
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarnessing trace water for enhanced photocatalytic oxidation of biomass-derived alcohols to aldehydes†
Wenhua
Xue
a,
Jian
Ye
a,
Zhi
Zhu
a,
Reeti
Kumar
a and
Jun
Zhao
 *ab
*ab
aDepartment of Biology, Hong Kong Baptist University, Kowloon Tong, Hong Kong SAR, China. E-mail: zhaojun@hkbu.edu.hk
bInstitute of Advanced Materials, Hong Kong Baptist University, Kowloon Tong, Hong Kong SAR, China
First published on 18th September 2024
Abstract
The photocatalytic conversion of biomass into valuable chemicals offers a more energy-efficient and low-carbon alternative to traditional thermal catalysis, representing a promising strategy for fine chemical production without relying on fossil fuels. While most studies have focused on catalyst design to improve efficiency, the precise tuning of solvents has been neglected. This study examines the conversion of 5-hydroxymethylfurfural (HMF) into 2,5-diformylfuran (DFF) as a representative biomass alcohol oxidation reaction, revealing a novel strategy for enhancing photocatalytic efficiency through solvent fine-tuning. The introduction of minute quantities of water into an acetonitrile solvent matrix during the HMF oxidation reaction was observed to engender pronounced enhancements in catalytic performance, with the degree of effect contingent on precise water concentration. Trace water is an active participant in the catalytic cycle, diminishing the activation energy barrier for bond cleavage and averting catalyst deactivation. This mechanism also proved applicable to other photocatalysts and various alcohol substrates, highlighting the potential of this strategy for enhancing photoenergy utilization and reaction efficiency.
Broader contextThis work on the photocatalytic conversion of biomass into valuable chemicals represents a significant advancement in the field of energy and environmental science. The study introduces a novel strategy for enhancing photocatalytic efficiency by focusing on the precise tuning of solvents in addition to catalyst design. Specifically, the discovery that the introduction of minute quantities of water into solvents can greatly improve catalytic performance offers a promising avenue for increasing the efficiency of biomass alcohol oxidation reactions. This finding not only sheds light on the underlying mechanisms at play during photocatalysis but also demonstrates a versatile approach that can be applied to various photocatalysts and alcohol substrates. The ability to lower activation energy barriers and prevent catalyst deactivation through the involvement of trace water in the catalytic system represents a significant step forward in the quest for sustainable and efficient chemical production from biomass feedstock. |
Introduction
The catalytic transformation of biomass is pivotal in renewable energy and resource utilization, offering a strategy to lessen the dependence on fossil fuels and curb environmental pollution.1–4 In contrast to traditional energy-intensive industrial processes, solar-driven biomass conversion has emerged as a promising approach for the sustainable production of high-value liquid-phase chemicals, characterized by its low energy consumption and minimal environmental footprint. Therefore, converting biomass alcohols into aldehydes is particularly valuable and industrially important since aldehydes serve as critical intermediates in producing fine chemicals and polymer precursors. However, the mechanism underlying the photocatalytic oxidation of these alcohols in the liquid phase remains largely obscure owing to the susceptibility of the catalytic activity of catalysts to reaction conditions (solvents and atmosphere). Therefore, a clear understanding of such photo-induced reactions is critical for advancing these industrial processes as it promises to reduce the dependence on fossil fuels in traditional chemical production and associated pollutant emissions, thereby achieving waste valorization in a more sustainable way.As a representative biomass-derived chemical, the oxidation of 5-hydroxymethylfurfural (5-HMF) to 2,5-diformylfuran (DFF) is a significant process in biomass valorization as DFF holds high industrial potential in furan-based polyesters, pharmaceuticals, electrooptical devices, fungicides, etc. To date, Cd-based sulfides, ZnIn2S4, g-C3N4, halide perovskites and some composites have been developed for catalyzing the oxidation of HMF to DFF in acetonitrile (ACN).5–9 However, this photocatalytic process is still at an early stage of development as there is a considerable lack of understanding and ongoing debate on the mechanisms governing the conversion of HMF into DFF in the liquid phase. Although certain photocatalytic systems have demonstrated commendable efficacy, and multiple putative reaction mechanisms involving charge transfer and radical interactions based on energy-level alignment have been postulated, the process remains yet to be elucidated owing to the mentioned condition-dependent reactivities of catalysts, as well as the obscure surface reaction in the organic phase.10–13 This gap in knowledge hampers its further development and highlights the imperative for further investigation to elucidate the dehydrogenation of alcohols to aldehydes in the liquid phase including DFF synthesis from HMF.
Currently, the principle of catalyst design is well-understood and acknowledged as an essential approach to improve reaction efficiencies within the domain of photocatalysis.14–16 As an indispensable part of heterogeneous photocatalysis, the reaction conditions of the catalyst also strongly affect the photo-synthesis reaction and even exert a more substantial impact on the reaction kinetics, product selectivity and the overarching efficiency of the reaction. However, understanding the influence of solvents on the process of analogous light-induced reactions at the molecular level still faces significant challenges and uncertainties. Accordingly, there is still very limited knowledge on the solvent refinement to improve the photo-synthesis reaction efficiency. In most cases, researchers often conduct tests by simply replacing solvents to identify the optimal one, without delving deeper into the finer and more comprehensive regulation of solvents, thus confining exploration of their potential benefits. The changes in reaction rates and product distribution in different solvents are often attributed to the diversities of produced reactive oxygen species, differences in redox potential, polarities and dielectric constants of solvents.17–21 In fact, solvents possess the ability to not only modify the surface chemistry by binding to active sites and (de)stabilizing surface intermediates but also to reduce reaction barriers, facilitate proton transfer, and even alter the reactivity of reactive oxygen species (ROS).22–26 In other words, it is imperative to disclose the concomitantly critical effect of solvent dynamics, which frequently exert a significant impact on biomass conversion.
Based on these considerations, HMF oxidation into DFF was employed as a typical process, and we decided to take a different approach by optimizing the solvents to overcome the challenges associated with substantial enhancement in the efficiency, which has proven difficult through catalyst design alone. In our reaction system where Cd0.75Zn0.25S nanoparticles were employed as the model catalyst and ACN (HPLC grade with water content <30 ppm) served as the solvent, it is observed that the reaction tended to stagnate after reaching a certain extent, despite the presence of a significant amount of unreacted HMF. Surprisingly, the addition of a small amount of water to the reaction system enabled continuous progression of the reaction, leading to a noticeable improvement in DFF yield. The product yield began to decline once beyond a certain proportion of water in the solvent. This intriguing and unique reaction phenomenon suggested that the role or mechanism of water or solvent in this reaction is far more complex than initially anticipated. Our results indicated that water molecules actively participated in the reaction process via the H-bonding interaction. Importantly, the same solvent refinement strategy was also applicable to other alcohol-to-aldehyde reactions and even other photocatalysts. This approach not only advances the photocatalytic reaction efficiency but also highlights the critical role of solvent grade in reducing energy consumption and promoting sustainable chemical processes.
Results
Photocatalytic HMF conversion into DFF and optimization
The characterization results of Cd0.75Zn0.25S nanoparticles are given in the ESI† (Fig. S1–S3). The photocatalytic reaction was initially performed in air atmosphere at room temperature with ACN (HPLC grade, water content <30 ppm) as the solvent under blue LED irradiation. Fig. 1(a) indicates that the DFF yield reached 23.5% with about 40.5% HMF conversion after 12 h reaction in ACN, and FFCA (5-formylfuran-2-carboxylic acid, one of the over-oxidation products) was the main over-oxidation product with a yield around 3%. The mineralization amount of HMF was about 12% in ACN. However, DFF generation in ACN ceased after about 6 hours of reaction (Fig. 1(b)), and extending the reaction time cannot further improve the DFF yield; instead, the prolonged reaction time only resulted in increased HMF conversion. It is affirmed that the portion deviating from the carbon balance for HMF conversion almost entirely converted into inorganic carbon (called mineralization in later text), which was reflected by the gas chromatography–mass spectrometry (GC–MS) results of the reacted solution, as shown in Fig. S4 (ESI†). Furthermore, the DFF yield remained at a low level from 20% to 25% by changing the Cd/Zn ratio in CdxZn1−xS (Fig. S5, ESI†), but the conversion and mineralization percent of HMF both decreased with the increase in Cd content, indicating that the CB/VB position may have a significant influence on HMF conversion, while DFF formation may be mainly limited by other factors. Similarly, co-catalysts such as Au, Ag, Pt, Pd, Ru, Co, and Ni were unable to further increase the DFF yield in ACN due to the stagnant reaction process (Fig. S6a and Table S1, ESI†). We believe that these reaction phenomena also exist when using similar photocatalysts.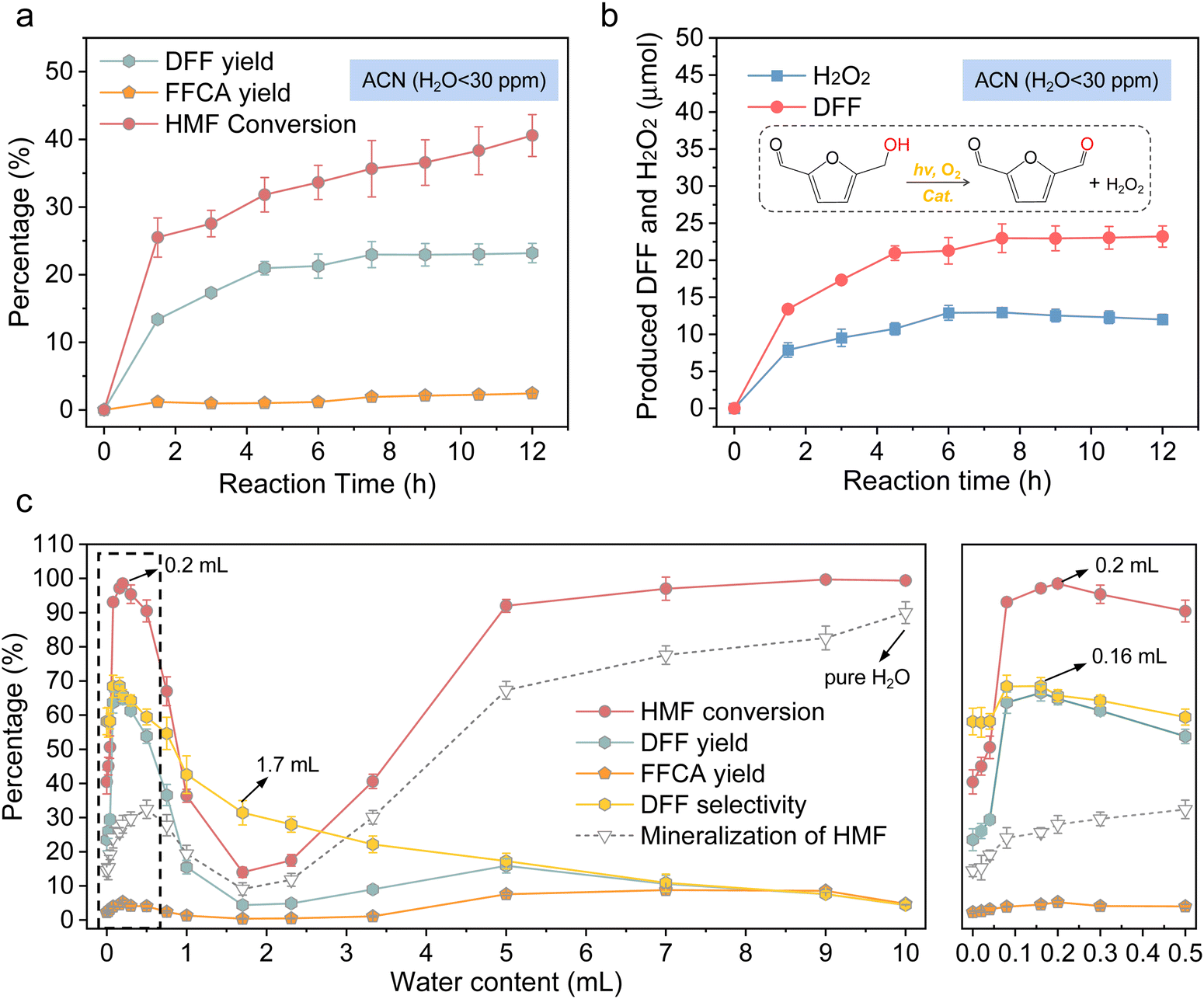 | ||
| Fig. 1 (a) Time-dependent HMF conversion, DFF yield and FFCA yield in ACN over Cd0.75Zn0.25S. (b) Time-dependent DFF and H2O2 production in ACN over Cd0.75Zn0.25S. (c) HMF oxidation activity of Cd0.75Zn0.25S in a mixed solution of ACN and H2O (VH2O + VACN = 10 mL; reaction time: 12 h). | ||
Interestingly, the addition of a small amount of water to the reaction solution can effectively overcome the issues of reaction stagnation and low product yield (original data are shown in Table S2, ESI†). As indicated in Fig. 1(c), starting from 0, the gradual increase in the amount of water in the reaction system led to an increase in both the HMF conversion and the DFF yield, reaching their maximum values at a water content of 0.16 mL (VH2O + VACN = 10 mL). The HMF conversion achieved an impressive 97%, while the DFF yield reached 66.6%. Subsequently, as the water content in the solvent further increased from 0.16 mL to 1.7 mL, both the HMF conversion and the DFF yield gradually declined. However, as the water content continued to increase, the HMF conversion started to rise again. Unfortunately, due to severe mineralization, the DFF yield remained consistently low. The fluctuating HMF conversion and DFF yield observed in this study indicate that water in the reaction system exhibited a more intricate influence and mechanism of action than initially anticipated. Control experiments (Fig. S7 and Table S3, ESI†) indicated that no DFF could be detected in the absence of catalyst, and the extra H2O addition and O2 are both essential for the high DFF yield. Under the atmosphere of N2 and vacuum condition, there was almost no DFF product but with high HMF mineralization percent and small H2 evolution after reaction, the O2 atmosphere slightly restrained the activity of Cd0.75Zn0.25S. Additionally, the above-mentioned metal co-catalysts could further improve the DFF yield or selectivity to more than 80% only in the presence of trace water in ACN (Fig. S6b and Table S1, ESI†). Therefore, the focus of this study is to elucidate the reasons behind the reaction stagnation in the reaction system with acetonitrile as the solvent, as well as to understand the mechanism by which the introduction of water into the reaction solution led to the fluctuating HMF conversion and DFF yield.
First and foremost, the issue of sulfide experiencing photocorrosion under irradiation should be highlighted, resulting in decreased reactivity after the initial operation, irrespective of the addition of water, such deactivation often frequently occurs on bare sulfide photocatalysts and can be alleviated by other reported strategies (see regeneration procedures and Fig. S8 in ESI†).10 Consequently, in situ surface-enhanced Raman spectroscopy (SERS) was initially employed to eliminate the ambiguous influence of surface changes of catalysts on our conclusions. The findings reveal the potential formation of an oxide layer on the surface, occurring regardless of the presence of additional water (as depicted in Fig. S8c and d, ESI†). This implies that analogous surface alterations occur irrespective of water addition, while the activity differs significantly. These observations refute the notion that variations in DFF yield and HMF conversion are attributed to the formation of a potentially active oxide layer caused by trace water in the solvent. The results strongly indicate that the fluctuations in faint water content do not modify the intrinsic nature of the catalyst, or to say, the physical properties of the catalyst changed in the same tendency; rather, they support the notion that performance disparities arise from the variation in the solvent composition.
Mechanistic study
To gain a deeper understanding of the chemical transformations occurring in the reaction system, the contents of the product and main byproducts were monitored. In line with previous studies, H2O2 was the main by-product in ACN (Fig. 1(b)). Nevertheless, the stoichiometric ratio between H2O2 and DFF in ACN was much lower than the theoretical value of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 according to the previously proposed reaction path (see Fig. S9, ESI†), indicating the partial decomposition of produced H2O2.12,27 What is more, the accumulated H2O2 concentration in ACN almost exhibited no decline during the reaction. Control experiment suggests that H2O2 alone cannot trigger HMF oxidation without catalyst under irradiation (Fig. S7b, ESI†). These results strongly implied that the formation and consumption of surface H2O2 on catalysts had reached a balanced state leading to the steady concentration of H2O2 in bulk ACN, and thus may block the active sites by preferentially reacting with active species due to the relatively strong affinity of the O–O bond in H2O2 with metal sites, and then leading to the cessation of DFF generation. This speculation was supported by DFT calculation results, H2O2 and HMF are both energetically favorable to be absorbed on the Zn sites of bare Cd0.75Zn0.25S (Fig. S10, ESI†), and the H2O2 absorption is more favorable than HMF (−1.19 eV of H2O2vs. −0.8 eV of HMF). Such detrimental effect of H2O2 was further experimentally demonstrated by the photocurrent response and pre-adding a small amount of oxalic acid (10 mg) in ACN during the reaction, as the current density of Cd0.75Zn0.25S (Fig. S11a, ESI†) was greatly restrained in the presence of H2O2, and a relatively high DFF yield (60%) was obtained (Fig. S11b, ESI†) owing to the accelerated H2O2 decomposition (H2C2O4 + H2O2 → CO2 + 2H2O) by oxalic acid.
:1 according to the previously proposed reaction path (see Fig. S9, ESI†), indicating the partial decomposition of produced H2O2.12,27 What is more, the accumulated H2O2 concentration in ACN almost exhibited no decline during the reaction. Control experiment suggests that H2O2 alone cannot trigger HMF oxidation without catalyst under irradiation (Fig. S7b, ESI†). These results strongly implied that the formation and consumption of surface H2O2 on catalysts had reached a balanced state leading to the steady concentration of H2O2 in bulk ACN, and thus may block the active sites by preferentially reacting with active species due to the relatively strong affinity of the O–O bond in H2O2 with metal sites, and then leading to the cessation of DFF generation. This speculation was supported by DFT calculation results, H2O2 and HMF are both energetically favorable to be absorbed on the Zn sites of bare Cd0.75Zn0.25S (Fig. S10, ESI†), and the H2O2 absorption is more favorable than HMF (−1.19 eV of H2O2vs. −0.8 eV of HMF). Such detrimental effect of H2O2 was further experimentally demonstrated by the photocurrent response and pre-adding a small amount of oxalic acid (10 mg) in ACN during the reaction, as the current density of Cd0.75Zn0.25S (Fig. S11a, ESI†) was greatly restrained in the presence of H2O2, and a relatively high DFF yield (60%) was obtained (Fig. S11b, ESI†) owing to the accelerated H2O2 decomposition (H2C2O4 + H2O2 → CO2 + 2H2O) by oxalic acid.
In contrast, no H2O2 was detected in the presence of 160 μL H2O after 12 hours of reaction. To clarify whether H2O2 was produced or not during the reaction, the time-dependent H2O2 concentration was determined. It was found that the accumulated H2O2 increased first and then decreased within all mixed solutions, and its variation tendencies were closely related with the H2O contents in ACN (Fig. S11c and d, ESI†), in accordance with the activity. The produced H2O2 had been completely decomposed in the presence of 160 μL H2O, and the highest apparent H2O2 decomposition rate (Fig. S11e, ESI†) was observed under the optimal condition of 160 μL H2O addition. In addition, the DFF yield and HMF conversion could still be maintained to be more than 63% and 90% respectively with nearly no H2O2 production in the presence of 160 μL H2O even though the initial HMF concentration was increased to 50 mM (Fig. S12a, ESI†); this result indicated the state-of-the-art performance in the current study with an average DFF productivity of about 27.3 μmol h−1 (see Table S4, ESI†). By comparison, the evolved H2O2 and DFF were almost linearly increasing with the increase in substrate concentration without water addition (Fig. S12b, ESI†), whereas the DFF yield and HMF conversion decreased, also implying that H2O2 was indeed produced by the hydroxymethyl dehydrogenation instead of two-electron water oxidation of the trace amount of H2O in ACN. Moreover, HMF conversion and DFF formation rate were both inhibited once 160 μL H2O containing H2O2 was pre-added into ACN (Fig. S12c and d, ESI†), indicating again that H2O2 is unfavorable to the reaction process, which was further supported by the subsequent in situ electron spin resonance (ESR) spectrum.
Although the introduction of water ensured the continuous progress of the catalytic reaction, the yield of DFF was also influenced by the reaction selectivity or, in other words, HMF mineralization. Therefore, before further investigating the mechanism of water's role, it is necessary to understand the types of reactive oxygen species and their roles in the reaction process. To investigate this, in situ ESR and trapping experiment were performed. The results evidenced that three active oxygen of ˙O2−, ˙OOH (protonated ˙O2−) and 1O2 existed (Fig. S13, ESI†), and no signal of ˙OH was detected in both ACN and the mixed solution with 160 μL H2O.28,29 The trapping experiment (Fig. S14, ESI†) indicated that both ˙O2− and 1O2 were the key active ROS during the reaction. Nevertheless, it was ˙O2− that led to persistent mineralization of HMF on account of nearly no mineralization after ˙O2− was trapped, and the mineralization of HMF still reached a high level close to the control after 1O2 was trapped. Moreover, a pretty low DFF yield was obtained once 1O2 was quenched, indicating that 1O2 was the practical active oxygen that was directly responsible for DFF formation.
With the increase in the amount of introduced water, its impact on the reaction was divided into three stages. The first stage occurred between 0 and 0.16 mL or 0.2 mL, where both the HMF conversion and DFF yield increased with the water content. The second stage took place between 0.2 and 1.7 mL, where both the HMF conversion and DFF yield decreased as the water content increased. The third stage spanned from 1.7 to 10 mL, where the HMF conversion increased with the water content, but the DFF yield did not show any significant increase. In order to conduct an in-depth investigation into the role of water in different stages, we first conducted a study on the form of water and HMF, and their interactions in the solution by Raman spectrum and molecular dynamics (MD) simulation. The spectrum (Fig. 2(a)–(c)) of the mixed solution with different H2O/ACN ratios indicated that H2O existed as a single molecule when the H2O concentration was low (<17 vol%), while the intermolecular hydrogen bond would be formed once the H2O content was further increased. This result was in good agreement with the molecular dynamics (MD) simulation (the overall and localized solvation structures of HMF in different solvents are given in Fig. S15, ESI†), no hydrogen bonds were observed in the system with 160 μL H2O and 10 mM HMF equivalent, and the average number of hydrogen bonds in the system increased to 3.3 when the water content was increased to 1.7 mL equivalent. The average number of H-bonding further increased to 33.9 in pure water (Table S5, ESI†). These results implied that H2O and HMF molecules are both isolated in bulk solvent when the water content is low, while H-bonding interaction occurs with the increased water addition. The radial distribution functions (RDFs; Fig. 2(d)) of oxygen atoms around the hydroxyl-H atom in hydroxymethyl indicate that there is a long distance (0.774 nm) between the H atom and the O atom of H2O, further revealing that the H-bonding interaction is nonexistent between HMF and H2O, while a peak at 0.328 nm attributed to H-bonding can be observed in the first coordination shell of the hydroxyl-H atom with the surrounding H2O molecule in the presence of 1.7 mL H2O equivalent. The peak of the first coordination shell of the hydroxyl-H atom further decreased to 0.226 nm in pure water, implying that the hydrogen bonding interaction between HMF and H2O gradually strengthens as the water content increases.
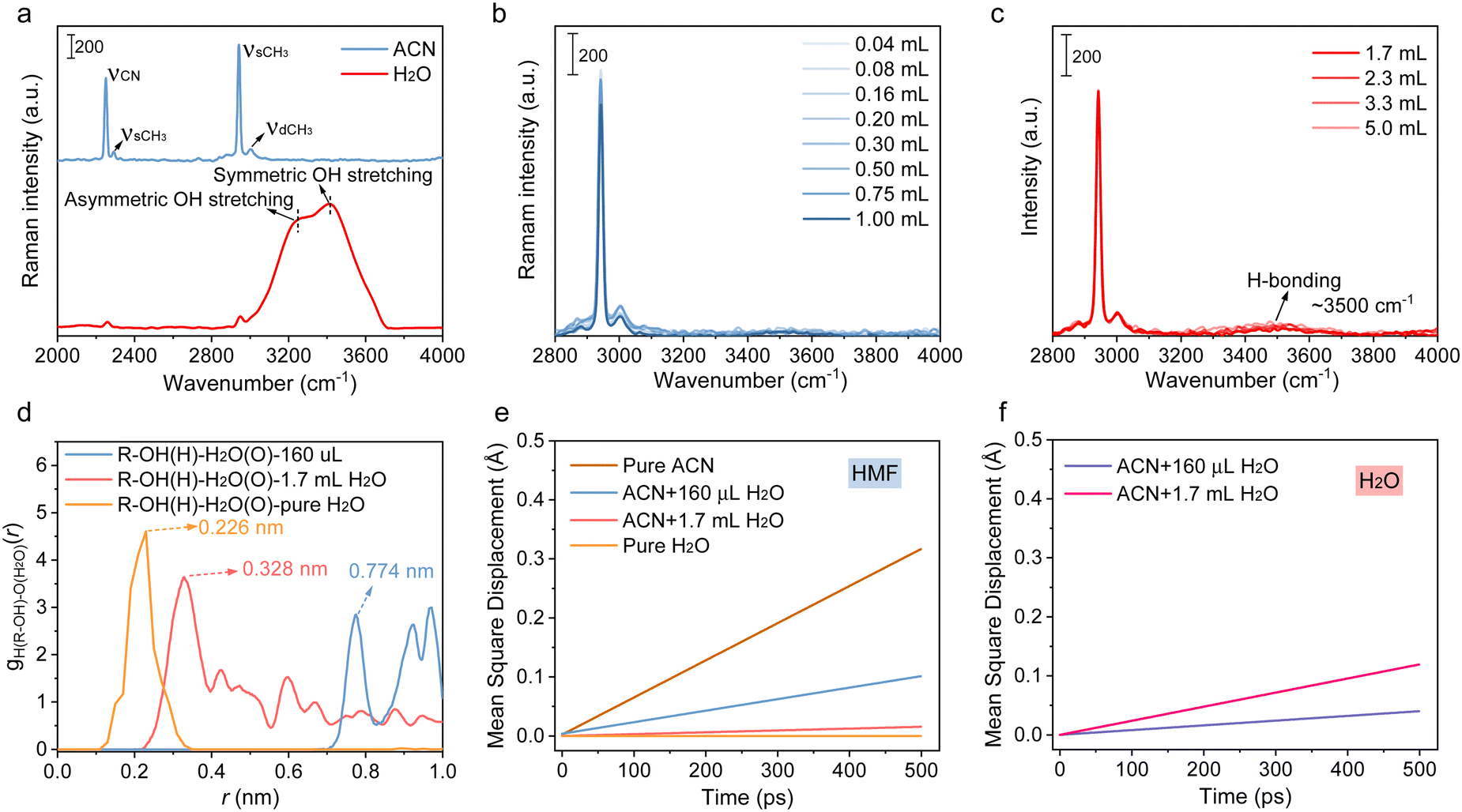 | ||
| Fig. 2 (a) Raman spectrum of ACN and water. (b) and (c) Raman spectrum of ACN with different H2O contents (water content: 0 to 50 vol%). The strong hydrogen interaction generates the asymmetric stretching vibration of OH, whereas weak hydrogen bonds cause the symmetric stretching vibration of OH. The result demonstrates that when water content is low, the hydrogen bonds between the water molecules are totally disrupted and the water molecule remains in the solution as a single molecule. (d) Radial distribution functions of oxygen atoms around the hydroxyl hydrogen of hydroxymethyl. (e) and (f) MSD curves of HMF and H2O in different solvent compositions. | ||
Since H2O can alter the amount of hydrogen bonds or the polarizability of reactants, both of which can stabilize the transition state of the reaction intermediate, H2O often plays a more substantial role when there is a variation in the dipole orientation between the reactant and the transition state.30 For instance, the polar effect of H2O can stabilize the accumulated negative charge on oxygen (shown in Fig. S16, ESI†), increasing the dipole moment of ˙OH, ˙O2− and ˙OOH. In this case, their reactivities were much higher than that in bare ACN owing to developed dipole moments.31 As demonstrated by previous kinetic studies, the hydrogen abstraction rate constants of ˙OH from organic substrates in aqueous solutions are often one to two orders of magnitude higher than that in ACN.32 These discussions give reason to why the mineralization of HMF becomes the overwhelming process in the mixed solvents with high water contents (>17 vol%) or in pure H2O, because water can act as a H-bond acceptor or donor to bond either with the hydroxyl group in HMF or with the ROS in solvent via weak interactions. On the one hand, the HMF molecules could be solvated due to hydrogen bonding with solvent molecules, making it difficult to be adsorbed on the catalyst surface. On the other hand, the hydrogen bond interaction not only enhanced the reactivity of ˙O2−, but also even converted it into ˙OH (˙O2− + H+ → *O + ˙OH) in the presence of massive water. As indicated in Fig. S17a (ESI†), only ˙OH can be detected in pure water. These species then reacted with solvated HMF non-selectively in the bulk solvent, resulting in its deep mineralization. Similar conclusions can also be extended to other solvents with a relatively strong hydrogen bonding capacity, such as DMSO and DMF, in which the product yield was much lower than that in ACN but displayed a high conversion and mineralization of HMF (Fig. S17b, ESI†). Therefore, controlling the water content at a lower level (<17%) can effectively reduce the mineralization of HMF. However, based on the reaction data, when the water content exceeded 0.2 mL, the excessive isolated water molecules near the surface of the catalyst may exhibit a strong competitive adsorption effect on HMF. This resulted in a deterioration of the reaction, as observed in the range from 0.2 mL to 1.7 mL in Fig. 1(c), where both the DFF yield and HMF conversion decreased with the increase in water content.
The above discussion can also be supported by further DFT and MD simulations of the different solvent compositions contenting 10 mM HMF equivalent. H2O was energetically favorable with a moderate absorption energy of −0.58 eV on the catalyst (Fig. S18, ESI†). Thus, the presence of excess isolated water was detrimental to the adsorption of HMF, as it engaged in competitive adsorption for the active sites on the catalyst's surface. What is more, as suggested by the mean square displacement (MSD) of HMF and H2O in Fig. 2(e), the diffusion rate of HMF from bulk solvent to catalyst surface decreased with the increase in water content, attributed to strengthened H-bonding interaction with H2O, which stabilizes HMF in bulk solvents, as indicated by the decreased H-bond length in RDFs. In pure water, HMF is totally solvated, its diffusion can hardly occur, and it is difficult to approach the catalyst surface. Moreover, the diffusion of H2O is easier than that of HMF in the mixed solvent (Fig. 2(f)), indicating that the excessive water may form a covering layer that impeded the access of HMF to the surface of the catalyst. In this case, the oxidation of the alcohol group is typically unfavorable, thus leading to reduced HMF conversion and DFF yield ranging from 0.2 mL to 1.7 mL H2O addition. The above-mentioned experiments and simulations together elucidate the experimental results, in which massive isolated water is unfavorable for the efficient oxidation of hydroxymethyl groups, whereas HMF is stabilized in the bulk solvent phase and then mineralized by free radicals once large amounts of water are present.
Besides, these analyses are proved by the experimental results obtained after adding 160 μL D2O instead of H2O. As displayed in Fig. 3(a)–(c), the enhancement of H-bonding interaction by introducing D2O slightly facilitated HMF conversion but imposed no enhancement on DFF formation. Accordingly, a secondary kinetic isotope effect (KIE) was observed for both HMF conversion and DFF formation, indicating that water molecules affect the DFF formation and HMF conversion via hydrogen bonding with other surface species. It was also found that the mineralization of HMF was dramatically accelerated in the presence of D2O (Fig. 3(c)), which displayed a solvent isotope effect (SIE) compared with that containing H2O, giving further evidences that the H-bonding interaction between the reactants and oxygen-containing radicals in the bulk phase of solvent only led to the deterioration of HMF mineralization.33 To gain more insights into the surface reaction, 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) was added as a radical scavenger (Fig. S19, ESI†), and it can be seen that the HMF conversion and DFF formation were completely suppressed, verifying that HMF conversion proceeds via a radical intermediate pathway.34 The carbon-central radical (R-˙CH(OH)) was affirmed as the intermediate from the C–H bond cleavage of hydroxymethyl (Fig. 3(d)), as the carbon-central scavengers of butylated hydroxytoluene (BHT) greatly suppressed DFF formation (Fig. S19, ESI†). The addition hole scavenger of triethanolamine (TEOA) almost completely blocked HMF conversion (Fig. S19, ESI†), implying that photoinduced holes are indispensable for DFF formation, which is further revealed by in situ ESR. In Fig. 3(e), no attenuation of the DMPO-R-˙CH(OH) signal was observed once ˙O2− and 1O2 were quenched, while the signal intensity of DMPO-R-˙CH(OH) sharply decreased in the presence of hole scavengers, suggesting that R-˙CH(OH) was not produced by the interaction between the C–H bond in hydroxymethyl and ˙O2− or 1O2 but the direct attacking by photogenerated holes. The result also showed that 1O2 was mainly produced by the electron transfer between ˙O2− and holes (˙O2− + h+ → 1O2), because the 1O2 signal in both ACN and ACN + H2O decreased significantly once p-benzoquinone (BQ, ˙O2− trapper) was added (Fig. 3(f)). What is more, the relative intensity of 1O2 linearly increased with the intensity of ˙O2− (Fig. 4(a)). It has been indicated that if the 1O2 were produced by the bi-molecular dimerization reaction of ˙O2− and ˙OOH (2˙O2− + 2H+ →1O2 + H2O2, 2˙OOH → 1O2 + H2O2), its signal would be much more intensive under higher ˙O2− concentration.35,36 As given in Fig. 4(b) and (c), the appropriate quantity of water addition also greatly enhanced the signals of ˙O2−/˙OOH, 1O2, and R-˙CH(OH), where their signals monotonically increased with the prolonged reaction time, rather than almost tended to be invariable after certain time irradiation in ACN due to the adverse effects caused by H2O2. It is worth noting that the relative amount of ˙O2−/˙OOH in ACN increased first and then decreased, attributed to the quick consumption of ˙O2−/˙OOH during irradiation (Fig. 4(d)).
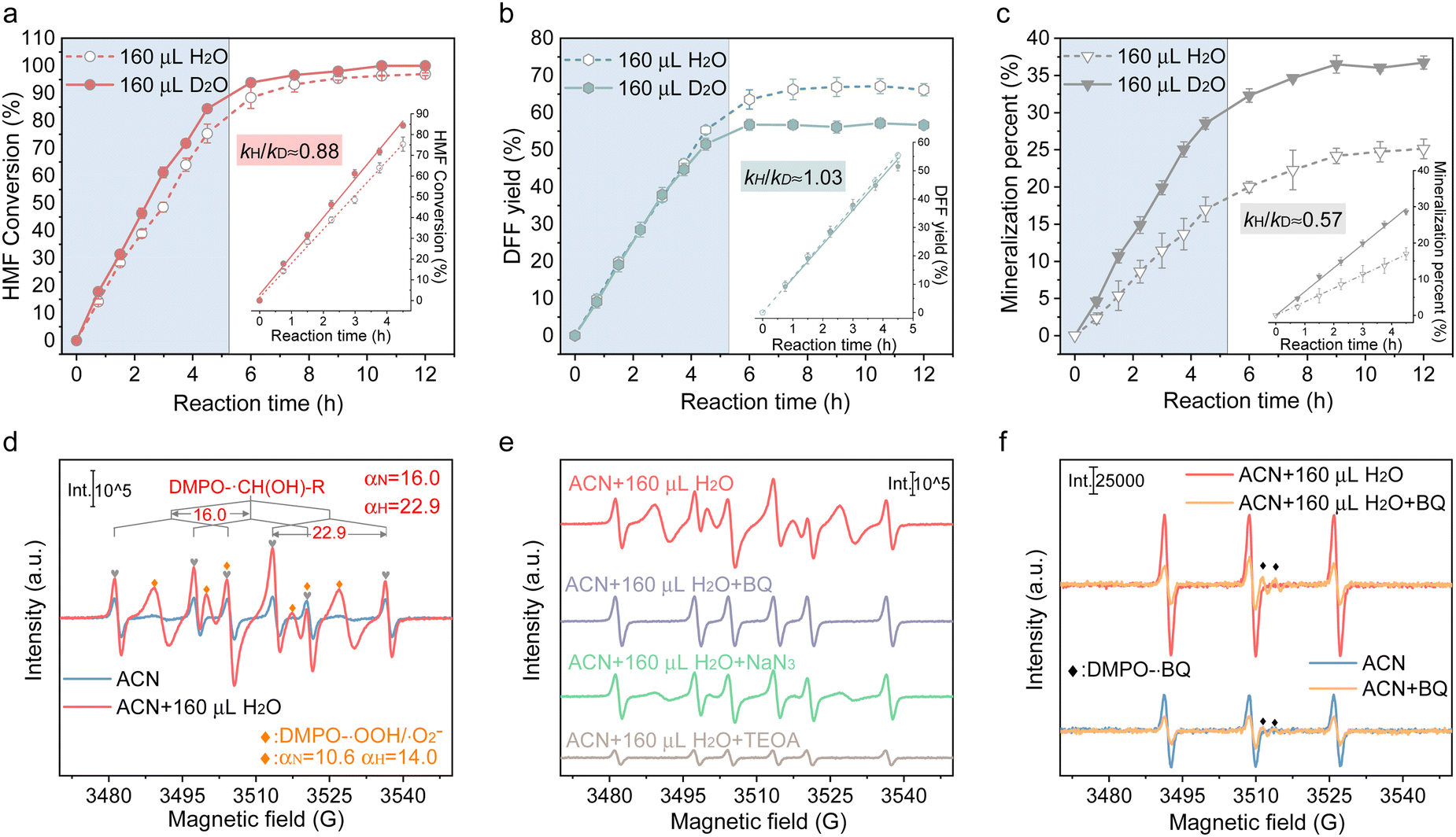 | ||
| Fig. 3 Time-dependent efficiency of HMF oxidation over Cd0.75Zn0.25S in H2O/ACN and D2O/ACN: (a) conversion of HMF; (b) yield of DFF; and (c) mineralization percent of HMF. Kinetic isotope effect (KIE): 0.88 for HMF conversion, 1.03 for DFF production, and 0.57 for mineralization. (d) and (e) in situ ESR signal of DMPO-R-CH(OH) over Cd0.75Zn0.25S after 20 min irradiation under different conditions (In ACN, ACN + H2O, ACN + H2O + BQ, ACN + H2O + NaN3, ACN + H2O + TEOA). (f) In situ ESR signal of TEMPO-1O2 over Cd0.75Zn0.25S in ACN and ACN + H2O with or without adding ˙O2− quencher (the original ESR spectrum is given in Fig. S13, S22 and S23, ESI†). | ||
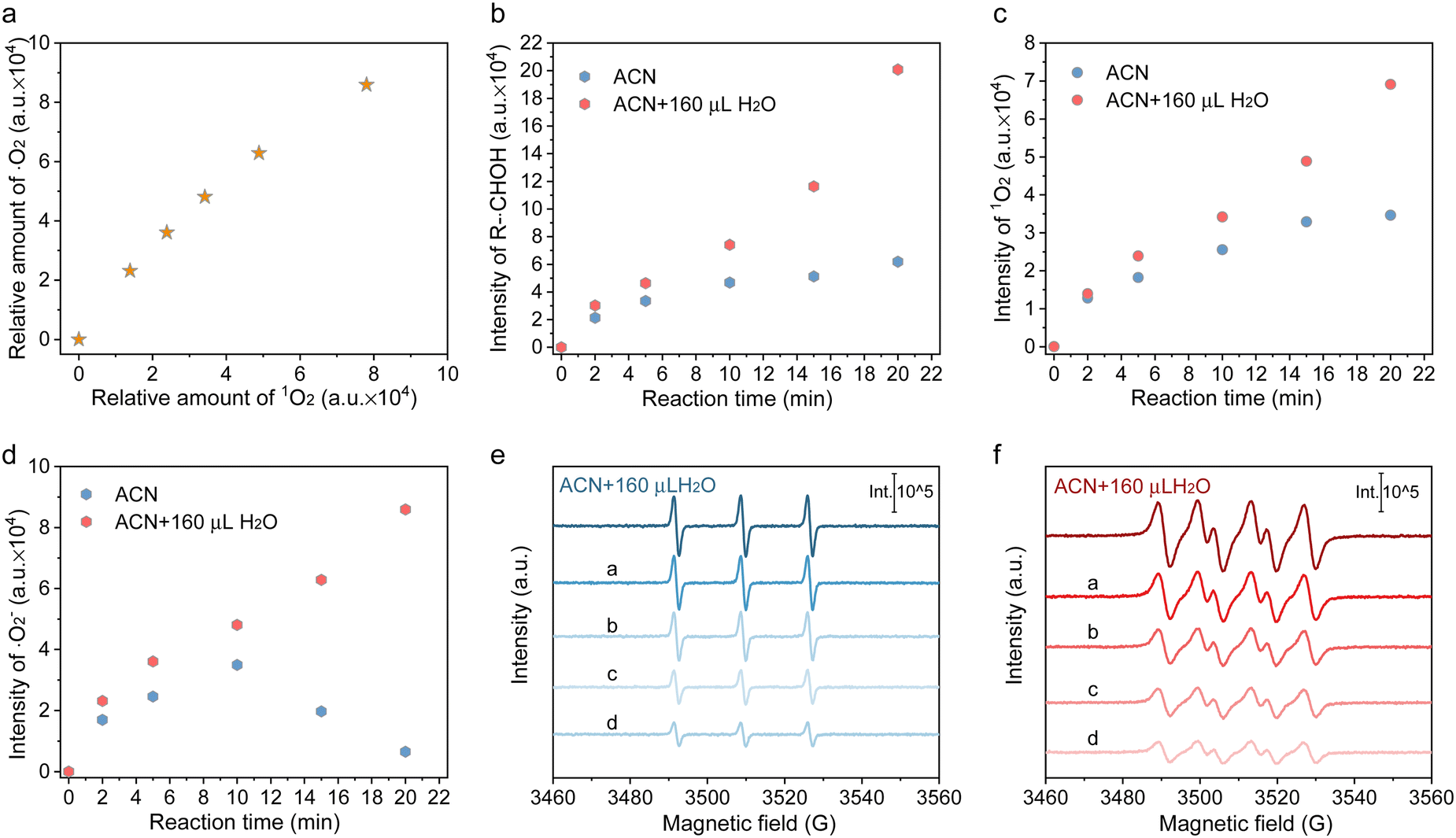 | ||
| Fig. 4 (a) Relative amount correlation between ˙O2− and 1O2 in ACN + H2O (the original data are given in Fig. S13, ESI†). (b)–(d) Time-dependent in situ ESR signal intensity of R-CH(OH), ˙O2− and 1O2 over Cd0.75Zn0.25S during 20 min irradiation (the original results are given in Fig. S13, ESI†). (e) and (f) ESR signals of 1O2 and ˙O2−/˙OOH in ACN + 160 μL H2O with pre-adding a certain amount of H2O2 after 20 min irradiation (a: 0.5 equiv.; b: 1 equiv.; c: 2 equiv;. d: 4 equiv.; the original spectrum is given in Fig. S24 and Fig. S25, ESI†). | ||
It is (Fig. S20, ESI†) indicated that the activation barrier for H2O2 decomposition by holes over the bare catalyst surface is as high as 183.7 kJ mol−1, whereas the activation barrier was relatively low (114.6 kJ mol−1) to produce surface *OH via the electron induced pathway. The presence of water can largely lower the activation barriers of the electron-induced pathway for H2O2 decomposition to only 6.6 kJ mol−1via H-bonding interaction, thus facilitating H2O2 removal. However, the H2O2 decomposition by holes was still as high as 129.7 kJ mol−1 even with H-bonding interaction, and thus H2O2 decomposition by holes was kinetically unfavorable. We therefore inferred that H2O2 decomposition mainly undergoes an electron-induced pathway (H2O2 + e− → *OH + OH−) in the presence of water, instead of the hole-induced pathway (H2O2 + h+ → H+ + *OOH). Moreover, the absorption energy of O2 on the Zn site of Cd0.75Zn0.25S (−1.14 eV; Fig. S21, ESI†) was proved to be quite close to that of H2O2 (−1.19 eV), thus the residual H2O2 in ACN also adversely affected oxygen activation, and as illustrated by the ESR results in Fig. 4(e) and (f), the signals of ˙O2− and 1O2 both decreased with the increase in H2O2 concentration once water containing equivalent H2O2 was pre-added into ACN before radiation. Overall, the occurrence of H2O2 is unfavorable for the normal progression of the reaction process as it blocks the active sites of HMF activation and also exhibits a negative impact on the O2 activation. Thus, a reasonable explanation for the suppressed activity under an O2 atmosphere is that the excessive O2 may restrain the electron-induced pathway of H2O2 decomposition by the competitive process of electron trapping for ˙O2− generation.
In situ ATR-SEIRAS and isotope labeling were carried out for further clarifying the key role of water and O2 during the DFF formation process. The in situ ATR-SEIRAS in ACN without H2O addition is shown in Fig. 5(a), and the peak located around at 1109 cm−1 corresponded to the ν(C–O) mode of bidentate bridged *CH2OH of the hydroxymethyl group in HMF.37–39 The peaks over 1363 cm−1 and 1443 cm−1 can be attributed to the deformation and symmetric stretching vibration of the C–H bond in the CH2OH group, which were further proved by the typical absorption bands located at 2867 cm−1, 2917/2932 cm−1 and 2971 cm−1 of the C–H bond.37 It can be concluded that HMF was anchored on the catalyst surface in the form of bidentate adsorption through C–H and C–O of the hydroxymethyl group. The downward peaks at 2360/2333 cm−1 are ascribed to the interface CO2 derived from the partial mineralization of HMF, and the band at 2259 cm−1 and 1285 cm−1 corresponded to the cyano group of interfacial ACN and over-oxidation product of HMF, respectively. No typical C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond located at ∼1700 cm−1 is observed, which suggested that the aldehyde group can hardly be absorbed on the surface of the catalyst, explaining the low yield of carboxyl-containing products such as FFCA and FDCA under current conditions.
O bond located at ∼1700 cm−1 is observed, which suggested that the aldehyde group can hardly be absorbed on the surface of the catalyst, explaining the low yield of carboxyl-containing products such as FFCA and FDCA under current conditions.
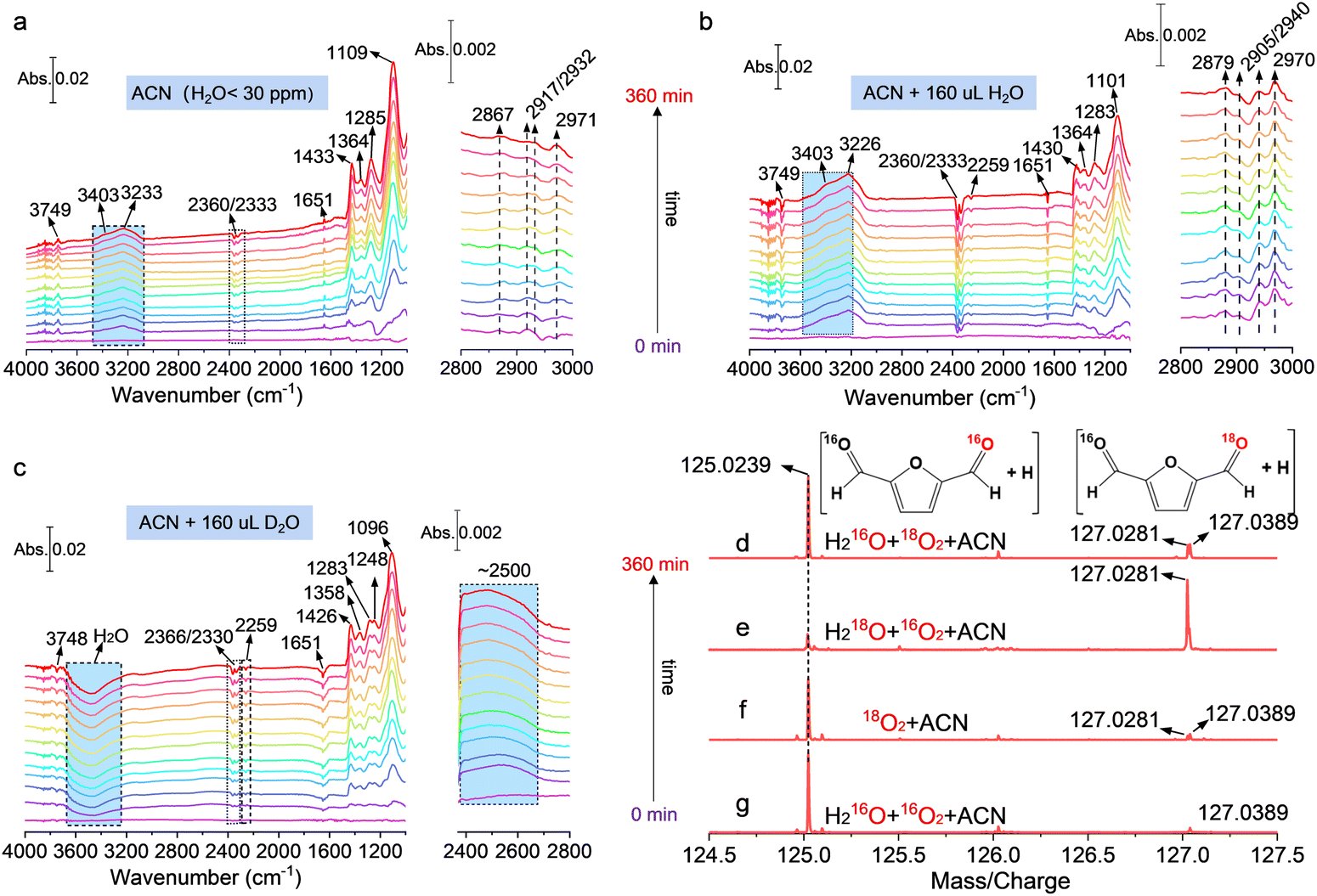 | ||
| Fig. 5 In situ ATR-SEIRAS measurement of HMF selective to DFF over Cd0.75Zn0.25S in different reaction mediums: (a) ACN; (b) ACN + H2O; and (c) ACN + D2O. (d)–(g) High-resolution mass spectrum of the products using different isotopes. | ||
The band located at 1651 cm−1 was assigned to the O–H bending vibration mode (δO–H), and the broad absorption bands from about 3100 cm−1 to 3800 cm−1 corresponded to the O–H stretching vibration mode (νO–H). The incremental frequency of νO–H originated from the decreased degree of hydrogen bonds on the catalyst, which led to the increased mobility of surface O–H. Specifically, the bands located near 3749, 3403, and 3233 cm−1 were attributed to the surface hydroxy, isolated water molecule, and H-bonded water.40,41 The increased vibration intensity of water at 3403 cm−1 and 3233 cm−1 with irradiation time suggested that H2O was actually one of the products from the dehydrogenation of hydroxymethyl, and this result is totally different from the previous report that H2O2 was generated as the only by-product from proton transfer. Moreover, the upward peaks of surface hydroxy at 3749 cm−1 in Fig. 5(a) more probably originated mainly from the dissociation of surface water, because the peak of surface hydroxy became downward and its vibration intensity increased with irradiation time after adding 160 μL H2O in ACN (Fig. 5(b)), corresponding to the ongoing lessening of surface hydroxy. Such observation indicated that water also participated in the dehydrogenation of hydroxymethyl as a reactant. This conclusion is supported by the results after replacing H2O with D2O (Fig. 5(c)), where the peaks of surface hydroxy, isolated water, and H-bonded water became downward, and a distinct upward vibration peak assigned to D–O bond stretching was observed at 2500 cm−1, giving direct evidence that D–O was generated due to D2O dissociation on the surface of the catalyst.12 Accordingly, the absorbed surface hydroxy could act as a proton-acceptor to form H2O with the proton from –CH2OH. However, electron-induced H2O2 dissociation into surface hydroxy can still not be excluded.
The high-resolution time-of-flight mass spectrum (TOF-MS) in Fig. 5(d)–(g) shows the results using labeled water (H218O) and oxygen (18O2), which further clarify the role of H2O and O2. The peaks located at a mass/charge ratio of 125.0239 correspond to DFF with no 18O incorporation, whereas the peak at 127.0281 belongs to the DFF with one 18O atom incorporation. As indicated in Fig. 5(e), the strong peak corresponding to one 18O atom incorporation into DFF was observed when H218O was used under an 16O2 atmosphere, as a result of rapid hydroxy exchange between surface CH2OH* with 18OH from H218O dissociation.30,42 The result here implied that there was almost no O–O bond cleavage during the reaction and the O2 play only as the proton-acceptor to form surface OOH species during hydroxymethyl dehydrogenation, which is in good agreement with the in situ ESR result that activated oxygen do not drive for C–H cleavage. As contrast, two very weak peaks of 18O incorporation were detected under 18O2 atmosphere when using ACN and ACN + H216O as the solvent (Fig. 5(e) and (f)). A possible reason for this was that the produced surface H218O2 undergoes dissociation into two 18OH*, which then participated in the surface reaction and as the proton acceptor to form H218O, leading to minor 18O incorporation.
It should also be noted that the identical IR absorption peak assignment and similar peak positions with or without water addition (Fig. 5(a) and (b)) implied the same reaction intermediates and identical intrinsic pathway of DFF formation in ACN or a mixed solution of ACN and water, which also demonstrated that the addition of water did not change the reaction pathway of DFF evolution, and the trace amount of water in ACN itself (<30 ppm) contributed a lot to HMF photocatalytic oxidation. Moreover, the much stronger peak intensity of H-bonded water in Fig. 5(a) and (b) also verified that the surface water was mainly hydrogen-bonded with each other or surface species such as –CH2OH, considering that the vibration frequency of *CH2OH slightly decreased with the enhanced H-bond degree of solutions (ACN < H2O + ACN < D2O + ACN).33,43 In light of the above-mentioned findings, a control experiment using anhydrous ACN (H2O < 10 ppm, absolute anhydrous ACN cannot be obtained) was performed (Fig. S26, ESI†), and the result showed that the HMF conversion and DFF yield are almost the same as that in ACN with 30 ppm H2O, but a higher H2O2 concentration (∼25% higher than that in HPLC-grade ACN) was observed. Even though about 40% H2O2 was still decomposed during the reaction in anhydrous ACN, giving rise to about 16 ppm H2O remained in the reaction medium, which was even higher than the water contained in anhydrous ACN itself. We therefore deduced that the H2O in the solvent itself and that originating from H2O2 decomposition both play a decisive role in the reaction process. Combining with the observed secondary KIE effect, it can be inferred that water had been involved in the dehydrogenation of hydroxymethyl process by promoting bond activation in hydroxymethyl and H2O2 removal via the surface hydrogen bond interaction. It should also be acknowledged that truly anhydrous polar solvents are typically nonexistent; that is to say, our work here indicates that the selection of solvent grade plays a crucial role in the outcomes of similar reactions and may even lead to misjudgments of the conclusions.
The DFT calculation provided further insights into the water-involved reaction process. As given in Fig. S27 (ESI†), the adsorption of HMF on CZS (002) was more energetically favorable with the help of moderate isolated water, which increased the adsorption energy from −0.8 eV to −1.38 eV, suggesting that surface water could facilitate HMF adsorption by H-bonding interaction with the O–H bond. Charge distribution analysis near surface showed a clear electron depletion region around the metal-O bond, whether water existed or not (Fig. S28, ESI†), demonstrating a hole-dependent bond cleavage path combining with the difference charge density of bare Cd0.75Zn0.25S, in line with our experimental results. The kinetic results (Fig. 6(a)) evidenced that the initial activation of hydroxymethyl can hardly occur on the bare catalyst surface. Specifically, the activation barriers for the dissociation of HMF via –CH2OH over CZS (002) without water are as high as 278.4 and 150.6 kJ mol−1 for C–H (Fig. S29a, ESI†) and O–H (Fig. S29b, ESI†) bonds, and thus, the bond cleavage by catalyst alone was unlikely. The presence of surface-bound water intermediates, however, can facilitate bond activation via H-bonding in much the same way as it occurred in the bare catalyst surface without H-bonding. This process lowered the barrier for the activation of the C–H bond by 108.9 kJ mol−1 (Fig. S29c, ESI†). The presence of adsorbed water intermediates also lowered the subsequent activation barrier of O–H to 88.1 kJ mol−1 (Fig. S29d, ESI†) for the ensuing carbon-central intermediate to form the aldehyde over the surface of the catalyst. The ability of adsorbed water to effectively activate both the O–H and C–H bonds helped to explain the overall increase in catalytic activity in the presence of trace amounts of water in the solvent. Note that the reaction path shown in Fig. 6(b) is nonexistent according to the in situ ATR-SEIRS results and the high C–H bond activation energy on the bare catalyst. Therefore, as shown in Fig. 6(c) and (d), this reaction will begin with 1O2 formation and H2O dissociation, generating surface hydroxy (energy barrier: 73.9 kJ mol−1; Fig. S30a, ESI†). The 1O2 and surface hydroxy could accept the protons originating from hole-driven C–H and O–H bond cleavage, respectively, to form *OOH species (53.0 kJ mol−1; Fig. S30b, ESI†) and regenerate surface H2O with nearly no energy barriers (3.8 kJ mol−1; Fig. S30c, ESI†). Then, the *OOH species can easily (23.7 kJ mol−1; Fig. S30d, ESI†) extract proton from adjacent H2O to form H2O2 and surface hydroxy, and thus, close the catalytic cycle. The overall reaction pathways are also shown in Fig. S31 (ESI†) step by step, where the first three steps refer to H2O dissociation and 1O2 formation, the fourth and fifth steps represent the dehydrogenation of the hydroxymethyl to form DFF, the sixth step is water regeneration, and the last two steps are H2O2 generation. Thereafter, the formed H2O2 would then decompose with nearly no energy barrier (6.6 kJ mol−1, vs. 114.6 kJ mol−1 without water) via H-bonding interaction (Fig. S32, ESI†). Overall, the water-involved pathway greatly reduced the energy barriers of C–H, O–H and H2O2 decomposition but hardly increased the proton migration barrier (Fig. S33, ESI†), and the energy barriers of *OOH and H2O2 formation in the presence of water increased by less than 10 kJ mol−1.
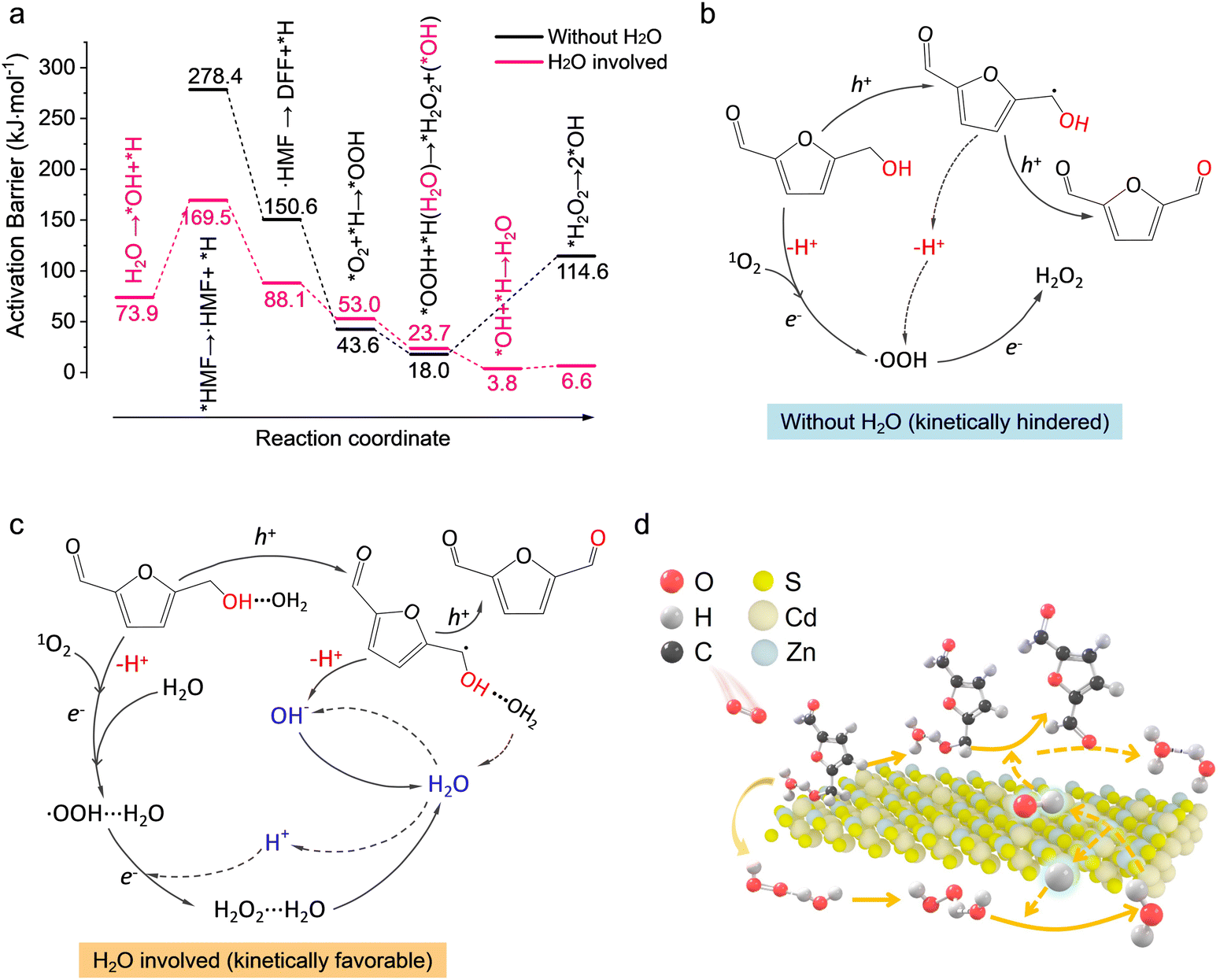 | ||
| Fig. 6 (a) Activation barriers of the elementary reaction during HMF dehydrogenation in the absence (dark line) and presence (pink line) of water. (b) Schematic of the dehydrogenation pathway without water participation. (c) and (d) Schematic of the water-involved reaction pathway of HMF dehydrogenation to DFF. | ||
Broadening applications of water-addition strategy to other alcohol oxidation reactions and catalysts
In light of the above, 14 kinds of different substrates containing hydroxyl groups were investigated under the same conditions (Table S7, ESI†). The results indicate that water-promoted alcohol oxidation in ACN could be further effectively extended to other aromatic and heterocyclic alcohols. The corresponding aldehyde yields of phenethyl alcohol, furfuryl alcohol, thenyl alcohol and pyridinemethanol, as well as benzyl alcohol and its various para-orientating derivatives, can be further improved by adding a trace amount of water into the reaction medium. For example, the yield is improved from 48.7% to 80.6% for benzaldehyde, 17.9% to 48% for furfural, and 26.2% to 53.4% for acetophenone, etc. Additionally, although the yields of Cyclohexanecarboxaldehyde (6.5%) and Cyclohexanone (8.8%) are relatively low even after adding water to ACN, they are still much higher than that in ACN alone.Sulfides and g-C3N4 are two kinds of most widely reported catalysts for HMF conversion for their visible light harvesting ability, thus we employed them for the conceptional proof of the proposed tactic in this work, including CdS, Mn-doped CdS (Mn0.2Cd0.8S), CdIn2S4, ZnIn2S4, and carbon-doped g-C3N4 (C-g-C3N4; pristine g-C3N4 has no DFF for its high-valence potential). Upon optimization, the results shown in Fig. S34 (ESI†) indicate a substantial enhancement in DFF yield for various photocatalysts: from 18.1% to 65.8% for CdS, from 12.2% to 35.9% for Mn0.2Cd0.8S, from 7.2% to 18.7% for CdIn2S4, and from 10.2% to 17.5% for C-g-C3N4, achieved solely by adding a small amount of water to ACN. However, the optimal water contents vary for different catalysts, possibly owing to their distinct structural nature. For CdS and Mn0.2Cd0.8S, the optimal water content remains at 160 μL. Conversely, for CdIn2S4, the optimal water content is 200 μL, while for C-g-C3N4, it decreases to 120 μL. Notably, fresh ZnIn2S4 exhibits high DFF selectivity (nearly 85%) and HMF conversion (over 80%) at a low initial HMF concentration (10 mM) in ACN, but as the HMF concentration increases, both the HMF conversion and the DFF yield decrease without water addition (50.3% HMF conversion and 28.8% DFF yield at 50 mM; Fig. S35a, ESI†). With the addition of 300 μL water in ACN, the HMF conversion and DFF yield over ZnIn2S4 remain at a high level of approximately 65% even if the initial HMF concentration increases to 50 mM (Fig S35b, ESI†), aligning well with the observed phenomenon in Cd0.75Zn0.25S.
However, it is noteworthy first that the solvent-mediated strategy proposed in this work for enhancing the activity of the reaction system should at least be based on one of the following two assumptions: (1) the catalyst demonstrates certain activity for the catalytic conversion of HMF in ACN; or (2) the catalyst has a low tolerance for H2O2. Moreover, the choices for photocatalysts capable of efficient aerobic oxidization of HMF are currently quite limited. Apart from the mentioned materials, other available catalysts reported in the literature are either based on these materials and overly complex to prepare or exhibit major deficiencies (mainly refers to broad-band gap, over high-valence potential, etc.) and not suitable for this conversion. Overall, it has been proved that the observed water-promoted efficiency extends beyond the analogous aerobic photocatalytic dehydrogenation of hydroxyl groups to corresponding aldehydes, and is applicable to a range of other catalysts. Nevertheless, there remain numerous materials that we are not able to verify individually in this work due to the cognitive limits of photocatalytic HMF conversion at current stage, which need further study.
Conclusions
In summary, the study starts from a very interesting phenomenon, i.e. the spontaneous termination reaction of HMF to DFF over Cd0.75Zn0.25S in ACN, while the HMF conversion was only 40.5%, with only 23.5% DFF yield. H2O2 was the main by-product as a result of hydroxymethyl dehydrogenation, which is unfavorable to the reaction. Miraculously, adding trace amounts of water into the reaction could effectively reverse the unfavorable circumstances of the reaction. The yield of DFF can reach about 66.6% with more than 97% HMF conversion. This optimization reduces energy losses associated with by-product formation, leading to a more efficient reaction pathway. It was found that both O2 and water were indispensable for efficient DFF production. The addition of water not only promoted H2O2 decomposition, but also facilitated hole-derived C–H and O–H bond activation of HMF via intermolecular H-bonding. Importantly, the extra water did not change the reaction path of DFF production, and the trace water contained in ACN itself (<30 ppm) had been directly involved in the dehydrogenation of hydroxymethyl and played a key role even without adding extra water. Our findings on the positive impacts of water could be applied to other photocatalytic processes, offering pathways to enhance the energy efficiency and sustainability in a wide range of applications, contributing to the further development of efficient and sustainable fine chemical production system.Methods
Materials
All the reagents were purchased and used without further purification. Self-made ultrapure water with an electrical resistivity of 18 MΩ cm was used in the experiments. Acetonitrile (ACN) (HPLC grade, water content <30 ppm) was obtained from Fisher Chemical (USA). N,N-Dimethylformamide (DMF, AR, 99.5%, water content <0.1%) and dimethyl sulfoxide (DMSO, AR, 99%, water content <0.2%), anhydrous acetonitrile (water content <10 ppm), CdCl2·2.5H2O, Zn(Ac)2·6H2O, NaOH, 5,5-dimethyl-1-pyrroline N-oxide (DMPO), oxalic acid (AO), benzoquinone (BQ), sodium azide (NaN3), butylated hydroxytoluene (BHT), HMF (98%), DFF, FFCA, H2O2, H218O, D2O, benzyl alcohol and its derivatives, 2-pyridinemethanol, 2-thiophenemethanol, cyclohexanol, etc., were all purchased from Aladdin Bio-Chem Technology Co., LTD, Shanghai.Physical characterization
An X-ray diffractometer (XRD, Rigaku Smartlab) with Cu Kα radiation (λ = 0.154 nm) was used to determine the crystal structure of samples. Energy-dispersive spectrum (EDS) and transmission electron microscopic images (Tecnai G2 F30) were acquired to analyze the morphology of photocatalysts. A UV/vis spectrophotometer (Shimadzu UV-3600) was employed to analyze the photoresponse properties of the catalysts. In order to further study the chemical composition of samples, X-ray photoelectron spectroscopy (XPS, Kratos AXIS NOVA spectrometer) was performed. The Brunauer–Emmett–Teller (BET) method was used to determine the specific surface area.Sample preparation
In detail, 1.713 g CdCl2·2.5H2O and 0.549 g Zn(Ac)2·2H2O were dispersed in 50 mL DI water and stirred for 5 min. After that, 10 mL of 2 M NaOH solution and 1.141 g of thiourea were added into the suspension. Then, the suspension was further stirred for 30 min and transferred into a 100 mL stainless-steel autoclave, maintained at 180 °C for 18 h. Finally, the obtained precipitates were collected and washed. The final products were dried at 60 °C for 6 h. The CdxZn1−xS nanoparticles with different elementary ratios were fabricated by using different amounts of CdCl2·2.5H2O and Zn(Ac)2·2H2O.For loading different metal co-catalysts: 100 mg Cd0.5Zn0.5S nanoparticles were dispersed into 40 mL water containing 50 μL of 2 M NaOH. After ultrasonication for 30 min, certain amounts of Co(NO3)2·6H2O, Ni(NO3)2·6H2O, Cu(NO3)2, PdCl2, AgNO3, HAuCl4, and H2PtCl6 solution were added into the suspension and stirred for 15 min. Subsequently, 10 mL fresh NaBH4 (2 M) solution was added into the suspension dropwise, and after further stirring for 30 min, the obtained green suspension was centrifuged and dried in vacuum.
Photocatalytic HMF oxidation
The photocatalytic HMF oxidation was carried out using a photochemical reactor equipped with 445 ± 10 nm LED (20 W). Typically, 20 mg catalyst was dispersed in a 15 mL quartz tube containing 10 mL reaction solution with an HMF concentration of 10 mM (unless otherwise noted). A mixed solution of water and ACN (HPLC-grade ACN with a H2O content of 30 ppm was employed unless stated otherwise) was used as the reaction solution (VACN + VH2O = 10 mL), in which the water addition volume was controlled to be 0.02 mL, 0.04 mL, 0.08 mL, 0.16 mL, 0.20 mL, 0.30 mL, 0.50 mL, 0.75 mL, 1.70 mL, 2.30 mL, 3.3 mL, 5.0 mL, 7.0 mL, 9.0 mL, and 10.0 mL. After ultrasonic treatment for 4 min, the suspension was employed under air atmosphere for the reaction, and sampling was carried out after a certain reaction time. The other solvents (DMF, DMSO, and water) were also employed for comparison under air atmosphere. For different atmospheres, the air in the reactor was extracted first using a vacuum pump, and then N2 or O2 was injected.For other alcohol substrates and catalysts, the procedures were the same as above HMF oxidation except that different feedstocks were added in the reaction solution of 10 mL ACN or ACN + H2O.
| Product yield = (amount of specific product/initial amount of HMF) × 100% |
| HMF conversion = (initial amount of HMF − residual amount of HMF)/initial amount of HMF × 100%, |
| Mineralization percentage = (initial amount of HMF − overall number of detected products)/initial amount of HMF × 100%. |
Product quantification
High-performance liquid chromatography (HPLC) was performed using WATERS 2695 equipped with an ultraviolet-visible detector for the quantification of HMF, DFF, FFCA and FDCA (Wavelength: 248 nm; Column: Bio-Rad, Aminex HXP-87P; Mobile phase: 5 mM H2SO4; Flow rate: 0.6 mL min−1). For other alcohol substrates, Agilent 7890A equipped with an FID detector and gas chromatograph–mass spectrometer (GC–MS, GC 7890A/5975C inert XL MSD) were used for quantification. The possible gas product through the photocatalytic process was quantified by gas chromatography (Agilent 7890 equipped with a Molecular Sieve packed column and a thermal conductivity detector). The identification and quantification of the products were carried out via the calibration curves by applying standard solutions with known concentrations of commercially purchased pure reactants, intermediates, and products.The produced H2O2 was quantified by a reagent color-developing method. First, 1 mL filtered reaction solution was attenuated by 2 mL DI water; after that, 1 mL of the attenuated sample was added into a mixed solution of 1 mL KI (0.4 M) aqueous solution and 1 mL commercial pH buffer with pH = 4 (potassium biphthalate). Finally, the solution was kept for 1 h in the darkness. The H2O2 concentration can be determined by the triiodide anion (I3−) concentration according to the following equation, where I3− can be estimated by Lambert–Beer's law due to their strong absorbance at 350 nm.
Author contributions
Wenhua Xue: conceptualization, formal analysis, investigation, methodology, writing – original draft. Jian Ye: software, resources, visualization. Zhi Zhu: resources, software, validation. Reeti Kumar: data curation, validation. Jun Zhao: conceptualization, investigation, supervision, writing – review & editing.Data availability
The data supporting this article have been included in this article or as part of the ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the General Research Fund of the University Grants Committee (grant no. 12202723), National Natural Science Foundation of China (NSFC 21908184).References
- N. Luo, T. Montini, J. Zhang, P. Fornasiero, E. Fonda, T. Hou, W. Nie, J. Lu, J. Liu, M. Heggen, L. Lin, C. Ma, M. Wang, F. Fan, S. Jin and F. Wang, Nat. Energy, 2019, 4, 575–584 CrossRef CAS.
- D. Wakerley, M. Kuehnel, K. Orchard, K. Ly, T. Rosser and E. Reisner, Nat. Energy, 2017, 2, 17021 CrossRef CAS.
- H. G. Cha and K. Choi, Nat. Chem., 2015, 7, 328–333 CAS.
- Z. Huang, N. Luo, C. Zhang and F. Wang, Nat. Rev. Chem., 2022, 6, 197–214 Search PubMed.
- J. DiMeglio, A. Breuhaus-Alvarez, S. Li and B. Bartlett, ACS Catal., 2019, 9, 5732–5741 CAS.
- X. Tan, S. Si, D. Xiao, X. Bao, K. Song, Z. Wang, Y. Liu, Z. Zheng, P. Wang, Y. Dai, B. Huang and H. Cheng, ACS Catal., 2023, 13, 14395–14403 CAS.
- E. García-López, F. Arcidiacono, A. Vincenzo, L. Palmisano, P. Meo and G. Marcì, Photochem. Photobiol. Sci., 2023, 22, 1517–1526 Search PubMed.
- M. Zhang, Z. Li, X. Xin, J. Zhang, Y. Feng and H. Lv, ACS Catal., 2020, 10, 14793–14800 CrossRef CAS.
- A. Kumar and R. Srivastava, ACS Appl. Nano Mater., 2021, 4, 9080–9093 CrossRef CAS.
- H. Zhao, D. Trivedi, M. Roostaeinia, X. Yong, J. Chen, P. Kumar, J. Liu, B. Su, S. Larter, M. Kibria and J. Hu, Green Chem., 2023, 25, 692–699 RSC.
- Y. Guo, B. Liu, J. Zhang, G. Wang, C. Pan, H. Zhao, C. Wang, F. Yu, Y. Dong and Y. Zhu, Appl. Catal., B, 2024, 340, 123217 CrossRef CAS.
- L. Yin, Y. Wu, X. Bao, X. Liu, D. Dai, M. Zhang, Z. Wang, Z. Zheng, Y. Liu, H. Cheng, Y. Dai, B. Huang and P. Wang, Chem. – Eur. J., 2023, 29, e202300999 CrossRef CAS.
- B. Yang, W. Hu, F. Wan, C. Zhang, Z. Fu, A. Su, M. Chen and Y. Liu, Chem. Eng. J., 2020, 396, 125345 CrossRef CAS.
- Y. Yang and T. Mu, Green Chem., 2021, 23, 4228–4254 RSC.
- C. Li, J. Li, L. Qin, P. Yang and D. Vlachos, ACS Catal., 2021, 11, 11336–11359 CrossRef CAS.
- C. Toe, C. Tsounis, J. Zhang, H. Masood, D. Gunawan, J. Scott and R. Amal, Energy Environ. Sci., 2021, 14, 1140–1175 RSC.
- T. Simon, N. Bouchonville, M. Berr, A. Vaneski, A. Adrović, D. Volbers, R. Wyrwich, M. Döblinger, A. Susha, A. Rogach, F. Jäckel, J. Stolarczyk and J. Feldmann, Nat. Mater., 2014, 13, 1013–1018 CrossRef CAS PubMed.
- H. Huang, Z. Fang, K. Yu, J. Lü and R. Cao, J. Mater. Chem. A, 2020, 8, 3882–3891 RSC.
- J. Cheng, L. Liu, G. Liao, Z. Shen, Z. Tan, Y. Xing, X. Li, K. Yang, L. Chen and S. Liu, J. Mater. Chem. A, 2020, 8, 5890–5899 RSC.
- Y. Zhu, Y. Zhang, L. Cheng, M. Ismael, Z. Feng and Y. Wu, Adv. Powder Technol., 2020, 31, 1148–1159 CrossRef CAS.
- T. Xia, W. Gong, Y. Chen, M. Duan, J. Ma, X. Cui, Y. Dai, C. Gao and Y. Xiong, Angew. Chem., Int. Ed., 2022, 61, e202204225 CrossRef CAS.
- N. Pfriem, P. Hintermeier, S. Eckstein, S. Kim, Q. Liu, H. Shi, L. Milakovic, Y. Liu, G. Haller, E. Baráth, Y. Liu and J. Lercher, Science, 2021, 372, 952–957 CrossRef CAS.
- Z. Liu, E. Huang, I. Orozco, W. Liao, R. M. Palomino, N. Rui, T. Duchoň, S. Nemšák, D. Grinter, M. Mahapatra, P. Liu, J. Rodriguez and S. Senanayake, Science, 2020, 368, 513–517 CrossRef CAS.
- M. Mellmer, C. Sanpitakseree, B. Demir, P. Bai, K. Ma, M. Neurock and J. Dumesic, Nat. Catal., 2018, 1, 199–207 CrossRef CAS.
- J. Saavedra, H. Doan, C. Pursell, L. Grabow and B. Chandler, Science, 2014, 345, 1599–1602 CrossRef CAS PubMed.
- J. Adams, A. Chemburkar, P. Priyadarshini, T. Ricciardulli, Y. Lu, V. Maliekkal, A. Sampath, S. Winikoff, A. Karim, M. Neurock and D. Flaherty, Science, 2021, 371, 626–632 CrossRef CAS PubMed.
- H. Sun, R. Xu, X. Jia, Z. Liu, H. Chen and T. Lu, Biomass Convers. Biorefin., 2023, 1–18 Search PubMed.
- Y. Jiang, S. Li, S. Wang, Y. Zhang, C. Long, J. Xie, X. Fan, W. Zhao, P. Xu, Y. Fan, C. Cui and Z. Tang, J. Am. Chem. Soc., 2023, 145, 2698–2707 CrossRef CAS.
- D. Zhang, P. Wang, J. Wang, Y. Li, Y. Xia and S. Zhan, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2114729118 CrossRef CAS PubMed.
- B. Zope, D. Hibbitts, M. Neurock and R. Davis, Science, 2010, 30, 74–78 CrossRef PubMed.
- G. Litwinienko and K. Ingold, Acc. Chem. Res., 2007, 40, 222–230 CrossRef CAS PubMed.
- S. Mitroka, S. Zimmeck, D. Troya and J. Tanko, J. Am. Chem. Soc., 2010, 132, 2907–2913 CrossRef CAS.
- Z. Gao, J. Mu, J. Zhang, Z. Huang, X. Lin, N. Luo and F. Wang, J. Am. Chem. Soc., 2022, 144, 18986–18994 CrossRef CAS PubMed.
- L. Luo, W. Chen, S. Xu, J. Yang, M. Li, H. Zhou, M. Xu, M. Shao, X. Kong, Z. Li and H. Duan, J. Am. Chem. Soc., 2022, 144, 7720–7730 CrossRef CAS PubMed.
- Y. Nosaka and A. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- H. Saito and Y. Nosaka, Catal. Commun., 2015, 61, 117–120 CrossRef CAS.
- G. Szijjártó, Z. Pászti, I. Sajó, A. Erdőhelyi, G. Radnóczi and A. Tompos, J. Catal., 2013, 305, 290–306 CrossRef.
- Q. Liu, J. Lin, H. Cheng, L. Wei and F. Wang, Angew. Chem., Int. Ed., 2023, 62, e202218720 CrossRef CAS PubMed.
- V. Augugliaro, H. Kisch, V. Loddo, M. López-Muñoz, C. Márquez-Álvarez, G. Palmisano, L. Palmisano, F. Parrino and S. Yurdakal, Appl. Catal., A, 2008, 349, 189–197 CrossRef CAS.
- K. Sun, X. Wu, Z. Zhuang, L. Liu, J. Fang, L. Zeng, J. Ma, S. Liu, J. Li, R. Dai, X. Tan, K. Yu, D. Liu, W. Cheong, A. Huang, Y. Liu, Y. Pan, H. Xiao and C. Chen, Nat. Commun., 2022, 13, 6260 CrossRef CAS.
- Q. Sun, N. Oliveira, S. Kwon, S. Tyukhtenko, J. Guo, N. Myrthil, S. Lopez, I. Kendrick, S. Mukerjee, L. Ma, S. Ehrlich, J. Li, W. Goddard III, Y. Yan and Q. Jia, Nat. Energy, 2023, 8, 859–869 CrossRef CAS.
- C. Meng, K. Yang, X. Fu and R. Yuan, ACS Catal., 2015, 5, 3760–3766 CrossRef CAS.
- T. Wang, Y. Zhang, B. Huang, B. Cai, R. Rao, L. Giordano, S. Sun and Y. Shao-Horn, Nat. Catal., 2021, 4, 753–762 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee02301c |
| This journal is © The Royal Society of Chemistry 2025 |