
Salt dissociation and localized high-concentration solvation at the interface of a fluorinated gel and polymer solid electrolyte†
Dechao
Zhang
ab,
Yuxuan
Liu
c,
Dedi
Li
a,
Shimei
Li
ab,
Qi
Xiong
ab,
Zhaodong
Huang
ab,
Shixun
Wang
 a,
Hu
Hong
a,
Jiaxiong
Zhu
a,
Haiming
Lv
*b and
Chunyi
Zhi
*abde
a,
Hu
Hong
a,
Jiaxiong
Zhu
a,
Haiming
Lv
*b and
Chunyi
Zhi
*abde
aHong Kong Center for Cerebro-Cardiovascular Health Engineering (COCHE), City University of Hong Kong, Shatin N. T. 999077, Hong Kong SAR, China. E-mail: cy.zhi@cityu.edu.hk
bDepartment of Materials Science and Engineering, City University of Hong Kong, Kowloon, 999077, Hong Kong SAR, China. E-mail: haimilyu@cityu.edu.hk
cGuangdong Provincial Key Laboratory of Advanced Energy Storage Materials, School of Materials Science and Engineering, South China University of Technology, Guangzhou, 510641, China
dHong Kong Institute for Advanced Study, City University of Hong Kong, Kowloon, Hong Kong 999077, China
eHong Kong Institute for Clean Energy, City University of Hong Kong, Kowloon, 999077, Hong Kong
First published on 12th November 2024
Abstract
Low salt dissociation and the unstable [Li(N,N-dimethylformamide (DMF))x]+ solvent structure in poly(vinylidene fluoride) (PVDF)-based solid polymer electrolyte (SPE) remarkably restricts the high throughput ion transport and interfacial stability. Here, we designed a hybrid electrolyte (denoted as HFGP-SE) composed of fluorinated gel solid electrolyte (FG-SE) and poly(vinylidene fluoride-co-hexafluoropropylene) (PVHF)-based solid polymer electrolyte (PVHF-SPE). We found that in the HFGP-SE, the interface of FG-SE and PVHF-SPE effectively promotes lithium salt dissociation and creates a localized high-concentration (LHC) solvation structure. The developed HFGP-SE shows high ionic conductivity (0.84 mS cm−1) and a remarkably improved lithium transference number (tLi+ = 0.87). Meanwhile, the controlled LHC solvation structure formed at the interface between FG-SE and PVHF-SPE supports the formation of inorganic-rich solid electrolyte interphases (SEIs) derived from anions, allowing for stable lithium deposition and ultra-stable plating/stripping performance for over 1200 hours at a current density of 0.5 mA cm−2. Additionally, HFGP-SE supported stable cycling in 4.5 V class Li||NCM811 full cells under practical conditions, with a 50 μm thick lithium metal anode and cathodes with a mass loading of 12 mg cm−2, achieving an areal capacity >2 mA h cm−2. This work proposes a novel strategy using interfaces existing in hybrid solid electrolytes to significantly enhance lithium salt dissociation, fast ion transport, and interfacial stability of solid-state electrolytes for lithium metal batteries.
Broader contextPoly(vinylidene fluoride) (PVDF)-based solid polymer electrolytes (SPEs) with the “salt in polymer with small amounts of residual solvent” configuration have become particularly attractive due to their high room temperature ionic conductivity. However, the low salt dissociation and unstable [Li(N,N-dimethylformamide (DMF))x]+ solvent structure in PVDF-SPE remarkably restricts the high throughput ion transport and interfacial stability. To improve the dissociation of the lithium salts and electrode/electrolyte interface stability, we designed a hybrid electrolyte (denoted as HFGP-SE) composed of fluorinated gel solid electrolyte (FG-SE) and poly(vinylidene fluoride-co-hexafluoropropylene) (PVHF)-based solid polymer electrolyte (PVHF-SPE). We found that the interfaces of FG-SE and PVHF-SPE in the HFGP-SE form abundant, stable, and effective ion transport pathways by promoting a high lithium salt dissociation and forming a local high-concentration (LHC) solvation structure. The developed HFPG-SE enables the stable cycling of lithium metal batteries. Therefore, this work proposes a novel strategy using interfaces existing in hybrid solid electrolytes to significantly enhance lithium salt dissociation, fast ion transport, and interfacial stability of solid-state electrolytes for lithium metal batteries. |
Introduction
The rapid development of sustainable economics raises the demand for high-energy-density and safe energy storage batteries for electric transportation and smart grids. Metallic Li is being pursued extensively as a promising anode candidate to fabricate high energy density batteries owing to its ultrahigh specific capacity (3860 mA h g−1), relatively low mass density (0.59 g cm−3), and lowest potential (−3.04 V vs. standard hydrogen electrode).1,2–4 The difficulty of management and safety issues also emerges with the organic liquid electrolyte-based lithium metal batteries, drawing significant public concern towards solid-state batteries.5–7 By replacing the organic liquid electrolytes with solid-state electrolytes (SSEs), liquid leakage and flammability of devices are supposed to be largely eliminated. In addition, SSEs with high mechanical properties and rigidity can inhibit lithium dendrite propagation and improve the cycling stability of batteries.8–10Among all SSEs, solid-state polymer electrolytes (SPEs) are accepted as promising candidates due to their desirable flexibility, low density, facile processability, and good interfacial stability with electrodes.11–13 Recently, vinylidene fluoride (VDF)-based SPEs with the “salt in polymer with small amounts of residual solvent” configuration have become particularly attractive due to their high room temperature ionic conductivity (10−4 S cm−1).14–16 However, this configuration faces two significant issues: low lithium salt dissociation and severe electrode/electrolyte interface side reactions. The ion transport characteristic of VDF-based SPEs is due to the unique “salt-polymer-trace residual solvent” configuration, where high concentration Li salts interact with residual N,N-dimethylformamide (DMF) solvent to form [Li(DMF)x]+ solvation molecules and transport along VDF chains based on the interactions between VDF and solvation molecules.17,18 However, the poor dissociation of the lithium salts in VDF-based polymer matrixes results in the low concentration of charge carriers, ionic conductivity, and lithium transference number. Additionally, the side reactions of [Li(DMF)x]+ with lithium metal result in the severe capacity decay of the solid-state lithium metal batteries during long-term cycling. Therefore, it is essential to improve the dissociation of the lithium salts and regulate the microcosmic solvation structure in VDF-based SPEs to ensure fast Li+ transport and create a stable interfacial reaction environment for practical applications. Conventional strategies include incorporating inorganic or organic fillers into PVDF-SPE to enhance salt dissociation and regulate electrode/electrolyte interface stability through interactions at the filler interface. However, the agglomeration of fillers significantly influences their efficiency.19–21 In contrast, the hybrid interface between the gel electrolyte and solid polymer electrolyte may exhibit a uniform structure and regulated Li+ binding energies. This characteristic may enhance the dissociation of lithium salts more efficiently.
In this work, we designed a hybrid electrolyte (denoted as HFGP-SE) composed of fluorinated gel solid electrolyte (FG-SE) and poly(vinylidene fluoride-co-hexafluoropropylene) (PVHF)-based solid polymer electrolyte (PVHF-SPE). We found that, in HFGP-SE, the interfaces between FG-SE and PVHF-SPE form fast ion transport pathways with high lithium salt dissociation and a local high-concentration (LHC) solvation structure. As a result, the developed HFGP-SE demonstrates improved ionic conductivity (0.84 mS cm−1) and a high lithium transference number (tLi+ = 0.87). Meanwhile, HFGP-SE exhibits enhanced stability with both the Li metal anode and high voltage cathode, which enables the Li||Li symmetric cells to achieve record cycling times of 1200 h at 0.5 mA cm−2 and 4.5 V class Li||NCM811 full cell stable cycling (testing conditions of 50 μm thin Li metal anode, and high areal cathode capacity >2 mA h cm−2). These findings indicate that this HFGP-SE holds significant potential for advancing the performance and durability of solid-state lithium metal batteries.
Results
Fig. 1a illustrates the structure and working mechanism of the designed HFGP-SE composed of PVHF-SPE and a small amount of FG-SE. Different from the conventional quasi-solid electrolyte with Celgard separators, the interfaces of PVHF-SPE and FG-SE form abundant 3D interconnected fast ion-conductive pathways with high lithium salt dissociation and LHC solvation structures. The FG-SE includes 1 M LiTFSI dissolved in a mixture of bis(2,2,2-trifluoroethyl) carbonate (TFEC)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :fluoroethylene carbonate (FEC) :2,2,3,4,4,4-hexafluorobutyl acrylate (HFBA) (6:3:1 by volume). After polymerization, this liquid FG-SE precursor can transform into a non-flowing white gel (Fig. S1a, ESI†). Meanwhile, the disappearance of the C
:fluoroethylene carbonate (FEC) :2,2,3,4,4,4-hexafluorobutyl acrylate (HFBA) (6:3:1 by volume). After polymerization, this liquid FG-SE precursor can transform into a non-flowing white gel (Fig. S1a, ESI†). Meanwhile, the disappearance of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C spectra (Fig. S1b, ESI†) in FG-SE indicates the completed polymerization of HFBA. Specific properties of poly(HFBA) in FG-SE are detailed in Table S1 (ESI†). As shown in Fig. 1b, the selected components of the FG-SE show much lower HOMO energy than DMF, demonstrating the superior oxidation resistance of these fluorine solvents owing to the strong electron-withdrawing effect of fluorine atoms on the core of solvent molecules. Meanwhile, these fluorinated components show a decreased binding energy with Li+ and increased binding energy with TFSI− compared with the DMF solvent (Fig. 1c and Fig. S2, ESI†). Therefore, introducing FG-SE in PVHF-SPE can dilute [Li(DMF)x]+ solvation molecules, promoting the dissociation of lithium salts and facilitating the formation of LHC solvation structures in the local interface regions. Furthermore, the LHC solvation structure, utilizing fluorinated solvents, maintains a weak-interaction solvation structure and enhances interface stability by forming anion-derived solid electrolyte interphases (SEIs). These SEIs kinetically inhibit electrolyte degradation by preventing direct contact between the electrode and the electrolyte, thereby enhancing the interface stability between the electrolyte and the Li metal anode.
C spectra (Fig. S1b, ESI†) in FG-SE indicates the completed polymerization of HFBA. Specific properties of poly(HFBA) in FG-SE are detailed in Table S1 (ESI†). As shown in Fig. 1b, the selected components of the FG-SE show much lower HOMO energy than DMF, demonstrating the superior oxidation resistance of these fluorine solvents owing to the strong electron-withdrawing effect of fluorine atoms on the core of solvent molecules. Meanwhile, these fluorinated components show a decreased binding energy with Li+ and increased binding energy with TFSI− compared with the DMF solvent (Fig. 1c and Fig. S2, ESI†). Therefore, introducing FG-SE in PVHF-SPE can dilute [Li(DMF)x]+ solvation molecules, promoting the dissociation of lithium salts and facilitating the formation of LHC solvation structures in the local interface regions. Furthermore, the LHC solvation structure, utilizing fluorinated solvents, maintains a weak-interaction solvation structure and enhances interface stability by forming anion-derived solid electrolyte interphases (SEIs). These SEIs kinetically inhibit electrolyte degradation by preventing direct contact between the electrode and the electrolyte, thereby enhancing the interface stability between the electrolyte and the Li metal anode.
 | ||
| Fig. 1 Designed hybrid fluorinated gel and polymer solid electrolyte (HFGP-SE). (a) Schematic illustration of the structure and fast ion-conductive mechanism of the designed HFGP-SE. (b) Calculated LUMO and HOMO energy values of the main components of DMF, PVHF, FEC, TFEC, and HFBA based on density functional theory. (c) Binding energies of DMF, PVHF, FEC, TFEC, and HFBA for a Li+ cation and a TFSI− anion. (d) SEM images of PVHF-SPE and (e) HFGP-SE. Combustion tests of HFGP-SE (f) and FG-SE (g). | ||
Fig. 1d and Fig. S3a (ESI†) show the SEM images of PVHF-SPE. The PVHF-SPE exhibits a porous structure and a solid-state whitening appearance (Fig. 1d inset). This porous structure is due to phase separation between the polymer and solvent during evaporation. After coupling with FG-SE, the pores in PVHF-SPE are filled, resulting in a smoother surface and denser structure (Fig. 1e and Fig. S3b, ESI†). The solvent content and FG-SE in HFGP-SE is approximately 22.7 wt% and 30.1 wt%, respectively (Fig. S4, ESI†). Furthermore, the burning tests with fire were applied to measure the flameproof properties of FG-SE and HFGP-SE. Fig. 1f and g show that no combustion phenomenon can be found when the fire is moved away from HFGP-SE and FG-SE, demonstrating the superior fireproof properties. This excellent safety feature is due to the use of non-combustible fluorinated solvents in FG-SE and the stable polymer matrices, which contribute to the safety and fire resistance of the battery.
Molecular dynamics (MD) simulations were performed to reveal the solvation structures in HFGP-SE. Fig. 2a shows the snapshot extracted from the interface area of PVHF-SPE and FG-SE in HFGP-SE, along with the average coordination number (CN) of Li–TFSI− derived from statistical length distribution analysis. The average CN of Li–TFSI− at the interface is 2.8, which is lower than that of PVHF-SPE (3.2) and FG-SE (3.4). This reduction in the coordination number suggests a higher degree of lithium salt dissociation at the interface, allowing more free Li+ ions to participate in ionic conduction. The calculated radial distribution functions (RDFs) and coordination numbers (CN) for Li+ in various electrolyte cases are shown in Fig. 2b and Fig. S5 (ESI†). In PVHF-SPE, TFSI− was the dominant solvent in the first Li+ solvation shell (∼2.65 Å), with a coordination number of 3.29. DMF is the dominant solvent in the second Li+ solvation shell (∼4.2 Å), with a coordination number of 1.27. In FG-SE, TFSI− and solvents are both dominant in the first Li+ solvation shell. The coordination numbers are 3.5 for TFSI−, 1.0 for FEC, and 0.9 for TFEC. However, the Li+ solvation structure was changed significantly at the interface. Specifically, TFSI− and DMF were the dominant solvents in the first Li+ solvation shell, with dropped coordination numbers of 2.98 and 1.06, respectively. The FEC and TFEC occupy the first to second Li+ solvation shell. The solvation structure is schematically shown in Fig. 2c. This locally concentrated solvent structure with TFEC–FEC outer layers and TFSI−–DMF inner layers can effectively suppress interface side reactions caused by DMF, thereby improving interface stability.
 | ||
| Fig. 2 Characteristics of HFGP-SE. (a) A snapshot extracted from the simulation of the PVHF-SPE/FG-SE system and the average coordination number (CN) of Li–TFSI− based on statistical length distribution. (b) Radial distribution function (RDF), g(r), and coordination number of the PVHF-SPE/FG-SE interface. (c) Schematic diagram of the solvation structure at the PVHF-SPE/FG-SE interface. (d) Raman spectra of PVHF-SPE, FG-SE, and HFGP-SE. (e) FT-IR spectra of the TFEC, FEC solvent FG-SE and HFGP-SE. (f) Arrhenius plots of PVHF-SPE, FG-SE, and HFGP-SE. (g) Li+ transference numbers (tLi+) of PVHF-SPE, FG-SE and HFGP-SE. (h) CV curves of Li||HFGP-SE||SS cell. | ||
Furthermore, Raman spectroscopy was used to investigate the coordination state of TFSI−-anions in PVHF-SPE, FG-SE, and HFGP-SE. The Raman peak for the TFSI− anion, located around 740 cm−1, can be resolved into four distinct peaks, as shown in Fig. 2d, which correspond to ion aggregates (AGGs), contact ion pairs (CIPs), and free TFSI− anions.22 The changes in Raman peaks when comparing PVHF-SPE and FG-SE with HFGP-SE suggest differences in their solvation structures. The Raman spectroscopy fitting results indicate that HFGP-SE is predominantly composed of CIPs and AGGs (Fig. S6a, ESI†), confirming the formation of localized high-concentration (LHC) solvation structures within HFGP-SE. Meanwhile, the higher ratio of free TFSI− and lower ratio of AGGs at the interface between FG-SE and PVHF-SPE (Fig. S6b and c, ESI†) compared to that of HFGP-SE, indicates the improved lithium salt dissociation at the interface. These results indicate that LHC solvation structures with improved lithium salt dissociation form at the interface between FG-SE and PVHF-SPE. FT-IR techniques were applied to investigate the solvent state in FG-SE and HFGP-SE. Fig. 2e displays the CO characteristic peaks of TFEC and FEC. The reduced peak intensity for free TFEC solvent molecules, along with the emergence of new bands for Li+-coordinated TFEC. Meanwhile, the peak intensity of Li+ coordinated FEC increased, which verifies that more solvent molecules are coordinated with Li+ and TFSI− anions in HFGP-SE.23Fig. 2f presents the temperature dependencies of the ionic conductivities for PVHF-SPE, FG-SE, and HFGP-SE. At room temperature, HFGP-SE shows an ionic conductivity of 0.84 mS cm−1, which is higher than PVHF-SEP (0.21 mS cm−1) but lower than FG-SE (1.47 mS cm−1). The improved ionic conductivity of HFGP-SE compared to PVHF-SPE is mainly due to the improved solvent content and enhanced dissociation of lithium salts.
As shown in Fig. 2g and Fig. S7 (ESI†), the HFGP-SE delivers a Li-ion transference number (tLi+) of 0.87, which is remarkably higher than PVHF-SPE (0.41) and FG-SE (0.64), indicating the weakest anion movement among HFGP-SE. The high tLi+ of HFGP-SE is highly related to the solvation structure and Li-ion transport mechanism. On the one hand, the interface between the FG-SE and PVHF-SPE with a high lithium salt dissociation increases the concentration of charge carriers. On the other hand, Li conduction along the FG-SE/PVHF-SPE interfaces with an LHC solvation structure obeys the “solvent-assisted lithium-ion diffusion” mechanism;24 the aggregation of anion-rich domains introduces the formation of a solvent-rich 3D percolating network, which limits TFSI− transport and enables fast Li+ transport. Therefore, the enhanced lithium salt dissociation and LHC solvation structure in HFPG-SE resulted in fast ion transport and a high Li+ transference number. The cyclic voltammetry (CV) curves of the HFGP-SE lithium metal electrode, shown in Fig. 2h, demonstrate that the 1st and 5th cycles are entirely repeatable, indicating that HFGP-SE has excellent electrochemical stability with a lithium metal anode. Meanwhile, the time-dependence electrochemical impedance spectroscopy (EIS) further confirms the improved interfacial chemical stability with Li metal of HFGP-SE compared to PVHF-SE (Fig. S8, ESI†). These results highlight the ideal compatibility of HFGP-SE with lithium metal.
The voltage variation of symmetric Li||Li cells was evaluated during the long-term lithium plating/stripping process to assess the interfacial stability of Li metal anodes between different electrolytes. As presented in Fig. 3a, the cell using HFGP-SE maintained a stable plating/stripping process for over 1200 hours at a current density of 0.5 mA cm−2. In contrast, the lithium plating/stripping curves for symmetric cells with PVHF-SPE and FG-SE demonstrated a gradual increase in overpotential after 120 hours and 400 hours, respectively. The increased overpotential could be attributed to the poor lithium compatibility of PVHF-SPE and FG-SE. Meanwhile, the symmetric Li||Li cell exhibited exceptional stability during the plating/stripping process, lasting over 2500 hours at a lower current density of 0.1 mA cm−2 (Fig. S9, ESI†). Furthermore, the HFGP-SE-based Li symmetric cell was tested from 0.1 to 1 mA cm−2 to explore the critical current density. As shown in Fig. 3b, the overpotentials increased with rising current density. However, despite these higher overpotentials, no short-circuiting occurred, indicating the excellent electrochemical stability of HFGP-SE even at elevated current densities. Furthermore, the morphology of electrodeposited Li metal in different electrolytes was investigated by SEM. The deposited lithium in the PVHF-SPE showed an uneven and corroded morphology, indicating severe side reactions between residual DMF molecules and lithium metal (Fig. 3c). Meanwhile, irregular lithium growth with high porosity can be observed in the case of the FG-SE (Fig. 3d). However, lithium deposited in the HFGP-SE displayed a uniform and compact morphology (Fig. 3e). Such favorable morphology is attributed to the greatly reduced side reactions against lithium metal and its high compatibility with the lithium compatibility of HFGP-SE.
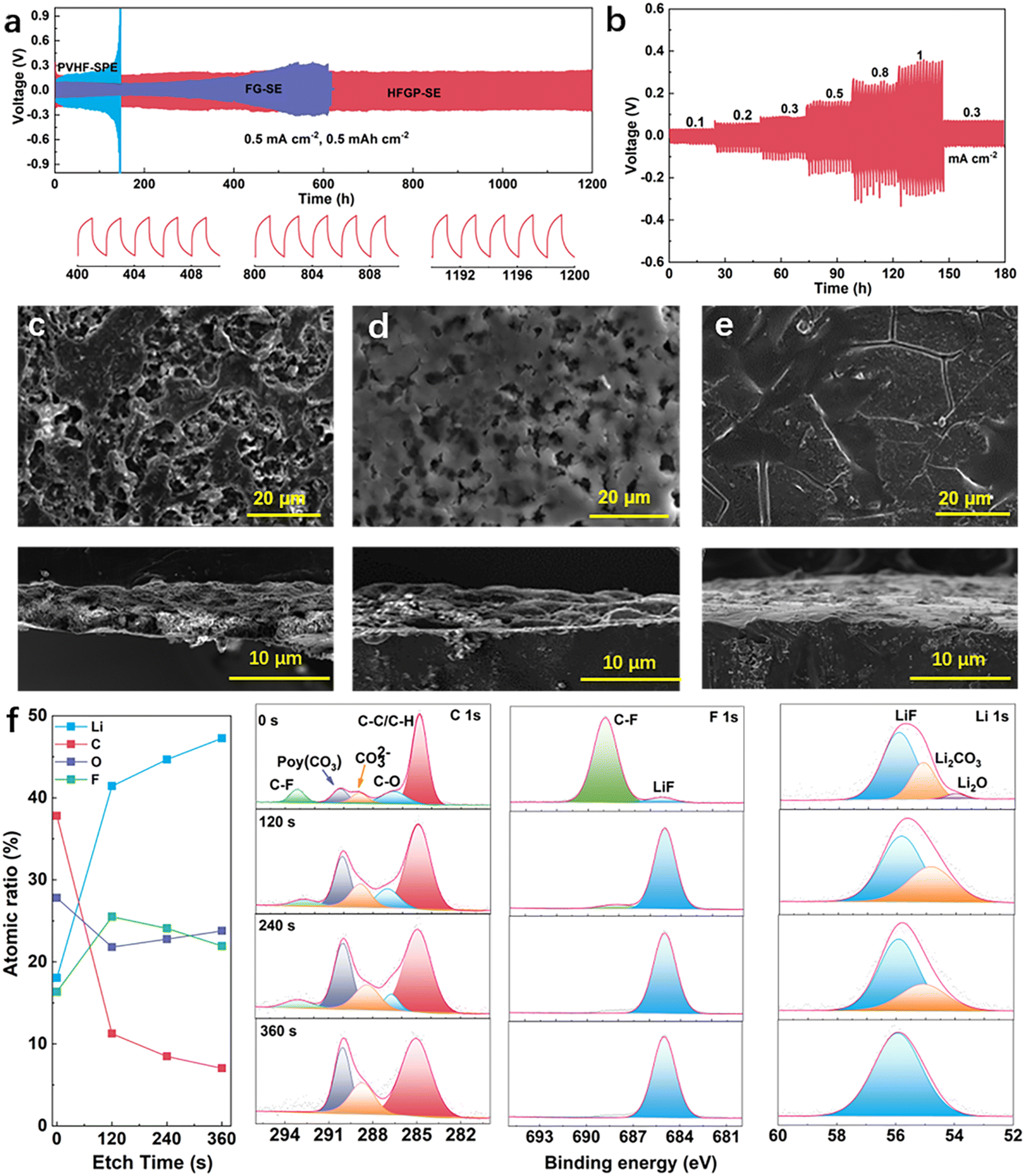 | ||
| Fig. 3 Evaluation of Li–metal plating/stripping performance and characterization of lithium deposition morphologies and SEI layers. (a) Voltage profiles of symmetrical Li||Li cells at 0.5 mA cm−2 and 0.5 mA h cm−2 using PVHF-SPE, FG-SE and HFGP-SE. (b) Voltage profiles of Li||HFGP-SE||Li cells at current densities from 0.1 to 1 mA cm−2. Surface and cross-section SEM images of the cycled Li metal using PVHF-SPE (c), FG-SE (d), and HFGP-SE (e). (f) XPS results of the SEI on cycled Li metal in HFGP-SE: quantified atomic ratios of the elements in SEI and C 1s, F 1s, and Li 1s spectra at different sputter times. | ||
X-Ray photoelectron spectroscopy (XPS) with an Ar+ sputtering depth profiling was used to analyze the solid electrolyte interphase (SEI) components on cycled lithium anodes. Fig. 3f illustrates the elemental composition and atomic ratios, along with the C 1s, F 1s, and Li 1s spectra observed in the SEI. The results indicate that the top surface of SEI mainly consists of C-containing organic species (C–F) and inorganic species (Li2CO3, Li2O, and LiF). These components are primarily attributed to the decomposition of the solvent and the TFSI− anion. As the sputtering depth increases, the organic signals decrease while the LiF signal remains consistent, ultimately becoming the dominant component throughout the entire depth profile. Based on these results, it is concluded that a robust bilayer SEI with a LiF-rich inner layer and organic-rich outer layer was formed. This formation plays a key role in regulating lithium ion deposition and helps prevent the growth of lithium dendrites, thereby enhancing the longevity of lithium batteries.
With the promising properties of HFGP-SE, we further elaborated the performance with a full cell configuration. Fig. 4a presents the rate performance of the Li||LiFePO4 (LFP) full cell assembled with HFGP-SE. This cell delivered discharge capacities of 159, 151, 148, 141, 127, 116, and 101 mA h g−1 at 0.1, 0.2, 0.3, 0.5, 1, 3, and 5C, respectively. Furthermore, when the current was returned to 0.1C, the cell retained over 99% of its capacity, indicating excellent stability and reversible performance. Fig. 4b illustrates the corresponding charge/discharge profiles at various rates. These profiles indicate that the Li||LiFePO4 (LFP) full cell exhibits two distinct voltage plateaus, corresponding to the delithiation and lithiation of the LFP cathode. Notably, there is no significant voltage polarization, even at the relatively high rate of 1C. Therefore, the long-term cycling stability of the Li||HFGP-SE||LFP full cell at 1C was evaluated. As shown in Fig. 4c and Fig. S10 (ESI†), the Li||LFP cell demonstrated stable cycling performance with a capacity retention of 70.5% after 2200 cycles. This significant endurance indicates that the cell has a stable interface against lithium metal, aligning with the interfacial stability observed in Li||Li symmetric cells. In addition, the broad electrochemical stability window of up to 4.7 V for HFGP-SE (Fig. S11, ESI†) indicates its compatibility with high-voltage LiNi0.8Co0.1Mn0.1O2 (NCM811) cathodes. As illustrated in Fig. S12 (ESI†), the Li||HFGP-SE||NCM811 cell displayed impressive rate performance and cycling performance within the voltage range of 2.8 to 4.3 V, indicating the excellent compatibility of the HFGP-SE with high-voltage cathodes.
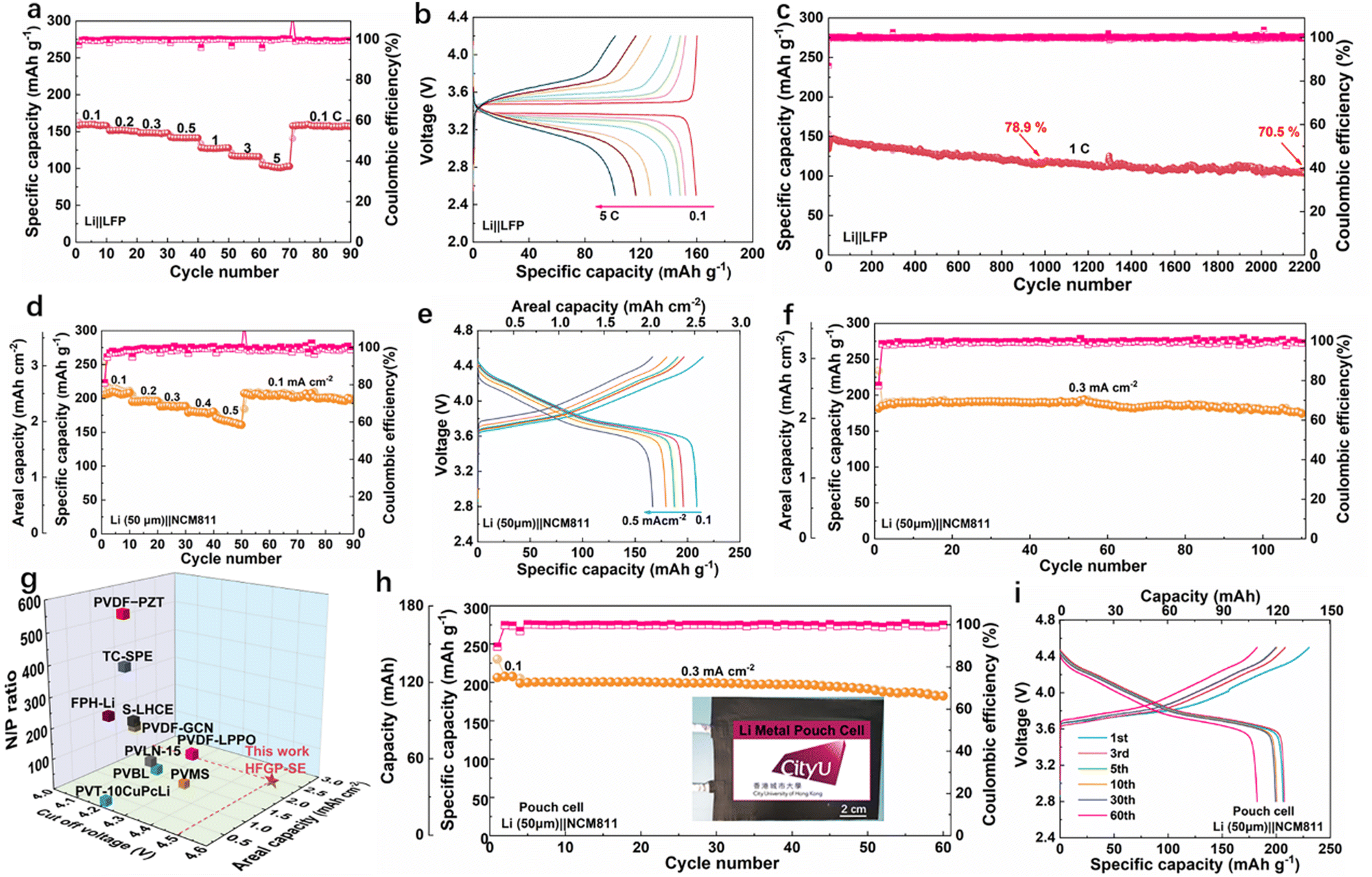 | ||
| Fig. 4 Electrochemical performance of HFGP-SE based solid-state lithium metal batteries. (a) Rate performance and (b) related charge/discharge profiles of Li||HFGP-SE||LFP. (c) Long-term cycling performance of Li||HFGP-SE||LFP at 1C. (d) Rate performance and (e) corresponding charge/discharge voltage profiles of the coin cells with high-loading NCM811 cathodes (12 mg cm−2) and 50 μm Li anode at 4.5 V cut-off voltage. (f) Cycling performance of Li||HFGP-SE||NCM811 coin cells. (g) Comparison of the electrochemical performance of the solid-state lithium batteries in this work with that of other recently reported solid-state batteries using VDF-based SPEs. (h) Cycling performance and (i) corresponding voltage profiles of the Li(50 μm)||HFGP-SE||NCM811 soft pouch type cell (inset of (h) shows the optical photograph of the pouch cell). | ||
To maximize the practicality, a high-loading cathode, high cut-off voltage, and restricted lithium metal anode should be used simultaneously. Therefore, we evaluated the electrochemical performance of Li||HFGP-SE||NCM811 full cells using 50μm Li foils as the anode and high-mass loading NCM811 of 12 mg cm−2 as the cathode. The negative/positive (N/P) ratio in these cells was approximately 4:1. Notably, the Li (50 μm)||NCM811 assembled with HFGP-SE exhibited a low interface impendence compared to that with PVHF-SPE, which was mainly attributed to an excellent wettability of FG-SE, ensuring good interface contact and rapid lithium ion migration in the electrode (Fig. S13, ESI†). Furthermore, Fig. 4d and e shows the rate performance and corresponding charge/discharge voltage profiles of the Li (50 μm)||HFGP-SE||NCM811 cell at various current densities, which achieved specific capacities of 208, 196, 187, 179, and 166 mA h g−1 at current densities of 0.1, 0.2, 0.3, 0.4 and 0.5 mA cm−2, respectively. This corresponds to areal capacities of 2.49, 2.36, 2.25, 2.13, and 1.95 mA h cm−2. Additionally, as shown in Fig. 4f, the Li (50 μm)||HFGP-SE||NCM811 cell delivered a high discharge capacity of 190 mA h g−1 (corresponding to an areal capacity of 2.27 mA h cm−2) and excellent cycling performance at a current density of 0.3 mA cm−2. It also retained 94% of its capacity after 110 cycles with an average coulombic efficiency exceeding 99%. Meanwhile, the charge/discharge curves (Fig. S14, ESI†) demonstrated a stable charge/discharge slope platform throughout cycling, and the voltage polarization remained nearly unchanged. This suggests that the HFGP-SE can support the operational requirements of the NCM811 cathode in 4.5 V class high-voltage lithium metal batteries. To the best of our knowledge, the electrochemical performance of our Li (50 μm)||HFGP-SE||NCM811 cell, including cut-off voltage, areal capacity, and negative/positive (N/P) ratio, significantly exceeds that of other solid-state batteries recently reported using VDF-based solid polymer electrolytes (Fig. 4g and Table S2, ESI†).25–34
To further assess the practicality of HFGP-SE, we assembled and tested sandwich-like soft pouch-type cells. As shown schematically in Fig. S15 (ESI†), a double NCM811 cathode (active material mass loading 24 mg cm−2, size 4.3 × 5.6 cm) was sandwiched by the HFGP-SE electrolyte membrane. Meanwhile, 50 μm thick Li–Cu composite foil was used as the anode. As shown in Fig. 4h and i, the pouch cell achieved capacities of 125 and 120 mA h (corresponding specific capacities of 208 and 200 mA h g−1) at current densities of 0.1 and 0.3 mA cm−2, respectively. Additionally, the pouch cell demonstrated robust cycling performance, retaining 91% of its capacity after 60 cycles. These results suggest that the HFGP-SE electrolyte has a remarkable potential for use in high-voltage Li||NMC811 batteries.
To investigate the source of improved high-voltage electrochemical performance, we examined the oxidation stability of different electrolytes on the NCM811 cathode. In leakage current tests conducted at 4.5 V over an 8 h holding period (Fig. 5a), the leakage current density in a Li||NCM811 cell with HFGP-SE was 5 μA, which is significantly lower than the 71 μA observed with PVHF-SPE. This lower leakage current in the HFGP-SE cell indicates suppressed side reactions on the cathode surface, confirming its superior oxidation stability compared to PVHF-SPE. Furthermore, high-resolution transmission electron microscopy (HRTEM) analysis revealed that the cathode from the Li||PVHF-SPE||NCM811 cell had an uneven CEI (cathode electrolyte interphase) layer with a thickness of 3 nm (Fig. 5b), while the cathode from the Li||HFGP-SE||NCM811 cell exhibited a well-defined and thin CEI layer with a thickness less than 1 nm (Fig. 5c). The X-ray photoelectron spectroscopy (XPS) results (Fig. 5d) for these cathodes present that the C–C, C–H, and C–F peaks are similar, which are attributed to the segments of the PVDF polymer. Additionally, the inorganic composition of Li2CO3 and LiF differed: XPS analysis of the CEI from the Li||PVHF-SPE||NCM811 cell showed a significant presence of Li2CO3. In contrast, the CEI from the Li||HFGP-SE||NCM811 cell had low levels of Li2CO3 and high levels of LiF, indicating an inorganic-rich composition. These results demonstrate that the HFGP-SE has superior oxidation stability, which is critical for high-voltage solid-state lithium batteries. It effectively passivates the catalytic cathode by forming a protective, inorganic LiF-rich CEI layer. This inhibition of side reactions between the electrolyte and cathode improves high-voltage performance.
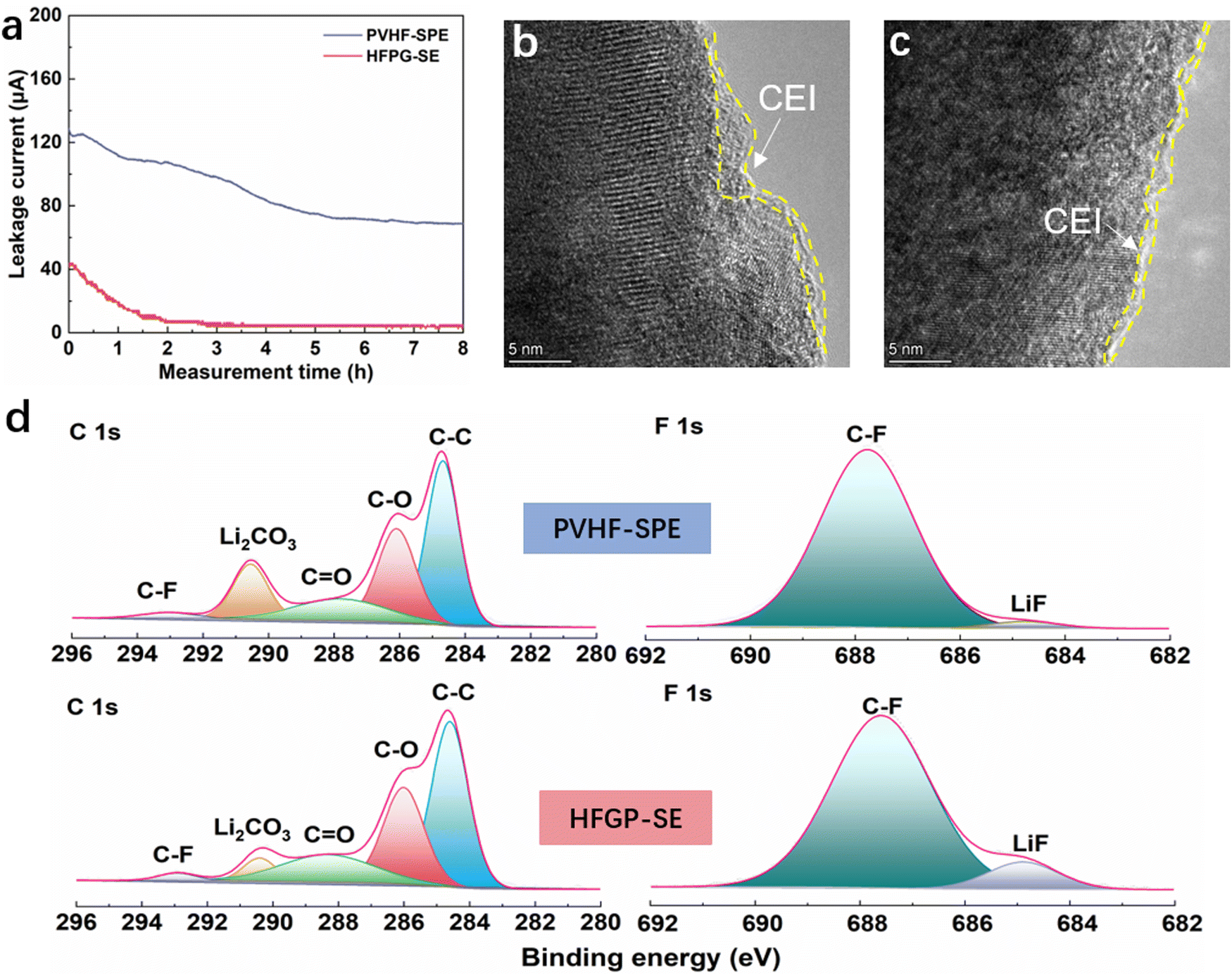 | ||
| Fig. 5 Oxidation stability of the HFGP-SE and interfacial chemistry of the NCM811 cathodes. (a) Leakage current during a constant-voltage (4.5 V vs. Li+/Li) floating test of the NCM811 cathodes in the indicated electrolyte. HRTEM images of NCM811 cathodes retrieved from (b) the Li||PVHF-SPE||NCM811 cell and (c) the Li||HFGP-SE||NCM811 cell after constant-voltage charge at 4.5 V for 8 h. (d) XPS profiles of the NCM811 cathode surface. | ||
Conclusion
This work found that properly designed interfaces between FG-SE and PVHF-SPE can effectively promote lithium salt dissociation. In the meanwhile, a local high-concentration (LHC) solvation structure with an ideal weak-interaction characteristic also formed on the interface, forming abundant ion transport pathways. As a result, the developed HFGP-SE exhibited a high ionic conductivity (0.84 mS cm−1) and a high lithium transference number (tLi+ = 0.87). The Li||Li symmetric cells and Li||LFP full cells with HFGP-SE achieved record long-term cycling performances. In addition, HFGP-SE realizes 4.5 V class Li (50 μm)||NCM81 (areal capacity >2 mA h cm−2) full cell stable cycling under practical testing conditions (N/P = 4:1). This work reveals the strategy of using a hybrid fluorinated gel and polymer solid electrolyte and the interfaces created can significantly enhance lithium salt dissociation, and induce fast ion transport and superior interfacial stability in solid-state electrolytes for lithium metal batteries.
Author contributions
C. Y. Z. and H. M. L. conceived the idea and supervised the project. C. Y. Z., H. M. L. and D. C. Z. designed the experiments and wrote the paper. D. C. Z. and Y. X. L. designed and drew the figures. D. C. Z., D. D. L., S. M. L., Q. X., Z. D. H., S. X. W. and H. H. performed the experiments. Y. X. L. and S. X. W. performed the DFT calculations. D. C. Z., S. M. L., Q. X., Z. D. H., and J. X. Z. discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The work described in this paper was partially supported by a grant from the Research Grants Council of the Hong Kong Special Administrative Region, China (Project No. CityU C4004-23GF). This research was supported by the National Key R&D Program of China under Project 2019YFA0705104. This work was supported in part by InnoHK Project on [Project 1.4 – Flexible and Stretchable Technologies (FAST) for monitoring of CVD risk factors: Soft Battery and self-powered, flexible medical devices] at Hong Kong Centre for Cerebro-cardiovascular Health Engineering (COCHE). All data supporting the findings of this study are available within the main text and the ESI.† All relevant data are available from the corresponding authors upon reasonable request.References
- H. Wan, J. Xu and C. Wang, Nat. Rev. Chem., 2024, 8, 30–40 CrossRef CAS PubMed.
- S. C. Kim, J. Wang, R. Xu, P. Zhang, Y. Chen, Z. Huang, Y. Yang, Z. Yu, S. T. Oyakhire, W. Zhang, L. C. Greenburg, M. S. Kim, D. T. Boyle, P. Sayavong, Y. Ye, J. Qin, Z. Bao and Y. Cui, Nat. Energy, 2023, 8, 814–826 CrossRef CAS.
- Y. C. Yin, J. T. Yang, J. D. Luo, G. X. Lu, Z. Huang, J. P. Wang, P. Li, F. Li, Y. C. Wu, T. Tian, Y. F. Meng, H. S. Mo, Y. H. Song, J. N. Yang, L. Z. Feng, T. Ma, W. Wen, K. Gong, L. J. Wang, H. X. Ju, Y. Xiao, Z. Li, X. Tao and H. B. Yao, Nature, 2023, 616, 77–83 CrossRef CAS.
- Y. Xie, Y. Huang, Y. Zhang, T. Wu, S. Liu, M. Sun, B. Lee, Z. Lin, H. Chen, P. Dai, Z. Huang, J. Yang, C. Shi, D. Wu, L. Huang, Y. Hua, C. Wang and S. Sun, Nat. Commun., 2023, 14, 2883 CrossRef CAS.
- G. V. Alexander, C. Shi, J. O'Neill and E. D. Wachsman, Nat. Mater., 2023, 22, 1136–1143 CrossRef CAS PubMed.
- S. Han, P. Wen, H. Wang, Y. Zhou, Y. Gu, L. Zhang, Y. Shao-Horn, X. Lin and M. Chen, Nat. Mater., 2023, 22, 1515–1522 CrossRef CAS PubMed.
- Y. Hou, Z. Huang, Z. Chen, X. Li, A. Chen, P. Li, Y. Wang and C. Zhi, Nano Energy, 2022, 97, 107204 CrossRef CAS.
- Q.-K. Zhang, X.-Q. Zhang, J. Wan, N. Yao, T.-L. Song, J. Xie, L.-P. Hou, M.-Y. Zhou, X. Chen, B.-Q. Li, R. Wen, H.-J. Peng, Q. Zhang and J.-Q. Huang, Nat. Energy, 2023, 8, 725–735 CrossRef CAS.
- P. Lennartz, B. A. Paren, A. Herzog-Arbeitman, X. C. Chen, J. A. Johnson, M. Winter, Y. Shao-Horn and G. Brunklaus, Joule, 2023, 7, 1471–1495 CrossRef CAS.
- P. Prakash, B. Fall, J. Aguirre, L. A. Sonnenberg, P. R. Chinnam, S. Chereddy, D. A. Dikin, A. Venkatnathan, S. L. Wunder and M. J. Zdilla, Nat. Mater., 2023, 22, 627–635 CrossRef CAS.
- D. Zhang, S. Li, Q. Xiong, Z. Huang, H. Hong, S. Yang, J. Zhu and C. Zhi, MetalMat, 2024, 1, e13 CrossRef.
- D. Zhang, Y. Liu, Z. Sun, Z. Liu, X. Xu, L. Xi, S. Ji, M. Zhu and J. Liu, Angew. Chem., Int. Ed., 2023, 62, e202310006 CrossRef CAS.
- Z. Chen, X. Ma, Y. Hou, H. Cui, X. Li, Q. Yang, Z. Huang, D. Wang, B. Dong, J. Fan and C. Zhi, Adv. Funct. Mater., 2023, 33, 2214539 CAS.
- H. Wang, Y. Yang, C. Gao, T. Chen, J. Song, Y. Zuo, Q. Fang, T. Yang, W. Xiao, K. Zhang, X. Wang and D. Xia, Nat. Commun., 2024, 15, 2500 CAS.
- X. Zhang, S. Wang, C. Xue, C. Xin, Y. Lin, Y. Shen, L. Li and C. W. Nan, Adv. Mater., 2019, 31, e1806082 Search PubMed.
- D. Xu, J. Su, J. Jin, C. Sun, Y. Ruan, C. Chen and Z. Wen, Adv. Energy Mater., 2019, 9, 1900611 Search PubMed.
- W. Liu, C. Yi, L. Li, S. Liu, Q. Gui, D. Ba, Y. Li, D. Peng and J. Liu, Angew. Chem., Int. Ed., 2021, 60, 12931–12940 CrossRef CAS.
- Q. Liu, G. Yang, X. Li, S. Zhang, R. Chen, X. Wang, Y. Gao, Z. Wang and L. Chen, Energy Storage Mater., 2022, 51, 443–452 CrossRef.
- C. Lai, C. Shu, W. Li, L. Wang, X. Wang, T. Zhang, X. Yin, I. Ahmad, M. Li, X. Tian, P. Yang, W. Tang, N. Miao and G. W. Zheng, Nano Lett., 2020, 20, 8273–8281 CrossRef CAS.
- Y. Wu, Y. Li, Y. Wang, Q. Liu, Q. Chen and M. Chen, J. Energy Chem., 2022, 64, 62–84 CrossRef CAS.
- K. Yang, J. Ma, Y. Li, J. Jiao, S. Jiao, X. An, G. Zhong, L. Chen, Y. Jiang, Y. Liu, D. Zhang, J. Mi, J. Biao, B. Li, X. Cheng, S. Guo, Y. Ma, W. Hu, S. Wu, J. Zheng, M. Liu, Y. B. He and F. Kang, J. Am. Chem. Soc., 2024, 146, 11371–11381 CAS.
- Q. Zhou, J. Ma, S. Dong, X. Li and G. Cui, Adv. Mater., 2019, 31, e1902029 CrossRef PubMed.
- J. Wu, X. Wang, Q. Liu, S. Wang, D. Zhou, F. Kang, D. Shanmukaraj, M. Armand, T. Rojo, B. Li and G. Wang, Nat. Commun., 2021, 12, 5746 CrossRef CAS PubMed.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- H. Wang, H. Cheng, D. Li, F. Li, Y. Wei, K. Huang, B. Jiang, H. Xu and Y. Huang, Adv. Energy Mater., 2023, 13, 2204425 CrossRef CAS.
- P. Shi, J. Ma, M. Liu, S. Guo, Y. Huang, S. Wang, L. Zhang, L. Chen, K. Yang, X. Liu, Y. Li, X. An, D. Zhang, X. Cheng, Q. Li, W. Lv, G. Zhong, Y. B. He and F. Kang, Nat. Nanotechnol., 2023, 18, 602–610 CrossRef CAS.
- Q. Wu, M. Fang, S. Jiao, S. Li, S. Zhang, Z. Shen, S. Mao, J. Mao, J. Zhang, Y. Tan, K. Shen, J. Lv, W. Hu, Y. He and Y. Lu, Nat. Commun., 2023, 14, 6296 CrossRef CAS.
- Y. F. Huang, J. P. Zeng, S. F. Li, C. Dai, J. F. Liu, C. Liu and Y. B. He, Adv. Energy Mater., 2023, 13, 2203888 CrossRef CAS.
- L. Chen, T. Gu, J. Ma, K. Yang, P. Shi, J. Biao, J. Mi, M. Liu, W. Lv and Y.-B. He, Nano Energy, 2022, 100, 107470 CrossRef CAS.
- J. Mi, J. Ma, L. Chen, C. Lai, K. Yang, J. Biao, H. Xia, X. Song, W. Lv, G. Zhong and Y.-B. He, Energy Storage Mater., 2022, 48, 375–383 CrossRef.
- K. Yang, L. Chen, J. Ma, C. Lai, Y. Huang, J. Mi, J. Biao, D. Zhang, P. Shi, H. Xia, G. Zhong, F. Kang and Y. B. He, Angew. Chem., Int. Ed., 2021, 60, 24668–24675 CrossRef CAS PubMed.
- S. Xu, R. Xu, T. Yu, K. Chen, C. Sun, G. Hu, S. Bai, H.-M. Cheng, Z. Sun and F. Li, Energy Environ. Sci., 2022, 15, 3379–3387 RSC.
- P. Zhai, Z. Yang, Y. Wei, X. Guo and Y. Gong, Adv. Energy Mater., 2022, 12, 2200967 CrossRef CAS.
- B. H. Kang, S. F. Li, J. Yang, Z. M. Li and Y. F. Huang, ACS Nano, 2023, 17, 14114–14122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee04078c |
| This journal is © The Royal Society of Chemistry 2025 |