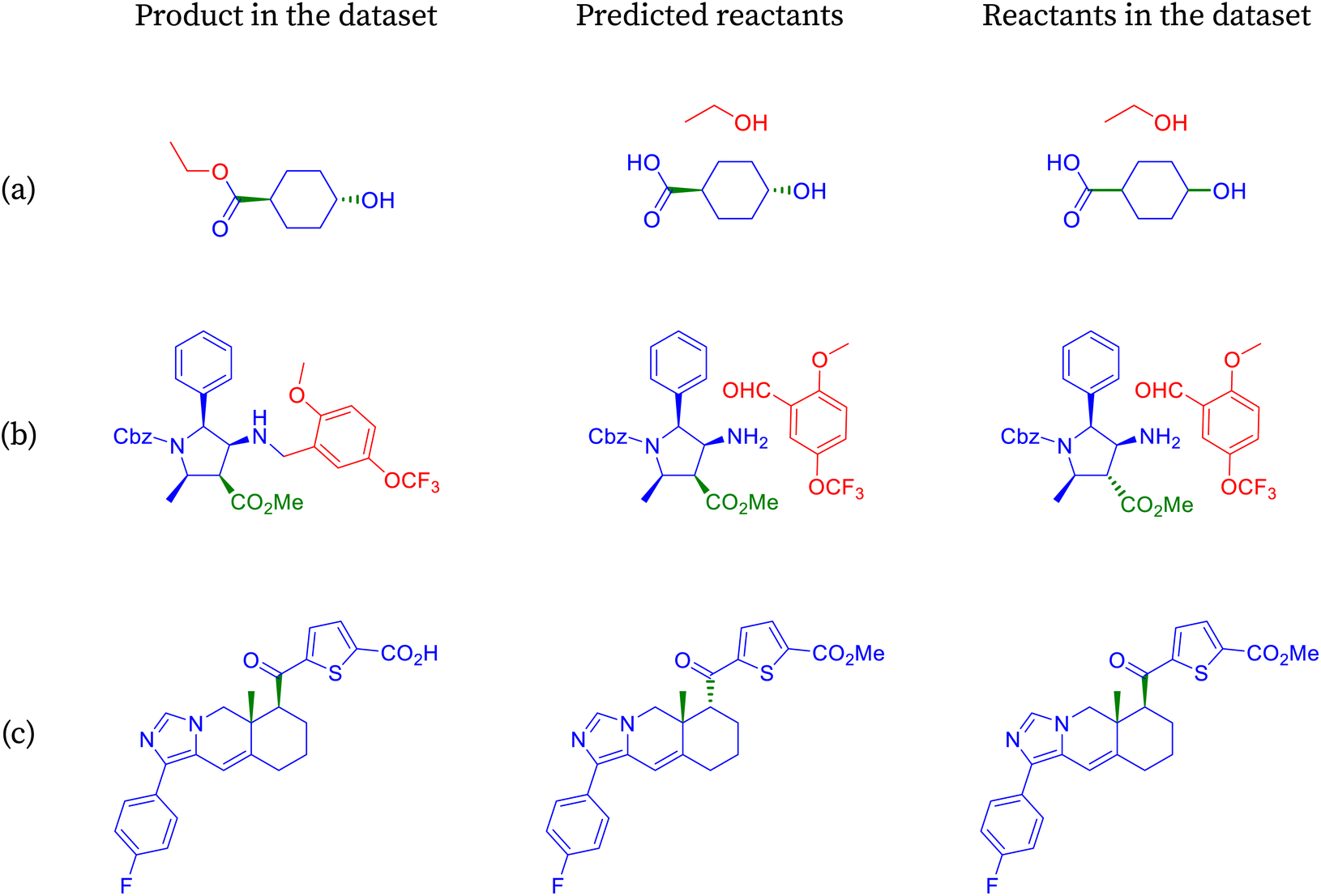
| Fig. 5 Reactions labeled as incorrect by accuracy are identified as correct by partial accuracy. | ||
The product depicted in Fig. 5a is an alkene, commonly synthesized using various methods, with one prominent approach being the Witting reaction.75 This reaction involves a ketone or an aldehyde and a phosphorus ylide, typically in a base-mediated process. Initially, the base abstracts a proton from the methylene group of the ylide, generating a nucleophilic carbon that reacts with the electrophilic carbon of the carbonyl compound. This results in the formation of an intermediate, which decomposes to yield the alkene and the oxidized form of the phosphorus. SynFormer accurately identifies the Witting method for alkene synthesis, correctly predicting the specific ketone reactant. However, it differs slightly in predicting the Wittig–Horner ylide instead of the Witting ylide. While both ylides are effective, the Witting ylide requires stronger bases and more stringent reaction conditions compared to the Wittig–Horner ylide.
The product molecule depicted in Fig. 5b is a biaryl compound, typically synthesized via a palladium-mediated coupling reaction between an aryl bromide and an arylboronic acid, known as Suzuki coupling.76 It contains various functional groups such as primary amine, methoxy, N-alkyl amide, and nitrogen-incorporated aromatic rings. SynFormer accurately identifies the biaryl nature of the molecule and predicts the presence of aryl bromide as one of the reactants, consistent with the dataset. However, there is a disparity in the form of the arylboronic acid; while SynFormer predicts a free arylboronic acid, the dataset suggests a pinacol derivative. Both reagents are viable, but the pinacol derivative in the dataset offers advantages such as increased solubility in organic solvents like dichloromethane and greater stability. Despite this difference, SynFormer effectively identifies the essential reactants, showcasing its utility in retrosynthetic analysis.
The product molecule depicted in Fig. 5c is a complex nitrogen heterocyclic compound featuring several distinct substructures, including N-acetyl azetidine, fused diazole, fused benzo[b][1,4]oxazepane, and 1,2,4-triazole rings.77 Upon retrosynthesis, SynFormer predicts the formation of azetidine and acetyl chloride as reactants, matching one of the reactants in the dataset. However, there is a discrepancy in the choice of reagent, with SynFormer predicting acetyl chloride while the dataset indicates the presence of acetic anhydride. Acetyl chloride is preferred due to its availability, reactivity, and cost-effectiveness, whereas acetic anhydride, present in the dataset, is restricted in many countries due to its association with illicit drug synthesis. Despite this disparity, SynFormer accurately identifies both the substrate and reagent, showcasing its effectiveness in retrosynthetic analysis.
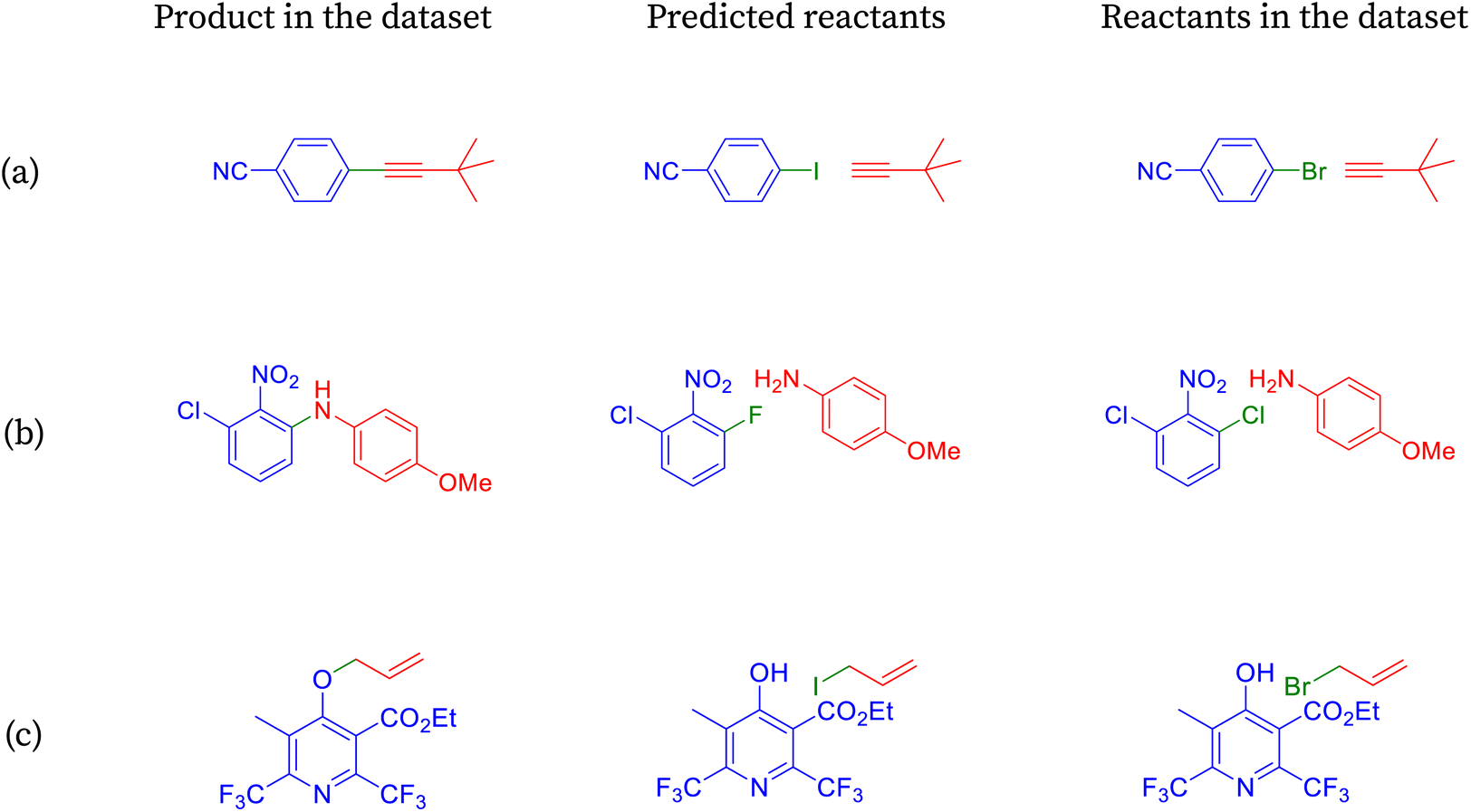 | ||
| Fig. 6 Reactions classified as incorrect by accuracy are shown to be correct under halogen-agnostic conditions. | ||
The product molecule from Fig. 6a can be prepared readily by coupling an appropriate aryl halide and alkyne in the presence of a palladium(0) catalyst, copper(I) cocatalyst, and an amine base.78 The reaction is known as Sonogashira coupling. The aryl iodides or aryl bromides can be employed as the coupling partners. Of the two, the aryl iodides participate in the reaction more efficiently but are more difficult to prepare. SynFormer predicted alkyne correctly, and the structure matches that of the reactants in the dataset. On the other hand, SynFormer predicted an aryl iodide instead of aryl bromide in the reactants from the dataset, possibly because of its higher reactivity. This discrepancy highlights SynFormer's focus on chemical reactivity rather than practical considerations like availability, ease of preparation, and cost, emphasizing the importance of considering all factors in retrosynthetic analysis.
The easiest and industrially feasible method for the synthesis of the product molecule from Fig. 6 is aromatic nucleophilic substitution of a halide in an electron deficient aromatic halide with 4-methoxyaniline.79 SynFormer predicted 1-chloro-3-fluoro-2-nitrobenzene instead of 1,3-dichloro-2-nitro-benzene as seen in the reactants in the dataset as one of the reacting partners. It has correctly identified 4-methoxyaniline as the second reacting partner. Although 1-chloro-3-fluoro-2-nitrobenzene is more difficult to prepare, it is more suitable for aromatic nucleophilic substitution compared to 1,3-dichloro-2-nitrobenzene. This discrepancy underscores the importance of considering both practical feasibility and reactivity in retrosynthetic analysis.
The synthesis of the product molecule from Fig. 6c could proceed through Williamson ether synthesis where the deprotonated alcohol (in this case a phenolic compound) reacts with a halide (in this case allyl bromide or allyl iodide) to form an ether.80 Apart from the ether functional group, the product molecule has a few other functional groups like ester, 2,6-disubstituted pyridine ring. Among many possibilities, SynFormer has correctly identified substituted 4-hydroxypyridine as one of the reacting partners, and this result agrees with reactants in the dataset. However, it has identified allyl iodide as the other reacting partner instead of allyl bromide, despite the latter being more commonly used and easier to handle. Although allyl iodide is more reactive, its preparation and handling are challenging. This discrepancy highlights the need for careful consideration of practical factors in retrosynthetic analysis.
4 Conclusion
In this paper, we introduced the Retro-Synth Score for fine-grained evaluation of reaction predictions and presented SynFormer, a transformer model designed for template-free organic reaction synthesis and retrosynthetic predictions. Through a detailed case-study, we illustrated how our introduced metrics relax evaluation conditions, enabling consideration of alternative valid chemical routes and addressing some limitations in the dataset. We discovered that many reactions deemed incorrect by accuracy metrics are, in fact, viable reaction mechanisms, as identified by our metrics, offering a more nuanced evaluation method for retrosynthetic analysis. Our analysis reveals that the predicted reactants differ in chemical reactivity, the time taken for the reaction to occur, availability, ease of preparation, and cost of the reactant molecules, which are overlooked in the dataset but crucial for practical applications. Through a detailed evaluation of various algorithms using R-SS, we challenge the belief that language models significantly outperform graph generative models in retrosynthesis. Our analysis shows only marginal differences in performance on the USPTO-50k dataset, questioning the necessity of pre-training. While prior language models benefit from extensive pre-training, SynFormer achieves comparable performance to Chemformer PT, leading by 0.17%, and shows a 5.61% advantage over Graph2SMILES in R-SS, all without requiring pre-training. However, it is worth noting that Graph2SMILES performs significantly better in Top-10 accuracy, highlighting its strength in broader output scenarios. With SynFormer, we observe comparable performance and a noticeable improvement efficiency through key changes in the original transformer architecture, serving as a drop-in replacement for existing transformer architectures in molecule transformation tasks.Data availability
The complete implementation code for this work is publicly available in our GitHub repository: https://github.com/devalab/SynFormer. To ensure long-term preservation and reproducibility, all models, code, and datasets have been archived on zenodo (https://zenodo.org/records/14725617). The dataset used in this study can be accessed from the MolecularAI/Chemformer repository: https://github.com/MolecularAI/Chemformer.Conflicts of interest
There are no conflicts to declare.Appendices
A Retro-synth score
The above metrics are computed on the predicted set of molecules against the ground truth set of reactant molecules for each reaction. We will consider G to be the ground truth set comprising of g1, g2, …, gn reactant molecules whose corresponding SMILES representation is represented by S1g, S2g, …, Sng and P to be the predicted set comprising p1, p2, …, pn product molecules whose corresponding SMILES representation is represented by S1p, S2p,…, Snp.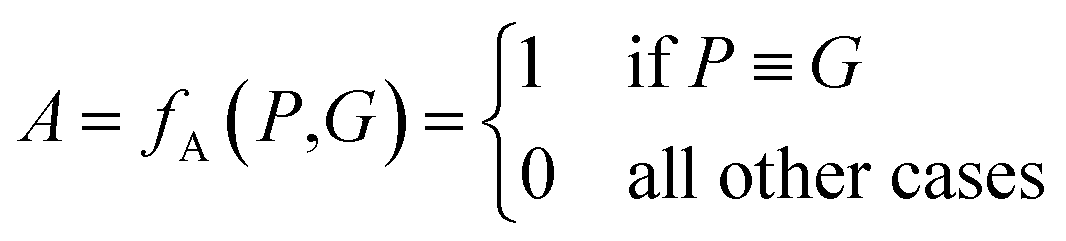 | (5) |
| P ≡ G ⇔ f(S1p○S2p○…Snp) ≡ f(S1g○S2g○…Sng) | (6) |
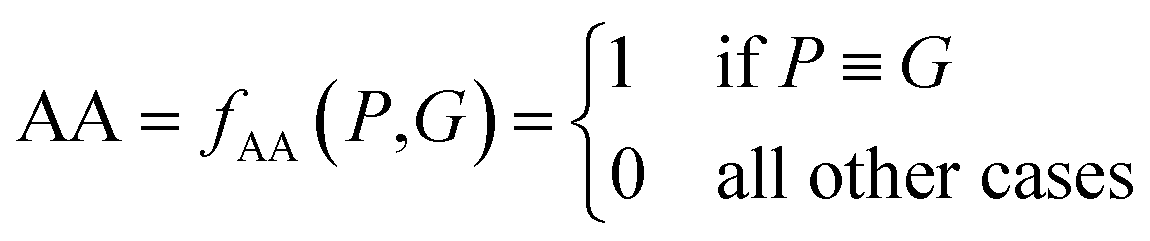
| (7) |
| P ≡ G ⇔ P ⊆ G and G ⊆ P | (8) |
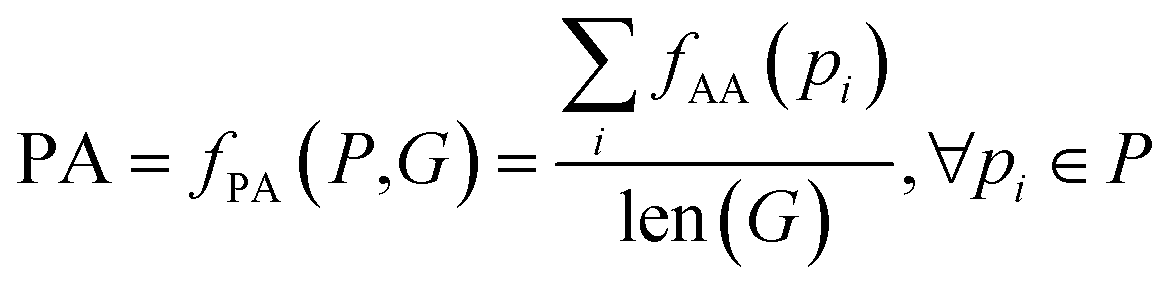
| (9) |
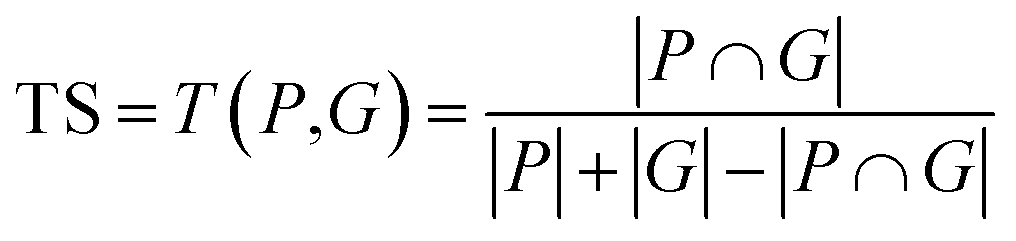
| (10) |
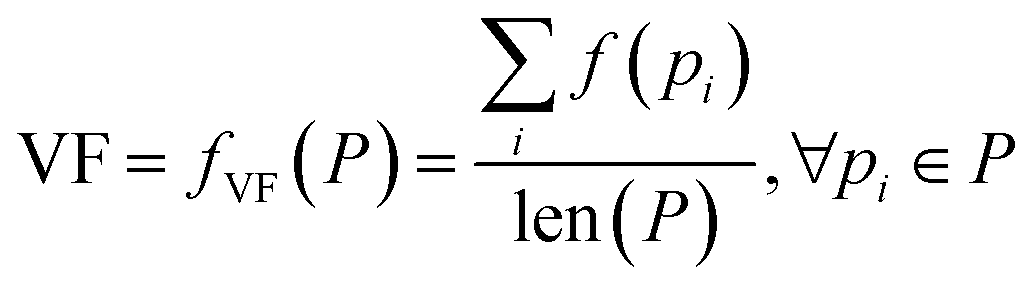
| (11) |
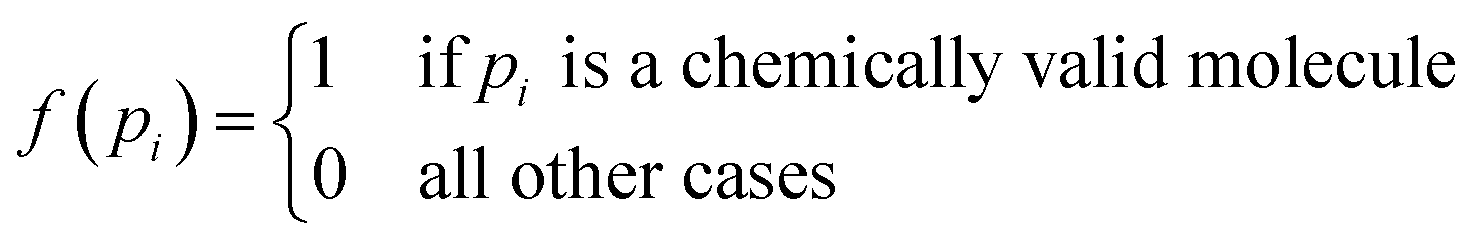
| (12) |
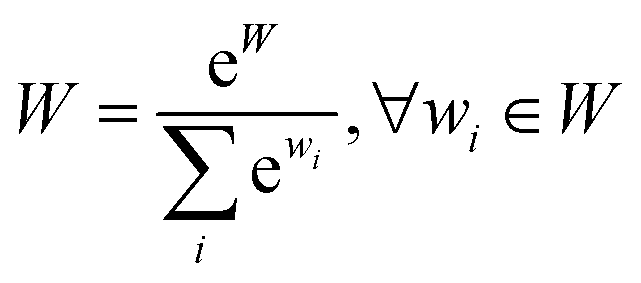 | (13) |
B SynFormer
The modifications to the network can be expressed as: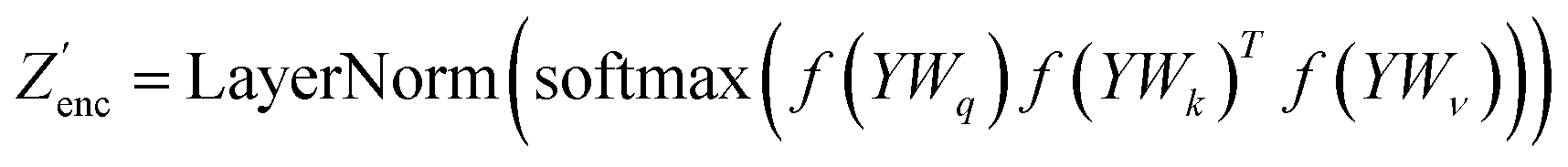 | (14) |
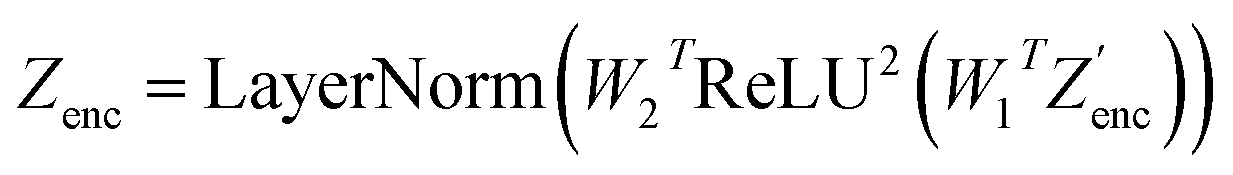 | (15) |
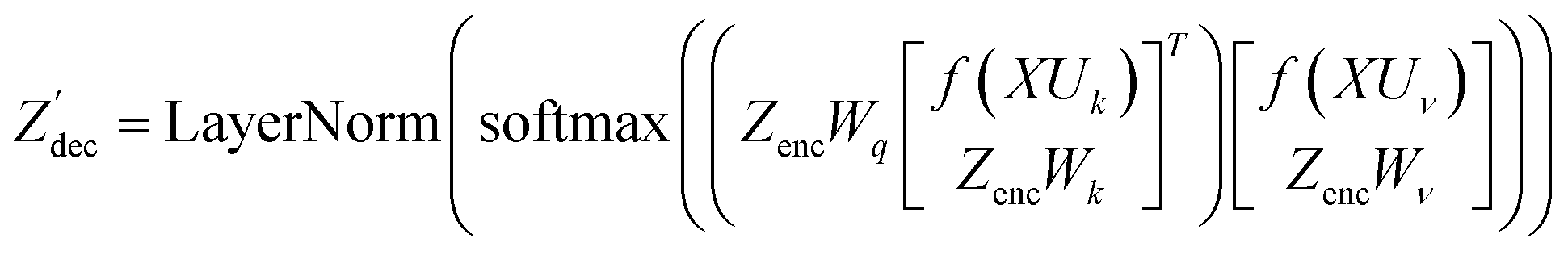 | (16) |
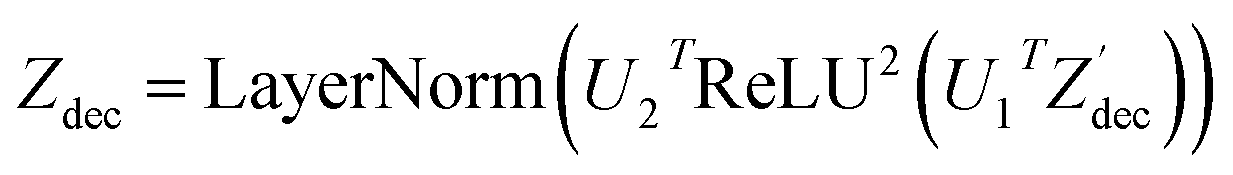 | (17) |
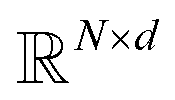 where N is the number of tokens and d is the feature vector dimension. Wq, Wk, and Wv are the query, key, and value projection parameters for the encoder, while Uq, Uk, and Uv are the query, key, and value projection parameters for the decoder in
where N is the number of tokens and d is the feature vector dimension. Wq, Wk, and Wv are the query, key, and value projection parameters for the encoder, while Uq, Uk, and Uv are the query, key, and value projection parameters for the decoder in 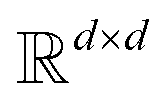 . The feed-forward network of the encoder is a multi-layer perceptron stacked with a linear layer with weights W1 in
. The feed-forward network of the encoder is a multi-layer perceptron stacked with a linear layer with weights W1 in 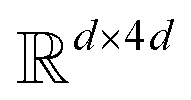 , non-linearity ReLU2, followed by a linear layer with weights W2 in
, non-linearity ReLU2, followed by a linear layer with weights W2 in 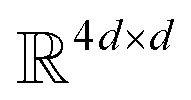 and likewise for the decoder where the linear layer weights are U1 and U2, respectively.
and likewise for the decoder where the linear layer weights are U1 and U2, respectively.
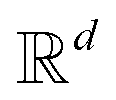 from the attention block. The function f is required to have the following properties:
from the attention block. The function f is required to have the following properties:| 〈f(q,m),f(k,n)〉 = g(q,k,m − n) | (18) |
In RoFormer, they formulate the function f as:
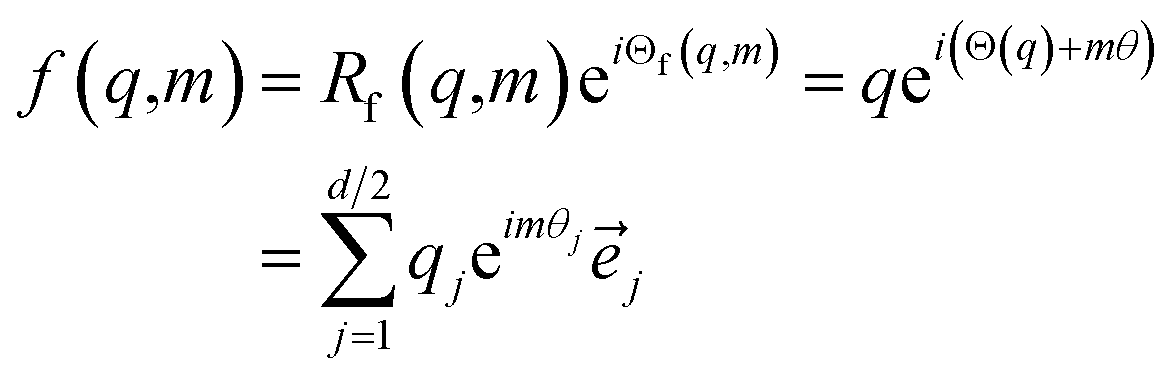 | (19) |
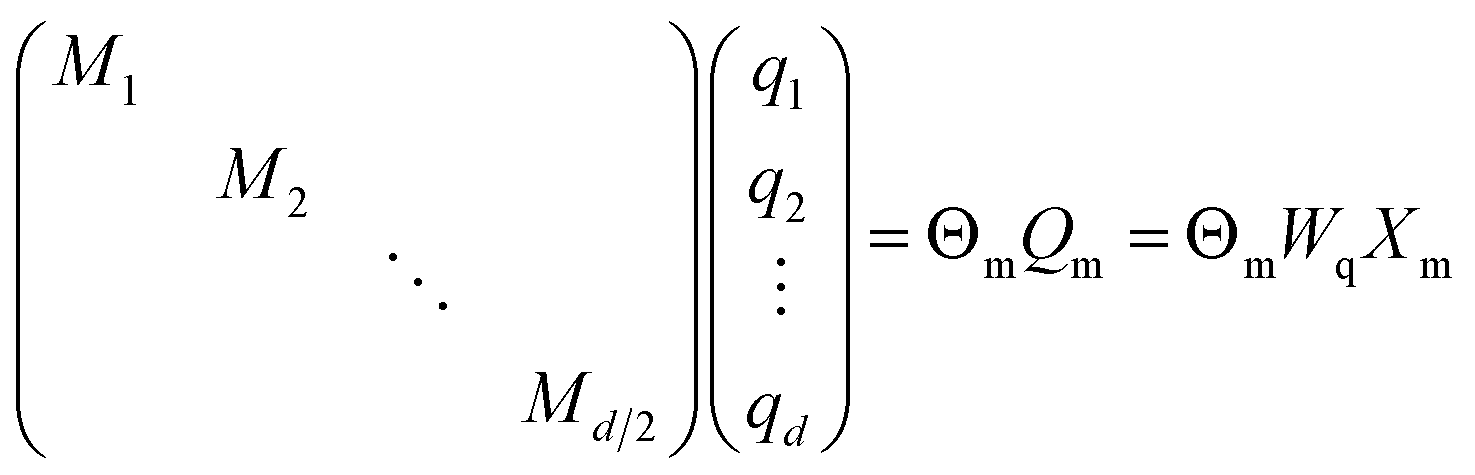 | (20) |
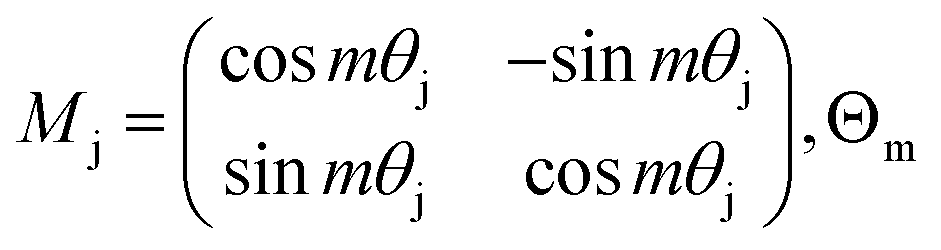 is the block diagonal rotation matrix, Wq is the learned query weights, and Xm is the embedding of the m token. We also have the corresponding equation for k.
is the block diagonal rotation matrix, Wq is the learned query weights, and Xm is the embedding of the m token. We also have the corresponding equation for k.
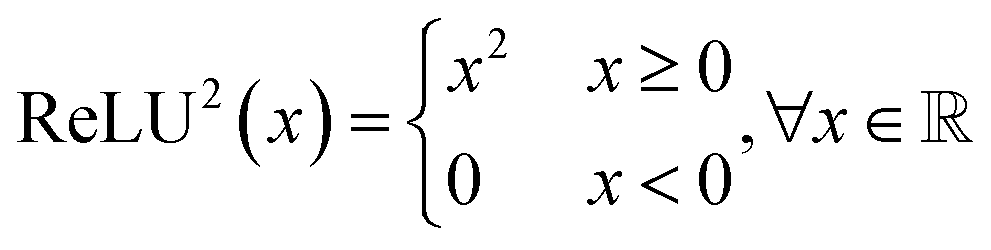 | (21) |
C Data augmentation
R-SMILES, or Root Aligned SMILES,81 demonstrated that reducing the entropy between the input (product) and target (reactants) SMILES improves the performance in sequence-to-sequence tasks. Here, entropy refers to the dissimilarity between SMILES, often measured using the edit distance.In this work, we use input-side augmentation, which shuffles the order of the input SMILES, generating different representations for the same molecule. Unlike R-SMILES, this method imposes no restrictions on the target SMILES. R-SMILES, however, finds the optimal target SMILES that minimizes the edit distance between the input and target SMILES.
D Effect of varying weights on R-SS
| Algorithm | w A | w AA | w PA | w TS | R-SS |
|---|---|---|---|---|---|
| Chemformer | 1 | 2 | 4 | 1 | 0.585 |
| Graph2SMILES | 0.572 | ||||
| Chemformer PT | 0.605 | ||||
| SynFormer | 0.606 | ||||
| Chemformer | 1 | 1 | 1 | 1 | 0.605 |
| Graph2SMILES | 0.592 | ||||
| Chemformer PT | 0.624 | ||||
| SynFormer | 0.624 | ||||
| Chemformer | 1 | 1 | 1 | 0.5 | 0.587 |
| Graph2SMILES | 0.575 | ||||
| Chemformer PT | 0.606 | ||||
| SynFormer | 0.607 |
E Improvement over language model without pre-training
| Algorithm | HA | A | AA | PA | TS | WA | R-SS |
|---|---|---|---|---|---|---|---|
| SynFormer (ours) vs. chemformer | ✗ | +4.670% | +3.832% | +3.512% | +1.020% | +3.512% | +3.465% |
| ✓ | +4.480% | +3.826% | +3.426% | +1.020% | +3.263% |
Acknowledgements
We extend our sincere gratitude to IHub-Data (IIIT Hyderabad, grant D4) for their generous support and to the Department of Science and Technology, Science and Engineering Research Board (DST-SERB) (Grant No. CRG/2021/008036) for their funding contributions, which made this research possible.References
- E. J. Corey, Chem. Soc. Rev., 1988, 17, 111–133 RSC.
- B. V. Badami, Retrosynthetic Analysis, Springer Nature, 2019, vol. 24, pp. 1071–1086 Search PubMed.
- E. J. Corey, R. D. I. Cramer and W. J. Howe, J. Am. Chem. Soc., 1972, 94, 440–459 CrossRef CAS.
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 CrossRef PubMed.
- M. H. S. Segler and M. P. Waller, Chem. - Eur. J., 2017, 23, 5966–5971 CrossRef CAS PubMed.
- J. L. Baylon, N. A. Cilfone, J. R. Gulcher and T. W. Chittenden, J. Chem. Inf. Model., 2019, 59, 673–688 CrossRef CAS PubMed.
- H. Dai, C. Li, C. W. Coley, B. Dai and L. Song, in Retrosynthesis prediction with conditional graph logic network, Curran Associates Inc., Red Hook, NY, USA, 2019 Search PubMed.
- S. Chen and Y. Jung, JACS Au, 2021, 1, 1612–1620 CrossRef CAS PubMed.
- C. W. Coley, L. Rogers, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2017, 3, 1237–1245 CrossRef CAS PubMed.
- W. Jin, C. W. Coley, R. Barzilay and T. Jaakkola, Proceedings of the 31st International Conference on Neural Information Processing Systems, Red Hook, NY, USA, 2017, pp. 2604–2613 Search PubMed.
- C. W. Coley, W. Jin, L. Rogers, T. F. Jamison, T. S. Jaakkola, W. H. Green, R. Barzilay and K. F. Jensen, Chem. Sci., 2019, 10, 370–377 RSC.
- K. Do, T. Tran and S. Venkatesh, Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, New York, NY, USA, 2019, pp. 750–760 Search PubMed.
- M. Sacha, M. Błaż, P. Byrski, P. Dąbrowski-Tumański, M. Chromiński, R. Loska, P. Włodarczyk-Pruszyński and S. Jastrzębski, J. Chem. Inf. Model., 2021, 61, 3273–3284 CrossRef CAS PubMed.
- W. W. Qian, N. T. Russell, C. L. W. Simons, Y. Luo, M. D. Burke and J. Peng, ChemRxiv, 2020, DOI:10.26434/chemrxiv.11659563.v1.
- C. Shi, M. Xu, H. Guo, M. Zhang and J. Tang, Proceedings of the 37th International Conference on Machine Learning, 2020 Search PubMed.
- C. Yan, Q. Ding, P. Zhao, S. Zheng, J. Yang, Y. Yu and J. Huang, Proceedings of the 34th International Conference on Neural Information Processing Systems, Red Hook, NY, USA, 2020 Search PubMed.
- V. R. Somnath, C. Bunne, C. W. Coley, A. Krause and R. Barzilay, Neural Information Processing Systems, 2020 Search PubMed.
- X. Wang, Y. Li, J. Qiu, G. Chen, H. Liu, B. Liao, C.-Y. Hsieh and X. Yao, Chem. Eng. J., 2021, 420, 129845 CrossRef CAS.
- J. Nam and J. Kim, arXiv, 2016, preprint, arXiv:1612.09529, DOI:10.48550/arXiv.1612.09529.
- P. Schwaller, T. Gaudin, D. Lányi, C. Bekas and T. Laino, Chem. Sci., 2018, 9, 6091–6098 RSC.
- B. Liu, B. Ramsundar, P. Kawthekar, J. Shi, J. Gomes, Q. Luu Nguyen, S. Ho, J. Sloane, P. Wender and V. Pande, ACS Cent. Sci., 2017, 3, 1103–1113 CrossRef CAS PubMed.
- R. Irwin, S. Dimitriadis, J. He and E. J. Bjerrum, Mach. Learn.: Sci. Technol., 2022, 3, 015022 Search PubMed.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, ACS Cent. Sci., 2019, 5, 1572–1583 CrossRef CAS PubMed.
- K. Lin, Y. Xu, J. Pei and L. Lai, Chem. Sci., 2020, 11, 3355–3364 RSC.
- A. A. Lee, Q. Yang, V. Sresht, P. Bolgar, X. Hou, J. L. Klug-McLeod and C. R. Butler, Chem. Commun., 2019, 55, 12152–12155 RSC.
- H. Duan, L. Wang, C. Zhang, L. Guo and J. Li, RSC Adv., 2020, 10, 1371–1378 RSC.
- I. V. Tetko, P. Karpov, R. Van Deursen and G. Godin, Nat. Commun., 2020, 11, 5575 CrossRef CAS PubMed.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- E. J. Bjerrum, arXiv, 2017, preprint, arXiv:1703.07076, DOI:10.48550/arXiv.1703.07076.
- J. Arús-Pous, S. Johansson, O. Prykhodko, E. J. Bjerrum, C. Tyrchan, J. Reymond, H. Chen and O. Engkvist, International Conference on Artificial Neural Networks, 2019 Search PubMed.
- S. Zheng, J. Rao, Z. Zhang, J. Xu and Y. Yang, J. Chem. Inf. Model., 2020, 60, 47–55 CrossRef CAS PubMed.
- S. Wang, M. Khabsa and H. Ma, Proceedings of the 58th Annual Meeting of the Association for Computational Linguistics, Online, 2020, pp. 2209–2213 Search PubMed.
- N. Schneider, N. Stiefl and G. A. Landrum, J. Chem. Inf. Model., 2016, 56, 2336–2346 CrossRef CAS PubMed.
- D. M. Lowe, PhD thesis, University of Cambridge, 2012.
- K. Maziarz, A. Tripp, G. Liu, M. Stanley, S. Xie, P. Gaiński, P. Seidl and M. Segler, arXiv, 2024, preprint, arXiv:2310.19796, 10.1039/D4FD00093E.
- P. Schwaller, Neural Information Processing Systems, 2019 Search PubMed.
- U. V. Ucak, I. Ashyrmamatov, J. Ko and J. Lee, Nat. Commun., 2022, 13, 1186 CrossRef CAS PubMed.
- D. Bajusz, A. Rácz and K. Héberger, J. Cheminf., 2015, 7, 20 Search PubMed.
- G. Landrum and The RDKit team, RDKit: Open-source cheminformatics, 2022, http://www.rdkit.org.
- A. Capecchi, D. Probst and J.-L. Reymond, J. Cheminf., 2020, 12, 43 CAS.
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, L. u. Kaiser and I. Polosukhin, Advances in Neural Information Processing Systems, 2017 Search PubMed.
- J. Devlin, M.-W. Chang, K. Lee and K. Toutanova, North American Chapter of the Association for Computational Linguistics, 2019 Search PubMed.
- M. Lewis, Y. Liu, N. Goyal, M. Ghazvininejad, A. rahman Mohamed, O. Levy, V. Stoyanov and L. Zettlemoyer, Annual Meeting of the Association for Computational Linguistics, 2019 Search PubMed.
- A. Radford and K. Narasimhan, Improving Language Understanding by Generative Pre-Training, 2018, https://api.semanticscholar.org/CorpusID:49313245.
- A. Radford, J. Wu, R. Child, D. Luan, D. Amodei and I. Sutskever, Language Models are Unsupervised Multitask Learners, 2019, https://api.semanticscholar.org/CorpusID:160025533.
- H. Touvron, T. Lavril, G. Izacard, X. Martinet, M.-A. Lachaux, T. Lacroix, B. Rozière, N. Goyal, E. Hambro, F. Azhar, A. Rodriguez, A. Joulin, E. Grave and G. Lample, arXiv, 2023, preprint, arXiv:2302.13971, DOI:10.48550/arXiv.2302.13971.
- A. Kazemnejad, I. Padhi, K. Natesan, P. Das and S. Reddy, Thirty-seventh Conference on Neural Information Processing Systems, 2023 Search PubMed.
- J. Su, M. Ahmed, Y. Lu, S. Pan, W. Bo and Y. Liu, Neurocomputing, 2024, 568, 127063 CrossRef.
- C. Raffel, N. Shazeer, A. Roberts, K. Lee, S. Narang, M. Matena, Y. Zhou, W. Li and P. J. Liu, J. Mach. Learn. Res., 2020, 21, 140 Search PubMed.
- O. Press, N. A. Smith and M. Lewis, arXiv, 2021, preprint, arXiv:2108.12409, DOI:10.48550/arXiv.2108.12409.
- D. R. So, W. Ma’nke, H. Liu, Z. Dai, N. M. Shazeer and Q. V. Le, arXiv, 2021, preprint, arXiv:2109.08668, DOI:10.48550/arXiv.2109.08668.
- N. M. Shazeer, arXiv, 2020, preprint, arXiv:2002.05202, DOI:10.48550/arXiv.2002.05202.
- K. Shen, J. Guo, X. Tan, S. Tang, R. Wang and J. Bian, arXiv, 2023, preprint, arXiv:2302.06461, DOI:10.48550/arXiv.2302.06461.
- Y. Dauphin, A. Fan, M. Auli and D. Grangier, International Conference on Machine Learning, 2016 Search PubMed.
- P. Ramachandran, B. Zoph and Q. V. Le, arXiv, 2018, preprint, arXiv:1710.05941, DOI:10.48550/arXiv.1710.05941.
- A. F. Agarap, arXiv, 2018, preprint, arXiv:1803.08375, DOI:10.48550/arXiv.1803.08375.
- Z. Tu and C. W. Coley, J. Chem. Inf. Model., 2022, 62, 3503–3513 CrossRef CAS PubMed.
- I. Loshchilov and F. Hutter, arXiv, 2017, preprint, arXiv:1711.05101, DOI:10.48550/arXiv.1711.05101.
- W. Falcon and The PyTorch Lightning team, PyTorch Lightning, 2019, https://github.com/Lightning-AI/lightning Search PubMed.
- P. Wang, X-Transformers, 2021, https://github.com/lucidrains/x-transformers.
- E. J. Bjerrum, T. Rastemo, R. Irwin, C. C. Kannas and S. Genheden, ChemRxiv, 2021, DOI:10.26434/chemrxiv-2021-kzhbs.
- T. Sterling and J. J. Irwin, J. Chem. Inf. Model., 2015, 55, 2324–2337 CrossRef CAS PubMed.
- T. Okuyama and H. Maskill, Organic Chemistry: A Mechanistic Approach, Oxford University Press, USA, 2013 Search PubMed.
- R. Valiulin, Organic chemistry: 100 must-know mechanisms, de Gruyter, 2nd edn, 2023 Search PubMed.
- R. B. Grossman, The art of writing reasonable organic reaction mechanisms the art of writing reasonable organic reaction mechanisms, Springer Nature, Cham, Switzerland, 3rd edn, 2021 Search PubMed.
- P. Chaloner, Organic Chemistry: A Mechanistic Approach, CRC Press, 2014 Search PubMed.
- P. Sykes, A guidebook to mechanism in organic chemistry, Hassell Street Press, 2021 Search PubMed.
- J. Clayden and S. Warren, Solutions Manual to accompany Organic Chemistry, Oxford University Press, London, England, 2nd edn, 2012 Search PubMed.
- M. B. Smith, March's advanced organic chemistry: Reactions, mechanisms, and structure, Wiley, 7th edn, 2013 Search PubMed.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry: Part A: Structure and Mechanisms, Springer Science & Business Media, 2007 Search PubMed.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry: Part B: Reactions and Synthesis, Springer US, Boston, MA, 2007 Search PubMed.
- H. Kim, M. K. Kim, H. Choo and Y. Chong, Bioorg. Med. Chem. Lett., 2016, 26, 3213–3215 CrossRef CAS PubMed.
- M. Ikunaka, Y. Shishido and M. Nakane, US Pat., US6083943A, Pfizer Inc, 2000 Search PubMed.
- T. M. Dhar and H. Y. Xiao, WO2009058944 2, Bristol-Myers Squibb Company, 2009.
- M. Takatani, Y. Shibouta, Y. Sugiyama and T. Kawamoto, WO9740051A1, Takeda Chemical Industries, Ltd, 1997.
- H. F. Chang, M. Chapdelaine, B. T. Dembofsky, K. J. Herzog, C. Horchler and R. J. Schmiesing, WO2008155572A2, AstraZeneca AB and AstraZeneca UK Limited, 2008.
- M.-G. Braun, K. Garland, E. Hanan, H. Purkey, S. T. Staben, R. A. Heald, J. Knight, C. Macleod, A. LU, G. Wu and S. K. Yeap, US Pat., US10065970B2, Genentech Inc, 2018 Search PubMed.
- V. Coltuclu, E. Dadush, A. Naravane and G. W. Kabalka, Molecules, 2013, 18, 1755–1761 CrossRef CAS PubMed.
- R. Tamai, U. Yukio, B. Takabe, K. Satoshi, T. Maruyama and R. Kobayashi, WO2020158925A1, Kumiai Chemical Industry Co., Ltd, 2020.
- L. F. Lee and M. L. Miller, US Pat., US4655816A, Rohm and Haas Co, 1987 Search PubMed.
- Z. Zhong, J. Song, Z. Feng, T. Liu, L. Jia, S. Yao, M. Wu, T. Hou and M. Song, Chem. Sci., 2022, 13, 9023–9034 RSC.
| This journal is © The Royal Society of Chemistry 2025 |