
Anti-freezing hydrogel electrolyte with a regulated hydrogen bond network enables high-rate and long cycling zinc batteries†
Shao-Jie
Guo
a,
Meng-Yu
Yan
b,
Dong-Ming
Xu
a,
Pan
He
c,
Kai-Jian
Yan
b,
Jie-Xin
Zhu
 b,
Yong-Kun
Yu
b,
Ze-Ya
Peng
a,
Yan-Zhu
Luo
*a and
Fei-Fei
Cao
*a
b,
Yong-Kun
Yu
b,
Ze-Ya
Peng
a,
Yan-Zhu
Luo
*a and
Fei-Fei
Cao
*a
aCollege of Chemistry, Huazhong Agricultural University, Wuhan 430070, P. R. China
bState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, International School of Materials Science and Engineering, Wuhan University of Technology, Wuhan 430070, P. R. China
cDepartment of Chemistry, University College London, London WC1H 0AJ, UK
First published on 12th November 2024
Abstract
Zinc-based batteries utilizing hydrogel electrolytes present significant promise as power sources for next-generation flexible devices owing to their stretchable nature and enhanced safety features. Nonetheless, the current hydrogel electrolytes require improvements in terms of cycling stability and rate capability. In this study, 1,2-propylene glycol is added as a cosolvent to polyacrylamide hydrogel electrolytes. The cosolvent effectively modulates the internal hydrogen bond network of the hydrogel through hydroxyl and terminal methyl groups, inhibits the activity of water while preventing the solvent from forming a “hand-in-hand” long-chain molecular structure, and enhances the stability of the electrode/electrolyte interface. Consequently, a symmetrical battery assembled with PAM-1,2-PG exhibited a stable cycling performance of over 490 h at 100 mA cm−2 and 50 mA h cm−2 (DOD of 86%). A change in the hydrogen bond network endows the battery with remarkable low-temperature performance of more than 3780 h under −30 °C at 1 mA cm−2. Furthermore, the resulting aqueous zinc-based devices showcase high capacity and outstanding cycling durability in a wide temperature range. This work provides valuable insights into the development of high-performance hydrogel electrolytes, paving the way for dendrite-free, fast-charging, and environmentally adaptable Zn-based energy storage systems.
Broader contextAqueous zinc-ion batteries (AZIBs) are known for their inherent safety and widespread use in wearable electronic devices. However, the risk of liquid electrolyte leakage remains a concern. Hydrogel electrolytes offer a promising solution to electrolyte leakage owing to their environmental friendliness, flexibility, and safety. Despite these advantages, hydrogels contain a large number of active water molecules, which not only freeze at low temperatures but also induce side reactions that compromise interfacial stability. To address this, we incorporated 1,2-propylene glycol (1,2-PG) with inert end groups into the polyacrylamide (PAM) gel electrolyte, modifying the internal hydrogen bond network to enhance antifreezing properties and rate performance. A symmetrical Zn||Zn battery assembled with the gel electrolyte exhibits a stable cycling of more than 490 hours at 100 mA cm−2 and 50 mA h cm−2 (86% DOD). Moreover, the battery maintained excellent cycling stability even at low temperatures of −30 °C. These remarkable results could also extend to Zn||VOH AZIBs and Zn||FSC aqueous zinc-ion capacitors, demonstrating the potential of this novel hydrogel electrolyte for fast charging and long-term cycling across a wide temperature range. This study provides valuable insights into the design of high-rate and low-temperature aqueous zinc-based batteries. |
Introduction
The rapid advancement of flexible and wearable electronic devices, such as motion and health monitors, has heightened the demand for rechargeable flexible batteries that offer superior safety, durability, and stretchability.1,2 In this context, aqueous zinc-ion batteries (AZIBs) have emerged as a highly promising battery system. They are characterized by intrinsic safety, environmental friendliness, and low cost.3,4 However, traditional liquid electrolytes have strong fluidity, are prone to leakage problems, and are thus unsuitable for wearable devices.5 Hydrogel electrolytes consist of intricate three-dimensional (3D) cross-linked networks of hydrophilic polymer chains, which provide free migration channels for Zn2+, ensuring excellent flexibility while effectively addressing the issue of electrolyte leakage.6,7 However, a large number of free water molecules within the electrolytes would cause freezing at low temperatures and easily induce hydrogen evolution and corrosion reactions, resulting in poor interfacial compatibility and rapid growth of zinc dendrites.8–10 Therefore, the reduction of water activity is crucial for improving the performance of zinc-ion batteries.By reconstructing a hydrogen bond network structure within hydrogel electrolytes, the content of free water molecules can be effectively reduced.8,11 The incorporation of salts, ionic liquids, and organic solvents has been explored as an effective pathway to address this challenge.12–15 In this regard, small-molecule alcohol solvents containing hydroxyl groups are considered the most efficient cosolvents to form new hydrogen bonds with water molecules, reduce the activity of water, and construct a stable electrode/electrolyte interface.12,15 In particular, glycerol (Gly) and ethylene glycol (EG) have mostly been studied in the context of AZIBs. For instance, Gly provides a large number of hydroxyl groups for the construction of strong hydrogen bonds with water molecules, which weakens the hydrogen bonds between water molecules in the hydrogel electrolyte.14 However, it is worth noting that owing to the abundant hydroxyl terminal groups in Gly and EG, the formation of long-chain structures with “hand-in-hand” additive molecules results in high viscosity in the gel-like liquid.16–19 This high viscosity would impede the transport of solvated Zn2+ ions, thereby reducing the ionic conductivity of the hydrogel electrolyte, resulting in a short cycle life and low-rate performance.18,19
Thus, the effect of solvent-solvent interaction on battery performance is necessary to consider.16 MeOH, characterized by its methyl (–CH3) group, has been employed by researchers to effectively disrupt the original interconnected chain structure of EG, leading to a drastically reduced viscosity of the electrolyte and a twofold increase in ionic conductivity.18 Similarly, another groundbreaking research reported that the design CH3O–(CH2–CH2–O)n–CH3 further underscores the significance of terminal groups. By replacing –OH terminal groups present in H–(O–CH2–CH2)n–OH with –OCH3, viscosity drops from 115 to 22.4 mPa s, whereas ionic conductivity rises from 0.8 to 1.8 mS cm−1.19 Although, the toxicity of MeOH and the complex modification process related to organic molecules for the modification of hydrogel electrolytes remains to be discussed, the proposed end-capping effect offers a significant reference point. Inspired by this principle, the end-capping effect was applied to a hydrogel electrolyte for the first time to effectively improve its ionic conductivity and interface stability.
Herein, utilizing polyacrylamide (PAM) and Zn(OTf)2 as the foundation for the hydrogel electrolyte, we have introduced 1,2-propylene glycol (1,2-PG) to develop a novel electrolyte named PAM-1,2-PG, which possesses a regulated hydrogen bond network, as illustrated in Fig. 1a. The incorporation of 1,2-PG serves a multifaceted role, effectively reducing the hydrogen bonds between free H2O molecules through the strong interaction between free H2O and 1,2-PG molecules. Furthermore, the terminal methyl groups present in 1,2-PG molecules play a pivotal role in breaking the interconnected “hand-in-hand” chains of PG, leading to a higher ionic conductivity of the hydrogel electrolyte. The regulation of 1,2-PG on the structure of the external solvation layer of Zn2+ results in reduced desolvation tendencies, and the formation of solid electrolyte interface with an organic-rich surface and an inorganic-rich bottom at the electrode/electrolyte interface leads to a reduction in side reactions. As a culmination of these improvements, a symmetrical Zn||Zn battery assembled with PAM-1,2-PG exhibited a stable cycling performance of over 2160 h at 10 mA cm−2 and of over 490 h at 100 mA cm−2 and 50 mA h cm−2 (DOD of 86%). Furthermore, the maximum discharge of depth can reach as high as 95%. Even under extreme conditions of −30 °C, the assembled Zn||Zn battery exhibits significant cycling stability of more than 3780 h at 1 mA cm−2 and 1 mA h cm−2. These exceptional outcomes also extend to Zn||VOH AZIBs and Zn||FSC aqueous zinc-ion capacitors (AZICs). This proof-of-concept demonstration introduces a novel strategy that holds the promise of enabling fast charging and long-term cycling capabilities for zinc-based devices across a wide temperature range.
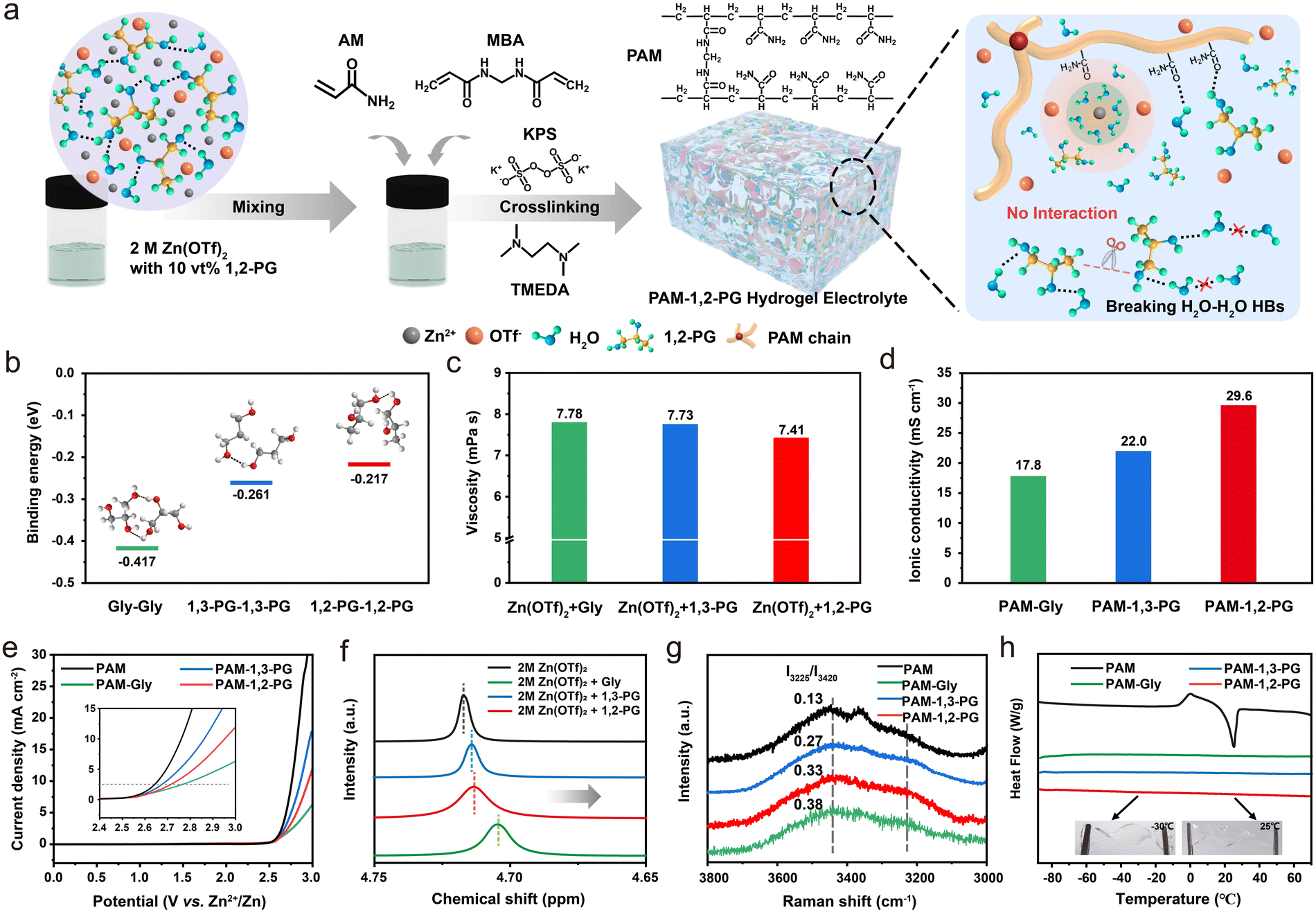 | ||
| Fig. 1 Preparation and properties of the PAM-1,2-PG hydrogel electrolyte. (a) Illustration of the polymerization processes and structure of PAM-1,2-PG. (b) Relative binding energies between different cosolvent molecules. (c) The viscosities of electrolytes with different cosolvents at RT. (d) Ionic conductivity of hydrogel electrolytes based on different cosolvents. (e) Linear sweep voltammograms of different hydrogel electrolytes. (f) 1H NMR spectrum of 2 M Zn(CF3SO3)2/D2O solution with different cosolvents. (g) Raman spectra of different hydrogel electrolytes in the range of 3000–3800 cm−1. (h) DSC test of different hydrogel electrolytes from −90 to 70 °C at a heating rate of 5 °C min−1. Insets: The flexibility of the PAM-1,2-PG hydrogel under 25 °C and −30 °C. | ||
Results and discussion
To explore the impact of terminal groups and the number of functional groups in electrolyte solvents on hydrogel electrolyte performance, we chose propyl alcohol as an ideal model for study owing to its structural diversity. Specifically, we examined glycerin with three hydroxyl groups (referred to as Gly), 1,3-propylene glycol with two terminal hydroxyl groups (referred to as 1,3-PG), and 1,2-propylene glycol with one central and one terminal hydroxyl group (referred to as 1,2-PG). The calculated relative interaction binding energies for Gly, 1,3-PG, and 1,2-PG are −0.417, −0.261, and −0.217 eV, respectively (Fig. 1b). Meanwhile, viscosity tests for a 2 M Zn(OTf)2 electrolyte with various cosolvents were carried out, and the results indicated that the presence of inert end groups can reduce the viscosity of the electrolyte (Fig. 1c). These values highlight the weakest intermolecular interactions among 1,2-PG molecules (Fig. S1, ESI†), which bode well for enhancing ionic conductivity. To verify the successful polymerization of the hydrogel electrolyte, we generated in situ Raman spectra for the 1,2-PG-based polymerization process at different reaction times, as presented in Fig. S2 (ESI†). The reaction started immediately following the addition of KPS and TMEDA. Over time, bands associated with C–N stretching, CH2 deformation, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching, CH2 symmetrical stretching, and CH2 asymmetrical stretching (located at 1289, 1439, 1636, 3050, and 3119 cm−1, respectively) gradually weakened.20,21 Simultaneously, signals for C–C–C deformation vibration at 350 cm−1 and alkyl C–H symmetric stretching at 2937 cm−1 consistently increased with time, thus being indicative of the successful polymerization of acrylamide (AM) monomers into polyacrylamide (PAM) after 45 minutes.22,23 It is noteworthy that a peak at 842 cm−1, attributed to C–C–O bending (characteristic of 1,2-PG), remained stable throughout the entire polymerization process. Hydrogel electrolytes with Gly and 1,3-PG cosolvents were also prepared and named PAM-Gly and PAM-1,3-PG, respectively, while the hydrogel electrolyte prepared without a cosolvent was named PAM.
C stretching, CH2 symmetrical stretching, and CH2 asymmetrical stretching (located at 1289, 1439, 1636, 3050, and 3119 cm−1, respectively) gradually weakened.20,21 Simultaneously, signals for C–C–C deformation vibration at 350 cm−1 and alkyl C–H symmetric stretching at 2937 cm−1 consistently increased with time, thus being indicative of the successful polymerization of acrylamide (AM) monomers into polyacrylamide (PAM) after 45 minutes.22,23 It is noteworthy that a peak at 842 cm−1, attributed to C–C–O bending (characteristic of 1,2-PG), remained stable throughout the entire polymerization process. Hydrogel electrolytes with Gly and 1,3-PG cosolvents were also prepared and named PAM-Gly and PAM-1,3-PG, respectively, while the hydrogel electrolyte prepared without a cosolvent was named PAM.
Fourier-transform infrared (FTIR) spectra also verified the successful polymerization of PAM in the presence of 1,2-PG (Fig. S3, ESI†).24 Fig. S4 (ESI†) illustrates that all hydrogel electrolytes are colourless and transparent, with their respective ionic conductivities tested, as displayed in Fig. 1d. Nyquist plots in Fig. S5 (ESI†) confirmed that PAM-1,2-PG exhibited the highest ionic conductivity of 29.6 mS cm−1 compared to PAM-Gly (17.8 mS cm−1) and PAM-1,3-PG (22.0 mS cm−1), which could be attributed to the reduced interactions between the additive molecules. Scanning electron microscopy (SEM) images of freeze-dried hydrogel electrolytes (Fig. S6, ESI†) reveal that the introduction of alcohols results in a three-dimensional network structure with enlarged pore sizes and a uniform porous configuration, thereby minimizing obstructions to ion transport pathways within the polymer skeleton walls.25 Simultaneously, owing to the increased number of functional groups, interactions involving the –OH functional group and H2O expand the electrochemical stability window (ESW) of the hydrogel electrolyte, which is confirmed by linear sweep voltammetry (LSV), as given in Fig. 1e. Specifically, compared to PAM-1,2-PG, PAM-Gly demonstrates a wider ESW owing to its greater overall hydrogen bonding capacity. In contrast, PAM-1,3-PG exhibits a narrower ESW, which is attributed to the reduced number of effective functional groups available after the formation of “hand-in-hand” hydrogen bonds within the cosolvent molecules.
The functional mechanism of the cosolvents in the hydrogel electrolyte was subjected to further investigation. The interaction between 1,2-PG and H2O was characterized using 1H nuclear magnetic resonance (NMR) analysis. As illustrated in Fig. 1f, the 1H signal of H2O in the presence of the cosolvents exhibits a chemical shift towards a higher field, indicative of increased electron density around the hydrogen (H) atoms.26,27 This shift arises from the formation of additional intermolecular hydrogen bonds between cosolvents and H2O, disrupting the preexisting hydrogen bond network among H2O molecules. This variation is beneficial for inhibiting side reactions at the electrolyte/electrode interface. To delve deeper into the hydration state of the hydrogel electrolyte, Raman spectroscopy was employed to examine the region between 3800 and 3000 cm−1 (Fig. 1g). Raman peaks at 3225 cm−1 and 3420 cm−1 correspond to O–H symmetric stretching vibration and O–H asymmetric stretching vibration of water, respectively.23,28 These peaks represent strong hydrogen bonds in a tetrahedral coordination (double donor-double acceptor, DDAA–OH) and weak hydrogen bonds in an incomplete tetrahedral structure (single donor-single acceptor, DA–OH). The relative intensity ratio of these two bands (I3225/I3420, the ratio of the intensity of the strong hydrogen bond at 3225 cm−1 to that of the weak hydrogen bond at 3420 cm−1) is used to determine the ratio of strong HBs to weak HBs. I3225/I3420 ratios for PAM-Gly, PAM-1,2-PG, and PAM-1,3-PG hydrogels were 0.38, 0.33, and 0.27, respectively, all of which are notably higher than that of the PAM hydrogel (0.13). This suggests that the initial H2O–H2O HBs, which are non-tetrahedral in nature, have been disrupted to varying degrees and replaced by more stable tetrahedral structures. This structural alteration contributes to reduced water activity and enhanced anti-freezing properties. Differential scanning calorimetry (DSC) was employed to investigate the crystallization temperature of hydrogel electrolytes, as shown in Fig. 1h. During the heating process from −90 °C to 70 °C, PAM gel electrolyte exhibited an exothermic peak at around −6 °C. This peak results from the conversion of ice into water as precipitated Zn(OTf)2 dissolves again. In contrast, no such peak is observed for PAM-Gly, PAM-1,2-PG, and PAM-1,3-PG gel electrolytes. This difference arises because the –OH functional group of cosolvents forms hydrogen bonds with H2O molecules, disrupting the original hydrogen network structure of H2O molecules in the solution and conferring superior anti-freezing properties upon the gel electrolytes. Based on these experimental results, the PAM-1,2-PG gel electrolyte demonstrates an effective regulation of the hydrogen bond network while reducing viscosity, thus emerging as a high-performance hydrogel electrolyte for zinc-ion batteries.
At the same time, an examination of the calculated number of hydrogen bonds between H2O molecules in the PAM-1,2-PG electrolyte using molecular dynamics (MD) simulations indicates a decrease in their number compared to those in PAM electrolyte (Fig. S7, ESI†). This result again show that 1,2-PG plays a role in regulating the initial hydrogen bond system of active water molecules through its hydroxyl functional groups, thereby reconstructing a stable hydrogen bond network. This network will limit the activity of water, reduce the freezing point of the hydrogel electrolyte and the degree of the hydrogen evolution reaction, and improve the stability of the zinc anode.27
Furthermore, PAM-1,2-PG displays enhanced physical properties owing to the formation of physical crosslinks driven by hydrogen bonding. The tensile stress–strain curves, shown in Fig. S8 (ESI†), demonstrate that PAM-1,2-PG possesses the highest ductility among all the gel electrolytes. Fig. S9 (ESI†) presents dendrite puncture simulation, which reveals that the mechanical strength of the PAM-1,2-PG hydrogel electrolyte is sufficient to withstand dendrite growth. This robustness is attributed to the interaction between the hydroxyl groups of 1,2-PG and PAM chains, which leads to the bundling of some PAM molecular chains.29 When subjected to external force, these bundles gradually break as a sacrificial mechanism, releasing hidden length to dissipate energy. Furthermore, the abundant hydrogen bonds in and high flexibility of the PAM-1,2-PG hydrogel endow it with excellent adhesive properties toward various substrates, including Zn sheets, Ti mesh, Cu sheets, and carbon cloths (Fig. S10a, ESI†). Even at −30 °C, PAM-1,2-PG exhibits the capability to twist and adhere to polymers weighing over 38 g (Fig. 1h and Fig. S10b, ESI†). Based on these findings, cost-effective and low-toxicity 1,2-PG is considered an ideal co-solvent for enhancing hydrogel electrolyte performance. By adjusting the hydrogen bond network structure, the PAM-1,2-PG hydrogel electrolyte, with its combination of high ionic conductivity, adequate mechanical strength, robust adhesive properties, and exceptional adaptability to low temperatures, hold promise for facilitating practical applications.
In pursuit of a deeper understanding of the distinctive solvation structures of Zn2+ within PAM and PAM-1,2-PG electrolytes, we conducted comprehensive MD simulations, radial distribution function (RDF) analysis, and coordination number (CN) calculations. Firstly, the MD simulation of the evolution of Zn2+ solvation structure in the presence or absence of the 1,2-PG co-solvent was studied (Fig. 2a and b), indicating that 1,2-PG was not involved in the inner solvation structure. Analysis of RDFs, as illustrated in Fig. 2c and d, revealed prominent coordination peaks for Zn–O (H2O) at approximately 2.1 Å in PAM and PAM-1,2-PG hydrogel electrolytes. This indicates that the chemical environment of the first solvation sheath at a radius of 2.1 Å remains unchanged and exists in the form of Zn(H2O)62+ coordination in both systems. The specific coordination numbers of O(OTf−), O(PAM), and O(H2O) with Zn2+ are shown in Fig. S11 (ESI†). Within the PAM-1,2-PG electrolyte, new peaks emerge at a distance of approximately 4.4 Å. These peaks can be attributed to interactions between Zn2+ ions and the oxygen atoms of the two hydroxyl groups of 1,2-PG molecules, affirming the existence of 1,2-PG molecules in the outer solvation structure.12,30
 | ||
| Fig. 2 Study on the solvation structure of Zn2+ in the hydrogel electrolyte. MD simulation snapshots of (a) PAM and (b) PAM-1,2-PG hydrogel electrolytes (PAM: n = 6). The RDFs of the oxygen atom in molecules around Zn2+ using MD simulations in (c) PAM and (d) PAM-1,2-PG hydrogel electrolytes. (e) Normalized Zn K-edge XANES spectra and enlarged Zn K-edge XANES spectra of metal Zn, PAM and PAM-1,2-PG hydrogel electrolytes. (f) The fitting of EXAFS spectra in R-space for PAM and PAM-1,2-PG hydrogel electrolytes. Wavelet transforms of Zn K-edge EXAFS signals for (g) PAM and (h) PAM-1,2-PG hydrogel electrolytes. | ||
To confirm the solvation layer structure, we employed X-ray absorption fine structure (XAFS) technique to elucidate the local environment surrounding Zn2+. Fig. 2e displays Zn K-edge X-ray absorption near-edge structure (XANES) spectra for zinc foil and the gel electrolyte. Thus, zinc foil exists in a zero-valent state, as evidenced by the lowest energy front peak in XANES. Upon introducing 1,2-PG, absorption energy shifts to a higher energy range (PAM < PAM-1,2-PG).31–33 This shift indicates distinct local structures for Zn2+, with the average electron density around it being lower in PAM-1,2-PG than in PAM.32 Further analysis of k-space data indicates that under the influence of 1,2-PG, the intensity of the k3-weight peak weakens (Fig. S12, ESI†). This weakening suggests a reduction in the coordination number of Zn–O in the inner solvation sheath. For a precise characterization of the coordination structure, we conducted Fourier transform of the Zn K-edge in R-space (FT-EXAFS). By comparing the gel electrolyte PAM with PAM-1,2-PG, we observed a slight decrease in the radial distance of the Zn–O bond and a decrease in the coordination number of the inner solvation sheath from 5.81 to 5.78 (Fig. 2f). These findings are in line with the results obtained through MD simulations. To visually highlight the variation in Zn2+ coordination states, we analysed the EXAFS spectra using wavelet transform (Fig. 2g and h). The results indicate that 1,2-PG exerts an influence on the inner solvent sheath. However, owing to steric hindrance effects, 1,2-PG predominantly resides in the outer cosolvation structure during the reaction.33 Taken together, our findings demonstrate that the strong interaction between 1,2-PG and water molecules not only disrupts hydrogen bonds between water molecules, thus reducing the freezing point, but also affects the solvation structure of Zn2+. This effect is conducive to enhance the electrochemical stability of the gel electrolyte.
The effect of PAM-1,2-PG on the electrode–electrolyte interface was studied in detail. Density functional theory (DFT) calculations compared the adsorption energies of H2O and 1,2-PG on the Zn (002) plane (Fig. 3a). The results showed that 1,2-PG has a lower adsorption energy (−2.730 eV) compared to H2O (−1.407 eV), indicating that 1,2-PG molecules preferentially adsorb on the Zn surface over H2O molecules.27,34,35 To verify the adsorption behaviour of 1,2-PG, PAM, and PAM-1,2-PG hydrogel electrolytes, these materials were contacted with zinc sheets for 14 hours, and the contact layer was analyzed using X-ray photoelectron spectroscopy (XPS). The total XPS spectra (Fig. S13, ESI†) of Zn, C, O, S, and F atoms were fitted and calculated (Fig. 3b). The elemental content of C in the contact layer with the PAM-1,2-PG hydrogel electrolyte was significantly higher than that with PAM, indicating the preferential adsorption of 1,2-PG molecules on the Zn surface. The effect of 1,2-PG was further evaluated using Tafel plots (Fig. S14, ESI†). Compared to the Zn electrode in the PAM electrolyte, the Zn electrode in the PAM-1,2-PG electrolyte exhibited a lower corrosion current (0.067 mA cm−2) and higher corrosion potential (0.031 V), suggesting that 1,2-PG adsorbed on the Zn surface effectively inhibits corrosion.24,36 SEM images of the Zn electrode after contact with hydrogel electrolytes for 14 hours support this finding. The surface morphology of the Zn electrode after PAM contact showed significant non-compact flakes (Fig. 3c), whereas that of the Zn electrode after PAM-1,2-PG contact was relatively flat (Fig. 3d).
 | ||
| Fig. 3 Characterization of the PAM-1,2-PG hydrogel electrolyte-derived SEI. (a) The adsorption energies of H2O and 1,2-PG onto the Zn metal surface. (b) The corresponding element content of Zn, C, and O calculated from the XPS spectra of the Zn anode after contact with PAM and PAM-1,2-PG hydrogel electrolytes for 14 h. SEM images of Zn morphologies obtained after contact with (c) PAM and (d) PAM-1,2-PG hydrogel electrolytes for 14 h. (e) F 1s, S 2p, O 1s, and C 1s XPS spectra of the Zn electrode surface with the PAM-1,2-PG hydrogel electrolyte after cycling at different Ar+ etching times. (f) 3D reconstruction maps of Zn electrodes retrieved from the PAM-1,2-PG hydrogel electrolyte measured through TOF-SIMS. | ||
The chemical composition of the interfacial layer on the cycled Zn surface with PAM and PAM-1,2-PG hydrogel electrolytes was analyzed using XPS with Ar+ sputtering (Fig. 3e and Fig. S15, ESI†). The depth profile of the F 1s spectra indicated that the peak could be deconvoluted into inorganic ZnF2 and organic –CF3. The dominance of ZnF2 with extended sputtering time largely arises from OTf− decomposition, and the ZnF2 content of the Zn anode with PAM-1,2-PG was higher than that of the anode with PAM at different sputtering times. A similar trend was observed in the S 2p spectra, which could be fitted into organic CF3SO3− and inorganic Zn–S. In the O 1s spectra, the interphase surface was dominated by organic species (CO or C–O) and inorganic Zn–O, with the Zn–O peak gradually dominating oxygen-containing components over etching. In the C 1s spectra, the peaks comprised C–F, C–O, and C–C/C–H groups, and organic species rapidly decreased with increased sputtering time.37–39 Based on the above analysis, it can be concluded that organic components predominate the outer layer of the PAM-1,2-PG assisted solid electrolyte interphase (SEI), whereas inorganic phase products such as ZnF2, ZnO, and ZnS predominate the inner layer. Time-of-flight secondary-ion mass spectrometry (TOF-SIMS) was conducted to acquire information on the chemical composition and spatial distribution of the SEI formed in the Zn anode with PAM-1,2-PG and PAM hydrogel electrolytes (Fig. 3f and Fig. S16, S17, ESI†). The signal of C was stronger at the surface for the PAM-1,2-PG-based sample, and the signal intensity of ZnO, ZnF, and ZnS showed apparent regularity compared to the PAM-assisted SEI. These results suggest that the 1,2-PG-containing hydrogel electrolyte is characterized by a more uniform SEI with rich organic species on the outer surface and primarily inorganic species in the inner region, which is beneficial for inducing homogeneous deposition of Zn metal and inhibiting side reactions.
In order to explore the effect of additives on zinc deposition behaviour, the gel electrolyte was subjected to electrochemical impedance test (EIS) at different temperatures, and activation energy (Ea) was obtained using Arrhenius equation fitting to determine the desolvation rate of hydrated zinc ions (Fig. S18, ESI†). The calculated Ea for Zn deposition within the PAM-1,2-PG electrolyte is 30.0 kJ mol−1, which is notably lower than that within the PAM electrolyte (44.6 kJ mol−1). This result confirms the enhanced desolvation kinetics of hydrated Zn2+ ions with the PAM-1,2-PG electrolyte.40,41 Additionally, cyclic voltammetry (CV) curves of Zn||Cu asymmetric batteries reveal that PAM-1,2-PG demonstrates a notably larger nucleation overpotential (237 mV) compared to PAM (203 mV) (Fig. S19, ESI†). This discrepancy suggests the formation of smaller zinc deposition grains during the initial deposition stage, which ultimately led to the development of a well-oriented crystal structure and optimized zinc deposition behaviour.42 The effect of 1,2-PG on Zn2+ diffusion behavior during growth was further investigated using chronoamperometry (CA) (Fig. S20, ESI†). The results indicate that the current in the symmetrical cell with the PAM-1,2-PG hydrogel electrolyte stabilized more rapidly, effectively preventing 2D Zn2+ diffusion and inhibiting dendrite growth.
In situ optical microscopy was employed to observe the metallic zinc plating process using PAM and PAM-1,2-PG hydrogel electrolytes, with a deposition duration of 60 minutes at 5 mA cm−2 (Fig. 4a). Within the initial 20 minutes, the growth of small, loosely-packed protrusions on the zinc foil was evident when utilizing PAM. However, at 40 minutes, these protrusions rapidly developed into dendrite-like structures, leading to irreversible zinc consumption. Remarkably, when PAM-1,2-PG was employed, the interface between the electrode and electrolyte remained uniformly smooth throughout the entire plating process. To demonstrate the superiority of the 3D network in the hydrogel electrolyte, an in situ optical microscope test of the liquid electrolyte was conducted (Fig. S21, ESI†). While 1,2-PG partially inhibits zinc dendrite growth, its effectiveness is inferior to that of PAM-1,2-PG. This is due to the 3D porous structure of the hydrogel electrolyte, which promotes uniform ion transport and exerts mechanical stress on the zinc anode, facilitating the formation of a dense and smooth Zn2+ deposition layer. Furthermore, the 3D surface morphology of the zinc anode was observed after 100 cycles using a laser scanning confocal microscope (LSCM). In the presence of PAM, significant depressions and protrusions on the zinc surface were attributed to non-directional deposition and zinc anode corrosion, resulting in a height difference of approximately 10 μm (Fig. 4b). In contrast, the surface roughness of PAM-1,2-PG-induced zinc surfaces was considerably reduced, indicating uniform zinc deposition facilitated by PAM-1,2-PG (Fig. 4c). The morphology of zinc anodes cycled after 100 cycles at 1 mA cm−2 and 1 mA h cm−2 was subsequently investigated. The zinc anodes cycled with PAM exhibited a profusion of zinc dendrites (Fig. 4d), whereas those based on PAM-1,2-PG displayed a denser and smoother morphology (Fig. 4e), thus corroborating the role of PAM-1,2-PG in regulating uniform zinc deposition and inhibiting dendrite growth.
 | ||
| Fig. 4 Analysis of Zn deposition behavior. (a) In situ optical images of Zn deposition in Zn||Zn symmetric cells with PAM and PAM-1,2-PG. Laser scanning confocal microscopy images of Zn plated on (b) PAM and (c) PAM-1,2-PG after 100 cycles at 1 mA cm−2 and 1 mA h cm−2. SEM images of Zn morphologies in Zn||Zn symmetric cells with (d) PAM and (e) PAM-1,2-PG electrolytes under plating/stripping states after 100 cycles at 1 mA cm−2 and 1 mA h cm−2. The contour patterns of in situ XRD test of the Zn electrode obtained with (f) PAM and (g) PAM-1,2-PG (the green line represents the discharge curve, and the red line represents the charge curve). Schematic of the Zn deposition mechanism in (h) PAM and (i) PAM-1,2-PG hydrogel electrolytes. | ||
To elucidate the functional mechanism of PAM-1,2-PG in achieving uniform zinc deposition, in situ XRD characterization was performed (Fig. 4f and g). During the in situ XRD test, the battery was discharged at 1 mA cm−2 for 2 hours and then charged to an upper limit cut-off potential of 0.5 V. The observed peaks at 36.3°, 38.9°, and 43.2° corresponded to the (002), (100), and (101) crystal planes of zinc, respectively. These peaks gradually increased with the deposition of zinc, reaching their maximum value during the discharge process, and subsequently decreased as zinc was stripped during charging (Fig. S22, ESI†). As the number of cycles increased, the ratio of (002) crystal planes to (101) crystal planes (I(002/101)) slightly increased from 0.67 to 0.71. Conversely, with the PAM-1,2-PG electrolyte, there was an increasing trend in the selection of the (002) crystal plane for zinc deposition, and the ratio of I(002/101) significantly increased from 0.87 to 1.06. These results further confirm that in the PAM-1,2-PG-based system, Zn2+ prefers horizontal and continuous growth along the (002) plane, contributing to tight zinc deposition and dendrite reduction, thereby enhancing the cycling stability of AZIBs.
Based on the experimental findings and theoretical analysis presented above, the functional mechanisms of PAM and PAM-1,2-PG are schematically depicted in Fig. 4h and i. The introduction of 1,2-PG molecules alters the outer solvation sheath structure of hydrated Zn2+, promoting the rapid dissolution and adsorption of Zn2+ at the interface. Simultaneously, an organic/inorganic composite SEI forms at the electrode/electrolyte interface, with rich organic species on its outer surface and mainly inorganic species in its inner region. The internal dense inorganic layer possesses excellent ionic conductivity, while the external organic layer provides good flexibility, effectively preventing the inorganic layer from cracking.38,43 This unique SEI layer facilitates Zn2+ conduction, prevents direct contact between the electrolyte and the Zn electrode, inhibits dendrite growth, and reduces the occurrence of various side reactions, thus enhancing the performance of high-rate and long-cycling batteries.
The assessment of Zn plating/stripping reversibility in enhancing Zn electrochemistry is depicted in Fig. S23 (ESI†), where Zn||Cu asymmetrical cells are employed. Impressively, Zn||PAM-1,2-PG||Cu batteries consistently achieve a high average coulombic efficiency (CE) of 99.1% over an impressive span of more than 3700 cycles operated at 1 mA cm−2. In stark contrast, Zn||PAM||Cu batteries exhibit erratic performance during testing and rapidly deteriorate after just 300 cycles. This decline can be attributed to dendritic deposition and parasitic reactions. Even at 5 mA cm−2 and 5 mA h cm−2, the Zn||PAM-1,2-PG||Cu batteries could cycle stably for over 300 cycles, maintaining an average coulombic efficiency of 99.6% (Fig. S24, ESI†). Furthermore, the influence of Gly, 1,3-PG, and 1,2-PG on the physical and chemical properties of PAM directly impacts its electrochemical performance. The rate performance of Zn||Zn symmetric cells, spanning from 1 to 50 mA cm−2 at a fixed capacity of 1 mA h cm−2, is depicted in Fig. 5a and Fig. S25. Notably, cells featuring the PAM-1,2-PG hydrogel electrolyte display lower overpotentials than those featuring PAM, PAM-1,3-PG and PAM-Gly hydrogel electrolytes. These findings provide further confirmation of the enhanced reaction kinetics and superior reversibility of the PAM-1,2-PG electrolyte.
 | ||
| Fig. 5 The electrochemical performance of symmetrical cells. (a) Rate performance of Zn||Zn symmetric cells based on PAM and PAM-1,2-PG from 1 to 50 mA cm−2. (b) Cycling performance of Zn||Zn symmetric cells at 10 mA cm−2 and 1 mA h cm−2. (c) Electrochemical performance of Zn||Zn symmetric cells with the PAM-1,2-PG hydrogel electrolyte at 100 mA cm−2 and 50 mA h cm−2 with a DOD of 86%. (d) Performance comparison of PAM-1,2-PG with previous studies in terms of current densities and accumulative capacities. (e) Cycling stability of Zn||Zn symmetric cells at 1 mA cm−2 and 1 mA h cm−2 at −30 °C. | ||
To further explore the effectiveness of PAM-1,2-PG in guiding uniform Zn deposition, a long-term cycling analysis of Zn||Zn symmetric cells at a current density of 10 mA cm−2 and 1 mA h cm−2 is presented in Fig. 5b. Remarkably, Zn||PAM||Zn cells succumb to dendrite-induced internal short circuits after 390 hours, whereas Zn||PAM-1,2-PG||Zn cells exhibit a stable voltage profile and an extended lifespan exceeding 2160 hours. The significantly improved cycling performance of the PAM-1,2-PG-based battery is also observed at 1.0 mA cm−2 (Fig. S26, ESI†). Higher depths of discharge (DOD) were also investigated to assess applicability under stringent conditions. Benefiting from the regulation of the PAM-1,2-PG hydrogel electrolyte in the solvation structure and SEI, the symmetric cell maintains stable cycling performance for 490 hour under an extraordinarily high current density of 100 mA cm−2 (50 mA h cm−2, DOD of 86%), as shown in Fig. 5c. Notably, the symmetric cell achieves a stable lifespan of 260 hours even at an extremely high DOD of 95% (60 mA cm−2, 55 mA h cm−2) (Fig. S27, ESI†). These results confirm the rapid reaction kinetics, outstanding stability, and effective Zn deposition/stripping reversibility enabled by PAM-1,2-PG. Importantly, the superior high DOD value, high rate capability, and stable performance of PAM-1,2-PG is impressive among the reported symmetrical cells employing hydrogel electrolyte engineering, as illustrated in Fig. 5d and Fig. S28 (ESI†), and Table S1 (ESI†). Notably, even under extreme conditions of −30 °C, batteries utilizing PAM-1,2-PG retain stability for over 3780 hours at 1 mA cm−2 for 1 mA h cm−2 (Fig. 5e). Variations in polarization voltage observed in the symmetric batteries under various cycling conditions can be attributed to changes in testing temperature, current density, and areal capacity. Collectively, these electrochemical findings underscore the pivotal role played by 1,2-PG in facilitating rapid charge/discharge, anti-freezing capabilities, and dendrite inhibition within the hydrogel electrolyte.
To showcase the exceptional reversibility of Zn plating and stripping in full cells, we employed VO2·0.4H2O (VOH) nanosheets as cathodes in AZIBs, as illustrated in Fig. S29 (ESI†).44 XRD analysis confirmed the pure phase of VO2, while SEM images revealed its nanosheet morphology. CV curves, as depicted in Fig. 6a, exhibited two pairs of redox peaks positioned at 0.65/0.51 V and 0.93/0.91 V for Zn||VOH AZIBs utilizing PAM and PAM-1,2-PG as electrolytes. These peaks correspond to the redox reactions of V4+/V3+ driven by the (de)intercalation processes of Zn2+ and irreversible structural changes in the active materials.45,46 Notably, the enhanced current response observed in PAM-1,2-PG-based full cells signifies superior dynamic kinetics, redox reaction reversibility, and increased capacity.46–48
 | ||
| Fig. 6 Electrochemical performance and application of full cells. (a) CV curves of Zn||VOH AZIBs at 0.1 mV s−1. (b) Rate performance of Zn||VOH AZIBs with PAM and PAM-1,2-PG at room temperature. (c) The electrochemical performance of full zinc-ion batteries with the PAM-1,2-PG hydrogel electrolyte at 0.5 A g−1 under different temperatures. (d) Cycling performance of the AZIBs at a N/P ratio of 3.5. (e) Cycling performance of the AZIBs at −30 °C. (f) Cycling performance of the assembled pouch cell with different folding angles from 0° to 180°. (g) Specific capacity and capacity retention of the pouch cell after different number of bending cycles. (h) Cycling performance of the Zn||VOH pouch cell at 1 A g−1 with the PAM-1,2-PG hydrogel. Insets are an optical photograph of the pouch cell. (i) Optical photographs of pouch cells at different working states: cold, hammer, fold, and cut. (j) Demonstration of the flexible pouch battery providing power to a 4 V watch. | ||
Furthermore, we conducted a comparative analysis of the rate performance of Zn||VOH cells by employing different hydrogel electrolytes and varying temperatures to elucidate the structural advantages of PAM-1,2-PG. Significantly, Zn batteries equipped with PAM-1,2-PG consistently demonstrated an enhanced rate performance and higher specific capacities across all conditions. At 25 °C, when subjected to current densities of 0.5, 1, 3, 5, and 10 A g−1, the Zn batteries exhibited stable discharge capacities of 380, 350, 312, 291, and 261 mA h g−1, respectively (Fig. 6b). The advantage of PAM-1,2-PG-based AZIBs is further confirmed by their cycling performance at 1 A g−1 (Fig. S30, ESI†). Even at a low N/P ratio of 3.5, PAM-1,2-PG-based AZIBs demonstrated an initial discharge specific capacity of 323 mA h g−1 with a retention rate of 73.5% after 200 cycles at 1 A g−1 (Fig. 6d). Performance improvement was also evident at a low temperature of −30 °C (Fig. S31, ESI†). The capacities of full batteries using PAM-1,2-PG at 0.5 A g−1 across varying temperatures are shown in Fig. 6c. Notably, even at an extremely low temperature of −30 °C, a substantial capacity of 97 mA h g−1 was achieved, highlighting the impressive low-temperature tolerance of the PAM-1,2-PG hydrogel electrolyte. Similarly, cycling stability at −30 °C was further studied (Fig. 6e). It is worth noting that the performance of the Zn||VOH full cell based on PAM-1,2-PG remains stable after a sharp temperature change (marked by the black box). However, PAM-based AZIBs suffer from rapid battery failure under the same conditions. Thus, the robust stability conferred by PAM-1,2-PG with its enhanced Zn2+ transport ability, superior anti-freezing properties, and a stable hydrogel electrolyte/electrode interface demonstrate the immense potential of PAM-1,2-PG in low-temperature applications. In addition to AZIBs, we evaluated the viability of PAM and PAM-1,2-PG in AZICs, where devices were constructed using biomass-derived FSC as the cathode material.49 Characterization through XRD revealed a low degree of graphitization and amorphous structure, while SEM imaging showcased a typical 3D interconnected porous structure with irregular honeycomb-like morphology (Fig. S32, ESI†). CV curves at 0.1 V s−1 for PAM and PAM-1,2-PG-based AZICs displayed a characteristic rectangular shape, indicative of capacitive behavior with fast faradaic reactions (Fig. S33, ESI†). The superior rate performance of Zn||PAM-1,2-PG||FSC compared to Zn||PAM||FSC at room temperature and −30 °C can be attributed to rapid charge-transfer kinetics, efficient charge/ion transport, and a relatively low diffusion barrier at the hydrogel/electrode interface (Fig. S34, ESI†). Notably, the PAM-1,2-PG-based AZICs exhibited outstanding cyclability, with an ultralong cycle life of up to 65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles and a specific capacity of 212 F g−1 (Fig. S35, ESI†). Even at −30 °C, Zn||PAM-1,2-PG||FSC maintained a high-capacity retention rate over 16470 cycles, retaining a remarkable specific capacity of 145 F g−1 (Fig. S35, ESI†). This superior electrochemical performance of PAM-1,2-PG-based AZIBs and AZICs underscores the substantial advantages of PAM-1,2-PG in terms of capacity, power, and cycling stability across a wide temperature range, highlighting its immense industrial application potential.
000 cycles and a specific capacity of 212 F g−1 (Fig. S35, ESI†). Even at −30 °C, Zn||PAM-1,2-PG||FSC maintained a high-capacity retention rate over 16470 cycles, retaining a remarkable specific capacity of 145 F g−1 (Fig. S35, ESI†). This superior electrochemical performance of PAM-1,2-PG-based AZIBs and AZICs underscores the substantial advantages of PAM-1,2-PG in terms of capacity, power, and cycling stability across a wide temperature range, highlighting its immense industrial application potential.
Given the exceptional electrochemical performance demonstrated by the PAM-1,2-PG hydrogel electrolyte in enhancing the reversibility, high-rate capability, and temperature adaptability of zinc-ion batteries, pouch cells were prepared and various performance tests were performed, as depicted in Fig. S36 (ESI†). Remarkably, these pouch cells exhibited stable discharge and charge functionality even when subjected to varying bending angles ranging from 0 to 180°, as illustrated in Fig. 6f. Even at a bending angle of 180°, capacity retention after 60 cycles remained at an impressive 97%, as showcased in Fig. 6g. In the cycling performance test, the pouch cells were subjected to repeated discharging and charging at a rate of 1 A g−1 (activated at 500 mA g−1 for the initial ten cycles). These cells consistently delivered a high reversible specific capacity of 210 mA h g−1 and maintained stability with a capacity retention of 76.3% after 1000 cycles, as indicated in Fig. 6h. Owing to the shape-tuneable nature of the PAM-1,2-PG, the zinc-ion batteries could be folded into distinctive shapes such as triangles, hearts, and squares. When three cells were connected in series, they effortlessly powered “HZAU” lights, enduring extreme conditions including freezing, hammering, folding, and cutting (Fig. 6i and Video S1, ESI†). Furthermore, the series connection of multiple stacked soft-pack batteries provided a dependable power supply for a 4 V electronic watch, as shown in Fig. 6j (Fig. S37, ESI†, Video S2). This comprehensive set of results underscores the potential of the PAM-1,2-PG hydrogel electrolyte in empowering zinc-ion batteries as promising power sources for wearable electronic devices.
Conclusions
In summary, 1,2-PG, with its end-capping effect, is used as a cosolvent to successfully enhance the performance of hydrogel electrolytes for AZIBs. The incorporation of the 1,2-PG cosolvent enables precise manipulation of intermolecular interactions within the gel network, reduces free water activity, and weakens interactions between cosolvents. Additionally, various tests show that 1,2-PG alters the structure of the Zn2+ external solvation layer and promotes the formation of a solid electrolyte interphase with rich organic species on the outer surface and primarily inorganic species in the inner region, leading to rapid ion diffusion and (002) parallel Zn deposition. The application of this innovative hydrogel electrolyte significantly improves reaction kinetics, Zn plating/stripping reversibility, and rate performance in symmetrical Zn||Zn cells across a wide temperature range. With the assistance of PAM-1,2-PG, the battery achieves a stable lifetime of over 2160 hours at 10 mA cm−2 and over 490 hours at 100 mA cm−2 and 50 mA h cm−2 (DOD of 86%) at room temperature. It also demonstrates an impressive cycle life of 3780 hours at 1 mA cm−2 and 1 mA h cm−2 at −30 °C. Furthermore, zinc-ion batteries paired with VOH cathodes and zinc-ion capacitors coupled with FSC exhibit ultra-long cycle life and high capacity retention at room temperature and −30 °C. These advancements hold significant promise for the further development of advanced Zn-based rechargeable devices.Author contributions
F. C. and Y. L. supervised the research. Y. L. and S. G. conceived the project, analyzed the data, and wrote the manuscript. S. G., K. Y., Y. Y., and P. Z. conducted experiments and collected data. M. Y. conducted a synchrotron radiation test. S. G. and J. Z. analyzed the synchrotron radiation data. F. C., D. X., and P. H. gave helpful advice on manuscript preparation.Data availability
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was financially supported by the National Natural Science Foundation Regional Innovation and Development Joint Fund (U23A20684), National Natural Science Foundation of China (22122902), the Nature Science Foundation of Hubei Province (2023AFB845), the Open Fund of the Hubei Longzhong Laboratory (2022KF-03), the Wuhan Dawn Plan Project (2023010201020345), the Fundamental Research Funds for the Central Universities of China (2662023PY016). The in situ XRD cell of Zn batteries was purchased and technically supported by EVASTAR, Suzhou, China.Notes and references
- L. Wang, Y. Zhang and P. G. Bruce, Nat. Sci. Rev., 2023, 10, nwac062 CrossRef.
- X. Xiao, Z. Zheng, X. Zhong, R. Gao, Z. Piao, M. Jiao and G. Zhou, ACS Nano, 2023, 17, 1764 CrossRef CAS.
- G. Weng, X. Yang, Z. Wang, Y. Xu and R. Liu, Small, 2023, 19, 2303949 CrossRef CAS.
- Z. Zheng, S. Guo, M. Yan, Y. Luo and F. Cao, Adv. Mater., 2023, 35, 2304667 CrossRef CAS.
- H. Ge, X. Xie, X. Xie, B. Zhang, S. Li, S. Liang, B. Lu and J. Zhou, Energy Environ. Sci., 2024, 17, 3270–3306 RSC.
- S. Lei, Z. Liu, C. Liu, J. Li, B. Lu, S. Liang and J. Zhou, Energy Environ. Sci., 2022, 15, 4911–4927 RSC.
- L. Hu, P. Chee, S. Sugiarto, Y. Yu, C. Shi, R. Yan, Z. Yao, X. Shi, J. Zhi, D. Kai, H. Yu and W. Huang, Adv. Mater., 2022, 35, 2205326 CrossRef PubMed.
- D. Sheng, X. Liu, Z. Yang, M. Zhang, Y. Li, P. Ren, X. Yan, Z. X. Shen and D. Chao, Adv. Funct. Mater., 2024, 34, 2402014 CrossRef CAS.
- L. Han, Y. Guo, F. Ning, X. Liu, J. Yi, Q. Luo, B. Qu, J. Yue, Y. Lu and Q. Li, Adv. Mater., 2024, 36, 2308086 CrossRef CAS.
- T. Liu, X. Du, H. Wu, Y. Ren, J. Wang, H. Wang, Z. Chen, J. Zhao and G. Cui, Angew. Chem., Int. Ed., 2023, 62, e202311589 CrossRef CAS.
- Z. Hou and B. Zhang, EcoMat, 2022, 4, e12265 CrossRef CAS.
- Q. Ma, R. Gao, Y. Liu, H. Dou, Y. Zheng, T. Or, L. Yang, Q. Li, Q. Cu, R. Feng, Z. Zhang, Y. Nie, B. Ren, D. Luo, X. Wang, A. Yu and Z. Chen, Adv. Mater., 2022, 34, 2207344 CrossRef CAS.
- S. Huang, L. Hou, T. Li, Y. Jiao and P. Wu, Adv. Mater., 2022, 34, 2110140 CrossRef CAS.
- R. Wang, M. Yao, S. Huang, J. Tian and Z. Niu, Sci. China Mater., 2022, 65, 2189 CrossRef CAS.
- N. Chang, T. Li, R. Li, S. Wang, Y. Yin, H. Zhang and X. Li, Energy Environ. Sci., 2020, 13, 3527–3535 RSC.
- Y. Wang, Z. Cao, Z. Ma, G. Liu, H. Cheng, Y. Zou, L. Cavallo, Q. Li and J. Ming, ACS Energy Lett., 2023, 8, 1477–1484 CrossRef CAS.
- D. Wu, W. Zhang, H. Feng, Z. Zhang, X. Chen and P. Cui, ACS Appl. Energy Mater., 2022, 5, 12067–12077 CrossRef CAS.
- C. Cui, D. Han, H. Lu, Z. Li, K. Zhang, B. Zhang, X. Guo, R. Sun, X. Ye, J. Gao, Y. Liu, Y. Guo, R. Meng, C. Wei, L. Yin, F. Kang, Z. Weng and Q. Yang, Adv. Energy Mater., 2023, 13, 2301466 CrossRef CAS.
- D. Dong, J. Xie, Z. Liang and Y. Lu, ACS Energy Lett., 2021, 7, 123–130 CrossRef.
- H. Lu, J. Hu, L. Wang, J. Li, X. Ma, Z. Zhu, H. Li, Y. Zhao, Y. Li, J. Zhao and B. Xu, Adv. Funct. Mater., 2022, 32, 2112540 CrossRef CAS.
- N. Jonathan, J. Mol. Spectrosc., 1961, 6, 205–214 CrossRef CAS.
- Y. Qin, H. Li, C. Han, F. Mo and X. Wang, Adv. Mater., 2022, 34, 2207118 CrossRef CAS.
- Y. Xu, L. Xing, X. Cao, D. Li, Z. Men, Z. Li, S. Wang and C. Sun, Spectrochim. Acta, Part A, 2023, 284, 121825 CrossRef CAS PubMed.
- W. Zhang, F. Guo, H. Mi, Z. Wu, C. Ji, C. Yang and J. Qiu, Adv. Energy Mater., 2022, 12, 2202219 CrossRef CAS.
- H. Hoshino, S. Okada, H. Urakawa and K. Kajiwara, Polym. Bull., 1996, 37, 237–244 CrossRef CAS.
- T. Wei, Y. Peng, L. E. Mo, S. Chen, R. Ghadari, Z. Li and L. Hu, Sci. China Mater., 2021, 65, 1156–1164 CrossRef.
- H. Wang, W. Ye, B. Yin, K. Wang, M. S. Riaz, B. Xie, Y. Zhong and Y. Hu, Angew. Chem., Int. Ed., 2023, 135, e202218872 CrossRef.
- C. Li, Q. Li, Z. Wu, Y. Wang, R. Zhang, H. Cui, Y. Hou, J. Liu, Z. Huang and C. Zhi, Adv. Mater., 2024, 36, 2304878 CrossRef CAS PubMed.
- J. Kim, G. Zhang, M. Shi and Z. Suo, Science, 2021, 374, 212–216 CrossRef CAS.
- K. Zhao, G. Fan, J. Liu, F. Liu, J. Li, X. Zhou, Y. Ni, M. Yu, Y. Zhang, H. Su, Q. Liu and F. Cheng, J. Am. Chem. Soc., 2022, 144, 11129–11137 CrossRef CAS.
- M. Li, X. Wang, J. Hu, J. Zhu, C. Niu, H. Zhang, C. Li, B. Wu, C. Han and L. Mai, Angew. Chem., Int. Ed., 2023, 62, e202215552 CrossRef CAS.
- B. Liu, C. Wei, Z. Zhu, Y. Fang, Z. Bian, X. Lei, Y. Zhou, C. Tang, Y. Qian and G. Wang, Angew. Chem., Int. Ed., 2022, 61, e202212780 CrossRef CAS PubMed.
- C. Xie, S. Liu, H. Wu, Q. Zhang, C. Hu, Z. Yang, H. Li, Y. Tang and H. Wang, Sci. Bull., 2023, 68, 1531–1539 CrossRef CAS.
- Z. Hu, Z. Song, Z. Huang, S. Tao, B. Song, Z. Cao, X. Hu, J. Wu, F. Li, W. Deng, H. Hou, X. Ji and G. Zou, Angew. Chem., Int. Ed., 2023, 62, e202309601 CrossRef CAS.
- S. Zhan, Y. Guo, K. Wu, F. Ning, X. Liu, Y. Liu, Q. Li, J. Zhang, S. Lu and J. Yi, Chem. – Eur. J., 2024, 30, e202303211 CrossRef CAS.
- S. Chen, C. Peng, D. Xue, L. Ma and C. Zhi, Angew. Chem., Int. Ed., 2022, 134, e202212767 CrossRef.
- R. Huang, J. Zhang, W. Wang, X. Wu, X. Liao, T. Lu, Y. Li, J. Chen, S. Chen, Y. Qiao, Q. Zhao and H. Wang, Energy Environ. Sci., 2024, 17, 3179–3190 RSC.
- W. Xu, J. Li, X. Liao, L. Zhang, X. Zhang, C. Liu, K. Amine, K. Zhao and J. Lu, J. Am. Chem. Soc., 2023, 145, 22456 CrossRef CAS.
- R. Chen, W. Zhang, C. Guan, Y. Zhou, I. Gilmore, H. Tang, Z. Zhang, H. Dong, Y. Dai, Z. Du, X. Gao, W. Zong, Y. Xu, P. Jiang, J. Liu, F. Zhao, J. Li, X. Wang and G. He, Angew. Chem., Int. Ed., 2024, 63, e202401987 CrossRef CAS.
- F. Wang, J. Zhang, H. Lu, H. Zhu, Z. Chen, L. Wang, J. Yu, C. You, W. Li, J. Song, Z. Weng, C. Yang and Q. Yang, Nat. Commun., 2023, 14, 4211 CrossRef CAS.
- X. Yang, Z. Zhang, M. Wu, Z. Guo and Z. Zheng, Adv. Mater., 2023, 35, 2303550 CrossRef CAS PubMed.
- J. Feng, D. Ma, K. Ouyang, M. Yang, Y. Wang, J. Qiu, T. Chen, J. Zhao, B. Yong, Y. Xie, H. Mi, L. Sun, C. He and P. Zhang, Adv. Funct. Mater., 2022, 32, 2207909 CrossRef CAS.
- S. Qin, J. Zhang, M. Xu, P. Xu, J. Zou, J. Li, D. Luo, Y. Zhang, H. Dou and Z. Chen, Angew. Chem., Int. Ed., 2024, 63, e202410422 CAS.
- Y. Tao, D. Huang, H. Chen and Y. Luo, ACS Appl. Mater. Interfaces, 2021, 13, 16576–16584 CrossRef CAS.
- W. Liu, X. Liu, F. Ning, S. Subhan, Y. Liu, Q. Li, J. Zhang, S. Lu and J. Yi, J. Mater. Chem. A, 2024, 12, 11883 RSC.
- H. Tang, C. Zuo, F. Xiong, C. Pei, S. Tan, P. Luo, W. Yang, Q. An and L. Mai, Sci. China Mater., 2022, 65, 2197–2206 CrossRef CAS.
- P. He, M. Yan, X. Liao, Y. Luo, L. Mai and C. Nan, Energy Storage Mater., 2020, 29, 113–120 CrossRef.
- Y. Luo, X. Xu, Y. Zhang, Y. Pi, Y. Zhao, X. Tian, Q. An, Q. Wei and L. Mai, Adv. Energy Mater., 2014, 4, 1400107 CrossRef.
- P. Wang, H. Ye, Y. Yin, H. Chen, Y. Bian, Z. Wang, F. Cao and Y. Guo, Adv. Mater., 2019, 31, 1805134 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee02772h |
| This journal is © The Royal Society of Chemistry 2025 |