
Binary all-polymer solar cells with 19.30% efficiency enabled by bromodibenzothiophene-based solid additive†
Haisheng
Ma
a,
Jiali
Song
b,
Jiawei
Qiao
c,
Bingyu
Han
d,
Qianqian
Wang
 e,
Min Hun
Jee
f,
Laju
Bu
d,
Donghui
Wei
e,
Han Young
Woo
f,
Xiaotao
Hao
c and
Yanming
Sun
*ab
e,
Min Hun
Jee
f,
Laju
Bu
d,
Donghui
Wei
e,
Han Young
Woo
f,
Xiaotao
Hao
c and
Yanming
Sun
*ab
aHangzhou International Innovation Institute, Beihang University, Hangzhou 311115, P. R. China. E-mail: sunym@buaa.edu.cn
bSchool of Chemistry, Beihang University, Beijing 100191, P. R. China
cSchool of Physics State Key Laboratory of Crystal Materials, Shandong University, Jinan 250100, P. R. China
dSchool of Chemistry, Xi’an Jiaotong University, Xi’an, Shaanxi 710049, P. R. China
eCollege of Chemistry, Zhengzhou University, Zhengzhou, Henan 450001, P. R. China
fDepartment of Chemistry, College of Science, KU-KIST Graduate School of Converging Science and Technology, Korea University, Seoul 136-713, Republic of Korea
First published on 15th November 2024
Abstract
All-polymer solar cells (all-PSCs) are thought to be the most promising candidates for the practical application of organic solar cells (OSCs). However, the efficiencies of all-PSCs remain lower than those of small molecule acceptor (SMA)-based OSCs due to their unfavorable active-layer morphology. The complicated molecular interaction and aggregation behavior involved in all-polymer blends make it highly challenging to achieve optimal morphology. Herein, two volatile solid additives named dibenzothiophene (DBTP) and 4-bromodibenzothiophene (4-BDBTP) were developed to finely modulate the morphology of all-PSCs. We clarify that the subtle bromine substitution enables 4-BDBTP to form enhanced intermolecular interactions with the host material, which is beneficial to control the molecular aggregation and crystallization, thus facilitating the formation of more ordered molecular stacking and well-defined fibril networks in the all-polymer blend. As a result, the 4-BDBTP-treated-PM6:PY-DT all-PSC achieved a high efficiency of 19.30% (certified as 18.82%). Moreover, three other all-polymer systems validate the broad applicability of 4-BDBTP, and these devices all showed enhanced efficiencies. Our work demonstrates the promising role of solid additive in regulating molecular aggregation and packing in all-polymer blends, offering valuable insight into fabricating high-performance all-PSCs.
Broader contextRecently, all-polymer solar cells (all-PSCs) have achieved significant progress, with power conversion efficiencies (PCEs) surpassing 19%. However, their efficiency still lags behind those of small-molecule-based organic solar cells due to challenges in optimizing the active layer morphology. In this study, two volatile solid additives, dibenzothiophene (DBTP) and 4-bromodibenzothiophene (4-BDBTP), were developed to finely modulate the morphology of all-PSCs. Consequently, the 4-BDBTP-treated PM6:PY-DT all-PSC achieved a PCE of 19.30% (certified as 18.82%). Additionally, the applicability of 4-BDBTP has been validated in PM6:PY-IT, PM6:PY-C11 and PBDB-T:PY-DT systems, which all show improved efficiencies. Our findings highlight the promising role of solid additive in regulating molecular aggregation and packing in all-polymer blends, providing valuable insights for the fabrication of high-efficiency and stable all-PSCs. |
Introduction
Polymer solar cells (PSCs) are attracting great attention as a promising alternative energy technology, because of their low cost, light weight, structural tunability, and mechanical flexibility.1–6 Over the past few years, significant advancements in material innovation and device engineering have propelled PSCs with power conversion efficiencies (PCEs) over 20%.7–10 As one of the crucial branches of PSCs, all-polymer solar cells (all-PSCs), which incorporate semiconducting polymers as both electron donors and electron acceptors, have recently garnered increasing interest.11–15 In particular, due to their compelling advantages of excellent mechanical properties and superior stability, all-PSCs are considered to be the most attractive candidates for realizing the practical application of PSCs.16–19 Unfortunately, the PCEs of all-PSCs are still much lower than those of PSCs based on a small-molecule acceptor, primarily attributed to the non-optimal active-layer morphology induced by the complicated molecular interactions and aggregation in all-polymer blends, making it highly difficult to achieve optimal morphology with appropriate phase separation and desirable molecular ordering.20–28 Therefore, reasonable morphology control is crucial for further improving the photovoltaic performance of all-PSCs.Currently, morphology control of all-PSCs still depends on the utilization of liquid additives, such as 1,8-diiodooctane (DIO) and 1-chloronaphthalene (1-CN).29–34 Nevertheless, such liquid additives generally feature high boiling points, which are difficult to completely remove during the film formation process.35,36 The residual solvent additive will induce severe detrimental implications on device stability and producibility, which presents a significant obstacle to the industrial manufacturing and widespread application of all-PSCs.30,37–39 To eliminate these limitations, the development of highly volatile solid additives (VSAs) has been proposed as a promising and effective alternative. Studies have revealed that the VSAs can manipulate the molecular crystallization and aggregation via the strong intermolecular interactions, thus optimizing film morphology and resulting in improved device performance.40–42 Moreover, VSAs can also be easily removed during the film-forming procedure, leading to better device stability and reproducibility.43,44 Despite these advantages, the application of solid additives in all-PSCs has been comparatively limited, and the constitutive relationship between the solid-additive structure, nanoscale morphology and photovoltaic performance of all-PSCs requires further exploration.
In this study, two volatile solid additives, dibenzothiophene (DBTP) and 4-bromodibenzothiophene (4-BDBTP), were developed to improve the active-layer morphology of all-PSCs. It is revealed that the bromine substitution strategy enables 4-BDBTP to form enhanced intermolecular interactions with the host materials. Compared to DBTP, the utilization of 4-BDBTP significantly increases molecular stacking and further optimizes aggregation of both the donor and acceptor, which contributes to the formation of a well-defined fibre network in an all-polymer blend, leading to improved exciton dissociation, efficient charge transport, and reduced charge recombination. Remarkably, the 4-BDBTP-treated all-PSC achieved an outstanding efficiency of 19.30% (certified as 18.82%). This significant improvement underscores the potential of 4-BDBTP as an effective additive for precisely controlling the aggregation behavior of polymer molecules. Our study provides valuable insights into the design and application of solid additives for the optimization of morphology and enhancement of photovoltaic performance in all-PSCs, providing a robust pathway for advancing the commercial viability of all-PSCs.
Results and discussion
Fig. 1a and b present the chemical structures of PM6, PY-DT,45 DBTP and 4-BDBTP solid additives. DBTP and 4-BDBTP are white powders with melting points of ∼97 °C and ∼85 °C, respectively. They can both be easily sublimated under the heating conditions. Fig. 1c shows the UV-vis absorption spectra of PM6, PY-DT, DBTP and 4-BDBTP neat films. It is seen that both DBTP and 4-BDBTP have negligible absorption in the wavelength range of 350–900 nm. The absorption schematic of PM6 and PY-DT neat films treated under different conditions are shown in Fig. 1d and e, respectively. When the additives were introduced into the blend films, the characteristic absorption peaks of PM6 were red-shifted from ∼620 nm to ∼623 nm (DBTP) and ∼625 nm (4-BDBTP), and those of PY-DT were red-shifted from ∼800 nm to ∼802 nm (DBTP and 4-BDBTP). The same tendency is also observed in the absorption spectra of the blend films (Fig. S1, ESI†), which demonstrates that the introduction of such additives is beneficial for improving molecular aggregation of PM6 and PY-DT. The density flood theory (DFT) simulations were further performed to theoretically calculate the binding energies (ΔE) of the two solid additives with PM6 and PY-DT. As shown in Fig. S2 (ESI†), the ΔE values of 4-BDBTP with PM6 and PYDT are 28.97 and 17.79 kcal mol−1, which are higher than those of DBTP. These results indicate that the introduction of bromine atoms enables 4-BDBTP to exhibit enhanced intermolecular interactions with the host materials, which is more favorable to regulate the molecular aggregation and crystallization during the film formation process.42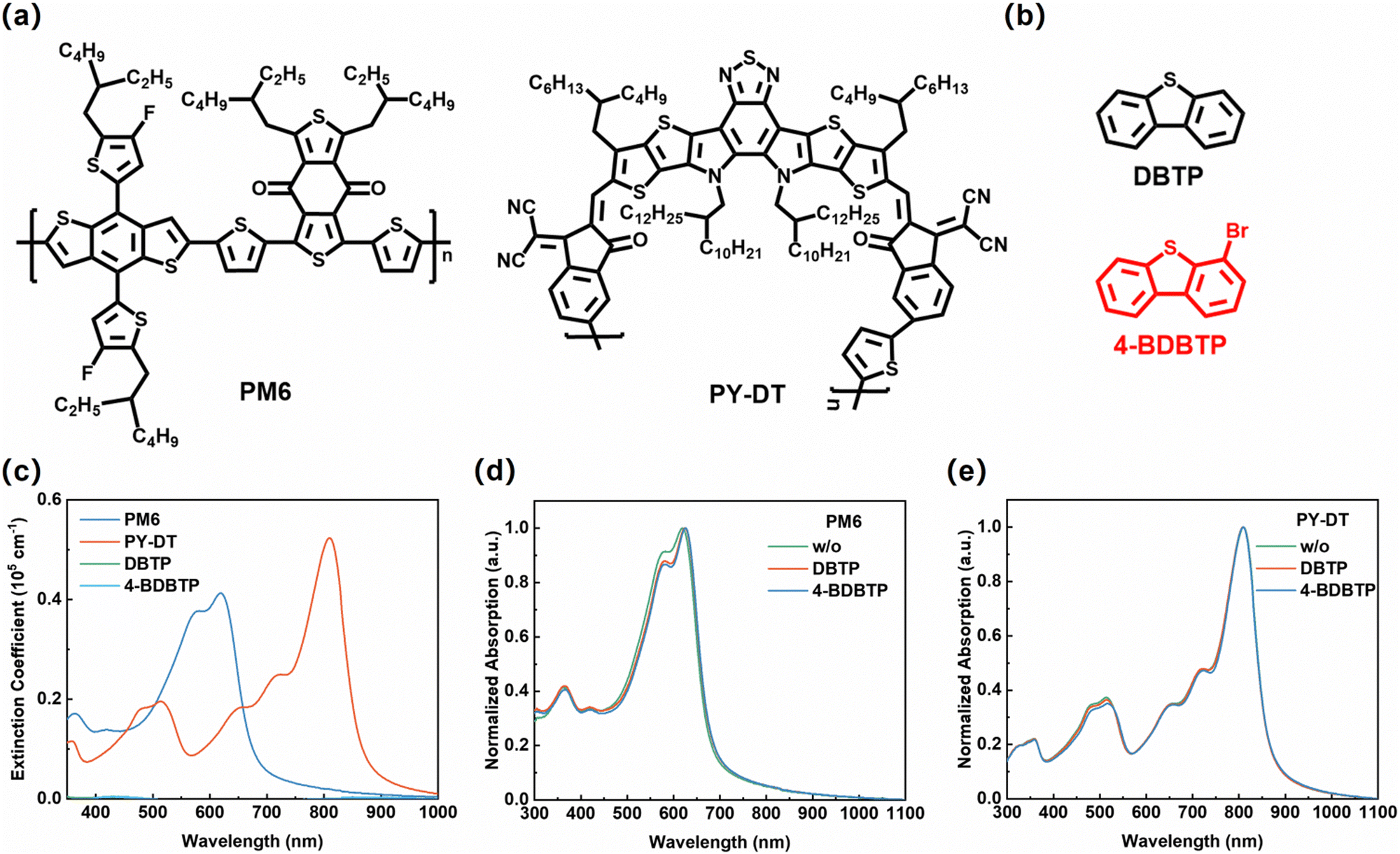 | ||
| Fig. 1 Chemical structures of (a) PM6 and PY-DT and (b) DBTP and 4-BDBTP. (c) The absorption coefficients of PM6, PY-DT, DBTP and 4-BDBTP neat films. Normalized absorption spectra of (d) PM6 and (e) PY-DT films with different treatments. | ||
Photovoltaic devices with the conventional structure of ITO/2PACz/PM6:PY-DT/PNDIT-F3N/Ag were fabricated to investigate the effect of DBTP and 4-BDBTP on the performance of all-PSCs. Table S1 (ESI†) summarizes the optimization process of all-PSCs with DBTP and 4-BDBTP. The current density–voltage (J–V) characteristics of all-PSCs fabricated under different conditions are shown in Fig. 2a. As presented in Table 1, the all-PSCs without any additives generated a PCE of 15.73%, with an open-circuit voltage (Voc) of 0.972 V, a short-circuit current (Jsc) of 24.56 mA cm−2, and a fill factor (FF) of 65.9%. When DBTP was added to the PM6:PY-DT blend, the PCE was improved to 18.36%, with a Voc of 0.952 V, a Jsc of 25.21 mA cm−2, and an FF of 76.5%. Incredibly, when employing 4-BDBTP as the solid additive, the PCE of all-PSCs was further improved to a high level of 19.30% (certified as 18.82%, Fig. S3, ESI†), with a Voc of 0.951 V, a Jsc of 25.85 mA cm−2, and a significantly increased FF of 78.5%. To the best of our knowledge, this PCE is among the highest efficiencies reported to date for binary all-PSCs (Fig. 2b and Table S2, ESI†).13,46–51 The external quantum efficiency (EQE) spectra of the studied all-PSCs are shown in Fig. 2c. It can be seen that the all-PSCs treated with 4-BDBTP produced significant enhancement in the EQE values at 400–850 nm compared to all-PSCs without additives, and the response values of all-PSCs treated with DBTP lies in the middle of them. The current densities of all-PSCs treated under different conditions were calculated to be 24.14 mA cm−2 (w/o), 24.67 mA cm−2 (DBTP), and 25.23 mA cm−2 (4-BDBTP), agreeing well with the results of the J–V measurements. Fig. 2d shows the efficiency-distributed box plots of all-PSCs for the 30 devices, which shows the good performance reproducibility of these devices. To verify the universality of the DBTP-series of additives in all-PSCs, three representative all-polymer systems, PM6:PY-IT, PM6:PY-C11 and PBDB-T:PY-DT, were further employed (Fig. S4, ESI†).52,53 The J–V and EQE spectra of the corresponding devices processed under different conditions are shown in Fig. S5 (ESI†). The detailed photovoltaic parameters are listed in Table S3 (ESI†). Consequently, the 4-BDBTP-treated devices all attained the highest PCEs compared to the additive-free, DBTP-treated devices. In detail, the PCEs of the 4-BDBTP-treated PM6:PY-IT, PM6:PY-C11 and PBDB-T:PY-DT devices reached 18.65%, 18.41% and 17.37%, respectively, and the EQE response of the corresponding devices were substantially improved in the wavelength range of 400–850 nm.
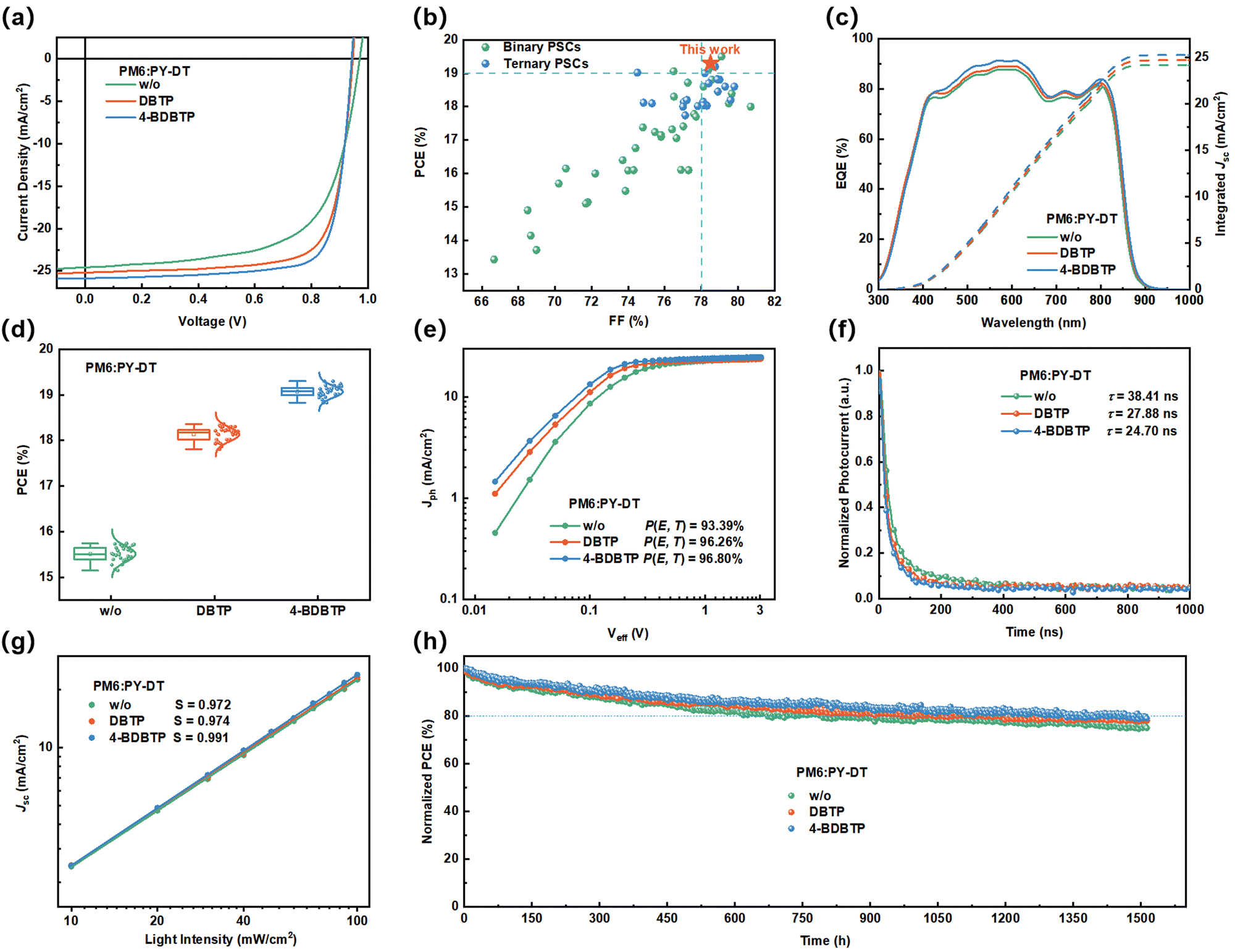 | ||
| Fig. 2 (a) J-V characteristics of all-PSCs processed with different treatments. (b) Plots of the PCE versus FF of the efficient all-PSCs reported in the literature. (c) EQE spectra (solid lines) and integrated current densities (dashed lines) of all-PSCs with different treatments. (d) Efficiency-distributed box plots of all-PSCs with different treatments. (e) Photocurrent density (Jph) versus effective bias (Veff) characteristics of all-PSCs with different treatments. (f) Transient photocurrent (TPC) decay kinetics of all-PSCs with different treatments. (g) Jscversus light intensity characteristics of all-PSCs with different treatments. (h) Normalized PCE degradation curves of all-PSCs processed with different conditions under continuous illumination by using MPP tracking mode. | ||
| Active layer | V oc (V) | J sc (mA cm−2) | FF (%) | PCEa (%) |
|---|---|---|---|---|
| a Average PCEs obtained from 30 devices. b Certified efficiency by the National Institute of Metrology (NIM), China. | ||||
| PM6:PY-DT (w/o) | 0.972 (0.970 ± 0.003) | 24.56 (24.31 ± 0.24) | 65.9 (65.3 ± 0.7) | 15.73 (15.48 ± 0.25) |
| PM6:PY-DT (DBTP) | 0.952 (0.953 ± 0.003) | 25.21 (24.98 ± 0.26) | 76.5 (75.8 ± 0.8) | 18.36 (18.16 ± 0.20) |
| PM6:PY-DT (4-BDBTP) | 0.951 (0.951 ± 0.002) | 25.85 (25.51 ± 0.36) | 78.5 (77.7 ± 0.9) | 19.30 (19.14 ± 0.16) |
| PM6:PY-DT (4-BDBTP)b | 0.946 | 25.47 | 78.1 | 18.82 |
To verify the reason for the 4-BDBTP-treated all-PSCs showing better photovoltaic performance, we measured the characteristics of the photocurrent density (Jph) versus the effective voltage (Veff) to probe the probability of charge dissociation (P(E, T)), defined as the ratio of Jsc/Jsat, where Jsat stands for the current saturation value of the all-PSCs.54 As shown in Fig. 2e, the P(E, T) of the all-PSCs without any additives, with DBTP and 4-BDBTP, are 93.39%, 96.26% and 96.80%, respectively. The higher P(E, T) values indicate more efficient charge dissociation and extraction in the 4-BDBTP-treated device. Besides, we further explored charge extraction in different devices through transient photocurrent (TPC) measurements under short-circuit conditions (Fig. 2f). We determined the charge extraction time (τext) by fitting a mono-exponential decay model. Compared with the all-PSC without any treatments, the τext of additive-treated all-PSCs was shortened from 38.41 ns to 27.88 ns (DBTP) and 24.70 ns (4-BDBTP), indicating that the introduction of solid additives effectively promotes the charge extraction after exciton dissociation, which creates the prerequisites for efficient charge transport.55 Additionally, the dependence of Voc on light intensity (Plight) under open-circuit conditions was determined to assess the charge recombination behavior of all-PSCs. In general, the trap-assisted recombination of the device is minimal when the slope is close to kT/q (where k is Boltzmann's constant, T is the temperature in Kelvin, and q is the elementary charge). As shown in Fig. S6 (ESI†), by linearly fitting the slopes of the Vocversus Plight curves, the slope of the 4-BDBTP-treated all-PSCs is determined to be 1.09kT/q, which is smaller than those of the all-PSCs treated without any additives (1.14kT/q) and DBTP (1.10kT/q). These results suggest that trap-assisted recombination in all-PSCs can be suppressed by adding 4-BDBTP to the all-PSCs.56 To further measure the charge recombination behavior of all-PSCs, we tested the dependence of Jsc on the light intensity (Plight). In general, there is an exponential relationship between Jsc and Plight, i.e., Jsc ∝ (Plight)S, where S is the exponential factor, and the closer S is to 1, the lower the degree of bimolecular recombination of the device. As shown in Fig. 2g, the S value of the 4-BDBTP-treated device (0.991) is higher than that of the comparison samples (0.972 and 0.974), which proves that the bimolecular recombination of the device containing 4-BDBTP is suppressed.57
Additionally, the space-charge-limited current (SCLC) method was applied to investigate the charge transport properties of all-PSCs. As shown in Fig. S7 and Table S4 (ESI†), the hole mobility (μh) and electron mobility (μe) of the PM6:PY-DT blend without any additives were 3.87 × 10−4 cm2 V−1 s−1 and 6.09 × 10−4 cm2 V−1 s−1, with a μe/μh ratio of 1.57. For the DBTP-treated blend, μh and μe increased to 4.70 × 10−4 cm2 V−1 s−1 and 7.26 × 10−4 cm2 V−1 s−1, respectively, with a μe/μh ratio of 1.55. When 4-BDBTP was introduced into the blend, μh and μe were further increased to 6.23 × 10−4 cm2 V−1 s−1 and 8.06 × 10−4 cm2 V−1 s−1, respectively, with a μe/μh ratio of 1.29. The increased and more balanced charge mobility well explains the higher Jsc and FF achieved in the 4-BDBTP-treated all-PSC.
Furthermore, we tested the photostability of all-PSCs under continuous LED illumination (one solar intensity) by using the maximum power point (MPP) tracking mode. As shown in Fig. 2h, the T80 (the time required to reach 80% of its initial efficiency) of the devices treated without any additives, with DBTP and 4-BDBTP, occurred at 673, 970, and 1315 h, respectively. The thermal stability of the devices shows a similar trend (Fig. S8, ESI†), and the 4-BDBTP-treated devices exhibited the best thermal stability, still retaining 80% after continuous heating at 70 °C for 350 h, superior to devices without additives and with DBTP treatment.
In order to reveal the effect of 4-BDBTP on the morphological characteristics of all-PSCs, we carried out atomic force microscopy (AFM) measurements on PM6:PY-DT blend films with different treatments. The height images of the hybrid films are shown in Fig. 3a, and the root mean square (RMS) roughness of the additive-free, DBTP-treated, and 4-BDBTP-treated PM6:PY-DT blends are 0.96, 1.08, and 1.11 nm, respectively. Furthermore, the corresponding phase images of the different blends are shown in Fig. 3b. It can be seen that all the blends formed a clear fibril structure, which is favorable for charge transport. Compared to the additive-free and DBTP-treated blends, the 4-BDBTP-treated blend exhibits a more visible and coarser fibril network. In addition, to quantify the fibril size of different blends, we analyzed the line-cut profiles of the AFM phase images. As shown in Fig. 3d and Fig. S9 (ESI†), the average fibril widths of the blends without any additives, with DBTP and 4-BDBTP, are 9.1, 13.0, and 20.7 nm, respectively, indicating that the 4-BDBTP-treated blend has a larger fibril size.
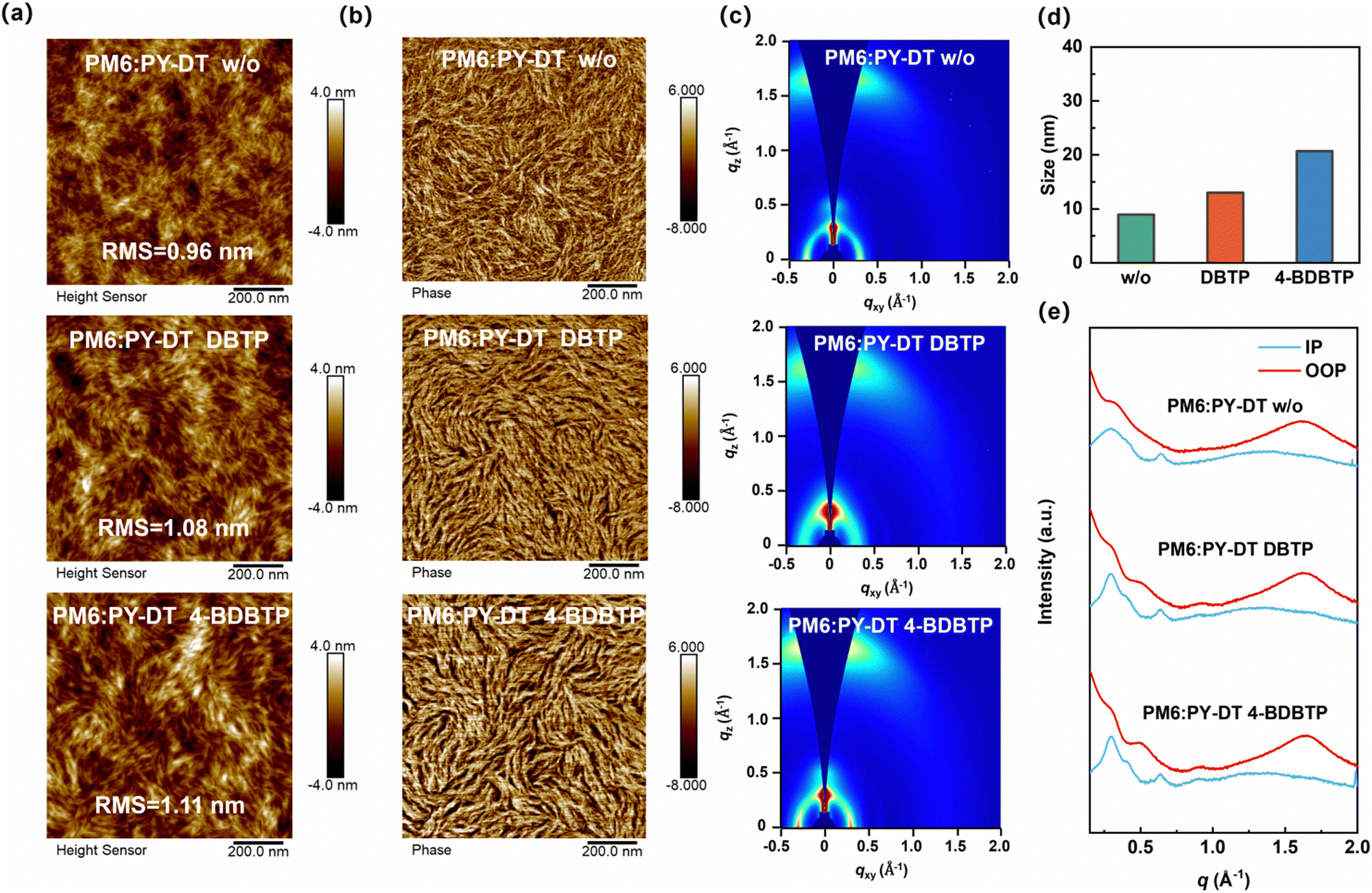 | ||
| Fig. 3 AFM (a) height images and (b) phase images of PM6:PY-DT blends processed under different conditions. (c) 2D GIWAXS patterns and (e) the corresponding in-plane and out-of-plane line-cuts of PM6:PY-DT blends processed under different conditions. (d) Histogram of fibre size based on line-cut profiles of AFM phase images for different PM6:PY-DT blends. | ||
The molecular stacking and crystallinity of the PM6:PY-DT blends were investigated by using grazing incidence wide-angle X-ray scattering (GIWAXS) measurements. The 2D GIWAXS plots of the PM6:PY-DT blends under different conditions are shown in Fig. 3c. The corresponding in-plane (IP) and out-of-plane (OOP) line-cuts are shown in Fig. 3e. The detailed parameters are listed in Table S5 (ESI†). Obviously, the additive-free blend has distinct (100) lamellar stacking peaks in the IP direction and (010) π–π stacking peak in the OOP direction. When 4-BDBTP was introduced into the blend, the diffraction intensities of both the laminar diffraction peaks and π–π stacking peaks were significantly enhanced, and the corresponding CCL values were increased from 47.39/18.33 Å to 88.76/24.10 Å, respectively. Meanwhile, the d-spacing distance obtained from π–π stacking peaks decreased from 3.91 Å to 3.86 Å. The same trend can be observed in the plots and parameters of PM6 and PY-DT neat films (Fig. S10, S11 and Table S5, ESI†). The above results show that the incorporation of 4-BDBTP into PM6:PY-DT results in more compact π–π stacking of the conjugated backbone and delicate regulation of the molecular aggregation behavior. At the same time, both PM6 and PY-DT maintain a good crystallinity in the blend, which creates an efficient pathway for charge transport and facilitates the devices achieving elevated efficiency.
To explore whether 4-BDBTP was completely removed from the active layer after thermal annealing, FT-IR measurements were performed. From the FT-IR spectra (Fig. S12, ESI†), it can be seen that DBTP and 4-BDBTP have three common characteristic peaks at 742, 1027 and 1308 cm−1, respectively. These characteristic peaks were still observed when DBTP and 4-BDBTP were added to the PM6:PY-DT blend. After thermal annealing, these characteristic peaks disappeared, indicating that DBTP and 4-BDBTP were completely removed from the blends. In order to verify the different volatility of the DBTP-series of additives, the same mass of DBTP and 4-BDBTP was spin-coated onto the silicon substrate, and then heated at 80 °C (Fig. S13, ESI†). It can be seen that DBTP was completely volatilized after three minutes, with little 4-BDBTP remaining; after another two minutes, 4-BDBTP was also completely volatilized, revealing that DBTP is easier to volatilize compared to 4-BDBTP.
In situ UV-Vis absorption measurements were utilized to investigate the mechanism behind the active layer morphology formation during the spin-coating and annealing process. Generally, the continuous volatilization of the additive molecules creates more free space for the aggregation of the acceptor molecules, resulting in a red-shift of the acceptor absorption peaks.41 As shown in Fig. 4 and Fig. S14 (ESI†), the primary absorption peaks of the blend without additives are located at 619 nm and 800 nm, belonging to PM6 and PY-DT, respectively. Before thermal annealing, the characteristic absorption peaks of PM6 and PY-DT in the 4-BDBTP-treated blend are located at 624 nm and 790 nm, respectively, demonstrating that 4-BDBTP may enhance the aggregation of PM6 molecules while inhibiting the aggregation of PY-DT. A similar conclusion could be obtained for the DBTP-treated blend. During the thermal process, the PY-DT absorption peak in the DBTP-treated blend progressively red-shifts from 795 nm to 802 nm, while for the 4-BDBTP-treated blend, the PY-DT absorption peak significantly red-shifts from 790 nm to 803 nm, revealing the aggregation process of PY-DT. Notably, the aggregation duration of PY-DT in the 4-BDBTP-treated blend is 120 s, much longer than that of 50 s in the DBTP-treated blend. The longer aggregation time may contribute to a more ordered stacking and arrangement of PY-DT. In summary, we can reasonably speculate the possible working mechanism of the two additives. During the film deposition process, the additives promote the aggregation of PM6, resulting in the formation of an organized fibril network where the donor serves as the structural framework (Fig. 4g). The additive molecules and PY-DT molecules are interspersed within this fibrous network. During the annealing process, the additive molecules start to volatilize, which can generate more space to promote the π–π stacking and crystalline growth of PY-DT, and eventually achieving a well-organized fiber network structure (Fig. 4h and i). This prediction aligns well with the AFM and GIWAXS data of 4-BDBTP-treated neat films before and after the thermal annealing process (Fig. S15, S16 and Table S6, ESI†).
 | ||
| Fig. 4 (a)–(c) In situ 2D UV-visible absorption of the PM6:PY-DT blend treated under different conditions during the thermal annealing process. (d)–(f) The time evolution of the peak location of PM6 and PY-DT in the blend treated under different conditions during the thermal annealing process. (g)–(i) Schematic diagram of the working mechanism of 4-BDBTP during film deposition and the thermal annealing process. | ||
Furthermore, transient absorption spectroscopy (TAS) measurements were carried out to further investigate the effect of additives on the charge-dynamic process of the blends. Fig. S15 (ESI†) exhibits the 2D transient absorption (TA) spectra of the PY-DT neat film. All the blend films were pumped at a low energy level flux of 8 μJ cm−2 to avoid exciton–exciton annihilation in the film. Evidently, the ground-state bleaching (GSB) signal of PY-DT is located between 500 and 820 nm, with the most distinct GSB signal peak at 805 nm. Thus, we investigated the hole transfer dynamics of the blend with a pump light centered at 800 nm. Fig. 5a–c show the 2D TA spectra of PM6:PY-DT blends treated under different conditions. The characteristic PM6 GSB signal can be detected in the wavelength range of 610–650 nm (Fig. 5e), indicating the occurrence of hole transfer from the polymer acceptor to the polymer donor in the blend. The decay properties of different blends were estimated by probing the GSB signal of the acceptor at 805 nm (Fig. 5d). As shown in Fig. 5f, by fitting the dynamic curves with a biexponential function, the carrier lifetimes obtained are divided into τ1 and τ2, where τ1 is related to the fast dissociation of the exciton at the donor–acceptor interface, and τ2 is related to the diffusion of the exciton in the donor–acceptor phase. Detailed fitting information is shown in Table S6 (ESI†). The τ1 and τ2 values of the PM6:PY-DT blends treated with 4-BDBTP are 0.51 ps and 7.60 ps, respectively, which are higher than those of 0.66 ps and 13.29 ps in the DBTP-treated blends. In contrast, the τ1 and τ2 values of the blends without any additives reached 1.54 ps and 16.37 ps, respectively, demonstrating that the introduction of 4-BDBTP facilitates efficient charge transfer on the donor–acceptor interfaces.
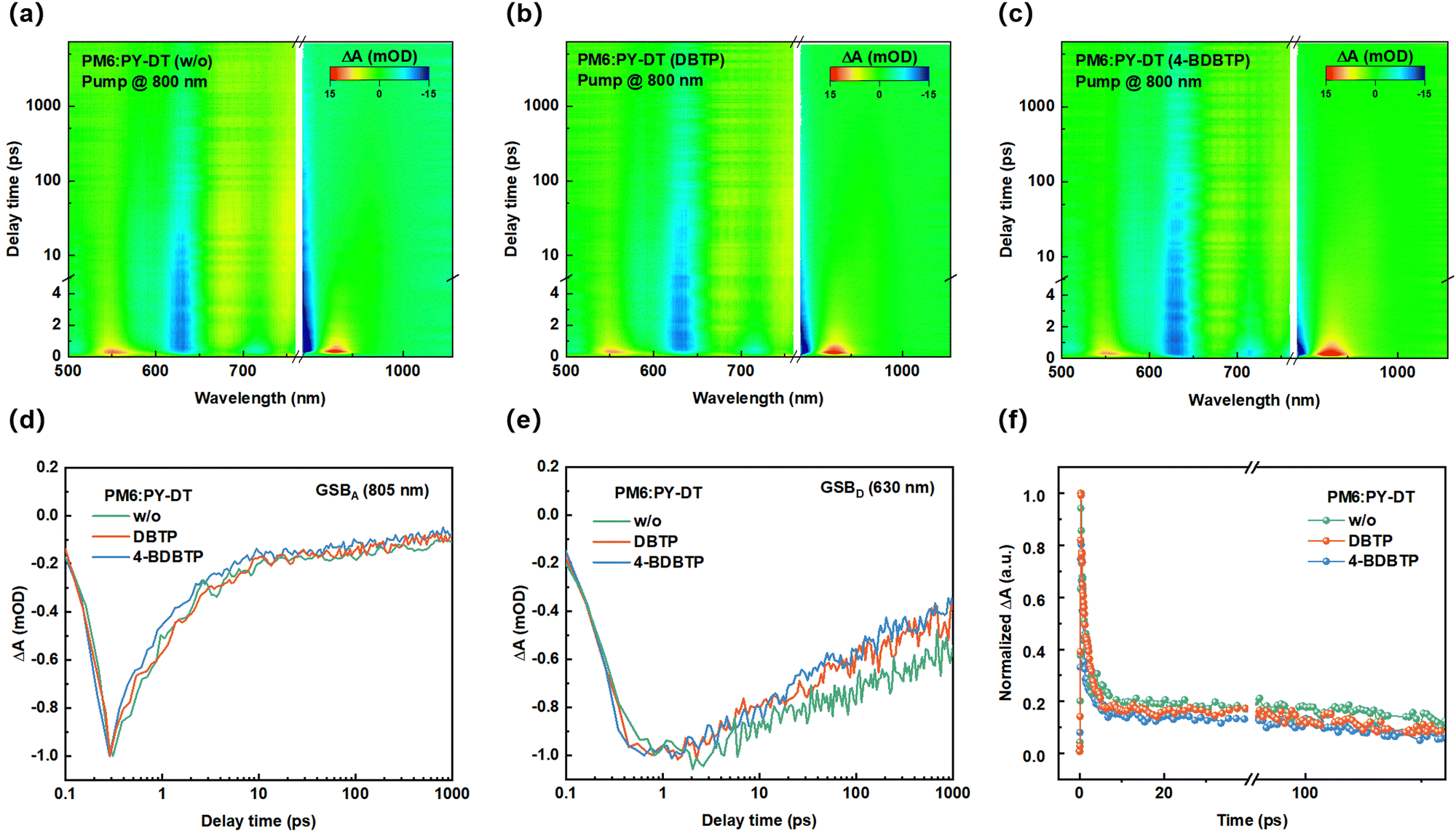 | ||
| Fig. 5 (a)–(c) 2D TAS images of the PM6:PY-DT blends treated under different conditions with the pump light at 800 nm. (d) PY-DT GSB and (e) PM6 GSB kinetic curves of PM6:PY-DT blends treated under different conditions. (f) Normalized TA kinetics probed at PY-DT GSB of PM6:PY-DT blends treated under different conditions. | ||
To explore the potential of 4-BDBTP for large-scale production, we fabricated large-area, non-halogenated solvent and thick-film devices, respectively. As shown in Fig. S18 and Table S8 (ESI†), the 1 cm2 devices of PM6:PY-DT treated with 4-BDBTP achieved a PCE of 17.27%, with a Voc of 0.968 V, a Jsc of 24.85 mA cm−2, and an FF of 71.8%. In addition, non-halogenated solvent is one of the key participants in green fabrication. Therefore, we prepared all-PSC devices with o-xylene (o-XY) as the active layer solvent. As shown in Fig. S19 and Table S9 (ESI†), the o-XY-processed PM6:PY-DT device with 4-BDBTP achieves a high PCE of 19.07%, with a Voc of 0.953 V, a Jsc of 25.66 mA cm−2, and an FF of 78.0%. Furthermore, the development of all-PSCs with thick active layers is essential for roll-to-roll printing of large-area solar cells. As shown in Fig. S20 and Table S10 (ESI†), the 4-BDBTP-treated PM6:PY-DT device with a thickness of 300 nm yielded a PCE of 17.46%, with a Voc of 0.941 V, a Jsc of 26.59 mA cm−2, and an FF of 69.8%. In conclusion, these results suggest 4-BDBTP shows promising potential for large-scale and green-process manufacture, which is expected to facilitate the practical application of all-PSCs.
Conclusions
In conclusion, we developed a novel solid additive to boost the efficiency and stability of all-PSCs. The addition of volatile 4-BDBTP to PM6:PY-DT can well regulate the aggregation of PM6 and PY-DT during film deposition and thermal annealing processes, which leads to the formation of well-defined fibril network morphology with appropriate phase separation scales, thus leading to effective enhancement of exciton dissociation and charge transport, and reduction of charge recombination in the devices. As a result, the PM6:PY-DT binary all-PSC achieved a high PCE of 19.30% (certified as 18.82%). In addition, higher PCE values were also achieved in PM6:PY-IT, PM6:PY-C11 and PBDB-T:PY-DT blends when employing 4-BDBTP as the solid additive, revealing the excellent universality of 4-BDBTP in enhancing the photovoltaic performance of all-PSCs. Our work highlights the importance of solid additives in modulating the morphology of all-PSCs, contributing to the further development of the photovoltaic performance and stability of all-PSCs in the future.Author contributions
Y. S. directed this work. H. M. fabricated the devices and performed the device characterization. J. Q. and X. T. performed the TAS measurement. B. H. performed the in situ UV-Vis absorption measurement. Q. W. and D. W. conducted the DFT theoretical calculations. H. Y. W. and M. H. J. performed GIWAX measurement. H. M., J. S. and Y. S. wrote the manuscript. All authors commented on the manuscript.Data availablility
The data are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 52333005), and the Beijing Natural Science Foundation (Z230018).References
- E. Zhou, J. Cong, Q. Wei, K. Tajima, C. Yang and K. Hashimoto, Angew. Chem., Int. Ed., 2011, 50, 2799–2803 CrossRef CAS.
- H. Kang, G. Kim, J. Kim, S. Kwon, H. Kim and K. Lee, Adv. Mater., 2016, 28, 7821–7861 CrossRef CAS.
- G. Wang, F. S. Melkonyan, A. Facchetti and T. J. Marks, Angew. Chem., Int. Ed., 2019, 58, 4129–4142 CrossRef CAS.
- C. Cui and Y. Li, Energy Environ. Sci., 2019, 12, 3225–3246 RSC.
- J. Song, L. Ye, C. Liu, Y. Cai, C. Zhang, G. Yue, Y. Li, M. H. Jee, Y. Zhao, D. Wei, H. Y. Woo and Y. Sun, Energy Environ. Sci., 2023, 16, 5371–5380 RSC.
- K. Zhou, K. Xian, R. Ma, J. Liu, M. Gao, S. Li, T. Liu, Y. Chen, Y. Geng and L. Ye, Energy Environ. Sci., 2023, 16, 5052–5064 RSC.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- J. Song, C. Zhang, C. Li, J. Qiao, J. Yu, J. Gao, X. Wang, X. Hao, Z. Tang, G. Lu, R. Yang, H. Yan and Y. Sun, Angew. Chem., Int. Ed., 2024, 63, e202404297 CrossRef CAS PubMed.
- S. Guan, Y. Li, C. Xu, N. Yin, C. Xu, C. Wang, M. Wang, Y. Xu, Q. Chen, D. Wang, L. Zuo and H. Chen, Adv. Mater., 2024, 36, 2400342 CrossRef CAS PubMed.
- H. Lu, D. Li, W. Liu, G. Ran, H. Wu, N. Wei, Z. Tang, Y. Liu, W. Zhang and Z. Bo, Angew. Chem., Int. Ed., 2024, 63, e202407007 CrossRef CAS.
- T. Kim, J.-H. Kim, T. E. Kang, C. Lee, H. Kang, M. Shin, C. Wang, B. Ma, U. Jeong, T.-S. Kim and B. J. Kim, Nat. Commun., 2015, 6, 8547 CrossRef CAS PubMed.
- H. Fu, Y. Li, J. Yu, Z. Wu, Q. Fan, F. Lin, H. Y. Woo, F. Gao, Z. Zhu and A. K. Y. Jen, J. Am. Chem. Soc., 2021, 143, 2665–2670 CrossRef CAS PubMed.
- B. Liu, W. Xu, R. Ma, J.-W. Lee, T. A. Dela Peña, W. Yang, B. Li, M. Li, J. Wu, Y. Wang, C. Zhang, J. Yang, J. Wang, S. Ning, Z. Wang, J. Li, H. Wang, G. Li, B. J. Kim, L. Niu, X. Guo and H. Sun, Adv. Mater., 2023, 35, 2308334 CrossRef CAS PubMed.
- Y. Xu, J. Wang, T. Zhang, Z. Chen, K. Xian, Z. Li, Y.-H. Luo, L. Ye, X. Hao, H. Yao and J. Hou, Energy Environ. Sci., 2023, 16, 5863–5870 RSC.
- J. Zhang, Q. Huang, K. Zhang, T. Jia, J. Jing, Y. Chen, Y. Li, Y. Chen, X. Lu, H. Wu, F. Huang and Y. Cao, Energy Environ. Sci., 2022, 15, 4561–4571 RSC.
- J. Choi, W. Kim, S. Kim, T.-S. Kim and B. J. Kim, Chem. Mater., 2019, 31, 9057–9069 CrossRef CAS.
- B. Lin, L. Zhang, H. Zhao, X. Xu, K. Zhou, S. Zhang, L. Gou, B. Fan, L. Zhang, H. Yan, X. Gu, L. Ying, F. Huang, Y. Cao and W. Ma, Nano Energy, 2019, 59, 277–284 CrossRef CAS.
- H. Yu, Y. Wang, X. Zou, J. Yin, X. Shi, Y. Li, H. Zhao, L. Wang, H. M. Ng, B. Zou, X. Lu, K. S. Wong, W. Ma, Z. Zhu, H. Yan and S. Chen, Nat. Commun., 2023, 14, 2323 CrossRef CAS PubMed.
- H. Fu, Z. Peng, Q. Fan, F. R. Lin, F. Qi, Y. Ran, Z. Wu, B. Fan, K. Jiang, H. Y. Woo, G. Lu, H. Ade and A. K.-Y. Jen, Adv. Mater., 2022, 34, 2202608 CrossRef CAS.
- Q. Zhang, Z. Chen, W. Ma, Z. Xie and Y. Han, J. Mater. Chem. C, 2019, 7, 12560–12571 RSC.
- C. R. McNeill, Energy Environ. Sci., 2012, 5, 5653–5667 RSC.
- Q. Wu, W. Wang, Y. Wu, Z. Chen, J. Guo, R. Sun, J. Guo, Y. Yang and J. Min, Adv. Funct. Mater., 2021, 31, 2010411 CrossRef CAS.
- K. Zhou, K. Xian and L. Ye, InfoMat, 2022, 4, e12270 CrossRef CAS.
- J. Song, C. Li, J. Qiao, C. Liu, Y. Cai, Y. Li, J. Gao, M. H. Jee, X. Hao, H. Y. Woo, Z. Tang, H. Yan and Y. Sun, Matter, 2023, 6, 1542–1554 CrossRef CAS.
- Q. Fan, Q. An, Y. Lin, Y. Xia, Q. Li, M. Zhang, W. Su, W. Peng, C. Zhang, F. Liu, L. Hou, W. Zhu, D. Yu, M. Xiao, E. Moons, F. Zhang, T. D. Anthopoulos, O. Inganäs and E. Wang, Energy Environ. Sci., 2020, 13, 5017–5027 RSC.
- Y. Wu, S. Schneider, C. Walter, A. H. Chowdhury, B. Bahrami, H.-C. Wu, Q. Qiao, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2020, 142, 392–406 CrossRef CAS PubMed.
- T. Zhang, Y. Xu, H. Yao, J. Zhang, P. Bi, Z. Chen, J. Wang, Y. Cui, L. Ma and K. Xian, Energy Environ. Sci., 2023, 16, 1581–1589 RSC.
- J. Wang, C. Han, S. Wen, F. Bi, Z. Hu, Y. Li, C. Yang, X. Bao and J. Chu, Energy Environ. Sci., 2023, 16, 2327–2337 RSC.
- J. K. Lee, W. L. Ma, C. J. Brabec, J. Yuen, J. S. Moon, J. Y. Kim, K. Lee, G. C. Bazan and A. J. Heeger, J. Am. Chem. Soc., 2008, 130, 3619–3623 CrossRef CAS.
- B. J. Tremolet de Villers, K. A. O’Hara, D. P. Ostrowski, P. H. Biddle, S. E. Shaheen, M. L. Chabinyc, D. C. Olson and N. Kopidakis, Chem. Mater., 2016, 28, 876–884 CrossRef CAS.
- A. Classen, T. Heumueller, I. Wabra, J. Gerner, Y. He, L. Einsiedler, N. Li, G. J. Matt, A. Osvet, X. Du, A. Hirsch and C. J. Brabec, Adv. Energy Mater., 2019, 9, 1902124 CrossRef CAS.
- C. V. Hoven, X.-D. Dang, R. C. Coffin, J. Peet, T.-Q. Nguyen and G. C. Bazan, Adv. Mater., 2010, 22, E63–E66 CrossRef CAS.
- R. Ma, Q. Fan, T. A. Dela Peña, B. Wu, H. Liu, Q. Wu, Q. Wei, J. Wu, X. Lu, M. Li, W. Ma and G. Li, Adv. Mater., 2023, 35, 2212275 CrossRef CAS.
- L. Chen, R. Ma, J. Yi, T. A. Dela Peña, H. Li, Q. Wei, C. Yan, J. Wu, M. Li, P. Cheng, H. Yan, G. Zhang and G. Li, Aggregate, 2024, 5, e455 CrossRef CAS.
- M. Dong, S. Chen, L. Hong, J. Jing, Y. Bai, Y. Liang, C. Zhu, T. Shi, W. Zhong, L. Ying, K. Zhang and F. Huang, Nano Energy, 2024, 119, 109097 CrossRef CAS.
- L. Ye, Y. Cai, C. Li, L. Zhu, J. Xu, K. Weng, K. Zhang, M. Huang, M. Zeng, T. Li, E. Zhou, S. Tan, X. Hao, Y. Yi, F. Liu, Z. Wang, X. Zhan and Y. Sun, Energy Environ. Sci., 2020, 13, 5117–5125 RSC.
- C. McDowell, M. Abdelsamie, M. F. Toney and G. C. Bazan, Adv. Mater., 2018, 30, 1707114 CrossRef.
- J. Fu, H. Chen, P. Huang, Q. Yu, H. Tang, S. Chen, S. Jung, K. Sun, C. Yang, S. Lu, Z. Kan, Z. Xiao and G. Li, Nano Energy, 2021, 84, 105862 CrossRef CAS.
- Y. Xie, H. S. Ryu, L. Han, Y. Cai, X. Duan, D. Wei, H. Y. Woo and Y. Sun, Sci. China: Chem., 2021, 64, 2161–2168 CrossRef CAS.
- X. Yang, B. Li, X. Zhang, S. Li, Q. Zhang, L. Yuan, D.-H. Ko, W. Ma and J. Yuan, Adv. Mater., 2023, 35, 2301604 CrossRef CAS.
- J. Song, Y. Li, Y. Cai, R. Zhang, S. Wang, J. Xin, L. Han, D. Wei, W. Ma and F. Gao, Matter, 2022, 5, 4047–4059 CrossRef CAS.
- L. Tu, H. Wang, W. Duan, R. Ma, T. Jia, T. A. Dela Peña, Y. Luo, J. Wu, M. Li, X. Xia, S. Wu, K. Chen, Y. Wu, Y. Huang, K. Yang, G. Li and Y. Shi, Energy Environ. Sci., 2024, 17, 3365–3374 RSC.
- J. Wang, Y. Wang, P. Bi, Z. Chen, J. Qiao, J. Li, W. Wang, Z. Zheng, S. Zhang, X. Hao and J. Hou, Adv. Mater., 2023, 35, 2301583 CrossRef CAS PubMed.
- H. Kang, Y. Jing, Y. Zhang, Y. Li, H. Zhang, H. Zhou and Y. Zhang, Sol. RRL, 2023, 7, 2201084 CrossRef CAS.
- Y. Li, J. Song, Y. Dong, H. Jin, J. Xin, S. Wang, Y. Cai, L. Jiang, W. Ma and Z. Tang, Adv. Mater., 2022, 34, 2110155 CrossRef CAS PubMed.
- T. Chen, Y. Zhong, T. Duan, X. Tang, W. Zhao, J. Wang, G. Lu, G. Long, J. Zhang, K. Han, X. Wan, B. Kan and Y. Chen, Angew. Chem., Int. Ed., 2024, e202412983, DOI:10.1002/anie.202412983.
- J. Song, C. Li, H. Ma, B. Han, Q. Wang, X. Wang, D. Wei, L. Bu, R. Yang, H. Yan and Y. Sun, Adv. Mater., 2024, 36, 2406922 CrossRef CAS PubMed.
- H. Yu, Y. Wang, C. H. Kwok, R. Zhou, Z. Yao, S. Mukherjee, A. Sergeev, H. Hu, Y. Fu and H. M. Ng, Joule, 2024, 8, 2304–2324 CrossRef CAS.
- X. Zhang, H. Gao, Y. Kan, X. Wang, W. Zhang, K. Zhou, H. Xu, L. Ye, R. Yang, Y. Yang, X. Hao, Y. Sun and K. Gao, Angew. Chem., Int. Ed., 2024, e202415583, DOI:10.1002/anie.202415583.
- Z. Wang, X. Wang, L. Tu, H. Wang, M. Du, T. Dai, Q. Guo, Y. Shi and E. Zhou, Angew. Chem., Int. Ed., 2024, 63, e202319755 CrossRef CAS.
- R. Zeng, L. Zhu, M. Zhang, W. Zhong, G. Zhou, J. Zhuang, T. Hao, Z. Zhou, L. Zhou and N. Hartmann, Nat. Commun., 2023, 14, 4148 CrossRef CAS PubMed.
- Y. Li, Q. Li, Y. Cai, H. Jin, J. Zhang, Z. Tang, C. Zhang, Z. Wei and Y. Sun, Energy Environ. Sci., 2022, 15, 3854–3861 RSC.
- Z. Luo, T. Liu, R. Ma, Y. Xiao, L. Zhan, G. Zhang, H. Sun, F. Ni, G. Chai, J. Wang, C. Zhong, Y. Zou, X. Guo, X. Lu, H. Chen, H. Yan and C. Yang, Adv. Mater., 2020, 32, 2005942 CrossRef CAS.
- P. W. M. Blom, V. D. Mihailetchi, L. J. A. Koster and D. E. Markov, Adv. Mater., 2007, 19, 1551–1566 CrossRef CAS.
- J. Song, C. Li, L. Zhu, J. Guo, J. Xu, X. Zhang, K. Weng, K. Zhang, J. Min, X. Hao, Y. Zhang, F. Liu and Y. Sun, Adv. Mater., 2019, 31, 1905645 CrossRef CAS.
- L. J. A. Koster, V. D. Mihailetchi, R. Ramaker and P. W. M. Blom, Appl. Phys. Lett., 2005, 86, 123509 CrossRef.
- A. K. K. Kyaw, D. H. Wang, V. Gupta, W. L. Leong, L. Ke, G. C. Bazan and A. J. Heeger, ACS Nano, 2013, 7, 4569–4577 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee02978j |
| This journal is © The Royal Society of Chemistry 2025 |