
Theoretical calculation-driven rational screening of d-block single-atom electrocatalysts based on d–p orbital hybridization for durable aqueous zinc–iodine batteries†
Jin
Yang‡
a,
Yuanhong
Kang‡
a,
Fanxiang
Meng
a,
Weiwei
Meng
b,
Guanhong
Chen
a,
Minghao
Zhang
a,
Zeheng
Lv
a,
Zhipeng
Wen
c,
Cheng Chao
Li
 *c,
Jinbao
Zhao
*a and
Yang
Yang
*a
*c,
Jinbao
Zhao
*a and
Yang
Yang
*a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, State-Province Joint Engineering Laboratory of Power Source Technology for New Energy Vehicle, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, P. R. China. E-mail: jbzhao@xmu.edu.cn; yangyang419@xmu.edu.cn
bKey Laboratory of Functional Materials and Devices for Special Environments of CAS, Xinjiang Key Laboratory of Electronic Information Materials and Devices; Xinjiang Technical Institute of Physics & Chemistry of CAS, Urumqi, 830011, P. R. China
cSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou, 510006, P. R. China. E-mail: licc@gdut.edu.cn
First published on 1st November 2024
Abstract
Aqueous Zn–iodine (Zn–I2) batteries, featuring intrinsically high-safety aqueous electrolytes and eco-friendly cathode/anode materials, however are restricted by the shuttling of polyiodide and sluggish redox kinetics of iodine redox. Although various single atom catalysts (SACs) have been proposed to improve the electrochemical performance, the underlying mechanisms of different SACs involved in iodine redox are not completely elucidated. Herein, the interaction between d-block SACs and polyiodide is demonstrated to follow d–p orbital hybridization theory, thus a series of SACs with different d-block transition metal sites are pre-screened using DFT calculations to assess the hybridization effectiveness. Among these, Nb–NC is selected due to its numerous unfilled antibonding orbitals, which facilitate effective d–p hybridization between Nb-d and I-p orbitals. Accordingly, Nb–NC with a low d-band center of 0.271 eV exhibits the highest binding energy for polyiodide and the lowest reaction barrier for the rate-determining step (I3− → I−). These theoretical predictions are well corroborated by various in/ex situ characterization studies, which confirm the suppressed shuttle effect and enhanced redox conversion of iodine species by using a free-standing Nb–NC/I2 cathode. Consequently, the Zn‖Nb–NC/I2 battery can maintain an exceptional capacity of 140 mA h g−1 over 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 10 A g−1, with only 0.00008% capacity decay per cycle.
000 cycles at 10 A g−1, with only 0.00008% capacity decay per cycle.
Broader contextAqueous Zn–I2 batteries, known for their high safety due to the use of aqueous electrolytes and environmentally friendly cathode and anode materials, face limitations caused by the polyiodide shuttle effect and the slow kinetics of iodine redox reactions. While various single-atom catalysts (SACs) have been suggested to enhance their electrochemical performance, the identification of optimal SACs remains unclear due to the diversity of d-block transition metals. Previous studies have primarily focused on binding energies and reaction free energies during discharge, with limited exploration of how the d-electron structure of SACs affects catalytic activity. Understanding this relationship is critical, as the d-electron structure influences chemical bonding and reactivity. In this study, comprehensive theoretical calculation results demonstrate that the primary interaction between iodine species and d-block SACs is mediated by d–p orbital hybridization. Nb–NC was identified as the optimal candidate due to its numerous unfilled antibonding orbitals, which promote effective d–p hybridization. As a result, Nb–NC, with a low d-band center of 0.271 eV, demonstrates the strongest polyiodide binding energy and the lowest reaction barrier for the rate-limiting step (I3− → I−). The assembled Zn‖Nb–NC/I2 battery delivers an outstanding stability. |
Introduction
The escalating global climate concerns have spurred intensive research into renewable and clean energy sources, such as wind and solar power.1 However, due to their inherent intermittence, effective energy storage devices are imperative to harness maximum energy. Aqueous zinc-ion batteries (AZIBs) are one of the most competitive options to conventional lithium-ion batteries due to their affordability, environmental compatibility, and non-flammability.2–5 Among various AZIB systems, zinc–iodine (Zn–I2) batteries stand out due to the abundance of iodine (55 mg per liter seawater), the relatively high theoretical capacity (211 mA h g−1 based on the I−/I0 redox couple), and a satisfactory working potential (1.38 V vs. Zn2+/Zn).6–12 Despite their potential advantages, the sluggish redox kinetics between iodine species and the shuttle effect of polyiodide significantly impede the actual electrochemical performance and cycling lifespan of Zn–I2 batteries.13–18 To address these problems, the most widely used strategy is to confine iodine with carbon-based hosts, which can accommodate I2 in their porous structure and improve electronic conductivity.19–21 Nevertheless, the weak physisorption of nonpolar carbon frameworks cannot completely prevent the formation and transfer of polyiodide, especially as the adsorption sites become saturated at high polyiodide concentrations. Therefore, enhancing the rate of iodine reduction reactions by electrocatalysts is a significant approach for realizing high-performance Zn–I2 batteries.Single-atom catalysts (SACs) are a unique class of electrocatalysts characterized by isolated individual metal atoms uniformly dispersed on a carrier without any interaction between the metal atoms.22 Due to the maximum atom utilization, SACs are extremely easy to chemically interact with reactive species, thus exhibiting ultra-high intrinsic activity.23,24 The d–p hybridization theory has been utilized in Li–S batteries to explain the efficient catalytic mechanism of SACs. When SACs adsorb sulfur intermediates, the hybridization state between the d orbitals of SACs and the p orbitals of sulfur will change the electronic structure of adsorbates, which will in turn lower the reaction energy barrier. Considering that both iodine and sulfur have unfilled p-orbitals (the outer electron arrangement of iodine is 5s25p5, while that of sulfur is 3s23p4), iodine can also hybridize with the d orbitals of transition metals to form d–p hybridized orbitals. Recent investigations have explored the applications of SACs including Ni, Fe, Co, and Cu to facilitate the iodine reduction reactions within the Zn–I2 battery framework, achieving good rate capability and improved cycling stability.25–29 It can be inferred that the active sites of these reported SACs are mainly focused on d-block traditional metals, which possess partially filled d bands, exhibiting great potential for chemical interactions and catalytic conversion of polyiodides. Considering the diversity of d-block traditional metal species, identifying the “right” SACs for achieving high-performance Zn–I2 batteries is still ambiguous. On the other hand, previous research has often evaluated the catalytic effects of SACs by examining binding energies and conversion reaction free energies during the discharging process, while the intrinsic interaction mechanisms between SACs and iodine species, particularly the influence of the d-electron structure of SACs on catalytic activity, have been less explored. Understanding this relationship is crucial, as the d-electron structure plays a crucial role in determining the chemical bonding and reactivity of the active sites. Therefore, establishing a correlation between the polyiodide conversion reaction and d-electron structure of SACs may unlock potential opportunities for developing high-performance electrocatalysts that are more efficient in inhibiting the shuttling effect in Zn–I2 batteries.
In this work, DFT (density functional theory) calculations are utilized as a preliminary strategy to explore how different d-block SACs in the form of M–N4 (M = Co, Cu, Fe, Nb, Ni, Zn, Mo, Re, Rh, W, Ru, Ti) configurations affect the polyiodide conversion reaction (Fig. 1a). Comprehensive theoretical calculation results reveal that the primary interaction between iodine species and transition metal active sites of SACs is mediated by d–p orbital hybridization (Fig. 1b). The d-electron structure of SACs will affect the interaction with polyiodide based on the different d–p orbital hybridization effectiveness, which is directly related to binding strengths and conversion reaction energy barriers. Among different d-block SACs studied, Nb possesses fewer d-orbital electrons and a rearrangement of the d-orbital energy levels is verified in Nb–NC. Accordingly, the increase in dxz/yz orbital energy levels and decrease in dxy and dx2−y2 orbital energy levels leads to prioritized bonding orbitals' electron occupation. Therefore, Nb–NC with abundant unfilled antibonding states and a d-band center of 0.271 eV near the Fermi level enables more effective d–p orbital hybridization, thus providing a strong iodine affinity and rapid iodine redox kinetics. Furthermore, Nb–NC exhibits the lowest reaction barrier for the rate-determining step (I3− → I−), which improves the redox reversibility of iodine species. Motivated by this theoretical proposal, Nb–NC is synthesized and adopted as an efficient electrocatalyst for Zn–I2 batteries. Various in/ex situ experimental characterization studies demonstrate that the shuttle effect of polyiodide is effectively suppressed and the electrocatalytic redox conversion of iodine is enhanced. Due to the high catalytic-active sites in the conductive network, iodine species can be well confined in Nb–NC electrocatalysts, allowing for the fabrication of self-standing I2 electrodes, which can further increase the battery energy density by eliminating the utilization of traditional current collectors. As a result, the Nb–NC/I2 cathode could exhibit an inspiring durability of 1700 h (an ultrahigh retention ratio of 96% at 10 A g−1 over 50000 cycles).
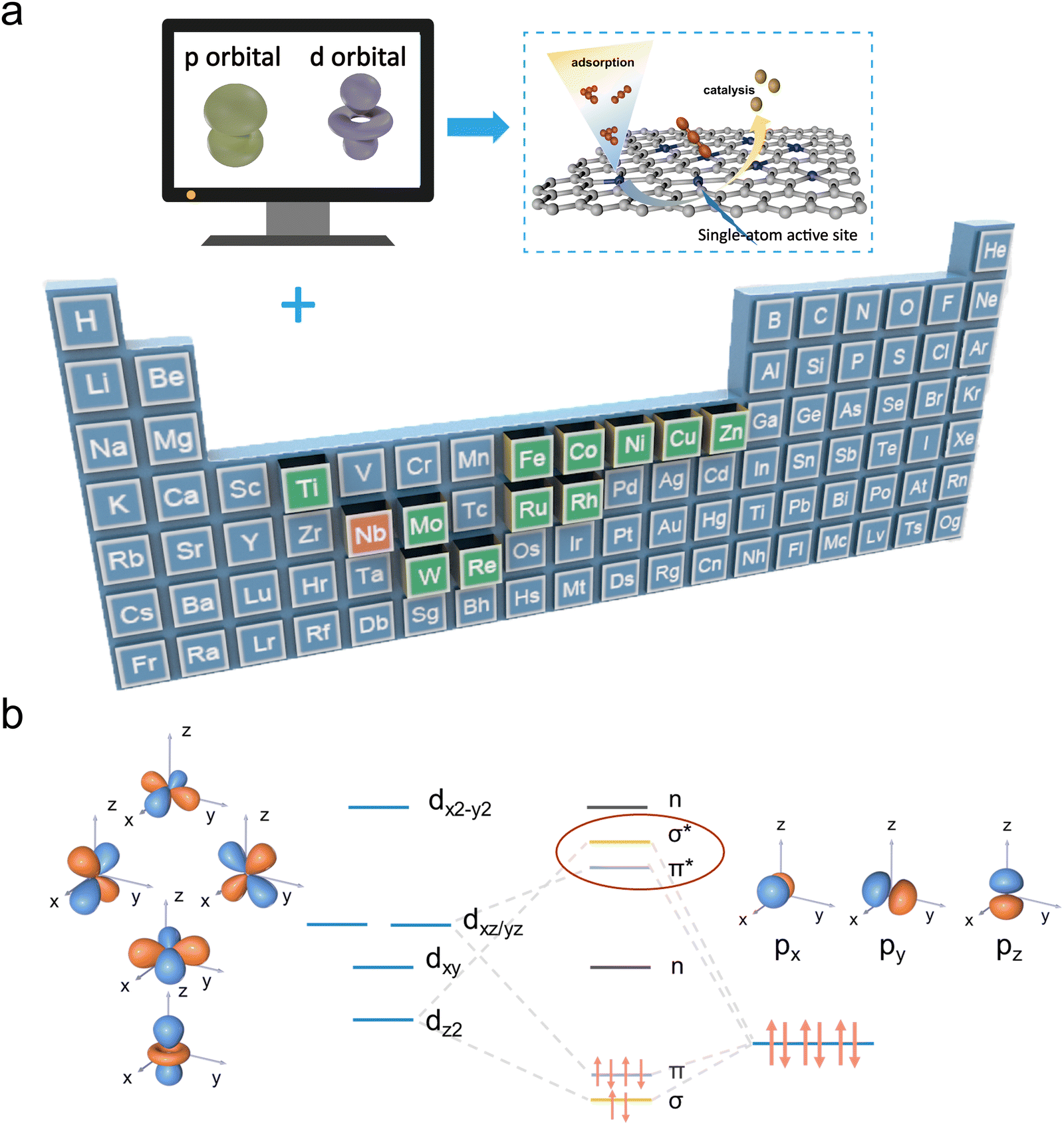 | ||
| Fig. 1 (a) Schematic illustration of iodine species adsorption and catalysis on M–NC (M = Co, Cu, Fe, Nb, Ni, Zn, Mo, Re, Rh, W, Ru, and Ti). (b) Scenario of d–p orbital hybridization. | ||
Results and discussion
A series of DFT computations were performed on twelve typical monatomic electrocatalysts with M–N4 configurations (M = Co, Cu, Fe, Nb, Ni, Zn, Mo, Re, Rh, W, Ru, Ti). These computations aim to evaluate the interaction between the electrocatalysts and various iodine species, which are crucial for enhancing the performance of Zn–I2 batteries. Firstly, as a good indicator, the adsorption energies of M–NCs on various iodine species were calculated and compared to those of metal-free nitrogen-doped carbon (NC) (Fig. 2a and Fig. S1, ESI†). The calculation results show that adsorption energies of M–NCs on iodine species are all higher than those of pure NC and Nb–NC possesses the largest values of −4.27 eV, −3.22 eV, −3.63 eV, and −3.92 eV for I−, I2, I3−, and I5− respectively, suggesting the potential of Nb–NC for adsorption catalysis on iodine species. Besides, total density of states (DOS) results reveal that the energy bands of Co, Nb, Re, Rh, Ru, Ti, W, and Zn extend across the Fermi energy level (0 eV), indicating that these corresponding M–NCs possess excellent electrical conductivity (Fig. 2b and Fig. S2, ESI†). In contrast, NC without metal active centers has a wide band gap, which is not conducive to efficient electron transfer. For effective electrocatalysts, the d orbitals of transition metal elements play a key role in hybridizing with adsorbed intermediates, which influences the potential hybridization between the M-d orbitals and the I-p orbitals. To reveal the intrinsic interaction mechanism between SACs and iodine species and to understand how the d-electron structure of SACs affects their catalytic activity, the electronic structures of the SAC-ZnI2 systems were analyzed. As displayed in Fig. 2c and Fig. S3 (ESI†), a higher degree of overlap between Nb-d orbitals and I-p orbitals is observed compared to other SACs, suggesting the efficient d–p hybridization. Coordination field theory was applied to further explore the electronic structure of Nb–NC. It has been demonstrated that dz2 and dxy/yz states of the SAC with the M–N4 configuration are more oriented towards the D4h crystal field.23 The five d orbitals are dxy, dx2−y2, dxz/yz (two-fold degenerate), and dz2 in descending order of energy level (Fig. 2e, I). Upon adsorption of iodine species, the energy levels of dxz/yz and dz2 increase, while those of dxy and dx2−y2 reduced (Fig. 2d and e, II). This energy level shift is contributed to the structural change of Nb–NC for iodine (Fig. S1, ESI†), where the Nb atom slightly protrudes from the substrate, facilitating effective d–p orbital hybridization.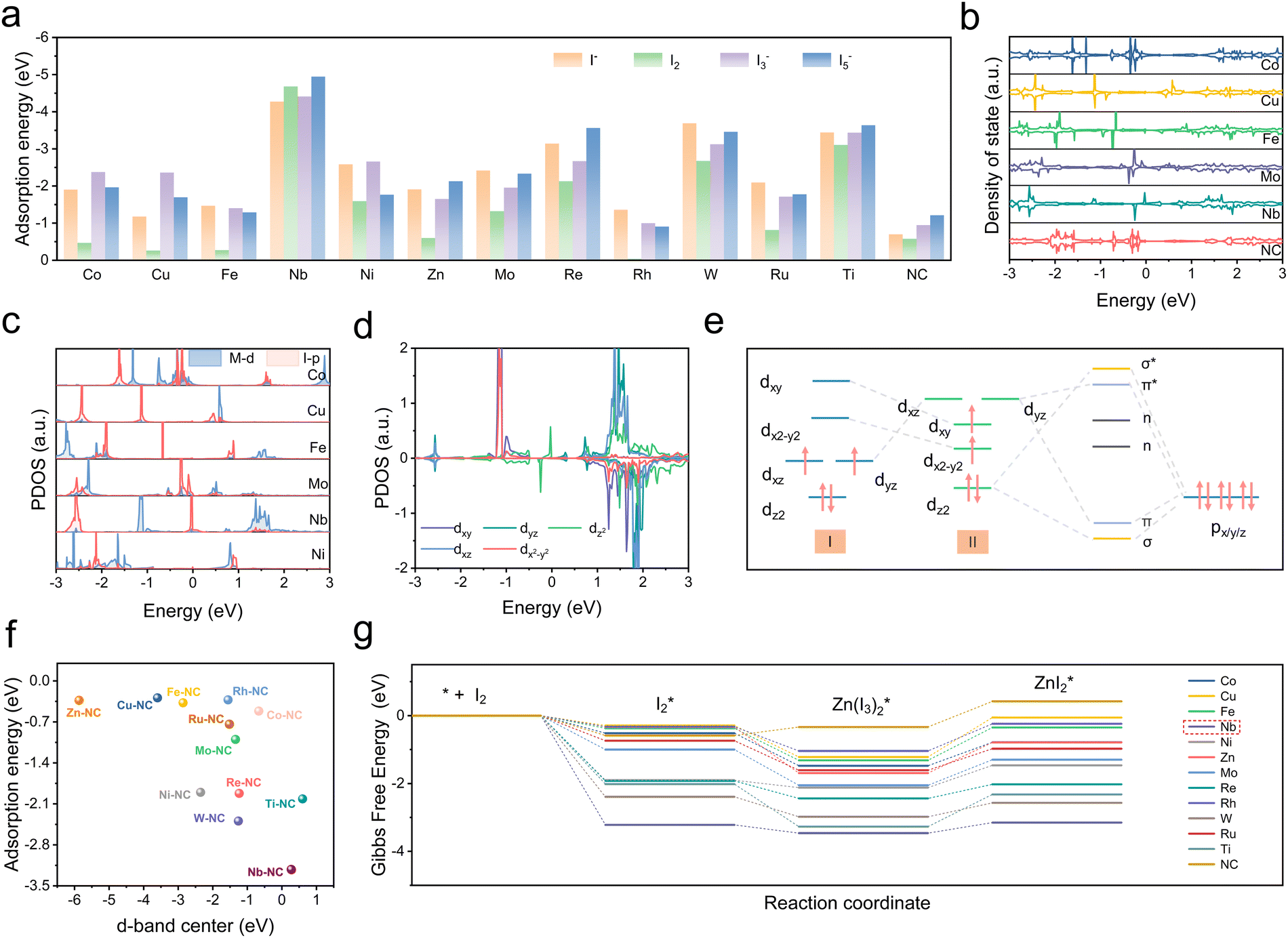 | ||
| Fig. 2 (a) Comparison of adsorption energy between M–NC (M = Co, Cu, Fe, Nb, Ni, Zn, Mo, Re, Rh, W, Ru, and Ti) and NC for I−, I2, I3−, and I5−. (b) DOS for the different M–NC (M = Co, Cu, Fe, Mo, and Nb) and NC. (c) PDOS of the p orbital of I and the d orbital of M–NC (M = Co, Cu, Fe, Mo, Nb, and Ni). (d) PDOS of Nb-d orbitals. (e) Scenario of d–p orbital hybridization. (f) The relationship between the d-band center of different SACs and the adsorption energies for I2*. (g) Gibbs free energy for reduction of iodine on M–NC (M = Co, Cu, Fe, Nb, Ni, Zn, Mo, Re, Rh, W, Ru, and Ti) and NC. | ||
More specifically, it is clear that dxz and dyz demonstrate the highest energy and are not electronically populated, while dz2, dxy, and dx2−y2 are electronically populated. The overlap of the pz orbital of the iodine atom with the dxz of Nb suggests the formation of σ and σ* states (Fig. S4, ESI†). Similarly, twofold π and π* states derived from the hybridization of the dxz/yz orbitals with the px/y orbitals, while dxy and dx2−y2 do not bond with the p orbitals (non-bonding states). Subsequently, after the rearrangement of energy levels in the d orbitals of Nb–NC, electrons preferentially occupy the bonding orbitals, leading to the effective d–p hybridization between Nb-d orbitals and I-p orbitals, thus enhancing the adsorption and redox kinetics of iodine species. It is noteworthy that elements with many electrons in the d orbitals will have their antibonding orbitals gradually filled, resulting in weak bonding strength. In addition to the efficient d–p orbital hybridization, the d-band center serves as a vital indicator for evaluating the electrical conductivity and catalytic properties of SACs. As depicted in Fig. 2f and Table S1 (ESI†), the d-band center of Nb–NC is only 0.271 eV close to the Fermi energy level, corresponding to the high adsorption energy for I2* of up to −3.22 eV. To further assess the catalytic effect of SACs on the I2 reduction reaction, the Gibbs free energies of I2 reduction pathways in different M–NCs and NC are given in Fig. 2g. The full reduction reaction pathway can be summarized as follows: I2 → I2* → Zn(I3)2* → ZnI2*, with two important intermediates of I2* and I3* being considered in this analysis. In the overall reduction reaction (I2 → ZnI2*), the more negative Gibbs free energy of Nb–NC suggests that the reaction is most likely to occur in the presence of Nb–NC. Moreover, for many electrocatalysts, the reaction path (I3− → I−) is identified as the rate-determining step in the reduction process. In the presence of Nb–NC, the reaction energy barrier for this decisive step is found to be the lowest among these M–NCs studied, further demonstrating that the Nb–N4 configuration, with Nb serving as the metal active center, is most favorable for catalyzing and accelerating redox kinetics of iodine. This combination of a low d-band center, strong adsorption energy of iodine species, and a low reaction energy barrier highlights Nb–NC as a highly effective electrocatalyst for Zn–I2 batteries, which is expected to significantly enhance their electrochemical performance and cycling stability.
To investigate the practical feasibility of Nb–NC as an efficient electrocatalyst for Zn–I2 batteries, Nb–NC and Fe–NC were successfully synthesized according to previous work.23 The microstructure of Nb–NC was examined by using transmission electron microscopy (TEM), revealing a dodecahedral morphology with an average size of approximately 160 nm (Fig. 3a). Moreover, the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image shows the atomic dispersion of the Nb metal on the carbon skeleton (Fig. 3b). The successful incorporation of Nb was also validated by the energy dispersive spectroscopy (EDS) elemental mapping result (Fig. 3c). Furthermore, the elemental composition and chemical environments of NC and Nb–NC were further characterized by X-ray photoelectron spectroscopy (XPS). A weak peak located at 207.1 eV in the Nb–NC spectrum corresponds to the Nb element (Fig. S5a, ESI†). Additionally, the C–N peaks at 285.6 eV confirm the introduction of electron-accepting N atoms, which allows the surrounding C atoms to form a more positive surface, thus enhancing iodine chemisorption through polarization interactions, rather than relying solely on the physical confinement offered by the porous structure (Fig. 3d and Fig. S5b, ESI†). The coordination form of Nb–N is also corroborated by the emergence of a peak at 399.6 eV in the N 1s spectrum (Fig. 3e and Fig. S5c, ESI†). Besides, the peaks at 204.3 and 206.9 eV in the Nb 3d spectrum are indicative of the presence of Nb4+ (Fig. 3f), which tentatively demonstrates that the synthesized Nb–NC adopts the Nb–N4 configuration.
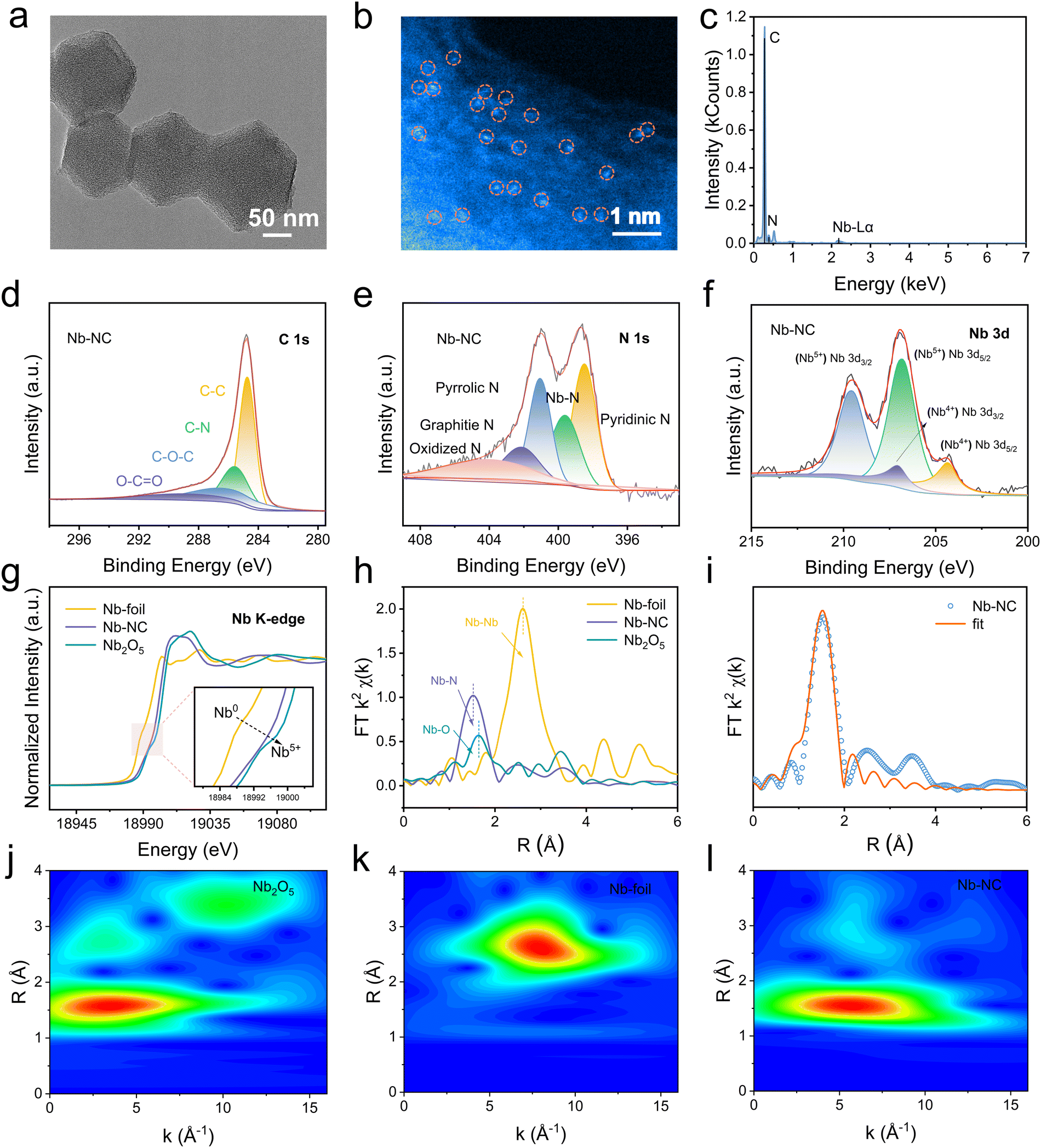 | ||
| Fig. 3 (a) TEM image, (b) HAADF-STEM image, and (c) STEM-EDS elemental mapping of Nb–NC. (d) XPS high-resolution spectra of C 1s, (e) N 1s, and (f) Nb 3d for Nb–NC. (g) Nb K-edge XANES spectra, (h) FT-EXAFS spectra for Nb-foil, Nb–NC, and Nb2O5. (i) EXAFS spectra and the curve-fit in R-space for Nb–NC. WT of EXAFS plots of (j) Nb2O5, (k) Nb-foil, and (l) Nb–NC. | ||
To further clarify the coordination number, valence, bond length, and electronic structure of the Nb single atoms, X-ray absorption near edge structure (XANES) measurements were carried out. The adsorption edge for Nb–NC in Nb–K edge XANES spectra is located between Nb foil and N2O5, indicating that the average valence state of Nb in Nb–NC is between 0 and +5 (Fig. 3g). The peak attributed to Nb–N (1.52 Å) is detected for Nb–NC in the extended X-ray absorption fine structure (EXAFS) spectra via Fourier transform (FT) in R-space, while peaks corresponding to Nb–O (1.65 Å) and Nb–Nb (2.62 Å) are notably absent (Fig. 3h). Furthermore, the EXAFS spectra of Nb–NC, Nb foil, and Nb2O5 were fitted to the R-space to analyze their coordination environments (Fig. 3i, Fig. S6, and Table S2, ESI†). The coordination number of Nb–N in Nb–NC is confirmed to be 4.0 ± 0.5 with a bond length of 2.08 Å, validating the Nb–N4 conformation as the predominant structure. Given the similarity between the Nb–N and Nb–O peaks in the EXAFS spectra, a high-resolution wavelet-transform (WT) analysis of EXAFS was performed. As can be seen in Fig. 3l, the WT plot of Nb–NC shows Nb–N scattering maximum at 5.59 Å−1, further validating the form of Nb–N and confirming the absence of Nb–Nb and Nb–O (Fig. 3j and k). These analytical results verify the successful synthesis of Nb–NC in the form of Nb–N4 conformation. In addition, the pore structures of Nb–NC and NC were analyzed using low temperature nitrogen adsorption and desorption tests (Fig. S7, ESI†). Both NC and Nb–NC exhibit isotherms with typical type IV characteristics, indicating a composition of microporous and mesoporous structures. Specifically, Nb–NC exhibits a large specific surface area (755.96 m2 g−1) and a relatively wide size distribution centered at 1.74 nm. The larger ID/IG ratio of Nb–NC (1.073) compared to NC (1.058) in Raman tests further indicates a higher density of defect/edge sites (Fig. S8, ESI†). Such a porous and defective structure enlarges contact areas between iodine and Nb for efficient electrocatalysis. The corresponding material characterization studies of Fe–NC are also shown in Fig. S9 and S10 (ESI†).
To verify the adsorption and catalytic effects of Nb–NC in Zn–I2 batteries, Nb–NC was mixed with I2 by a dry-cast process to fabricate a self-standing and flexible Nb–NC/I2 composite cathode with a thickness of ∼100 μm (Fig. S11, ESI†). The electrochemical performances of Zn–I2 cells were tested using cathodes made from Nb–NC/I2, NC/I2, and Fe–NC/I2 to compare their effectiveness. One critical issue in Zn–I2 batteries is the self-discharge phenomenon caused by the repeated shuttling of polyiodides. Therefore, suppressing the shuttle effect during resting time is crucial to enable the good storage performance of batteries. As shown in Fig. 4a, the improved voltage maintenance after 100 h for the cell with the Nb–NC/I2 cathode indicates that Nb–NC can effectively adsorb polyiodides, thereby inhibiting the self-discharge behavior of Zn–I2 batteries. In situ electrochemical impedance spectroscopy (EIS) was conducted during the initial charge–discharge cycle to assess the charge-transfer behavior of different cathodes. The charge-transfer resistance of the Nb–NC/I2 electrode (28.34 Ω) at the pristine state is notably smaller than that of the NC/I2 electrode (111.46 Ω). Throughout the charging and discharging process, the Nb–NC/I2 electrode consistently maintains a much smaller interfacial resistance (Fig. 4b and Fig. S12, ESI†), demonstrating that the incorporation of Nb alters the electronic state of the carbon framework, improving the electrical conductivity and enhancing the reaction kinetics of iodine species.
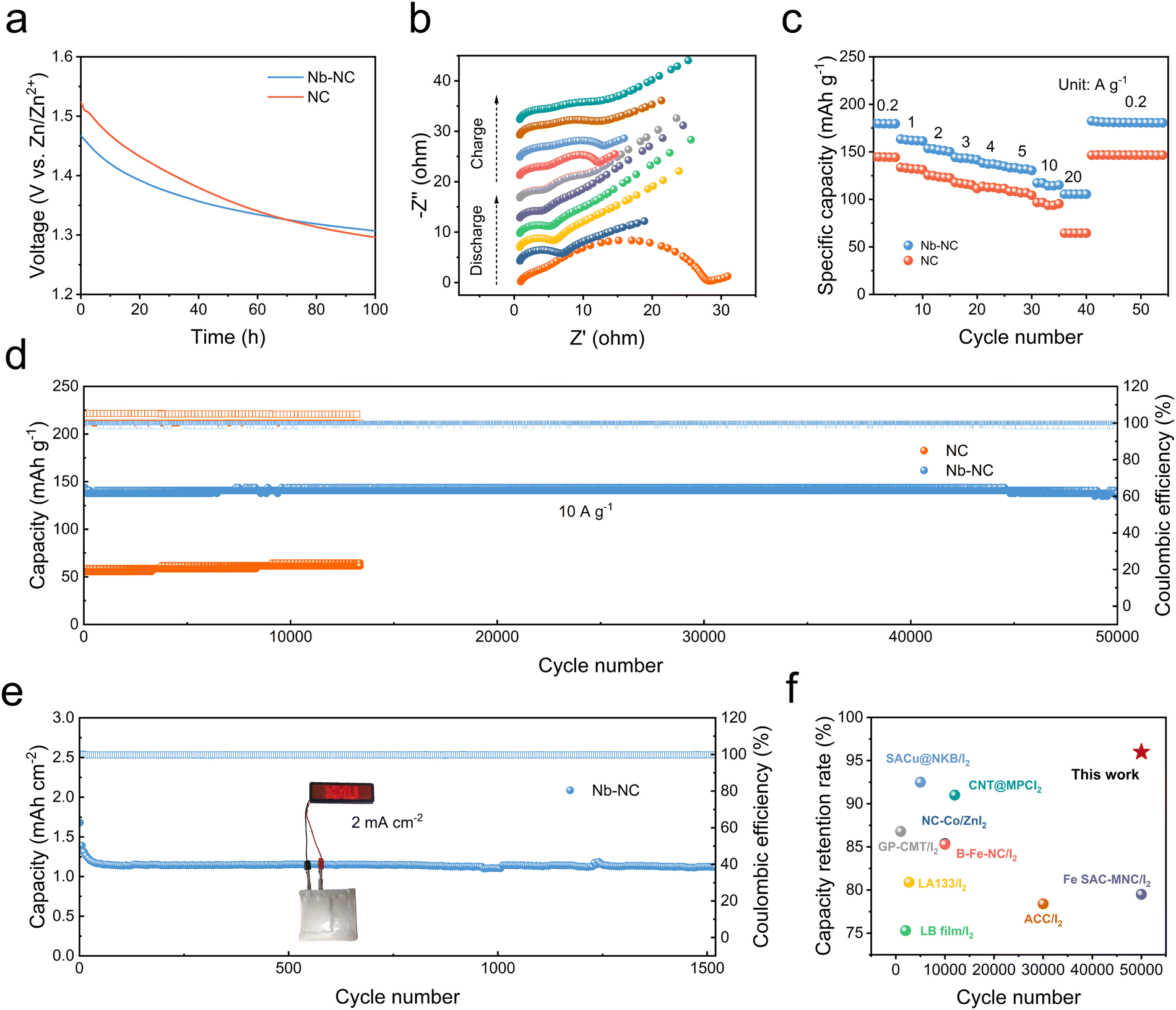 | ||
| Fig. 4 (a) Self-discharge curves of Zn–I2 batteries with NC/I2 and Nb–NC/I2 electrodes. (b) In situ EIS characterization of Zn–I2 batteries with the Nb–NC/I2 electrode. (c) Rate performance of Zn–I2 batteries with NC/I2 and Nb–NC/I2 electrodes. (d) Long cycle performance test of Zn–I2 batteries with NC/I2 and Nb–NC/I2 electrodes at a current density of 10 A g−1. (e) Long cycle performance test of pouch batteries with the Nb–NC/I2 electrode at 2 mA cm−2. (f) Comparison of the Nb–NC/I2 electrode with other representative I2 electrodes. | ||
The rate performance of both Nb–NC/I2 and NC/I2 electrodes was further tested under varying current densities ranging from 0.2 to 20 A g−1 (Fig. 4c). The Nb–NC/I2 cathode demonstrates a specific capacity of up to 250 mA h g−1 at 0.2 A g−1 and maintains a high specific capacity of 106 mA h g−1 when the current density is increased to 20 A g−1. When the current density returns to 0.2 A g−1, a specific capacity of 216 mA h g−1 can be recovered. Such excellent rate performance and reversibility of the Nb–NC/I2 cathode are far superior to those of NC/I2 and widely adopted Fe–NC/I2 cathodes (Fig. S13, ESI†). Notably, even at a high cycling rate of 10 A g−1, the Nb–NC/I2 electrode could still deliver a specific capacity of ∼140 mA h g−1 and stable cycling over 50000 cycles with only 0.00008% capacity decay per cycle, whereas the NC/I2 electrode only delivers a specific capacity of ∼60 mA h g−1 under the same conditions. This excellent cycling stability of Nb–NC/I2 is superior to many representative I2 cathodes reported previously (Fig. 4d and f).30–38 Furthermore, it is often overlooked but especially crucial to maintain stable long-term cycling at a low cycling rate (0.5–1C) and high loading conditions for practical applications.39,40 Therefore, the Zn‖Nb–NC/I2 cell was assembled for long cycling tests at 0.2 A g−1 (1C) (Fig. S14, ESI†). Even at such a relatively low cycling rate, the Zn‖Nb–NC/I2 cell could still demonstrate a stable long cycle with a specific capacity of 170 mA h g−1, while the Zn‖Fe–NC/I2 cell exhibits a slightly lower capacity. The Zn‖Nb–NC/I2 pouch cell with a loading mass of ∼9 mg cm−2 could also perform up to 1500 cycles, maintaining a capacity of 1.12 mA h cm−2 at 2 mA cm−2 (Fig. 4e).
Based on the excellent electrochemical performance of Nb–NC/I2 electrodes achieved in Zn–I2 batteries, further ex/in situ characterization studies were conducted to get deeper insights into the underlying adsorption and catalysis mechanisms of Nb–NC. As the I2 electrode generates polyiodide dissolved in the electrolyte during the charging and discharging process, which results in deterioration of electrochemical performance, the adsorption capacities of NC and Nb–NC on polyiodide were first verified. To assess this, 5 mg of NC powder and 5 mg of Nb–NC powder were each placed in 4 mL of Zn(I3)2 solution (prepared by mixing I2 and ZnI2 in deionized water at a molar ratio of 2:1 with continuous stirring to obtain a 4 mM Zn(I3)2 solution). The color changes of solutions were observed over time: the solution containing Nb–NC powder shows a lighter color after the same resting time (Fig. S15, ESI†). To make quantitative comparisons, the corresponding standard plots revealing the relationship between concentration and absorbance were obtained by measuring the absorbance of Zn(I3)2 solution with different concentrations (Fig. S16, ESI†). The concentration of I3− in solution containing Nb–NC and NC powder after 24 h was calculated by substituting the absorbance at this point in the standard curve to be 0.208 and 0.344 mM respectively, indicating that Nb–NC had a significantly higher absorption capacity compared to NC.
The impact of differences in adsorption capacity on the formation of polyiodide during electrochemical processes was preliminarily investigated using in situ ultraviolet-visible (UV-vis) spectra using a homemade electrochemical cell (Fig. 5a). The characteristic absorption wavelengths of two peaks located at 288 and 350 nm are attributed to I3−. During the discharging process, these spectral peaks of I3− gradually diminish, attributed to the transition from I3− to I−. Conversely, during the charging process, the peaks associated with I3− reappear and intensity increases with the charging potential, reaching their strongest level at 1.6 V. The obvious weaker peak intensity of I3− in the electrolyte of the Zn‖Nb–NC/I2 cell throughout the process (Fig. 5b) compared with that of the Zn‖NC/I2 cell (Fig. 5c) indicates that there is relatively less polyiodide dissolved in the electrolyte during cycling, proving that Nb–NC possesses a stronger ability to adsorb I3− compared to NC. It has demonstrated that the shuttle effect of polyiodide will also result in the corrosion of the zinc anode and the generation of by-products. Therefore, the XRD patterns of zinc foils after cycling were obtained to confirm the suppressed shuttle effect (Fig. S17, ESI†). The Zn4SO4(OH)6·5H2O by-product was detected on the zinc foil after 50 cycles in the Zn‖NC/I2 cell, while a weaker by-product signal was observed in the Zn‖Nb–NC/I2 cell. Furthermore, the Nb–NC/I2 cathode is coupled with a thinner Zn anode (30 μm) to prove the feasibility (Fig. S18, ESI†). Even at a small current density of 0.2 A g−1, the Nb–NC/I2 cathode still remains stable for more than 500 cycles, further verifying that the corrosion of Zn metal anodes can be suppressed through the inhibited polyiodide shuttling in the Zn‖Nb–NC/I2 cell.
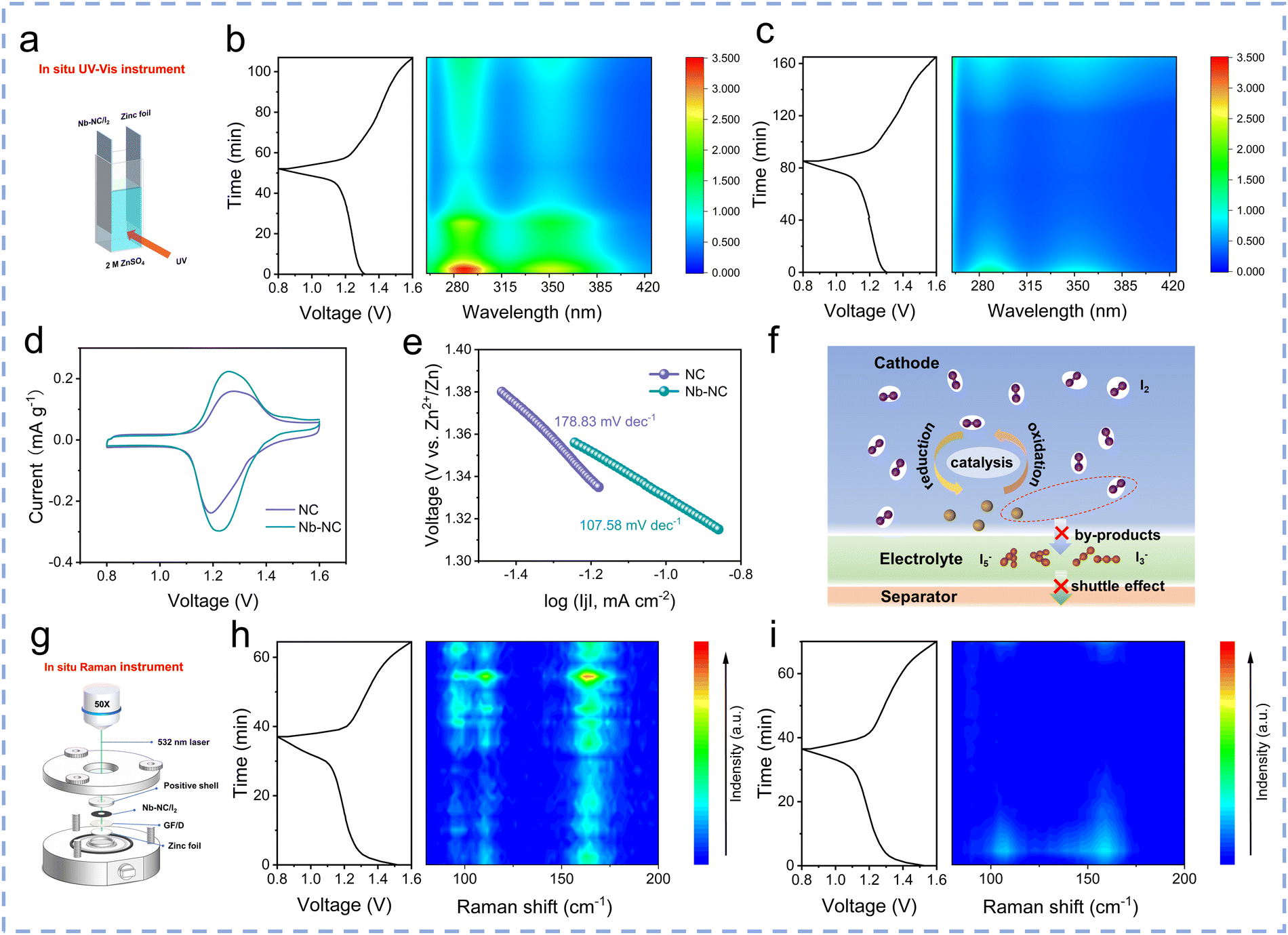 | ||
| Fig. 5 (a) Schematic diagram of an in situ UV-Vis experiment. In situ UV-Vis spectra of (b) NC/I2 and (c) Nb–NC/I2 electrodes during the charge–discharge process. (d) The CV curves for Nb–NC/I2 and NC/I2 electrodes at 0.1 mV s−1. (e) Tafel curves for Nb–NC/I2 and NC/I2 electrodes at 0.1 mV s−1. (f) Schematic illustrations of adsorption and catalysis effects of Nb–NC on polyiodide. (g) Schematic diagram of an in situ Raman experiment. In situ Raman spectra of (h) NC/I2 and (i) Nb–NC/I2 electrodes. | ||
The catalytic effect of Nb–NC on the kinetics of the reaction between iodine species during the redox process was investigated by cyclic voltammetry (CV) measurements. The redox peaks in the CV curves are attributed to the conversion of I2/I−, and the higher peak currents and lower polarization of Nb–NC/I2 prove that Nb–NC possesses a better catalytic activity (Fig. 5d). It has been demonstrated that the low Tafel slope (η) corresponds to fast reaction kinetics and better catalytic activity.41 As can be obtained from the data in Fig. 5e, the Nb–NC/I2 electrode exhibited a lower η of 107.58 mV dec−1, compared with 178.83 mV dec−1 for the NC/I2 electrode, indicating the faster reaction kinetics of the Nb–NC/I2 electrode. To demonstrate the effective restrain of polyiodide generation by the Nb–NC host with high adsorption capacity and catalyst activity, in situ Raman measurements were conducted (Fig. 5g). The peaks initially located at 110 and 166 cm−1 are attributed to I3− and I5−, which are consistently weak during the whole charging and discharging process in the Zn‖Nb–NC/I2 cell (Fig. 5h). In the case of the Zn‖NC/I2 cell, the peak intensities of polyiodide remain strong (Fig. 5i), indicating weak adsorption of polyiodide by NC and incomplete conversion of polyiodide during cycling. In summary, the free-standing Nb–NC/I2 electrode can efficiently adsorb polyiodide and enhance iodine redox conversion, thereby ensuring outstanding rate capability and durable cycling stability (Fig. 5f).
Conclusions
In summary, DFT calculations are utilized as a preliminary strategy to explore how different d-block SACs in the form of M–N4 (M= Co, Cu, Fe, Nb, Ni, Zn, Mo, Re, Rh, W, Ru, Ti) configurations affect the polyiodide conversion reaction following the d–p orbital hybridization theory. Among different d-block SACs studied, Nb possesses fewer d-orbital electrons, and a rearrangement of the d-orbital energy levels is verified in Nb–NC. Nb–NC with abundant unfilled antibonding states and a d-band center of 0.271 eV near the Fermi level enables more effective d–p orbital hybridization, thus providing a strong iodine affinity and rapid iodine redox kinetics. Calculations further reveal that Nb–NC possesses the highest binding energy for polyiodide and the lowest reaction barrier for the rate-determining step (I3− → I−). Motivated by this theoretical proposal, Nb–NC is synthesized and adopted as an efficient electrocatalyst for Zn–I2 batteries. Various in/ex situ experimental characterization studies demonstrate that the shuttle effect of polyiodide is effectively suppressed and the electrocatalytic redox conversion of iodine is enhanced. Consequently, the Zn‖Nb–NC/I2 battery delivered a long-cycling stability of 50000 cycles even at a high current density of 10 A g−1 (140 mA h g−1 with a capacity retention of 96%).
Author contributions
J. Y, and Y. K. contributed equally to this work. Conceptualization: J. Y, Y. K, J. Z and Y. Y. Experimental design and investigation: J. Y and Y. K. Data analyses: J. Y, Y. K, F. M, W. M, G. C, M. Z, Z. L, Z. W, C. L, J. Z, and Y. Y. Calculation: Y. K. Writing – original draft: J. Y. Writing – review & editing: J. Y, C. L, J. Z, and Y. Y.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 22379125, 22109030, and 22021001), the Fundamental Research Funds for the Central Universities (20720220073), the Fujian Industrial Technology Development and Application Plan (2022I0002), and the Key Research and Development Program Project of the Xinjiang Autonomous Region (2023B01025-1). The numerical calculations in this paper have been done at the Hefei advanced computing center.Notes and references
- Chinese Society of Electrochemistry, J. Electrochem., 2024, 30(1), 2024121, DOI:10.61558/2993-074X.3444.
- L. Lin, Z. Lin, J. Zhu, K. Wang, W. Wu, T. Qiu and X. Sun, Energy Environ. Sci., 2023, 16, 89–96 RSC.
- Y. Song, P. Ruan, C. Mao, Y. Chang, L. Wang, L. Dai, P. Zhou, B. Lu, J. Zhou and Z. He, Nano-Micro Lett., 2022, 14, 218 CrossRef CAS PubMed.
- J. Ke, Z. Wen, Y. Yang, R. Tang, Y. Tang, M. Ye, X. Liu, Y. Zhang and C. Li, Adv. Funct. Mater., 2023, 33, 2301129 CrossRef CAS.
- S. Zheng, L. Wei, Z. Zhang, J. Pan, J. He, L. Gao and C. Li, Nano Lett., 2022, 22, 9062–9070 CrossRef CAS.
- S. Zhang, J. Hao, H. Wu, Q. Chen, C. Ye and S. Qiao, Adv. Mater., 2024, 2404011 CrossRef CAS.
- T. Liu, C. Lei, H. Wang, J. Li, P. Jiang, X. He and X. Liang, Adv. Mater., 2024, 2405473 CrossRef CAS.
- J. Ma, M. Liu, Y. He and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 12636–12647 CrossRef CAS PubMed.
- M. Wang, Y. Meng, M. Sajid, Z. Xie, P. Tong, Z. Ma, K. Zhang, D. Shen, R. Luo, L. Song, L. Wu, X. Zheng, X. Li and W. Chen, Angew. Chem., Int. Ed., 2024, e202404784 CAS.
- Y. Liu, F. Li, J. Hao, H. Li, S. Zhang, J. Mao, T. Zhou, R. Wang, L. Zhang and C. Zhang, Adv. Funct. Mater., 2024, 34, 2400517 CrossRef CAS.
- J. Hao, S. Zhang, H. Wu, L. Yuan, K. Davey and S. Qiao, Chem. Soc. Rev., 2024, 53, 4312–4332 RSC.
- S. Huang, R. Tang, X. Liu, Y. Zhang, Y. Tang, Z. Wen, M. Ye, Y. Yang and C. C. Li, Energy Environ. Sci., 2024, 17, 591–601 RSC.
- L. Zhang, H. Guo, W. Zong, Y. Huang, J. Huang, G. He, T. Liu, J. Hofkens and F. Lai, Energy Environ. Sci., 2023, 16, 4872–4925 RSC.
- L. Ma, G. Zhu, Z. Wang, A. Zhu, K. Wu, B. Peng, J. Xu, D. Wang and Z. Jin, Nano Lett., 2023, 23, 5272–5280 CrossRef CAS.
- J. Yang, Z. Yu, J. Wu, J. Li, L. Chen, T. Xiao, T. Xiao, D. Cai, K. Liu, P. Yang and H. J. Fan, Adv. Mater., 2023, 35, 2306531 CrossRef CAS.
- W. Li, H. Xu, H. Zhang, F. Wei, T. Zhang, Y. Wu, L. Huang, J. Fu, C. Jing, J. Cheng and S. Liu, Energy Environ. Sci., 2023, 16, 4502–4510 RSC.
- J. L. Yang, H. H. Liu, X. X. Zhao, X. Y. Zhang, K. Y. Zhang, M. Y. Ma, Z. Y. Gu, J. M. Cao and X. L. Wu, J. Am. Chem. Soc., 2024, 146, 6628–6637 CrossRef CAS PubMed.
- F. Wang, W. Liang, X. Liu, T. Yin, Z. Chen, Z. Yan, F. Li, W. Liu, J. Lu, C. Yang and Q. Yang, Adv. Energy Mater., 2024, 14, 2400110 CrossRef CAS.
- W. Liu, P. Liu, Y. Lyu, J. Wen, R. Hao, J. Zheng, K. Liu, Y. Li and S. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 8955–8962 CrossRef CAS PubMed.
- Q. Zhao, Y. Lu, Z. Zhu, Z. Tao and J. Chen, Nano Lett., 2015, 15, 5982–5987 CrossRef CAS PubMed.
- X. Guo, H. Xu, Y. Tang, Z. Yang, F. Dou, W. Li, Q. Li and H. Pang, Adv. Mater., 2024, 2408317 CrossRef CAS.
- L. Zhang, D. Liu, Z. Muhammad, F. Wan, W. Xie, Y. Wang, L. Song, Z. Niu and J. Chen, Adv. Mater., 2019, 31, 1903955 CrossRef CAS.
- Y. Zhang, C. Kang, W. Zhao, Y. Song, J. Zhu, H. Huo, Y. Ma, C. Du, P. Zuo, S. Lou and G. Yin, J. Am. Chem. Soc., 2023, 145, 1728–1739 CrossRef CAS PubMed.
- Z. Han, S. Zhao, J. Xiao, X. Zhong, J. Sheng, W. Lv, Q. Zhang, G. Zhou and H. Cheng, Adv. Mater., 2021, 33, 2105947 CrossRef CAS.
- W. Qu, J. Zhu, G. Cao, S. Chen, Y. Tan, B. Chen and M. Zhang, Small, 2024, 20, 2310475 CrossRef CAS.
- X. Yang, H. Fan, F. Hu, S. Chen, K. Yan and L. Ma, Nano-Micro Lett., 2023, 15, 126 CrossRef CAS.
- F. Yang, J. Long, J. A. Yuwono, H. Fei, Y. Fan, P. Li, J. Zou, J. Hao, S. Liu, G. Liang, Y. Lyu, X. Zheng, S. Zhao, K. Davey and Z. Guo, Energy Environ. Sci., 2023, 16, 4630–4640 RSC.
- M. Liu, Q. Chen, X. Cao, D. Tan, J. Ma and J. Zhang, J. Am. Chem. Soc., 2022, 144, 21683–21691 CrossRef CAS PubMed.
- C. Guo, Y. Cao, Y. Gao, C. Zhi, Y. Wang, Y. Luo, X. Yang and X. Luo, Adv. Funct. Mater., 2024, 34, 2314189 CrossRef CAS.
- L. Zhang, J. Ge, T. Wang, H. Guo, S. Chen, Y.-E. Miao, E. Debroye, G. He, I. P. Parkin, J. Hofkens, F. Lai and T. Liu, Nano Lett., 2024, 24, 3036–3043 CrossRef CAS.
- X. Yang, H. Fan, F. Hu, S. Chen, K. Yan and L. Ma, Nano-Micro Lett., 2023, 15, 126 CrossRef CAS.
- F. Yang, J. Long, J. A. Yuwono, H. Fei, Y. Fan, P. Li, J. Zou, J. Hao, S. Liu, G. Liang, Y. Lyu, X. Zheng, S. Zhao, K. Davey and Z. Guo, Energy Environ. Sci., 2023, 16, 4630–4640 RSC.
- K. Wang, H. Li, Z. Xu, Y. Liu, M. Ge, H. Wang, H. Zhang, Y. Lu, J. Liu, Y. Zhang, Y. Tang and S. Chen, Adv. Energy Mater., 2024, 2304110 CrossRef CAS.
- J. Ma, A. Azizi, E. Zhang, H. Zhang, A. Pan and K. Lu, Chem. Sci., 2024, 15, 4581–4589 RSC.
- M. Liu, Q. Chen, X. Cao, D. Tan, J. Ma and J. Zhang, J. Am. Chem. Soc., 2022, 144, 21683–21691 CrossRef CAS PubMed.
- Y. Kang, G. Chen, H. Hua, M. Zhang, J. Yang, P. Lin, H. Yang, Z. Lv, Q. Wu, J. Zhao and Y. Yang, Angew. Chem., 2023, 135, e202300418 CrossRef.
- J. He, H. Hong, S. Hu, X. Zhao, G. Qu, L. Zeng and H. Li, Nano Energy, 2024, 119, 109096 CrossRef CAS.
- S. Chai, J. Yao, Y. Wang, J. Zhu and J. Jiang, Chem. Eng. J., 2022, 439, 135676 CrossRef CAS.
- C. Li, S. Jin, L. A. Archer and L. F. Nazar, Joule, 2022, 6, 1733–1738 CrossRef.
- Z. Xing, G. Xu, J. Han, G. Chen, B. Lu, S. Liang and J. Zhou, Trends Chem., 2023, 5, 380–392 CrossRef CAS.
- L. Ma, Y. Ying, S. Chen, Z. Huang, X. Li, H. Huang and C. Zhi, Angew. Chem., 2021, 133, 3835–3842 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: XPS, BET, electrochemical tests and calculation results. See DOI: https://doi.org/10.1039/d4ee04119d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |