
A review on organic nanoparticle-based optoelectronic devices: from synthesis to applications
Zhe
Liu
 a,
Chen
Xie
b,
Thomas
Heumueller
cd,
Iain
McCulloch
e,
Christoph J.
Brabec
cd,
Fei
Huang
a,
Yong
Cao
a and
Ning
Li
*af
a,
Chen
Xie
b,
Thomas
Heumueller
cd,
Iain
McCulloch
e,
Christoph J.
Brabec
cd,
Fei
Huang
a,
Yong
Cao
a and
Ning
Li
*af
aInstitute of Polymer Optoelectronic Materials & Devices, Guangdong Basic Research Center of Excellence for Energy and Information Polymer Materials, State Key Laboratory of Luminescent Materials & Devices, South China University of Technology, Guangzhou 510640, China. E-mail: ningli2022@scut.edu.cn
bCollege of New Materials and New Energies, Shenzhen Technology University, Shenzhen 518118, China
cInstitute of Materials for Electronics and Energy Technology (i-MEET), Department of Materials Science and Engineering, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Martensstr. 7, 91058 Erlangen, Germany
dHelmholtz Institute Erlangen-Nürnberg for Renewable Energy (HI-ERN), Forschungszentrum Jülich GmbH, Immerwahrstr. 2, 91058 Erlangen, Germany
eDepartment of Chemistry, University of Oxford, Oxford, UK
fGuangdong Provincial Key Laboratory of Luminescence from Molecular Aggregates, South China University of Technology, Guangzhou, 510640, P. R. China
First published on 12th November 2024
Abstract
Over the past few decades, organic optoelectronic devices have shown broad application prospects. However, the solubility of organic materials requires the use of toxic solvents, hindering the fabrication of these devices in an air environment. Furthermore, the common approaches such as exploring non-halogenated solvents and side-chain modification do not address the root of this problem. In this case, considering the increasing focus on the subject of nanoparticles, dispersing organic materials as nanoparticles in non-toxic water/alcohol solvents provides a perfect and universally applicable solution to address the toxicity problem. In addition, the unique properties of nanoparticles can provide new ideas for the optimization and tuning of organic optoelectronic devices. In this review, we present an up-to-date overview of the water/alcohol-based organic nanoparticles applied in optoelectronic devices, encompassing the entire journey from nanoparticles to practical applications, which is partitioned into four vital segments: nanoparticle intrinsic property, from organic semiconducting materials to nanoparticles, organic thin films, and finally device applications.
Broader contextWith the emerging development of organic optoelectronics technologies, organic optoelectronic materials and devices have shown broad application prospects. However, the use of toxic organic solvents in the fabrication of these devices poses a serious environmental hazard. In this case, the generation of aqueous/alcohol-based organic nanoparticles effectively mitigates the issue of toxicity during the manufacturing process, significantly promoting the industrialization of organic optoelectronic devices. Meanwhile, the special properties of nanoparticles, which differ from bulk organic materials, are beneficial for the improvement of organic optoelectronic devices. The presence of “globular/granular” domains within nanoparticle films has captivated researchers, sparking investigations into the film formation mechanisms of these nanoparticles. Simultaneously, the separation of the domain sizes can be precisely controlled by varying the nanoparticle size, thereby introducing fresh perspectives for research into morphology and kinetics. Additionally, the high surface energy of nanoparticles is conducive to the improvement of catalytic ability, which makes them attractive in the field of photocatalytic hydrogen catalysis. We anticipate that this review will promote the development of conjugated organic nanoparticles for versatile applications. |
1. Introduction
Organic semiconductors continue to attract increasing attention from wide disciplines due to their unique advantages such as strong designability,1 low fabrication cost,2 light weight,3 and flexibility,4,5 enabling novel organic optoelectronic devices beyond the possibilities of classical inorganic semiconductors. However, organic semiconductors are mostly non-polar or weakly polar molecules having very low solubility in polar solvents, such as water/alcohol, typically dissolving in chlorinated and aromatic solvents, such as chloroform (CF) and chlorobenzene (CB).6 However, the use of toxic solvents for device fabrication has hazardous effects on human health and the environment, which extremely limits the large-scale production and commercialization of organic optoelectronic devices.In view of this, researchers have focused their efforts on reducing the toxicity in the solution processing of organic semiconductors or even achieving non-toxic processes. In the early stage, some non-halogenated/aromatic solvents such as toluene, o-xylene, trimethylbenzene, tetrahydrofuran (THF), and 2-methyltetrahydrofuran (Me-THF) were used to decrease the toxicity of the solution process. Most organic semiconductors remain soluble in these low-toxic solvents, and their good solubility can be attributed to the formation of π–π interactions between the aromatic nuclei of solvents and the organic semiconductors.7 Therefore, the performance of non-halogenated solvent-based devices can achieve comparable performances to that of the devices prepared with toxic solvents.8,9 However, the use of low-toxicity solvents in the fabrication of organic optoelectronic devices as a phased approach from toxicity to non-toxicity only mitigates their environmental accumulation, still having the risk of explosion and carcinogenesis.
Thus, to increase the solubility of these materials in water/alcohol high-polar solvents, researchers have been focused on the decoration of organic semiconductors, such as side/branched chain extension and side chain modification.10–16 A greater volumetric proportion of side chains can enhance the solubility of organic molecules owing to the less interactions within the polymer main chain. Thus, extending the alkyl chain or introducing branched alkyl chain/olefin linkages into the main chain of organic semiconductors can effectively increase their solubility in water/alcohol.10–13 Side chain modification using polar functional groups, especially molecularly engineered side chains containing oxygen, can form intermolecular hydrogen bonds with the hydrogen atom of the polar solvent, resulting in an enhancement of solubility in water/alcohol solvents.14–16 Based on the enhanced solubility of organic semiconductors in polar solvents, fully solution-processed organic optoelectronic devices have been fabricated using aqueous/alcohol solvents, achieving a non-toxic process.17,18 However, the performance of organic optoelectronic devices prepared through molecular engineering is less competitive than that with materials dissolved in toxic solvents, which can be explained by the changes in the inherent properties of organic semiconductors due to the polar modification, such as the lack of well-ordered lamellar stacking and charge trapping effects originating from the polarity of the side chain. In addition, the structural modification strategy for specific materials is not universal. Thus, to address the issue of environmental toxicity, dispersing organic semiconductors as nanoparticles (NPs) in non-toxic water/alcohol solvents provides a perfect and universally applicable solution to achieve non-toxicity and sustainability for industrial production. Meanwhile, from the aspect of maintenance of processing equipment, water/alcohol nanosuspensions can avoid corrosion caused by organic solvents. For example, the Epson inkjet L110 printer only uses water-based inks to avoid the damaging tendency of n-hexane to the expensive printer.19 The prepared aqueous/alcohol-based nanosuspensions are indistinguishable from a normal organic toxic solution to the naked eye and can be deposited to homogenous films using common solution deposition techniques, exhibiting excellent properties in the non-toxicity fabrication of organic optoelectronic devices. Furthermore, the unique properties of nanoparticles can provide new ideas and concepts for the optimization and tuning of organic optoelectronic devices.
In this review, we aim to provide an up-to-date overview of this field by focusing on the water/alcohol-based nanoparticles applied in optoelectronic devices. As exhibited in Fig. 1, our exploration encompasses the entire journey from nanoparticles to practical applications, partitioned into four vital segments: nanoparticle intrinsic property, from organic semiconducting materials to nanoparticles, from nanoparticles to organic thin films, and finally device applications. Firstly, we focus on the intricate formation and stabilization mechanisms of nanoparticles, serving as the foundational introduction to organic nanoparticles. Following the detailed description of the common techniques that enable the precise measurement of the properties of nanoparticles, we introduce the processes from organic materials to nanoparticles. In addition to traditional methods such as precipitation and emulsion, we provide a comprehensive summary of the advanced techniques employed to synthesize nanoparticles without the need for surfactants. Subsequently, we study the stage that transitions from nanoparticles to thin films, which although often overlooked, is of utmost significance. We analyse the challenges encountered during the formation of nanoparticle films, as well as their inherent advantages. From organic thin films to functional device applications, we concentrate on the myriad of applications of these films in the realm of organic optoelectronic technologies, including organic solar cells (OSCs), organic light-emitting diodes (OLEDs), organic field effect transistors (OFETs), and photocatalytic hydrogen evolution, showing their vast potential and wide-reaching impact.
 | ||
| Fig. 1 Overview outline of this review from organic semiconducting materials to organic nanoparticles, organic thin films, and functional device applications. | ||
2. Organic nanoparticles
Presently, organic nanoparticles play a crucial role across diverse fields, including chemistry, biology, physics, and other interdisciplinary areas due to their unique properties, which differ from that of bulk materials.20,21 Understanding the basic theory and characterization of nanoparticles is the basis for further research in targeted nanoparticles design or transformative application. Before introducing the theory of nanoparticle formation, a few definitions that are confusing or inconsistent in the literature are explained. The dispersion of one or more substances in another substance constitutes a dispersion system (often abbreviated as dispersion). The substance that is dispersed in the dispersion system is called the dispersion phase, whereas the other substance is called the dispersion medium (a continuous phase).22 Depending on the size of the dispersed phase particles (r), dispersion systems are often divided into molecular (or ionic) dispersion systems (r < 1 nm, also called solutions), colloidal dispersion systems (also colloids, called 1 nm < r < 100 nm), and coarse dispersion systems (r > 100 nm). In fact, the international definition of colloidal is not absolutely strict in terms of particle size.23 Most textbooks and organizations define it as 1–100 nm, and a few define it as 1–1000 nm.24–27 In this review, we will not discuss which definition is more accurate, and we review the particle size in the range of 1–1000 nm (named nanoparticle, NP), given that particles with a size in the range of a few hundred nm can also be stable as well as having colloidal properties. When organic nanoparticles are smaller than 10 nm, they are called nanodots (NDs). According to the type of dispersing medium, suspensions are a class of dispersed system in which the dispersing medium is liquid, and nanosuspensions are nanosized colloidal dispersion systems (also called nano-dispersions).282.1 Theory for the formation of nanoparticles
In this part, we focus on the theory for the formation of nanoparticles covering two sequential steps of classical nucleation theory (CNT), nucleation and growth. (Fig. 2a). This concept was first proposed by Gibbs29 and most commonly used to describe the formation of nanoparticles. Initially, a system with multiple components is regarded as a single phase, and the components also can be called monomers, referring to atoms, ions, or molecules. The decrease in Gibbs free energy serves as the driving force for the formation of nanoparticles. When changing the boundary conditions such as altering temperature or adding additive/dopant, the free energy initiates a change, thus making phase separation more energetically favourable. The monomers coalesce (forming clusters), resulting in the formation of nuclei because of the phase separation. Subsequently, growth occurs as the monomers attach to the nuclei through merging or aggregating, inducing larger size. According to theory, the change in the Gibbs energy (ΔG) during the formation of nanoparticles process can be defined as follows:| ΔG = ΔGs + ΔGv = 4πr2γ + 4/3πr3Δgv | (1) |
 | (2) |
 the energy barrier for heterogeneous nucleation
the energy barrier for heterogeneous nucleation 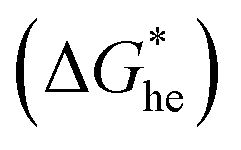 is lower due to correction coefficient ϕ:33,34
is lower due to correction coefficient ϕ:33,34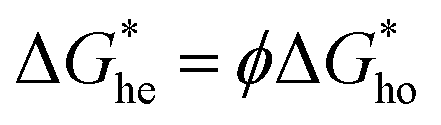 | (3) |
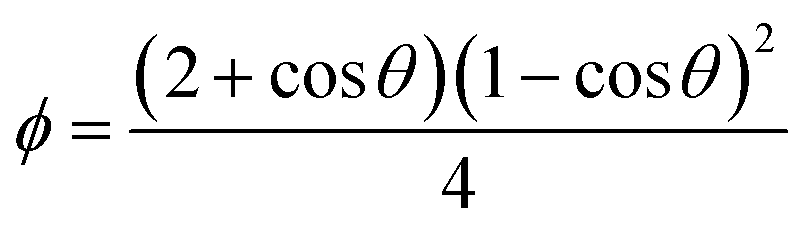 | (4) |
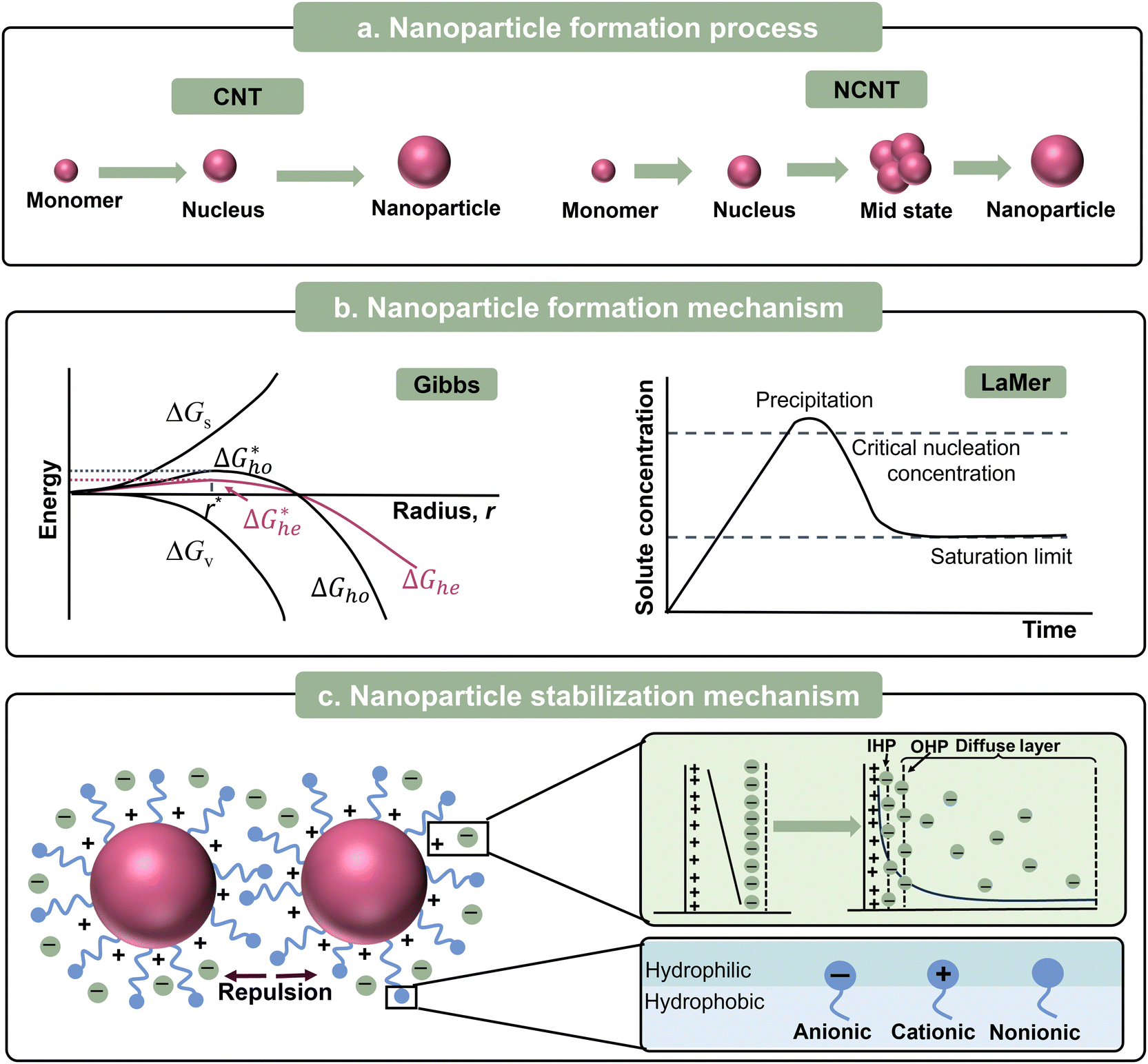 | ||
| Fig. 2 (a) Formation process, (b) formation mechanism, and (c) stabilization mechanism of organic nanoparticles. | ||
Later in 1950, LaMer and Dinegar expanded the CNT framework by proposing that the process of nucleation and growth should be separated by a burst of nucleation (Fig. 2b). This approach ensures that all the nanoparticles grow consistently, achieving monodispersity.36,37 The core principle of the LaMer model is that nanoparticle formation occurs in two distinct stages, firstly, “instantaneous nucleation” (or “burst nucleation”), and secondly, “diffusion-controlled growth”. As the solute is continuously supplied into the dispersion medium, the concentration of dissolved solute is proportional to the release time before the solute reaches the critical supersaturation. When the concentration exceeds the critical supersaturation, nucleation starts. Consequently, the concentration temporarily drops below the critical threshold, preventing the formation of new nuclei. The existing nuclei keep on growing until the dissolved solute concentration decreases to the equilibrium level.
With the development of nanoparticles, non-classical nucleation theories (NCNTs) have been developed to describe the complex behaviours that cannot be explained by CNT. In CNT, nucleation occurs in a single concise step including the formation of thermodynamically stable nuclei and their spontaneous growth. In contrast, NCNTs involve multiple steps to describe the intermediate states, as depicted in Fig. 2a.38–42 The most common mechanisms of NCNTs are the two-step nucleation mechanism43 and the pre-nucleation cluster (PNC) mechanism.44,45
2.2 Mechanism for the stabilization of nanoparticles
A nanosuspension is a highly dispersed multi-phase system, having large phase interfaces, and thus is a thermodynamically unstable system. Dispersed-phase nanoparticles have a tendency to automatically aggregate owing to their large specific surface area, which is called agglomeration instability.46 Thus, to ensure the long shelf life of these colloidal dispersions, high stability against aggregation is desirable. The stability of nanoparticles depends on the equilibrium between the repulsive and attractive forces that arise as the particles come close to each other. The attractive force between nanoparticles is essentially the same as the van der Waals forces between molecules. Electrostatic repulsion and/or steric stabilization acts as repulsive forces to stabilize nanoparticles (Fig. 2c).46 Furthermore, the concepts of electrostatic repulsion and steric stabilization are crucial for the successful synthesis of nanoparticles.Electrostatic stabilization is mainly effective in aqueous solutions, which originates from the repulsive electrostatic force between the surface charges of the nanoparticles. Under the action of an external electric field, the nanoparticles with an electric charge under directional migration, which is called electrophoresis. The electrophoresis of nanoparticles proves that nanoparticles are electrically charged. The electric property of nanoparticles was clearly understood after the formulation of the theory of double electric layers. At the interface between the nanoparticles and the liquid dispersion medium, the surface of nanoparticles and neighbouring liquid dispersion medium carry two layers with the same number of ions but opposite electrical properties, respectively, thus forming an electric double layer.22 In 1987, the electrical double layer (EDL) model was proposed by Helmholtz, which is also called the Helmholtz or parallel-plate condenser model.47 This model is comprised of two parallel layers carrying equal amounts but opposite charges. As shown in Fig. 2c, the charges near the surface (solid black line) are called surface charges and the other free charges in the dispersion medium are counterions. However, this model ignores the thermal motion of the counterions. Accordingly, Gouy and Chapman modified the above-mentioned model (Gouy–Chapman, GC theory), proposing the concept of the diffusion layer.48,49 Only some of the counterions with opposite charges are tightly packed on the surface, which was later named the Stern layer, whereas the other counterions can diffuse from the surface.50 Subsequently, Graham expanded the Stern model by introducing the concepts of inner and outer Helmholtz planes (IHP and OHP, respectively) in the diffusion region based on their respective distances from the surface, as shown in Fig. 2c.51
The relation between the van der Waals attractive forces and repulsive forces between two nanoparticles can be expressed as the Derjaguin–Landau–Verwey–Overbeek (DLVO) theory.52–55 When the distance between nanoparticles is large, the double layer of electric charges do not overlap, while the attractive force causes the net potential energy to be negative. When the nanoparticles become so close together that the electric double layers overlap, the repulsive force plays a major role, thus the potential energy increases significantly. However, at the same time, the attractive force between the nanoparticles also increases with the shortening of the distance. When the nanoparticles keep getting close to each other, the attraction dominantly induces a decrease in the net potential energy. A deep negative energy trap is formed during this process, which can result in irreversible aggregation.
Unlike electrostatic stabilization, steric stabilization is suitable for both aqueous and non-aqueous media. The principle of steric stabilization relies on the repulsion between molecules adsorbed on neighbouring nanoparticles, such as surfactants and dopants. Molecules with long chains (e.g. surfactants) adsorbed on the nanoparticles act as protective layer, which function like springs, preventing their aggregation (Fig. 2c). It should be noted that the practical stabilization principle is driven by changes in the configurational entropy/enthalpy in the isothermal surroundings, rather than the stiffness of the long chains.56–58
2.3 Characterization methods
Compared with bulk materials, the characterization of nanoparticles requires more precise and credible analytical techniques to capture their behaviour at the nanoscale, which inversely promotes the development of emerging test techniques.If all the particles that make up a particle system have the same or approximately the same size, the particle system is monodisperse. Although the aim of nanoparticle synthesis is to obtain a monodisperse population of nanoparticles, in most cases the samples always display a certain degree of size variation.59 This variation can be represented by the polydispersity index (PDI), while the particle size distribution (PSD) refers to the distribution pattern of the size of all nanoparticles in the sample, as illustrated in Fig. 3a. Various techniques relying on different physical principles and data processing methods are available to determine particle size, which can be categorized as sieving, sedimentation, light scattering, and electrical induction.60 Dynamic light scattering (DLS) is the most commonly used technique to determine the hydrodynamic diameter of particles. The size of a nanoparticle during the test defaults to a uniform hard sphere. A monochromatic beam is transmitted to a nanosuspension, and then a detector records the change in the intensity of the scattered light caused by the random thermal motion of particles. In the DLS test, a relatively low concentration of nanosuspension is needed to avoid the multiple scattering effect.61 Autocorrelation analysis is employed to calculate the diffusion coefficient of the particles, which is proportional to the scattering correlation time. Subsequently, the diameter is inferred from the diffusion coefficient via the Stokes–Einstein equation.62 It is worth noting that after conducting DLS experiments, the instrument will give multiple types of data regarding nanoparticles size, making it confusing which one is the most accurate.63Z-Average and PDI are obtained by cumulant analysis of the measured correlation curve.64,65 The sample defaults to monomodal (only one peak), spherical, and monodisperse when calculating the results of Z-average, and thus it can be comparable with other methods only when the sample is monomodal or the PDI is very small (<0.1). Intensity PSD is obtained in another way from multi-index analysis, allowing multi-peak distribution. Intensity PSD is the pristine distribution results obtained from correlation equations, but through the Mie theory, it can be converted into volume PSD and number PSD. When the PDI is very small (<0.3), the size value derived from different analytical methods does not significantly differ (Fig. 3a). If the PDI is very large (0.3–1), the Z-average and intensity PSD usually give much larger values, while number PSD gives the lowest values (Fig. 3a). This can be explained by the considerably higher proportion of small sized nanoparticles for number PSD. Thus, to simply diminish the influence of a few large nanoparticles on the overall size of nanoparticles, Number PSD is mostly adopted.
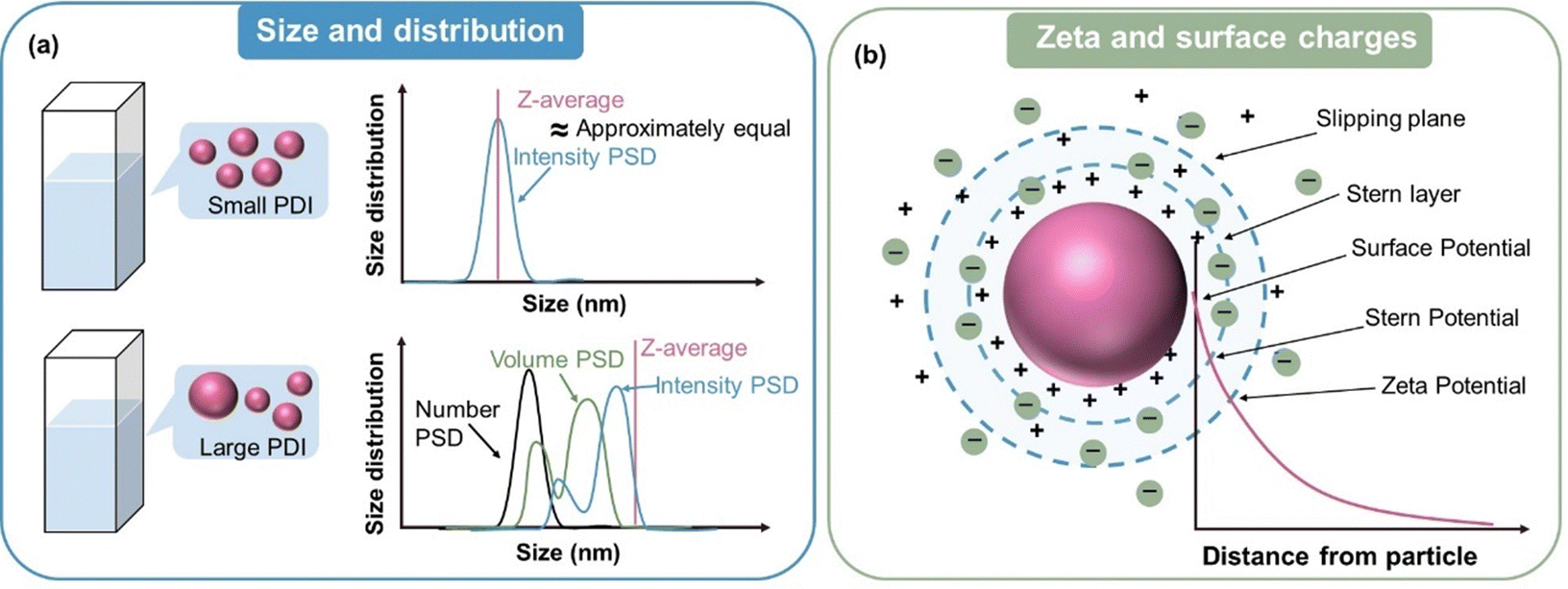 | ||
| Fig. 3 (a) Schematic diagram of the size and size distribution tested by DLS measurement. (b) Surface charge and zeta potential of nanoparticles in a suspension. | ||
In addition to the light scattering technique, the size distribution and PDI of nanoparticles can also be roughly determined from some morphological characterization techniques, such as TEM and SEM, which can afford sufficient resolution to visualize individual nanoparticles down to the smallest sizes and determine the nanoparticle dimensions based on the projection of the particle outline onto a two-dimensional image plane.66 The micrograph usually contains images of several hundred particles, and software such as Image J can be used to analyse and calculate the size of the nanoparticles.67–70 Coleman et al.68 compared the size distributions of nanoparticles obtained from DLS and TEM measurement, and found that the DLS measurement showed higher values (∼42 nm) than TEM (∼25 nm). The different results can be explained by the different sensitivity to large-sized nanoparticles between the two test techniques. Only a very small number of large or even agglomerated particles are in the field of the electron microscope, and thus they can be excluded during the calculation. According to Rayleigh's law, the intensity of the scattered light is proportional to the sixth power of the nanoparticle size. Therefore, the measurement of particle size by DLS requires the distribution of the sample to be as narrow as possible, otherwise the signal from small particles can easily be overshadowed by large particles. In the case of nanoparticles with large differences in size, it is necessary to segment the nanoparticle region by conducting data analysis to determine the accurate size or select other measurement techniques such as differential centrifugal sedimentation (DCS) and microchannel resonator (MCR) that can overcome the limitations of narrow PDI.68
 | ||
| Fig. 4 SEM images of (a) perylene nanocrystals in the absence of DMPBI (left) and presence of 30% DMPBI additive (right). Scale bars represent 100 nm. Reproduced with permission from ref. 32. Copyright 2010, Elsevier Ltd. (b) Hollow (half-moon) NPs, scale bars represent 200 nm.75 (c) STEM images of PFODTBT nanodots at low magnification (top) and high magnification (bottom). Reproduced with permission from ref. 76. Copyright 2019, The Royal Society of Chemistry. (d) Cryo-TEM images of pure PDPP5T nanoparticles, pure PCBM nanoparticles, and PDPP5T:PCBM nanoparticles (below). Reproduced with permission from ref. 84. Copyright 2017, the American Chemical Society. (e) Cryo-TEM images of PTQ10:Y6 nanoparticles.86 (f) Color-coded elemental EFTEM images of PTB7:PC71BM Janus nanoparticles (left), and core–shell nanoparticles (right), the PTB7 part was yellow, containing both of carbon (red) and sulphur (green) elements.75 (g) STXM images of the fractional composition of P3HT in P3HT:PCBM nanoparticles (left) and radially averaged compositional particle cross-sections for an average of P3HT:PCBM imaged by STXM. Reproduced with permission from ref. 87. Copyright 2015, Elsevier Ltd. | ||
In the case of organic nanoparticles with more than one component, it is necessary to characterize their internal shape and composition. To explore their internal morphologies, Du et al.75 utilized the energy-filtered TEM (EFTEM) technique for PTB7:PC71BM nanoparticles. This technique can provide material contrast by detecting the differences between elements.88 Sulphur was present in PTB7 but not in PC71BM, and thus the contrast in the elemental sulphur map could characterize the internal shape of the nanoparticles, as shown in Fig. 4f. By combining the chemical sensitivity of near-edge X-ray absorption fine structure (NEXAFS),89 scanning transmission X-ray microscopy (STXM) can be used to quantitatively calculate the composition of nanoparticles.90 Further details of the STXM experimental methods and data analysis methods were reported in the literature.91–93Fig. 4g shows the STXM composition maps of P3HT:PCBM nanoparticles.87 Lighter colour indicates higher concentration, with the colour scale ranging from black (0%) to white (100%). According to the colour contrast, P3HT was resolved to be mainly distributed in the shell, with a few molecules in the core, while that for PCBM was the opposite. The position of the interface between the core and shell was determined to be around 0.58 of the nanoparticle radius from the radial compositional averaging plot, indicating that the PCBM-rich core region constituted approximately 20% by volume of the P3HT:PCBM nanoparticles.
When nanoparticles are synthesized using surfactants, the polar head of the surfactant determines whether their zeta potential is positive or negative. As shown in Table 1, Bellacanzone et al.69 prepared P3HT:PCBM nanoparticles with three different types of surfactants including SDS, CTAB, and AOT, and all three blended nanosuspensions presented a high zeta potential. The nanoparticles synthesized with SDS and AOT exhibited a negative zeta potential due to the sulphate/sulphonate group in these surfactants. In contrast, the nanoparticles with CTAB showed a positive zeta potential value because of the positive quaternary ammonium group.69 Xie et al.101 investigated various residual surfactants in P3HT:IDTBR systems subjected to centrifugation for different number of times. To a certain extent, the absolute value of surface charge can reflect the residue of surfactant attached to the nanoparticles. The surface charge value decreased distinctly after washing repeated times for the nanoparticles synthesized with F127 surfactant, and then saturated at the lowest value of ∼10 mV. In contrast, the nanoparticles synthesized using other surfactants showed higher zeta potential values, retaining a certain amount of surfactant attached them, which could not be completely eliminated. In the case of water/alcohol-based surfactant-free nanosuspensions, the zeta potential of the nanoparticles is closely related to the properties of the dispersion systems, such as the structure of the organic material, solvent polarity, and pH value.91,94,102 Marlow et al.94 tested surfactant-free P3HT ethanol-based nanosuspensions, and the ELS results showed the mobility of 10−8 m2 V−1 s−1 with a zeta potential of +80 mV. The positive charge of P3HT could be explained by the role of protonation. Polythiophenes can undergo protonation by acids, with their conjugated π-electron systems stabilizing their protonated form through charge delocalization.103,104 Ethanol can be regarded as a weak acid providing protons, while in the case of water as a dispersant, Nagai et al.105 observed a negative zeta potential for P3HT nanoparticles. According to Kamogawa et al.,106 the stability of aqueous surfactant-free nanoparticles can be influenced by their polarity. Molecules containing highly polar groups, such as carbonyl groups, tend to induce a significantly negative zeta potential. Xie et al.91 prepared P3HT:ICBA nanosuspensions using five different alcohols as the dispersion medium, and all the batches of nanoparticles demonstrated negatively charged surfaces in the zeta potential measurements, as summarized in Table 1. The surface charge density (σ0) of the five batches nanoparticles was quantified using a simplified equation, as follows:107
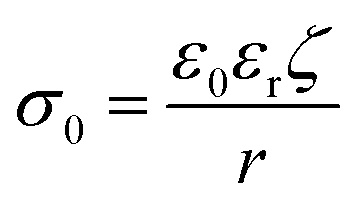 | (5) |
| Sample | Surfactant | Synthesis method | Disperse medium | Size of nanoparticles | Zeta potential (mV) | Ref. | |
|---|---|---|---|---|---|---|---|
| Before centrifugation | After centrifugation | ||||||
| P3HT:PCBM | SDS | Miniemulsion | Water | 134.0 | −57.7 | −42.8 | 69 |
| CTAB | 150.6 | +56.2 | +32.3 | ||||
| AOT | 148.9 | −54.5 | −40.3 | ||||
| P3HT:IDTBR | F127 | Miniemulsion | Water | −40.5 | −10.3 | 101 | |
| SDS | −69.0 | −34.6 | |||||
| DTAB | +50.8 | +21.8 | |||||
| Brij | −39.5 | −17.0 | |||||
| P3HT:ICBA | None | Nanoprecipitation | Ethanol | 104 | −20.2 | 91 | |
| 2-Propanol | 95 | −16.2 | |||||
| 2-Butanol | 90 | −11.6 | |||||
| 3-Hexanol | 80 | −6.2 | |||||
| cis-3-Hexen-1-ol | 79 | −5.6 | |||||
| P3HT:ICBA + P3HT-Py | None | Nanoprecipitation | Ethanol + AcOH | 77–84 | Positive | 108 | |
| P3HT | None | Pre-emulsion + nanoprecipitation | Water + ethanol | 65 nm | −33.6 | 105 | |
| 33 nm | −29.4 | ||||||
| Water + i-propanol | −12.5 | ||||||
| P3HT | None | Nanoprecipitation | Ethanol | 77 nm | +80 | 94 | |
| P3HT | None | Nanoprecipitation | Water | Negative | 102 | ||
3. Production of water/alcohol-based nanosuspensions from organic semiconducting materials
Nanoparticle synthesis techniques can be broadly categorized into physical techniques (top-down technique), which involve extensively breaking down bulk materials into nanoparticles through mechanical means, and chemical techniques (bottom-up technique), where nanoparticles are synthesized from monomers through the nucleation and growth process.109 In the case of the bottom-up technique, different procedure have been used to synthesize organic conjugated nanoparticles via polymerization in direct polymerization or post-polymerization methods.Direct polymerization is a one-pot approach in which polymerization and nanoparticle formation are achieved concurrently, focusing on synthesis from monomers to organic conjugated materials and nanoparticles. It includes radical polymerization and anionic, cationic, oxidative, and catalytic polymerization, while post-polymerization is an effective synthetic approach to form nanoparticles using organic conjugated materials. Readers interested in the direct polymerization strategy can refer to the review article.110 Here, we focus on the post-polymerization preparation methods, which involve the production of nanoparticles from preformed polymers excluding any polymerization processes. In these methods, organic nanoparticles are prepared using universal polymers and the corresponding non-solvent. They are very versatile and enable the use of organic conjugated materials, where a variety of organic nanoparticles can be prepared depending on the choice of materials and control of the synthesis parameters. Several methods to synthesize nanoparticles from a preformed polymer have been well-established.
3.1 Miniemulsions
The miniemulsion method is the most commonly used and universal way to synthesize highly stable organic conjugated nanoparticles. An emulsion is a mixture where one or more immiscible liquids is finely dispersed within another as droplets.111,112 According to the size of droplets, a macroemulsion refers to that with a size in the range of 1–10 μm.113 Alternatively, a miniemulsion is defined as droplets with a size of less than 100 nm (or 500 nm and 1000 nm), with no universal definition. The formation of emulsions requires an external energy supply, such as agitation, ultrasonic, and grinding, between the dispersed phase and dispersed medium. Agitation splits the two immiscible liquid phases, leading to emulsification. Alternatively, ultrasonication is often chosen to obtain stable miniemulsions, which is performed in an ice bath. When sound waves propagate through a mixture of two immiscible solvents, they induce the formation of microbubbles.114 However, the increased interfacial area of miniemulsions makes them thermodynamically unstable systems, according to ΔG = γΔA.115–118 Thus, a surfactant is required to reduce the surface tension, thereby inducing the formation of a stable miniemulsion. Increasing the surfactant concentration decreases the surface/interfacial tension. When the γ value approaches zero, it reaches a critical point, which is named the critical micelle concentration (CMC). When the concentration of surfactant is greater than the CMC (Fig. 5a), surfactant forms micelles in the dispersed medium, which encapsulate the droplets in the dispersed phase when it is formed. | ||
| Fig. 5 Schematic illustration of organic conjugated nanoparticle synthesis through: (a) emulsification, (b) nanoprecipitation, (c) self-assembly method, (d) electrostatic method, and factors influencing the synthesis process. | ||
In the post-emulsification process for organic conjugated nanoparticles, ultrasonication and surfactants are combined to synthesize stable miniemulsions. Considering the environmentally friendly nature of water, dispersing the organic phase in an aqueous phase is the preferred choice for the synthesis of organic conjugated nanoparticles. Given that surfactants are amphiphilic molecules with a hydrophilic head and a hydrophobic tail, they can dissolve in both the aqueous phase and organic phase. When surfactants are dissolved in the aqueous phase, their hydrophobic groups are close together facing inward away from the environment, whereas their hydrophilic groups face outward in contact with water, forming surfactant micelles. As shown in Fig. 5a, an organic solution is added to the aqueous phase containing surfactant micelles, which can encapsulate the organic phase, forming stable nanoparticles. Chloroform (CF) is mostly used as the precursor organic solvent due to its low-boiling point and good general solubility for organic materials. Although high-boiling solvents are rarely selected, toluene,119,120ortho-dichlorobenzene,121 and o-xylene122 have been reported as good solvents for synthesizing nanosuspensions. A pre-mixing process before ultrasonication for an hour can form more homogeneous nanoparticles with a lower PDI. Following the ultrasonication process in ice bath, their dispersion results in the formation of small organic droplets, and non-toxic aqueous nanosuspensions are obtained after evaporation of the organic solvent. The organic conjugated nanoparticles synthesized using this method are typically nanospheres with diameters ranging from 5 to 250 nm (Table 2). Additionally, nanocapsules can be synthesized by adding a certain amount of oil to the organic phase, such as triglycerides.123
| Number | Material | Solvent | Anti-solvent | Surfactant | Particle size (nm) | Year | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | Me-LPPP | CF | Water | SDS | 154 | 2002 | 124 |
| 2 | F8BT:PFB | CF | Water | SDS | 53 | 2003 | 125 |
| PF2/6:m-LPPP | CF | 64–149 | |||||
| 3 | PFB:F8BT | CF | Water | SDS | 49–53 | 2004 | 126 |
| 4 | POPPV:PFO | CF | Water | SDS | 59 | 2009 | 127 |
| 5 | PFFO | CF | Water | SDS | 60 | 2010 | 128 |
| 6 | P3HT | CF | Water | SDS | 60 | 2011 | 129 |
| 7 | P1:PC60BM | CF | Water | SDS + FSO-100 | 130 | 2011 | 130 |
| P2:PC60BM | 32 | ||||||
| P3:PC60BM | 87 | ||||||
| 8 | F8BT:PFB | CF | Water | SDS | 51.9 | 2012 | 131 |
| 9 | P3HT:PCBM | CF | Water | SDS | 81–102 | 2012 | 132 |
| 10 | P3HT | CF | Water | SDS | 122 | 2012 | 119 |
| Toluene | 110 | ||||||
| Toluene + CF | 73 | ||||||
| 11 | P3HT:PCBM | CF | Water | SDS | 38.0 | 2013 | 99 |
| 12 | P3HT:ICBA | CF | Water | SDS | 33.8 | 2013 | 133 |
| 13 | P3HT | CF | Water + 1-propanol | SDS | 15–125 | 2014 | 134 |
| PCBM | CF | Water | CTAB | 42–100 | |||
| 14 | P3HT:PCBM | CF | Water | SDS | 28–42 | 2014 | 135 |
| 15 | P3HT:PCBM | CF | Water + ethanol | FSO-100 | 100 | 2014 | 136 |
| 16 | P3HT:PCBM (mNPs) | CF | Water | SDS | 74 | 2014 | 137 |
| P3HT:PCBM (bNPs) | 80 | ||||||
| 17 | P3HT:PCBM | CF | Water | SDS | 38–150 | 2015 | 138 |
| 18 | P(TBT-DPP):ICBA | CF | Water | SDS | 80–110 | 2015 | 139 |
| 19 | TQ1:PC71BM | CF | Water | SDS | 21–130 | 2016 | 140 |
| 20 | PBTTT | CF | Water | SDS | 83 | 2016 | 141 |
| 21 | PCDTBT:PC71BM | CF | Water | SDS | 30–330 | 2017 | 142 |
| 22 | PDPP-TNT:PC71BM | CF | Water | SDS | 113 | 2017 | 143 |
| 23 | PDPP5T:PC60BM | CF | Water | SDS | 62–34 | 2017 | 84 |
| 24 | PCDTBT:PC71BM | o-DCB | Water | SDS | 36–74 | 2017 | 121 |
| 25 | P3HT:ICxA | CF | Water | SDS/FSO | 41 | 2018 | 144 |
| 26 | P3HT:ICxA | CF | Water | SDS | 32–38 | 2018 | 145 |
| CF | Water | SDS | 32 | 2018 | 122 | ||
| o-Xylene | Water | SDS | 27 | ||||
| 28 | P3HT | CF | Water | SDS | 150 | 2018 | 90 |
| PCBM | 118 | ||||||
| P3HT:PCBM | 150 | ||||||
| 29 | P3HT:PCBM | CF | Water | SDS (P3HT), CTAB (PCBM) | 151 | 2019 | 146 |
| 30 | P3HT:PC60BM | CF | Water | SDS | 29 | 2019 | 147 |
| 31 | P3HT:PCBM | CH2Cl2 | Water | SDS | 114.9 | 2020 | 69 |
| CTAB | 136.3 | ||||||
| AOT | 142.5 | ||||||
| 32 | PTB7-Th:EH-IDTBR | CF | Water | SDS/TEBS | 50–80 | 2020 | 79 |
| 33 | MEH-PPV:PS | Toluene | Water | SDS | 65 | 2020 | 120 |
| SY-PPV:PS | CF | 1250 | |||||
| 34 | P3HT:PCBM | CF | Water | SDS | 27.9–56.4 | 2021 | 148 |
| 35 | P3HT:N2200 | CF | Water | SDS | 233 | 2021 | 149 |
| TQ1:PNDIT10 | CF | Water | SDS | 233 | |||
| P3HT:o-IDTBR | CF | Water | SDS | 212 | |||
| P3HT:eh-IDTBR | CF | Water | SDS | 170 | |||
| TQ1:N2200 | CF | Water | SDS | 172 | |||
| 36 | PM6:Y6 | CF | Water | TEBS | 83.03–101.6 | 2022 | 150 |
| PM6:PCBM | 83.09–91.64 | ||||||
| 37 | IDTBT:oIDTBR | CF | Water | SDS | 70.43 | 2022 | 151 |
| gIDTBT:oIDTBR | 68.96 | ||||||
| gPTB7-Th:oIDTBR | 55.81 | ||||||
| PTB7-Th:oIDTBR | 56.32 | ||||||
| FgBT:PC70BM | 53.84 | ||||||
| F8BT:PC70BM | 61.99 | ||||||
| 38 | PTB7-Th:EH-IDTBR | CF | Water | TEBS | 60.00 | 2023 | 152 |
| 39 | PTQ10:PCBM | CF | Water | SDS/SDBS/F127 | 78 | 2023 | 86 |
| PTQ10:Y6 | |||||||
| 40 | PTQ10:Y6 | CF | Water | SDS | 90 | 2024 | 153 |
This method was first developed by Vanderhoff et al. in 1979 for producing latexes.154,155 In the following years, this method was mainly used to synthesize biodegradable nanoparticles, such as poly(L-lactide) (PLLA), poly(D,L-lactide-co-glycolide) (PLGA), and poly(ε-caprolactone) (PCL).156,157 Song et al.158 adopted the double-emulsion method employing dichloromethane and acetone as binary precursor organic solvents and polyvinyl alcohol (PVA) as a surfactant to prepare PLGA nanoparticles with diameter in the range of 60–200 nm. In 2002, Landfester et al.124 first fabricated organic conjugated aqueous nanosuspensions via the miniemulsion method using three different conjugated polymers including Me-LPPP, PF, and PCPDT. Hard spherical nanoparticles with controllable particle sizes ranging from 70–250 nm were obtained. In addition to single-component nanoparticles, Kietzke et al.125 first synthesized blended nanoparticles (bNPs), PFB:F8BT, which contained electron-donating and electron-accepting materials, using sodium dodecyl sulfate (SDS) as the surfactant. By adjusting the weight ratio of PFB![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :F8BT in the nanoparticles, their sizes ranged from 40 to 155 nm.126 They also prepared mixed nanoparticles (mNPs) from two nanosuspensions, hole-accepting M3EH-PPV nanosuspensions (diameter of 54 nm) and electron-accepting CN-Ether-PPV nanosuspensions (diameter of 36 nm), with equal ratios.159 As summarised in Table 2, P3HT and fullerene-based materials have been frequently used to synthesize nanoparticles with different surfactants, and the diameter of nanoparticles was less than 150 nm.129,138 With the development and modernization of organic conjugated materials, ranging from fullerene acceptors to non-fullerene acceptors, synthesized organic nanoparticles are evolving. Due to the good encapsulation properties of the commonly used surfactants, this method is universal, having no structural limitations for organic conjugated materials.
:F8BT in the nanoparticles, their sizes ranged from 40 to 155 nm.126 They also prepared mixed nanoparticles (mNPs) from two nanosuspensions, hole-accepting M3EH-PPV nanosuspensions (diameter of 54 nm) and electron-accepting CN-Ether-PPV nanosuspensions (diameter of 36 nm), with equal ratios.159 As summarised in Table 2, P3HT and fullerene-based materials have been frequently used to synthesize nanoparticles with different surfactants, and the diameter of nanoparticles was less than 150 nm.129,138 With the development and modernization of organic conjugated materials, ranging from fullerene acceptors to non-fullerene acceptors, synthesized organic nanoparticles are evolving. Due to the good encapsulation properties of the commonly used surfactants, this method is universal, having no structural limitations for organic conjugated materials.
As shown in Fig. 6, the most used surfactants can be categorized into three types including anionic, cationic, and non-ionic.160 As described in Section 2.2, non-ionic surfactants stabilize nanoparticles by steric hindrance, while ionic surfactants can stabilize nanoparticles through electrostatic repulsions and steric repulsions.161–163 Depending on the head group of anionic surfactants, they can be classified as sulfonate, carboxylate, sulphate, and phosphate. Among them, SDS is the most universal anionic surfactant employed in the synthesis of organic nanoparticles. According to the position of the nitrogen atom, cationic surfactants can be categorized as straight-chain amine salts, quaternary ammonium salts, and cyclic pyridine types. The emulsification effectiveness of surfactants can be influenced by their hydrophilic/lipophilic balance (HLB) value.164–167 Specifically, w/o (water-in-oil) emulsions are formed when HLB < 10, while inverse o/w (oil-in-water) emulsions are formed when HLB > 10. Therefore, the type and dosage of surfactants have a great influence on the size of nanoparticles.84,140,142,168 Cho et al.169 studied 18 different surfactants to generalize rules regarding the choice of a suitable surfactant for the preparation of miniemulsions. They found that surfactants with linear chains and charged hydrophilic groups both produced stably dispersed colloidal particles due to their efficient van der Waals interactions with the linear alkyl chains of polymers. In contrast, ionic surfactants with large aromatic hydrophobic groups did not interact efficiently with the polymer, resulting in very large particles with a size of several hundreds of micrometres. These results were also confirmed by Zhang et al.,170 who found that ionic surfactants with large aromatic hydrophobic groups were not ideal, whereas surfactants with longer alkyl chains were better for forming stable miniemulsions. Tan et al.171 compared three different cationic surfactants, where an increase in the conjugation of the surfactant from SDS to SDBS and DOBS led to an increase in the chain order and conjugation-length of P3HT and a red-shifted absorbance spectrum. Bellacanzone et al.69 reported that the AOT surfactant, which had branched alkyl chains, could be employed to synthesize P3HT nanoparticles with a higher degree of crystallinity than SDS or CTAB possessing linear aliphatic chains. According to the literature, the emulsion effect of non-ionic surfactants is lower than that of ionic surfactants. Cho et al.169 found that the non-ionic surfactants C12E4 and Triton X-100 were insufficient to produce stable aqueous PNDI-TVT nanosuspensions, which had a tendency to under intermolecular aggregation. Given that surfactants are insulating and can act as defects or traps, which hinder the application of organic optoelectronic devices, there is usually an additional step to eliminate surfactants by centrifugation or dialysis.140 In the case of large volumes of inks used in the process for the fabrication of large-area devices, such as roll-to-roll (R2R) production, which requires 50–100 mL ink just to fill the reservoir, repeated centrifugal filtering is not suitable.172 Alternatively, the crossflow ultrafiltration technique with a flow-cartridge has been proven to be an effective method for purifying large volumes of nanoparticle inks from excess-surfactant.130,173 Although both techniques can produce the same purification effect in nanoparticle inks, centrifugal ultrafiltration proceeds faster than using a crossflow Millipore filtration system.144,145
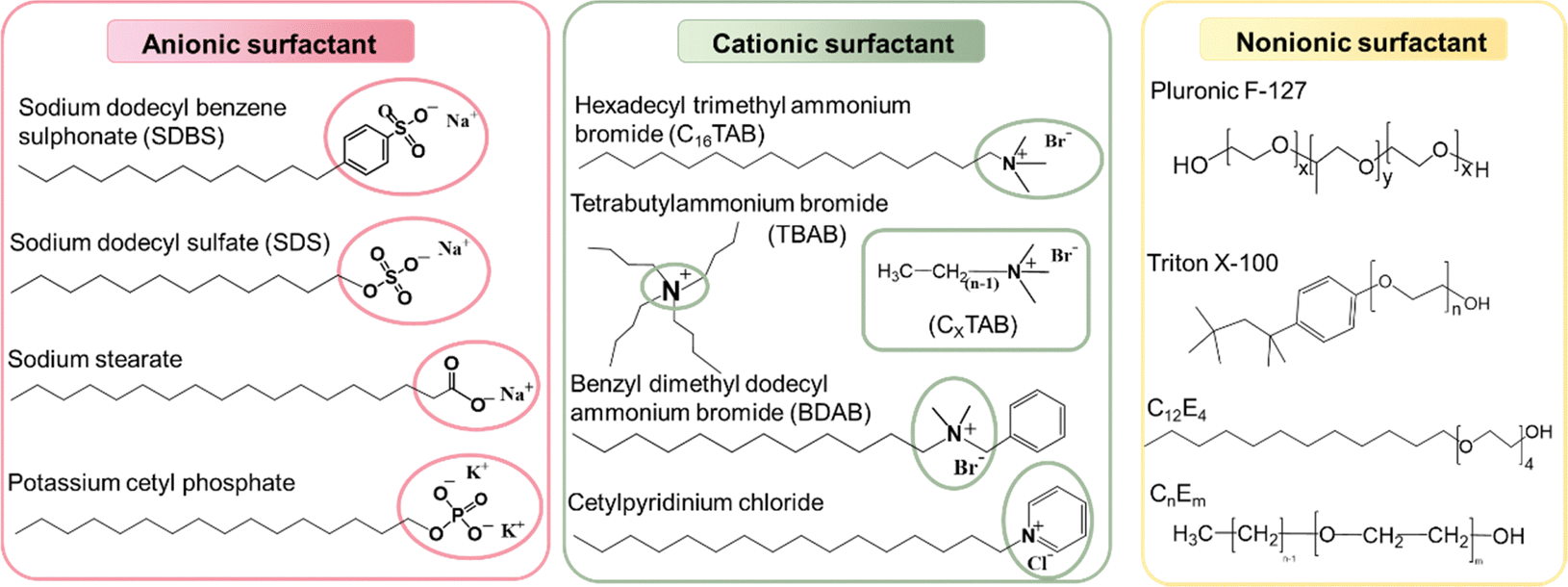 | ||
| Fig. 6 Classification of common surfactants: anionic, cationic, and non-ionic surfactants. | ||
In the case of the miniemulsion method, the particle size can be modified based on the precursor molecular weight/concentration, the ratio between the organic solvent and non-solvent, ultrasonication intensity/time and type/concentration of surfactant. Holmes et al.135 synthesized P3HT:PCBM nanoparticles incorporating P3HT with a range of molecular weights (Mw = 5–72 kDa). The nanosuspensions synthesized with higher molecular weight (72 kDa) P3HT possessed nanoparticles with a larger diameter of 42 nm compared with that with a lower molecular weight (5 kDa), showing a diameter of 28 nm. Ulum et al.148 studied the effect of varying the polymer concentration from 36 mg mL−1 to 179 mg mL−1, showing that the particle size systematically increased with an increase in the concentration of the polymer in chloroform. A higher concentration of organic solution led to a larger micelle core, thus resulting in larger nanoparticles. Assuming a constant number of droplets in the aqueous phase, a larger volume of organic solvent led to bigger droplet sizes. The size could be theoretically estimated through the total volume of the organic solution, showing an almost linear correlation.174 During the sonication process, an increase in the sonication time and amplitude resulted in further droplet fission, causing a smaller droplet size.110 It was reported that increasing the sonication time from 2 min to 5 min resulted in the formation of smaller nanoparticles (from 140 nm to 52 nm),142 and increasing the intensity from 150 W to 225 W resulted in a decrease in the nanoparticle size from 140 nm to 4 nm for PFFO nanoparticles.174 Increasing the amount of surfactant could decrease the size of the nanoparticles because of the higher surface coverage propensity.142 Colberts et al.84 increased the SDS concentration from 10 to 40 mM, and the nanoparticle size decreased from 65 to 30 nm. The reduction in the nanoparticle volume was very pronounced at high surfactant concentrations until it reached a threshold limit.
3.2 Nanoprecipitation
Nanoprecipitation is also called solvent displacement or interfacial deposition. Three components make up nanoprecipitation systems including an organic material (hydrophobic solute), organic solvent, and non-solvent. Different from the miniemulsion method, the organic solvent and the non-solvent should be well-soluble in each other for the nanoprecipitation method. The solute is dissolved in an organic solvent, forming organic solutions. The rapid injection of an organic solution into the majority of the non-solvent results in a sharp drop in solubility, inducing the solute to reach a state of supersaturation in the organic solution, finally leading to solute precipitation (Fig. 5b).175 The nanosuspensions can be obtained via the evaporation of the organic solvent.175–177 The rapid diffusion of the organic solution to plenty of non-solvent makes it sufficiently dilute to be located in the metastable region in the phase diagram, which explains the instability of nanosuspensions synthesized using the nanoprecipitation method.110,178,179 The metastable region is also called the ‘‘ouzo’’ region, named by Vitale and Katz.180,181 In the ouzo region, nanoparticles follow the mechanism of the Lamer model according to the classical nucleation theory, as shown in Fig. 2b.182 Nucleation behaviour occurs when the concentration of the organic solution reaches the critical nucleation concentration.183,184 In the later concentration dropping stage, nucleation and growth generate competition. High supersaturation will result in rapid precipitation, which favours nucleation over growth, resulting in the formation of a large number of small nanoparticles.183,185This method was first patented by Fessi et al. in 1989.176 The organic solvent needs to be miscible with a non-solvent (alcohol/water), and tetrahydrofuran or acetone is mostly selected. P3HT is acknowledged as a polymer that plays a significant role in stabilizing the particle dispersion without the need for a surfactant.186 The initial application of P3HT in the preparation of colloids via the precipitation method originated from the investigation of its thermochromic and solvatochromic effects.187,188 In 1988, Inganäs et al.189 discovered that solutions of P3HT in CF formed clear light red-orange solutions, while a colour change in the transmission of the polymer to blue-violet was observed when a poor solvent was added. The addition of a larger amount of good solvent restored the polymer to its original colour. Sandstedt et al.190 reported that the addition of the non-solvent ethanol to a P3HT chloroform solution afforded nanosuspensions. Later, Yamamoto et al. observed that the addition of methanol to a CF solution of P3HT produced a stable colloidal solution across a wide range of CF/CH3OH ratios.191,192 Subsequently, pure P3HT and P3HT:PCBM nanosuspensions were developed with the advantage of being surfactant-free.193,194
Table 3 shows different examples of the polymers, solvents, and non-solvents used in nanoprecipitation formulations and particle size achieved in the field of optoelectronics. Although a wide range of polymers are theoretically applicable for this method and successful research has been reported, only a few are applied in practice.195,196 Based on the traditional nanoprecipitation principle, Nagai et al.105 developed a two-step reprecipitation method, which divided the solution splitting and particle formation processes. Uniform size droplets were formed by mixing an organic solution with water in the absence of a surfactant. A nanosuspension was formed using these droplets in the same way by adding a poor solvent.
| Number | Material | Solvent | Anti-solvent | Surfactant | Particle size | Year | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | R-P3HT | THF | Water | Tween 80 | 2004 | 193 | |
| 2 | P3HT:PCBM | THF | Water | 37.4–67.7 nm | 2009 | 194 | |
| 3 | P3HT:PCBM | THF | Water | 41–68 nm | 2010 | 186 | |
| 4 | P3HT:PCBM | CF | Ethanol | 130 nm | 2014 | 197 | |
| 5 | P3HT:ICBA | CF | Ethanol/methanol (MeOH) | 160 nm | 2014 | 198 | |
| 6 | PFBT | THF | Water | PS-PEG-COOH | 30–50 nm | 2016 | 199 |
| 7 | P3HT:ICBA | CF | Ethanol | 100 nm | 2016 | 200 | |
| 8 | P3HT | CF + water | Ethanol | 10–65 nm | 2017 | 105 | |
| 9 | PCDTBT:PC71BM | THF | Water | 40–133 nm | 2017 | 196 | |
| 10 | P3HT:ICBA | CF | Ethanol | 104 nm | 2018 | 91 | |
| P3HT:ICBA | CF | 2-Propanol | 95 nm | ||||
| P3HT:ICBA | CF | 2-Butanol | 90 nm | ||||
| P3HT:ICBA | CF | 3-Hexanol | 83 nm | ||||
| P3HT:ICBA | CF | cis-3-Hexen-1-ol | 79 nm | ||||
| 11 | P3HT:o-IDTBR | THF | Water | F127 | 60–80 nm | 2018 | 101 |
| PBQ-QF:ITIC | THF | Water | F127 | 60–80 nm | 2018 | ||
| 12 | P3HT | THF | Water | Poly(styrene-co-maleic anhydride) | 40–189 nm | 2020 | 201 |
| F8BT | THF | Water | Poly(styrene-co-maleic anhydride) | 40–189 nm | 2020 | ||
| 13 | PM6:BTP-eC9 | THF | Water | F127 | 90 nm | 2022 | 202 |
| 14 | CNP | THF | Water | Spheres (100 nm), fibers (40 nm) | 2023 | 203 |
Nanosuspensions synthesized using precipitation methods are in metastable ouzo domains. The absence of a surfactant typically results in the formation of less stable nanosuspensions than that synthesized using the miniemulsion method. Thus, to improve the stability of nanoparticles, surfactants or additives are also added in the nanoprecipitation method. In 2004, Morgera et al.193 adopted a non-ionic biodegradable commercial surfactant (Tween 80), synthesizing stable R-P3HT aqueous nanosuspensions. Xie et al.101 selected a unique surfactant, poloxamer (Pluronic F127), with temperature sensitivity to synthesize polymer and non-fullerene blended nanoparticles. Lowering the nanosuspensions temperature to nearly 0 °C resulted in the conversion of the surfactant micelles back to linear poloxamers in the aqueous phase. This ensured the elimination of the residual surfactant in the centrifugal filtration process, achieving almost surfactant-free nanosuspensions.
The characteristics of nanoparticles prepared using the nanoprecipitation process are influenced by the polymer molecular weight/concentration, the ratio between the organic solvent and miscible non-solvent, and stirring speed. The inherent property and concentration of the polymer are the most influential parameters on the nanoparticle size.204,205 Higher molar masses or concentrations of organic materials can produce larger nanoparticles, given that more polymer chains in a single nanodroplet enlarge the diameter of the nanoparticles.102,196,206 Also, different proportions of organic solvent and water can affect the shape and size of nanoparticles.207,208 The larger the amount of water, the higher the supersaturation level, resulting in a smaller particle size. Prunet et al.196 reported an increase in the size of nanoparticles with an increase in the solvent/non-solvent volume ratio. Yang et al.203 reported the synthesis of an organic conjugated molecule that can exhibit two aggregate states in aqueous nanosuspensions. THF/H2O ratios lower than 1/25 resulted in the formation of amorphous nanosphere suspensions. The addition of more THF to the nanosuspension could promote the transformation into nanofibers, which was driven by the switch to π–π stacking. During the process of adding the organic phase to the non-solvent phase, an increase in the stirring rate induced rapid nucleation, thus producing smaller nanoparticles and a narrow size distribution.206,209,210 However, the order of the addition of the organic phase and aqueous phase had no effect on the formation of nanoparticles. Even by reversing this order and adding the aqueous phase to the organic phase led to the formation of polymeric nanoparticles (PNP).91 Chambon et al.211 reported that homogeneous nanoparticles could be prepared by quickly adding a large amount of water to the organic phase. To finely control the mixing process of the organic solution and non-solvent, Fischer et al.212 proposed a scalable high-throughput continuous-flow microfluidic system, which consisted of two syringe pumps, one of which was filled with the organic solvent, while the other was filled with the non-solvent. By adjusting the differential speed of the syringe pumps and the irradiation of the microfluidic chip, the formation of surfactant-free nanoparticles could be tuned. After the mixing process, some approaches to shorten the evaporation time were developed. Kumar et al.213 introduced a rapid solvent removal technique based on flash evaporation, and the results showed that two flash stages reduced the THF concentration by over 95%.214 Marks et al.147 also shortened the evaporation time under vacuum and heating rotation, and during this process the system needed to be vented periodically to prevent foaming (if a surfactant is present) and loss of material.
3.3 Other methods for the synthesis of surfactant-free organic nanoparticles
According to the comparison of the miniemulsion and nanoprecipitation methods, nanosuspensions synthesized from miniemulsion method are stable while existing the issue of residual surfactant, precipitated nanosuspensions without the need for surfactants are usually unstable. Therefore, some novel approaches have been developed to synthesize surfactant-free organic conjugated nanoparticles in water/alcohol dispersion medium, including electrostatic stabilization, amphiphilic copolymer self-assembly, hydrothermal method, and laser stabilization.According to the electrostatic stabilization mechanism described in Section 2.2, nanoparticles in liquid medium have surface charges and stabilized by the repulsion forces between them. The charges surrounding the nanoparticles play an important role in their electric stability. Based on the principle of solvent displacement by nanoprecipitation, that the polymer collapses into nanoparticles, and introducing the appropriate amount of electrical dopant can charge the nanoparticles, forming electrically stable surfactant-free nanosuspensions (Fig. 5d). Excessive electrical doping or the introduction of some salts (electrolytes) can cause side effects, resulting in the coagulation of the nanoparticles.215 The electrical doping effect can effectively help to stop nanoparticle growth, reducing the nanoparticle size. The common methods for charging nanoparticles include the adsorption of ions from the non-solvent or dopants/surfactants, proton transfer reactions, and charge transfer reactions.216 Manger et al.217 synthesized electrostatically stabilized nanoparticles using the p-doping agent F4TCNQ. A small amount of F4TCNQ acetonitrile solution was added to the P3HT chloroform solution prior to nanoprecipitation. Doping with F4TCNQ resulted in the formation of an ion pair, P3HT+ and F4TCNQ−. In the case of non-polar solvents such as chloroform, the ion pair remained bound owing to the Coulomb attraction forces. When a chloroform solution was rapidly injected into ethanol, P3HT nuclei were formed, maintaining the charge distribution. The F4TCNQ− anions dissociated from the nanoparticles due to the high permittivity of ethanol, leaving a positive charge on the P3HT nanoparticles. Manger et al.218 found that the addition of iodine (I2) could induce the oxidation (p-doping) of the polymer, promoting electrostatic repulsion between the nanoparticles, and thus forming stable surfactant-free J71:Y6 nanoparticles. Due to the high ionization potential of Y6, the polarons of I2 only acted on J71 for charging J71:Y6 composite nanoparticles. Liu et al.219 found that the conjugated polymer BBL with remarkable electron mobility and high electron-affinity (EA) was not soluble in any common solvents and quickly aggregated in water. Mutual electrical doping occurred when a water-soluble donor polymer PACT-K with a low ionization energy (IE) was mixed with BBL, inducing the formation of stable aqueous nanosuspensions. A spontaneous ground-state electron transfer (GSET) in the blended solutions promoted the dispersion of BBL, and the nanoparticle size decreased from ∼530 nm for pure BBL to about 30 nm for BBL:PCAT-K. Li et al.220 also found that pristine single-walled carbon nanotubes (SWCNTs) exhibited hydrophobic characteristics, which precipitated immediately in most organic solvents and water. A well-dispersed nanosuspension could be obtained by adding SWCNTs to an aqueous nanosuspension of PC61BM nanoparticles. The PC61BM nanoparticles could directly attach to the surface of SWCNTs, and the zeta potential of the PC61BM:SWCNT (1:1) nanosuspension was −30.7 mV lower than that of the pure PC61BM nanosuspension of −41.4 mV. Based on the electrostatic effect, Saxena et al.108 synthesized pyridine end-capped P3HT (P3HT-Py), which could stabilize the P3HT:ICBA dispersions as an additive. When using acetic acid (AcOH) as the non-solvent, the P3HT-PyH+AcO− ion pair was formed. This ion pair could effectively stabilize the nanosuspensions and AcOH could be evaporated by thermal annealing at 120 °C or above after deposition.221
As shown in Fig. 5c, nanoparticles can also be synthesized through the self-assembly of amphiphilic rod–coil block copolymers (BCPs). BCPs have been widely used to achieve self-assembled nanoparticles with a variety of peculiar nano morphologies, such as lamellar, spherical, cylindrical, vesicular, and microporous structures.222–225 The hydrophilic block in these copolymers includes polyethylene oxide (PEO) or polyethylene glycol (PEG), polyvinyl pyrrolidone (PVP), and polyvinyl alcohol (PVA).226 In 2018, Zappia et al.227 synthesized an amphiphilic rod–coil block copolymer (BCP5). The hydrophobic rod was a p-type semiconductor (PCPDTBT), while the hydrophilic coil was a short chain of poly-4-vinylpyridine. The hydrophilic coil interacted with water, serving as a surfactant during the nanoparticle synthesis process. A BCP5:PC61BM chloroform solution was poured into water to obtain macro suspensions, and then the macro suspensions were sonicated for 10 min. A stable surfactant-free aqueous nanosuspension was obtained after the removal of the organic solvent. Ganzer et al.228 investigated the effect of coil length (from 2 to 100 hydrophilic coil units) on the formation of free charges in organic solar cells through ultrafast transient absorption (TA) spectroscopy. Using conjugated rods (PCPDTBT) coupled with various 4-vinylpyridine (4VP) based flexible coils, a thin asymmetric carbon-rich shell structure was observed in the neat BCP2 nanoparticles films, while the BCP100-based nanoparticles did not exhibit this well-defined core–shell structure, possessing a less defined shape. In 2022, Diterlizzi et al.229 selected PTB7 as the rod block coupled with a segment of P4VP composed of around 15 repeating units because of the higher polymer crystallinity of PCPDTBT. The obtained blended aqueous nanosuspensions were spherical with a Janus structure, featuring PTB7-b-P4VP mainly on a half of the sphere, while PC71BM was mostly on the other half.
Other promising methods such as hydrothermal processes and laser stabilization have been reported for the synthesis of organic conjugated nanoparticles. Cho et al.230 rapidly injected perylene THF solutions into water using the nanoprecipitation method to form pristine perylene nanoparticles. In the hydrothermal process, the nanosuspensions were placed in a hydrothermal chamber and heated in an oven at various temperatures ranging from 110 °C to 160 °C for 10 h. The physical shape of the perylene nanoparticles varied from rectangular to polyhedral, ellipsoidal, and finally spherical with an increase in the hydrothermal temperature. Jeon et al.231 added PTCDI-C13 to acetonitrile solutions and mixed them under sonication for 1 h. The PTCDI-C13/acetonitrile mixture together with a magnetic stirring bar was placed in a quartz cell, and an Nd:YAG nano-second pulse laser was used for irradiation. After laser irradiation for 10 min, PTCDI-C13 was fragmented into nanoparticles, obtaining a dilute nanosuspension. As discussed above, novel synthesis methods can be employed to synthesize surfactant-free nanoparticles, which can remain stable for a certain time, while the shortcoming of these methods are not widely universalized.
3.4 Structures of synthesized organic nanoparticles
Nanoparticles containing two organic conjugated materials (A and B) can be divided into two types according to mixing before ultrasonication/stirring or mixing after ultrasonication/stirring (Fig. 7). Mixed nanoparticles based on pure nanoparticle A and pure nanoparticle B can be named mNPs. The nanoparticles synthesized by injecting an A:B blended organic solution to a non-solvent are called bNPs or composite nanoparticles. mNPs are obtained from preformed nanoparticles, and thus their shape is decided by the shape of pure nanoparticles A/B. The shape of nanoparticles is not always spherical. In addition to spherical nanoparticles, non-spherical organic nanoparticles have been previously reported.232–235 The shape anisotropy was linked to the internal arrangement of the polymer chains within the nanoparticle.233 In the case of composite nanoparticles, they are simply regarded as nanospheres for research inside the nanoparticle. According to the distribution of A and B inside one nanoparticle, the structures of composite nanoparticles are classified as core–shell,211,236 Janus,237 intermixed or more complex structures (Fig. 7).238 The core/shell type of nanoparticles refers to multicomponent nanoparticles consisting of one component mainly distributed in the core of the sphere and the other component mainly in its shell. In addition to the common core/shell structures, eccentric, uniform, and occluded can be possibly formed based on the method of nanoparticle preparation.238 Janus nanoparticles are named after the two-faced Roman god, Janus, given that they have asymmetric structures and different properties of ‘‘two faces’’. The intermixed structure represents a high degree of mixing of the two materials inside the nanoparticle. The different distributions of the two components depends on two main aspects, i.e., the surface energy difference between the two materials and the synthesis method.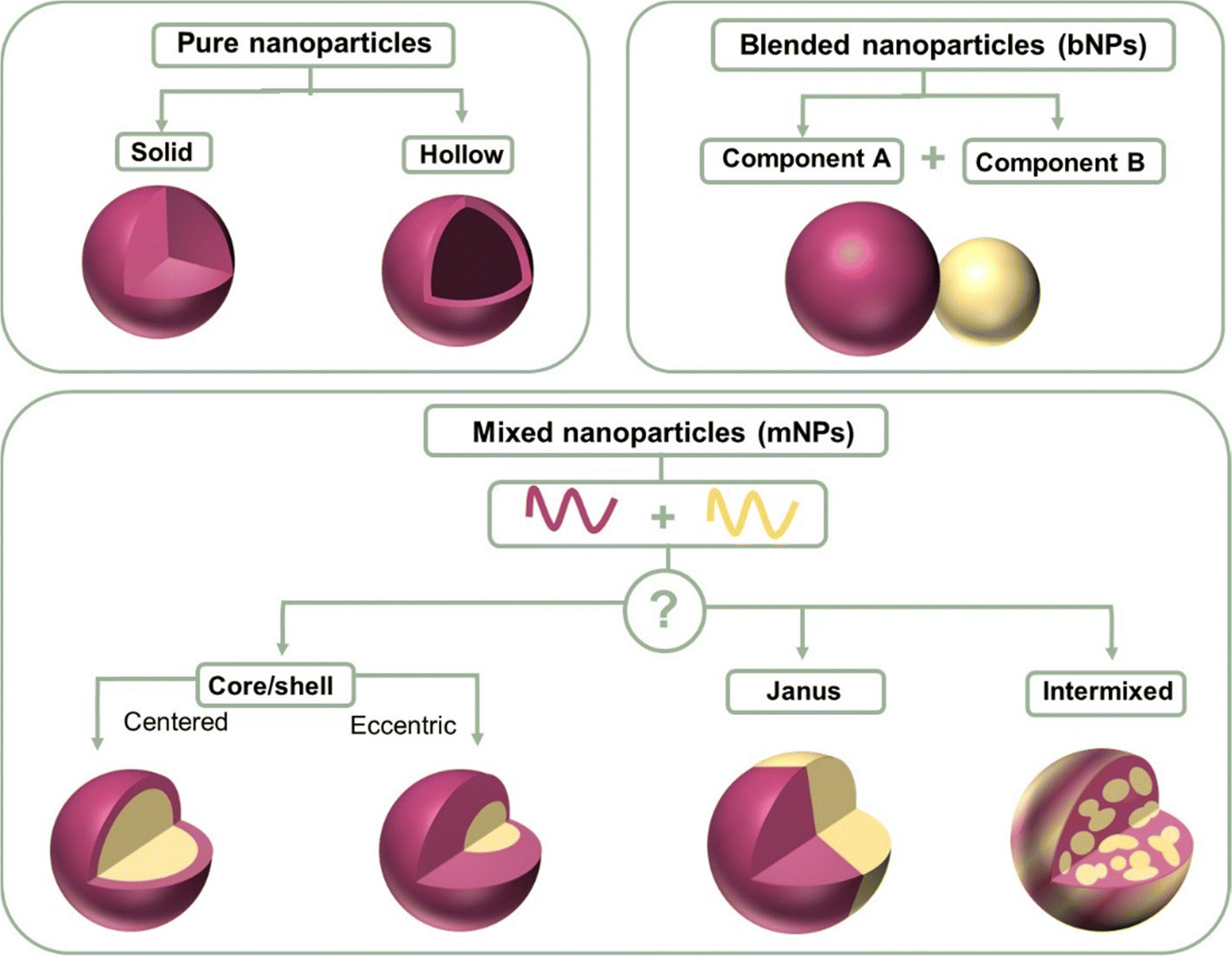 | ||
| Fig. 7 Structure schematic illustration of pure nanoparticles, blended nanoparticles, and mixed nanoparticles. | ||
The surface energy between two components plays a more important role in affecting the parameters of component distribution inside nanoparticles. Due to the higher surface energy of fullerene derivatives, (38.2 mJ m−2 for PC61BM and 39 mJ m−2 for PC71BM), polymer (donor):fullerene (acceptor) the composite nanoparticles synthesized using the miniemulsion method usually exhibited a core–shell structure with a polymer shell and fullerene core.239 As reported, P3HT:PC61BM,240 TQ1:PC71BM,140 PDPP5T:PC60BM,84 and P3HT:ICxA (a novel indene-adducted fullerene acceptor mixture)145 all showed a fullerene-rich core/polymer-rich shell. Richards et al.238 prepared P3HT:PC61BM nanosuspensions with varying P3HT/PCBM ratios. At a small percentage of P3HT, PCBM was still enriched in the core. In the case of P3HT-rich particles, they were eccentric, and the center of the particle was displaced from ∼10% of its radius. When the fullerene phase was composed of 5 wt% C60 and 95 wt% PCBM, the P3HT:fullerene composite nanoparticle showed an eccentric or anisotropic structure.
The material transition from fullerene to non-fullerene acceptor materials (NFA) expands the synthesis versatility of NFAs, allowing a broader range of surface energy. The preparation of core–shell structures with a donor-rich core/acceptor-rich shell has also been reported using the miniemulsion method, such as P3HT:eh-IDTBR,149 P3HT:N2200,149 and TQ1:N2200 systems.149 The converse core/shell structure could be explained by the lower surface energies of NFAs than fullerene derivatives. In addition, composite nanoparticles can also exhibit an intermixed structure or Janus structure using the miniemulsion method. Laval et al.86 chose two different organic systems, low surface energy difference PTQ10:Y6 (4 mN m−1) and large surface energy difference PTQ10:PC61BM (9.5 mN m−1). No clear phase separation was observed within the PTQ10:Y6 nanoparticle that was intermixed. Alternatively, the PTQ10:PC61BM nanoparticle exhibited the classical core–shell structure. The lower the surface energy difference, the closer the nanomorphology was to an intermixed structure.149 As shown in Fig. 4f, Du et al.75 using the different solubility properties between donor and acceptor materials converted the PTB7:PC71BM core–shell structure into a Janus structure.
In the case of the nanoprecipitation method, in addition to the common intermixed structure, nanoparticles can also form a core–shell/Janus structure.151,241,242 Chambon et al.211 conducted a sequential double nanoprecipitation technique. Initially, a P3HT:PC61BM THF solution was dropped into dimethyl sulfoxide (DMSO) with P3HT precipitating as the core, where PC61BM remained soluble. Subsequently, the dispersion was poured into the water phase, forming the PC61BM shell of the nanoparticle. Holmes et al.243 found that that P3HT:PC61BM composited nanoparticles precipitated in water exhibited a Janus structure. According to the measurement results of cryo-TEM, a Janus structure was observed, with P3HT (lighter) and PC61BM (darker) clearly phase segregated from each another with or without the presence of surfactants. The crystallinity of the P3HT domains could be observed in the bright face with striped areas characteristic of lamellar stacking of regioregular P3HT polymer chains.244 The generation of the Janus structure was found to be closely linked to the interaction parameters between two organic materials, as well as the choice of non-solvent.245
4. Fabrication of organic thin films from nanosuspensions
In this section, we focus on the process of transforming organic nanoparticles from nanosuspensions into thin films. Different from the deposition from organic solutions, the hydrophobic nature of the substrate induces the incompatibility between the substrate and the aqueous nanosuspension, and the high boiling point of water makes the deposition process challenging. Therefore, a pre-treatment, such as oxygen plasma/UV-ozone treatment, is needed prior to the deposition of organic nanosuspensions to improve the wettability of the substrate (Fig. 8).246,247 Nanoparticle films can also be formed using common solution processing methods, including spin-coating, doctor-blading, spray-coating, and ink-jet printing. The hydrogen bonds and van der Waals forces between surrounding nanoparticles are crucial in the formation of films, leading to diverse microstructures in nanoparticle-based films.248 The characterization for nanoparticle-based films is similar to that of solution-cast films, focusing more on surface roughness and phase separation. Due to the inherent properties of nanoparticles, the morphology of organic thin films formed from organic nanosuspensions differs from that processed from organic solutions, which create “globular/granular” domains with interstitial gaps between nanoparticles.143,249 Therefore, a post-treatment is performed to fill the interstitial gaps between nanoparticles and eliminate the insulating residual surfactants in the films. | ||
| Fig. 8 Schematic illustration of the process from nanosuspensions to nanoparticle-based thin films. | ||
4.1 Pre-treatment before nanosuspension deposition
To solve the incompatibility between the substrate and nanosuspension, several methods have been developed. The most common approach is using oxygen plasma/UV-ozone-based surface treatments to change the surface chemistry of the substrate, which can modify the interfacial surface energy (Fig. 8).246,247 Substrates are treated with UV-ozone for a long period before the deposition of the nanosuspension to increase the hydrophilicity of the surface.86 Almyahi et al.250 discovered that ozone treatment reduced the sodium and sulphur levels on the surface of a film, while increasing the oxygen content. This was consistent with previous reports, indicating the generation of hydroxyl and carboxyl groups on the sample surface following the ozone process. Taking silicon wafers as an example, the contact angle decreased from 49° (bare silicon wafer) to 24° and 6° after being treated with UV-O3 and O2-plasma, respectively. UV-ozone treatment has been proven to be effective for small-scale thin films. Alternatively, in the case of R2R-printed films, ozone surface treatment could be achieved through corona discharge, and the conditions were optimized by coating water-based nanosuspensions using a single stripe slot-die head onto substrates treated with varying corona intensities in the range of 0 to 100 W m−2 min−1.130,250Controlling the appropriate surfactant content is essential for the deposition of organic nanoparticle films. The residual surfactant in nanosuspensions can reduce their surface tension, making it easier to spread over the substrate for deposition. However, too much residual surfactant as impurities reduces the quality of the film, increasing its roughness. Almyahi et al.144 found that a spin-cast nanoparticle-based film was not fully covered over the whole substrate and formed aggregates, resulting in uneven films with high roughness. As reported, the surface tension of organic nanosuspensions containing SDS that is suitable for deposition is around 38–48 mN m−1.101,144 Other methods, such as mixing water with a certain proportion of alcohol, have also been developed to decrease the total surface tension of nanosuspensions, forming homogenous films. Yamamoto et al.139 added 20% ethanol in an aqueous nanosuspension to decrease its surface tension, and the contact angle between the nanosuspension and substrate changed from 59° to 45°. The optimized binary nanosuspension improved the wettability and formed a more homogeneous film.
4.2 Process methods used in the deposition of nanoparticle-based films
Similar to solution cast films, spin-coating is the most commonly used method for the deposition of nanoparticle-based films in the laboratory. The nanosuspensions synthesized through the miniemulsion route have high concentrations that can reach 60 mg mL−1, which is sufficient to achieve the normal thickness of functional layers in the fabrication of optoelectronic devices.143,251 When nanosuspensions were synthesized without the addition of a surfactant, multiple repetitions of deposition were required to achieve a normal thicknesses due to the low concentration of the suspensions. Manger et al.218 deposited J71:Y6 nanosuspensions via spin-coating on a PEDOT:PSS substrate at 800 rpm. Repeating the deposition steps 50 times was required to achieve about 80 nm-thick films for dispersion with 20% I2. As the mass ratio of I2 increased to 80%, only 23 repetitive coating steps were needed to obtain 90 nm-thick layers. In the case of diluted nanosuspensions, Hernandez et al.249 deposited nanoparticle-based films by drop-casting and observed the coffee ring effect. Holmes et al.143 compared the film morphology of nanoparticle-based and organic solutions cast through spin-coating, as shown in Fig. 9a. The morphology of the PDPP-TNT:PC71BM cast from chloroform solutions was cobblestone like, consisting of PC71BM-rich islands (200–500 nm in size) surrounded by PDPP-TNT polymer-rich valleys. The P3HT:PC61BM films coated from chloroform solution possessed a finely blended morphology. In contrast, the morphology of the PDPP-TNT:PC71BM or P3HT:PC61BM films formed from nanoparticle nanosuspensions showed “globular/granular” domains. The domain size could be adjusted by tuning the size of the nanoparticles. | ||
| Fig. 9 (a) SEM images of PDPP-TNT:PC71BM and P3HT:PC61BM films deposited from nanosuspensions or organic solutions. Reproduced with permission from ref. 143. Copyright 2017, Materials Research Society. (b) Double slot-die coating equipment: simultaneous coating of P3HT:PCBM nanosuspensions (red) and PEDOT:PSS (blue). Reproduced with permission from ref. 252. Copyright 2011, Elsevier B.V. (c) Schematic representation of the electroplating method used for coating a conductive substrate with a layer of organic polymer nanoparticle. Reproduced with permission from ref. 253. Copyright 2004, Elsevier Ltd. (d) Digital photos of the PTB7:PC71BM nanoparticle alignment from net-like structure to a close-packed structure at air/water interface. Transferred nanoparticle film on various substrates: (e) quartz glass, (f) silicon wafer, (g) filter paper, (h) flexible PDMS, and (i) patterned PTF.254 | ||
The spin-coating method is widely used due to its simple operation and advantage of realizing uniform films with a wide range of thicknesses, while this method is not suitable for deposition on large areas or flexible/curved substrates. To realize the deposition of water/alcohol nanosuspensions on large areas or flexible/curved substrates, doctor blading or other printable deposition techniques have also been investigated and developed. Sankaran et al.200 reported the feasibility of blading P3HT:IC60BA nanoparticles dispersed in ethanol. After repeatedly blading two times, the obtained film thickness was similar to that by spin-coating. The blading speed was inversely proportional to the obtained thickness, and a faster blading speed led to the more rapid accumulation of nanosuspensions, resulting in the formation of thick films.255 In addition, Yamamoto et al.139 printed aqueous P(TBT-DPP):ICBA nanosuspensions onto flexible ITO-free substrates. The printed nanoparticle film thicknesses ranged from 100 to 150 nm. Different from doctor blading, ink-jet printing or spray coating has strict requirements regarding the volatility of the non-solvent and temperature. Low boiling non-solvents, such as the common ethanol, can easily cause clogging of the print nozzles due to their high volatility. Therefore, binary solvent blends are often used to solve this problem, through adding part of the high boiling point non-solvent to increase the overall boiling point.256,257 Sankaran et al.200 optimized the temperature and ink composition for inkjet printing. Ethanol was mixed with the high-boiling point 2-butoxyethanol as a binary solvent blend (3:1 v/v) to deposit P3HT:IC60BA nanosuspensions. The optimized P3HT:IC60BA layers exhibited similar homogeneity for both spin-coated and doctor-bladed films.
Aqueous organic nanosuspensions were also applied in R2R large-scale production, as reported in the literature.130,250 Andersen et al.130 carried out R2R slot-die coating on flexible substrates using aqueous nanosuspensions. They found that the shear force produced by the coating process could cause depletion of the surfactant on the nanosuspension surface, and therefore the surface tension of the nanosuspensions gradually increased to dewetting limit during coating. Therefore, the coating speed must be reduced to allow the surfactant sufficient time to diffuse to the sample surface and maintain a low surface tension. Larsen-Olsen et al.252 adopted the simultaneous formation of two layers by double slot-die coating from P3HT:PCBM aqueous nanosuspensions and PEDOT:PSS aqueous dispersions. As shown in Fig. 9b, the aqueous P3HT:PCBM nanosuspension was pumped into the first chamber, while the aqueous PEDOT:PSS dispersion was pumped into the second chamber. Two films could be deposited simultaneously, and the TOF-SIMS depth profiling results proved that a double layer was indeed formed without detrimental interlayer mixing. Compared to small-area nanoparticle-based films, there are fewer reports on the large-area deposition of water/alcohol nanosuspensions. The deposition of large-area nanoparticle-based films is currently based on the R2R blading technique, and its application is in the preliminary stage. The obtained film roughness is higher than that of small-area nanoparticle-based films prepared using the spin-coating method, and thus improvements in the deposition approaches are required in the future to realize the non-toxic industrial fabrication of high-performance devices.
In addition to the common solution processing techniques, nanosuspensions can also be deposited via the water transferring technique or electroplating. Snaith et al.253 developed an electroplating method for the formation of a thin film of organic nanospheres. They deposited PFB:F8BT aqueous nanosuspensions onto ITO connected to the negative output of a power supply, with another slide connected to the positive output (Fig. 9c). A continuous monolayer was formed after 30 s of electroplating, and the film thickness was almost the same as the nanoparticle diameter of 80 nm. Du et al.254 introduced an air/water interfacial transferring technique, which could easily deposit nanosuspensions onto desired substrates, whether hard (quartz glass, silicon wafer), soft (filter paper, flexible PDMS), or curved (Fig. 9d–i). During this deposition approach, the aqueous PTB7:PC71BM nanosuspensions were blended with alcohols (e.g. ethanol/isopropanol/butanol) and injected slowly onto the surface of water. The surface tension difference caused the nanoparticles to migrate toward the higher tension area and pack from a net-like structure to sparse hexagonal arrays, and then the nanoparticle-films were transferred to the desired substrates.258–260
4.3 Nanoparticle stacking pattern in nanoparticle-based thin films
During the deposition of single or multi-component nanoparticle inks, the nanoparticles can assemble into ordered or randomly packed particles, depending on their particle–particle interactions, size dispersity, and size ratio (Fig. 10a and b).261–263 In the case of inorganic nanoparticles, the stacking between surrounding nanoparticles contacts is hindered by ligands that are strongly bound to the nanoparticle surface, resulting in poor charge transport.264 In organic nanoparticles, neighboring particles are connected by strong van der Waals interactions between them because the weakly bound surfactants can be dislodged to a certain extent, leading to jammed disordered assemblies with intimate nanoparticle contacts.265,266 Regarding the repeated deposition of nanosuspensions, a good agreement between the actual metrical layer thickness and the predicted model has been reported, suggesting that the nanoparticle film is constructed from sequential close-packed monolayers (Fig. 10c).131 | ||
| Fig. 10 Nanoparticle stacking 2D pattern of (a) blended nanoparticles (bNPs), (b) mixed nanoparticles (mNPs), and (c) 3D pattern nanoparticle-based films. | ||
Vaughan et al.267 found that the nanoparticulate active layers were deposited in a hexagonal close packed (HCP) structure for both single and double layers through SEM images when the size distribution of the nanoparticles was low. Holmes et al.90 observed that different organic polymer nanoparticles were arranged in different ways. Pure P3HT nanoparticles were packed in a mostly randomly close packed (RCP) array, whereas pristine PC61BM nanoparticles formed both RCP arrays and hexagonally close packed (HCP) arrays. They also prepared P3HT:PC61BM mNPs by mixing pure P3HT nanoparticles and pure PC61BM nanoparticles. A two-phase microstructure film was generated by depositing mNPs, and the two types of nanoparticles were evenly mixed throughout the mNP-film. mNPs formed jammed assemblies with particle–particle contacts, and the packing of the P3HT:PC61BM mNP-film was mostly an RCP array.90 Gehan et al.137 dipped P3HT:PCBM nanoparticle films into dichloromethane to experimentally differentiate the P3HT and PCBM nanoparticles. The binary-scale SEM images showed that nearly half of the film was P3HT nanoparticles, while the other half was voids, given that PCBM was removed (Fig. 11a). The results showed that P3HT and PCBM were closely packed in the composite nanoparticle-based films before treatment. Alternatively, Pedersen et al.268 observed that the spherical particles in a dispersion collapsed into oblate nanoparticles upon film deposition. The differences in the two film structures were speculated to be due to the variations in the nanoparticle synthesis.
 | ||
| Fig. 11 (a) Top view SEM images of P3HT:PCBM blended nanoparticles without treatment (left) and after being dipped in DCM for 15 min (right). Binary scale SEM images of P3HT:PCBM blended nanoparticles without treatment (left) and after being dipped in DCM for 15 min (right). Reproduced with permission from ref. 137. Copyright 2014, the American Chemical Society. (b) TEM images of TQ1:PC71BM nanoparticle film following thermal annealing treatment. Reproduced with permission from ref. 140. Copyright 2016, Elsevier Ltd. (c) SEM images and (d) 3D AFM images of PM6 nanoparticle films with or without thermal annealing treatment. (e) Cross-sectional SEM images and EDS mapping of PM6:BTP-eC9 layer-by-layer films under different thermal annealing treatments. Reproduced with permission from ref. 269. Copyright 2024, The Royal Society of Chemistry. (f) Optical microscopy images of IDT-BT nanoparticle films under different post-treatments. Reproduced with permission from ref. 249. Copyright 2020, Elsevier B.V. (g) X-ray photoelectron spectroscopy analysis of PIDTBT and PDPPTBT nanoparticle films before (red lines) and after the post-washing process (green line for PIDTBT and blue line for PDPPTBT).270 | ||
4.4 Post-treatment after nanosuspension deposition
Different from solution-cast films, nanoparticle-based films consist of packed nanoparticles with interstitial gaps and limited contact (Fig. 10a and b). Accordingly, precise control of the post-treatment is crucial for achieving a homogenous nanoparticle film as well as removing the moisture and avoiding large phase separation.A drying step (sometimes omitted) that involves the gentle sintering of the particles is also essential for nanoparticle-based films to fill their interstitial gaps.126,131,132,232 The drying temperature is usually less than 150 °C and conducted in an air environment. Alternatively, a high-temperature thermal annealing process (mostly higher than 150 °C) allows the diffusion of the materials in the interparticle voids, leading to the formation of more compact films (Fig. 8). In the case of multi-layered nanoparticle films, annealing will lead to a slight reduction in their thickness, indicating nanoparticle sintering and filled void spaces.131,147 The optimal annealing conditions are usually affected by the glass transition temperature (Tg) and crystallinity of the organic materials. When the annealing temperature is higher than Tg, materials transition from the glassy state to the rubbery state with the movement of the polymer chains. If the material Tg is too high, it will cause the difficulty in film annealing without degrading other components. Meanwhile, Tg should not be too low (higher than operating temperature of OSCs, which can reach to 80 °C) to avoid excessive phase separation and ensure that mild drying treatments can be performed to remove the residual casting/printing solvent without affecting the film morphology.271 Obvious phase segregation was observed for annealed P3HT:PCBM nanoparticle films due to the relatively low Tg of P3HT.272 Amorphous materials more easily achieve viscous flow during sintering given that their viscosity quickly decreases above Tg.273 Holmes et al.140 confirmed that the high Tg (100 °C) and non-crystalline polymer TQ1 was an interesting candidate to conduct mild thermal annealing treatments in the range of its Tg, making delicate morphology modifications.274,275 As shown in Fig. 11b, thermal treatments at 110 °C and 120 °C (above the Tg of the polymer) effectively sintered the TQ1:PC71BM composite particles. Between 110–160 °C, the PC71BM-rich cores remained largely glassy, resulting in limited material mobility. Above 160 °C, the PC71BM-rich nanoparticle core domains transitioned to the viscous state (Tg of PC71BM is 163 °C),276 leading to the free movement of the materials within the bulk film, and consequential gross phase separation. As shown in Fig. 11c, Xie et al.269 found that thermal treatment from 100 °C to 200 °C gradually sintered PM6 nanoparticles and blurred the mesopores. The PM6 aqueous nanoparticle-based films without thermal annealing showed discrete spheres and cavities, while the films after annealing at 200 °C exhibited a tightly interconnected film with a smoothened surface, as shown in Fig. 11d. According to the cross-sectional SEM images together with energy dispersive spectroscopy (EDS) mapping (Fig. 11e), higher temperature thermal treatment of the PM6 nanoparticle film could inhibit the interdiffusion of further deposited layers. Holmes et al.153 proposed that high thermal treatment can pose challenges for large-scale production. Therefore, they synthesized “soft” PTQ10:Y6 nanoparticles using the nanoprecipitation method, with a lower crystallinity than their counterparts synthesized via miniemulsion. The required annealing treatment was reduced to 130 °C for 5 min, increasing the corresponding device performance.
Most commonly, the morphology of composite nanoparticles can be changed owing to the diffusion of their components during thermal annealing.133,273 Thermal annealing could change the structure of P3HT:ICBA nanoparticles from core–shell structure to intermixed structure.133 Upon annealing, diffusion from the shell to outside the particle was activated and ICBA continued to move from the core to the shell to become homogenously blended. This is because ICBA remained miscible in P3HT at all weight fractions and was not affected by P3HT crystallization, avoiding phase segregation. Holmes et al.135 determined the extent of polymer movement in nanoparticles of a given radius (“threshold mobility radius”) during thermal annealing, using the Stokes–Einstein continuum model.236 In the case of low molecular weight P3HT (less than 44 kDa), the core of the nanoparticle could move freely inside the nanoparticle due to its large threshold mobility radii (>1 μm). Alternatively, for P3HT (Mw, 72 kDa), the threshold mobility radius was less than the nanoparticle radius, and thus the core was immobile.
The residual surfactant that has not been eliminated by the centrifugation process acts as impurities or insulating traps. As reported, high temperature annealing above the boiling point of the surfactant could remove it.131 Cho et al.277 removed the SDS surfactant from a film by annealing it at a high temperature of 270 °C. However, due to the high boiling point of surfactants, it is not ideal to remove them via thermal evaporation, which will exceed the limit temperature of organic films. During thermal treatment of the nanoparticle-based layer, the nanoparticles coalesce and exclude SDS to the film-air interface, where they take a preferential orientation.278,279 Based on the solubility of the used surfactant, one can select a solvent that can highly solubilize the used surfactants but not organic materials to wash or soak the formed films (Fig. 8), such as water/alcohol.84,269 Hernandez et al.249 dropped IDT-BT nanosuspensions on a substrate and observed the films using optical microscopy. Given that IDT-BT was an amorphous polymer, the crystals observed under optical microscopy were the residual SDS surfactant (Fig. 11f). They reproduced the same drop-casting procedure with annealing at 250 °C, observing smaller crystal sizes. After washing the films with water, empty spaces were generated where the crystals were previously found, and a sintered surface was observed upon annealing at 250 °C for 10 min (Fig. 11f). Rahmanudin et al.270 dropped ethanol on an organic nanoparticle thin film, leaving it for 60 s, and then spinning the film to remove SDS. The post-washing step was proven to be effective by X-ray photoelectron spectroscopy (XPS) (Fig. 11g). The intensity of the sulphate peak was reduced after the post-washing step, and the samples after washing showed a similar intensity to that of DCB solution-cast thin films. However, it is important to note that prolonged immersion to ensure adequate surfactant removal may result in poor connectivity between the nanoparticle film and the substrate, leading to subsequent degradation of the device performance. In this case, SDS could be efficiently removed by immersing the substrate in a water–ethanol mixture (1:1), followed by drying at 110 °C. In contrast, it was observed that when ZnO was used as the electron transport layer, the washing process in water–ethanol was unsuccessful because ZnO was not resistant to the washing step.84
5. Applications of organic nanoparticles in optoelectronic devices
With the emerging development of organic optoelectronic technologies, organic optoelectronic devices have shown broad application prospects. In this case, the use of toxic organic solvents in the fabrication of devices poses a serious environmental hazard; however, common approaches such as exploring non-halogenated solvents and side-chain modification do not address the root of this problem. Accordingly, dispersing organic materials as nanoparticles in non-toxic water/alcohol solvents provides a perfect and universally applicable solution to the toxicity problem. In addition, the unique properties of nanoparticles provide new ideas for the optimization and tuning of organic optoelectronic devices. Thus far, organic nanoparticles have gradually attracted attention in the field of optoelectronics. Here, we focus on the representative applications of organic optoelectronics, including organic solar cells (OSCs), organic electroluminescent devices (OLEDs), organic thin film transistors (OTFTs), and organic photocatalysts for the hydrogen evolution reaction (HER).5.1 Organic solar cells (OSCs)
Water/alcohol-based organic nanosuspensions have been applied in the deposition of active layers or interface layers of OSCs. Aqueous organic nanosuspensions were introduced as active layers in OSCs in 2003,125 with low current, while after about 20 years of development, the efficiency and current density of nanoparticle-based OSCs increased to more than 10%,86,153,202,218 as shown in Fig. 12a and b. | ||
| Fig. 12 (a) Efficiency development, and (b) current density development of water/alcohol-based nanoparticle OSCs (organic nanosuspensions applied as active layers). | ||
In the following years, the development of water-based nanoparticle OSCs mainly focused on P3HT and fullerene acceptors. Lee et al.132 prepared P3HT:PCBM nanoparticle OSCs using the miniemulsion method and demonstrated the photovoltaic effect using conducting AFM under illumination. Larsen-Olsen et al.173 reported the fabrication of R2R-printed OSCs based on P3HT:PCBM nanoparticles with a PCE of 0.3%. When annealed, the nanoparticle P3HT:PCBM OSCs underwent extensive phase segregation, leading to a decrease in the device performance. Ulum et al.133 demonstrated OSCs based on stabilized P3HT:ICBA nanoparticles using the miniemulsion method, which yielded a PCE of 2.5%. In 2014, Gärtner et al.198 synthesized P3HT:ICBA nanoparticles using a surfactant-free precipitation method, and the nanosuspensions were stable for a couple of days without any visible particle sedimentation. Without the limitations imposed by utilizing surfactant-stabilized nanoparticles, OSCs were obtained from a surfactant-free alcoholic dispersion with a PCE of 4.1%, which can almost match the PCE of 4–6% for inverted P3HT:ICBA bulk-heterojunction (BHJ) OSCs in the literature that were fabricated from chlorinated solvents.198,280–283 Additionally, Sankaran et al.200 fabricated P3HT:ICBA nanoparticle OSCs via the doctor blading and ink-jet printing process. The yielded ethanol nanoparticle-based OSCs achieved a PCE of 3.9% for 0.105 cm2 small area devices, which is comparable with that of spin-cast devices.
According to the above-mentioned reported results, the efficiencies of P3HT:PC61BM nanoparticle OSCs fabricated using the nanoprecipitation method are usually higher than that using the miniemulsion method with surfactant. This can be explained by the formed core–shell structure when using the miniemulsion method, which is believed to hinder charge generation, and especially transport in these materials.91,284–286 Darwis et al.287 studied the difference between nanoprecipitated and SDS-stabilized nanoparticle OSCs in response to extended thermal annealing. Under 140 °C thermal annealing for different times, the performance of the precipitated nanoparticle OSCs increased systematically as the annealing time increased. In contrast, the performance of the SDS-stabilized nanoparticle OSCs reached the maximum after only 4 min of annealing, and then their performance dropped rapidly until the devices no longer functioned.
To avoid the use of surfactant, several attempts were made to synthesize organic donor copolymers, and then couple them with an acceptor to form D:A aqueous nanosuspensions. The BCP5:PC61BM blended nanoparticle aqueous suspension prepared by Zappia et al.227 achieved a PCE of 0.52%, which increased to 2.53% with the deposition of a top thin PC61BM layer from a chlorinated solution. Ganzer et al.228 investigated that increasing the coil length resulted in a decrease in the charge formation efficiency. In 2022, Diterlizzi et al.229 prepared PTB7-b-P4VP:PC71BM blended nanoparticles with a Janus structure. The nanoparticle OSCs showed a better device performance with a PCE of 0.85% compared with that of organic solution cast OSCs of 0.63%.
000× higher electrical conductivities than the pristine polymers. Therefore, the devices based on the BBL:PCAT-K films as the electron transport layers and PM6:Y6:PC71BM ternary films as the active layers achieved a high PCE of 16.03%.
For the application of nanosuspensions in the active layers, repeated deposition in the fabrication of multilayered BHJ structures of NP-OSCs has been widely adopted due to the relatively low concentration of nanosuspensions or the need to fill the gaps generated during repeated deposition.218 Due to the unique shape and stacking of nanoparticles, repeated deposition can help fill in the gaps between adjacent nanoparticles and decrease pinholes.198,200 Stapleton et al.131 demonstrated that the surface roughness consistently decreased with sequential layer deposition, given that particles filled the vacancies or depressions in the underlying film. Darwis et al.197 performed multiple depositions of P3HT:PCBM nanoparticles. The rms roughness of the nanoparticle films increased from 9.5 nm for one layer to 13.2 nm for five continuous layers. However, the relation between device performance and nanoparticle film thickness is still not clear according to different material systems. According to Darwis et al.,287 the J–V curves of P3HT:PC61BM nanoparticle OSCs changed from ‘‘S’’ shape (3 layers) to ‘‘J’’ shape (5 layers), and then converted back to ‘‘S’’ shape (10 layers), showing a distinct change in charge recombination.
In addition to BHJ nanoparticle OSCs, owing to the unique properties of nanoparticles, the sequential deposition of monolayers with varying composition nanoparticles provides a unique opportunity to fabricate OSCs with graded nanoparticles (GNPs) to directly control the vertical morphology at different length scales appropriate for efficient charge separation within the active layer. As reported in solution-cast OSCs, the formation of a vertical phase distribution enhance the Voc and Jsc. This phenomenon can be explained by the reduced recombination and enhanced charge collection efficiency.295,296 Bag et al.136 spin-coated a thin PCBM buffer layer from dichloromethane solutions on top of a P3HT:PCBM nanoparticle active layer, and probed the effect of the buffer layer on the charge transport through the nanoparticle assembly. Despite the slightly improved efficiency, the selected buffer layer still needed to be dissolved in chlorinated/benzene solvent, contradicting the notion of non-toxicity. Gärtner et al.297 inserted P3HT nanoparticle layers (ethanol dispersion) on top of a nanoparticulate P3HT:ICBA layer (ethanol dispersion). An increase in Voc was observed in the GNP nanoparticle OSCs compared with the P3HT:ICBA BHJ nanoparticle structures, leading to an increase in the PCEs to 4.2% compared with that of the P3HT:ICBA BHJ nanoparticle OSCs (PCE, 3.7%). Later, several research groups adopted the strategy of using buffer layers between the nanoparticle active layer and the electrode to increase the charge extraction and reduce the leakage current at the polymer/electrode interface.136,137,227 Vaughan et al.267 fabricated organic nanoparticle-based active layers with an engineered vertical morphology through the sequential deposition of varying donor–acceptor concentration monolayers. Successive layers of PFB:F8BT nanoparticles with varying PFB weight ratios ranging from 100% to 0% were deposited on an ITO substrate. The gradient concentration enabled the pure material to be located at the electrode, and the prepared GNP OSCs showed a systematically higher Voc than their non-graded counterparts, indicating the reduced electron–hole recombination in the graded devices.298,299 Recently, Xie et al.269 combined water-processing with the LBL strategy for the fabrication of high-performance OSCs. A mesoporous film with discrete spheres and cavities was first deposited using a donor PM6 aqueous nanosuspension (Fig. 11e). In the mn-LBL film under mild temperature annealing, the nanoparticles showed a certain degree of collapse, which prevented the removal of the pure donor layer underneath the nanoparticle film, leading to an improved PCE of over 19% for the PM6:BTP-eC9 OSCs.
The efficiency of nanoparticle OSCs prepared using P3HT:fullerene derivative nanosuspensions and the mini-emulsion method is limited to around 2% due to the residual surfactant and classical core–shell structure. Alternatively, the nanoparticle OSCs prepared from P3HT:fullerene derivative surfactant-free dispersions with an intermixed structure showed a higher efficiency of 4%.198,200,297,300 Laval et al.86 showed that intermixed PTQ10:Y6 nanoparticle OSCs without large phase separation showed a higher efficiency than PTQ10:PCBM nanoparticle OSCs with a core/shell structure. Holmes et al.243 prepared P3HT:PC61BM nanoparticles featuring Janus morphology nanoparticles (JNPs) using a one-step surfactant-assistant nanoprecipitation method. They concluded that a high exciton dissociation rate occurred within JNPs, which opens the way for the use of these dispersions in photovoltaic technologies. Du et al.75 discovered a potent electron transfer from the polymer toward the fullerene acceptor in aqueous D:A Janus nanoparticles. The PTB7:PC71BM Janus nanoparticle OSCs yielded a higher PCE value of 2.03% than that of the core–shell structured nanoparticle OSCs (1.43%), indicating the efficient charge transfer of JNPs at the D:A interface.
Introducing organic nanoparticles in OSCs is a facile way to finely regulate their domain (grain) size by controlling the particle size of the nanoparticles.119,137,198,236,238,301,302 To avoid exciton deactivation, the ideal phase separation regions need to be less than the effective diffusion length of excitons. Given that the effective diffusion length of the excitons in organic materials is about 10–20 nm,303 the ideal nanoparticle size is expected to be about 20 nm to achieve effective exciton dissociation and bicontinuous percolation pathways for both holes and electrons. It has been reported for PFB:F8BT solution-cast devices that the domain size of the unannealed devices is sub-10 nm, resulting in a poor performance due to the enhanced geminate recombination of the separated excitons.301 On the contrary, annealing above 160 °C produces domains larger than 40 nm, resulting in reduced charge separation. Based on the optimized domain range, Stapleton et al.131 controlled the mean diameter of PFB:F8BT composite nanoparticles to about 50 nm, and the core–shell morphology of the nanoparticles confined each domain of PFB and F8BT to around 20–25 nm, resulting in an increase in Jsc and concomitant enhanced efficiency of the nanoparticle OSCs. The size of donor and acceptor domains in the PDPP-TNT:PC71BM nanoparticles formed by Holmes et al. approached the exciton diffusion length (20–50 nm for PDPP-TNT-rich shells and 50–110 nm for PC71BM-rich cores).143 It is ideal for the PDPP-TNT-rich nanoparticle shells to transport holes to the anode after annealing, and the small amount of PC71BM (15–30%) in the nanoparticle shell layer facilitated electron transport to the cathode. To control semiconductors at the nanoscale and probe the effect of domain size on the OPV performance, Gehan et al.137 demonstrated multiscale control of the active layer morphology by the changing particle composition, particle radius, and ratio of P3HT:PCBM particles. They prepared blended P3HT:PCBM (1:1) bNPs and mNPs with diameters of 115 nm, 90 nm, 80 nm, and 70 nm. Both the bNP and mNP OSCs showed an optimal domain size of 80 nm, which was proposed to support the increasing belief that the optimal domain size can be larger than 10 nm.304
5.2 Organic light-emitting devices
In addition to addressing the hazardous environmental problem from lab to fab in the production of organic photoelectronic devices, organic nanoparticles have emerged as extraordinarily bright fluorescent tags with potential applications in organic light-emitting diodes (OLEDs). Their numerous advantageous properties, such as small particle size, size-tuneable, and amplified energy transfer, can be exploited to achieve exceptional fluorescence brightness, high quantum efficiency, and long-time photostability. | ||
| Fig. 13 (a) Structures of fluorene-based π conjugated polymers applied in the synthesis of water/alcohol fluorescence nanoparticles. (b) Schematic drawing of absorption/PL spectra compared with water/alcohol nanoparticle system and organic solution system. (c) Changes in the number of reported organic nanoparticle-based OLEDs over the years. | ||
In the case of nanoparticle-based films deposited from nanosuspensions, a blue-shift is typically observed compared with organic solution cast films owing to the reduced inter-chain interactions, and the films can exhibit a high fluorescence quantum efficiency. Wu et al.310 synthesized a series of conjugated nanoparticle dots with diameters as low as 4 nm, and among them, the PFO nanoparticles showed the highest fluorescence quantum yield of 40%. Based on the experiment results, they stated that the fluorescence brightness of the organic nanoparticle dots exceeded that of inorganic/metal nanoparticles with the same size. Yu et al.311 found that PFBT nanoparticles with a diameter of 15 nm had a fluorescence quantum efficiency of 7% and fluorescence cross section of about 2.0 × 10−14 cm2, which exceeded that of <20 nm-diameter fluorescent nanoparticles by more than one times. Huyal et al.307 discovered that a nanoparticle-based film of PF with a smaller diameter (30 nm) showed a high quantum efficiency of 68%, which was about two-times higher than that of the solution cast films of 23–44%. Meanwhile, the PL peak of the PF nanoparticle-based film was blue-shifted to around 20 nm compared with the solution-cast films.
In addition, D:A blended organic nanosuspensions have efficient energy transfer inducing a higher quantum efficiency and easy to control emission color.307,312 Wu et al.313 chose blue-emitting conjugated PF as the host polymer, adding green (PFPV), yellow (PFBT), and red (MEH-PPV) emitting fluorene-based polymers separately to synthesize blended nanoparticles. The pure aqueous PF nanosuspension exhibited a blue colour, while the blue colour was quenched and replaced by the corresponding dopant emitting colour when the percentage of dopant increased to 6%. The change in the emitted colour indicated the efficient energy transfer from PF to the doped conjugated polymer.
In 2003, Piok et al.314 synthesized m-LPPP nanoparticles with a mean size of 94 nm using the miniemulsion method, and firstly applied the aqueous organic nanosuspensions as a functional layer in OLEDs. The nanoparticle OLEDs showed improved opto-electronic characteristics, with a lower onset and slightly higher efficiency than solution-cast OLEDs. The improved performance was explained by the increased electron injection due to the formation of a “stalactite” morphology during the evaporation of the nanoparticle-based devices. Later, Fisslthaler et al.315 and Mauthner et al.316 fabricated aqueous-based nanoparticle OLEDs through inject printing, although the devices showed a relatively poor performance with extremely high current and light emission onset voltages. In addition to ITO/glass substrate, Sarrazin et al.317 applied PFFO photoluminescent aqueous nanosuspension deposition on commercial tracing paper.
The use of aqueous organic nanoparticles can also handle the inherent lack of solubility of most p-conjugated polymers in solvents. Based on the low solubility of POPPV in organic solvents, which inhibits its potential electroluminescence (EL) characteristics, Huebner et al.127 synthesized POPPV:PFO aqueous composite nanoparticles using SDS and deposited them as the emitting layer in OLEDs. Efficient energy transferred from PFO to the POPPV within the nanoparticle, and a wide range of CIE colour maps was observed due to Förster resonance energy transfer. Similar to the active layers in OSCs, the residual ionic surfactant can lead to considerable performance losses, given that mobile ionic species typically exhibit much slower transport than electronic charge carriers. Alves et al.318 compared three different types of surfactants, cationic (CTAB), anionic (SDS), and non-ionic (Triton X-405–TX-405), for synthesizing stable PANI nanoparticles based on chemical oxidative polymerization. By changing the pH values in the range of 1.3 to 12.4, the colour of the PANI nanoparticles (SDS as the surfactant) changed from green to blue, and finally violet. The PANI aqueous suspensions presented a surprisingly high fluorescence quantum yield value (ranging from 1.9 × 10−3 to 6.9 × 10−3) using different surfactants and controlled pH values. To determine the optimal SDS concentration, Ribeiro et al.120 monitored the surface tension of SY-PPV:PS and MEH-PPV:PS nanosuspensions, as well as the equilibrium (ionic) conductivity as a function of dialysis time. The surface tension increased sub-linearly from ∼38 to ∼60 mN m−1 within 96 h compared with pure water (72 mN m−1). Consequently, the green nanoparticle-based OLEDs produced a low leakage current of |V| < 2 V. However, the luminous efficiency of the nanoparticle OLEDs was lower than that of the solution-cast OLEDs by a factor of about 2.5. In addition to the emitting layer, Mauro et al.319 synthesized dark-green aqueous PANI–PSS nanosuspensions and deposited them as hole-injection layers (HILs) in OLEDs. The devices based on the PANI–PSS nanoparticle layer showed a low switch-on voltage and improved efficiency of up to 1.20 cd A−1.
5.3 Organic thin-film transistors (OTFTs)
OTFTs are transistors that connect organic materials between their source and drain electrodes. Based on the mechanism modulating the current between the source and drain electrodes, OTFTs can be classified as organic field effect transistors (OFETs), electrolyte-gated organic field effect transistors (EGOFETs), water-gated organic field-effect transistors (WGOFETs) and organic electrochemical transistors (OECTs). Despite the high performance of OTFTs obtained by vacuum-deposition, this method is not cost-effective, making their commercial production challenging. In this case, the fabrication of OTFTs using water/alcohol organic nanosuspensions not only solves the problem of cost but also achieves non-toxic production, which is favorable for commercialization.In 2005, Ong et al.320 fabricated nanoparticle OTFTs using PTQ12 dichlorobenzene dispersions obtained under ultrasonic agitation as channel layers. The nanoparticle-based devices showed a high performance with a mobility of 0.12 cm2 V−1 s−1 and 107–108 on/off ratio, indicating the potential of nanoparticle-based devices; however, dichlorobenzene is still a toxic organic solvent. In 2007, Dionigi et al.321 synthesized aqueous nanosuspensions of H4T6, a semiconductive oligomer. The aqueous nanosuspensions were firstly deposited into the channel of a bottom-contact field effect transistor, and the device exhibited a charge mobility of 4 × 10−3 cm2 V−1 s−1, with a threshold voltage close to 0 V, and on/off ratios in excess of 102. Later, Darwis et al.129 used P3HT aqueous nanosuspensions prepared using the miniemulsion method to deposit the semiconducting channel layer of OTFTs with a thickness of about 60 nm. The obtained devices operated at a low voltage and showed output characteristics comparable to that fabricated using chloroform.322 Surfactant-free P3HT nanosuspensions were also applied in the fabrication of nanoparticle OTFTs, and the devices showed better performances than surfactant-containing devices.323,324 In 2012, Jeon et al.231 prepared PTCDI-C13 nanoparticles in acetonitrile by adopting a surfactant-free strategy of laser ablation. The increased grain size after post-annealing treatment was several hundred nanometres, which resulted in an increase in mobility to 0.027 cm2 V−1 s−1. However, compared with the devices fabricated via vacuum deposition with a mobility of 2.1 cm2 V−1 s−1,325 there is still large room for the optimization of solution nanoparticle OTFTs.
To improve the performance of nanoparticle OTFTs, researchers have been focused on copolymers and the elimination of surfactants to increase the charge carrier mobility and on/off ratio. Cheon et al.326 synthesized alcohol-based nanosuspensions of a conjugated random copolymer with low-molecular-weight, using ethanol, propanol, and butanol. It was proven that the DPP-TT/DPP-SVS nanoparticle films still maintained their crystalline order to some degree, and the fabricated alcohol-based nanoparticle OTFTs showed a high mobility of 1.03 cm2 V−1 s−1. Cho et al.327 fabricated devices using nanosuspensions of surfactant-free donor–acceptor copolymers DPP–TT in butyl acetate (BA) or ethyl acetate (EA). Both the BA-based devices and EA-based devices showed the typical p-channel transfer/output characteristics, and the BA-based nanoparticle OFETs showed a high mobility of 2.7 cm2 V−1 s−1, which is quite close to the realistic minimum requirement for driving the FETs of commercial OLEDs.328 To effectively eliminate the residual SDS in PBTTT nanoparticle OTFTs, Cho et al.141 conducted a thermal annealing process at the optimized temperature of 270 °C, which is higher than the thermal degradation temperature of SDS. At this temperature, SDS could be removed by thermal degradation, while PBTTT was proven to still be orderly π–π stacked. The obtained devices showed the maximum mobility of 0.15 cm2 V−1 s−1, indicating the severe damage caused by surfactants to organic photoelectric devices despite the great efforts to eliminate them. Instead of using SDS, Cho et al.277 investigated three amphiphilic oligomers non-ionic surfactants, C12E4, C16E10, C18E20. Owing to the weak inter surfactant interactions, the non-ionic surfactants could be more easily eliminated from the deposited thin films by post-washing procedure. The films deposited from the synthesized aqueous DPP-SVS nanosuspensions possessed a relatively good crystalline orientation, and the corresponding nanoparticle OTFT devices showed a high charge carrier mobility of up to 2.5 cm2 V−1 s−1, which was comparable to that of the devices fabricated from DPP-SVS chlorinated solvents, and due to their surface tension difference, they have been considered candidate electrets.
In addition, transistor-type memory, due to its advantages of non-destructive readout and multibit storage, has been considered a promising candidate for next-generation non-volatile memories. Shih et al.329 fabricated PFBT nanoparticle-based transistor memory devices utilizing aqueous nanosuspensions as the discrete trapping sites. The nanoparticle-based memories exhibited a threshold voltage shift, which demonstrated an enhanced electron-trapping effect, and the devices exhibited an increased performance with a 35 V memory window and an on/off ratio greater than 104, indicating the potential of organic nanoparticle-based transistor memory. As summarised in Table 4, there are few reports about organic nanoparticle-based transistor devices that use water/alcohol nanoparticles as functional layers. Furthermore, the mobility and other key parameters of nanoparticle-based OFTFs are lagging behind the performance of vacuum-deposited devices and organic-solution cast devices. Addressing the hazards posed by residual surfactants and maintaining stability are crucial to increase the mobility of nanoparticle-based devices, indicating a large space for the improvement of nanoparticle transistor-based devices.
| Year | Material | Surfactant | Threshold voltage (|VT|) | Mobility (cm2 V−1 s−1) | On/off ratio | Ref. |
|---|---|---|---|---|---|---|
| 2007 | H4T6:PS | None | 0.26 | 4.0 × 10−3 | >102 | 321 |
| 2010 | P3HT | None | 1.4 × 10−3 | 323 | ||
| 2012 | PTCDI-C13 | None | 47 | 2.7 × 10−2 | 231 | |
| 2013 | P3HT | None | 2.8 | 324 | ||
| 2015 | P1 | None | 1.03 | 326 | ||
| 2015 | DPP-TT | None | 2.7 | 327 | ||
| 2015 | DPP-SVS | C16E10 | 8.5 | 2.5 | 104–105 | 277 |
| 2016 | PBTTT | SDS | 2 | 1.9 × 10−1 | 105–106 | 141 |
| 2020 | IDT-BT | SDS | 60 | 2.5 × 10−1 | 105 | 249 |
| DPPT-BT | 20 | 4.8 × 10−1 | 104 |
5.4 Photocatalytic hydrogen
H2 as the simplest ‘solar fuel’, can be effectively produced from water through the photocatalysis process using photocatalysts.79,330–334 The effectiveness of photocatalysis largely depends on the design of photocatalysts, which can be strategically adjusted for efficient light harvesting, enhanced carrier transport, and faster surface reactions. Photocatalysts primarily based on UV-active inorganic semiconductors have been the most widely studied thus far. However, less than 5% of solar energy lies in the UV spectrum.335 Thus, owing to the easily adjustable light absorption spectrum of organic materials, they hold significant potential for the synthesis of high-performance photocatalysts.336,337 The earliest reported organic polymer photocatalyst was carbon nitride (g-C3N4),338 and types of organic photocatalysts developed broadly in the following years.339–342 However, most systems have been reported to suffer from low dispersibility and solubility of the conjugated polymers in water. Therefore, organic nanoparticles have emerged as a new generation of photocatalysts, owing to their good dispersibility in water and large surface area to provide a greater number of active sites, resulting in a better photocatalytic performance (Fig. 14).134,146,343,344 | ||
| Fig. 14 Schematic drawing and features of different types of organic nanoparticle photocatalysts. | ||
Without the assistance of cocatalysts, it is challenging for all-organic systems to function as efficient photocatalysts.345,346 Sachs et al.201 suggested that more efficient organic nanoparticle photocatalysts must target materials that combine both rapid reductive quenching and rapid charge transfer in a metal-based cocatalyst. They stated that electrons with a long life are located on the nanoparticle instead of the Pd clusters, indicating efficient reductive quenching but slow electron transfer to the residual Pd clusters. Based on single conjugated F8BT nanoparticles as a photocatalyst, Kosco et al.347 stated that the residual metal during the polymerization reaction can act as a cocatalyst. Consequently, the hydrogen evolution activity decreased when the residual Pd content in the F8BT nanoparticles was less than 100 ppm.
6. Summary and outlook
In this review, we explored the entire journey from organic nanoparticles to practical optoelectronic applications. Firstly, we focused on the intricate formation and stabilization mechanisms of organic nanoparticles, which form the basis and premise of nanoparticle research and applications. Based on the classical nucleation theory, the process of nanoparticle formation includes two sequential steps, nucleation and growth. The surface charges and/or long chains of the surfactant on the formed nanoparticles act as repulsive forces to stabilize the nanoparticles to achieve a long shelf-life. From organic materials to nanoparticles, the most adopted synthesis methods are miniemulsion and nanoprecipitation. However, considering the limitations of residual surfactant in the miniemulsion method and unstable property of the nanoprecipitation method, novel methods such as self-assembly and electrostatic stabilization have been developed to synthesize stable surfactant-free organic nanoparticles. The synthesized nanoparticles can have arbitrary shapes and sizes according to the material selection and conditions for their synthesis. Due to the hydrophobic nature and “globular/granular” domains formed in nanoparticle-based films, additional pre-treatment and post-treatment procedures are essential for forming smooth and homogenous films that are similar to solution-cast films. From thin films to functional devices, we concentrated on the myriad of applications of these films in the realm of organic optoelectronic technologies, showcasing their vast potential and wide-reaching impact.The most attractive feature of water/alcohol organic nanosuspensions is the non-toxic fabrication of organic optoelectronic devices. Meanwhile, the unique features of nanoparticles are beneficial for the optimization of organic optoelectronic applications. As illustrated in Fig. 15, size-controllable nanoparticles can be used to tune the D–A domains within the range of the exciton diffusion length to avoid the deactivation of excitons. In the case of nanoparticles with a small size of around a few nanometres, also called nanodots, they exhibit a large relative surface area and energy, which are beneficial for efficient luminescence and hydrogen evolution reactions. The non-toxic and unique properties of water/alcohol-based organic nanoparticles have gradually attracted increasing attention, which are promising in the field of organic optoelectronics devices. However, the reported performance of water/alcohol-based nanoparticle optoelectronic devices still lags behind that of the devices fabricated from organic solutions, which can be explained by the residual surfactant, inhibiting charge transport, and the inadequate contact with the adjacent layer.
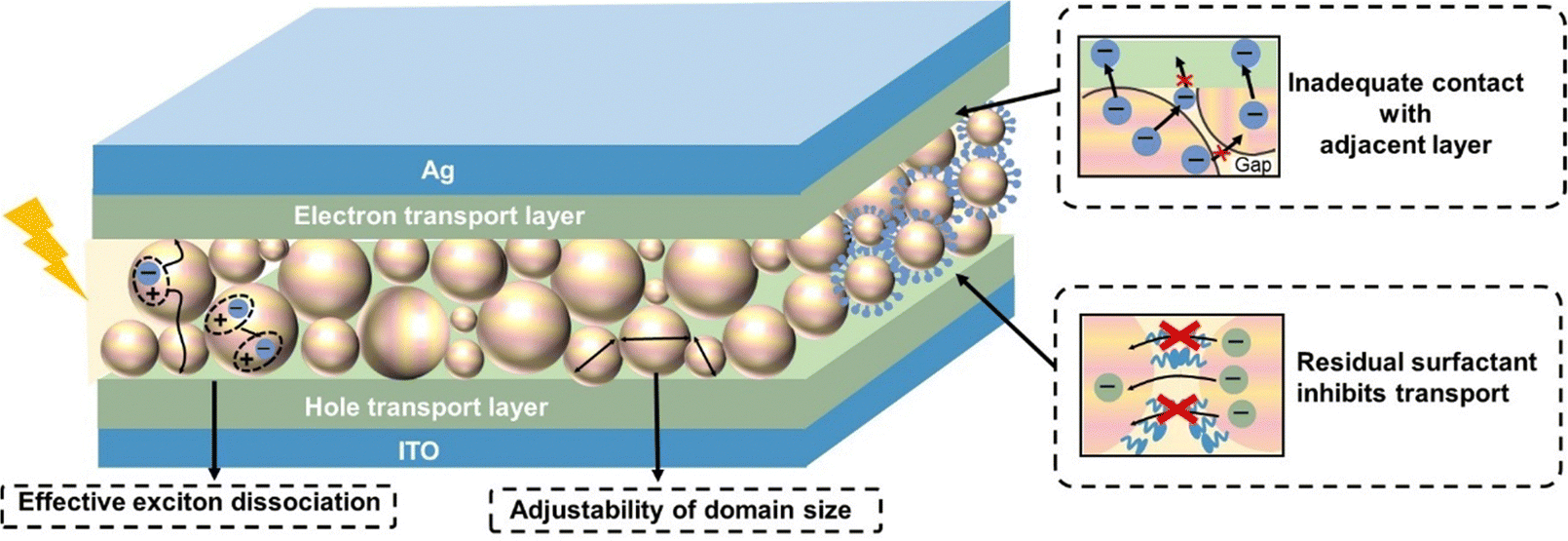 | ||
| Fig. 15 Summary of water/alcohol-based nanoparticles applied in organic optoelectronic devices. | ||
To overcome the limitations and further improve the performance of water/alcohol-based nanoparticle optoelectronic devices, modifications can focus on the synthesis of nanoparticles and nanoparticle film deposition (Fig. 16). (1) Establishing material-processing-property relationships. There is currently no clear reported relation between the chemical structure or property of organic materials with the corresponding formed nanoparticles, which is essential to promote the development of synthesis methods and functional devices. The emergence of material genome engineering, which combines high-throughput experimentation, artificial intelligence (AI), big-data, etc., is expected to accelerate material screening, establishing material-processing-property relationships in the development of emerging water/alcohol-based organic optoelectronic devices.348 (2) Accelerating the development of novel synthesis methods. From the aspect of nanoparticle synthesis, ideal organic nanoparticles applied in optoelectronic devices should be surfactant-free, long-term stable and intermixed for bNPs, promoting the carrier transport. The current common synthesis methods are the classical miniemulsion and nanoprecipitation methods. However, the residual surfactant in the emulsion is difficult or impossible to completely eliminate, and nanosuspensions are unstable without the use of a surfactant. In this case, methods such as the electrostatic method and self-assembly method can overcome these limitations, but they are not broadly generalizable. A universal strategy that can synthesize ideal nanoparticles with surfactant-free, long-term stability, and intermixed morphology is expected to overcome these bottlenecks. (3) Simplifying the deposition process of nanoparticle thin films. From the aspect of nanoparticle film deposition, the ideal nanoparticle-based thin films should be smooth, homogenous, and continuous. To increase the wetting ability of the substrate, a pre-treatment such as long-time UV/O3 or rinsing procedure is conducted. After the deposition process, high-temperature thermal annealing helps to compact the nanoparticle-forming continuous films. However, the additional pre-treatment and post-treatment procedures increase the time/cost for device fabrication, and high-temperature thermal annealing is not conducive to large-scale production. Meanwhile, regarding the deposition process, more approaches for large-area nanoparticle-based films need to be developed in the future to facilitate non-toxic industrial device fabrication.
 | ||
| Fig. 16 Prospects of water/alcohol-based nanoparticles applied for organic optoelectronic devices. | ||
Overall, the application of water/alcohol-based nanosuspensions provides a practical way to realize non-toxic nanoparticle optoelectronic devices. Meanwhile, the unique properties of nanoparticles give new ideas for the optimization and tuning of organic optoelectronic devices. Although there is a lag in the performance of organic nanoparticle devices, they are promising in the field of organic optoelectronics.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. L. acknowledges the financial support by the National Key Research and Development Program of China (2022YFB4200400), the National Natural Science Foundation of China (52394273 and 52373179), the Guangdong Basic and Applied Basic Research Foundation (2023B1515040026), the TCL science and technology innovation fund, and the Fund of Guangdong Provincial Key Laboratory of Luminescence from Molecular Aggregates (2023B1212060003). C. X. acknowledges the financial support by the National Natural Science Foundation of China (grant No. 62104158), Natural Science Foundation of Top Talent of SZTU (grant No. GDRC202113), Development of new perovskite optoelectronics (contract No. HT20231107001).References
- M. Granström, K. Petritsch, A. C. Arias, A. Lux, M. R. Andersson and R. H. Friend, Nature, 1998, 395, 257–260 CrossRef
.
- X. J. Li, F. Pan, C. K. Sun, M. Zhang, Z. W. Wang, J. Q. Du, J. Wang, M. Xiao, L. W. Xue, Z. G. Zhang, C. F. Zhang, F. Liu and Y. F. Li, Nat. Commun., 2019, 10, 519 CrossRef CAS
- Y. N. Chen, Y. Zhao and Z. Q. Liang, Energy Environ. Sci., 2015, 8, 401–422 RSC
- Y. Qian, X. W. Zhang, D. P. Qi, L. H. Xie, B. K. Chandran, X. D. Chen and W. Huang, Sci. China Mater., 2016, 59, 589–608 CrossRef CAS
- Y. Qian, X. W. Zhang, L. H. Xie, D. P. Qi, B. K. Chandran, X. D. Chen and W. Huang, Adv. Mater., 2016, 28, 9243–9265 CrossRef CAS
-
F. Campana, D. Lanari, A. Marrocchi and L. Vaccaro, in Sustainable Strategies in Organic Electronics, ed. A. Marrocchi, Woodhead Publishing, 2022, pp. 425–462 Search PubMed
- S. Zhang, L. Ye, H. Zhang and J. Hou, Mater., 2016, 19, 533–543 CAS
- Z. Liu, Y. Song, K. An, L. Hong, W. Zhong, Y. Cao, K. Zhang, N. Li, F. Huang, Y. Ma and Y. Cao, Appl. Phys. Lett., 2023, 123, 141104 CrossRef CAS
- R. Ma, X. Jiang, J. Fu, T. Zhu, C. Yan, K. Wu, P. Müller-Buschbaum and G. Li, Energy Environ. Sci., 2023, 16, 2316–2326 RSC
- L. Z. Zhang, X. Y. Zhou, X. W. Zhong, C. Cheng, Y. Q. Tian and B. M. Xu, Nano Energy, 2019, 57, 248–255 CrossRef CAS
- Y. Chen, S. Q. Zhang, Y. Wu and J. H. Hou, Adv. Mater., 2014, 26, 2744–2749 CrossRef CAS PubMed
- Y. Zhao, Z. Y. Xie, C. J. Qin, Y. Qu, Y. H. Geng and L. X. Wang, Sol. Energy Mater. Sol. Cells, 2009, 93, 604–608 CrossRef CAS
- A. Yassar, L. Miozzo, R. Gironda and G. Horowitz, Prog. Polym. Sci., 2013, 38, 791–844 CrossRef CAS
- F. Tang, K. L. Wu, Z. J. Zhou, G. Wang, B. Zhao and S. T. Tan, ACS Appl. Energy Mater., 2019, 2, 3918–3926 CrossRef CAS
- S. Ding, Z. J. Ni, M. X. Hu, G. G. Qiu, J. Li, J. Ye, X. T. Zhang, F. Liu, H. L. Dong and W. P. Hu, Macromol. Rapid Commun., 2018, 39, 1800225 CrossRef
- H. H. Choi, J. Y. Baek, E. Song, B. Kang, K. Cho, S. K. Kwon and Y. H. Kim, Adv. Mater., 2015, 27, 3626–3631 CrossRef CAS
- R. Søndergaard, M. Helgesen, M. Jørgensen and F. C. Krebs, Adv. Energy Mater., 2011, 1, 68–71 CrossRef
- B. Schmatz, A. W. Lang and J. R. Reynolds, Adv. Funct. Mater., 2019, 29, 1905266 CrossRef CAS
- C. Wang, M. J. Park, Y. W. Choo, Y. Huang, S. Phuntsho and H. K. Shon, Desalination, 2023, 565, 116841 CrossRef CAS
- C. L. Cleveland, U. Landman, T. G. Schaaff, M. N. Shafigullin, P. W. Stephens and R. L. Whetten, Phys. Rev. Lett., 1997, 79, 1873–1876 CrossRef CAS
- A. P. Alivisatos, Science, 1996, 271, 933–937 CrossRef CAS
-
X. C. Fu, W. X. Shen and T. Y. Yao, Physical Chemistry, Higher Education Press, Beijing, 2006 Search PubMed
-
P. Dobson, S. King and H. Jarvie, Encyclopedia Britannica, Britannica Academic, 2024 Search PubMed
-
N. Strambeanu, L. Demetrovici, D. Dragos and M. Lungu, Nanoparticles' Promises and Risks: Characterization, Manipulation, and Potential Hazards to Humanity and the Environment, Springer, 2014, pp. 3–8 Search PubMed
-
I. Bryukhovetskiy, O. Pak, Y. Khotimchenko, A. Bryukhovetskiy, A. Sharma and H. S. Sharma, in International Review of Neurobiology, ed. I. Bryukhovetskiy, A. Sharma, Z. Zhang and H. S. Sharma, Academic Press, 2020, pp. 67–98 Search PubMed
- M. Namakka, M. R. Rahman, K. A. M. B. Said, M. Abdul Mannan and A. M. Patwary, Environ. Nanotechnol., Monit. Manage., 2023, 20, 100900 CAS
- D. R. Boverhof, C. M. Bramante, J. H. Butala, S. F. Clancy, M. Lafranconi, J. West and S. C. Gordon, Regul. Toxicol. Pharmacol., 2015, 73, 137–150 CrossRef CAS
-
H. A. Lieberman, M. M. Rieger and G. S. Banker, Pharmaceutical dosage forms: disperse systems, CRC Press, Boca Raton, 1988 Search PubMed
- J. Gibbs, Trans. Conn. Acad. Arts Sci., 1874, 3(108–248), 343–524 Search PubMed
- P. E. Wagner and R. Strey, J. Phys. Chem., 1981, 85, 2694–2698 CrossRef CAS
- X. Y. Liu, J. Chem. Phys., 2000, 112, 9949–9955 CrossRef CAS
- D. Oliveira, K. Baba, J. Mori, Y. Miyashita, H. Kasai, H. Oikawa and H. Nakanishi, J. Cryst. Growth, 2010, 312, 431–436 CrossRef CAS
- K. J. Wu, E. C. M. Tse, C. X. Shang and Z. X. Guo, Prog. Mater. Sci., 2022, 123, 100821 CrossRef CAS
- M. Volmer and A Weber, Z. Phys. Chem., 1926, 119U, 277–301 CrossRef
- Y. Liu, K. Kathan, W. Saad and R. K. Prud’homme, Phys. Rev. Lett., 2007, 98, 036102 CrossRef PubMed
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS
- T. Sugimoto, F. Shiba, T. Sekiguchi and H. Itoh, Colloids Surf., A, 2000, 164, 183–203 CrossRef CAS
- D. Erdemir, A. Y. Lee and A. S. Myerson, Acc. Chem. Res., 2009, 42, 621–629 CrossRef CAS PubMed
- A. S. Myerson and B. L. Trout, Science, 2013, 341, 855–856 CrossRef CAS PubMed
- L. Fornaro, D. Ferreira, H. B. Pereira and A. Olivera, J. Cryst. Growth, 2020, 533, 125454 CrossRef CAS
- A. N. Naik, S. Patra, D. Sen and A. Goswami, Phys. Chem. Chem. Phys., 2019, 21, 4193–4199 RSC
- J. J. Li, Y. P. Li, Q. Li, Z. C. Wang and F. L. Deepak, Nanoscale Horiz., 2019, 4, 1302–1309 RSC
- P. R. T. Wolde and D. Frenkel, Science, 1997, 277, 1975–1978 CrossRef
- D. Gebauer, A. Völkel and H. Cölfen, Science, 2008, 322, 1819–1822 CrossRef CAS
- E. M. Pouget, P. H. H. Bomans, J. A. C. M. Goos, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Science, 2009, 323, 1455–1458 CrossRef CAS
- A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757–3778 CrossRef CAS
- H. Helmholtz, Ann. Phys., 1879, 243, 337–382 CrossRef
- M. Gouy, C. R. Acad. Sci., 1909, 149, 654–657 CAS
- D. L. Chapman, Philos. Mag., 1913, 25, 475–481 Search PubMed
- O. Stern, Z. Elektrochem. Angew. Phys. Chem., 1924, 30, 508–51613 CrossRef CAS
- D. C. Grahame, Chem. Rev., 1947, 41, 441–501 CrossRef CAS PubMed
-
J. H. Adair, E. Suvaci and J. Sindel, in Encyclopedia of Materials: Science and Technology, ed. K. H. J. Buschow, R. W. Cahn, M. C. Flemings, B. Ilschner, E. J. Kramer, S. Mahajan and P. Veyssière, Elsevier, Oxford, 2001, pp. 1–10 Search PubMed
- B. V. Derjaguin and L. D. Landau, Zh. Eksp. Teor. Fiz., 1945, 15, 663 Search PubMed
- E. J. W. Verwey, J. Phys. Chem. C, 1947, 51, 631–636 CrossRef CAS
-
W. Zhang, Nanomaterial: Impacts on Cell Biology and Medicine, Springer, Newark, 2014 Search PubMed
-
J. Dutta and H. Hofmann, Encyclopedia of nanoscience and nanotechnology, American Scientific Publishers, California, 2004, pp. 617–640 Search PubMed
- J. Polte, CrystEngComm, 2015, 17, 6809–6830 RSC
- M. Prause, H.-J. Schulz and D. Wagler, Biotechnol. Acta, 1984, 4, 143–151 CrossRef
- A. Zielinska, F. Carreiró, A. M. Oliveira, A. Neves, B. Pires, D. N. Venkatesh, A. Durazzo, M. Lucarini, P. Eder, A. M. Silva, A. Santini and E. B. Souto, Molecules, 2020, 25, 3731 CrossRef CAS
- P. Hirschle, T. Preiß, F. Auras, A. Pick, J. Völkner, D. Valdepérez, G. Witte, W. J. Parak, J. O. Rädler and S. Wuttke, CrystEngComm, 2016, 18, 4359–4368 RSC
-
H. Kato, Nanomaterials: Processing and Characterization with Lasers, 2012 Search PubMed
- J. Ugelstad, M. S. El-Aasser and J. W. Vanderhoff, J. Polym. Sci., Part B: Polym. Lett. Ed., 1973, 11, 503–513 CAS
- R. A. Sperling, T. Liedl, S. Duhr, S. Kudera, M. Zanella, C. A. J. Lin, W. H. Chang, D. Braun and W. J. Parak, J. Phys. Chem. C, 2007, 111, 11552–11559 CrossRef CAS
- N. Farkas and J. A. Kramar, J. Nanopart. Res., 2021, 23, 120 CrossRef
-
S. S. Leong, W. M. Ng, J. Lim and S. P. Yeap, in Handbook of Materials Characterization, ed. S. K. Sharma, Springer International Publishing, Cham, 2018, pp. 77–111 Search PubMed
- M. A. Aronova, A. A. Sousa and R. D. Leapman, Microsc. Microanal., 2016, 22, 1154–1155 CrossRef
- V. Kestens, G. Roebben, J. Herrmann, A. Jamting, V. Coleman, C. Minelli, C. Clifford, P.-J. de Temmerman, J. Mast, L. Junjie, F. Babick, H. Colfen and H. Emons, J. Nanopart. Res., 2016, 18, 171 CrossRef PubMed
-
V. A. Coleman, A. K. Jamting, H. J. Catchpoole, M. Roy and J. Herrmann, Instrumentation, Metrology, and Standards for Nanomanufacturing, Optics, and Semiconductors V, SPIE, 2011, pp. 13–19 Search PubMed
- C. Bellacanzone, M. Prosa, M. Muccini, G. Ruani, M. Seri and M. Bolognesi, Part. Part. Syst. Charact., 2021, 38, 2000219 CrossRef CAS
- K. Y. Giottonini, R. J. Rodríguez-Córdova, C. A. Gutiérrez-Valenzuela, O. Peñuñuri-Miranda, P. Zavala-Rivera, P. Guerrero-Germán and A. Lucero-Acuña, RSC Adv., 2020, 10, 4218–4231 RSC
-
J. Goldstein, D. E. Newbury, J. R. Michael, N. W. M. Ritchie, J. H. J. Scott and D. C. Joy, Scanning Electron Microscopy and X-Ray Microanalysis, Springer, New York, 2017 Search PubMed
- S. Rades, V.-D. Hodoroaba, T. Salge, T. Wirth, M. P. Lobera, R. H. Labrador, K. Natte, T. Behnke, T. Gross and W. E. S. Unger, RSC Adv., 2014, 4, 49577–49587 RSC
- E. Buhr, N. Senftleben, T. Klein, D. Bergmann, D. Gnieser, C. G. Frase and H. Bosse, Meas. Sci. Technol., 2009, 20, 084025 CrossRef
- S. Mourdikoudis, R. M. Pallares and N. T. K. Thanh, Nanoscale, 2018, 10, 12871–12934 RSC
- Y. Du, Y. Li, O. Aftenieva, T. Tsuda, P. Formanek, T. A. F. König and A. Synytska, Adv. Opt. Mater., 2022, 10, 2101922 CrossRef CAS
- A. Liu, C.-W. Tai, K. Holá and H. Tian, J. Mater. Chem. A, 2019, 7, 4797–4803 RSC
-
D. B. Williams, C. B. Carter, D. B. Williams and C. B. Carter, Transmission electron microscopy: A textbook for materials science, Springer, 2009, pp. 115–126 Search PubMed
- A. Surrey, D. Pohl, L. Schultz and B. Rellinghaus, Nano Lett., 2012, 12, 6071–6077 CrossRef CAS PubMed
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. Gonzalez Lopez, L. Zou, A. Wadsworth, W. Zhang, L. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Nat. Mater., 2020, 19, 559–565 CrossRef CAS
- P. Hirschle, T. Preiß, F. Auras, A. Pick, J. Völkner, D. Valdepérez, G. Witte, W. J. Parak, J. O. Rädler and S. Wuttke, CrystEngComm, 2016, 18, 4359–4368 RSC
- E. Nakamura, N. A. J. M. Sommerdijk and H. Zheng, Acc. Chem. Res., 2017, 50, 1795–1796 CrossRef CAS
- S. T. Skowron, T. W. Chamberlain, J. Biskupek, U. Kaiser, E. Besley and A. N. Khlobystov, Acc. Chem. Res., 2017, 50, 1797–1807 CrossRef CAS
- J. P. Patterson, Y. Xu, M.-A. Moradi, N. A. J. M. Sommerdijk and H. Friedrich, Acc. Chem. Res., 2017, 50, 1495–1501 CrossRef CAS
- F. J. M. Colberts, M. M. Wienk and R. A. J. Janssen, ACS Appl. Mater. Interfaces, 2017, 9, 13380–13389 CrossRef CAS PubMed
- W. Zhou and H. F. Greer, J. Inorg. Chem., 2016, 2016, 941–950 CAS
- H. Laval, A. Holmes, M. A. Marcus, B. Watts, G. Bonfante, M. Schmutz, E. Deniau, R. Szymanski, C. Lartigau-Dagron, X. X. Xu, J. M. Cairney, K. Hirakawa, F. Awai, T. Kubo, G. Wantz, A. Bousquet, N. P. Holmes and S. Chambon, Adv. Energy Mater., 2023, 13, 2300249 CrossRef CAS
- H. F. Dam, N. P. Holmes, T. R. Andersen, T. T. Larsen-Olsen, M. Barr, A. L. D. Kilcoyne, X. Zhou, P. C. Dastoor, F. C. Krebs and W. J. Belcher, Sol. Energy Mater. Sol. Cells, 2015, 138, 102–108 CrossRef CAS
- U. Schlötzer-Schrehardt, K.-H. Körtje and C. Erb, Curr. Eye Res., 2001, 22, 154–162 CrossRef
-
J. Stöhr, NEXAFS Spectroscopy, Springer, Berlin, 1992 Search PubMed
- N. P. Holmes, M. Marks, J. M. Cave, K. Feron, M. G. Barr, A. Fahy, A. Sharma, X. Pan, D. A. L. Kilcoyne, X. Zhou, D. A. Lewis, M. R. Andersson, J. van Stam, A. B. Walker, E. Moons, W. J. Belcher and P. C. Dastoor, Chem. Mater., 2018, 30, 6521–6531 CrossRef CAS
- C. Xie, X. Tang, M. Berlinghof, S. Langner, S. Chen, A. Späth, N. Li, R. H. Fink, T. Unruh and C. J. Brabec, ACS Appl. Mater. Interfaces, 2018, 10, 23225–23234 CrossRef CAS PubMed
- C. R. McNeill, B. Watts, L. Thomsen, W. J. Belcher, N. C. Greenham and P. C. Dastoor, Nano Lett., 2006, 6, 1202–1206 CrossRef CAS PubMed
- B. A. Collins and H. Ade, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 119–128 CrossRef CAS
- P. Marlow, F. Manger, K. Fischer, C. Sprau and A. Colsmann, Nanoscale, 2022, 14, 5569–5578 RSC
-
R. J. Hunter, Zeta potential in colloid science: principles and applications, Academic Press, London, 1981 Search PubMed
- M. M. Modena, B. Rühle, T. P. Burg and S. Wuttke, Adv. Mater., 2019, 31, 1901556 CrossRef
-
V. S. Kulkarni, Handbook of Non-Invasive Drug Delivery Systems: Science and Technology, William Andrew, Oxford, 2009 Search PubMed
-
R. H. Müller, Colloidal Carriers for Controlled Drug Delivery and Targeting: Modification, Characterization and In Vivo Distribution, CRC Press, Boca Raton, FL, 1992, p. 57 Search PubMed
- S. Ulum, N. Holmes, D. Darwis, K. Burke, A. L. D. Kilcoyne, X. J. Zhou, W. Belcher and P. Dastoor, Sol. Energy Mater. Sol. Cells, 2013, 110, 43–48 CrossRef CAS
- L. D'Olieslaeger, G. Pirotte, I. Cardinaletti, J. D'Haen, J. Manca, D. Vanderzande, W. Maes and A. Ethirajan, Org. Electron., 2017, 42, 42–46 CrossRef
- C. Xie, T. Heumüller, W. Gruber, X. Tang, A. Classen, I. Schuldes, M. Bidwell, A. Späth, R. H. Fink, T. Unruh, I. McCulloch, N. Li and C. J. Brabec, Nat. Commun., 2018, 9, 5335 CrossRef CAS PubMed
- H. Shimizu, M. Yamada, R. Wada and M. Okabe, Polym. J., 2008, 40, 33–36 CrossRef CAS
- D. Valencia, G. T. Whiting, R. E. Bulo and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2016, 18, 2080–2086 RSC
- A. Knežević and Z. B. Maksić, New J. Chem., 2006, 30, 215–222 RSC
- M. Nagai, J. Huang, D. Cui, Z. L. Wang and W. Huang, Colloid Polym. Sci., 2017, 295, 1153–1164 CrossRef CAS
- K. Kamogawa, H. Akatsuka, M. Matsumoto, S. Yokoyama, T. Sakai, H. Sakai and M. Abe, Colloids Surf., A, 2001, 180, 41–53 CrossRef CAS
- S. N. Clafton, D. A. Beattie, A. Mierczynska-Vasilev, R. G. Acres, A. C. Morgan and T. W. Kee, Langmuir, 2010, 26, 17785–17789 CrossRef CAS
- S. Saxena, P. Marlow, J. Subbiah, A. Colsmann, W. W. H. Wong and D. J. Jones, ACS Appl. Mater. Interfaces, 2021, 13, 36044–36052 CrossRef CAS
- N. Toshima and T. Yonezawa, New J. Chem., 1998, 22, 1179–1201 RSC
- S. J. Kang, T. W. Yoon, G. Y. Kim and B. S. Kang, ACS Appl. Nano Mater., 2022, 5, 17436–17460 CrossRef CAS
- B. Abismaıl, J. P. Canselier, A. M. Wilhelm, H. Delmas and C. Gourdon, Ultrason. Sonochem., 1999, 6, 75–83 CrossRef
- J. M. Asua, Prog. Polym. Sci., 2002, 27, 1283–1346 CrossRef CAS
- C. S. Chern, Prog. Polym. Sci., 2006, 31, 443–486 CrossRef CAS
- S. P. Siva, K.-W. Kow, C.-H. Chan, S. Y. Tang and Y. K. Ho, Ultrason. Sonochem., 2019, 55, 348–358 CrossRef PubMed
-
C. E. P. Silva and W. Loh, in Developments in Clay Science, ed. F. Wypych and R. A. de Freitas, Elsevier, 2022, pp. 37–59 Search PubMed
-
T. F. Tadros and B. Vincent, in Encyclopaedia of Emulsion Technology, ed. P. Ed Becher, Marcel Dekker, New York, 1980 Search PubMed
- J. T. G. Overbeek, Faraday Discuss. Chem. Soc., 1978, 65, 7–19 RSC
-
J. Eastoe, Surfactant Chenmistry, Wuhan University Press, Wuhan China, 2005 Search PubMed
- G. Nagarjuna, M. Baghgar, J. A. Labastide, D. D. Algaier, M. D. Barnes and D. Venkataraman, ACS Nano, 2012, 6, 10750–10758 CrossRef CAS PubMed
- A. H. Ribeiro, A. Fakih, B. van der Zee, L. Veith, G. Glaser, A. Kunz, K. Landfester, P. W. M. Blom and J. J. Michels, J. Mater. Chem. C, 2020, 8, 6528–6535 RSC
- L. D’Olieslaeger, M. Pfannmöller, E. Fron, I. Cardinaletti, M. Van Der Auweraer, G. Van Tendeloo, S. Bals, W. Maes, D. Vanderzande, J. Manca and A. Ethirajan, Sol. Energy Mater. Sol. Cells, 2017, 159, 179–188 CrossRef
- X. Pan, A. Sharma, D. Gedefaw, R. Kroon, A. D. de Zerio, N. P. Holmes, A. L. D. Kilcoyne, M. G. Barr, A. Fahy, M. Marks, X. J. Zhou, W. Belcher, P. C. Dastoor and M. R. Andersson, Org. Electron., 2018, 59, 432–440 CrossRef CAS
- A. Zielinska, F. Carreiró, A. M. Oliveira, A. Neves, B. Pires, D. N. Venkatesh, A. Durazzo, M. Lucarini, P. Eder, A. M. Silva, A. Santini and E. B. Souto, Molecules, 2020, 25, 3731 CrossRef CAS
- K. Landfester, R. Montenegro, U. Scherf, R. Güntner, U. Asawapirom, S. Patil, D. Neher and T. Kietzke, Adv. Mater., 2002, 14, 651–655 CrossRef CAS
- T. Kietzke, D. Neher, K. Landfester, R. Montenegro, R. Güntner and U. Scherf, Nat. Mater., 2003, 2, 408–412 CrossRef CAS PubMed
- T. Kietzke, D. Neher, M. Kumke, R. Montenegro, K. Landfester and U. Scherf, Macromolecules, 2004, 37, 4882–4890 CrossRef CAS
- C. F. Huebner, R. D. Roeder and S. H. Foulger, Adv. Funct. Mater., 2009, 19, 3604–3609 CrossRef CAS
- P. Sarrazin, D. Beneventi, A. Denneulin, O. Stephan and D. Chaussy, Int. J. Polym. Sci., 2010, 2010, 612180 Search PubMed
- D. Darwis, D. Elkington, S. Ulum, A. Stapleton, G. Bryant, X. J. Zhou, W. Belcher and P. Dastoor, AIP Conf. Proc., 2011, 1415, 124–127 CrossRef CAS
- T. R. Andersen, T. T. Larsen-Olsen, B. Andreasen, A. P. L. Böttiger, J. E. Carlé, M. Helgesen, E. Bundgaard, K. Norrman, J. W. Andreasen, M. Jorgensen and F. C. Krebs, ACS Nano, 2011, 5, 4188–4196 CrossRef CAS PubMed
- A. Stapleton, B. Vaughan, B. F. Xue, E. Sesa, K. Burke, X. J. Zhou, G. Bryant, O. Werzer, A. Nelson, A. L. D. Kilcoyne, L. Thomsen, E. Wanless, W. Belcher and P. Dastoor, Sol. Energy Mater. Sol. Cells, 2012, 102, 114–124 CrossRef CAS
- Y. B. Lee, S. H. Lee, K. Kim, J. W. Lee, K. Y. Han, J. Kim and J. Joo, J. Mater. Chem., 2012, 22, 2485–2490 RSC
- S. Ulum, N. Holmes, M. Barr, A. L. D. Kilcoyne, B. Bin Gong, X. J. Zhou, W. Belcher and P. Dastoor, Nano Energy, 2013, 2, 897–905 CrossRef CAS
- S. Satapathi, H. S. Gill, L. Li, L. Samuelson, J. Kumar and R. Mosurkal, Appl. Surf. Sci., 2014, 323, 13–18 CrossRef CAS
- N. P. Holmes, S. Ulum, P. Sista, K. B. Burke, M. G. Wilson, M. C. Stefan, X. Zhou, P. C. Dastoor and W. J. Belcher, Sol. Energy Mater. Sol. Cells, 2014, 128, 369–377 CrossRef CAS
- M. Bag, T. S. Gehan, L. A. Renna, D. D. Algaier, P. M. Lahti and D. Venkataraman, RSC Adv., 2014, 4, 45325–45331 RSC
- T. S. Gehan, M. Bag, L. A. Renna, X. B. Shen, D. D. Algaier, P. M. Lahti, T. P. Russell and D. Venkataraman, Nano Lett., 2014, 14, 5238–5243 CrossRef CAS PubMed
- O. Ghazy, Macromol. Symp., 2015, 352, 25–32 CrossRef CAS
- N. A. D. Yamamoto, M. E. Payne, M. Koehler, A. Facchetti, L. S. Roman and A. C. Arias, Sol. Energy Mater. Sol. Cells, 2015, 141, 171–177 CrossRef CAS
- N. P. Holmes, M. Marks, P. Kumar, R. Kroon, M. G. Barr, N. Nicolaidis, K. Feron, A. Pivrikas, A. Fahy, A. D. D. Mendaza, A. L. D. Kilcoyne, C. Milller, X. J. Zhou, M. R. Andersson, P. C. Dastoor and W. J. Belcher, Nano Energy, 2016, 19, 495–510 CrossRef CAS
- J. Cho, K. H. Cheon, J. Ha and D. S. Chung, Chem. Eng. J., 2016, 286, 122–127 CrossRef CAS
- L. Parrenin, G. Laurans, E. Paylopoulou, G. Fleury, G. Pecastaings, C. Brochon, L. Vignau, G. Hadziioannou and E. Cloutet, Langmuir, 2017, 33, 1507–1515 CrossRef CAS
- N. P. Holmes, B. Vaughan, E. L. Williams, R. Kroon, M. R. Anderrson, A. L. D. Kilcoyne, P. Sonar, X. J. Zhou, P. C. Dastoor and W. J. Belcher, MRS Commun., 2017, 7, 67–73 CrossRef CAS
- F. Almyahi, T. R. Andersen, N. A. Cooling, N. P. Holmes, M. J. Griffith, K. Feron, X. J. Zhou, W. J. Belcher and P. C. Dastoor, Beilstein J. Nanotechnol., 2018, 9, 649–659 CrossRef CAS PubMed
- F. Almyahi, T. R. Andersen, N. Cooling, N. P. Holmes, A. Fahy, M. G. Barr, D. Kilcoyne, W. Belcher and P. C. Dastoor, Org. Electron., 2018, 52, 71–78 CrossRef CAS
- S. Rajasekar, P. Fortin, V. Tiwari, U. Srivastva, A. Sharma and S. Holdcroft, Synth. Met., 2019, 247, 10–17 CrossRef CAS
- M. Marks, N. P. Holmes, A. Sharma, X. Pan, R. Chowdhury, M. G. Barr, C. Fenn, M. J. Griffith, K. Feron, A. L. D. Kilcoyne, D. A. Lewis, M. R. Andersson, W. J. Belcher and P. C. Dastoor, Phys. Chem. Chem. Phys., 2019, 21, 5705–5715 RSC
- M. S. Ulum, E. Sesa and W. Belcher, J. Phys.: Conf. Ser., 2021, 1763, 012073–012077 CrossRef CAS
- M. G. Barr, S. Chambon, A. Fahy, T. W. Jones, M. A. Marcus, A. L. D. Kilcoyne, P. C. Dastoor, M. J. Griffith and N. P. Holmes, Mater. Chem. Front., 2021, 5, 2218–2233 RSC
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, T. Fei, Y. F. Dong, R. Sougrat, G. T. Harrison, Y. Firdaus, R. Sheelamanthula, B. Purushothaman, F. Moruzzi, W. D. Xu, L. Y. Zhao, A. Basu, S. De Wolf, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Nat. Energy, 2022, 7, 340–351 CrossRef CAS
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, W. M. Zhang, M. Moser, R. Sheelamanthula, L. Y. Zhao, B. Willner, T. C. Hidalgo, H. Faber, B. Purushothaman, M. Sachs, H. Cha, R. Sougrat, T. D. Anthopoulos, S. Inal, J. R. Durrant and I. McCulloch, Adv. Mater., 2022, 34, 2105007 CrossRef CAS PubMed
- M. M. O'Connor, T. J. Aubry, O. G. Reid and G. Rumbles, Adv. Mater., 2023, 2210481, DOI:10.1002/adma.202210481
- A. Holmes, H. Laval, M. Guizzardi, V. Maruzzo, G. Folpini, N. Barbero, E. Deniau, M. Schmutz, S. Blanc, A. Petrozza, G. M. Paternò, G. Wantz, S. Chambon, C. Lartigau-Dagron and A. Bousquet, Energy Environ. Sci., 2024, 17, 1107–1116 RSC
-
J. W. Vanderhoff, M. S. El-Aasser and J. Ugelstad, Polymeric emulsification process, US4177177, 1979 Search PubMed
- J. Ugelstad, M. S. El-Aasser and J. W. Vanderhoff, J. Polym. Sci., Polym. Lett. Ed., 1973, 11, 503–513 CrossRef CAS
- M. C. Venier-Julienne and J. P. Benoit, Pharm. Acta. Helv., 1996, 71, 121–128 CrossRef
- J. Vandorpe, E. Schacht, S. Stolnik, M. C. Garnett, M. C. Davies, L. Illum and S. S. Davis, Biotechnol. Bioeng., 1996, 52, 89–95 CrossRef CAS
- C. Song, V. Labhasetwar, H. Murphy, X. Qu, W. Humphrey, R. Shebuski and R. Levy, J. Controlled Release, 1997, 43, 197–212 CrossRef
- T. Kietzke, D. Neher, R. Montenegro, K. Landfester, U. Scherf and H.-H. Hoerhold, Organic Photovoltaics IV, SPIE, 2004, vol. 5215, pp. 206–210.
- M. Rammal, P. Lévêque, G. Schlatter, N. Leclerc and A. Hébraud, Mater. Chem. Front., 2020, 4, 2904–2931 RSC
-
Y. Nakama, Surfactants. Cosmetic Science and Technology, Elsevier, Amsterdam, The Netherlands, 1st edn, 2017, pp. 231–244 Search PubMed
-
K. Kosswig, Surfactants. Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KgaA, Weinheim, Germany, 2000, p. a25_747 Search PubMed
-
K. Holmberg, B. Jonsson, B. Kronberg and B. Lindman, Surfactants and Polymers in Aqueous Solution, John Wiley & Sons, Ltd, Chichester, UK, 2002, pp. 451–471 Search PubMed
-
M. J. Rosen and J. T. Kunjappu, Surfactants and Interfacial Phenomena, John Wiley & Sons, 2012, pp. 336–337 Search PubMed
- W. L. Griffin, J. Soc. Cosmet. Chem., 1949, 1, 311–326 Search PubMed
- J. T. Davies, Proc. 2nd Int. Congr. Surface Activity, 1957, vol. 1, pp. 426–438.
- M. Nollet, H. Boulghobra, E. Calligaro and J.-D. Rodier, Int. J. Cosmet. Sci., 2019, 41, 99–108 CrossRef CAS
- S. Satapathi, H. S. Gill, L. Li, L. Samuelson, J. Kumar and R. Mosurkal, Appl. Surf. Sci., 2014, 323, 13–18 CrossRef CAS
- J. Cho, S. Yoon, K. M. Sim, Y. J. Jeong, C. E. Park, S. K. Kwon, Y. H. Kim and D. S. Chung, Energy Environ. Sci., 2017, 10, 2324–2333 RSC
- S. W. Zhang, H. Zhang, S. Yang, X. Zhang, S. L. Li, L. Q. Huang, Y. N. Jing, L. E. Xiao, Y. Zhang, B. Han, J. J. Kang and H. Q. Zhou, Nano Res., 2023, 16, 13419–13433 CrossRef CAS
- B. Tan, Y. C. Li, M. F. Palacios, J. Therrien and M. J. Sobkowicz, Colloids Surf., A, 2016, 488, 7–14 CrossRef CAS
- T. R. Andersen, N. A. Cooling, F. Almyahi, A. S. Hart, N. C. Nicolaidis, K. Feron, M. Noori, B. Vaughan, M. J. Griffith, W. J. Belcher and P. C. Dastoor, Sol. Energy Mater. Sol. Cells, 2016, 149, 103–109 CrossRef CAS
- T. T. Larsen-Olsen, B. Andreasen, T. R. Andersen, A. P. L. Böttiger, E. Bundgaard, K. Norrman, J. W. Andreasen, M. Jørgensen and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2012, 97, 22–27 CrossRef CAS
- O. Pras, D. Chaussy, O. Stephan, Y. Rharbi, P. Piette and D. Beneventi, Langmuir, 2010, 26, 14437–14442 CrossRef CAS PubMed
- E. Lepeltier, C. Bourgaux and P. Couvreur, Adv. Drug Delivery Rev., 2014, 71, 86–97 CrossRef CAS
- H. Fessi, F. Puisieux, J. P. Devissaguet, N. Ammoury and S. Benita, Int. J. Pharm., 1989, 55, R1–R4 CrossRef CAS
- B. Mishra, B. B. Patel and S. Tiwari, Nanomedicine, 2010, 6, 9–24 CrossRef CAS
-
M. Mulder, Basic Principles of Membrane Technology, Springer, Dordrecht, 1996, pp. 89–106 Search PubMed
-
L. Rozelle, J. Cadotte, R. Corneliussen, E. Erickson, K. Cobian and C. Kopp Jr, Encyclopedia of separation science, Academic Press, 2000, pp. 3331–3346 Search PubMed
- S. A. Vitale and J. L. Katz, Langmuir, 2003, 19, 4105–4110 CrossRef CAS
- F. Ganachaud and J. L. Katz, ChemPhysChem, 2005, 6, 209–216 CrossRef CAS
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS
- W. S. Saad and R. K. Prud’homme, Nano Today, 2016, 11, 212–227 CrossRef CAS
- J. W. Cahn, Acta Metall., 1961, 9, 795–801 CrossRef CAS
- J. Tao, S. F. Chow and Y. Zheng, Acta Pharm. Sin. B, 2019, 9, 4–18 CrossRef
- Z. J. Hu, D. Tenery, M. S. Bonner and A. J. Gesquiere, J. Lumin., 2010, 130, 771–780 CrossRef CAS
- O. Inganäs, W. R. Salaneck, J. E. Österholm and J. Laakso, Synth. Met., 1988, 22, 395–406 CrossRef
- K. Faied, M. Frechette, M. Ranger, L. Mazerolle, I. Levesque, M. Leclerc, T.-A. Chen and R. D. Rieke, Chem. Mater., 1995, 7, 1390–1396 CrossRef CAS
- O. Inganäs, W. R. Salaneck, J. E. Österholm and J. Laakso, Synth. Met., 1988, 22, 395–406 CrossRef
- C. A. Sandstedt, R. D. Rieke and C. J. Eckhardt, Chem. Mater., 1995, 7, 1057–1059 CrossRef CAS
- T. Yamamoto, D. Komarudin, M. Arai, B.-L. Lee, H. Suganuma, N. Asakawa, Y. Inoue, K. Kubota, S. Sasaki, T. Fukuda and H. Matsuda, J. Am. Chem. Soc., 1998, 120, 2047–2058 CrossRef CAS
- T. Yamamoto, D. Komarudin, K. Kubota and S. Sasaki, Chem. Lett., 1998, 235 CrossRef CAS
- A. Fraleoni-Morgera, S. Marazzita, D. Frascaro and L. Setti, Synth. Met., 2004, 147, 149–154 CrossRef CAS
- Z. J. Hu and A. J. Gesquiere, Chem. Phys. Lett., 2009, 476, 51–55 CrossRef CAS
-
J.-P. Devissaguet, H. Fessi and F. Puisieux, Process for the preparation of dispersible colloidal systems of a substance in the form of nanocapsules, US Pat., 5049322, 1991 Search PubMed
- G. Prunet, L. Parrenin, E. Pavlopoulou, G. Pecastaings, C. Brochon, G. Hadziioannou and E. Cloutet, Macromol. Rapid Commun., 2018, 39, 1700504 CrossRef PubMed
- D. Darwis, N. Holmes, D. Elkington, A. L. D. Kilcoyne, G. Bryant, X. J. Zhou, P. Dastoor and W. Belcher, Sol. Energy Mater. Sol. Cells, 2014, 121, 99–107 CrossRef CAS
- S. Gärtner, M. Christmann, S. Sankaran, H. Röhm, E.-M. Prinz, F. Penth, A. Pütz, A. E. Türeli, B. Penth, B. Baumstümmler and A. Colsmann, Adv. Mater., 2014, 26, 6653–6657 CrossRef
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. A. Fernandes, L. Tian, H. Chen and H. N. Tian, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 CrossRef CAS PubMed
- S. Sankaran, K. Glaser, S. Gärtner, T. Rödlmeier, K. Sudau, G. Hernandez-Sosa and A. Colsmann, Org. Electron., 2016, 28, 118–122 CrossRef CAS
- M. Sachs, H. Cha, J. Kosco, C. M. Aitchison, L. Francàs, S. Corby, C. L. Chiang, A. A. Wilson, R. Godin, A. Fahey-Williams, A. I. Cooper, R. S. Sprick, I. McCulloch and J. R. Durrant, J. Am. Chem. Soc., 2020, 142, 14574–14587 CrossRef CAS
- C. Xie, S. Q. Liang, G. Y. Zhang and S. P. Li, Polymers, 2022, 14, 4229 CrossRef CAS
- H. F. Yang, C. Li, T. Liu, T. Fellowes, S. Y. Chong, L. Catalano, M. Bahri, W. W. Zhang, Y. J. Xu, L. J. Liu, W. Zhao, A. M. Gardner, R. Clowes, N. D. Browning, X. B. Li, A. J. Cowan and A. I. Cooper, Nat. Nanotechnol., 2023, 18, 307–315 CrossRef CAS PubMed
- I. L. Blouza, C. Charcosset, S. Sfar and H. Fessi, Int. J. Pharm., 2006, 325, 124–131 CrossRef
- V. Ferranti, H. Marchais, C. Chabenat, A. M. Orecchioni and O. Lafont, Int. J. Pharm., 1999, 193, 107–111 CrossRef CAS PubMed
- S. X. Wang, A. Singh, N. Walsh and G. Redmond, Nanotechnology, 2016, 27, 245601 CrossRef
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36, 887–913 CrossRef CAS
- A. P. Valera, C. Schatz, E. Ibarboure, T. Kubo, H. Segawa and S. Chambon, Front. Energy Res., 2019, 6, 146 CrossRef
- S. R. Acharya and P. R. V. Reddy, Asian J. Pharm. Sci., 2016, 11, 427–438 CrossRef
- R. Botet and K. Roger, Curr. Opin. Colloid Interface Sci., 2016, 22, 108–112 CrossRef CAS
- S. Chambon, C. Schatz, V. Sébire, B. Pavageau, G. Wantz and L. Hirsch, Mater. Horiz., 2014, 1, 431–438 RSC
- K. Fischer, P. Marlow, F. Manger, C. Sprau and A. Colsmann, Adv. Mater. Technol., 2022, 7, 2200297 CrossRef CAS
- V. Kumar and R. K. Prud'homme, Chem. Eng. Sci., 2009, 64, 1358–1361 CrossRef CAS
-
W. L. McCabe, J. Smith and P. Harriott, Unit Operations of Chemical Engineering, McGraw-Hill, New York, 2004, pp. 663–665 Search PubMed
-
J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, Waltham, 2011 Search PubMed
- J. B. Rosenholm, Adv. Colloid Interface Sci., 2018, 259, 21–43 CrossRef CAS PubMed
- F. Manger, P. Marlow, K. Fischer, M. Nöller, C. Sprau and A. Colsmann, Adv. Funct. Mater., 2022, 32, 2202566 CrossRef CAS
- F. Manger, K. Fischer, P. Marlow, H. Röhm, C. Sprau and A. Colsmann, Adv. Energy Mater., 2023, 13, 2202820 CrossRef CAS
- T. F. Liu, J. Heimonen, Q. L. Zhang, C. Y. Yang, J. D. Huang, H. Y. Wu, M. A. Stoeckel, T. P. A. van der Pol, Y. X. Li, S. Y. Jeong, A. Marks, X. Y. Wang, Y. Puttisong, A. Y. Shimolo, X. J. Liu, S. L. Zhang, Q. F. Li, M. Massetti, W. M. Chen, H. Y. Woo, J. Pei, I. McCulloch, F. Gao, M. Fahlman, R. Kroon and S. Fabiano, Nat. Commun., 2023, 14, 8454 CrossRef CAS
- Z. Li, P. He, H. Chong, A. Furube, K. Seki, H.-H. Yu, K. Tajima, Y. Ito and M. Kawamoto, ACS Omega, 2017, 2, 1625–1632 CrossRef CAS PubMed
- T. Rashid, C. F. Kait and T. Murugesan, Chin. J. Chem. Eng., 2017, 25, 1266–1272 CrossRef CAS
- M. Hufnagel, M. Fischer, T. Thurn-Albrecht and M. Thelakkat, Macromolecules, 2016, 49, 1637 CrossRef CAS
- H. J. Kim, J.-H. Kim, J.-H. Ryu, Y. Kim, H. Kang, W. B. Lee, T.-S. Kim and B. J. Kim, ACS Nano, 2014, 8, 10461 CrossRef CAS
- A. Braunová, L. Kostka, L. Sivák, L. Cuchalová, Z. Hvězdová, R. Laga, S. Filippov, P. Černoch, M. Pechar, O. Janoušková, M. Šírová and T. Etrych, J. Controlled Release, 2017, 245, 41–51 CrossRef PubMed
- B. D. Olsen and R. A. Segalman, Mater. Sci. Eng., R, 2008, 62, 37–66 CrossRef
- K. Kuperkar, D. Patel, L. I. Atanase and P. Bahadur, Polymers, 2022, 14, 4702 CrossRef CAS PubMed
- S. Zappia, G. Scavia, A. M. Ferretti, U. Giovanella, V. Vohra and S. Destri, Adv. Sustainable Syst., 2018, 2, 1700155 CrossRef
- L. Ganzer, S. Zappia, M. Russo, A. M. Ferretti, V. Vohra, M. Diterlizzi, M. R. Antognazza, S. Destri and T. Virgili, Phys. Chem. Chem. Phys., 2020, 22, 26583–26591 RSC
- M. Diterlizzi, A. M. Ferretti, G. Scavia, R. Sorrentino, S. Luzzati, A. C. Boccia, A. A. Scamporrino, R. Po, E. Quadrivi, S. Zappia and S. Destri, Polymers, 2022, 14, 1588 CrossRef
- E. H. Cho, M. S. Kim, D. H. Park, H. Jung, J. Bang, J. Kim and J. Joo, Adv. Funct. Mater., 2011, 21, 3056–3063 CrossRef CAS
- H. G. Jeon, N. Oguma, N. Hirata and M. Ichikawa, Org. Electron., 2013, 14, 19–25 CrossRef CAS
- B. Vaughan, E. L. Williams, N. P. Holmes, P. Sonar, A. Dodabalapur, P. C. Dastoor and W. J. Belcher, Phys. Chem. Chem. Phys., 2014, 16, 2647–2653 RSC
- Z. Yang, W. T. S. Huck, S. M. Clarke, A. R. Tajbakhsh and E. M. Terentjev, Nat. Mater., 2005, 4, 486–490 CrossRef CAS
- S. Haseloh, C. Ohm, F. Smallwood and R. Zentel, Macromol. Rapid Commun., 2011, 32, 88–93 CrossRef CAS PubMed
- R. H. Staff, I. Lieberwirth, K. Landfester and D. Crespy, Macromol. Chem. Phys., 2012, 213, 351–358 CrossRef CAS
- N. P. Holmes, K. B. Burke, P. Sista, M. Barr, H. D. Magurudeniya, M. C. Stefan, A. L. D. Kilcoyne, X. J. Zhou, P. C. Dastoor and W. J. Belcher, Sol. Energy Mater. Sol. Cells, 2013, 117, 437–445 CrossRef CAS
- T. Kietzke, D. Neher, M. Kumke, O. Ghazy, U. Ziener and K. Landfester, Small, 2007, 3, 1041–1048 CrossRef CAS
- J. J. Richards, C. L. Whittle, G. Shao and L. D. Pozzo, ACS Nano, 2014, 8, 4313–4324 CrossRef CAS
- N. P. Holmes, S. Chambon, A. Holmes, X. Xu, K. Hirakawa, E. Deniau, C. Lartigau-Dagron and A. Bousquet, Curr. Opin. Colloid Interface Sci., 2021, 56, 101511 CrossRef CAS
- N. P. Holmes, N. Nicolaidis, K. Feron, M. Barr, K. B. Burke, M. Al-Mudhaffer, P. Sista, A. L. D. Kilcoyne, M. C. Stefan, X. J. Zhou, P. C. Dastoor and W. J. Belcher, Sol. Energy Mater. Sol. Cells, 2015, 140, 412–421 CrossRef CAS
- X. Yan, J. Bernard and F. Ganachaud, Adv. Colloid Interface Sci., 2021, 294, 102474 CrossRef CAS PubMed
- T. Higuchi, A. Tajima, H. Yabu and M. Shimomura, Soft Matter, 2008, 4, 1302–1305 RSC
- A. Holmes, H. Laval, M. Schmutz, S. Blanc, J. Allouche, B. Watts, G. Wantz, N. P. Holmes, K. Hirakawa, E. Deniau, S. Chambon, C. Lartigau-Dagron and A. Bousquet, Mater. Today Chem., 2022, 26, 101229 CrossRef CAS
- V. Chaudhary, R. K. Pandey, R. Prakash, N. Kumar and A. K. Singh, Synth. Met., 2019, 258, 116221 CrossRef CAS
- N. N. Li, A. Z. Panagiotopoulos and A. Nikoubashman, Langmuir, 2017, 33, 6021–6028 CrossRef CAS
- P. Lukes, M. Clupek, V. Babicky, V. Janda and P. Sunka, Phys. D, 2005, 38, 409 CAS
- S. T. Summerfelt, Aquacult. Eng., 2003, 28, 21–36 CrossRef
- L. X. Du, Z. M. Yu, J. W. Wang, M. P. Wolcott, Y. Zhang and C. S. Qi, Cellulose, 2020, 27, 6921–6933 CrossRef CAS
-
R. M. Hernández, Preparation and Synthesis of Conjugated Polymer Nanoparticles in Aqueous Dispersion Via Miniemulsion Polymerisation for Organic Field Effect Transistors, The University of Manchester, United Kingdom, 2020 Search PubMed
- F. Almyahi, T. R. Andersen, A. Fahy, M. Dickinson, K. Feron, W. J. Belcher and P. C. Dastoor, J. Mater. Chem. A, 2019, 7, 9202–9214 RSC
- C. Xie, A. Classen, A. Späth, X. F. Tang, J. Min, M. Meyer, C. H. Zhang, N. Li, A. Osvet, R. H. Fink and C. J. Brabec, Adv. Energy Mater., 2018, 8, 1702857 CrossRef
- T. T. Larsen-Olsen, B. Andreasen, T. R. Andersen, A. P. L. Böttiger, E. Bundgaard, K. Norrman, J. W. Andreasen, M. Jørgensen and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2012, 97, 22–27 CrossRef CAS
- H. J. Snaith and R. H. Friend, Synth. Met., 2004, 147, 105–109 CrossRef CAS
- Y. Du, Y. Wang, V. Shamraienko, K. Pöschel and A. Synytska, Small, 2023, 19, 2206907 CrossRef CAS
- H. T. Yang and P. Jiang, Langmuir, 2010, 26, 13173–13182 CrossRef CAS
- Y. N. Liang, B. K. Lok, L. B. Wang, C. G. Feng, A. C. W. Lu, T. Mei and X. Hu, Thin Solid Films, 2013, 544, 509–514 CrossRef CAS
- A. Olziersky, A. Vilà and J. R. Morante, Thin Solid Films, 2011, 520, 1334–1340 CrossRef CAS
- V. Sashuk, K. Winkler, A. Zywocinski, T. Wojciechowski, E. Gorecka and M. Fiałkowski, ACS Nano, 2013, 7, 8833 CrossRef CAS
- T. Udayabhaskararao, T. Altantzis, L. Houben, M. Coronado-Puchau, J. Langer, R. Popovitz-Biro, L. Liz-Marzán, L. Vuković, P. Král, S. Bals and R. Klajn, Science, 2017, 358, 514 CrossRef CAS PubMed
- J. Zhang, Y. Sun, R. Feng, W. Liang, Z. Liang, W. Guo, I. Abdulhalim, J. Qu, C. W. Qiu and L. Jiang, Nanoscale, 2019, 11, 23058 RSC
- J. A. Labastide, M. Baghgar, I. Dujovne, Y. Yang, A. D. Dinsmore, B. G. Sumpter, D. Venkataraman and M. D. Barnes, J. Phys. Chem. Lett., 2011, 2, 3085–3091 CrossRef CAS
- E. V. Shevchenko, D. V. Talapin, N. A. Kotov, S. O'Brien and C. B. Murray, Nature, 2006, 439, 55–59 CrossRef CAS
- C. L. Phillips and S. C. Glotzer, J. Chem. Phys., 2012, 137, 104901 CrossRef
- J.-Y. Kim and N. A. Kotov, Chem. Mater., 2014, 26, 134–152 CrossRef CAS
- M. Bag, T. S. Gehan, D. D. Algaier, F. Liu, G. Nagarjuna, P. M. Lahti, T. P. Russell and D. Venkataraman, Adv. Mater., 2013, 25, 6411–6415 CrossRef CAS
- X. Han, M. Bag, T. S. Gehan, D. Venkataraman and D. Maroudas, Chem. Phys. Lett., 2014, 610–611, 273–277 CrossRef CAS
- B. Vaughan, A. Stapleton, E. Sesa, N. P. Holmes, X. Zhou, P. C. Dastoor and W. J. Belcher, Org. Electron., 2016, 32, 250–257 CrossRef CAS
- E. B. L. Pedersen, M. C. Pedersen, S. B. Simonsen, R. G. Brandt, A. P. L. Böttiger, T. R. Andersen, W. Jiang, Z. Y. Xie, F. C. Krebs, L. Arleth and J. W. Andreasen, J. Mater. Chem. A, 2015, 3, 17022–17031 RSC
- C. Xie, X. Zeng, C. Li, X. Sun, S. Liang, H. Huang, B. Deng, X. Wen, G. Zhang, P. You, C. Yang, Y. Han, S. Li, G. Lu, H. Hu, N. Li and Y. Chen, Energy Environ. Sci., 2024, 17, 2441–2452 RSC
- A. Rahmanudin, R. Marcial-Hernandez, A. Zamhuri, A. S. Walton, D. J. Tate, R. U. Khan, S. Aphichatpanichakul, A. B. Foster, S. Broll and M. L. Turner, Adv. Sci., 2020, 7, 2002010 CrossRef CAS
- M. O. Reese, S. A. Gevorgyan, M. Jørgensen, E. Bundgaard, S. R. Kurtz, D. S. Ginley, D. C. Olson, M. T. Lloyd, P. Morvillo, E. A. Katz, A. Elschner, O. Haillant, T. R. Currier, V. Shrotriya, M. Hermenau, M. Riede, K. R. Kirov, G. Trimmel, T. Rath, O. Inganäs, F. Zhang, M. Andersson, T. Tvingstedt, M. Lira Cantu, D. Laird, C. McGuiness, S. Gowrisankerm, M. Pannone, M. Xiao, J. Hauch, R. Steim, D. M. DeLongchamp, R. Rösch, H. Hoppe, N. Espinosa, A. Urbina, G. Yaman-Uzunoglu, J.-B. Bonekamp, A. J. J. M. van Breemen, C. Girotto, E. Voroshazi and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2011, 95, 1253–1267 CrossRef CAS
- A. Sharma, X. Pan, J. A. Campbell, M. R. Andersson and D. A. Lewis, Macromolecules, 2017, 50, 3347–3354 CrossRef CAS
- D. Leman, M. A. Kelly, S. Ness, S. Engmann, A. Herzing, C. Snyder, H. W. Ro, R. J. Kline, D. M. DeLongchamp and L. J. Richter, Macromolecules, 2015, 48, 383–392 CrossRef CAS
- R. Kroon, R. Gehlhaar, T. T. Steckler, P. Henriksson, C. Müller, J. Bergqvist, A. Hadipour, P. Heremans and M. R. Andersson, Sol. Energy Mater. Sol. Cells, 2012, 105, 280–286 CrossRef CAS
- E. Wang, L. Hou, Z. Wang, S. Hellström, F. Zhang, O. Inganäs and M. R. Andersson, Adv. Mater., 2010, 22, 5240–5244 CrossRef CAS PubMed
- D. Leman, M. A. Kelly, S. Ness, S. Engmann, A. Herzing, C. Snyder, H. W. Ro, R. J. Kline, D. M. DeLongchamp and L. J. Richter, Macromolecules, 2015, 48, 383–392 CrossRef CAS
- J. Cho, K. H. Cheon, H. Ahn, K. H. Park, S. K. Kwon, Y. H. Kim and D. S. Chung, Adv. Mater., 2015, 27, 5587–5592 CrossRef CAS PubMed
- D. Scalarone, M. Lazzari, V. Castelvetro and O. Chiantore, Chem. Mater., 2007, 19, 6107–6113 CrossRef CAS
- C. Arnold, F. Thalmann, C. Marques, P. Marie and Y. Holl, J. Phys. Chem. B, 2010, 114, 9135–9147 CrossRef CAS PubMed
- P.-P. Cheng, L. Zhou, J.-A. Li, Y.-Q. Li, S.-T. Lee and J.-X. Tang, Org. Electron., 2013, 14, 2158–2163 CrossRef CAS
- R. Lampande, G. W. Kim, D. C. Choe, J. Hoon Kong and J. H. Kwon, Sol. Energy Mater. Sol. Cells, 2014, 125, 276–282 CrossRef CAS
- S.-H. Lin, S. Lan, J.-Y. Sun and C.-F. Lin, Org. Electron., 2013, 14, 26–31 CrossRef CAS
- A. R. b Mohd Yusoff, H. P. Kim and J. Jang, Org. Electron., 2013, 14, 858–861 CrossRef
- M. Ameri, M. F. Al-Mudhaffer, F. Almyahi, G. C. Fardell, M. Marks, A. Al-Ahmad, A. Fahy, T. Andersen, D. C. Elkington, K. Feron, M. Dickinson, F. Samavat, P. C. Dastoor and M. J. Griffith, ACS Appl. Mater. Interfaces, 2019, 11, 10074 CrossRef CAS PubMed
- M. F. Al-Mudhaffer, M. J. Griffith, K. Feron, N. C. Nicolaidis, N. A. Cooling, X. Zhou, J. Holdsworth, W. J. Belcher and P. C. Dastoor, Sol. Energy Mater. Sol. Cells, 2018, 175, 77–88 CrossRef CAS
- D. Darwis, N. Holmes, D. Elkington, A. L. David Kilcoyne, G. Bryant, X. Zhou, P. Dastoor and W. Belcher, Sol. Energy Mater. Sol. Cells, 2014, 121, 99 CrossRef CAS
- D. Darwis, E. Sesa, S. Ulum, N. P. Holmes, K. Feron, M. Thameel, R. Chowdhury, D. Farhamsah, L. Tegg, X. J. Zhou, P. C. Dastoor and W. J. Belcher, J. Electron. Mater., 2020, 49, 4168–4179 CrossRef CAS
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS
- J. Wang, Y. Xie, K. Chen, H. Wu, J. M. Hodgkiss and X. Zhan, Nat. Rev. Phys., 2024, 6, 365–381 CrossRef CAS
- P. Cheng, G. Li, X. Zhan and Y. Yang, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS
- J. Wang, P. Xue, Y. Jiang, Y. Huo and X. Zhan, Nat. Rev. Chem., 2022, 6, 614–634 CrossRef
- I. McCulloch, M. Chabinyc, C. Brabec, C. B. Nielsen and S. E. Watkins, Nat. Mater., 2023, 22, 1304–1310 CrossRef CAS PubMed
- J. Xu, A. Späth, W. Gruber, A. Barabash, P. Stadler, K. Gubanov, M. Wu, K. Forberich, E. Spiecker, R. H. Fink, T. Unruh, I. McCulloch, C. J. Brabec and T. Heumüller, Nano Energy, 2023, 118, 108956 CrossRef CAS
- S. Heutz, P. Sullivan, B. M. Sanderson, S. M. Schultes and T. S. Jones, Sol. Energy Mater. Sol. Cells, 2004, 83, 229–245 CrossRef CAS
- R. Pandey and R. J. Holmes, IEEE J. Sel. Top. Quantum Electron., 2010, 16, 1537–1543 CAS
- S. Gärtner, S. Reich, M. Bruns, J. Czolk and A. Colsmann, Nanoscale, 2016, 8, 6721–6727 RSC
- L. J. A. Koster, V. D. Mihailetchi, R. Ramaker and P. W. M. Blom, Appl. Phys. Lett., 2005, 86, 123509 CrossRef
- M. D. Perez, C. Borek, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2009, 131, 9281–9286 CrossRef CAS
- S. Gärtner, A. J. Clulow, I. A. Howard, E. P. Gilbert, P. L. Burn, I. R. Gentle and A. Colsmann, ACS Appl. Mater. Interfaces, 2017, 9, 42986–42995 CrossRef
- C. R. McNeill, S. Westenhoff, C. Groves, R. H. Friend and N. C. Greenham, J. Phys. Chem. C, 2007, 111, 19153–19160 CrossRef CAS
- K. B. Burke, A. J. Stapleton, B. Vaughan, X. J. Zhou, A. L. D. Kilcoyne, W. J. Belcher and P. C. Dastoor, Nanotechnology, 2011, 22, 265710 CrossRef
- J. J. M. Halls, K. Pichler, R. H. Friend, S. C. Moratti and A. B. Holmes, Appl. Phys. Lett., 1996, 68, 3120–3122 CrossRef CAS
- M.-H. Ham, G. L. C. Paulus, C. Y. Lee, C. Song, K. Kalantar-zadeh, W. Choi, J.-H. Han and M. S. Strano, ACS Nano, 2010, 4, 6251–6259 CrossRef CAS PubMed
- G. Heliotis, P. N. Stavrinou, D. C. C. Bradley, E. Gu, C. Griffin, C. W. Jeon and M. D. Dawson, Appl. Phys. Lett., 2015, 87, 103505 CrossRef
- F. Kong, J. Liu, X. F. Li, Y. An and T. Qiu, Dyes Pigm., 2010, 84, 165–168 CrossRef CAS
- I. O. Huyal, T. Ozel, D. Tuncel and H. V. Demir, Opt. Express, 2008, 16, 13391–13397 CrossRef CAS
- A. J. R. Son, H. Lee and B. Moon, Synth. Met., 2007, 157, 597–602 CrossRef CAS
- C.-Y. Yu and A. S. Godana, Eur. Polym., 2018, 99, 165–171 CrossRef CAS
- C. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeill, ACS Nano, 2008, 2, 2415–2423 CrossRef CAS
- J. Yu, C. Wu, S. P. Sahu, L. P. Fernando, C. Szymanski and J. McNeill, J. Am. Chem. Soc., 2009, 131, 18410–18414 CrossRef CAS
- X. Gong, J. C. Ostrowski, G. C. Bazan, D. Moses, A. J. Heeger, M. S. Liu and A. K.-Y. Jen, Adv. Mater., 2003, 15, 45–49 CrossRef CAS
- C. Wu, H. Peng, Y. Jiang and J. McNeill, J. Phys. Chem. B, 2006, 110, 14148–14154 CrossRef CAS
- T. Piok, S. Gamerith, C. Gadermaier, H. Plank, F. P. Wenzl, S. Patil, R. Montenegro, T. Kietzke, D. Neher, U. Scherf, K. Landfester and E. J. W. List, Adv. Mater., 2003, 15, 800–804 CrossRef CAS
- E. Fisslthaler, S. Sax, U. Scherf, G. Mauthner, E. Moderegger, K. Landfester and E. J. W. List, Appl. Phys. Lett., 2008, 92, 183305 CrossRef
- G. Mauthner, K. Landfester, A. Köck, H. Brückl, M. Kast, C. Stepper and E. J. W. List, Org. Electron., 2008, 9, 164–170 CrossRef CAS
- P. Sarrazin, D. Beneventi, A. Denneulin, O. Stephan and D. Chaussy, Int. J. Polym. Sci., 2010, 2010, 612180 Search PubMed
- K. G. B. Alves, E. F. de Melo, C. A. S. Andrade and C. P. de Melo, J. Nanopart. Res., 2013, 15, 1339 CrossRef
- A. De Girolamo Del Mauro, G. Nenna, V. Bizzarro, I. A. Grimaldi, F. Villani and C. Minarini, J. Appl. Polym., 2011, 122, 3618–3623 CrossRef CAS
- B. S. Ong, Y. Wu, P. Liu and S. Gardner, Adv. Mater., 2005, 17, 1141–1144 CrossRef CAS
- C. Dionigi, P. Stoliar, W. Porzio, S. Destri, M. Cavallini, I. Bilotti, A. Brillante and F. Biscarini, Langmuir, 2007, 23, 2030–2036 CrossRef CAS
- H. G. O. Sandberg, T. G. Bäcklund, R. Österbacka, M. Shkunov, D. Sparrowe, I. McCulloch and H. Stubb, Org. Electron., 2005, 6, 142–146 CrossRef CAS
- J. E. Millstone, D. F. J. Kavulak, C. H. Woo, T. W. Holcombe, E. J. Westling, A. L. Briseno, M. F. Toney and J. M. J. Fréchet, Langmuir, 2010, 26, 13056–13061 CrossRef CAS PubMed
- D. Darwis, D. Elkington, S. Ulum, G. Bryant, W. Belcher, P. Dastoor and X. Zhou, J. Colloid Interface Sci., 2013, 401, 65–69 CrossRef CAS
- S. Tatemichi, M. Ichikawa, T. Koyama and Y. Taniguchi, Appl. Phys. Lett., 2006, 89, 112108 CrossRef
- K. H. Cheon, H. Ahn, J. Cho, H. J. Yun, B. T. Lim, D. J. Yun, H. K. Lee, S. K. Kwon, Y. H. Kim and D. S. Chung, Adv. Funct. Mater., 2015, 25, 4844–4850 CrossRef CAS
- J. Cho, K. H. Cheon, K. H. Park, S.-K. Kwon, Y.-H. Kim and D. S. Chung, Org. Electron., 2015, 24, 160–164 CrossRef CAS
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS
- C.-C. Shih, Y.-C. Chiu, W.-Y. Lee, J.-Y. Chen and W.-C. Chen, Adv. Funct. Mater., 2015, 25, 1511–1519 CrossRef CAS
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC
- G. Zhang, Z.-A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed
- H. L. Hou, X. K. Zeng and X. W. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed
- Y. Shiraishi, T. Takii, T. Hagi, S. Mori, Y. Kofuji, Y. Kitagawa, S. Tanaka, S. Ichikawa and T. Hirai, Nat. Mater., 2019, 18, 985–993 CrossRef CAS PubMed
- L. H. Lin, Z. Y. Lin, J. Zhang, X. Cai, W. Lin, Z. Y. Yu and X. C. Wang, Nat. Catal., 2020, 3, 649–655 CrossRef CAS
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS
- J. Kosco, F. Moruzzi, B. Willner and I. McCulloch, Adv. Energy Mater., 2020, 10, 2001935 CrossRef CAS
- C. H. Dai and B. Liu, Energy Environ. Sci., 2020, 13, 24–52 RSC
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed
- Y. Guo, Q. Zhou, J. Nan, W. Shi, F. Cui and Y. Zhu, Nat. Commun., 2022, 13, 2067 CrossRef CAS
- Z. Lin and J. Guo, Macromol. Rapid Commun., 2022, 44, 2200719 CrossRef PubMed
- G. F. Liao, Y. Gong, L. Zhang, H. Y. Gao, G. J. Yang and B. Z. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS
- M. Xiao, Z. L. Wang, M. Q. Lyu, B. Luo, S. C. Wang, G. Liu, H. M. Cheng and L. Z. Wang, Adv. Mater., 2019, 31, 1801369 CrossRef
- P. B. Pati, G. Damas, L. Tian, D. L. A. Fernandes, L. Zhang, I. B. Pehlivan, T. Edvinsson, C. M. Araujo and H. Tian, Energy Environ. Sci., 2017, 10, 1372–1376 RSC
- S. Ye, R. Wang, M. Z. Wu and Y. P. Yuan, Appl. Surf. Sci., 2015, 358, 15–27 CrossRef CAS
- J. Kosco, M. Sachs, R. Godin, M. Kirkus, L. Francas, M. Bidwell, M. Qureshi, D. Anjum, J. R. Durrant and I. McCulloch, Adv. Energy Mater., 2018, 8, 1802181 CrossRef
- Y. Shang, Z. Xiong, K. An, J. A. Hauch, C. J. Brabec and N. Li, MGE Adv., 2024, 2, e28 Search PubMed
| This journal is © The Royal Society of Chemistry 2025 |