
Fine-tuning the hierarchical morphology of multi-component organic photovoltaics via a dual-additive strategy for 20.5% efficiency†
Shitao
Guan‡
a,
Yaokai
Li‡
ab,
ZhaoZhao
Bi
c,
Yi
Lin
d,
Yuang
Fu
e,
Kangwei
Wang
f,
Mengting
Wang
a,
Wei
Ma
 c,
Jianlong
Xia
f,
Zaifei
Ma
d,
Zheng
Tang
d,
Xinhui
Lu
e,
Lijian
Zuo
*ab,
Hanying
Li
a and
Hongzheng
Chen
*ab
c,
Jianlong
Xia
f,
Zaifei
Ma
d,
Zheng
Tang
d,
Xinhui
Lu
e,
Lijian
Zuo
*ab,
Hanying
Li
a and
Hongzheng
Chen
*ab
aState Key Laboratory of Silicon and Advanced Semiconductor Materials, International Research Center for X Polymers, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, P. R. China. E-mail: zjuzlj@zju.edu.cn; hzchen@zju.edu.cn
bZJU-Hangzhou Global Scientific and Technological Innovation Center, Hangzhou 310022, P. R. China
cState Key Laboratory for Mechanical Behavior of Materials, Xi’an Jiaotong University, Xi’an 710049, P. R. China
dState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, Center for Advanced Low-dimension Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, P. R. China
eDepartment of Physics, The Chinese University of Hong Kong, New Territories, Hong Kong 999077, People's Republic of China
fState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Center of Smart Materials and Devices, Wuhan University of Technology, Wuhan 430070, P. R. China
First published on 25th November 2024
Abstract
The multiple-component strategy shows great potential in optimizing the performance of organic photovoltaics (OPVs), while the addition of extra components does not usually guarantee a positive effect on the already-perfected morphology of the binary blend, and thus results in an inferior device performance. To address this issue, we develop a facile dual-additive strategy to compensate the negative effect caused by extra components, and this allows the full exploitation of the multiple-component strategy toward a breakthrough in device performance. Specifically, by employing the dual additives of liquid additive 1,8-diiodooctane and solid additive 1,4-diiodobenzene, the film formation kinetics are optimized, and an optimal hierarchical morphology is formed with balanced crystallization, phase separation and prominent vertical distribution. Therefore, this dual-additive strategy leads to efficient exciton dissociation, charge transport, reduced exciton recombination and suppressed energy loss, offering great promise for high performance OPVs. Consequently, we reach a high efficiency of 20.52% (certified 19.92%) in single-junction OPVs. This work highlights the importance of morphology control for multi-component OPVs and sets a benchmark to accelerate their commercialization.
Broader contextOrganic photovoltaics (OPVs) are emerging as a promising candidate for the next-generation renewable energy technology, featuring the merits of light weight, tunable energy levels and absorption, etc. In particular, a bulk-heterojunction blend with balanced crystallinity and refined nanoscale phase-separation morphology is the priority for an efficient OPV device. In recent years, the multiple-component strategy has shown great potential in optimizing the optoelectronic properties of organic photovoltaics (OPVs), while the addition of extra components usually results in a negative effect on the already-perfected morphology of the binary blend, reducing the device performance. However, directly casting the precursor solution to solid films can hardly form an ideal morphology, which typically involves balanced crystallinity and a hierarchical donor:acceptor (D:A) phase-separated morphology ranging from nanometers to hundreds of nanometers. Herein, we develop a facile dual-additive strategy by employing both liquid additive 1,8-diiodooctane (DIO) and solid additive 1,4-diiodobenzene (DIB) to regulate the complicated morphology in a multiple-component system. The film formation kinetics and an optimal hierarchical morphology are formed with balanced crystallization and phase separation. Consequently, we reach a record efficiency of 20.52% (certified 19.92%) in single-junction organic photovoltaic devices. |
Introduction
Organic photovoltaics (OPVs) are emerging as a promising candidate for the next-generation renewable energy technology, featuring the merits of light weight, tunable energy levels and absorption, etc.1–5 Benefiting from the rapid progress in molecular designs and morphology control, the current best certified power conversion efficiency (PCE) of OPVs has exceeded 19%.6–15 The performance of OPVs is lagging behind their inorganic counterparts, due to the insufficient photon harvesting and complexity in morphology manipulation.16–22 In particular, a bulk-heterojunction blend with balanced crystallinity and refined nanoscale phase-separation morphology is the priority for an efficient OPV device.It is generally observed that directly casting the precursor solution to a solid film can hardly form an ideal morphology, which typically involves balanced crystallinity and a hierarchical donor:acceptor (D:A) phase-separated morphology ranging from nanometers to hundreds of nanometers.21,23–25 Therefore, a variety of regulation methods, including additive and post-annealing strategies are developed in the state-of-the-art OPVs.26–28 Among all the protocols, additive engineering has been demonstrated as an indispensable ingredient for high-performance OPVs. For example, the low volatile 1,8-diiodooctane (DIO) has been a classic liquid additive in OPVs since 2008, and exhibits marvelous ability to optimize the molecular packing and phase-separation structure, as well as the optoelectronic properties.29,30 Most recently, tremendous attention has been paid to the development of solid additives, which show great potential in optimizing the morphology via solidifying the active layer. For example, the introduction of a solid additive, i.e., 1,4-diiodobenzene (DIB), leads to faster film drying kinetics, which pronouncedly enhances the intermolecular stacking and molecular arrangement, with advanced device performance.31,32
Recently, the multiple-component strategy has been demonstrated to be one of the most effective ways to achieve high performance due to simultaneously extended absorption range, optimized morphology, reduced charge recombination and improved charge transport properties.33–36 Meanwhile, considering that most of the high-performance binary OPVs deliver optimized morphology, blending extra components can usually change the optimal phase-separation and crystallinity or the molecular packing in the multiple-component active layer.15 However, the superposition of extra components does not universally promise a higher PCE, which is caused by the deterioration of the bulk morphology. As a result, a delicate balance between the benefits of multiple-component blend and morphology optimization is critical.37–39 In particular, a facile manipulation strategy is needed to eliminate the negative effect and regulate the complex multiple-component morphology. This essentially involves kinetical and thermodynamical control of the multi-scale morphology, i.e., crystallinity, molecular orientation, and desired phase separation.40,41
In this work, we proposed a dual-additive strategy, which involves both liquid additive DIO and solid additive DIB to simultaneously optimize the crystalline and phase-separation features, respectively. We observe that the dual additives help to elongate the acceptor solidification kinetics while shortening that of the donor's. Consequently, an optimal hierarchical morphology is achieved during the organization process of a quaternary D:A blend, where the molecular packing is strengthened, the phase-separation domain is optimized, and favorable vertical phase-separation is formed. As a result, a high PCE of 20.52% (19.92% certified) is reached in the single-junction OPVs, due to the efficient charge transport and reduced energy loss. Therefore, this study provides a facile but effective dual-additive strategy for manipulating the complicated morphology, especially in multiple-component bulk heterojunctions (BHJs) for high performance OPVs.
Results and discussion
Interaction between additives and active materials
The chemical structures of polymer donors of poly((4,8-bis(5-(2-ethylhexyl)-4-fluoro-2-thienyl)benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl)-2,5-thiophenediyl(5,7-bis(2-ethylhexyl)-4,8-dioxo-4H,8H-benzo[1,2-c:4,5-c′]dithiophene-1,3-diyl)-2,5-thiophenediyl)) (PM6) and (poly[(2,6-(4,8-bis(5-(2-ethylhexyl-4-chloro-2-thienyl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(2-butyloctyl)thiophen-2-yl)-8-(4-(2-butyloctyl)-5-methylthiophen-2-yl)dithieno[3′,2′:3,4;2′′,3′′:5,6]benzo[1,2-c][1,2,5]thiadiazole)]) (D18-Cl), small molecule acceptors 2,2′-((2Z,2′Z)-((3,9-bis(2-butyloctyl)-12,13-bis(2-ethylhexyl)-12,13-dihydro-[1,2,5]thiadiazolo [3,4-e]thieno[2′′,3′′:4′,5′]thieno[2′,3′:4,5]pyrrolo[3,2-g]thieno[2′,3′:4,5]thieno[3,2-b]indole-2,10-diyl)bis(methaneylylidene))bis(5,6-difluoro-3-oxo-2,3-dihydro-1H-indene-2,1-diylidene))dimalononitrile (L8-BO) and 2,2′-[[12,13-bis(2-butyloctyl)-12,13-dihydro-3,9-dinonylbisthieno[2′′,3′′:4′,5′]thieno[2′,3′:4,5]pyrrolo[3,2-e:2′,3′-g][2,1,3]benzothiadiazole-2,10-diyl]bis[methylidyne(5,6-chloro-3-oxo-1H-indene-2,1(3H)-diylidene)]]bis[propanedinitrile] (BTP-eC9), and the additives (i.e.DIO and DIB) are shown in Fig. 1a and b.42–45 UV-vis absorption spectra of the donors and the acceptors with various additives are presented in Fig. 1c. The PM6 shows an absorption peak and absorbing edge at 618 and 683 nm, respectively, while those of D18-Cl locate at 578 and 638 nm, respectively. The additive-processed PM6 and D18-Cl films almost overlap with the pristine films without additives, suggesting that these additives have a slight influence on the donors.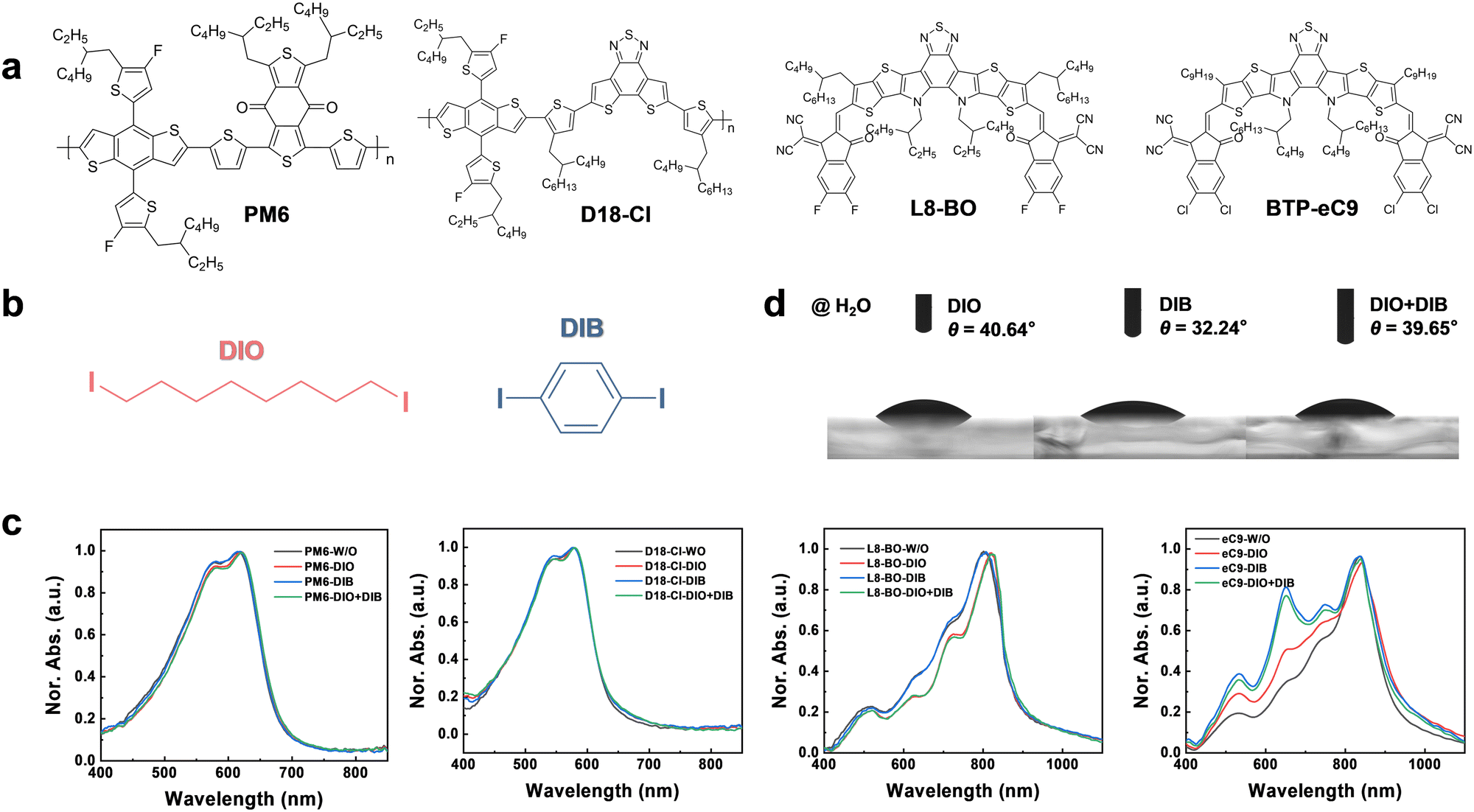 | ||
| Fig. 1 Chemical structures and UV-vis absorption. Chemical structures of (a) photo-active materials PM6, D18-Cl, L8-BO and BTP-eC9 and (b) additives DIO and DIB. (c) Normalized absorption spectra of PM6, D18-Cl, L8-BO and BTP-eC9 films with varied additives. (d) Contact angles of H2O on DIO, DIB, and DIO + DIB films. | ||
However, the additives significantly affect the absorption of the acceptors. As shown in Fig. 1c, the absorption peaks of the L8-BO film significantly shift from 798 to 819, 798, and 825 nm with DIO, DIB, and DIO + DIB as additives, respectively. Therefore, the addition of DIO red-shifts the L8-BO absorption peak by over 20 nm, while the DIB almost does not affect the absorption. Meanwhile, a notable shift of BTP-eC9 absorption is observed, e.g. the edges shift from 942 to 970, 965, and 960 nm for films that possess DIO, DIB, and DIO + DIB additives, respectively, with DIO extending the absorption by 28 nm. Moreover, the rather broadened absorption in the range of 450–750 nm with a shoulder peak at 650 nm indicates strong interaction of the BTP core unit of BTP-eC9 with the introduction of DIB due to close π–π interactions.46 The relative absorption intensity of 0–1 and 0–0 (I0–1/I0–0) shows an increasing trend of 0.345, 0.503, 0.770, and 0.811, suggesting that the compact molecular packing of BTP-eC9 has been disturbed with the addition of DIB.47
To examine the interaction between the photo-active materials and additives, surface tensions are measured via the Owens and Wendt theory and Young's equation, and show values of 27.13, 26.58, 45.18, and 41.72 mJ cm−2 for PM6, D18-Cl, L8-BO, and BTP-eC9, and 173.58, 191.91, and 175.88 mJ cm−2 for DIO, DIB, and DIO + DIB, respectively (Fig. 1d and Table S1, Fig. S1, S2, ESI†). The surface tensions of the acceptors are larger than those of the donors, yet much smaller than both additives, which indicates that additives are more likely to interact with acceptors, and contribute to the differences in absorption profiles (Fig. S3 and S4, ESI†). Furthermore, the Flory–Huggins interaction parameter χD/A–Add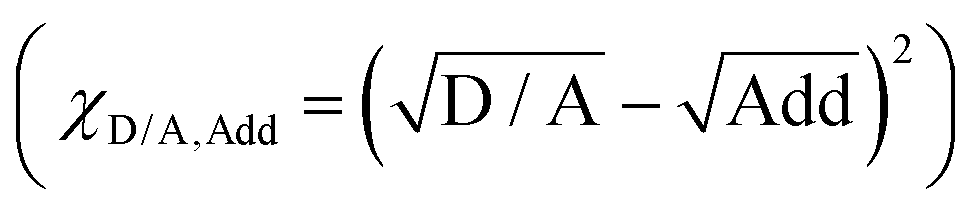 is used to identify the miscibility between the additive and the donor or acceptor materials (Table S2, ESI†).48–50 As shown, the acceptors:additives show a smaller χD/A–Add value of ∼40, while the donors show higher χD/A–Add of ∼60, confirming that acceptors are more miscible with both DIO and DIB additives. These results are consistent with the absorption variation trends of polymer donors and small molecule acceptors with different additives, i.e. the absorption spectra with different additives remain almost unchanged for donors, while they varied for acceptors (Fig. 1c).
is used to identify the miscibility between the additive and the donor or acceptor materials (Table S2, ESI†).48–50 As shown, the acceptors:additives show a smaller χD/A–Add value of ∼40, while the donors show higher χD/A–Add of ∼60, confirming that acceptors are more miscible with both DIO and DIB additives. These results are consistent with the absorption variation trends of polymer donors and small molecule acceptors with different additives, i.e. the absorption spectra with different additives remain almost unchanged for donors, while they varied for acceptors (Fig. 1c).
Photovoltaic performance
Furthermore, the effect of different additives on the performance of the quaternary OPV is examined, via a conventional bulk-heterojunction structure, i.e., indium tin oxide (ITO)/2-(9H-carbazol-9-yl) (2PACz)/PM6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D18-Cl:L8-BO:BTP-eC9 (1:0.25:0.75:0.75, wt%)/aliphatic amine-functionalized perylene-diamide (PDINN)/Ag (Fig. 2a). The current-density–voltage (J–V) curves and photovoltaic parameters of the OPV devices are shown in Fig. 2a–c and Table 1. The additive-free device exhibits a PCE of 19.03%, with an open-circuit voltage (VOC) of 0.901 V due to band alignments of the four materials (Fig. S5, ESI†), a short-circuit current density (JSC) of 27.80 mA cm−2, and a fill factor (FF) of 75.79%. With the introduction of a single additive of DIO or DIB, the device efficiency gets increased, and champion PCEs of 19.42% (VOC of 0.863 V, JSC of 28.43 mA cm−2, and FF of 78.77%) and 19.25% (VOC of 0.884 V, JSC of 27.55 mA cm−2, and FF of 78.67%) are obtained, respectively. A reduction in VOC and an increase in FF are observed with both DIO and DIB as additives, and the improved balance between VOC and FF determines the increment in PCE. Surprisingly, the dual-additive based OPV devices deliver a maximum PCE of 20.52%, with a VOC of 0.879 V, a JSC of 28.55 mA cm−2, and an FF of 81.33%. This champion device is transferred to and certified by the National Photovoltaic Industry Metrology and Testing Center of China, and exhibits a PCE of 19.92% (Fig. S6, ESI†). To our knowledge, it currently represents one of the best among all certified single-junction OPVs ever reported in the literature (Fig. S7 and Table S3, ESI†).7–9
:D18-Cl:L8-BO:BTP-eC9 (1:0.25:0.75:0.75, wt%)/aliphatic amine-functionalized perylene-diamide (PDINN)/Ag (Fig. 2a). The current-density–voltage (J–V) curves and photovoltaic parameters of the OPV devices are shown in Fig. 2a–c and Table 1. The additive-free device exhibits a PCE of 19.03%, with an open-circuit voltage (VOC) of 0.901 V due to band alignments of the four materials (Fig. S5, ESI†), a short-circuit current density (JSC) of 27.80 mA cm−2, and a fill factor (FF) of 75.79%. With the introduction of a single additive of DIO or DIB, the device efficiency gets increased, and champion PCEs of 19.42% (VOC of 0.863 V, JSC of 28.43 mA cm−2, and FF of 78.77%) and 19.25% (VOC of 0.884 V, JSC of 27.55 mA cm−2, and FF of 78.67%) are obtained, respectively. A reduction in VOC and an increase in FF are observed with both DIO and DIB as additives, and the improved balance between VOC and FF determines the increment in PCE. Surprisingly, the dual-additive based OPV devices deliver a maximum PCE of 20.52%, with a VOC of 0.879 V, a JSC of 28.55 mA cm−2, and an FF of 81.33%. This champion device is transferred to and certified by the National Photovoltaic Industry Metrology and Testing Center of China, and exhibits a PCE of 19.92% (Fig. S6, ESI†). To our knowledge, it currently represents one of the best among all certified single-junction OPVs ever reported in the literature (Fig. S7 and Table S3, ESI†).7–9
 | ||
| Fig. 2 Photovoltaic performance. (a) Device structure and the corresponding J–V curves of devices based on PM6:D18-Cl:L8-BO:BTP-eC9 quaternary blend. (b) PCE variation of quaternary OPVs without additive and with additives of DIO, DIB, and DIO + DIB. (c) VOC, JSC, and FF variations. (d) EQE spectra of quaternary OPVs without additives and with additives of DIO, DIB, and DIO + DIB. | ||
mWcm−2
| Additive | V OC (V) | J SC (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|
| wo | 0.901 (0.896 ± 0.003) | 27.80 (27.66 ± 0.10) | 75.79 (74.79 ± 1.00) | 19.03 (18.57 ± 0.33) |
| DIO | 0.863 (0.865 ± 0.04) | 28.43 (28.21 ± 0.29) | 78.77 (78.70 ± 1.11) | 19.42 (19.30 ± 0.31) |
| DIB | 0.884 (0.884 ± 0.04) | 27.55 (27.54 ± 0.29) | 78.67 (77.91 ± 0.79) | 19.25 (19.05 ± 0.36) |
| DIO + DIB | 0.879 (0.874 ± 0.01) | 28.55 (28.54 ± 0.18) | 81.33 (80.91 ± 0.44) | 20.52 (20.31 ± 0.12) |
Statistically, the dual-additives strategy of both DIO and DIB better compromises the benefits of individual additives, and enables the OPV device to achieve a maximum JSC and FF, as well as a balanced VOC. And a similar enhancement of efficiency can be obtained by replacing D18-Cl with D18 due to energy alignment and complementary absorption (Table S4, ESI†). To examine the effect of additives on photo-carrier generation, the external quantum efficiency (EQE) spectra are measured and shown in Fig. 2d. The DIB-based device exhibits a higher EQE response in the whole range, with a slight 8 nm red-shifted edge compared with additive-free device (from 908 nm to 916 nm). Yet, the DIO-based device shows a red-shift of 26 nm (934 nm) and an EQE increase in the near-infrared region, which contributes to the much-improved JSC. Encouragingly, the dual-additive device shows an extended EQE range to 938 nm, and this contributes to the enhanced photocurrent. The calculated JSC values from EQE spectra are 27.36 28.03, 27.60, and 28.19 mA cm−2 for quaternary OPVs without and with additives of DIO, DIB, and DIO + DIB, in line with the values extracted from the J–V curves. The increase in JSC of the devices with DIO and DIO + DIB additives can be attributed to the extended absorption of acceptors in the near infrared region (Fig. S8, ESI†). The higher response in 550–610 nm and 750–900 nm is further attributed to preferred molecular stacking and extended absorption onset.51
The hierarchical morphology
The nano-scaled surface morphology and internal morphology of the quaternary films based on different additive addition are investigated. The surface topological structures of the photo-active layer with different additives are recorded via atomic force microscopy (AFM) measurements (Fig. S9, ESI†). The roughnesses of PM6:D18-Cl:L8-BO:BTP-eC9 blend films without and with DIO, DIB, and DIO + DIB additives are measured to be 0.81, 0.82, 0.76, and 0.78 nm, respectively. Meanwhile, distinct fibril network structures have been observed in all cases, and this is demonstrated to be beneficial to balance the exciton dissociation and charge transport.6 The detailed phase structure is further characterized in the infrared AFM (IR-AFM) plots (Fig. 3a and Fig. S10, S11, ESI†), which show the calculated phase ratio maps of BTP-eC9 or L8-BO (in red) in comparison to the host donor PM6 (in blue) by over-layering the two components. The dominance of acceptors on top indicates the hierarchical separation morphology. However, the films with no additive and with DIO or DIB are more evenly-distributed.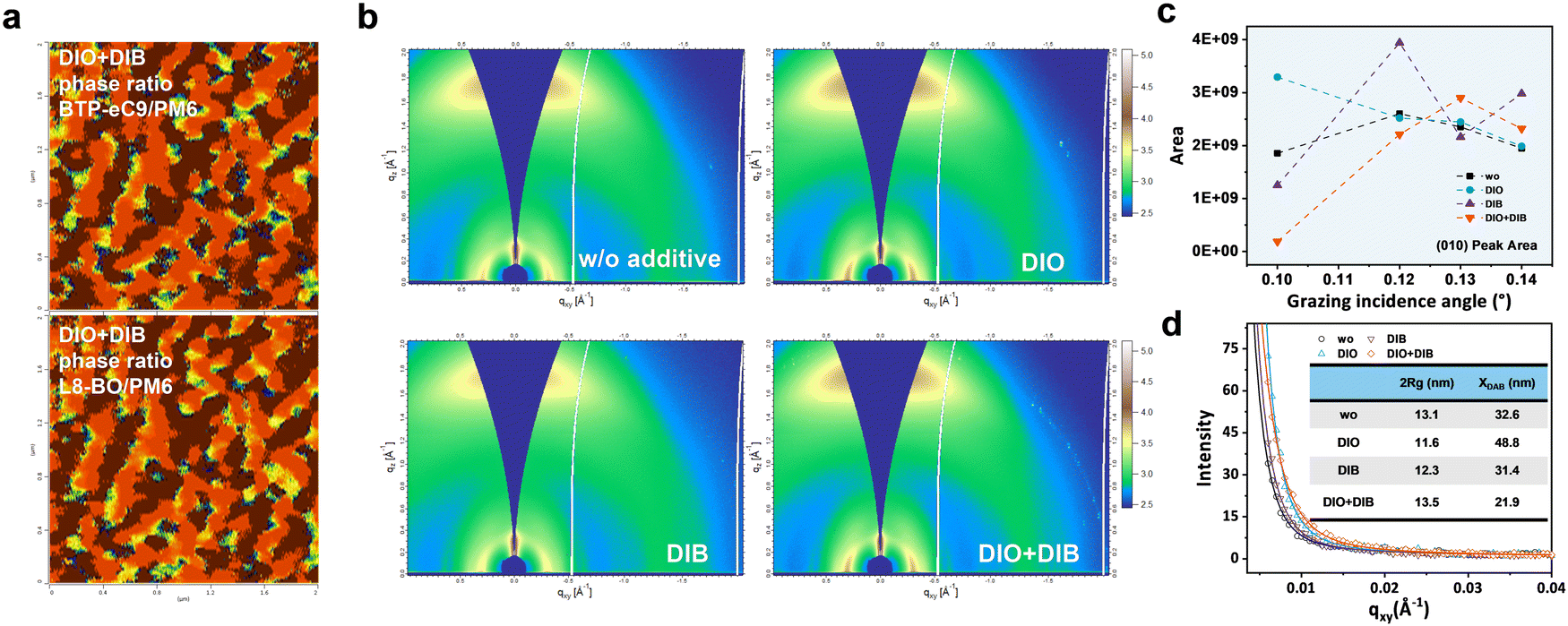 | ||
| Fig. 3 Self-organized hierarchical morphology. (a) IR-AFM images of the calculated phase ratio of L8-BO and BTP-eC9 in comparison to PM6 with DIO + DIB additive. (b) 2D-GIWAXS patterns. (c) (010) peak area calculated from the GIWAXS measurement with beam incidents at a grazing angle of 0.10°–0.14° of the additive-free, DIO, DIB, DIO + DIB based quaternary blends. (d) GISAXS plots of the in-plane line-cut plots and fitting lines. | ||
Grazing-incidence wide-angle X-ray scattering (GIWAXS) measurement with beam incidents at grazing angles of 0.10°–0.14° is employed to investigate the effect of additives on the crystallinity and orientation of the active layers at different depths. 2D diffraction patterns and the corresponding line-cuts profiles are presented in Fig. 3b, c and Fig. S12–S17, Tables S5–S12 (ESI†). All the blend films show pronounced face-on orientation and π–π stacking diffraction in the out-of-plane (OOP) direction, which is beneficial to vertical charge transport. The π–π stacking peaks at a grazing angle of 0.10° are located at 1.709, 1.715, 1.705 and 1.710 Å−1, corresponding to the π–π stacking distance of 3.68, 3.66, 3.69, and 3.67 Å for the films without additive, with DIO, DIB, and DIO + DIB, respectively. Furthermore, it shows a similar trend at a deeper position: the DIO based-film exhibits the strongest crystallinity, while the DIB based film exhibits the weakest crystallinity. This indicates that DIO is favorable for crystallization, and subsequently, the DIO + DIB based film shows moderate crystallinity.
The (010) peak area and crystalline coherence lengths (CCL) are calculated to compare the π–π stacking and determine the relative degree of crystallinity of the films (Fig. 3c and Fig. S18, ESI†). From bottom to top, the amount of π–π stacking increases for the DIB and DIO + DIB based films, suggesting that DIB favors the vertical hierarchical structure. The CCL length of the DIO + DIB based-film near the upper surface (2.46 nm) is much higher than that of the films with no additive, DIO, and DIB (∼2.25 nm), and this benefits the charge transport at the interface and the charge recombination reduction at the photo-active layer/PDINN surface. As a supplement of the crystallinity near the surface, the similar CCL length at 0.12, 0.13 and 0.14° confirms identical crystallization properties across the film, especially the film with DIO + DIB additive, which indicates the enhancement in crystallinity. This is favorable for the hierarchical morphology and charge transport in the photo-active layer, thus promoting JSC and FF.
The grazing incidence small-angle X-ray scattering (GISAXS) measurements are used to examine the effect of additives on the phase separation characteristics (Fig. 3d). The sizes of the mixed phase (XDAB) and pure phase (2Rg) are shown in Fig. S19 (ESI†), where the derived XDAB values are 32.6, 48.8, 31.4 and 21.9 nm and the 2Rg values are 13.1, 11.6, 12.3 and 13.5 nm for the additive-free, DIO, DIB, and DIO + DIB based quaternary blends, respectively. These results suggest that DIB facilitates the formation of a purer phase. The increase of the length scale in the pure phase and the decrease in the mixed phase indicate the low content of the mixed domain in the dual additive-based blend, which tends to contribute to a low charge recombination, efficient exciton dissociation and fast charge transport.52 These are in line with the increased FF and JSC. Furthermore, when we continue exploring other factors that influence the device performance, the details including material properties, operating temperature, and stability should also be systematically explored (Fig. S20–S22, ESI†).
Morphology evolution kinetics
In order to investigate the origin of hierarchical morphology formation with the dual additives, the film formation kinetics was investigated via in situ time-resolved UV-vis absorption spectroscopy measurements (Fig. 4a and Fig. S23, ESI†). The temporal peak position evolutions of donors and acceptors are plotted in Fig. 4b and c. The spin-casting processes undergo a transition from a solution state to a solid state, where the absorption intensity begins to get decreased and the absorption peak red-shifts before a stable stage is reached. As for the film without additive, the absorption peaks of both donors (621 nm) and acceptors (800 nm) become stable simultaneously at 0.950 s. Interestingly, for the DIB based film, the absorption peaks of acceptors (0.825 s) get saturated before that of donors (1.075 s), contributing to a reduced molecular packing for acceptors due to the insufficient organization duration. For DIO and DIO + DIB based films, an opposite trend happens, with an elongated acceptor solidification but shortened donor solidification, which will quickly precipitate the PM6 and D18-Cl at the bottom, while the BTP-eC9 and L8-BO either penetrate the donors and/or stack on top at a moderate rate. As a result, a desired morphology with self-organized hierarchical distribution with balanced crystallinity and desired phase separation is reached. On the contrary, too fast solidification (DIB-based blend) and too slow solidification (DIO-based blend) lead to severe aggregation, which prohibits the ordered stacking and vertical distribution. Finally, we observe that the absorption peaks of donors are similar, ranging between 621 and 624 nm, while the absorption peaks of acceptors red-shift to 803 nm for DIO and DIB-based films and 806 nm for the DIO + DIB based film (800 nm for the pristine film without additive). The diverse film organization and formation kinetics based on different additive treatments will further influence the hierarchical distribution feature. | ||
| Fig. 4 Film organization and formation kinetics. (a) The color mapping of in situ UV-vis absorption spectra as a function of spin-coating time for the quaternary OPVs without additive and with DIO, DIB, and DIO + DIB additives. (b) Temporal evolution of the peak positions of acceptors in the quaternary blend films. (c) Temporal evolution of the peak positions of donors in the quaternary blend films. (d) 3D diagrams of the I− distribution in quaternary blend films based on additive-free, DIO, DIB, and DIO + DIB. (e) F− and Cl− distribution in quaternary blend films. (f) Illustration diagram of the material distribution based on PM6:D18-Cl:L8-BO:BTP-eC9 with varied additives. | ||
The hierarchical morphology is further characterized by film depth-dependent light absorption spectroscopy (FLAS) and time-of-flight secondary ion mass spectrometry (ToF-SIMS) measurements in Fig. 4d–f and Fig. S24 (ESI†). By plotting the upper surface, the peak intensities of D:A are 1.82, 1.09, 1.81, and 1.50 for additive-free, DIO, DIB and DIO + DIB based films, respectively, indicating the upper surface of the active layer with an acceptor-rich feature. At the bottom, the peak intensities of D:A are 1.38, 1.40, 1.42, and 1.47, respectively, which implies the dominance of the donors, especially for the DIO + DIB based film. Furthermore, the ion intensity versus sputtering time of varied films on elements I−, F− and Cl− is characterized, where the sputter time can be correlated with the film depth. From the molecular structure of the active layers, the I− originates from the additives (both DIO and DIB), F− belongs to PM6 and L8-BO, and Cl− belongs to D18-Cl and BTP-eC9. In the film without additives, there is no I− detected. Interestingly, for the sample with DIB additive, a trace amount of I− residue is observed in the bulk, while the films with DIO and DIO + DIB additives exhibit higher I− intensity (Fig. S24 and S25, ESI†). As a result, by delicately manipulating the iodine element, molecular packing can be regulated correspondingly, which correlates with the pathways for charge formation and transport.
From the above results, the vertical distribution of the components can be identified, which is possibly assisted by the different vapour pressures of the DIO and DIB additives (Fig. 4e and f). First, the Cl− concentration of all films shows a relatively flat trend throughout the bulk, indicating an even vertical spreading of D18-Cl and BTP-eC9 from bottom to top. However, the F− signals show a diversified trend, where the films without additives show a steeper intensity variation as the sputter time ranges from 50–180 s. Due to the low intensity of absorption at the bottom, both PM6 and L8-BO ought to accumulate on top. Similar behaviors have been observed for films with DIO or DIB as additives. In contrast, the DIO + DIB based film shows a relatively stable F− signal, affirming the perfect vertical distribution, where PM6 enriches at the ITO side, while L8-BO moves upward. Such vertical hierarchical structure favours the charge generation, separation and transport.
Carrier properties and energy loss analysis
The competition between charge extraction and recombination ultimately determines the device performance. First, transient absorption spectroscopy (TAS) measurements were carried out to investigate the exciton dissociation dynamics of blend films with various additives. To selectively excite the acceptors, an exciton wavelength at 800 nm is imposed (Fig. 5a and Fig. S26, ESI†). With the ground state bleaching decay of the acceptors, the photo-bleach signal of the donors rises, which proves the hole transfer process occurring from acceptor to donor. The kinetics at ∼600 nm are fitted with a biexponential decay function to quantitatively analyze the carrier dynamics. τ1 values of 1.51, 1.47, 1.55, 1.22 ps and τ2 values of 15.41, 15.24, 14.49, 10.40 ps are extracted for the additive-free, DIO, DIB, and DIO + DIB based films. Smaller τ1 and τ2 values of the films with dual additives suggest a faster exciton dissociation as well as a faster diffusion.53 The smallest τ1 is attributed to a clear D:A interface and the lowest τ2 is associated with the enhanced intermolecular packing, which benefits the exciton diffusion.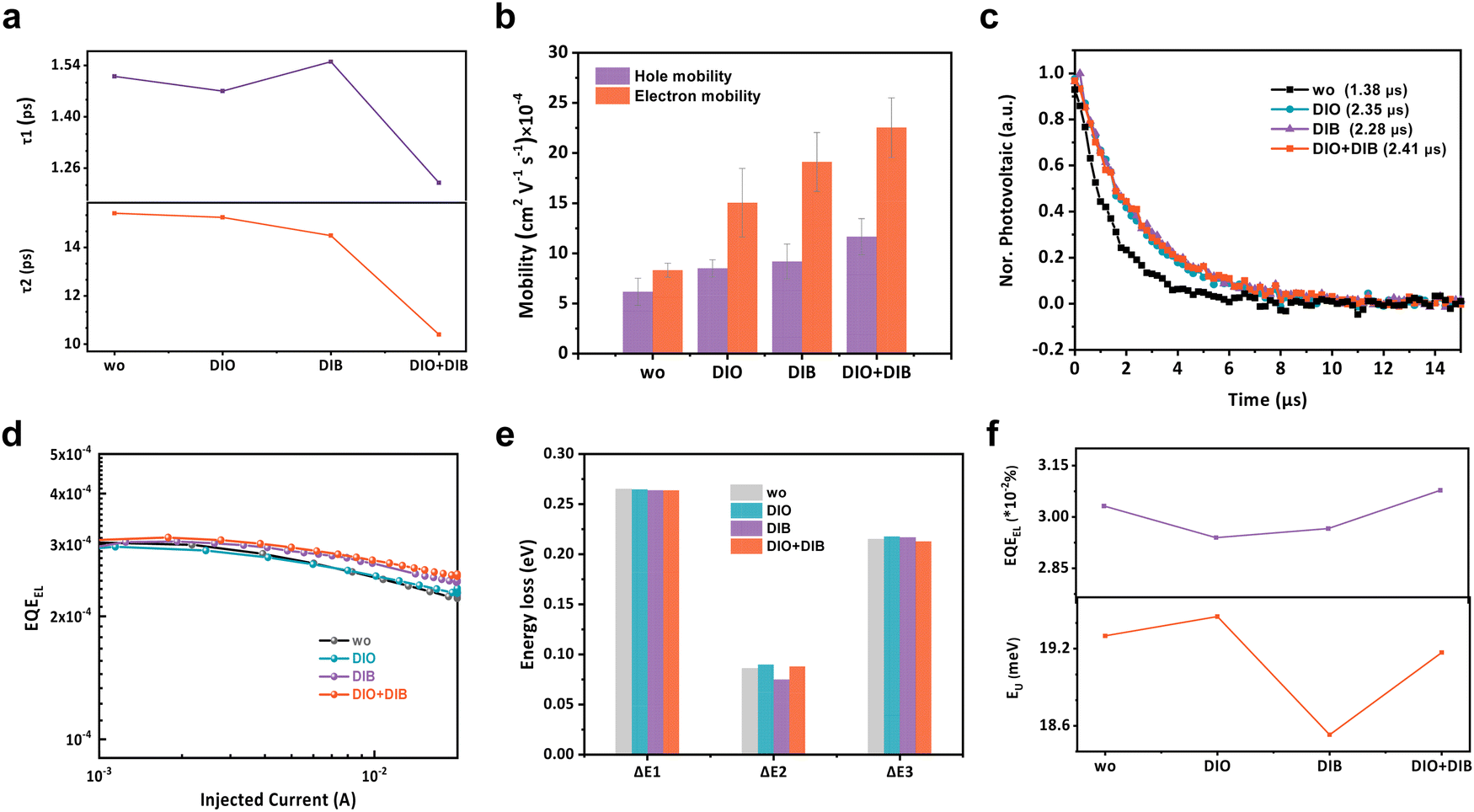 | ||
| Fig. 5 Carrier properties and energy loss analysis. (a) τ1 and τ2 values extracted from the TAS measurements. (b) Hole and electron mobilities of devices with different additives. (c) TPV measurements of the corresponding quaternary OPVs. (d) EQEEL of devices at various injected currents. (e) Detailed energy loss in additive-free, DIO, DIB, and DIO + DIB based quaternary devices. (f) Variations of EQEEL and EU in the corresponding OPV devices. | ||
The charge transport is further measured by the space-charge limited current (SCLC) method.54 The hole (μh) and electron (μe) mobilities exhibit an increasing trend from films without additive to that of DIO, DIB, and DIO + DIB, with the dual-additive based film exhibiting the highest mobility of 1.23 × 10−3 cm2 V−1 s−1 and 1.05 × 10−3 cm2 V−1 s−1 for μe and μh, respectively (Fig. 5b and Fig. S27, Table S13, ESI†), which are one order higher than its counterparts. The addition of DIB contributes to both higher μh and μe, which leads to suppressed recombination and efficient charge extraction. The photocurrent (Jph) versus effective voltage shows improved exciton dissociation (ηdiss) and charge collection (ηcoll) properties, where the ηdiss and ηcoll values are 0.989, 0.984, 0.967, 0.990 and 0.891, 0.916, 0.884, 0.937 for the additive-free, DIO, DIB, and DIO + DIB based devices (Fig. S28 and Table S14, ESI†).
Transient photovoltaics (TPV) is employed to investigate the impact of additives on carrier lifetime (Fig. 5c).55 The additive-free devices exhibit the shortest carrier lifetime of 1.38 μs, whereas the lifetimes of the DIO, DIB, and DIO + DIB based devices are 2.35, 2.28, and 2.41 μs. This is consistent with the reduced charge recombination from the light intensity-dependent J–V measurements. The twofold increase in carrier lifetime validates suppressed charge recombination with dual additives. Charge recombination via the light intensity-dependent VOC (Fig. S29, ESI†) is also investigated. The slopes of these curves are 1.31kT/q, 1.12kT/q, 1.27kT/q and 1.16kT/q for additive-free, DIO, DIB, and DIO + DIB based devices, where k, T, and q are the Boltzmann constant, absolute temperature in Kelvin, and elementary charge, respectively, while the light intensity-dependent JSC shows almost unity α values. This implies that charge recombination is similar under short-circuit conditions, while trap-assisted monomolecular recombination is suppressed in the DIO and DIO + DIB based devices. It can be concluded that improved morphology contributes to efficient exciton dissociation, charge transport and reduced recombination.
Energy loss in these four systems is also analyzed (Fig. 5d–f and Fig. S30, S31, Tables S15–S17, ESI†).56,57 According to previous work, the energy loss can be classified into three channels (ΔEloss = ΔE1 + ΔE2 + ΔE3). For these four representative devices with the sole difference in the active-layer morphology, ΔE1 is similar due to the band gap. However, for the radiative loss below the band gap, i.e. the ΔE2, the values are 0.086, 0.090, 0.075 and 0.088 eV for additive-free, DIO, DIB, and DIO + DIB based devices. The DIB-based device shows the lowest ΔE2, indicating the smallest energetic disorder or reorganization energy, while the DIO + DIB based-device lies in between. The degree of energetic disorder can also be quantified by the Urbach energy (EU). By plotting the high-resolution Fourier transform photocurrent spectroscopy EQE spectra (FTPS-EQE), the EU values are measured to be 19.3, 19.45, 18.53, and 19.17 meV for additive-free, DIO, DIB, and DIO + DIB based devices, which is consistent with ΔE2. As a result, the ΔE3 values for these devices are calculated to be 0.215, 0.218, 0.217, and 0.213 eV, respectively. Additionally, ΔE3 can also be obtained by calculating the electroluminescence quantum efficiency (EQEEL), where the corresponding values of 0.209, 0.210, 0.210, and 0.209 eV are calculated. These results suggest that the DIO + DIB based device shows the lowest non-radiative recombination loss. Furthermore, the total energy losses are 0.566, 0.572, 0.556, and 0.565 eV for additive-free, DIO, DIB, and DIO + DIB based OPV devices, respectively. The compromised radiative loss via suppressed electron vibration and non-radiative loss via elongated electroluminescence efficiency eventually contribute to the decrease in VOC loss.
Conclusion
In conclusion, we proposed a dual-additive strategy to manipulate the morphology of a multiple-component system. The dual-additive strategy with the combination of liquid additive DIO and solid additive DIB enables a self-organized hierarchical morphology, in which enhanced crystallinity and phase separation are concurrently achieved via the favorable film formation kinetics. As a result, we have demonstrated a high efficiency of 20.52% (certified 19.92%) in single-junction OPVs. This work elucidates a dual-additive strategy, which regulates the morphology intelligently toward efficient exciton dissociation, charge transport, reduced exciton recombination and suppressed energy loss, offering great promise for high performance OPVs.Author contributions
S. G., L. Z., H. L. and H. C. conceived and designed the research. S. G. and Y. L. designed the experiments and performed device fabrications. B. Z. and W. M. performed the GIWAXS measurements. Y. F. and X. L. performed the GISAXS measurements. Y. L. and Z. M. and Z. T. performed the energy loss measurements. K. W. and J. X. performed the TAS measurements. M. W. performed the contact angle measurements.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank the National Natural Science Foundation of China (No. 52321165650, 52173185, 52394274, and 52127806) for financial support. We also acknowledge gratefully the Fundamental Research Funds for the Central Universities (No. 226-2022-00133, 226-2024-00005).References
- P. Cheng, G. Li, X. Zhan and Y. Yang, Nat. Photon., 2018, 12, 131 CrossRef CAS
.
- Y. Li, C. He, L. Zuo, F. Zhao, L. Zhan, X. Li, R. Xia, H.-L. Yip, C.-Z. Li, X. Liu and H. Chen, Adv. Energy Mater., 2021, 11, 2003408 CrossRef CAS
- X. Zheng, L. Zuo, F. Zhao, Y. Li, T. Chen, S. Shan, K. Yan, Y. Pan, B. Xu, C.-Z. Li, M. Shi, J. Hou and H. Chen, Adv. Mater., 2022, 34, 2200044 CrossRef CAS
- Y. Li, J. Wang, C. Yan, S. Zhang, N. Cui, Y. Liu, G. Li and P. Cheng, Joule, 2024, 8, 527 CrossRef CAS
- L. Feng, J. Yuan, Z. Zhang, H. Peng, Z.-G. Zhang, S. Xu, Y. Liu, Y. Li and Y. Zou, ACS Appl. Mater. Interfaces, 2017, 9, 31985 CrossRef CAS
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656 CrossRef CAS PubMed
- Y. Jiang, S. Sun, R. Xu, F. Liu, X. Miao, G. Ran, K. Liu, Y. Yi, W. Zhang and X. Zhu, Nat. Energy, 2024, 9, 975–986 CrossRef CAS
- Y. Sun, L. Wang, C. Guo, J. Xiao, C. Liu, C. Chen, W. Xia, Z. Gan, J. Cheng, J. Zhou, Z. Chen, J. Zhou, D. Liu, T. Wang and W. Li, J. Am. Chem. Soc., 2024, 146, 12011–12019 CrossRef CAS
- S. Guan, Y. Li, C. Xu, N. Yin, C. Xu, C. Wang, M. Wang, Y. Xu, Q. Chen, D. Wang, L. Zuo and H. Chen, Adv. Mater., 2024, 36, 2400342 CrossRef CAS
- L. Wang, C. Chen, Y. Fu, C. Guo, D. Li, J. Cheng, W. Sun, Z. Gan, Y. Sun, B. Zhou, C. Liu, D. Liu, W. Li and T. Wang, Nat. Energy, 2024, 9, 208–218 CrossRef CAS
- Z. Chen, S. Zhang, J. Ren, T. Zhang, J. Dai, J. Wang, L. Ma, J. Qiao, X. Hao and J. Hou, Adv. Mater., 2024, 36, 2310390 CrossRef CAS
- M. Wang, T. Chen, Y. Li, G. Ding, Z. Chen, J. Li, C. Xu, A. Wupur, C. Xu, Y. Fu, J. Xue, W. Fu, W. Qiu, X. Yang, D. Wang, W. Ma, X. Lu, H. Zhu, X. Chen, X. Wang, H. Chen and L. Zuo, Energy Environ. Sci., 2024, 17, 2598 RSC
- T. Chen, S. Li, Y. Li, Z. Chen, H. Wu, Y. Lin, Y. Gao, M. Wang, G. Ding, J. Min, Z. Ma, H. Zhu, L. Zuo and H. Chen, Adv. Mater., 2023, 35, 2300400 CrossRef CAS
- C. He, Y. Pan, Y. Ouyang, Q. Shen, Y. Gao, K. Yan, J. Fang, Y. Chen, C.-Q. Ma, J. Min, C. Zhang, L. Zuo and H. Chen, Energy Environ. Sci., 2022, 15, 2537 RSC
- Y. Li, Y. Guo, Z. Chen, L. Zhan, C. He, Z. Bi, N. Yao, S. Li, G. Zhou, Y. Yi, Y. (Michael) Yang, H. Zhu, W. Ma, F. Gao, F. Zhang, L. Zuo and H. Chen, Energy Environ. Sci., 2022, 15, 855 RSC
- J. Fu, P. W. K. Fong, H. Liu, C.-S. Huang, X. Lu, S. Lu, M. Abdelsamie, T. Kodalle, C. M. Sutter-Fella, Y. Yang and G. Li, Nat. Commun., 2023, 14, 1760 CrossRef CAS
- R. Yu, H. Yao, L. Hong, Y. Qin, J. Zhu, Y. Cui, S. Li and J. Hou, Nat. Commun., 2018, 9, 4645 CrossRef PubMed
- G. Ding, T. Chen, M. Wang, X. Xia, C. He, X. Zheng, Y. Li, D. Zhou, X. Lu, L. Zuo, Z. Xu and H. Chen, Nano-Micro Lett., 2023, 15, 92 CrossRef CAS
- X. Xu, L. Yu, H. Meng, L. Dai, H. Yan, R. Li and Q. Peng, Adv. Funct. Mater., 2022, 32, 2108797 CrossRef CAS
- S. Lai, Y. Cui, Z. Chen, X. Xia, P. Zhu, S. Shan, L. Hu, X. Lu, H. Zhu, X. Liao and Y. Chen, Adv. Mater., 2024, 36, 2313105 CrossRef CAS
- X. Xu, W. Jing, H. Meng, Y. Guo, L. Yu, R. Li and Q. Peng, Adv. Mater., 2023, 35, 2208997 CrossRef CAS
- S. Chen, L. Hong, M. Dong, W. Deng, L. Shao, Y. Bai, K. Zhang, C. Liu, H. Wu and F. Huang, Angew. Chem., 2023, 135, e202213869 CrossRef
- C. McDowell, M. Abdelsamie, M. F. Toney and G. C. Bazan, Adv. Mater., 2018, 30, 1707114 CrossRef PubMed
- X.-K. Chen, M. K. Ravva, H. Li, S. M. Ryno and J.-L. Brédas, Adv. Energy Mater., 2016, 6, 1601325 CrossRef
- Y. Fu, T. H. Lee, Y.-C. Chin, R. A. Pacalaj, C. Labanti, S. Y. Park, Y. Dong, H. W. Cho, Y. Kim, D. Minami, J. R. Durrant and J.-S. Kim, Nat. Commun., 2023, 14, 1 CAS
- E. Meighen and R. Yue, Biochim. Biophys. Acta, 1975, 412, 262 CrossRef CAS PubMed
- L. Xie, D. He and F. Zhao, J. Mater. Chem. C, 2024, 12, 819 RSC
- C. Cui and Y. Li, Aggregate, 2021, 2, e31 CrossRef CAS
- J. Peet, N. S. Cho, S. K. Lee and G. C. Bazan, Macromolecules, 2008, 41, 8655 CrossRef CAS
- J. K. Lee, W. L. Ma, C. J. Brabec, J. Yuen, J. S. Moon, J. Y. Kim, K. Lee, G. C. Bazan and A. J. Heeger, J. Am. Chem. Soc., 2008, 130, 3619 CrossRef CAS
- J. Fu, H. Chen, P. Huang, Q. Yu, H. Tang, S. Chen, S. Jung, K. Sun, C. Yang, S. Lu, Z. Kan, Z. Xiao and G. Li, Nano Energy, 2021, 84, 105862 CrossRef CAS
- Y. Xie, H. S. Ryu, L. Han, Y. Cai, X. Duan, D. Wei, H. Y. Woo and Y. Sun, Sci. China: Chem., 2021, 64, 2161 CrossRef CAS
- P. P. Khlyabich, M. Sezen-Edmonds, J. B. Howard, B. C. Thompson and Y.-L. Loo, ACS Energy Lett., 2017, 2, 2149 CrossRef CAS
- L. Zuo, S. B. Jo, Y. Li, Y. Meng, R. J. Stoddard, Y. Liu, F. Lin, X. Shi, F. Liu, H. W. Hillhouse, D. S. Ginger, H. Chen and A. K.-Y. Jen, Nat. Nanotechnol., 2022, 17, 53 CrossRef CAS
- S. Li, C. He, T. Chen, J. Zheng, R. Sun, J. Fang, Y. Chen, Y. Pan, K. Yan, C.-Z. Li, M. Shi, L. Zuo, C.-Q. Ma, J. Min, Y. Liu and H. Chen, Energy Environ. Sci., 2023, 16, 2262 RSC
- K. Jiang, J. Zhang, C. Zhong, F. R. Lin, F. Qi, Q. Li, Z. Peng, W. Kaminsky, S.-H. Jang, J. Yu, X. Deng, H. Hu, D. Shen, F. Gao, H. Ade, M. Xiao, C. Zhang and A. K.-Y. Jen, Nat. Energy, 2022, 7, 1076 CrossRef
- L. Arunagiri, Z. Peng, X. Zou, H. Yu, G. Zhang, Z. Wang, J. Y. Lin Lai, J. Zhang, Y. Zheng, C. Cui, F. Huang, Y. Zou, K. S. Wong, P. C. Y. Chow, H. Ade and H. Yan, Joule, 2020, 4, 1790 CrossRef CAS
- R. Zeng, M. Zhang, X. Wang, L. Zhu, B. Hao, W. Zhong, G. Zhou, J. Deng, S. Tan, J. Zhuang, F. Han, A. Zhang, Z. Zhou, X. Xue, S. Xu, J. Xu, Y. Liu, H. Lu, X. Wu, C. Wang, Z. Fink, T. P. Russell, H. Jing, Y. Zhang, Z. Bo and F. Liu, Nat. Energy, 2024, 9, 1117–1128 CAS
- Y. Cai, Q. Li, G. Lu, H. S. Ryu, Y. Li, H. Jin, Z. Chen, Z. Tang, G. Lu, X. Hao, H. Y. Woo, C. Zhang and Y. Sun, Nat. Commun., 2022, 13, 2369 CrossRef CAS PubMed
- F. Liu, C. Wang, J. K. Baral, L. Zhang, J. J. Watkins, A. L. Briseno and T. P. Russell, J. Am. Chem. Soc., 2013, 135, 19248 CrossRef CAS PubMed
- L. Ye, H. Hu, M. Ghasemi, T. Wang, B. A. Collins, J.-H. Kim, K. Jiang, J. H. Carpenter, H. Li, Z. Li, T. McAfee, J. Zhao, X. Chen, J. L. Y. Lai, T. Ma, J.-L. Bredas, H. Yan and H. Ade, Nat. Mater., 2018, 17, 253 CrossRef CAS
- M. Zhang, X. Guo, W. Ma, H. Ade and J. Hou, Adv. Mater., 2015, 27, 4655 CrossRef CAS PubMed
- J. Qin, L. Zhang, C. Zuo, Z. Xiao, Y. Yuan, S. Yang, F. Hao, M. Cheng, K. Sun, Q. Bao, Z. Bin, Z. Jin and L. Ding, J. Semicond., 2021, 42, 010501 CrossRef CAS
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma, C. An, C. He, Z. Wei, F. Gao and J. Hou, Adv. Mater., 2020, 32, 1908205 CrossRef CAS
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605 CrossRef CAS
- X. Song, K. Zhang, R. Guo, K. Sun, Z. Zhou, S. Huang, L. Huber, M. Reus, J. Zhou, M. Schwartzkopf, S. V. Roth, W. Liu, Y. Liu, W. Zhu and P. Müller-Buschbaum, Adv. Mater., 2022, 34, 2200907 CrossRef CAS PubMed
- C. Li, X. Gu, Z. Chen, X. Han, N. Yu, Y. Wei, J. Gao, H. Chen, M. Zhang, A. Wang, J. Zhang, Z. Wei, Q. Peng, Z. Tang, X. Hao, X. Zhang and H. Huang, J. Am. Chem. Soc., 2022, 144, 14731 CrossRef CAS PubMed
- L. Ye, H. Hu, M. Ghasemi, T. Wang, B. A. Collins, J.-H. Kim, K. Jiang, J. H. Carpenter, H. Li, Z. Li, T. McAfee, J. Zhao, X. Chen, J. L. Y. Lai, T. Ma, J.-L. Bredas, H. Yan and H. Ade, Nat. Mater., 2018, 17, 253 CrossRef CAS PubMed
- N. D. Treat, A. Varotto, C. J. Takacs, N. Batara, M. Al-Hashimi, M. J. Heeney, A. J. Heeger, F. Wudl, C. J. Hawker and M. L. Chabinyc, J. Am. Chem. Soc., 2012, 134, 15869 CrossRef CAS PubMed
- L. Ye, B. A. Collins, X. Jiao, J. Zhao, H. Yan and H. Ade, Adv. Energy Mater., 2018, 8, 1703058 CrossRef
- N. T. Shewmon, D. L. Watkins, J. F. Galindo, R. B. Zerdan, J. Chen, J. Keum, A. E. Roitberg, J. Xue and R. K. Castellano, Adv. Funct. Mater., 2015, 25, 5166 CrossRef CAS
- S. R. Cowan, W. L. Leong, N. Banerji, G. Dennler and A. J. Heeger, Adv. Funct. Mater., 2011, 21, 3083 CrossRef CAS
- Y. Tamai, Y. Murata, S. Natsuda and Y. Sakamoto, Adv. Energy Mater., 2023, 14, 2301890 CrossRef
- J. A. Bartelt, D. Lam, T. M. Burke, S. M. Sweetnam and M. D. McGehee, Adv. Energy Mater., 2015, 5, 1500577 CrossRef
- S. Wood, J. C. Blakesley and F. A. Castro, Phys. Rev. Appl., 2018, 10, 024038 CrossRef CAS
- Y. Wang, D. Qian, Y. Cui, H. Zhang, J. Hou, K. Vandewal, T. Kirchartz and F. Gao, Adv. Energy Mater., 2018, 8, 1801352 CrossRef
- X. Xu, H. Wu, S. Liang, Z. Tang, M. Li, J. Wang, X. Wang, J. Wen, E. Zhou, W. Li and Z. Ma, Acta Phys. -Chim. Sin., 2022, 38, 2201039 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee03778b |
| ‡ S. G. and Y. L. contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |