
A multifunctional zeolite film enables stable high-voltage operation of a LiCoO2 cathode†
Zezhou
Lin‡
 ab,
Yiran
Ying‡
a,
Zhihang
Xu
a,
Gao
Chen
a,
Xi
Gong
a,
Zehua
Wang
b,
Daqin
Guan
b,
Leqi
Zhao
b,
Mingyang
Yang
c,
Ke
Fan
a,
Tiancheng
Liu
a,
Hao
Li
a,
Honglei
Zhang
a,
Huangxu
Li
a,
Xi
Zhang
d,
Ye
Zhu
a,
Zhouguang
Lu
c,
Zongping
Shao
*b,
Peiyu
Hou
*e and
Haitao
Huang
*a
ab,
Yiran
Ying‡
a,
Zhihang
Xu
a,
Gao
Chen
a,
Xi
Gong
a,
Zehua
Wang
b,
Daqin
Guan
b,
Leqi
Zhao
b,
Mingyang
Yang
c,
Ke
Fan
a,
Tiancheng
Liu
a,
Hao
Li
a,
Honglei
Zhang
a,
Huangxu
Li
a,
Xi
Zhang
d,
Ye
Zhu
a,
Zhouguang
Lu
c,
Zongping
Shao
*b,
Peiyu
Hou
*e and
Haitao
Huang
*a
aDepartment of Applied Physics, The Hong Kong Polytechnic University, Hong Kong 999077, China. E-mail: aphhuang@polyu.edu.hk
bWA School of Mines: Minerals, Energy and Chemical Engineering, Curtin University, Bently, WA 6102, Australia. E-mail: zongping.shao@curtin.edu.au
cDepartment of Materials Science and Engineering, Shenzhen Key Laboratory of Interfacial Science and Engineering of Materials, Southern University of Science and Technology, Shenzhen 518055, China
dInstitute of Nanosurface Science and Engineering, Guangdong Provincial Key Laboratory of Micro/Nano Optomechatronics Engineering, Shenzhen University, Shenzhen 518060, China
eSchool of Physics and Technology, University of Jinan, Jinan, Shandong 250022, China. E-mail: sps_houpy@ujn.edu.cn
First published on 13th November 2024
Abstract
Increasing the upper cut-off voltage is a useful way to enhance the specific capacity of the LiCoO2 (LCO) cathode and the energy density of the corresponding lithium-ion batteries (LIBs), while the main challenge is concurrent phase transition associated with the oxygen evolution reaction that results in a quick decay in electrochemical performance. Here, we report a significant improvement in both capacity and durability at high voltage by simply growing an AlPO4-5 zeolite protecting layer over LCO, with good crystallinity, ordered porous channels and full surface coverage. Such a coating, realized by using triethylamine as a template, acts multifunctionally to remarkably alleviative phase transition via suppressing the oxygen release at high voltage, enable fast Li+ diffusion through its nanoporous structure, accelerate the Li+-desolvation on the cathode/electrolyte interface, and boost the redox kinetics, as supported by various in situ and ex situ measurements of the LCO@AlPO4-5 zeolite (LCO@Z) cathode at a high cut-off voltage of 4.6 V (vs. Li/Li+) and density functional theory (DFT) calculations. As a result, the surface engineered LCO@Z electrode exhibits outstanding cycling stability (capacity retention of 90.3% after 200 cycles) and high-rate capability (108.2 mA h g−1 at 10C). Such a zeolite coating strategy provides a new way for developing high-energy-density LIBs with great application potential.
Broader contextThe development of high-voltage and durable LiCoO2 (LCO) cathode materials has become an urgent requirement to extend the battery life of portable electronics. Surface coating modification plays a crucial role by serving as a physical barrier, preventing electrolyte erosion on the cathode surface, and improving stability at high voltages. However, the conventional amorphous coating potentially hinders Li+ diffusion, leading to sluggish Li+ kinetics. To overcome these issues, a simple method of growing a multifunctional AlPO4-5 zeolite coating is employed to protect the LCO cathode. The AlPO4-5 zeolite coating exhibits a unique porous structure that fully covers the cathode to prevent its direct contact with the liquid electrolyte, establish a stable Li+ diffusion pathway, accelerate the Li+-desolvation process, and suppress irreversible phase transition. The well-designed structure of AlPO4-5 zeolite coated LCO exhibits outstanding cycling stability and opens up new possibilities of using zeolite coatings for developing high-energy-density Li-ion batteries. |
Introduction
Lithium-ion batteries (LIBs) utilizing the LiCoO2 (LCO) cathode and graphite anode, operating at 3.7 V (vs. Li/Li+), have seen widespread adoption in personal electronic equipment owing to their favorable volumetric energy density, high cyclability, and high safety. With the quickly rising demand for smart portable electronic devices, there is an urgent need for further significant improvement in the volumetric energy density of portable power sources.1–4 The cathode material is widely recognized for its pivotal influence on the energy density, cycling lifespan, and cost of LIBs. Among various cathode materials explored within the contemporary battery community, the first commercialized LCO oxide electrode with ultrahigh compaction density still dominates the market of high-volumetric-energy-density LIBs because of its numerous benefits.5,6 Currently, only about half of the theoretical capacity of LCO has been utilized, leaving a lot of room for improvement of its reversible capacity.A simple and efficient approach to improve the energy density of a LIB involves raising the upper cut-off voltage of the cathode. Over recent years, significant endeavors have been dedicated to increasing the upper cut-off voltage of LCO, aiming at increasing its reversible specific capacity and the energy density of the corresponding LIBs. Unfortunately, numerous challenges exist for the high-voltage operation of the commercial LCO cathode, including irreversible phase transitions, structural collapse, interfacial side reactions, and oxygen escape, all intertwined with the inherent high-voltage instability of the LCO structure. These factors collectively pose negative impacts on the reversible capacity, cycling durability and safety.7,8
Surface and/or bulk modification of LCO is necessary to make it applicable for operation at high voltages. Surface coating stands out as a promising strategy to improve the structural stability of the high-voltage LCO cathode by preventing the direct interaction between the cathode and electrolyte.9,10 An ideal coating layer should exhibit high Li+ conductivity to ensure the high-rate performance of the electrode while minimizing electrode–electrolyte contact to reduce side reactions. Traditional coating matrixes mainly include oxides, phosphates, fluorides, and carbonaceous materials. Previous studies demonstrated that stable phosphate-based coatings can mitigate the detrimental side reactions and enhance the electrochemical performance of cathode materials.11–14 However, in traditional aqueous preparation methods, aluminum salts and phosphate salts, due to their weak surface adsorption capability on the cathode material, tend to form an amorphous coating in the form of non-uniform island structures with large areas of fragmented phosphate salts. The previously reported coating layers of the AlPO4 matrix on layered cathodes are normally amorphous in nature.14–16 These traditional amorphous AlPO4 coatings exhibit irregular growth on substrate cathode materials, leading to uneven coating thickness, and the amorphous coating itself is poor in Li+ or electron conduction.16–21 Therefore, it is crucial to take both the crystallinity and surface coverage of the coating layer into consideration, as excessive thickness or an amorphous structure will hinder Li+ or electron diffusion and partial surface coverage cannot fully protect the cathode, leading to diminished electrochemical performance.9 Moreover, sluggish Li+-desolvation may also occur at the cathode/electrolyte interface upon Li+ intercalation into the host structure during the discharging process, which further hampers the redox kinetics of LIBs.22–25 Although the traditional phosphate-based coating matrixes can enhance cathode stability, they also create an intrinsic physical barrier that blocks the Li+ diffusion pathways. This barrier exacerbates the already slow Li+-desolvation process, particularly at low temperatures below 0 °C. Therefore, an ideal coating should exhibit the following features simultaneously, (1) good crystallinity for fast Li+ diffusion, (2) appropriately sized and ordered porous channels for efficient Li+-desolvation, and (3) full surface coverage for complete electrochemical and mechanical protection of the cathode.
Here, we propose a multifunctional AlPO4-5 zeolite coating layer on the LCO cathode. We applied a triethylamine template to facilitate the full coverage of a uniform crystalline phosphate-based zeolite coating on the LCO grain surface by increasing the number of nucleation sites via decreasing the surface adsorption energy. The AlPO4-5 zeolite exhibits a unique porous structure that establishes a stable diffusion pathway for Li+ ion transport, where an appropriate pore size efficiently accelerates the Li+-desolvation process. At the same time, the zeolite coating effectively minimizes the direct contact between the electrode and the organic liquid electrolyte, thereby improving the durability of the cathode. In addition, the full coverage of the highly porous coating layer acts as a robust elastic matrix to provide mechanical clamps on the cathode during the charge–discharge process, thus avoiding the delamination of cathode grains during cycling. As a result, high performance LCO with a long cycle life can be realized at a high voltage.
Results and discussion
As shown in Fig. S1 (ESI†), the pristine LCO exhibits a relatively smooth surface and a layered structure with a lattice spacing of 0.472 nm. Fig. 1a schematically illustrates the two different paths for AlPO4 surface engineering of LCO, which are prepared with/without a template agent. In the traditional path 1 without a template, amorphous and inhomogeneous AlPO4 coated LCO (LCO@A) is achieved (Fig. 1c, d and Fig. S2, ESI†), which is similar to those reported phosphate-based coatings.26–28 In path 2 with a triethylamine (TEA) template, crystalline and homogeneous AlPO4-5 zeolite fully coated LCO (LCO@Z) is successfully synthesized (Fig. 1e, f and Fig. S3, ESI†), where the introduction of a template induces the phase transition from an amorphous to crystalline structure and the uniform formation of a AlPO4-5 zeolite coating layer. High resolution transmission electron microscopy (HRTEM) images combined with fast Fourier transform (FFT) images demonstrate the presence of a crystalline AlPO4-5 zeolite coating layer on the surface of LCO. The coating layer exhibits a uniform thickness of approximately 10 nm. The atomic structure of the (210) slab for the AlPO4-5 zeolite exhibits open-framework structures for Li+ diffusion in different directions, as shown in Fig. S4 (ESI†). The organic structure directing agent plays a pivotal role in shaping the framework topology of the zeolite material.29,30 The presence of TEA in the synthesis process prompts a significant structural transformation into the crystalline AlPO4-5 zeolite due to the following two reasons. First, the TEA template acts as a structure directing agent in the gel by forming hydrogen bonds with phosphoric acid (P–OH). This interaction facilitates the incorporation of P–OH into the zeolite framework.31,32 Second, TEA also acts as a highly active ligand and forms stable chelation complexes with Al3+ ions and PO43− ions.33 The reduced free concentrations of Al3+ and PO43− ions in the gel decrease the nucleation rates of zeolite. Under the action of the structural orientation and low nucleation rate by employing TEA, crystalline zeolite is gradually formed.34–36 The utilization of TEA as a template promotes the phase transformation of amorphous AlPO4 into the crystalline AlPO4-5 zeolite state, as confirmed by X-ray diffraction (XRD) results (Fig. S5, ESI†), demonstrating the important role played by TEA in the induced phase transformation of the AlPO4-5 zeolite.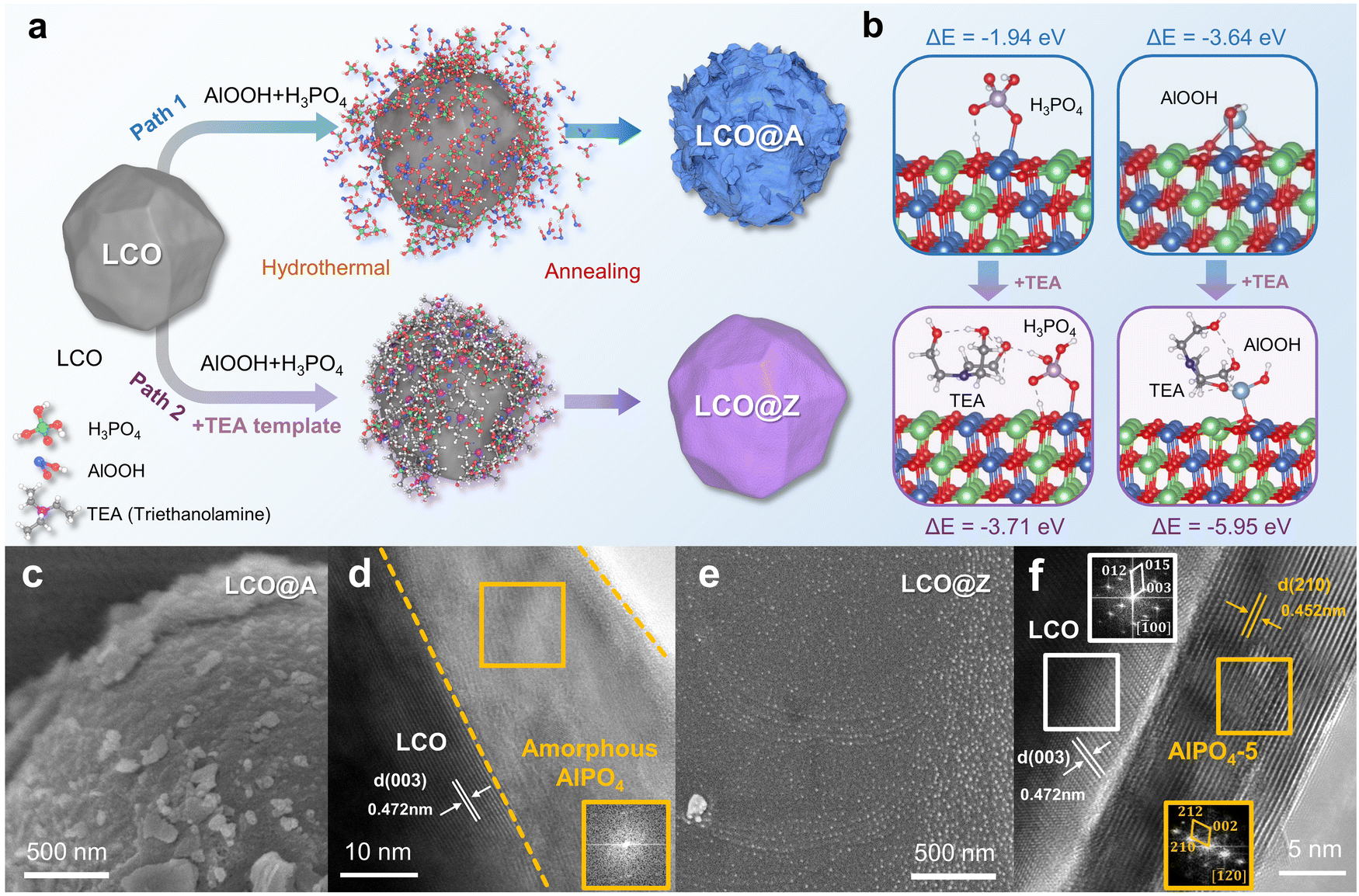 | ||
| Fig. 1 Conceptual design and preparation. (a) Schematic illustration of the synthesis design of LCO@A and LCO@Z. (b) Adsorption configurations and the corresponding adsorption energies (ΔE) of H3PO4 and AlOOH on LCO (upper panel) and TEA-modified LCO (lower panel). (c) SEM and (d) HRTEM images of LCO@A. (e) SEM and (f) HRTEM images of LCO@Z. Insets indicate the FFT patterns of the regions enclosed by rectangles. | ||
Density functional theory (DFT) calculations are employed to investigate the changes in the adsorption energy between the LCO surface and these reactants. From the adsorption energy (ΔE) calculations, it can be inferred that surface-modified TEA significantly reduces the adsorption energy of both H3PO4 (−1.94 eV to −3.71 eV) and AlOOH (−3.64 eV to −5.95 eV) reactants on the surface of LCO (Fig. 1b). The stronger adsorption energy benefits the uniform distribution of Al and P atoms on the cathode surface, which facilitates the homogeneous nucleation and growth of a uniform AlPO4-5 zeolite crystal coating layer. Moreover, TEA as a surfactant reduces the surface tension of LCO and thereby improves the wettability of the coating, allowing for better coverage of the LCO surface by Al3+ and PO43− ions.37–39 The XRD patterns (Fig. S6, ESI†) of LCO@A and LCO@Z samples can be indexed to a layered hexagonal α-NaFeO2 phase (space group: R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m),40 indicating that surface engineering does not alter the lattice structure of pristine LCO.
m),40 indicating that surface engineering does not alter the lattice structure of pristine LCO.
In the LCO@Z used in the study, the optimized concentration of AlPO4-5 was 0.5%. The contents of Al and P were confirmed from inductively coupled plasma optical emission spectroscopy (ICP-OES) results provided in Table S1 (ESI†). In the XRD patterns, at an ultralow content of 0.5%, no peaks of AlPO4-5 can be observed. Consequently, we conducted additional comparative studies by increasing the AlPO4-5 concentration to 2%, 10%, and 20% to prepare LCO@Z coatings of different thicknesses. As shown in Fig. S7 (ESI†), XRD results revealed that as the thickness of the LCO@Z coating increased, the (002) peak intensity of the AlPO4-5 phase also gradually increased. This further confirms that the AlPO4-5 zeolite is coated on the surface of LCO@Z. As shown in Fig. S8 (ESI†), the SEM images show that the coating prepared from a 0.25% precursor is thin and uneven. As the concentration increases, a smooth LCO surface progressively gets covered with a thicker layer of AlPO4-5. Brunauer–Emmett–Teller (BET) results (Fig. S9, ESI†) reveal an increase in the specific surface area from 0.23 m2 g−1 for LCO to 4.25 m2 g−1 for LCO@Z after coating by the AlPO4-5 zeolite with a high specific surface area of 17.28 m2 g−1.
The electrochemical performance of LCO, LCO@A, and LCO@Z was investigated through galvanostatic charge/discharge (GCD) measurements. Fig. 2a–c provide an intuitive comparison of the specific capacity and energy density of LCO, LCO@A, and LCO@Z at the same rate of 0.1C in the voltage range of 3.0–4.6 V (vs. Li/Li+). In the initial cycle, LCO@Z delivers a larger specific capacity of 228.2 mA h g−1 with a higher initial Coulombic efficiency of 96.7% than those of LCO@A (208.4 mA h g−1, 94.4%) and LCO (223.6 mA h g−1, 94.1%). The minor plateaus at around 4.1 V and 4.2 V can be ascribed to two first-order phase transitions occurring at around x = 0.5 in LixCoO2, which are related to the transformation between hexagonal II and monoclinic phases.41 The AlPO4-5 zeolite coating layer, acting as a distinctive elastic matrix, improves the Li+ kinetic,42,43 which can be attributed to the more uniformly Li+ distribution on the surface structure (confirmed by analysis of Fig. 4d and e). Moreover, the AlPO4-5 zeolite coating mitigates unfavorable phase transitions from O3 to H1–3 at high voltages for LCO@Z (confirmed by the following in situ XRD analysis). This enhanced structural stability is accompanied by improved redox reversibility and a higher Coulombic efficiency in the initial cycle of LCO@Z, resulting in a higher discharge capacity than the original LCO. The LCO@Z also exhibits a record high energy density of 918.8 W h kg−1 (based on the cathode mass only), higher than those of LCO (900.4 W h kg−1) and LCO@A (835.6 W h kg−1). Moreover, in the initial charge–discharge cycle, the uniform and crystalline AlPO4-5 zeolite coating alleviates the electrochemical polarization in LCO@Z while the amorphous AlPO4 coating aggravates the polarization in LCO@A. Notably, LCO@Z delivers higher specific capacities than those of the LCO and LCO@A at all C-rates (Fig. 2d and Fig. S10, ESI†), indicating enhanced rate capability. Specifically, LCO@Z still delivers a large capacity of 108.2 mA h g−1 even at 10C (6 min charging/discharging time), which is much higher than those of LCO@A (57.1 mA h g−1) and LCO (6.3 mA h g−1).
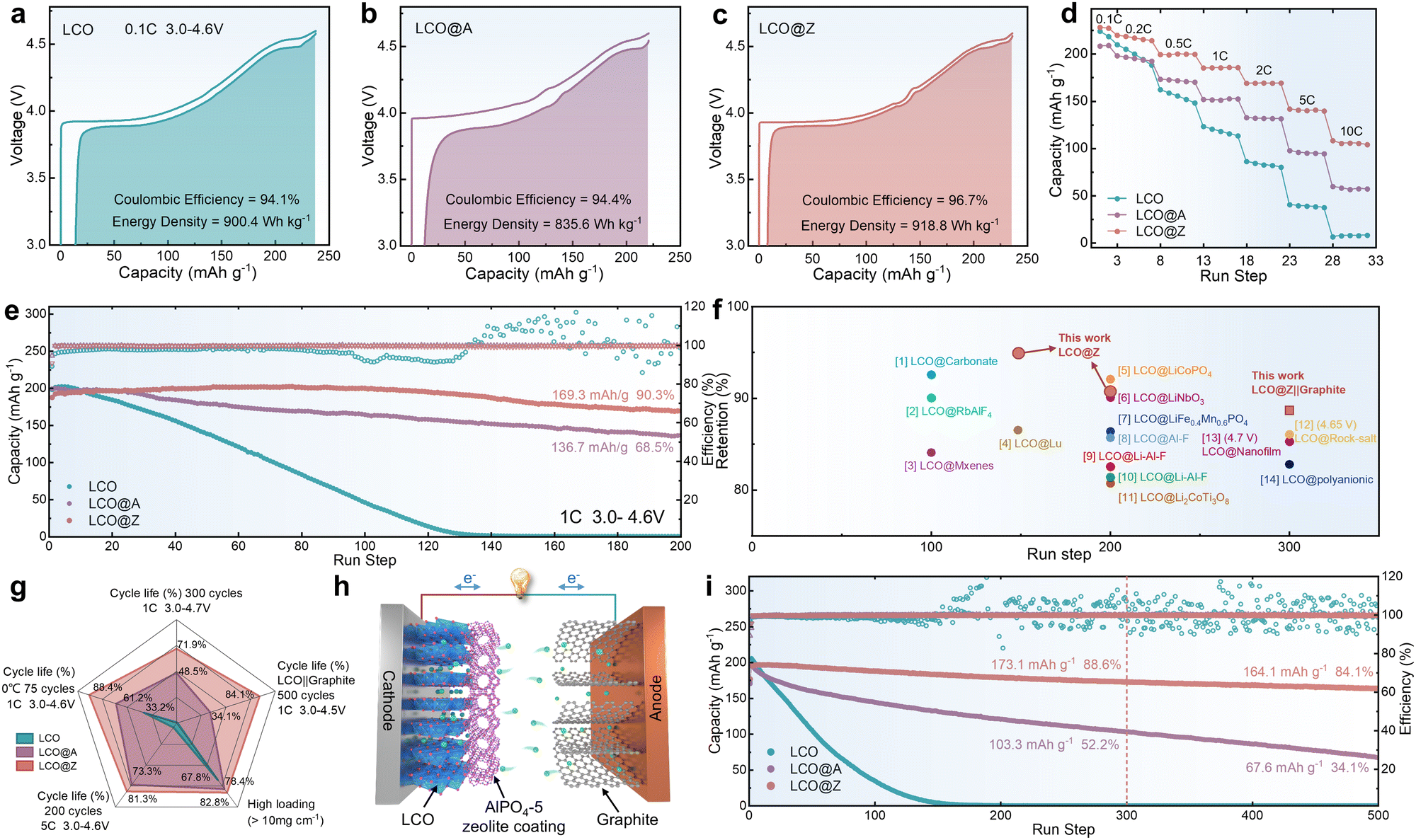 | ||
| Fig. 2 Electrochemical performance. The initial charge–discharge cycle of (a) LCO, (b) LCO@A and (c) LCO@Z at 0.1C. (d) Rate capability. (e) Cycling stability at 1C and a high upper cut-off voltage of 4.6 V (vs. Li/Li+) in a half cell. (f) Comparison of capacity retention of various surface-modified commercial LCO cathodes at a high voltage. The specific details of each modification are listed in Table S2 (ESI†). (g) Radar summary chart for comprehensive performance comparison. (h) Schematic illustration of the LCO@Z real battery. (i) Cyclic stability of bare LCO and LCO@Z in coin-type full cells at 0.5C in the voltage range of 3.0–4.5 V (vs. graphite). | ||
The cycling stability of LCO, LCO@A and LCO@Z at a high upper cut-off voltage of 4.6 V (vs. Li/Li+) is measured, as shown in Fig. 2e and Fig. S11 (ESI†). In general, the high charging voltage is accompanied by the irreversible phase transition and structural degradation of pristine LCO,44–46 leading to a rapid decline of reversible capacity and almost zero capacity after 200 cycles. The cycling stability of LCO under high-voltage operation is remarkably improved by coating it with the AlPO4-based matrix. LCO@Z maintains a large reversible capacity of 169.3 mA h g−1 with a superior capacity retention of ∼90% after 200 cycles, which is better than 136.7 mA h g−1 and 68% of LCO@A, respectively. The capacity increase in the initial cycles can be attributed to the gradual activation process of the LCO@Z cathode. As the LCO cathode particles are not perfectly monodispersed single crystals, they may contain agglomerated secondary particles with a certain amount of grain boundaries within the bulk structure. During charge–discharge cycling, the anisotropic volume expansion and contraction of the single crystal primary particles gradually open up these grain boundaries, leading to a temporary increase in capacity. In general, a formation process is needed to charge and discharge LIB in the initial cycles under a low current density. This process can accelerates the activation process of LCO@Z electrode and promotes its practical applications in high-energy LIBs. The uniform crystalline AlPO4-5 zeolite coating also relieves the voltage decay and enhances the stability of the mid-point voltage of LCO@Z as shown in Fig. S11d (ESI†).
The cycling stability of LCO, LCO@A and LCO@Z is further evaluated at a higher rate of 5C, a higher temperature of 60 °C, a lower temperature of 0 °C, a higher upper cut-off voltage of 4.7 V (vs. Li/Li+) and a high mass loading (>10 mg cm−2), as shown in Fig. S12–S16 (ESI†). The LCO@Z electrode exhibits a capacity retention of 81.7% (112.5 mA h g−1) after 200 cycles at 5C (Fig. S12, ESI†). Compared with LCO@A and LCO, LCO@Z also maintains improved cycling stability at 60 °C and 0 °C (Fig. S13 and S14, ESI†), indicating great application potential in a wide temperature range. We further evaluate the electrochemical performance of LCO, LCO@A, and LCO@Z cathodes at a higher cut-off voltage of 4.7 V (vs. Li/Li+), as presented in Fig. S15 (ESI†). LCO@Z maintains a capacity retention of 71.1% (157.9 mA h g−1) after 300 cycles, compared with LCO (0 mA h g−1, 0%) and LCO@A (107.5 mA h g−1, 48.5%). The high mass loading (>10 mg cm−2) performances were tested as shown in Fig. S16 (ESI†), and LCO@Z showed a high capacity retention of 82.8% at 0.5C after 100 cycles. The electrochemical performances of pristine LCO, LCO@A, and LCO@Z cathodes were compared and are summarized in the radar chart as shown in Fig. 2g. LCO@Z demonstrates enhanced overall electrochemical performance, including cycling stability at 4.7 V, 5C, 0 °C, high mass loading and full cell. The long-term cycling stability provided by LCO@Z stands competitively with those of recently reported various surface-modified cathodes47–60 for high voltage LIBs, as shown in Fig. 2f (detailed information listed in Table S2, ESI†). Additionally, an excessively thick AlPO4-5 zeolite coating may impede Li+ transport, leading to a continuous decrease in initial capacity with an increase in thickness (2%: 166.4 mA h g−1, 10%: 136.3 mA h g−1), as shown in Fig. S17 (ESI†). To further evaluate the electrochemical performance of LCO cathodes in practical applications, LCO‖graphite full cells (Fig. 2h) were tested. The capacity ratio of graphite/LCO is about 1.1 for the designed full cells. As shown in Fig. 2i, LCO@Z‖graphite full cells exhibit a higher energy density and retention (164.1 mA h g−1, 84.1%) after 500 cycles than those of LCO‖graphite full cells (0 mA h g−1, 0%) and LCO@A‖graphite full cells (67.6 mA h g−1, 34.1%). Moreover, LCO@Z‖graphite pouch cells based on a higher mass loading (>20 mg cm−2) cathode were assembled, which maintained high capacity and retention (60.6 mA h and 80.9%) within 50 cycles (Fig. S18, ESI†) without gas generation, showing the great potential of LCO@Z for practical application in energy storage and consumer electronic products.
The Li+ diffusion kinetics is closely linked to the Li+-desolvation at the electrode/electrolyte interface, subsequently influencing the overall redox kinetics of LIBs. The ordered porous framework channels have a lot of potential for Li+-desolvation.61–65 To elucidate the improved redox kinetics of LCO@Z, DFT calculations are employed to analyze the energy barriers of interface Li+-desolvation. We conducted adsorption energy (Ead) calculations for the Li-EC-DMC-PF6 complex at three different channel sites: 3.8 Å, 5.1 Å, and 10.2 Å. Based on the Ead calculations presented in Fig. 3a–d, the Li-EC-DMC-PF6 complex exhibits a preference for adsorption at the top of the 10.2 Å pore of AlPO4-5, as indicated by a negative Ead value. In contrast, adsorption on other smaller pores is not energetically favorable. As shown in Fig. 3e, we modelled the Li+-desolvation process of the Li-EC-DMC-PF6 complex on the surfaces of LCO, amorphous AlPO4 and AlPO4-5. DFT results show that the AlPO4-5 zeolite layer lowers the Li+-desolvation energy barrier from 3.20 eV (for LCO) to 1.17 eV, indicating easier Li+-desolvation on the AlPO4-5 zeolite layer than on LCO. Moreover, DFT results also revealed that amorphous AlPO4 lacks suitable adsorption sites, resulting in the need to overcome a higher energy barrier (2.98 eV) during the adsorption process. The low energy barriers of LCO@Z can be partly attributed to the porous and chemically inert structure of AlPO4-5 (space group P6/mcc, Fig. 3g). This porous structure allows for easier diffusion of the desolvated Li+ without the creation of chemical bonds between the surface and Li+ or solvent molecules.
 | ||
| Fig. 3 Li+-desolvation and diffusion. Adsorption energies of Li-EC-DMC-PF6 complex on top of the (a) 3.8 Å, (b) 5.1 Å, and (c) 10.2 Å pores of AlPO4-5. (d) Adsorption energy comparison. (e) DFT-calculated energy barriers for the Li+-desolvation of Li-EC-DMC-PF6 complex on LCO amorphous AlPO4 and the AlPO4-5 zeolite. (f) DFT-calculated energy barriers for the Li+ diffusion in LCO and AlPO4-5. Initial state (IS), transition state (TS), and final state (FS) structures are shown as insets. (g) Lattice structure of the AlPO4-5 zeolite. In situ EIS of symmetrical cells for (h) LCO, (i) LCO@A and (j) LCO@Z in the range of 0–3.6 V at 0.2C. The corresponding resistances and calculated Li+ diffusion coefficients of (k) LCO, (l) LCO@A and (m) LCO@Z. | ||
To better explain the Li+-desolvation effect on Li+ kinetics (eliminating the structural transformation impact on Li+ kinetics at high voltages), we further measured the rate performance of these LCO cathodes at a low cut-off voltage of 4.2 V, as shown in Fig. S19 (ESI†). LCO@A shows poor rate capability compared with uncoated LCO, confirming the amorphous coating itself exhibits poor Li+ or electronic conduction.
To assess whether the 10.2 Å pore of AlPO4-5 supports the diffusion of Li+ with a primary solvation sheath, we compared the diffusion energy barriers of the Li-EC-DMC-PF6 complex and Li+-desolvation energy barriers in AlPO4-5, as shown in Fig. S20 (ESI†). Our results indicate a notably high energy barrier of 2.88 eV for the Li-EC-DMC-PF6 complex diffusing into the pores of AlPO4-5 (even the largest 10.2 Å pore), whereas Li+-desolvation on the AlPO4-5 surface only requires 1.17 eV. The results indicate that Li+ ions with a primary solvation sheath are more energetically favorable to undergo Li+-desolvation on the surface of AlPO4-5 rather than diffuse directly into the pores.
Furthermore, after Li+-desolvation on the AlPO4-5 layer, the Li+ ions can efficiently diffuse into the large 10.2 Å pore of AlPO4-5 with a minimal 0.001 eV energy barrier (Fig. 3f). After structure relaxation, the IS of Li+ ions in AlPO4-5 will automatically transform into the FS of Li+ ions in LCO, indicating that the Li+ diffusion from AlPO4-5 into LCO is spontaneous (almost zero energy barrier). This finding implies that after Li+-desolvation, Li+ ions can spontaneously diffuse from AlPO4-5 into LCO, and the resistance during diffusion can be ignored. In contrast, LCO without an AlPO4-5 zeolite layer lacks a diffusion pathway for the solvated Li+. Additionally, Li+-desolvation is challenging to occur on LCO due to a large energy barrier of 3.20 eV. Our calculation results indicate that Li+-desolvation predominantly takes place on the AlPO4-5 zeolite layer, facilitating easy diffusion of desolvated Li+ ions through the porous structure of AlPO4-5 onto LCO.
To investigate the effects of surface engineering on the overall redox kinetics of LCO, in situ electrochemical impedance spectroscopy (EIS) of the first cycle is performed for symmetrical cells with LCO, LCO@A and LCO@Z electrodes (Fig. 3h–j), and the charge/discharge curves are shown in Fig. S21 (ESI†). The equivalent circuits are shown in Fig. S22 (ESI†), in which Rsf and Rct refer to the resistances of surface film and charge transfer, respectively, while W1 indicates the semi-infinite Warburg diffusion impedance in the bulk. For LCO@A and LCO@Z, a new interface (Rin in parallel with CPE2) is added to the equivalent circuit, which gives better fitting results than the equivalent circuit without this interface. The fitted electrochemical parameters are shown in Table S3 (ESI†). The LCO@Z electrodes exhibit relatively lower Rin values than the LCO@A do. The reduced values suggest the higher electronic conductivity induced by the AlPO4-5 zeolite coating layer. The LCO@Z electrodes also exhibit relatively lower Rct values than the pristine LCO and LCO@A do. These reduced values suggest the great electronic conductivity induced by the AlPO4-5 zeolite coating layer. From the density of states (DOS) curves in Fig. S23 (ESI†), LCO has a band gap around the Fermi level, while the DOS values are larger than zero for LCO@Z at the Fermi level, indicating that the AlPO4-5 zeolite can enhance the electronic conductivity of LCO, changing its characteristics from semiconducting to metallic. Moreover, the high Rct and Rin values of the LCO@A electrode are probably related to the poor electronic conductivity of the amorphous AlPO4 coating layer. As for Li+ ion conductivity, the Li+ diffusion coefficient (DLi+) can be calculated using the following equation,
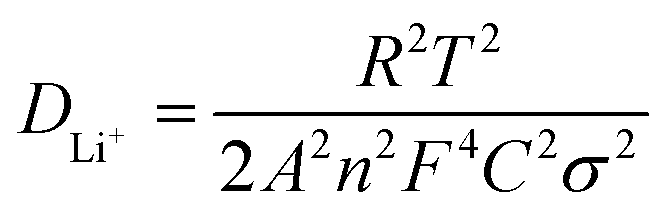 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 C mol−1), A is the apparent area of the electrode (0.503 cm2), n is the number of electrons transferred in the reaction, C is the molar concentration of Li+ ions, and σ is the Warburg factor, which satisfies the following equation:
500 C mol−1), A is the apparent area of the electrode (0.503 cm2), n is the number of electrons transferred in the reaction, C is the molar concentration of Li+ ions, and σ is the Warburg factor, which satisfies the following equation: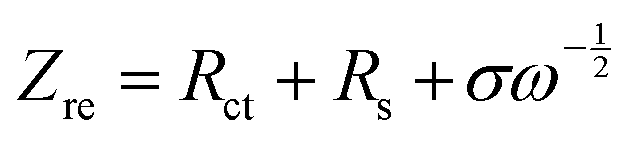 | (2) |
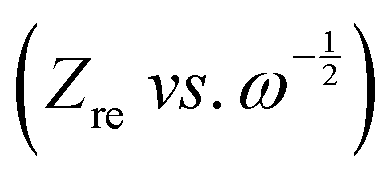 plot as exhibited in Fig. S24 (ESI†), the σ term in eqn (2) can be determined as the slope of the fitted line. In the charge/discharge process, the DLi+ at different voltages are shown in Fig. 3k–m and Table S3 (ESI†). LCO@Z shows larger average diffusion coefficient (3.58 × 10−12 cm2 s−1) than those of LCO (1.22 × 10−12 cm2 s−1) and LCO@A (6.16 × 10−13 cm2 s−1). The lower Rsf, Rct and Rin values and higher Li+ diffusion coefficient of the LCO@Z electrode demonstrate that the surface engineering with the AlPO4-5 zeolite effectively enhances the overall redox kinetics.
plot as exhibited in Fig. S24 (ESI†), the σ term in eqn (2) can be determined as the slope of the fitted line. In the charge/discharge process, the DLi+ at different voltages are shown in Fig. 3k–m and Table S3 (ESI†). LCO@Z shows larger average diffusion coefficient (3.58 × 10−12 cm2 s−1) than those of LCO (1.22 × 10−12 cm2 s−1) and LCO@A (6.16 × 10−13 cm2 s−1). The lower Rsf, Rct and Rin values and higher Li+ diffusion coefficient of the LCO@Z electrode demonstrate that the surface engineering with the AlPO4-5 zeolite effectively enhances the overall redox kinetics.
The electron conductivity of the cathodes was evaluated using the four-point probe resistivity measurement method at a pressure of 10 MPa.66 As shown in Fig. S25 and Table S4 (ESI†), the conductivity of amorphous AlPO4 (1.34 × 10−7 S m−1) was lower than that of LCO (8.07 × 10−5 S m−1). Consequently, LCO@A exhibited relatively low conductivity (4.56 × 10−5 S m−1). On the other hand, the conductivity of AlPO4-5 powder (1.21 × 10−4 S m−1) was higher than that of LCO, leading to the superior conductivity performance of LCO@Z (9.59 × 10−4 S m−1). The higher conductivity of LCO@Z than those of LCO and AlPO4-5 may be due to improved contact among grains after uniform zeolite coating. These findings are consistent with the EIS results discussed earlier (Fig. 3h–m), especially in terms of the reduction in Rct after coating with the AlPO4-5 zeolite.
Unveiling the mechanism behind the improved structural stability resulting from surface AlPO4-5 zeolite engineering is crucial. The phase transition from O3 to H1–3, accompanied by notable alterations in the c-axis lattice and structural deterioration, occurs at voltages exceeding 4.5 V (vs. Li/Li+).46In situ XRD measurements were performed to investigate the phase transition behaviors of LCO, LCO@A and LCO@Z upon Li+ de/intercalation. Although LCO, LCO@A and LCO@Z possess the same O3-type crystal structure before cycling, distinct differences in structural evolution are observed during charging and discharging, as seen in Fig. 4a. The dramatic shift of (003) diffraction peak at the high voltage of ∼4.6 V (vs. Li/Li+) is observed in LCO and LCO@A, which is attributed to the O3 to H1–3 phase transition. A relatively weak shift of the (003) diffraction peak in LCO@Z suggests the suppressed phase transition from O3 to H1–3 at the high voltage of ∼4.6 V (vs. Li/Li+). As shown in Fig. 4b, the c-axis lattice parameter variation (Δc) is smaller for LCO@Z (3.20%) than that of pristine LCO (6.58%) and LCO@A (6.57%). These results suggest that the full coverage of the crystallized coating layer (AlPO4-5 zeolite) acts as a robust elastic matrix to provide mechanical clamps on the LCO cathode grains during charge–discharge processes, which can effectively restrict the volume changes and further inhibit the O3 to H1–3 phase transition at a high voltage of 4.6 V (vs. Li/Li+).
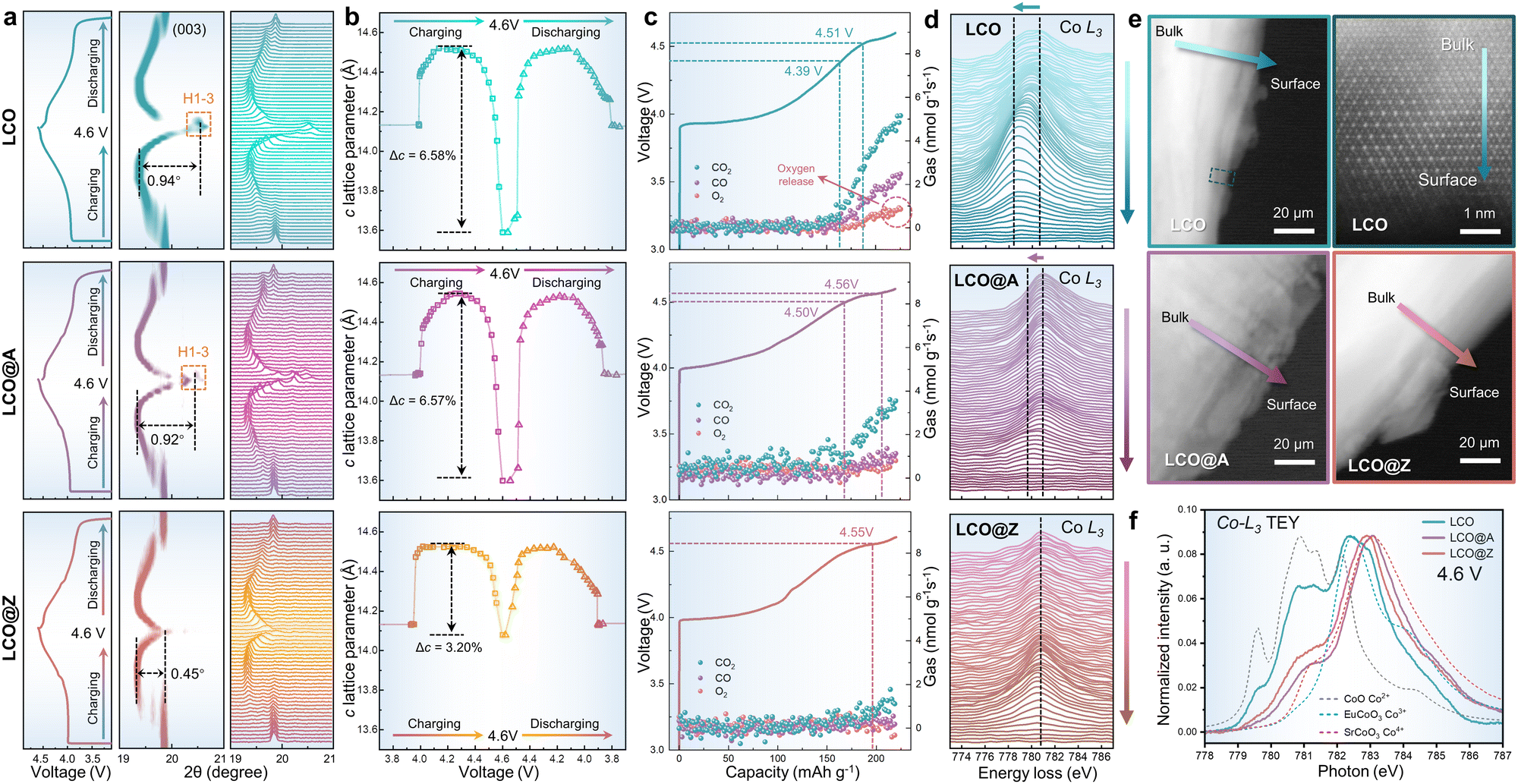 | ||
| Fig. 4 Structural evolutions at a high voltage of 4.6 V (vs. Li/Li+). (a) Charge/discharge curves and in situ XRD results of LCO, LCO@A and LCO@Z in the initial cycle. (b) The c lattice evolution of LCO, LCO@A and LCO@Z as a function of charge/discharge voltage, fitted from the in situ XRD patterns. Δc represents the variation of c lattice parameter from O3 to H1–3 phase transition during Li+ de-intercalation. (c) In situ DEMS profiles of LCO, LCO@A and LCO@Z half cells obtained during the initial charge process to 4.6 V (vs. Li/Li+) at 0.2C. (d) EELS line scan of Co-L3 edge and (e) TEM images from bulk to surface in the STEM-HAADF mode. (f) Co-L3 edge XAS data of LCO, LCO@A and LCO@Z charging to 4.6 V (vs. Li/Li+). | ||
In addition to the changes in the LCO bulk structure at a high voltage measured by in situ XRD, the side reactions on the surface are also characterized by in situ differential electrochemical mass spectrometry (DEMS) (Fig. 4c). O2 as a signature gas product indicates an irreversible oxygen evolution reaction on the particle surface,67 while carbonaceous gas is generated through the side reactions between the LCO surface and electrolyte.58,68 The carbonaceous gas occurs at above 4.5 V for LCO@Z and LCO@A during charging, in contrast to the 4.4 V for pristine LCO. The gas production rates of CO2 and CO of the pristine LCO reach 5.16 and 2.49 nmol g−1 s−1, respectively, at 4.6 V (vs. Li/Li+), much higher than those of 3.83 and 1.09 nmol g−1 s−1 for LCO@A, and 1.31 and 0.41 nmol g−1 s−1 for LCO@Z, respectively. The LCO shows an onset voltage of oxygen release at ∼4.5 V and a high oxygen production rate of 0.98 nmol g−1 s−1 at 4.6 V (vs. Li/Li+). The LCO@A shows delayed onset voltage and reduced amount of oxygen production while the LCO@Z exhibits almost zero oxygen production during charging. The delayed onset voltage and reduced amount of gas production validate the effectiveness of the AlPO4-5 zeolite coating in inhibiting oxygen release and side reactions on the cathode surface.
To establish a comprehensive understanding of the structure–performance relationship emerging from the coating layer, we conducted electron energy loss spectroscopy (EELS) to investigate the valence variation of Co from their bulk to the surface at a high voltage of 4.6 V using the STEM-HAADF mode, as shown in Fig. 4d. EELS results clearly demonstrate that LCO exhibits surface instability at a high voltage of 4.6 V (vs. Li/Li+), where the Co species on the surface are in a lower valence state (indicated by the shift of Co-L3 edge to lower energy) than those in the bulk. As shown in Fig. 4e, the TEM image reveals the corresponding irreversible phase transition from layered to spinel and/or rock-salt phase, which causes uneven Li+ de-intercalation from the bulk to the surface and results in sluggish kinetics. In contrast, LCO@A and LCO@Z exhibit a relatively stable structure even at the high voltage due to the protective coating layers. For LCO@A, the scanned spectral lines of the Co-L3 edge show only a minor shift from bulk to the surface and LCO@Z shows almost no shift of the Co-L3 edge.
During the charging and discharging processes, a series of corresponding phase transitions occur in LCO as Li ions are extracted/inserted. In these processes, the phase transitions in LCO and the two-phase coexistence are primarily governed by Li+ kinetics. Taking the charging process as an example, for LCO, there is an apparent two-phase coexistence at 4.2 V and a gradual shift of the X-ray diffraction peak (Fig. 4a) due to the slow Li+ kinetics. While for LCO@Z, the phase transition is more abrupt or discontinuous. This corresponds to the more uniform Li+ distribution, where the phase equilibrium can be more quickly achieved due to the fast Li+ kinetics facilitated by the effective AlPO4-5 zeolite coating. The uniform AlPO4-5 zeolite coating aids in the establishment of stable pathways for Li+ diffusion, as evidenced by EELS data demonstrating uniform Li+ distribution from the surface to the bulk during delithiation (Fig. 4d). This exceptional Li+ kinetics facilitate an abrupt two-phase transition in LCO during charging/discharging processes.
Additionally, soft X-ray absorption spectroscopy (XAS) spectra at the Co-L3 edge collected by the total electron yield (TEY) mode are sensitive to the valence state of near surface Co in the cathodes,69 revealing the role of the AlPO4 coating at a high voltage (4.6 V vs. Li/Li+), as shown in Fig. 4f. Upon charging to 4.6 V (vs. Li/Li+), the Co-L3 spectrum of LCO exhibited a shift to lower energy, which indicates a gradual decrease in the Co valence state of surface structure. The Co2+/Co3+ mixed phases correspond to an irreversible phase transition from layered to spinel and/or rock-salt phases on the surface (Fig. 4e). In contrast, LCO@A and LCO@Z exhibit a favorable Co3+/Co4+ mixture, showcasing surface stability induced by the AlPO4 coating in the deep delithiated state. Moreover, the uniform zeolite structure aids in stabilizing surface Co dissolution, thereby restraining excessive generation of Co4+ valence states at a high voltage.
The structural evolution after long-term cycling is also analyzed by TEM, cross-sectional SEM and time-of-flight secondary ion mass spectrometry (TOF-SIMS), as shown in Fig. 5 and Fig. S26 (ESI†). The parasitic side reactions between oxidative species and organic electrolytes cause the creation and accumulation of a cathode electrolyte interphase (CEI) on the LCO surface during repeated cycles.70,71Fig. 5a illustrates TEM images of the cycled LCO, LCO@A, and LCO@Z after 200 cycles. The LCO@A and LCO@Z exhibit a thin CEI layer on the grain surface, in contrast to a thick CEI layer for LCO. Remarkably, even after long-term cycling, the AlPO4-5 zeolite coating layer still maintains its original crystalline behavior on the surface of LCO@Z, as evidenced by the FFT pattern.
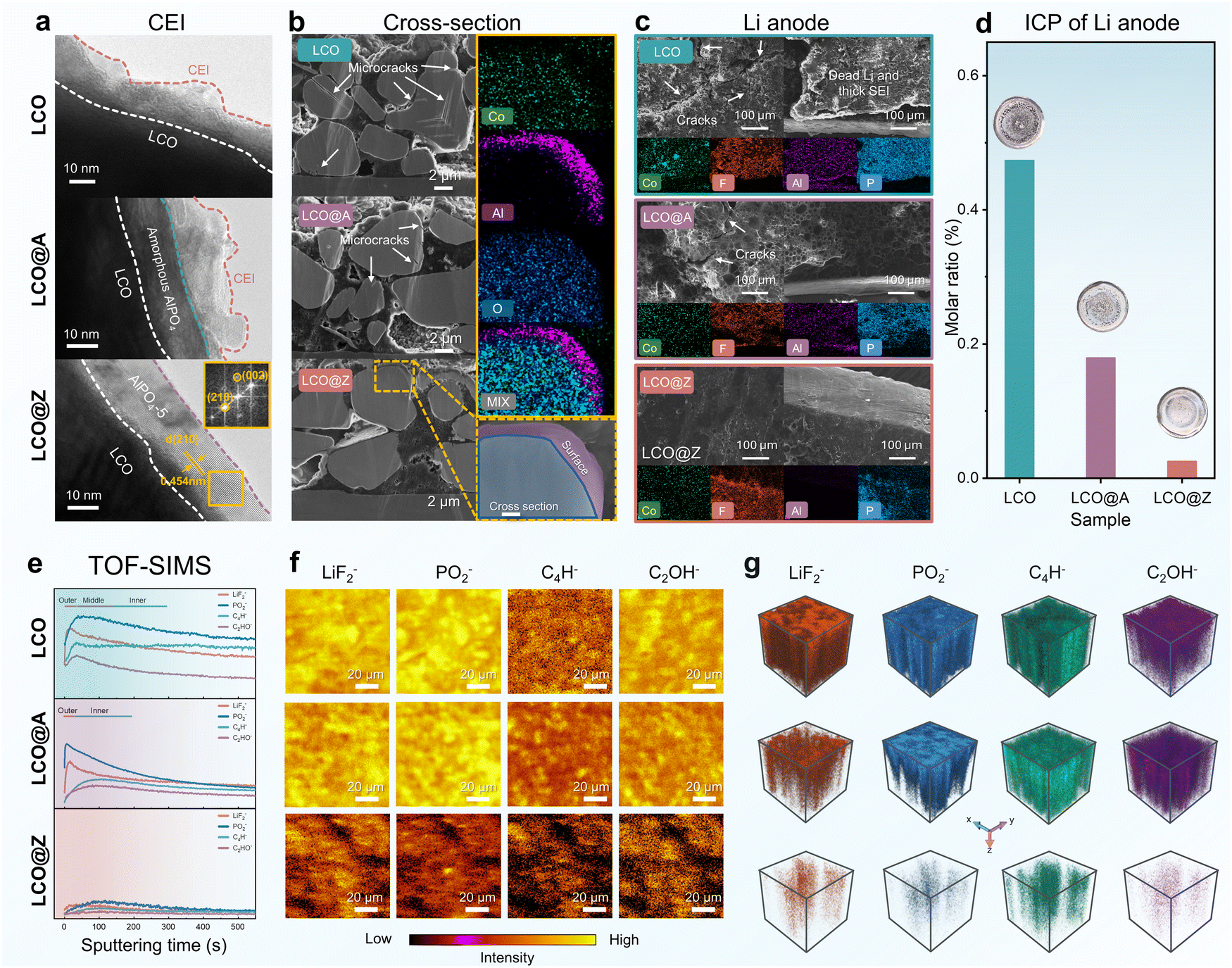 | ||
| Fig. 5 Structural evolution during long-term cycling. (a) TEM images of surface morphology of LCO, LCO@A and LCO@Z after 200 cycles. (b) Cross-sectional SEM and the corresponding EDS image of LCO@Z after 200 cycles. (c) Plane-view SEM, cross-sectional SEM and EDS images of the Li metal anodes paired with LCO, LCO@A, and LCO@Z cathodes after 50 cycles. (d) ICP-OES concentration of Co on Li metal anode after 50 cycles. (e) Intensity depth profiles, (f) surface species distributions and (g) 3D rendering TOF-SIMS fragments of LiF2−, PO2−, C4H− and C2OH− on the surface of LCO, LCO@A and LCO@Z after 50 cycles. | ||
The repeated phase transitions between O3 and H1–3 may lead to the occurrence of severe anisotropic mechanical strains in the crystal structure, resulting in microcracks within the grain.44,72 Cross-sectional SEM images of LCO, LCO@A and LCO@Z grains after 200 charge–discharge cycles are shown in Fig. 5b. The cycled LCO grains exhibit severe microcracks within the bulk and surface, while the cycled LCO@A shows only some small microcracks near the surface. The cycled LCO@Z has no observable microcracks. These cracks expose fresh LCO surfaces to electrolyte species and readily damage the grains with a substantial decrease in elastic modulus, hardness, and fracture toughness, further enhancing side reactions and creating autocatalytic scenarios.6,73 The energy-dispersive spectroscopy (EDS) mapping confirms that the Al element still enriches on the surface of LCO@Z, indicating the good electrochemical and mechanical stability of the AlPO4-5 zeolite coating layer.
As shown in Fig. 5c, the rough interface of the Li anode coupled with LCO is evident in the SEM image, displaying an uneven layer consisting of the solid–electrolyte interphase (SEI) and inactive Li compounds. While LCO@A reduces side reactions, it still leads to crack formation and a thick SEI on the Li anode surface. In contrast, the interface of the Li anode appears smooth when paired with LCO@Z, exhibiting only an intact SEI layer in the cross-section image. Furthermore, Co dissolution from the cathode would significantly deteriorate the electrochemical performance of the Li metal anode.74,75 Concerning coating dissolution and Co dissolution, SEM-EDS and ICP-OES analyses were conducted on the cycled anode Li metal (Fig. 5c, d and Table S5, ESI†). SEM-EDS mapping profiles revealed significantly lower levels of Co, Al, and P in the anode of LCO@Z compared to LCO and LCO@A. ICP-OES tests unveiled that LCO@Z showed a lower Co dissolution (0.249‰) than those of LCO (4.736‰) and LCO@A (1.793‰). These findings suggest that the AlPO4-5 coating can maintain a stable structure and high crystallinity after long-term cycling, effectively inhibiting the dissolution of transition metals at a high voltage, thereby enhancing cycling stability.
TOF-SIMS is further employed to investigate the chemical composition of CEI components after cycling. The relative intensities of negative secondary ion species and TOF-SIMS patterns are shown in Fig. 5e–g and Fig. S26 (ESI†). For the negative secondary ion mode, a higher proportion of side reaction fragments76 is determined from the surface of LCO, including LiF2−, PO2−, C4H− and C2OH−. Those results show that the CEI induced by side reactions was mitigated in LCO@Z, in contrast with the side reaction fragments enriched surface in the cycled LCO and cycled LCO@A. The 3D reconstruction and TOF-SIMS chemical images (Fig. 5g) revealed a more gradual concentration gradient of interface degradation-generated species within the AlPO4-5 zeolite-modified cathode. The analysis revealed that the surface CEI film of LCO@Z is relatively thinner than those of LCO and LCO@A, which demonstrated that the AlPO4-5 zeolite coating serves as an effective protective layer to mitigate the side reactions between the cathode and electrolyte. Besides, the cycled LCO@Z exhibits a much reduced amount of C-containing species, as evidenced in X-ray photoelectron spectroscopy (XPS) results (Fig. S27, ESI†). These results demonstrate that the AlPO4-5 zeolite coating serves as an effective protective layer that mitigates the side reactions between the cathode and electrolyte, leading to a thin CEI layer on the LCO surface.
The effect of structural evolutions of LCO within cycling on the Li+ diffusion kinetics is further investigated by the galvanostatic intermittent titration technique (GITT), as depicted in Fig. S28 and S29 (ESI†). These results confirm that the AlPO4-5 zeolite coating maintains the stable Li+ diffusion coefficients in the LCO@Z upon cycling, in contrast to gradually decreased Li+ diffusion coefficients in the LCO and LCO@A. As shown in Fig. S30 (ESI†), the cyclic voltammetry (CV) curve indicates better redox reversibility and weaker electrochemical polarization of LCO@Z after 200 cycles than those of LCO and LCO@A.
Based on the above results and discussion, the functional mechanism of the AlPO4-5 zeolite coating in enhancing the electrochemical performance of LCO is schematically depicted in Fig. 6, and summarized as follows. (1) The electrochemically and mechanically stable AlPO4-5 zeolite coating acts as a protective layer to prevent the direct contact between the organic liquid electrolyte and LCO grains, reduce the lattice oxygen loss and the side reactions on the LCO surface, and thereby relieve the surface decay and the excessive accumulation of CEI upon cycling. (2) The full coverage of the robust elastic AlPO4-5 zeolite coating layer on LCO effectively provides mechanical reinforcements to suppress the phase transition from O3 to H1–3, leading to the absence of microcracks and good structural stability. (3) The crystalline AlPO4-5 zeolite with a unique ordered nanoporous structure establishes a stable diffusion pathway for Li+ ion transport, whose appropriate pore size efficiently accelerates the Li+-desolvation process and further improves the Li+ kinetics. As a result, a multifunctional AlPO4-5 zeolite coating layer enables stable high-voltage operation of the LCO cathode in high-energy LIBs.
 | ||
| Fig. 6 Schematic illustration of the mechanism of the AlPO4-5 zeolite layer in electrochemical performance enhancement. | ||
Conclusions
The current work introduces a novel coating approach utilizing the AlPO4-5 zeolite, which leads to enhanced stability of the LCO cathode at a high cut-off voltage of 4.6 V (vs. Li/Li+). The improved stability observed in LCO@Z can be attributed to the multifunctional coating layer which effectively establishes fast Li+-desolvation and diffusion pathways, mitigates irreversible phase transitions at a high voltage, and protects against structure/surface degradation during long-term cycling. The well-designed surface engineering in LCO@Z successfully suppresses the phase transition from O3 to H1–3, thereby preserving the stability of the surface lattice oxygen at a high voltage. Remarkably, LCO@Z exhibits an outstanding capacity retention of 90.3% after 200 cycles at a rate of 1C. These remarkable achievements provide valuable insight into the rational design of advanced cathodes with zeolite-based coatings, opening new possibilities for future developments in surface engineering strategies.Author contributions
H. T. Huang, P. Y. Hou and Z. P. Shao conceived the experiments and supervised this project. Z. Z. Lin, X. Gong, T. C. Liu, L. Q. Zhao and H. Li performed the preparation, characterization and performance measurement of electrodes, electrolytes and coin cells. Y. R. Ying and K. Fan performed the DFT calculation. Z. H. Xu and Y. Zhu performed the STEM-EELS characterization. M. Y. Yang and Z. G. Lu performed in situ XRD. Z. H. Wang performed TOF-SIMS. G. Chen, D. Q. Guan, H. L. Zhang, H. X. Li and X. Zhang provided experimental advice. Z. Z. Lin, H. T. Huang, P. Y. Hou and Z. P. Shao wrote the manuscript. All the authors discussed the results and commented on the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Research Grants Council of the Hong Kong Special Administrative Region, China (PolyU152178/20E), the Innovation and Technology Commission of Hong Kong (MHP/080/22), the Hong Kong Polytechnic University (G-SAC1), the National Natural Science Foundation of China (22379052), the Science and Technology Program of Guangdong Province of China (2021B1515130010), and the Shenzhen Excellent Youth Basic Research Fund (No. RCYX20231211090249068).Notes and references
- C.-Y. Wang, T. Liu, X.-G. Yang, S. Ge, N. V. Stanley, E. S. Rountree, Y. Leng and B. D. McCarthy, Nature, 2022, 611, 485–490 CrossRef CAS PubMed.
- H. W. Ji, J. P. Wu, Z. J. Cai, J. Liu, D. H. Kwon, H. Kim, A. Urban, J. K. Papp, E. Foley, Y. S. Tian, M. Balasubramanian, H. Kim, R. J. Clément, B. D. McCloskey, W. L. Yang and G. Ceder, Nat. Energy, 2020, 5, 213–221 CrossRef CAS.
- W. D. Li, E. M. Erickson and A. Manthiram, Nat. Energy, 2020, 5, 26–34 CrossRef CAS.
- Q. Wang, M. Zhu, G. Chen, N. Dudko, Y. Li, H. Liu, L. Shi, G. Wu and D. Zhang, Adv. Mater., 2022, 34, 2109658 CrossRef CAS PubMed.
- Z. Lin, K. Fan, T. Liu, Z. Xu, G. Chen, H. Zhang, H. Li, X. Guo, X. Zhang, Y. Zhu, P. Hou and H. Huang, Nano-Micro Lett., 2023, 16, 48 CrossRef.
- C. Lin, J. Li, Z. W. Yin, W. Huang, Q. Zhao, Q. Weng, Q. Liu, J. Sun, G. Chen and F. Pan, Adv. Mater., 2024, 36, e2307404 CrossRef.
- J. X. Wang, K. Jia, J. Ma, Z. Liang, Z. F. Zhuang, Y. Zhao, B. H. Li, G. M. Zhou and H. M. Cheng, Nat. Sustainability, 2023, 6, 797–805 CrossRef.
- P. Xiao, Y. Zhao, Z. Piao, B. Li, G. Zhou and H.-M. Cheng, Energy Environ. Sci., 2022, 15, 2435–2444 RSC.
- U. Nisar, N. Muralidharan, R. Essehli, R. Amin and I. Belharouak, Energy Storage Mater., 2021, 38, 309–328 CrossRef.
- S. Kalluri, M. Yoon, M. Jo, S. Park, S. Myeong, J. Kim, S. X. Dou, Z. Guo and J. Cho, Adv. Energy Mater., 2017, 7, 1601507 CrossRef.
- C. Yang, X. Liao, X. Zhou, C. Sun, R. Qu, J. Han, Y. Zhao, L. Wang, Y. You and J. Lu, Adv. Mater., 2023, 35, e2210966 CrossRef.
- Q. Hu, Y. F. He, D. S. Ren, Y. Z. Song, Y. Z. Wu, H. M. Liang, J. H. Gao, G. Xu, J. Y. Cai, T. Y. Li, H. Xu, L. Wang, Z. H. Chen and X. M. He, Nano Energy, 2022, 96, 107123 CrossRef CAS.
- Y. Wang, Q. Zhang, Z. C. Xue, L. Yang, J. Wang, F. Meng, Q. Li, H. Pan, J. N. Zhang, Z. Jiang, W. Yang, X. Yu, L. Gu and H. Li, Adv. Energy Mater., 2020, 10, 2001413 CrossRef CAS.
- J. Gu, L. Chen, X. Li, G. Luo, L. Fan, Y. Chao, H. Ji and W. Zhu, J. Energy Chem., 2024, 89, 410–421 CrossRef CAS.
- Y.-H. Du, H. Sheng, X.-H. Meng, X.-D. Zhang, Y.-G. Zou, J.-Y. Liang, M. Fan, F. Wang, J. Tang, F.-F. Cao, J.-L. Shi, X.-F. Cao and Y.-G. Guo, Nano Energy, 2022, 94, 106901 CrossRef CAS.
- Y. Zhang, Y. Pei, W. Liu, S. Zhang, J. J. Xie, J. Xia, S. Nie, L. Liu and X. Y. Wang, Chem. Eng. J., 2020, 382, 122697 CrossRef CAS.
- F. Wu, X. Zhang, T. Zhao, L. Li, M. Xie and R. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 3773–3781 CrossRef CAS.
- J. Song, Y. Wang, Z. Feng, X. Zhang, K. Wang, H. Gu and J. Xie, ACS Appl. Mater. Interfaces, 2018, 10, 27326–27332 CrossRef CAS PubMed.
- G. Nava, J. Schwan, M. G. Boebinger, M. T. McDowell and L. Mangolini, Nano Lett., 2019, 19, 7236–7245 CrossRef CAS.
- K. S. Tan, M. V. Reddy, G. V. S. Rao and B. V. R. Chowdari, J. Power Sources, 2005, 141, 129–142 CrossRef CAS.
- Y. Wu, A. Vadivel Murugan and A. Manthiram, J. Electrochem. Soc., 2008, 155, A635 CrossRef CAS.
- Y. Chen, Q. He, Y. Zhao, W. Zhou, P. Xiao, P. Gao, N. Tavajohi, J. Tu, B. Li, X. He, L. Xing, X. Fan and J. Liu, Nat. Commun., 2023, 14, 8326 CrossRef CAS.
- Z. Chang, H. Yang, Y. Qiao, X. Zhu, P. He and H. Zhou, Adv. Mater., 2022, 34, 2201339 CrossRef CAS.
- X. Zhou, Y. Huang, B. Wen, Z. Yang, Z. Hao, L. Li, S.-L. Chou and F. Li, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2316914121 CrossRef CAS.
- Z. Sun, J. Zhao, M. Zhu and J. Liu, Adv. Energy Mater., 2023, 14, 2303498 CrossRef.
- X. Wang, Q. Wu, S. Y. Li, Z. M. Tong, D. Wang, H. L. L. Zhuang, X. Y. Wang and Y. Y. Lu, Energy Storage Mater., 2021, 37, 67–76 CrossRef.
- F. L. Yang, W. Zhang, Z. X. Chi, F. Q. Cheng, J. T. Chen, A. M. Cao and L. J. Wan, Chem. Commun., 2015, 51, 2943–2945 RSC.
- B. Kim, J. G. Lee, M. Choi, J. Cho and B. Park, J. Power Sources, 2004, 126, 190–192 CrossRef CAS.
- M. Moliner, F. Rey and A. Corma, Angew. Chem., Int. Ed., 2013, 52, 13880–13889 CrossRef CAS.
- S. Li, J. Li, M. Dong, S. Fan, T. Zhao, J. Wang and W. Fan, Chem. Soc. Rev., 2019, 48, 885–907 RSC.
- K. Gotoh, S. I. Ishimaru and R. Ikeda, Phys. Chem. Chem. Phys., 2000, 2, 1865–1869 RSC.
- K.-H. Schnabel, G. Finger, J. Kornatowski, E. Löffler, C. Peuker and W. Pilz, Microporous Mater., 1997, 11, 293–302 CrossRef CAS.
- G. Scott, R. W. Thompson, A. G. Dixon and A. Sacco, Zeolites, 1990, 10, 44–50 CrossRef CAS.
- Z. Li and Y.-W. Yang, Adv. Mater., 2022, 34, 2107401 CrossRef CAS.
- D. Fu, J. E. Schmidt, Z. Ristanović, A. D. Chowdhury, F. Meirer and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2017, 56, 11217–11221 CrossRef CAS PubMed.
- J. Grand, H. Awala and S. Mintova, CrystEngComm, 2016, 18, 650–664 RSC.
- B. You, Z. Wang, F. Shen, Y. Chang, W. Peng, X. Li, H. Guo, Q. Hu, C. Deng, S. Yang, G. Yan and J. Wang, Small Methods, 2021, 5, 2100234 CrossRef CAS.
- S. Ding, L. Chen, J. Liao, Q. Huo, Q. Wang, G. Tian and W. Yin, Small, 2023, 19, 2300341 CrossRef CAS PubMed.
- G. Dantelle, S. Beauquis, R. Le Dantec, V. Monnier, C. Galez and Y. Mugnier, Small, 2022, 18, 2200992 CrossRef CAS.
- J. Chen, H. Chen, S. Zhang, A. Dai, T. Li, Y. Mei, L. Ni, X. Gao, W. Deng, L. Yu, G. Zou, H. Hou, M. Dahbi, W. Xu, J. Wen, J. Alami, T. Liu, K. Amine and X. Ji, Adv. Mater., 2022, 34, e2204845 CrossRef PubMed.
- P. Hou, G. Chu, J. Gao, Y. Zhang and L. Zhang, Chin. Phys. B, 2016, 25, 016104 CrossRef.
- M. Zhu, Y. Huang, G. Chen, M. Lu, A. A. Nevar, N. Dudko, L. Shi, L. Huang, N. V. Tarasenko and D. Zhang, Chem. Eng. J., 2023, 468, 143585 CrossRef CAS.
- C. A. Marianetti, G. Kotliar and G. Ceder, Nat. Mater., 2004, 3, 627–631 CrossRef CAS PubMed.
- Z. H. Chen and J. R. Dahn, Electrochim. Acta, 2004, 49, 1079–1090 CrossRef CAS.
- J. Li, C. Lin, M. Weng, Y. Qiu, P. Chen, K. Yang, W. Huang, Y. Hong, J. Li, M. Zhang, C. Dong, W. Zhao, Z. Xu, X. Wang, K. Xu, J. Sun and F. Pan, Nat. Nanotechnol., 2021, 16, 599–605 CrossRef CAS PubMed.
- G. G. Amatucci, J. M. Tarascon and L. C. Klein, J. Electrochem. Soc., 2019, 143, 1114–1123 CrossRef.
- H. Liao, M. Cai, W. Ma, Y. Cao, S. Zhao, Y. Dong and F. Huang, Adv. Mater., 2024, e2402739 CrossRef.
- T. Fan, Y. Wang, V. K. Harika, A. Nimkar, K. Wang, X. Liu, M. Wang, L. Xu, Y. Elias, H. Sclar, M. S. Chae, Y. Min, Y. Lu, N. Shpigel and D. Aurbach, Adv. Sci., 2022, 9, e2202627 CrossRef PubMed.
- C. Sun, B. Zhao, J. Mao, K. H. Dai, Z. Y. Wang, L. B. Tang, H. Z. Chen, X. H. Zhang and J. C. Zheng, Adv. Funct. Mater., 2023, 33, 2300589 CrossRef CAS.
- J. Xia, N. Zhang, Y. Yang, X. Chen, X. Wang, F. Pan and J. Yao, Adv. Funct. Mater., 2022, 33, 2212869 CrossRef.
- X. R. Yang, C. W. Wang, P. F. Yan, T. P. Jiao, J. L. Hao, Y. Y. Jiang, F. C. Ren, W. G. Zhang, J. M. Zheng, Y. Cheng, X. S. Wang, W. Yang, J. P. Zhu, S. Y. Pan, M. Lin, L. Y. Zeng, Z. L. Gong, J. T. Li and Y. Yang, Adv. Energy Mater., 2022, 12, 2200197 CrossRef CAS.
- Y. Yan, Q. Fang, X. Kuai, S. Zhou, J. Chen, H. Zhang, X. Wu, G. Zeng, Z. Wu, B. Zhang, Y. Tang, Q. Zheng, H. G. Liao, K. Dong, I. Manke, X. Wang, Y. Qiao and S. G. Sun, Adv. Mater., 2024, 36, e2308656 CrossRef.
- Y. Yan, S. Zhou, Y. Zheng, H. Zhang, J. Chen, G. Zeng, B. Zhang, Y. Tang, Q. Zheng, C. Wang, C. W. Wang, H. G. Liao, I. Manke, X. Kuai, K. Dong, Y. Sun, Y. Qiao and S. G. Sun, Adv. Funct. Mater., 2023, 34, 2310799 CrossRef.
- W. Huang, Q. Zhao, M. Zhang, S. Xu, H. Xue, C. Zhu, J. Fang, W. Zhao, G. Ren, R. Qin, Q. Zhao, H. Chen and F. Pan, Adv. Energy Mater., 2022, 12, 2200813 CrossRef CAS.
- T. Fan, W. Kai, V. K. Harika, C. Liu, A. Nimkar, N. Leifer, S. Maiti, J. Grinblat, M. N. Tsubery, X. Liu, M. Wang, L. Xu, Y. Lu, Y. Min, N. Shpigel and D. Aurbach, Adv. Funct. Mater., 2022, 32, 2204972 CrossRef CAS.
- J. Qian, L. Liu, J. Yang, S. Li, X. Wang, H. L. Zhuang and Y. Lu, Nat. Commun., 2018, 9, 4918 CrossRef PubMed.
- S. Mao, Z. Shen, W. Zhang, Q. Wu, Z. Wang and Y. Lu, Adv. Sci., 2022, 9, e2104841 CrossRef.
- W. Ding, H. Ren, Z. Li, M. Shang, Y. Song, W. Zhao, L. Chang, T. Pang, S. Xu, H. Yi, L. Zhou, H. Lin, Q. Zhao and F. Pan, Adv. Energy Mater., 2024, 14, 2303926 CrossRef CAS.
- J. Yao, Y. Li, T. Xiong, Y. Fan, L. Zhao, X. Cheng, Y. Tian, L. Li, Y. Li, W. Zhang, P. Yu, P. Guo, Z. Yang, J. Peng, L. Xue, J. Wang, Z. Li, M. Xie, H. Liu and S. Dou, Angew. Chem., Int. Ed., 2024, 63, e202407898 CrossRef CAS.
- W. Zheng, G. Liang, H. Guo, J. Li, J. Zou, J. A. Yuwono, H. Shu, S. Zhang, V. K. Peterson, B. Johannessen, L. Thomsen, W. Hu and Z. Guo, Energy Environ. Sci., 2024, 17, 4147–4156 RSC.
- Z. Chang, Y. Qiao, H. Deng, H. Yang, P. He and H. Zhou, Joule, 2020, 4, 1776–1789 CrossRef CAS.
- Y. He, Y. Qiao, Z. Chang and H. Zhou, Energy Environ. Sci., 2019, 12, 2327–2344 RSC.
- Z. Chang, H. Yang, X. Zhu, P. He and H. Zhou, Nat. Commun., 2022, 13, 1510 CrossRef CAS.
- Z. Chang, Y. Qiao, H. Yang, H. Deng, X. Zhu, P. He and H. Zhou, Energy Environ. Sci., 2020, 13, 4122–4131 RSC.
- Z. Zhang, J. Wang, X. Sun, C. Sheng, M. Cheng, H. Liu, Y. Wang, H. Zhou and P. He, Adv. Funct. Mater., 2024, 2411409 CrossRef CAS.
- F. Pouraghajan, A. I. Thompson, E. E. Hunter, B. Mazzeo, J. Christensen, R. Subbaraman, M. Wray and D. Wheeler, J. Power Sources, 2021, 492, 229636 CrossRef CAS.
- J. Wandt, A. T. S. Freiberg, A. Ogrodnik and H. A. Gasteiger, Mater. Today, 2018, 21, 825–833 CrossRef CAS.
- B. L. D. Rinkel, D. S. Hall, I. Temprano and C. P. Grey, J. Am. Chem. Soc., 2020, 142, 15058–15074 CrossRef CAS.
- D. Guan, G. Ryu, Z. Hu, J. Zhou, C.-L. Dong, Y.-C. Huang, K. Zhang, Y. Zhong, A. C. Komarek, M. Zhu, X. Wu, C.-W. Pao, C.-K. Chang, H.-J. Lin, C.-T. Chen, W. Zhou and Z. Shao, Nat. Commun., 2020, 11, 3376 CrossRef CAS PubMed.
- L. Wang, J. Ma, C. Wang, X. Yu, R. Liu, F. Jiang, X. Sun, A. Du, X. Zhou and G. Cui, Adv. Sci., 2019, 6, 1900355 CrossRef.
- F. C. Zhang, N. Qin, Y. Z. Li, H. Guo, Q. M. Gan, C. Zeng, Z. Q. Li, Z. Y. Wang, R. Wang, G. Y. Liu, S. Gu, H. Huang, Z. L. Yang, J. Wang, Y. H. Deng and Z. G. Lu, Energy Environ. Sci., 2023, 16, 4345–4355 RSC.
- Z. Zhu, H. Wang, Y. Li, R. Gao, X. Xiao, Q. Yu, C. Wang, I. Waluyo, J. Ding, A. Hunt and J. Li, Adv. Mater., 2020, 32, 2005182 CrossRef CAS.
- J. G. Swallow, W. H. Woodford, F. P. McGrogan, N. Ferralis, Y.-M. Chiang and K. J. Van Vliet, J. Electrochem. Soc., 2014, 161, F3084 CrossRef CAS.
- I. Kochetkov, T.-T. Zuo, R. Ruess, B. Singh, L. Zhou, K. Kaup, J. Janek and L. Nazar, Energy Environ. Sci., 2022, 15, 3933–3944 RSC.
- Z. Dai, Z. Li, R. Chen, F. Wu and L. Li, Nat. Commun., 2023, 14, 8087 CrossRef CAS PubMed.
- S. Zhao and F. Huang, ACS Nano, 2024, 18, 1733–1743 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee04370g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |