
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
La2(CN2)3 – the missing link of rare-earth carbodiimides, prepared through an efficient synthetic route and its Ce3+ activated photoluminescence†
Philipp
Schneiderhan
a,
Elaheh
Bayat
 a,
Markus
Ströbele
a,
David
Enseling
b,
Thomas
Jüstel
b and
H.-Jürgen
Meyer
*a
a,
Markus
Ströbele
a,
David
Enseling
b,
Thomas
Jüstel
b and
H.-Jürgen
Meyer
*a
aSection for Solid State and Theoretical Inorganic Chemistry, Institute of Inorganic Chemistry, University of Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: juergen.meyer@uni-tuebingen.de
bFH Münster, University of Applied Science, Stegerwaldstraße 39, 48565 Steinfurt, Germany
First published on 24th February 2025
Abstract
Rare-earth (RE) carbodiimides according to the composition RE2(CN2)3 have been reported for the whole series of RE elements, all prepared by solid-state metathesis (SSM) reactions. Only one compound, La2(CN2)3, could not be made by this way of synthesis. Herein, we report the preparation of La2(CN2)3 by using lanthanum cyanurate as a single-source precursor. The conversion of the precursor is analyzed by thermoanalytical studies. The crystal structure of the precursor and the novel La2(CN2)3 are characterized by X-ray diffraction techniques. La2(CN2)3 is represented by a distinct crystal structure with a dodecahedral environment of the La3+ ion. Having the knowledge of the last missing rare-earth carbodiimide, we herein present a summary of all existing RE2(CN2)3 compounds, including their structural relationships. Doping with Ce3+ leads to the La2(CN2)3:Ce3+ phosphor, which is reported with its photoluminescence properties.
Introduction
Metal dinitridocarbonates, commonly denoted as metal cyanamides (N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–N2−) and metal carbodiimides (–N
C–N2−) and metal carbodiimides (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CN–), have been reported by different ways of synthesis. The first method for synthesizing a carbodiimide involves reacting calcium carbide (CaC2) with nitrogen gas (N2) at elevated temperatures, approximately 1000 °C, in a process known as the Frank-Caro method.1
CN–), have been reported by different ways of synthesis. The first method for synthesizing a carbodiimide involves reacting calcium carbide (CaC2) with nitrogen gas (N2) at elevated temperatures, approximately 1000 °C, in a process known as the Frank-Caro method.1
Following the discovery of calcium carbodiimide, numerous metal carbodiimide or metal cyanamide compounds were developed. An early synthetic approach to lithium carbodiimide (Li2(CN2)) was achieved in the 1970s by heating Li3N and Li2C2 together at about 600 °C.2 Another proposed method for the synthesis of Li2(CN2) is ammonolysis, which involves the reaction of lithium carbonates (Li2CO3) with ammonia (NH3).3 Alternatively, lithium carbodiimide (Li2(CN2)) can be synthesized using lithium nitride (Li3N)4 (eqn (1)) or hydride (LiH)5 and melamine. For synthesizing other alkali metal carbodiimides such as sodium6 and potassium7 carbodiimides an alternative route was employed. Alkaline-earth metal carbodiimides such as Mg(CN2), Sr(CN2), and Ba(CN2) can be produced similarly utilizing the reaction of alkaline-earth metal nitrides with melamine at temperatures ranging from 740 to 850 °C.8
One of the most effective approaches for synthesizing many other metal carbodiimides is the solid-state metathesis (SSM), which allows for the production of relatively pure carbodiimides under moderate heating conditions, typically at 450–600 °C.9,10 Within this process, a metal halide (MX2 or MX3) is converted with lithium carbodiimide in a salt-balanced reaction (eqn (2)) to yield a metal carbodiimide.
| 2Li3N + C3H6N6 → 3Li2(CN2) + 2NH3 | (1) |
| 2MX3 + 3Li2(CN2) → M2(CN2)3 + 6LiX | (2) |
To produce calcium carbodiimide of high purity, another synthetic route was developed as shown in eqn (3). This methodology was first proposed by Seifer11 and then further refined in 2023.12 The study done by Seifer suggests briefly the preparation of lead, barium, and strontium carbodiimide in addition to calcium carbodiimide. These carbodiimides are prepared by the reaction of cyanuric acid with metal chlorides in the presence of sodium hydroxide in water to produce the corresponding cyanurate salts which are subsequently pyrolyzed to give the desired carbodiimides. This synthetic route shows the promising route of using triazine-derived precursors, for example, melamine,5,13 and derivatives of cyanuric acid in the production of carbodiimides.
| Ca(HO3C3N3) → Ca(CN2) + HOCN + CO2↑ | (3) |
It is noteworthy to mention that some transition metal carbodiimides were also synthesized using aqueous solution-based methods for instance, Zn(CN2),14 Co(CN2),15 Ni(CN2),15 Cu(CN2),16 Cd(CN2),17 Ag2(CN2),18 Hg(CN2).19 Other transition metal carbodiimides, such as Mn(CN2),20 Cr2(CN2)3,21 Zr(CN2)2,22 and Hf(CN2)2,22 can be synthesized using the similar solid-state metathesis (SSM) reactions between a metal halide with Li2(CN2) or Zn(CN2).14 p-Block metal carbodiimides, like Pb(CN2),23 Bi2(CN2)3,24 Tl2(CN2),25 and In2.24(CN2)3,13,26 are synthesized by reacting metal salts with cyanamide or cyanide compounds. For tin carbodiimides, the conventional SSM reaction of Li2(CN2) with metal halides such as SnCl2 or SnF2, yields Sn(CN2) and Sn4Cl2(CN2)3.3,27
SSM reactions have been extensively utilized by our research group to synthesize rare-earth (RE) carbodiimides, essentially by reacting lithium carbodiimide with a rare-earth metal chloride.28,29 Most prominent is the series of RE2(CN2)3 compounds that have been reported for RE elements from Sc to Lu, except for RE = La (and the radioactive element Pm).28–30 Lanthanum carbodiimide was first reported 76 years ago from reactions of La2O3 with HCN.31 However, the products were poorly characterized, with no structural or spectroscopic data available.31 It is worth mentioning that compounds of smaller rare-earth ions like RE = Sc,32 Tm, Yb, and Lu28,33 crystallize with the trigonal rhombohedral space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c (Z = 6), and the coordination number (CN) six of the RE3+ ion.28 Corresponding compounds with RE = Y and Ce–Er (except Pm) crystallize monoclinically with the space group C2/m (Z = 2) and the CN of the RE3+ being seven.28,33,34 Tm2(CN2)3 has been shown to undergo a pressure transformation from Rc into C2/m with a significant volume reduction.28
c (Z = 6), and the coordination number (CN) six of the RE3+ ion.28 Corresponding compounds with RE = Y and Ce–Er (except Pm) crystallize monoclinically with the space group C2/m (Z = 2) and the CN of the RE3+ being seven.28,33,34 Tm2(CN2)3 has been shown to undergo a pressure transformation from Rc into C2/m with a significant volume reduction.28
Through this route, not only pseudobinary but also pseudoternary, rare-earth (RE) compounds have been synthesized. These compounds cover a wide range of mixed cation and mixed anion carbodiimides such as RE2O2(CN2) (RE = Ce, Pr, Nd, Sm, Eu, Gd, Dy–Yb),35,36 RECl(CN2) (RE = La–Pr),37 Sc2O(CN2),32 RE2Cl(CN2)N (RE = La, Ce),38 RE2Br(CN2)N (RE = La, Pr),39 REI(CN2)N (RE = La, Gd),40 and Eu2I2(CN2),41 Eu4F5(CN2)2.42 Furthermore, some pseudoquaternary NCN rare-earth (RE) compounds containing three different cations have been also developed by adding the third reactant to the conventional SSM reactions. Examples of these compounds are rare-earth carbidonitridosilicates,34,43,44 and tetracyanamidogermanates.45,46 For lanthanum, mixed anion compounds La2O2(CN2),47 La2S2(CN2),48 La2O(CN2)2,37 La3(CN2)3N49 or LaCl(CN2)50 have been so far reported.
The photoluminescence properties of lanthanide (Ln) doped RE2(CN2)329 such as Gd2(CN2)3:Ce or Tb, Ce, and Tb29 have been thoroughly studied, leading to the development of a pc-LED prototype-based on Y2(CN2)3:Ce.51 In Ce3+ doped materials luminescence usually occurs due to electronic transition from ground state levels of (2F5/2 and 2F7/2) of the [Xe]4f1 configuration to lowest crystal-field components of the [Xe]5d1. By tuning the crystal-field strength and covalent character of Ce3+, the material's luminescence properties can be adjusted.29
Herein we explore the preparation of lanthanum carbodiimides through this efficient way of synthesis. We describe the formation and structural characterization of intermediate lanthanum cyanurates obtained from aqueous solution and the thermal conversion into the novel La2(CN2)3. The La2(CN2)3 represents the missing compound among the series of RE2(CN2)3 compounds with a new crystal structure. The dodecahedral coordination of the lanthanum ion in La2(CN2)3 parallels the coordination pattern of yttrium in the structure of yttrium aluminum garnet (YAG), which resembles the host structure for the most prominent YAG:Ce phosphor in todaýs phosphor converted light-emitting diodes (pc-LED).
Results and discussion
Preparation
Rare-earth carbodiimides were successfully prepared by means of solid-state metathesis reaction (see eqn (2)). However, this way of synthesis has been unsuccessful for La2(CN2)3, because all reactions have led to the formation of LaCl(CN2).37Herein, we report the preparation of La2(CN2)3via a precursor route. The compound was synthesized through the thermal decomposition of a lanthanum cyanurate precursor. This precursor was obtained by reacting lanthanum chloride with cyanuric acid and sodium hydroxide in an aqueous solution, yielding an insoluble precipitate eqn (4).
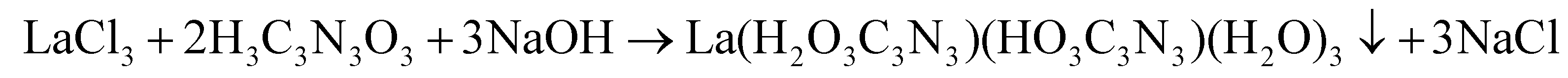 | (4) |
It is noteworthy that two distinct phases of lanthanum cyanurates were obtained as insoluble precipitates. We found out that the formation of these two phases was dependent on the pH of the solution which can be controlled by varying the amount of water content. Lower amounts of water and thus higher pH values led to the formation of a lanthanum cyanurate composed of a single deprotonated and a double deprotonated cyanurate (La(HC3N3O3)(H2C3N3O3)(H2O)3). In contrast, higher amounts of water and thus lower pH values led to the formation of a lanthanum cyanurate with two single deprotonated cyanurates along with a hydroxide ion (La(H2C3N3O3)2(OH)(H2O)4·H2O). These results can be explained by the pKa values of the first two deprotonation steps of cyanuric acid (pKa1 = 6.88 and pKa2 = 11.40).52 Only at higher pH values, the solution becomes sufficiently basic to allow the second deprotonation of the cyanuric acid and thus the formation of the lanthanum cyanurate, whereby the cyanuric acid unit is present in its double deprotonated form. However, thermoanalytical studies in the next section, will demonstrate that both lanthanum cyanurate phases could be converted to lanthanum carbodiimide at 770 °C under a flow of argon. The lanthanum carbodiimide demonstrated air and water stability over a period of four weeks, as confirmed by X-ray powder diffraction.
Thermoanalytic studies
The thermal decomposition of lanthanum cyanurates was investigated using thermal analysis methods such as thermogravimetric analysis (TGA) combined with differential thermal analysis (DTA). The measurements were made in an argon flow within a temperature range from room temperature up to 770 °C at a rate of 2 K min−1 for both heating and cooling. Fig. 1 represents the TGA of La(HC3N3O3)(H2C3N3O3)(H2O)3 which shows that this material undergoes a decomposition in three steps, ultimately leading to the formation of lanthanum carbodiimide. Based on the measured and theoretical mass losses at each stage, the first step at 187 °C, with a mass loss of −12.04% (theory: −12.15%), is attributed to the loss of water (eqn (5)). The second step at 405 °C, with a mass loss of −28.38% (theoretical: −29.03%), can be corresponded to the decomposition of La(HC3N3O3)(H2C3N3O3) to La(C3N3O3) which is a completely deprotonated unit (eqn (6)). The final step, occurring at 737 °C with a mass loss of −14.82% (theory: −14.85%), is associated with the release of carbon dioxide, resulting in the formation of lanthanum carbodiimide, La2(CN2)3 (eqn (7)). Heating the lanthanum carbodiimide to even higher temperatures (1000 °C), resulted in the formation of lanthanum nitride (LaN). This sequence can be summarized in the following reactions: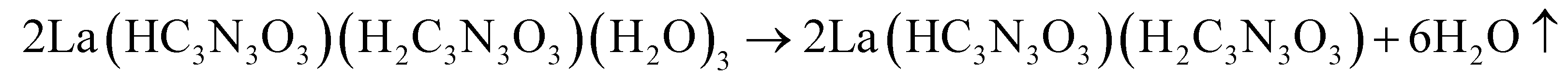 | (5) |
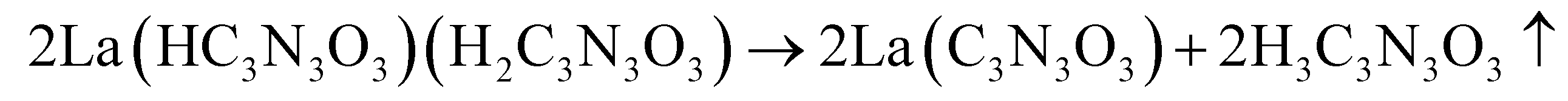 | (6) |
| 2La(C3N3O3) → La2(CN2)3 + 3CO2↑ | (7) |
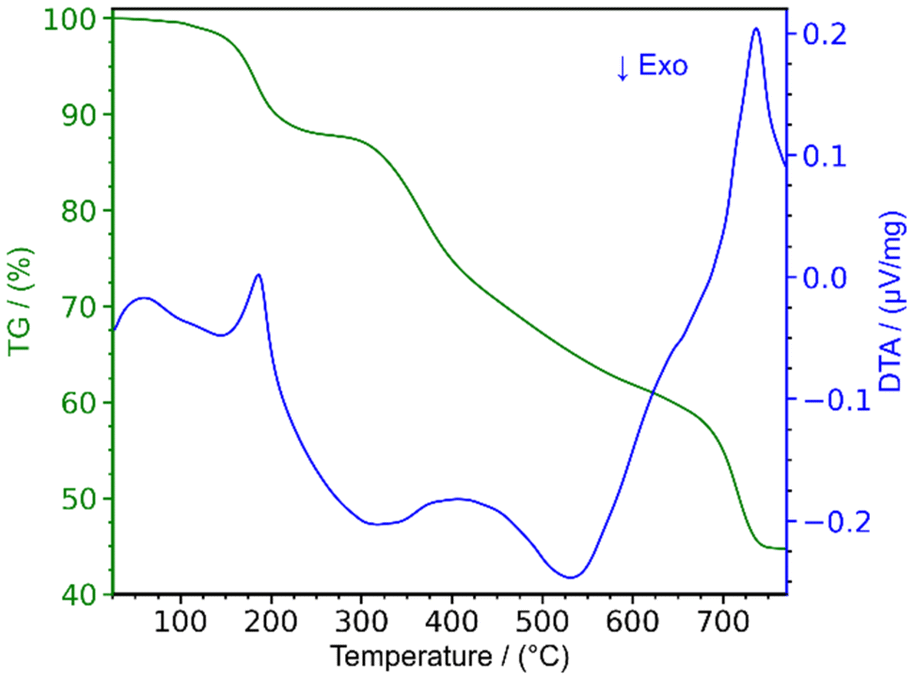 | ||
| Fig. 1 Thermogravimetric analysis (TGA) combined with differential thermal analysis (DTA) of La(HC3N3O3)(H2C3N3O3)(H2O)3. | ||
Crystal structures
The synthesis of La2(CN2)3 can be achieved by the decomposition of a single source precursor, which can be either La(HC3N3O3)(H2C3N3O3)(H2O)3(1) or La(H2C3N3O3)2(OH)(H2O)4·H2O (2). Crystal structures of both precursors were refined based on single-crystal X-ray diffraction, with relevant data summarized in Table 1. Both compounds contain one type of La3+ in the structure. The first compound (1) contains mono- and divalent cyanurate anions and three water molecules in the coordination environment of the La3+, the latter (2) monovalent cyanurate anions, bridging hydroxide and four water molecules as shown in Fig. 2 and 3. There are at least two more lanthanum cyanurate structures being reported in the literature. The crystal structure of La[H2N3C3O3]3·8.5 H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 53 was refined with the space group P1, containing monovalent cyanurate anions. Another structure, described as La(H2C3N3O3)2·OH·2H2O,54 leaves a precise assignment of hydrogen atoms behind.
53 was refined with the space group P1, containing monovalent cyanurate anions. Another structure, described as La(H2C3N3O3)2·OH·2H2O,54 leaves a precise assignment of hydrogen atoms behind.
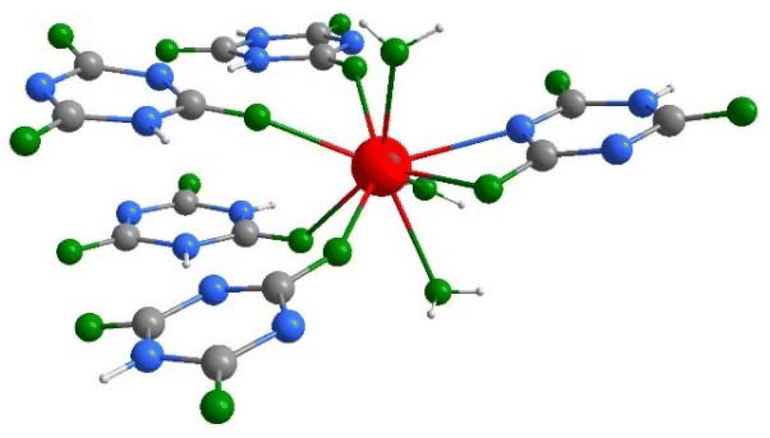 | ||
| Fig. 2 Coordination environment of the La3+ in the structure of La(HC3N3O3)(H2C3N3O3)(H2O)3(1) (gray: C, blue: N, green: O, red: La, white: H). | ||
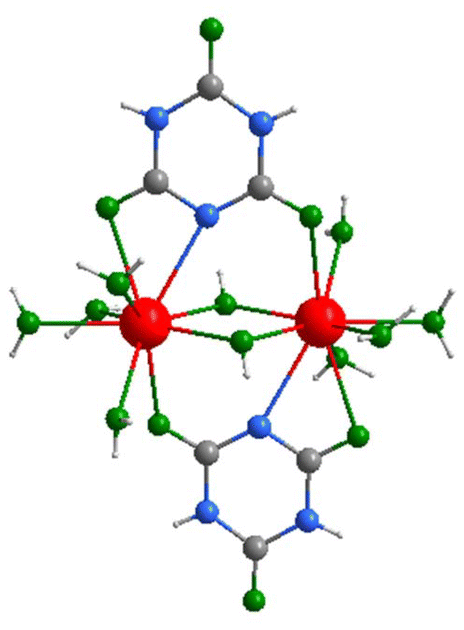 | ||
| Fig. 3 Coordination environment of the La3+ ion in the structure of La(H2C3N3O3)2(OH)(H2O)4·H2O (2) (gray: C, blue: N, green: O, red: La, white: H). | ||
| (1) | (2) | La2(CN2)3 | |
|---|---|---|---|
| CCDC code | 2403637 | 2409323 | 2393819 |
| Empirical formula | C6H9LaN6O9 | C6H15LaN6O12 | C3La2N6 |
| Formula weight (g mol−1) | 448.10 | 502.15 | 397.88 |
| Crystal system | Monoclinic | Triclinic | Monoclinic |
| Space group | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
I2/a |
| a/Å | 8.0793(4) | 6.3215(3) | 8.69003(6) |
| b/Å | 17.1808(7) | 11.0993(6) | 6.88968(5) |
| c/Å | 8.5107(4) | 11.8544(5) | 10.30517(9) |
| α/° | 66.312(5) | ||
| β/° | 99.223(5) | 88.178(3) | 105.0711(6) |
| γ/° | 75.768(4) | ||
| V/Å3 | 1166.09(9) | 736.20(7) | 595.764(8) |
| Z | 4 | 2 | 4 |
| μ/mm−1 | 29.027 | 23.237 | 108.706 |
| D c/g cm−3 | 2.552 | 2.265 | 4.436 |
| Crystal size | 0.05 × 0.03 × 0.01 | 0.04 × 0.04 × 0.01 | Powder |
| Θ range/° | 5.149 to 74.464 | 4.084 to 72.102 | 2.5 to 60 |
| Reflections collected | 41457 |
22199 |
488 |
| Parameters | 235 | 251 | 45 |
| R Bragg | — | — | 4.0955 |
| χ 2 | — | — | 1.0244 |
| Wavelength (Cu-Kα) (Å) | 1.54184 | 1.54184 | 1.54184 |
| R 1, wR2 (I > 2σ(I)) | 0.0162, 0.0385 | 0.0333, 0.0837 | — |
| R indices (all data) | 0.0175, 0.0389 | 0.0360, 0.0854 | — |
| GOOF | 1.041 | 1.076 | 1.012 |
The appearance of a number of different lanthanum cyanurate compounds emphasizes the influence of pH conditions during precipitation in aqueous solution and by a varying extent of hydration. However, so far there is no indication that the decomposition of any of these precursors would not lead to La2(CN2)3.
The crystal structure of La(HC3N3O3)(H2C3N3O3)(H2O)3(1) is characterized by one type of lanthanum, being surrounded by three coordinated water molecules and five cyanurate ions showing a bridging functionality (Fig. 2).
The crystal structure of La2(CN2)3 was solved and refined on the basis of X-ray powder diffraction data by Rietveld refinement with the space group I2/a (Table 1 and Fig. 4). Crystal structures of RE carbodiimides follow a characteristic structure pattern in which the [NCN]2− ions are arranged in layers, following the motif of a hexagonal closed packing of sticks. Cations are situated in between these layers to form an alternating arrangement.
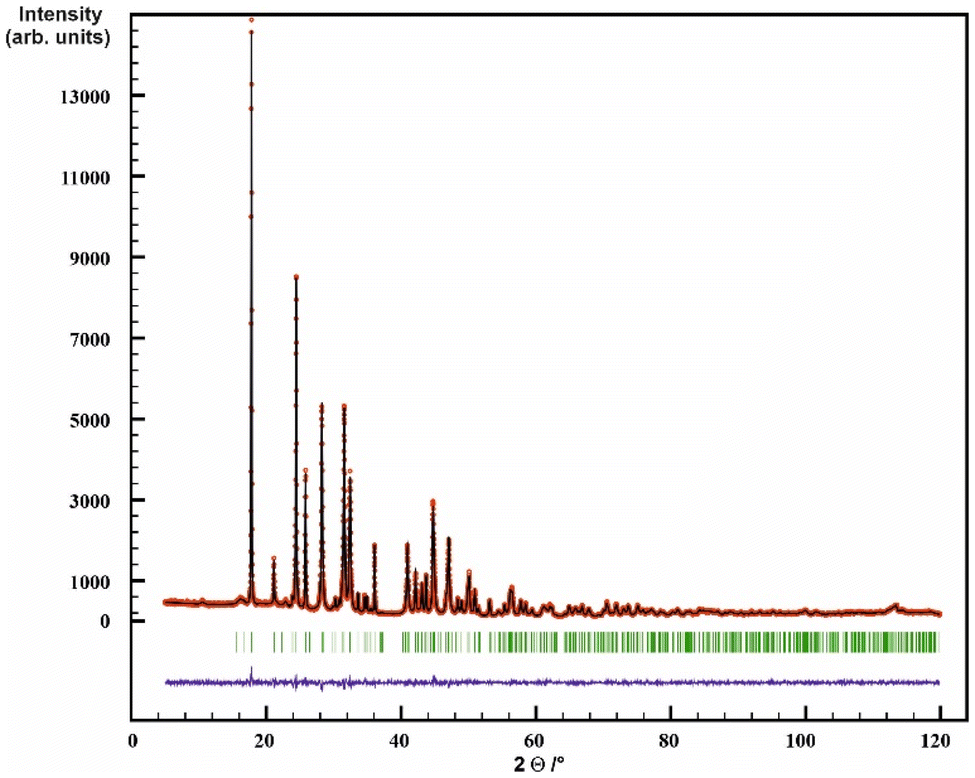 | ||
| Fig. 4 Rietveld refinement of the XRD powder pattern of La2(CN2)3. Observed intensities are marked as red circles, and calculated intensities as black lines. Bragg positions are marked as green lines, the difference curve Iobserved − Icalculated as blue line. | ||
This pattern is apparent also in the structure of La2(CN2)3, displayed in Fig. 5. Major differences among the structures of RE2(CN2)3 compounds are the tilting of NCN ions within layers relative to each other, and the exact position of RE ions in structures. The crystal structure of La2(CN2)3 contains one type of lanthanum in the structure and two distinct carbodiimides ions. Lanthanum ions in the structure have the coordination number eight, shown in Fig. 6, together with the pattern of the corresponding dodecahedron.
 | ||
| Fig. 5 The layered appearance of the crystal structure of La2(CN2)3 with lanthanum ions is shown in red. | ||
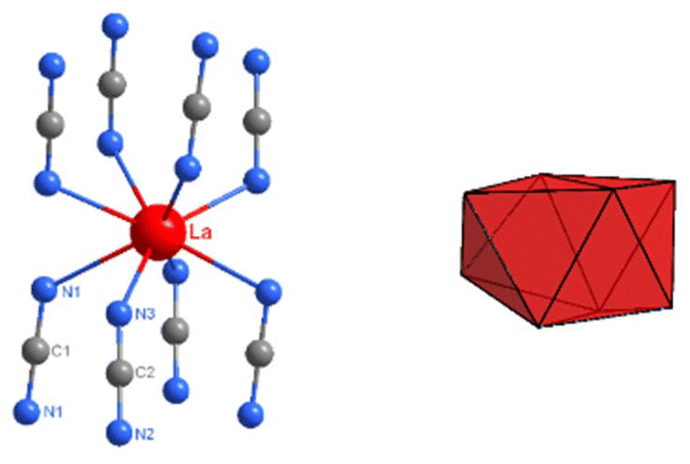 | ||
| Fig. 6 The environment of lanthanum with two crystallographically distinct [NCN]2− ions (distances: C1–N1 = 124.41(6) pm, ∠N1–C1–N1 = 177.82(4)°; C2–N2 = 123.95(9) pm, C2–N3 = 124.17(9) pm, ∠N2–C2–N3 = 178.55(7)°) and a dodecahedron representing the coordination environment of La3+ in the structure of La2(CN2)3. | ||
The characterization of La2(CN2)3 completes the series of binary rare-earth carbodiimides, which is represented by three distinct structures, respectively, coordination patterns (Fig. 7), with their unit volumes displayed in Fig. 8. The general trend of molar unit cell volumes represents the lanthanide contraction for the series from Ce to Tm with the space group C2/m (Z = 2) with the coordination number (CN) of the RE3+ being seven. Compounds of Tm, Yb, and Lu follow the same trend, however with a trigonal structure with the CN of the RE being six. Tm2(CN2)3 is dimorphic and undergoes a pressure transformation from Rc into C2/m. Thereby, the coordination number of the Tm3+ ion increases from six to seven. Lanthanum, as the largest lanthanide ion, appears with the coordination number eight.
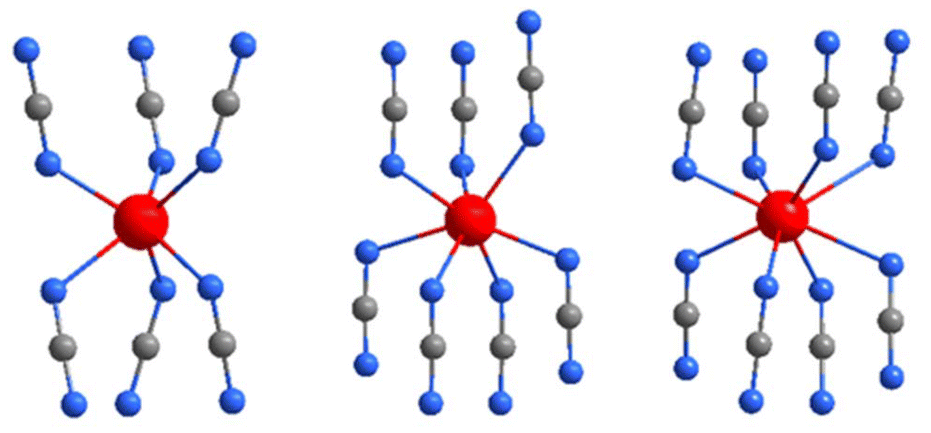 | ||
| Fig. 7 Coordination environments of rare-earth ions (red) in the trigonal (Rc), monoclinic (C2/m), and (I2/a) structures, from left to right. | ||
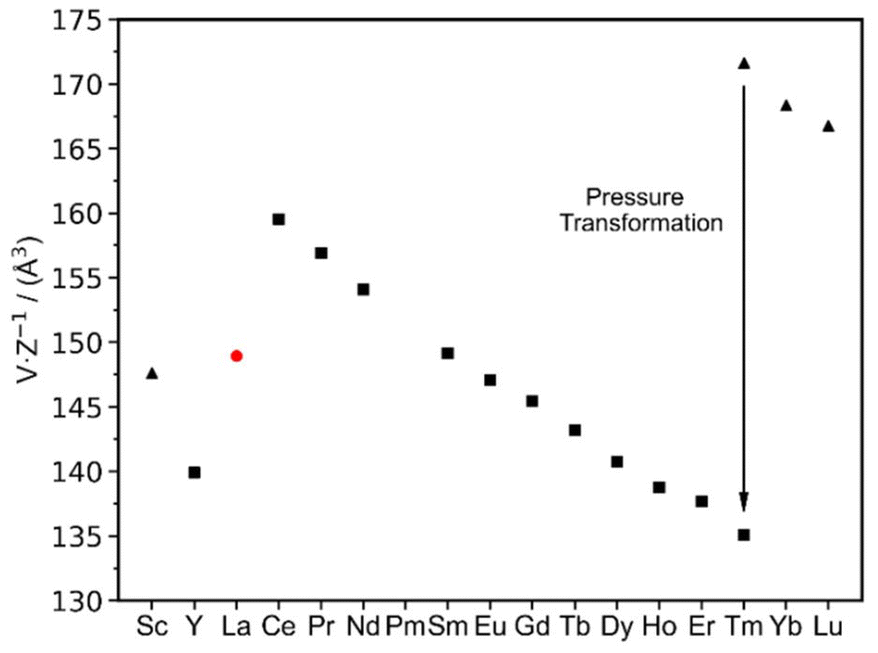 | ||
| Fig. 8 Molar volume (V/Z) of rare-earth carbodiimides, RE2(CN2)3, with the monoclinic (C2/m) structure for the series RE = Y, and Ce–Tm (displayed as squares), the trigonal (Rc) structure for RE = Sc (displayed as triangles) and Tm–Lu, and monoclinic (I2/a) La2(CN2)3 (displayed as a red dot). Tm2(CN2)3 has been shown to be dimorphic with the monoclinic (C2/m) structure and the trigonal (Rc) structure of the high-pressure phase. | ||
The lanthanum site with its dodecahedral environment in La2(CN2)3 appears to be an interesting site for doping with a photoluminescence activator. A most prominent example of such a dodecahedral environment is apparent in the pc-LED phosphor Y3Al5O12:Ce. Consequently, we conducted photoluminescence studies on La2(CN2)3:Ce, discussed later in this work.
Infrared spectroscopy
The infrared (IR) spectrum of La2(CN2)3 has also been reported to support its carbodiimide character (see Fig. 6 for distances and angles). As can be observed in Fig. 9 the characteristic vibrational bands of the [NCN]2− ion at 2029 and 1929 cm−1 can be ascribed to the asymmetric stretching vibrations (νas). Furthermore, the corresponding bending vibrations (δ) are observed at 685, 660, and 621 cm−1. The absence of a symmetric vibration (νs) in the IR spectrum supports the identification of the compound as a carbodiimide rather than a cyanamide.37 Furthermore, the results align well with previously reported data on Table 2 for two other rare-earth metal carbodiimide modifications.55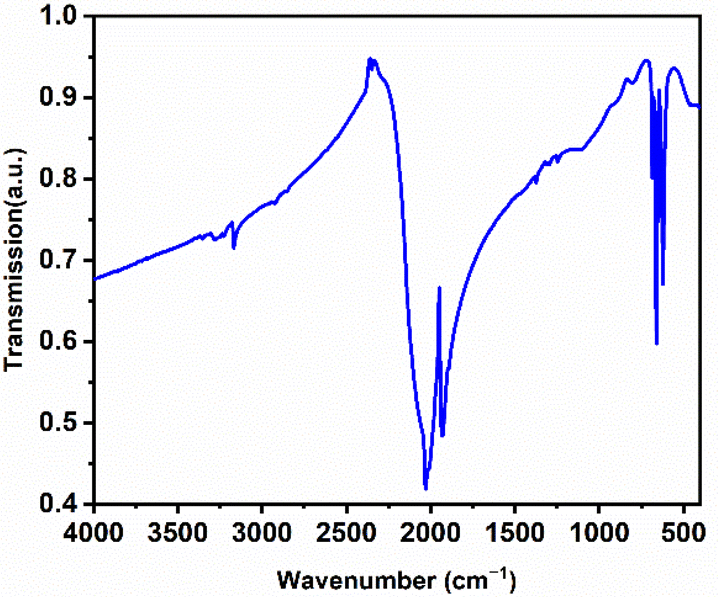 | ||
| Fig. 9 FT-IR spectrum of La2(CN2)3. | ||
| ν as (CN2)2− | δ (CN2)2− | |||||
|---|---|---|---|---|---|---|
| Lu2(CN2)3 | 2080 | 2009 | — | 680 | 640 | — |
| Sm2(CN2)3 | 2023 | 1955 | 705 | 668 | 634 | 616 |
| La2(CN2)3 | 2029 | 1929 | 685 | 660 | 621 | — |
Photoluminescence spectroscopy
The photoluminescence (PL) spectrum of La2(CN2)3 doped with Ce3+ (5 atom% w.r.t La) has been studied to evaluate the potential of this novel material for application in a luminescent screen. Ce3+ is already well-known as an efficient emitter in various applications of luminescent materials.56–59 For instance, Ce3+ is one of the most important emitters in LED phosphors, such as Ln3(Al,Ga,Sc)5O12:Ce (Ln = Y, Gd, Tb, Lu), or acts as a primary activator ion in scintillators, including Lu2SiO5:Ce, LuAlO3:Ce, and Lu3Al5O12:Ce (LuAG:Ce3+) (see Table 3). Ce3+-activated materials also play an important role in UV lamps, such as YPO4:Ce, LaPO4:Ce, LaMgAl11O19:Ce, and YMgB5O10:Ce.60| Ce3+ phosphor or scintillator | Emission max. nm | Density g cm−3 | Decay time ns |
|---|---|---|---|
| LaBr3:Ce | 358 | 5.3 | 35 |
| YAlO3:Ce | 360 | 5.6 | 20–30 |
| LuAlO3:Ce | 365 | 8.3 | 18 |
| Lu2SiO5:Ce | 390 | 7.4 | 30 |
| Gd2SiO5:Ce | 420 | 6.7 | 60 |
| Lu3Al5O12:Ce | 525 | 6.7 | 54 |
The photoluminescence of materials doped with Ce3+ ions originates from interconfigurational transitions, i.e. of transitions between different electronic configurations. In the ground state, Ce3+ has the configuration [Xe]4f1 which, upon excitation is promoted to the configuration [Xe]5d1. The energy required for this transition depends on the crystal field splitting of the 5d levels, which is influenced by the surrounding coordinated anions and centroid shift caused by the local environment. The emission process caused by the relaxation of the 5d1 to the 4f1 configuration yields two broad emission bands due to the ground state term spin-orbit splitting yielding the terms 2F5/2 and 2F7/2. This kind of transition is spin and parity allowed, therefore, resulting in a high oscillator strength and a short decay constant. Since a 5d orbital is involved, cerium-activated materials may show UV, blue, green, or yellow emission depending on the crystal-field-determined energetic position of absorption and emission bands. The 5d orbitals are more spatially extended and thus strongly interact with the crystal field around the atom, therefore, splitting into different levels.
Fig. 10a shows the broad emission band of La2(CN2)3 doped with 5% of Ce3+, with two maxima at 450 nm (2222 cm−1) and 500 nm (20000 cm−1) upon 400 nm excitation, at various temperatures. The excitation spectrum monitored for 495 nm is also shown in Fig. 10b and exhibits a strong excitation band at 400 nm (25000 cm−1), thus the Stokes shift is just 2800 cm−1, which points to little relaxation in the excited state. As mentioned above, the crystal field strength and thus chemical environment such as ligand type, symmetry, and metal-to-ligand distance determines the energy gap between the [Xe]4f1 and [Xe]5d1 configuration. Therefore, the PL spectra will change if Ce3+ is located onto different crystallographic sites. A weaker crystal field increases the energy gap and results in a shift of the PL spectra towards higher energy such as UV or blue light. A comparison with the earlier published Gd2(CN2)3:Ce3+ with a broad emission band at 575 nm (17391 cm−1) under 415 nm excitation,29 and Y2(CN2)3:Ce3+51 with a emission band range of 570–577 nm under 415 nm excitation, confirms that the emission bands in La2(CN2)3:Ce3+ are strongly blue-shifted. This can be explained by the weaker crystal field in La2(CN2)3 than Gd2(CN2)3, and Y2(CN2)3 due to the larger metal-to-ligand distances onto the La3+ site. The material shows at low temperature a single exponential decay curve, while the calculated decay time of 26 ns at 77 K (Fig. 10c) is typical for blue-emitting Ce3+ activated luminescent materials or scintillators.
 | ||
| Fig. 10 (a) Emission spectra at several temperatures and (b) excitation spectra, and (c) decay curve upon 375 nm excitation of La2(CN2)3:Ce3+(5%). | ||
Conclusions
The vast majority of rare-earth carbodiimide compounds have been prepared by solid-state metathesis (SSM) reactions. However, the preparation of La2(CN2)3 has yet failed by SSM, because its formation is hindered in favour of the formation of La2Cl(CN2). An alternative way of preparation of metal carbodiimides is the employment of metal cyanurate precursors, showing the formation of pure La2(CN2)3 by decomposition of lanthanum cyanurate hydrate as a single-source precursor.Lanthanum cyanurate compounds can appear with mono- or divalent cyanurate anions and with different amounts of water molecules. The thermal conversion into lanthanum carbodiimide appears in three basic steps, by release of water, the loss of cyanuric acid, and finally the loss of CO2.
The crystal structure of lanthanum carbodiimide completes the series of rare-earth carbodiimides that is represented by three distinct structure types and coordination numbers of the rare-earth ions. The structure of La2(CN2)3 contains La3+ with the coordination number eight and represents an attractive host lattice for doping with rare-earth activators. Doping with Ce3+ leads to the luminescent material La2(CN2)3:Ce which shows a blue photoluminescence on excitation with 375 nm. Due to its high density and short decay time, this phosphor can be considered as a perspective szintillator material.
Experimental section
Materials and methods
The following starting materials cyanuric acid (Sigma-Aldrich, 98%), lanthanum(III) chloride heptahydrate (Fluka AG, 99.9%), sodium hydroxide (Chemsolute, 98.8%), and cerium(III) chloride heptahydrate (Merck, 98.5%) were used without further purification and were handled under air and standard conditions.The thermal decomposition was carried out in a Carbolite HST 12/300 furnace equipped with a 1 m long quartz glass furnace tube. The decomposition product was handled under an inert argon atmosphere and transferred into a glovebox with maintained moisture and oxygen levels below 1 ppm.
Synthesis
The resulting precipitate was collected by filtration (yield w.r.t. La 64%), washed three times with 20 mL of deionized water to remove sodium chloride and dried in an oven at 87.5 °C.
Thermoanalytic studies
Thermogravimetric analysis and differential thermal analysis were performed using a Netzsch Jupiter STA 449 F3 thermal analyzer. The synthesized samples were measured in an open corundum crucible under a continuous argon flow.X-ray powder diffraction
Compounds were investigated by powder X-ray diffraction (XRD) on a Stadi-P (STOE, Darmstadt) diffractometer with germanium monochromated Cu-Kα1 radiation. The powder XRD pattern of La2(CN2)3 was indexed in the space group I2/a (No. 15) using N-Treor0963 and the structure was solved by direct methods (both included in the program EXPO64). Other possible candidate space groups suggested by EXPO were lower in symmetry (structure solutions with space groups Ia or I2 were discarded by using the progam missym included in the program package PLATON65) or gave no reasonable structure solution (I2/m, Im). The crystal structure refinement was carried out with Winplotr (Fullprof), with the final full refinement plot displayed in Fig. 4.
Single-crystal X-ray diffraction
Suitable crystals were selected and mounted on XtaLAB Synergy, Dualflex, HyPix diffractometer. The crystals were kept at a steady T = 150.0(2) K during data collection. The structure was solved, and the space group was determined with the ShelXT 2018/2 (Sheldrick, 2018) solution program using dual methods and by using Olex2 1.5-ac5-02466 as the graphical interface. The model was refined with ShelXL 2018/367 using full matrix least squares minimization on F2. All non-hydrogen atoms were refined anisotropically. Hydrogen atom positions were found in the electron difference map and refined.
Infrared spectroscopy
The infrared spectroscopy was performed using a Bruker VERTEX 70 Fourier-transform infrared (FT-IR) spectrometer, with the measurement recorded in the spectral range of 400–4000 cm−1. The samples were prepared as potassium bromide (KBr) pellets, with a pure KBr pellet serving as the reference for baseline correction.Photoluminescence spectroscopy
Emission and excitation spectra of La2(CN2)3:Ce3+ were measured using an FLS920 fluorescence spectrometer, Edinburgh Instruments, equipped with a 450 W xenon arc lamp OSRAM. The sample chamber was fitted with a mirror optic, specifically designed for powder samples. Detection was carried out with a R2658P single-photon counting photomultiplier tube Hamamatsu. The luminescence spectra were recorded at a spectral resolution of 1 nm, dwell time 0.5 seconds per 1 nm step, and the measurement was repeated three times. The correction file for the emission spectra was obtained from calibration with a tungsten incandescent lamp certified by NPL (National Physics Laboratory, UK). For recording temperature dependent emission spectra, a cryostat “MicrostatN” from Oxford Instruments was introduced in the above described spectrometer. The decay curves were measured using an Edinburgh Instruments FLS920 fluorescence spectrometer with a photomultiplier from Hamamatsu H74422-40. The samples were excited with an Edinburgh EPL655 pulsed diode laser (65 ps pulse width).Author contributions
H.-J. M.: conceptualization, supervision, funding acquisition, writing, review, and editing. P. S.: synthesis, PXRD, IR, TGA, writing. E. B.: writing, preparation, experimental analysis. M. S.: X-ray diffraction refinements and structure solutions. T. J. and D. E.: photoluminescence spectroscopy. All authors have read and agreed to the published version of the manuscript.Data availability
Crystallographic data have been deposited at the CCDC under 2403637(1), 2409323(2), and 2393819 (La2(CN2)3).†Data are available within the article.
The data that support the findings of this study are available on request from the corresponding author, H.-J. Meyer.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledge the support provided by the Deutsche Forschungsgemeinschaft (DFG-Bonn) for this research project (ME 914/34-1).References
- M. Kastens and W. McBurney, Ind. Eng. Chem., 1951, 43, 1020–1033 CrossRef CAS.
- M. G. Down, M. J. Haley, P. Hubberstey, R. J. Pulham and A. E. Thunder, J. Chem. Soc., Dalton Trans., 1978, 1407–1411 RSC.
- M. Löber, Doctoral dissertation, Universität Tübingen, 2022.
- M. Becker, M. Jansen, A. Lieb, W. Milius and W. Schnick, Z. Anorg. Allg. Chem., 1998, 624, 113–118 CrossRef CAS.
- M. Ströbele, E. Bayat and H.-J. Meyer, Inorg. Chem., 2024, 63, 16565–16572 CrossRef PubMed.
- M. Becker, J. Nuss and M. Jansen, Z. Anorg. Allg. Chem., 2000, 626, 2505–2508 CrossRef CAS.
- M. Becker and M. Jansen, Solid State Sci., 2000, 2, 711–715 CrossRef CAS.
- U. Berger and W. Schnick, J. Alloys Compd., 1994, 206, 179–184 CrossRef CAS.
- H.-J. Meyer, Dalton Trans., 2010, 39, 5973–5982 RSC.
- K. Gibson, M. Ströbele, B. Blaschkowski, J. Glaser, M. Weisser, R. Srinivasan, H. J. Kolb and H.-J. Meyer, Z. Anorg. Allg. Chem., 2003, 629, 1863–1870 CrossRef CAS.
- G. Seifer, Russ. J. Coord. Chem., 2002, 28, 301–324 CrossRef CAS.
- A. Klimek, J. Yount, D. Wozniak, M. Zeller and D. G. Piercey, Inorg. Chem., 2023, 62, 16280–16282 CrossRef CAS PubMed.
- E. Bayat, M. Ströbele, D. Enseling, T. Jüstel and H.-J. Meyer, Dalton Trans., 2024, 53, 10912–10918 RSC.
- M. Becker and M. Jansen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 347–348 CrossRef CAS PubMed.
- M. Krott, X. Liu, B. P. Fokwa, M. Speldrich, H. Lueken and R. Dronskowski, Inorg. Chem., 2007, 46, 2204–2207 CrossRef CAS PubMed.
- X. Liu, M. A. Wankeu, H. Lueken and R. Dronskowski, Z. Naturforsch., B:J. Chem. Sci., 2005, 60, 593–596 CrossRef CAS.
- G. Baldinozzi, B. Malinowska, M. Rakib and G. Durand, J. Mater. Chem., 2002, 12, 268–272 RSC.
- M. Becker, J. Nuss and M. Jansen, Z. Naturforsch., B:J. Chem. Sci., 2000, 55, 383–385 CrossRef CAS.
- J. Peng, Y. Wang, M. Fecčík, L. Bayarjargal and R. Dronskowski, ACS Appl. Mater. Interfaces, 2024, 61946–61956 CrossRef CAS PubMed.
- X. Liu, M. Krott, P. Müller, C. Hu, H. Lueken and R. Dronskowski, Inorg. Chem., 2005, 44, 3001–3003 CrossRef CAS PubMed.
- X. Tang, H. Xiang, X. Liu, M. Speldrich and R. Dronskowski, Angew. Chem., Int. Ed., 2010, 49, 4738–4742 CrossRef CAS PubMed.
- K. Dolabdjian, A. Kobald, C. P. Romao and H.-J. Meyer, Dalton Trans., 2018, 47, 10249–10255 RSC.
- X. Liu, A. Decker, D. Schmitz and R. Dronskowski, Z. Anorg. Allg. Chem., 2000, 626, 103–105 CrossRef CAS.
- X. Qiao, K. Chen, A. J. Corkett, D. Mroz, X. Huang, R. Wang, R. Nelson and R. Dronskowski, Inorg. Chem., 2021, 60, 12664–12670 CrossRef CAS PubMed.
- L. Stork, X. Liu, B. P. Fokwa and R. Dronskowski, Z. Anorg. Allg. Chem., 2007, 633, 1339–1342 CrossRef CAS.
- R. Dronskowski, Z. Naturforsch., B:J. Chem. Sci., 1995, 50, 1245–1251 CrossRef CAS.
- M. Löber, K. Dolabdjian, M. Ströbele, C. P. Romao and H.-J. Meyer, Inorg. Chem., 2019, 58, 7845–7851 CrossRef PubMed.
- M. Neukirch, S. Tragl and H.-J. Meyer, Inorg. Chem., 2006, 45, 8188–8193 CrossRef CAS PubMed.
- J. Glaser, L. Unverfehrt, H. Bettentrup, G. Heymann, H. Huppertz, T. Jüstel and H.-J. Meyer, Inorg. Chem., 2008, 47, 10455–10460 CrossRef CAS PubMed.
- O. Reckeweg and F. J. DiSalvo, Z. Anorg. Allg. Chem., 2003, 629, 177–179 CrossRef CAS.
- H. Hartmann and W. Eckelmann, Z. Anorg. Allg. Chem., 1948, 257, 183–194 CrossRef CAS.
- P. Kallenbach, M. Ströbele and H.-J. Meyer, Z. Anorg. Allg. Chem., 2020, 646, 1281–1284 CrossRef CAS.
- O. Reckeweg, T. Schleid and F. J. DiSalvo, Z. Naturforsch., B:J. Chem. Sci., 2007, 62, 658–662 CrossRef CAS.
- D. Dutczak, M. Ströbele, D. Enseling, T. Jüstel and H.-J. Meyer, Eur. J. Inorg. Chem., 2016, 2016, 4011–4016 CrossRef CAS.
- Y. Hashimoto, M. Takahashi, S. Kikkawa and F. Kanamaru, J. Solid State Chem., 1996, 125, 37–42 CrossRef CAS.
- M. Li, W. Yuan, J. Wang, C. Gu and H. Zhao, Powder Diffr., 2007, 22, 59–63 CrossRef CAS.
- R. Srinivasan, J. Glaser, S. Tragl and H.-J. Meyer, Z. Anorg. Allg. Chem., 2005, 631, 479–483 CrossRef CAS.
- R. Srinivasan, M. Ströbele and H.-J. Meyer, Inorg. Chem., 2003, 42, 3406–3411 CrossRef CAS PubMed.
- R. Srinivasan, Doctoral dissertation, Universität Tübingen, 2004.
- D. Dutczak, A. Siai, M. Ströbele, D. Enseling, T. Jüstel and H.-J. Meyer, Eur. J. Inorg. Chem., 2020, 2020, 3954–3958 CrossRef CAS.
- W. Liao, U. Englert and R. Dronskowski, Eur. J. Inorg. Chem., 2006, 2006, 4233–4236 CrossRef.
- L. Unverfehrt, M. Ströbele, J. Glaser, T. Langer, R.-D. Hoffmann, R. Pöttgen and H.-J. Meyer, Inorg. Chem., 2011, 50, 6010–6018 CrossRef CAS PubMed.
- J. Glaser and H.-J. Meyer, Angew. Chem., Int. Ed., 2008, 47, 7547–7550 CrossRef CAS PubMed.
- J. Glaser, H. Bettentrup, T. Jüstel and H.-J. Meyer, Inorg. Chem., 2010, 49, 2954–2959 CrossRef CAS PubMed.
- M. Kalmutzki, D. Enseling, J. E. Wren, S. Kroeker, V. V. Terskikh, T. Jüstel and H.-J. Meyer, Inorg. Chem., 2013, 52, 12372–12382 CrossRef CAS PubMed.
- K. Dolabdjian, C. Schedel, D. Enseling, T. Jüstel and H.-J. Meyer, Z. Anorg. Allg. Chem., 2017, 643, 488–494 CrossRef CAS.
- Y. Hashimoto, M. Takahashi, S. Kikkawa and F. Kanamaru, J. Solid State Chem., 1995, 114, 592–594 CrossRef CAS.
- A. T. Schwarz, M. Ströbele and H.-J. Meyer, Z. Anorg. Allg. Chem., 2024, 650, e202400038 CrossRef CAS.
- L. Unverfehrt, Doctoral dissertation, Universität Tübingen, 2011.
- N. Asakuma, M. Iijima, T. Tamura, S. Honda, D. Urushihara, T. Asaka, S. Bernard and Y. Iwamoto, Inorg. Chem., 2024, 63(43), 20380–20387 CrossRef CAS PubMed.
- Y.-C. Wu, T.-M. Chen, C.-H. Chiu and C.-N. Mo, J. Electrochem. Soc., 2010, 157, J342 CrossRef CAS.
- W. Haynes, T. Bruno and D. Lide, CRC handbook of chemistry and physics, CRC Press, Boca Raton, 95th edn, 2014, pp. 5–94 Search PubMed.
- O. Reckeweg, F. Lissner and T. Schleid, Z. Naturforsch., B:J. Chem. Sci., 2021, 76, 733–738 CrossRef CAS.
- X. Hao, M. Luo, C. Lin, D. Lin, L. Cao and N. Ye, Dalton Trans., 2019, 48, 12296–12302 RSC.
- M. Neukirch, Doctoral dissertation, Universität Tübingen, 2007.
- P. Dorenbos, J. Lumin., 2002, 99, 283–299 CrossRef CAS.
- Z. Xia and A. Meijerink, Chem. Soc. Rev., 2017, 46, 275–299 RSC.
- A. C. Berends, M. A. van de Haar and M. R. Krames, Chem. Rev., 2020, 120, 13461–13479 CrossRef CAS PubMed.
- S. Wang, Z. Song and Q. Liu, J. Mater. Chem. C, 2023, 11, 48–96 RSC.
- A. Zukauskas, M. Shur and R. Gaska, Solid state lighting, John Wiley & Sons, 2002 Search PubMed.
- C. W. Van Eijk, Phys. Med. Biol., 2002, 47, R85 CrossRef PubMed.
- P. A. Rodnyi, Physical processes in inorganic scintillators, CRC, New York, 1997 Search PubMed.
- A. Altomare, G. Campi, C. Cuocci, L. Eriksson, C. Giacovazzo, A. Moliterni, R. Rizzi and P.-E. Werner, J. Appl. Crystallogr., 2009, 42, 768–775 CrossRef CAS.
- A. Altomare, C. Cuocci, C. Giacovazzo, A. Moliterni, R. Rizzi, N. Corriero and A. Falcicchio, J. Appl. Crystallogr., 2013, 46, 1231–1235 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2403637, 2409323 and 2393819. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00060b |
| This journal is © The Royal Society of Chemistry 2025 |