
An electric double layer regulator empowers a robust solid–electrolyte interphase for potassium metal batteries†
Xueyu
Lian‡
a,
Liang
Xu‡
b,
Zhijin
Ju‡
c,
Ziang
Chen
a,
Xiaopeng
Chen
a,
Yuyang
Yi
d,
Zhengnan
Tian
e,
Tao
Cheng
 *b,
Shixue
Dou
f,
Xinyong
Tao
*g and
Jingyu
Sun
*ah
*b,
Shixue
Dou
f,
Xinyong
Tao
*g and
Jingyu
Sun
*ah
aCollege of Energy, Soochow Institute for Energy and Materials Innovations, Key Laboratory of Advanced Carbon Materials and Wearable Energy Technologies of Jiangsu Province, Soochow University, Suzhou 215006, China. E-mail: sunjy86@suda.edu.cn
bInstitute of Functional Nano & Soft Materials, Jiangsu Provincial Key Laboratory for Carbon-Based Functional Materials & Devices, Joint International Research Laboratory of Carbon-Based Functional Materials and Devices, Soochow University, Suzhou 215123, China. E-mail: tcheng@suda.edu.cn
cCollege of Chemistry and Materials Engineering, Wenzhou University, Wenzhou 325035, China
dDepartment of Industrial and Systems Engineering, The Hong Kong Polytechnic University, Hong Kong 999077, China
eCollege Physical Sciences and Engineering Division, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia
fInstitute of Energy Materials Science, University of Shanghai for Science and Technology, Shanghai 200093, China
gCollege of Materials Science and Engineering, Zhejiang University of Technology, Hangzhou 310014, China. E-mail: tao@zjut.edu.cn
hBeijing Graphene Institute, Beijing 100095, China
First published on 6th November 2024
Abstract
The electric double layer (EDL) plays a key role in constructing a solid electrolyte interphase (SEI) for high-energy metal anodes. Nevertheless, the significance of the EDL and its associated influence remain elusive especially in the potassium metal battery realm. Here we propose an EDL regulation strategy via separator modification targeting durable and longevous potassium metal batteries. We employ a universal metal hydroxide combined with a sulfur-doped graphene layer and show that the H-bond effect exerted by the metal hydroxide could overcome the EDL repulsion and thus rearrange the anode interface to enrich the anion population. In this sense, a robust inorganic-rich SEI is generated, which manages to sustain dynamic evolutions not only in the initial formation stage but also during the cycling stage. Consequently, uniform and stable potassium electroplating occurs even under harsh conditions, enabling high-rate capability at 10 mA cm−2 and elongated lifespan over 6000 h at 8.0 mA cm−2/8.0 mA h cm−2. Our separator modification concept with vast explored design space offers an appealing path for fast-charging and long-lifespan potassium metal batteries.
Broader contextThe potassium metal battery is regarded as a promising candidate for large-scale energy storage applications due to its abundant resource, high operation voltage and favourable ion kinetics in electrolytes. However, the rampant dendrite growth at the anode side accompanied by unstable interface chemistry impedes its practicability, which is closely related to the vulnerable properties of the native solid–electrolyte interphase (SEI). The electric double layer (EDL) plays a decisive role in the SEI construction. It could repel the anions away from the anode, leaving a large amount of solvent congesting the anode interface and decomposing into an organic-dominated SEI. In this study, an EDL regulation strategy is proposed based on separator modification to construct an inorganic-enriched SEI. The metal hydroxide combined with sulfur-doped graphene brings about H-bond interactions between anions, solvent and metal hydroxide, which suppress the EDL repulsion toward the anion and rearrange the anode interface species, rendering a robust SEI. In addition, the metal hydroxide serves as an anion transport hub to continuously deliver anions from the electrolyte for the anode during the cycling stage, thereby guaranteeing the SEI stability and ensuring longevous potassium anodes. This work delineates an EDL regulator to help construct an inorganic-enriched SEI, offering opportunities to develop next-generation energy storage technologies. |
Introduction
Following the boom of lithium metal batteries, potassium metal batteries (PMBs) have been highly sought after because of their abundant resource reserve (2.09 wt%), low electrode potential (−2.93 V vs. standard hydrogen electrode) and favourable K+ transport in electrolytes.1–3 However, the rampant dendrite growth on the anode side arising from the high reactivity of the K metal markedly impedes its implementation.4,5 Recent research endeavours readily highlighted the key role of interfacial chemistry in guiding metal deposition behaviour, which is closely related to the presence of a solid electrolyte interphase (SEI).6,7 A typical SEI could restrain excessive electrolyte decomposition, expanding the electrochemical stability window and facilitating smooth ion transport in between the electrolyte/electrode.8,9 Unfortunately, the native SEI typically exhibits poor mechanical robustness via its uneven mosaic patterns comprising inorganic and organic granules, which experience continuous rupture, dissolution and regeneration during the electrochemical process, ultimately forming a loose interphase. In contrast, inorganic-rich phases with a dense structure are conducive to uniform ion conduction and metal deposition.10–13 It is therefore imperative to modulate the constituent and architecture of the SEI to realize the enrichment of inorganic phases. Note that the SEI could undergo dynamic evolutions, involving not only formation at the initial stage but also regeneration during the cycling stage.14 In view of this, interface modification between the metal anode and electrolyte is a top priority.The electric double layer (EDL) refers to the regime formed at the interface between the electrode and electrolyte due to electrostatic interactions and particle thermal motion.15,16 Its properties largely dictate the competitive reduction reaction at the electrode interface during SEI generation.17 Specifically, the negatively charged nature of the metal anode tends to attract cation aggregation and repel anions away from the interface, giving rise to an unfavourable organic phase SEI, as shown in Fig. 1(a). At present, adopted strategies based on electrolyte modification via tailoring the solvation shell have proven their effectiveness.18,19 However, inherent disparity might exist between the bulk electrolyte and interface chemistry, which accordingly leads to sluggishness in passivating the EDL.20,21 It is meaningful to promptly “break” the EDL repulsion toward anions at the anode side. To the best of our knowledge, the significance of the EDL and its associated influence remain elusive in the PMB realm.
 | ||
| Fig. 1 Separator design concept. (a) Conventional EDL repels anions and induces aggregation of solvent molecules at the anode interface, thus forming an inhomogeneous organic-rich SEI to induce rampant dendrite growth. (b) The designed MOH-SG@GF separator assists anions to resist the electrostatic repulsion and gather at the anode surface, resulting in a durable inorganic-rich SEI to guide uniform metal deposition. | ||
The evolution of the SEI in subsequent cycles is governed by the ion solvation configuration at the interface, which dynamically varies during migration from the bulk electrolyte to the metal anode.22–24 As such, separator modification is of great feasibility in regulating the transmission and transformation of the solvation shell. Prolonging the Sand's time by an anion anchor was proposed to optimize the service life of anodes.25–27 Nevertheless, if anion migration is inhibited without appropriate solvent management, numerous solvent-separated ion pairs (SSIPs) would approach the anode, resulting in the formation of an undesirable organic-phase enriched SEI.28,29 Apart from that, there exists a contradiction between anion capture to suppress their motion and anion liberation to construct an inorganic SEI.30 Indeed, the significance of solvent modulation is on par with anion regulation in achieving an inorganic-rich SEI. Ideally, an anode surface saturated with contact-ion pairs (CIPs) and aggregate ion pairs (AGGs) is favourable.31,32 Since the component and configuration of the SEI closely correlate with the EDL and solvation states at the anode surface, modifying interfacial chemistry has become an important driver toward developing a robust SEI dominated by inorganic phases.
In this contribution, we showcase a glass fibre separator modification strategy targeting dendrite-free K metal anodes via customizable metal hydroxide flakes combined with sulfur-doped graphene carpets (MOH-SG@GF; M = Co, Fe, or Ni). The presence of hydrogen bonding between graphene-supported MOH, solvent molecules and anions effectively suppresses the EDL repulsion and reconfigures its structure, rendering an anion-enriched interface and thus an inorganic-dominated SEI. Alongside, the MOH acts as an anion transport hub without consumption during long-term cycling. It also realizes the modulation of the solvation structure via concurrent interaction with anions and solvent molecules, thereby ensuring the SEI stability (Fig. 1(b)). We validate the EDL rearrangement toward realizing an inorganic-enriched SEI by molecular dynamics (MD) simulations and density functional theory (DFT) calculations. High-resolution cryo-electron microscopy confirms the stable inorganic-phase SEI formation affording large-sized crystalline granules. Through a spectrum of instrumental characterization, we further reveal the initial formation and dynamic evolution of the tailored SEI. The thus-obtained dendrite-free K metal electrode shows collective merits of low overpotential, facile charge-transfer kinetics and durable cycling stability. The K‖MOH-SG@GF‖K symmetric cell can sustain under elevated current density and deposition capacity (enduring 6000 h at 8 mA cm−2/8 mA h cm−2 and 4000 h at 10 mA cm−2/10 mA h cm−2), surpassing its state-of-the-art counterparts.
Results and discussion
Separator design
The fabrication route to our MOH-SG@GF separator mainly encompasses plasma-enhanced chemical vapor deposition (PECVD) growth of S-doped graphene and subsequent hydrothermal synthesis of metal hydroxide (Co element as a representative) on a commercial glass fiber separator (Fig. S1, ESI†). Note that the directly synthesized S-doped graphene separator was subject to a gentle air plasma treatment (80 W, 40 s) prior to the hydrothermal treatment.33 This would ensure the eradication of one-side carbon species to avoid any short circuits.The as-prepared MOH-SG@GF separator features tan-colored Co(OH)2 on one side and black-colored Co(OH)2-SG on the other side visible to the naked eye, in contrast to the bare GF separator (Fig. 2(a)). To gain insight into the fabrication design, the interactionsof S-doped graphene with glass or Co(OH)2 were investigated by theoretical computations. The MD simulations indicate that the directly grown graphene over glass benefits from the formation of C–O bonds at elevated temperatures, which promotes strong adhesion of graphene to the GF separator without easy detachment (Fig. S2, ESI†). Fig. 2(b) presents geometrical configurations of non-doped or S-doped graphene interacting with Co(OH)2. The binding energy values for the four model systems reach 1.92, −2.60, −1.26 and −2.03 eV, respectively (Fig. 2(c)). It is evident that S-doped graphene affords strong affinity for MOH, exerting a positive impact upon the uniform formation of MOH over the SG carpet.
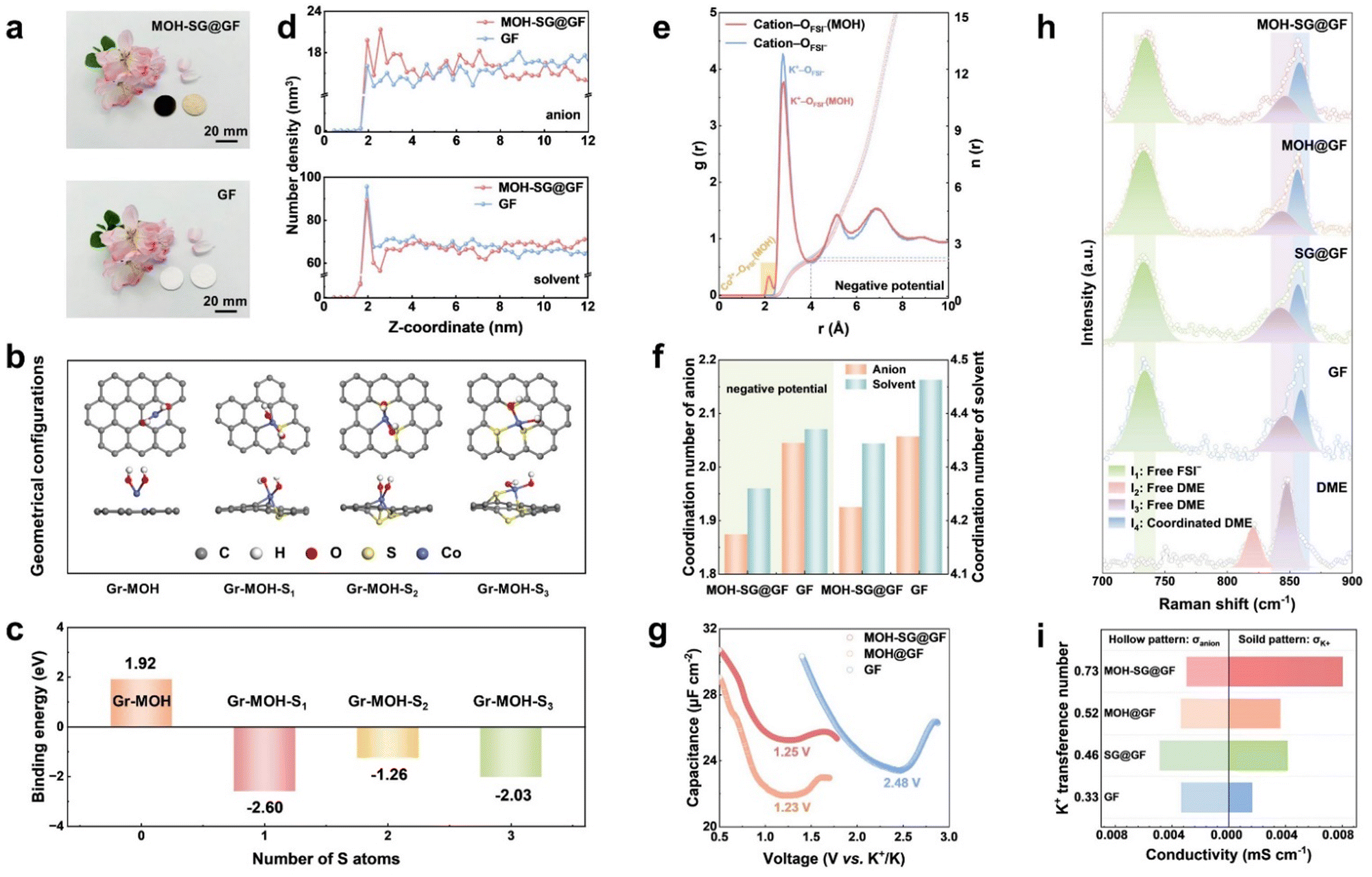 | ||
| Fig. 2 Interface chemistry regulation. (a) Photograph of modified and pristine GF separators. (b) and (c) Geometrical configurations and corresponding binding energy values of MOH on Gr and S-doped Gr. (d) Number density profiles of anions and solvents at the anode interface as a function of the distance from the K anode to electrolyte. (e) Radical distribution functions [RDFs, g(r), solid line] and cumulative distribution functions [CDFs, n(r), dashed line] of interactions between cations (K+ and Co2+) and anions as a function of distance (r) from MD simulations with the applied negative potential. (f) Comparison of the coordination number for anions and solvents based on different separators with/without applying negative potential. (g) Differential capacitance–potential curves of the K electrode. (h) Raman spectra of the DME solvent and the separator surface. (i) Comparison between the K+ transference number (tK+) and ionic conductivity (σK+ and σanion) of all samples. | ||
Top-view scanning electron microscopy (SEM) images reveal the dense and uniform MOH nanoflakes grown on the SG carpet in MOH-SG@GF, in contrast to the sparse and uneven growth for MOH@GF without graphene coating (Fig. S3 and S4, ESI†). This is in good agreement with the computational results. The obtained Co(OH)2 shows a hexagonal flake texture (Fig. S5 and S6, ESI†), with homogeneous distributions of Co and O elements across the energy-dispersive spectroscopy (EDS) maps (Fig. S7, ESI†). High-resolution transmission electron microscopy (HRTEM) inspection and X-ray diffraction (XRD) analysis confirm the successful synthesis of Co(OH)2 (Fig. S8 and S9, ESI†). The MOH-SG@GF material presents improved electrolyte uptake and retention, where a 1.2-fold increase in electrolyte uptake and 12 wt% greater electrolyte retention could be gained as compared to the bare GF (Fig. S10, ESI†). In addition, the obtained material exhibits favorable mechanical stability, implying its potential for long-term operation (Fig. S11, ESI†).
Interface chemistry regulation
The design principle of separator modification to disrupt EDL repulsion and achieve an anion-enriched interface relies on the H-bond build-up between MOH and salt/solvent molecules in the electrolyte.30 In this sense, anions can resist electrostatic repulsion from the EDL and redistribution on the anode surface via the H-bond effect because of the direct contact between the separator and anode. MD simulations were carried out to dissect the interface chemical differences of the EDL when applying GF and MOH-SG@GF. Fig. 2(d) shows the number density profiles comparing anions and solvents in a pristine and modified separator.30 Apparently, the intensity of the anion belonging to MOH-SG@GF is higher than that of GF at the interface, illustrating the successful capture of anions via the H-bond of MOH. Moreover, a negative potential was imposed to simulate the interface states during electrochemical operation, which is related to the SEI evolution (Fig. S12, ESI†). As expected, anions and solvents accumulate with the aid of a H-bond, suggesting a weakened solvation sheath and accelerated desolvation dynamics (Fig. S13 and S14, ESI†). As for the control sample, anions are excluded by the EDL, leaving surplus of solvents at the interface to form undesirable organics. Radial distribution functions (RDFs) and cumulative distribution functions (CDFs) were also used to explore the impact of the H-bond on solvation sheath.34,35 The intensities representing solvents and anions in the MOH sample are obviously lower, indicative of impaired K+–solvent and K+–anion interactions (Fig. S15, ESI†). Intriguingly, a signal peak appears at ∼2.16 Å in addition to the 2.82 Å peak of K+–ODME and K+–OFSI−, which is associated with the coordination of Co2+–OFSI− and Co2+–ODME. The intensity profile of anions also elevates visibly when a negative potential was applied, manifesting a marked impact of the H-bond with respect to collecting anions in the EDL (Fig. 2(e)). The coordination numbers of solvents and anions are summarized in Fig. 2(f), clearly indicating that the formation of H-bonds weakens the association between K+ and the solvent sheath. This phenomenon becomes pronounced when applying a negative potential, suggestive of improved desolvation kinetics as the solvation structure approaches the anode.To decipher the ion management behavior of the EDL over the K anode, alternating current voltammetry (ACV) was carried out, with the potential of zero charge shown in Fig. 2(g). Obviously, the tested systems affording MOH both present conspicuous negative shifts in comparison with that employing a pristine separator, verifying the anion capture on the anode surface.15,35 Raman spectroscopy profiles were recorded to unveil the interaction between the solvation sheath and the H-bond. As shown in Fig. 2(h), two Raman peaks appearing at 821 and 847 cm−1 are ascribed to free DME molecular contributions (I2 and I3) in a pure solvent reference system.36 The other bands pertaining to the vibration of free FSI− (I1) at 734 cm−1 and coordinated DME (I4) at 856 cm−1 could also be observed in different separator systems under the identical electrolyte scenario. The intensity ratios of I1/I3 (2.50, 2.47, 2.41 and 2.38) decrease and those of I4/I3 (2.04, 2.09, 2.18 and 2.31) increase in the sequence of GF, SG@GF, MOH@GF and MOH-SG@GF, respectively. This variation trend corroborates those anions and solvent molecules (including free DME and K+-coordinated DME) have formed new coordination configurations with MOH throughout the H-bond, which not only regulates the EDL by anchoring the anion at the interface but also stabilizes the SEI framework by grabbing free solvents near the anode.
Electrochemical impedance spectroscopy (EIS) was performed to evaluate the initial interfacial states of all systems (Fig. S16, ESI†), showing reduced interfacial impedance of the modified separators, especially for the MOH-SG@GF. The ion transference number (tK+) was measured to probe the K-ion transport properties of separators (Fig. S17 and S18, ESI†). tK+ was derived from EIS and potentiostatic polarization tests, where the ion transmission capability of MOH-SG@GF (0.73) is substantially higher than that of the control sample (0.33). As for the ion conductivity, the value of MOH-SG@GF (0.011 mS cm−1) increases two-fold as compared to that of bare GF (0.005 mS cm−1). Moreover, the conductivity of K+ (σK+) and the anion (σanion) could be deduced from these results (ion conductivity × tK+/anion). Fig. 2(i) illustrates the anionic and K+ conductivities of each separator-equipped system. σK+ (0.0081 mS cm−1) in the MOH-SG@GF system possesses a significantly enhanced value compared with its counterparts, suggesting that the K+ migration dominates the ion transportation in the bulk electrolyte. In stark contrast, the σanion value is conspicuously higher than that of σK+ in the GF system, which may pose a risk of dendrite growth originating from concentration polarization based on the space-charge theory. Taken together, the optimized tK+ and σK+ values are beneficial to ion-transfer kinetics and dendritic suppression, expecting to prolong the anode lifespan.
SEI microstructure and configuration
We next carried out cryogenic TEM inspection to atomically visualize the morphology and composition of the formed SEI. EDS maps with element intensity profiles for the MOH-SG@GF and GF enabled systems were collected, showcasing the presence of C, K, F, S, N and O elements (Fig. S19–S21, ESI†).37 Among them, the element intensities of F and S are noticeable in the former but almost vanish in the latter, plus the C intensity is pronounced in the GF system, suggesting the formation of the organic SEI. HRTEM observations were performed to reveal the specific SEI components for different separator systems. Fig. 3(a) and (b) show the nano-patchwork structure of the SEI in the modified separator system, comprising numerous inorganic crystal domains. The distinct spots in the corresponding FFT patterns reflect the presence of large-area crystalline components, including K2O, KHSO4, K2CO3 and K2SO3 along with their derivatives.38 In stark contrast, as for the GF system, only a KHSO4 island emerges within many amorphous regions (Fig. 3(c)). The related FFT pattern featuring few clear spots also verifies the uneven mixture of the SEI texture.39 In detail, representative components affording crystal characteristics were carefully identified for MOH-SG@GF. For instance, KF nanograins with well matched (220) planes could be observed, showing a lattice spacing of 1.88 Å (Fig. 3(d)). The lattice aligning along the (102), (200) and (204) planes of K2CO3 and the (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 32), (24) and (210) planes of its analogues KHCO3 could also be identified (Fig. 3(e) and Fig. S22, ESI†). Similarly, the lattice planes of (002) and (130) for K2SO4 and (230) for KHSO4 were indexed by cryo-TEM (Fig. S23, ESI†). As shown in Fig. 3(f) and Fig. S24 (ESI†), the KOH (200) plane with a lattice spacing of 2.80 Å and the K2O (202) plane with a lattice fringe of 2.07 Å were found. These crystalline regions forcefully manifest the feasibility of breaking the EDL electrostatic repulsion and the significance of collecting the anions at the anode interface, which gives rise to the construction of a robust inorganic-rich SEI. In terms of the bare GF without EDL regulation, turbulent textures interspersed by a few nanocrystal fragments constitute an inhomogeneous SEI (Fig. S25, ESI†). Fig. 3(g) and (h) show a vivid comparison of the key discrepancy in the cryo-imaged SEI between the designed and control systems. Apparently, the SEI derived from MOH-SG@GF enabled cells displays a homogeneous and compact nature even enduring long-term cycling, which effectively protects the anode from the electrolyte corrosion and readily facilitates uniform ion transfer during repeated K plating/stripping. In contrast, many of the heterogeneous organic species generated in the GF system during continuous cycling ultimately turn into a loose and porous SEI architecture, posing a high threat toward rampant dendrite growth (Fig. 3(i)).
32), (24) and (210) planes of its analogues KHCO3 could also be identified (Fig. 3(e) and Fig. S22, ESI†). Similarly, the lattice planes of (002) and (130) for K2SO4 and (230) for KHSO4 were indexed by cryo-TEM (Fig. S23, ESI†). As shown in Fig. 3(f) and Fig. S24 (ESI†), the KOH (200) plane with a lattice spacing of 2.80 Å and the K2O (202) plane with a lattice fringe of 2.07 Å were found. These crystalline regions forcefully manifest the feasibility of breaking the EDL electrostatic repulsion and the significance of collecting the anions at the anode interface, which gives rise to the construction of a robust inorganic-rich SEI. In terms of the bare GF without EDL regulation, turbulent textures interspersed by a few nanocrystal fragments constitute an inhomogeneous SEI (Fig. S25, ESI†). Fig. 3(g) and (h) show a vivid comparison of the key discrepancy in the cryo-imaged SEI between the designed and control systems. Apparently, the SEI derived from MOH-SG@GF enabled cells displays a homogeneous and compact nature even enduring long-term cycling, which effectively protects the anode from the electrolyte corrosion and readily facilitates uniform ion transfer during repeated K plating/stripping. In contrast, many of the heterogeneous organic species generated in the GF system during continuous cycling ultimately turn into a loose and porous SEI architecture, posing a high threat toward rampant dendrite growth (Fig. 3(i)).
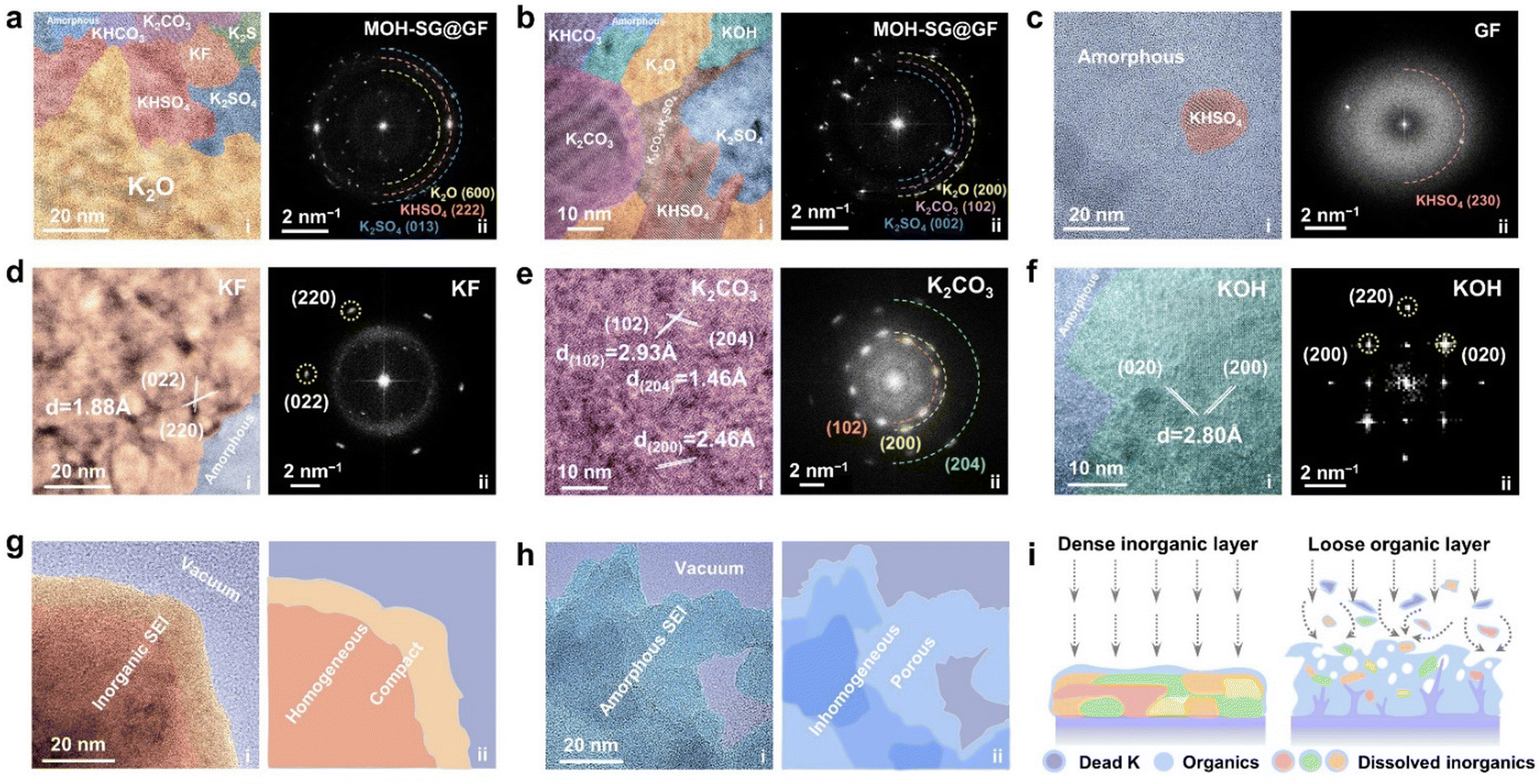 | ||
| Fig. 3 Cryo-TEM examination of the formed SEI. (a)–(c) Typical TEM views and the corresponding FFT patterns of the SEI formed over MOH-SG@GF (a) and (b) and the GF system (c). (d)–(f) Representative HRTEM images and the corresponding FFT patterns of KF (d), K2CO3 (e) and KOH (f). (g) and (h) TEM views showing the SEI morphology and the corresponding schematic illustration of MOH-SG@GF (g) and the GF system (h). (i) Schematic depicting the differences in SEI formation and evolution between MOH-SG@GF and the GF system. | ||
X-ray photoelectron spectroscopy (XPS) analysis was performed to gain insight into the chemical configuration and bonding characteristics of the SEI derived from the formation stage under our separator modification. As depicted in Fig. 4(a), the high-resolution F 1s spectra present C–F (687.5 eV) and K–F (684.6 and 682.6 eV) contributions.40 Intriguingly, the intensity of these profiles displays a clear trend of attenuation or proliferation, especially for the K–F located at 682.6 eV, where almost no signal could be captured in the GF system in contrast to the scenario with the involvement of MOH. Two deconvoluted peaks located at 399.3 and 397.8 eV in the N 1s spectra indicate the presence of NxOy and N–S species (Fig. S26, ESI†). Analogous to the F 1s spectrum, the N–S bond signal merely appears in the sample employing MOH, demonstrating the effect of MOH upon promoting the transformation of FSI− into inorganics. In the O 1s profiles, four signals including O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (533.3 eV), C–O (532.4 eV), CO (531.3 eV) and K–O (530.1 eV) could be observed.41 Note that a new signal with respect to K–O could be detected in MOH samples, which corresponds to K2O. This finding suggests an additional inorganic component variation induced by MOH, given that most O species originate from the solvent decomposition. The existence of K–C (295.3 and 292.5 eV) and K–O (296.1 and 293.3 eV) bonding is supported by the K 2p spectra. Fig. 4(b) shows a content comparison between representative inorganic- (K2O and KF) and organic-configurations (O–CO and NxOy). Apparently, the ratio of KF decreases in the order of MOH-SG@GF (53.9%), MOH@GF (32.0%), SG@GF (21.3%) and GF (16.7%), harvesting a 3.2-fold elevation from the target material to the control sample (Fig. S27, ESI†). It should be noted that K2O only presents in the MOH system, suggestive of a favorable environment to generate the inorganic-based SEI. In contrast, the proportion of organics displays a remarkable rise without MOH modification, where the content of O–CO in GF (44.3%) is 3.7-fold higher than that of MOH-SG@GF (12.0%). The elemental ratio of (F + N + S)/C further reveals the correlation between inorganics and organics since the FSI− anion is the only source of F, N and S, while the C is mainly derived from the solvent. Obviously, the value of MOH samples exhibits superiority to the control systems, again validating the importance of MOH in regulating the SEI architecture via the H-bond effect. As shown in Fig. 4(c), despite the inevitable introduction of O by MOH (Fig. S28 and Table S1, ESI†), the inorganic O species occupy the majority. K2O, for instance, constitutes over half of the K–O configurations in MOH-SG@GF, while the O-containing ingredients are basically organics in the GF system.
O (533.3 eV), C–O (532.4 eV), CO (531.3 eV) and K–O (530.1 eV) could be observed.41 Note that a new signal with respect to K–O could be detected in MOH samples, which corresponds to K2O. This finding suggests an additional inorganic component variation induced by MOH, given that most O species originate from the solvent decomposition. The existence of K–C (295.3 and 292.5 eV) and K–O (296.1 and 293.3 eV) bonding is supported by the K 2p spectra. Fig. 4(b) shows a content comparison between representative inorganic- (K2O and KF) and organic-configurations (O–CO and NxOy). Apparently, the ratio of KF decreases in the order of MOH-SG@GF (53.9%), MOH@GF (32.0%), SG@GF (21.3%) and GF (16.7%), harvesting a 3.2-fold elevation from the target material to the control sample (Fig. S27, ESI†). It should be noted that K2O only presents in the MOH system, suggestive of a favorable environment to generate the inorganic-based SEI. In contrast, the proportion of organics displays a remarkable rise without MOH modification, where the content of O–CO in GF (44.3%) is 3.7-fold higher than that of MOH-SG@GF (12.0%). The elemental ratio of (F + N + S)/C further reveals the correlation between inorganics and organics since the FSI− anion is the only source of F, N and S, while the C is mainly derived from the solvent. Obviously, the value of MOH samples exhibits superiority to the control systems, again validating the importance of MOH in regulating the SEI architecture via the H-bond effect. As shown in Fig. 4(c), despite the inevitable introduction of O by MOH (Fig. S28 and Table S1, ESI†), the inorganic O species occupy the majority. K2O, for instance, constitutes over half of the K–O configurations in MOH-SG@GF, while the O-containing ingredients are basically organics in the GF system.
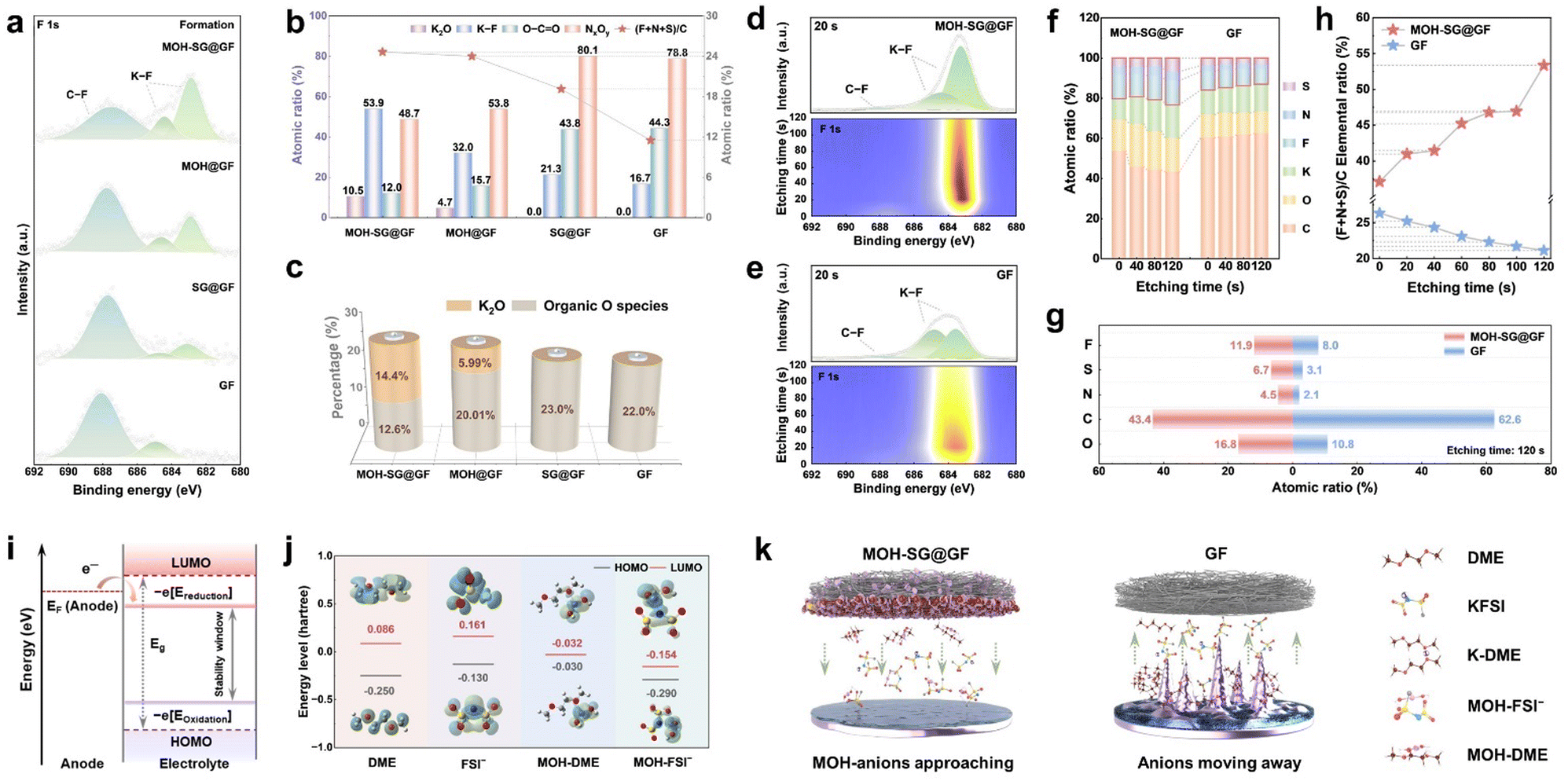 | ||
| Fig. 4 SEI compositional analysis and mechanistic investigation. (a) XPS F 1s spectra at the formation stage. (b) Percentages of representative inorganic/organic phases and the atomic ratio of (F + N + S)/C in the SEI formed in different separator systems. (c) The inorganic/organic O species ratio in the SEI. (d), (e) XPS F 1s depth-profiles of the MOH-SG@GF (d) and GF (e) systems. Each plot comprises two individual profiles, i.e., the spectrum at the initial etching stage (upper panel) and the depth profile (lower panel). (f), (g) Atomic ratio profiles from MOH-SG@GF and GF systems with respect to different etching times (f) and the final etching stage (g). (h) The comparison of the elemental ratio of (F + N + S)/C with the increase of the etching time for MOH-SG@GF and GF systems. (i) Schematic diagram showing the orbital energy levels for SEI formation at the anode interface. (j) The molecular orbital energies of primary components (DME, FSI−, MOH-DME and MOH-FSI−). Insets show the corresponding geometrical structures. (k) Schematic illustrating the significance of MOH species in the stabilization of the anode interface. | ||
XPS depth profiling was employed to further dissect the dynamic evolution of the SEI composition over 100 cycles. The F 1s, N 1s and O 1s spectra collectively reveal that those organic phases (C–F, NxOy and O–CO) tend to distribute at the outer layer of the SEI, where their intensities gradually diminish in the course of Ar+ etching (Fig. 4(d), (e) and Fig. S29, ESI†).
As for the MOH-SG@GF system, the K–F and N–S intensities show a clearly enhanced trend during etching.42 This is in stark contrast to the GF case, implying that plenty of anions appear at the anode interface owing to the H-bond interaction and thereafter transform into inorganic species. In further contexts, elemental percentages at different etching stages for MOH-SG@GF and GF systems were collected to track the SEI compositional evolution with the depth variation.43 As presented in Fig. 4(f), the contents of C and O elements decrease whereas F, N, and S contents increase in the target sample. Fig. 4(g) shows a visual representation at an etching time of 120 s. More specifically, the elemental ratio of (F + N + S)/C elevates from 37.1% to 53.4% for MOH-SG@GF but reduces from 26.3% to 21.1% for GF (Fig. 4(h)).
SEI formation mechanism and interfacial dynamics
By foregoing MD simulation and Raman inspection, we have investigated the anchoring effect of H-bonds on anions and solvent molecules, which mitigates the electrostatic repulsion from the EDL, reduces the population of free solvent molecules at the interface and weakens the solvation sheath. This would ultimately lead to an inorganic-enriched SEI formation, as evidenced by cryo-TEM and XPS analyses. One question remains: why could an inorganic-rich (rather than organic-rich) SEI generate at both the formation/cycling stages when MOH concurrently coordinates with anions and solvents? In this sense, the solvation structure at the interface plays a decisive role. The SEI generation is intrinsically governed by the electron transport between the Fermi level of the electrode and the molecular orbital energy-level of the electrolyte (Fig. 4(i)). As for the anode side, a lower electrolyte reduction potential indicates a higher priority for reduction.18,44 In our case, MD simulations were performed to unravel the potential SEI formation mechanism via calculating LUMO energy levels of representatively dominant solvation states. Fig. 4(j) presents the LUMO and HOMO energy levels of four primary components at the interface: DME, FSI−, MOH-DME and MOH-FSI−, along with their matching configurations shown as the insets. Based on this, MOH-FSI− becomes the first species to be reduced due to the lowest LUMO energy level (−0.154 eV) compared to the others (DME: 0.086 eV, FSI−: 0.161 eV and MOH-DME: −0.032 eV). Note that the MOH acts as an anion transport hub without participating in the SEI construction even during long cycles (Fig. S30, ESI†). In particular, MOH captures both the anion and solvent in the electrolyte and rearranges EDL. The coordinated anions preferentially participate in the reduction reaction to produce inorganic species, while the decomposition of the coordinated solvents is inhibited. Therefore, anions consistently retain priority for the reaction even when the anode interface was saturated with various electrolyte components, thus enabling the generation of an inorganic-dominated SEI. Taken together, the H-bond effect of MOH shows a valuable influence to form a durable and robust inorganic-rich SEI benefiting from EDL regulation, which could offer rapid ion conduction, provide uniform K deposition and afford long-term cycling. In contrast, a typical SEI without EDL regulation still contains considerable organic phases, which would suffer from repeated dissolution and reconstruction, leading to a loose and porous morphology that fails to inhibit the dendrites (Fig. 4(k)).To further probe the crucial role of the regulated inorganic-rich SEI in interfacial dynamics, temperature-dependent EIS was carried out to evaluate the ion transport through the SEI and desolvation at the interface. The interfacial impedance of the MOH-SG@GF system greatly improves across a range of temperatures as compared to the GF counterpart (Fig. S31, ESI†). The SEI resistance (RSEI) and charge-transfer resistance (Rct) of the GF system remain nearly constant at different temperatures, where Rct accounts for a considerable proportion, suggestive of a sluggish desolvation process (Fig. S32 and Tables S2, S3, ESI†). In contrast, the proportion of Rct in MOH-SG@GF displays a conspicuous attenuation, implying a rapid desolvation behavior via a weakly solvated structure influenced by the H-bond effect. The activation energy (Ea) was accordingly derived to assess the mass transfer kinetics at the anode interface, where a low Ea value represents favorable interfacial dynamics.45,46Fig. 5(a) illustrates that the MOH-SG@GF system harvests the minimum Ea value of the SEI (29.98 kJ mol−1) compared to its counterparts (SG@GF: 36.42 kJ mol−1, MOH@GF: 51.20 kJ mol−1 and GF: 91.40 kJ mol−1), showing conducive K+ transport properties originating from the homogeneous inorganic-rich SEI. Likewise, the target system also realizes an optimal Ea for charge transfer, revealing the accelerated electrochemical reactions at the interface (Fig. S33, ESI†).47,48 In addition, the prominent response current and exchange current density of the MOH-SG@GF system echo well with these results (Fig. 5(b) and Fig. S34, ESI†).49 This kind of separator design affords improved interfacial electrochemical behavior, where the presence of SG renders an enhanced electron conductivity and the existence of MOH enables a facilitated charge transfer and desolvation dynamics.
 | ||
| Fig. 5 Interfacial dynamics and electrochemical performances via different separators. (a) Activation energy values for K+ diffusion through the SEI. (b) Tafel plots of all samples. (c) Average Coulombic efficiency tests for different separator systems. (d) Galvanostatic plating/stripping profiles for half-cells at 0.5 mA cm−2/0.5 mA h cm−2. (e) Rate performances of symmetric cells equipped with different separators. (f), (g) Cyclic performances of different symmetric cells at 0.5 mA cm−2/0.5 mA h cm−2 (f) and 10 mA cm−2/10 mA h cm−2 (g). (h) Comparison of cumulative capacity and current density between this work and other related reports involving LMBs, SMBs and PMBs. (i) Rate performances of the PTCDA‖K full cell. | ||
Half-cell and full-cell performance
We next tested the electrochemical performances of half- and full-cells assembled using modified separators. Average coulombic efficiency (CE) was employed to evaluate the correlation between the SEI properties and battery lifespan.50,51 The MOH-SG@GF system has the highest value of 99.3%, while the pristine GF system gains only 96.3% (Fig. 5(c)). This discrepancy reveals that an inorganic-dominated SEI enables reversible K stripping/plating during cycling, whereas an organic-rich SEI incurs dendritic formation and “dead K” generation, resulting in a reduced CE value. Galvanostatic voltage profiles of Al‖K half cells assembled with different separators were analyzed to investigate the impact of the SEI on interface chemistry.52,53 Among all systems, MOH-SG@GF enables a minimum nucleation barrier (μtip) of −0.2 mV, a nucleation overpotential (μnuc) of 6.0 mV and a polarization of 147 mV, in contrast to MOH@GF (−0.32, 48 and 178 mV), SG@GF (−0.21, 52 and 155 mV) and GF (−0.25, 43 and 164 mV) (Fig. S35 and S36, ESI†). In addition, the shorter relaxation time of the target system reflects advanced nucleation dynamics (Fig. S37, ESI†).Fig. 5(d) presents voltage–time profiles of asymmetric Al‖K half cells based on different separators under a current density of 0.5 mA cm−2 and an areal capacity of 0.5 mA h cm−2. The MOH-SG@GF-based cell readily holds a stable operation for more than 2000 h. However, its counterparts manifest apparent voltage fluctuations (Fig. S38, ESI†), which are evidenced by the corresponding CE profiles (Fig. S39, ESI†). As for the symmetric K‖K cells, rate performances were evaluated for all separators at varying current densities ranging from 0.5 to 10 mA cm−2 with a fixed capacity of 1.0 mA h cm−2. As shown in Fig. 5(e), the overpotential values for the MOH-SG@GF based cell gradually enlarge in a stepwise manner in response to the increased rates.54,55 The stable voltage profiles (Fig. 5(e) inset) illustrate the admirable interface quality to enable steady operation under repetitively alternated current densities. When switched back to 0.5 mA cm−2, a slightly decreased overpotential could be gained, implying the formation of a favorable SEI with an ion conduction pathway. In contrast, considerably enlarged polarization and suddenly undermined overpotential appear at certain points for the counterparts when the current density rises toward 5 and 10 mA cm−2, indicative of possible short-circuits because of dendrite proliferation (Fig. S40, ESI†).
Pertaining to the long-term cycling stability tests, the symmetric cells equipped with MOH-SG@GF exhibit durable K plating/stripping behaviors to sustain 6000, 1000 and 2000 h under 0.5 mA cm−2/0.5 mA h cm−2, 1.0 mA cm−2/1.0 mA h cm−2 and 2.0 mA cm−2/2.0 mA h cm−2 with the overpotentials of 27, 62 and 52 mV, respectively (Fig. 5(f) and Fig. S41, ESI†). Nevertheless, the performances of all other cells deteriorate substantially with premature short-circuits, implying inferior interface stability. Encouragingly, the MOH-SG@GF cell manages to deliver stable longevous cycling under relatively challenging conditions (Fig. 5(g) and Fig. S42, ESI†). The separator modified by the synergy of SG and MOH affords a uniform inorganic-rich SEI, enduring 6000 h with an overpotential of 125 mV under 8.0 mA cm−2/8.0 mA h cm−2 and 4000 h with a voltage hysteresis of 318 mV under 10 mA cm−2/10 mA h cm−2. Moreover, the smooth time–voltage profile along with the corresponding EIS curves upon cycling indeed demonstrate no occurrence of (soft) short-circuits (Fig. S43, ESI†).56,57 As such, the electrochemical properties of the MOH-SG@GF cell surpass those of the state-of-the-art K metal energy storage systems and compare favorably with representative Na and Li metal batteries reported by far in terms of current density and cumulative capacity (Fig. 5(h) and Table S4, ESI†).58,59 It is worth-noting that the separator decorated with MOH other than Co(OH)2 [such as MOH(Fe)-SG@GF and MOH(Ni)-SG@GF] was prepared to demonstrate the universality of our strategy. As shown in Fig. S44 (ESI†), the cells assembled with MOH(Fe)-SG@GF and MOH(Ni)-SG@GF separators all showcase impressive cycling stability.
Linear sweep voltammetry (LSV) was performed to probe the oxidation stability of cells based on different separators (Fig. S45, ESI†).60,61 MOH-SG@GF harvests an elevated stable voltage up to 4.35 V, suggesting potential practicability for full cells. The GF counterpart only sustains certain stability below 3.26 V. The full cell was assembled by pairing a pre-potassiated PTCDA cathode and a metallic K anode with different separators.25,62 As shown in Fig. 5(i), PTCDA‖MOH-SG@GF‖K presents negligible capacity attenuation below 0.5 A g−1, where an initial capacity reaches 142.8 mA h g−1 at 0.02 A g−1 and a capacity output of 75.7 mA h g−1 can be harvested at 2.0 A g−1. When the rate returns to 0.05 A g−1, a reversible capacity of 132.9 mA h g−1 is achieved, indicative of favorable rate capability. In sharp contrast, the PTCDA‖GF‖K full cell equipped with bare GF displays inferior rate performance: the capacity falls to 42.4 mA h g−1 at 2.0 A g−1 and fails to retain operation when returned to 0.05 A g−1, primarily owing to SEI pulverization/passivation and “dead K” accumulation. In terms of cycling performances, PTCDA‖MOH-SG@GF‖K displays an initial discharge capacity of 122.3 mA h g−1 and a reversible capacity of 90.3 mA h g−1 at 0.5 A g−1 after 300 cycles, managing a capacity retention of 75.6%, which is two-fold higher than that of PTCDA‖GF‖K (36.8%) (Fig. S46, ESI†). Similarly, the PTCDA‖MOH-SG@GF‖K full cell maintains a 76.1% capacity retention at 1.0 A g−1 after 500 cycles. The smaller polarization voltage observed in PTCDA‖MOH-SG@GF‖K across different cycle numbers as compared to PTCDA‖GF‖K demonstrates the profitable ion conductivity and effective dendrite suppression via the inorganic-rich SEI, accordingly elongating the lifespan of PMBs (Fig. S47, ESI†).
Electroplating stability mediated by the designed separator
We employed operando Raman spectroscopy to monitor the dynamic evolution of solvation structures over the anode surface during the electrodeposition process. The counter map within a complete plating/stripping stage depicts obvious response signals at ∼734/856 cm−1 corresponding to free FSI− and K+-coordinated DME (I1 and I3) and a relatively weak signal at 847 cm−1 ascribed to free DME (I2) (Fig. S48, ESI†).As shown in Fig. 6(a) and (b), the intensity ratios of I1/I3 increase and I2/I3 decrease during the plating, manifesting the stepwise enrichment of components at the anode side that are in favor of the inorganic-dominated SEI formation. Of note, the anion aggregation could be associated with the transport hub function of MOH, which does not participate in the reduction reaction, while it could coordinate with solvents to inhibit the generation of organic species.
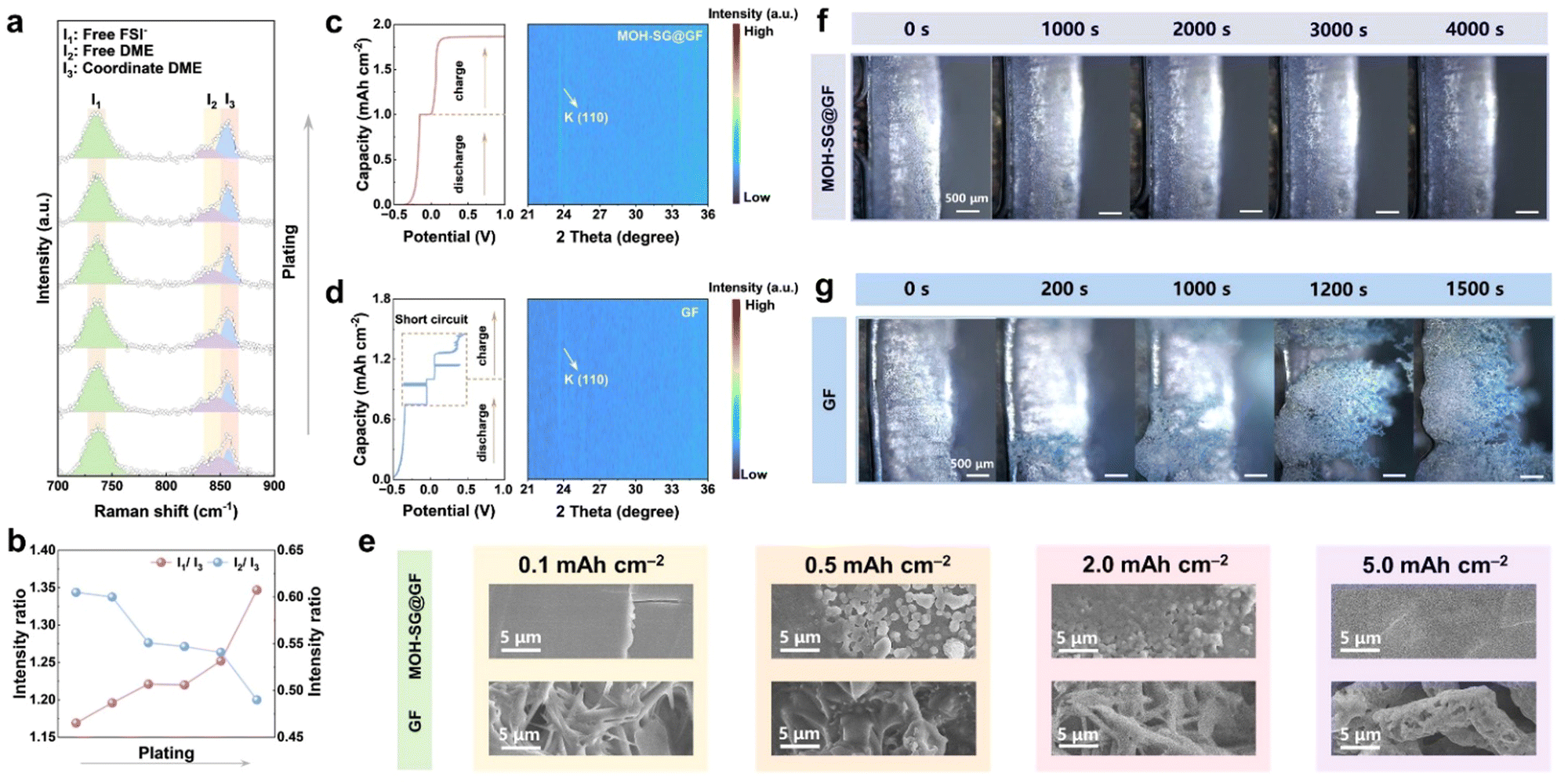 | ||
| Fig. 6 Probing the potassium deposition behavior. (a) and (b) Operando Raman spectroscopy (a) and corresponding peak identity ratio (b) during the plating stage for the MOH-SG@GF system. (c) and (d) Contour plots of operando XRD tests for K deposition based on MOH-SG@GF (c) and GF (d) separators along with the charge–discharge profiles. (e) Ex situ SEM inspections of K metal (0.1, 0.5, 2.0 and 5.0 mA h cm−2) deposited on Al foil through modified and pristine separators. (f), (g) In situ optical microscopy observations of K deposition based on MOH-SG@GF (f) and GF (g) separators. | ||
The Raman results from the initial and plating states manifest that MOH plays an essential role in facilitating the continuous construction of an inorganic-enriched SEI during both the formation and cycling process. These are in good agreement with the XPS depth profiles, revealing the vertical distribution of inorganic species in the SEI, again demonstrating that the H-bond effect enables anions to overcome the electrostatic repulsion from the EDL.
Operando XRD was carried out to trace the discrepancy in K plating/stripping behavior based on modified and pristine separators across one selected discharge–charge cycle.40 The diffraction signal appearing at 23.6° in both intensity counter maps is related to the (110) plane of metal K. Interestingly, this signal arises in the course of plating and thereafter disappears during the late stages of charge for the MOH-SG@GF system, manifesting the distinguished reversibility of K deposition/dissolution without the generation of “dead K” (Fig. 6(c)). Nevertheless, the signal persistently exists throughout the entire charge stage for the GF case, implying the failure in stripping (Fig. 6(d)). Corresponding charge/discharge profiles indeed suggest the smooth operation of the MOH-SG@GF based cell. In contrast, the pristine GF enabled cell suffers a sudden cliff-like voltage drop to 0 V possibly originating from massive dendrite formation, indicative of the fragility of the organic-dominated SEI.
The K deposition morphology over the Al current collector mediated by separators under different capacities was further explored. The SEM from the top view shows that the K deposits with a GF separator exhibit sharp needle-like dendrites even at a small capacity of 0.1 mA h cm−2 (Fig. 6(e)), which become thicker and more porous in response to elevated deposition capacities of 0.5, 2.0 and 5.0 mA h cm−2 (Fig. S49 and S50, ESI†). In comparison, K deposits from the cell equipped with a modified separator display a spherical-cluster morphology, which is prone to merge into a smooth and flat layer without apparent protrusions (Fig. S51 and S52, ESI†). Moreover, a polypropylene membrane was utilized to attach the modified/pristine separator and then assembled into K‖Al cells for 200 h cycling. The membrane dismantled from the MOH-SG@GF system shows a clean state, whereas considerable K dendritic residues appear in the case of pristine GF (Fig. S53, ESI†).
We lastly employed in situ optical microscopy to witness the dynamic K deposition through assembling a K‖K symmetric cell based on different separators. As presented in Fig. 6(f) and (g), both electrodes equipped with MOH-SG@GF or bare GF exhibit flat surface textures prior to the K deposition. During electroplating, K dendritic clusters suddenly appear and quickly grow merely after 200 s for the GF system. In stark contrast, the electrode with MOH-SG@GF affords a smooth surface without any protrusions and dendrites even after 4000 s, illustrating the stable electroplating behavior mediated by separator regulation.
Conclusions
In this study, we have demonstrated a multifunctional separator design to realize the EDL regulation and SEI tailoring in potassium metal anodes. The H-bond effect between anions, solvents and MOH suppresses the electrostatic repulsion from the EDL, weakens the solvation structure and enriches the anion population at the anode interface. The versatile MOH-SG@GF promotes the formation of a homogeneous, compact and robust inorganic-enriched SEI that enables dendrite-free K anodes, which is substantiated through computational simulations and cryo-TEM analysis. Using this design strategy, the K metal anode manifests a high-rate capability at 10 mA cm−2 and a stable plating/stripping performance to sustain over 6000 h at 8.0 mA cm−2/8.0 mA h cm−2, possessing considerable competitiveness in the alkali metal energy storage realm. Our work also offers a vast unexplored design space of separator modification and could be extended widely to many MOH selections. The results demonstrated here pave the way toward the future design of advanced PMBs capable of long lifespan and fast charging for effective operation under practical conditions.Author contributions
J. Sun conceived the idea. X. Lian, Z. Chen, and X. Chen performed the separator synthesis. X. Lian, Y. Yi, and Z. Tian carried out the material characterization and electrochemical measurements. Z. Ju and X. Tao carried out the cryo-TEM measurements. L. Xu and T. Cheng conducted the computational simulations. X. Lian and X. Chen carried out the in situ optical microscopy and XRD analysis. The manuscript was written by X. Lian, T. Cheng, S. Dou, X. Tao and J. Sun with input from all authors. All authors contributed to the analysis and discussion of the results leading to the manuscript. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Key R&D Program of China (2019YFA0708201), the National Natural Science Foundation of China (T2188101, 22179089), and the Science Fund for Distinguished Young Scholars of Jiangsu Province (BK20211503). The authors also acknowledge support from the Suzhou Key Laboratory for Advanced Carbon Materials and Wearable Energy Technologies, Suzhou, China.References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- H. Wang, D. Yu, C. Kuang, L. Cheng, W. Li, X. Feng, Z. Zhang, X. Zhang and Y. Zhang, Chem, 2019, 5, 313–338 CAS.
- Z. G. Yang, J. L. Zhang, M. C. W. Kintner-Meyer, X. C. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 Search PubMed.
- X. Gao, Y.-N. Zhou, D. Han, J. Zhou, D. Zhou, W. Tang and J. B. Goodenough, Joule, 2020, 4, 1864–1879 CrossRef CAS.
- W. Liu, P. Liu and D. Mitlin, Adv. Energy Mater., 2020, 10, 2002297 CrossRef CAS.
- Q. Zhao, S. Stalin and L. A. Archer, Joule, 2021, 5, 1119–1142 CrossRef CAS.
- B. Li, Y. Chao, M. Li, Y. Xiao, R. Li, K. Yang, X. Cui, G. Xu, L. Li, C. Yang, Y. Yu, D. P. Wilkinson and J. Zhang, Electrochem. Energy Rev., 2023, 6, 7 CrossRef CAS.
- M. Yang, X. Zeng, M. Xie, Y. Wang, J.-M. Xiao, R.-H. Chen, Z.-J. Yi, Y.-F. Huang, D.-S. Bin and D. Li, J. Am. Chem. Soc., 2024, 146, 6753–6762 CrossRef CAS.
- J. Tan, J. Matz, P. Dong, J. Shen and M. Ye, Adv. Energy Mater., 2021, 11, 2100046 CrossRef CAS.
- H. Wu, H. Jia, C. Wang, J. G. Zhang and W. Xu, Adv. Energy Mater., 2020, 11, 2003092 CrossRef.
- J. Zhang, J. Wen, W.-D. Liu, X. Cui and Y. Chen, Sci. China Mater., 2022, 65, 2613–2626 CrossRef.
- H. Yuan, J. Nai, H. Tian, Z. Ju, W. Zhang, Y. Liu, X. Tao and X. W. D. Lou, Sci. Adv., 2020, 6, eaaz3112 CrossRef CAS.
- Y. Tian, Y. An and B. Zhang, Adv. Energy Mater., 2023, 13, 2300123 CrossRef CAS.
- C. Yan, H.-R. Li, X. Chen, X.-Q. Zhang, X.-B. Cheng, R. Xu, J.-Q. Huang and Q. Zhang, J. Am. Chem. Soc., 2019, 141, 9422–9429 CrossRef CAS PubMed.
- D. Rakov, M. Hasanpoor, A. Baskin, J. W. Lawson, F. Chen, P. V. Cherepanov, A. N. Simonov, P. C. Howlett and M. Forsyth, Chem. Mater., 2021, 34, 165–177 CrossRef.
- Q. Wu, M. T. McDowell and Y. Qi, J. Am. Chem. Soc., 2023, 145, 2473–2484 CrossRef CAS.
- Z. Wen, W. Fang, F. Wang, H. Kang, S. Zhao, S. Guo and G. Chen, Angew. Chem., Int. Ed., 2024, 63, e202314876 CrossRef CAS PubMed.
- M. Fang, B. Du, X. Zhang, X. Dong, X. Yue and Z. Liang, Angew. Chem., Int. Ed., 2023, 63, e202316839 CrossRef PubMed.
- X. Shen, S. Shi, B. Li, S. Li, H. Zhang, S. Chen, H. Deng, Q. Zhang, J. Zhu and X. Duan, Adv. Funct. Mater., 2022, 32, 2206388 CrossRef CAS.
- W. Zhang, Y. Lu, L. Wan, P. Zhou, Y. Xia, S. Yan, X. Chen, H. Zhou, H. Dong and K. Liu, Nat. Commun., 2022, 13, 2029 CrossRef CAS PubMed.
- H. Adenusi, G. A. Chass, S. Passerini, K. V. Tian and G. Chen, Adv. Energy Mater., 2023, 13, 2203307 CrossRef CAS.
- B. Wu, C. Chen, L. H. J. Raijmakers, J. Liu, D. L. Danilov, R.-A. Eichel and P. H. L. Notten, Energy Storage Mater., 2023, 57, 508–539 CrossRef.
- S. Kim, K. Y. Cho, J. Kwon, K. Sim, D. Seok, H. Tak, J. Jo and K. Eom, Small, 2023, 19, 2207222 CrossRef CAS.
- Y. Yang, S. Yao, Z. Liang, Y. Wen, Z. Liu, Y. Wu, J. Liu and M. Zhu, ACS Energy Lett., 2022, 7, 885–896 CrossRef CAS.
- T. Kang, C. Sun, Y. Li, T. Song, Z. Guan, Z. Tong, J. Nan and C. S. Lee, Adv. Energy Mater., 2023, 13, 2204083 CrossRef CAS.
- W. Yu, L. Shen, X. Lu, J. Han, N. Geng, C. Lai, Q. Xu, Y. Peng, Y. Min and Y. Lu, Adv. Energy Mater., 2023, 13, 2204055 CrossRef CAS.
- M. S. Kim, Z. Zhang, P. E. Rudnicki, Z. Yu, J. Wang, H. Wang, S. T. Oyakhire, Y. Chen, S. C. Kim, W. Zhang, D. T. Boyle, X. Kong, R. Xu, Z. Huang, W. Huang, S. F. Bent, L. W. Wang, J. Qin, Z. Bao and Y. Cui, Nat. Mater., 2022, 21, 445–454 CrossRef CAS PubMed.
- H. Yang, J. Li, Z. Sun, R. Fang, D.-W. Wang, K. He, H.-M. Cheng and F. Li, Energy Storage Mater., 2020, 30, 113–129 CrossRef.
- M. Mao, X. Ji, Q. Wang, Z. Lin, M. Li, T. Liu, C. Wang, Y.-S. Hu, H. Li, X. Huang, L. Chen and L. Suo, Nat. Commun., 2023, 14, 1082 CrossRef CAS.
- N. von Aspern, G. V. Röschenthaler, M. Winter and I. Cekic-Laskovic, Angew. Chem., Int. Ed., 2019, 58, 15978–16000 CrossRef CAS PubMed.
- Z. W. Zhang, Y. Cui, R. Vila, Y. B. Li, W. B. Zhang, W. J. Zhou, W. Chiu and Y. Cui, Acc. Chem. Res., 2021, 54, 3505–3517 CrossRef CAS.
- C. Li, Z. Sun, T. Yang, L. Yu, N. Wei, Z. Tian, J. Cai, J. Lv, Y. Shao, M. H. Rummeli, J. Sun and Z. Liu, Adv. Mater., 2020, 32, 2003425 CrossRef CAS.
- Z. Li, H. Rao, R. Atwi, B. M. Sivakumar, B. Gwalani, S. Gray, K. S. Han, T. A. Everett, T. A. Ajantiwalay, V. Murugesan, N. N. Rajput and V. G. Pol, Nat. Commun., 2023, 14, 868 CrossRef CAS.
- S. Liu, C. Shu, Y. Yan, D. Du, L. Ren, T. Zeng, X. Wen, H. Xu, X. Wang, G. Tian and Y. Zeng, Energy Environ. Mater., 2024, 7, e12635 CrossRef CAS.
- L. Tu, Z. Zhang, Z. Zhao, X. Xiang, B. Deng, D. Liu, D. Qu, H. Tang, J. Li and J. Liu, Angew. Chem., Int. Ed., 2023, 135, e202306325 CrossRef.
- Y. Liu, Z. Ju, B. Zhang, Y. Wang, J. Nai, T. Liu and X. Tao, Acc. Chem. Res., 2021, 54, 2088–2099 CrossRef CAS PubMed.
- Q. Zhang, B. Han, Y. C. Zou, S. C. Shen, M. H. Li, X. Z. Lu, M. Wang, Z. P. Guo, J. Q. Yao, Z. Chang and M. Gu, Adv. Mater., 2021, 33, 2102666 CrossRef CAS PubMed.
- H. Yuan, J. Nai, H. Tian, Z. Ju, W. Zhang, Y. Liu, X. Tao and X. Lou, Sci. Adv., 2020, 6, eaaz3112 CrossRef CAS PubMed.
- S. W. Li, H. L. Zhu, Y. Liu, Z. L. Han, L. F. Peng, S. P. Li, C. Yu, S. J. Cheng and J. Xie, Nat. Commun., 2022, 13, 4911 CrossRef CAS PubMed.
- L. Qin, Y. Lei, H. Wang, J. Dong, Y. Wu, D. Zhai, F. Kang, Y. Tao and Q. H. Yang, Adv. Energy Mater., 2019, 9, 1901427 CrossRef.
- S. Zhang, Y. Li, L. J. Bannenberg, M. Liu, S. Ganapathy and M. Wagemaker, Sci. Adv., 2024, 10, eadl4842 CrossRef.
- Y. Xia, P. Zhou, X. Kong, J. Tian, W. Zhang, S. Yan, W.-H. Hou, H.-Y. Zhou, H. Dong, X. Chen, P. Wang, Z. Xu, L. Wan, B. Wang and K. Liu, Nat. Energy, 2023, 8, 934–945 CrossRef CAS.
- P. Peljo and H. H. Girault, Energy Environ. Sci., 2018, 11, 2306–2309 RSC.
- W. Tang, T. Zhao, K. Wang, T. Yu, R. Lv, L. Li, F. Wu and R. Chen, Adv. Funct. Mater., 2024, 34, 2401281 CrossRef.
- C. Li, S. Liu, C. Shi, G. Liang, Z. Lu, R. Fu and D. Wu, Nat. Commun., 2019, 10, 1363 CrossRef PubMed.
- Q. Zhang, Z. Yang, X. Gu, Q. Chen, Q. Zhai, J. Zuo, Q. He, H. Jiang, Y. Yang, H. Duan, P. Zhang, P. Zhai and Y. Gong, Energy Storage Mater., 2023, 61, 102900 CrossRef.
- Y. Yang and J. Zhang, Energy Storage Mater., 2021, 37, 135–142 CrossRef.
- J. Ryu, D.-Y. Han, D. Hong and S. Park, Energy Storage Mater., 2022, 45, 941–951 CrossRef.
- D. Lee, A. Jung, J. G. Son and B. Yeom, Energy Storage Mater., 2023, 61, 102902 CrossRef.
- D. Lee, A. Jung, P. Liu and B. Yeom, Energy Storage Mater., 2024, 65, 103107 CrossRef.
- Y. Wen, J. Ding, J. Liu, M. Zhu and R. Hu, Energy Environ. Sci., 2023, 16, 2957–2967 RSC.
- L. Zuo, Q. Ma, P. Xiao, Q. Guo, W. Xie, D. Lu, X. Yun, C. Zheng and Y. Chen, Adv. Mater., 2023, 36, 2311529 CrossRef.
- J. Wang, Z. Xu, Q. Zhang, X. Song, X. Lu, Z. Zhang, A. J. Onyianta, M. Wang, M. M. Titirici and S. J. Eichhorn, Adv. Mater., 2022, 34, 2206367 CrossRef CAS.
- J. Wang, Y. Gao, D. Liu, G. Zou, L. Li, C. Fernandez, Q. Zhang and Q. Peng, Adv. Mater., 2024, 36, 2304942 CrossRef CAS.
- P. C. Liu, H. C. Hao, H. Celio, J. L. Cui, M. Q. Ren, Y. X. Wang, H. Dong, A. R. Chowdhury, T. Hutter, F. A. Perras, J. Nanda, J. Watt and D. Mitlin, Adv. Mater., 2022, 34, 2105855 CrossRef CAS PubMed.
- H. Liu, X. Zheng, Y. Du, M. C. Borrás, K. Wu, K. Konstantinov, W. K. Pang, S. Chou, H. Liu, S. Dou and C. Wu, Adv. Mater., 2024, 36, 2307645 CrossRef CAS.
- G. Yu, Y. Cui, S. Lin, R. Liu, S. Liu, Y. Zhu and D. Wu, Adv. Funct. Mater., 2024, 34, 2314935 CrossRef CAS.
- M. Li, G. Lu, W. Zheng, Q. Zhao, Z. Li, X. Jiang, Z. Yang, Z. Li, B. Qu and C. Xu, Adv. Funct. Mater., 2023, 33, 2214759 CrossRef CAS.
- T. Wang, Q. Liu, J. Zhou, X. Wang and B. Lu, Adv. Energy Mater., 2022, 12, 2202357 CrossRef CAS.
- P. Mou, L. Zhang, C. Lu, Y. Luo, W. Yuan, L. Li, Y. Chen, Z. Shen, J. Xu and J. Shu, Adv. Energy Mater., 2023, 13, 2300734 CrossRef CAS.
- Y. Liu, Z. Tai, I. Rozen, Z. Yu, Z. Lu, A. P. LaGrow, O. Bondarchuk, Q. Chen, G. Goobes, Y. Li and L. Liu, Adv. Energy Mater., 2023, 13, 2204420 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee03978e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |