
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual acid/glutathione-responsive core-degradable/shell-sheddable block copolymer nanoassemblies bearing benzoic imines for enhanced drug release†
Xiaolei
Hu
 ,
Kevin
Larocque
,
Alisa
Piekny
and
Jung Kwon
Oh
*
,
Kevin
Larocque
,
Alisa
Piekny
and
Jung Kwon
Oh
*
Department of Chemistry and Biochemistry, Concordia University, Montreal, Quebec, Canada H4B 1R6. E-mail: john.oh@concordia.ca
First published on 26th November 2024
Abstract
The development of amphiphilic block copolymer (ABP)-based nanoassemblies that degrade in response to dual stimuli at dual locations (e.g. hydrophobic cores and core/corona interfaces) offers a promising platform for controlled drug delivery. This work harnesses the features of an acid-labile benzoic imine (BzIm) bond and a glutathione (GSH)-cleavable disulfide linkage. We synthesized a poly(ethylene glycol) (PEG)-based dual location dual acid/GSH-degradable ABP with BzIm pendants in a hydrophobic polymethacrylate block and a disulfide at the block junction. The acid-catalyzed hydrolysis rate of BzIm depends on substituents attached to its para-position. Hydrolysis is faster with electron-donating substituents (methoxy), and slower with electron-withdrawing ones (bromo and nitro). Well-defined ABP synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization of a methoxy-substituted BzIm methacrylate in the presence of a disulfide-labeled PEG-based chain transfer agent, enables self-assembly to form colloidally-stable, monomodal, and spherical nanoassemblies. These nanoassemblies are capable of encapsulating cancer drug doxorubicin (Dox) and exhibit enhanced release of Dox through core degradation upon the cleavage of BzIm bonds in acidic pH and shell detachment upon the cleavage of disulfide bonds in the presence of GSH. Moreover, Dox-loaded nanoassemblies show excellent uptake by HeLa cell multi-tumor spheroids, demonstrating their potential as drug delivery nanocarriers. This study highlights the importance of substituent effects on the hydrolysis of BzIm and the dual acid/GSH-responsive strategy for developing a promising drug delivery system with precise control over drug release.
Introduction
Amphiphilic block copolymers (ABPs) that exhibit stimuli-responsive degradation (SRD) have emerged as a promising platform for controlled drug delivery.1–3 The general synthetic approach for SRD-exhibiting ABPs involves polymerization in the presence of cleavable bond-functionalized initiators or monomers via reversible-deactivation radical polymerization (RDRP), typically by atom transfer radical polymerization (ATRP) and reversible addition–fragmentation chain transfer (RAFT).4–8 This enables the incorporation of cleavable bonds into polymer precursors at various positions, such as the backbone, junction, and side chain.9 The intrinsic amphiphilicity of ABPs allows them to self-assemble to form stable aqueous nanoassemblies.10–12 In response to specific stimuli, the cleavage of SRD linkages causes the disassembly of nanoassemblies through changes in hydrophobic/hydrophilic balance, main chain degradation, or corona detachment, leading to site-specific and enhanced release of encapsulated drugs.13–15 When SRD-based polymeric nanoassemblies are designed for intracellular delivery, the selection of cleavable linkages is critical. Acidic pH is a stimulus found in subcellular and tissue microenvironments, such as endosomes and lysosomes (pH = 4–5.5), and tumors (pH = 6.5–6.8), respectively.16–18 Various covalent bonds that are cleavable in acidic pH ranges have been explored, including ketal, acetal, 2,3-dialkylmaleamidic amide, β-thiopropionate, oxime, hydrazone, and benzoic imine (BzIm) linkages.19 Among these pH-sensitive linkers, BzIm (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) bonds have attracted particular interest due to their facile synthesis via a catalyst-free click-type reaction between carbonyl and amine groups.2 In an acidic environment, the CN bond is cleaved into the corresponding carbonyl and amine precursors. As acid-responsive degradation directly regulates drug release process via degradation-induced disassembly, it is critical to control their cleavage kinetics by chemical design. Mechanistic studies show that CN hydrolysis is initiated by protonation of the N atom and followed by nucleophilic attack of H2O. Consequently, the rate of acid catalyzed imine cleavage depends on electrophilicity, which can be tuned by substituent effects on the CN bond and phenyl ring for BzIm via inductive, resonance, and steric effects.20
N) bonds have attracted particular interest due to their facile synthesis via a catalyst-free click-type reaction between carbonyl and amine groups.2 In an acidic environment, the CN bond is cleaved into the corresponding carbonyl and amine precursors. As acid-responsive degradation directly regulates drug release process via degradation-induced disassembly, it is critical to control their cleavage kinetics by chemical design. Mechanistic studies show that CN hydrolysis is initiated by protonation of the N atom and followed by nucleophilic attack of H2O. Consequently, the rate of acid catalyzed imine cleavage depends on electrophilicity, which can be tuned by substituent effects on the CN bond and phenyl ring for BzIm via inductive, resonance, and steric effects.20
Built on these promising acid-responsive properties, the development of BzIm-based (co)polymers has been extensively explored as acid-responsive drug delivery nanocarrier in various forms including self-assembled nanoassemblies,21,22 core-crosslinked nanogels,23,24 and polymer–drug conjugates.2,25 These systems are generally fabricated by post-polymerization modification of either amine or aldehyde-labelled (co)polymer precursors. Recently, our group demonstrated direct polymerization22 and post-polymerization modification21 approaches as effective means to synthesize a well-defined poly(ethylene glycol)-based ABP containing hydrophobic poly(methacrylate) with pendant BzIm groups. Despite these advances, BzIm-bearing nanoassemblies suffered from slow release and aggregation. More precise control over degradation behavior of imine and resulting drug release is highly demanded. Inspired by the structure-dependent hydrolysis of BzIm bonds,26 we hypothesized that para-substituents on BzIm groups with varying electronic properties (electro-denoting or withdrawing) could tune their acid-catalyzed cleavage kinetics and thus drug release profiles.
Further studies have explored polymers with dual or multiple stimuli-responsive degradation mechanisms.27,28 Glutathione (GSH), a tripeptide with a cysteine residue and a pendant thiol group, is a reducing agent that is found at millimolar concentrations in the cytosol of cells, and can be elevated in cancer cells.29–32 GSH-responsive disulfide (SS) group has been explored together with a BzIm linkage for the construction of dual acid/GSH-responsive degradable ABP nanoassemblies.33–38 However, these nanoassemblies were designed with both BzIm and disulfide linkages in a single location, either at core/corona interface or in hydrophobic core. We postulated that the integration of SS bonds into the block junction of PEG-based ABP-bearing pendant BzIm bonds could facilitate synergistic effects for controlled drug release. This strategy known as dual location dual acid/glutathione-responsive degradation causes dual responses at dual locations as in the cores and at core/corona interfaces, thus enabling to not only independently regulate the release of encapsulated molecules but also facilitate synergistic/accelerated release at dual locations.39–41
In this work, we explored tunable imine hydrolysis and dual location dual acid/GSH response to construct dual location dual acid/GSH-degradable ABPs and their nanoassemblies for enhanced control over drug release (Scheme 1). A series of BzIm-bearing methacrylate monomers with various para substituents (RBzImMA) (R = MeO, H, NO2, and Br) were synthesized to investigate the effect of their electronic properties on acid-catalyzed hydrolysis rates. Using a new imine methacrylate with MeO substituent (e.g.MBzImMA) with enhanced acid-responsive hydrolysis, well-defined PEG-based ABPs with pendant BzIm groups in the hydrophobic block and a SS positioned at the block junction was synthesized by a controlled radical polymerization technique. The copolymer was characterized for aqueous self-assembly, dual acid/GSH-responsive degradation and drug release, as well as cellular uptake with a HeLa multi-cellular tumor spheroid model. Our results show that the dual acid/GSH-responsive nanoassemblies enabled synergistic control over drug release and minimized degradation products. Drug-loaded nanoassemblies demonstrated superior uptake by HeLa spheroids compared to free drug.
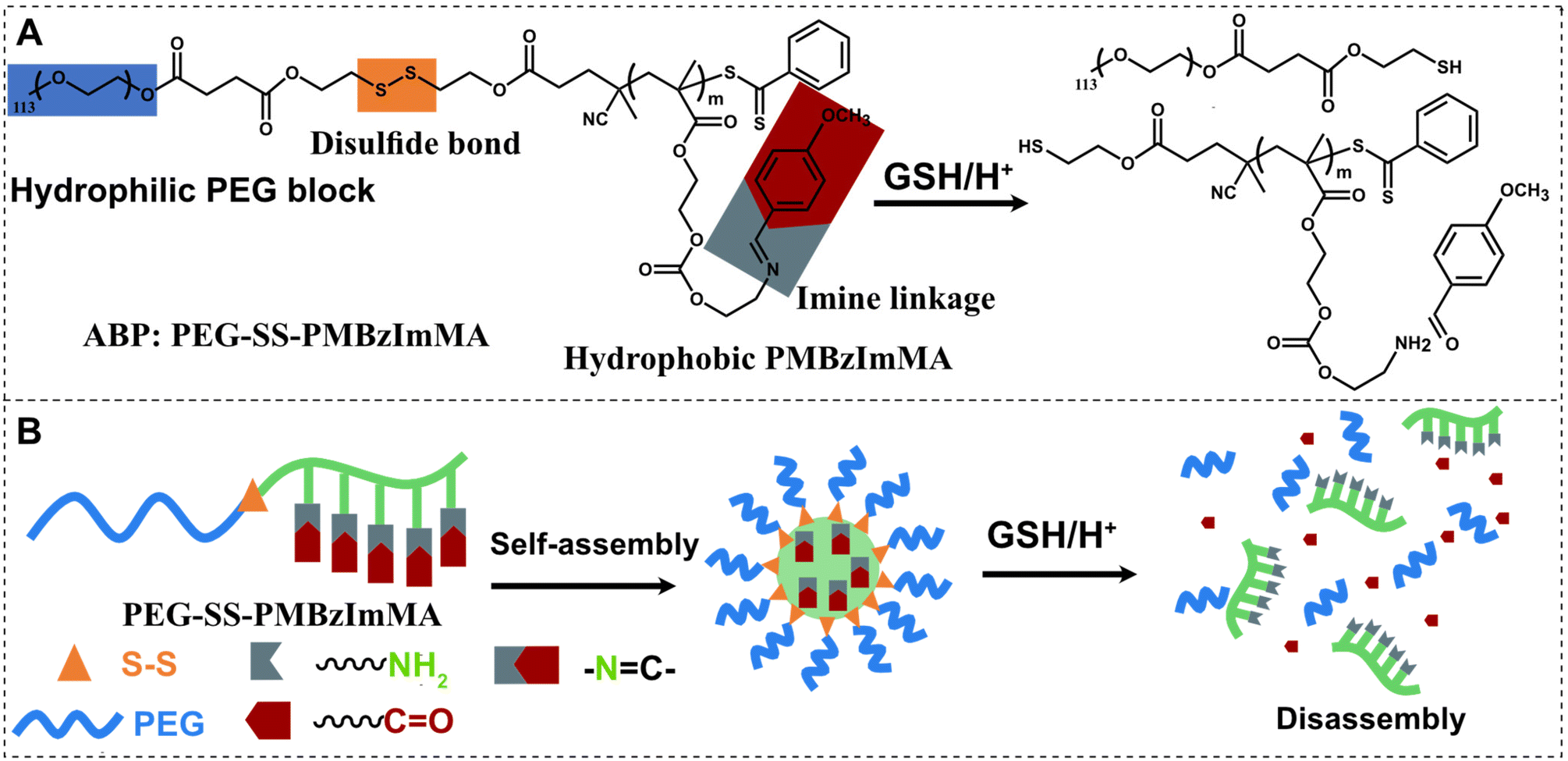 | ||
| Scheme 1 Illustration of dual location dual acid/GSH-degradable ABP with BzIm pendants in the hydrophobic block and a disulfide bond at the block junction (A) and its nanoassemblies exhibiting synergistic/enhanced release of encapsulated drug molecules in dual acid/GSH responses at dual locations as in cores and at core/corona interfaces (B). | ||
Experimental
Instrumentation and materials
NMR spectra were recorded using 500 MHz Varian spectrometer. The CDCl3 singlet at 7.26 ppm was selected as the reference standard. Molecular weight and molecular weight distribution were determined using gel permeation chromatography (GPC) with Malvern equipped with a Viscotek VE1122 pump and a refractive index detector. Tetrahydrofuran (THF) was used as eluent at room temperature at a flow rate of 1.0 mL min−1. Molecular weight and molecular weight distribution were calculated relative to linear poly(methyl methacrylate) standards from Fluka. Aliquots of the polymer samples dissolved in THF were filtered through a 0.45 μm PTFE filter to remove any THF-insoluble species and injected into the GPC with a drop of anisole as a flow rate marker.Dynamic light scattering (DLS) with a Malvern Instruments Nano S ZEN1600, transition electron microscope (TEM) using a Talos L120C TEM operated at 80 kV electrons, fluorescence spectroscopy with Varian Cary Eclipse fluorescence spectrometer, and UV/vis spectroscopy with Agilent Cary 60 UV/vis spectrometer were used as described in our previous publications.42,43
Most reagents including triethylamine (Et3N, 99.5%), carbonyl diimidazole (CDI, ≥90%), 2-hydroxyethyl methacrylate (HEMA, ≥99%), ethanolamine (EA, ≥98%), 4-methoxybenzaldehyde (98%), 4-nitrobenzaldehyde (98%), 4-bromobenzaldehyde (99%), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 98%), succinic anhydride (SA, ≥99%), 4-cyano-4-(phenylcarbonothioylthio) pentanoic acid (CTA, 97%), 4-(methylamino)pyridine (DMAP, 99%), 1-ethyl-3-(3 dimethylaminopropyl) carbodiimide·HCl (EDC), acetyl chloride (≥98%), ethylene glycol vinyl ether (≥97%), pyridinium p-toluenesulfonate (PPTS, 98%), doxorubicin (Dox, –NH3+Cl− form, >98%), and deuterium chloride solution (DCl, 35 wt% in D2O) from Sigma-Aldrich as well as 2,2′-azodi(2-methylbutyronitrile) (AMBN) from Wako Chemicals were purchased and used as received. Poly(ethylene glycol) monomethyl ether (PEG, MW = 5000 g mol−1, EO# = 113) purchased from Sigma-Aldrich was dried by azeotropic distillation with toluene to remove residual moisture. A carbonylimidazole (CI)-activated 2-hydroxyethylmethacrylate (HEMA-CI) were synthesized as described in our previous publication.22,44
Synthesis of RBzImOH precursors
RBzImOH intermediates were synthesized based on our previous report with R-substituted benzaldehyde.22 A solution of EA (1.22 g, 0.02 mol) dissolved in DCM (5 mL) was mixed with a solution containing R-benzaldehyde dissolved in DCM (50 mL) in presence of sodium sulfate (5 g) under stirring at room temperature for 13 h. Solvent was removed by rotary evaporation and residue was dried in a vacuum oven at room temperature for 13 h.![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) 6
6![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 4OCH3), 3.85 (t, 2H, HOCH22NCH–), 3.8 (S, 3H, –C6H4O3) 3.66 (t, 2H, HO2CH2NCH–).
C6H4NO2), 8.22 and 7.88 (d, 4H, –64NO2), 3.93 (t, 2H, HOCH22–), 3.81 (t, 2H, HO2CH2–).
C6H4Br), 7.54 (q, 4H, – 64Br), 3.89 (t, 2H, HOCH22–), 3.71 (t, 2H, HO2CH2–).
4OCH3), 3.85 (t, 2H, HOCH22NCH–), 3.8 (S, 3H, –C6H4O3) 3.66 (t, 2H, HO2CH2NCH–).
C6H4NO2), 8.22 and 7.88 (d, 4H, –64NO2), 3.93 (t, 2H, HOCH22–), 3.81 (t, 2H, HO2CH2–).
C6H4Br), 7.54 (q, 4H, – 64Br), 3.89 (t, 2H, HOCH22–), 3.71 (t, 2H, HO2CH2–).
Synthesis of RBzImMA
Substituted benzoic imine methacrylates (RBzImMAs) were synthesized based on our previous report using RBzImOH.22 The dried RBzImOH precursor was mixed with a solution consisting of HEMA-CI and DBU dissolved in DCM under stirring at room temperature for 13 h. The reaction mixture was washed with saturated NaHCO3 solution three times and dried over sodium sulfate. After the removal of solvent, the product was dried in a vacuum oven for 13 h.C6H4–), 8.2 and 7.9 (d, 4H, –NCH64NO2), 6.1 and 5.5 (s, 2H, 2C(CH3)–), 4.5 (t, 2H, –2CH2OC(O)O), 4.3 (m, 2H, –CH22OC(O)O), 4.3 (m, 2H, –2CH2NCH–), 3.9 (t, 2H, –CH22NCH–), 1.9 (s, 3H, CH2C(3)–).
C6H4–), 7.6 and 6.9 (d, 4H, –NCH64OCH3), 6.1 and 5.5 (s, 2H, 2C(CH3)–), 4.4 (t, 2H, –2CH2OC(O)O), 4.3 (m, 2H, –CH22OC(O)O), 4.3 (m, 2H, –2CH2NCH–), 3.8 (m, 5H, –CH22NCHC6H4O3), 1.9 (s, 3H, CH2C(3)–).
C6H4–), 7.54 (q, 4H, – 64Br), 6.1 and 5.6 (s, 2H, 2C(CH3)–), 4.5 (t, 2H, –2CH2OC(O)O), 4.3 (m, 2H, –CH22OC(O)O), 4.3 (m, 2H, –2CH2NCH–), 3.85 (m, 5H, –CH22NCHC6H4O3), 1.9 (s, 3H, CH2C(3)–).
Synthesis of PEG-COOH
SA (0.22 g, 2.2 mmol) dissolved in anhydrous DCM (5 mL) was mixedwith an organic solution containing PEG (2 g, 0.4 mmol) and DMAP (49 mg, 0.4 mmol) in DCM (15 mL) in an ice-bath under stirring for 48 h. The resultant mixture was washed with brine solution three times, dried over sodium sulfate, and then precipitated from cold diethyl ether to remove excess succinic anhydride. The product, white solid, was isolated by vacuum filtration and dried in a vacuum oven at room temperature for 13 h. Yield = 1.35 g (65%). 1H-NMR (CDCl3, ppm): 4.25 (t, 2H, –2OC(O)–), 3.5–3.8 (m, –22O– of PEG main chain), 3.36 (s, 3H, CH3–), 2.6 (m, 4H, –22COOH).
Synthesis of PEG-SS-CTA
In the first step to synthesize CTA-ss-OH, CTA (0.2 g, 0.72 mmol) dissolved in anhydrous DCM (15 mL) was mixed with an organic solution containing ss-diol (166 mg, 1.1 mmol), EDC (270 mg, 1.4 mmol) and TEA (145 mg, 2 mmol) in DCM (85 mL) in an ice-bath under stirring. After proceeding at room temperature for 48 h, the solvent was evaporated, and the product was purified by silica gel column chromatography. The product, a red oil, was collected as the third band of total five bands. Yield = 0.06 g (30%); Rf = 0.35 (ethyl acetate/hexane = 1/1, v/v). 1H-NMR (CDCl3, ppm): 7.9–7.3 (m, 5H, –SC(S)65), 4.4 (t, 2H, –2OC(O)–), 3.9 (t, 2H, –CH22OH), 2.94 (t, 2H, –2SSCH2CH2OH), 2.88 (t, 2H, –SS2CH2OH), 2.7 (t, 2H, –2CH2(CH3)C(CN)–), 2.4 (t, 2H, –CH22(CH3)C(CN)–), 1.94 (s, 3H, –(3)C(CN)–).
In the second step to synthesize PEG-ss-CTA, EDC (79 mg, 0.41 mmol) dissolved in anhydrous DCM (20 mL) was mixed with an organic solution containing PEG-COOH (0.5 g, 98 μmol), DMAP (3.6 g, 29 μmol) and CTA-ss-OH (0.16 g, 0.39 mmol) in DCM (80 mL) in an ice-bath under stirring for 24 h. The resultant mixture was washed with brine solution three times, dried over sodium sulfate, and then precipitated from cold diethyl ether to remove excess CTA. The pink solid product was isolated by vacuum filtration and dried in a vacuum oven at room temperature for 13 h. Yield = 0.46 g (70%). 1H-NMR (CDCl3, ppm): 7.9–7.3 (m, 5H, –SC(S)C6H5), 4.34 (m, 4H, –CH2OC(O)–), 4.24 (t, 2H, –OCH2CH2OC(O)–), 3.5–3.8 (m, –CH2CH2O– of PEG main chain), 3.38 (s, 3H, CH3O–), 2.92 (t, 4H, –CH2SSCH2), 2.7 (m, 2H, –CH2CH2(CH3)C(CN)–), 2.69 (m, 4H, –OC(O)CH2CH2 C(O)O–), 2.4 (t, 2H, –CH2CH2(CH3)C(CN)–), 1.94 (s, 3H, –(CH3)C(CN)–).
RAFT polymerization to synthesize PEG-SS-PMBzImMA (ImSSP)
MBzImMA (0.55 g, 1.64 mmol), PEG-SS-CTA (0.12 g, 22 μmol), AMBN (1.08 mg, 7 μmol) and anhydrous anisole (0.92 mL) were mixed in a 5 mL Schlenk flask. The mixture was deoxygenated by purging with nitrogen for 45 min, and then immersed into an oil bath at 70 °C to initiate polymerization. For kinetics study, aliquots were taken out periodically to follow monomer conversion by 1H-NMR and molecular weight by GPC. Polymerization was stopped by cooling the reaction mixture in an ice-bath and exposing to air.Investigation of acid-catalyzed imine degradation using 1H-NMR spectroscopy
Hydrolysis in acidic conditions were carried out based on our previous report.22 For the hydrolysis of monomers, RBzImMA (10.0 μmol) dissolved in CD3CN (0.3 mL) was mixed with the as-prepared PB solutions in D2O (0.1 mL, 0.2 M, pD = 5.0, 6.8 and 7.4). For PEG-SS-pMBzImMA block copolymer, the purified ImSSP (10 mg) dissolved in CD3CN (0.3 mL) was mixed with the PB solutions in D2O (0.1 mL, 0.2 M, pD = 5). The resultant mixtures were subjected to 1H-NMR analysis at 25 °C for given periods of time.Aqueous micellization of ImSSP by nanoprecipitation method
PBS solution (10 mL, pH = 7.4) was dropwise added to an organic solution of ImSSP (10 mg) dissolved in THF (2 mL) at a rate of 1 mL min−1 using a syringe pump under stirring. The resultant mixture was kept stirring at room temperature for 40 h to remove THF, yielding an aqueous solution of nanoassemblies at 1 mg mL−1.Colloidal stability and acid/GSH responsive disassembly of aqueous nanoassemblies
Aliquots of empty nanoassemblies (1 mL, 1 mg mL−1) were incubated with acetate buffer solution (3 mL, 0.2 M, pH = 5) or GSH (10 mM) under stirring at room temperature. In parallel, empty nanoassemblies were kept on bench at room temperature. DLS analysis was followed for given periods of time.Preparation of Dox-loaded NPs by dialysis
PBS solution (10 mL, pH = 7.4) was added dropwise to an organic solution consisting of Dox (2 mg), Et3N (5 uL) and ImSSP (20 mg) dissolved in DMF (2 mL) at a rate of 1 mL min−1 using a syringe pump under stirring for 1 h. The resulting mixture was transferred to a dialysis tubing (MWCO = 11–13 kDa) for dialysis against PBS solution (1 L, pH = 7.4) at room temperature for 24 h to remove free (not encapsulated) Dox molecules and residual DMF. The resulting dispersion was passed through a 0.45 μm PES filter, yielding an aqueous Dox-loaded nanoparticles (Dox-NPs) at 1.6 mg mL−1.To determine the loading level of Dox in micelles, aliquots of aqueous Dox-NPs (1 mL) were mixed with DMF (5 mL). The resultant mixture was subjected to filtration using a 0.45 μm PTFE filter to remove insoluble salts and analyzed by UV/vis spectroscopy.
Acid/GSH-responsive release of Dox from Dox-NPs
Aliquots of Dox-NPs (1.7 mg mL−1, 2 mL) were mixed with buffer solutions (2 mL) at different pHs = 5, 6.8 and 7.4 as well as 10 mM GSH. The mixtures were transferred to a dialysis tubing (MWCO = 11–13 kDa) and immersed into corresponding buffer solutions (40 mL) at 25 °C under stirring. Aliquots of outer solutions (3 mL) were taken out at given interval to record fluorescence spectra excited at λex = 480 nm. Equivalent amounts of fresh buffer solutions were added back to keep sink condition.Cellular uptake with HeLa multi-cellular tumor spheroids
HeLa cells were maintained in DMEM (Wisent) supplemented with 10% cosmic calf serum (Hyclone) with 100 U penicillin (Wisent), and 0.1 mg mL−1 streptomycin (Wisent). Cells were maintained in humidified incubators at 37 °C with 5% CO2. Cells were split using 0.5% trypsin (Wisent) as per standard protocols. Multi-cellular tumor spheroids were generated from HeLa cells in 96-well plates (BioLite, Thermo Scientific). Wells were coated with 1.5% agarose (Biotechnology Grade, BioShop), then seeded with 500–1000 cells in 150 μl of media, which were left to aggregate with gravity at 37 °C and 5% CO2. Spheroids were grown for 6 to 10 days and were monitored daily using an Invertoskop 40 C light microscope. Once spheroid formation was confirmed, they were transferred into 24-well plates coated with agarose in 2 mL of media. Spheroids were incubated with Dox or nanoassemblies with encapsulated Dox for 6 hours to compare Dox penetration. Spheroids were imaged using a Nikon TiE microscope epifluorescence microscope using the 4× objective, an LED Heliophor and filters for excitation at 488 nm and emission at 561 nm, and a Photometrics Evolve EMCCD camera with NIS Elements acquisition software. The images were exported as TIFFs and analyzed using Image J (NIH). The average intensity of Dox fluorescence in a defined surface area was measured for each spheroid and normalized to their initial values at time = 0.Results and discussion
Synthesis of substituted BzIm methacrylates
Our synthesis of dual location dual acid/GSH-degradable ABP began with BzIm-bearing methacrylates with varying substituents. Scheme 2 illustrates our approach with two steps to synthesize a series of BzIm-bearing methacrylates with different R substituents on the para position of phenyl group (RBzImMAs). The R groups were chosen based on their electronic properties: electron-donating groups (methoxyl group) and electron-withdrawing groups (bromo and nitro groups). Note R = H (HBzImMA), p-methoxy (MBzImMA), bromo (BrBzImMA), and nitro group (NBzImMA). The synthesis and NMR analysis of HBzImMA are described in our previous publication.22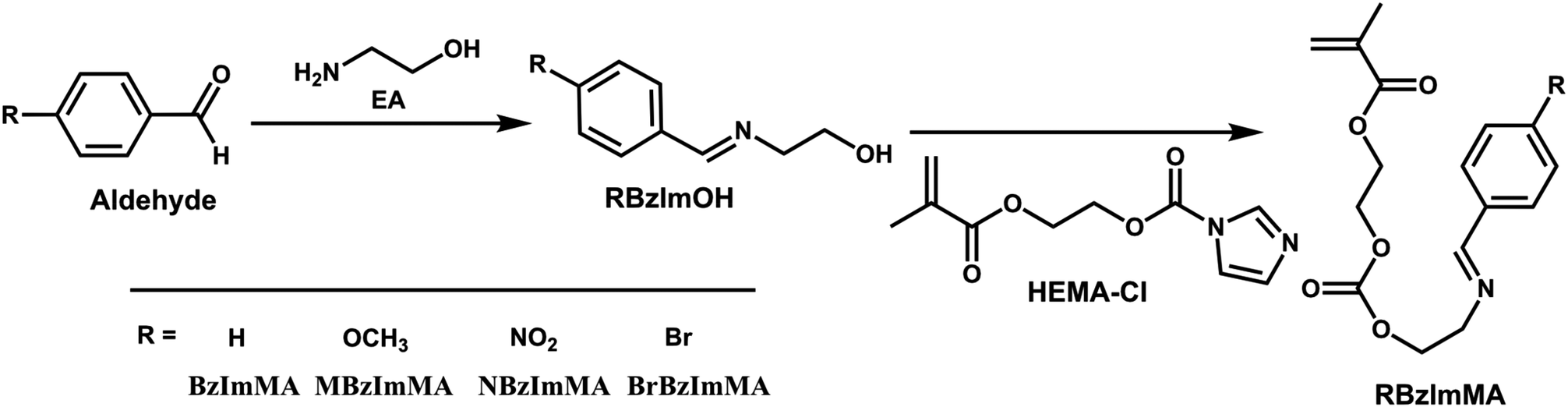 | ||
| Scheme 2 Our approach to synthesize a series of methacrylates bearing BzIm group with different para substituents (RBzImMAs). | ||
The first step was the facile coupling reaction of EA with various substituted benzaldehydes to synthesize their RBzImOH precursors bearing a BzIm linkage. The mole ratio of EA to aldehydes was kept at 1/1. Sodium sulfate was used to remove water, a biproduct formed during reactions, to promote forward reactions to form BzIm bonds. 1H-NMR analysis confirms the successful synthesis of MBzImOH (Fig. S1†), BrBzImOH (Fig. S2†), and NBzImOH (Fig. S3†). Next step was the conjugation of the formed RBzImOH precursors with HEMA-CI at a 1/1 mole ratio in the presence of DBU as a base catalyst, yielding RBzImMAs. 1H-NMR spectra of the purified RBzImMAs (Fig. 1) show their characteristic peaks, including the peak at ∼8.2 ppm (g) corresponding to imine proton as well as two peaks at 5.5–6.1 ppm (a) presenting methacrylate protons. The 1H-NMR analysis, together with 13C-NMR results (Fig. S4–S6†), confirms the successful synthesis of BzIm methacrylates.
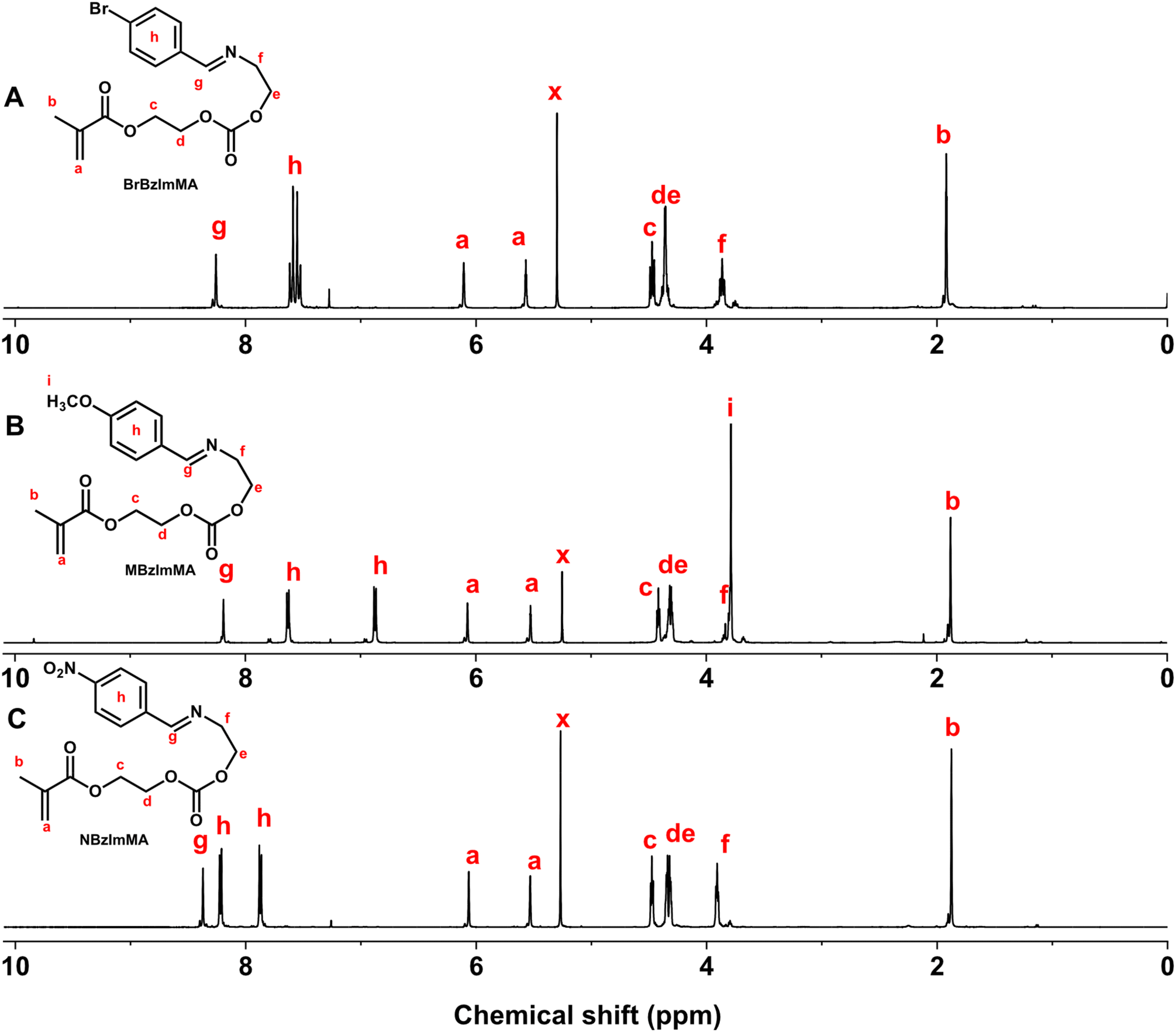 | ||
| Fig. 1 1H-NMR spectra of RBzImMAs, including BrBzImMA (R = Br, A), MBzImMA (R = Me, B), and NBzImMA (R = NO2, C) in CDCl3. | ||
Impact of substituents on acid-catalyzed hydrolysis of BzIm bond
Given the synthesis of RBzImMAs, the effect of the substituents (R = H, MeO, Br, and NO2) on acid-catalyzed hydrolysis of BzIm bonds was examined by 1H-NMR analysis to estimate their acid-catalyzed hydrolysis kinetics in their corresponding polymers. Aliquots were dissolved in a mixture of CD3CN/PB D2O (3/1, v/v) adjusted to pD 6.8 and 5.0, mimicking tumoral and endo/lysosomal pH, respectively. Their 1H-NMR spectra were recorded over incubation times at given pDs (Fig. S7–S9† for MBzImMA, BrBzImMA, and NBzImMA, and Fig. 2 in our previous publication22 for HBzImMA). As illustrated in Fig. 2A, acid-catalyzed hydrolysis of RBzImMAs results in the cleavage of BzIm bonds, generating amine-bearing methacrylates and R-aldehydes. 1H-NMR spectra show that the imine proton peak at ∼8.2 ppm decreased, while a new peak at ∼10 ppm equivalent to aldehyde proton appeared and increased over incubation time. Their integral ratios as aldehyde/(aldehyde + imine) were used to determine the percentage hydrolysis of BzIm bonds. This quantity, along with the time when a half concentration of BzIm linkages is cleaved (t1/2), were used for quantitative evaluation and comparison of acid-catalyzed hydrolysis rate of RBzImMAs with different substituents. The results are shown in Fig. 2B at pD = 6.8 and Fig. 2C at pD = 5.0. At both pDs, the percentage hydrolysis of BzIm bonds increased over incubation time for all methacrylates.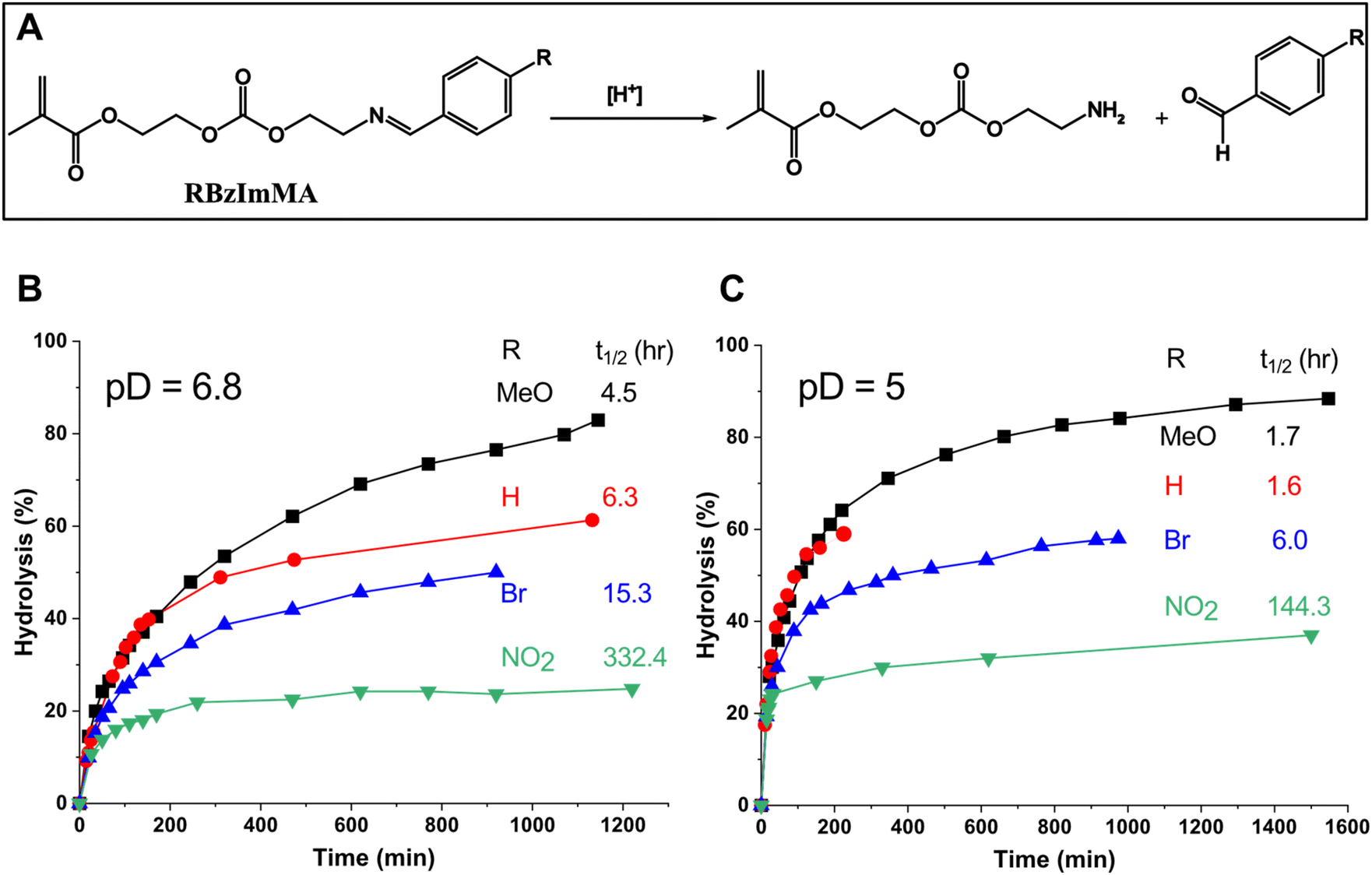 | ||
| Fig. 2 Schematic illustration of acid-catalyzed hydrolysis of RBzImMAs upon the cleavage of BzIm bonds (A) and evolution of their percentage hydrolysis incubated at pD = 6.8 (B) and 5.0 (C) with their t1/2 values. | ||
At pD = 6.8, the hydrolysis rate turned to increase in the order of R = MeO > H > Br > NO2 group (Fig. 2B). Compared with unsubstituted HBzImMA (t1/2 = 6.3 h), the electron-denoting MeO group enhanced the hydrolysis rate (MBzImMA, t1/2 = 4.5 h), while the electron-withdrawing Br (BrBzImMA, t1/2 = 15.3 h) and NO2 (NBzImMA, t1/2 = 332.4 h) groups slowed down the hydrolysis rate. The acid-catalyzed hydrolysis of imine bonds can be initiated with the protonation of the imine nitrogen, generating an electrophilic carbocation intermediate that facilitates nucleophilic attack by water, leading to the formation of an unstable tetrahedral intermediate and ultimately resulting in imine bond dissociation.45 The plausible reason of this observed substituents effect could be due to their ability to stabilize generated carbocation intermediates. For example, electron-donating MeO group could stabilize the formed carbocation through resonance in phenyl ring, thus increasing hydrolysis rate; while electron-withdrawing Br and NO2 groups could destabilize it, impeding acid-catalyzed hydrolysis.
When being incubated at pD = 5, a similar trend of substituent effect on imine hydrolysis rate was observed, following the order of R = MeO ≈ H > Br > NO2 group (Fig. 2C). Interestingly, no significant difference in t1/2 is observed for MBzImMA and HBzImMA, likely due to their rapid hydrolysis at lower pD = 5. As expected, the hydrolysis rates of BzIm bonds were greater at pD = 5.0, compared with pD = 6.8 for all methacrylates. For example, the t1/2 was 1.8 h (4.5 h at pD = 6.8) for MBzImMA and 6 h (15.9 h at pD = 6.8) for NBzImMA. These results suggest that the acid-catalyzed hydrolysis of BzIm bonds is significantly influenced by the electronic properties of their para substituents.
Given its rapid hydrolysis, MBzImMA was chosen to explore the synthesis of amphiphilic block copolymer for further experiments, e.g. aqueous micellization, acid/GSH-responsive disassembly and Dox release as well cellular uptake.
Synthesis of PEG-SS-PMBzImMA (ImSSP)
As depicted in Scheme 3, RAFT polymerization of MBzImMA was explored to synthesize ABP bearing a disulfide junction for hydrophilic PEG block and hydrophobic polymethacrylate block bearing pendant BzIm groups. The approach began with three-step synthesis of a disulfide-labeled PEG-based chain transfer agent (PEG-SS-CTA). The first step involved the reaction of CTA with excess SS-Diol to synthesize a disulfide-labelled dithiocarbonyl CTA with a terminal hydroxyl group (CTA-SS-OH). The desired mono-substituted product was purified and collected as the third band out of total five during silica gel column chromatography with an eluent solution (ethyl acetate/hexane = 1/1, v/v) at a yield of 30%. The second step involved the reaction of PEG (MW = 5000) with excess succinic anhydride, yielding a PEG functionalized with carboxylic acid (PEG-COOH). The last step was an EDC-mediated coupling reaction between CTA-SS-OH and PEG-COOH to synthesize PEG-SS-CTA. Their chemical structures were confirmed by 1H-NMR analysis (Fig. 3). Specifically, 1H-NMR spectrum of PEG-SS-CTA (Fig. 3C) shows the characteristic peaks at 7.3–8.0 ppm (m) corresponding to phenyl protons in CTA moiety and the peak at 3.2 ppm (a) corresponding to methoxy protons in PEG moiety. The integrals of all peaks were consistent with the numbers of their protons. From the integral ratio of peaks (a and h), the conjugation efficiency was estimated to be >95%.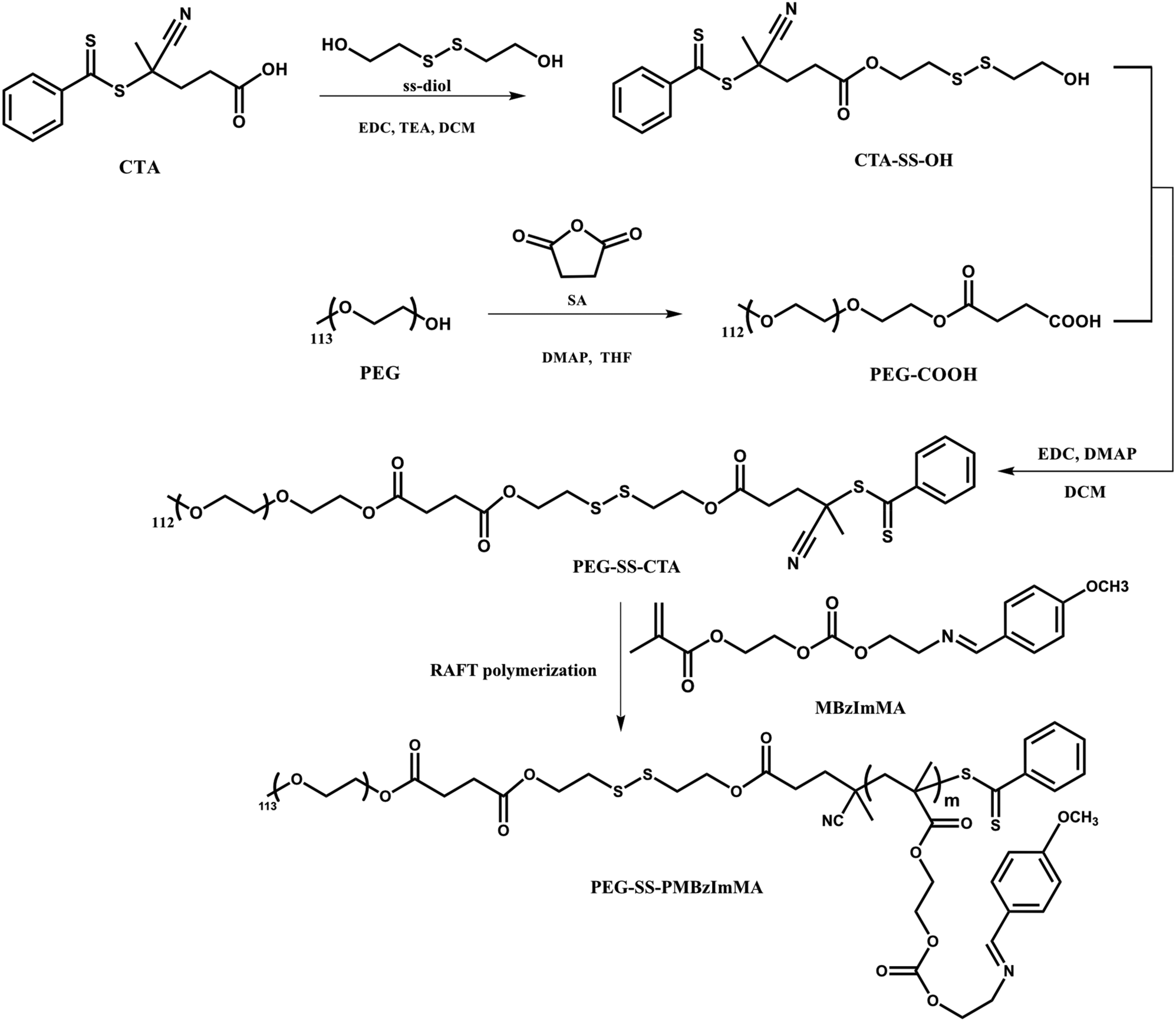 | ||
| Scheme 3 Synthesis of PEG-SS-CTA macromediator and PEG-SS-PMBzImMA (ImSSP) block copolymer by RAFT polymerization. | ||
 | ||
| Fig. 3 1H-NMR spectra of CTA-SS-OH (A), PEG-COOH (B), and PEG-SS-CTA (C) in CDCl3. x denotes residual solvents or impurities. | ||
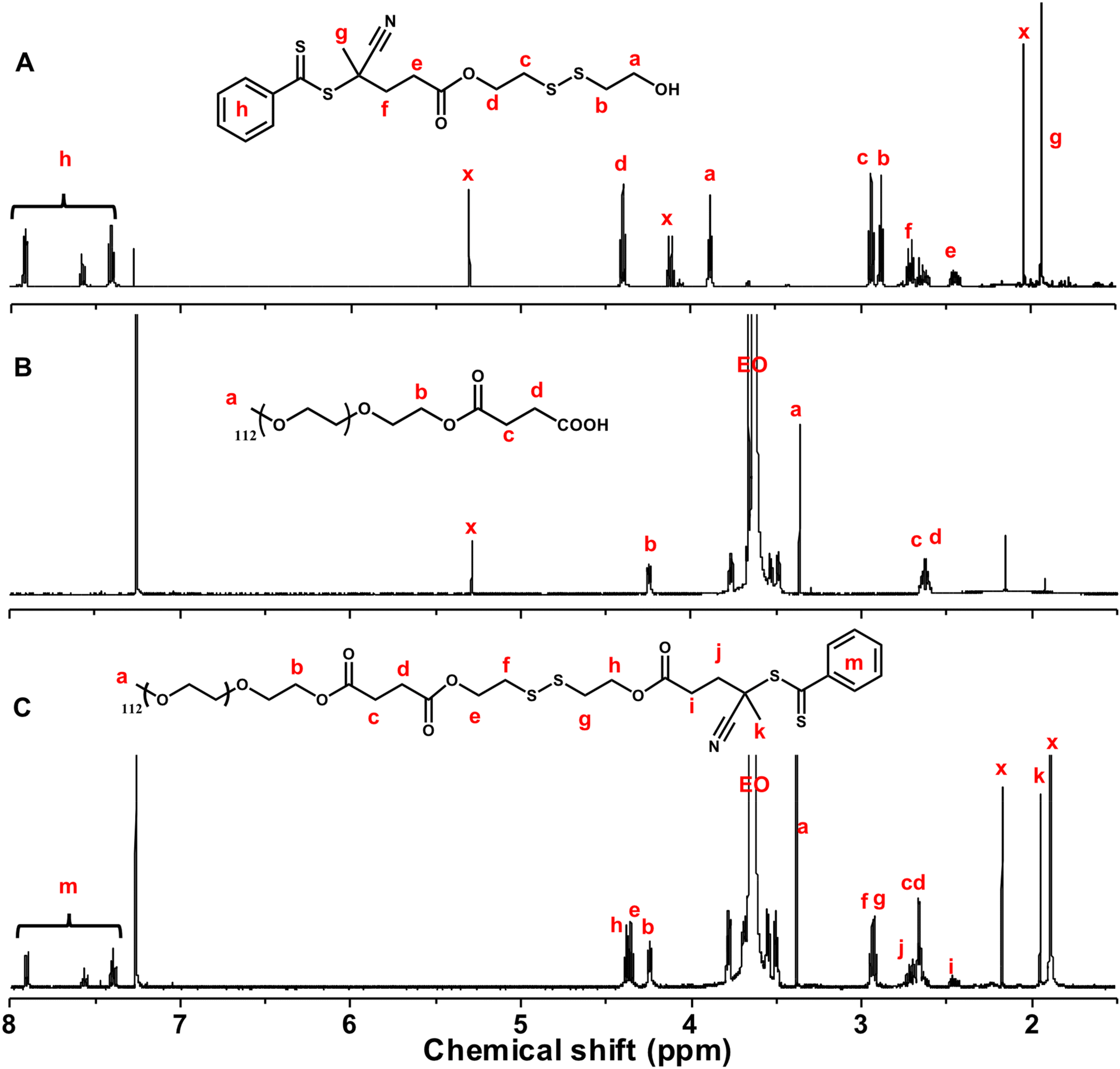
With the successful synthesis of PEG-SS-CTA and optimal acid-cleavable monomer MBzImMA, RAFT polymerization for MBzImMA was conducted in the presence of PEG-SS-CTA as a macromediator, initiated with AMBN (an azo-initiator) at 70 °C. This yielded a shell-sheddable/core-degradable PEG-SS-PMBzImMA (ImSSP), consisting of hydrophilic PEG block conjugated via a disulfide bond to hydrophobic PMBzImMA block having pendant BzIm bonds. Kinetic analysis revealed that monomer conversion reached 65% within 4 h after an induction period of ca. 165 min (Fig. 4A). The crude polymer was purified by precipitation in diethyl ether. The 1H-NMR spectrum of the purified PEG-SS-PMBzImMA in Fig. 4B shows the characteristic peaks at 8.3 ppm (g) for the imine bond, 7.4–7.7 ppm (h) for the phenyl group, 0.7–1.1 ppm (b) for the backbone methyl group of PMBzImMA block as well as 3.6 ppm (EO) for PEG block. The presence of imine peak in 1H-NMR spectrum after purification confirms its stability during RAFT polymerization. Based on their integral ratio, the degree of polymerization (DP) was estimated to be 33, forming PEG-SS-PMBzImMA33. Its GPC trace was shifted to higher molecular weight region after polymerization (Fig. 4D), indicating successful chain extension from PEG-SS-CTA with acid-cleavable PMBzImMA block. A small shoulder in low molecular weight region appeared to overlap with PEG-SS-CTA, suggesting that the block copolymer could contain a small portion of unreacted PEG-SS-CTA. The block copolymer had a low dispersity of Đ = 1.12 and a number average molecular weight of Mn = 20.3 kg mol−1, which was in good agreement with the theoretical Mn = 20.1 kg mol−1 calculated based on the targeted DP of PMImBzMA blocks and monomer conversion.
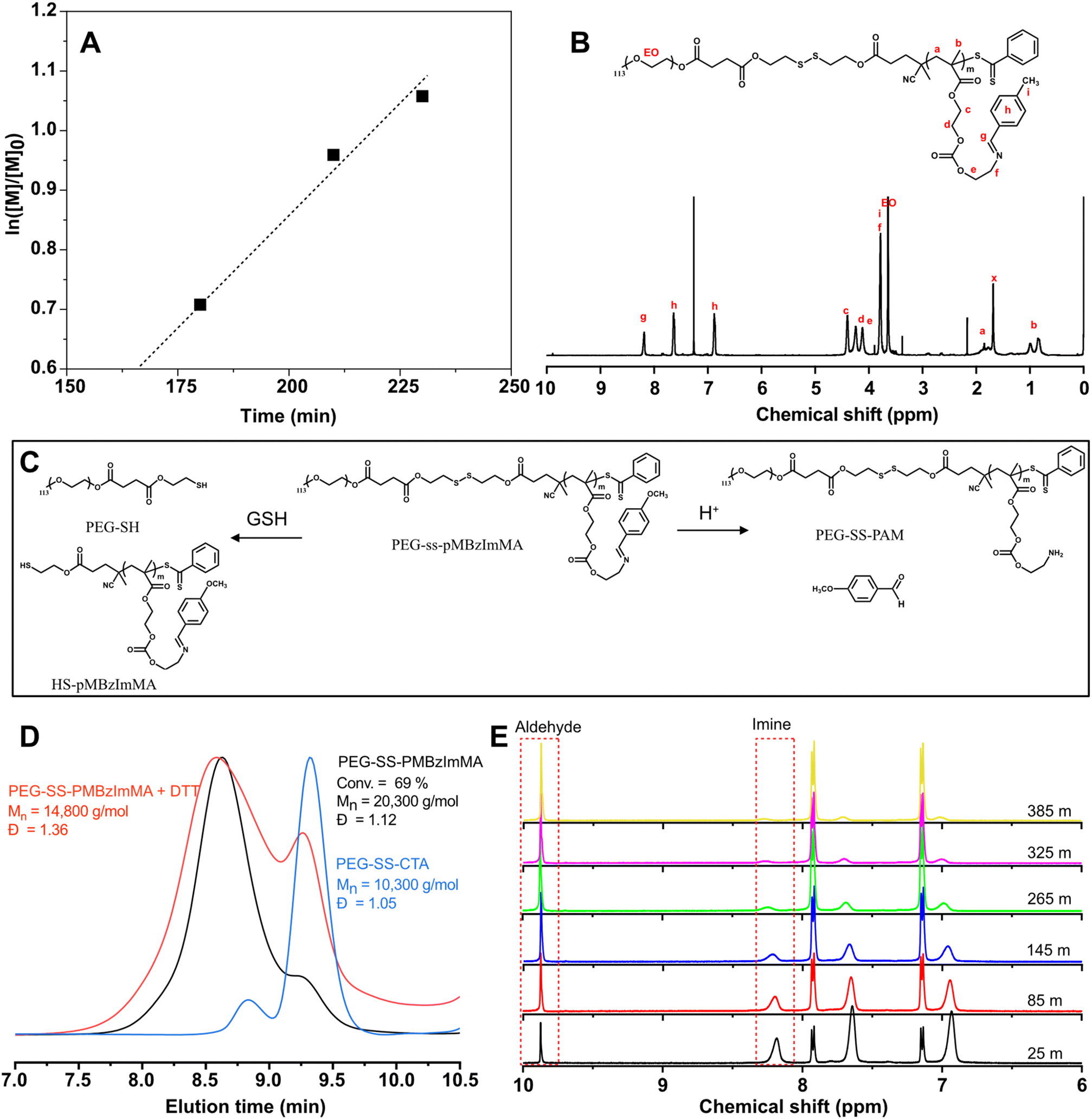 | ||
| Fig. 4 Synthesis and acid/GSH-responsive cleavage of ImSSP. First-order kinetic plot for RAFT polymerization of MBzImMA in presence of PEG-SS-CTA (A); 1H-NMR spectrum of purified PEG-SS-PMBzImMA at 65% monomer conversion (B); conditions for RAFP polymerization: [MBzImMA]0/[PEG-SS-CTA]0/[AMBN]0 = 75/1/0.3 in anisole at 70 °C and [BzImMA]0/anisole = 0.6 wt/wt; degradation of ImSSP upon the cleavage of junction disulfide linkage and pendant BzIm bond in the presence of GSH or in acidic pHs (C). Overlaid GPC diagrams before and after GSH-induced cleavage, compared with PEG-ss-CTA (D); and overlaid 1H-NMR spectra at pD = 5.0 over incubation time (E). | ||
Acid/GSH-responsive degradation of ImSSP
The synthesized PEG-SS-PMBzImMA contains a reductively cleavable disulfide linkage at the block junction and acid-labile BzIm pendants in the hydrophobic block. Fig. 4C shows schematic illustration of its degradation in response to reduction and acidic pH. To investigate reductive degradation using GPC, the copolymer was incubated with DTT, a reducing agent (5 equivalent excess to disulfide bonds). As seen in Fig. 4D, the Mn decreased from 20.3 kg mol−1 to 14.8 kg mol−1 as a consequence of the cleavage of junction disulfide bond to generate PEG-SH and SH-PMBzImMA. The GPC diagram of the degraded products shows two peaks: a pronounce shoulder peak at lower MW region corresponding to PEG-SS-CTA and a peak in higher MW region corresponding to SH-PMBzImMA.To investigate acid-responsive degradation using 1H-NMR, the copolymer was incubated at pD = 5.0. Similar to its MBzImMA monomer counterpart, the peak at 8.3 ppm corresponding to BzIm proton in the copolymer decreased over incubation time, while the peak at 9.8 ppm corresponding to aldehyde proton in p-methoxybenzaldehyde as a degraded product increased (Fig. 4E). The t1/2 was determined to be 1.9 h at pD = 5 for the copolymer, which is slightly higher than that (1.77 h) for MBzIMMA monomer under the similar condition. Further, the degraded products were isolated by centrifugation for their structural analysis. 1H-NMR spectrum in Fig. S10† confirms the generation of methoxybenzaldehyde and PEG-SS-PAM as degraded products upon the cleavage of pendant BzIm bonds.
Aqueous micellization and acid/GSH-responsive disassembly
The synthesized diblock copolymer consists of a hydrophilic PEG block and a hydrophobic pMBzImMA block with multiple imine pendants, connected via a disulfide linkage. Its amphiphilicity was utilized for fabricating polymer micelles at 1 mg mL−1. The formed micellar aggregates are composed of imine-containing hydrophobic PMBzImMA cores, surrounded with sheddable hydrophilic PEG corona. They had a z-average diameter of 94 ± 0.1 nm with a monomodal distribution by DLS analysis (Fig. 5A).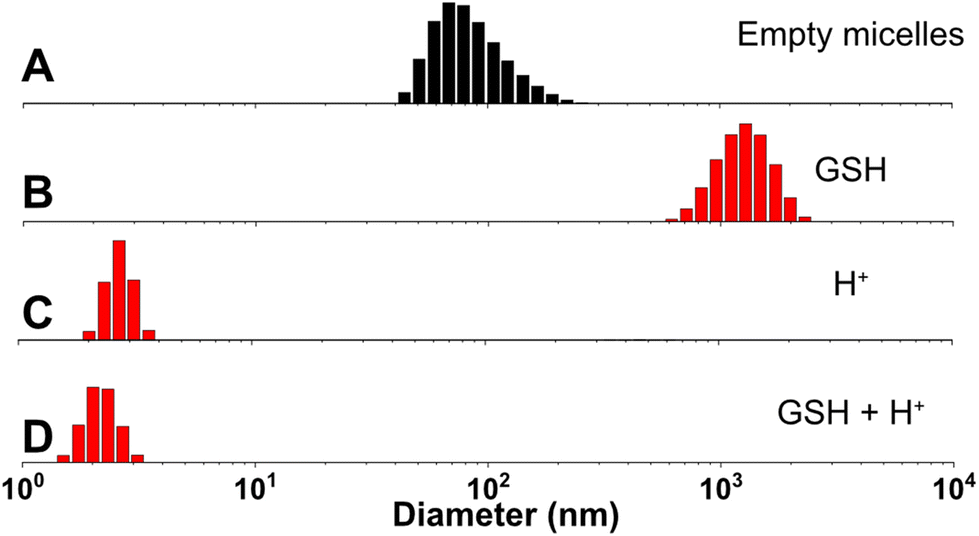 | ||
| Fig. 5 DLS diagrams of nanoassemblies formed through self-assembly of PEG-SS-PMImBzMA before (A) and after being incubated with 10 mM GSH (B), in acidic pH = 5 (C), and both (D). | ||
Up incubation with GSH, both z-average diameter and polydispersity (PDI) of the nanoassemblies increased rapidly, with the occurrence of large aggregates (diameter > 1 μm) within 24 h and PDI rising from 0.1 to 0.3. Such change is likely due to the destabilization and aggregation of formed hydrophobic cores after the loss of their hydrophilic coronas upon the cleavage of disulfide linkages at core/corona interfaces. In acidic pH = 5, their diameter significantly decreased to 3.5 nm. This is probably attributed to the cleavage of imine bonds in hydrophobic cores to generate primary amine pendants, which could be further protonated to form highly water-soluble PEG-SS-PAM. A similar decrease in their diameter to 2.2 nm was observed when the nanoassemblies were treated with both H+ and GSH simultaneously.
Loading and acid/GSH-responsive release of Dox
To explore the potential of ImSSP-based nanoassemblies as an intracellular delivery nanocarrier for chemotherapy, Dox, a clinically approved cancer drug, was encapsulated in the nanoassemblies using a dialysis method. The resulting Dox-loaded nanoparticles (Dox-NPs) had a diameter of 160.6 ± 0.1 nm as measured by DLS analysis (Fig. 6A), which is larger than their empty counterparts (94 ± 0.1 nm). The increase in size could be due to the formation of larger hydrophobic cores upon incorporation of hydrophobic Dox. TEM analysis (Fig. 6B) revealed that Dox-NPs had a uniform spherical morphology with an average diameter of 48 ± 7 nm. This smaller size determined by TEM, compared to DLS, is attributed to the shrinkage of nanoparticles in the dried state, which is commonly observed for block copolymer-based nanoassemblies. Using the extinction coefficient of Dox (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 M−1 cm−1) in DMF/water mixture (1/5, v/v), the loading efficiency of Dox was determined to be 11% with a loading content of 18.6 μg mL−1 (Fig. S11†).
400 M−1 cm−1) in DMF/water mixture (1/5, v/v), the loading efficiency of Dox was determined to be 11% with a loading content of 18.6 μg mL−1 (Fig. S11†).
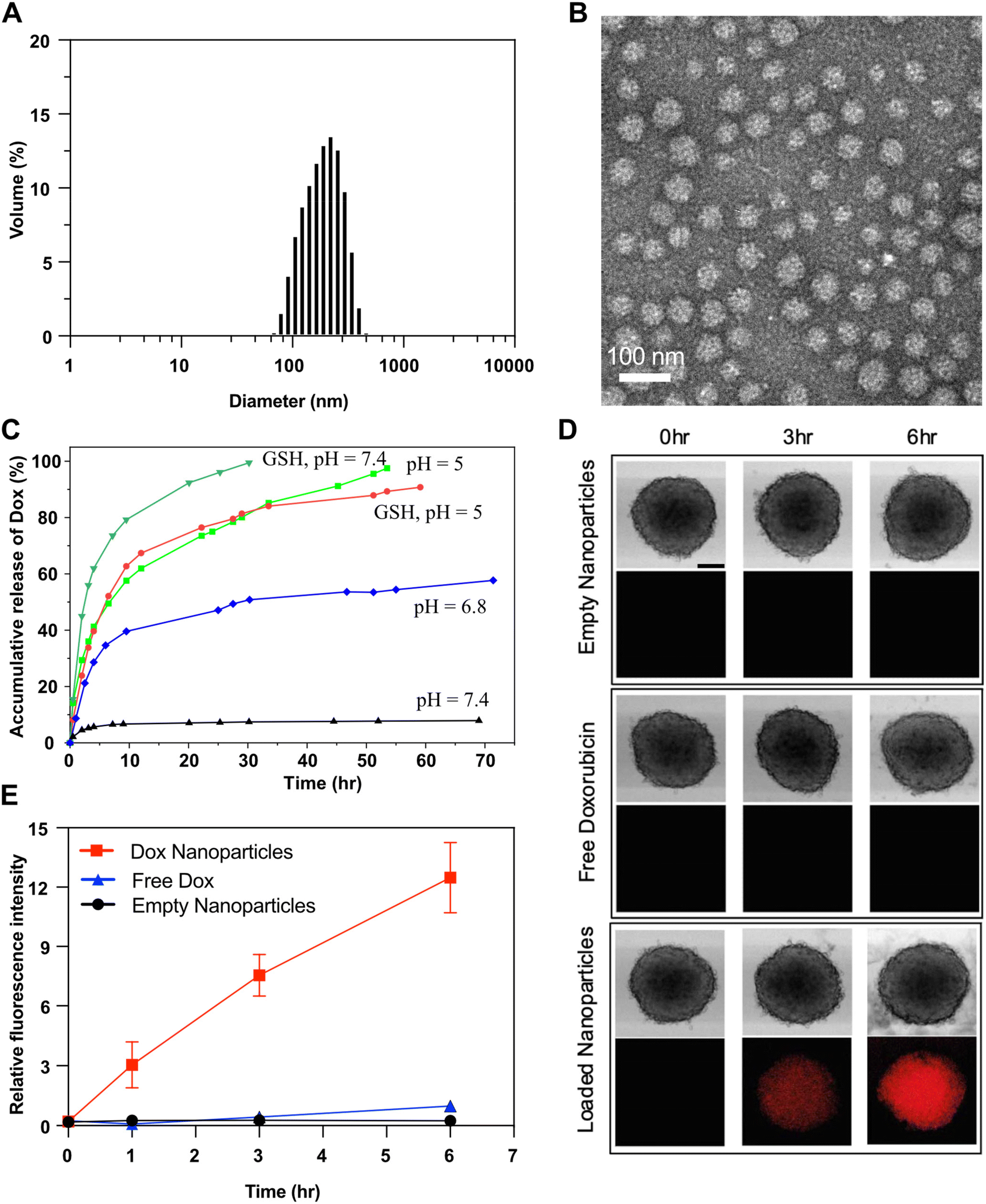 | ||
| Fig. 6 DLS diagram (A) and TEM image (B, scale bar = 100 nm) of Dox-loaded NPs. %Dox release over incubation time at acidic pHs = 5.0 and 6.8 and GSH, compared with in absence of external stimuli at physiological pH = 7.4 (C). Fluorescence microscope images of HeLa multicellular tumor spheroids incubated for 6 hours with empty nanoparticles, free Dox and Dox-loaded nanoparticles. n = 4, scale bar = 200 μm. (D). Quantitative analysis of florescence intensity in the spheroids (E). | ||
Self-assembled Dox-NPs could disintegrate upon the acid-catalyzed hydrolysis of pendant imine linkages in their hydrophobic cores, disrupting hydrophilic/hydrophobic balance. Alternatively, NPs could also disassemble in the presence of GSH due to the loss of the hydrophilic PEG corona upon the cleavage of junction disulfide bonds. As shown in Fig. 6C, Dox release was significantly enhanced in acidic pH or in the presence of GSH, compared to that in physiological pH at 7.4. For instance, only 8% of Dox was released in neutral pH after >80 h, while 50% and 80% of Dox were released at pH = 6.8 and pH = 5, respectively. These enhanced Dox release results are consistent with previously observed pH-dependent hydrolysis kinetics of BzIm bonds. Additionally, Dox release was also higher in the presence of GSH, which is attributed to the disintegration of Dox-NPs upon the cleavage of disulfide bonds at interfaces.
Biological evaluation of the release of Dox by HeLa multi-cellular tumor spheroids
Next, we evaluated the ability of the dual-responsive nanoparticles to penetrate and release Dox in tumors. Multi-cellular tumor spheroids grown from cultured HeLa cells in vitro were used to a mimic solid tumors in vivo. HeLa cells were induced to form spheroids, which were then treated with Dox-NPs or with free Dox and empty NPs as controls. As shown in Fig. 6D, bright field (top) and fluorescence images (bottom) of HeLa spheroids after treatment revealed fluorescence signal with Dox-NPs after 3 h, but not the controls (free Dox and empty NPs). The fluorescence intensity in spheroids treated with Dox-NPs increased further after 6 h. Quantitative analysis of fluorescence intensity (Fig. 6E) indicated that spheroids treated with Dox-NPs had a pronounced increase in fluorescence, whereas there was only small increase for free Dox and no increase for the spheroids incubated with empty nanoparticles. These results suggest that the formed Dox-NPs can effectively penetrate and release Dox in the HeLa spheroids. Combined with their dual-responsive nature in the tumor microenvironment (acidic pH and elevated GSH), these results highlight the potential of Dox-NPs as a promising drug delivery system for cancer therapy in vivo.Conclusion
In this study, we have developed and optimized BzIm-based dual acid/GSH-responsive degradable ABPs and their nanoassemblies for enhanced drug release. The ABPs were designed with acid-cleavable BzIm pendants in hydrophobic blocks and GSH-cleavable disulfides at PEG hydrophilic/hydrophobic block junctions. Various methacrylate monomers bearing BzIm groups (RBzImMAs) with different para-substituents (R = MeO, H, NO2, and Br) were synthesized. Their acid-catalyzed hydrolysis rate by 1H-NMR analysis was revealed in the increasing order of MeO > H > Br > NO2, which could be attributed to the electronic properties of their para substituents. Thus, tunable hydrolysis kinetics of BzIm bonds could be useful for designing distinct pH-responsive delivery systems.Given that MBzImMA exhibited an enhanced acid-catalyzed hydrolysis rate, a dual acid/GSH-responsive ImSSP (PEG-SS-PMBzImMA) was synthesized through direct RAFT polymerization in the presence of PEG-SS-CTA. 1H-NMR and GPC results confirmed that disulfide linkage and BzIm pendants in synthesized ImSSP were cleaved in response to acid and GSH. The resulting ImSSP ABP self-assembled to form colloidally stable, monomodal, and spherical nanoassemblies, as confirmed by both TEM and DLS analysis. They were capable of Dox encapsulation and exhibited enhanced release of Dox in response to acidic pH and GSH, mimicking the tumor acidic microenvironment and intracellular cytosol, respectively. This enhanced release could be attributed to the degradation of hydrophobic cores to hydrophilic polymers upon the cleavage of BzIm bonds as well as the detachment of the sheddable PEG corona upon the cleavage of SS bonds at core/corona interfaces. Moreover, biological evaluation using HeLa multi-cellular tumor spheroids confirmed that the penetration of Dox-loaded nanoassemblies and their release is greater compared to free Dox.
These results show that ImSSP-based nanocarriers are robust acidic pH and GSH-responsive drug release systems, leveraging the tunable hydrolysis kinetics of BzIm bonds by varying substituents. Further biological evaluation in cell and animal models of these dual-responsive nanoassemblies could pave the way their application as delivery system for cancer therapies.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported from Natural Science and Engineering Research Council (NSERC) in Canada through Discovery Grant, Collaborative Research and Training Experience Training (CREATE) Grant entitled Polymer Nanoparticles Drug Delivery (PoND), and Canada Research Chair (CRC) Award. JKO was entitled Tier II CRC in Nanobioscience (2011-2021). XH thanks the NSERC CREATE grant for financial support (stipend and travel) to his MSc program.References
- C. Li, Z. Deng and E. R. Gillies, Curr. Opin. Biomed. Eng., 2023, 25, 100437 CrossRef CAS.
- X. Hu, A. M. Jazani and J. K. Oh, Polymer, 2021, 230, 124024 CrossRef CAS.
- M. A. Beach, U. Nayanathara, Y. Gao, C. Zhang, Y. Xiong, Y. Wang and G. K. Such, Chem. Rev., 2024, 124, 5505–5616 CrossRef CAS PubMed.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- X. Hu, G. Szczepaniak, A. Lewandowska-Andralojc, J. Jeong, B. Li, H. Murata, R. Yin, A. M. Jazani, S. R. Das and K. Matyjaszewski, J. Am. Chem. Soc., 2023, 145, 24315–24327 CrossRef CAS PubMed.
- J. Jeong, X. Hu, R. Yin, M. Fantin, S. R. Das and K. Matyjaszewski, J. Am. Chem. Soc., 2024, 146, 13598–13606 CrossRef CAS PubMed.
- X. Hu, R. Yin, J. Jeong and K. Matyjaszewski, J. Am. Chem. Soc., 2024, 146, 13417–13426 CrossRef CAS PubMed.
- A. M. Jazani, H. Murata, M. Cvek, A. Lewandowska-Andralojc, R. Bernat, K. Kapil, X. Hu, F. De Luca Bossa, G. Szczepaniak and K. Matyjaszewski, Chem. Sci., 2024, 15, 9742–9755 RSC.
- Q. Zhang, N. R. Ko and J. K. Oh, Chem. Commun., 2012, 48, 7542–7552 RSC.
- A. Harada and K. Kataoka, Prog. Polym. Sci., 2006, 31, 949–982 CrossRef CAS.
- A. S. Mikhail and C. Allen, J. Controlled Release, 2009, 138, 214–223 CrossRef CAS PubMed.
- X.-B. Xiong, A. Falamarzian, S. M. Garg and A. Lavasanifar, J. Controlled Release, 2011, 155, 248–261 CrossRef CAS PubMed.
- A. M. Jazani and J. K. Oh, Polym. Chem., 2020, 11, 2934–2954 RSC.
- K. K. Bawa, A. M. Jazani, Z. Ye and J. K. Oh, Polymer, 2020, 194, 122391 CrossRef CAS.
- J. K. Oh, Polym. Chem., 2019, 10, 1554–1568 RSC.
- I. F. Tannock and D. Rotin, Cancer Res., 1989, 49, 4373–4384 CAS.
- P. Watson, A. T. Jones and D. J. Stephens, Adv. Drug Delivery Rev., 2005, 57, 43–61 CrossRef CAS PubMed.
- S. Bazban-Shotorbani, M. M. Hasani-Sadrabadi, A. Karkhaneh, V. Serpooshan, K. I. Jacob, A. Moshaverinia and M. Mahmoudi, J. Controlled Release, 2017, 253, 46–63 CrossRef CAS PubMed.
- S. Binauld and M. H. Stenzel, Chem. Commun., 2013, 49, 2082–2102 RSC.
- K. Neuvonen, F. Fülöp, H. Neuvonen, A. Koch, E. Kleinpeter and K. Pihlaja, J. Org. Chem., 2003, 68, 2151–2160 CrossRef CAS PubMed.
- K. Bairagi, J. T. Liu, A. Thinphang-nga and J. K. Oh, Macromolecules, 2023, 56, 4307–4317 CrossRef CAS.
- X. Hu and J. K. Oh, Macromol. Rapid Commun., 2020, 41, 2000394 CrossRef CAS PubMed.
- Y. Li, Q. N. Bui, L. T. M. Duy, H. Y. Yang and D. S. Lee, Biomacromolecules, 2018, 19, 2062–2070 CrossRef CAS PubMed.
- J. Xue and P. Liu, Eur. Polym. J., 2024, 217, 113313 CrossRef CAS.
- J. Li, Y. Zhou, C. Li, D. Wang, Y. Gao, C. Zhang, L. Zhao, Y. Li, Y. Liu and X. Li, Bioconjugate Chem., 2015, 26, 110–119 CrossRef CAS PubMed.
- N. Kongkatigumjorn and D. Crespy, Polym. Chem., 2024, 15, 4491–4518 RSC.
- N. Chan, B. Khorsand, S. Aleksanian and J. K. Oh, Chem. Commun., 2013, 49, 7534–7536 RSC.
- X. Fu, L. Hosta-Rigau, R. Chandrawati and J. Cui, Chem, 2018, 4, 2084–2107 CAS.
- A. Russo, W. DeGraff, N. Friedman and J. B. Mitchell, Cancer Res., 1986, 46, 2845–2848 CAS.
- F. G. Ottaviano, D. E. Handy and J. Loscalzo, Circ. J., 2008, 72, 1–16 CrossRef CAS PubMed.
- C. Hwang, A. J. Sinskey and H. F. Lodish, Science, 1992, 257, 1496–1502 CrossRef CAS PubMed.
- G. Wu, Y.-Z. Fang, S. Yang, J. R. Lupton and N. D. Turner, J. Nutr., 2003, 134, 489–492 CrossRef PubMed.
- Y. Wang, Q. Luo, W. Zhu, X. Li and Z. Shen, Polym. Chem., 2016, 7, 2665–2673 RSC.
- J. Zhang, H. Tang, Y. Shen, Q. Yu and Z. Gan, Macromol. Rapid Commun., 2018, 39, 1800139 CrossRef PubMed.
- Y. Zhang, J. Ding, M. Li, X. Chen, C. Xiao, X. Zhuang, Y. Huang and X. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 10673–10682 CrossRef CAS PubMed.
- Z. Su, Y. Xu, Y. Wang, W. Shi, S. Han and X. Shuai, Biomater. Sci., 2019, 7, 3821–3831 RSC.
- L. Sun, H. Wei, X. Zhang, C. Meng, G. Kang, W. Ma, L. Ma, B. Wang and C. Yu, Polym. Chem., 2020, 11, 4469–4476 RSC.
- X. Hu, H. Li, S. Luo, T. Liu, Y. Jiang and S. Liu, Polym. Chem., 2013, 4, 695–706 RSC.
- A. M. Jazani, N. Arezi, C. Shetty, S. H. Hong, H. Li, X. Wang and J. K. Oh, Polym. Chem., 2019, 10, 2840–2853 RSC.
- Y. Ding, C. Du, J. Qian, L. Zhou, Y. Su, R. Zhang and C.-M. Dong, Polym. Chem., 2018, 9, 3488–3498 RSC.
- Z. Su, Y. Xu, Y. Wang, W. Shi, S. Han and X. Shuai, Biomater. Sci., 2019, 7, 3821–3831 RSC.
- D. Biswas, S. Y. An, Y. Li, X. Wang and J. K. Oh, Mol. Pharmaceutics, 2017, 14, 2518–2528 CrossRef CAS PubMed.
- S. Y. An, S. H. Hong, C. Tang and J. K. Oh, Polym. Chem., 2016, 7, 4751–4760 RSC.
- G. Zhang and J. K. Oh, ACS Appl. Polym. Mater., 2020, 2, 2319–2326 CrossRef CAS.
- E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1963, 85, 2843–2848 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lp00299g |
| This journal is © The Royal Society of Chemistry 2025 |