
Advancing micro-nano supramolecular assembly mechanisms of natural organic matter by machine learning for unveiling environmental geochemical processes†
Ming
Zhang
 a,
Yihui
Deng
b,
Qianwei
Zhou
c,
Jing
Gao
b,
Daoyong
Zhang
*a and
Xiangliang
Pan
*b
a,
Yihui
Deng
b,
Qianwei
Zhou
c,
Jing
Gao
b,
Daoyong
Zhang
*a and
Xiangliang
Pan
*b
aCollege of Geoinformatics, Zhejiang University of Technology, Hangzhou, 310014, P. R. China. E-mail: zhangdaoyong@zjut.edu.cn
bCollege of Environment, Zhejiang University of Technology, Hangzhou, 310014, P. R. China. E-mail: panxl@zjut.edu.cn
cCollege of Computer Science and Technology, Zhejiang University of Technology, Hangzhou 310023, P. R. China
First published on 19th December 2024
Abstract
The nano-self-assembly of natural organic matter (NOM) profoundly influences the occurrence and fate of NOM and pollutants in large-scale complex environments. Machine learning (ML) offers a promising and robust tool for interpreting and predicting the processes, structures and environmental effects of NOM self-assembly. This review seeks to provide a tutorial-like compilation of data source determination, algorithm selection, model construction, interpretability analyses, applications and challenges for big-data-based ML aiming at elucidating NOM self-assembly mechanisms in environments. The results from advanced nano-submicron-scale spatial chemical analytical technologies are suggested as input data which provide the combined information of molecular interactions and structural visualization. The existing ML algorithms need to handle multi-scale and multi-modal data, necessitating the development of new algorithmic frameworks. Interpretable supervised models are crucial owing to their strong capacity of quantifying the structure–property–effect relationships and bridging the gap between simply data-driven ML and complicated NOM assembly practice. Then, the necessity and challenges are discussed and emphasized on adopting ML to understand the geochemical behaviors and bioavailability of pollutants as well as the elemental cycling processes in environments resulting from the NOM self-assembly patterns. Finally, a research framework integrating ML, experiments and theoretical simulation is proposed for comprehensively and efficiently understanding the NOM self-assembly-involved environmental issues.
Environmental significanceSupramolecular self-assembly of NOM profoundly influences the fate of NOM and pollutants in large-scale complex environments. The difficulty in recognizing NOM self-assembly and its environmental processes keeps increasing due to the diversity of NOM molecular structures, the growing involvement of pollutants in self-assemblies and the complexity of geochemical conditions. ML is essentially advanced in environmental research through fast analyzing a large amount of input data and customizing the services of algorithms to different circumstances. With the assistance of ML, the environmental process mechanisms of emerging pollutants, bioavailability of pollutants/NOM and related global issues can be approached and unveiled. Research frameworks coupling ML, experiments and theoretical simulation are proposed to comprehensively, efficiently and innovatively understand the NOM self-assembly-involved environmental processes. |
1. Introduction
Natural organic matter (NOM) is a complex supramolecular mixture widely distributed in environments, including water, sediments and living organisms.1,2 One of the remarkable features of NOM is its ability to form self-ordered structures by virtue of high convergence and spontaneous assembling of relatively simple NOM subunits.3 Pollutants such as heavy metals and organic micropollutants may participate in or mediate the assembling processes and structures of NOM through non-covalent interactions.4–8 Existing and functioning at and beyond the nanoscale,9 those self-assemblies significantly change geochemical processes and effects of supramolecular self-assemblies in environments (Fig. 1(a) and (b)). Deciphering those complex nanoscale structures has always been crucial for evaluating and predicting the fate of NOM and the eco-impact of pollutants. In environmental geochemistry, elucidating supramolecular assembly patterns and structures has long depended on experimental exploration, instrumental analyses, (i.e., spectroscopic characterization and microscopic observation) and theoretical simulation methods (typically, quantum chemical calculations such as molecular dynamics (MD) simulations) (Fig. 1(c)).7,8,10–14 However, the complexity of NOM components and the diverse chemical conditions of environmental media result in vastly varied structural patterns and properties of supramolecular assemblies, which need substantial experiment studies and structure interpretations at the nanometer or submicron scale. Thus, it is difficult to thoroughly understand NOM supramolecular assemblies merely with the above-mentioned approaches, which could be either time-consuming or lack universality.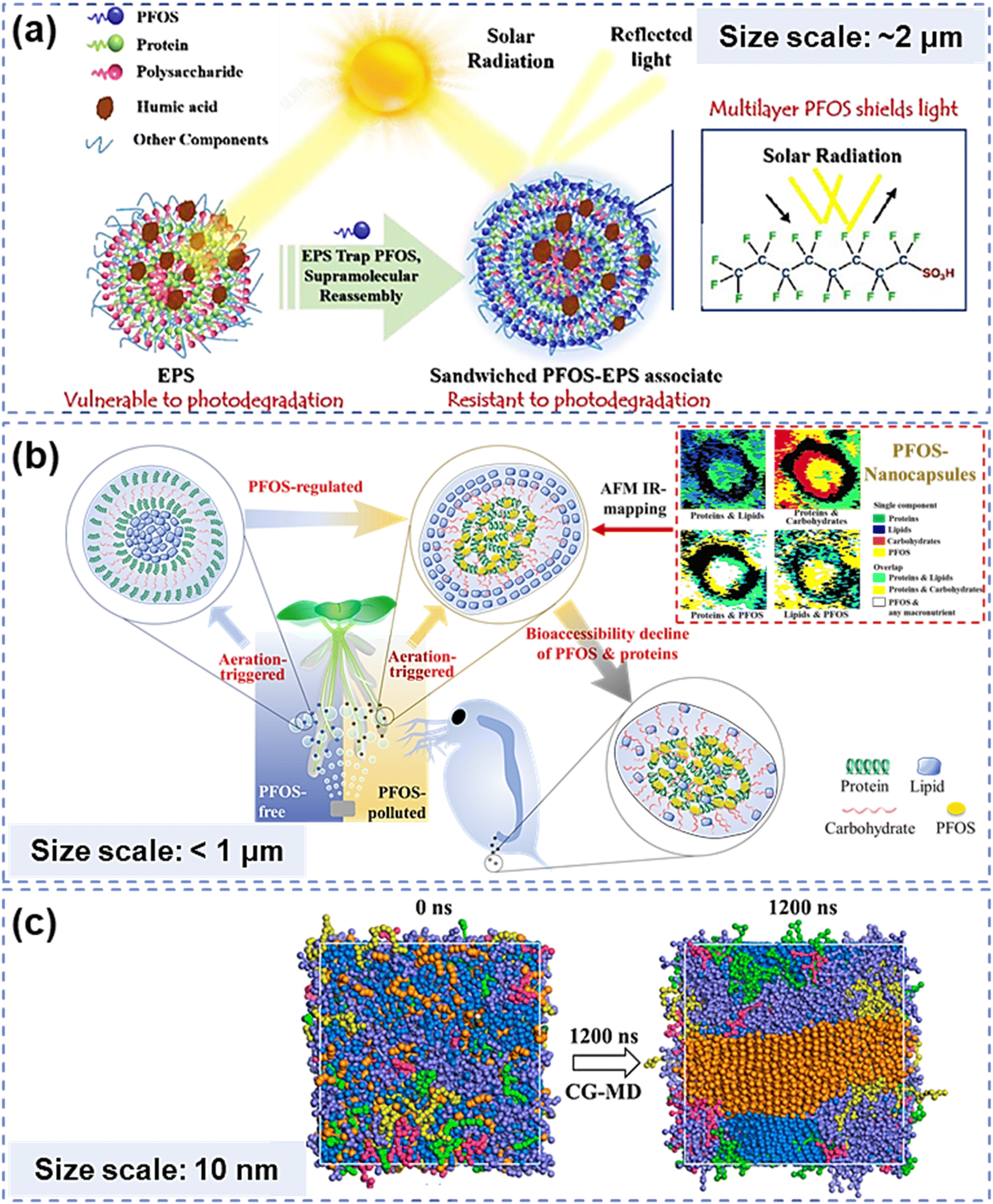 | ||
| Fig. 1 NOM self-assembly structures and environmental behaviors obtained from experiments, instrumental analyses or simulation approaches. (a) Sandwiched supramolecular assembly between exopolymers and PFOS obtained by AFM IR-mapping and relevant photo-shield effects.13 (b) Aeration-triggered and PFOS-reconstructed NOM assembly and relevant bioaccessibility decline effects.14 (c) Orderly multilayer structure of NOM self-assembly obtained by coarse-grained dynamic simulation. The bead types and color: water – blue, LHA – purple, lipid – orange, peptide – red, carbohydrate – green, and lignin – yellow.10 | ||
With computational capabilities and versatile algorithms, machine learning (ML) may become an essential alternative through fast analyzing a large amount of input data (i.e., analytical results of NOM self-assembly structures attained from multi-techniques) and thus customizes the services of algorithms to the circumstances (specifically, prioritizing the environmental behaviors of assemblies).15 Integrating ML into the existing research frameworks holds the promise of substantially improving data analysis efficiency, elaborating complex chemical processes, and revealing new mechanisms of interaction between supramolecular self-assembly components. Currently, supramolecular assembly studies by ML predominantly focus on biological macromolecules such as proteins, amino acids, DNA and RNA, particularly for behavior prediction, component interaction and gene expression (Fig. 2). In the natural environment, the NOM components involved in supramolecular self-assembly cover a wide range of types, which include but are not limited to extracellular polymers13,16 or biological macromolecules such as polysaccharides, proteins, lipids, nucleic acids17–20 and humic substances.21–23 These NOM components spontaneously interact with one another and aggregate under thermodynamic driving forces through non-covalent interactions such as hydrogen bonding, hydrophobic interactions, electrostatic interactions, π–π stacking and van der Waals force.9,21,24–27 The environmental geochemistry field shares commonalities with other disciplines (i.e., materials, biology, medicine and so forth) in terms of universal self-assembly mechanisms, warranting the adoption of cross-disciplinary research approaches particularly such as ML in common. With the assistance of ML, fundamental understanding will be extended which helps close the gap between the experimental or simulation recognition of NOM supramolecular self-assemblies and their performance in real environments. Relevant studies also confirm the development tendency of supramolecular self-assembly research and applications (Fig. 3).
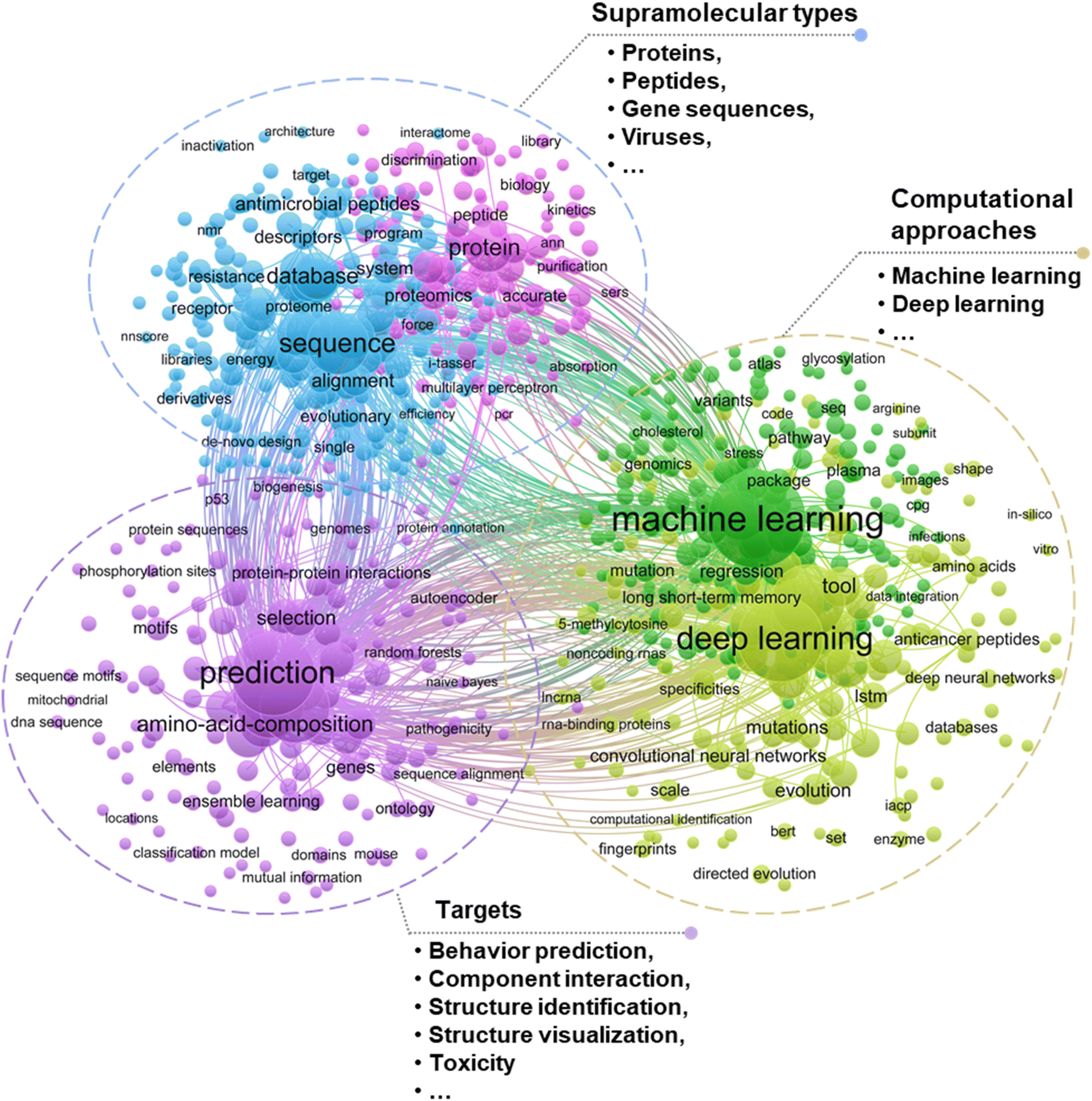 | ||
| Fig. 2 Literature cluster analysis using ML in supramolecular assembly research based on the Web of Science Core Collection database. Bubble size reflects the number of occurrences, and bubble colors indicate different clusters classified according to co-occurrence analysis. Search details are provided in Text S1.† | ||
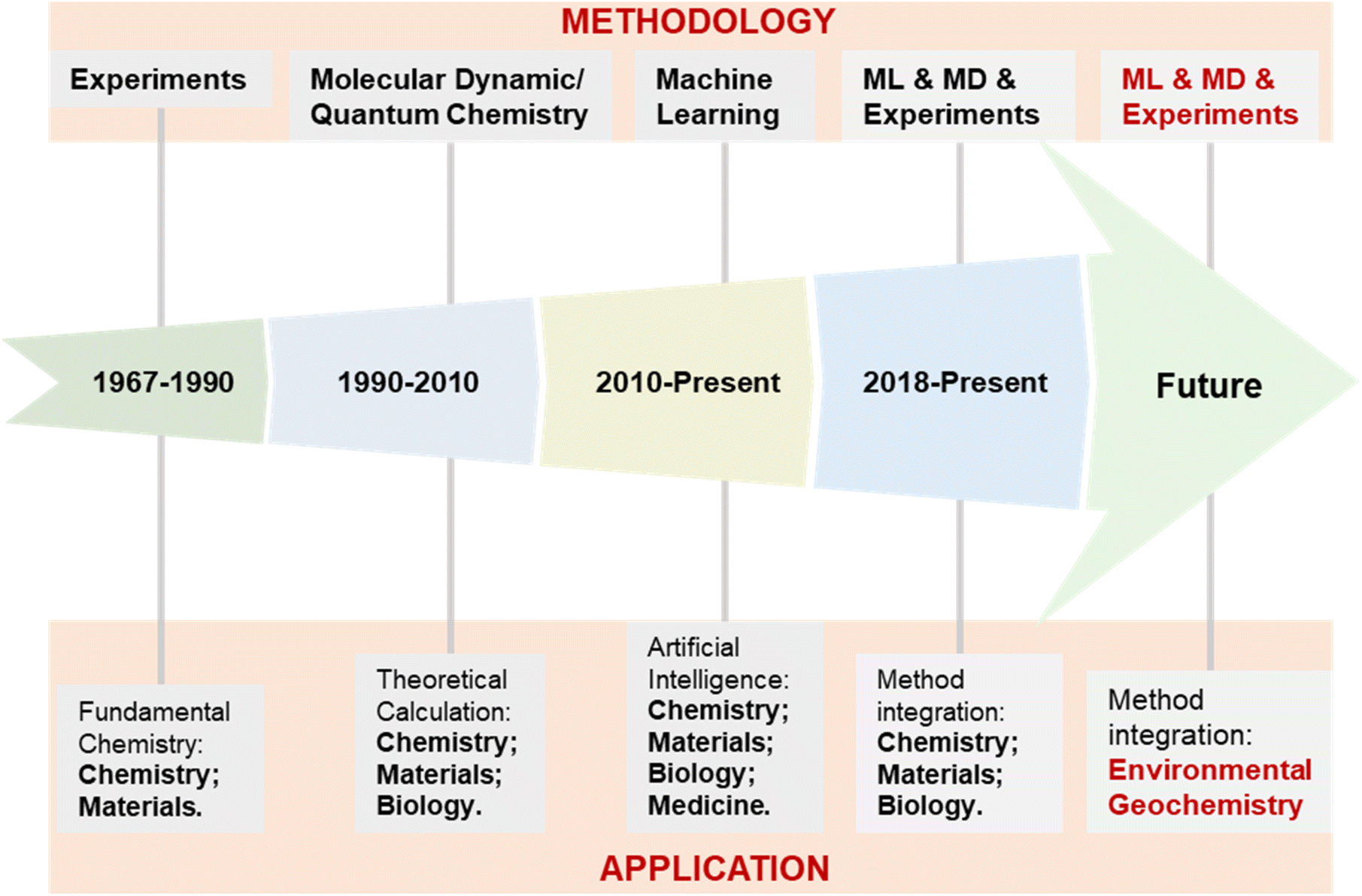 | ||
| Fig. 3 Development process of the exploration methodology and application fields of supramolecular self-assembly. | ||
Given the context mentioned above, this critical review first suggested the input data sources applicable to developing predictive models especially for micro-nanoscale NOM supramolecular self-assemblies. Then, algorithm selection, model construction and model interpretability analyses were depicted and interpretable supervised ML was particularly focused on. Recent studies on supramolecular self-assembly involving ML from biology, materials, chemistry or relevant fields as well as their commonalities within environmental geochemistry were discussed. Subsequently, prominent applications and major challenges were proposed for ML-interpreting or predicting the geochemical behaviors and eco-impacts involving NOM or NOM–pollutant assemblies in environments. Finally, a combination framework of ML, theoretical simulation and experimental research was suggested for the NOM self-assembly study in the environmental fields.
2. Selection of data sources from micro-nano scale spatial chemical analyses for ML of supramolecular micro-nano self-assembly
2.1. Guide for data source selection
To interpret or predict supramolecular self-assembly processes, structures and properties by ML, the featured input data must provide multi-dimensional and multi-perspective information depicting organized nano-structures. For this purpose, ML models can accept the input data formats including (i) descriptor data calculated from physicochemical parameters which indirectly reflect the molecular properties of supramolecular assembly systems, (ii) commonly adopted inputs of spectroscopic and mass spectrometric data which directly characterize the vibration of functional groups and their molecular interactions in supramolecular assemblies, and (iii) nanometer-submicron-scale spatial spectral-imaging data which accurately depict the distribution of each component in assembled structures.Notably, not all data types are equally informative for ML in this context, and it is crucial to integrate heterogeneous data from multiple sources and choose input data types rationally based on specific research requirements. For instance, while being valuable for providing morphological and elemental distribution information, the data from electron microscopy and energy spectroscopy imaging cannot directly depict supramolecular structures. As such, these data types can serve as supplementary data to support ML of NOM supramolecular assembly but should not be used as sole data sources. Besides, the models require substantial and sufficient training datasets which are particularly important when the ML tasks are subjected to environmental or geochemical condition changes. Adequate data should be available for each condition not only to enable the model to capture subtle changes in assemblies but also to enhance the model's generalization ability.28
2.2. Molecular descriptors as widely used input data for compound assembly ML
Molecular descriptors, being essential for characterizing molecular structures, physicochemical properties and other relevant information, have been widely applied in cheminformatics, drug design and materials science as input features for ML models. To make them machine-readable during the modeling process, chemical structures need to be converted into numerical or binary vectors.29 These descriptors encompass geometric descriptors based on three-dimensional conformational information,30 physicochemical property descriptors directly representing molecular attributes (i.e., molecular weight, hydrogen bond donors and acceptors, polar surface area and ionization potential),31,32 topological descriptors describing topological structures and branching of molecules (i.e., molecular connectivity index)29 and quantum chemical descriptors derived from high-precision calculations (i.e., frontier orbital energies, condensed Fukui index, electrostatic potential, polarizability and electronic structure parameters obtained through Density Functional Theory (DFT)).33,34 Functioning as digital signatures, molecular fingerprints are capable of mapping molecular structures to binary bit strings or encoding information of atomic composition, chemical bonds and spatial arrangements.35–38Data extracted from MD simulations can also be considered descriptors in a broader sense. While conventional molecular descriptors are mostly computed based on static structural information, MD simulations capture the dynamic behavior of molecules. By constructing computational models with assembly units and applying appropriate force fields and boundary conditions, MD simulates and reconstructs the spatiotemporal evolution trajectories of self-assembling systems. From the simulation trajectories, detailed information can be obtained such as atomic-level coordinate data, energy, temperature and pressure.30,39,40
When molecular descriptors are utilized as input data for ML, we should be aware of two critical factors. The potential redundancy and irrelevance among descriptors possibly introduce unwanted complexity to ML models. Feature selection needs to be performed to remove unimportant features.41–43 Furthermore, the presence of missing values among descriptors poses challenges that must be addressed through appropriate data preprocessing.42 For numerical descriptors, missing values can be handled either through simple statistical measures (mean or median imputation) or through similarity-based imputation where values are inferred from molecules based on their structural or property similarities. For categorical molecular descriptors (i.e., atom types and functional group types), there are three main preprocessing approaches: mode imputation where missing entries are replaced with the most frequent category, introducing a new “missing” category as a meaningful category in itself to treat the missing values,44 and applying the similarity-based imputation approach as used for numerical descriptors. Those categorical descriptors need to be converted into numerical formats through encoding techniques such as one-hot encoding or label encoding before being fed into ML models.45
2.3. Spectral information as a fundamental chemical data source
Comprehensive information on molecular structures and components can be obtained from various spectroscopic techniques, which lay the foundation for recognizing self-assembling molecules and their aggregation behaviors. Mass spectrometry, such as liquid chromatography-mass spectrometry (LC-MS) and gas chromatography-mass spectrometry (GC-MS), enables precise identification and quantitative analysis of molecular components in self-assembling systems in terms of molecular weight, molecular formulae and structures.46–48 MS demonstrates robust performance in detecting single compounds, but complete signal assignment in natural complex mixtures remains challenging. Nonetheless, ML is capable of grouping isotope peaks, determining molecular formulae and, subsequently, analyzing the chemical composition of NOM.49 Researchers have successfully elucidated the structure–activity relationships between peptide fragments and their biological activities via applying ML to the LC-MS analysis results of peptide assemblies.46 Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR-MS) detects various organic molecules in unknown compounds and their change trends (Fig. 4(a)).50,51 Based on the feature data of elemental ratios, double bond equivalents and aromaticity indices, it is possible to interpret and predict the NOM behaviors with respect to spontaneous combination, photochemical processes and redox reactions under specific environmental conditions.52,53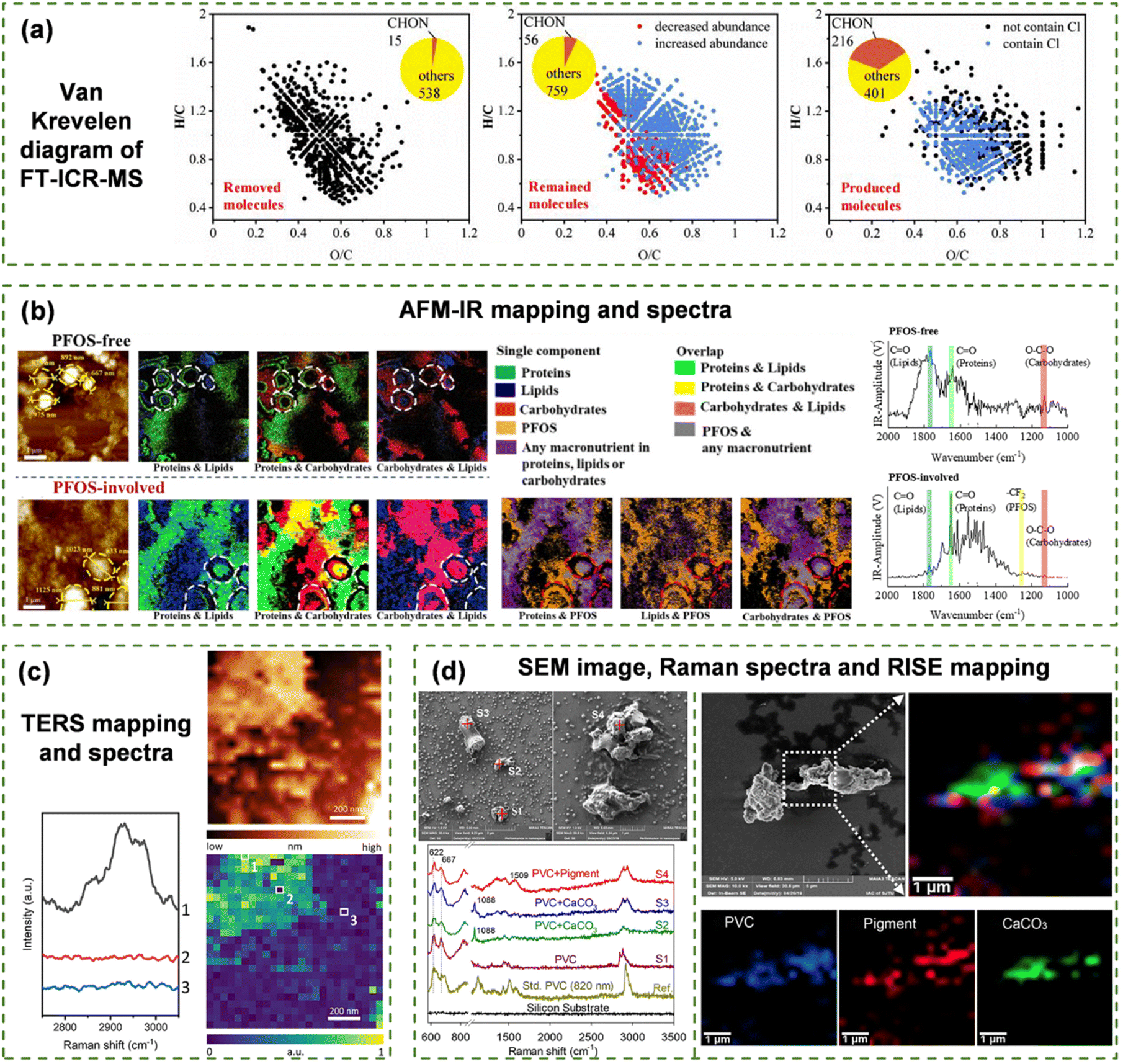 | ||
| Fig. 4 Feature data for supramolecular assemblies. (a) Van Krevelen diagrams of Carbonyl molecule distribution derived from FT-ICR-MS analysis.51 (b) Morphology and structures of PFOS-free NOM and PFOS-involved NOM assemblies shown by AFM images and IR mapping.14 (c) TERS analysis of phospholipid assemblies, showing C–H stretching intensity images and corresponding discontinuity spectra.58 (d) RISE analysis of RPP, showing SEM images, Raman spectra of corresponding points (left) and Raman mapping (right).59 | ||
As a non-invasive analytical method, nuclear magnetic resonance (NMR) spectroscopy characterizes the three-dimensional structure of supramolecular assemblies, internal molecular dynamic processes and intermolecular interactions.48,54–56 Based on the NMR data, ML may establish spectral-structure relationships between the components of supramolecular self-assemblies. For instance, 1329 NMR data sets from 100 proteins along with their sequences were used as deep learning (an important branch of ML) input data for the assignment of protein backbones and side-chain chemical shifts. This chemical shift assignment provided atomic-level insights into protein assembly, subsequently enabling the reconstruction of structures for these protein assemblies.57
Infrared (IR) and Raman spectroscopy, owing to their high or even extreme sensitivity to both intra- and intermolecular bonds, have been regarded as powerful tools to both qualitatively and quantitatively identify supramolecular assembly structures and chemical compositions.60–62 The change in dipole moment of molecules during a given normal mode of vibration is always detected by IR spectroscopy whilst the total symmetric vibrations of large molecules having a symmetry center need to be resorted to Raman spectroscopy.63 Through tracking characteristic or fingerprint peaks of organic compounds in complex biopolymers (e.g., carbohydrates, lipids, and proteins),64 IR or Raman spectroscopy is used to analyse the biochemical changes in the structure (in particular, secondary structures) and content of bio-macromolecules such as proteins and glucose.65 In the domain of environmental science, those spectral data may serve as input data for ML models to identify organic polymers.66,67
Before being input into ML models, those spectral data must undergo preprocessing to ensure data quality and consistency, including polynomial fitting for baseline correction, smoothing algorithms for noise reduction, normalization to adjust spectral data to the same intensity level and first-order derivative processing to highlight characteristic peaks in the spectra.66–69 For data implementation in the convolutional neural network (CNN) which is typically designed for image processing, two approaches are often used for converting spectral data into images by which feature extraction can be well enhanced: transforming spectra into spectrograms using short-time Fourier transforms70 or converting spectra into scalogram images using wavelet transforms.71
The ML models that were built according to the near-IR reflectance spectroscopic results of pectin, cellulose, lignin, and phenols in cuticles and cell walls have been reported to accurately capture the content and structure variation of these assembled macromolecules.72 The corresponding spectral features, when being extracted by ML, can establish correlations between characteristic spectra and the chemical context.73,74 In addition, ultraviolet-visible spectroscopy can observe the transition process of supramolecular self-assemblies from monomeric to aggregated states as well.75,76 It should be noted that mere spectroscopic information will never depict the spatial organization and configuration of the assembled organic compounds not to mention at the nanometer or submicron scale, which, however, is key to the ML of NOM self-assembly.
2.4. Micro-nano scale spatial imaging as an indispensable data source
Morphological imaging techniques reveal the structure of supramolecular assemblies at the nanometer or submicron scale. While traditional optical microscopy techniques are unable to resolve features below 200 nm due to the light diffraction limit, some ultrahigh-resolution techniques enable the detailed characterization of supramolecular assemblies below 100 nm.77,78 Transmission electron microscopy (TEM) has significantly contributed to studying small molecules, polymers, and biomolecules self-assembly by facilitating the discovery, visualization, and quantification of structures and dynamic processes.79 Integrating these imaging data with ML enables in-depth analysis of, for instance, quantifying nanoparticle distribution in polymer aggregates.80 Liquid-phase TEM videos with ML unveiled dynamic self-assembly processes of nanoparticles in solution.81 The assemblies mediated by metal ions or metal complexes through non-covalent bonds with organic ligands have been characterized by scanning tunneling microscopy (STM) which tunnels current to image conductive sample surfaces under ultra-high vacuum conditions.26,82,83 STM generates a series of imaging data illustrating the self-assembly structures formed by molecules with identical functional groups (i.e., polycyclic aromatic hydrocarbons) adsorbing on the metal (i.e., Ag(1 1 1)) surfaces.84,85 These data sets are suitable for ML applications aimed at predicting supramolecular assembly structures involving heavy metals. As the scanning probe technique is primarily used for analyzing the topology and surface morphology at the nanometer or submicron scale, atomic force microscopy (AFM) has been successfully adopted to predict the novel self-assembled structures of nanocomposite films via training the ML models with 595 AFM images.86 In particular, with the data from the high-speed AFM (HS AFM) characterization of protein self-assembly processes, ML could extract the information of protein spatial positions and orientations and finally elaborate the self-assembly and dynamic behaviors on solid surfaces.87,88 Spatial imaging data require specific preprocessing steps before being fed into CNNs, including image normalization, noise reduction and standardization of image dimensions and pixel values. However, the abovementioned spatial imaging data can only be used for ML when the self-assembly systems are composed of single or simple components due to the lack of chemical or fingerprint evidence for compound recognition. The limitations hinder the ML study of NOM self-assembly under real and complicated geochemical and environmental conditions that may be overcome by simultaneously utilizing spectroscopic and spatial imaging techniques.2.5. Micro-nano scale spatial chemical analyses as suggested data sources of NOM self-assembly ML with environmental relevance
Considering the heterogeneity of NOM structures and the complexity of inter-molecular interactions at different environmental chemistries, the input data of ML models must reflect nanometer or submicron-scale spatial chemical features. Molecular descriptors, spectroscopic analyses or morphological imaging techniques can only provide partial information as previously discussed. The integration of chemical and micro-spatial analytical techniques can provide the most appropriate data sources of ML for multidimensional and comprehensive mechanism interpretation of NOM self-assembly. Advanced spectroscopic mapping techniques, such as IR scattering-type scanning near-field optical microscopy,89 AFM-IR spectroscopy (Fig. 4(b)),90 tip-enhanced Raman spectroscopy (TERS) (Fig. 4(c))91 and Raman imaging and scanning electron (RISE) microscopy (Fig. 4(d)),92 allow the in situ visualization of spatial distribution of chemical groups in supramolecular self-assembled systems at the nanoscale (Fig. 1(b)). The as-attained data are sufficient to develop ML models for understanding and predicting NOM supramolecular assembly processes and behaviors. The energy-dispersive X-ray spectroscopy (EDS) mapping technique coupled with electron microscopy (typically, SEM-EDS and STEM-EDS)93,94 rapidly qualitatively and quantitatively detects the elemental compositions on the surface of samples, showing elemental migration and enrichment behaviors during the assembly processes.14 Nevertheless, the analytical results from those techniques can only be adopted as supportive data for ML input due to the lack of molecular chemical information.In particular, to develop ML models for elucidating NOM self-assembly behaviors involving micropollutants (i.e., per- and polyfluoroalkyl substances, PFAS), the main data source is suggested to include molecular chemical micro-spatial results of AFM-IR-mapping. AFM-IR-mapping presents the nanoscale distribution of NOM and PFAS in self-assembly aggregates by mapping the characteristic IR peak of each NOM component and the fingerprint C–F bond of PFAS. In this scenario, the SEM-EDS-mapping data could be adopted to map the F element in samples to further verify the PFAS spatial distribution. When the supramolecular assembly involves inorganic pollutants (i.e., heavy metal nanoparticles), the combination of spectroscopic (mapping) analyses is needed to obtain the input data depicting chemical interactions between organics and inorganics as well as their spatial distribution. Then, the input data for ML may be collected from single-particle inductively coupled plasma mass spectrometry,95 or high-resolution imaging of elemental and isotopic distributions using nanoscale secondary ion mass spectrometry96 in addition to analytical methods for identifying typical NOM components. It is worth noting that the NOM self-assembly is dynamic and environmentally dependent since self-assembly processes and structures are highly influenced by environmental geochemical conditions such as temperature, pH and ionic strength. Consequently, the input data for ML models are supposed to be acquired from the analytical techniques in the targeted geochemical circumstances such that the output of ML could be of environmental relevance and significance.
3. Algorithms, models and interpretability analyses favored by ML of NOM self-assembly
3.1. Guide for algorithm selection
When selecting an appropriate ML algorithm for supramolecular self-assembly research, two key factors need to be considered. On the one hand, the choice among regression, classification, clustering and dimensionality reduction algorithms is made given the specific issues of supramolecular assembly to be solved (Table 1). On the other hand, a suitable algorithm is determined based on the input data characteristics including scale, dimensionality, quality and types (i.e., image data, molecular graphs, molecular sequences and tabular data). To be specific, traditional ML algorithms such as logistic regression (LR), random forest (RF) and support vector machine (SVM) are more sensitive to numerical data; convolutional neural networks (CNNs) are better suited for reading and processing image data; graph neural networks (GNNs) are adept at handling molecular graph data (Table 1). These neural networks are better suited for large-scale datasets.| Self-assembly component | Input | Outputa | Data pointsb | ML task | Type of task | Algorithmc |
|---|---|---|---|---|---|---|
| a EAC: electron accepting capacity; EDC: electron donating capacity; ETC: electron transfer capacity. b —: not mentioned. c GBT: gradient boosting tree; DT: decision tree; KRR: kernel ridge regression; PCA: principal component analysis; NMF: non-negative matrix factorization; GMM: Gaussian mixture model; PLS: partial least squares; MLR: multiple linear regression; BPNN: back propagation neural network. | ||||||
| Dipeptide hydrogels100 | Molecular descriptors | Whether or not a hydrogel is formed | 2304 | Prediction of hydrogel formation | Classification | RF, GBT, and LR |
| Blends of nanoparticles, polymers and small molecules86 | Assembly nature and environmental conditions | Parameters measured from AFM images describing assembly morphology | 595 | Prediction of new nanocomposite morphology | Regression | GBT, RF, GBR, SVM, LGER, and KRR |
| Proteins87 | HS AFM images | Protein location and orientation | — | Protein identification and localization | Classification | CNN (U-Net) |
| Protein spatial distribution in assembly | Protein identification in the assembly | Clustering | PCA, NMF, and GMM | |||
| Proteins57 | NMR spectra and protein sequences | Signal positions, resonance assignments, structure proposals, and protein structures | 100 protein sequences, 1329 NMR spectra | Prediction of protein structures | Regression and classification | ARTINA (ResNet, GNN, and GBT) |
| Assemblies of metals with organic ligands85 | Molecular diagrams of organics and metal substrate types | Whether or not there is a target attribute associated with the assembly | 30 | Prediction of molecular self-assembly structures | Classification | GNN, SVM, RF, and DT |
| DNA oligonucleotides and melamine cyanurate107 | Fluorescence microscopy images | Mg2+ concentration | 7000 | Analysis of correlation between the supramolecular structure and Mg2+ concentration | Classification | CNN (VGG16, VGG19, and ResNet50) |
| Humic substances108 | Element contents, chemical group contents, and experimental optical parameters | Redox activity of humic substances: EAC, EDC, and ETC | 144 | Relationship between the structure and redox activity | Regression | PLS, MLR, and BPNN |
| NOM53 | FT-ICR-MS results | Reactive or unreactive | — | Understanding whether the constituent molecules of NOM react after UV irradiation | Classification | LR, SVM, RF, and XGBoost |
Based on the mapping relationships to be established and the data availability, the widely-known three categories of ML algorithms – supervised, unsupervised and semi-supervised may be adopted to interpret the mechanisms and structures of NOM self-assembly. Supervised learning aims to learn the mapping relationship between input and output data. The output can either be continuous numerical values (regression algorithms) such as predicting the aggregation propensity (AP) of peptides (Fig. 5(a))97 or discrete or categorical (classification algorithms) such as the reactivity53,98,99 or formability97,100 of NOM under specific environmental conditions (Fig. 5(a)). Most of the issues of NOM supramolecular self-assembly can be addressed by supervised learning, including but not limited to predicting assembly structures from the input data of elemental and component composition, the thermal stability, photoconductivity and redox capacity from the input data of assembly structures and environmental chemistry, as well as the vertical transportation rate from the input data of assembly size and density. Unsupervised learning is not associated with specific prediction tasks but focuses on identifying trends, patterns or clusters of input data alone. Dimensionality reduction algorithms were used to reduce the high-dimensional descriptors obtained from MD simulations with a large amount of spatiotemporal details to the low-dimensional data which were conducive for visualization and analysis.101–103 Cluster analysis divides data into multiple groups which can be used to identify data association patterns. For example, clustering techniques were used to identify ordered and disordered regions in the phase transition process from MD simulation data of lipids at different temperatures. The results revealed the temperature dependence of lipid phase changes.104 Unsupervised learning should contribute to data exploration and pattern recognition when the input data of ML for interpreting NOM self-assembly structures are of high dimensionality. Typically, it can be used to identify self-assembly pathways using MD simulation data or discover clustering relationships in high-dimensional IR-mapping data which directly correspond to assembly structures. Semi-supervised learning can be useful when there are abundant input data but limited corresponding output data though its applications are currently not extensive. To further improve the prediction performance, ensemble methods that combine multiple individual models have been widely adopted, such as bagging (i.e., RF), boosting (i.e., XGBoost and LightGBM) and stacking. Ensemble modeling techniques can effectively reduce overfitting and improve model generalization by aggregating predictions from multiple base models.105 For example, integrating various simple weak ML algorithms such as extremely randomized trees, adaptive boosting, RF, cascade forest, LightGBM and XGBoost is used to predict whether small molecule compounds would interact with proteins.106
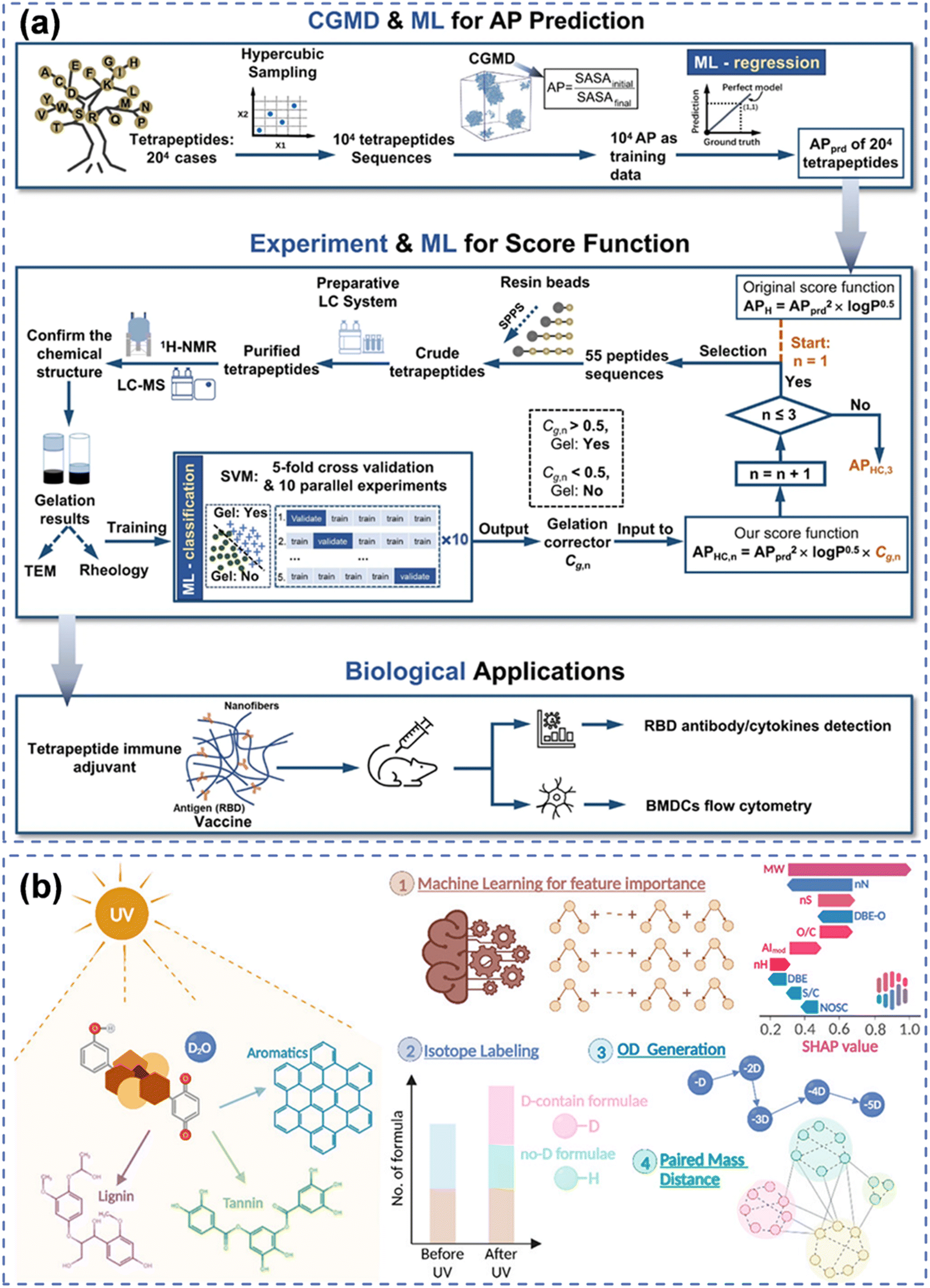 | ||
| Fig. 5 ML methods. (a) An integrated framework adopting MD simulation and experimentation results as data sources of ML for predicting and discovering tetrapeptide hydrogels.97 (b) Using isotope-labeled FT-ICR-MS as input data for interpretable ML and paired mass distance networks to reveal photo-reactivity and photo-transformation of NOM.53 | ||
3.2. Optimal model construction
Supervised learning facilitates the establishment of relationship models between variables and results. These models predict the assembly structures and quantify structure–property–effect relationships, which enhance researchers' understanding of NOM supramolecular assembly patterns. Herein, we specifically focus on discussing strategies for building supervised learning models. The model training process begins with dataset partitioning, where the training set is used for model fitting and the validation set aids in hyperparameter tuning and overfitting prevention. K-Fold cross-validation ensures the robustness of models across different data subsets and mitigates overfitting issues.86,98,108,109 Monte Carlo cross-validation (MCCV) offers a more flexible approach by randomly partitioning the dataset multiple times, enabling arbitrary validation iterations and potentially better handling imbalanced datasets.110,111 However, the limitations of MCCV might be potential sample utilization imbalance, reduced result reproducibility without fixed random seeds and higher computational costs.112,113Apart from cross-validation, several complementary techniques are essential to prevent the overfitting of ML models. Regularization methods (in particular, L1 and L2 regularization) add penalty terms to the loss function such that the model complexity is controlled. L1 regularization promotes sparse solutions by potentially setting some weights to zero.114 L2 regularization prevents excessive weight magnitudes by penalizing large weights quadratically, ensuring even consideration of all features by models.114 Dropout randomly deactivates neurons during training, forcing the network to learn robust features.115 Early stopping monitors the validation performance during training and halts the process when performance begins to degrade, which effectively prevents overtraining.114,116
The optimal average performance across validation iterations guides hyperparameter selection. With the optimal values corresponding to the best model, hyperparameters are external configurations whose slight adjustments can significantly improve or impair the model performance. However, manual hyperparameter optimization is computationally expensive and not optimal. More advanced methods employ automatic optimization algorithms to find the best hyperparameter configurations such as Bayesian optimization,117,118 random search,119,120 grid search120–122 and particle swarm optimization.123,124
Transfer learning shows great potential for leveraging the knowledge gained from the large and related datasets in enhancing the predictive performance based on comparatively small target datasets, through which the data scarcity problems could be solved. A pre-trained model refers to a deep neural network that has been trained on a large dataset for a general task typically using architectures such as GoogLeNet or ResNet.125 The early convolutional layers of that model learn basic visual feature extractors (i.e., edges, textures, etc.).126 Fine-tuning enables the adaptation of CNN-based models to new scenarios in two main ways: retraining the last few layers to recognize new types of patterns while keeping the early feature-detecting layers unchanged126 and using a smaller learning rate to carefully update the models without losing their previously learned knowledge.127
The success of transfer learning relies on finding the underlying similarities between the source and target domains. As demonstrated in Raman spectroscopy applications, the fundamental basis for transfer learning is built upon the similar hierarchical features between those Raman spectral patterns and the image patterns learned and recognized by pre-trained CNNs.71 The early layers of these models, which detected basic features such as edges and textures, could be effectively repurposed for analyzing spectral patterns and molecular interactions. This principle of leveraging similarities can be applied to molecular systems. For instance, an initial model was trained on a large non-transmembrane protein dataset and then transferred to a small transmembrane protein dataset to predict the contact points between protein chains.128 The premise of that transfer was the structural similarities between those two types of proteins. Here is another example. A pre-trained model was obtained using a large protein dataset calculated with low-precision quantum mechanical methods and then transferred to a small fragmented protein dataset calculated with high-precision methods to accelerate quantum mechanical calculations.129 It is the universality of quantum mechanical calculation principles that made the transfer learning possible. For fine-tuning, optimization algorithms such as stochastic gradient descent,130 stochastic gradient descent with momentum,131 adaptive moment estimation,132 adadelta133 and adagrad134 are commonly employed to effectively transfer knowledge while preserving relevant features. This approach may be useful when NOM assembly data are limited. It allows the model to gain insights into the influence of non-covalent intermolecular interactions on the assembly structures via first learning from a broader range of supramolecular systems. The knowledge from the pre-trained models can then be transferred to specific NOM assembly datasets through fine-tuning.
The choice of model evaluation metrics depends on specific problem types. Generally, for regression problems, the coefficient of determination (R2) and root mean square error are commonly used. For classification problems, the F1 score, area under the curve (AUC) and accuracy are typical metrics. For supramolecular assembly structure prediction, specific evaluation metrics exist, which are root-mean-square deviation and root-mean-square fluctuation. The former measures the degree of structural change and the latter measures atomic fluctuations relative to the average structure.135 Benchmark evaluations compare the performance of different models using unified datasets and metrics, providing references for algorithm comparison on specific tasks. For example, the XGBoost model outperformed LR, SVM and RF in AUC-based model evaluation to predict NOM reactivity under UV irradiation.53
3.3. Model interpretability analyses
Predictive accuracy is not the only criterion of model evaluation, and the interpretability of the model is equally important.136 Interpretable ML enhances the model credibility while bridging the gap between data-driven ML and NOM assembly principles. Model interpretability encompasses three key aspects: data interpretability, intrinsic interpretability and post hoc interpretability.137,138 Data interpretability aims at understanding the degree of association between input data and prediction results, as well as the quality, distribution and representativeness of data. Taking molecular descriptor as an example, data interpretability should comprehend the physicochemical significance of each descriptor, identifying correlations between descriptors and assessing the quality and completeness of data.137 The chemical composition, environmental parameters and assembly behaviors of NOM supramolecular self-assemblies inherently have strong correlations which, however, might not be considered to have a direct causal relationship due to hidden variables (such as metal ions in the solvent environment promoting bridging between NOM molecules)139,140 or indirect influences (pH changes affecting the deprotonation of carboxylic and phenolic functional groups in NOM, altering the chemical properties of these functional groups and in turn influencing the NOM assembly structure).141 Therefore, input variables need to be selected as broadly as possible, allowing the model to demonstrate the potential impact mechanisms of NOM assembly at the data level. Common data interpretability methods consist of exploratory data analysis techniques, including traditional statistical methods and dimensionality reduction approaches such as PCA and t-SNE.142,143The intrinsic model interpretability refers to ML models with inherently interpretable structures and mechanisms that are easy to understand and interpret. The interpretability stems from the model design, which allows us to understand how ML works such that the trust in the models increases.144 Nevertheless, a model's intrinsic interpretability often comes at the cost of accuracy,145 and specifically, interpretable models such as decision trees and linear regression have transparent decision processes but limited ability to solve complex nonlinear problems.146 In contrast, deep neural network models excel at capturing complex nonlinear relationships but their internal decision processes are often viewed as black boxes. As for post hoc interpretability, it is a method to complement the intrinsic interpretability of models for understanding how specific decisions are made after a black-box model is trained. Methods such as Shapley additive explanations (SHAP), locally interpretable model-agnostic explanations and partial dependence plots (PDPs) are used to understand the molecular properties, structures, environmental conditions and other features influencing the processes or properties of assembly.53,98,147 SHAP provides local explanations and helps to understand feature interaction effects while PDPs show the global relationship between a single feature and the target variable across the entire dataset.148–150 SHAP enables the quantification of feature importance in complex models by calculating each feature's contribution to specific predictions. Dwinandha et al.53 discovered that larger, unsaturated NOM containing S and N atoms was more susceptible to photodegradation through using interpretable ML (Fig. 5(b)). SHAP can theoretically be used as a feature reduction technique via ranking the features based on their absolute SHAP values. Some studies have shown that the SHAP-based feature selection can achieve better results than traditional methods such as mutual information or ANOVA.148 However, post-training approaches have two main limitations. Firstly, SHAP values are calculated based on a specific trained model whose feature interactions, that is to say, importance ranking, may change with different model architectures or training runs; secondly, the computational cost is relatively high.
However, the explanations provided by model interpretability only estimate the working mechanism of a specific ML model and may not correspond to real-world patterns.144,151,152 The interpretations from ML need to be further validated through prior knowledge or experimental results.
4. Applications of interpretable ML in elucidating processes, architectures and properties of NOM supramolecular self-assembly in real environments
4.1. NOM supramolecular self-assembling processes
Supramolecular assembly processes are primarily driven by intrinsic properties of assembling units and external environmental conditions. For the former, the inter-component forces and energy barriers at various stages of assembly significantly influence the entire self-assembly pathway and conformation. Protein–peptide and protein–protein interactions play crucial roles in regulating supramolecular functions, which can be predicted by current ML based on protein sequences, peptide sequences or binding domain data.153–155 Binding affinities between proteins are thus elucidated through recognizing the specific protein domains bound to other peptide segments or analyzing descriptors of protein complexes.155,156 These algorithms have been extended to predict interactions in protein–nanoparticle complexes as well.156The thermodynamic factors in the environment such as temperature, pH, and ionic strength directly affect the non-covalent interactions between assembling units, thereby impacting the kinetic pathways. Thermal responses and sensitivities of NOM are influenced by inherent thermodynamic properties of molecules across different climatic regions and nutrient levels.157 Therefore, researchers developed data-driven predictive models to forecast supramolecular assembly processes under various conditions. These models were trained using data obtained from high-throughput experiments, learning features from experimental designs (such as components, solvent conditions, temperature, and pH) to predict the phase transition and morphology of supramolecular assemblies.158,159 Typically, temperature changes are reported to induce the phase transitions of lipids and thus regulated their self-assembly process. These transitions could alter the permeability of biological membranes, influence the sedimentation of lipid assemblies in the environment and affect the adsorption and desorption behaviors of pollutants on lipid self-assembled structures.
4.2. NOM supramolecular self-assembly architectures
A central challenge in ML of NOM self-assembly is to interpret or predict the architectures of assembled systems with diverse functionality. Conventional approaches rely on structure preselection followed by experimental validation, importantly dependent on intuition and trial-and-error methods. While these approaches have uncovered numerous groundbreaking supramolecular materials, severe limitations hinder further discovery: first, human intuition is constrained by inductive reasoning from known systems and unable to fill in the knowledge gap in the unknown areas; then, traditional intuition-based and trial-and-error methods are time-consuming and resource-intensive. Promising approaches could be computational methods.Taking the prediction of protein assembly structures as an instance, homology modeling could be adopted based on existing sequences in the Protein Data Bank (PDB).160 However, many sequences lack homologous proteins, necessitating ab initio prediction. Ab initio methods segment the sequence into fragments, search for similar known structures for each fragment, and finally assemble these fragment structures using energy functions to predict the final protein structure.160,161 To improve precision using energy functions, researchers have begun decomposing the three-dimensional structure prediction problem into secondary structure prediction and contact map prediction. Secondary structure prediction identifies helices, sheets and other structural elements, facilitating the discovery of more suitable structural fragments. Contact maps serve as constraints for energy functions, with both approaches guiding more accurate tertiary structure predictions.162,163 AlphaFold, developed by DeepMind, represents a series of artificial intelligence-based deep learning methods for three-dimensional protein structure prediction, which has undergone continuous iterations and updates. AlphaFold 1 predicts continuous angle distributions and distances between amino acid pairs from protein sequences, transforming these into protein-specific energy functions to infer three-dimensional structures.164 AlphaFold 2 employs multiple sequence alignments and deep learning algorithms, incorporating physical and biological knowledge about protein structures. However, it can only effectively predict structures of isolated proteins, not those interacting in the real-world context.165 AlphaFold 3 addresses the limitations of its predecessor, accurately predicting structures and interactions of proteins, DNA, RNA, and ligands among other biomolecules.166
In recent years, ML was applied to establish relationships between the chemical structures of peptides and their self-assembly, helping to identify peptide sequences prone to form stable supramolecular structures through aggregation.100 Descriptors for each compound were calculated using PaDEL for ML100 to predict the formation potential of urea hydrogels30 and nucleotide hydrogels43 and also to determine the key structural influencing factors. This approach assisted researchers in designing and predicting peptide hydrogels with novel chemical structures. For the prediction of tetrapeptide hydrogels, researchers employed MD simulations and ML-trained regression models to predict the aggregation propensity of peptide self-assembly.97 The selected 55 peptide sequences were used to refine the ML model through iterative processes which were applied to predict the likelihood of hydrogel formation for 8000 peptide sequences. Additionally, specific peptide hydrogels underwent biological testing (Fig. 5(b)).
5. Necessities and challenges of unveiling NOM-self-assembly-involved environmental geochemical processes by ML
5.1. Environmental geochemical processes of pollutants influenced by NOM self-assembly
5.2. Bioavailability of pollutants in NOM self-assemblies in real environmental scenarios
NOM supramolecular self-assemblies may concentrate certain bioactive components and pollutants. As a result, the acquiring efficiency of energy and nutrients from assembly components by organisms can be impacted, and meanwhile, the toxic effects of pollutants may be amplified or mitigated, which even influence lethal doses.23 As a typical example, the bioavailability of metals dependent on their solubility and oxidation state can be influenced by NOM complexation.184,185 Predictive models based on pollutant structures revealed molecular structural features relating to the bioavailability of pollutants. Taking the micropollutants of PFAS as instances, featuring high persistence and bioaccumulation,6 PFAS present complicated ecotoxicological traits due to the diversity of PFAS categories. ML-based quantitative structure–activity relationship (QSAR) models were employed to predict the bioactivity of PFAS with various biological targets from the perspective of PFAS molecular structures.186,187Besides, the complex interactions in pollutant–water–plant root systems challenge traditional QSAR predictions. To address this issue, ML captured nonlinear relationships between molecular structures of organic pollutants and root concentration factors in complex systems.188–190 This ML framework can be extended to assess the bioaccumulation of any emerging organic pollutant in plants. It should be emphasized that common experimental studies using high-dose exposures may not accurately reflect the ecological risks of pollutants in real environments. ML is capable of uncovering the real bioavailability and effectiveness of NOM and pollutants through training model learning with the data relevant to environmental dose levels and real environmental scenarios. Nanoparticles in natural environments can adsorb biomolecules onto their surfaces, forming a protein corona (PC). The bio-nano interactions of nanoparticle–PCs complexes then alter their bioavailability and toxicity.191 Predicting interactions and interaction sites will help figure out the specific protein species or structures more likely to adsorb on nanoparticle surfaces, and thus, the behaviors of these nanoparticles in organisms can be understood.156
5.3. Correlation between NOM self-assembly and global environmental issues
The dynamic turnover of NOM has a major impact on element cycling (typically, cycling of carbon, nitrogen and phosphorus), which further results in a series of environmental issues such as global climate change, water eutrophication, harmful algal blooms and so forth.192,193 With the reactive oxygen species generation from structural mineral oxygenation, the association between NOM and minerals in supramolecular self-assemblies may either help to store carbon by decelerating NOM decomposition or release a fraction of carbon to the atmosphere as CO2 and CH4via degrading NOM.194 Disaggregation and decomposition of particulate organic matter (typically, NOM assemblies), along with exogenous input, causes steep and prolonged increases of total nitrogen, ammonium nitrogen and total phosphorus in the bottom waters of reservoirs.195To explore the process mechanisms and causative geochemical factors, long-term and large-scale sampling, observation and analyses are needed albeit only partial truths can be derived due to the insurmountable spatial and temporal restriction of those studies. Alternatively, the applications of ML, remote sensing technologies and supramolecular self-assembly analysis methods are promising in facilitating relevant research. The eXtreme Gradient Boosting ML framework has been developed using the data obtained from remote sensing technology for spatio-temporal estimation of the key nutrient – P in eutrophic lakes.196 Similarly, satellite variables were used to train the RF model to predict the emission of greenhouse gas – CH4.197 It is well known that ML is capable of estimating the intermediates and products of NOM (i.e., types and quantities of greenhouse gases) during the (bio-)mineralization process using the input data of NOM structures, quantitative analyses of NOM components, microbial community and environmental parameters. Nonetheless, ML frameworks for the correlation between NOM supramolecular self-assemblies and large-scale geochemical processes have not been systematically conducted, leaving a knowledge gap in NOM-assembly-driven geochemical effects.
6. Remarks and perspectives
The molecule-, nanometer- or submicron-scale supramolecular self-assembly of NOM is capable of profoundly impacting large-scale geochemical processes. To unveil a great ocean behind a drop of water, ML-based computational modeling is among the most promising and reliable approaches in the current big data era. Accordingly, this review presented specific strategies for adopting ML in interpreting the assembling processes, structures and environmental behaviors of NOM nano-assembly, including data source recommendation, algorithm selection, model construction and interpretability for ML as well as applications of interpretable ML. Considering the importance of datasets, the data obtained from the advanced nanoscale spatial chemical analyses are suggested (i.e., AFM-IR-mapping, Raman-mapping, and SEM-Raman-mapping) which provide the combined information of molecular interactions, features and structural visualization. Thus, the existing ML algorithms may struggle to handle multi-scale and multi-modal data. With an appropriate algorithm chosen from regression, classification, clustering and dimensionality reduction, ML models can be constructed based on the mapping relationships to be established and the data availability. In particular, supervised models with interpretability are crucial for the understanding of NOM supramolecular assembly patterns owing to their strong capacity of predicting assembly structures and quantifying structure–property–effect relationships as well as bridging the gap between simply data-driven ML and complicated NOM assembly practice.It is worth noting that the difficulty of recognizing NOM self-assembly also comes from the diversity of NOM molecular structures, the growing appearance and involvement of emerging pollutants in assemblies as well as the complicated and varied environmental geochemical conditions. The ultimate tasks of studying NOM supramolecular self-assembly in environments are to understand how different components (i.e., pollutants) regulate NOM supramolecular assembly processes and how the resulting assembly structures regulate the geochemical behaviors, bioavailability and elemental cycling processes. Given the complexity of geochemical conditions where the NOM self-assembly happens and exists, the combination of experimental investigations, molecular theoretical simulation and ML computation modeling is highly advised. The experiments conducted under the targeted environmental geochemical conditions and the accompanied advanced chemical analytical technologies are always indispensable, which provide ML with the contextualized input data and in turn validate the ML output in practical scenarios. Besides, DFT, a quantum mechanical simulation tool, denotes the NOM–pollutant interactions and electronic properties; in particular, MD reveals the structure, thermodynamics and kinetics of NOM supramolecular assemblies at the molecular level.7,198 The theoretical simulation results uncover dynamic behaviors and mechanisms of supramolecular assembly processes specifically at the molecular and atomic levels. As another powerful assisting tool, DFT and MD simulation are suitable to examine the reliability and precision of ML models and experimental observations as well as to guide the subsequent experimental designs. Research frameworks coupling ML, experiments and theoretical simulation may provide a comprehensive, efficient and innovative view of understanding NOM nano-assembly-involved environmental issues (Fig. 6).
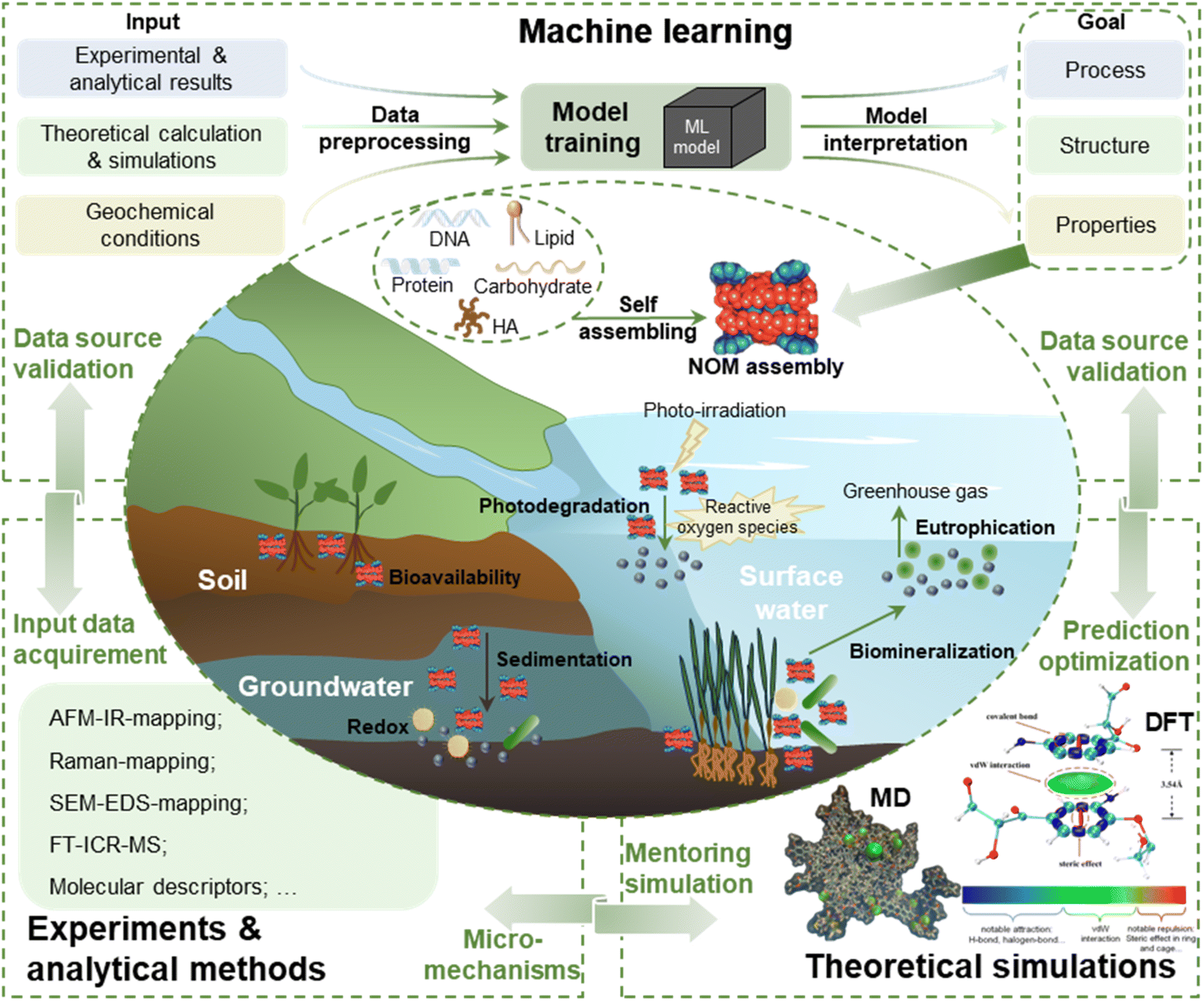 | ||
| Fig. 6 Research proposal of coupling ML, experiments and theoretical simulation to elucidate or predict NOM supramolecular nano-assembly and relevant environmental geochemical processes (figures of MD and DFT reproduced with permission from ref. 11 and 7). | ||
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 42177442) and Research and Development Plan of “Leading Goose” in Zhejiang Province (No. 2023C03128).References
- H. Feng, Y. N. Liang and X. Hu, Natural organic matter (NOM), an underexplored resource for environmental conservation and remediation, Mater. Today Sustain., 2022, 19, 100159 Search PubMed.
- J. Hur and G. Kim, Comparison of the heterogeneity within bulk sediment humic substances from a stream and reservoir via selected operational descriptors, Chemosphere, 2009, 75, 483–490 Search PubMed.
- M. J. M. Wells and H. A. Stretz, Supramolecular architectures of natural organic matter, Sci. Total Environ., 2019, 671, 1125–1133 Search PubMed.
- Y. K. Mouvenchery, J. Kučerík, D. Diehl and G. E. Schaumann, Cation-mediated cross-linking in natural organic matter: a review, Rev. Environ. Sci. Biotechnol., 2012, 11, 41–54 Search PubMed.
- I. Ali, X. Tan, J. Li, C. Peng, I. Naz, Z. Duan and Y. Ruan, Interaction of microplastics and nanoplastics with natural organic matter (NOM) and the impact of NOM on the sorption behavior of anthropogenic contaminants – a critical review, J. Cleaner Prod., 2022, 376, 134314 Search PubMed.
- M. G. Evich, M. J. B. Davis, J. P. McCord, B. Acrey, J. A. Awkerman, D. R. U. Knappe, A. B. Lindstrom, T. F. Speth, C. Tebes-Stevens, M. J. Strynar, Z. Wang, E. J. Weber, W. M. Henderson and J. W. Washington, Per- and polyfluoroalkyl substances in the environment, Science, 2022, 375, eabg9065 Search PubMed.
- Z. Zhou, C. Zhang, M. Xi, H. Ma and H. Jia, Multi-scale modeling of natural organic matter–heavy metal cations interactions: aggregation and stabilization mechanisms, Water Res., 2023, 238, 120007 Search PubMed.
- Y. Liu, T. Yue, L. Liu, B. Zhang, H. Feng, S. Li, X. Liu, Y. Dai and J. Zhao, Molecular assembly of extracellular polymeric substances regulating aggregation of differently charged nanoplastics and subsequent interactions with bacterial membrane, J. Hazard. Mater., 2023, 457, 131825 Search PubMed.
- D. Philp and J. F. Stoddart, Self-assembly in natural and unnatural systems, Angew. Chem., Int. Ed. Engl., 1996, 35, 1154–1196 Search PubMed.
- Q. Xue, Z. Jiao, X. Liu, W. Pan, J. Fu and A. Zhang, Dynamic behavior and interaction mechanism of soil organic matter in water systems: a coarse-grained molecular dynamics study, Environ. Sci. Technol., 2024, 58, 1531–1540 Search PubMed.
- D. Devarajan, L. Liang, B. Gu, S. C. Brooks, J. M. Parks and J. C. Smith, Molecular dynamics simulation of the structures, dynamics, and aggregation of dissolved organic matter, Environ. Sci. Technol., 2020, 54, 13527–13537 Search PubMed.
- C. Li, X. Zhang, Y. Guo, F. Seidi, X. Shi and H. Xiao, Naturally occurring exopolysaccharide nanoparticles: formation process and their application in glutathione detection, ACS Appl. Mater. Interfaces, 2021, 13, 19756–19767 Search PubMed.
- S. Xu, P. Zhu, C. Wang, D. Zhang and X. Pan, Environmental concentration PFOS as a light shield for lake exopolymers against photodegradation by formation of sandwiched supramolecular nanostructures, Water Res., 2022, 227, 119345 Search PubMed.
- M. Zhang, W. Qiu, R. Nie, Q. Xia, D. Zhang and X. Pan, Macronutrient and PFOS bioavailability manipulated by aeration-driven rhizospheric organic nanocapsular assembly, Water Res., 2024, 253, 121334 Search PubMed.
- M. I. Jordan and T. M. Mitchell, Machine learning: trends, perspectives, and prospects, Science, 2015, 349, 255–260 Search PubMed.
- S. Ikeda, D. Murayama, A. Tsurumaki, S. Sato, T. Urashima and K. Fukuda, Rheological characteristics and supramolecular structure of the exopolysaccharide produced by Lactobacillus fermentum mtcc 25067, Carbohydr. Polym., 2019, 218, 226–233 Search PubMed.
- S. Madrigal-Carballo, S. Lim, G. Rodriguez, A. O. Vila, C. G. Krueger, S. Gunasekaran and J. D. Reed, Biopolymer coating of soybean lecithin liposomes via layer-by-layer self-assembly as novel delivery system for ellagic acid, J. Funct. Foods, 2010, 2, 99–106 Search PubMed.
- C. M. Runnels, K. A. Lanier, J. K. Williams, J. C. Bowman, A. S. Petrov, N. V. Hud and L. D. Williams, Folding, assembly, and persistence: the essential nature and origins of biopolymers, J. Mol. Evol., 2018, 86, 598–610 Search PubMed.
- C. S. Swenson, A. Velusamy, H. S. Argueta-Gonzalez and J. M. Heemstra, Bilingual peptide nucleic acids: encoding the languages of nucleic acids and proteins in a single self-assembling biopolymer, J. Am. Chem. Soc., 2019, 141, 19038–19047 Search PubMed.
- T. Roversi and L. Piazza, Supramolecular assemblies from plant cell polysaccharides: self-healing and aging behavior, Food Hydrocolloids, 2016, 54, 189–195 Search PubMed.
- A. Piccolo, The supramolecular structure of humic substances, Soil Sci., 2001, 166, 810 Search PubMed.
- G. Chilom, A. Baglieri, C. A. Johnson-Edler and J. A. Rice, Hierarchical self-assembling properties of natural organic matter's components, Org. Geochem., 2013, 57, 119–126 Search PubMed.
- H. Xu and L. Guo, Intriguing changes in molecular size and composition of dissolved organic matter induced by microbial degradation and self-assembly, Water Res., 2018, 135, 187–194 Search PubMed.
- H.-J. Schneider, Binding mechanisms in supramolecular complexes, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 Search PubMed.
- D. S. Lawrence, T. Jiang and M. Levett, Self-assembling supramolecular complexes, Chem. Rev., 1995, 95, 2229–2260 Search PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Supramolecular coordination: self-assembly of finite two- and three-dimensional ensembles, Chem. Rev., 2011, 111, 6810–6918 Search PubMed.
- X. Kong, T. Jendrossek, K.-U. Ludwichowski, U. Marx and B. P. Koch, Solid-phase extraction of aquatic organic matter: loading-dependent chemical fractionation and self-assembly, Environ. Sci. Technol., 2021, 55, 15495–15504 Search PubMed.
- A. Vogelsang and M. Borg, in 2019 IEEE 27th International Requirements Engineering Conference Workshops (REW), 2019, pp. 245–251 Search PubMed.
- Y. Zhao, R. J. Mulder, S. Houshyar and T. C. Le, A review on the application of molecular descriptors and machine learning in polymer design, Polym. Chem., 2023, 14, 3325–3346 Search PubMed.
- R. V. Lommel, J. Zhao, W. M. D. Borggraeve, F. D. Proft and M. Alonso, Molecular dynamics based descriptors for predicting supramolecular gelation, Chem. Sci., 2020, 11, 4226–4238 Search PubMed.
- P. W. Kenny, Hydrogen Bonding, Electrostatic Potential, and Molecular Design, J. Chem. Inf. Model., 2009, 49, 1234–1244 Search PubMed.
- G. Caron and G. Ermondi, Molecular descriptors for polarity: the need for going beyond polar surface area, Future Med. Chem., 2016, 8, 2013–2016 Search PubMed.
- Y. Lyu, T. Huang, W. Liu and W. Sun, Unveil the quantum chemical descriptors determining direct photodegradation of antibiotics under simulated sunlight: batch experiments and model development, J. Environ. Chem. Eng., 2022, 10, 108086 Search PubMed.
- L. Wang, J. Ding, L. Pan, D. Cao, H. Jiang and X. Ding, Quantum chemical descriptors in quantitative structure–activity relationship models and their applications, Chemom. Intell. Lab. Syst., 2021, 217, 104384 Search PubMed.
- L. Pattanaik and C. W. Coley, Molecular representation: going long on fingerprints, Chem, 2020, 6, 1204–1207 Search PubMed.
- S. Riniker and G. A. Landrum, Similarity maps – a visualization strategy for molecular fingerprints and machine-learning methods, J. Cheminf., 2013, 5, 43 Search PubMed.
- F. Sandfort, F. Strieth-Kalthoff, M. Kühnemund, C. Beecks and F. Glorius, A structure-based platform for predicting chemical reactivity, Chem, 2020, 6, 1379–1390 Search PubMed.
- T. J. Wills, D. A. Polshakov, M. C. Robinson and A. A. Lee, Impact of chemist-in-the-loop molecular representations on machine learning outcomes, J. Chem. Inf. Model., 2020, 60, 4449–4456 Search PubMed.
- S. Jamal, A. Grover and S. Grover, Machine learning from molecular dynamics trajectories to predict caspase-8 inhibitors against Alzheimer’s disease, Front. Pharmacol., 2019, 10 DOI:10.3389/fphar.2019.00780.
- P. Panwar, Q. Yang and A. Martini, PyL3dMD: Python LAMMPS 3D molecular descriptors package, J. Cheminf., 2023, 15, 69 Search PubMed.
- L.-Y. Xia, Q.-Y. Wang, Z. Cao and Y. Liang, Descriptor selection improvements for quantitative structure–activity relationships, Int. J. Neural Syst., 2019, 29, 1950016 Search PubMed.
- Y. Liu, D. Zhang, Y. Tang, Y. Zhang, Y. Chang and J. Zheng, Machine learning-enabled design and prediction of protein resistance on self-assembled monolayers and beyond, ACS Appl. Mater. Interfaces, 2021, 13, 11306–11319 Search PubMed.
- W. Li, Y. Wen, K. Wang, Z. Ding, L. Wang, Q. Chen, L. Xie, H. Xu and H. Zhao, Developing a machine learning model for accurate nucleoside hydrogels prediction based on descriptors, Nat. Commun., 2024, 15, 2603 Search PubMed.
- H. Moriwaki, Y.-S. Tian, N. Kawashita and T. Takagi, Mordred: a molecular descriptor calculator, J. Cheminf., 2018, 10, 4 Search PubMed.
- D. S. Wigh, J. M. Goodman and A. A. Lapkin, A review of molecular representation in the age of machine learning, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1603 Search PubMed.
- C. T. Madsen, J. C. Refsgaard, F. G. Teufel, S. K. Kjærulff, Z. Wang, G. Meng, C. Jessen, P. Heljo, Q. Jiang, X. Zhao, B. Wu, X. Zhou, Y. Tang, J. F. Jeppesen, C. D. Kelstrup, S. T. Buckley, S. Tullin, J. Nygaard-Jensen, X. Chen, F. Zhang, J. V. Olsen, D. Han, M. Grønborg and U. de Lichtenberg, Combining mass spectrometry and machine learning to discover bioactive peptides, Nat. Commun., 2022, 13, 6235 Search PubMed.
- J. Cox, Prediction of peptide mass spectral libraries with machine learning, Nat. Biotechnol., 2023, 41, 33–43 Search PubMed.
- G. Hu and M. Qiu, Machine learning-assisted structure annotation of natural products based on MS and NMR data, Nat. Prod. Rep., 2023, 40, 1735–1753 Search PubMed.
- D. A. Boiko, K. S. Kozlov, J. V. Burykina, V. V. Ilyushenkova and V. P. Ananikov, Fully automated unconstrained analysis of high-resolution mass spectrometry data with machine learning, J. Am. Chem. Soc., 2022, 144, 14590–14606 Search PubMed.
- Y. Qi, Q. Xie, J.-J. Wang, D. He, H. Bao, Q.-L. Fu, S. Su, M. Sheng, S.-L. Li, D. A. Volmer, F. Wu, G. Jiang, C.-Q. Liu and P. Fu, Deciphering dissolved organic matter by Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS): from bulk to fractions and individuals, Carbon Res., 2022, 1, 3 Search PubMed.
- S. Yu, S. Tang, J. Lv, F. Li, Z. Huang, L. Zhao, D. Cao and Y. Wang, High throughput identification of carbonyl compounds in natural organic matter by directional derivatization combined with ultra-high resolution mass spectrometry, Water Res., 2024, 258, 121769 Search PubMed.
- J. Liu, C. Wang, Z. Hao, G. Kondo, M. Fujii, Q.-L. Fu and Y. Wei, Comprehensive understanding of DOM reactivity in anaerobic fermentation of persulfate-pretreated sewage sludge via FT-ICR mass spectrometry and reactomics analysis, Water Res., 2023, 229, 119488 Search PubMed.
- D. Dwinandha, M. Elsamadony, R. Gao, Q.-L. Fu, J. Liu and M. Fujii, Interpretable machine learning and reactomics assisted isotopically labeled FT-ICR-MS for exploring the reactivity and transformation of natural organic matter during ultraviolet photolysis, Environ. Sci. Technol., 2024, 58, 816–825 Search PubMed.
- J. A. Purslow, B. Khatiwada, M. J. Bayro and V. Venditti, NMR methods for structural characterization of protein-protein complexes, Front. Mol. Biosci., 2020, 7, 9 Search PubMed.
- J. Peuravuori, NMR spectroscopy study of freshwater humic material in light of supramolecular assembly, Environ. Sci. Technol., 2005, 39, 5541–5549 Search PubMed.
- M. Weingarth and M. Baldus, Solid-state NMR-based approaches for supramolecular structure elucidation, Acc. Chem. Res., 2013, 46, 2037–2046 Search PubMed.
- P. Klukowski, R. Riek and P. Güntert, Rapid protein assignments and structures from raw NMR spectra with the deep learning technique ARTINA, Nat. Commun., 2022, 13, 6151 Search PubMed.
- S. Bonhommeau, G. S. Cooney and Y. Huang, Nanoscale chemical characterization of biomolecules using tip-enhanced Raman spectroscopy, Chem. Soc. Rev., 2022, 51, 2416–2430 Search PubMed.
- W. Zhang, Z. Dong, L. Zhu, Y. Hou and Y. Qiu, Direct observation of the release of nanoplastics from commercially recycled plastics with correlative Raman imaging and scanning electron microscopy, ACS Nano, 2020, 14, 7920–7926 Search PubMed.
- M. Li, A. Wu, L. Li, Z. Li and H. Zang, Three stages of dynamic assembly process of dipeptide-based supramolecular gel revealed by in situ infrared spectroscopy, ACS Biomater. Sci. Eng., 2024, 10, 863–874 Search PubMed.
- R. Chevigny, E. D. Sitsanidis, J. Schirmer, E. Hulkko, P. Myllyperkiö, M. Nissinen and M. Pettersson, Nanoscale probing of the supramolecular assembly in a two-component gel by near-field infrared spectroscopy, Chem.–Eur. J., 2023, 29, e202300155 Search PubMed.
- S. Y. Schmid, K. Lachowski, H. T. Chiang, L. Pozzo, J. De Yoreo and S. Zhang, Mechanisms of biomolecular self-assembly investigated through in situ observations of structures and dynamics, Angew. Chem., Int. Ed., 2023, 62, e202309725 Search PubMed.
- H. Schulz and M. Baranska, Identification and quantification of valuable plant substances by IR and Raman spectroscopy, Vib. Spectrosc., 2007, 43, 13–25 Search PubMed.
- K. B. Beć, J. Grabska and C. W. Huck, Biomolecular and bioanalytical applications of infrared spectroscopy – a review, Anal. Chim. Acta, 2020, 1133, 150–177 Search PubMed.
- S. Yang, Q. Zhang, H. Yang, H. Shi, A. Dong, L. Wang and S. Yu, Progress in infrared spectroscopy as an efficient tool for predicting protein secondary structure, Int. J. Biol. Macromol., 2022, 206, 175–187 Search PubMed.
- L. Xie, S. Luo, Y. Liu, X. Ruan, K. Gong, Q. Ge, K. Li, V. K. Valev, G. Liu and L. Zhang, Automatic identification of individual nanoplastics by Raman spectroscopy based on machine learning, Environ. Sci. Technol., 2023, 57, 18203–18214 Search PubMed.
- Y. Liu, W. Yao, F. Qin, L. Zhou and Y. Zheng, Spectral classification of large-scale blended (micro)plastics using FT-IR raw spectra and image-based machine learning, Environ. Sci. Technol., 2023, 57, 6656–6663 Search PubMed.
- W. Cowger, Z. Steinmetz, A. Gray, K. Munno, J. Lynch, H. Hapich, S. Primpke, H. De Frond, C. Rochman and O. Herodotou, Microplastic spectral classification needs an open source community: open specy to the rescue!, Anal. Chem., 2021, 93, 7543–7548 Search PubMed.
- S. Zinchik, S. Jiang, S. Friis, F. Long, L. Høgstedt, V. M. Zavala and E. Bar-Ziv, Accurate characterization of mixed plastic waste using machine learning and fast infrared spectroscopy, ACS Sustain. Chem. Eng., 2021, 9, 14143–14151 Search PubMed.
- Y. Qi, L. Yang, B. Liu, L. Liu, Y. Liu, Q. Zheng, D. Liu and J. Luo, Accurate diagnosis of lung tissues for 2D Raman spectrogram by deep learning based on short-time Fourier transform, Anal. Chim. Acta, 2021, 1179, 338821 Search PubMed.
- T.-Y. Huang, J. Wang, Q. Liu and J. Yu, The application of wavelet transform of Raman spectra to facilitate transfer learning for gasoline detection and classification, Talanta Open, 2022, 5, 100106 Search PubMed.
- M. Li, L. Zhang, L.-L. Jiang, Z.-B. Zhao, Y.-H. Long, D.-M. Chen, J. Bin, C. Kang and Y.-J. Liu, Label-free Raman microspectroscopic imaging with chemometrics for cellular investigation of apple ring rot and nondestructive early recognition using near-infrared reflection spectroscopy with machine learning, Talanta, 2024, 267, 125212 Search PubMed.
- N. M. Ralbovsky and I. K. Lednev, Towards development of a novel universal medical diagnostic method: Raman spectroscopy and machine learning, Chem. Soc. Rev., 2020, 49, 7428–7453 Search PubMed.
- L. Huang, H. Sun, L. Sun, K. Shi, Y. Chen, X. Ren, Y. Ge, D. Jiang, X. Liu, W. Knoll, Q. Zhang and Y. Wang, Rapid, label-free histopathological diagnosis of liver cancer based on Raman spectroscopy and deep learning, Nat. Commun., 2023, 14, 48 Search PubMed.
- M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. de Greef, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, How to distinguish isodesmic from cooperative supramolecular polymerisation, Chem.–Eur. J., 2010, 16, 362–367 Search PubMed.
- M. Wehner, M. I. S. Röhr, M. Bühler, V. Stepanenko, W. Wagner and F. Würthner, Supramolecular polymorphism in one-dimensional self-assembly by kinetic pathway control, J. Am. Chem. Soc., 2019, 141, 6092–6107 Search PubMed.
- S. Dhiman, T. Andrian, B. S. Gonzalez, M. M. E. Tholen, Y. Wang and L. Albertazzi, Can super-resolution microscopy become a standard characterization technique for materials chemistry?, Chem. Sci., 2022, 13, 2152–2166 Search PubMed.
- D. V. Chapman, H. Du, W. Y. Lee and U. B. Wiesner, Optical super-resolution microscopy in polymer science, Prog. Polym. Sci., 2020, 111, 101312 Search PubMed.
- A. Rizvi, J. T. Mulvey, B. P. Carpenter, R. Talosig and J. P. Patterson, A close look at molecular self-assembly with the transmission electron microscope, Chem. Rev., 2021, 121, 14232–14280 Search PubMed.
- E. Z. Qu, A. M. Jimenez, S. K. Kumar and K. Zhang, Quantifying nanoparticle assembly states in a polymer matrix through deep learning, Macromolecules, 2021, 54, 3034–3040 Search PubMed.
- L. Yao, Z. Ou, B. Luo, C. Xu and Q. Chen, Machine learning to reveal nanoparticle dynamics from liquid-phase TEM videos, ACS Cent. Sci., 2020, 6, 1421–1430 Search PubMed.
- Z. Zhu, J. Lu, F. Zheng, C. Chen, Y. Lv, H. Jiang, Y. Yan, A. Narita, K. Müllen, X.-Y. Wang and Q. Sun, A deep-learning framework for the automated recognition of molecules in scanning-probe-microscopy images, Angew. Chem., Int. Ed., 2022, 61, e202213503 Search PubMed.
- J. Shi, Y. Li, X. Jiang, H. Yu, J. Li, H. Zhang, D. J. Trainer, S. W. Hla, H. Wang, M. Wang and X. Li, Self-assembly of metallo-supramolecules with dissymmetrical ligands and characterization by scanning tunneling microscopy, J. Am. Chem. Soc., 2021, 143, 1224–1234 Search PubMed.
- A. Jeindl, J. Domke, L. Hörmann, F. Sojka, R. Forker, T. Fritz and O. T. Hofmann, Nonintuitive surface self-assembly of functionalized molecules on Ag(111), ACS Nano, 2021, 15, 6723–6734 Search PubMed.
- F. Zheng, J. Lu, Z. Zhu, H. Jiang, Y. Yan, Y. He, S. Yuan and Q. Sun, Predicting molecular self-assembly on metal surfaces using graph neural networks based on experimental data sets, ACS Nano, 2023, 17, 17545–17553 Search PubMed.
- E. Vargo, J. C. Dahl, K. M. Evans, T. Khan, P. Alivisatos and T. Xu, Using machine learning to predict and understand complex self-assembly behaviors of a multicomponent nanocomposite, Adv. Mater., 2022, 34, 2203168 Search PubMed.
- M. Ziatdinov, S. Zhang, O. Dollar, J. Pfaendtner, C. J. Mundy, X. Li, H. Pyles, D. Baker, J. J. De Yoreo and S. V. Kalinin, Quantifying the dynamics of protein self-organization using deep learning analysis of atomic force microscopy data, Nano Lett., 2021, 21, 158–165 Search PubMed.
- S. Zhang, R. Sadre, B. A. Legg, H. Pyles, T. Perciano, E. W. Bethel, D. Baker, O. Rübel and J. J. De Yoreo, Rotational dynamics and transition mechanisms of surface-adsorbed proteins, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2020242119 Search PubMed.
- A. Centrone, Infrared imaging and spectroscopy beyond the diffraction limit, Annu. Rev. Anal. Chem., 2015, 8, 101–126 Search PubMed.
- A. Dazzi and C. B. Prater, AFM-IR: technology and applications in nanoscale infrared spectroscopy and chemical imaging, Chem. Rev., 2017, 117, 5146–5173 Search PubMed.
- X. Wang, S.-C. Huang, T.-X. Huang, H.-S. Su, J.-H. Zhong, Z.-C. Zeng, M.-H. Li and B. Ren, Tip-enhanced Raman spectroscopy for surfaces and interfaces, Chem. Soc. Rev., 2017, 46, 4020–4041 Search PubMed.
- F. Liu, X. Zou, N. Yue, W. Zhang and W. Zheng, Correlative Raman imaging and scanning electron microscopy for advanced functional materials characterization, Cell Rep. Phys. Sci., 2023, 4, 101607 Search PubMed.
- X.-T. Zheng, Y.-Q. Dong, X.-D. Liu, Y.-L. Xu and R.-K. Jian, Fully bio-based flame-retardant cotton fabrics via layer-by-layer self assembly of laccase and phytic acid, J. Cleaner Prod., 2022, 350, 131525 Search PubMed.
- A. M. Kobaisy, M. F. Elkady, A. A. Abdel-Moneim and M. E. El-Khouly, Surface-decorated porphyrinic zirconium-based metal–organic frameworks (MOFs) using post-synthetic self-assembly for photodegradation of methyl orange dye, RSC Adv., 2023, 13, 23050–23060 Search PubMed.
- C. Jiang, S. Liu, T. Zhang, Q. Liu, P. J. J. Alvarez and W. Chen, Current methods and prospects for analysis and characterization of nanomaterials in the environment, Environ. Sci. Technol., 2022, 56, 7426–7447 Search PubMed.
- Q. Li, J. Chang, L. Li, X. Lin and Y. Li, Research progress of nano-scale secondary ion mass spectrometry (NanoSIMS) in soil science: evolution, applications, and challenges, Sci. Total Environ., 2023, 905, 167257 Search PubMed.
- T. Xu, J. Wang, S. Zhao, D. Chen, H. Zhang, Y. Fang, N. Kong, Z. Zhou, W. Li and H. Wang, Accelerating the prediction and discovery of peptide hydrogels with human-in-the-loop, Nat. Commun., 2023, 14, 3880 Search PubMed.
- J. Li, W. Qin, B. Zhu, T. Ruan, Z. Hua, H. Du, S. Dong and J. Fang, Insights into the transformation of natural organic matter during UV/peroxydisulfate treatment by FT-ICR MS and machine learning: non-negligible formation of organosulfates, Water Res., 2024, 256, 121564 Search PubMed.
- C. Zhao, K. Wang, Q. Jiao, X. Xu, Y. Yi, P. Li, J. Merder and D. He, Machine learning models for evaluating biological reactivity within molecular fingerprints of dissolved organic matter over time, Geophys. Res. Lett., 2024, 51, e2024GL108794 Search PubMed.
- F. Li, J. Han, T. Cao, W. Lam, B. Fan, W. Tang, S. Chen, K. L. Fok and L. Li, Design of self-assembly dipeptide hydrogels and machine learning via their chemical features, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11259–11264 Search PubMed.
- A. Gardin, C. Perego, G. Doni and G. M. Pavan, Classifying soft self-assembled materials via unsupervised machine learning of defects, Commun. Chem., 2022, 5, 1–15 Search PubMed.
- A. Cardellini, M. Crippa, C. Lionello, S. P. Afrose, D. Das and G. M. Pavan, Unsupervised data-driven reconstruction of molecular motifs in simple to complex dynamic micelles, J. Phys. Chem. B, 2023, 127, 2595–2608 Search PubMed.
- C. S. Adorf, T. C. Moore, Y. J. U. Melle and S. C. Glotzer, Analysis of self-assembly pathways with unsupervised machine learning algorithms, J. Phys. Chem. B, 2020, 124, 69–78 Search PubMed.
- R. Capelli, A. Gardin, C. Empereur-mot, G. Doni and G. M. Pavan, A data-driven dimensionality reduction approach to compare and classify lipid force fields, J. Phys. Chem. B, 2021, 125, 7785–7796 Search PubMed.
- X. Dong, Z. Yu, W. Cao, Y. Shi and Q. Ma, A survey on ensemble learning, Front. Comput. Sci., 2020, 14, 241–258 Search PubMed.
- M. Gao, L. Zhao, Z. Zhang, J. Wang and C. Wang, Using a stacked ensemble learning framework to predict modulators of protein–protein interactions, Comput. Biol. Med., 2023, 161, 107032 Search PubMed.
- T. A. Aliev, A. A. Timralieva, T. A. Kurakina, K. E. Katsuba, Y. A. Egorycheva, M. V. Dubovichenko, M. A. Kutyrev, V. V. Shilovskikh, N. Orekhov, N. Kondratyuk, S. N. Semenov, D. M. Kolpashchikov and E. V. Skorb, Designed assembly and disassembly of DNA in supramolecular structure: from ion regulated nuclear formation and machine learning recognition to running DNA cascade, Nano Sel., 2022, 3, 1526–1536 Search PubMed.
- J. Ou, J. Wen, W. Tan, X. Luo, J. Cai, X. He, L. Zhou and Y. Yuan, A data-driven approach for understanding the structure dependence of redox activity in humic substances, Environ. Res., 2023, 219, 115142 Search PubMed.
- S. Lise, C. Archambeau, M. Pontil and D. T. Jones, Prediction of hot spot residues at protein-protein interfaces by combining machine learning and energy-based methods, BMC Bioinf., 2009, 10, 365 Search PubMed.
- Q. Xu and Y. Liang, Monte Carlo cross validation, Chemom. Intell. Lab. Syst., 2001, 56, 1–11 Search PubMed.
- P. Vormittag, T. Klamp and J. Hubbuch, Ensembles of hydrophobicity scales as potent classifiers for chimeric virus-like particle solubility – an amino acid sequence-based machine learning approach, Front. Bioeng. Biotechnol., 2020, 8, 395 Search PubMed.
- J. Allgaier and R. Pryss, Cross-validation visualized: a narrative guide to advanced methods, Mach. Learn. Knowl. Extr., 2024, 6, 1378–1388 Search PubMed.
- D. K. Barrow and S. F. Crone, Cross-validation aggregation for combining autoregressive neural network forecasts, Int. J. Forecast., 2016, 32, 1120–1137 Search PubMed.
- X. Ying, An overview of overfitting and its solutions, J. Phys.: Conf. Ser., 2019, 1168, 22022 Search PubMed.
- N. Srivastava, G. Hinton, A. Krizhevsky, I. Sutskever and R. Salakhutdinov, Dropout: a simple way to prevent neural networks from overfitting, J. Mach. Learn. Res., 2014, 15, 1929–1958 Search PubMed.
- M. Vilares Ferro, Y. Doval Mosquera, F. J. Ribadas Pena and V. M. Darriba Bilbao, Early stopping by correlating online indicators in neural networks, Neural Netw., 2023, 159, 109–124 Search PubMed.
- S. A. Bonke, G. Trezza, L. Bergamasco, H. Song, S. Rodríguez-Jiménez, L. Hammarström, E. Chiavazzo and E. Reisner, Multi-variable multi-metric optimization of self-assembled photocatalytic CO2 reduction performance using machine learning algorithms, J. Am. Chem. Soc., 2024, 146, 15648–15658 Search PubMed.
- Q. Dong, X. Gong, K. Yuan, Y. Jiang, L. Zhang and W. Li, Inverse design of complex block copolymers for exotic self-assembled structures based on Bayesian optimization, ACS Macro Lett., 2023, 12, 401–407 Search PubMed.
- J. A. Mysona, P. F. Nealey and J. J. De Pablo, Machine learning models and dimensionality reduction for prediction of polymer properties, Macromolecules, 2024, 57, 1988–1997 Search PubMed.
- Y. Ge, X. Wang, Q. Zhu, Y. Yang, H. Dong and J. Ma, Machine learning-guided adaptive parametrization for coupling terms in a mixed united-atom/coarse-grained model for diphenylalanine self-assembly in aqueous ionic liquids, J. Chem. Theory Comput., 2023, 19, 6718–6732 Search PubMed.
- Y. Ishiwatari, T. Yokoyama, T. Kojima, T. Banno and N. Arai, Machine learning prediction of self-assembly and analysis of molecular structure dependence on the critical packing parameter, Mol. Syst. Des. Eng., 2024, 9, 20–28 Search PubMed.
- T. Inokuchi, N. Li, K. Morohoshi and N. Arai, Multiscale prediction of functional self-assembled materials using machine learning: high-performance surfactant molecules, Nanoscale, 2018, 10, 16013–16021 Search PubMed.
- A. Ghosh, R. Sahu and S. K. Reddy, Constructing one-dimensional supramolecular polymer structures using particle swarm optimization technique, Theor. Chem. Acc., 2024, 143, 24 Search PubMed.
- K. K. Bejagam, S. Singh, Y. An and S. A. Deshmukh, Machine-learned coarse-grained models, J. Phys. Chem. Lett., 2018, 9, 4667–4672 Search PubMed.
- X. Han, Z. Zhang, N. Ding, Y. Gu, X. Liu, Y. Huo, J. Qiu, Y. Yao, A. Zhang, L. Zhang, W. Han, M. Huang, Q. Jin, Y. Lan, Y. Liu, Z. Liu, Z. Lu, X. Qiu, R. Song, J. Tang, J.-R. Wen, J. Yuan, W. X. Zhao and J. Zhu, Pre-trained models: past, present and future, AI Open, 2021, 2, 225–250 Search PubMed.
- N. Tajbakhsh, J. Y. Shin, S. R. Gurudu, R. T. Hurst, C. B. Kendall, M. B. Gotway and J. Liang, Convolutional neural networks for medical image analysis: full training or fine tuning?, IEEE Trans. Med. Imaging, 2016, 35, 1299–1312 Search PubMed.
- X. Yin, W. Chen, X. Wu and H. Yue, in 2017 12th IEEE Conference on Industrial Electronics and Applications (ICIEA), 2017, pp. 1310–1315 Search PubMed.
- P. Lin, Y. Yan, H. Tao and S.-Y. Huang, Deep transfer learning for inter-chain contact predictions of transmembrane protein complexes, Nat. Commun., 2023, 14, 4935 Search PubMed.
- Y. Han, Z. Wang, A. Chen, I. Ali, J. Cai, S. Ye, Z. Wei and J. Li, A deep transfer learning-based protocol accelerates full quantum mechanics calculation of protein, Briefings Bioinf., 2023, 24, bbac532 Search PubMed.
- S. Ruder, arXiv, 2017, preprint, arXiv:1609.04747, DOI:10.48550/arXiv.1609.04747v2.
- I. Sutskever, J. Martens, G. Dahl and G. Hinton, in Proceedings of the 30th International Conference on International Conference on Machine Learning, JMLR.org, Atlanta, GA, USA, 2013, vol. 28, p. III Search PubMed.
- D. P. Kingma and J. Ba, arXiv, 2017, preprint, arXiv:1412.6980, DOI:10.48550/arXiv.1412.6980v9.
- M. D. Zeiler, arXiv, 2012, preprint, arXiv:1212.5701, DOI:10.48550/arXiv.1212.5701v1.
- J. Duchi, E. Hazan and Y. Singer, Adaptive subgradient methods for online learning and stochastic optimization, J. Mach. Learn. Res., 2011, 12, 2121–2159 Search PubMed.
- H. Zhou, Z. Dong and P. Tao, Recognition of protein allosteric states and residues: machine learning approaches, J. Comput. Chem., 2018, 39, 1481–1490 Search PubMed.
- X. Liu, D. Lu, A. Zhang, Q. Liu and G. Jiang, Data-driven machine learning in environmental pollution: gains and problems, Environ. Sci. Technol., 2022, 56, 2124–2133 Search PubMed.
- D. V. Carvalho, E. M. Pereira and J. S. Cardoso, Machine learning interpretability: a survey on methods and metrics, Electronics, 2019, 8, 832 Search PubMed.
- S. Ali, T. Abuhmed, S. El-Sappagh, K. Muhammad, J. M. Alonso-Moral, R. Confalonieri, R. Guidotti, J. Del Ser, N. Díaz-Rodríguez and F. Herrera, Explainable artificial intelligence (XAI): what we know and what is left to attain trustworthy artificial intelligence, Inf. Fusion, 2023, 99, 101805 Search PubMed.
- J. Zhao, R. A. Mathew, D. S. Yang, P. G. Vekilov, Y. Hu and S. M. Louie, Natural organic matter flocculation behavior controls lead phosphate particle aggregation by mono- and divalent cations, Sci. Total Environ., 2023, 866, 161346 Search PubMed.
- L. Xie, Q. Lu, X. Mao, J. Wang, L. Han, J. Hu, Q. Lu, Y. Wang and H. Zeng, Probing the intermolecular interaction mechanisms between humic acid and different substrates with implications for its adsorption and removal in water treatment, Water Res., 2020, 176, 115766 Search PubMed.
- C. Zhang, S. Mo, Z. Liu, B. Chen, G. Korshin, N. Hertkorn, J. Ni and M. Yan, Interpreting pH-dependent differential UV/vis absorbance spectra to characterize carboxylic and phenolic chromophores in natural organic matter, Water Res., 2023, 244, 120522 Search PubMed.
- A. del V. Turina, M. V. Nolan, J. A. Zygadlo and M. A. Perillo, Natural terpenes: self-assembly and membrane partitioning, Biophys. Chem., 2006, 122, 101–113 Search PubMed.
- R. Romero, A. Ramanathan, T. Yuen, D. Bhowmik, M. Mathew, L. B. Munshi, S. Javaid, M. Bloch, D. Lizneva, A. Rahimova, A. Khan, C. Taneja, S.-M. Kim, L. Sun, M. I. New, S. Haider and M. Zaidi, Mechanism of glucocerebrosidase activation and dysfunction in Gaucher disease unraveled by molecular dynamics and deep learning, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 5086–5095 Search PubMed.
- Z. C. Lipton, The mythos of model interpretability: in machine learning, the concept of interpretability is both important and slippery, Queue, 2018, 16, 31–57 Search PubMed.
- A. Nguyen and M. R. Martínez, On quantitative aspects of model interpretability, ArXiv, 2020, preprint, arXiv:2007.07584, DOI:10.48550/arXiv.2007.07584.
- M. H. Chehreghani, A review on the impact of data representation on model explainability, ACM Comput. Surv., 2024, 56, 1–21 Search PubMed.
- Z. Ye, W. Yang, Y. Yang and D. Ouyang, Interpretable machine learning methods for in vitro pharmaceutical formulation development, Food Front., 2021, 2, 195–207 Search PubMed.
- W. E. Marcilio and D. M. Eler, in 2020 33rd SIBGRAPI Conference on Graphics, Patterns and Images (SIBGRAPI), IEEE, Recife/Porto de Galinhas, Brazil, 2020, pp. 340–347 Search PubMed.
- D. Sreedev, S. Kurukkal Balakrishnan and N. Kalarikkal, A machine learning aided yield prediction model for the preparation of cellulose nanocrystals, ACS Appl. Eng. Mater., 2024, 2, 1561–1571 Search PubMed.
- J. H. Friedman, Multivariate adaptive regression splines, Ann. Stat., 1991, 19, 1–67 Search PubMed.
- J.-J. Zhu, M. Yang and Z. J. Ren, Machine learning in environmental research: common pitfalls and best practices, Environ. Sci. Technol., 2023, 57, 17671–17689 Search PubMed.
- S. Jiang, L. Sweet, G. Blougouras, A. Brenning, W. Li, M. Reichstein, J. Denzler, W. Shangguan, G. Yu, F. Huang and J. Zscheischler, How interpretable machine learning can benefit process understanding in the geosciences, Earth's Future, 2024, 12, e2024EF004540 Search PubMed.
- B. Mewara and S. Lalwani, Sequence-based prediction of protein–protein interaction using auto-feature engineering of RNN-based model, Res. Biomed. Eng., 2023, 39, 259–272 Search PubMed.
- Y. Lei, S. Li, Z. Liu, F. Wan, T. Tian, S. Li, D. Zhao and J. Zeng, A deep-learning framework for multi-level peptide–protein interaction prediction, Nat. Commun., 2021, 12, 5465 Search PubMed.
- J. M. Cunningham, G. Koytiger, P. K. Sorger and M. AlQuraishi, Biophysical prediction of protein–peptide interactions and signaling networks using machine learning, Nat. Methods, 2020, 17, 175–183 Search PubMed.
- M. Cha, E. S. T. Emre, X. Xiao, J.-Y. Kim, P. Bogdan, J. S. VanEpps, A. Violi and N. A. Kotov, Unifying structural descriptors for biological and bioinspired nanoscale complexes, Nat. Comput. Sci., 2022, 2, 243–252 Search PubMed.
- A. Hu, K.-S. Jang, A. J. Tanentzap, W. Zhao, J. T. Lennon, J. Liu, M. Li, J. Stegen, M. Choi, Y. Lu, X. Feng and J. Wang, Thermal responses of dissolved organic matter under global change, Nat. Commun., 2024, 15, 576 Search PubMed.
- T. C. Le and N. Tran, Using machine learning to predict the self-assembled nanostructures of monoolein and phytantriol as a function of temperature and fatty acid additives for effective lipid-based delivery systems, ACS Appl. Nano Mater., 2019, 2, 1637–1647 Search PubMed.
- Y. Lu, D. Yalcin, P. J. Pigram, L. D. Blackman and M. Boley, Interpretable machine learning models for phase prediction in polymerization-induced self-assembly, J. Chem. Inf. Model., 2023, 63, 3288–3306 Search PubMed.
- K. A. Dill and J. L. MacCallum, The protein-folding problem, 50 years on, Science, 2012, 338, 1042–1046 Search PubMed.
- R. Pearce and Y. Zhang, Deep learning techniques have significantly impacted protein structure prediction and protein design, Curr. Opin. Struct. Biol., 2021, 68, 194–207 Search PubMed.
- S. M. Mortuza, W. Zheng, C. Zhang, Y. Li, R. Pearce and Y. Zhang, Improving fragment-based ab initio protein structure assembly using low-accuracy contact-map predictions, Nat. Commun., 2021, 12, 5011 Search PubMed.
- Y. Zhou, T. Litfin and J. Zhan, 3 = 1 + 2: how the divide conquered de novo protein structure prediction and what is next?, Natl. Sci. Rev., 2023, 10, nwad259 Search PubMed.
- A. W. Senior, R. Evans, J. Jumper, J. Kirkpatrick, L. Sifre, T. Green, C. Qin, A. Žídek, A. W. R. Nelson, A. Bridgland, H. Penedones, S. Petersen, K. Simonyan, S. Crossan, P. Kohli, D. T. Jones, D. Silver, K. Kavukcuoglu and D. Hassabis, Improved protein structure prediction using potentials from deep learning, Nature, 2020, 577, 706–710 Search PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596, 583–589 Search PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C.-C. Hung, M. O'Neill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Žemgulytė, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve, A. I. Cowen-Rivers, A. Cowie, M. Figurnov, F. B. Fuchs, H. Gladman, R. Jain, Y. A. Khan, C. M. R. Low, K. Perlin, A. Potapenko, P. Savy, S. Singh, A. Stecula, A. Thillaisundaram, C. Tong, S. Yakneen, E. D. Zhong, M. Zielinski, A. Žídek, V. Bapst, P. Kohli, M. Jaderberg, D. Hassabis and J. M. Jumper, Accurate structure prediction of biomolecular interactions with AlphaFold 3, Nature, 2024, 630, 493–500 Search PubMed.
- O. C. Zafiriou, J. Joussot-Dubien, R. G. Zepp and R. G. Zika, Photochemistry of natural waters, Environ. Sci. Technol., 1984, 18, 358A–371A Search PubMed.
- E. M.-L. Janssen, P. R. Erickson and K. McNeill, Dual roles of dissolved organic matter as sensitizer and quencher in the photooxidation of tryptophan, Environ. Sci. Technol., 2014, 48, 4916–4924 Search PubMed.
- C. Song, K.-X. Zhang, X.-J. Wang, S. Zhao and S.-G. Wang, Effects of natural organic matter on the photolysis of tetracycline in aquatic environment: kinetics and mechanism, Chemosphere, 2021, 263, 128338 Search PubMed.
- A. P. S. Batista, A. C. S. C. Teixeira, W. J. Cooper and B. A. Cottrell, Correlating the chemical and spectroscopic characteristics of natural organic matter with the photodegradation of sulfamerazine, Water Res., 2016, 93, 20–29 Search PubMed.
- H. Xu, W. J. Cooper, J. Jung and W. Song, Photosensitized degradation of amoxicillin in natural organic matter isolate solutions, Water Res., 2011, 45, 632–638 Search PubMed.
- H. Wang, M. Wang, H. Wang, J. Gao, R. A. Dahlgren, Q. Yu and X. Wang, Aqueous photochemical degradation of BDE-153 in solutions with natural dissolved organic matter, Chemosphere, 2016, 155, 367–374 Search PubMed.
- P. Yu, Z. Guo, T. Wang, J. Wang, Y. Guo and L. Zhang, Insights into the mechanisms of natural organic matter on the photodegradation of indomethacin under natural sunlight and simulated light irradiation, Water Res., 2023, 244, 120539 Search PubMed.
- B. Gu, Y. Bian, C. L. Miller, W. Dong, X. Jiang and L. Liang, Mercury reduction and complexation by natural organic matter in anoxic environments, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1479–1483 Search PubMed.
- B. Li, P. Liao, L. Xie, Q. Li, C. Pan, Z. Ning and C. Liu, Reduced NOM triggered rapid Cr(VI) reduction and formation of NOM-Cr(III) colloids in anoxic environments, Water Res., 2020, 181, 115923 Search PubMed.
- P. Herzsprung, V. Wentzky, N. Kamjunke, W. Von Tümpling, C. Wilske, K. Friese, B. Boehrer, T. Reemtsma, K. Rinke and O. J. Lechtenfeld, Improved understanding of dissolved organic matter processing in freshwater using complementary experimental and machine learning approaches, Environ. Sci. Technol., 2020, 54, 13556–13565 Search PubMed.
- C. Zhao, X. Xu, H. Chen, F. Wang, P. Li, C. He, Q. Shi, Y. Yi, X. Li, S. Li and D. He, Exploring the complexities of dissolved organic matter photochemistry from the molecular level by using machine learning approaches, Environ. Sci. Technol., 2023, 57, 17889–17899 Search PubMed.
- J. R. Helms, J. Mao, K. Schmidt-Rohr, H. Abdulla and K. Mopper, Photochemical flocculation of terrestrial dissolved organic matter and iron, Geochim. Cosmochim. Acta, 2013, 121, 398–413 Search PubMed.
- L. Sun, W.-C. Chin, M.-H. Chiu, C. Xu, P. Lin, K. A. Schwehr, A. Quigg and P. H. Santschi, Sunlight induced aggregation of dissolved organic matter: role of proteins in linking organic carbon and nitrogen cycling in seawater, Sci. Total Environ., 2019, 654, 872–877 Search PubMed.
- J. T. K. Quik, M. C. Stuart, M. Wouterse, W. Peijnenburg, A. J. Hendriks and D. Van De Meent, Natural colloids are the dominant factor in the sedimentation of nanoparticles, Environ. Toxicol. Chem., 2012, 31, 1019–1022 Search PubMed.
- J. T. K. Quik, I. Lynch, K. V. Hoecke, C. J. H. Miermans, K. A. C. D. Schamphelaere, C. R. Janssen, K. A. Dawson, M. A. C. Stuart and D. V. D. Meent, Effect of natural organic matter on cerium dioxide nanoparticles settling in model fresh water, Chemosphere, 2010, 81, 711–715 Search PubMed.
- S. Qian, X. Qiao, W. Zhang, Z. Yu, S. Dong and J. Feng, Machine learning-based prediction for settling velocity of microplastics with various shapes, Water Res., 2024, 249, 121001 Search PubMed.
- S. Dittmar, A. S. Ruhl, K. Altmann and M. Jekel, Settling velocities of small microplastic fragments and fibers, Environ. Sci. Technol., 2024, 58, 6359–6369 Search PubMed.
- Y. Gao, J. Zhu and A. He, Effect of dissolved organic matter on the bioavailability and toxicity of cadmium in zebrafish larvae: determination based on toxicokinetic–toxicodynamic processes, Water Res., 2022, 226, 119272 Search PubMed.
- E. E. Daugherty, B. Gilbert, P. S. Nico and T. Borch, Complexation and redox buffering of Iron(II) by dissolved organic matter, Environ. Sci. Technol., 2017, 51, 11096–11104 Search PubMed.
- W. Cheng and C. A. Ng, Using machine learning to classify bioactivity for 3486 per- and polyfluoroalkyl substances (PFASs) from the OECD list, Environ. Sci. Technol., 2019, 53, 13970–13980 Search PubMed.
- H. Kwon, Z. A. Ali and B. M. Wong, Harnessing semi-supervised machine learning to automatically predict bioactivities of per- and polyfluoroalkyl substances (PFASs), Environ. Sci. Technol. Lett., 2023, 10, 1017–1022 Search PubMed.
- F. Gao, Y. Shen, J. B. Sallach, H. Li, C. Liu and Y. Li, Direct prediction of bioaccumulation of organic contaminants in plant roots from soils with machine learning models based on molecular structures, Environ. Sci. Technol., 2021, 55, 16358–16368 Search PubMed.
- F. Gao, Y. Shen, J. Brett Sallach, H. Li, W. Zhang, Y. Li and C. Liu, Predicting crop root concentration factors of organic contaminants with machine learning models, J. Hazard. Mater., 2022, 424, 127437 Search PubMed.
- L. Xiang, J. Qiu, Q.-Q. Chen, P.-F. Yu, B.-L. Liu, H.-M. Zhao, Y.-W. Li, N.-X. Feng, Q.-Y. Cai, C.-H. Mo and Q. X. Li, Development, evaluation, and application of machine learning models for accurate prediction of root uptake of per- and polyfluoroalkyl substances, Environ. Sci. Technol., 2023, 57, 18317–18328 Search PubMed.
- J. Wang, J. Cong, J. Wu, Y. Chen, H. Fan, X. Wang, Z. Duan and L. Wang, Nanoplastic-protein corona interactions and their biological effects: a review of recent advances and trends, TrAC, Trends Anal. Chem., 2023, 166, 117206 Search PubMed.
- N. J. Anderson, H. Bennion and A. F. Lotter, Lake eutrophication and its implications for organic carbon sequestration in Europe, Global Change Biol., 2014, 20, 2741–2751 Search PubMed.
- M. W. I. Schmidt, M. S. Torn, S. Abiven, T. Dittmar, G. Guggenberger, I. A. Janssens, M. Kleber, I. Kögel-Knabner, J. Lehmann, D. A. C. Manning, P. Nannipieri, D. P. Rasse, S. Weiner and S. E. Trumbore, Persistence of soil organic matter as an ecosystem property, Nature, 2011, 478, 49–56 Search PubMed.
- H. Dong, Q. Zeng, Y. Sheng, C. Chen, G. Yu and A. Kappler, Coupled iron cycling and organic matter transformation across redox interfaces, Nat. Rev. Earth Environ., 2023, 4, 659–673 Search PubMed.
- X. Gao, H. Chen, B. Gu, E. Jeppesen, Y. Xue and J. Yang, Particulate organic matter as causative factor to eutrophication of subtropical deep freshwater: role of typhoon (tropical cyclone) in the nutrient cycling, Water Res., 2021, 188, 116470 Search PubMed.
- J. Xiong, C. Lin, Z. Cao, M. Hu, K. Xue, X. Chen and R. Ma, Development of remote sensing algorithm for total phosphorus concentration in eutrophic lakes: conventional or machine learning?, Water Res., 2022, 215, 118213 Search PubMed.
- H. Duan, Q. Xiao, T. Qi, C. Hu, M. Zhang, M. Shen, Z. Hu, W. Wang, W. Xiao, Y. Qiu, J. Luo and X. Lee, Quantification of diffusive methane emissions from a large eutrophic lake with satellite imagery, Environ. Sci. Technol., 2023, 57, 13520–13529 Search PubMed.
- P. Anil Kumar Reddy, T. G. Senthamaraikannan, D.-H. Lim, M. Choi, S. Yoon, J. Shin, K. Chon and S. Bae, Unveiling the positive effect of mineral induced natural organic matter (NOM) on catalyst properties and catalytic dechlorination performance: an experiment and DFT study, Water Res., 2022, 222, 118871 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4em00662c |
| This journal is © The Royal Society of Chemistry 2025 |